Submitted:
20 October 2025
Posted:
21 October 2025
You are already at the latest version
Abstract
Prediction of polypeptide chain fragmentation of protein substrates during proteolysis by trypsin was performed for globular β-lactoglobulin (β-LG) and micellar β-casein (β-CN). Despite significant differences in the protein structures of these substrates, the concentra-tions of peptide fragments were calculated as functions of the time or degree of hydrolysis using the same equations derived from a general proteolysis model. This model consid-ered the opening of protein substrate, the so-called demasking, and the subsequent hy-drolysis of specific peptide bonds at different rates determined by the amino acid sequence of the hydrolyzed sites. The potential of this model for in silico proteolysis and the possi-bility of quantifying the rate constants for an arbitrary protein substrate are discussed. An algorithm for calculating demasking rate constants based on the experimental distribu-tion of peptide fragments is presented. The calculated concentration dependences on the degree of hydrolysis were compared with experimental data for the intermediate and final peptide fragments of β-LG and β-CN. The predicted and experimental concentration curves for the final products were compared based on their curvatures. For both sub-strates, the predicted distribution of peptide fragments was consistent with the experi-mental one.
Keywords:
proteolysis mechanisms
; trypsin
; β-lactoglobulin
; β-casein
; demasking
; peptide release
; rate constants
1. Introduction
Enzymatic hydrolysis of proteins by proteases (proteolysis) is widespread in living systems and is also used in the food industry, biotechnology, pharmaceuticals, cosmetology etc. Proteolysis is quite difficult to study, since it is a continuous process of interconversion of various peptide fragments, i.e. the composition of the reaction mixture changes significantly during the hydrolysis of peptide bonds. The concentrations of the final products of proteolysis and of sufficiently long intermediate peptides containing not yet hydrolyzed enzyme-specific bonds change with the time of hydrolysis and it is difficult to predict these concentration dependences [1,2,3].
A practically important task is to optimize the production of biopeptides from proteins using proteolysis. To obtain the final products of proteolysis, proteolysis must be carried out to the end. In contrast, intermediate biopeptides can be obtained by limited proteolysis during a given time interval, after which the process should be stopped to avoid further hydrolysis of peptide bonds. To determine this time interval, or the corresponding degree of hydrolysis of peptide bonds, it is necessary to use proteolysis modeling to assist the experimental path, which requires a large number of experiments [4,5,6,7,8,9,10,11,12].
Using the rate constants of hydrolysis of peptide bonds and other kinetic parameters, the time course of final and intermediate products can be calculated using the methods of chemical kinetics [4,9,10]. To simplify this task, it is proposed to use several approaches that both reduce the number of rate constants and the number of peptide fragments that are taken into consideration when modeling the proteolysis process [9].
Unlike low-molecular protease substrates, many peptide bonds in proteins are initially inaccessible to the enzyme, since they are located inside the protein globule or, for example, in the casein micelle. The destruction of the protein globule or micelle at the first stage of proteolysis initiates the process of opening the internal peptide bonds for a successful enzymatic attack [1,4,13]. This process, called demasking, eliminates steric hindrances for the enzyme so that the active center of the enzyme and the amino acid residues of the peptide substrate can fully interact. After demasking, hydrolysis of various peptide bonds occurs with the different hydrolysis rate constants. At this stage of proteolysis, the peptide bonds are hydrolyzed in accordance with their specificity, determined by the amino acid sequence of the polypeptide chain of the substrate.
Previously, we described methods for determining the demasking rate constants using fluorescence spectroscopy, which allows one to estimate the rate of substrate opening during proteolysis by the red shift of fluorescence accompanying proteolysis [13]. The use of this spectral method in addition to the traditional determination of the rate of hydrolysis of peptide bonds allowed us to analyze the interdependence of the demasking and hydrolysis processes within the framework of the two-step proteolysis model [4,13]. The demasking rate constants obtained by the fluorescence method were used to model the release of peptides during proteolysis of β-lactoglobulin (β-LG) by trypsin [9].
Modeling of peptide release during proteolysis of β-LG by trypsin was recently completed using a proteolysis model that takes into account both one- and two-stage demasking of peptide bonds without their secondary masking [9]. In addition to simplifications regarding the proteolysis mechanism, some simplifications of the calculation procedure were also made. It was assumed that there is no secondary masking [14], the size of the intermediate peptide fragments is limited by two enzyme-specific peptide bonds within them, which can be subjected to further hydrolysis. The formation of some minor peptide fragments, which are formed very slowly, was neglected [9].
It is believed that the hydrolysis of proteins depends on their spatial structure, at least at the initial stage of the process [1,15,16]. Traditionally, proteolysis of globular milk proteins, including β-LG, and structurally disordered caseins, most often β-casein (β-CN), is compared to reveal these dependencies [1,14,17].
The major protein in bovine whey is the small globular protein β-LG (55–60%) [18,19]. The monomeric β-LG is made up of 162 amino acid residues (∼18.3 kDa) and is stabilized by 2 disulfide bonds [19]. Its secondary structure was predicted mainly as β-sheet (50%) [20]. β-LG and its enzymatic hydrolysates have high functional and nutritional properties. The hydrolysates of β-LG contain a lot of biopeptides that can be used as biologically active additives in therapeutic nutrition and cosmetic [21].
Casein micelles are colloidal complexes of proteins and salts, and their main biological function is to transport sparingly soluble calcium phosphate in liquid form to infants [22]. They have four major protein species, termed αs1-, αs2-, β- and κ-caseins [22]. β-CN is the most hydrophobic one within the group of caseins, which constitutes about 45% of the casein of bovine milk [18]. It is a ∼24 kDa single polypeptide chain which consists of 209 amino acid residues [23]. Since β-CN does not contain disulfide bonds, it has no tertiary structure, but some regions of the polypeptide chain have secondary structure. β-CN has a hydrophobic C-terminus and a hydrophilic negative N-terminal region [24]. Caseins are known to be easily hydrolysed by proteases due to their conformational flexibility and the abundance of the enzyme-accessible peptide bonds. β-CN is of interest because of its nutritional importance and utility as a drug delivery vehicle [25,26]. The hydrolysates of β-CN, as well as β-LG, are rich in biologically active peptides [27,28].
Since demasking of peptide bonds predetermines their hydrolysis, it can be assumed that the demasking rate constants can be calculated from the distribution of the resulting peptide fragments. Previously, the sum of the concentrations of the peptides obtained during the hydrolysis of the analyzed peptide bond was used to determine the selectivity parameter of this bond in the hydrolyzed protein [29,30,31]. Herein, we expand this approach and present a method for estimating the demasking rate constants by analyzing changes in the sums of the concentrations of some peptides over time. Grouping of such peptides and calculation of the sum of their concentrations corresponds to the proposed model of proteolysis [9].
Computer search for bioactive peptides in enzymatic hydrolysates of proteins of various origins has recently become increasingly popular, requiring the development of reliable algorithms for in silico proteolysis [32,33,34]. Existing approaches are primarily aimed at predicting peptide fragments that can potentially be released during enzymatic hydrolysis of various protein substrates. Prediction of concentration dependences for peptide fragments and the degree of hydrolysis required to obtain target peptides was not performed. These aspects of in silico proteolysis are of particular interest to us, as the proteolysis model we are developing is capable of solving such problems.
A simpler method for determining demasking rate constants than using fluorescence spectroscopy is described here for a proteolysis model formulated earlier [9]. Previously, demasking rate constants were estimated from the fluorescence shift during proteolysis, but now this is done using experimental data on peptide release. As a proteolysis substrate, along with β-LG, studied earlier [9], β-CN is also considered. In addition to a detailed demonstration of the method for calculating kinetic curves, our goal is to compare the results predicted by the model with experiment for these two substrates with different protein structures.
2. Results
2.1. Proteolysis model
The same scheme of polypeptide chain cleavage was used here as in the previous work devoted to modeling the release of peptides during proteolysis [9]. The main principles of the model are presented in Figure 1. The hydrolysis of masked peptide bonds is preceded by their two-stage demasking, associated with conformational changes in the polypeptide chains and leading to the accessibility of peptide bonds for the enzyme. The illustration shows the peptide fragment ABC formed by the hydrolysis of the most specific peptide bonds at the beginning of proteolysis. It consists of three relatively short peptides A, B and C, which include only peptide bonds that are nonspecific for the enzyme. These peptides are linked by enzyme-specific peptide bonds A-B and B-C, indexed i and j, respectively.
The left side of Figure 1 shows that these bonds are not hydrolyzed until their demasking is complete. The right side of Figure 1 shows how these demasked bonds are hydrolyzed to form the corresponding peptide fragments.
Thus, Figure 1 shows the transformation of the peptide chain -ABC- into a partially demasked fragment -ABC at the first stage of demasking. Then, at the second stage of demasking, a completely demasked peptide fragment ABC is obtained. After demasking, the fragmentation of the ABC trimer occurs as a result of hydrolysis of the peptide bonds i and j with the formation of peptide fragments AB, BC, A, B, and C. The model determines that hydrolysis of the most specific bonds occurs at the beginning of proteolysis together with demasking, and hydrolysis of less specific bonds is possible only after the completion of demasking.
2.2. Application of the fragmentation scheme to the proteolysis of β-CN by trypsin
To specify the general scheme (Figure 1) for the proteolysis of β-CN by trypsin, we collected data that were previously published for this enzyme-substrate pair (Table 1). These parameters include the selectivity and initial rate of hydrolysis of the enzyme-specific peptide bonds Lys-X and Arg-X in β-CN. Both of these characteristics correlate fairly well with each other, and they allow ranking various peptide bonds by their rate of hydrolysis. From these data it follows that peptide bonds with indexes 99, 105, and 169 are the most rapidly hydrolyzed bonds, while bonds 1, 25, 32, 48, and 202 are the most slowly hydrolyzed bonds, the hydrolysis of which is neglected within the framework of the considered model.
The remaining bonds (28/29, 97, 107, 113, 176, 183) are considered initially masked, and the model must quantitatively determine how these peptide bonds are demasked and hydrolyzed. Thus, during proteolysis of β-CN by trypsin, the ABC fragments are f(1-99), f(106-169), and f(170-209), and the fragment f(100-105) is a rapidly forming peptide that does not contain enzyme-specific bonds.
Peptide bonds assigned to the group of the most slowly hydrolyzed bonds are considered non-hydrolyzed in our simplified approximation, therefore the concentrations of peptide fragments obtained during their hydrolysis are added to the concentrations of the peptides from which they originated. For example, the concentrations of fragments 184-202 and 202-209 are also added to the concentration of peptide 184-209, since bond 202 is considered the most slowly hydrolyzed bond (Table 1). The sum obtained in this way is considered the concentration of fragment 184-202.
In the previous analysis of β-LG proteolysis by trypsin, demasking of two ABC fragments occurred by the two-stage demasking mechanism, and of one trimer by the one-stage demasking mechanism [9]. Proteolysis of β-CN by trypsin is a multistep process, including association of intermediate peptides and formation of nanoparticles during proteolysis [35]. Due to the complexity of this process, we consider demasking all three trimers f(1-99), f(106-169) and f(170-209) using the two-stage demasking scheme.
2.3. Estimation of rate constants for demasking and hydrolysis
Like the selectivity parameter [29], was determined using a simple exponential equation for the most rapidly hydrolyzed peptide bond (Equation (9)).
In this work, a new approach to estimate the demasking rate constants was used, different from the fluorescence spectroscopy method [13]. This approach requires additional calculations using the dependences of the sum of concentrations of some peptide fragments on the hydrolysis time. To calculate the demasking rate constant kd the equation following from the material balance equations was used. This Equation (10) is given in Section 4 for the sum of the concentrations [ABC], [AB], and [A], corresponding to balance of A units. To track balance of C units, it is necessary to calculate the sum [ABC]+[BC]+[C]. As a result of fitting according to Equation (10), kd value was obtained for each of ABC trimer. The found values of kd then allowed us to determine the rate constants of hydrolysis of peptide bonds using the Equation (11) we had previously derived for the concentrations of peptide bonds [1,14].
A new approach to estimate the rate constants required to model the peptide release according to Scheme 1 was applied to the proteolysis of β-CN by trypsin (Table 2). For comparison with the previous result on peptide release [9], this method was also applied to the proteolysis of β-LG by trypsin (Table 2).
2.4. Simulation of peptide release during proteolysis by trypsin
Figure 2a shows an example of the calculated dependences of peptide concentrations on the hydrolysis time for the intermediate and final peptide fragments. The intermediate peptide products (ABC, AB, and BC) are first formed and then disappear due to the hydrolysis of internal enzyme-specific peptide bonds (Figure 2a). The final products (A, B, and C) only accumulate, since they do not contain internal enzyme-specific peptide bonds. Demasking of the peptide bonds leads to the fact that the concentrations of proteolysis products may increase not immediately with the onset of proteolysis, but after a lag phase [1].
The concentrations of peptide fragments can also be represented as functions of the degree of hydrolysis achieved by a given time (Figure 2b), since the dependence of the degree of hydrolysis on time is known. Representation of concentration curves as functions of the degree of hydrolysis is more convenient for determining the mechanisms by which peptides are released during proteolysis. The transition from time to the degree of hydrolysis does not change the shape of the curves for intermediate products, but changes the dependences for the final peptides. For them, the curves for rapidly released peptides remain convex, and the curves for slowly released peptides become concave.
The parameter dr, the degree of hydrolysis at which the major part of a given peptide is released, was calculated for each of the intermediate products using the following simple equation:
where C(di) are the concentrations of peptide fragments determined at six hydrolysis degrees di (0, 1.5, 3, 4.5, 6, 7.9% for β-LG and 0, 1, 2, 3, 4, 4.3 for β-CN). Thus, dr is defined as the weighted average of 6 degrees of hydrolysis, and the relative concentrations of intermediate peptides corresponding to these degrees of hydrolysis are the weighting factors.
For the final peptides, the parameter n, which characterizes their concentration dependence on the degree of hydrolysis C(di), was calculated using the following equation:
where a is a constant factor, dmax is the maximal hydrolysis degree, and n is the exponent of the power function. Thus, we compared the kinetic curves for the final products simply by comparing the parameter n, which reflects the curvature of the curves.
Examples of the calculated and experimental concentration dependences for intermediate (Figure 3a) and final (Figure 3b) peptide fragments are given. Figure 3a shows the release of the intermediate trimeric peptide ABC during proteolysis and the subsequent formation of the intermediate dimeric peptide AB from it. Theoretically, one would expect that the dr for the ABC fragment is less than that for the AB or BC fragment. Figure 3b shows the concentration curves for the final peptides. For a rapidly forming final peptides the curve is convex and n is less than 1, while for a slowly forming final peptides the curve is concave and n should be greater than 1.
Table 3 presents the parameters of the concentration curves for all studied peptide fragments, namely dr for the intermediate peptides and n for the final peptides. The parameters dr and n were calculated using Equations (1, 2) from the simulated curves obtained using the rate constants from Table 2 and the experimental concentration curves. The published peptide concentrations were for tryptic proteolysis of β-LG and β-CN, and the maximal hydrolysis degree (dmax) was 7.9 and 4.3% for β-LG and β-CN, respectively [7]. The concentrations of the peptide fragments were calculated using Equations (3-8) for the same degrees of hydrolysis as in the experiment.
A comparison of the n parameter for the simulated and experimental concentration curves obtained for both β-LG and β-CN proteolysis is shown in Figure 4a. The coefficient of proportionality between the experimental and calculated n values was 0.999 ± 0.038 (r2=0.9998) for β-LG (Figure 4a) with the expected coefficient of 1. Thus, the agreement between the simulation results and the experiment was very good when using the n parameter for comparison. The proportionality coefficient for the dr values was 0.68 ± 0.07 (r2=0.87), indicating an underestimation of this parameter in the simulation for intermediate peptide fragments with high dr.
For the parameter n, the proportionality coefficient was 0.77±0.10 (r2=0.892) with previous quantification of rate constants [9], while in this study it is 0.96±0.04 (r2=0.989) for all points in Figure 4a. Thus, the use of a new method for estimating the rate constants of demasking and hydrolysis has led to a more accurate prediction of the release of proteolysis products using the same proteolysis model that we proposed earlier [9]. This confirms the correctness of the main ideas of the model based on taking into account the processes of the peptide bond demasking before their hydrolysis and the difference in hydrolysis rate for the various peptide bonds.
Despite the differences in the protein structure of the substrates, calculations of the concentration dependences and parameters of dr and n were performed using the same algorithm and yielded results consistent with the experiment.
2.5. Prospects for in silico proteolysis
The possibility of predicting the hydrolysis constants ki for various protein substrates based on the analysis of their amino acid sequences is illustrated by Figure 4b. The total number of charged amino acid residues in the positions P2 and P-2 was chosen as the determining parameter. It was previously shown that the presence of charged residues (E, D, R, K) in these positions significantly reduces the selectivity parameters for proteolysis with trypsin [7]. Figure 4b shows that lg(ki) decreases with the number of charged groups in the positions P2 and P-2. To construct more reliable correlation equations, a larger volume of experimental data obtained for a number of protein substrates is required. It is also possible to take into account the amino acid residues in the positions P1 and P-1, as was done to predict the Michaelis parameters for the hydrolysis of peptide bonds in low-molecular substrates. Within the framework of our approach, the structure-reactivity correlation equations must be sought specifically for the hydrolysis rate constants ki, since these parameters reflect the hydrolysis step of proteolysis. According to the model, all demasked regions of the polypeptide chain and their corresponding ki, regardless of their affiliation with specific protein substrates, must be considered together to find the correlation equation corresponding to a given proteolytic enzyme. Therefore, we plotted the data for β-LG and β-CN on a single graph (Fig. 4b).
The demasking rate constant =0.46 for β-LG, determined by the opening of protein globule, is expectedly lower than that of β-CN (=3.5), which is a disordered coil of the polypeptide chain with more accessible peptide bonds. Consequently, the ratio of the demasking rate constants of β-CN and β-LG determined here is 7.6 under the proteolysis conditions published in [7]. It makes sense to compare proteolysis processes carried out at the same concentrations of substrate and enzyme, if the temperature and other environmental conditions remain unchanged. Previously, the ratio of the demasking rate constants during proteolysis of these two substrates by trypsin was determined from the fluorescence shift [13], and the value kII(β-CN)/kII(β-LG) = 0.0244/0.0189/0.1 = 12.9 was obtained [13]. The proteolysis conditions in that study differed from those used in the present calculation, which may explain the difference in the estimates of this ratio. The secondary demasking rate constants kd are apparently more complexly related to the structure of the polypeptide fragments. For β-LG proteolysis, the lowest kd = 0.32 was observed for the f(76-100/101) fragment, which participates in the formation of the core resistant to trypsin hydrolysis [36]. For β-CN proteolysis, the lowest kd = 1 was determined for the central part and the hydrophobic C-terminus of β-CN f(106-209). A significant contribution of the hydrophobic C-terminal fragment to the demasking process was previously noted, explaining the formation of C-terminal peptides of β-CN with a lag [4].
The dependence of the model parameters on enzyme and substrate concentrations is essential for the comparison and correct interpretation of experiments performed under various kinetic conditions. We have demonstrated how the contribution of secondary masking increases during proteolysis and how the concentration dependences change with decreasing enzyme concentration at a constant substrate concentration [14]. However, experimental data are not yet sufficient to accurately determine the dependence of the model parameters on the conditions of proteolysis, mainly on the substrate concentration.
3. Discussion
The algorithm for calculating the release of peptides during proteolysis that we used is a new method that considers the need to demask peptide bonds before their hydrolysis [1,9]. The phenomenon of peptide bond demasking was first discovered based on the analysis of non-monotonic changes in the rate of hydrolysis of peptide bonds during enzymatic hydrolysis of milk protein substrates [4]. Then, the kinetics of demasking was studied by monitoring the fluorescence of tryptophan residues in the comparative analysis of β-LG and β-CN proteolysis by trypsin [13,14,17]. The importance of considering demasking processes in studying proteolysis was shown by other analytical and physicochemical methods [37,38].
It was shown previously that the precise determination of the demasking parameters may be even more important than the precise determination of the rate constants of peptide bond hydrolysis [9]. In the previous work, the demasking rate constants were determined using the spectral method, which itself could yield additional errors. In order to exclude possible inaccuracies in determining the demasking rate constants, in this work the demasking parameters were determined precisely using the published experimental data [7] on the distribution of peptide fragments during proteolysis. Here, more precise values of kd were used for each of the ABC trimer under consideration, but the calculation algorithm of fragment concentrations was the same. The results of modeling the release of peptides gave better agreement with the experiment than in the previous work [9].
The focus here is on the calculation algorithm. It turned out that the simulation results using the same equations for the proteolysis of β-LG and β-CN proteins provide good agreement with experiment. Thus, the proposed calculation algorithm with more precise parameters correctly accounts for the key features of proteolysis even for structurally different protein substrates.
According to AFM, FTIR spectroscopy and static light scattering data, proteolysis of β-CN by trypsin is accompanied not only by degradation of the initial β-CN micelles, but also by parallel formation of new nanoparticles, which then degrade to form peptide proteolysis products [1,35,39]. When viewed at the macromolecular level, there is a parallel degradation and association of intermediate polypeptide products with degradation gradually becoming predominant as the process progresses. New nanoparticles formed during proteolysis are denser than the initial micelles, and the content of secondary protein α-, β-structures in them does not decrease during proteolysis, but is comparable to that in the initial β-CN micelles. In this regard, during proteolysis at low enzyme concentrations, the content of secondary protein structures changes non-monotonically. It first decreases due to hydrolysis of the original micelles, then increases due to the formation of these new nanoparticles, and finally slowly decreases as they are hydrolyzed [35,39]. The kinetics of degradation of β-CN micelles by trypsin was described by analytical functions in the case of modeling the proteolytic system using linear differential equations [35]. This process was also described by numerical modeling methods using nonlinear differential equations [1]. In this case, the original micelles, nanoparticles of different sizes and their aggregates, as well as the change in the concentration of the active enzyme during proteolysis were considered [1].
The macromolecular description of β-CN proteolysis by trypsin [35,39] and the model considered here represent sequential three-stage processes. A fairly rapid hydrolysis of micelles is the first stage in the macromolecular model and it is initiated by the hydrolysis of the most specific peptide bonds [35]. Hydrolysis of the most specific bonds also initiates the process of the first stage of demasking with rate constant . The ratio of the rate constant of micelle hydrolysis to the rate constant of peptide product formation in the macromolecular model was 33:1 [35]. A close value of 35 was obtained for the ratio of to kj for the peptide bonds with j=107 and 113.
Relatively small β-CN and β-LG are used as model reactions of proteolysis with various proteases, allowing the kinetic features of proteolysis to be studied [1]. The number of peptide fragments is not very large (several dozen) and they are easily identified and quantified by HPLC-MS [40,41,42,43,44,45]. Among these studies, those in which the concentrations of peptide fragments are determined at several time points during proteolysis are especially important. Such data are suitable for verifying proteolysis models and determining the numerical values of kinetic parameters. In particular, such studies have been carried out for trypsin proteolysis of β-CN [7] and β-LG [7,8,10]. Accurate determinations of the concentrations of individual peptides made it possible to determine the change in the concentrations of peptide bonds over the course of proteolysis and then determine the selectivity parameters for various peptide bonds [7]. In this study, summing the concentrations of individual peptides ABC, AB, and A was used to determine the demasking rate constants.
Although spectroscopic methods were not used in this work, these methods have great potential for the monitoring of protein opening during proteolysis and hence for the monitoring of demasking. The possibility of the monitoring of protein structure changes in course of proteolysis was demonstrated using fluorescence [13], infrared (FTIR) [35], ultrasound [46,47,48] and other physicochemical methods. Undoubtedly, further development of these methods is of great importance for successful modeling of proteolysis.
In practical-oriented proteolysis models, the peptide bonds and their hydrolysis rate constants are assumed to be identical, and the main model variables are the degree of peptide bond hydrolysis and the total hydrolysis rate [49,50,51,52,53,54,55]. Such models can be called total proteolysis models, since they do not consider the hydrolysis processes of different hydrolysis sites (different peptide bonds). The model we are developing is more accurate because it considers the differences in the hydrolysis of various peptide bonds in proteins, as well as the limited accessibility of the enzyme to some temporarily masked regions of the polypeptide chain [1,9]. To optimize the production of protein hydrolysate with the required degree of hydrolysis, it is necessary to use total proteolysis models, while to optimize the yield of target biopeptides, it is necessary to use our approach.
Although the relationship between the structural features of β-LG and β-CN and the demasking parameters is demonstrated in this study, it is still difficult to formulate specific correlation dependences that could be used for the quantitative prediction. Since in silico proteolysis of various protein substrates is impossible without successful prediction of model parameters, a hybrid approach can be proposed in which some parameters are determined experimentally. Thus, in the calculations presented here, the demasking parameter dr was determined by analyzing experimental data with Equation (10), and the identification of rapidly hydrolyzed bonds was also based on the experimental data (Table 1). Apparently, the success of hybrid approach is possible if the number of experimentally determined parameters is minimized. We hope that the use of our approach will bridge the gap between the experimental study of specific proteolytic processes and the development of a general model of proteolysis.
4. Materials and Methods
4.1. Quantitative modelling of proteolysis
The solutions of the differential equations corresponding to the proteolysis scheme (Fig. 1) were found in the form of analytical functions of time t, min. The relative concentrations C(t) of the peptide fragments ABC, AB, BC, A, B, and C are:
4.2. Estimation of the rate constants
The parameter was the largest value of the rate constant for hydrolysis of various peptide bonds corresponding to the following simple equation for dependence of the peptide bond concentration on the hydrolysis time:
By summing the concentrations of components ABC, AB and A from Equations (3), (4) and (6), we obtained the following equation for the sum [ABC]+[AB}+[A]:
A similar equation was obtained for the sum [ABC]+[BC}+[C]. The dependences of the sums of the components [ABC]+[AB}+[A] and [ABC]+[BC}+[C] on the hydrolysis time were calculated from published data [7] and used to estimate the parameter kd with Equation (10).
The parameters and kd were then used to calculate the rate constants for the hydrolysis of different peptide bonds using the following equation expressing the dependence of the peptide bond concentration on the hydrolysis time [1,14]:
The experimental concentrations of peptide fragments taken from Table S-4 [7] were divided by a factor of 50 to obtain the relative concentrations of the peptide fragments that were used in the calculations. The values of model parameters dr and n were independent of this factor.
Author Contributions
Not applicable.
Funding
This research was supported by Ministry of Science and Higher Education of the Russian Federation (Contract/agreement No. 075-00276-25-00).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Vorob’ev, M.M. Towards a quantitative description of proteolysis: Contribution of demasking and hydrolysis steps to proteolysis kinetics of milk proteins. Foods 2025, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Linderstrom-Lang, K.U. Lane Medical Lectures. Volume 6, Stanford: Stanford University Press, 1952; pp. 53–72.
- Adler-Nissen, J. Enzymatic Hydrolysis of Food Proteins; Elsevier Applied Science Publishers: London, UK, 1986. [Google Scholar]
- Vorob’ev, M.M. Kinetics of peptide bond demasking in enzymatic hydrolysis of casein substrates. J. Mol. Catal. B 2009, 58, 146–152. [Google Scholar] [CrossRef]
- Muñoz-Tamayo, R.; De Groot, J.; Wierenga, P.A.; Gruppen, H.; Zwietering, M.H.; Sijtsma, L. Modeling peptide formation during the hydrolysis of β-casein by Lactococcus lactis. Process Biochem. 2012, 47, 83–93. [Google Scholar] [CrossRef]
- Deng, Y.; Gruppen, H.; Wierenga, P.A. Comparison of protein hydrolysis catalyzed by bovine, porcine, and human trypsins. J. Agric. Food Chem. 2018, 66, 4219–4232. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; van der Veer, F.; Sforza, S.; Gruppen, H.; Wierenga, P.A. Towards predicting protein hydrolysis by bovine trypsin. Process Biochem. 2018, 65, 81–92. [Google Scholar] [CrossRef]
- Leeb, E.; Stefan, T.; Letzel, T.; Hinrichs, J.; Kulozik, U. Tryptic hydrolysis of b-lactoglobulin: A generic approach to describe the hydrolysis kinetic and release of peptides. Int. Dairy J. 2020, 105, 104666. [Google Scholar] [CrossRef]
- Vorob’ev, M.M. Modeling of the peptide release during proteolysis of β-lactoglobulin by trypsin with consideration of peptide bond demasking. Int. J. Mol. Sci. 2023, 24, 11929. [Google Scholar] [CrossRef]
- Fernandez, A.; Riera, F. b-Lactoglobulin tryptic digestion: A model approach for peptide release. Biochem. Eng. J. 2013, 70, 88–96. [Google Scholar] [CrossRef]
- Beaubier, S.; Framboisier, X.; Fournier, F.; Galet, O.; Kapel, R. A new approach for modelling and optimizing batch enzymatic proteolysis. Chem. Eng. J. 2021, 405, 126871. [Google Scholar] [CrossRef]
- Ozols, M.; Eckersley, A.; Platt, C.I.; Stewart-McGuinness, C.; Hibbert, S.A.; Revote, J.; Li, F.; Griffiths, C.E.M.; Watson, R.E.B.; Song, J.; et al. Predicting proteolysis in complex proteomes using deep learning. Int. J. Mol. Sci. 2021, 22, 3071. [Google Scholar] [CrossRef]
- Vorob’ev, M.M.; Vogel, V.; Güler, G.; Mäntele, W. Monitoring of demasking of peptide bonds during proteolysis by analysis of the apparent spectral shift of intrinsic protein fluorescence. Food Biophys. 2011, 6, 519–526. [Google Scholar] [CrossRef]
- Vorob’ev, M.M. Modeling of proteolysis of β-lactoglobulin and β-casein by trypsin with consideration of secondary masking of intermediate polypeptides. Int. J. Mol. Sci. 2022, 23, 8089. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; de Laureto, P.P.; Spolaore, B.; Frare, E.; Picotti, P.; Zambonin, M. Probing protein structure by limited proteolysis, Acta Biochimica Polonica 2004, 51, 299–321. [CrossRef]
- Hubbard, S.J. The structural aspects of limited proteolysis of native proteins, Biochim. Biophys. Acta 1998, 1382, 191–206. [Google Scholar] [CrossRef]
- Vorob’ev, M.M. Proteolysis of β-lactoglobulin by trypsin: Simulation by two-step model and experimental verification by intrinsic tryptophan fluorescence. Symmetry 2019, 11, 153. [Google Scholar] [CrossRef]
- Farrell, H.M.; Jimenez-Flores, R.; Bleck, G.T.; Brown, E.M.; Butler, J.E.; Creamer, L.K.; Hicks, C.L.; Hollar, C.M.; Ng-Kwai-Hang, O.F.; Swaisgood, T.H.E. Nomenclature of the proteins of cows’ milk, 6th rev. J. Dairy Sci. 2004, 87, 1641–1674. [Google Scholar] [CrossRef]
- Hambling, S.G.; McAlpine, A.S.; Sawyer, L. β-Lactoglobulin. In: P.F. Fox (Ed.), Adv. Dairy Chem. Proteins, vol. 1, 1992, pp. 141–190, Essex: Elsevier.
- Creamer, L.K.; Parry, D.A.; Malcolm, G.N. Secondary structure of bovine beta-lactoglobulin B. Arch. Biochem. Biophys. 1983, 227, 98–105. [Google Scholar] [CrossRef]
- Rama, G.R.; Saraiva Macedo Timmers, L.F.; Volken de Souza, C.F. In silico strategies to predict anti-aging features of whey peptides. Mol. Biotechnol. 2024, 66, 2426–2440. [Google Scholar] [CrossRef]
- Fox, P.F.; McSweeney, P.L.H. (Eds.), Advanced dairy chemistry—volume 1: proteins (parts A and B), Kluwer Academic/Plenum Publishers: New York, USA, 2003.
- Grosclaude, F.; Mahé, M.F.; Ribadeaudumas, B. Primary structure of alpha casein and of bovine beta casein Eur. J. Biochem. 1973, 40, 323–324. [Google Scholar] [CrossRef]
- Holt, C. Structure and stability of bovine casein micelles. Adv. Protein Chem. 1992, 43, 63–151. [Google Scholar] [CrossRef]
- Shapira, A.; Assaraf, Y.G.; Livney, Y.D. Beta-casein nanovehicles for oral delivery of chemotherapeutic drugs, Nanomedicine: NBM 2010, 6, 119–126. [CrossRef]
- McClements, D.J.; Decker, E.A.; Park, Y.; Weiss, J. Structural design principles for delivery of bioactive components in nutraceuticals and functional foods, Crit. Rev. Food Sci. Nutr. 2009, 49, 577–606. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; FitzGerald, R.J. Enhancing bioactive peptide release and identification using targeted enzymatic hydrolysis of milk proteins. Anal. Bioanal. Chem. 2018, 410, 3407–3423. [Google Scholar] [CrossRef]
- Nielsen, S.D.H.; Liang, N.; Rathish, H.; Kim, B.J.; Lueangsakulthai, J.; Koh, J.; Qu, Y.; Schulz, H.J.; Dallas, D.C. Bioactive milk peptides: an updated comprehensive overview and database, Crit. Rev. Food Sci. Nutr. 2023, 1–20. [Google Scholar] [CrossRef]
- Butre, C.I.; Sforza, S.; Gruppen, H.; Wierenga, P.A. Introducing enzyme selectivity: a quantitative parameter to describe enzymatic protein hydrolysis. Anal. Bioanal. Chem. 2014, 406, 5827–5841. [Google Scholar] [CrossRef] [PubMed]
- Butré, C.I. Introducing enzyme selectivity as a quantitative parameter to describe the effects of substrate concentration on protein hydrolysis. PhD thesis, Wageningen University, 2014. ISBN 978-94-6257-023-8.
- Butre, C.I.; Sforza, S.; Wierenga, P.A.; Gruppen, H. Determination of the influence of the pH of hydrolysis on enzyme selectivity of Bacillus licheniformis protease towards whey protein isolate. Int. Dairy J. 2015, 44, 44–53. [Google Scholar] [CrossRef]
- Turalić, A.; Hegedüs, Z.; Altieri, F.; Martinek, T.A.; Đeđibegović, J. Discordance between in silico and in vitro results on proteolytic stability of milk protein-derived DPP-4 inhibitory peptides. Eur. Food Res. Technol. 2025, 251, 3199–3214. [Google Scholar] [CrossRef]
- Gomez, H.L.R.; Peralta, J.P.; Tejano, L.A.; Chang, Y.-W. In silico and in vitro assessment of Portuguese Oyster (Crassostrea angulata) proteins as precursor of bioactive peptides. Int. J. Mol. Sci. 2019, 20, 5191. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Pino, F.; Idowu, C.; Malfroy, H.; Rueda, D.; Lester, H. In silico analyses as a tool for regulatory assessment of protein digestibility: Where are we? Computational Toxicology 2025, 35, 100372. [Google Scholar] [CrossRef]
- Vorob'ev, M.M.; Açıkgöz, B.D.; Güler, G.; Golovanov, A.V.; Sinitsyna, O.V. Proteolysis of micellar β-casein by trypsin: secondary structure characterization and kinetic modeling at different enzyme concentrations. Int. J. Mol. Sci., 2023, 24, 3874. [Google Scholar] [CrossRef]
- Cheison, S.C.; Leeb, E.; Letzel, T.; Kulozik, U. Influence of buffer type and concentration on the peptide composition of trypsin hydrolysates of β-lactoglobulin. Food Chem. 2011, 125, 121–127. [Google Scholar] [CrossRef]
- Rivera-Burgos, D.; Regnier, F.E. Disparities between immobilized enzyme and solution based digestion of transferrin with trypsin. J. Sep. Sci. 2013, 36, 454–460. [Google Scholar] [CrossRef]
- Melikishvili, S.; Dizon, M.; Hianik, T. Application of high-resolution ultrasonic spectroscopy for real-time monitoring of trypsin activity in β-casein solution. Food Chem. 2021, 337, 127759. [Google Scholar] [CrossRef]
- Vorob'ev, M.M.; Sinitsyna, O.V. Degradation and assembly of β-casein micelles during proteolysis by trypsin. Int. Dairy J. 2020, 104, 104652. [Google Scholar] [CrossRef]
- Chobert, J.-M.; Briand, L.; Tran, V.; Haertle, T. How the substitution of K188 of trypsin binding site by aromatic amino acids can influence the processing of b-casein. Biochem. Biophys. Res. Commun. 1998, 246, 847–858. [Google Scholar] [CrossRef]
- Vreeke, G.J.C.; Vincken, J.-P.; Wierenga, P.A. The path of proteolysis by bovine chymotrypsin. Food Res. Int. 2023, 165, 112485. [Google Scholar] [CrossRef]
- Mamone, G.; Picariello, G.; Caira, S.; Addeo, F. , Ferranti, P. Analysis of food proteins and peptides by mass spectrometry-based techniques. J. Chrom. A 2009, 1216, 7130–7142. [Google Scholar] [CrossRef]
- Vreeke, G.J.C.; Lubbers, W.; Vincken, J.-P.; Wierenga, P.A. A method to identify and quantify the complete peptide composition in protein hydrolysates. Anal. Chim. Acta 2022, 1201, 339616. [Google Scholar] [CrossRef]
- Butré, C.I.; Buhler, S.; Sforza, S.; Gruppen, H.; Wierenga, P.A. Spontaneous, non-enzymatic breakdown of peptides during enzymatic protein hydrolysis, Biochim. Biophys. Acta - Proteins Proteomics 2015, 1854, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Vreeke, G.J.C.; Vincken, J.-P.; Wierenga, P.A. Quantitative peptide release kinetics to describe the effect of pH on pepsin preference. Proc. Biochem. 2023, 351–362. [Google Scholar] [CrossRef]
- Buckin, V.; Altas, M.C. Ultrasonic monitoring of biocatalysis in solutions and complex dispersions. Catalysts 2017, 7, 336. [Google Scholar] [CrossRef]
- Dizon, M.; Buckin, V. Ultrasonic monitoring of enzymatic hydrolysis of proteins. 1. Effects of ionization. Food Hydrocol. 2023, 144, 108866. [Google Scholar] [CrossRef]
- Dizon, M.; Buckin, V. Ultrasonic monitoring of enzymatic hydrolysis of proteins. 2. Relaxation effects. Food Hydrocol. 2025, 158, 110221. [Google Scholar] [CrossRef]
- Sopade, P.A. Computational characteristics of kinetic models for in vitro protein digestion: A review. J. Food Eng. 2024, 360, 111690. [Google Scholar] [CrossRef]
- Le Feunteun, S.; et al. Mathematical modelling of food hydrolysis during in vitro digestion: From single nutrient to complex foods in static and dynamic conditions. Trends Food Sci. Technol. 2021, 116, 870–883. [Google Scholar] [CrossRef]
- Margot, A.; Flaschel, E.; Renken, A. Empirical kinetic models for tryptic whey-protein hydrolysis. Process Biochem. 1997, 32, 217–223. [Google Scholar] [CrossRef]
- Martinez-Araiza, G.; Castano-Tostado, E.; Amaya-Llano, S.L.; Regalado-Gonzalez, C.; Martinez-Vera, C.; Ozimek, L. Modeling of enzymatic hydrolysis of whey proteins. Food Bioprocess Technol. 2012, 5, 2596–2601. [Google Scholar] [CrossRef]
- Valencia, P.; Pinto, M.; Almonacid, S. Identification of the key mechanisms involved in the hydrolysis of fish protein by Alcalase. Process Biochem. 2014, 49, 258–264. [Google Scholar] [CrossRef]
- Valencia, P.; Espinoza, K.; Astudillo-Castro, C.; Salazar, F. Modeling tool for studying the influence of operating conditions on the enzymatic hydrolysis of milk proteins. Foods 2022, 11, 4080. [Google Scholar] [CrossRef] [PubMed]
- Beaubier, S.; Framboisier, X.; Fournier, F.; Galet, O.; Kapel, R. A new approach for modelling and optimizing batch enzymatic proteolysis. Chem. Eng. J. 2021, 405, 126871. [Google Scholar] [CrossRef]
Figure 1.
General scheme of proteolysis, including two stages of demasking and subsequent hydrolysis of the demasked peptide bonds.
Figure 1.
General scheme of proteolysis, including two stages of demasking and subsequent hydrolysis of the demasked peptide bonds.
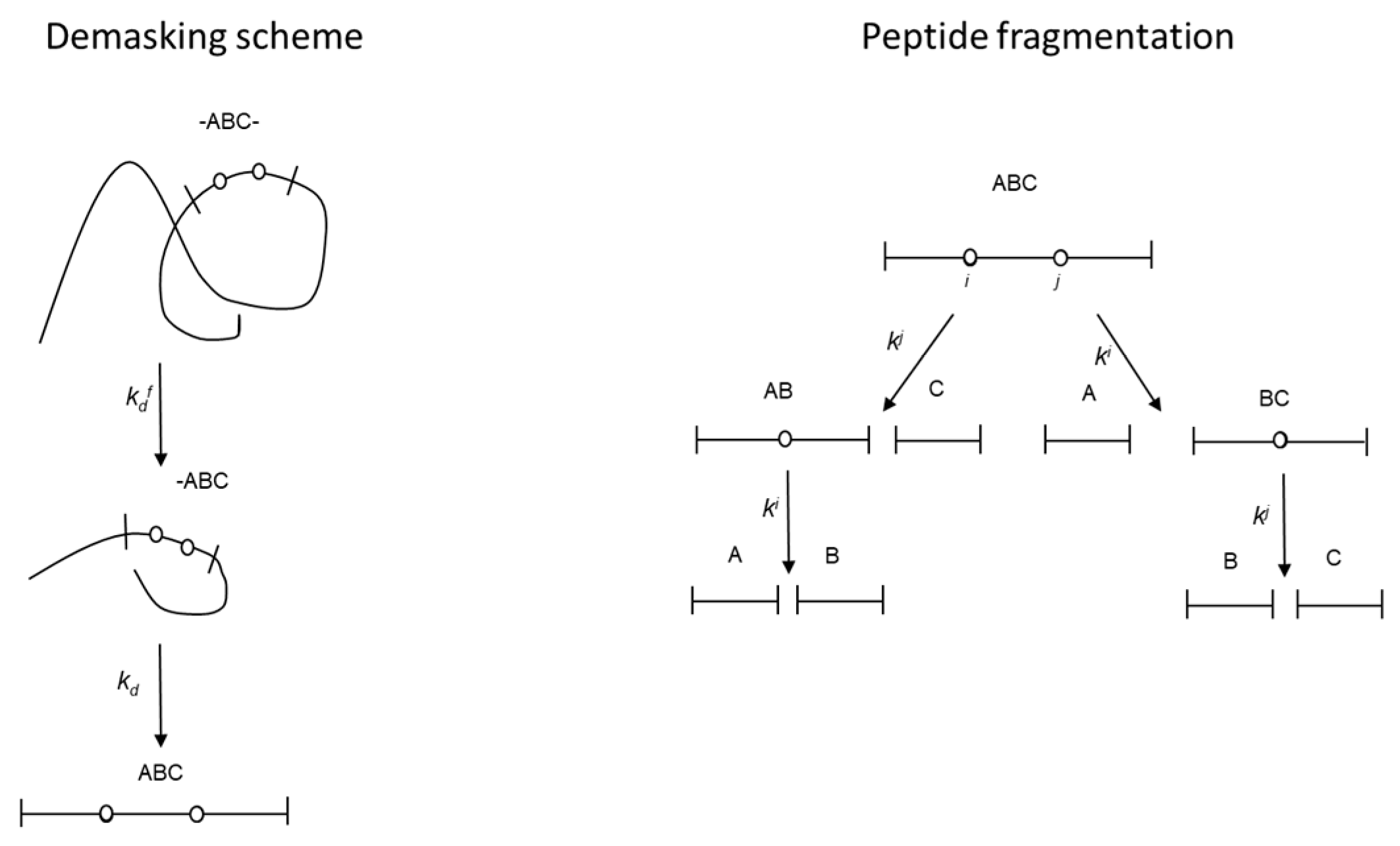
Figure 2.
Simulation of peptide release during tryptic hydrolysis of β-CN: (a) calculated dependences of peptide concentrations on the hydrolysis time for the intermediate peptides f(1-99) (■) and f(28/29-99) (▲), and for the final products of proteolysis f(170-176) (●) and f(106-107) (○); (b) calculated dependences of peptide concentrations on the degree of hydrolysis for the same peptide fragments.
Figure 2.
Simulation of peptide release during tryptic hydrolysis of β-CN: (a) calculated dependences of peptide concentrations on the hydrolysis time for the intermediate peptides f(1-99) (■) and f(28/29-99) (▲), and for the final products of proteolysis f(170-176) (●) and f(106-107) (○); (b) calculated dependences of peptide concentrations on the degree of hydrolysis for the same peptide fragments.
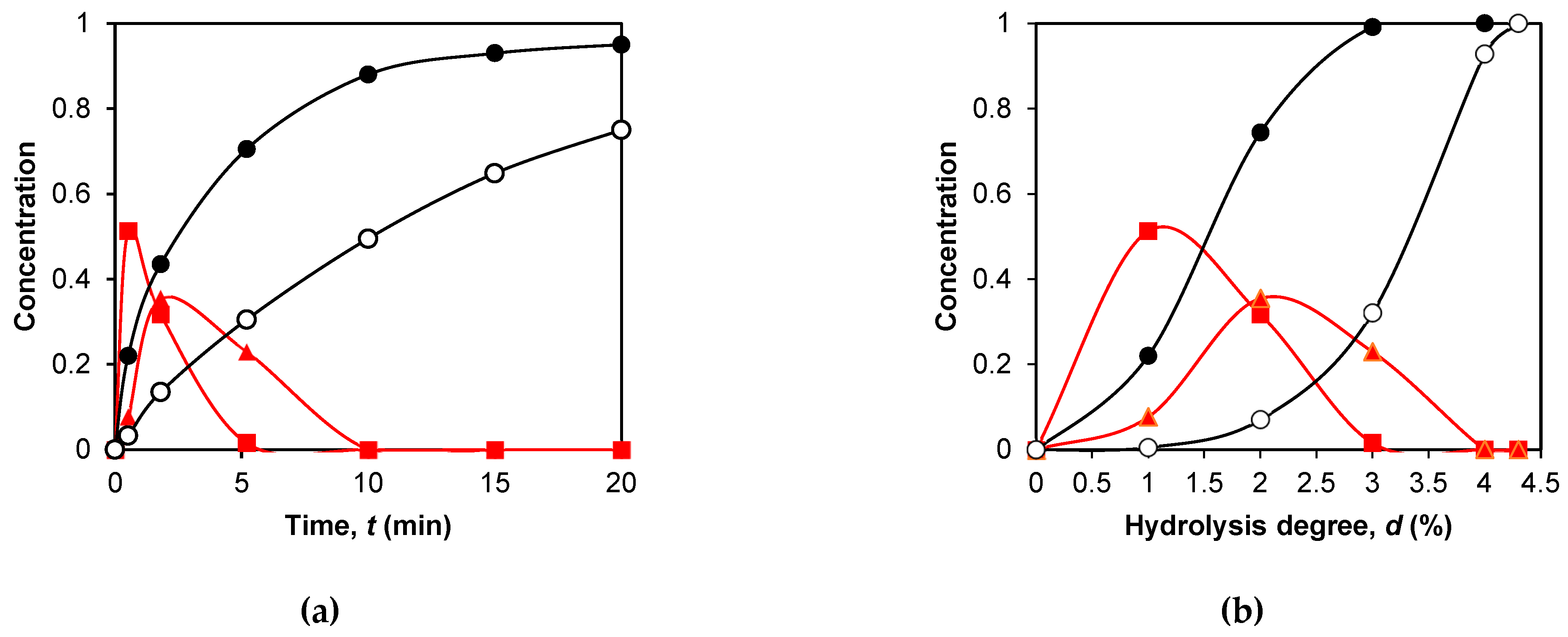
Figure 3.
Comparison of the calculated release of β-CN peptides with the experimental one: (a) calculated (■) and experimental (●) curves for peptide fragment f(106-169), ABC. Calculated (□) and experimental (○) curves for peptide fragment f(106-113), AB; (b): calculated (■) and experimental (□) concentration dependences for peptide f(170-176), A. Calculated (○) and experimental (●) concentration dependences for peptide f(114-169), C. Solid lines correspond to Equation (2) with n=0.55 for peptide f(170-176) (red) and n=2.98 for peptide f(114-169) (black).
Figure 3.
Comparison of the calculated release of β-CN peptides with the experimental one: (a) calculated (■) and experimental (●) curves for peptide fragment f(106-169), ABC. Calculated (□) and experimental (○) curves for peptide fragment f(106-113), AB; (b): calculated (■) and experimental (□) concentration dependences for peptide f(170-176), A. Calculated (○) and experimental (●) concentration dependences for peptide f(114-169), C. Solid lines correspond to Equation (2) with n=0.55 for peptide f(170-176) (red) and n=2.98 for peptide f(114-169) (black).
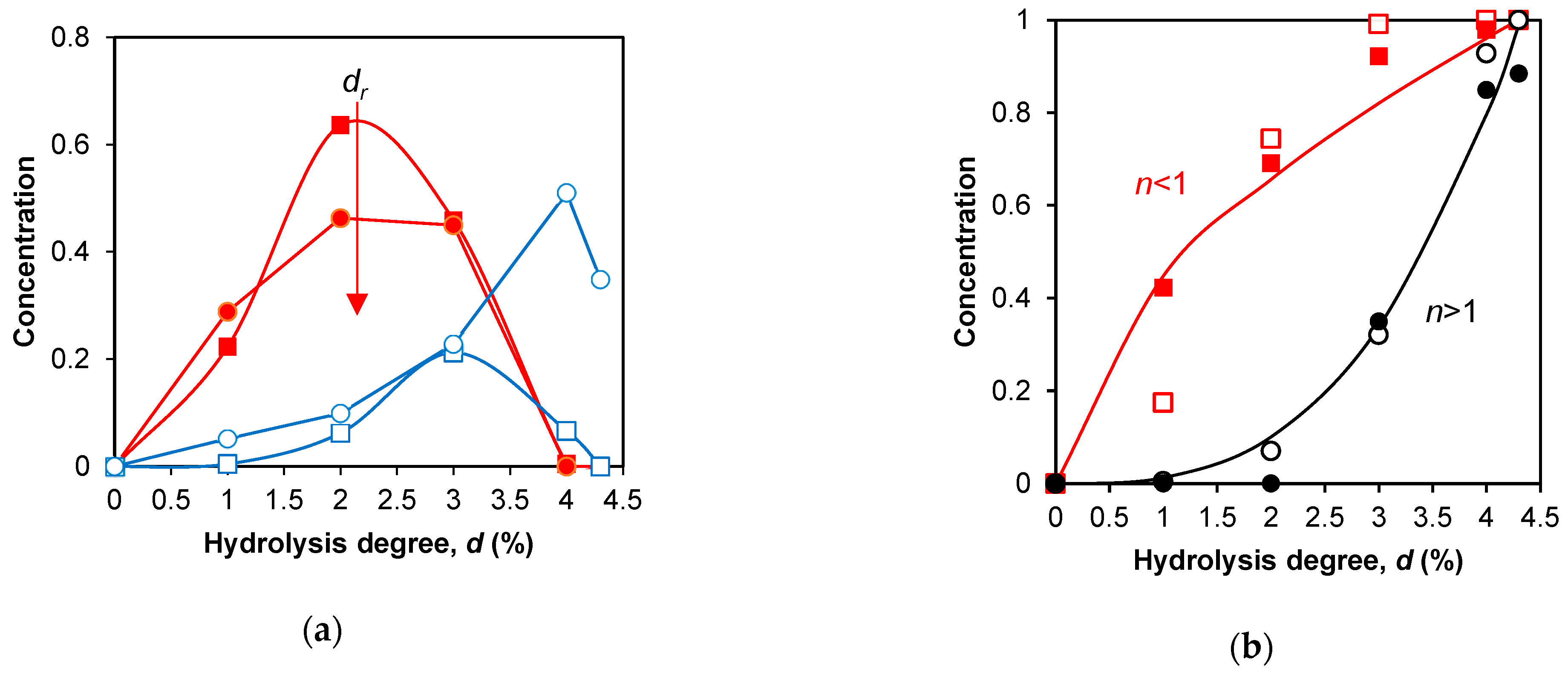
Figure 4.
Estimation of the model parameters: (a) correlation between experimental and calculated parameters n for the final products of β-LG (●) and β-CN (■). The straight line shows the expected proportionality coefficient of 1; (b) Estimation of the hydrolysis rate constants ki based on the number of charged amino acid residues in positions P2 and P-2 for peptide bonds in β-LG 14 (●), 40 (▲), 83 (■), 91 (○), 124 (∆), 135 (□) and in β-CN 28/29 (●), 97 (▲), 107 (■), 113 (○), 176 (∆),183 (□) . A linear correlation between lg(ki) and number of polar groups with slope of -0.76±0.18 (r2=0.632) is shown.
Figure 4.
Estimation of the model parameters: (a) correlation between experimental and calculated parameters n for the final products of β-LG (●) and β-CN (■). The straight line shows the expected proportionality coefficient of 1; (b) Estimation of the hydrolysis rate constants ki based on the number of charged amino acid residues in positions P2 and P-2 for peptide bonds in β-LG 14 (●), 40 (▲), 83 (■), 91 (○), 124 (∆), 135 (□) and in β-CN 28/29 (●), 97 (▲), 107 (■), 113 (○), 176 (∆),183 (□) . A linear correlation between lg(ki) and number of polar groups with slope of -0.76±0.18 (r2=0.632) is shown.
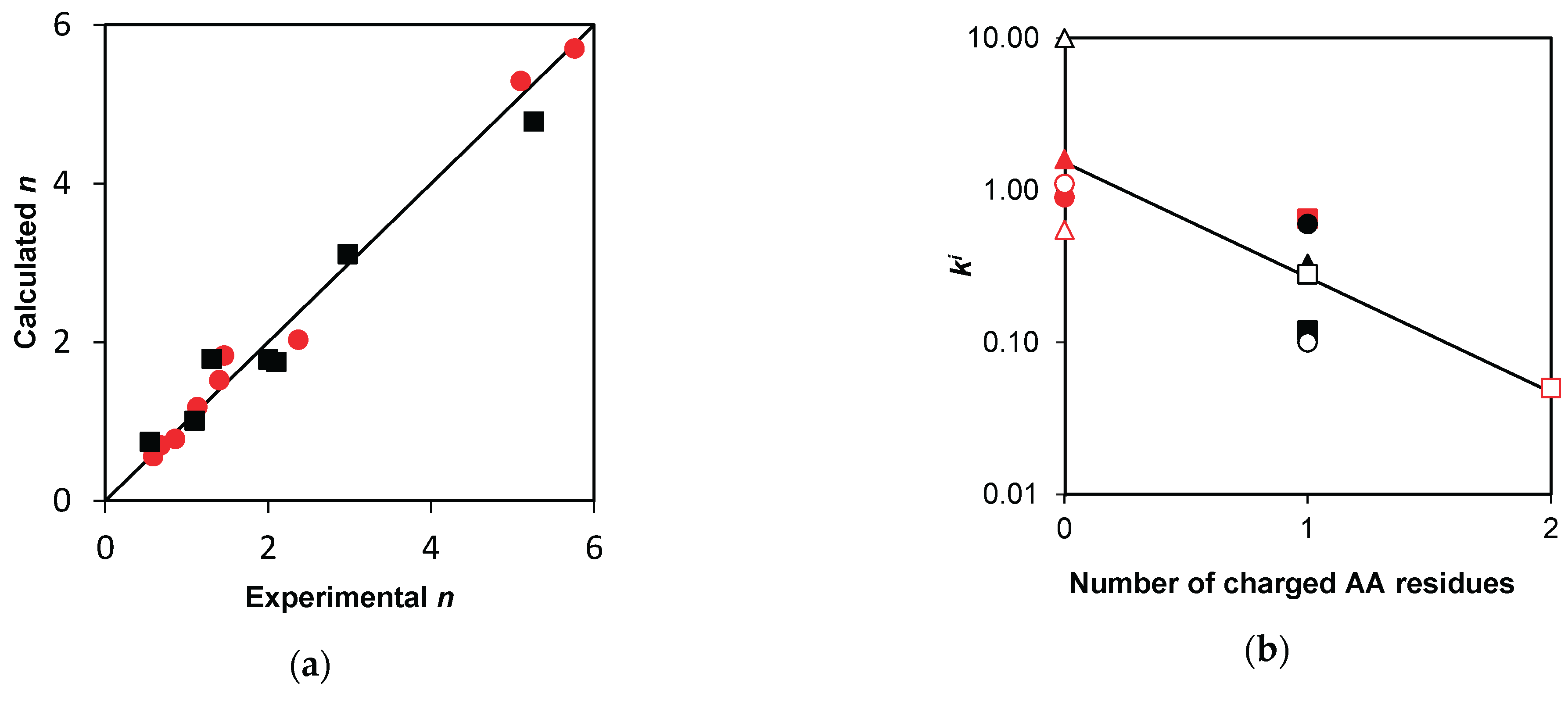

Table 1.
Kinetic characterization of tryptic proteolysis of β-CN.
| Bond index i | Cleavage site1 | Selectivity2 (%) | Initial hydrolysis rate2 | Most rapidly hydrolyzed bonds | Most slowly hydrolyzed bonds | Peptide fragments in trimer |
|---|---|---|---|---|---|---|
| 1 | R-EI | 0 | 0 | + |
1-28/29, 30-97, 98-99 |
|
| 25 | TR-IN | 0.7 | 0 | + | ||
| 28/29 | NK-KI/KK-IE | 8.2/0.9 | 0.1 | |||
| 32 | EK-FQ | 0.7 | 0 | + | ||
| 48 | DK-IH | 0.02 | 0 | + | ||
| 97 | EK-TK | 0.6 | 0.1 | |||
| 99 | VK-EA | 15.3 | 0.8 | + | ||
| 105 | PK-HK | 23.4 | 0.8 | + | 106-107, 108-113, 114-169 | |
| 107 | HK-EM | 2.7 | 0.3 | |||
| 113 | PK-YP | 1.0 | 0.05 | |||
| 169 | SK-VL | 32.4 | 1 | + | ||
| 176 | QK-AV | 11.4 | 0.6 | 170-176, 177-183, 184-209 | ||
| 183 | QR-DM | 2.8 | 0.2 | |||
| 202 | VR-GP | 0.2 | 0 | + |
1 Peptide bonds 28 and 29 with amino acid sequence Lys-Lys- were considered as one hydrolysis site with single Lys bond in P1 position Lys-X and denoted as cleavage site 28/29. 2Values of enzyme selectivity and initial hydrolysis rate were from [7].
Table 2.
Demasking and hydrolysis rate constants of tryptic proteolysis of β-CN and β-LG.
| Substrate | Bond index i | kd2 | ki3 |
3 |
|
|---|---|---|---|---|---|
| 28/29 | 3.5 | 5 | 0.6±0.2 | 70±3 | |
| 97 | 3.5 | 5 | 0.3±0.1 | 32±2 | |
| 99 | Most rapidly hydrolyzed bond | ||||
| 105 | Most rapidly hydrolyzed bond | ||||
| β-CN | 107 | 3.5 | 1 | 0.10±0.01 | 19±1 |
| 113 | 3.5 | 1 | 0.10±0.01 | 75±3 | |
| 169 | Most rapidly hydrolyzed bond | ||||
| 176 | 3.5 | 1 | 10±1 | 78±5 | |
| 183 | 3.5 | 1 | 0.3±0.1 | 80±8 | |
| ________________________________________________________________________________ | |||||
| 8 | Most rapidly hydrolyzed bond | ||||
| 14 | 0.46 | >>1 | 0.9±0.2 | 93±7 | |
| 40 | 0.46 | >>1 | 1.6±0.4 | 102±5 | |
| 69/70 | Most rapidly hydrolyzed bond | ||||
| β-LG1 | 75 | Most rapidly hydrolyzed bond | |||
| 83 | 0.46 | 0.32 | 0.6±0.1 | 115±8 | |
| 91 | 0.46 | 0.32 | 1.1±0.3 | 96±3 | |
| 100/101 | Most rapidly hydrolyzed bond | ||||
| 124 | 0.46 | 1.1 | 0.55±0,14 | 80±6 | |
| 135 | 0.46 | 1.1 | 0.05±0.01 | 19±1 | |
| 138 | Most rapidly hydrolyzed bond | ||||
| 141 | Most rapidly hydrolyzed bond | ||||
| 148 | Most rapidly hydrolyzed bond | ||||
1 The β-LG cleavage sites were taken to be the same as in [9]. 2The rate constants kd were determined using Equation (10). 3The rate constants ki and amplitudes N0 were determined using Equation (11).
Table 3.
Modeling of peptide release during tryptic proteolysis of β-CN and β-LG in comparison with experiment.
Table 3.
Modeling of peptide release during tryptic proteolysis of β-CN and β-LG in comparison with experiment.
| Substrate | Peptide | Type of fragment | Calculated dr (%)1 | Experimental dr (%)1 | Calculated n2 | Experimental n2 | |
|---|---|---|---|---|---|---|---|
| f(1-99), ABC | Intermediate | 1.41 | 1.00 | ||||
| f(1-97), AB | Intermediate | 2.04 | 1.77 | ||||
| f(30-99), BC | Intermediate | 2.23 | 2.28 | ||||
| f(106-169), ABC | Intermediate | 2,18 | 2.14 | ||||
| f(106-113), AB | Intermediate | 2.98 | 3.61 | ||||
| f(108-169), BC | Intermediate | 2.98 | 1.96 | ||||
| f(170-209), ABC | Intermediate | 1.32 | 1.33 | ||||
| f(170-183), AB | Intermediate | 1.40 | 1.48 | ||||
| f(177-209), BC | Intermediate | 2.14 | 1.61 | ||||
| f(1-28/29), A | Final | 1.01 | 1.10 | ||||
| β-CN | f(30-97), B | Final | 1.75 | 2.10 | |||
| f(98-99), C | Final | 1.42 | -3 | ||||
| f(106-107), A | Final | 3.11 | -3 | ||||
| f(108-113), B | Final | 4.78 | 5.26 | ||||
| f(114-169), C | Final | 3.11 | 2.98 | ||||
| f(170-176), A | Final | 0.74 | 0.55 | ||||
| f(177-183), B | Final | 1.79 | 1.31 | ||||
| f(184-209), C | Final | 1.78 | 2.00 | ||||
| ____________________________________________________________________________________________________ | |||||||
| f(9-69/70), ABC | Intermediate | 2.45 | 1.503 | ||||
| f(9-40), AB | Intermediate | 2.90 | 3.60 | ||||
| f(15-69/70), BC | Intermediate | 2.65 | -3 | ||||
| f(76-100/101) ABC | Intermediate | 3.62 | 3.40 | ||||
| f(76-91), AB | Intermediate | 4.29 | 4.40 | ||||
| f(84-100/101), BC | Intermediate | 4.06 | 4.70 | ||||
| β-LG | f(101/102-138), ABC | Intermediate | 3.41 | 3.40 | |||
| f(101/102-135), AB | Intermediate | 4.20 | -3 | ||||
| f(125-138), BC | Intermediate | 4.85 | 6.10 | ||||
| f(9-14), A | Final | 0.70 | 0.68 | ||||
| f(15-40), B | Final | 0.78 | 0.86 | ||||
| f(41-69/70), C | Final | 0.56 | 0.59 | ||||
| f(76-83), A | Final | 1.83 | 1.46 | ||||
| f(84-91), B | Final | 2.03 | 2.37 | ||||
| f(92-100/101), C | Final | 1.52 | 1.40 | ||||
| f(101/102-124), A | Final | 1.18 | 1.13 | ||||
| f(125-135), B | Final | 5.70 | 5.76 | ||||
| f(136-138), C | Final | 5.29 | 5.10 | ||||
1 The parameter dr was determined using Equation (1). 2The exponent of the power function n was determined using Equation (2). 3These peptide fragments were not found [7].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



