Submitted:
23 October 2025
Posted:
27 October 2025
You are already at the latest version
Abstract
We describe a patient with post-essential thrombocythemia myelofibrosis treated with intermittent hydroxyurea (Hu) therapy (20mg/Kg, given as a single dose, thrice weekly), achieving sustained disease control and regression of bone marrow fibrosis; and, discuss the efficacy of and rationale for use of intermittent Hu therapy in patients with myeloproliferative neoplasms, including those deemed to be Hu-resistant or intolerant on the commonly used continuous therapy.
Keywords:
essential thrombocythemia
; myelofibrosis
; intermittent hydroxyurea therapy
; fibrosis regression
Introduction
Essential thrombocythemia (ET) is one of four chronic myeloproliferative neoplasms (MPN), characterised by clonal thrombocytosis [1]; approximately 85% of patients will have a pathognomonic driver mutation at presentation – JAK-2 (about 50%), CALR (about 30%) or MPL (about 5%) [2]. CALR-positive ET patients generally have a good prognosis, the median survival being 20 years; patients less than 65 years old have a better median survival at 32 years [3]. However, these patients are at a higher risk of progression to myelofibrosis than those with JAK-2 mutation [4,5,6], especially if they are resistant or intolerant to hydroxyurea (Hu) therapy [7]. The resulting clinical entity, referred to as post-ET myelofibrosis [8], has an adverse impact on patient survival – even “low risk” patients (according to the Dynamic International Prognostic Scoring System plus -DIPSS+) have a median survival of only 15 years [3]. Current therapeutic guidelines for post-ET myelofibrosis patients comprise initiation of a JAK-2 inhibitor (e.g. ruxolitinib) and consideration of stem cell transplant as definitive treatment [9,10]. In this report we describe a patient with post-ET myelofibrosis treated with intermittent Hu, achieving effective disease control including regression of bone marrow fibrosis from grade 3 to grade 1[11]..
Case Report
In December 2022, a 56 year old female presented to our Centre for management of post-ET myelofibrosis. Five years earlier, she had been diagnosed with type 2 CALR-positive ET at another centre: Hb 112g/l, WBC 6.3x109/L and platelets 1200x109/l. She had been treated with HU 500mg daily but this was discontinued after four months because of lack of response and neutropenia. She remained under regular reviews without any further treatment. In November 2022, she was found to be anaemic; Hb 104g/l, WBC 9.1 109/land platelets 921 109/l. Bone marrow examination showed marked hyper-cellularity, reduced erythropoiesis, hypo-granular neutrophils, dysplastic megakaryocytes and markedly increased fibrosis (grade 3) [11] - Figure 1. She was assessed to be be in the “low risk” prognostic category and “not-a-candidate” for allogenic stem cell transplantation because of potential procedure-related morbidity.
At presentation to our Centre, she was clinically well but anxious about the possibility of thrombotic events and the progression of thrombocythemia to myelofibrosis. Physical examination was unremarkable; in particular, there was no splenomegaly. whole blood platelet aggregation studies showed features of platelet hyperactivity; repeat tests on aspirin 100mg daily dose showed optimum anti-platelet effect [12]. She was considered to be a candidate for cytoreductive therapy to control the disease activity (i.e. thrombocytosis) and to slow down its progression. She was commenced on hydroxyurea 1.5g (20g/Kg) given as a single dose, thrice weekly. She tolerated the treatment well and remained under 2-3 monthly reviews. Twelve months later, blood counts showed Hb 128g/l, WBC2.7x109/l (Neutrophils 1.1) and platelets 662x109/l. These numbers remained stable over the next 18 months. A repeat bone marrow examination in May 2025 showed mild hyper-cellularity, adequate erythropoiesis and normal myelopoiesis with occasional hypo-granular neutrophils and increased number of megakaryocytes with hyper-/hypo-lobulated nuclei; reticulin stain showed grade 1 of 3 fibrosis [11] – Figure 2. Cytogenetic studies were normal. Gene panel studies showed variants in TET 2, CALR (type 2) and DNMT3 A, consistent with myeloproliferative neoplasm (MPN). At the time of writing (October 2025), she remains on Hu and aspirin therapies and leads an active life, with a performance status score of 100% [13].
Discussion
The aim of treatment in patients with ET is to prevent thrombosis and to slow down or delay disease progression. Our patient’s concern for thrombotic events is common among ET patients. A survey by Mesa et al in the United States has found thrombosis prevention to be the most important goal of treatment for 35% of patients with ET and 57% of physicians treating ET patients [14]. At our Centre, we routinely use whole blood platelet aggregation studies in all MPN patients for risk assessment and to tailor anti-platelet therapy in individual patients. In a study of 132 patients (including 98 ET patients) we have documented varying degrees of platelet hyperactivity in 115 (87 ET patients) and thrombosis- free survival of 1-23 years (median 8 years) whilst on the individualised anti-platelet therapy [12]; no significant differences were noted in terms of aspirin dose requirements (ranging from 100mg twice or thrice weekly to 400mg daily) between JAK-2, CALR or MPL positive patients and among the four IPSET-thrombosis [15,16] sub-groups.
Hu is the commonly used first-line therapeutic agent for cytoreduction in patients with MPN [17,18,19], the most used schedule being 500mg twice daily and titrated on the basis of response and blood counts [20,21,22]. However, several studies have reported resistance , intolerance or disease progression in 10-30% of patients receiving Hu as continuous therapy [23,24]. A recent Spanish study of 1080 ET patients has reported inferior clinical outcomes in those with resistance or intolerance to first-line Hu therapy [7].
At our Centre, we have used Hu as intermittent therapy, (akin to schedules used in patients with solid tumours) at 20-30mg/kg doses, given as a single dose, twice or thrice weekly. During the past 30 years, we have treated 118 MPN patients (polycythaemia vera-29, ET-84, primary myelofibrosis-5) and have observed (median follow-up 8.5years) sustained responses without troublesome cytopenias or the need for treatment interruptions [25]. The total weekly dose of intermittent Hu used at our Centre is comparable to those commonly recommended in the continuous regimen. Based on the review of pharmaco-kinetics [26,27] of Hu and our experience, we hypothesize that the better clinical outcome with intermittent therapy is attributable to i) higher plasma level of Hu achieved with the single dose intake ( more than 80% of orally administered dose of Hu is readily absorbed, reaching peak plasma levels in 1-4 hrs); ii) the preferential uptake of Hu by the mitotically more active clonal proliferative cells; and, iii) the unhindered, normal haemopoietic activity on treatment-free days each week ( the plasma half-life of Hu is short - 2-4 hrs).
Recent review articles on the management of patients with post-ET myelofibrosis recommend prognostic stratification using a clinical-molecular prognostic model to predict survival [28,29], treatment with a JAK-2 inhibitor agent (ruxolitinib, fedratinib, pacritinib, momelotinib or jaktinib) and early consideration of stem cell transplant, especially for patients in the “low risk” category [9,10,30]. JAK-2 inhibitor therapy can be effective in reducing splenic volume and minimise disease-related symptoms (thus improving the quality of life) even in patients with CALR- positive post-ET myelofibrosis [31]; however, these agents have no effect on the underlying clonal proliferation, nor can change the course of the disease. The patient described in this report did not have splenomegaly or any disease-related symptoms. Her clinical picture comprised persistent thrombocytosis, anaemia and dense bone marrow fibrosis. Intermittent Hu therapy, through its action on the proliferating clone, has been effective, achieving improvement of all these parameters.
Bone marrow fibrosis in patients with MPN is a reactive process [32], attributable to overproduction of abnormal megakaryocytes and excessive release of cytokines that stimulate fibroblasts to deposit collagen, resulting in fibrosis [33]. Reversibility of this reactive process has been well documented with effective chemotherapy, including Hu [34,35,36,37]. Continuous Hu therapy at a starting dose of 500mg daily, however, is commonly associated with worsening anaemia or cytopenias, necessitating change of treatment in 80% of patients at 12 months [19]. The patient described in this report has had no clinical problems with the intermittent therapy over a 2½ year period. Regression of fibrosis with effective chemotherapy suggests that the decrease in the number of megakaryocytes and also possibly the release of pathogenic cytokines, enable normal collagenase activity in the marrow micro-environment to gradually reduce fibrosis.
The efficacy and tolerability of intermittent Hu therapy in MPN patients need to be confirmed in large, randomized studies. In the interim, we hope our publications will encourage clinicians to consider the intermittent dosage schedule i) in the Hu resistant or intolerant patients on continuous therapy , before changing over to more invasive and/or expensive second-line therapies; and, ii) in “low risk” post-ET myelofibrosis patients before embarking on stem cell transplantation, as definitive therapy.
Author Contributions
A.M. treated the patient and wrote the manuscript; I.T. reviewed the pre- and post- treatment bone marrow slides and provided the figures for the manuscript.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Illawarra Private Cancer Care Centre Institutional Review Board (Project identification code: 22-006) on 6 July 2022
Informed Consent Statement
Informed consent for publication was obtained from all identifiable human participants.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; et al. The International Consensus Classification of Myeloid Neoplasms and Acute Leukemia: Myeloproliferative Neoplasms. Am J Hematol. 2023, 98, 166–179. [Google Scholar] [CrossRef]
- Tefferi, A. Myeloproliferative Neoplasms: A Decade of Discoveries and Treatment Advances. Am J Hematol. 2016, 92, 50–58. [Google Scholar] [CrossRef]
- Tefferi, A.; Wassie, E.A.; Lasho, T.L.; et al. Calreticulin Mutations and Long-term Survival in Essential Thrombocythemia. Leukemia, 2014. [CrossRef]
- Barbui, T.; Thiele JPassamonti, F.; et al. Survival and Disease Progression in Essential Thrombocythemia Are Significantly Influenced by Accurate Morphologic Diagnosis: An International Study. J Clin Oncol. 2011, 29, 3179–84. [Google Scholar] [CrossRef]
- Al Assaf, C.; Van Olbergh, F.; Billiet, J.; et al. Analysis of Phenotype and Outcome in Essential Thrombocythemia with CALR or JAK2 Mutations. Haematologica 2015, 100, 893–897. [Google Scholar] [CrossRef]
- Erdos, K.; Lee, N.; Lebbe, A.; et al. Low Thrombosis Risk with CALR Mutation Confer Higher Risk of ET Progression. Blood 2023, 142, 1819. [Google Scholar] [CrossRef]
- Santaliestra, M.; Garrote, M.; Noya, M.S.; et al. Prognostic Value of Response to IPSET Stratification in Essential Thrombocythemia. Leukemia 2024, 38, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G.; Mesa, R.A.; Thiele, J.; et al. Proposed Criteria for the Diagnosis of Post-Polycythemia Vera and Post-essential Thrombocythemia Myelofibrosis: A consensus Statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008, 22, 437–8. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Mora, B. Myelofibrosis. Blood 2023, 141, 1954–70. [Google Scholar] [CrossRef] [PubMed]
- Puglianini, O.C.; Peker, D.; Zhang, L.; et al. Essential Thrombocythemia and Post-essential Thrombocythemia Myelofibrosis: Updates on Diagnosis, Clinical Aspects and Management. Laboratory Medicine 2023, 54, 13–22. [Google Scholar] [CrossRef]
- Thiele, J.; Krosnicka, H.M.; Facchetti, F.; et al. European Consensus on Grading of Bone Marrow Fibrosis and Assessment of Cellularity. Haematologica 2005, 90, 1128–1132. [Google Scholar]
- Manoharan, A.; Gemmel, R.; Cavanaugh, L.; et al. Thrombosis in Myeloproliferative Neoplasms: A Single Center Experience of Using Whole Blood Platelet Aggregation Studies for Risk Assessment and Thromboprophylaxis. Clin Appl Thr/Hemo. 2022, 28, 1076029622111. [Google Scholar] [CrossRef]
- Schag, C.C.; Heinrich, R.L.; Gang, P.A. Kornofsky Performance Status Revisited: Reliability, Validity and Guidelines. J Clin Oncol 1984, 2, 187–193. [Google Scholar] [CrossRef]
- Mesa, R.A.; Miller, C.B.; Thyne, M.; et al. Differences in Treatment Goal and Perception of Symptoms Burden between Patients with Myeloproliferative Neoplasms (MPNs) and Hematologist/Oncologists in the United States. Findings from the MPN Landmark Survey. Cancer 2017, 123, 449–58. [Google Scholar]
- Barbui, T.; Vannucchi, A.M.; Buxhofer-Ausch, V.; et al. Practice -relevant revision of IPSET=thrombosis Based on 1019 Patients with WHO-Defined Essential Thrombocythemia. Blood Cancer J. 2015, 5, e369. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Gangat, N.; Lasho, T.; et al. Validation of the Revised International Prognostic Score of Thrombosis for Essential Thrombocythemia (IPSET-thrombosis) in 585 Mayo Clinic Patients. Am J Hematol. 2016, 91, 390–4. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Vannucchi, A.; Finazzi, G.; et al. A Reappraisal of the Benefit-Risk Profile of Hydroxyurea in Polycythemia Vera: A Propensity-matched Study. Am J Hematol. 2017, 92, 1131–1136. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Essential Thrombocythemia: 2024 Update on Diagnosis, Risk Stratification and Management. Am J Hematol. 2024, 99, 697–718. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Trillos, A.; Gaya, A.; Maffioli, M.; et al. Efficacy and Tolerability of Hydroxyurea in the Treatment of Hyperproliferative Manifestations of Myelofibrosis: Results in 40 Patients. Ann Hematol. 2010, 89, 1233–1237. [Google Scholar] [CrossRef]
- Buyukasik, Y.; Ali, R.; Turgut, M.; et al. Patterns of Hydroxyurea Prescription and Use in Routine Clinical Management of Polycythemia Vera: A Multicentre Chart Review Study. Turkish J Hematol. 2020, 37, 177. [Google Scholar] [CrossRef]
- Grunwald, M.; Kutes, D.J.; Altomare, I.; et al. Treatment Patterns and Blood Counts in Patients with Polycythemia Vera Treated with Hydroxyurea in the United States: An Analysis from the REVEAL Study. Clin Lymphoma Myeloma and Leuk. 2020, 20, 219–225. [Google Scholar] [CrossRef]
- Ferrer-Marin, F.; Hernandez-Boluda, J.C.; Alvarez-Larran, A. Essential Thrombocythemia: A Contemporary Approach with New Drugs on the Horizon. Br J Haematol. 2024, 204, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Kuykendall, A.T. Treatment of Hydroxyurea-Resistant/Intolerant Polycythemia Vera: A Discussion of Best Practices. Ann Hematol. 2023, 102, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Nejadnik, B.; Mascarenhas, J.; Rappaport, K.M.; et al. Treatment of Essential Thrombocythemia Patients Intolerant/Resistant to Hydroxyurea. J Clin Oncol. 2017, 35, e. 18565. [Google Scholar] [CrossRef]
- Manoharan, A.; Enggist, S.I. Hydroxyurea: An Old Drug in Need of New Clinical Trials in Myeloproliferative Neoplasms? Int Internal Med J. 2024, 2, 1–6. [Google Scholar]
- Tracewell, W.G.; Trump, D.L.; Vaughan, W.P.; et al. Population Pharmacokinetics of Hydroxyurea in Cancer Patients. Cancer Chemotherapy and Pharmacology 1995, 35, 417–422. [Google Scholar] [CrossRef]
- Gwilt, P.R.; Tracewell, W.G. Pharmacokinetics and Pharmacodynamics of Hydroxyurea. Clin Pharmacokinetics 1998, 34, 347–358. [Google Scholar] [CrossRef]
- Passamonti, F.; Giorgino, T.; Mora, B.; et al. A Clinical-Molecular Prognostic Model to Predict Survival in Patients with Post Polycythemia Vera and Post Essential Thrombocythemia Myelofibrosis. Leukemia 2017, 31, 2726–31. [Google Scholar] [CrossRef]
- Hernandez-Boluda, J.-C.C.; Pereira, A.; Correa, J.-G.G.; et al. Performance of the Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM) in A Series of 262 Patients from the Spanish Registry of Myelofibrosis. Leukemia 2018, 32, 553–5. [Google Scholar] [CrossRef]
- Martino, M.; Pitea, M.; Sgarlata, A.; et al. Treatment Strategies Used in Treating Myelofibrosis: State of the Art. Hematol Rep. 2024, 16, 698–713. [Google Scholar] [CrossRef] [PubMed]
- Thaw, K.; Harrison, C.N.; Srikandarajah, P. JAK Inhibitors for Myelofibrosis: Strengths and Limitations. Curr Hematol Malig Rep. 2024, 19, 264–275. [Google Scholar] [CrossRef]
- Jacobson, R.J.; Salo, A.; Fialkow, P.J. Agnogenic Myeloid Metaplasia: A Clonal Proliferation of Hemopoietic Stem Cells with Secondary Myelofibrosis. Blood 1978, 51, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Zahr, A.A.; Salama, M.E.; Correau, N.; et al. Bone Marrow Fibrosis in Myelofibrosis: Pathogenesis, Prognosis and Targeted Strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, A.; Pitney, W.R. Chemotherapy Resolves Symptoms and Reverses Marrow Fibrosis in Myelofibrosis. Scand J Haematol. 1984, 33, 453–459. [Google Scholar] [CrossRef]
- Editorial. Reversible Myelofibrosis?. Lancet, 1985; 325: 497-498.
- Manoharan, A.; Chen, C.F.; Wilson, L.S.; et al. Ultrasonic Characterization of Splenic Tissue in Myelofibrosis: Further Evidence for Reversal of Fibrosis with Chemotherapy. Eur J Haematol. 1988, 40, 149–154. [Google Scholar] [CrossRef]
- Lofvenberg, E.; Wahlin, A.; Roos, G.; et al. Reversal of Myelofibrosis by Hydroxyurea. Eur J Haematol. 1990, 44, 33–38. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pre-treatment bone marrow biopsy. (A) (H&E x10) showing marked hyper-cellularity and numerous abnormal megakaryocytes; (B) (Reticulin stain x10) showing dense (Grade 3) fibrosis.
Figure 1.
Pre-treatment bone marrow biopsy. (A) (H&E x10) showing marked hyper-cellularity and numerous abnormal megakaryocytes; (B) (Reticulin stain x10) showing dense (Grade 3) fibrosis.
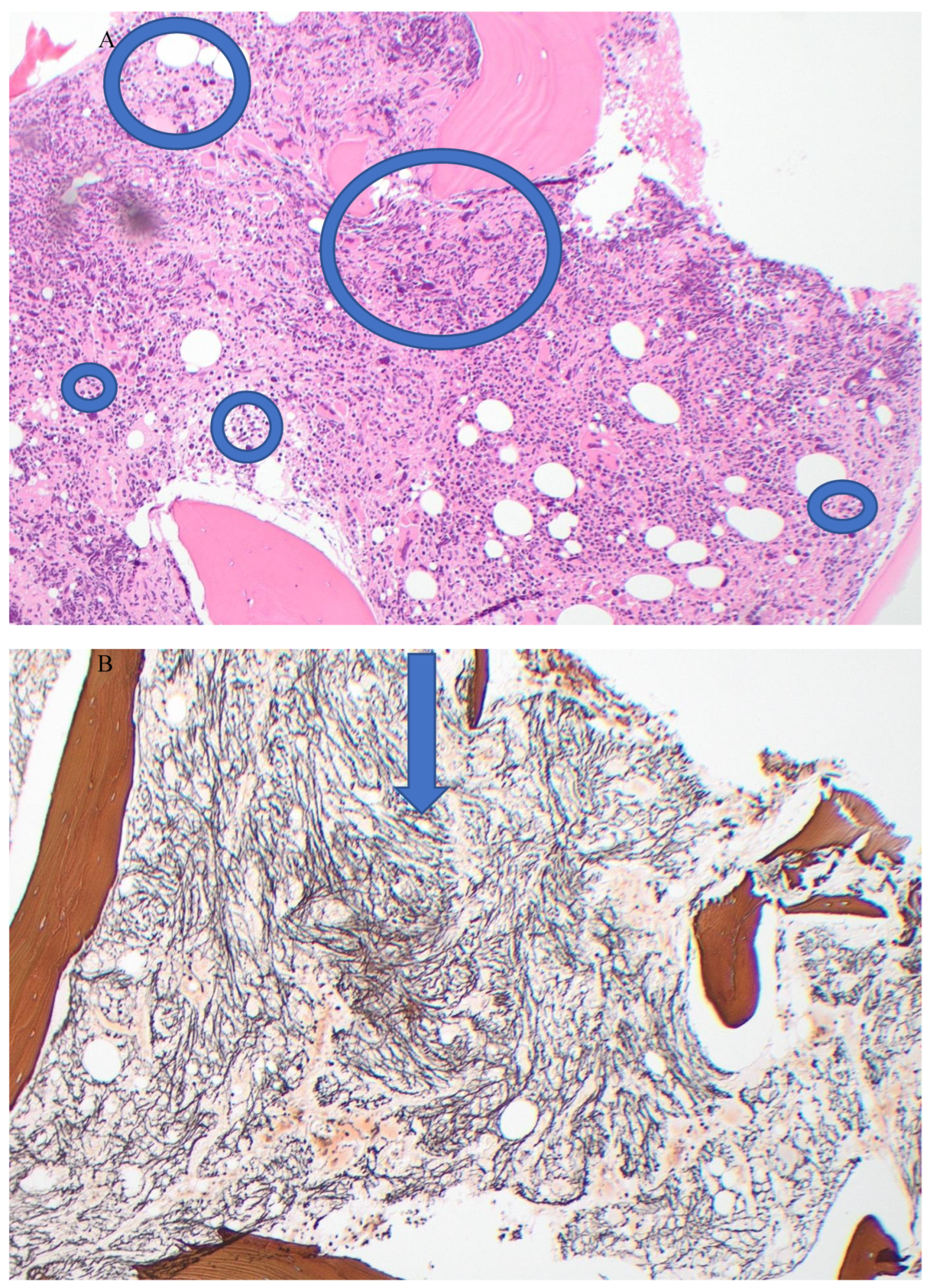
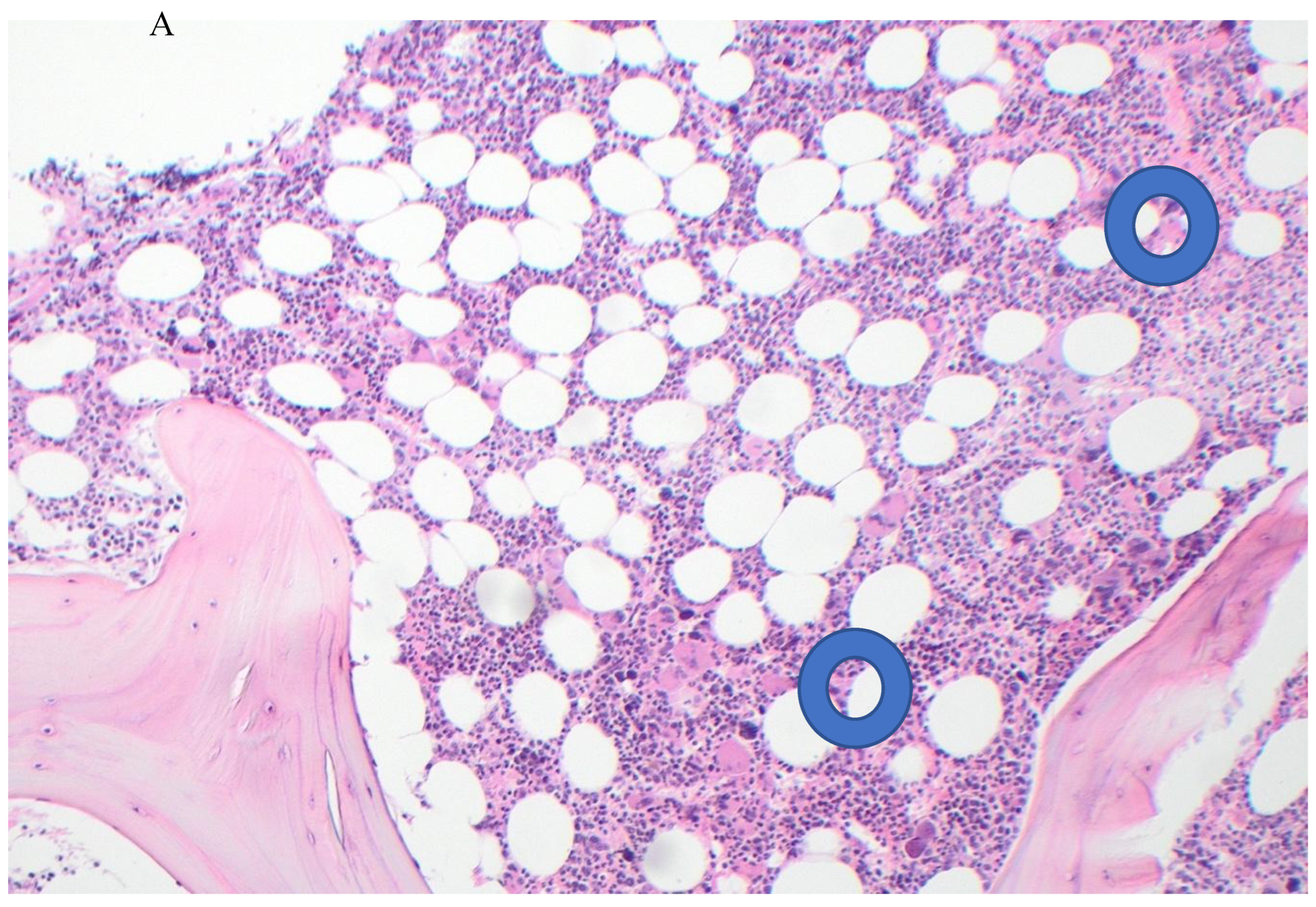
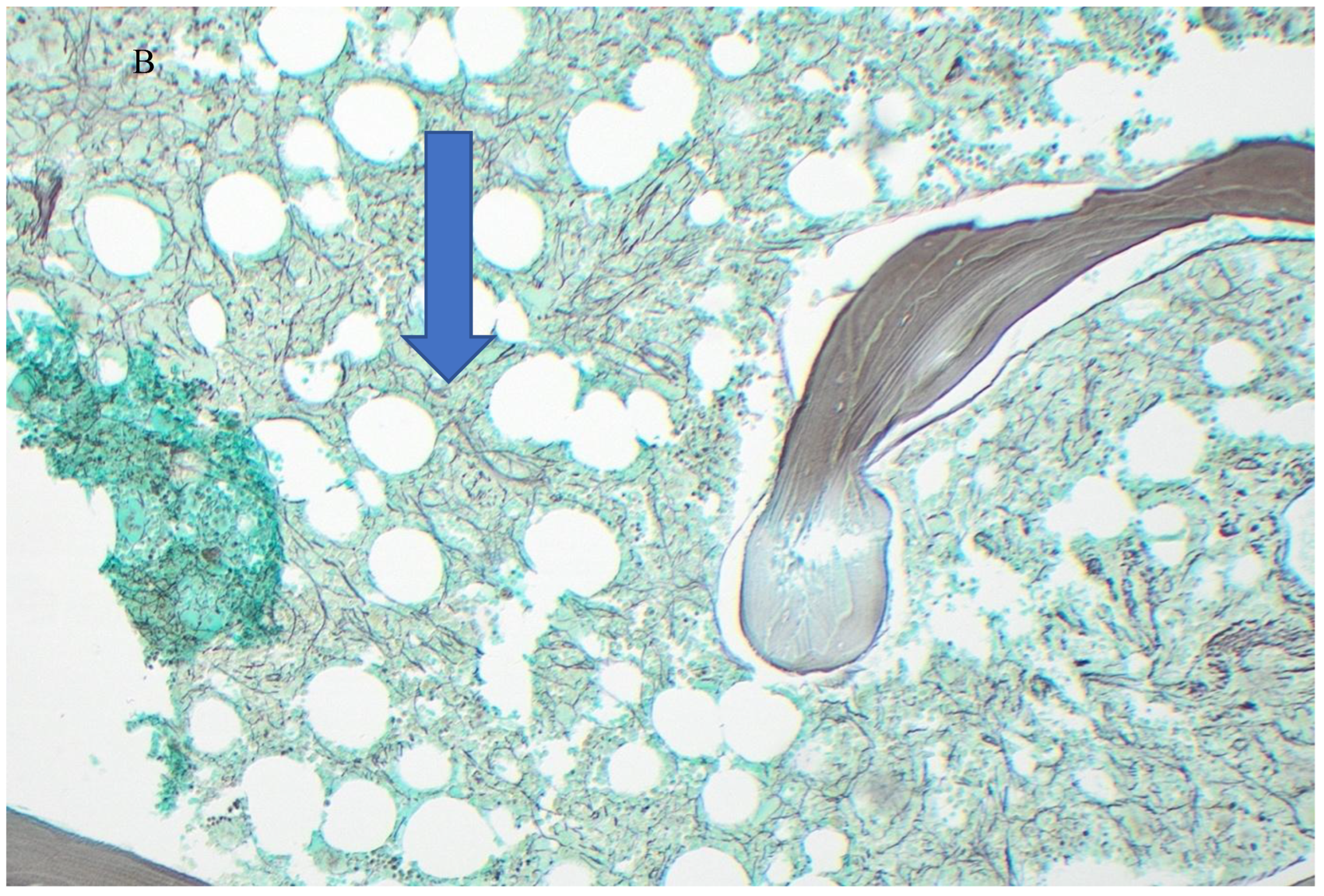
Figure 2.
Post-treatment bone marrow biopsy. A. (H&Ex10) showing mild hyper-cellularity and fewer abnormal megakaryocytes; B. (Reticulin stain x!0) showing mild (Grade 1) fibrosis.
Figure 2.
Post-treatment bone marrow biopsy. A. (H&Ex10) showing mild hyper-cellularity and fewer abnormal megakaryocytes; B. (Reticulin stain x!0) showing mild (Grade 1) fibrosis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



