
Submitted:
23 October 2025
Posted:
27 October 2025
You are already at the latest version
Abstract
Indolent lymphoproliferative diseases or disorders (LPD) derived from T cells or Natural Killer (NK) cells may be neoplastic or non-neoplastic, which are often difficult to distinguish from each other and from the aggressive counterparts. The etiology and pathogenesis are mostly nebulous and may be related to infections or immune dysfunction. Indolent lymphomas differ from the high-grade aggressive counterparts by a prolonged clinical course of persistent or relapsing disease, histology, immunophenotype and genetics. In the recent decades, indolent lymphomas or LPD of T or NK cell derivation have been increasingly recognized, causing diagnostic and nosologic confusion. The issue is particularly challenging in the arena of indolent intestinal lymphomas and LPD, as evidenced by the myriad of names given to the indolent intestinal T- and NK-cell lymphomas and LPD. Confounding the picture are also reports of Epstein-Barr virus (EBV)-positivity in various indolent non-intestinal LPD and rarely even in indolent intestinal T-cell lymphoma, which have been widely accepted to be typically EBV-negative. This review aims to curate current information and understanding of these diseases with the goal to resolve these issues. The recently described indolent T-lymphoblastic proliferation (iTLBP) and the re-classified indolent primary cutaneous CD4-positive small or medium T-cell LPD and primary cutaneous acral CD8-positive T-cell LPD also require greater awareness and recognition. It is important to diagnose these indolent entities in order to avoid over-treatment and unnecessary therapeutic intervention, and to provide for accurate prognostic prediction and appropriate follow up.
Keywords:
indolent
; T-/NK- cell
; lymphoma
; lymphoproliferative diseases
; intestinal
; EBV
1. Introduction
Most primary
gastrointestinal (GI) NK- or T-cell lymphomas are aggressive high-grade
lymphomas
[1]
. In the recent 3 decades, there have been reports of indolent T- and
NK-cell LPD of the GI tract, designated by a plethora of names. The myriad
names given to these entities reflect the enigma of their biology, behavior and
nosology. These are clonal proliferations and widely accepted to be
EBV-negative
[2–49]
. This compares with their high grade
counterparts which may be EBV+ (in extranodal NK/T-cell lymphoma of the GI
tract - ENNKTL)
[50]
or EBV- (in monomorphic epitheliotropic
intestinal T-cell lymphoma- MEITCL and enteropathy-associated T-cell
lymphoma-EATCL)
[51,52]
. Interestingly, there are also rare reports of
EBV-positivity in indolent intestinal T-cell lymphoma (iITCL)
[8,27,30]
. This
subjects the widely accepted belief that indolent T-cell GI lymphomas are
EBV-negative to question. Furthermore, EBV-positive indolent non-intestinal NK-
or T-cell LPD have also been reported
[54–62]
, despite also isolated reports of indolent
EBV-negative NK-cell LPD in two non-intestinal sites, the nasopharynx
[49]
and the
vagina
[63]
. The reported molecular features of indolent GI LPD also differ from
those of ENNKTL, MEITCL and EATCL
[2–4,8,20,23,26–29,31,32,48–50]
, though there is some overlap in alterations of
genes related to the JAK/STAT pathway. There are other recently recognized or
re-categorized indolent T-cell LPD, including indolent T-lymphblastic
proliferation (iTLBP), primary cutaneous CD4+ small or medium T-cell LPD
(PcutCD4+TLPD) and primary cutaneous acral CD8+ T-cell LPD (PcutacCD8+TLPD)
that require greater attention
[32,64–71]
. This review summarizes the hitherto reported
clinical, morphologic and immunophenotypic features, the evolving genetic
landscape and the dubious possible role of EBV in these entities.
2. Indolent Intestinal Lymphoma and LPD
These received
attention in the past 3 decades. This review retrieved papers on these entities
by searching Scopus and Google Scholar for the period 1990 to 2025 with the
keywords ”indolent, intestinal, T-cell, NK-cell, lymphoma, lymphoproliferative
disease, lymphomatoid gastropathy and enteropathy”. A total of 81 cases of
indolent intestinal T-cell lymphoma (iITCL) and 56 cases of indolent intestinal
NK-cell LPD (iINKLPD) were identified as a single case report or case series
and were analyzed .
2.1. Indolent Intestinal T-cell Lymphoma
This was first
described by Carbonnel et al in 1994 under the name of “extensive small
intestinal lymphoma of low-grade malignancy associated with a new chromosomal
translocation”
[4]
. The reported case manifested non-specific
abdominal symptoms with insignificant intestinal endoscopic findings. The
patient eventually developed acute small bowel obstruction and the resected
small bowel featured dilatation, thickened wall and multiple 1-2 cm diameter
serosal tumors associated with enlarged mesenteric lymph nodes. Small bowel
histology revealed massive involvement that was limited to the mucosa by a
diffuse infiltrate of small atypical lymphoid cells, associated with multiple
non-caseating granulomas. The mesenteric lymph nodes and the liver portal
tracts were involved by similar atypical lymphoid infiltrate.
Immunophenotypically, the lymphoid cells are T-cells expressing CD3, CD4,
betaF1, but negative for CD8. There was negligible Ki67 staining. A chromosomal
translocation t(4;16)(q26;p13) involving the interleukin-2 (IL-2) gene
was also detected.
This report was
followed by many other similar reports under designations of “small intestinal,
gut, GI tract, stomach, indolent T-cell, CD4-positive lymphoma,
lymphoprolferative disease” in varying word combinations
[4–32]
. This
great variability in the use of designations reflects uncertainty within the
then academia about the nature of this entity. This review collected 81 cases
of iITCL. There were 55 males and 26 females with an age range 14-80 years and
median age of 50 years. The reported cases had wide geographic distribution
from Europe, Asia to Africa. As in the case reported in 1994
[4]
, all
presented with non-specific abdominal symptoms including bloating, pain,
diarrhea and weight loss. One was complicated by intestinal obstruction
[4]
and another
by intestinal perforation
[13]
. Extraintestinal involvement or metastasis were
reported in mesenteric lymph nodes
[4,8,13,15,16,27–32]
, liver
[4,8]
peripheral blood
[4,8,13,32]
,
bone marrow
[14,15,19,23,32]
, tonsil
[15]
, lungs
[4,8]
, skin
[4,8,32]
and epiglottis
[32]
. In 2 cases, inguinal lymph node involvement was
reported
[23]
, which was unusual as metastasis to peripheral lymph nodes has been
regarded to be very rare or absent in iITCL. The histology of the reported
iITCL cases was similar, namely non-destructive, diffuse infiltrates of small
or medium atypical lymphoid cells often limited to the mucosa, though some
cases show focal involvement of the muscularis mucosae and submucosa. There
were differences in immunophenotypic expression of CD4 and CD8, in varying
combinations. The Ki67 positivity rate was usually low
[1,2,4–29,31,32]
.
2.1.1. Involved Sites and Gross Presentation
iITCL involves
mostly the small intestine and the colon. However, other GI sites including the
stomach, esophagus and oral cavity may also be involved
[5,7,9,11,13–15,17,19,23–25,32]
. Multifocal involvement of the GI tract is
common
[1–32]
, with gross presentations of polyps, folds, nodules, fissures or ulcers
[1–32]
.
2.1.2. Histology
iITCL is
characterized by non-destructive, partial involvement of the intestinal wall,
often restricted to the lamina propria of the mucosa. A proportion of cases,
however, may also show involvement of the muscularis mucosae or submucosa.
There is usually no epitheliotropism and the surface and glandular epithelium
are usually spared. In some cases, there are non-caseating granulomas or
conspicuous eosinophils
[2,4–32]
. The infiltrate is typically composed of
atypical small to medium lymphoid cells with irregular dark nuclei and
infrequent mitotic activity. There is no angiocentricity or angioinvasion (
Figure 1
)
[2,4–32]
.
Figure 1.
(A) Male, 46 years, vague abdominal symptoms. Multifocal GI involvement. Colonic biopsy. (A) Non-destructive mucosal atypical lymphoid infiltrates. H&E, X50. (B) & (C) Mucosal small atypical lymphoid cells infiltrate. Focal intraepithelial involvement present. H&E, (B) X200, (C) X400. (D) CD3+, X100. (E) Ki67 low, X100. (F) Aberrant CD20+, X100.
Figure 1.
(A) Male, 46 years, vague abdominal symptoms. Multifocal GI involvement. Colonic biopsy. (A) Non-destructive mucosal atypical lymphoid infiltrates. H&E, X50. (B) & (C) Mucosal small atypical lymphoid cells infiltrate. Focal intraepithelial involvement present. H&E, (B) X200, (C) X400. (D) CD3+, X100. (E) Ki67 low, X100. (F) Aberrant CD20+, X100.
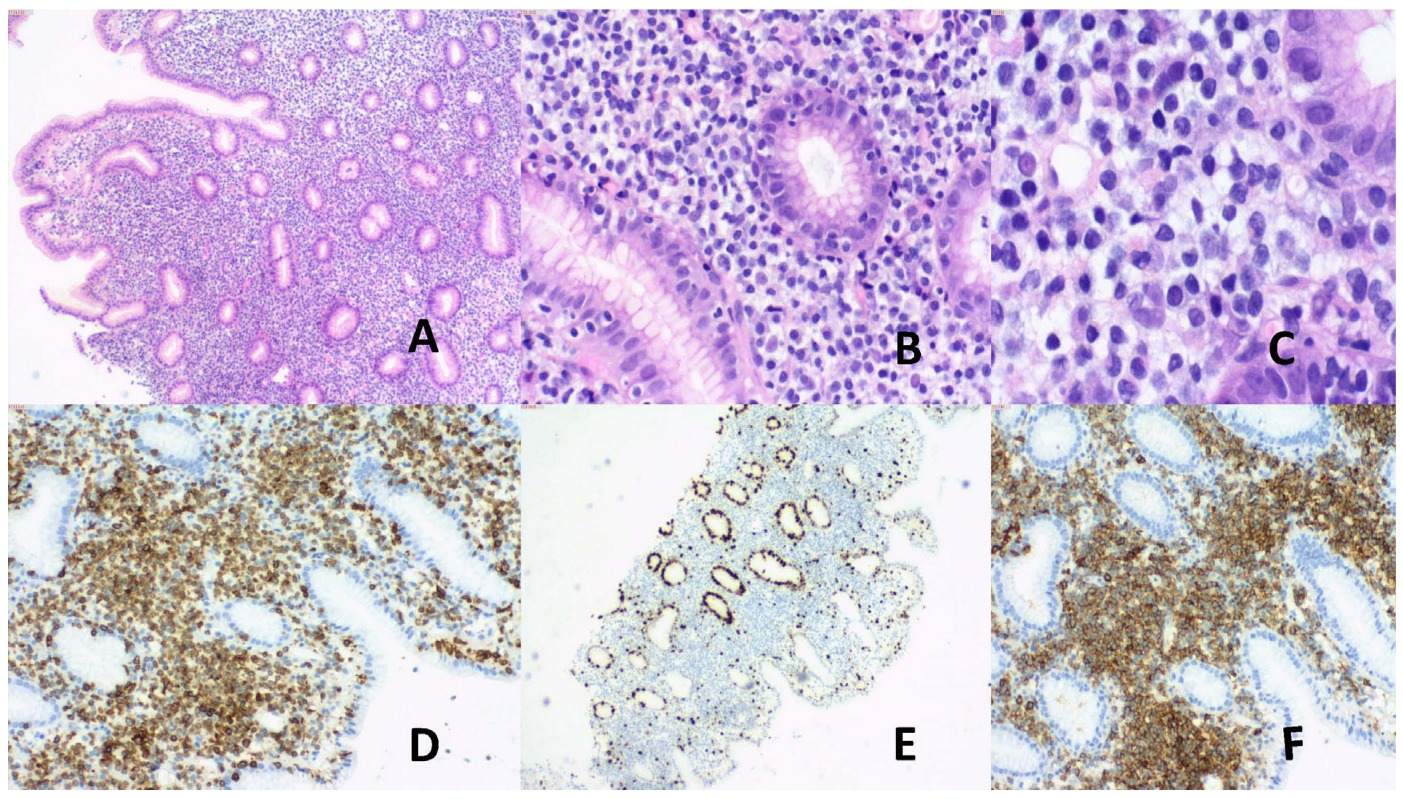
2.1.3. Immunophenotype
The atypical
lymphoid cells exhibit T-cell phenotype and are positive for T-cell receptor
(TCR, mostly beta and uncommonly gamma), CD3, CD2, CD4, CD5, CD7 and CD8. There
are more CD4+ than CD8+ iITCL. According to the pattern of expression of CD4
and CD8, iITCL can be categorized as CD4+/CD8-, CD4-/CD8+, CD4+/CD8+ and
CD4-/CD8-
[2,4–32]
. This immunophenotypic categorization appears to
correlate well with non-neoplastic T cells. CD8+ iITCL more frequently express
TIA1 (but not granzyme B or perforin)
[14,22,23,25–30]
. These different immunophenotypes of iITCL show
different genetic features (see subsection 2.1.4), and may have bearing on
pathogenetic mechanisms. Though there had been reports of CD4+ iITCL running
higher risks of disease progression
[8,13,16,19,20]
, this association is no longer documented in the
5th edition of the WHO Classification
[2]
. Ki67
proliferation index is low and mostly <10%.
2.1.4. Genetic Alterations
(a)
TCR
rearrangement
Clonal TCR
rearrangement is present in all iITCL, more commonly TCR beta and less
commonly TCR gamma
[1,2,4–17,19–32]
.
(b) JAK/STAT
and NF-KB pathway related genes
This is more frequent in CD4+/CD8-, CD4+/CD8+ and
CD4-/CD8- iITCL
[20,23,26–29,31]
. Using Next Generation Sequencing (NGS),
alterations in STAT3 and JAK2, resulting in JAK2::STAT3
fusion have been demonstrated
[20,23,26–29,31]
. Other genes with demonstrated alterations
include STAT3 SH2 mutation, STAT5 alteration and SOCS1
deletion. Mutations in epigenetic modifier genes DNMT3A, EZH2, KMT2D,
TET2 and TNFAIP3 that contribute to JAK/STAT pathway activation are
also identified, more commonly in CD4+/CD8- and CD4-/CD8- iITCL
[2,28,29,31]
.
Activation of the NF-KB pathway through histone modification of KMT2D
and methylation of TET2 may also contribute to pathogenesis.
(c) IL2 gene
IL2
gene alterations occur more commonly in CD8+ than CD4+ cases
[2,23,29,31]
.
Implication of the IL2 gene in iITCL was first demonstrated by
traditional G-banding cytogenetic study
[4]
, and later also by NGS. NGS uncovered IL2::RHOH
(Ras homolog family member H) fusion due to inversion of chromosome 4 and IL2:TNIP3
(TNFAIP3 interacting protein 3) fusion
[23,29]
. RHOH deficiency has been shown to be associated
with T helper 17 cell polarization and T-cell defects
[72,73]
. In
CD4-/CD8+ iITCL, deletion of 3’ untranslated regions of IL2 and mutation
of MCM5 (minichromosomal maintenance complex 5) have been demonstrated
[23]
.
(d) Other genetic alterations
Alterations in DIS3, MAPK1, TP53, POLE, SMAD4,
SFB1 and CDKN2A have also been reported in some cases of CD4+ iITCL
[23]
.
The
number of studies and reported cases, however, are hitherto small and it is
immature for definitive conclusion on distinctive pathogenetic and genetic
differences among the different immunophenotypic categories of iITCL to be
made. Furthermore, the number of genes, gene panels so far studied were limited
and the technological methods used vary from study to study. With future
evolution in molecular methodology, discovery of more genetic alterations in
these indolent T-cell lymphomas may be made. It remains to be seen with more
future work for better correlation of immunophenotype, genetics, pathogenesis
and prognosis in iITCL.
2.1.4
Etiology and Role of EBV
The etiology of iITCL has remained nebulous. It
has been etiologically associated with persistent antigenic stimulation
[23]
, Crohn’s
disease
[11,74]
, autoimmune enteropathy
[15]
and coeliac disease
[8]
. None of
these postulations, however, has been substantiated by further evidence. It is
widely accepted that iITCL is not associated with EBV and is typically
EBV-negative. However, among the 81 cases identified by this review, only 22
cases were reported to have been tested for EBV (22/81, 27%). Among those
tested for EBV, 19 cases were EBV-negative (19/22, 86%) and three were
EBV-positive (3/22,13%)
[8,27,30]
. It is notable that the status of EBV infection
in the remaining reported iITCL represented the majority of those reported
cases (59/81,73%). The methodology of EBV detection was not mentioned in a
significant number of reports, but in-situ hybridization on tissue sections for
EBV-encoded small RNA (EBER) was the most frequently used in reports where the
methodology of EBV detection was mentioned, including the 3 EBV-positive cases
[8,27,30]
.
Though EBER is regarded as the preferred method for detecting latent EBV
infection in tissue sections
[75–78]
, there are alternative methods of EBV detection
[54,55,57,75,76]
.
By using these alternative methods, reclassification of the so far reported
EBV-negative cases to EBV-positive remains possible. Testing the remaining
EBV-unknown status cases may also skew the hitherto reported EBV-status in
iITCL. Since there were documented cases of EBV+ iITCL (3 cases, 13% among
tested cases)
[8,27,30]
, the role of EBV in iITCL remains to be
explored. Furthermore, in one case of CD8+EBV+ iITCL, there was a high Ki67
proliferation index (90%)
[30]
, which may potentially portend more aggressive
behavior. The possibility of 2 groups of iITCL, namely EBV- and EBV+, therefore
stays to be further studied. The possible groups may be different in
immunophenotype, genetics and clinical behavior.
2.1.5
Neoplastic Nature of iITCL
The true biologic nature of indolent intestinal
T-cell proliferations had been enigmatic, as reflected by the plethora of names
ranging from “proliferative disorders, diseases to lymphomas” given to these
entities by multiple researchers and authors in the literature in the past 3
decades.
[4–32]
. The body of cumulative information and evidence hitherto on this entity
is in favor of its neoplastic nature, as follows:
1. Ability to disseminate. There have been multiple reports of dissemination to various non-intestinal distant sites including mesenteric lymph nodes, liver, lungs, skin, tonsils, epiglottis, peripheral blood and bone marrow, as described in Section 2.1 above [4,8,13,14,15,16,19,23,27,28,29,30,31,32]. More importantly, dissemination to inguinal lymph nodes had also been reported in 2020 in 2 cases [23]. This is in striking contrast to the former belief that lymph nodes spread from iITCL should be limited within the abdomen.
2. Disease Progression. Progression or transformation to higher grade lymphoma was documented in 5 of 81 cases (6%) [20,21,23]. Three cases transformed to overt T-cell lymphoma [20,21], with one demonstrated to share the same STAT3::JAK2 fusion as the pre-existing iITCL.
3. Frequent Genetic Mutations. Alterations in TCR genes, genes of the JAK/STAT signaling pathway and epigenetic modifier genes that contribute to JAK/STAT activation, IL2 gene and DIS3, MAPK1, TP53, POLE, SMAD4, SFB1 and CDKN2A gene alterations in many cases as discussed in Subsection 2.1.3 [2,4,17,19,20,21,22,23,24,25,26,27,28,29,30,31,32].
In the Fifth edition of the WHO Classification of
Haematolymphoid Tumours
[1,2]
, the neoplastic nature of iITCL is recognized
due to the significant morbidities related to the disease and its ability to
disseminate. This entity is redesignated to “Indolent T-cell Lymphoma of the
Gastrointestinal Tract” from the former revised Fourth edition designation of
“Indolent T-cell Lymphoproliferative Disorder of the Gastrointestinal Tract”.
2.1.6. Treatment
Among the 81 reported cases, 19 cases did not
receive any specific treatment, 1 was treated with surgery, 17 with
chemotherapy, 4 with steroids, 7 with gluten free diet, 1 with mogamulizumab,
and whether treatment was instituted was not mentioned in the remaining 32
patients. Though the initial response was favorable in a handful of cases where
it was reported, the clinical course of iITCL was indolent with persistent or
relapsing disease, irrespective of the treatment modality or whether treatment
was instituted or not.
[4–32]
.
2.2. Indolent Intestinal NK-cell Lymphoproliferative Disease (iINKLPD)
iINKLPD is a more recently recognized entity than
iITCL
[2,33–49]
, and was initially designated lymphomatoid gastropathy
(in the stomach) and NK-cell enteropathy (in the
remaining intestine tract). It behaves in indolent fashion as persistent or
relapsing disease over long durations. The common presentations include vague
abdominal symptoms
or may be asymptomatic, only to be discovered
incidentally on endoscopy. This review uncovered 56 cases of iINKLPD from 17
case reports or case series. There were 22 males, 32 females, and 2 cases with
unidentified sex. The age ranged from 14 to 76 years (median 52 years). One
case showed also concomitant involvement of the glans penis
[47]
, while all
the remaining 55 cases showed only involvement of the GI tract.
[33–46,48,49]
.
2.2.1. Sites of Involvement and Gross Findings
The stomach and small/large intestines are more
commonly involved. The gall bladder was also rarely implicated
[31,44,46,49]
.
The disease may present as single or multiple elevated lesions or ulcers with
hemorrhage and edema, measuring 1-2 cm in size
[33–49]
.
2.2.2. Histology
The disease is usually limited to the lamina
propria featuring expansion by confluent generally non-destructive infiltrates
of mostly medium-sized atypical lymphoid cells with irregular nuclei and pale
granular cytoplasm. This contrasts with iITCL where small atypical lymphoid
cells are more common and the cytoplasm is agranular. Epithelial invasion with
destruction and displacement of intestinal glands can occur. Mucosal ulceration
and focal necrosis are occasionally present. The muscularis mucosae is usually
intact, but may be involved. There is no angiocentricity or angioinvasion.
Admixture with other inflammatory cells is usually present (
Figure 2
)
[3,33–49]
.
Figure 2.
Male, 48 years, abdominal pain. Jejunal biopsy. (A) & (B) Non-destructive mucosal medium-sized atypical lymphoid cell infiltrate with pale granular cytoplasm. Involvement of muscularis mucosae in (B). H&E, X200. (C) CD56+, X100. (D) CD3+, X100. (E) Ki67 high (about 60%), X100.
Figure 2.
Male, 48 years, abdominal pain. Jejunal biopsy. (A) & (B) Non-destructive mucosal medium-sized atypical lymphoid cell infiltrate with pale granular cytoplasm. Involvement of muscularis mucosae in (B). H&E, X200. (C) CD56+, X100. (D) CD3+, X100. (E) Ki67 high (about 60%), X100.
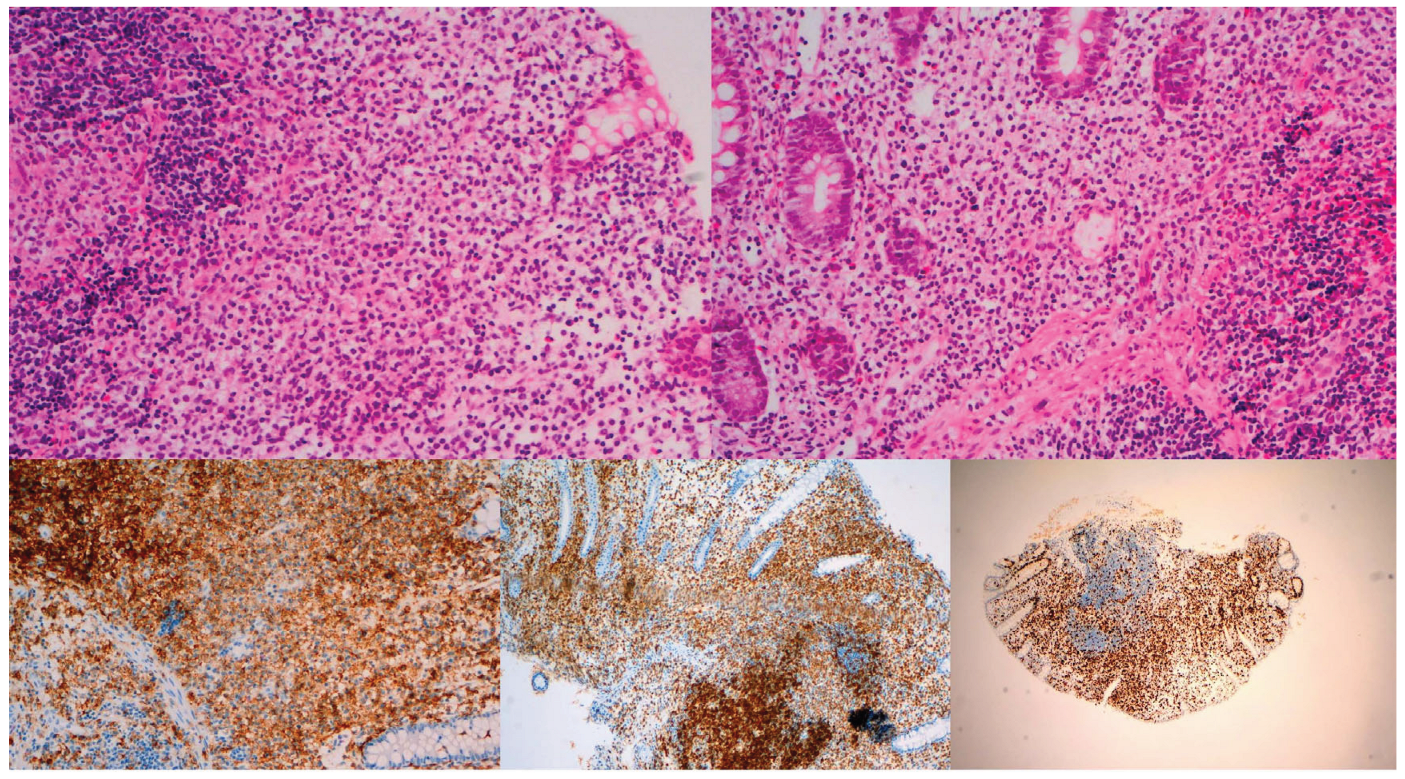
2.2.3. Immunophenotype
By definition, the atypical lymphoid cells do not
express TCR antigens and surface CD3. They frequently express CD56 and
cytotoxic effector proteins TIA1 and granzyme B. CD2, CD7, CD8 and cytoplasmic
CD3 may be expressed. The Ki67 positivity rate is usually low (<10%) but may
be higher and up to 80%
[3,33,35–37,41,43,45,48,49]
. The significance of this difference in Ki67
positivity rate from iITCL, which is mostly low in the latter, is unknown.
2.2.4. Etiology and EBV Status
The etiology of iINKLPD is unknown, though
Helicobacter pylori (HP) infection has been suggested to play a role, as
response of iINKLPD to anti-HP treatment has been reported
[3,29,34,39,43]
.
This, however, was not universally observed in the reported cases. In the 56
reported cases, all were EBV-negative. Though the number of cases reported was
small for a definitive conclusion on the role of EBV in iINKLPD to be made, it
appears that EBV is probably not etiologically or pathogenetically related to
iINKLPD.
2.2.5. Neoplastic or Non-Neoplastic?
Unlike iITCL, the biologic nature of iINKLPD has
not been clearly defined. Among the 56 reported cases, there was no observation
of distant dissemination, progression or transformation of the disease.
However, genetic alterations including mutations in JAK3, RUNX1, CIC, ERBB4
and SETD5 had been reported in iINKLPD
[3,29,48,49]
. Phosphorylated STAT3
[2,49]
and STAT5
[9]
proteins had also been demonstrated in some cases of iINKLPD. These genetic and
epigenetic anomalies point to a possible neoplastic nature of iINKLPD. The
involvement of the glans penis in one case
[47]
also suggests dissemination of the disease
outside of the intestine. However, the small number of reported cases with
these changes preclude making definitive conclusions. The biologic nature of
iINKLPD requires further study on a larger number of cases for clarification.
2.2.6. Treatment
Among the 56 reported cases, 34 were not given
any specific treatment, 4 received surgery, 7 chemotherapy (with 2 given
concomitant bone marrow transplant), 1 anti-HP therapy, and 10 cases without
reported treatment modality. Despite the treatment modality, the indolent
course of persistent or relapsing disease appeared unaffected
[33–49]
. More
work is required on a greater number of cases for better understanding of the
clinical outcome and optimal management of iINKLPD.
2.3. Differential Diagnoses of iITCL and iINKLPD from Other Intestinal NK- or T-Cell Lymphomas
The major differential diagnoses (DDX) are
monomorphic epitheliotropic intestinal T-cell lymphoma (MEITCL),
enteropathy-associated T-cell lymphoma (EATCL) and primary GI extranodal
NK/T-cell lymphoma (ENNKTL) of the intestine. Intestinal metastasis from primary
non-GI ENNKTL may also cause diagnostic confusion, but can be distinguished by
presence of a primary (nasal or other non-nasal sites) in another location.
MEITCL, EATCL and GI ENNKTL can be distinguished by their distinctive clinical
manifestations, aggressive behavior, high grade morphology, immunophenotypic
and genetic differences. In EATCL, there is often histologic evidence of
residual coeliac disease. Primary GI ENNKTL may be distinguished by
characteristic histological evidence of frequent angiocentricity,
angioinvasion, and extensive geographic necrosis. Further, EBV is always
positive in ENNKTL, in contrast to the usual EBV-negativity in iITCL and almost
always EBV-negativity in iINKLPD
[50–53]
. A summary of the differences among these
entities is depicted in
Table 1
.

Table 1.
Comparative Features of Gastrointestinal NK-/T-cell Lymphomas and LPD.
| iITCL | iINKLPD | ENNKTL | MEITL | EATL | |
| Age (years) | CD4+: Median 51 CD8+: Median 45 |
30-90 | 35-58 | Median 54-67 | Median 61 |
| Sex | M>F | M>F | M>F | M>F | M>F |
| Predominant sites involved |
Small intestine, colon | Stomach, small intestine, colon, gall bladder | GI tract | Small intestine | Small intestine (mostly jejunum) |
| Multifocality | Common | Yes | 27% | 20-35% | 32-54% |
| Enteropathy association | No | No | No | No | 80% |
| Metastasis | BM, PB, tonsil, mesenteric LN | Rarely mesenteric LN | Multiple extraintestinal sites, frequently Stage IV | LN, lung, liver, brain, skin | LN, BM, lung, liver |
| Transformation | Reported | NR | NA | NA | NA |
| Depth involved | Mucosa, sometimes also MM and SM | Mucosa | Full thickness | Full thickness | Often full thgickness |
| Histology | Small /medium atypical cells, non-destructive dense infiltrate, no/rare epithelial invasion, no angioinvasion | Medium atypical cells, small nucleoli, pale granular cytoplasm, circumscribed confluent infiltrate, glands displaced, epithelial invasion +/-, necrosis +/- | Range of atypical cells, geographic necrosis, epithelial invasion, angiocentricity, angioinvasion, angiodestruction | Monotonous atypical cell infiltrates, necrosis, epithelial invasion, severe inflammatory backdrop, “starry sky” appearance | Range of atypical cells, epithelial invasion, angioinvasion, angiodestruction, features of CD |
| Molecular/genetic alterations | STAT3, JAK2,, JAK2::STAT3 fusion, STAT5, SOCS1, KMT2D, TET2, DNMT3A, EZH2, TNFAIP3, IL2, RHOH, TNIP3, TCR | JAK3, RUNX1T1, CIC, ERB4, SETD5 | PRDM1, PTPRK, HACE1, FOXO3, STAT3, JAK3, STAT5B, BCOR, KMT2D, ARID1A, EP300, TCR (in T-cell type) | Myc, SETD2, STAT3, STAT5, JAK1, KAK3, TCR | JAK1, STAT3, TET2, KMT2D, DOX3X, TNFA1P3, TNIP3, POT1, TP53, CD58, FAS, B2M, TCR |
| Immunophenotype | CD3+, CD4+CD8-, CD4-CD8+,CD4+CD8+, CD4-CD8-, TCR+, KI67 low* | CD56+, CD2+, cCD3+, CD7+, TIA1+, GZB+, TCR-, Ki67 high | CD56+ (NK-cell type), cCD3+, sCD3+ (T-cell type), CD2+, TIA1+, GZB+, perforin+, TCR- (in NK-cell type), TCR+ (in T-cell type), Ki 67 high | CD2+, sCD3+, CD7+, CD8+, CD56+/-, TIA1+, TCR+, Ki67 high | CD3+, CD7+, TIA1+, GZB+, perforin+, Ki67>50%, CD30+/EMA+ (in anaplastic cases) |
| EBV | Negative@ | Negative | Positive | Negative | Negative |
Footnote: *Some cases reported Ki67 high, @Some cases reported EBV+, +: positive, -: negative, +/-: weak or focal positive, cCD3: cytoplasmic CD3, CD: Coeliac disease, BM: bone marrow, EATL: enteropathy associated T-cell lymphoma, ENNKTL: extranodal NK-/T-cell lymphoma, F: female, GZB: granzyme B, iINKLPD: indolent intestinal NK-cell lymphoproliferative disorder, iITCL: indolent intestinal T-cell lymphoma, LN: lymph node, M: male, MEITL: monomorphic epitheliotropic intestinal T-cell lymphoma, MM: muscularis mucosae, NA: not applicable, NR: not reported, PB: peripheral blood, sCD3: surface CD3, SM: submucosa, TCR: T-cell receptor.
3. Indolent Non-Intestinal NK- or T-Cell LPD
This review uncovered 11 cases of indolent NK- or T-cell LPD that arose from non-intestinal sites. Four cases occurred in the nose, 1 in the nasopharynx (NP), 3 in the skin, 1 in lymph nodes, 1 in the pericardium and 1 in the vagina [49,54,55,56,57,58,59,60,61,62,63]. All cases were characterized by indolent clinical behavior of persistent or relapsing disease over long periods. Four cases were of T-cell lineage (2 nose, 1 pericardium, 1 lymph node) and 7 were of NK-cell lineage (2 nose, 3 skin, 1 NP and 1 vagina) [49.54-63]. Apart from the EBV-status unreported NK-cell NP case [49] and EBV-negative vaginal [63] case, the remaining 4 T-cell (1 lymph node, 2 nose and 1 pericardium) and 5 NK-cell (2 nose and 3 skin) were all EBV-positive, including an indolent NKLPD of the skin which was not initially tested for EBV but progressed to an EBV+ ENNKTL after a prolonged relapsing disease [56]. The rate of EBV-positivity among indolent non-intestinal NK- or T-cell LPD was therefore 82% (9/11).
The majority (5/6 tested cases, 83%) [57,58,59,60,61,62] of these cases were associated with a high Ki67 positivity rate with a reported rate of over 90% in one pericardial case [62]. Two NK-lineage EBV-positive cases of the skin where outcome information was available revealed high grade transformation to ENNKTL (2/2, 100%) after long durations of indolent disease [55,56]. This compares with the lower rate of transformation in iITCL (5/81, 6%), where the EBV status of all 5 cases were not reported [19,20,23], and that none of the 19 tested EBV-negative iITCL cases resulted in transformation [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32]. There was also no disease progression or transformation in all 56 NK-cell EBV-negative iINKLPD [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49].
It appears that EBV-positivity is more prevalent in indolent non-intestinal NK- or T-cell LPD, and may be associated with higher KI67 proliferation index [57,58,59,60,61,62] and high grade transformation of disease [55,56]. It may be argued that indolent EBV-positive non-intestinal NK- or T-cell LPD should not be compared with other indolent EBV-negative intestine counterparts and should be grouped together with the high grade primary GI EBV-positive ENNKTL [50]. However, ENNKTL is clearly different from indolent EBV+ NK- or T-cell LPD in clinical behavior, histology, immunophenotype, genetics and outcome and should be separately categorized [50,53]. Despite easy distinction from those ENNKTL with a conspicuous large cell component, prominent geographic necrosis, angiocentricencity and angioinvasion, indolent EBV+ NK- or T-cell LPD can mimic some ENNKTL with a predominant small cell component [50,53] morphologically and immunophenotypically, thus causing great diagnostic difficulty without clinical follow up. The indolent EBV+ NK- or T-cell LPD may also cause confusion with chronic active EBV disease (CAEBV), but could be distinguished by its lack of severe systemic symptoms and deranged liver functions at presentation which are characteristic of CAEBV [53]. It should also be mentioned that indolent EBV+ T-cell LPD of the skin may be confused with various other cutaneous lymphomas or LPD which are also often indolent but distinguishable by being always negative for EBV. Since the number of reported indolent EBV+ non-intestinal NK- or T-cell LPD is so far very small, it is not possible to define this as a distinctive biologic entity. Further work on many more cases is required for elucidation on the biologic nature and role of EBV in these entities. Table 2 summarizes the miscellaneous features of the 9 reported EBV+ indolent non-intestinal NK- or T-cell LPD.
4. Indolent T-Lymphoblastic Proliferation (iTLBP)
iTLBP was first described in 1999 in the upper aerodigestive tract of a male subject which remained indolent and stable for over 16 years without treatment [32,64,65]. iTBLP is rare and about 50 cases have been reported in various nodal and non-nodal sites. The latter includes mandibular, cervical, supraclavicular, parotid, inguinal, abdominal, liver, adrenal, retroperitoneal, nasopharyngeal and mediastinal locations [32,64,65,66]. iTBLP may affect reactive lymph nodes [66], occurs in Castleman disease, myasthenia gravis, paraneoplastic autoimmune multiorgan syndrome, and may be associated with neoplasms (hepatocellular carcinoma, dendritic follicular cell sarcoma and acinic cell carcinoma) [32,64,65,66]. The clinical course is benign and indolent and does not require treatment. It is characterized histologically as non-destructive presenting as tumor-like, tumor infiltrating lymphocyte-like or focal cellular cluster (>50 cells) lesions [64]. The cells are small to medium-sized that lack significant atypia. Immunophenotypically, they are positive for Tdt and CD3, with variable positivity for CD1a, CD4, CD7, CD8, CD10, CD34 and CD99. This immunophenotype is in keeping with that of a common lymphoid precursor [64,65,66]. Genotypically, clonal TCR rearrangement is not present. The most important DDX is T-cell lymphoblastic lymphoma (TLBL). TLBL is distinguishable by tissue destruction, larger lymphoblastic cells with significant atypia, aberrant immunophenotypic features including expression of LMO2 and presence of clonal TCR rearrangement. The rarity of TLBL in adults should also be helpful in distinction from iTBLP in adult patients. It is important to recognize the association of iTBLP in a variety of malignant tumors, in order to avoid confusion and misinterpretation [64,65,66].
5. Indolent Cutaneous LPD
Many cutaneous T-cell lymphomas or LPD are clinically indolent and include primary cutaneous CD4-positive small or medium T-cell LPD (PcutCD4+TLPD), primary cutaneous acral CD8-positive T-cell LPD (PcutacCD8+TLPD), mycosis fungoides, lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. PcutCD4+TLPD was a provisional entity in the 2017 WHO Classification and PcutacCD8+TLPD was originally designated as a lymphoma. These are recently reclassified as LPD, due to the benign indolent clinical course. These 2 entities are highlighted below.
5.1. PcutCD4+TLPD
This was a provisional entity in the 2017 revised fourth edition of the WHO Classification [32,67]. It is currently designated an LPD because of the benign indolent clinical behavior, with spontaneous regression in about 30% of cases [32,67,68,70]. It is a clonal disease and TCR gene rearrangement is demonstrable in the majority of tested cases [68,69]. It affects a wide age range with a median of 59 years. Both sexes are affected almost equally. Clinically, it affects most commonly the neck and neck and presents as an asymptomatic erythematous solitary nodule and less frequently as a tumor, papule, macule or plaque with size ranging from 4-60 mm. Histologically, two patterns were reported. The entire dermis is involved in one pattern, sometimes with extension to the subcutis. The other pattern features a subepidermal band-like infiltrate of the superficial dermis. Epidermotropism and folliculotropism may be present, rendering mycosis fungoides a differential diagnosis. The infiltrate consists mostly of small to medium atypical lymphoid cells and contains no more than 40% large cells. Immunophenotypically, the lymphoid cells are CD3+, CD4+, CD8-, CD30-, follicular T-helper cell markers+ (mostly PD-1+) and low Ki67 proliferation index. Reactive B cells and CD8+ T cells may be present. The prognosis is excellent. Treatment with intralesional steroids, excision or radiotherapy are effective, and spontaneous remission without treatment occurs in 30% cases. The major DDX are mycosis fungoides, primary cutaneous marginal zone lymphoma and cutaneous lymphoid hyperplasia [32,68,70].
5.2. PcutacCD8+TLPD
It was designated as a lymphoma in the 2017 revised fourth edition of the WHO Classification and has been recently regarded as an LPD in view of the indolent benign clinical behavior. It is a slow growing solitary reddish purple skin nodule or plaque measuring up to several centimeters. It affects acral sites including the face, ear, nose, hands or feet. It is histologically characterized by a dermal dense monotonous atypical lymphoid infiltrate consisting of medium-sized cells. Extension into the subcutis is common. Immunophenotypically, the lymphoid cells are CD3+, CD4-, CD8+, CD30-, cytotoxic cell phenotype+ (TIA1+ mostly), CD56- and PD1-. CD68 is positive, showing the characteristic Golgi-dot-like positivity. Ki67 proliferation index is low. Clonal TCR rearrangement is present in nearly all cases. The prognosis is excellent, with complete remission after excision or local irradiation [32,69,71].
6. Conclusions
Indolent NK- or T-cell LPD are less recognized and characterized, compared to the B-cell counterparts. The latter including B-cell lymphocytic lymphoma/leukemia, marginal zone lymphoma of mucosa-associated lymphoid tissue, lymphoplasmacytic lymphoma, indolent mantle cell lymphoma (leukemic type) and follicular lymphoma are well delineated morphologically, immunophenotypically, genotypically and nosologically [79]. iITCL was first described 3 decades ago, followed almost a decade later by reports of iINKLPD. The indolent clinical behavior of these entities has been the major confounding factor in delineating their biologic nature. The significant morbidities, ability to disseminate, potential for disease progression and transformation and multiple genetic alterations identified in iITCL have led to its recent recognition as a lymphoma [2,28,31]. iINKLPD, on the other hand, appears less definitive. However, the frequent presence of JAK3 and other genetic mutations, and expression of the phosphorylated STAT3 and STAT5 proteins point to the possible neoplastic nature of iINKLPD.
The role of EBV in these indolent NK- or T-cell lymphomas or LPD is also perplexing. Though it is commonly believed that EBV-negativity is a feature of iITCL and iINKLPD, the reports of EBV+ iITCL challenge this commonplace conviction [8,27,30]. The more frequent reports of EBV-positivity in indolent non-intestinal NK- or T-cell LPD, provides more conjectural aura to this belief [57,58,59,60,61,62]. The small number of tested reported cases of iITCL that were confirmed to be EBV-negative, amid an even larger number of cases with no reports of their EBV status (59/81,73%), raises the need for accumulation of further evidence and work before a firm dogma to be made on the role of EBV in these entities. Ki67 proliferation index is low in most iITCL. However, it is higher in iINKLPD and indolent non-intestinal EBV+ NK- or T-cell LPD. This finding is interesting, but the significance is unknown.
The recognition of iTBLP is eye-opening since the presence of lymphoblasts often conveys the notion of proliferation and neoplasia. The indolent behavior of iTLBP warrants greater awareness of the entity, in order to avoid unnecessary radical therapy. Though many cutaneous T-cell lymphomas are indolent in behavior, PcutCD4+LPD and PcutacCD8+LPD have recently been reclassified as LPD instead of lymphoma due to the uniformly excellent clinical behavior. Such redesignation is important for better recognition of their biological nature and avoidance of over-treatment.
Conflict of interest: The author declares no conflict of interest.
Ethics Committee Approval
This is not required for a Review Article.
Funding
This work received no external funding.
Acknowledgment
The authors thank Dr. Xingen Wang, Department of Pathology, Peking University Shenzhen Hospital, for supplying images of Figures 1 and 2.
References
- Chan J; Alaggio, R. Intestinal T-cell and NK-cell lymphoid proliferations and lymphomas. Introduction. In: WHO Classification of Haematolymphoid Tumours, 5th ed. IARC: Lyons, France, 2025. Available online: https://tumourclassification.iarc.who.int/chapters/63 (accessed on 23 October 2025).
- Bhagat, G; Takeuchi, K; Naresh, KN; Dave, SS. tract. In WHO Classification of Haematolymphoid Tumours, 5th ed; Chan, J, Washington, MK, Eds.; de Jong, D, Eds. IARC: Lyons, France, 2025. [Google Scholar]
- Xiao, W; Takeuchi, K; Ferry, JA; Chang, CL. tract. In WHO Classification of Haematolymphoid Tumours, 5th ed; Chan, J, Ed.; Lee Wood, B, Eds. IARC: Lyons, france, 2025. [Google Scholar]
- Carbonnel, F; Lavergne, A; Messing, B; Tsapis, A; Berger, R; Galian, A; Nemeth, J; Brouet, JC; Rambaud, JC. Extensive Small Intestinal T-Cell Lymphoma of Low-Grade Malignancy Associated with a New Chromosomal Translocation. Cancer, 1994, 73, 1286–1291.
- Egawa, N; Fukayama, N; Kawaguchi, K; Hishima, Y; Funata, N; Ibuka, T; Koike, M; Miyashita, H; Tajima, T. Relapsing Oral and Colonic Ulcers with Monoclonal T-cell Infiltration. Caner, 1995, 75, 1728–1733. [CrossRef]
- Tsutsumi, Y; Inada, K; Morita, K; Suzuki, T. T-cell Lymphomas Diffusely Involving the Intestine: Report of Two Rare Cases. Jpn J Clin Oncol 1996, 26, 264–272. [CrossRef]
- Hirakawa, K; Fuchigam, T; Nakamura, S; Daimaru, Y; Ohshima, K; Sakai, Y; Ichimaru, T. Primary Gastrointestinal T-cell Lymphoma Resembling Multiple Lymphomatous Polyposis. Gastroenterol 1996,778-782. [CrossRef]
- Carbonnel, F; d’Almagne, H; Lavergne, A; Matuchansky, C; Brouet, JC; Sigaux, F; Beaugerie, L; Nemeth, J; Coffin, B; Cosnes, J etal. The Clinicopathological Features of Extensive Small Intestinal CD4 T Cell Infiltration. Gut 1999, 45, 662–667.
- Zivny, J; Banner, BF; Agrawal, S; Pihan, G; Barnard, G. CD4+ T-cell Lymphoproliferative Disorder of the Gut Clinically Mimicking Coeliac Sprue. Dig Ds Sci 2004, 49, 551–555.
- Svrcek, M; Garderet, L; Sebbagh, V; Rosenzwajg, M; Parc, Y; Lagrange, M; Bennis, M; Lavergne-Slove, A; Flejou, J; Fabiani, B. Small Intestinal CD4+ T-cell Lymphoma: A Rare Distinctive Clinicopathological Entity Associated with Prolonged Survival. Virchows Arch 2007, 451, 1091–1093. [CrossRef]
- Perry, AM; Warnke, RA; Hu, Q; Gaulard, P; Copie-Bergman, C; Alkan, S; Wang, H; Cheng, JX; Bacon, CM; Delabie, J etal. Indolent T-cell Lymphoproliferative Disease of the Gastrointestinal Tract. Blood 2013, 122, 3599–2606. [CrossRef]
- Ranheim, EA; Jones, C; Zehnder, JL; Warnke, RA; Yuen, A. Spontaneously Relapsing Clonal, Mucosal Cytotoxic T-cell Lymphoproliferative Disorder: Case Report and Review of the Literature. Am J Surg Pathol 2000, 24, 296–301.
- Margolskee, E; Jobanputra, V; Lewis, SK; Alobeid, B; Green, PHR; Bhagat, G. Indolent Small Intestinal CD4+ T-cell Lymphoma is a Distinct Entity with Unique Biologic and Clinical Features. Plos One 2013, 8, e68343. [CrossRef]
- Leventaki, V; Manning, JT Jr; Luthra, R; Mehta, P; Oki, Y; Romaguera, J; Medeiros, LJ; Vega, F. Indolent Peripheral T-cell Lymphoma Involving the Gastrointestinal Tract. Hum Pathol 2014, 45, 421–426. [CrossRef]
- Malamut, G; Meresse, B; Kaltenbach, S; Derrieux, C; Verkarre, V; Macintyre, E; Ruskone-Fourmestraux, A; Fabiani, B; Radford-Weiss, I;Brousse, N; Hermine, O etal. Small Intestinal CD4+ T-cell Lymphoma is a Heterogeneous Entity with Common pathology features. Clin gastroenterol Hepatol 2014, 12, 599–608. [CrossRef]
- Mendes, LST; Attygalle, AD; Cunningham, D; Benson, M; Andreyev, J; Gonzales-de-Castro, D; Wootherspoon, A. CD4-positive Small T-cell Lymphoma of the Intestine Presenting with Severe Bile-acid Malabsorption: A Supportive Symptom Approach. Br J Haematol 2014, 167, 265–269. [CrossRef]
- Wang, X; Ng, CS; Chen C; Yu, G, Yin, W. An Unusual Case Report of Indolent T-cell Lymphoproliferative Disorder with Aberrant CD20 Expression Involving the Gastrointestinal Tract and Bone Marrow. Diagn Pathol 2018, 13, 82.
- Nagaishi, T; Yamada, D; Suzuki, K; Fukuyo, R; Saito, E; mFukuda, M; Watanabe, T; Tsugawa, N; Takeuchi, K; Yamaoto, K. Indolent T Cell Lymphoproliferativ Disorder with Villous Atrophy in Small Intestine Diagnosed by Single-Balloon Enteroscopy. Clin J Gastroenterol 2019, 12, 434–440. [CrossRef]
- Perry, AM; Bailey, NG; Bonnett, M; Jaffe, ES; Chan, WC. Disease Progression in a Patient with Indolent T-cell Lymphoproliferative Disease of the Gastrointestinal Tract. Int J Surg Pathol 2019, 27, 102–107.
- Sharma, A; Oishi, N; Boddicker, RL; Hu, G; Benson, HK; Ketterling, RP; Greipp, PT; Knutson, DL; Kloft-Nelson, SM; Eckloff, BW etal. Recurrent STAT3-JAK2 Fusions in Indolent T-cell Lymphoproliferative Disorder of the Gastrointestinal Tract. Blood 2018, 131, 2262–2266.
- Guo, L; Wen, Z; Su, X; Xiao, S; Wang, Y. Indolent T-cell Lymphoproliferative Disease with Synchronous Diffuse Large B-cell Lymphoma. A Case Report. Medicine 2019, 98, e15323.
- Wu, J; Li, LG; Zhang, XY; Wang, LL; Zhang, L; Xiao, YJ; Xing, XM; Lin, DL. Indolent T-cell Lymphoproliferative Disorder of the Gastrointestinal Tract: An Uncommon Case with Lymph Node Involvement and the Classic Hodgkin’s Lymphoma. J Gastroenterol Oncol 2020, 11, 812–819.
- Soderquist, CR; Patel, N; Murty, VV; Betman, S; Aggarwai, N; Young, KH; Xerri, L; Leeman-Neill, R; Lewis, SK; Green, PH etal. Genetic and Phenotypic Characterization of Indolent T-cell Lymphoproliferative Disorder of the Gastrointestinal Tract. Haematologica 2020, 105, 1895–1906. [CrossRef]
- Zanelli, M; Zizzo, M; Sanguedolce, F; Martino, G; Soriano, A; Ricci, S; Ruiz, CC; Anessi, V; Ascani, S. Indolent T-cell Lymphoproliferative Disorder of the Gastrointestinal Tract: A Tricky Diagnosis of a Gastric Case. Gastroenterol 2020, 20, 336.
- Takahashi, N; Tsukasaki, K; Kohri, M; Akuzawa, Y; Saeki, T; Okamura, D; Ishikawa, M; Maeda, T; Kawai, N; Matsuda, A etal. Indolent T-cell Lymphoproliferative Disorder of the Stomach Successfully Treated by Radiotherapy. J Clin Exp Hematopathol 202o,60,7-10. [CrossRef]
- Nishimura, MF; Nishimura, Y; Nishikori, A; Yoshino, T; Sato, Y. Primary Gastrointestinal T-cell Lymphoma and Indolent Lymphoproliferative Disorders: Practical Diagnostic and Treatment Approaches. Caners 2021, 13, 5774.
- Montes-Moreno, S; King, RL; Oschlies, I; Ponzoni,M; Goodlad, JR; Dotlic, S; Traverse-Glehen, A; Ott, G; Ferry, JA; Calaminici, M. Update on Lymphoproliferative Disorders of the Gastrointestinal Tract: Disease Spectrum from Indolent lymphoproliferations to Aggressive Lymphomas. Virchows Archiv 2020, 476, 667–681.
- Miranda, RN; Amador, C; Chan, JKC; Guitart, J; Rech, KL; Medeiros, LJ; Naresh, KN. Fifth Edition of the World Health Organization Classification of Tumors of the Hematopoietic and Lymphoid Tissues: Mature T-cell, NK-cell, and Stroma-Derived Neoplasms of Lymphoid Tissues. Mod Pathol 2024, 37, 100512.
- Wang, X; Yin, W; Wang, H. Indolent T-cell/Natural Killer-Cell Lymphomas/Lymphoproliferative Disorders of the Gastrointestinal Tract – What Have We Learned in the Last Decade? Lab Invest 2024, 104, 102028.
- Chuang, S; Tzioni, M; Chen, Z; Feng, Y; Shih, C; Casa, C; Du, M. Indolent EBV-positive T-cell Lymphoma of the Gastrointestinal with Metachronous Lesions Involved by Different Neoplastic Clones. Pathology 2024, 56, 431–434. [CrossRef]
- Attygalle, AD; Karube, K; Jeon, YK; Cheuk, W; Bhagat, G; Chan, JKC; Naresh, KN. The Fifth Edition of the WHO Classification of Mature T Cell, NK Cell and Stroma-Derived Neoplasms. J Clin Pathol 2025, 78, 217–232. [CrossRef]
- Quintanilla-Martinez, L; Bosch-Schips, J; Gasljevic, G; van den Brand, M; Balague, O; Anagnostopoulos, I; Ponzoni, M; Cook, JR; Dirnhofer, S; Sander, B etal. Exploring the Boundaries between Neoplastic and Reactive Lymphoproliferations: Lymphoid Neoplasms with Indolent Behavior and Clonal Lymphoproliferations – A Report of the 2024 EA4HP/SH Lymphoma Workshop. Virchows Archiv 2025, 487, 327–347.
- Vega, F; Chang, C; Schwartz, MR; Preti, HA; Younes, M; Ewton, A; Verm, R, Jaffe, ES. Atypical NK-cell Proliferation of the Gastrointestinal Tract in a Patient with Antigliadin Antibodies but not Coeliac Disease. Am J Surg Pathol 2006, 30, 539–544.
- Takeuchi, K; Yokoyama, M; Ischizawa, S; Terui, Y; Nomura, K; Marutsuka, K; Nunomura, M; Fukushima, N; Yagyuu, T; Nakamine, H et al. Lymphomatoid Gastropathy: A Distinct Clinicopathologic Entity of Self-limited Pseudomalignant NK-cell Proliferation. Blood 2010, 116, 5631–5637.
- Mansoor, A; Pittaluga, S; Beck, PL; Wilson, WH; Ferry, JA; Jaffe, ES. NK-cell Enteropathy: A Benign NK-cell Lymphoproliferative Disease Mimicking IntestinaL Lymphoma: Clinicopathologic Features and Follow-up in a Unique Case Series. Blood, 117, 1447-1452. [CrossRef]
- Tanaka, T; Megahed, N; Takata, K; Asano, N; Niwa, Y; Hirooka, Y; Goto, H. A Case of Lymphomatoid Gastropathy: An Indolent CD56-positive Atypical Gastric Lymphoid Proliferation, Mimicking Aggressive NK/T Cell Lymphomas. Pathol Res Pract 2011, 207, 786–789. [CrossRef]
- Yamamoto, J; Fujishima, F; Ichinohasama, R; Imatani, A; Asano, N; Harigae, H. A Case of Benign Natural Killer Cell Proliferative Disorder of the Stomach (Gastric Manifestation of Natural Killer Cell Lymphomatoid Gastropathy) Mimicking Extranodal Natural Killer/T-cell Lymphoma. Lek Lymphoma 2011, 52, 1803–1805. [CrossRef]
- McElroy, MK; Read, WL; Harmon, GS; Weidner, N. A Unique Case of an Indolent CD56-positive T-cell Lymphoproliferative Disorder of the Gastrointestinal Tract: A Lesion Potentially Misdiagnosed As Natural Killer/T-cell Lymphoma. Ann Diagn Pathol 2011, 15, 370–375. [CrossRef]
- Terai, T; Sugimoto, M; Uozaki, H; Kitagawa, T; Kinoshita, M; Baba, S; Yamada, T; Osawa, S; Sugimoto, K. Lymphomatoid Gastropathy Mimicking Extranodal NK/T Cell Lymphoma, Nasal Type: A case Report. World J Gastroenterol 2102, 18, 2140–2144.
- Ishibashi, Y; Matsuzono, E; Yokoyama, F; Ohara, Y; Sugai, N; Seki, H; Miura, A; Fujita, J; Suzuki, J; Fujisawa, T etal. A Case of Lymphomatoid Gastropathy: A Self-limited Pseudomalignant Natural Killer (NK)-Cell Proliferative Disease Mimicking NK/T-Cell Lymphomas. Clin J Gastroenterol 2013, 6, 287–290. [CrossRef]
- Koh, J; Go, H; Lee, WA; Jeon, YK. Benign Indolent CD56-positive NK-cell Lymphoproliferative Lesion Involving Gastrointestinal Tract in an Adolescent. Korean J Pathol 2014, 48, 73–76.
- Hong, M; Kim, WS; Ko, YH. Indolent CD56-positive Clonal T-cell Lymphproliferative Disease of the Stomach Mimicking Lymphomatoid Gastropathy. Korean J Pathol 2014, 48, 430–433.
- Takata, K; Noujima-Harada, M; Miyata-Takata, T; Ichimura, K; Sato, Y; Miyata, T; Naruse, K; Iwamoto, T; Tari, A; Masunari, T etal. Clinicopathologic Analysis of 6 Ly,phomatoid Gastropathy Cases. Expanding the Disease Spectrum to CD4-CD8- Cases. Am J Surg Pathol 2015, 39, 1259–1266.
- Hwang, SH; Park, JS; Jeong, SH; Yim, H. Indolent NK Cell Proliferative Lesion mimicking NK/T Cell Lymphoma in the Gallbladder. Hum Pathol Case Rep 2016, 5, 39–42. [CrossRef]
- Wang, R; Kariappa,S; Toon, C; Varikatt, W. NK-cell Enteropathy, a Potential Diagnostic Pitfall of Intestinal Lymphoproliferative Disease. Pathology 2019, 51, 338–340. [CrossRef]
- Xia, D; Morgan, EA; Berger, D; Pinkus, GS; Ferry, JA; Zukerberg, LR. NK-cell Enteropathy and Similar Indolent Lymphoproliferative Disorders. A Case Series with Literature Review. Am J Clin Pathol 2019, 151, 75–85. [CrossRef]
- Panigraphi, MK; Patra, S; Kumar, C; Kumar Nayak, H; Ayyanar, P; Bhat, SJ; Chouhan, MI; Samal, SC. Natural Killer Cell Enteropathy with Extraaintestinal Involvement: Presenting as Symptomatic Anemia. ACG Case Rep J 2021, 8, e00599. [CrossRef]
- Xiao, W; Gupta, GK; Yao, J; Yang, YJ; Xi, L; Baik, J; Sigler, A; Kumar, A; Moskowitz, AJ; Arcila, ME et al. recurrent Somatic JAK3 mutations in NK-cell Enteropathy. Blood 2019, 134, 986–991. [CrossRef]
- Yi, H; Li, A; Ouyang, B; Da, Q; Dong, L; Liu, Y; Xu, H; Zhang, X; Zhang, W; Jin X etal. Clinicopathological and Molecular Features of Indolent Naturall Killer-Cell Lymphoproliferative Disorder of the Gastrointestinal Tract. Histopathology 2023, 82, 567–575. [CrossRef]
- Chuang, SS; Li, GD; Cheng, CL; Ng, SB; Huang, Y; Zhao, S; Yamaguchi, M; Zhao, WL; Kwong, YL. Lymphoma. In WHO Classification of Haematolymphoid Tumours, 5th ed; Chan, J, Ed.; Lee Wood, B, Eds. IARC: Lyons, France, 2025; Available online: https://tumourclassification.iarc.who.int/chapters/63 (accessed on 23 October 2025).
- Bhagat, G; Naresh, KN; Dave, SS; Cerf-Benussan, N. Lymphoma. In WHO Classification of Haematolymphoid Tumours, 5th ed; Chan, J, Washington, NK, Eds.; de Jong, D, Eds. IARC: Lyons, France, 2015. [Google Scholar]
- Chan, J; Bhagat, G; Chuang, SS; Naresh, KN; Dave, SS; Nakamura, N; Kato, S; Tan, SY. Lymphoma. In WHO Classification of Haematolymphoid Tumours, 5th ed; de Jong, D, Ed.; Washington, NK, Eds. IARC: Lyons, France, 2015. [Google Scholar]
- Ng, CS. From the Midfacial Destructive Drama to the Unfolding EBV Story: A Short History of EBV-positive NK-cell and T-cell Lymphoproliferative Diseases. Pathology 2024, 56, 773–785. [Google Scholar] [CrossRef]
- Rahemtullah, A; Longtine, JA; Harris, NL; Dorn, M; Zembowicz, A; Quintanilla-Fend, L; Preffer, FI; Ferry, JA. CD20+ T-cell Lymphoma. Clinicopathologic Analysis of 9 Cases and a Review of the Literature. Am J Surg Pathol 2008, 32, 1593–1607.
- Watabe, D; Kanno, H; Inoue-Narita, T; Onodera, H; Izumida, W; Kowata, S; Sawai, T; Akasaka, T. A Case of primary Cutaneous Natural Killer/T-cell Lymphoma, Nasal Type, with Indolent Clinical Course: Monoclonal Expansion of Epstein-Barr Virus Genome Correlating with the Terminal Aggressive Behaviour. Br J Haematol 2009, 160, 197–228. [CrossRef]
- Seishima, M; Yuge, M; Kosugi, H; Nagasaka, T. Extranodal NK/T-Cell lymphoma, Nasal Type, Possibly arising from Chronic Epstein-Barr Virus Infection. Acta Derm Venerol 2010, 90, 102–103. [CrossRef]
- Jiang, Q; Liu, S; Yang, Y; Tan,X; Peng, J; Xiong, Z; Li, Z. CD20-positive NK/T-cell Lymphoma with Indolent Clinical Course: Report of Case and Review of Literature. Diagn Pathol 2012, 7, 133. [CrossRef]
- Zuriel, D; Fink-Puches, R; Cerroni, L. A Case of Primary Cutaneous Extranodal Natural Killer/T-cell Lymphoma, Nasal Type, with a 22-Year Indolent Clinical Course. Am J Dermatopathol 2012, 34, 194–197. [Google Scholar] [CrossRef]
- Tabanelli, V; Valli, R; Zanelli, M; Righi, S; Gazzola, A; Mannu, C; Pileri, S; Sabattini, E. A Case of Sinonasal Extranodal NK/T-cell Lymphoma with Indolent Behaviour and Low-Grade Morphology. Case Rep Clin Med 2014, 3, 596–600.
- Zhang, QF; Hau, YN; Sun, LM; Tian, Y; Qiu, XS. Nasal Extranodal NK/T-cell Lymphoma Mimicking Inflammatory Polyps: a Case with Indolent Clinical Behavior. Int J Clin Exp Pathol 2016, 9, 336–340.
- Devins, K; Schuster, SJ; Caponetti, GC; Bogusz, M. Rare Case of Low-Grade Extranodal NK/T-cell Lymphoma, Nasal Type, Arising in the Setting of Chronic Rhinosinusitis and Harboring a Novel N-Terminal KIT Mutation. Diagn Pathol 2018, 13, 92.
- Wang, Z; Gong, Q; Zhao, Y; Xu, H; Hu, S; Zhang, Z. Indolent EBV-positive T-cell Lymphoproliferative Disorder Arising in a Chronic Pericardial Hematoma: the T-cell Counterpart of Fibrin-associated Diffuse Large B-cell Lymphoma? Haematologica 2020, 105, e437. [CrossRef]
- Krishman, R; Ring, K; Williams, E; Portell, C; Jaffe, ES; Gru, AA. An Enteropathy-like Indolent NK-cell proliferation Presenting in the Female Genital Tract. Am J Surg Pathol 2020, 44, 561–565.
- Saglam, A; Singh, K; Gollapudi, S; Kumar, J; Brar, N; Butzmann, A; Warnke, R; Ohgami, RS. Indolent T-lymphoblastic Proliferation: A Systemic Review of the Literature Analyzing the Epidemiologic, Clinical, and Pathologic Features of 45 Cases. Int J Lab Hematol 2022, 44, 700=711.
- Ohgami, RS; Arber, DA; Zehnder, JL; Nathunam, Y; Warnke, R. Indolent T-lymphoblastic Proliferation (iT-LBP): a Review of Clinical and Pathologic Features and Distinction from Malignant T-lymphoblastic Lymphoma. Adv Anat Pathol 2013, 20, 137–140.
- Pizzi, M; Brignola, S; Righi, S; Algostinelli, C; Bertuzzi, C; Pillon, M; Semenzato, G; Rugge, M; Sabattini, E. Benign Tdt-positive Cells in Pediatric and Adult Lymph Nodes: a Potential Diagnostic Pitfall. Hum Pathol 2018, 81, 131–137.
- Goodlad, JR; Cerroni, L; Swerdlow, SH. Recent Advances in Cutaneous Lymphoma – Implications for Current and Future Classifications. Virchows Archiv 2023, 482, 281–298.
- Beltzung, F; Ortonne, N; Pelletier, L; Beylot-Berry, M; Ingen-Housz-Oro, S; Franck, F; Pereira, B; Godfraind, C; Delfau, M; D’Incan, M etal. Primary Cutaneous CD4+ Small/Medium T-cell Lymphoproliferative Disorders: a Clinical, Pathologic, and Molecular Study of 60 Cases Presenting with a Single Lesion: a Multicenter Study Group. Am J Surg Pathol 2020, 44, 862–872.
- Greenblatt, D; Ally, M; Child, F; Scarisbrick, L; Whittaker, S; Morris, S; Calonje, E; Petrella, T; Robson, A. Indolent CD8(+) Lymphoid Proliferation of Acral Sites: a Clinicopathologic Study of Six Patients with Some Atypical Features. J Cutan Pathol 2013, 40, 248–258.
- Vergier,B; Jansen, PM; Ortonne, N; Pulitzar, M; Williemze, T; Mitteldorf, C. Disorders. In WHO Classificarion of Haematolymphoid Tumours, 5th ed; Lazar, AJ, Coupland, SE, Eds.; Akkari, Y, Eds. IARC: Lyons, France, 2025; Available online: https://tumourclassification.iarc.who.int/chapters/63 (accessed on 23 October 2025).
- Kemp, W; Pulitzer, M; Robson, A; Mitteldorf, C. Primary Cutaneous Acral CD8-positive T-cell Lymphoproliferative Disorders. In: WHO Classification of Haematolymphoid Tumours, 5th ed; Lazar, AJ; Coupland, SE; Akkar, Y, Eds. IARC, Lyons. France, 2025. Available online: https://tumourclassification.iarc.who.int/chapters/63 (accessed on 23 October 2025).
- Tamehiro, N; Nishida, K; Sugita, Y; Hayakawa, K; Oda, H; Nitta, T; Nakano, M; Nishioka, A; Yanobu-Takanashi, R; Goto, M etal. Ras Homolog Gene Family H (RhoH) Deficiency Induces Psoriasis-like Chronic Fermatitis by Promoting TH17 Cell Polarization. J Allergy Clin Immunol 2019, 143, 1878–1891. [CrossRef]
- Crequer, A; Troeger, A; Patin, E; Ma, CS; Picard, C; Pederana, V; Fieschi, C; Lim, A; Abhyankar, A; Gineau, L etal. Human RHOH Deficiency Causes T Cell Defects and Susceptibility to EV-HPV Infection. J Clin Invest 2012, 122, 3239–3247. [CrossRef]
- Edison, N; Belhanes-Peled, H; Eitan, Y; Gutthmann, Y; Yeremenko, Y; Raffeld, M; Elmalah, I; Troughuboff, P. Indolent T-cell Lymphoproliferative Disease of the Gastrointestinal Tract after a Treatment with adaloimumab in Resistant Crohn’s Colitis. Hum Pathol 2016, 5, 45–50. [CrossRef]
- Chen, K; Wang, M; Zhang, R; Li, J. Detection of Epstein-Barr Virus Encoded RNA in Fixed Cells and Tissues using CRISPR/Cas-mediated R Cas FISH. Anal Biochem 2021, 625, 114211.
- Qi, Z; Han, X; Hu, J; Wang, G; Gao, J; Wang, X; Liang, D. Comparison of Three Methods for Detection of Epstein-Barr Virus in Hodgkin’s Lymphoma in Paraffin-Embedded Tissues. Mol Med Rep 2013, 7, 89–92. [CrossRef]
- Leenman, EE; Panzer-Grumayer, RE; Fischer, S; Leitch, HA; Horsman, DE; Lion, T; Gadner, H; Ambros, PF; Leston, VS. Rapid Determination of Epstein-Barr Virus Latent or Lytic Infection in Single Human Cells Using In situ Hybridization. Mod Pathol 2 0024, 17, 1564–1572. [CrossRef]
- Sun, Y; Ling, S; Tang, D; yang, M; Shen, C. Advances in Epstein-Barr Virus Detection: From Traditional Methods to Modern Technologies. Viruses 2005, 17, 1026.
- Naresh, KN; Ferry, JA; Du, M. Introduction to B-cell Lymphproliferative Disorders and Neoplasms. In: WHO Classfication of Haematolymphoid Tumours, 5th ed. IARC: Lyons, France, 2025. Available online: https://tumourclassification.iarc.who.int/chapters/63 (accessed on 23 October 2025).
Table 2.
Summary of Reported EBV+ non-intestinal indolent T- and NK-cell LPD.
|
Series (reference, year) |
Sex | Age |
LPD Duration (years) |
Site(s) |
Disease progression |
Px of progressed disease |
Clinical outcome |
Histology of LPD |
Immunophenotype |
Geneotype | Lineage |
EBV/EBER of LPD |
Ki67 |
| Rahemtullah et al[54] (2008) |
M | 71 | 4 | neck LN |
EBV+ BCL with plasmacytic differentiation | CT | DOD (66 m) |
LG | CD2+, CD3+, CD5+, CD7+, CD4+, CD20+, CD30+(dim), CD56+(dim) |
TCR: GL Ig: polyclonal |
T | + (EBV clonal) |
NR |
| Watabe et al[55] (2009) |
F | 39 | 10 | skin (legs) |
ENNKTL, nasal type (skin) |
CT | DOD (128 m) |
LG* | CD3+, CD56+, GZB+, CD8+/-, perforin +/- |
TCR:GL | NK | + (LMP+) (EBV biclonal) |
NR |
| Seishima FM et al[56] (2010) |
F | 60 | 11.5 | skin (lip and cheek) |
EBV+ ENNKTL, nasal type (nose & multiple skin sites) |
CT | DOD (146m) |
LG* | CD56-, CD4+/-, CD8+/-, cytotoxic (ND) |
NR | NK (CD56 turned + on progression) |
NR | NR |
| Jiang QP et al [157] (2012) |
F | 78 | 10 | nose | NP | NA | AWD | LG | CD3+, CD56+, Cytotoxic+, CD20+ |
TCR:GL Ig : GL |
NK | + (EBV genome) |
60% |
| Zuriel D et al[58] (2012) |
F | 55 | 22 | skin (recurrent, right upper arm) |
NP | NA | AWD (recurrence 192 m & 264 m, skin right upper arm) | LG | CD2+, CD3+, cytotoxic+, CD56+ (at 264 m) Ki67 >90% |
TCR: GL | NK | + | 90% |
| Tabeanelli V et al[59] (2014) |
F | 52 | 13 | nose | NP | NA | AWD at 156 m |
LG | CD2+, CD3+, CD5+, CD7+, CD56+, βF1+, TCR α/δ+, cytotoxic+, Ki67 (moderately high) |
TCR: clonal | T | + | Moderate high |
| Zhang QF et al[60] (2016) |
M | 53 | 20 | nose | NR | NA | AWD at 242 m |
LG | CD3+, CD56+, cytotoxic+, Ki67(80%) |
NR | NK | + | 80% |
| Devins K et al[61] (2018) |
F | 71 | long standing | nose | NR | NA | AWD (many years) |
LG | CD2+, CD3+, CD3+/-, CD5+/-, CD7+/-, CD56- cytotoxic+, Ki67 (<1%) |
TCR: clonal KIT mutation+ |
T | + | <1% |
| Wang Z et al [62] (2020) | M | 64 | 19 | Pericardium | NP | NA | AW | LG | CD3+,CD30+,CD43+, TIA1+, MUM1+, BCL2+ |
TCR:clonal | T | + | >90% |
Footnote: AW: alive and well; AWD: alive with disease; BCL: B-cell lymphoma; CT: chemotherapy; DOD: died of disease; ENNKTL: extranodal NK/T-cell lymphoma; GZB: Granzyme B; IgG: immunoglobulin gene; LG: low grade; LN: lymph node; LPD: lymphoproliferative disease: MG*: medium grade histology with lymphoepithelial lesion; MG@: medium grade histology with angiocenctricity and focal necrosis, NA: not applicable; NP: no progression of disease; NR: not reported; NED: no evidence of disease; Px: treatment modality; RT: radiotherapy; TCR: T-cell receptor gene; *: interpreted from published images; +: positive, +/-: weak or focal positive.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



