Submitted:
13 October 2025
Posted:
15 October 2025
You are already at the latest version
Abstract
A versatile family of functional soft materials with a wide range of applications, electrostatically coassembled micelles forms when neutral, hydrophilic coronas in aqueous solution are microphase-separated from a core containing related polycations and polyanions. Because of their hydrated state and structural and chemical adaptability, complex coacervate core micelles (C3Ms) are a desirable solution for distribution and basic research on polymer physics. Fundamental structure-property relationships can be established by utilizing block copolymer design with controlled self-assembly to precisely tune the size, morphology, and stability of C3Ms in pursuit of tailored nanocarriers that ultimately provide active ingredient storage, protection, transport, and delivery. The chemical structure and physical characteristics of the micellar building blocks, such as charge density, block length (ratio), and hydrophobicity, have a significant impact on the nanostructures that result from the mixing of specific oppositely charged block copolymers (BCPs) and other ionic species. The structure and characteristics of the steady-state association colloids have been significantly clarified by over thirty years of research since the discovery of this novel class of polymer micelles. Dynamics and out-of-equilibrium processes have been receiving increasing attention. Examples of these processes include reaction-assembly networks, (dis)assembly pathways, and the exchange kinetics of the micellar constituents. I anticipate that the expanded scope will aid in the planning and design of hitherto unachievable buildings with emergent features and functions. The process of BCPs self-assembly in solution has been the subject of extensive scientific investigation for a number of years because of the remarkable variety of morphologies and achievable complexity of the resulting nanoassemblies, which include vesicles, lamellae, spheres, cylinders, and many other complex, bicontinuous, or even hierarchical structures. A vast array of macromolecules with different chemical compositions, structures, characteristics, and properties are now accessible due to the ever-improving sophistication of synthetic chemistry methodologies and procedures. These diverse properties have thus given rise to an abundance of fascinating self-organized polymeric nanostructures, offering a multitude of potential uses in various nanotechnological domains associated with physics, chemistry, material science, nanomedicine, and biomaterials. Here, I provide a summary of the current hypotheses on block polymer micelles. I discuss in brief the association behavior of triblock terpolymer and concentrate on the equilibrium structure of nanoaggregates generated by solvophobic/solvophilic diblock copolymers in a diluted solution. I present several difficult issues for theoretical advancements as well as recent discoveries in the subject. Through illustrative examples from the modern era, the current Review seeks to shed light on the significance and intriguing possibilities of BCPs solution self-assembly. It does this by highlighting recent developments and developing trends in the area as well as noteworthy application-oriented accomplishments. With an emphasis on (i) structure-property interactions to target precise nanoscale dimensions and shapes and (ii) measurement of C3M dynamics largely utilizing time-resolved scattering techniques, this Review focuses on recent initiatives to investigate these dynamic, out-of-equilibrium phenomena in more spatiotemporal detail. I explore important prospects for C3M design to promote precision medicine and offer many vignettes from these two new fields of C3M research. I provide many methods and talk about how they explain and expose parallels and discrepancies in the behavior of mixed micelles made from distinct polymeric building blocks and manufactured under varied circumstances.
Keywords:
complex coacervate core micelles (C3Ms)
; block copolymers (BCPs)
; dynamics
; out-of-equilibrium
; exchange kinetics
; triblock terpolymer
Table of Content
1. INTRODUCTION
2. UTILIZING NANOTECHNOLOGY FOR DRUG ADMINISTRATION: METHOD OF DRUG DELIVERY AND NANOTECHNOLOGY OF POLYMER MICELLES
3. STANDARD POLYMER MICELLES
4. AN ALTERNATIVE KIND OF POLYMER MICELLE NANOPARTICLES
5. POLYMER MICELLE NANOPARTICLES THAT ARE ACID-CLEAVABLE AND pH SENSITIVE
6. MICELLE NANOPARTICLES OF CROSS-LINKED POLYMERS
7. INNOVATIVE APPROACHES TO POLYMER MICELLE NANOTECHNOLOGY
8. C3M STRUCTURE-PROPERTY RELATIONSHIPS
8.1. Steady-State Properties.
8.2. Morphological Transitions.
8.3. Sturdy, Long-Lasting, and Stimuli-Sensitive Pharmaceutical Micelles.
8.3.1. Longevity.
8.3.1.1. Steric Stabilization.
8.3.1.1.1. Poly(ethylene glycol). 8.3.1.1.2. Substitute Coatings. 8.3.1.1.2.1. Poly(N-vinylpyrrolidone). 8.3.1.1.2.2. Polysaccharides. 8.3.1.1.2.3. Additional Hydrophilic Blocks.
8.3.1.2. The Size of Micellar Particles.
8.3.1.3. Additional Methods to Enhance Circulation Times.
8.3.1.4. Longevity of Polymeric Micelles with Active Targeting.
8.3.2. Stability of Micellar Matter.
8.3.2.1. Lowering the CMC. 8.3.2.2. Physical Interactions. 8.3.2.3. Crosslinking of Covalent Bonds. 8.3.2.3.1. Crosslinking of Shells. 8.3.2.3.2. The Crosslinking of Interfaces. 8.3.2.3.3. Crosslinking at the Core. 8.3.2.3.4. Cleavable Crosslinks. 8.3.2.3.5. Crosslinking’s effects on drug release and loading. 8.3.2.4. Drug Compatibility with Micellar Core. 8.3.3. Sensitivity to Stimuli. 8.3.3.1. Polymeric Micelles with Thermosensitivity. 8.3.3.2. Polymeric Micelles with pH-Sensitivity. 8.3.3.3. Micellar Disintegration Induced by Chemical Hydrolysis. 8.3.3.3.1. Chemical Hydrolysis of the Polymeric Framework. 8.3.3.3.2. Cleavable Side Chains. 8.3.3.3.3. Breakdown of Polymer-Drug Complexes. 8.4.3.3. Polymeric Micelle Destabilization Induced by Enzymes. 8.5.3.3. Polymeric Micelles Susceptible to Oxidation and Reduction. 8.6.3.3. Micellar Deformation Caused by Light. 8.6.3.3.1. Irreversible Reactions that Occur on Illumination (photolysis). 8.6.3.3.2. Light-Triggered Reversible Alterations. 8.7.3.3. Other Physical Triggers that cause Polymeric Micelles to become Unstable. 8.8.3.3. Multi-trigger-Responsive Polymeric Micelles. 8.8.3.3.1. Temperature and pH Sensitivity. 8.8.3.3.2. Temperature-Sensitive Biodegradable Polymers. 8.8.3.3.2. Diverse. 8.9.3.3. Drug Delivery Guided by Imaging. 8.4.3. Combining Stability, Longevity, and Stimulus Sensitivity.
9. SELF-ASSEMBLY, SYNTHESIS AND THEORY OF BLOCK COPOLYMERS (BCP) SOLUTION
9.1. Self Assembly. 9.1.1. General Aspects.
9.1.2. Morphology of Micellar Structures. 9.1.2.1. Spherical, Cylindrical Micelles and Polymersomes. 9.1.2.2. Complex Supramolecular Structures. 9.1.3. Alternative Self-Assembly Routes.9.1.3.1. Polymerization-Induced and Electrostatic Self-Assembly. 9.1.3.2. Self-assembly in Confinement and in other Media.
9.2. Synthesis and Theory. 9.2.1. Amphiphilic block copolymer (AmBC) synthesis using a mixture of selective post-polymerization functionalization and anionic polymerization. 9.2.1.1. Amphiphilic Diblock Copolymers. 9.2.1.2. Double Hydrophilic Diblock Copolymers. 9.2.2. Theory of Nonionic and Ionic Diblock Copolymer Micelles. 9.2.3. Synthesis of Linear Triblock and Multiblock Copolymers. 9.2.3.1. Sequential RAFT and ATRP. 9.2.3.2. Sequential AP, AROP and CROP. 9.2.3.3. Macroinitiators Available Commercially. 9.2.3.4. Combination of Various Methods for Polymerization: To Combine AB and C by Click Reactions. 9.2.4. Theory of Triblock Co- and Terpolymer Self-Assembly. 9.2.4.1. Soluble C Block ABC Polymers. 9.2.4.2. Soluble A and C Blocks in ABC Polymers. 9.2.5. Non-Linear Architectures.
10. MECHANISMS OF C3M FORMATION: MECHANISM OF MICELLE ASSEMBLY AND DISASSEMBLY
10.2. Mechanism of Aggregation: Factors Influencing and Impact of External Factors, Polymer Architecture, the Length of the Core-Forming Block and Charged Functionality’s Structure towards C3M Formation. 10.2. Mechanism of Micelle Assembly and Disassembly.
11. KINETICS OF MICELLIZATION AND KINETICS OF EXCHANGE
11.1. Kinetics of Micellization. 11.2. Kinetics of Exchange. 11.3. Time-Resolved in situ Polyelectrolyte Complex Micelle Formation Kinetics Uncovered by Small-Angle X-ray Scattering.
12. BALANCE OF MICELLAR FREE ENERGY
13. COMPLEX COACERVATE DROPLETS AND MICELLES: DNA DYNAMICS
14. C3M IN DILUTE SOLUTIONS: INTERPARTICLE INTERACTIONS
15. METHODS 15.1. ζ-potential and Viscosimetry.
15.2. Conductometry and Static Light Scattering (SLS). 15.3. Dynamic Light Scattering (DLS) and Other Methods.
16. APPLICATIONS 16.1. Biomedical Applications. 16.1.1. Control of Enzymatic Activity: Optimizing Enzyme Encapsulation Stability and Efficiency in Complicated Coacervate Core Micelles. 16.1.2. C3M-based Biomolecule Delivery. 16.1.2.1. Nucleic Acid Delivery. 16.1.2.2. Brain Delivery. 16.1.2.2.1. Passing the Brain-Blood Barrier. 16.1.2.2.2. Potential Applications of C3M in Glioblastoma Treatment. 16.1.2.2.2.1. The Importance of Micelle-based Glioblastoma Multiforme (GBM) therapy implementation hindrances. 16.1.2.2.2.2. Glioma-Specific Targeting Moieties. 16.1.2.2.2.3. Therapeutic Micelle Delivery to Brain Tumors. 16.1.2.3. Drug Delivery. 16.1.2.3.1. Polymeric Micelles and Vesicles: Their Characteristics. 16.1.2.3.2. The Building Blocks. 16.1.2.3.3. Polymeric Micelles: Loading, Retention, and Release of Drugs. 16.1.2.4. Delivery of Therapeutic Proteins. 16.1.3. Diagnostics, Imaging and Theranostics: Combination of Diagnosis and Treatment. 16.2. Nanofabrication. 16.3. BCP Self-Assembly Applications in Ionic Liquids (ILs). 16.3.1. Soft Actuators. 16.3.2. Electrochemical Applications and Devices. 16.3.3. Lithium-Ion Batteries. 16.3.4. The Electrolyte-Gated Transistors. 16.4. Other Applications.
17. MICELLAR FORMULATIONS IN CLINICAL TRIALS 17.1. Genexol-PM and NK105. 17.2. SP1049C AND NK911. 17.3. NC-6004 AND NC-4016. 17.4. NC-6300 and NK102.
CONCLUSION AND PERSPECTIVE
REFERENCES
1. Introduction
Biological macromolecules including proteins, peptides, and nucleic acids can be used as therapeutic agents to treat diseases like cancer, infectious and immunological diseases, and metabolic problems. This is becoming a more and more effective treatment option. This new class of medications’ great specificity, which significantly reduces off-target effects, is one of the keystones of its efficacy.[1,2,3] Significant advancements in molecular biology have made it possible to produce such fragile biomolecules on a massive scale, which has led to the development of new medicines based on such biological macromolecules. Traditional tiny synthetic compounds, which frequently exhibit both a low selectivity and hazardous side-effects in healthy tissues, now have an alternative due to their growing availability.[4] One of the most promising areas of research in biochemical and pharmaceutical science is the application of biomacromolecular pharmaceuticals in novel treatment approaches, due to their distinct advantages over small molecule therapeutic agents. Biomacromolecular therapeutic agents (BTAs) have a great deal of potential, but very few examples of these therapies have been effectively implemented in clinical settings. Successful examples include the use of insulin to treat diabetes, the use of somatropin in growth hormone therapy, or the approval of monoclonal antibodies to treat cancer and other illnesses.[5,6,7] In actuality, less than 2% of the more than 20,000 medications that the FDA has currently approved are BTAs.[8] Successful therapeutic outcomes are uncommon, despite the fact that recent scientific advancements have increased the effectiveness of treatment based on the usage of BTAs. A significant obstacle that numerous clinical studies are currently facing is the insufficient and misdirected administration of these medicinal substances.[3,9,10] Specifically, a number of studies blamed the supposedly inadequate delivery mechanism of BTAs for the failure of treatments based on their use.[11,12,13] More than ever, BTA delivery systems must be designed with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in mind. This is especially important in light of the current global health crisis. The latest COVID-19 vaccines, such as those created by Moderna and Pfizer/BioNTech, provide the host cell with the genetic sequence of the viral protein in the form of mRNA.[14] The expression of the virus protein is induced by the mRNA, which also results in immunity against the original virus. However, utilizing mRNA alone would be ineffective since mRNA is too big to effectively traverse cellular membranes and is readily broken down by RNAses during blood circulation.[15] The utilization of lipid nanoparticles as the delivery method, a remarkable feat of nanotechnological engineering in and of itself, may have contributed to the success of the mRNA vaccine formulation.[16] The mRNA is shielded from extracellular RNases by being encapsulated in cationic lipid nanoparticles, which also makes it easier for the gene to be absorbed and released endosomally in the intended cells. However, the ionizable lipid nanoparticles that are used as the mRNA delivery vehicle are primarily responsible for both the vaccine’s poor stability and the challenges in scaling up manufacturing.[17] It is evident that the strong cooperation between bio and nanotechnology is the reason for the scientific success of these vaccinations, which offer a way out of the current world issue. This achievement not only demonstrates the promise and advantages of biomacromolecular therapies, but it also emphasizes how urgently effective, reliable, and adaptable delivery mechanisms for these compounds must be created. The usage of medication delivery systems based on polymers is growing.[18] The first instances of these systems were successfully transferred from laboratory settings to clinical settings in the past few decades. These applications included the delivery of small molecules (such as the antibiotic Atridox®, the lung and breast cancer treatments Abraxanes®, and the opioid addiction treatment Sublocades®) as well as biomacromolecular therapeutic agents (such as the leukemia treatment Oncaspar®).[19,20,21,22] The design of novel polymer DDSs for bio-macromolecular medicinal agents presents some obstacles that are similar to those encountered in the design of DDSs for small molecules. The development of more potent and efficient DDSs as well as the standardization of manufacturing procedures are essential to the ongoing progress in advancing polymer DDSs toward clinical applications.[23] While lipid-based nanocarriers were used to achieve the notable success of mRNA distribution in COVID-19 vaccines, a number of clever polymer nanostructures are emerging as viable substitutes. These drug delivery systems (DDSs) offer unparalleled design flexibility and diversity.
Compaction of two aqueous solutions containing macromolecules with opposing charges can result in complicated coacervation, a liquid-liquid phase separation.[24] The concentration of both polyelectrolytes in the two resultant liquids is different: the dense, complex coacervate phase is enriched in both, while the dilute phase is deficient in the macromolecules. The coupling of the attractive Coulombic contacts with the entropic gain resulting from the counterion release drives this phase separation.[25] Physical and chemical characteristics of the constituents, such as the concentration of salt, have a significant impact on the relative contributions of these driving factors. It is advantageous to limit macroscopic phase separation to the nanometric scale in order to conjugate an uncharged, soluble polymer block to one or both of the charged macroions, resulting in mixed association colloids.[26] The resulting hydrocolloids are frequently micelles with a neutral corona encircling a complicated coacervate core (Figure 1). The terms polyion complex (PIC)[26] and interpolyelectrolyte complex (IPEC)[27] have also been used to refer to these so-called C3Ms.[28] We present a schematic image of such a micelle in Figure 2.
Because of their intriguing potential in biomedical applications, polymer-based micellar assemblies are attracting more and more attention.[31,32,33,34,35,36,37,38] As shown in Figure 3, C3Ms are a type of polymer-based micelle that are primarily formed through electrostatic interactions between a charged-neutral block copolymer (such as PMVP-PEO) and an oppositely charged polymer. The micellar core is composed of the oppositely charged polymers. Organic or metal-to-ligand coordination polymers are examples of charged core polymers; more recently, highly-charged monomeric units have also been used.
Nanotechnology has long aimed to achieve controlled self-assembly and compartmentalization on the 1 - 1000 nm length scale in solution. This research is starting to tackle new difficulties in energy management,[40] green catalysis,[41] surfactant compatibilizers,[42] and human health.[43] Due to their ability to undergo microphase separation, polymeric micelles have produced a wide range of hierarchical nanoaggregates that are widely acknowledged as excellent options to deal with these problems. These nanoparticles make it possible to package cargo into distinct domains that can move molecules through otherwise impenetrable barriers and endure harsh environments. Typically, charged polymer interaction in an aqueous solution or amphiphilic polymer association in certain solvents induce the construction of micelles. Our basic understanding of amphiphilic materials has significantly advanced thanks to foundational efforts in modeling and simulation,[44,45,46] scaling theories,[47,48,49] self-consistent mean field theory,[50,51,52] experiments, and so on.[53,54,55] Micellar size, shape, aggregation number, and chain exchange dynamics can be precisely tuned for intended applications by utilizing the synthetic versatility of block copolymers to precisely tune the energetic components of (i) chain stretching in the core, (ii) excluded volume of the corona, and (iii) interfacial energy of the micelle in solvent. Other noncovalent association driving mechanisms have surfaced to further customize self-assembly and broaden the range of complex nanostructures available, beyond hydrophobic effects in polymers. With attention from the interface and colloid science, biology, polymer physics, and other interdisciplinary domains, complex coacervation has become a possible path toward self-assembled materials.[56] The entropy gain from counterion release[57] is primarily responsible for the assembly of oppositely charged polyelectrolytes, which leads to phase-separated complex assemblies of polyelectrolytes that display a variety of fundamentally distinct static and dynamic properties. Polyelectrolyte complex materials can be synthesized into hydrophilic corona external nanoparticles or C3Ms. C3Ms generally use the coassembly of oppositely charged polymers in systems where at least one polymer has a block architecture, as seen in Figure 4.
C3Ms have a more complex thermodynamic framework due to various underlying properties, making them significantly less quantitatively known at the molecular levelthan amphiphilic block copolymer micelles.[59,60,61] The ionic core, for instance, is made up of two different polyelectrolytes that, when their charges are matched stoichiometrically, generate intrinsic ion pairs that serve as physical cross-links between repeat units of polycation and polyanion. Because these pairs can be broken by heating or adding salt, C3Ms are extremely sensitive to environmental changes in their immediate surroundings. Due to the intrinsic multicomponent character of complex coacervates, the low interfacial tension and water solubility of polyelectrolyte chains result in the presence of water in both the core and corona, which further complicates attempts to understand the fundamental physics of these nanoparticles.[62] There are few systematic investigations on the stability and morphology of C3Ms, despite their great interest. The influence of salt is the subject of most investigations on the stability of C3Ms. Pioneers in this field, Kabanov et al. observed a drop in light-scattering intensity as salt content increased, which they attributed to C3M breakdown.[63] Later, it was discovered that adding salt causes a decrease in the mass and quantity of C3Ms.[13,14] An rise in the critical micelle concentration (cmc) directly correlates with a decrease in the number of C3Ms. All micelles dissociate when the cmc surpasses the total polymer concentration, which happens at a critical salt concentration that varies greatly throughout C3M types. Yan et al. and Wang et al. discovered that the cmc of C3Ms increases exponentially with the square root of the salt concentration by utilizing the dependence of the critical salt concentration on the polymer concentration.[64,65] The polymer chain length has an impact on the critical salt concentration as well. Gaucher et al. conducted a qualitative study on this chain length dependence and found that as the homo-polymer chain length increases, so does the resistance of C3Ms against salt-induced disintegration, or salt stability.[66] Sadly, they only looked at three homopolymer lengths in their study. The influence of salt on the hydrodynamic radius (Rh) is the main focus of experimental studies on the morphology of C3Ms. There have been reports of both a decrease[67,68,69] and an increase[64,65,70,71,72,73,74,75,76,77,78] in Rh following the addition of salt; however, the processes underlying these opposing trends are not well understood. It was initially demonstrated by Yan et al. that salt can occasionally alter the morphology of C3Ms.[64] They discovered the presence of sizable aggregates at high salt concentrations using cryogenic transmission electron microscopy, some of which seemed to be worm-like micelles. Not too far ahead of the critical concentration of salt, they also noticed a peak in the intensity of light scattering. A maximum in Rh and a local low in the polydispersity index seemed to correspond with this peak. A previous report tackled two unanswered questions: how does the length of the polymer chain impact the salt stability of C3Ms, and why do certain C3Ms expand while others contract when salt is added? For the first question, the authors employed light-scattering salt titrations; for the second, they employed a mix of small-angle X-ray scattering and other light-scattering approaches. The authors selected anionic homopolymer poly(acrylic acid) (PAA) and cationic−neutral diblock copolymer poly(N-methyl-2-vinylpyridinium)-b-poly-(ethylene oxide) (PM2VP-b-PEO) micelles as model system due to their good characterization.[79,80] According to their measurements, for short polymer chains, the salt stability of C3Ms increases with chain length and levels out for longer chains. The authors provided a polyelectrolyte complexation model based on Overbeek and Voorn’s mean-field theory to corroborate these findings.[81] Moreover, they discovered that when salt is added, short homopolymers cause C3Ms to grow in average size while long homopolymers cause C3Ms to shrink. There is always a peak in the intensity of scattering along with the increase in size. They contended that the morphological change from spherical to wormlike micelles, which enlarge with increasing polymer concentration, is the cause of both the apparent expansion and the intensity rise. The insights the report offers here enable the stability and shape of these common coassembled nanostructures to be fine-tuned. It was further reported that the fundamental ideas are more universal and can be applied to other charge-driven complexes as well, like complex coacervate membranes and triblock copolymer hydrogels with complex coacervate junction points[82,83]
Many different types of (bio)molecules are encapsulated by C3Ms.[27,61] Associative liquid-liquid phase separation of oppositely charged polyelectrolytes from the water phase is the basis for the development of these C3Ms. A neutral, hydrophilic block that is bonded to at least one of the two polyelectrolytes prevents macroscopic phase separation. The micelle core is made up of polyelectrolytes, whereas these neutral blocks comprise the micelle corona. Charged or hydrophilic chemicals can be incorporated into the hydrophilic environment of the core and then be shielded from outside substances by the micelle corona. The C3Ms can react to variations in the concentration of salt and, in certain situations, even variations in pH because the core creation is dependent on electrostatic attraction. The C3Ms are attractive agents for medication and gene delivery because of their capacity to respond to external stimuli and protect the corona.[84,85] Studies on C3Ms have thus far mostly concentrated on their average static properties in various environmental settings, such as various pH levels and ionic strengths. These average static features, however, conceal the C3Ms’ underlying molecular interaction. The C3Ms are a dynamic system where molecular exchange can continue even when they are fully equilibrated and the average static properties do not vary over time. The C3M exchange dynamics have been the subject of only a few number of research,[86,87,88,89,90] some of which have offered suggestions for the C3M exchange processes and the associated regulating parameters. Although their exchange dynamics can significantly affect their encapsulation efficiency, the precise C3M exchange mechanisms remain unclear as of this writing. Ultimately, the exchange dynamics dictate the speed at which the cargo is exposed to the environment and, thus, the degree of protection provided by the C3M. Moreover, the preparation pathways of the C3Ms sometimes control their ultimate structures.[91,92,93,94,95] This shows that the C3M characteristics and, thus, their encapsulation efficiency, can be determined by kinetic processes. The two primary types of exchange mechanisms are typically used to interpret C3M exchange experiments. The first involves the insertion into a different micelle after the evacuation of a single polymer or a tiny cluster of polymers. In the latter scenario, the micelle divides into two components of both significant sizes, which may then recombine with other micelles. The distinction between the expulsion and insertion exchange and this kind of exchange, known as fission and fusion, is that all produced clusters retain a significant micelle corona. Because this necessitates significant remodeling of the micelle corona polymers, the merging of the micelles is therefore thought to be the rate-limiting phase for fission and fusion, whereas the expulsion from the core is thought to be the rate-limiting step for the expulsion and insertion case. It was demonstrated using Langevin dynamics simulations that both exchange mechanisms may take place during the first micellization of C3Ms.[89] It was found that the expulsion/insertion exchange is strongly favored for oppositely charged polyelectrolytes with matched lengths and weak nonelectrostatic attraction, whereas the fission/fusion mechanism may become more significant for unmatched chain lengths and stronger nonelectrostatic interactions. A recent study using small-angle X-ray scattering (SAXS) revealed that a delayed rearrangement of the micelles can happen following the very quick initial micellization.[88] This rearrangement did not depend on concentration, indicating that ejection or insertion accounts for the majority of the exchange during these rearrangements. Because of the variations in micelle size at each stage, the exchange processes during initial C3M production and rearrangement may differ from the exchange of equilibrated C3Ms.[96] Determining the exchange kinetics of equilibrated micelles is crucial as a result. Because of the absence of structural rearrangements and the comparatively long equilibration durations, respectively, neither dynamics simulations nor SAXS can be utilized to analyze the exchange in this equilibrated state. Time-resolved small-angle neutron scattering (TR-SANS) measurements have been utilized to track the exchange of equilibrated micelles for amphiphilic diblock copolymer micelles.[55,97,98,99,100,101,102] Nevertheless, this calls for the synthesis of deuterated polymers as well as the utilization of sophisticated, non-commercial equipment. Utilizing Fischer resonance energy transfer (FRET), a nonradiative energy transfer from an excited donor fluorophore to a neighboring acceptor fluorophore, is a more user-friendly method of tracking the exchange dynamics of equilibrated micelles. In the aforementioned studies (Figure 5), micelles containing donor and acceptor fluorophores are combined. When the micelles interchange, the donor and acceptor may merge into a single micelle core, indicating that their proximity is sufficient for FRET to transpire. Consequently, the micelle exchange rate can be determined by tracking the rise in FRET efficiency over time. The generation and exchange dynamics of C3Ms containing proteins as well as the exchange dynamics of C3Ms at various charge stoichiometry ratios have previously been studied using the FRET method.[86,87] Both investigations ignored any other factors that might have affected the normalized FRET rise and instead used the increase in FRET efficiency normalized to the final FRET efficiency as a direct indicator of the micelle exchange rate. This method is sufficient to provide a general understanding of the exchange time scales and shown that the C3Ms including proteins exchanged at a significantly faster rate than the C3Ms made entirely of polymers. Nevertheless, further quantitative comparisons of the exchange rates are necessary to clarify the exchange mechanisms in greater detail, and in that case these additional aspects cannot be disregarded. As the exchanged chain fraction increases, the FRET effectiveness actually varies depending on other factors such as the size of the micelle core and the characteristics of the donor and acceptor fluorophores. To connect the observed normalized FRET rise to the underlying micelle exchange rates, a more sophisticated explanation is thus required.
As mentioned above, C3M or PE complex micelles form when oppositely charged homo- and diblock copolymers, or mixes of them, come together by electrostatically induced self-assembly.[28] A scaling theory was created in recent work[59] to forecast the development and characteristics of spherical and nonspherical aggregates, building on an earlier effort to characterize spherical micelles with PECC cores.[67] By adding salt, the micellar core density is lowered, which results in the core swelling and more ionic block extension, according to the method described in Reference 59. With nonspherical micelle morphologies, the energy of the core block stretching is small,[104,105] so salt-induced changes from spheres to worm-like aggregates and finally to lamellae/vesicles are anticipated.[59] The van der Gucht’s group’s observations of salt-induced sphere-to-cylinder transitions[106] are in agreement with this theoretical prediction. According to Lueckheide et al., the oppositely charged PLL-b-PEG diblock copolymer’s hybridization with DNA controls the shape of PE complex micelles made of the latter.[107] Micelles with single-stranded DNA (ssDNA) have a spherical shape, while dsDNA complexation results in a cylindrical aggregate shape. It's interesting to note that 22-bp DNA duplexes within the core show hexagonal packing and coaxial end-to-end stacking of the resulting rod-like aggregates,[107] which are representative of the columnar LC phase seen in dsDNA solutions.[108] Theoretically, orientational ordering of dsDNA in macroscopic PECCs is understood,[109] but the competing roles of ensuing LC character of the core and high stiffness of rod-like dsDNA aggregates in determining the wormlike shape of micelles are not understood. Micelles shaped like ellipsoids have also been discovered when the stiff polyfluorene derivative is present in the center as the polyanion.[110] The goal of recent modeling and experimental efforts has been to better understand the dynamics of chain exchange and micelle generation between equilibrium aggregates. According to these studies, the diblock copolymer architecture can be used to tune both the dominant formation mechanism (micelle fusion/fission versus single-chain insertion/expulsion), which creates a dynamic equilibrium between them, and the time required to obtain equilibrium aggregates.[89,92] The in-depth explanation of the thermodynamic characteristics of complicated coacervation has been thoroughly discussed by others,[37,111,112,113,114,115,116,117,118,119,120,121] to whom we direct interested readers. In summary, the ionic strength of the solution has a major impact on both the enthalpic and entropic contributions, which can both be substantial. A cloud of counterions envelops a polyelectrolyte chain. The concentration of ions surrounding the polyion is higher than the concentration of bulk ions when the ionic strength is low. As a trade-off between the counterion entropy and Coulombic attraction, the long Debye length results in diluted counterion clouds. When an oppositely charged polymer is added, the counterions are released into the solution (entropy gain) and the charges are brought closer together (Coulombic attraction; exothermic) via tight complexation. However, the counterion clouds get denser at high ionic strength, making the addition of and complexation with the second polymer endothermic. Nevertheless, complex creation experiences a net energy gain due to the entropy gain resulting from counterion release. But, the net driving force for complexation disappears and complex coacervation stops occurring over a certain salt concentration. The chemical makeup of the constituents can impact the energy balance in addition to the concentration of salt. Hydrophobic interactions and the creation of hydrogen bonds are frequently additional driving forces in addition to electrostatic interactions.[122] When polyelectrolytes exhibiting these extra interactions are exacerbated, the entropic and enthalpic contributions of complexation are amplified, leading to an increased concentration of critical salt.[123,124] As long as the polyelectrolyte chains are connected to sufficiently long neutral solubilizing chains, the formation of micellar complex coacervates is constrained to mesoscopic dimensions. Shorter neutral blocks than the charged blocks they are conjugated to typically result in precipitation rather than micellization.[27] The same physical rules that determine the dimensions of other micelles also determine the size of the resulting C3Ms. The expansion of the core is driven by the free energy of the surface formed by the phase separation, whereas the micellar dimensions are often limited by the simultaneous stretching of the corona- and core-forming blocks. The size-dependent free energy gain linked to micellization and, consequently, the average aggregation number and size of the C3Ms in equilibrium are determined by the interaction of these parameters. Numerous studies conducted over the past few decades have provided insight into the various forms of coassembled micelles that result from the intricate coacervation of oppositely charged components and (block) copolymers.[27,61] Ionic-neutral diblock copolymers (dbp) can be combined with a wide range of oppositely charged species, such as polysaccharides, DNA, proteins, dendrimers, peptides, branched (synthetic) polyelectrolytes, dendrimers, multivalent ions, and metallic complexes, to create C3Ms.[107,125,126,127,128,129,130,131,132] Their potential for use varies greatly depending on the chemical makeup of the constituent and embedded building blocks, and it extends from materials science to nanomedicine. The encapsulation of biomolecules, including proteins, RNA, and DNA, for the goals of controlled release and protected delivery is one of the most active areas of basic and applied research on C3Ms.[115,116,133,134] Supramolecular compartmentalization has been shown to provide precise targeting, greater cellular absorption, and improved stability in the circulation.[37,135,136,137] In order to create nanoreactors and templates for the creation of inorganic nanoparticles and nanogels, for instance, C3Ms have also been employed as restricted reaction environments.[138,139] C3Ms are an intriguing class of aqueous polymer materials because of their inherent hydrophilicity, responsiveness, and adaptability. The water content of complex coacervates has been observed to reach up to 77%.[140,141,142] This is significantly higher than the water content of amphiphilic micelle cores, which are just slightly present. Such high hydration creates an ideal environment for charged, brittle, and water-soluble molecules to be encapsulated. It is possible to program the micellar carriers to release their payload in response to variations in temperature, ionic strength, pH, and chemical triggers like sugars.[80,143,144] Numerous physicochemical and functional qualities, including dimensions, shape, and stability, can be adjusted based on environmental signals and composition. A great deal of mapping has been done on the phase behavior of C3Ms, explaining how the final assembly is affected by the length of the diblock copolymer (dbp) and homopolymer (hp), charge mixing ratio, total concentration, and ionic strength.[30,63,106,110,145,146] The processes governing C3M attachment and dissociation are of tremendous interest to explore in addition to steady-state features. The formation of structures as soon as components are mixed and tracking their temporal evolution to the final associated state can help design (nano)structured functional materials and offer fundamental insights into (self- and co)assembly processes in synthetic and living systems. Before relaxing to the lowest energy level, complexes go through a number of intermediary stages. Similar to direct preparation at low (salt) concentration, coassembly at high concentration (salt) followed by dilution can yield different colloidal objects.[147] Furthermore, the assembly and equilibration pathway of complex coacervates are altered by the sequence of dbp/hp addition and incomplete or sluggish mixing, which may have a substantial effect on the compounds’ structure and characteristics.[148,149,150] Although it was previously impossible to obtain the spatial and temporal resolution needed to see these processes, new developments in experimental instruments now make it possible to examine reaction-assembly networks, equilibration, and (dis)assembly pathways in greater detail. By taking advantage of this possibility, we can enhance our ability to regulate the creation of kinetically trapped states and use these building blocks to create a larger library of (nano)structured materials. Conventional amphiphilic micelles typically cannot incorporate or protect hydrophilic substrates like enzymes and nucleic acids due to their water-insoluble (yet hydrated) coacervate core. This is not the case with C3Ms. C3Ms have a high degree of biocompatibility and a high degree of design flexibility. This may make it possible to include targeting moieties and a variety of drug release mechanisms in their structure. Currently, a variety of micelle formulations are being employed in clinical settings to target various cancer types.[27,126,151,152,153] C3Ms can be adorned with targeted molecules and have been demonstrated to significantly increase physiological circulation times, bioavailability, cellular absorption, and therapeutic efficacy.[154,155,156,157,158] They can be used to filter wastewater by flocculating charged impurities. In solution, they are possible transporters of charged molecules, including enzymes, DNA, RNA, antibodies, nanoparticles, dendritic photosensitizers, and metal ions.[27,84,159,160,161,162,163,164] C3Ms function as antifouling agents at surfaces.[165,166] Understanding the stability and shape of these micelles is crucial for these objectives. These structures can be tailored to fulfill the required parameters after the precise effects of polymer concentration, polymer chain length, and salt concentration on the stability and morphology of C3Ms are understood. The use of nanoparticles and nanotechnology in medicine is known as nanomedicine. This suggests that diseases may be detected, tracked, controlled, prevented, and treated with the use of nanomaterials.[167] Given that it has the ability to address many issues with traditional medicine, including solubility, targeting, and drug release, it might be viewed as a significant development in the personalization and advancement of therapy.[168] While there is still debate about whether it reached its full potential, numerous breakthroughs have undoubtedly been made.[169,170] Numerous disorders, including cardiovascular,[171] cancer targeting,[172] diagnostic and therapy,[173] HIV,[174] and Alzheimer’s disease,[175] have been greatly impacted by nanomedicine. However, the advancements in nanobiotechnology and nanomedicine have had the greatest impact on cancer. In this vein, even with the advancements in technology and medicine over the last few decades, cancer remains a major worldwide concern. The traditional methods of treating cancer consist of radiotherapy, surgery, and anti-cancer medications (chemotherapy). Chemotherapy becomes necessary for stage III and IV malignancies since radiation and surgery are insufficient in their efficacy for early stage tumors.[176] In this regard, the use of drug delivery-either targeted or passive-shows great promise for lowering side effects and raising the therapeutic index of chemotherapy. For instance, patients with brain tumors still have extremely poor prognoses and inadequate treatment options, making them the tenth most common cause of death among cancer patients globally. After diagnosis, the disease has a median survival span of roughly 14 months.[177] Brain tumor therapies are ineffective due to a number of factors, such as the blood brain barrier (BBB), which restricts drug penetration into the brain, and the difficulty of surgically removing tumors because they are located in the central nervous system (CNS) without negatively impacting survivors’ quality of life.[178] The most common and deadly type of primary brain tumors found in adult patients are malignant gliomas, which include astrocytomas, oligodendrogliomas, and ependymomas.[179] GBM is a grade IV astrocytoma, and after receiving adjuvant temozolomide for radiation treatment, patients with GBM have a median survival of 14.6 months and a 5-year survival rate of less than 10%.[180] Following radiation therapy, patients with GBM have a median survival of 14.6 months, and after five years, the survival rate is less than 10%.[180] Not much has changed in the standard-of-care for these patients over the last ten years. A few other therapies, including oncolytic viruses, liposomal doxorubicin, and anti-angiogenic drugs like bevacizumab, have also been used with varying degrees of clinical success.[181,182,183,184] To boost the dispersion of a medicine injected intratumorally, patients have also been treated using novel delivery techniques such convection-enhanced delivery (CED).[185,186] However, there is currently no known “cure” for this illness, highlighting the need for both innovative drug delivery approaches and an improved knowledge of the underlying disease process in GBM. Chemotherapy drugs and other tiny biomolecules have a lot of potential when delivered via nanomedicines. Depending on the system, these tiny particles can perform a range of tasks, including longer drug circulation durations, more targeted administration, and increased penetration into solid tumors.[187,188] A growing number of clinically implemented nanoparticle systems are being made available to cancer patients. For instance, liposomal doxorubicin formulations (e.g., Doxil®, Caelyx®) are being tested for efficacy in patients with GBM and brain metastases from solid tumors[189,190] and are currently being used for patients with a variety of cancers.[191,192,193,194] Furthermore, it may be able to overcome the blood-brain barrier and customize treatment in order to improve anticancer medication delivery and distribution.[195] Figure 6 makes it clear that while the majority of chemotherapeutic medications have shown a notable therapeutic benefit, they have also been associated with unfavorable side effects.[196] Moreover, they do not work well in treating ovarian cancer recurrences. Crucially, women with ovarian cancer frequently respond well to various therapeutic approaches at first but subsequently develop resistance to treatment. Consequently, the greatest treatment challenge for ovarian cancer is still medication resistance. Utilizing nanotechnology-based formulations (encapsulated, conjugated, or entrapped/loaded forms in nanocarriers or drug delivery vehicle/vectors) is one approach to increase the efficacy and specificity of chemotherapeutic medicines.
In comparison to conventional drug delivery systems, nanoparticle drug delivery systems, or nanodrug delivery systems,[198] offer several advantages, such as: (i) protecting drugs in their nano-cores from degradation in biological fluids; (ii) improving targeting efficacy to specific tissues; and (iii) controlling the release of drugs in response to specific signals.[199,200] Furthermore, C3Ms have demonstrated tremendous promise for boosting cancer immunotherapy, in addition to the quick advancements in immunology and science.[201,202,203] By delivering drugs to specific cancer sites, C3Ms can, on the one hand, effectively improve the pharmacokinetic characteristics and minimize the negative effects of therapeutic or imaging agents.[204,205] To enhance tumor immunotherapy, however, they can also target immune cells and organs in order to alter the immunological microenvironment.[206,207] Many factors, such as micelle properties like size and surface charge density, polymer properties like molecular weight, linear charge density, and molecular geometry, polymer to micelle stoichiometry, ionic strength, and temperature, can easily affect polyelectrolyte-micelle coacervation.[208,209,210] Because of their special qualities that allow for prospective uses, such as the hydrophobic core of micelles that can load and carry drugs[211] and their intelligent response to CO2/N2[212] light[213,214] or temperature[215,216] stimuli, polyelectrolyte-micelle complexes are of significant interest and importance. While all polyelectrolyte-surfactant mixtures are sometimes called complexes regardless of whether a phase separation has place,[217,218] coacervates are the compounds created when the combination of surfactant and polyelectrolyte causes a phase separation. One way to segregate chemical reactants, nanoparticles, and proteins in the microscale, water-filled environments is by complex coacervation.[219,220,221,222] This partitioning or compartmentalization can be useful for a number of purposes, including the loading of pharmaceutical compounds into pH-sensitive coacervates for drug delivery,[222] the concentration and removal of diluted contaminants from aqueous solutions,[219,221,223] the selective purification of proteins,[224] the use as a non-membrane bound protocell for studies on the origin of life, and more.[225] Membrane-bounded microcompartments have been extensively studied as a means of compartmentalizing solutes; as a result, they may be suitable for use as microreactors or as models for protocells. Examples of these microcompartments include self-assembled bilayer vesicles, polymer capsules, and inorganic vesicles.[226,227,228,229,230,231] However, the low permeability of the membrane may restrict some important processes, such as chemical or enzymatic reactions within the membrane-bounded microcompartments.[232,233] Because of the restricted mass transfer of reagents to those microcompartments, this transport limitation may prevent the ongoing chemical activity within the microcompartments. Hence, spontaneous complex coacervation offers a straightforward and adaptable substitute method for compartmentalization; yet, it lacks a membrane, potentially leading to increased permeability. Furthermore, the transfer of small molecules into the coacervate phase may be aided by the low surface tension that separates the phases that are rich in water and macromolecules.[234] The capacity to partition solutes into distinct complex coacervate phases is significantly influenced by unique intermolecular interactions between the solutes and macromolecules in the coacervate droplets, including hydrophobic interactions, hydrogen bonding, π-π stacking, and electrostatic interactions. For instance, an earlier research on the sequestration of methylene blue (MB), a cationic dye, into complex coacervates made of polyelectrolytes with opposite charges demonstrated how important electrostatic and π-π interactions are to this process. More specifically, compounds that are complex coacervate and can form both π-π and electrostatic connections with the solute exhibit a substantially higher sequestration of aromatic dye molecules compared to compounds that can only generate electrostatic interactions.[219,235] Furthermore, a different study on a hydrogen-bonding coacervate system suggests that the uptake of solutes into coacervates is facilitated by the creation of hydrogen bonds between solutes and polymers or by an increase in hydrophobicity inside the coacervate droplets.[219] Micelles are renowned for having a nonpolar center that increases the solubility of hydrophobic substances.[236,237,238] The process of polyelectrolyte-surfactant complexation has been thoroughly studied, with an emphasis on the variables that affect the complexes’ size and phase behavior. These variables include the molecular weight of the polyelectrolyte, its charge density, concentration, micelle surface charge density, surfactant chain length, ratio of the polyelectrolyte to surfactant, ionic strength, and temperature.[209,239,240,241,242,243,244,245,246,247] Furthermore, there are examples of work pertaining to the complexation of polyethylenimine (PEI) with SDS in the literature. For instance, Mezei et al. found that the size distribution and phase behavior of the PEI-SDS complexes can be significantly affected by various PEI and SDS mixing techniques.[217] An additional investigation into the interaction between PEI and SDS at a concentration of SDS below its critical micelle concentration (CMC) shows that the complexes may precipitate due to a rise in hydrophobicity and a fall in zeta potential.[248] A moderate dose of salt lowers the composition range over which BPEI-SDS complexes are kinetically stable, according to research on the effect of NaCl concentration on the phase behavior of PBEI and SDS.[249] Dye removal from solutions has received more attention recently as a result of industrial need to reduce the amount of color in effluent. According to a study on the removal of dyes from solutions using polyelectrolyte-surfactant complexes, the system’s charge is the primary factor influencing flocculation behavior and, consequently, dye sequestration.[250] An further investigation utilizing carbon nanotube-impregnated chitosan hydrogel beads demonstrates that the addition of carbon nanotubes considerably improves the elimination of congo red from solution.[251] C3Ms exhibit a number of desirable characteristics, including extended blood circulation, minimal cytotoxicity, high gene transfection efficiency, and adjustable imaging.[153,252,253,254,255,256,257] Significant understanding of the assembly of charged polymers into coacervate micelles has been gained over the last few decades. For instance, it was demonstrated that altering the lengths of the individual (block) polymers may be used to adjust the micellar size and stability.[27] By substituting charged polymers with charged metal-to-ligand coordination complexes, it was possible to regulate the stability and core structure of the micelle (polymeric versus oligomeric), as well as its final properties (fluorescence or magnetic properties for multimodal, like MR, imaging applications) by employing different metals.[65,256,257,258,259] Encasing distinct charged dendrimers from various generations into coacervate micelles-henceforth referred to as dendrimicelles-provided new information on the aggregation numbers and the minimum quantity of charges per core-unit.[260,261,262] The term “core-units” for coacervate dendrimers was used. Cyclodextrin-adamantane host-guest interactions were recently introduced into the core of C3Ms to form Cyclodextrin-based Complex Coacervate Core Micelles (C4Ms), in which monomeric europium(III) complexes can form core-units of the micelles, achieving an even more precise control on the minimum number of charges per core-unit, required for coacervation.[263] While smaller numbers of charges per monomeric unit, like six, were insufficient to form well-defined C4Ms and instead produced polydisperse aggregates, nine negative charges per monomeric core-unit resulted in spherical and well-monodispersed micelles with a 45 nm diameter. When a guest adamantane crosslinker (Ad-Glu-Ad) is mixed with low-charged monomeric units, which are unable to form well-defined stable micelles, dimeric and oligomeric core-units form, increasing the charge per core-unit and producing micelle formation with an increased stability against salt, pH, and competing free βCD. With the very subtle control over micelle formation and stability achieved by tuning the charge in monomeric and oligomeric core-units, we conjectured that supramolecular stimuli-responsive bridging linkers could be incorporated to enable reversible assembly and disassembly with coacervate-core micelles based on cyclodextrin. The most current developments in the realm of nanoscale PECs and their biological applications will be covered in this article. The terms PEC micelles (shown in Figure 7), polyion complex micelles, interpolyelectrolyte complex micelles, block ionomer complex micelles, and C3Ms are frequently used to describe these nanoscale PECs.[264] At now, these unique vesicular and micellar structures are referred to by four distinct labels. All of them originate from the makeup of the micellar microphase, which is made up of (i) block ionomer chains or (ii) complexed polyion chains; in other words, a microphase made up of what are known as (iii) interpolyelectrolyte complexes or (iv) a coacervate (if the microphase is liquid-like). There has been a surge in research on the theoretical and practical aspects of this unique class of nanoparticles since the early reports on micelles created by the electrostatic interaction of two oppositely charged polymers in the mid-1990s.[26,63,145] Reviews of polyelectrolyte-containing polymers in general,[265,266] double hydrophilic block copolymers,[267,268] block copolymer micelles,[269,270] and reviews of particular applications, like drug delivery,[43,84,85,271,272,273,274,275,276,277] have covered some of the work.
Fundamental concepts were established by Kataoka et al., who were pioneers in the field. They employed a slightly different micellar topology than the one shown in Figure 2, in which a neutral PEG block (poly(ethylene glycol)) was connected to both oppositely charged polyelectrolyte blocks (poly-(L-lysine) and poly(α,β-aspartic acid)).[26] The resulting monodisperse micelles have a hydrodynamic radius of 15 nm. The mixing ratio’s function in this investigation has previously been established. There was electroneutrality in the stoichiometric micelles. The ζ-potential values under stoichiometric circumstances were extremely low. They further demonstrated that mixtures of BCPs and homopolymers (HPs) form larger micelles than mixtures of two oppositely charged BCPs (double BCP micelles). Additionally, they demonstrated that double BCP micelles form only when the lengths of the oppositely charged blocks match, a phenomenon known as chain length recognition.[113] In the diblock copolymer, Kabanov et al.[63] employed PVP (Poly(N-ethyl-4-vinylpyridinium bromide)), a quenched polyelectrolyte, and an annealed ionic block. They demonstrated that micelles break down beyond 0.35 M NaCl. Over an extensive pH range, the micelles remained stable. The physics of the micelles is unaffected by the substitution of biological species for synthetic polyelectrolytes. The micelles’ spherical morphology was demonstrated by dynamic light scattering experiments conducted at various angles.[278] Cohen Stuart et al. made additional pioneering contributions by examining the impact of charge stoichiometry, ionic strength, and block length ratios on C3Ms generation and morphology (worm-like and spherical).[27] Since then, a great deal of fascinating work has been done in the field, which has been covered elsewhere, on the creation of novel C3Ms and their uses. A transitory phase separation that reorganizes into micelles after a specific relaxation period may occur when combining solutions of poly(acrylic acid) and poly(dimethylamino)ethyl methacrylate)-co-poly(glyceryl methacrylate), as demonstrated by Cohen Stuart et al.[279] These relaxation periods varied by as much as 104 over a salt range of up to 0.3 M NaCl, demonstrating how sensitive they were to salt. For micellar disintegration, the threshold ionic strength was 0.5 M NaCl, which is consistent with the value reported by Kabanov et al.[63] Harada and Kataoka have published two more publications that go into great depth into the core-shell architecture of the micelles utilizing both static and dynamic light scattering.[280,281] They used chicken egg white lysozyme and PEG-Pasp (poly(ethylene glycol)-poly(aspartic acid)) in their system. There were 273 apartments in the PEG block. Expressed as the number of aspartic acid groups over the total number of lysine and arginine groups in the lysozyme, micellar stability was seen over a broad range of mixing ratios, ranging from 1.0 to 2.67. Molecular weights ranging from 1 to 2 × 106 g/mol were discovered by static light scattering tests, and these values increased as the mixing ratio increased. According to the estimated aggregation numbers, when the mixing ratio increases, the number of PEG-Pasp molecules per micelle (62–122) increases while the number of lysozyme units per micelle (56–40) decreases. The micellar size increased in tandem with this, increasing from 23.6 to 32.9 nm hydrodynamic radius. Based on the micelle molecular weights and macroscopic loading ratios, they inferred that the micellar core remained constant (≈7 nm) across the whole mixing range, indicating that the increased corona chain stretching would be the cause of the micellar radius increase. According to theoretical and practical study on polymers grafted on a curved surface in a good solvent, the corona thickness was determined to be halfway between theoretical estimates on completely stretched and coil conformation.[282] They come to the conclusion that the electrostatics in the core are more important in the mechanism of micelle generation than the thermodynamic penalty of corona stretching. In the micellar core, Harada and Kataoka[283] also show chain length recognition. Their system consists of two diblock copolymers with oppositely charged ionic blocks (poly(aspartic acid) and poly(L-lysine)) and similar neutral blocks (PEG) of identical molecular weight (5K). For the ionic blocks, samples with chain lengths of 18 and 78 were made. Micellization could only occur in blocks with matching lengths. Block lengths 18 and 78 of polycation combined with block length 78 of polyanion, or three component mixtures, demonstrated that the eighteen unit species were not involved in the micellization process. Harada and Kataoka[284] established the suitability of these complicated coacervation core micelles as nanoreactors using a PEG-P-L-lysine/lysozyme system. They changed the ionic strength around 0.15 M NaCl, which is the threshold value, several times. Alongside these shifts in ionic strength, there was a corresponding and reversible variation in the lysozyme's enzymatic activity. By encapsulting anti-sense oligonucleotide molecules in the micellar core, Kataoka et al.[285] extended the concept of employing these micelles as biologically active nanoreactors. Cammas and Kataoka[286] highlighted a possible role in drug delivery. Numerous experimental parameters, such as block lengths, the homopolymer’s molecular weight, pH, ionic strength, mixing ratio, overall concentration, and the chemical makeup of the (diblock) copolymers, can be changed while working with complicated coacervation core micelles. The majority of these variables affect how the oppositely charged blocks interact with one another and ultimately the aggregation mechanism. The impact of block length variations on micellar stability is the main topic of this investigation. Two systems that differed in their chemical makeup were examined. Titrations of the mole fraction were carried out in a light scattering cell. Particle size, pH, and light scattering intensity were all measured during these titrations. The pH at which these titrations are carried out is around 6.7. Essentially, the mixing ratio may be adjusted at a nearly constant pH since at this pH, complexation only marginally alters the bulk pH. Over the last twenty years, substantial advancements have been made in our comprehension of block copolymer self-assembly in specific solvents.[270,287] When insoluble macromolecular blocks come together, multichain aggregates are created, and the soluble blocks guarantee the aggregates’ thermodynamic stability in the solution. The dimensions and form of assembled aggregates are determined by the length, solubility, and copolymer topology of the building pieces. A spherical micelle created by diblock AB copolymer in a solvent of choice is a common example. The corona of blocks A swelled in solvent S surrounds the micellar core of insoluble blocks B in such an aggregate, and the core-corona boundary is small in relation to the diameters of the core and coronal domains. The current state of the theory of polymer micelles is also highlighted in this Review, with an emphasis on recent developments and ongoing difficulties, where, I mostly talk about the theory of micelles created in a diluted solution by solvophobic/solvophilic diblock copolymers, and I just touch on the structures created by triblock terpolymers. Self-consistent field (SCF) approaches have advanced the theoretical modeling of polymer micelles.[50,51,288,289,290,291,292,293,294,295,296,297] The numerical SCF technique utilizes the mean-field Flory-Huggins theory of polymer solutions and integrates Edward’s formalism[298] to account for chain conformations.[299] The SCF models gave numerical dependences on the sizes of the coronal and core domains and the aggregation number in an equilibrium micelle as a function of the polymerization degree of the blocks (NA and NB) and the polymer-solvent and polymer-polymer Flory-Hugginson interaction parameters (χAB, χAS, and χBS). This method supported the thin interface approximation, which has been frequently utilized in future theoretical modeling, and anticipated the trends in the behavior of a polymer micelle. Subsequently, the numerical SCF approaches were expanded to include charged block copolymersand they are now a valuable resource for theoretical research on polymer self-assembly.[300,301,302,303] The scaling theory of polymer solutions encouraged further developments in the theory of polymer micelles.[304] The scaling model introduced the idea of correlation blob as an effective unit of a semidilute solution and accounted for the polymer density correlations, in contrast to a mean-field Flory-Huggins theory. A densely packed system of correlation blobs with size ξ(c) and interaction free energy ≃ kBT per blob is envisioned as a solution of flexible polymer chains with concentration c of monomer units, where kB is the Boltzmann constant and T is the temperature. (The sign "≃" here and below denotes a numerical coefficient’s correctness of equality.) In the case of theta and excellent solvent conditions, respectively, exponent ν = 1/2 and ν ≈ 3/5 define the dependency ξ(c) as ξ ∼ c−v/(3ν−1). At length scales far longer than ξ(c), interactions between distal regions of the chain are screened by other chains, whereas inside the correlation blob, a polymer chain's structure is mostly unaffected by its interactions with nearby chains. Consequently, every macromolecule becomes a Gaussian chain of n(c) blobs, with an average size of ≃ ξ(c)[n(c)]1/2. The interaction free energy of each chain is proportional to the number of correlation blobs, n(c), and may be expressed as ≃ kBTn(c). Semiflexible polymer solutions behave in a more complicated way. Furthermore, the theory[305,306] predicts an intermediate (mean-field) regime in a marginally excellent solvent in addition to the scaling regime of semidilute solution (with dense packing of the correlation blobs under good solvent conditions). Here, the power law dependences of the mean-field exponents for the solution properties are obtained. Here, we also summarize the concepts that led to the first scaling models of diblock copolymer micelles and concentrate on flexible macromolecules with Kuhn segment length on the order of monomer size.[49,307,308,309]
In this Review, I address a number of recent research publications that provide fresh insights into the design approaches for C3M dilution solutions through integrated measurement, analysis, and prediction from computational and experimental instruments. The evolution of (i) scaling relationships controlling C3M size, shape, and morphological transitions, as well as (ii) micellization dynamics in C3M formation/growth, chain exchange, and disassembly routes, are given particular focus. Focusing on dynamic and out-of-equilibrium phenomena, including as association and dissociation kinetics, pathway complexity, exchange dynamics, and reaction-assembly networks, I highlight recent advances and explore fascinating new research directions in this review. Furthermore, I explore future prospects and give concrete examples of how to apply polyelectrolyte structure-property principles to impart desirable physiochemical properties for delivery applications. The research described here use fully ionized strong polyelectrolytes with stoichiometric charge ratios, unless otherwise noted. I shall be using the term C3Ms throughout this review. A C3M is defined as a core-shell structure that forms in aqueous solutions and is maintained by the water-insoluble core, which is made up of complexed oppositely charged units, and its surrounding shell of neutral, water-soluble units (Figure 8). Thus, this definition excludes onion-type micelles-which are made up of a hydrophobic core, a coacervate inner corona, and a charged or neutral outer corona-as well as soluble (core-shell) complexes made up of a polyelectrolyte and an oppositely charged molecule stabilized by excess charge alone.[310,311,312] These micelles can also be formed by co-assembly of polymers, such as I-b-A and C(-b-S), as well as non-aqueous systems.[312,313,314,315,316,317,318,319,320,321,322] In strict terms, the word C3M denotes that the aggregate is a micelle, meaning that vesicles (for which the term C3Vs may be used) are not included, and that the C3M core is a coacervate, which is characterized as liquid-like in nature.[24] In this review, I use a definition of C3Ms that goes beyond what the name itself suggests for practical reasons. Therefore, vesicles and structures with a solid or crystal-like core will also be referred to by the term C3M; in other words, their macroscopic counterparts would be a precipitate and a crystal, respectively.
2. Utilizing Nanotechnology for Drug Administration: Method of Drug Delivery and Nanotechnology of Polymer Micelles
When administered orally or intravenously, chemotherapy drugs in solution or polymer solution have poor pharmacokinetics and a limited therapeutic window (Figure 9A). These substances quickly attain the highest concentration that may be tolerated before being removed from the circulation. A medication formulation that maximizes patient benefits should release at the lowest possible effective concentration gradually. As a drug delivery vector or carrier, nanotechnology is expected to be crucial in meeting these requirements (Figure 9B). Drug carriers based on nanotechnology, such as carbon nanotubes, lipid/solid nanoparticles, polymer nanoparticles, dendrimers, polymer micelles, and polymer-drug conjugates, provide many advantages over traditional techniques. Therapeutics based on nanotechnology have been shown to increase patient compliance, lessen toxicity in healthy tissue, and increase drug efficacy. Nowadays, a lot of these nanoparticles are being used in cancer treatments.[323] Quan et al.[324] have examined a list of these nanotechnology-based formulations’ clinical and preclinical investigations. It is vitally important to construct a universal nanotechnology formulation that combines chemotherapy drugs. The following characteristics would characterize a successful formulation that serves as an effective therapeutic carrier for cancer therapies: (a) stability in the physiological environment; (b) longer circulation life time; (c) avoidance of opsonization and the reticuloendothelial system (RES) process; (d) promotion of endocytosis; and (e) enhancement of tumor uptake. The coupling of antibodies to the nanoformulations can further improve the specificity of these formulations, and the immunoconjugated formulations will exhibit superior therapeutic efficacy compared to other drug formulations (Figure 9C).
There are two ways to deliver chemotherapy agents or anti-cancer drugs to tumors: passive delivery and active delivery. Figure 10 provides a visual illustration of these methods. Diffusion into tumors or angiogenic tumor vasculatures, which contain leaky arteries with narrower gaps of 100-2000 nm, is how passive targeting occurs. Medicine-loaded nanoparticles, or nanoformulations, are better at retaining their medicine within tumors due to their increased interstitial access. Because of the compromised and inadequate lymphatic drainage, the leaky vasculature facilitates the uptake of nanoformulations by the tumors, which become trapped inside, and enhances the Enhanced Permeation and Retention (EPR) index. Furthermore, the passive targeting of nanoparticles to tumors is determined by their size and charge.[324,325,326,327] On the other hand, the coupling of nanoparticles to immunogens (antibodies or targeting moieties) is employed in the active targeting mode. Compared to a standard drug-loaded nanoparticle system (passive targeting), a tumor-specific antibody conjugated nanoparticle system (active targeting) can improve medication delivery. First, longer circulation due to the EPR effect increases the transit of nanoparticles absorbed by the tumor site. Furthermore, the targeting moiety aids in the process of endocytosis, which generally boosts the uptake of nanoparticles for a better therapeutic outcome.[328,329] By significantly increasing the uptake of nanoparticles in cancer cells, this targeting strategy has demonstrated improved therapeutic effects in animal models.[330,331] The internalization of nanoparticles plays a significant role in gene, siRNA, DNA, and biomacromolecular delivery in addition to anti-cancer medication delivery. Therefore, the effectiveness of delivering medications, genes, and biomolecules is increased when controlled and targeted deliveries are combined. In this review contemporary nano-formulations are also highlighted, particularly polymer micelle nanosystems, which have been identified for their unique properties.
The administration of chemotherapeutic drugs, imaging agents, biomacromolecules, and radionuclides in a tumor-targeted manner by polymer micelle nanotechnology may improve cancer therapy outcomes and detection.[332] A few clinical trials of various polymer micelle nanotechnology therapeutics are now being developed in this direction.[333] To put it simply, block or graft copolymer spontaneous self-assembly forms the hydrophobic core of polymer micelles, which is then coated with hydrophilic chains[43,334] Protecting hydrophobic (lipophilic) medications and enhancing their solubility and stability is the main purpose of polymer micelles. A 30,000-fold improvement in aqueous solubility has been demonstrated.[335,336] A research has shown that curcumin’s stability can be increased by a factor of 6-8 through the use of β-cyclodextrin, poly (β-cyclodextrin), and polymer nanoparticle curcumin assemblies.[337,338,339] When the drug molecules split into the hydrophobic polymer micelle core and an exterior medium, a protective hydrophilic shell interface, takes over, the stability increases. Many hydrophobic core-forming, biocompatible, and biodegradable polymer micelles are being used in drug delivery applications, including poly(ethylene-co-propylene-co-ethylene oxide) (PEO-b-PPO-b-PPO) or poly(ethylene-co-propylene oxide) (PEO-b-PPO), poly(lactic acid) (PLA), poly(D,L-lactide) (PDLLA), poly(lactic-co-glycolic acid) (PLGA), poly(ε-caprolactone) (PCL), poly(hydroxybutyrate) (PHB), and poly(β-benzyl L-asparate).[340,341,342,343,344,345] These polymers can only form micelles at a certain concentration (also known as the critical micelle concentration, or CMC). For these uses, the polymer micelle with a lower CMC value is a preferable option. Different models of the creation of polymer micelles based on their self-assembly mechanisms are schematically presented in Figure 11.
3. Standard Polymer Micelles
Many di-block or tri-block copolymers, either synthetic or natural, that are biodegradable and biocompatible have been used to load different medications and biological molecules. Among these, micelles produced by poly(lactic-co-glycolic acid) (PLGA) are widely recognized. Furthermore, the FDA has cleared the parent PLGA polymer for usage in both industry and medicine. For drug delivery applications, a variety of structurally diverse nanoformulations are available, including comb-like amphiphilic PLGA-b-poly(ethylene glycol) methacrylate (PLGA-b-PEGMA) copolymer, PLGA-b-poly(ethylene glycol)-b-PLGA (PLGA-b-PEG-b-PLGA) tri-block copolymer, and three- and four-arm star-shaped PLGA-b-PEG block copolymer micelles.[346,347,348] Furthermore, using a vinyl pyrrolidone shell layer, Park et al.[349] recently created a surface cross-linking PLGA-b-PEG copolymer to increase the general stability of polymer micelles. By conjugating to PLGA polymer, a naturally occurring carbohydrate polymer, such as hyaluronic acid (HA) copolymer, can be used as target-specific micelle carriers for doxorubicin (DOX).[350] This formulation demonstrated a 5.2-fold increase in cytotoxicity in the cancer cells over free DOX (IC50 value of DOX-HA-g-PLGA = 0.67 mg.mL-1 and free DOX = 3.48 mg.mL-1). This allowed loading of 4.8-7.2 wt.% DOX (i.e., DOX-HA-g-PLGA). Similarly, increased cellular absorption of DOX has been demonstrated in a mixed micelle nanoformulation of drug-resistant cancer cells loaded TPGS/PLGA-b-PEG-b-FOL (TPGS = α-tocopheryl succinate esterified to polyethylene glycol 1000 and FOL = folate). This has led to a higher degree of death in these cells. By using folate receptor-mediated intracellular delivery, PLGA coated with poly(L-lysine)-PEG-folate conjugates has been nanoformed and has demonstrated improved cellular uptake.[53,351]. According to a research, curcumin’s therapeutic benefits were enhanced in cisplatin-resistant ovarian (A2780CP) and metastatic breast (MDA-MB-231) cancer cells when PLGA formulations with poly (vinyl alcohol) (PVA) were added (Figure 12).[339]
4. An Alternative Kind of Polymer Micelle Nanoparticles
The stability, solubility, surface charge, and kind of functional groups that enable the release of the encapsulated drug and its targeting qualities to tumor cells define the attributes of all drug delivery carriers. Pluronic polymers, often referred to as PEO-b-PPO-b-PEO or poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide), are recognized as 40 nm-diameter micelle drug carriers that form readily. These micelle nanocarriers allow passive targeting to the solid tumor and can also improve the solubility of several hydrophobic anti-cancer medications. Research has additionally shown that pluronic micelles boost the lethal effects of a variety of anti-cancer medications by sensitizing cancer cells to P-glycoprotein (P-gp) activity inhibition caused by ATP depletion.[352] One excellent example is the poly(ethylene oxide)-linked poly(ethylene imine) (PEO-l-PEI) micelle gel, which binds oligonucleotide (ODN) molecules and improves their distribution via receptor-mediated delivery. ODNs are generally effective therapeutic agents that are severely enzymatically degraded by nucleases. ODN encapsulation in PEG/PEI micelles significantly suppressed tumor growth in vivo, decreased ODN concentrations, and controlled the growth of ovarian cancer cells (A2780).[352,353] Similarly, it has been demonstrated that stable micelle formulations of the 5’-triphosphates of gemcitabine (dFdCTP), floxuridine (FdUTP), and cytarabine (araCTP) in PEG-l-PEI networks accumulate more quickly and stop tumor growth in vivo.[354] At a dosage of 30 μM curcumin, curcumin-casein micelle complexes not only showed increased cytotoxicity against HeLa cells but also had the ability to cause damage to the cell nucleus through apoptosis.[355] Additionally, these complexes were taken up by the cells more quickly. It has been demonstrated in recent studies[337,338] that curcumin, a natural anti-cancer and cancer-prevention drug, works well in therapies involving self-assembly or nano self-assembly formulations of poly(b-cyclodextrin) or b-cyclodextrin. In human ovarian cancer cell line (OVCAR-3) PEG/PDLLA-Taxol combination (Genexol®-PM) is a formulation with high anti-tumor effectiveness.[356] In a different study, it was demonstrated that triptolide (TP) loaded PDLLA/PEG nanocarriers, administered intravenously at doses of 0.0375, 0.075, and 0.15 mg/kg, effectively inhibited tumor development with respective inhibition rates of 42.5%, 46.0%, and 49.9%. Drugs such as doxorubicin, ellipticine, and paclitaxel can be encapsulated using hydrolyzable polyesters of PCL and PDLLA.[357,358,359] Additionally, new biodegradable triblock copolymer micelles made of poly(ethyl ethylene phosphate) (PPE, polypho- sphorus ether) and PCL were created as drug carriers.[360] With an increase in PPE molecular weight, these micelles exhibit enhanced drug loading efficiency and are cytocompatible, biodegradable, and compact in size. These micelles' sophisticated characteristics provide them greater flexibility, and by varying the side group conjugation to phosphorus, one can modify their physico-chemical characteristics.[361] Tamoxifen and paclitaxel uptake were shown to increase 2.5-fold when a different biocompatible micelle, poly[2-(methacryloyloxy)ethyl phosphorylcholine] or MPC, was coupled with a folate targeting moiety to poly[2-(diisopropylamino)ethyl methacrylate] (DPA) (i.e., MPC-DPA-FA)].[362] Furthermore, cationic polymer micelles can be efficiently mediated by endosomal disruption or disintegration (also known as the “proton sponge” effect); yet, in vivo investigations frequently failed because of the micelles’ quick excretion from the bloodstream. Consequently, cationic polymer micelles known as polymerosomes were created, which wrap neutral polymers like PEG to block the positive charge. For instance, on LHRH receptor-overexpressing ovarian cancer cells (A2780), polyelectrolyte complex (PEC) micelles containing luteinizing hormone-releasing hormone (LHRH) peptide demonstrated improved cellular uptake by raising VEGF siRNA gene silencing efficiency via receptor-mediated endocytosis.[363] MePEG-b-PCL micelles conjugated with epidermal growth factor (EGF) can be administered at a concentration 13 times more effective than free EGF.[364]
5. Polymer Micelle Nanoparticles that Are Acid-Cleavable and pH Sensitive
These micelles’ primary benefit is that the medications they contain burst when placed in acidic intracellular compartments like lysosomes or endosomes. By using a pH-sensitive intracellular drug delivery system, these formulations increased anti-tumor activity.[35] Furthermore, it has been demonstrated that their folate conjugation increases in vivo anti-tumor activity at lower effective levels.[365] Furthermore, compared to free and typical polymer micelles, pH-sensitive micelles poly(L-hystidine)-b-PEG and PLA-b-PEG-l-FOL (PHSM-f) were superior.[366] After 27 days from the initial intravenous injection, the in vivo studies using a sensitive micelle system similarly show particle accumulation at the tumor site and tumor regression that is 4-5 times larger than free DOX. According to one study, the half-life of DOX in pH-sensitive micelles rose from free DOX in PBS and plasma media by approximately six times. Their absorption at pH 6.8 was five times more than at pH 7.4, suggesting that the drug release caused by the lower tumor pH was successful following the EPR effect’s accumulation of micelles. In comparison to normal pH 7.4, another unique tetra-block copolymer, poly(ethylene glycol)-b-poly(L-histidine)-b-poly(L-lactic acid)-b-poly(ethylene glycol), can cause the release of DOX at pH 6.8 (tumor acidic pH) or pH 6.4 (endosomal pH).[367] The molecular weight of the PLA block present in the tetra polymer determines this triggering or burst release effect, which may be a useful treatment for solid tumors or for delivering cytoplasmic cargo in vivo. In order to speed up the internalization process, a novel formulation consisting of DOX in PDLLA-b-PEG-b-poly(L-histidine)-TAT (transactivator of transcription) micelle was only able to expose TAT at a slightly acidic tumor extracellular pH.[368] In a nude mouse model, these micelles were tested with xenograft models of human lung tumor A549, human breast tumor drug-sensitive MCF-7, and human ovarian tumor drug-resistant A2780/AD. All tumors showed minimal weight loss and significant regress in size following three bolus injections at a dose of 10 mg DOX per kg body weight, spaced three days apart. Long-term drug release was made possible by the coupling of pharmaceuticals to acid-cleavable micelle polymers.[369] Because doxorubicin-conjugated PLLA-mPEG micelles were absorbed by cells and simultaneously released cleaved doxorubicin into the cytoplasm from acidic endosomes, they were more effective.[369] According to a triblock copolymer conjugated with DOX through the copolymer’s end OH groups, hydrazone linkage was broken in an acidic environment.[370] The degree of cellular uptake of micelle conjugated DOX and its distribution in the cytoplasm, endosomal/liposomal vesicles, and nucleus, as well as the localization of the free drug within the nucleus, were validated by flow cytometry and confocal imaging, which also confirmed this behavior.
6. Micelle Nanoparticles of Cross-Linked Polymers
Different polymer micelle nanoparticles have different ways of controlling the triggered release of the active medicinal ingredients; nonetheless, as delivery vehicles, the majority of these polymer micelle nanoparticles have disadvantages. For instance, shortly after being injected into the bloodstream, paclitaxel was easily separated from the micelle nanoparticle. [371] The breakdown of α- and β-globulin micelles and the transfer of paclitaxel to the many lipid components and carriers in blood may be the cause of this separation.[372] Creating cross-linked biodegradable micelles is one potential solution to this problem with polymer micelle nanoparticles.[373] The cross-linked corona of these micelles can firmly cover drug molecules, and biodegradable cross-linking can release the drug from the micelles in a regulated way. High stability is shown by core micelles cross-linked with divalent metal cations, although they also show pH-dependent swelling/collapse behavior.[374] These systems demonstrate a delayed and persistent release of platinum from the cisplatin-loaded cross-linked micelles in physiological saline, with an impressive platinum loading efficiency of approximately 22% weight/weight. A new formulation based on poly(N-isopropylacrylamide), N-hydroxysuccinimidyl esters (NHS), a-methoxypoly(ethylene oxide)-b-poly[N- (3-aminopropyl)methacrylamide]-b-poly[2-(diisopropylamino)ethyl methacrylate] (mPEO-PAPMA-PDPAEMA), and poly(N-isopropylacrylamide) prevented micelle dissolution due to dilution effects.[375]
7. Innovative Approaches to Polymer Micelle Nanotechnology
Compared to core-shell block copolymers, double-hydrophilic block copolymer-based micelles exhibit more exterior hydrophilic behavior that resembles biological fluid.[30,145] The chemotherapeutic drug attaches to the first hydrophilic charged block copolymer, while the second hydrophilic block enables steric stability. Excellent examples of copolymers that can bind oppositely charged species are lysozymes, proteins or peptides, nucleic acids, medicines (such doxorubicin and cisplatin), and polyaminoacid-b-polyethylene oxide.[281,376,377,378] Polymer micelle nanoparticles with ultra-sound sensitivity represent an additional approach for localized medication delivery to malignancies. When exposed to ultrasonic waves, these micelles break down into unimers, which intensifies the disruption of the cell membrane.[379] A brief exposure to high-frequency ultrasound, lasting only 15 to 30 seconds, significantly boosts the intracellular uptake of DOX from pluronic micelles.[380] In cancer therapeutic treatments, magnetic nanoparticle-based micelles serve as both external magnetic field guides and drug carriers.[381] Another option is to create micelle-magnetic nanoparticles. Such formulations have recently been designed to achieve many biological activities using a single formulation.[382] Along with drug administration, these formulations can be used for magnetic resonance imaging (MRI), visual targeting, magnetically targeted photodynamic therapy, targeted thermosensitive chemotherapy, and applications involving luminescence, near-infrared light, and multi-model imaging.[383,384,385,386,387,388,389,390] Regarding drug administration, imaging, and hyperthermia properties, a new formulation consisting of an iron oxide nano-core supported by a multi-layer covering may prove more feasible. However, its usage in cancer treatment applications is limited by the larger hydrodynamic diameter (> 200 nm) in aqueous medium.[391] As a result, Chauhan’s group have been creating a unique formulation of magnetic nanoparticles with an iron oxide core coated in β-cyclodextrin (CD) and pluronic F127 polymer (F-127), which has the ability to conjugate antibodies and load anti-cancer drugs, making it useful for a variety of multifunctional applications (Figure 13).[197] Smaller particle size, comparatively reduced protein binding, higher drug loading efficiency, and improved particle uptake in cancer cells without compromising intrinsic magnetic properties are some of the benefits of this formulation.
8. C3M Structure-Property Relationships
For C3Ms to be effective as medicinal delivery vehicles, their size and structure must be carefully regulated. Nanoparticles larger than 200 nm are more likely to accumulate nonspecifically in the spleen and liver, while those smaller than ∼10 nm may be eliminated from the bloodstream by the kidneys.[37] Apart from preventing renal clearance, the design of nanoparticles can significantly influence their biodistribution and cellular absorption. Recently, Ridolfo et al. compared the cellular uptake of amphiphilic spherical, worm-like, vesicular, and tubular nanoparticles to investigate the implications of shape in biological environments.[392] Because higher aspect ratio nanoparticles diffused more quickly than lower aspect ratio nanoparticles, they discovered that higher aspect ratio particles-such as worms and tubes-performed better than spheres and vesicles. Since these observations do not depend on the assembly mechanism, they should hold true for both C3Ms and amphiphilic micelles. For these reasons, creating effective C3M encapsulants requires exact control over C3M size and morphology.
Much less is known, from both experimental and theoretical viewpoints, concerning the connection between the C3M structure and the molecular/environmental design factors, despite a great deal of investigation on the structure and behavior of C3Ms.[59,67,393,394] However, in order to forecast and create the C3Ms for improved transportation and cellular absorption efficiency, this link is essential.[137,395,396] Similar to how amphiphilic molecules self-assemble, chain lengths, the ratio of core to corona blocks (Ncore to Ncorona), and the interfacial tension between coacervates and aqueous media are the main factors influencing the structure and morphology of C3Ms, including core radius (Rcore) and aggregation number (Nagg).[32,106,107,394,397,398,399] A scaling theory for C3Ms was put forth by Rumyantsev et al. using two diblock copolyelectrolytes that are weakly and oppositely charged (i.e., an AB + AC system, where A is the neutral hydrophilic block and B and C are the oppositely charged blocks). By taking into account two free energies contributed by the corona chain crowdedness and the excess energy at the core-corona interface, they were able to demonstrate the morphological transition and the structure of C3Ms that was dependent on ionic strength.[59] Marras et al. conducted an investigation of C3Ms from an experimental standpoint. They found that C3Ms, which are composed of diblock copolyelectrolytes and oppositely charged biomolecules (AB + C system), exhibit scaling relationships that are consistent across all AB + C systems.[394] While these research offer light on the scaling relationship for C3Ms, there is still a lack of a quantitative comparison between experimental data and theoretical explanation. Until far, the AB + C system has been mostly investigated experimentally as a delivery vehicle model; nevertheless, an extra degree of flexibility to ascertain the C3M structure is produced by the conformation and spatial distribution of C homopolyelectrolytes.[25,393] Furthermore, the theoretical forecast for the applications of the AB + AC systems and scientific comprehension is dubious due to the dearth of experimental data on them.
8.1. Steady-State Properties
Positively and negatively chargeable monomer concentrations (n+, n−) must be balanced for complex coacervate core micelles to form at (near-)charge stoichiometric compositions. PCC stands for preferred micellar composition, which is the composition in which C3Ms are most prevalent. A PMC of
is generally associated with polyelectrolytes that have a same degree of dissociation, α. When the C3Ms are made using weak polyelectrolytes with a pH-dependent charge density, the PMC, which is pH-dependent, may differ greatly from f+ = 0.5. The phosphate-to-amine molar ratio P/N, which should not be confused with the molar ratio of negative to positive charges, N/P, is a frequently used alternative notation for the composition of DNA-carrying C3Ms. We shall utilize f+ throughout this Review for clarity’s sake. The architecture of the polymeric components, their (relative) lengths, the mixing fraction, and a number of other parameters all affect the morphology and size of C3Ms.[27,106] Ncorona/Ncore, the ratio of the lengths of the corona- and core-forming blocks of the dbp, and solution ionic strength,[106] can be effectively utilized as handles to adjust the coassembled colloids’ morphology. Under otherwise identical conditions, a transition from spherical (Ncorona/Ncore ≥ 0.9−1.0) to wormlike micelles (WLM) to vesicular structures (Ncorona/Ncore ≤ 0.1−0.2) is expected with decreasing Ncorona/Ncore.[27,400] Diblock copolymers with extremely short neutral blocks relative to the length of the charged blocks can be used to create vesicles. The size of spherical C3Ms increases as the total molecular weight of the component diblock copolymers rises. The length of the corona-forming block is just slightly responsible for this; the majority of the cause is the longer length of the core-forming block.[30,106] A single dbp and an oppositely charged homopolymer (S-C3Ms) make up C3Ms, and their size is mostly unaffected by the length of the homopolymer[106] and dendrimer production in dendrimicelles.[261] This knowledge has been used to individually adjust the quantity of dendrimers contained in each C3M.[261] Micellar stability is influenced by the constituent polymers' chemical makeup and nature, and it is weakened at high ionic strengths. This is due to the fact that with higher salt concentrations, the cohesive connections between chains that are oppositely charged diminish. In addition to serving as a plasticizer, the salt also causes the core to melt at higher salt concentrations, even if it could otherwise be glassy.[117] Thus, once a specific ionic strength is exceeded, the majority of C3Ms breakdown entirely. The critical ionic strength (Icr, also known as cs,cr) is dependent on the (relative) length of the polyions in addition to their kinds.[106,122,401] Large C3Ms with a distinct core and corona were generated by pairs of oppositely charged block copolymers with equal cationic and anionic block lengths, as reported by Harada and Kataoka in 1999. Instead, only the smallest feasible neutral complexes were created by mixing dbps of unmatched polyelectrolyte block lengths.[283] Van der Burgh et al. discovered that in order to prevent macroscopic phase separation, the core-forming blocks shouldn't be very long in relation to the corona-forming blocks.[30] Precipitation mitigation was recommended for a Ncorona/Ncore > 3. Double dbp C3Ms (D-C3Ms) and S-C3Ms were compared by Hofs et al.[25] Compared to the D-C3Ms (RH = 18 nm, Icr = 50 mM NaNO3), the S-C3Ms were found to be larger (RH = 26 nm) and more resistant to salt (Icr > 50 mM NaNO3). Since the cationic homopolymers were longer than the cationic blocks of the positively charged dbp, there are a number of possible explanations for the observed discrepancies, including shorter core-forming blocks in the D-C3Ms and higher crowding. According to Van der Kooij et al., longer (anionic) homopolymer chains result in an increase in the critical ionic strength for S-C3Ms until a plateau value is attained.[106] Higher dendrimer production[261] and cationic block length of cationic-neutral dbps[106] also result in increased salt resistance. One useful method for keeping an eye on the (dis)appearance of C3Ms is light scattering. The weight concentration and mean mass of the scattering particles are directly correlated with the static scattering intensity. A prominent peak appears around charge stoichiometric conditions as f+ is changed, indicating the PMC where the greatest amount of micelles is present. Based on these findings, Van der Burgh et al. created a speciation diagram that illustrates how C3Ms form and break down as a function of f+ into individual (co)polymers and tiny complexes known as soluble complex particles (SCPs).[30] Recently, Cingil et al. used the optical response of mechanochromic polymers with noticeably different fluorescence profiles in monomeric, complexed, and condensed states to study in detail the creation and evolution of these SCPs into micelles (Figure 14).[29] In order to achieve this, a cationic-neutral dbp and a conjugated anionic homopolymer were combined at different mixing ratios ranging from f+ = 0 to f+ = 0.65 (Figure 14A). The molecularly dissolved mechanochromic polyanion emitted blue light (I in Figure 14C) when the copolymer was absent at f+ = 0. The polyanion is stretched when it binds the additional cationic-neutral dbp in a bottlebrush-like fashion when f+ increases from 0 to slightly less than 0.3. A drop in photoluminescence intensity (Figure 14C) and an increase in hydrodynamic radius and scattering intensity (Figure 14B) indicate the creation of these complexes. New vibronic bands (II in Figure 14C) indicated that multiples of these SCPs condensed into larger micelles at a further increase beyond f+ = 0.3. These are the result of many mechanochromic polymer chains in the multimolecular micellar core being in close proximity to one another. I anticipate more in-depth research employing (single-molecule) fluorescence tools and other cutting-edge characterisation techniques on the micellar state diagram, which has gotten little attention up to this point.
8.2. Morphological Transitions
C3M morphology may be determined by the length of each polymer block (A, B, or C in Figure 15). For both the (AB + AC) and (AB + C) systems, self-assembly produces spheroidal micelles approximately if the length of the neutral block is greater than that of the charged block, or if the neutral/charged length ratio (N/C) > 1 [26,30,110] Other morphologies can arise when N/C < 1. Since the modest amount of neutral polymer does not drive microphase separation, these assemblies are aggregates and complexes similar to bulk assemblies for the majority of (AB + C) systems.[30] Nonetheless, in the N/C < 1 regime, intriguing morphologies can arise for (AB + AC) systems. In contrast to spheres for the same system with N/C ∼ 2, the Kataoka group discovered polyelectrolyte complex vesicles when N/C ∼ 0.5.[398,402,403] When N/C < 1 with a very low poly(ethylene oxide) (PEO, commonly known as poly(ethylene glycol, or PEG) weight fraction, cylindrical and planar assemblies have also been seen.[403] According to C3M theory, morphology scales with degree of ionization (f) for N/C << 1.[59] Lamellae, cylinders, crew-cut spherical micelles (corona thickness << core size), and eventually star-like spherical micelles (corona thickness >> core size) are the morphological changes that occur when f grows. Micellarization is prevented and a solution of unimers arises at very low f, where the free energy gain of complexation is on the order of thermal energy. Similarly, limited complexation is observed in the experimental data for very short charged blocks.[30,404] Nonlinear polymer topologies, nonstoichiometric charge ratios,[405,406] salt concentration,[106,110] chirality,[407] and stimuli-responsive polymers are additional factors that can impact micelle morphology.[408,409] The concept of block length ratio suggests that despite the radically divergent driving mechanisms of self-assembly, there are commonalities, akin to the classic packing parameter arguments in hydrophobically driven systems.[410,411]
Based on N/C, morphological patterns are evident in the works reviewed above; however, due to inaccurate polymer synthesis, it is challenging to examine systems experimentally at the exact transition (N/C = 1). According to recent simulations by the Sing group 48, the length ratio is not the only factor influencing the separation of the macro- and microphases in a (AB + C) system with precisely matched block lengths in the block copolymer. They predict that for a N/C = 1 system, macrophase separation happens at shorter polymer lengths, but that in low salt circumstances, microphase separation (micelles) is expected when length goes past a critical point. Although it is difficult to duplicate experimentally, poly(allyl glycidyl ether) (PAGE)[82,397] is a reactive polymer system that is a good candidate to do so since reactive polymers are effective instruments for creating neutral, cationic, or anionic polymers that are architecturally equivalent. Although the macromolecules under consideration are primarily synthetic polymers, biomolecules serve as the inspiration and driving force behind the creation of hydrophilic PCMs and can provide new levels of complexity. One such example is the way that polypeptide chirality regulates the phase of bulk polyelectrolyte complexes.[407] PCMs that incorporate nucleic acids-often referred to as “polyplexes”-are much sought after for the therapeutic delivery of payloads such as plasmid DNA or small interfering RNA (siRNA).[412] Since nucleic acids are phosphate-backed biopolymers that are densely negatively charged, they can readily take the place of a charged block in the above-described generic systems design. Although double-stranded nucleic acids have a lot greater charge density and are substantially more rigid (about 50 times longer persistence length) than single-stranded nucleic acids, this is because of the creation of a double helix and the existence of the complementary strand. Single-stranded nucleic acids behave similarly to flexible hydrophilic polymers.[413,414] A morphological shift inside PCMs is driven by the conformational variations between single- and double-stranded nucleic acids. Due to variations in stiffness and charge density,[415,416] DNA hybridization in a bulk system of DNA + poly-L-lysine (pLys) drives a phase transition between solid precipitates for double-stranded DNA (dsDNA) and liquid-like coacervates for single-stranded DNA (ssDNA).[415] Spheroidal micelles containing different charged polymers are created when block copolymers combine with single-stranded DNA[107,404] or RNA.[417] On the other hand, the double-stranded version can cause a shape change to worm-like cylinders[107,404,418] with DNA or interfere with micellization as observed with RNA.[417] dsDNA micelles are generated as worm-like cylinders with DNA lengths ranging from 10 base pairs (bp) to 1000s of bp when N/C ≫ 1.37, [107,418] nonetheless, globular micelles can still form with dsDNA when N/C is approximately 1, since there is insufficient density in the PEO corona to compel the formation of long cylinders.[418] Because hybridization can drive therapeutic action, understanding the difference between single-stranded and double-stranded nucleic acids is crucial for the delivery of medicines.
Time-resolved studies are helpful in understanding how one micellar morphology changes into another upon fluctuations in salinity concentration, temperature, and composition, among other factors. They are particularly effective in studying the formation and dissociation of C3Ms. Such transitions are generally produced for micelles built from amphiphiles upon addition of a cosolvent and temperature variations, which impact the solvency and therefore the effective packing parameter of the amphiphiles. Changes in the mixing fraction and concentration of salt can further adjust the morphology of C3M by affecting the electrostatic interactions between the constituent building blocks and, consequently, the micellization free energy. For double diblock copolymer (dbp) C3Ms (D-C3Ms) with the ionic blocks poly(sodium 2-acrylamido-2-methylpropanesulfo-nate)293 (PAMPS) and poly(3-methacryloylaminopropyl tri-methylammonium chloride)215 (PMAPTAC) in the micellar core and the short zwitterionic block poly(2-methacryloyloxy ethyl phosphorylcholine)22 (PMPC) in the corona, Takahashi et al. have demonstrated how a dbp excess can cause a vesicle-to-micelle transition for D-C3Ms.[419] Vesicles form (d = 200 nm in 0.1 M NaCl) close to charge stoichiometry because Ncorona/Ncore ∼ 0.1. The vesicles become smaller spherical micelles (d = 44 nm) when one of the dbps is supplied in excess (f+ = 0.4 or 0.8). When the minority dbp is added, the transition can be reversed and charge stoichiometry is restored. The authors created wormlike D-C3Ms from PAMPS198-b-PMPC22 and PMAP-TAC206-b-PMPC22 at f+ = 0.55 (d = 35 nm in 0.01 M NaCl) for a follow-up investigation.[419]According to TrSAXS, these cylinders disintegrate into smaller, identically sized micelles over a 150-second period when extra PAMPS198-b-PMPC22 is added to attain f+ = 0.35. For such worm-to-sphere migrations, two possibilities have been put up. The WLM may fragment randomly at any point, or the end of the cylindrical micelles may pinch off to release spherical micelles. The first case has been seen before using amphiphilic polymer cylindrical micelles.[420] However, Takahashi et al. discover that these WLMs split into spherical C3Ms via random scission along the cylinders’ length since they see a sharp decrease in intensity that is in line with the cylinders breaking up into smaller, spherical micelles. In order to reestablish f+ = 0.55, the insertion of PMAPTAC206-b-PMPC22 reverses the structural transition. Furthermore, compared to the cylinder-to-sphere transition, the sphere-to-cylinder transition happens more slowly. First, short cylindrical micelles form in coexistence with the preexisting spheres during the 2.5 ms experimental dead period. Then, the growth of these young cylinders is a gradual process that takes up to a day to achieve a stable state, with hardly any change seen for the first 10 minutes. The reason for the noticeable variation in the reverse transition is a high activation energy linked to a sluggish expansion of the cylinder. Adjusting the ionic strength offers an additional handle for morphology transition since salt affects the interaction strength of C3Ms. The morphological changes seen for mixed micelles containing PAA and poly(N-methyl-2-vinylpyridinium iodide)41-block-poly(ethylene oxide)204 (PM2VP41-b-PEO204) at elevated salt concentrations (cNaCl > 0.1 M) were reported by Van der Kooij et al.[106] The micelles exhibit a change from spherical to more elongated shapes for relatively short homopolymer chains (NPAA < 50), which is followed by their eventual disintegration over the threshold ionic strength. The authors explained that the entropic penalty resulting from the stretching of the polyanion chain is the reason for the lack of this morphological transition in C3Ms with long homopolymer chains (NPAA < 130). Because C3Ms’ building blocks are thermoresponsive, variations in temperature can also cause morphologies to transition. The usage of poly(N-isoproylacrylamide) (PNIPAM or PNIPAAm) as a neutral block is a well-established example, with an approximate critical solution temperature of 32 0C. Therefore, a (partially) reversible transition into core-shell-corona structures was generated by raising the temperature of aqueous solutions of D-C3Ms of PAA49-b-PNIPAM70 and PLL50-b-PEO113 above this LCST.[408] On the other hand, the PAA49-b-PNIPAM70/PLL50 system experiences the (wormlike) S-C3Ms aggregating. These then reorganized into spherical C3Ms with bigger diameters than those seen before heating when they cooled. In contrast to a previous investigation by Voets et al. on D-C3Ms of PAA55-b-PNIPAM88 and PM2VP38-b-PEO211,[421] Shah and Leon concluded that the coacervate was still present in the core that collapsing PNIPAM chains covered at T > LCST.[408] Voets et al. described core-shell-corona structures that featured a PNIPAM core with a coacervate shell.
8.3. Sturdy, Long-Lasting, and Stimuli-Sensitive Pharmaceutical Micelles
For the treatment of life-threatening conditions like cancer, targeted drug delivery is especially crucial because cytostatic medicines can have extremely harmful side effects.[422,423,424] In order to meet the needs for selective and tissue-specific drug administration, polymeric micelles are currently the subject of intensive research as drug delivery systems[43,66,376,425,426,427,428,429,430,431,432,433,434,435,436] Their hydrophobic core, which has a comparatively great capacity to accommodate hydrophobic agents-which are typically challenging to formulate-is their most alluring property. Numerous highly effective but hydrophobic medications, including doxorubicin (DOX)[436,437,438,439,440,441,442] paclitaxel (PTX),[336,356,358,443,444,445,446,447,448] amphotericin B,[449] and photosensitizers (used for the photodynamic treatment of cancer)[450,451,452] have all been encapsulated in polymeric micelles. Several of these micellar formulations have previously been tested in clinical settings, and the findings look good in terms of the therapeutic index they provide for cancer patients.[448,453,454,455] A drug delivery system must meet a number of (pharmaceutical) conditions, including the ability to scale up the manufacture of the micellar formulation, a notable increase in therapeutic benefit relative to the free drug, and good biocompatibility. Additionally, the ideal micellar system i) has a high drug-loading capacity, ii) has long-circulating properties and adequate blood-stream stability, iii) can selectively accumulate at the target site, and iv) allows for the possibility of controlling the release of drugs at the target site-for instance, through external stimuli.[66,376,426,427,428,429,430,433,434,435,456] The capacity to be (degraded and) eliminated from the body upon drug release, as well as the ability to monitor and trace the micellar structure through the co-encapsulation of an imaging agent,[456] are additional advantageous characteristics of polymeric micelles. Here I primarily focus on the stability and lifespan of drug-loaded polymeric micelles following intravenous injection, as well as the potential for controlled release of the payload in response to external or local stimuli.
8.3.1. Longevity
Drug delivery systems, like polymeric micelles, must remain in the bloodstream for a certain amount of time in order to release their payloads selectively at the target areas. A long-circulating nanocarrier will sustain the blood level of its loaded drug for a longer period of time, thereby enhancing the therapeutic effect of the drug due to the prolonged interactions in the target organ,[433,434,435] as long as the encapsulated drug stays associated with the nanocarrier. Furthermore, the so-called increased permeability and retention (EPR) effect enables the accumulation of polymeric micelles in diseased tissue for an extended period of circulation. Maeda et al. first presented the idea of this EPR effect in the 1980s. They relate it to the increased permeability of the vasculature in sick areas as a result of decreased lymphatic drainage and discontinuous endothelium.[66,434,457,458,459] These two characteristics allow colloidal particles to enter and remain in the tumor and inflammatory areas after passing through the “leaky” endothelium membrane. On the other hand, opsonic proteins that are adsorbed onto the surface of foreign substances are quickly recognized and eliminated by the human immune system. Therefore, lowering the rate and degree of this opsonization and the detection by reticuloendothelial system (RES) cells is crucial for the sustained circulation of colloidal drug carriers. Steric stabilization has been demonstrated to effectively decrease the interaction with opsonic proteins and, consequently, the uptake by the RES cells of the liver, spleen, and bone marrow by coating the particle surface with hydrophilic polymers (e.g., poloxamer, poly(ethylene glycol)[460,461,462,463,464,465]). As will be discussed in this section (vide infra), in addition to surface properties, the biodistribution of polymeric micelles is influenced by a wide range of other parameters, principally particle size[66,376,455,466] and particle stiffness.[466,467]
8.3.1.1. Steric Stabilization
- 8.3.1.1.1. Poly(ethylene glycol)
The most widely utilized hydrophilic segment of amphiphilic micelle-forming copolymers is poly(ethylene glycol) (PEG), also known as poly(ethylene oxide) (PEO). Examples of PEG-b-poly(propylene glycol) (PPO),[428] PEG-b-polymers,[468,469,470,471,472] PEG-b-phospholipids,[473,474] and distinct PEG-b-poly(meth)acrylamide derivatives[475,476,477,478] are some examples of these hydrophilic segments. PEG is widely used because of its low immunogenicity and toxicity as well as FDA approval for usage in a variety of pharmaceutical formulations. Additionally, it possesses special physicochemical qualities that give it good “stealth” qualities, including great flexibility, excellent water solubility, and a large exclusion volume.[425,465,479,480,481,482] The mechanisms and factors responsible for the “stealth” properties and the beneficial effect of a PEG-coating on the circulation kinetics of colloidal drug delivery systems have been thoroughly studied since the early nineties,[483] but they have not yet been fully elucidated, as reviewed recently by Vonarbourgh et al.[466] PEG is generally thought to have reduced opsonization because it shields surface charge and increases the hydrophilic surface, which inhibits the two primary processes that drive protein adsorption: hydrophobic and electrostatic interactions. Moreover, the protective effect is thought to be caused by a decrease in Van der Waals interactions, an increase in repulsive forces, the creation of an impermeable polymeric layer on the particle surface, but also by the binding of dysopsonins, which are naturally occurring compounds that are known to prevent phagocytic ingestion.[435,463,466,482,484] It is only possible to attain prolonged circulation periods due to effective blocking of opsonization when the protective polymer layer is thick enough. For the best defense against immune system recognition, however, the PEG-chains must maintain their flexibility. PEG molecular weight, conformation, and surface chain density[66,463,466,484,485] are all correlated with these two parameters. Higher PEG molecular weights have been shown to diminish protein adsorption (Figure 16) and extend the circulation durations of colloidal particles. This was exemplified, among other things, in PEG-b-p(Asp) micelles labeled with 14C-benzylamine and covalently attached doxorubicin. A five-fold rise in the blood level of the micelles after 4 hours post-injection (13 vs 68% of the injected dose) and a decrease in hepatosplenic absorption were seen when the molecular weight of PEG was increased from 5000 to 12000 Da.[486,487] A series of Pluronic® (PEG-PPO-PEG) block copolymers[488] was also found to benefit from a longer PEG-chain with regard to blood circulation timings. Smaller PEG-chains may also be able to produce a protective layer, even though PEG with a molecular weight between 1000 and 15000 Da is typically utilized to build polymeric micelles for drug administration. For example, covering lipid nanocapsules with PEG (molar weight 660 Da) led to decreased protein opsonization and steric stabilization; this was explained by a high surface chain density.[489] Surprisingly, in terms of PEG chain conformation on the surface, it was demonstrated that, even with reduced chain mobility, attachment of both PEG chain ends to the surface produced more protection than attachment of only one chain end.[490,491] It was proposed that, in comparison to double chain end attachment, single chain end attachment facilitates easier protein penetration of the PEG-layer (Figure 16B), leading to a less dense PEG-layer.[490,491]
Despite being widely believed to be the gold standard currently for steric stabilization of nanoparticles, PEG is not as inert as people think. PEGylated liposomes have been used in several investigations that show blood protein binding[492] and the loss of stealth characteristics at low lipid dosages and/or after repeated delivery.[493,494,495,496] Numerous investigations have documented the repeated administration of polymeric micelles loaded with drugs. Nevertheless, these investigations concentrated more on the antitumor effect of a cytostatic drug's micellar formulation than on the drug’s loaded form or the carrier’s biodistribution.[356,444,450,497,498,499] It is expected that the phenomena of PEGylated liposomes losing their long circulation behavior after repeated administration or at low doses may also affect micelles with a hydrophilic PEG-shell. This justifies looking for other protective coatings, as the following section will explain.
- 8.3.1.1.2. Substitute Coatings
Alternative hydrophilic polymers (Table 1) must be biocompatible, hydrophilic, water soluble, and extremely flexible in order to meet the aforementioned mechanical requirements. Poly(oxazoline),[500] Poly(glycerol),[501] Poly(N-vinyl pyrrolidone) (PVP)[502,503] Poly(acrylamide) (PAAm),[503] Poly(vinyl alcohol) (PVA),[504] Poly(N-(2-hydroxypropyl) methacrylamide) (pHPMAm),[505] and Poly(amino acids)[506,507] are the polymeric coatings that effectively extended the circulation times of liposomal carriers. A number of these polymers have also been employed in the creation of polymeric micelles, which will be discussed in this section, as the hydrophilic component of amphiphilic block copolymers.
- 8.3.1.1.2.1. Poly(N-vinylpyrrolidone)
Poly(N-vinylpyrrolidone) (PVP), a highly hydrophilic, flexible, and biocompatible substitute for PEG, is appealing. A number of amphiphilic PVP-containing block copolymers have been reported to form micelles recently, including copolymers containing poly(lactic acid) (PLA),[450,513,517] PCL,[511,514,516] and pNIPAAM.[515] Additionally, it has been shown that these copolymers can effectively encapsulate hydrophobic medications, including indomethacin,[513] chloro aluminum phthalocyanine,[515] and paclitaxel (PTX).[450] PVP has been demonstrated to efficiently extend the liposomes’ circulation time;[502,503] however, micellar systems have not yet been the subject of these studies. The biodistribution and tumor accumulation of PVP-coated micelles loaded with PTX[450] or chloro aluminium phthalocyanine[515] were the main subjects of in vivo investigations. Comparing both medications to a similar dose synthesized in cremophor EL®, no improvement was seen in blood circulation times, tumor accumulation, or therapeutic impact. This could be connected to either the drug’s rapid release from the micelles or the micelles’ own dissolution. However, compared to Taxol® (PTX formulated in cremophor EL®), the maximum tolerated dose of PTX-loaded PVP-b-PLA micelles in mice was more than five times greater. A stronger anti-tumor effect could be obtained by employing a larger dose.[450]
- 8.3.1.1.2.2. Polysaccharides
The polysaccharide group, which is another class of polymers utilized as a PEG substitute, is involved in the surface features of several cells, including red blood cells. The fact that these cells successfully elude the immune system may be due to the oligosaccharide groups that are present on their surface.[433,520] Dextran-g-PCL and dextran-coated PLA nanoparticles were shown to have reduced protein adsorption in comparison to naked polyester nanoparticles.[518,519] Extended circulation durations were reported for poly(methyl methacrylate) (pMMA) nanoparticles coated with dextran or heparin, compared to naked pMMA nanoparticles,[521] despite the fact that these studies did not examine the circulation kinetics of these dextran-coated nanoparticles. It was recently revealed that hydroxyethyl starch (HES) micelles grafted with acyl chains could potentially avoid the RES.[522] Nevertheless, no research has been done on the pharmacokinetics of these HESylated nanoparticles in vivo. It has been shown that, in order to minimize interactions with plasma proteins, polysaccharide shape is crucial.[520,523] Compared to PEG (see section 8.3.1.1.2.1 above), a brush-like arrangement provided a higher level of protection than did the surface presence of dextran loops. Several cell membranes include saccharide receptors,[520] even though poly- and oligosaccharides on the surface of the nanoparticles may prevent RES-uptake. The detection of galactose-presenting nanoparticles by hepatocytes[524] demonstrates how this permits their application in active targeting techniques.
- 8.3.1.1.2.3. Additional Hydrophilic Blocks
Other biocompatible hydrophilic polymers, such as poly(N,N,dimethylamino-2-ethyl methacrylate) (pDMAEMA),[525] poly(ethylene imine) (PEI),[526] poly(acrylic acid) (pAAc)[426,527] and poly(asparagine) (pAsp),[528] have also been employed as the shell-forming element in polymeric micelles. However, due to the charges on the micellar shell surface, the lifespan of these micelles in vivo is uncertain. Furthermore, block[529] was proposed to be the shell-forming compound p(NIPAAM-co-N,N-dimethylacrylamide (pDMAAm)). As will be covered later in this review, this copolymer can be employed for temperature-triggered release of pharmaceuticals that are encapsulated because of its thermosensitive behavior. Micelles made of poly(2-ethyl-2-oxazoline)-b-polyester,[508,509,510,511,512] pHPMAm-b-PCL,[516] and poly(acrylamide) (pAAm)-b-PVA, palmitate[426,482] and PVA-b-oleylamine[530] may have longer circulation durations in vivo since these hydrophilic blocks successfully shielded liposomes from fast RES-uptake (see above), although there is currently insufficient data.
8.3.1.2. The Size of Micellar Particles
Another important factor that determines a particle’s destiny after intravenous injection is its size. Particles with a molecular weight less than 50 kDa (a hydrodynamic diameter of 5-10 nm) are prone to renal excretion, while particles bigger than 200 nm are eliminated via mechanical filtration by the spleen’s interendothelial cell slits.[430,433,439,531] Although elimination through these routes is hindered by the size of polymeric micelles (10 - 100 nm), it has been demonstrated that the size of the micelles also affects the amount of RES absorption and tumor penetration.[66,376,430,455,488] Shell crosslinked poly(tert- butylacrylate)-b-polystyrene (PBA-b-PSt) micelles, for instance, exhibited much longer blood residence durations at one hour following injection (50 % versus 5% of the injected dose) than their two times larger counterparts. 306 and PEG-b-PHDCA micelles measuring 80 and 170 nm showed the same pattern, respectively 59. It should be noted, nonetheless, that due to their smaller size in the latter study, the longer circulation durations of the 80 nm micelles may be associated with a greater PEG surface density. As was mentioned earlier, better shielding is produced by a greater PEG-density. Moreover, it was proposed that particle size matters since smaller particles have a larger curvature, which hinders protein adsorption.[466] On the other hand, Weissig and colleagues showed that in mice with subcutaneously developed Lewis lung cancer, intraperitoneally injected PEG5000-b-distearoyl phosphatidyl ethanolamine (PEG-b-DSPE) micelles measuring 15 nm circulated shorter than 100 nm liposomes. Interestingly, compared to liposomes,[474] the tumor accumulation of the tiny micelles was significantly higher. This is explained by the low cut-off size of the tumor vascular wall, which varies depending on the kind of tumor and controls how well nanoparticles can enter the tumor.[532,533] The small size of micelles is in fact an extra advantage over, say, liposomal and other larger nanoparticulate systems, especially in solid tumors as Lewis lung carcinoma. Doxorubicin-loaded PEG-b-poly(aspartate hydrazone adriamycin) micelles measuring 65 nm and liposomes measuring 150 nm were also found to have distinct tumor penetration.[376,534] Whereas the 150 nm liposomes were only detected on the peripheral,[376] the micelles were discovered inside tumoral spheroids. Because small-sized nanoparticles can more easily enter tumor tissue, they gain greater advantage from the EPR effect.
8.3.1.3. Additional Methods to Enhance Circulation Times
In addition to surface properties and particle size, the stiffness of the particle affects the kinetics of circulation. Long circulation periods are observed in liposomes with a stiff lipid bilayer even in the absence of a protective PEG-layer, as reported in studies.[535,536] Sun et al.[467] showed that a more rigid, glassy micellar core leads to a longer blood retention by comparing the biodistribution of (PBA-b-PSt) micelles with a high Tg, glassy PSt core to that of PBA-b-poly(methyl acrylate) (PMA) micelles with a low Tg fluid-like PMA core. Predosing with empty micelles to saturate the elimination processes may be another tactic to improve the circulation times of nanoparticles, in addition to adjusting particle-related properties including surface features, size, and rigidity. Although it hasn’t been tried with micellar systems yet, this was actually the first method to lengthen the liposomes’ circulation time.[537]
8.3.1.4. Longevity of Polymeric Micelles with Active Targeting
Long circulation periods facilitate the passive targeting of polymeric micelles, which in turn facilitates the delivery of the drug contained at the site of action. However, active targeting may enhance this delivery even more. To encourage cellular recognition and internalization of the drug carrier, certain ligands have been attached to the micellar shell, including internalizing antibodies,[448,538] sugar moieties,[539] transferrin, RGD sequences, and folate.[540,541] Although the features of such carriers are paradoxical, the distribution of pharmaceuticals by actively targeted long circulating micelles is a potential strategy to improve their site-specific impact. One way that a targeting ligand, such as an antibody or another protein, can help the immune system recognize and remove the micellar surface from the bloodstream is if it is present on the surface. However, the ligand’s ability to bind to its target may be hindered by the presence of a protective polymer like PEG. The application of a “sheddable” coating-that is, a coating that may be removed after reaching the target site-is one tactic to address the aforementioned conundrum. It was recently discovered that a TAT-peptide attached to the surface of PEG-b-PLLA micelles might deshield in a pH-sensitive manner. The anionic pH-sensitive poly(methacryloyl-sulfadimethoxine) (PSD) block of a PEG-b-PSD copolymer protected the cationic TAT ligand at pH 7.4. The complex was broken and TAT became visible when the pH was lowered to 6.6, which led to improved cellular absorption and localization at the nucleus’ surface.[542]
8.3.2. Stability of Micellar Matter
The ability of C3M to withstand nanoparticle disintegration in the face of rising ionic strength in solution is one indicator of C3M stability. Complication is disrupted by adding extra counterions from salt, which interfere with ion pairing between polymers. Increasing the length of either charged polymer increases the stability in bulk systems (B + C), which is easily testable using optical microscopy.[415] For nanoparticles smaller than the diffraction-limited resolution of optical microscopes, this is more challenging to analyze; however, light scattering and small-angle scattering techniques have been used to demonstrate a comparable effect for PCMs.[106,395] Similarly, in PEC systems containing charged biomolecules, a rise in charge density can promote micellization and boost complex stability.[115,396] The structure and stability of complex formation are also influenced by the molecular characteristics of each charged group, even though PCMs are commonly understood as two charged, flexible chains joining together in an entropically advantageous process.[142] Using thiolene click chemistry to attach each of the desired side groups onto otherwise identical polymers, the Choi group directly compared C3Ms comprising charged ammonium (pKa = 11), guanidinium (pKa = 14), carboxylate (pKa = 4), and sulfonate (pKa = 1) groups on a functionalizable PEO-b-PAGE. As the strength of ion pairing interactions increases, the neutron scattering results show an increase in both the aggregation number and core radius.[397] In order to compare two cationic charged monomers for complexing DNA at different lengths, a research examined lysine (primary amine, pKa = 10) and vinylbenzyltrimethylammonium (VBTMA, which consists of a permanently charged ammonium).[404] PVBTMA complexed less strongly with DNA, attributed to the steric barrier in ion pairing, despite the permanent charge and extra hydrophobicity supplied by the aromatic moiety, which have been previously shown to strengthen various PEC systems. [124,543,544] After micelle production, cationic polymers can be reversibly cross-linked using glutaraldehyde,[545] disulfide linkages,[546,547] or other methods[548] to increase stability. These examples show that robust C3Ms may be formed from both synthetic polymers and polypeptides, and that molecular features are important to take into account throughout the design process since they affect complexation characteristics and, ultimately, functionality.
Drug accumulation can be selective even in the case of a lengthy circulation, provided that the drug is released slowly in the initial hours following administration or that premature drug molecule leakage from the micelles is avoided. Thus, a great deal of research has been done on core-forming polymers that are primarily hydrophobic (Table 2), as these polymers are essentially responsible for determining the micellar stability, drug loading capacity, and drug release profile of the material.[336,426,430,434,439,451,469,498,549,550]
One might think of a polymeric micelle’s stability from a thermodynamic or kinetic perspective. When the concentration of polymer in the water above the critical micelle concentration (CMC), which is sometimes referred to as the critical aggregation concentration (CAC), polymeric micelles become thermodynamically stable. Amphiphilic block copolymers in water exist as single chains at the air-water contact and in the bulk below the CMC. Because of the hydrophobic contacts between the hydrophobic blocks,[430,557] the self-assembly of the amphiphiles reduces the system’s Gibbs free energy (G) as the concentration rises above the CMC. Knowing the CMC of polymeric micelles and administering a high enough dose are crucial because they are prone to dilution in the circulation after intravenous injection.[426,432] A micellar system’s kinetic stability is determined by the rate at which single polymer chains are exchanged between the micelles and the bulk; this rate can be maintained even when the system is diluted below the critical mass concentration (CMC).[426,430,557,558] The strength of the interactions within the micellar core, which is dependent on a number of factors, including the crystalline or amorphous state of the core-forming polymer, the presence of solvent (such as residues of methanol or dioxane from the preparation process) within the micellar core, the ratio between the hydrophilic and hydrophobic block lengths of the copolymer, and the encapsulation of hydrophobic compounds.[66,426,430,498] A number of other parameters, such as the length of the core-forming polymer segment and the amount of loaded drug,[559] affect the release of the loaded drug in addition to micellar stability. Crucially, the drug loading and release are impacted by the core-forming polymer and drug compatibility.[66,426] The compatibility of the medication was optimized for different micellar systems through appropriate block copolymer selection, which is anticipated to promote in vivo drug retention.[335,434,444,445,450,486,560,561,562,563,564] Nevertheless, blood components frequently cause early drug release through the extraction of encapsulated drug from intact micelles or by inducing micelle destabilization.[565,566,567,568,569] Consequently, there is a lot of work being done to enhance the kinetic and thermodynamic stability of drug-loaded micelles. Numerous approaches have been studied, such as reducing the CMC of the micelle-forming polymers, enhancing the drug-polymer compatibility, and achieving physical and covalent crosslinking.
8.3.2.1. Lowering the CMC
Lowering the amphiphilic block copolymer’s CMC can increase the thermodynamic stability of polymeric micelles. Blocks can be readily resized to do this.[66,426] A higher overall hydrophobicity is the product of both a larger hydrophobic block and a smaller hydrophilic block, which lowers the CMC.[430,439,450,468,570] Apart from its size, the hydrophobic block’s nature plays a crucial role in establishing the CMC. It has been shown that chemically altering the hydrophobic block-for instance, by adding aromatic groups-effectively lowers the CMC.[468,552,553,571,572] For example, a ten-fold reduction in the CMC was obtained by increasing the number of benzyl groups in the modified poly(-benzyl L-aspartate) (PBLAmod) block of PEG-b-PBLAmod copolymer from 44% to 75%.[572] By adding a benzoyl or naphthoyl moiety to the end group of mPEG750-b-oligo(-caprolactones), a 60-200-fold decrease was achieved.[468] In a similar vein, a decrease in the CMC was seen when the amount of hydrophobic oligolactates[476] or fatty acids[573] bound to the polymer backbone increased.
8.3.2.2. Physical Interactions
In addition to increasing the thermodynamic stability by lowering the CMC, the addition of aromatic groups may also increase the kinetic stability of the micelles by enhancing stacking-induced interactions inside the micellar core. Using fluorescence spectroscopy, Mahmud et al. investigated the viscosity of the core of PEG-b-PCL and PEG-b-poly(α-benzyl-ε-caprolactone) micelles. They discovered that the presence of aromatic groups[571] improved the stiffness of the micellar core in addition to decreasing the CMC (see the previous section). When compared to their amorphous counterparts, micelles’ stability was demonstrated to be increased upon the introduction of crystallinity or stereocomplex formation.[574,575] Hydrogen bonds also resulted in an increase in physical interactions.[576] Figure 17 summarizes the physical interactions that may contribute to the kinetic stability of micelles. The micellar stability may be improved by the addition of a hydrophobic medication in addition to interactions between the core-forming polymers. For instance, Yokoyama et al. showed that the quantity of doxorubicin (DOX) that was physically entrapped as well as chemically bound enhanced the stability of PEG-b-p(Asp) micelles.[498]
In order to generate a so-called polyion complex (PIC) or C3M, the electrostatic ionic contact forces of oppositely charged block copolymers may also induce the creation of micelles (Figure 18).[265,283,578]
Poly(N-ethyl-4-vinylpyridinium) (PEVP),[63] PEG-b-poly(2-vinylpyridinium) (p2VP), PEG-b-poly(L-lysine) (p(Lys)), and PEG-b-poly(-aspartic acid)[26,283] PEG-b-poly(methacrylic acid) (PMAa) are examples of polyion-couples. C3Ms with thermosensitive shells and C3Ms made of PEG-b-PMAa and Ca2+ with a crosslinked PMAa core have both been reported.[73,265] To create micelles with cationic medicines or peptides, polymers with negatively charged units, including PMAa and p(Asp) (co)polymers, were employed, and siRNA/DNA was formed with polycations, like PEG-p(Lys).[85] C3Ms have the benefit of being straightforward to prepare, requiring only a simple mixing of drug and polymer aqueous solutions. Their low stability in physiological saline and the drug’s requirement to be hydrophilic, however, limit their applicability. This might be addressed by copolymerizing phenylalanine in the polymer backbone, which would improve the hydrophobic/aromatic interactions.[563] Conversely, the destabilization of micelles caused by saline can be employed to regulate the release of the medicine that is loaded. As will be covered in the upcoming section, this idea was illustrated for cisplatin-complexed PEG-b-p(glutamic acid).[497]
8.3.2.3. Crosslinking of Covalent Bonds
Apart from the physical methods indicated above to improve the stability of micelles, stable particles with a micellar morphology have been prepared by chemical crosslinking of the micelle’s shell, interfacial layer, or core (Figure 19).[85,373,578,579] Among other methods, the insensitivity of covalently crosslinked micelles to a destabilizing agent (such as sodium dodecyl sulphate, or SDS)[580,581] demonstrated the higher stability of these micelles compared to their non-crosslinked counterparts. The crosslinking process maintained the micellar shape, but it also preserved the response to stimuli (such as pH, temperature, and salt concentration) and allowed for control over drug release.[374,582,583] But unlike physical crosslinking, the covalent crosslinking method can negatively impact the drug’s encapsulating structure and micelle’s overall degradability (if the crosslinking process is carried out with the drug present).
- 8.3.2.3.1. Crosslinking of Shells
Through chemical or photoinduced processes, the hydrophilic shell of polymeric micelles has been covalently crosslinked[85,373,467,584,585,586,587,588,589] For instance, glutaraldehyde or a diamine were used as crosslinking agents in polypeptide-b-polydiene micelles to create covalent linkages between the carboxylic acid or amine groups in the hydrophilic polypeptide block. An activating agent, such as a water-soluble carbodiimide, was added to cause crosslinking via amide bond formation.[589] The poly(N,N-dimethylaminoethyl methacrylate) (pDMAEMA) shell was crosslinked through alkylation using a bifunctional alkyl iodide,[590] while the poly(4-vinylpyridine) (p4VP)-b-polystyrene shell was crosslinked through the addition of a water-soluble radical initiator, which was then exposed to UV light at 50°C.[591] In addition to fixing shape, shell crosslinking gives medication molecules[592] another way to regulate the micellar shell’s permeability. In stimuli-sensitive micelles, surface stabilization can also be used to further regulate drug release.[582,588,590] The requirement for high polymer dilution in all operations to selectively crosslink the micellar shell and prevent the development of intermicellar crosslinking is a significant drawback of crosslinking the shell segments.[373] Moreover, the steric stabilization may be hampered by the shell fixation, which could reduce the shell-forming polymers' chain flexibility.
- 8.3.2.3.2. The Crosslinking of Interfaces
As an alternative, a crosslinkable spacer can be introduced between the hydrophobic and the hydrophilic block[585,593,594] to crosslink the interfacial layer between the micellar core and shell. This method may offer a means of controlling drug release, but it will not alter the micellar core or shell, and as a result, the loaded drug or steric stabilization, respectively. Poly(glycerol monomethacrylate) (pGMA) and poly(2-hydroxyethyl methacrylate) (pHEMA) are two examples of spacers that are employed. These spacers can be crosslinked by adding divinyl sulphone[583,593] or by derivatizing pGMA with cinnamoyl chloride and then UV-irradiating the aqueous micellar solution.[585]
- 8.3.2.3.3. Crosslinking at the Core
Functional groups located at the end of the chain or along the core-forming block can be used to prepare core crosslinked (CCL) micelles. (Meth)acrylate groups[358,580,595] are frequently used to functionalize the hydroxyl moiety in the hydrophobic block. Following micelle production, heat[580,595] or photo-induced polymerization[581,596] crosslinks the hydrophobic blocks. Using Michael addition with multifunctional thiol compounds[597] or hydrophobic polyfunctional acrylate[598] to construct an interpenetrating network are two further methods to produce CCL micelles from (meth)acrylate functional block copolymers. An other instance are C3Ms, which consist of the cationic protein trypsin and the anionic PEG-b-p(Asp) crosslinked via the Schiff-base production of glutaraldehyde with the protein. In the core, an interpenetrating network of crosslinked trypsin was formed, and it remained stable even at high ionic strength (0.6 M NaCl). This suggests that covalent bonds were formed in addition to protein-protein crosslinks between the aldehyde groups in glutaraldehyde and the primary amino groups at the ω-end of the p(Asp) segments. Furthermore, the protein maintained its enzymatic activity.[599] In the presence of a carbodiimide, the carboxylic acid groups in PMAa reacted with 1,2-diethylenediamine to form C3Ms made of PEG-b- PMAa/Ca2+.[374,548]
- 8.3.2.3.4. Cleavable Crosslinks
One potential disadvantage of crosslinked micelles could be the potential for covalent bonds within the interfacial layer, shell, or core to adversely impact the polymeric assemblies’ biodegradability. Reversible or degradable crosslinks could (partially) get around this. The thiol groups were added to the lysine units of PIC micelles and then oxidized to create disulfide linkages, forming reversible crosslinked micelles.[600] Furthermore, the interfacial layer of PEG-b-(poly(N,N-dimethylacrylamide)-stat-(N-acryloxysuccinimide))-b-pNIPAAM micelles was stabilized by disulfide bonds. The resultant particles could be reduced by substances like dithiothreitol or glutathione. Consequently, another possible catalyst for drug release and particle breakdown in the cytoplasm of cells is the reducing environment.[588] Methacrylate moieties were introduced at degradable oligolactate grafts of a core-forming polymer backbone recently to create hydrolyzable CCL micelles. In comparison to their non-crosslinked counterparts, these crosslinked micelles demonstrated greater physical stability, and their crosslink density allowed for control over the rate of deterioration.[601]
- 8.3.2.3.5. Crosslinking’s Effects on Drug Release and Loading
The primary obstacle to the practical use of C3Ms is their tendency to disintegrate readily, especially when exposed to high ionic strengths, despite their ease of manufacture.[27,80,112,134,602] A higher concentration of salt reduces the forces that drive the production of C3Ms, which makes the C3Ms less stable. C3Ms totally disintegrate above a specific salt concentration, sometimes referred to as the critical salt concentration (CSC). Moreover, the pH is a crucial factor for the stability of C3Ms in the case of weak polyelectrolytes.[27,602] Reversing the undesirable disintegration of C3Ms can be achieved by crosslinking the polymers. This can be carried out in the micelle’s corona or core, reversibly or irreversibly.[602,603,604,605,606] Crosslinking the corona/shell, however, may have an impact on the surface properties and decrease the hydrophilicity and, thus, the solubility of the micelles.[605] However, we anticipate that the surface properties of the micelles will not change when using the core-crosslinking approach. By physically or covalently attaching one or both of the polymer types found in the core of C3Ms together, core-crosslinking of C3Ms can be accomplished.[607,608,609] A more stable network will result from the constituent parts of the core being chemically crosslinked. Chemical crosslinks can be created through enzyme reactions, photo-polymerization from radiation, or “click” chemistry.[608,609] Reactive functional groups, such as amines, thiols, carboxylates, hydroxyls, acetoacetyl groups, acetal groups, acrylamide derivatives, or carbonyl groups, are required for the polymers to crosslink C3Ms.[610,611] A previous report[612] ascertained how well the core of C3Ms can be covalently crosslinked to increase their stability in the face of pH variations and salt addition. Poly(2-vinylpyridine)128-b-poly(ethylene oxide)477 (P2VP128-b-PEO477), a cationic-neutral diblock copolymer, and poly(acrylic acid)118 (PAA118), an anionic homopolymer, were used to create C3Ms. Prior to this, quaternization was used to functionalize the diblock copolymer containing P2VP with primary amine groups. Protection chemistry was needed during the quaternization process because primary amines are known to take part in quaternization reactions themselves. It is convenient to utilize a number of amine-protecting groups, such as phthalimides, t-butyl carbamate (BOC), and 9-fluorenylmethyl carbamate (FMOC).[613,614,615] N-(2-bromoethyl)phthalimide was the choice due to its stability in the quaternization reaction's necessary conditions, which include heating to 150 0C. Two types of crosslinkers were used in a previous study: dimethyl 3,3/-dithiopropionimidate dihydrochloride (DTBP) and 1-ethyl-3-(3/ dimethylaminopropyl)carbodiimide hydrochloride (EDC).[612] Whereas DTBP joins two amine groups together, EDC creates permanent crosslinks between amine and carboxylic groups. Moreover, reducing agents can be added to DTBP crosslinks to break them anew because they include disulphide bridges. In order to crosslink the core of C3Ms (Figure 20), the authors have looked into these two kinds of crosslinkers. The various core-crosslinking techniques-network creation between one or both types of polymers in the core and permanent versus reversible cross-linking have then been compared.
In addition to correcting the micellar morphology, the crosslinking procedures of the shell, interfacial layer, or micellar core mentioned above also delayed the release of the loaded medication.[588,592,616,617] Crosslinking may also affect a drug’s ability to load. After the chain ends were polymerized, eight times more triclosan was encapsulated in PEG-lipid micelles. This was attributed to a better stability of core crosslinked compared to unmodified micelles.[618] It goes without saying that one should constantly be mindful that after the chemical crosslinking of the core, the structural integrity of the loaded drug molecules should be maintained. In order to prevent unintentional alteration of drug molecules that are entrapped, the micelles can be loaded with pharmaceuticals after first being crosslinked. For instance, the thermally induced polymerization of the methacrylate groups in the core of mPEG-b-PLA micelles resulted in their crosslinking. Paclitaxel (PTX) was then injected into these CCL micelles using a microemulsion technique and the organic solvent was then evaporated. A loading capacity equivalent to non-crosslinked micelles was attained, ranging from 3 to 6 weight percent (% w/w).[581] Cisplatin was added to crosslinked PEG-b-poly(methacrylic acid) micelles through a 48-hour incubation period with an aqueous drug solution. After that, the unbound cisplatin was extracted by ultracentrifugation, yielding a drug loading of 22% (w/w).[374]
8.3.2.4. Drug Compatibility with Micellar Core
It is not assured that the loaded medicine will remain intact, even at high micellar stability. Blood components may cause the drug to be extracted and redistributed between the micellar core and drug upon contact.[565,566] Drug retention and release are influenced by the amount of drug loaded, the size of the micellar core,[559] the drug’s compatibility with the core, and the impact of environmental stimuli (vide infra). The Flory-Huggings interaction parameter will quantify and predict how well the polymer and medication will work together:
where Vsis the drug’s molar volume, χsp is the interaction parameter between the drug (solubilisate, s) and the core-forming polymer (p), and δs and δp are the drug’s and polymer’s Scatchard-Hildebrand solubility parameters, respectively.[426,434] χsp should be as low as possible to achieve the best possible compatibility between the medication and the core-forming polymer. This indicates that no single micellar system is suitable for all medications; rather, a mixture that works best for each drug must be identified in order to increase retention.[66,335,426,434,444,445,450,486,487,498,552,553,560,561,562,563,564,619] Liu et al. provided evidence for this method by comparing the interaction characteristics of several core-forming polymers with ellipticine, a medication. It was shown that a good selection may be made in this manner, leading to slow release and large drug loading capacities.[335] In a similar vein, it is possible to alter the core-forming polymer to improve its drug compatibility. For instance, adding more aromatic groups to the polymer backbone increased the loading efficiency in PEG-b-poly(-benzyl L-aspartate) (PBLA) micelles and the in vivo therapeutic effect of the aromatic drug camptothecin. This improvement was attributed to aromatic interactions between the benzyl groups and the drug.[552,553] A micellar system for doxorubicin paclitaxel (PTX)[444] and (DOX)[620] was designed using a similar methodology. A further strategy to improve the drug's compatibility with the micellar core is to chemically modify it. Prodrugs of the anticancer medication geldanamycin were synthesized by Forrest et al., who also showed that a greater encapsulation could be achieved by matching the prodrug's chemical structure with the segment core.[560] When the medication is chemically bonded to the micelle-forming polymer, it is steadily held within the micellar core. By using this method, Yokoyama et al. covalently bonded DOX to the PEG-b-p(Asp) copolymer’s p(Asp) block. Micelles were produced by the resultant PEG-b-p(Asp)-DOX conjugate.[486,498,619] Large volumes of free DOX could also be loaded through in addition to the bound drug; the amount of conjugated drug determined how well the drug stacked in these micelles and the efficiency of encapsulation.[498] Following an intraperitoneal administration of DOX, in vivo investigations revealed that these DOX-loaded micelles exhibited significantly more anti-tumour action in C26-bearing mice than free DOX.[441] This formulation (NK911, Figure 21) was used in a phase I clinical trial including 23 patients who had solid tumors that had spread or returned and were resistant to traditional chemotherapy. The toxicity profile of NK911 was discovered to be comparable to that of free DOX. In contrast to free DOX, NK911 showed longer half-lives, a lower clearance, and a bigger AUC, all of which suggested longer circulation durations. There is presently a phase II clinical investigation underway.[454]
Ideally, the drug should remain loaded in the micellar core of the micelles as they travel long and straight to the target site. But this is not the same as totally inert, non-degrading, and non-releasing micelles, as they would accumulate over time in the body, particularly following repeated administration. Furthermore, the medication needs to interact with the therapeutic target before it is released. Consequently, in order to facilitate clearance through the renal pathway, the encapsulated drugs should preferably be released selectively at the target site and the micelles should split into single block copolymer chains or even chain fragments with a molecular weight of less than 50 kDa.[431,621] Drug release from the carrier system can occur at the target site, but it is preferable to occur through a more regulated mechanism-that is, in response to particular stimuli, as this review’s subsequent parts will address.
8.3.3. Sensitivity to Stimuli
As was said in the earlier parts, in a perfect micellar system, the drug is stable while in circulation and only releases when environmental cues cause it to accumulate in the intended tissue. In order to attain a high concentration of the medicine in the target tissue, a release mechanism that takes advantage of the regionally distinct circumstances in diseased tissue as opposed to healthy tissue is appealing. In addition, an external trigger such as light, ultrasound, or temperature might cause the loaded medication to release. “Stimuli-sensitive micelles” are defined as those that become destabilized due to physiological or extrinsic variables. Stimulus-sensitive micelles develop and only deconstruct in response to specific triggers, such as alterations in the polymer’s characteristics (such as polarity). Furthermore, it is anticipated that the drug that was initially stably encapsulated will be released concurrently with the micelle breakdown. Many triggers, such as temperature (section 8.3.3.1), pH (section 8.3.3.2), hydrolysis (section 8.3.3.3), enzymatic reactions (section 8.3.3.4), redox processes (section 8.3.3.5), light (section 8.3.3.6), others (e.g., ultrasound in section 8.3.3.7), as well as combinations hereof (section 8.3.3.8) have been studied to destabilize drug-loaded polymeric micelles (Figure 22). These methods have been reported for micelle-forming amphiphilic block copolymers[578] and peptide amphiphiles[622] Co-loading an imaging agent results in a complex stimuli-sensitive release system that allows for in vivo tracking of the micelles, as will be covered in section 8.3.3.9.
8.3.3.1. Polymeric Micelles with Thermosensitivity
A thermosensitive polymer’s cloud point (CP) is a characteristic of the polymer’s aqueous solution. The polymer becomes water soluble below the CP because it is hydrated and intra- and interpolymer interactions are inhibited. Water is released from the polymer chains as a result of the disruption of the hydrogen bonds that hold water molecules to the polymer chain when the polymer solution is heated over the critical point. At this point, interactions between the hydrophobic moieties in the polymer chain can occur, leading to the polymer’s collapse and eventual phase separation (polymer aggregation or precipitation). The creation of polymeric micelles for medicinal applications is now being studied using a variety of thermosensitive block polymers.[488,624,625,626] With a CP of 32 0C poly(N-isopropylacrylamide) (pNIPAAm) is the most researched thermosensitive polymer.[627,628,629] Copolymerization with hydrophobic or hydrophilic comonomers can be used to modify the CP of a thermosensitive polymer, giving rise to a decreased or increased CP, respectively.[476,554,630,631,632,633] By using this technique, polymers with a CP close to body temperature were created, making them appropriate for micelle dissociation caused by temperature changes. Thermosensitive copolymers can be employed as a hydrophobic, core-forming segment of block copolymers (pNIPAAm and pHPMAm-Lacn [477,478]) or as a hydrophilic, shell-forming segment (p(NIPAAM-co-DMAAm) and poly(2-isopropyl- 2-oxazoline)[73,529]) (Figure 23). The benefit of thermosensitive core-forming segments is that no organic solvents are needed; instead, micelles can be easily created by heating an aqueous polymer solution (above the CMC) until it reaches the CP of the thermosensitive portion. The final size of the generated nanoparticles is largely dependent on the heating rate; show that micelles are produced at a faster rate as compared to a slower one.[475,634,635] The requirement for thermal treatment (hyperthermia or hypothermia) for the destabilization and concurrent drug release of the first generation of thermosensitive polymeric micelles based on non-degradable polymers (e.g., pNIPAAm) is a significant drawback, as it is not always practical in clinical practice. As a result, thermosensitivity is commonly coupled with other systems that respond to stimuli, like degradability and pH or light sensitivity.
8.3.3.2. Polymeric Micelles with pH-Sensitivity
A pH-sensitive carrier may become unstable due to the slightly acidic pH found in tumor and inflammatory tissues (pH ∼6.8) as well as the endosomal and lysosomal compartments of cells (pH ∼5-6).[636,637] Changes in charges within (polyion complex) micelles are the main mechanism causing pH-sensitivity; section 8.3.3.3 will address pH-dependent cleavage and destabilization. pH is usually a determining factor for block copolymeric micelles containing basic groups, like L-histidine (His)[638,639] pyridine,[640] and tertiary amine groups.[641,642] At a pH one unit above the pKa of the amines, the block copolymers assemble into micelles, with the pH-sensitive block forming the core of the micelles and being essentially uncharged and hydrophobic. The polymer becomes protonated when the pH falls below the pKa, which causes an increase in hydrophilicity and electrostatic repulsions, which ultimately cause the micelles to become unstable. It is possible to regulate the transition pH by combining several block copolymers. For example, depending on the ratio of the two block copolymers in the micelles, a mixture of PEG-b-p(His) and PEG-b-PLA generated stable micelles at pH 7.4 and dissociated at pH 6.0 to 7.2. [638,639] The sulphonamide-containing nanoparticles, which collapsed following protonation of the sulphonamide units (pKa = 6.1), releasing the loaded doxorubicin at pH < 7 are another example of pH-sensitive micelles.[643,644] Drugs can be readily loaded into block copolymers that are in their protonated (= water-soluble) state at pH < pKa. The PDPA block (pKa 6-7) became deprotonated (i.e. hydrophobic) and self-assembly occurred when the pH of an aqueous solution of 2-(methacryloyloxy)ethyl phosphorylcholine (PMPC)-b-2-(diisopropylamino)ethyl methacrylate (PDPA) block copolymers was raised from pH 3 to pH 7. The creation of micelles containing dipyridamole, a model drug that is only soluble below pH 5.8, was the outcome of this neutralization.[645] Moreover, at pH 7.4, paclitaxel and tamoxifen could be steadily encapsulated in these PMPC-b-PDPA micelles. Drug release was accelerated when the pH was lowered below the PDPA block’s pKa. When loaded micelles enter moderately acidic tumor tissue or are absorbed through the endocytotic route, this pH-triggered release may be beneficial.[646]
8.3.3.3. Micellar Disintegration Induced by Chemical Hydrolysis
Chemical hydrolysis, which includes breaking down the polymer backbone (discussed in 8.3.3.3.1), cleaving side groups (discussed in 8.3.3.3.2), and hydrolyzing covalent bonds between drug and polymer in micelle-forming polymer-drug conjugates (discussed in 8.3.3.3.3), can be used to demonstrate micelle disintegration and concurrent drug release.
- 8.3.3.3.1. Chemical Hydrolysis of the Polymeric Framework
One commonly utilized technique to destabilize drug delivery micelles is backbone hydrolysis of the hydrophobic block of an amphiphilic block copolymer (Table 3).[647] For instance, variations in particle size, indicative of micelle destabilization, were linked to the chemical degradation of the polyester block in PEG-b-poly(DL-lactic-co-glycolic acid) (PLGA),[648] PCL-b-PEG-b-PCL,[649] and mPEG-b-oligocaprolactones.[650] Furthermore, it was found that the hydrolysis of the ester linkages in PCL block caused the PEG-b-PCL worm-like micelles to transform into spherical micelles.[651] The optimal stability of ester bonds in oligolactates[652] and oligocaprolactones[650] was seen at pH ~4-5, indicating that the hydrolysis of ester is dependent on pH. Yet, the chemical hydrolysis of caprolactone-based polymers and oligomers is sluggish and will rarely be significant in vivo, even at physiological pH and temperature. It is expected that enzymatic degradation (see below)[650] will break these polymers mostly in the body. Furthermore, other types of polymers, including poly(ortho esters) (POE), have a better degradation profile[653] when the medication is introduced into the slightly acidic tumor tissue and endosomal compartments of cells, where micelle disintegration is needed at relatively low pH. It is true that PEG-b-POE micelles were more stable at pH 7.5 than they were at pH 5.5.[654] Although it is widely accepted that micelle destabilization causes the released medicines, there is a paucity of experimental data about the relationship between degradation and drug release. This is because hydrolytic degradation has a significant impact on micelle stability. One of the few instances was reported by Geng et al., who linked the release of the loaded paclitaxel (PTX) with the degradation-induced transition of PEG-b-PCL worm-to-sphere micelles. Due to the fact that spherical micelles had a lower volume-to-surface ratio than worm-like micelles, PTX release from these micelles was the result of a reduction in drug carrying capacity.[655]
- 8.3.3.3.2. Cleavable Side Chains
Micelles become destabilized when the hydrophilicity of the micellar core increases due to the hydrolysis of hydrophobic side chains in the core-forming block that are maintaining the micelle’s stability. An excellent illustration is PEG-b-p(Asp), which is stabilized via π-π stacking in the hydrophobic core by cyclicbenzylidene acetals. While the more polar diols were produced at pH 5 by the hydrolysis of the acetal linkages, the micelles remained stable at physiological pH (Table 3). The polymer's overall hydrophilicity increased, causing micellar disintegration and the release of a hydrophobic dye that was encapsulated.[656] For linear-dendritic block copolymers, a similar approach was used. Micelle breakdown caused by these polymeric micelles in vitro resulted in an accelerated release of encapsulated DOX at acidic pH.[437,658] Further section will cover thermosensitive (block co)polymers with biodegradable side chains.
- 8.3.3.3.3. Breakdown of Polymer-Drug Complexes
When it comes to polymer-drug conjugates, the cleavage of a labile bond that holds the pharmaceuticals to the polymer can be catalyzed by acid to release the drug from polymeric micelles. For instance, an increased release of DOX at an acidic pH was observed in vitro due to the acid labile hydrazone connection between DOX and PEG-b-p(Asp) (Table 3). These pH-sensitive DOX-hydrazone-micelles demonstrated a 15-fold increase in AUCblood, a larger level of anti-tumor efficacy, and a decreased level of toxicity in vivo when compared to free DOX. Furthermore, there was no anti-tumor efficacy exhibited by micelles containing DOX bound by a non-degradable amide link.[35,534,657] An acid labile ester linkage was utilized in a recent work to couple PTX via a triblock copolymer ((PLA-co-glycolic acid-alt-glutamic acid)-b-PEG-b-(PLA-co-glycolic acid-alt-glutamic acid)), and the resultant micelles showed a three-fold greater release of PTX at pH 4.2 than at 7.4.[659]
8.4.3.3. Polymeric Micelle Destabilization Induced by Enzymes
The destabilization of drug-loaded polymeric micelles has been attributed to the high concentration of certain enzymes seen in diseased tissues acting as an environmental trigger. Enzymes can break the bonds holding the medication to the polymer or the hydrophobic block, as well as the block’s side chains, in a manner similar to hydrolytic degradation. Polyesters were shown to be vulnerable to enzymatic degradation, such as that caused by lipases, in addition to hydrolytic breakdown. PEG-b-poly(3-hydroxybutyrate)(PHB)-b-PEG micelles,[344] PEG-b-PCL nanoparticles,[660] which were accompanied by the release of encapsulated pyrene, and PEG-b-oligo(-caprolactone) micelles[650] were demonstrated to undergo lipase-catalyzed degradation. Proteases can also cleave the peptide bonds in poly(amino acid)s, which are employed as hydrophobic blocks; poly(-glutamic acid)-g-L-phenylalanine (PGA-g-L-PEA) micelles are one example of this.[661]
8.5.3.3. Polymeric Micelles Susceptible to Oxidation and Reduction
Intracellular glutathione’s reduction of disulphide bonds in polymeric assemblies can be employed for complete destabilization[588,600,662,663] or micellar decrosslinking (see supra). Moreover, the appealing trigger for changing the charge density and, consequently, the solubility of polymers containing viologen or ferrocene is the reversible redox reactions of organometal compounds, such as viologen and ferrocene.[664] Redox-active micelles with a hydrophobic ferrocenylalkyl moiety in a block copolymer were shown to oxidize and break down into water-soluble unimers, altering the hydrophobic/hydrophilic balance. Zero-order kinetics might be achieved by carefully controlling the release of a model hydrophobic drug (perylene) from these micelles through a selective electrochemical oxidation of the ferrocenylalkyl moiety.[665] It is possible to achieve selective medication release at pathogenic areas by using an electric current applied externally or by capitalizing on the build-up of activated macrophages in inflammatory tissues and certain tumors. The production of oxygen-reactive substances by these macrophages has the potential to initiate the transformation of redox-sensitive micelles. These redox-sensitive polymeric micelles haven’t, however, been studied in vivo as of yet.
8.6.3.3. Micellar Deformation Caused by Light
Only when exposed to ultraviolet (UV), visible (VIS), or (near-) infrared ((N)IR) light do light-responsive polymeric micelles ideally release their confined guest molecules. Because NIR has a deeper tissue penetration and fewer harmful effects on healthy cells, it is very interesting for use in biomedical applications.
- 8.6.3.3.1. Irreversible Reactions that Occur on Illumination (Photolysis)
It is possible to apply light-induced micellar disruption by cleaving photolabile hydrophobic side chains under UV or IR illumination. Synthesis was carried out on an amphiphilic block copolymer consisting of PEG and a polymethacrylate with photolabile pyrene methyl esters (PPy) on the side chain as the hydrophobic core-forming domain.[616,666] The light caused these ester side groups to separate, which in turn changed the hydrophobic micellar block into a hydrophilic poly(methacrylic acid) (PMAa) block and caused the micelles to dissociate. Given that the dissociation kinetics were regulated by light intensity, a controlled release of encapsulated Nile Red may be achieved.[666] Furthermore, once these micelles underwent core crosslinking, the crosslinks stopped the micellar destabilization caused by photolysis. Even yet, the polymer’s overall hydrophilicity rose in response to illumination, and the micelle grew, releasing the hydrophobic guests that were loaded, albeit more slowly.[616] Furthermore, the hydrophobic micellar contact forces are eliminated by means of irreversible rearrangements during illumination. As an illustration, alkyl-PEG chains were linked to hydrophobic 2-diazo-1,2-naphthoquinone derivatives, which self-assembled into micelles. When these chromophores are illuminated, a process known as the Wolff rearrangement (Figure 24) occurs, which significantly alters the polarity of the chromophores, destabilizes the micelles, and releases the contained Nile red.[667]
- 8.6.3.3.2. Light-Triggered Reversible Alterations
In addition to irreversible disintegration of micellar structures, ambient light can cause destabilization that is both reversible and non-destructive. Amphiphilic block copolymers feature a number of photoactive groups linked to them that undergo reversible structural changes upon light, primarily causing a shift in the hydrophobic/hydrophilic balance. Azolebenzenes (dipole moment change),[668] cinnamoyl (isomerization into a more hydrophilic residue or photodimerization),[584] spyrobenzopyran (formation of zwitterionic species),[669] and triphenylmethane leucohydroxide (generation of charges)[670] are examples of chemical entities that exhibit photochemically induced transitions, as recently reviewed in detail.[623] For example, when methacrylate-b-(tert-butyl acrylate-co-acrylic acid) polymers containing azobenzenes are exposed to UV light, a trans-to-cis isomerization occurs and a more hydrophilic polymer is produced, which leads to the dissolution of the assemblies.[668] An further illustration is the reversible trans-to-cis photoisomerization that occurs when PEG-b-poly(methacrylate) polymers containing cinnamonoyl are exposed to UV light. This process produces molecules with heightened hydrophilicity or triggers reversible photodimerization (Figure 25).[584,585] As will be covered in the further section, some of these photosensitive moieties were also integrated into thermosensitive block copolymers.
8.7.3.3. Other Physical Triggers that cause Polymeric Micelles to Become Unstable
In addition to the temperature, pH, hydrolysis, and light triggers listed above, other methods of inducing drug release have included ultrasonic and ion exchange. Rapoport et al. conducted a thorough investigation of the use of ultrasound as an external trigger to release medication from Pluronic® micellar systems both in vitro and in vivo.[671,672,673] When combined with ultrasound, the cellular cytotoxicity of DOX-loaded micelles was 66%; in the absence of ultrasound, it was 53%; and when free DOX was used without applying ultrasound, the only effect seen was 15% cell death.[672] For sonicated tumor cells in vivo, there was an enhanced absorption of both free medication (in PBS) and DOX-loaded Pluronic® micelles.[671,673] The ultrasound effect may be caused by one of the following mechanisms: i) increased blood vessel permeability, which causes the carriers to extravasate; ii) micelle dissociation into unimers with concurrent drug release; iii) accelerated diffusion in the interstitial and tumor; or iv) enhanced membrane permeability. All of these mechanisms increase the amount of drug that is taken up by cells.[671,672,673] By complexing cisplatin with the carboxylate groups of a PEG-b-p(glutamic acid) block copolymer, ion-sensitive polymer–metal micelles were created. Ion exchange processes took place in 0.15 M NaCl, which led to the breakup of the micellar structure and the gradual release of cisplatin from the micelles (Figure 26). Compared to the free drug,[497] intravenous administration of these micelles resulted in a considerably higher plasma level and tumor accumulation of cisplatin. Recently, cisplatin-loaded crosslinked C3M micelles were used to demonstrate the cellular absorption and release of cisplatin.[374]
By encapsulating iron oxide nanoparticles with peptide-based polymers (polybutadiene-b-poly(glutamic acid)), magnetic micelles (220-430 nm) were produced in water. Although there are currently no experimental data available, it is expected that the micellar shape can be altered under a magnetic field. As discussed in section 8.9.3.3., these superparamagnetic self-assembled hybrids may also be useful as contrast agents in MRI in the future.[674,675]
8.8.3.3. Multi-Trigger-Responsive Polymeric Micelles
- 8.8.3.3.1. Temperature and pH Sensitivity
NIPAAm, N,N-dimethylacrylamide, and 10-undecenoic acid-based random copolymers showed a CP that depended on both the copolymer composition and pH. The hydrophobic undecenoic acid side chains of the polymer clustered together to create core-shell morphologies of around 200 nm when the polymer was engineered to be below its CP and hence highly hydrated at pH 7.4 and 20 0C. A dialysis technique could be used to load the hydrophobic medication DOX into the undecenoic acid core. The undecenoic carboxylate group was protonated when the pH was lowered to 6.6, which also resulted in a drop in the CP below 20 0C. The encapsulated DOX were released as a result of the micelles disintegrating.[676,677] At pH 7.4, raising the temperature over the CP of 40 0C resulted in a similar effect.[677] Moreover, a wide variety of other block copolymeric assemblies have been documented in the literature,[678,679,680,681,682] which employ this pH dependence to regulate the temperature sensitivity.
- 8.8.3.3.2. Temperature-Sensitive Biodegradable Polymers
There have been several descriptions of dual sensitive micellar systems based on biodegradable thermosensitive (block co) polymers.[476,477,554,683,684,685] In aqueous solution, the polymers self-assemble into micelles above their critical micelle temperature (CMT), which is the temperature at which the thermosensitive block phases out. The micellar core underwent a “hydrophobic-to-hydrophilic” transformation as a result of pH-dependent cleavage of hydrophobic side chains. As a result, the CMT progressively rises, leading to micelle destabilization and polymer disintegration in the end. As an example, a study reported 2-hydroxypropyl methacrylamide lactate (HPMAm- Lacn, where n is the number of lactic acid units in the oligolactate chain).[476,633,683] These biodegradable thermosensitive polymers have methacrylamide backbones and oligolactates attached via hydrolytically sensitive ester bonds. For these methacrylamide-oligolactate copolymers, the monomer feed ratio accurately determines the CMT. A temperature range of 10 to 63 0C (or 0 to 100% HPMAm-Lac1 correspondingly) is covered by the CMT of copolymers of HPMAm-Lac1 with HPMAm-Lac2. Copolymerization with HEMAm-Lac4 could lower the somewhat more hydrophilic homopolymer poly(N-(2-hydroxyethyl)methacrylamide-dilactate) (p(HEMAm-Lac2))’s CP of 22 0C. Generally speaking, the polymers’ CMT can be adjusted to be lower than the desired micelle temperature (e.g., ambient or body temperature). When lactate side chains were hydrolyzed to a sufficient degree to raise the CMT above body temperature, the block copolymer mPEG-b-p(HPMAm-Lac2) (CMT 8 0C) showed a transient stability at physiological conditions. This led to polymer dissolution and the micelles disintegrating (Figure 27).[686] Because HEMAm-Lacn hydrolyzes much more quickly than HPMAm-Lacn does, mPEG-b-p(HEMAm-Lacn) micelles resulted in a significantly quicker destabilization time of 8 hours.
Other thermosensitive biodegradable polymers with distinct hydrolysable groups have been found recently. In poly(NIPAAm-co-dimethyl-butyrolactone acrylate),[687] the degradable moiety is a cyclic ester; as acid-labile groups, hydrazone bonds have been employed in poly(NIPAAm-hydrazone-alkyln),[688] or orthoesters in poly(N-(2-(m)ethoxy-1,3-dioxan-5-yl)methacrylamide).[689] It is expected that a second generation of controlled biodegradable thermosensitive micelles can be produced by utilizing these kinds of polymers in block copolymer designs.
- 8.8.3.3.2. Diverse
In addition to reacting to temperature, photoresponsive thermosensitive copolymers show a change in CP caused by light. When a thermosensitive copolymer containing light-sensitive chemicals (such those listed in section 8.6.3.3.2.) is exposed to UV light, it can enhance its hydrogen bonding capacity, which in turn increases CP.[690,691,692] The application of light alone was insufficient to promote dissociation in the instance of poly(2-(dimethylamino)ethylmethacrylate)-b-poly(6-(4-phenylazo)phenoxy)-hexyl-methacrylate) micelles because the hydrophobic contacts were not overcome by the light-induced trans-to-cis isomerization.[693] Additionally, carboxylic acid units could be added to provide pH sensitivity.[694] In order to create crosslinked polymeric micelles that reacted to pH, temperature, and ionic strength, cinnamoyl units were photodimerized.[695] Armes et al. produced so-called schizophrenic micelles, which are based on the pH-induced micellar inversion of assemblies of zwitterionic diblock copolymers. Because the neutral pMEMA is hydrophilic and the protonated pDEA is protonated, a diblock copolymer consisting of 2-(diethylamino)ethyl methacrylate (pDEA) and 2-(N-morpholino)ethyl methacrylate (pMEMA) is completely dissolved at pH 6 and 20 °C. The pDEA block deprotonates at pH 8.5, generating micelles where pDEA serves as the core-forming component. To produce pMEMA-core micelles, the pMEMA block is selectively salted out by lowering the pH and adding enough electrolyte.[696] A recent publication[697] provided a summary of these particular micelles that exhibit dependence on pH and ionic strength. Enzyme and temperature sensitivity were combined in a polymer based on pNIPAAm containing side chains of peptides. The hydrophilization of the peptide chains caused the CP of the copolymers to rise (from 36.7 to 40 [0]C) when the peptide was phosphorylated by protein kinase A.[698] Because of this enzyme’s activity and physiological circumstances, the polymers eventually disintegrated. Nevertheless, responsive micelles have not yet been designed using this idea.
8.9.3.3. Drug Delivery Guided by Imaging
The goal of encapsulating medications in stimulus-sensitive polymeric micelles is to get the best possible therapeutic outcome, such as drug release in response to external stimuli once the loaded nanocarrier accumulates at the site of action. An essential new feature would be the ability to use in vivo imaging to identify the presence of drug-laden micelles at their intended site of action and to then initiate the release of the loaded drug.[699] Polymeric micelles were filled with heavy elements (like I, Br, and Ba) for CT imaging, -emitting radioactive labels (like 111indium or 99mtechnetium),[700] magnetic resonance imaging (MRI) contrast agents (primarily Fe, Mn, or Gd), or quantum dots (QDs)[455,456,701] for imaging purposes. Encapsulating these compounds in micelles is advantageous because, in that form, they are less likely to be cleared by the kidneys, and their accumulation in tumor tissue (through the EPR-effect) will increase the specificity and strength of the signal, respectively.[702,703] Fe3O4 and a photosensitizer-a medication that is triggered by light-co-loading diacyllipid micelles allowed for real-time monitoring of in vitro cellular absorption.[384] Moreover, putting these magnetic nanoparticle-incubated cells under an external magnetic field produced a phenomenon known as “magnetophoretic control” of the cellular uptake.[384,704] Thus, the concentration of the medication at the target region may be increased by future in vivo delivery of iron and drug-loaded micelles in conjunction with a local magnetic field. Reddy et al. created yet another complex drug delivery vehicle using PEGylated poly(acrylamide) nanoparticles loaded with iron oxide and photofrin (a photosensitizer) together with a targeting ligand attached to the micellar shell that directed the drug’s flow into the tumor vasculature. When compared to free photofrin or non-targeted nanoparticles, a greater photodynamic therapeutic effect-that is, the elimination of tumors-was seen in vivo. The multifunctional targeted nanoparticles internalized while the iron oxide allowed for real-time MRI monitoring of the micelles' tissue localization, allowing for the selection of the best lighting timing.[705,706] Using polymeric micelles, semiconductor quantum dots may also be used for image-guided medication administration. Since QDs absorb throughout a very wide spectral range, many markers can be tested at once, and their extremely high photostability allows for real-time monitoring over extended periods of time.[701,707] Moreover, QDs containing micelles can be employed for both (photodynamic) therapeutic and diagnostic applications since they produce extremely reactive free radicals when illuminated (Figure 28).[708] 218. CdSe, which has been encapsulated in phospholipids and antibody-decorated multiblock copolymeric micelles, is the most thoroughly researched QD in biology.[709] The latter kind of micelles allowed for extremely accurate tracking of the active tumor uptake and provided a distinct imaging signal in vivo.[710]
8.4.3. Combining Stability, Longevity, and Stimulus Sensitivity
To summarize, polymeric micelles have a distinctive shape, great adaptability, and a high drug loading capacity, making them particularly appealing drug delivery vehicles for hydrophobic medicines. Drugs ought to be stably encapsulated by the ideal micellar system, even when they are circulating in vivo. Numerous approaches have been looked into to increase the micellar carriers’ stability, retention of the pharmaceuticals stored in the micellar core, and circulation times (or “longevity”). Conflicting demands are placed on the micellar building blocks in order to achieve the desired release of the medicine that has been encapsulated once the polymeric micelle has arrived at the intended target site. The development of transiently stable polymeric micelles, from which the time and site of encapsulated drug release may be precisely tuned, is made possible by internal or external triggers. Using building blocks with a hydrophilic PEG-block and a degradable or stimuli-sensitive hydrophobic block, which account for both desired features, is the most widely used method of combining longevity and triggered release properties. Few systems, nevertheless, integrate stimuli-triggered drug release with further stabilizing techniques. For instance, either core crosslinking[358] or lowering the CMC[468,571] stabilized biodegradable PEG-b-PCL micelles. Additionally, thermosensitive PEG-b-p(HEMA-Lacn) micelles showed temporary stability under degrading[601] that was dependent on pH and were core crosslinked. PEG-b-p(Asp) micelles with covalently attached doxorubicin serve as a third illustration. These micelles improved the micellar core’s compatibility with extra physically encapsulated doxorubicin.[498] Notwithstanding all of the encouraging features, only a small number of micellar formulations have advanced to clinical trials to date. Two well-known concepts are PEG-b-p(Asp) micelles with covalently bound and physically entrapped doxorubicin (NK911) and Genexol-PM® (paclitaxel-loaded mPEG-b-PLA micelles).[446,454,711] Comparing these formulations to the standard formulations[454,711] or finding a better toxicity profile that permits higher dosing, phase I trials showed longer circulation durations.[446] In summary, it is expected that the right mix of long circulating qualities and advanced stabilizing techniques would produce highly stable micelles that can travel to the target site without breaking down. While adding targeted ligands will further improve the release behavior, the introduction of stimuli-sensitive building blocks will govern the release behavior. By using imaging agents, it is possible to identify the drug-loaded micelles at the target location and apply the external trigger at the right moment. Synthetic polymers, which are the fundamental components of polymeric micelles, provide nearly infinite options for customizing and optimizing the micellar structures to achieve the required shape, drug compatibility, and drug release profile.
9. Self-Assembly, Synthesis and Theory of Block Copolymers (BCP) Solution
9.1. Self Assembly
9.1.1. General Aspects
Generally speaking, amphiphilic AB diblock copolymers self-assemble into aggregated structures to avoid unfavorable thermodynamic interactions when they are diluted in a selective solvent for one of the blocks and at a concentration higher than the critical micelle concentration (CMC). Minimization of free energy between the two blocks in solution (polymer-polymer interaction parameter, χAB) and between each block and the surrounding solvent (polymer-solvent interaction parameters, χAS and χBS) is the primary force governing the self-assembly process.[712] These interactions are significantly correlated with the packing parameter p, which was initially proposed by Israelachvili to characterize surfactant micellization. Typically, these interactions are controlled by the relative volume fractions (f), the solvophobicity, and the degree of polymerization of each block.[713] The relationship between the curvature of the hydrophobic/hydrophilic interface, the resulting particle morphology, and the surfactant molecular structure (i.e., head group area, a0, hydrophobic tail length, lc, and volume of the hydrophobic segment, v) is elegantly described by the dimensionless packing parameter, as illustrated in Figure 29.
To be more precise, p = v / a0lc and spherical micelles form when p ≤ 1=3, cylindrical micelles-also referred to as “worm-like” or “rod-like” micelles-form when 1/3 < p ≤ 1=2, and vesicles or polymersomes for 1/2<p≤1. While this idea is based on low molecular weight surfactants, it can also be applied to macromolecular amphiphilic BCPs. In this scenario, the hydrophobic block functions as the surfactant tail, while the hydrophilic block acts as the polar head group. The area between the hydrophobic and hydrophilic interfacials within each BCP chain replaces the surfactant head group area. Similar micellar morphologies, such as spheres, cylinders, and vesicles, are built in this manner; the soluble solvophilic form creates the micelle’s corona, or shell, and the insoluble solvophobic block forms the micelle’s core, providing colloidal stability of the assembly in solution.[714] However, in this instance, in addition to the negative entropic contribution resulting from the solvophobic chains within the micellar core stretching and the decrease in interfacial energy between the solvent and the core-forming block, it is also necessary to take into account the repulsive interactions between the solvophilic corona chains that cause stretching. In actuality, the volume fractions of the two blocks-which are connected to their molar masses and chemistries-largely determine the micelle morphology that results. This is particularly true for the solvophilic block, where the solvent’s quality has a significant impact. Due to the increased number of interaction characteristics, the addition of a third (or even more) block(s) considerably complicates the accurate prediction of particle shape and block layout. However, comprehensive phase diagrams that describe all potential self-assembled morphologies for particular multiblock copolymers and solvent systems have been created, primarily based on experimental data.[715,716] The exchange between individual BCP chains and micelles is typically much slower than the assembly process, which means that thermodynamic equilibrium may not always be reached and intermediate kinetically frozen structures may form. This is another factor that must be taken into account.[714] Because of this, it has become normal practice to generate BCP micelles by using a cosolvent that is the same solvent for both blocks. This helps to simplify the transition to the selective solvent and raises the likelihood of obtaining structures that are closer to equilibrium. The key takeaway from this is that a wide variety of self-assembled structures may be created by carefully selecting the building pieces, carefully planning the macromolecular chain, and fine-tuning the interactions between the polymer and the solvent. The concept has been demonstrated in practice on multiple occasions. One such instance is the groundbreaking research conducted by Eisenberg’s group on the micellization of asymmetric polystyrene-poly(acrylic acid) (PS-PAA) BCPs with different PS/PAA ratios and polymerization degrees under various solution conditions. This work produced a broad range of thermodynamically controlled morphologies, including spheres, rods, bicontinuous rods, and bilayers (lamellae and vesicles) as well as more intricate inverse structures like large compound micelles and hexagonally packed hollow hoops.[717] An extensive range of structural characterization techniques is available and has been effectively used in pertinent experimental investigations to clarify various aspects of the micellization process and extract useful information about the morphology and properties of the formed micelles.[718,719,720,721] It is possible to separate them into three primary groups: spectroscopic techniques, microscopy, and scattering techniques. Static and dynamic light (SLS and DLS), small angle X-ray (SAXS), and small angle neutron scattering (SANS) are the most widely used scattering techniques for soft materials in solution. These methods examine micellar morphology and have the benefit of providing a mean value computed over a large number of micelles. The MW, the size, shape, and polydispersity of the scattering species in solution, as well as their interactions with one another, are all revealed by these kinds of observations. In a similar vein, the zeta potential and consequently the effective charge of the particles are obtained by electrophoretic light scattering (ELS). Direct visualization through atomic force, scanning, and transmission electron microscopy (AFM, SEM, and TEM) measurements is a useful way to examine the size and distribution of size, shape, morphology, internal structure, aggregation, and dispersion of polymeric assemblies when it comes to microscopy techniques. The constraints of conventional approaches related to surface deposition and solvent evaporation are also overcome by state-of-the-art environmental SEM and cryogenic TEM techniques, which provide observation of the solutes in their hydrated condition. Finally, among the many spectroscopic methods that are accessible, nuclear magnetic resonance (NMR) spectroscopy and fluorescence spectroscopy have been used extensively in the research of micellar solutions and are thought to be particularly useful in the examination of local processes within the micelles. The determination of CMC, the investigation of chain exchange between micelles and the unimer-micelle equilibrium, and even chain conformation dynamics are made easier by the use of appropriate fluorescent probes, such as pyrene. Measurements using NMR spectroscopy can also reveal conformational changes and dynamics, providing important information about the structure and composition of micellar materials. Extensive study has been done on theoretical modeling and simulations of BCP self-assembly in solution, in addition to experimental studies.[720,722,723] While theoretical methods have shown to be effective in analyzing the kinetics and thermodynamics of copolymer self-assembly in bulk, the presence of solvents complicates self-assembly in solution, making simulation modeling more difficult. Fortunately, it is now possible to mimic the self-assembly of copolymer solutions thanks to recent advancements in simulation techniques (such as the mesoscopic approaches) and an increase in processing capacity. Consequently, we are able to discern between the thermodynamically stable and meta-stable structures and comprehend the impact of individual parameters on the self-assembly of BCPs in solution by theoretical modeling and simulation.[723] The micellization process and the dependence of fundamental structural parameters of the micelles, such as CMC, aggregation number, overall size, core radius, and corona thickness, on the molecular characteristics of the block copolymer, with respect to the degrees of polymerization of the constituent blocks, as well as the Flory-Huggins interaction parameters between the blocks and between the blocks and the solvent, have been described by a number of theories (i.e., scaling theories, semi-analytical and/or lattice self-consistent mean-field theories) that have developed over the years as well as primarily Monte Carlo simulations.
9.1.2. Morphology of Micellar Structures.
9.1.2.1. Spherical, Cylindrical Micelles and Polymersomes
The size, aggregation number, and stability of a spheroidal micelle can all be impacted by the length of each polymer block within it. The C3M core radius (Rcore) in (AB + C) systems is primarily independent of the length of the homopolymer (NC), at least below a significant critical length about N ∼ 5000,[30,106,394] and is directly proportional to the length of the charged block in the block copolymer (NB),[107,394,404] Although it has been observed that the size of the neutral block (NA), which forms the corona, has a negligible impact on the size of the core, it significantly influences the thickness of the corona (H) and, consequently, the hydrodynamic radius (Rh), or micelle overall size, which is an important parameter for regulating biodistribution.[146,724] On the other hand, it is demonstrated that for a (AB + C) system, the aggregation number (P), or the total number of chains in a given micelle, decreases as the neutral block size grows.[30,146,394,724] A previous research used PCMs containing PEO-block-pLys (PEO-b-pLys) combined with ssDNA or pGlu to construct experimental scaling laws for Rcore, Rh, H, and P in order to quantify these physical trends.[394] In Figure 30, these scaling principles are displayed as black lines superimposed on top of a collection of published (AB + C) C3M data that reflect a range of synthetic and biological polymers. Plotting the results against the third length variable, the data were normalized using the scaling rules for the first two polymer lengths. This chart compares the normalization, which reduced all the data to a single trend, to the matching scaling law. It was shown that NC had no discernible impact on any physical parameter, which is useful for developing flexible delivery systems in which the C component is frequently a medicinal substance or biomolecule. Since it is taken into account for physical scaling, polyelectrolyte length is typically given as degree of polymerization; nevertheless, contour length or physical size are probably a little more accurate component. For two geometrical extremes of C3M structure, more accurate theoretical work on C3Ms suggests scaling relationships for physical properties (see below).[59,725] Equations 2 through 4 present predictions for Rcore, P, and H for PCMs with fully ionized chains in a “good” aqueous solvent at the star-like limit, where (Rcore << H).
Equations 5-7 provide scaling theory for the identical conditions, but at the crew-cut limit where (Rcore ≫ H).
As is the case for most experimental work, the C3Ms in Figure 30 are between the star-like and crew-cut regimes and are composed of fully ionized chains in excellent solvents. The experimental scaling laws[394] displayed in Figure 30 are in agreement with predictions for the two structural limits when taking into account the intermediate regime of these C3Ms. Additional theoretical predictions (see below), indicate dependency on solvent quality, salt content, and degree of ionization.[59,67,715,722,726] Designing customized carriers can be sped up by knowing how polymer structure affects C3M structural properties.
The three most prevalent types of micelle morphologies are spherical, cylindrical, and polymersomes, as was discussed in the preceding section. Since spherical micelles are the most common type of nanoassemblies, they were the first to be noticed and thoroughly investigated. PS-polyisoprene (PS-PI), PS-poly(ethylene oxide) (PS-PEO), poly(propylene oxide)-PEO (PPO-PEO), and their triblock equivalents known as poloxamers or Pluronics (PEO- PPO-PEO), polybutadiene-PEO (PBD-PEO), and many more are typical examples of related amphiphilic BCPs.[727,728] Despite their apparent simplicity from a morphological perspective, spherical micelles still exhibit a high degree of variability. The respective lengths (or, alternatively, polymerization degrees) of the two blocks and the matching sizes of the micellar sections determine the first level of classification for AB amphiphilic linear diBCPs. Micelles that exhibit a wide corona and a small core are referred to as “crew-cut” micelles, whereas those that exhibit the reverse pattern are called “star-like” or “hairy” micelles. The form of spherical micelles produced by linear triblock copolymers is also influenced by the chemical makeup of the blocks and how soluble they are in the chosen solvent. For instance, in a solvent where the outer A blocks are soluble, ABA copolymers produce typical core-corona micelles. However, in a solvent that is suitable for the center B block, the macromolecular chain loops, causing the A blocks to segregate in the core and generating “flower-like” micelles. When a third block with a distinct chemical makeup is added, as in the case of ABC triblock terpolymers, things become much more difficult. In particular, dissolving the outer A and C blocks in a suitable solvent produces core-corona micelles, which are characterized by a corona that is either mixed or compartmentalized based on how well the A and C blocks work together. The latter type of micelle is also referred to as “Janus” micelle. Otherwise, three-layered or core-shell-corona micellar morphologies with a soluble or insoluble shell arise when there is only one insoluble outer block A, depending on whether the solvent is selective for both B and C or only C blocks. Figure 31 provides a schematic overview of the many spherical micelle types that have been described.
Cylindrical micelles resulting from the direct dissolution of an amphiphilic BCP are less common, as indicated by the relatively restricted range of packing parameter values that correspond to their production. “Worm-like” micelles are typically anisotropic, one-dimensional structures with lengths several times longer than their cross-sectional diameters. They do, however, also display rich structural polymorphism, with the two extremities being elongated (reaching the micrometer length scale) thin flexible structures called “thread-like” micelles and relatively short (finite length) rigid cylinders. Enclosed cylindrical ring-shaped/toroidal structures have also been observed.[717,729] This class of nanoassemblies is mostly derived from BCPs including at least one crystallizable block, which is typically the micellar core. Consequently, crystallization is a crucial factor in determining the micellar morphology during the production of BCP micelles, which is why the name “crystallization-driven self-assembly” (or CDSA) was coined.[714,730,731,732,733] The self-assembly of BCPs with the metal-containing poly(ferrocenyldimethylsilane) (PFDMS or PFS)[734,735,736] or the regioregular semiconducting poly(3-hexylthiophene) (P3HT)[737,738,739] polymers as the crystallizable core-forming blocks that led to cylindrical/“fiber-like” micelles is one of the most extensive collective works in this field, according to Manners and Winnik et al.[733,734] Furthermore, since the exposed crystalline faces at each micelle core terminus are available for subsequent polymer crystallization, this group was the first to report on the elongation of these pre-existing micelles upon further addition of unimer chains via an epitaxial growth process, establishing the notion of “living” CDSA.[714,734] Two distinct seeding methods, namely “seeded growth” and “self-seeding,” were proposed to generate monodisperse cylinders (Figure 32). To put it succinctly, “seeded growth” is the process of creating very short micelle seeds by sonicating preformed polydisperse “fiber-like” micelles vigorously, then elongating those seeds by adding more unimers. However, in the “self-seeding” process, micelle fragments remain and serve as seeds for the epitaxial growth of released unimers upon cooling or removal of the good solvent. This is because free unimers are released from micelles through a partial dissolution process that involves either thermal annealing or the addition of a small amount of a good solvent for both the core- and corona-forming blocks.
Dove, O’Reilly, and their colleagues have also conducted a thorough investigation into CDSA as a technique for generating cylindrical micelles derived from BCPs that primarily contain biocompatible and biodegradable crystallizable blocks such as poly(ε-caprolactone) (PCL) and poly(L-lactide) or poly(D-lactide) (PLLA or PDLA).[741,742,743,744,745] They discovered, among other intriguing things, that while homochiral PLLA-PAA and PDLA-PAA BCPs separately form cylinders with crystalline cores in THF/H2O at 65 0C, an unimer-exchange process forms small stereocomplex spherical micelles upon heating a 1:1 mixture of both BCPs in solution or a mixture of the respective cylinders under the same conditions.[741] Other than these notable instances, a number of additional crystallizable polymers have been employed to create cylindrical or “fiber-like” micelles with crystalline cores; a thorough description of this process can be found in the most recent review by MacFarlane et al.,[733] while Pearce et al.,[746] has recently published an equally fascinating review on the preparation of anisotropic nanoparticles, including cylindrical micelles, and their uses.
Since the labs of Eisenberg and Meijer made the first observation of BCP vesicles, also known as polymersomes, in 1995, there has been a significant amount of research conducted on them because of their prospective applications as well as academic interest.[717,728,747] Generally speaking, polymersomes are hollow spheres with hydrophilic internal and external coronas and a hydrophobic wall. Primarily, their water-containing core has a significant deal of potential for compartmentalization, which is an essential process for life. The tight relationship between these materials and biological, biocatalytic, and biomimetic applications has been largely determined by their similarity to cellular structure, as demonstrated by the abundance of comprehensive literature reviews.[748,749,750,751,752,753,754,755,756,757,758] In terms of shape and function, polymersomes are essentially the macromolecular equivalent of liposomes; nevertheless, they are more stable and possess better mechanical qualities than vesicles based on lipids. Additionally, because they are made up of BCPs with theoretically greater MWs, they have thicker walls and more bending rigidity, which increases their capacity to withstand tension and area strain before rupturing but simultaneously decreases the fluidity and permeability of the membrane.[717] Their size varies from several tenths of nanometers to several microns; the preparation technique and the total interfacial energy between the solvent and the polymersome wall, which is highly dependent on the water content, are important factors. The content of the copolymer, the usage of a common solvent, the environment's pH and ionic strength, and other variables are also crucial. By adjusting these parameters, it is also feasible to regulate the pressure differential between the vesicle's exterior and interior, causing deformation that can result in structures that collapse completely or partially. Moreover, this manipulation can create more complex structures like entrapped vesicles, uniform and large polydisperse bilayer vesicles, hollow concentric vesicles, and “onion-like” vesicles.[717] As previously stated, a key factor in controlling the final vesicular morphology is the polymersome preparation process. There are many different preparation techniques that fall into two main categories: solvent-free and solvent-displacement. The first category consists of electroformation, in which amphiphilic copolymers are produced directly in aqueous solution, polymerization-induced self-assembly (PISA), and rehydration of thin polymer films. Conversely, solution displacement techniques such as direct injection, emulsion phase transfer, and microfluidics require dissolving the BCPs with an organic cosolvent, adding the polymer solution to an aqueous solution, and then eliminating the organic solvent. The most recent evaluations by Lefley et al.[747] and Iqbal et al.[756] provide a thorough summary of these various preparation techniques. The right approach can be chosen to modify not only the size and distribution of the polymersomes but also their corresponding shapes, potentially leading to the development of nonspherical structures. As with the other micellar morphologies, the ability to acquire the desired vesicle morphology has been greatly aided by the synthesis of BCPs with customized chemical compositions, degrees of polymerization, and macromolecular architectures made possible by modern controlled polymerization techniques such as RAFT, ATRP, and ROMP. Two different classes of polymers can be selected from the large selection pool of accessible polymers based on their biodegradability and, consequently, biocompatibility, which is an essential need for biological applications. The hydrophobic portion of nondegradable polymersomes is often composed of conjugated/aromatic hydrocarbon blocks, such as PBD, PS, poly(ethylethylene) (PEE), and poly(dimethylsiloxane) (PDMS), that lack any linkages that may be broken down by hydrolytic cleavage. On the other hand, ester and carbonate linkages in water-soluble polymers, such as PCL, poly(lactic acid) (PLA), and poly(lactic-co-glycolic acid) (PLGA), are hydrolytically broken with the addition of water, making them biodegradable. Polypeptide-based polymer systems are being used to produce polymersomes as a result of the need for biocompatibility.[759] Moreover, stimuli-responsive qualities are highly desired since they give the polymersomes more usefulness, particularly when they are meant for applications that are important to biology.[748,749,750,752,760] The availability of many BCP topologies is a final element that contributes to the diversity. AB diblock, ABA and ABC triblock linear and grafted copolymers, as well as the inclusion of a third block, are widely exploited and result in novel asymmetric membrane compostitions.[761]
9.1.2.2. Complex Supramolecular Structures
Either by secondary assembly of precursor micelles, usually triggered by an external stimulus, or by self-assembly of BCPs with enhanced architectural complexity, even more fascinating multicompartment nanostructures and hierarchical superstructures have been achieved.[714,727,762] Multicompartment micelles (MCMs), often called “patchy” nanoparticles, are typically created when linear triBCPs self-assemble by adjusting how the various blocks interact with the solvent and how miscibly they mix with one another. However, other sources of such morphologies include mixes of linear diBCPs or miktoarm starter-polymers, and another option is the utilization of crystallizable core-forming blocks. All of these nanostructures have in common the compartmentalization of either the micelle’s core or corona, which results in complex morphologies. These have piqued scientific interest due to their intriguing potential applications as well as their fascinating self-assembly case studies.[763,764,765,766,767,768] The work of Muller and his group, which carefully examined the self-assembly of linear ABC terpolymers with PS as the “sticky” A core block, PBD or poly(3-butenyl[dodecyl]sulphane) as the inert B core block, and poly(methyl methacrylate) (PMMA), poly(tert-butyl methacrylate) (PtBMA) or poly (2-vinylpyridine) (P2VP) as the repulsive C corona block segments, is one telling example of hierarchical superstructures created from MCMs.[769,770] By varying the volume ratios of the core-forming blocks and the solvent composition, the initial triBCP self-assembly formed monovalent and divalent compartmentalized colloidal building blocks that either co-assembled into a mixed superstructure or self-assembled into spherical clusters or linear supracolloidal chains. Moreover, they produced cylindrical micelles and vesicles with tunable “patchy” shape by fine-tuning the composition or suitable post-polymerization alterations of the same triblock terpolymers.[771,772] Similar to this, Gröschel et al.[773] reported on the creation of supracolloidal chains of patchy micelles using a terpolymer made up of a P2VP block that can flip between core and corona depending on pH, a hydrophobic PS core block, and a water-soluble nonionic PEO corona block.Following the sequential treatment of each block with the appropriate solvents (a mixture of acetone and isopropanol), precursor micelles with a collapsed PS core and a mixed corona were formed. The P2VP corona collapsed when the organic solvent was exchanged for water, which led to the patchy micelles assembling into supracolloidal chains (Figure 33). The propagation of extensive chains and networks was triggered by the colloidal solution being heat-treated and the pH being adjusted to the proper values. Another thorough example of triblock terpolymer-based “patchy” nanoparticles is found in the work of Moreno et al.,[774] who used PS-PBD-PMMA, PS-PI-PMMA, and PS-PI-P2VP linear triBCPs with different compositions to study the basic mechanisms that generate various types of “patchy” nanoparticles and create a complete morphological phase diagram of the resulting nanoassemblies.
Microarm star terpolymers, in addition to linear copolymers, are also favorable for the creation of multicompartment nanoparticles (MCNPs) because of the topological constraint on the block connection point. The self-assembly of μ-A(BC)n miktoarm and miktobrush star terpolymers, which results in the creation of “hamburger” micelles, patchy micelles, segmented “worm-like” micelles, and multicompartment vesicles, was first demonstrated by the Hillmyer and Lodge group.[775,776] Since then, the sector has seen several noteworthy examples of activity.[777,778,779,780] For instance, Huo et al.[779] recently used the self-assembly of a specially created PEG(PAA-PS)20 star polymer to make MCNPs with hydrophilic PAA and poly(ethylene glycol) (PEG) and hydrophobic PS subdomains. Micelles, vesicles, and threads made up of the single-star polymers’ 1D alignment were seen, as given in Figure 34, depending on the PS/PAA volume ratio, water content, polymer concentration, and pH value.
The self-assembled MCNPs produced from copolymers including crystallizable blocks via CDSA represent a different and particularly remarkable class.[714,727,762] The morphological diversity and complexity achieved in the case of cylindrical micelles with crystalline core and “patchy” surface are astounding. Examples include AB cylinder co-micelles, BABAB pentablock co-micelles,[781] PS-PE- PMMA patchy core-crystalline cylinder micelles with high control over the location of the patches,[782] concentric lenticular block co-micelles with fluorescent red, green, and blue colors,[783] “star-like” supermicelles of PFDMS-PI cylindrical arms on the PFDMS seed,[784] or even “windmill-like” supermicelles,[785] as outlined in the thorough review by Gröschel and colleagues.[765] Surface-compartmentalized cylindrical micelles can be further subdivided into micelles with a microphase-separated corona that is either “Janus”-type or “patch-like,” with multiple compartments of varying polarity or chemistry. One example of this is block co-micelles, which have multiple surface compartments arranged in a “block-like” fashion along the cylindrical long axis, as shown in Figure 35.[766]
In this vein, the Schmalz group has conducted a great deal of research on the self-assembly of linear terpolymers of PS, PE, and PMMA into micelles that resemble “worms” and have a crystalline PE core and a “patchy” PS/PMMA corona.[782,786,787] Comparable morphologies were discovered by Manners et al.[788,789] using the CDSA of a PS-PFS-PMMA triBCP. These researchers also looked at an alternative method for creating “patchy” nano-cylinders, which involves co-assembly of AB and BC diblock copolymers in which the B block forms the crystalline insoluble core, as in a number of instances involving a PFS core block.[736,790,791,792] Micellar nanostructures that are equally complex can be self-assembled from BCPs with intricate architectures, such as graft, comb, and branched copolymers,[793,794,795] as well as multiblock[796] and cyclic.[797] Meanwhile, a new class of BCPs known as bottlebrush copolymers has recently surfaced, offering an unparalleled capacity for architectural intricacy that can result in nanoassemblies that are equally unique.[731,798,799,800,801,802,803] Lastly, noteworthy examples of inverted bicontinuous morphologies are conetworks formed by the self-assembly of cross-linked star BCPs[804,805] and cubosomes and hexosomes that arise due to strong block asymmetry, which corresponds to p values above 1.[806,807,808]
9.1.3. Alternative Self-Assembly Routes
9.1.3.1. Polymerization-Induced and Electrostatic Self-Assembly
A number of commercial applications involving industrial scale-up are limited by the fact that BCP solution self-assembly is traditionally a post-polymerization process carried out in diluted solution conditions (<1% w/w).[714] PISA has been proposed as an alternative pathway for the in situ facile production of BCP nanoparticles with varying morphologies at high concentrations (up to 50% w/w) during the last ten years. The basic idea behind this polymerization process is that a monomer that corresponds to a homopolymer that is insoluble in the particular polymerization medium is used to chain extend a soluble macromolecular precursor (the stabilizing block), either in emulsion or dispersion polymerization conditions. In this manner, at a certain critical polymerization degree, the expanding second block eventually becomes insoluble, causing microphase separation and the creation of BCP self-assembled nanoparticles. Similar to conventional BCP micelles, these nanoassemblies undergo a diversity of morphologies as the core block grows and the packing parameter varies (Figure 36).
The most frequently found species are spherical, cylindrical (sometimes known as “worm-like”), and vesicular; other mixed phase and transitory nanostructures may also be present. The volume ratio between the solvophilic and solvophobic segments, copolymer concentration, solvent quality, and other reaction parameters control the equilibrium nanoparticle morphologies during chain extension polymerization.[809,810] PISA has previously been used with success for a wide range of functional monomers in a wide range of solvents, including as water, silicone fluids, supercritical CO2, polar solvents like ethanol, and nonpolar solvents like n-alkanes.[714,810] Aqueous emulsion polymerization and aqueous dispersion polymerization are the two types of RAFT polymerization that are suitable, and these two types account for the majority of the numerous published works on PISA that are available in the literature.[811,812] Water-miscible vinyl monomers are utilized to create the core-forming blocks in RAFT aqueous dispersion polymerization, whereas water-immiscible monomers are used to start the RAFT aqueous emulsion process from a water-soluble macromolecule. As outlined in Armes’ crucial review, Figure 37 displays a representative list of steric stabilizers that have been employed in both kinds of RAFT-mediated polymerizations, arranged according to charge.[811]
Of course, there are many other polymerization techniques available for PISA processes, such as ATRP, NMP, ROMP, ring-opening polymerization (ROP), living anionic polymerization (LAP), iodine-mediated polymerization (IMP), and organotellurium-mediated radical polymerization (TERP). The group of O'Reilly[813] recently summarized ROMP mediated PISA, and Pan and his group[814] recently published a thorough review with examples of each technique. Additionally, PISA approaches have recently been combined with CDSA in an attempt to use crystallizable core-forming blocks to reach nonspherical morphologies including cylinders/fibers and platelets. Manners’ group specifically used anionic polymerization to induce the crystallization of the structure-directing PFDMS block to drive the PISA synthesis of monodisperse cylindrical and “fiber-like” micelles of regulated length.[815,816] Similar to this, the Tang group used ferrocene-containing BCPs in combination with synchronous ROMP, PISA, and CDSA (ROMPI-CDSA) to produce uniform crystal-line lenticular platelet nano-objects,[817] while the Patterson group reported on the ring opening polymerization-induced CDSA (ROPI- CDSA) of PLLA-PEG copolymers.[818] Aside from varying the polymerization methods, choosing the right initiation approach is another way to manage the PISA process. To date, thermally initiated controlled/living polymerizations have been used in the majority of PISA syntheses published. However, other novel starting mechanisms including visible light, microwaves, enzymes, electrochemistry, or ultrasound are being investigated at the moment, with photoinitiated PISA being more common.[819] Vesicles are always of great interest among the multitude of morphologically different nano-structures created using PISA, primarily because of their biological application potential connected to drug delivery.[820,821] The ability of the nanocarrier to react to particular stimuli (e.g., temperature, pH, light, redox potential, reactive oxygen species, enzymes) is another essential component for such activities, as previously indicated. Therefore, a particularly fascinating area of research consists of stimuli-responsive, primarily vesicular structures assembled using PISA.[809,810,822] The ability of these particles to undergo order–order and order–disorder transitions in response to external stimuli is one of their most intriguing characteristics. This ability allows the particles to modify the packing parameter and the self-organized morphology post–synthesis, which in turn allows for transitions between various morphologies.[809,810] Cross-linking is a final way to further stabilize these nano-objects, preserving their structural integrity and boosting their potential for bioapplication.[810,820,823]
The main force behind all of the self-assembly mechanisms that have been covered thus far is hydrophobic contact. Using electrostatic interactions as a means of promoting self-organization is an alternate approach, particularly when considering BCPs that contain charged blocks.[61,824,825] A variety of oppositely charged species, such as linear homo- and block-polyelectrolytes, biopolymers, DNA and RNA, proteins, surfactants, metallic complexes, nanoparticles, etc., can interact electrostatically with these double hydrophilic block copolymers (DHBCs). The interacting charged species or segments become less soluble as a result of this interaction, generating the core of core-shell structured nanoassemblies that are supported by the hydrophilic blocks that comprise the shell. These electrostatically constructed micelle equivalents have been referred to by a number of names, including C3Ms, block ionomer complexes (BICs), (inter)polyelectrolyte complexes (PECs and IPECs), and polyion complex (PIC) micelles. The complexation of two or more suitable ionic-neutral di- and/or triBCPs can also produce multi-compartment micelles with a (partially) segregated core or corona, “Janus” micelles (or vesicles) with a laterally phase segregated corona, or even “onion-type” micelles, which are composed of a hydrophobic core, a coacervate inner corona, and a neutral outer shell. These morphologies are accessible in addition to simple core-corona morphologies.[61] The internal structure and morphology of these aggregates are determined by numerous important parameters associated with polymer composition, architecture, cohesive interactions, miscibility, and solubility. Furthermore, because these structures are determined by electrostatics, they are susceptible to variations in pH and salt concentration and type. They also exhibit a stimuli-responsive characteristic when the proper environmental conditions are met. The formation and characteristics of the resultant C3Ms are undoubtedly significantly influenced by additional driving forces, such as hydrophobic interactions, that are present in the system. In an analogy to classical micelles, one final significant factor influencing morphology is the composition of the interacting BCPs, particularly the block length ratios. Specifically, relatively long neutral soluble blocks result in spherical “star-like” micelles, whereas shorter corona-forming blocks typically produce “worm-like” micelles or vesicles. Anionic polyacrylic acid (PAA), poly(methacrylic acid) (PMA), cationic poly (2-dimethylamino ethyl methacrylate) (PDMAEMA), poly(ethyleneimine) (PEI), biocompatible poly (L-lysine) (PLL), poly(aspartic acid) (PAsp), and poly(L-glutamamic acid) (PGlu) are some examples of polymers that are commonly used for such formulations. On the other hand, PEO/PEG is the most commonly chosen polymer for corona-forming blocks.[61,824] C3Ms have remained highly attractive to scientists since the groundbreaking work of Harada and Kataoka in 1995.[113] This is mainly due to their potential biotechnological uses, particularly in nanomedicine where they can be used to control the release and delivery of therapeutic and bioactive molecules, such as biomacromolecules. It is feasible to select charged biocompatible and biodegradable blocks, such as polyesters, polysaccharides, and polypeptides, for the interacting segments of such systems.[824] As schematically shown in Figure 38, these BCPs complex with low- or high-MW active pharmaceutical ingredients (APIs) of relevant importance (i.e., drugs, proteins, peptides, genes), forming PIC micelles that are capable of stimuli-responsive destabilization/degradation (usually related to pH or ionic strength) that releases their pharmaceutical payload.
9.1.3.2. Self-assembly in Confinement and in other Media
Recently, a unique technique for producing soft matter-based colloidal particles with well-defined size, shape, surface patterns, and interior morphologies has emerged: solvent evaporation-driven self-assembly of BCPs in emulsions.[826] Control over the nanostructure of the resulting BCP nanoparticles is made possible in these systems by the soft and mobile emulsion interface, which causes a spontaneous deformation of the particle shape as the solvent evaporates. Consequently, it is essential to carefully manage the interfacial activity of the nanostructures using the right surfactants, and stimuli-responsive surfactants can provide even more control over the entire process of self-assembly. In this manner, a reversible shape transition can be facilitated and the interfacial activity of the particles can be changed in response to particular stimuli.[826] As was previously mentioned, the bulk morphology of amorphous BCPs is primarily determined by the volume fraction, number, and enthalpic interactions of the blocks; in the emulsion confinement, the degree of confinement, the rate of solvent evaporation, and the interfacial tension between the blocks and the surfactant/water interface further control the morphology.[827] The work of Gröschel’s group is one of the most common examples of BCPs restricted self-assembly in emulsion droplets.[827,828,829] By cross-linking the hemispherical lamellar nanostructures created by the evaporation-induced confinement assembly of a PS-PB-PtBMA triBCP in spherical oil-in-water droplets stabilized by cetyltrimethylammonium bromide (CTAB) surfactant, they have reported on the formation of shape- and surface-anisotropic “Janus” nanocups.[828] Similarly, they prepared “Janus” nanorings from a PS-PB-PMMA terpolymer.[829] In toluene/water/CTAB emulsions, a PS-PE-PMMA triBCP with a crystallizable middle PE block was contained, and protocols for evaporation and crystallization were followed at temperatures that favor either evaporation or crystallization first. This was their most recent similar investigation.[827] Not surprisingly, the morphology of the final nanostructures was dictated by the preparation process. In particular, the PS-PE-PMMA triBCP first microphase-separated into microparticles with lamellar morphology when evaporation was performed well above the bulk crystallization temperature of the PE block (Tevap > Tc). This was followed by crystallization into a variety of frustrated morphologies (e.g., “bud-like”, double staircase, spherocone). As shown in Figure 39, the PS-PE-PMMA terpolymer underwent CDSA into patchy crystalline-core micelles by evaporating at much lower temperatures that permit the PE block to crystallize from solution (Tevap < Tc). This was followed by confinement assembly into lenticular microparticles with compartmentalized hexagonal cylinder lattices.
Studies have also been conducted on BCP self-assembly in media other than water and typical organic solvents, including as ionic liquids (ILs).[830,831] ILs are a unique class of promising liquids that may find use in energy devices, reaction media, separation materials, and other applications. They are room-temperature molten salts that are typically constituted of an asymmetric organic cation and an inorganic or organic anion. Because of their special qualities-such as low vapor pressure, exceptional thermal stability, high ion conductivity, and noncombustibility-they are widely regarded as “green” solvents. As such, they are used for a variety of purposes, such as the extraction, partitioning, and separation of different target molecules as well as as electrolytes for electrochemical devices. Furthermore, by carefully matching anions and cations, it is possible to fine-tune characteristics like hydrophilicity and hydrophobicity, Lewis acidity and basicity, and hydrogen bonding capacity. With regard to BCPs self-assembly media with adjustable solvating capability, either at low or high polymer concentration, all these characteristics make ILs attractive candidates. In the second scenario, ion gels are typically created with intriguing potential uses in electroactive soft actuators, lithium-ion batteries, and electric double layer capacitors.[830,831] Lodge and colleagues were the first to report on the self-assembly of a PB-PEO BCP in a 1-butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6]) IL in diluted solutions. Depending on the length of the PEO block, this process resulted in the formation of spherical and “worm-like” micelles as well as bilayered vesicles with a PB core and a PEO corona.[832] Since then, Lodge’s group has continued to work in the field. Their most recent research focuses on spherical micelles with varying core sizes made of poly(methyl methacrylate)-block-poly(n-butyl methacrylate) (PMMA-PnBMA) in a combination of ILs (1-ethyl-3-methylimidazolium bis(-trifluoromethylsulfonyl)imide, [EMIM][TFSI], and 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)-imide, [BMIM][TFSI]).[833] Bhushan et al.’s work provides another example of a recent example of polymorphism that is currently available. Through the use of different ILs and suitable changes in polymer concentration and/or solvent, they were able to induce shape transformation of self-assembled PEG-PCL nanostructures and produce nonspherical polymersomes, such as “worm-like” aggregates, stomatocytes, nanotubes, large hexagonal, and tubular-shaped polymersomes.[834] The Alexandridis group also investigated the effects of various protic and aprotic ILs on the micellization of PEO-PPO-PEO triBCP in comparison to corresponding inorganic classic salts. Based on thermodynamic changes, the intermolecular interactions that propel the micellization process in each case were thoroughly discussed.[835]
9.2. Synthesis and Theory.
Non-covalent intermolecular interactions are the primary mechanism driving the development of the nanocarrier in both polymersomes and micelles. When amphiphilic polymers come together, the hydrophobic components are removed from the aqueous medium, which raises the system’s overall entropy and releases solvent molecules. Stability against dissociation is a critical feature in the production of self-assembled polymer micelles, particularly in the very diluted circumstances found following systemic injection. It is this parameter that the critical micelle concentration (CMC) describes.[427] The amphiphilic polymers are only found as single individual chains below the CMC. The polymer chains begin to link and form micelles when the concentration of the polymer rises and approaches the CMC. When block copolymers are compared to low-molecular-weight surfactants, they often show a lower CMC and therefore greater stability. Stronger interactions are caused by the larger polymer segments, which is why this feature exists. For example, the CMC of small molecules surfactants such as sodium dodecyl sulfate is approximately 0.2%, whereas the CMC of block copolymers is generally between 0.0005-0.002%.[427] Because they are unstable in a diluted aqueous environment and readily dissociate due to the substantial dilution encountered after injection, amphiphiles with higher CMC values might not be appropriate for use in the design of DDSs. But the process of creating self-assembling structures, like micelles and polymersomes, isn't always simple. A suspension of self-assembled structures can be created from a block copolymer solution using a variety of techniques.[836] Several of the most popular tactics entail a “solvent exchange.” After the preparation of micelles and polymersome for the encapsulation of BTAs, the block copolymer is first dissolved in an organic solvent (selected because it works well for all the blocks). The solution is then transferred to the selective solvent, often water. Water can be added to the organic solution step-by-step, dialyzed against water gradually, or the block copolymer solution in the organic solvent can be introduced straight to water while being vigorously stirred. After that, a thorough dialyzation of the suspension against water is performed to eliminate any remaining organic solvent.[837] Extrusion of the resultant dispersion through nanoscale pores after the polymer aggregate forms produces size uniformity and can also be utilized to cause the transition of micelle geometries from spherical to cylindrical.[838] Large polymersome formation, however, can be difficult, necessitating the use of more intricate formation techniques.[839] The majority of micelles and polymersomes are formed by carefully balancing segments that are hydrophilic and hydrophobic. It’s interesting to note that the creation of micelles can be accelerated by the block copolymer’s contact with the BTAs. A polyplex can arise via the complexation of a charged BTA with an oppositely charged polymer. On the other hand, if the polymer is a block copolymer made up of a neutral hydrophilic segment and a poly(ionomer) segment, the formation of the ionic complex is linked to the loss of hydration, and the micelles of the polyion complex are formed as a result of neutralizing the charged polymer block. In this scenario, the copolymer's ionomer block and the BTA combine to form the hydrophobic segment, which is encircled by the neutral hydrophilic block. The necessary amphiphilicity for the micelle assembly is induced by a charged payload neutralizing the charge of the block copolymers.[37,283] Typically, a polymer micelle or polymersome uses a passive loading technique to encapsulate the payload.[840] A hydrophobic agent may be encased in the solid micelle core during the nanocarrier manufacturing process, or the hydrophilic BTA may be immersed in the inner watery core of the polymersome. Although various studies have effectively encapsulated oxygen transport proteins, such as hemoglobin and myoglobin, into polymersomes while maintaining their bioactivity, the encapsulation efficiencies have not exceeded 30%. The loading efficiency of this approach is often low.[756,841,842] On the other hand, compared to passive approaches, active loading procedures yield a more efficient payload encapsulation because they often take use of diffusion features that follow a gradient of concentration across the bilayers. The enormous hydrodynamic radius of BTAs, however, causes low diffusivity of the molecules across the bilayer structures, hindering the efficacy of this approach.
9.2.1. Amphiphilic Block Copolymer (AmBC) Synthesis Using a Mixture of Selective Post-Polymerization Functionalization and Anionic Polymerization
Two fundamental requirements must be satisfied for the synthesis of amphiphilic block copolymers from precursor copolymers produced by anionic polymerization:[843,844] (a) The process that would result in the first block copolymer’s chemical alteration shouldn’t change the precursor’s molecular properties in any way, such as by chain scission or intra- or intermolecular crosslinking. In contrast, materials that do not match the intended functionality are obtained, such as those with a significantly changed structure and a widened molecular weight distribution. Block copolymers with the same number of monomeric segments, chain topology, and narrow molecular weight distribution as the original block copolymer should ideally result after the chemical alteration. The only modifications that ought to occur are those in the functional groups that are joined to the monomer segments of the chosen block. (b) The functionalization reactions that are selected should be moderate and efficient, meaning that they should almost entirely functionalize the block of choice with the intended functionality. This criterion is not met, and even with chemical modifications of less than 100%, the properties of the materials may still change significantly. Therefore, careful and comprehensive characterization of the modified product is necessary to determine the extent and type of functionalization (this is also true for 100% modification since the success of the modification should always be verified). Chemical modification of block copolymers prepared by anionic polymerization typically entails three steps: nucleophilic addition to C=C double bonds of the main or side chains or on aromatic rings of side chains; epoxidation of C=C double bonds with subsequent nucleophilic substitution; and deprotection of protected functionalities already present on the polymerized monomers (e.g. hydrolysis of ester bonds on monomer side chains). When amine or carboxylic functionalities are present on the copolymer chain, for example, a straightforward protonation/deprotonation equilibrium in solution can often render a block copolymer amphiphilic. If the reaction conditions used permit successive modification steps on distinct blocks, chemical functionalization can occur selectively on one of the blocks, on many blocks simultaneously, or in phases. It is clear that there is a significant deal of flexibility and variability in the final AmBC structures that can be achieved by carefully selecting the monomers and block sequences as well as the overall chemical modification synthesis method. The sections that follow provide some particular instances of synthesis.
9.2.1.1. Amphiphilic Diblock Copolymers
Thus far, reports have been made of the synthesis of a range of amphiphilic diblock copolymers through post-polymerization functionalization of precursor diblocks produced anionically. Several reaction types that vary in complexity and number of steps needed can be used to realize the functionalization. Simple pH variations in solutions can occasionally cause basic or acidic copolymer blocks to be protonated or deprotonated, which causes the system to become amphiphilic. The poly(2-vinylpyridine)-b-poly((dimethylamino)ethyl methacrylate) diblock copolymers (P2VP-b-PDMAEMA), which Jerome and colleagues manufactured using live anionic polymerization, are a nice example of such behavior. The di-block copolymers have been examined in connection to the constitutive homopolymers’ pH-dependent behavior, taking into account the copolymer composition and solution pH. Despite the two blocks being protonated, polydisperse loose aggregates form in low pH regimes. A protonated PDMAEMA corona and a hydrophobic P2VP core combine to create spherical micelles at pH 5. A hydrophobic P2VP core encircled by a PDMAEMA corona that is essentially uncharged makes up micelles at higher pH regimes.[845] The quaternization of tertiary nitrogens of various monomers, which can be polymerized by anionic polymerization via a regulated pathway, is one of the most frequently utilized functionalization reactions. Consequently, blocks containing nitrogen, such as poly(2-vinyl-pyridine) (P2VP), poly(4-vinylpyridine) (P4VP), and poly (2-(dimethylamino) ethyl methacrylate) (PDMAEMA), can be used to carry out the process. Several other alkyl or aryl halides can be used as quaternizing agents, making the reaction extremely adaptable even if extended reaction periods are typically required to get good yields. As a result, cationic sites arise on the polar basic block’s side chains. Depending on how long the halide's carbon chain is, these sites may or may not be hydrophobic. The production of quaternized poly(2-(dimethylamino) ethyl methacrylate)-b-poly(methyl methacrylate) diblock copolymers (PDMAEMA-b-PMMA) was described by Jerome and colleagues relatively recently.[846] The PDMAEMA block was quaternized using a variety of alkyl bromides (RBr) (R = ethyl, pentyl, heptyl, n-propyl, n-butyl, and tert-butyl). By titrating the non-quaternized basic groups with standard HCl solution, the degree of quaternization was ascertained, and it was discovered that the reaction yield was almost always substantially quantitative (Figure 40.1). An asymmetric poly(ferrocenyldimethylsilane-b-2-(dimethyl- amino) ethyl methacrylate) (PFS-b-PDMAEMA) diblock copolymer was quaternized by Manners et al. using extra CH3I in THF.[847] With a long hydrophilic, cationic block (QPDMAEMA) and a short hydrophobic organometallic block (PFS), they were able to create an amphiphilic block copolymer. As demonstrated by 1H NMR in D2O, which selectively solubilizes the QPDMAEMA blocks, the degree of quaternization was quantifiable. It was discovered that quaternization of the PDMAEMA block affected the self-assembly of the diblock copolymer, particularly the micellar structures in water. Schlaad et al.[320] studied the generation of amphiphilic polyion complex vesicles from mixtures of oppositely charged block polyelectrolytes. The cationic component of their investigation was a quaternized polystyrene-b-pol(4-vinyl pyridine) (PS-b-QP4VP). Anionic polymerization was used to create the precursor poly-styrene-b-poly(4-vinyl pyridine) (PS-b-QP4VP) diblock copolymer, and methyl iodide was then used to quaternize its P4VP block in a nearly quantifiable yield. The molecular weight distribution of the unfunctionalized diblock copolymer is narrow (Mw/Mn is close to 1.2), which is likewise indicative of the molecular weight distribution of the functionalized copolymer. Since it can typically be successfully applied to copolymers comprising polystyrene (PS), polyisoprene (PI), and polybutadiene (PB) as one of the blocks, sulfonation is a particularly popular reaction for the chemical modification of diblock copolymers generated by anionic polymerization techniques. Extremely severe circumstances can cause the phenyl side rings of PS to sulfonate, which can then cause the polydiene blocks to sulfonate. Because of this, PS sulfonation is primarily used to create amphiphilic diblock copolymers on precursor diblocks that have a poly(tert-butyl styrene) or hydrogenated polydiene block as the second block, which has a sterically protected phenyl ring that is resistant to sulfonation.[848,849,850,851,852,853] In-depth research has been done on the micelle structure created in aqueous conditions by such amphihilic block copolymers.[850,851,852,853] By employing anhydrous sulfuric acid in dioxane, block copolymers of polystyrene and sulfonated poly-isoprene, or PS-b-SPI, were effectively created. As a mild sulfonation agent for the PI block, the resulting sulfur trioxide-dioxane complex functions.[854] There is no sulfonation of the PS block under these circumstances. The asymmetric, amphiphilic PS-b-SPI diblock copolymers that were produced had varying degrees of polymerization, featuring a small PS block and a much longer SPI block (Figure 40.2). For the synthesis of amphiphilic block copolymers of poly(ethylene oxide) (PEO) and sulfonated polybutadiene (SPB), a similar sulfonation process was used.[855] The elemental analysis of the modified copolymers showed that the degree of sulfonation in the PB block could be adjusted from 5% to 80%. If the sulfonation level reached 15% or more, the sulfonated block copolymers became soluble in water. These new amphiphilic block copolymers were employed in emulsion polymerization as macromolecular stabilizers. Schlaad et al.[856] have demonstrated a highly effective and adaptable alteration of the poly-butadiene block in PB-b-PEO and PB-b-PS diblock copolymers with ω-functional mercaptans. The C = C double bonds of the PB-1,2 side chains can be filled with a wide range of functional mercaptans to create amphiphilic and double hydrophilic block copolymers with several distinct polar functional groups. Size exclusion chromatography (SEC) and MALDI-TOF MS analysis revealed that the resultant diblock copolymers have narrow polydispersity, typically between 1.05 and 1.15. Since some cyclization of two nearby groups results in some loss of the desired chemical functionality, the degree of functionalization of the diblock copolymer was in the range of 50-85%, frequently 70-80%, and double bond conversion was usually complete (Figure 40.3). Through the previously reported free-radical addition procedure, which involved peptide ω-functionalized mercaptans, novel hybrid peptide amphiphiles were created. These amphiphiles consisted of a PB-b- PEO copolymer as the backbone and grafted (L,L)-cysteine-phenylalanine hydrophobic dipeptides on the PB block.[857] It was demonstrated that the reaction was just as successful as those utilizing mercaptans with lower molecular weight. The discovered self-assembled structures exhibit a greater degree of sophistication and functionality as a result of the hybridization of synthetic copolymers with peptides. A variety of self-assembled nanostructures, including wormlike micelles, spherical micelles, and vesicles, were seen in the morphologies of this novel hybrid peptide (Figure 40.4). First, a PS-b-PI precursor was reacted with chlorosulfonyl isocyanate to create the amphiphilic polystyrene-b-poly((sulfamate-car-boxylate)isoprene) diblock copolymer. Next, the lactam groups that had formed on the PI blocks in the first step were hydrolyzed alkaline.[858] The presence of weak COO- and strong SO3- acidic groups in the poly(sulfamate-carboxylate)isoprene) block increases the solubility of the SCPI block. By using solid state 13C NMR spectroscopy to determine the functionalization of the PI block, it was discovered that 89% of the modified isoprene units were present in the molar fraction. Using the Wilkinson catalyst system, polybutadiene-b-poly (tert-butyl methacrylate) copolymers (PB-b-PtBMA) were selectively hydrogenated to create polyethylene-b-poly(tert-butyl methacrylate) (PE-b-PtBMA) diblock copolymers.[859,860] The combination of the semicrystalline PE block with the amorphous PtBMA block produces a group of materials with intriguing physical characteristics. PE-b-PMAA, or polyethylene-b-poly(methacrylic acid) copolymers, were created by hydrolyzing the PtBMA block of PE-b-PtBMA. Utilizing a stoichiometric excess of HCl over the ester groups, the hydrolysis reaction was achieved. Furthermore, the PB-b-PtBMA diblock copolymers were hydrolyzed, and the COOH groups were then neutralized with NaOH to create the polybutadiene-b-poly(methacrylic acid) and polybutadiene-b-poly(sodium methacrylate) copolymers. Muller et al. reported on a poly(acrylic acid)-b-poly(N,N-diethylacrylamide) di-block copolymer (PAA-b-PDEAAm). In contrast to the ester groups of the PtBA block, the diblock copolymer was created by hydrolyzing the PtBMA block of a PtBMA-b-PDEAAm copolymer, which had been prepared by anionic polymerization, in dichloromethane with a five-fold molar excess of trifluoroacetic acid.[861] In an aqueous solution, the PAA-b-PDEAAm block copolymer generated micelles with a pH-dependent and thermoresponsive structure. From the corresponding PS-b-PDEVP diblock copolymer, polystyrene-b-poly(vinylphosphonic acid) diblock copolymers (PS-b-PVPA) were synthesized. Styrene and diethyl vinyl phos-phonate were sequentially anionic polymerized in THF to provide PS-PDEVP precursors with varying comonomer compositions. The PVPA blocks were created by hydrolyzing the phosphonate ester groups of the PDEVP block in dichoromethane with hydrochloric acid.[862] Through acidic hydrolysis and subsequent neutralization, a poly(4-tert-butoxystyrene-b-4-vinylpyridine) precursor diblock copolymer (PtBOS-b-P4VP) was changed to poly(vinylphenol-b-4-vinylpyridine) (PVP-b-P4VP). Dioxane/DMF was hydrolyzed using a ten-fold excess of HCl, and the pH was neutralized to 6-7 using a 10-weight percent NaOH solution. DSC and solid-state NMR studies revealed strong hydrogen-bonding interactions between the pyridine group of P4VP and the hydroxyl group of PVPh in the bulk phase.[863] From polyisoprene-b-poly(ethylene oxide) diblock copolymers, amphiphilic block copolymers with β-lactam groups on the polyisoprene block were created.[864] By reacting the polyisoprene block with chlorosulfonyl isocyanate in dry diethyl ether, β-lactam functionalization was accomplished. It was discovered that the degree of change exceeded 70% when too much CSI was applied. The solubility of the β-lactam functionalized polyisoprene (LPI) blocks was found to be higher due to the presence of hydrophilic lactam groups, and the ability of lactam groups to form hydrogen bonds with water molecules in solution and the PEO blocks in the bulk phase was also attributed to the observed changes in the self-assembly behavior after functionalization. Anionic ring opening polymerization was used to create amphihilic poly(allyl glycidy ether)-b-poly(ethylene oxide) diblock copolymers (PAGE-b-PEO), which were then chemically altered by hydrogenating the PAGE block. Palladium served as the catalyst and cyclohexa-1,4-diene as the hydrogen donor to achieve the hydrogenation process in THF.[865] Methyl sulfanyl acetate in THF was used to create an addition reaction on the double bonds in the PAGE blocks of the identical diblocks, totally functionalizing the PAGE double bonds. Ultimately, radical copolymerization of the copolymer double bonds with 4-methoxystyrene and 2-hydroxy-2-methyl-1-phenylpropan as a photoinitiator produced crosslinked nanoparticles, which were then used to radical crosslink the micelle core of PAGE-b-PEO micelles in water.
9.2.1.2. Double Hydrophilic Diblock Copolymers
Under specific solution conditions, both blocks in a double hydrophilic diblock copolymer can be hydrophilic. Simple pH variations in solutions can cause basic or acidic copolymer blocks to be protonated or deprotonated in a number of systems, which can alter the system’s solubility and amphiphilicity. The poly(2-vinylpyridine)-b-(poly ((dimethylamino)ethyl methacrylate) diblock copolymers (P2VP-b-PDMAEMA) created by Jerome and associates serve as an excellent illustration of this behavior. By employing diphenyl methyl potassium as an initiator and live anionic polymerization in THF at 78 0C with LiCl present, these copolymers were created. First, 2VP was polymerized, and then DMAEMA was added. The two blocks form polydisperse loose aggregates in aqueous solutions at low pH levels, despite being protonated and water soluble. At pH 5, P2VP becomes hydrophobic as a result of deprotonation, while PDMAEMA remains protonated. Under this condition, protonated PDMAEMA corona and P2VP core combine to create spherical micelles. Higher pH regimes result in the presence of micelles in solution, which are made up of a basically uncharged PDMAEMA corona around a hydrophobic P2VP core. The adsorption of the specific block copolymers on solid surfaces as well as the structures that formed at various pH regimes were impacted by this pH-dependent behavior.[845,866] Another very typical example are poly(2-vinyl pyridine)-b-poly(ethylene oxide) block copolymers (P2VP-b-PEO),[867] which are likewise produced by anionic polymerization. As previously mentioned, the PEO block is soluble in water at all pH values, whereas the P2VP block’s solubility varies with pH. The structural diversity of block copolymers that may be produced by combining anionic polymerization and post-polymerization functionalization techniques is demonstrated by further instances of double hydrophilic block copolymers that follow. By selectively reacting the polyisoprene block with chlorosulfonyl isocyanate in dry diethyl ether, with a 10% molar excess over the double bonds in the PI block, the poly (sodium(2-sulfamate-3-carboxylate)isoprene)-b-poly(ethylene oxide) (SCPI-b-PEO) diblock copolymer was created.[868] Sulfamate and carboxylate groups are present at adjacent carbons of the PI double bonds in the resultant block copolymer. The degree of functionalization reported for functionalized diblock copolymers was approximately 75%, and they exhibit narrow molecular weight distributions (Figure 40.5). The precursor poly(p-tert-butoxystyrene-b-tert-butyl methacrylate) (PtBOS-b-PtBMA) diblocks was hydrolyzed acidically to create poly(p-hydroxystyrene-b-methacrylic acid) copolymers (PHOS-b-PAA).[869] With a five-fold molar excess of HCl in dioxane, the reaction was achieved. The hydrophilic block copolymers in aqueous solutions exhibited a complicated self-assembling activity due to the pH responsive nature provided by the presence of carboxyl and hydroxyl functional groups (Figure 40.6). Similar to this, premade PtBOS-b-PEO diblock copolymers were hydrolyzed acidically to create poly(p-hydroxystyrene-b-ethylene oxide) copolymers (PHOS-b-PEO).[870] A Mannich type procedure was then used to convert the PtBOS-b-PEO copolymers into poly[3,5-bis(dimethylamino- methylene) hydroxyl styrene-b-ethyleneoxide] copolymers (NPHOS-b-PEO). Dimethylamine and formaldehyde (12-fold molar excess over the hydroxystyrene units) were added to achieve the reaction. Lastly, methyl iodide treatment of the copolymers allowed the dimethylammino groups of the NPHOS blocks to become quaternized. The resulting copolymers have an acidic hydroxyl group and a QNPHOS block with two quaternized amino groups in almost every repeat unit (Figure 40.7). Using poly(ethylene oxide)-b-polyglycidol diblock copolymers, DHBC including phosphorylated, sulfonylated, and carboxymethylated polyglycidol blocks has been created.[871] POCl3 was used to phosphorylate dry triethyl phosphate; the degree of phosphorylation varied and was dependent on the initial molar ratio and reaction time. Ethyl bromoacetate and pure sodium hydride were used to do carboxymethylation in THF. Sulfonylation was achieved by reacting sultone with NaH powder in dry THF. There were reported substitution levels of between 50% and 100% for phosphorylation, between 50% and 50% for sulfonylation, and between 50% and 50% for carboxymethylation.
9.2.2. Theory of Nonionic and Ionic Diblock Copolymer Micelles
A limited interface between the core and the corona implies that the micellar corona can be thought of as a polymer brush made of soluble blocks A. The foundational works of Alexander[873] and de Gennes[48] laid the groundwork for the notion of polymer brushes. These investigations paved the way for the development of spherical scaling models that replicate the coronal domains present in nonplanar diblock copolymer aggregates.[874,875] Blob size ξ depends on distance r from the micelle center when the polymer density profile in a concave polymer brush (micellar corona) decays to the brush periphery. For a particular brush shape and chain grafting density, the blob size ξ(r) is determined by the condition of dense packing of the correlation blobs. The total number of blobs multiplied by the thermal energy, kBT, yields the brush free energy. Formulating the free energy per chain Fchain in a diblock copolymer aggregate is simple with these components. For lamellas, cylinders, and spheres, respectively, i = 1, 2, and 3 are the three generic geometries i that are taken into consideration (see Figure 41.1). In accordance with de Gennes,[304] the total of three contributions is the free energy per chain, or Fchain.
Fchain =Finterface+FB+FA
Here,
where γ is the free energy per unit area of the core-corona interface and s is the area per chain. Given the low concentration of monomer units A at the core-corona contact, B-S interactions (described by the Flory-Huggins interaction parameter χBS) mostly govern the value of γ, with γ ≈ γBS. The contributions FA and FB are resulting from blocks B and A in the micelle’s coral and coronal domains. The free energy of interaction between monomers and the elastic free energy of extended blocks B make up the coral contribution, which is expressed as FB = FB,interaction + FB,elastic. With a volume percentage of, the polymer density distribution in the micelle core created by a copolymer with relatively long blocks (NA, NB ≫ 1) is nearly uniform. φB is almost coincident with that of a polymer B precipitate in weak solvent S. Despite making the biggest contribution to the Fchain, free energy FB,interaction ∼ kBTNB is not affected by the size or shape of the core and is therefore ignored. The free energy FB,elastic is roughly represented as
where a is the monomer unit size (for simplicity, it is assumed that the sizes of monomer units A and B are equal), bi is the geometry-dependent numerical coefficient, and R = iNBa3/(φBs) is the radius of the core. In the seminal study of Semenov,[104] the values of b1 = π2/8, b2 = π2/16, and b3 = 3π2/80 were computed in the limit when blocks B are notably expanded with regard to Gaussian size, that is, when R > a(NB)1/2. The free energy per chain in the brush created by blocks A is known as the coronal contribution (FA). The latter are “tethered” (refer to Figure 41.2) to the micellar core surface with radius R by grafting area s.
Finterface = γs
The blob size ξ(r) rises with distance r from the micelle center due to dense blob packing inside the corona as
as long as blocks A’s free ends are concentrated inside the outermost coronal blobs. At a distance r from the micelle center, the volume fraction φA(r) of monomers A is proportional to ξ(r), as in a semidiliute polymer solution, ξ(r) ≃ aφA(r)−v/(3ν−1). The density profile φA(r) in the micellar corona of shape i is specified by the latter relationship and equation 4. The conservation condition yields the scaling expression for coronal thickness Hi.
The amount of blobs per coronal block, or free energy FA, is calculated using
The equilibrium parameters of diblock copolymer aggregates are surface area s and diameters Hi and R of the coronal and core domains, which are provided by the balance energy, Fchain ≈ Finterface + FA. A spherical micelle (i = 3), a cylindrical aggregate (i = 2), and a lamella (i = 1) are the aggregates' characterizations. The total number of chains (aggregation number) in a spherical micelle (p ≃ R2/s) and the number of chains per unit length (linear density) ≃ R/s are the aggregates’ characteristics. Micelle properties in these two limitations can be asymptotically dependent upon their division into starlike and crew-cut micelles. Equation 12 shows that a spherical corona (i = 3) has a substantially smaller free energy than a cylindrical or planar coronae when Hi/R ≫ 1. For this reason, in relation to lamellar and cylindrical aggregates with broad coronal domains, starlike micelles are always thermodynamically stable. The number of aggregations in a star-like, spherical, equilibrium micelle
is mostly determined by the insoluble block’s degree of polymerization (NB), and it only slightly (logarithmically) declines as coronal block A lengthens. Much like the numerical coefficient, eq. 13’s logarithmic prefactor is left out.
When crew-cut aggregates are used as the opposite limit, expanding FA in equation 12 with regard to small parameter Hi/R << 1 and taking Hi’’s curvature dependence into consideration yields[105]
As can be seen from equation 14, the coronal free energy FA in the crew-cut aggregate of morphology i is composed of the geometry-dependent adjustment (the second term in equation 14) in addition to the planarlike term F1(s). Consequently, the crew-cut aggregates of all geometries show universal power law dependence for area per chain s, and the primary contribution to the chain free energy, Fchain ≈ Finterface + F1, is the same for i = 1, 2, and 3. This leads, in particular, to the scaling expression of aggregation number for the spherical crew-cut micelles
having a correlation to the length NA of the coronal block that is much stronger (power law) than in the case of starlike micelles (eq 13). The curvature-induced correction ΔFA and the elastic contribution FB,elastic are assumed to be responsible for the difference δFi in the free energy of crew-cut aggregates with different morphologies.[725] As the micellar core’s curvature increases, conformational constraints on the core blocks also rise, causing blocks B to grow more elongated and the coronal free energy FA to drop (blocks A becoming less crowded, ΔFA < 0). Micelle polymorphism results from the elastic stretching of coral blocks B, even if in an equilibrium aggregate FB,elastic is always smaller than the surface free energy Finterface. An equilibrium diblock copolymer micelle would always be spherical in shape in the absence of its contribution. A crew-cut aggregate’s equilibrium morphology (i) is determined by the smallest free energy increment (δFi = FB,elastic + ΔFA). The coexistence line (binodal) for the morphological transformation i → i − 1 (i = 2, 3) is determined by the requirement δFi = δFi-1. For micelles of nonionic block copolymer, the critical micelle concentration (CMC) is calculated as
lnCMCn ≈ −Finterface (p=1)/kBT
The nonionic micelle is denoted by the subscript “n” in this instance, the surface free energy of a single condensed block B in an unimer is represented by Finterface(p = 1), and the contribution of coronal blocks A is not included. A review contains a more thorough explanation of micellization’s thermodynamics.[105] The scaling model described above proved to be a helpful tool to understand the experimental results on diblock copolymer micelles, despite the fact that it only takes into consideration the major power law dependences and ignores the numerical coefficients. The thermodynamic equilibration or “frozen” state of polymer micelles could be contingent upon the characteristics of coral block B. Conditions of equilibrium suggest that polymer molecules in aggregates and unimers interchange. Such micelles resemble star- or comb-like polymers with chemically set branch densities when seen in “frozen” aggregates, when this exchange is stopped. However, in other investigations (e.g., the discussion in ref 49), there were large differences between apparent and theoretical exponents.[876,877] Some experimental systems have shown reasonable agreement with the expected scaling exponents. Reference 878 presents a methodical examination of the scaling dependencies of the aggregation number p and the hydrodynamic radius of micelles created by the diblock copolymer poly(styrene)-b-poly(4-vinylpyridine) in the selective solvent toluene. It was shown that although the hydrodynamic radius for starlike micelles behaves as predicted theoretically, the value of exponent in the aggregation number p dependency on coral block NB length is about as expected for crew-cut micelles. Although notably smaller than that provided by eq 15 for crew-cut micelles, the observed relationship of aggregation number p on length of coronal block NA was stronger than that expected for starlike micelles (eq 13). Significant differences between the apparent and expected values of exponents are usually ascribed to several factors such as sample polydispersity, system parameter selection in the crossover zone between the crew-cut and starlike regimes, and incomplete thermodynamic equilibration. The scaling hypothesis of nonionic block polymer micelles was further improved in the following ways:
(i) For micelle parameters, power law asymptotic dependences hold true in the bounds Hi/R << 1 (crew-cut aggregates) and Hi/R >> 1 (starlike aggregates). Most experimental systems, however, reside in the intermediate region that lies between these two boundaries. Therefore, it is essential to keep all of the factors in FA, including the logarithmic dependency, eq. 12, and to take into consideration FB,elastic in reducing the chain free energy Fchain, in order to perform a thorough comparison between theory and experiment. The study of differences between complete and asymptotic expressions for aggregation number p and other micelle characteristics was made possible by this theoretical advancement.[725]
(ii) Two of the scaling model’s numerical coefficients are still unknown, even given defined values of bi. These coefficients could be added as prefactors to a planar brush’s thickness H1 and free energy F1 dependences.[725] They can be found by fitting the spherical micelles’ experimental data (with ), and they can then be applied to various aggregate morphologies and arbitrary values of NA and NB.
(iii) A scaling theory that has numerical coefficients that are not defined can forecast the polymorphism of crew-cut diblock co-polymer micelles and can give the values of exponents in power law binodal dependences. With respect to the sphere-to-cylinder and cylinder-to-lamella transitions, respectively, the model specifically predicts that the degrees of polymerization of coral block B, NBs−c and NBc−l, will show power law asymptotic dependences with comparable values of exponents.
It is possible to measure the relative breadth ΔNB/ NBc−l of the thermodynamic stability corridor of cylindrical micelles using numerical coefficients bi. Exact placement of the binodals in the NA, NB parameter space and direct comparison with the experiment are made possible by the full set of numerical coefficients being implemented. Figure 41.3 shows the diblock copolymer poly(styrene)-b-poly(isoprene) in n-heptane diagram of states in NA, NB coordinates. Using the complete set of numerical coefficients produced as previously discussed, the binodals separating zones of thermodynamic stability of spherical (S), cylindrical (C), and lamellar (L) aggregates (shown by solid lines) were computed.[725] Dense packing and correlated blobs in micellar corona (Figure 41.2) can be linked to the starlike polymer Daoud-Cotton model.[874] A more recent research,[879] however, has shown that this kind of concave brush arrangement is not really equilibrium. Blob organization becomes more loose compared to dense packing as the chains stretch further to reduce monomer-monomer interactions at the brush periphery, resulting in a lower free energy. The effect reaches its maximum in a starlike corona of spherical micelles, however the coronal free energy decreases to ≲10% while the values of exponents predicted by the original scaling model remain intact. Interestingly, neither eq 14 nor the binodal positions for the sphere-to-cylinder and cylinder-to-lamella transitions change. We briefly review a few open issues in the theory of nonionic polymer micelles to wrap up this discussion:
For monomer units of core and corona forming blocks, the values of the Flory-Huggins interaction parameters χBS and χAS are commonly used to account for the effect of solvent characteristics on micellar structure. Mixtures of low molecular weight solvents are frequently employed in experimentation to create micelles. A binary combination of compatible solvents can be effectively characterized by an effective Flory-Huggins interaction parameter, which varies depending on the composition.[880,881] Admixture of a thermodynamically superior solvent, however, may cause a significant rearrangement in a brush-like corona for incompatible solvents. The predicted behavior of a planar brush in contact with mixed solvents was abrupt collapse and reswelling.[882,883] Vertical brush segregation in two separate sublayers, each enriched in one of the solvents, occurs concurrently with the collapse. When a concave micelle’s corona comes into touch with mixed solvents that are also subpar for block B, similar phenomena could occur. The copolymer components’ preferential solvation by immiscible solvents may result in an even more intricate solvent distribution inside a micelle.[884,885] However, the analytical theory of polymer micelles has not yet adequately addressed the consequences associated with solvent complexity, particularly the various solubilities of the constituents in solvent/cosolvent mixtures. The models presented in this section can be applied to nonionic copolymers with water-soluble block (poly(ethylene oxide)) in the temperature interval away from LSCT or UCST, or co-polymers with organo-soluble blocks (poly(styrene)-b-poly(isoprene) in n-heptane). In a range of temperatures, copolymers consisting of flexible non-ionic biopolymers (polypeptides)[886,887,888] can also form stable spherical micelles or cylindrical aggregates. Thermoresponsive polypeptide blocks may display secondary structural components and experience intramolecular conformational changes (refer to Figure 41.4). For planar brushes, theoretical modeling of the helix-coil transition in tethered polypeptides has recently been carried out.[889] Incorporating the complete conformational complexity of the constituents is still a difficult issue in the theory of diblock copolymer micelles, though. The aggregates with an elastomeric (amorphous) core domain are the main focus of nonionic micelle theories. Numerous experiments suggest that self-assembled aggregates with adjustable shape could be produced by linear[890,891,892,893,894] and branching[895] block polymers containing crystallizable insoluble block B. There haven't been many theoretical advancements made for these kinds of structures.[896,897] The theory of morphological transitions developed for micelles with an elastomeric core might be extended to copolymer aggregates with a crystallizable coral domain by calculating the conformational entropy loss for block B consistent with crystallization.
Diblock copolymers including hydrophobic and polyelectrolyte (PE) blocks result in a range of nanostructures, much like nonionic macromolecules.[898] When compared to nonionic polymers, the behavior of charged amphiphilic macromolecules is more complex due to the interaction of van der Waals forces and long-range electrostatic interactions.[899,900] Block copolymer self-assembled aggregates of weak (annealing) and strong (quenched) PE coronal blocks may exhibit significantly distinct characteristics. The proportion of charged monomers (α) in a weak PE corona, such as poly(acrylic acid), is controlled by the local (coronal) pH and can be adjusted by adjusting the solution’s pH and ionic strength. In a robust PE coronal block (such as sulfonated poly(styrene)), the degree of chain sulfonation fixes α chemically and is independent of the surrounding environment. Micelles of both sorts respond to stimuli. That is, by adjusting the intensity of electrostatic contacts, for example, by adding salt ions or, in the case of a weak PE coronal block, by adjusting pH, aggregation size and shape can be controlled. Here, we concentrate on the circumstances under which the ionization of the PE block falls below the Manning condensation threshold,[901] αlB/a < 1, where lB = e2/εkBT, e denotes elementary charge, ε denotes the dielectric constant, and kBT represents the thermal energy. In these circumstances, counterions do not condense on ionized blocks A and maintain their translational mobility within the micellar corona (see to review 105 for a more thorough explanation). As of yet, no analytical theory of a charged corona micelle has been produced to the precision level attained for a neutral system. The theoretical ideas and current models of diblock copolymer micelles with ionized corona are summed up in a recent review.[105] Equation 8 can be used to represent the free energy per chain in such a micelle using the coronal free energy
including the contributions from electrostatic (ionic) interactions (Fion) and excluded volume (Fev). The latter results in further ionized block extension and a breach of the coronal correlation blobs’ dense packing. The equilibrium self-assembly of a copolymer with an ionizable coronal block is controlled by the balance of the surface and coronal free energy, Finterface ≃ FA, just as the association of nonionic macromolecules. Ionic groups have essentially no effect on the surface contribution Finterface, which is still determined by equation 9 with γ ≈ γBS. The Poisson-Boltzmann technique is necessary to predict the spatial distributions of mobile ions in several geometries (spherical, cylindrical, and planar) and the electrostatic potential Ψ in a consistent analytical theory of a micelle with PE corona in contact with a salt solution. For a planar PE brush that simulates the corona of a lamella (i = 1) in contact with a monovalent salt solution, this problem has been analytically solved.[902,903,904] Within the (negatively) charged planar corona of strong PE blocks A, the dimensionless electrostatic potential eΨ/kBT has a parabolic shape.[902]
where x is the distance from the A/B interface and H1is the coronal thickness.The characteristic electrostatic length is H0 = a[8α/(3π2)]1/2NA. The electrostatic potential outside the corona is equal to the charge number density H1/(2πlBH02) of a flat surface. The planar corona's thickness H1 is determined by the concentration of salt in the solution, cs, as well as the molecular characteristics of the diblock copolymer. However, the addition of salt ions does not alter the parabolic form of Ψ(x) (eq 12). The Poisson−Boltzmann equation for the electrostatic potential Ψ(x) might be solved numerically for additional geometries of ionized corona (i = 2, 3). Numerous internal micelle organization details were brought to light by the numerical SCF models that treated electrostatic interactions using the Poisson-Boltzmann framework.[300,301,905] The examination of ion distributions in the vicinity of micelles revealed that mobile ions are preferentially positioned within the coronal domain and nearly entirely offset the “bare” charge αpNA of the coronal blocks in salt-free solutions of charged micelles with aggregation number p ≲ 10. The two states of the ionized coronal domain—one with counterions condensed in micellar corona and the other with “bare” Coulomb repulsions between charged monomers-were distinguished by groundbreaking theoretical research.[906,907] The majority of the research that followed focused on micelles that had virtually completely neutralized net coronal charge and ions confined in micellar corona. Here, we concentrate on the scenario of aggregates with compensated coronal charge, which is relevant to experiments (see Figure 41.5).
FA =Fev +Fion
Such micelles are thought to have completely and locally electroneutral polymer brushes with entrapped mobile ions as their coronae. The local electroneutrality approximation (LEA) postulates that the local excess number density of mobile counterions is about equal to the local number density of ionized monomers in a (negatively) charged micelle corona.
In this case, cp(r) and α(r) represent the concentration of the monomer units and the degree of ionization in the corona at a distance r from the micelle center, respectively, while cj(r) is the local concentration of (monovalent) ions of type j. All cationic species (salt ions, cNa+, and hydrogen ions, cH+) are included in the summation on the right side of equation 20, while all anionic species (salt ions, cCl−, and hydroxyl ions, cOH−) are included in the summation on the left side of the same equation. In bulk solution, all mobile ion concentrations (cbj) are taken to be constant, and the osmotic pressure is determined by
The Boltzmann law governs how mobile ions are distributed.
In the micelle’s coronal region, surplus electrostatic potential is represented by ΔΨ(r) = Ψ(r) − Ψbulk. Entropy of ions disproportionate to the bulk solution and the interior of the corona is how the electrostatic interactions in the corona are represented in the LEA framework. Given the coronal free energy FA, the ionic contribution Fion is given by
as integration is carried out across the coronal volume VA, free energy density is represented by fion(r). If the degree of ionization is constant and the PE is strong (quenched), then α = αb.
For weak polyacid (PE) that is pH-sensitive, an extra contribution
explains the increase in free energy brought about by the ionization of the coronal blocks and produces
Here, the equation yields the degree of ionization, α(r).
where Ka is the (acidic) dissociation constant and αb is the degree of ionization of an isolated monomer in the bulk solution at a certain pH. Asymptotic dependences for fion are obtained by expanding equations 23, 25, and 26 in relation to the ratio α(r)cp(r)/cs, in the two limits α(r)cp(r)/cs >> 1 (low salt circumstances) and α(r) cp(r)/cs << 1 (high salt conditions). The osmotic (counterion dominated) and salt dominated regimes of polyelectrolyte brush are other names for these two regimes in the literature.[908,909,910] For low salt α(r)cp(r)/cs >> 1), the coronal free energy density
and degree of ionization
The degree of ionization, α(r), approaches the bulk value for high salt α(r) cp(r)/cs << 1, α(r) ≈ αb, and
The averaged values of cp(r) and α can be substituted for the gradients in polymer density and degree of ionization inside corona when formulating asymptotic power law dependences for micelle equilibrium parameters. Equations 27, 28, and 29 can be used to obtain the electrostatic contribution to the coronal free energy by changing cp(r) → cp and α(r) → α.
Fion =VAfion {cp,α}
Asymptotic power law dependences of micelle parameters are possible in the limit when short-range nonelectrostatic forces in the corona are negligible compared to long-range electrostatic interactions (FA ≈ Fion) due to the scaling models of ionic spherical micelles with strong[726,906,907,911,912] and pH-sensitive PE coronae.[913,914,915] The concentration of counterions in the micelle’s corona clearly surpasses the concentration cs of salt ions in the solution when the system is in the osmotic regime. In these circumstances, counterions control the differential ion pressure that tends to increase the coronal domain’s volume. The characteristics of an ionic micelle at low salt are found by balancing the coronal free energy FA ≈ Fion (eqs. 30 and 27) with the surface contribution Finterface (eq. 9). The aggregation number p in a micelle with a strong PE corona dramatically drops with increasing length of coronal block NA, in contrast to the situation of a nonionic micelle (eq. 13).
Surprisingly, equation 31 holds true for “osmotic” micelles that are crew-cut and starlike, and are created by diblock copolymer with a strong PE coronal block at low ionic strength. Critical micelle concentration (CMC) is significantly higher for ionized corona at low salt concentrations (cs) due to the entropic cost for counterion localization, as opposed to CMCn for nonionic micelles.
The concentrations of co- and counterions in the micelle’s coronal domain and in the solution gradually level out as cs increases, and this results in a drop in differential ion pressure. Local coronal pH is approaching that of the bulk solution at the same time. The interactions between charged monomers are screened at distances r > rD ≃ (lBcs)−1/2 at high ionic strength, when the salt concentration cs is significantly greater than the concentration of ionized monomers in micellar corona. rD is the salt regulated Debye screening length. Here, the distinction between strong and pH-sensitive PE block vanishes, and the behavior of an ionic micelle approaches that of a micelle with a nonionic corona. Coronal monomer electrostatic interactions are expressed as an effective salt controlled second virial coefficient, denoted as υA,eff = (υA + αb2/2cs), where υA represents the unit's “bare” excluded volume parameter when there is no charge present (see to equation 29). The aggregation number p’s dependence on the salt concentration cs is given by in the two limits of crew-cut and starlike micelles.
The addition of salt ions gradually lowers the critical micelle concentration (CMC) to CMCn.
According to Equation 33, there should be a qualitative difference in the dependency of micelle hydrodynamic radius on salt concentration cs for aggregates that are crew-cut and star-like. The extension of the coronal blocks determines the total size of a starlike micelle. With increasing ionic strength, Rmicelle ≈ H ≃ a2/5NA3/5(pυA,eff)1/5 ∼ υA,eff1/11. This is caused by two competing trends: the crowding of the coronal blocks due to salt-induced increase in p (first line in eq 33) and the decreasing binary interactions between monomers ∼ υA,eff (eq 29). The experimental results and this theoretical prediction agree quantitatively in a fair way.[916,917] Conversely, the radius of the micellar core, which rises with increasing ionic strength as Rmicelle ≈ R ≃ a(NBp)1/3 ∼ υA,eff−2/5, regulates the overall size of a crew-cut micelle. If a micelle has a strong PE corona, its behavior may be very different from that of a micelle with a pH-sensitive corona when tiny additions of salt ions are made.[913,914] pH-sensitive spherical micelles, in particular, are susceptible to abrupt changes in pH around pK or modest variations in solution salinity at pH ≃ pK. In micelles with polyacidic (polybasic) coronal blocks, the localization of counterions in the PE corona results in a locally lower (higher) coronal pH. Block A’s ionization in the corona is therefore less than that in the solution. Two populations of spherical micelles may emerge as a result of the linkage between the coronal block’s degree of ionization (α) and aggregation number (p): big micelle populations with weakly ionized coronas and tiny micelle populations with strongly charged coronas.[913] Electrostatic interactions between monomer units are subordinated to excluded volume interactions in the corona of a large (quasi-neutral) micelle. Smaller surface free energy Finterface per chain promotes the assembly of such deionized aggregate. Coronal block A ionization results in the gain of free energy per chain in a tiny charged micelle. In the bulk solution, the latter is comparable to that of a single block A. When the free energy (per chain) in the large quasi-neutral and small strongly charged micelles equalize, a jumplike larger-to-smaller micelle transition takes place. Changes in the solution’s pH and ionic strength can both cause this shift. The SCF numerical model[905] supported the expected abrupt micelle changes, which were also verified by experiment.[918] The pH-induced reorganizations in spherical micelles of the poly(dimethylaminoethyl methacrylate-block-poly(N-isopropylacrylamide), or PDMAEMA-b-PNIPAM, block copolymer, are shown in Figure 41.6.
Theoretically,[913] a rise in pH causes the (polybasic) PDMAEMA coronal block to deionize, and a narrow pH interval is where the abrupt change from smaller to larger micelles takes place. It is important to note that in spherical micelles with strong PE corona, such rearrangements are forbidden. The aggregates created by asymmetric ionic/hydrophobic block copolymer have the same morphological flexibility as neutral copolymer micelles when they take on a crew-cut shape. Since both variables regulate the intensity of repulsive interactions in the coronal domain, changes in either the ionic strength or pH (for pH-sensitive block copolymers) can cause the morphological transformations. The physical genesis of polymorphism in diblock copolymers including ionizable blocks is identical to that of nonionic copolymers.[919] By contrasting the increments of free energy δFi = Felastic,B + ΔFA The binodals for sphere-to-cylinder and cylinder-to-lamella transitions in ionic aggregates are found here, where Felastic,B is still given by eq 10 and ΔFA is now dictated by both nonionic and electrostatic interactions.[105] Under low and high salt circumstances, the values of exponents in the dependences of binodals NB c−s ∼ NBl−c on the system characteristics differ. Specifically, when ionic strength is high and υA,eff >> υA, the binodal lines that represent the transitions from sphere - to - cylinder to - lamella caused by salt are as follows:
for diblock copolymers with coronal blocks that are pH-sensitive and strongly dissociating.[915] (Take note that the degree of ionization α of pH-sensitive block A achieves its maximum value α = αb at high ionic strength.) Micelles possessing a pH-sensitive corona may exhibit an atypical series of morphological transformations from lamella to cylinder to sphere when exposed to low salt circumstances in the osmotic regime. However, micelles with a strong PE corona are forbidden from exhibiting this sequence. These inverted transitions have binodals that are defined as[915]
The flow chart for equilibrium aggregates with pH-sensitive corona in Figure 41.7(a) shows the succession of morphological transformations. The block copolymer’s theoretical state diagram, featuring a pH-sensitive coronal block that is short (NA = 50), is shown in Figure 41.7(b).[915] Solid lines represent the binodals that were computed taking into consideration the excluded volume as well as the electrostatic interactions between the coronal monomers. Dotted lines represent asymptotic dependences derived with the only dominant monomer-to-monomer interaction type (excluded volume or electrostatic ones, eq. 35) taken into consideration. The sequence of inverted (L→C→S) and direct (S→C→L) morphological transitions shown in Figure 41.7(a) are represented by the diagram of states whose intersection is indicated by a green arrow.
Figure 41.
Diagram representing the three general forms of self-assembled diblock copolymers: lamella (i = 1), spherical micelle (i = 2), and cylindrical aggregate (i = 3). (41.1) Diagram showing the coronal domain’s blob structure in a nonionic micelle. (41.2). Diagram of states in n-heptane in NA, NB coordinates for the nonionic block copolymer poly(styrene)-b-poly(isoprene). AFM pictures of spherical and cylindrical micelles are shown in the insets. (41.3) Diagram of a micelle polypeptide. (41.4) Diagram showing a spherical micelle in contact with a monovalent salt solution and an ionized corona. (41.5) The hydrodynamic radius R of unimers (open symbols) and micelles produced by PDMAEMA42-b-PNIPAM52 (filled squares) and PDMAEMA57-b-PNIPAM97 (filled circles) varies with solution pH. The number of monomer units in the appropriate copolymer block is indicated by subscripts. (41.6) Schematic representation of the sequence of morphological transformations caused by salt in diblock copolymer aggregates with pH-sensitive corona (41.7) (a), and the theoretical diagram of states in NB, cs coordinates (b). Adapted with permission from ref [722]. Copyright 2012, American Chemical Society.
Figure 41.
Diagram representing the three general forms of self-assembled diblock copolymers: lamella (i = 1), spherical micelle (i = 2), and cylindrical aggregate (i = 3). (41.1) Diagram showing the coronal domain’s blob structure in a nonionic micelle. (41.2). Diagram of states in n-heptane in NA, NB coordinates for the nonionic block copolymer poly(styrene)-b-poly(isoprene). AFM pictures of spherical and cylindrical micelles are shown in the insets. (41.3) Diagram of a micelle polypeptide. (41.4) Diagram showing a spherical micelle in contact with a monovalent salt solution and an ionized corona. (41.5) The hydrodynamic radius R of unimers (open symbols) and micelles produced by PDMAEMA42-b-PNIPAM52 (filled squares) and PDMAEMA57-b-PNIPAM97 (filled circles) varies with solution pH. The number of monomer units in the appropriate copolymer block is indicated by subscripts. (41.6) Schematic representation of the sequence of morphological transformations caused by salt in diblock copolymer aggregates with pH-sensitive corona (41.7) (a), and the theoretical diagram of states in NB, cs coordinates (b). Adapted with permission from ref [722]. Copyright 2012, American Chemical Society.
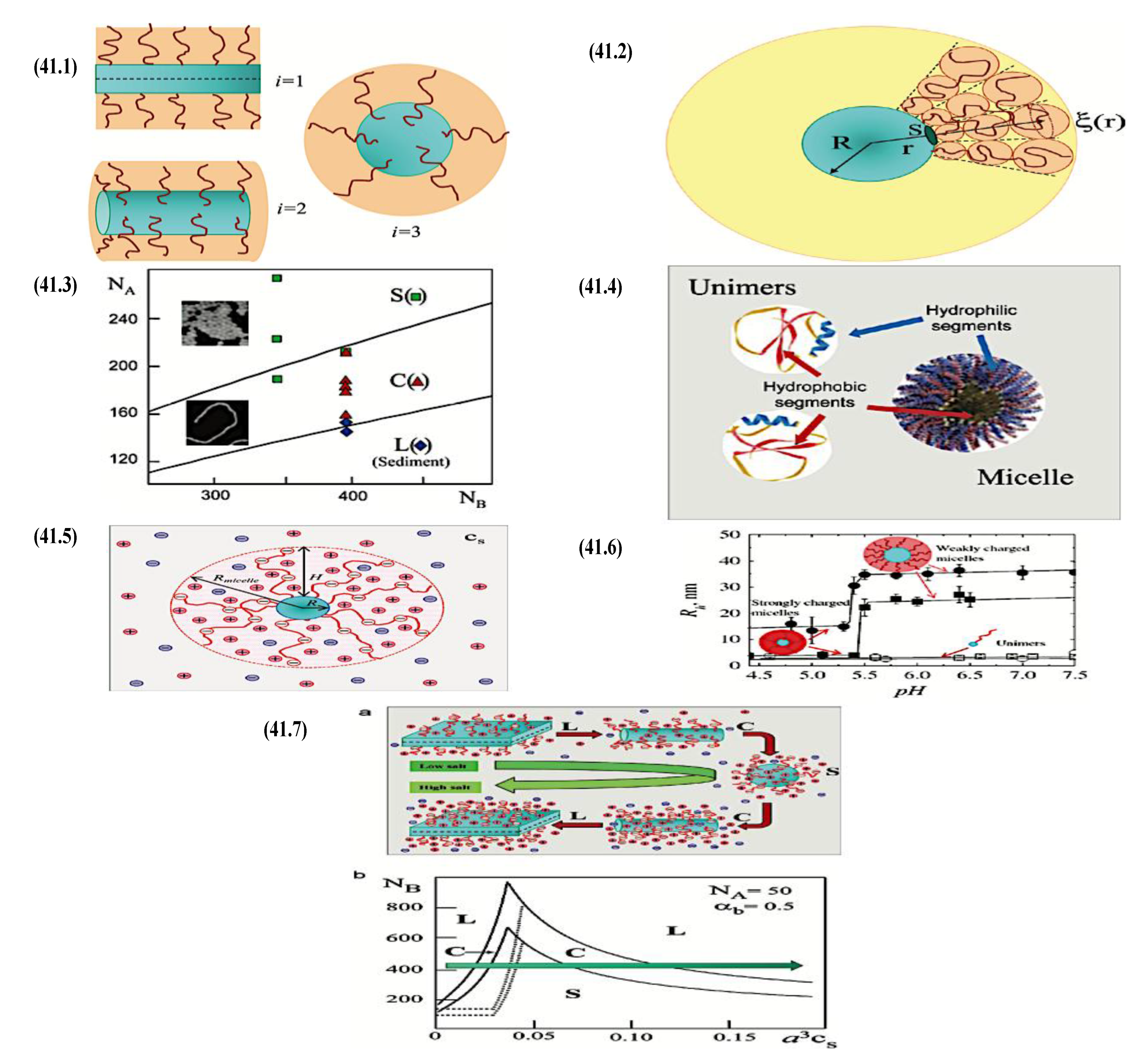
In order to study the general morphologies of micelles with pH-sensitive corona and some new structures (branching cylindrical aggregates), a sophisticated semianalytical SCF model has recently been created.[920] When the free energy of the branching (saddle-shaped) segment in a cylindrical micelle is less than that of the spherical end-caps, the branching cylinders are predicted to be thermodynamically stable. For block copolymers with strong PE blocks, branching topologies have also been proposed.[921] Nevertheless, there was no in-depth analysis done on these structures. It has also been investigated how polymer concentration affects the morphological transitions between aggregates and the self-assembly of diblock copolymer with strong PE block.[922] Reference 923 has proposed a theory for the micellization of a diblock copolymer with non-ionic hydrophilic coronal block A and weakly ionizable (pH-sensitive) hydrophobic coral block B. The spherical shape of the micelle and the collapsed state of the coral block were both preserved by weak ionization. The Poisson-Boltzmann method made it possible to approximate the electrostatic potential Ψ(x) in the micelle's coronal and coral domains. Power law dependences for the properties of such micelles have not yet been found, despite the fact that the formalism has highlighted significant questions regarding polydispersity and system equilibration and allowed for a thorough characterization of a micelle with an ionizable core.[923]
I summarize a few difficult theoretical issues for micelles with PE corona to wrap up this section. The stalled molecular exchange between micelles and unimers frequently makes it difficult to compare experimental results with theoretical predictions for ionic micelles. The core domain's glassy or entangled dynamics or the high incompatibility of monomer B with water are blamed for the absence of micelle equilibration.[54,924,925] In order to attain micelle responsiveness, copolymers including “soft” hydrophobic blocks-such as poly(isobutylene) connected to pH-sensitive PE blocks-have been used.[917] Inducing a specific percentage of ionizable comonomers in the core-forming block B is an alternate method for producing “dynamic” micelles. There has been evidence of a reversible pH-controlled relationship in block copolymers with statistical[926,927] or gradient[928,929] sequences of comonomers in block B associated with pH-responsive block A. The ionic strength and pH of the solution exhibited reversible changes in the aggregation quantity and dimensions of micelles. Block copolymer assemblies of this kind could be excellent candidates for quantitative comparison with the theory. Asymptotic dependences for micelle characteristics are absent, and the theoretical analysis of the self-assembly of block copolymers with ionizable monomers and comonomers in the associating block is restricted.[923] Micelles and vesicles that respond to stimuli are produced using hybrid polypeptide-synthetic block copolymers and amphiphilic block copolypeptides.[930,931,932,933] Reversible pH-induced secondary structure development in polypeptide blocks is linked to the ability of aggregates to change size and shape in response to fluctuations in the pH of the solution. The impact of secondary structure on association behavior and morphological changes in pH-responsive peptide-based copolymers has not yet been taken into account in any theory. For self-assembled vesicles of amphiphilic copolymer with oligonucleotide-based coronal block, a related theoretical issue emerges.[934] Such a vesicle's corona is made up of single-stranded oligonucleotide “probes,” or “microarrays,” that can preferentially hybridize with complementary free DNA fragments in the solution, or “targets.” The majority of the current ideas concentrate on solid-liquid interfaces, like “DNA chips,” and the hybridization that occurs between probes and targets in the bulk solution phase (see, e.g., review 935). The structure and even the morphology of the copolymer aggregate are predicted to be impacted by the hybridization on the self-assembled interface (vesicle surface). A difficult theoretical task is to analyze the link between hybridization and association of the oligonucleotide-based block copolymer. Counterion valence was shown to have a significant impact on the mechanical characteristics and structure of PE brushes in studies.[936,937] Although current theories adequately describe systems containing monovalent salt ions, the presence of di- and trivalent ions causes a more significant shrinkage of PE brush than anticipated by osmotic scaling models.[938,939] The usual explanation for this result is ion-ion correlations. Obviously, the self-assembly of the copolymer with the PE block would also be impacted by ion valence. Nevertheless, the impact of multivalent ions on equilibrium nanostructures containing PE coronae has not yet been the subject of theoretical discussion. The differentiation between intra- and intermolecular repulsions among charged coronal monomers is not evident in the SCF models or current scaling theories of micelles with ionized corona. In contrast to what is predicted by standard osmotic models, a scaling model of strong starlike PE has explained intramolecular electrostatic repulsions inside branches and predicted a more complex behavior of such a macromolecule.[940] The revised scaling model[940] predicted many subregimes in the salt-induced contraction of the branching PE, instead of a single exponent in the power law dependence of polymer size on ionic strength in the salt-dominated regime (H ∼ cs−1/5). A micellar corona of any geometry i may be predicted to undergo similar changes. It is important to take into account the effect on self-assembly.
9.2.3. Synthesis of Linear Triblock and Multiblock Copolymers
The synthesis of an ABC polymer is never simple: selecting the right solvent is essential, the monomer must be extremely pure, and frequently a modification to the end group or a shift in polymerization process is needed to synthesise the subsequent block. To overcome these obstacles and create a narrowly distributed polymer with the required block sequence, the polymerization method must be carefully designed. We will now provide a review of various synthetic approaches that can be used to produce linear ABC structures that are pertinent to research on self-assembly. Examples of ABC polymers are listed in Table 4 along with subscripts denoting the block length, average molecular weight (Mn), dispersity (ĐM), and polymerization method. Each one of them has a limited and monomodal molecular weight distribution, supporting the regulated nature of the polymerization process. When deciding which method will produce the desired ABC polymer, there are two basic ways to consider. The sequential approach involves polymerizing the monomers one at a time using the same polymerization procedure. When one monomer has been completely consumed, the polymerization process moves on to the next one with the right initiator under the right circumstances. The second method involves combining many polymerization processes, necessitating the purification of intermediate products and occasionally the modification of end-groups to act as a precursor to the subsequent stage.
The functionalization of linear triblock and multiblock copolymers, which are primed by anionic polymerization, has also attracted a lot of attention in an attempt to produce linear macromolecules with various functional block configurations. Through acidic hydrolysis of the PtBA middle block, a precursor polystyrene-b-poly(ter-butyl acrylate)-b-polystyrene triblock copolymer (PS-b-PtBA-b-PS), which was created using a difunctional anionic initiator in THF, was transformed into the corresponding polystyrene-b-poly (acrylic acid)-b-polystyrene (PS-b-PAA-b-PS) amphiphilic triblock. It was discovered that the very asymmetric triblock-which has extremely short PS end blocks-had a degree of hydrolysis that was about 98%. The PS-b-NaPAA-b-PS telechelic electrolyte was obtained by neutralizing the copolymer with NaOH, which changed it into the sodium salt form.[953,954] Tsitsilianis and colleagues synthesized an ABA type triblock copolymer with outside blocks of poly(-acryl acid) and a middle block of poly(2-vinyl pyridine). Another asymmetric copolymer created by acidic hydrolysis of protected acrylate ester segments is the double hydrophilic PAA-b-P2VP-b-PAA triblock. The degree of hydrolysis was determined to be 95% by 1H NMR and 93.3% by potentiometric titration for the tri-block copolymer, which displayed a narrow molecular weight distribution.[955,956,957] Anionic polymerization has also been used to create a double hydrophilic block copolymer known as poly(ethylene oxide)-b-poly(2-vinyl pyridine)-b-poly(ethylene oxide) (PEO-b-P2VP-b-PEO). By simply protonating the P2VP block in this system at a pH lower than 5, it is possible to make it hydrophilic. Based on the hydrogen bonding relationship between the PEO and PAA blocks, the complexation of PEO-P2VP-PEO with PAA has been investigated. As the molar masses of the two associating polymer components decrease, complexation becomes less strong.[958] The poly(N,N-diethylacrylamide)-b-poly(tert-butyl acrylate)-b-poly(N,N-diethylacrylamide) (PDEAAm-b-PAA-b-PDEAAm) precursor,[959] which was created by sequential anionic polymerization of tert-butyl acrylate and N,N-diethylacrylamide in that order, was hydrolyzed acidically to yield a thermoresponsive double hydrophilic triblock copolymer of the type poly(N,N-diethylacrylamide)-b-poly(acrylic acid)-b-poly(N,N-diethylacrylamide) (PDEAAm-b-PAA-b-PAAm). To achieve a low molecular weight distribution and regulated polymerization, LiCl had to be present. The resultant copolymer was found to have 96.6% hydrolysis degree. The temperature-sensitive solubility of the PDEAAM block and the pH sensitivity of the PAA block are responsible for the thermoassociative nature of the PDEAAm-b-PAA-b-PDEAAm triblock, which permits a rich self-assembly behavior in aqueous solutions. The anionically synthesized PI-b-PS-b-PEO triblock was reacted with chlorosulfonyl isocyanate to create the amphiphilic triblock terpolymer poly((sulfamate- carboxylate)isoprene)-b-polystyrene-b-poly(ethylene oxide) (SCPI-b-PS-b-PEO).[960] This reaction changed the PI block and produced a pH-sensitive polyelectrolyte. A neutral hydrophilic PEO block, a hydrophobic PS block, and a SCPI block with strong and weak acidic groups were all present in the multifunctional terpolymer. The type of functional groups and level of functionalization were determined by FTIR and solid state 13C NMR spectroscopy (Figure 42).
By acidically hydrolyzing the poly(2-vinyl pyridine)-b-poly(tert-butyl acrylate)-b-poly (butyl methacrylate) (P2VP-b-PtBA-b-PBMA) terpolymer, the triblock terpolymer poly(2-vinyl pyridine)-b-poly (acrylic acid)-b-poly(butyl methacrylate) (P2VP-b-PAA-b-PBMA) was created.[961] As per normal, the reaction was carried out using a five-fold molar excess of HCl in dioxane. Depending on the pH, this terpolymer that is doubly hydrophilic and responsive to stimuli has intriguing self-assembling characteristics. A physical gel formed at high pH, while three compartment microscopes were observed at low pH. The compartments had two concentric inner parts and a positively charged corona. By employing benzyl potassium as the initiator during successive anionic polymerization in THF, poly(isoprene-b-2-vinylpyridine-b-ethylene oxide) (PI-b-P2VP-b-PEO) is an amphiphilic linear triblock terpolymer that has also been created.[962] In aqueous conditions, the terpolymers formed spherical three-layer micelles. Because of the P2VP block’s protonation/deprotonation equilibrium, the middle layer’s characteristics, as well as the relative shell and corona widths, could be altered by the pH of the solution.[963] Eventually, the precursor P2VP-b-PMMA-b-PtBA triblock terpolymer was hydrolyzed to create a double hydrophilic ABC linear terpolymer with ampholytic/amphiphilic character, that is, including a basic A block, a hydrophobic B block, and an acidic C block. Micelles were produced in aqueous solutions by the P2VP-b-PMMA-b-PAA linear triblock terpolymer that was obtained. Depending on the pH of the solution, this double hydrophilic/amphiphilic terpolymer displayed distinct nanoscale self assemblages.[964] A linear pentablock terpolymer has been hydrolyzed in another investigation. A difunctional anionic polymerization initiator and the sequential monomer addition method were used to synthesize the precursor pentablock terpolymer poly(methyl methacrylate)-b-poly(tert-butyl acrylate)-b-poly(2-vinyl pyridine)-b-poly(tert-butyl acrylate)-b-poly(methyl methacrylate) (PMMA-b-PtBA-b-P2VP-b-PtBA-b-PMMA) through living anionic polymerization in THF. By hydrolyzing PMMA-b-PtBA-b-P2VP-b-PtBA-b-PMMA pentablock with an acid catalyst in dioxane, the amphiphilic pentablock terpolymer poly(methyl methacrylate)-b-poly(acrylic acid)-b-poly(2-vinyl pyridine)-b-poly(acrylic acid)-b-poly(methyl meth-acrylate) (PMMA-b-PAA-b-P2VP-b-PAA-b-PMMA) was created.[965,966] The resulting copolymer’s degree of hydrolysis was found to be greater than 97% molar. Two outside hydrophobic blocks (PMAA), two inner pH-sensitive acidic blocks (PAA), and a center pH-sensitive basic block (P2VP) make up this particular pentablock copolymer. A hydrogel is created at low pH levels. It is composed of a three-dimensional network of positively charged P2VP and non-ionic PAA segments joined by intricate bridging chains. The network is made up of PMMA hydrophobic cores. The hydrogel reversibly changes from a positively charged to a negatively charged network at high pH. In this instance, hydrophobic P2VP blocks separate ionized PAA segments to form the bridging chains. We will now discuss the second approach using a variety of polymerization approaches after going through the sequential strategy and providing numerous techniques with examples.
9.2.3.1. Sequential RAFT and ATRP
It is common practice to employ reversible addition-fragmentation chain transfer (RAFT)[967] polymerization for the manufacture of triblock copolymers with different topologies.[968,969] A multitude of vinylic monomers, including butadiene, acrylonitrile, styrene, vinyl acetate, N-vinylpyrrolidone, (meth)acrylates, and (meth)acrylamides, are available, which makes this polymerization favorable. Additionally, RAFT is particularly flexible because it may be used with a wide range of functional groups and in a variety of solvents, including water. However, the synthesis of an appropriate initiator can be difficult, and because of the possibility of chain recombination followed by a drop in active chain ends, this radical polymerization is controlled but not entirely living. This implies that each step requires the intermediate polymers to be purified, which necessitates a reinitiation in order to synthesize the subsequent block. Skandalis and Pispas[941] synthesized a sequence of poly[2-(dimethylamino)ethyl methacrylate] by employing sequential RAFT. The polymers shown in Figure 43 are poly[2-(dimethylamino)ethyl methacrylate]-block-poly(lauryl methacrylate)-block-poly-(oligo ethylene glycol)methacrylate (PDMAEMA-b-PLMA-b-POEGMA). Using azo-bisisobutyronitrile (AIBN) as an initiator and 4-cyano-4-[(dodecylsulfanylthiocarbonyl) sulfanyl] pentanoic acid as a chain transfer agent (CTA), they first polymerized DMAEMA in 1,4-dioxane. Then, under the identical circumstances, the resultant PDMAEMA homopolymer was employed as the macro-CTA for the addition of LMA. As a result, the AB diblock copolymer was produced, which functioned as the macro-CTA for the polymerization of OEGMA in comparable circumstances. After that, the ABC triblock copolymer was quaternized using methyl iodide to create an amphiphilic ABC polyelectrolyte.
Similar to RAFT, atom transfer radical polymerization (ATRP)[970] is a controlled radical polymerization method that provides access to a large variety of vinylic monomers. ATRP can be carried out in a variety of solvents, including water, and can polymerize a variety of styrenes, (meth)acrylates, (meth)acrylamides, and acrylonitrile.[971] However, a metal catalyst is needed for this polymerization, and Cu(I) is the most effective and adaptable one.[972] However, despite the fact that remedies have been offered, its toxicity may restrict the biomedical applications of polymers synthesized using ATRP.[973] Sequential ATRP was utilized to create ABC polymers since it is a live polymerization. For instance, using PEO-Br as a macroinitiator, Jin et al.[942] synthesized poly(ethylene oxide)-block-poly(2-hydroxyethyl methacrylate)-block-poly(tert-butyl acrylate) (PEO-b-PHEMA-b-PtBA) using a two-step ATRP of HEMA and tBA (Figure 44). CuBr/bipyridin was used as a catalyst in methanol for the first stage involving HEMA. The final tBA process was ligating pentamethyldiethylenetriamine (PMDETA) in dimethylformamide (DMF) to address the copolymer’s solubility. CuBr was present as a catalyst once more. Afterwards, sodium azide was added to the PHEMA blocks by reacting with the OH side groups of 2-bromoisobutyryl bromide. Reactive micelles with cross-linked cores and PtBA/PEOcorona were produced by the click reaction between the azido-modified PHEMA blocks and dialkynetrithiocarbonate.
9.2.3.2. Sequential AP, AROP and CROP
Another significant live polymerization process for polymers made from vinyl monomers is anionic polymerization (AP).[974] This method, which is less flexible than RAFT and ATRP, requires monomers including styrene, dienes, vinyl pyridine, and vinyl epoxide that have substituents that stabilize the negative charge by charge delocalization. A series of polystyrene-block-polybutadiene-block-poly(tert-butyl methacrylate) (PS-b-PB-b-PtBMA) triblock copolymers were synthesized by Muller and colleagues[943] using sequential AP (Figure 45). First, styrene and sec-Butyllithium (sec-BuLi) were combined in THF. Following the completion of styrene polymerization, 1,3-butadiene was added. The resulting 1,1-diphenylethylene (DPE) and PS-b-PB diblock copolymer were combined to provide the precursor for tBMA polymerization. The bulk phase behavior of such ABC polymers was then mapped by carefully examining their bulk morphologies.
An essential polymerization method for creating polyesters, polycarbonates, polyamides, polyurethanes, and polyphosphates in a living way is anionic ring-opening polymerization (AROP).[975,976] AROP is less impervious to contaminants than RAFT and ATRP, and it can’t be used in protic solvents. Among other things, Barthel et al.[944] used sequential AROP to synthesize poly(ethylene oxide)-block-poly(allyl glycidyl ether)-block-poly(tert- butyl glycidyl ether) (PEO-b-PAGE-b-PtBGE) (Figure 46). AGE and tBGE were sequentially polymerized to create ABC polymer after sodium hydride was used to activate a PEO macroinitiator containing a terminal OH group. PEO-b-PAGEGal-b-PtBGE was the outcome of post-polymerization addition of 2,3,4,6-tetra-O-acetyl-1-thio-β-D-galactopyranose by thiolene chemistry to the middle block PAGE. In order to create new micellar morphologies, this step was taken to further alter the weight fraction of the hydrophobic portions (PAGE, PtBGE).
Different monomers, such as cyclic amines and cyclic ethers, can be polymerized via cationic ring-opening polymerization (CROP).[975] Most notably, polyoxazolines are made with it.[977] Similar to AROP, CROP is restricted to particular solvents such as nitromethane, chlorobenzene, and acetonitrile and is very susceptible to contaminants. Hoogenboom et al.[978] synthesized a library of 30 triblock copolymers from 2-methyl, 2-ethyl, 2-nonyl, and 2-phenyl-2-oxazoline by means of the sequential addition approach. Kempe et al.[945] created novel amphiphilic triblock copoly(2-oxazoline)s with a fluorinated region in response to this trend. Utilizing methyl tosylate as an initiator, a series of triblock copolymers PODFOx-b-PEPOx-b-PEtOx were produced by sequential addition of 2-(2,6-diflurophenyl)-2-oxazoline (ODFOx), 2-(1-ethylheptyl)-2-oxazoline (EPOx), and 2-ethyl-2-oxazoline (EtOx) during microwave polymerization in nitromethane (Figure 47). This ABC triblock copolymer formed vesicular and aggregated cylindrical micellar structures by self-assembly in water.[979] These examples all demonstrate the effectiveness of sequential polymerization as a method for producing ABC polymers. By executing ten chain extensions, the Luxenhofer group even succeeded in pushing the boundaries of live nucleophilic ROP of N-substituted glycine N-carboxyanhydrides (NCAs) towards polypeptoids.[980] However, the controlled synthesis of longer and well-defined triblock copolymers using this approach may be hampered by side reactions such as chain termination and chain transfer. This issue is lessened by using commercially available macroinitiators since fewer chain extensions are required.
9.2.3.3. Macroinitiators Available Commercially
If a commercially accessible starting block such as poly(ethylene oxide), polystyrene, or poly-ethylene may be employed as a macroinitiator, the process of synthesizing ABC polymers can become less demanding. For instance, PEO’s favorable qualities (hydrophilic, non-inflammatory, and biocompatible) and widespread commercial availability make it a popular choice.[981] For a ROP of NCAs towards polypeptides, Sun et al. employed a PEO macroinitiator called PEO-b-PLLys-b-PLGlu (PLLys = poly(L)lysine, PLGlu = poly(L)glutamic acid).[982] During the polymerization process, the glutamic acid and lysine units were both shielded. In acidic water (pH 2.2, 0.5 M NaCl), the final ABC polymer self-assembled to form vesicles with symmetric (ABC-CBA) membranes.[982] A series of polyethylene-block-poly(ethylene oxide)-block-polycaprolactone (PE-b-PEO-b-PCL) polymers were synthesized by Wang et al.[946] All that was needed for the synthesis of the ABC polymer was ROP of ε-caprolactone (ε-CL), catalyzed by stannous(II) octoate (Sn(Oct)2) in DMF, using the commercially available AB (PE-b-PEO) diblock copolymer as a macroinitiator. The primary method for obtaining polycaprolactone is coordination-insertion ROP, which is the Sn(Oct)2-catalyzed ROP of ε-CL. However, most of the time, especially when using different polymerization processes, a properly designed macroinitiator is needed for the corresponding polymerization of the desired monomer.
9.2.3.4. Combination of Various Methods for Polymerization: To Combine AB and C by Click Reactions
The intended ABC polymer is obtained by a variety of polymerization processes in this synthetic route. For instance, Petrova et al.[947] used p-toluenesulfonyl chloride (TsCl) to endcap α-methoxy-ω-hydroxy-poly(ethylene oxide) and use it as a macroinitiator for the CROP of 2-ethyl-2-oxazoline (EtOx) in acetonitrile. A KOH/methanolic solution was used to conclude the later phase and add hydroxyl units as chain ends. The ROP of ε-CL, which produced PEO-b-PEtOx-b-PCL, was then catalyzed by the resultant AB diblock copolymer. The terminal functionality on AB required for the subsequent polymerization process was introduced in this instance by a well-chosen termination step. However, in order to facilitate the polymerization of the C block, modifications must be made to the AB diblock copolymer if it is not readily functional. The synthesis of poly(ethylene oxide)-block-poly(dimethylsiloxane)-block-poly(2-methyl-oxazoline) (PEO-b-PDMS-b-PMOXA) is one instance where this technique was used.[949,983] Using PEO as a macroinitiator, PEO-b-PDMS is first synthesized by AROP. On this resultant AB, PEO-b-PDMS was ω-functionalized with trifluoromethanesulfonic anhydride (triflate) in order to polymerize 2-methyl-2-oxazoline (MOXA) using CROP. The resultant PEO-b-PDMS-b-PMOXA self-assembled formed asymmetric polymersomes that caused transmembrane proteins to insert themselves in a guided manner. The synthesis of poly (ethylene oxide)-block-polycaprolactone-block-(poly-2-methyl-2-oxazoline) (PEO-b-PCL-b-PMOXA) was also carried out using a similar concept.[948,984] Using PEO as a macroinitiator, Sn(Oct)2-catalyzed ROP of ε-CL was used to synthesize PEO-b-PCL. PEO-b-PCL was ω-functionalized with TsCl in a manner similar to that of PEO-b-PDMS in order to polymerize MOXA by microwave-assisted CROP on this resultant AB (Figure 48).
The resulting PEO-b-PCL-b-PMOXA self-assembled into various microscale structures, most notably asymmetric membrane polymersomes, which we will go into further detail about in the review’s next section. Access to a larger pool of monomers and functionalities is made possible by the use of several polymerization processes. Having more adaptability is highly beneficial when designing ABC polymers. For instance, using a combination of coordination-insertion ROP and ATRP, Bian et al.[950] synthesized poly(ethylethylene phosphate)-block-poly-caprolactone-block-poly[2-(dimethylamino)ethyl methacrylate] (PEEP-b-PCL-b-PDMAEMA). In a similar fashion, Matter et al.[951] synthetized poly(ethylene glycol)-block-poly(γ-methyl-ε-caprolactone)-block-poly[2-(dimethylamino)-ethyl methacrylate] (PEO-b-PMCL-b-PDMAEMA) via a combination of coordination-insertion ROP and ATRP producing ABC polymers (Figure 49).
Additionally, AB diblock copolymer and C homopolymer can be covalently bound to create designed ABC polymers. Pre- and post-polymerization modification processes are necessary in this strategy in order to acquire the correct building blocks with the necessary functional end-groups. For example, He et al.[952] produced poly(ethylene oxide)-block-polystyrene-block-polycaprolactone (PEO-b-PS-b-PCL; Figure 50) by combining ATRP, coordination-insertion ROP, and click chemistry. In order to prepare the diblock precursor PEO-b-PS with terminal Br group, styrene was subjected to ATRP on a PEO macroinitiator that was produced by esterifying PEO with 2-bromo-2-methylpropionyl bromide. The terminal bromine group on PEO-b-PS was then nucleophilically substituted with sodium azide to generate an azide group. Propargyl-terminated PCL, the third block, was made via ROP of ε-caprolactone in the presence of Sn(Oct)2, starting with propargyl alcohol. The copper-catalyzed azide alkyne click (CuAAC) reaction of those two precursors in the presence of a CuBr/PMDETA catalytic system in DMF was then used to synthesize the final ABC polymer, PEO-b-PS-b-PCL. In conclusion, a variety of polymerization processes are available to construct ABC polymers. Every system has a unique set of response conditions that it needs. I shall talk about their fascinating self-assembly behavior next, which demonstrates how much work went into their synthesis.
9.2.4. Theory of Triblock Co- and Terpolymer Self-Assembly
On a scaling level, the self-assembly of the diblock AB with degree of polymerization NB/2 of the core block B and the linear triblock copolymer ABA are comparable.[897] On the other hand, when compared to diblock AB with degree of polymerization NA/2 of the coronal block A, linear triblock copolymer BAB shows new characteristics. Although a “flowerlike” micelle with a corona made up of loops A in a very diluted solution is similar to its diblock counterpart, an increase in polymer concentration causes aggregates to be attracted to one another and eventually causes solution gelation.[985] Comparable to spherical micelles, networks and mesogels[986] are formed by aggregates of cylindrical and lamellar forms, in which soluble blocks A create loops and bridges between condensed cores B. The third component’s introduction (C), when compared to AB or BC diblock copolymers, greatly increases the behavioral diversity of the linear terpolymer ABC. A range of multicompartment micelles (MCMs) are produced by the three components’ varying solubilities and the competition to maximize the structure of the self-assembling aggregate. A new review presents a thorough description of these structures.[775] Here, I differentiate MCMs that are corona- and core-compartmentalized. Corona-compartmentalized “Janus” micelles are produced by terpolymers with two incompatible solvophilic terminal blocks (A and C) and a solvophobic middle block (B). In the corona of the Janus micelle, lateral segregation of soluble blocks A and C has been seen experimentally[987,988,989] and theoretically investigated using the two-gradient SCF technique.[990] Two linear chains of length NB/2 can be thought of as making up a loop of block B in the Janus micelle core in an analytical procedure. Blocks A and C segregation in the coronal domain approximates FB,elastic, and the aggregation number p in the Janus micelle may be obtained by balancing the coronal free energy and the surface free energy Finterface. Based on the blob model of starlike polymer, a segregation threshold may be derived for a nonionic terpolymer with equal lengths of terminal blocks, NA = NC >> NB, and nonselective (athermal) solvent for A and C.[874] Each coronal block (A or C) in a starlike micelle with mixed corona of thickness H >> R and aggregation number p results in ≃ p1/2 ln(H/R) correlation blobs (eq 5, i = 3). Blocks A and C become incompatible when the interaction free energy resulting from A-C contacts ≃ kBTχACp1/2 ln(H/R) approaches the thermal energy order, or when χAC ∼ p−1/2(ln NA)−1 with p ∼ NB4/5 as determined by equation 6. When blocks A and C separate into different domains over the segregation threshold, more A/C interfaces and related free energy losses occur within the micellar corona. Still, the A/C interface-related free energy increase is less than the ∼kBTp3/2 dominating coronal contribution. Consequently, in a starlike Janus micelle with segregated corona, the power law asymptotic dependency for aggregation number p (eq 6) remains unchanged. Coronal segregation occurs for weakly incompatible blocks A and C by the creation of many A- and C-rich coronal domains with highly changing geometry. The application of an external field or the adsorption of micelles at the liquid-liquid interface may cause the segregation into the two hemispheres, A and C, with their distinct interfaces. Terpolymers with one soluble and two insoluble blocks generate core-compartmentalized MCMs with a variety of morphologies, such as core-shell spheres,[987,991] spherical aggregates with “hamburger”, “clover”, or “football” cores,[769,775,992] core-shell or core segmented cylinders,[769,775,993,994] etc. The theoretical assessment of the free energy per chain in an aggregate containing the MCM core microsegregated into domains with distinct components is considerably more complex than in a micelle consisting of a diblock copolymer. A manageable simplification of the MCM model is possible with the lens[995] or spherical segment[769] approximation of the domain shape. For instance, a star-shaped MCM with a “football” core is produced when terpolymer with two insoluble components (B and C) has an asymmetric composition (NB << NC). In such a micelle, the surface free energy Finterface can be expressed as a function of the number n of domains B on the surface of core C (shown as spherical segments). It is possible to formulate the free energy per chain in such an MCM as a function of the number of compartments (n), aggregation number (p), surface tensions (γBS and γCS at the core-corona boundary), surface tension (γBC) at the interface between B and C domains, and molecular weights of the blocks (NA, NB, and NC) by ignoring the confinement free energies of blocks B and C and decomposing the corona of soluble blocks A into n spherical sub-brushes adjacent to domains B and a laterally homogeneous sub-brush at larger distances. In such a micelle, the surface free energy Finterface can be expressed as a function of the number n of domains B on the surface of core C (shown as spherical segments). It is possible to formulate the free energy per chain in such an MCM as a function of the number of compartments (n), aggregation number (p), surface tensions (γBS and γCS at the core-corona boundary), surface tension (γBC) at the interface between B and C domains, and molecular weights of the blocks (NA, NB, and NC) by ignoring the confinement free energies of blocks B and C and decomposing the corona of soluble blocks A into n spherical sub-brushes adjacent to domains B and a laterally homogeneous sub-brush at larger distances. Power law dependences for the MCM parameters are possible when the free energy is minimized with respect to n and p. The number n of compartments in a starlike MCM with a “football” core is predicted to be with accuracy of the logarithmic prefactor.[769]
Equation 36 indicates that the number of compartments n grows as terpolymer asymmetry, NC/NB, increases. Additionally, the number of compartments can be controlled by changes in the free energy of the core-to-corona interface. In this case, the contact angle is θ = arccos[(γCS - γBC)/γBS], and BS ≃ γBS(1−cosθ)2/3(1+0.5cosθ)1/3. Recent experimental observations are in good qualitative accord with this theoretical prediction.[769]
It is not possible, however, to ignore the confinement free energy of core blocks for any given terpolymer composition. Like the generic morphologies of diblock copolymers, it is reasonable to assume that the free energy increment, δF = FB,elastic + FC,elastic + ΔFA, determines the geometry of a crew-cut terpolymer aggregate in a diluted solution. In this case, all three contributions- FB,elastic, FC,elastic, and ΔFA-depend on the internal organization of the MCM core, such as the number and shape of the core compartments. A set of difficult difficulties for further theoretical research is the accurate formulation of δF and the description of MCM characteristics and morphology as a function of molecular weights and solubilities of components A, B, and C. Several new self-assembled structures can be produced by switching the terpolymer architecture from linear to 3-miktoarm star.[996,997,998] The majority of the theoretical research on 3-miktoarm terpolymer[999,1000,1001,1002] was devoted to the examination of melt state morphologies, such as lamella, polygonal cylinders, perforated layers, lamella-in-spheres, etc. In all these phases, junctions between the blocks in a 3-miktoarm terpolymer localize on lines where three interfaces-A/B, A/C, and B/C-merge, according to computer simulation of such systems.[1002] For the 3-miktoarm terpolymer with two insoluble blocks, Monte Carlo simulations in a diluted solution[1003] revealed the presence of spherical micelles with dumble-like cores and modulated core segmentation cylinders (refer to Figure 51). A scaling model[1004] that describes the confinement of the chains in a dumble-like core uses simplifying approximations and permits power law asymptotic dependences for micelle parameters only under specific bounds. Currently, there is a lack of a thorough theoretical modeling of such designs at arbitrary compositions and solubilities of the terpolymer components.
The construction of complex structures is made possible by linear ABC triblock copolymers in a way that is not possible with traditional linear AB diblock copolymers. ABC polymers can have one soluble block (terminal A/C or middle B) and two soluble blocks (adjacent blocks A/C and B, or terminal blocks A and C), depending on the solvent in which self-assembly is carried out. Since these are the most well-studied kinds of ABC polymers, we concentrate on their self-assembly in this section: ABC polymers with one soluble C block and two soluble A and C blocks.
9.2.4.1. Soluble C Block ABC Polymers
These kinds of triblock copolymers are useful for creating multicompartment self-assembled structures, or structures with domains at their center. When the insoluble blocks A and B phase split in the core, compartments may result. By dissolving the polymers in a non-solvent for B and gradually swapping them via dialysis to a non-solvent for A and B, Müller and colleagues have shown how to elegantly create multicompartment structures using ABC polymers.[769,771,1005] A core containing A, domains forming B, and corona containing C are the likely near-equilibrium structures that arise from such a preparation procedure. Transmission electron microscopy (TEM) can be used to visualize domains by selectively staining particular blocks. RuO4 for poly (2-(cinnamoyloxy)ethyl methacrylate) (PCEMA), I2 for poly(2-vinylpyridine) (PVP), and OsO4 for polybutadiene are a few examples. As demonstrated for the polysterene-block-polybutadiene-block-poly(methyl methacrylate) (PS-b-PB-b-PMMA) model system in acetone/isopropanol mixtures, one approach to influence the number of domains being generated is the ratio between the various blocks. PMMA (C) is always soluble in this polymer/solvent combination, PB (B) is insoluble, and PS (A) collapse can be controlled by converting the solvent mixture from N,N-dimethylacetamide to acetone/isopropanol (Figure 52).[769] A collapsing A block is the outcome of B domains forming during the solvent exchange from a non-solvent for B to a non-solvent for A and B. A constant near-equilibrium rearrangement of the A/C corona is made possible by slow solvent exchange, and this results in trappable intermediate structures (Figure 52b and 52c). The solvophobic A block is exposed when the C block is unable to stabilize these micelles, which leads to the secondary aggregation of intermediate structures into multicompartment micelles with different numbers of domains (Figure 52d). A core with more than five B domains, called “football,” “clover,” or “hamburger,” is an example of a spherical micelle that is produced by a Janus-type (Figure 52c) intermediate structure with one exposed A-side (Figure 52d). A B core with two exposed A domains is present in “inverse hamburger” intermediate micelles formed by some polymers (Figure 52b). These “inverse hamburgers” have the potential to dimerize into “double-burgers,” which can then further aggregate into mesoscale colloidal chains (Figure 52e and 52f). The final multicompartment structure’s form (spherical or linear) is determined by the ratio of the volume fractions V of the blocks that constitute the core, A and B.[716] Spherical structures occur when VA/VB > 1. The number of domains in this instance is (for star-like micelles where dcorona >> Rcore):
where
N denotes the degree of polymerization; v, l, the volume and length of a monomer unit; τC, the solvent strength for the C block; and γ, the interfacial tension between a block or blocks and solvent S.
≅ γBS(1-Cosθ)⅔ (1+Cosθ/2)⅓ and Cosθ = (γAS – γAB) / γBS - B domains’ contact angle with the A core.
The “inverse hamburger” form, which has two A domains and a B core in contrast to “hamburgers,” is present in intermediate structures when VA/VB ≤ 1 and γAS < γBS (Figure 52b). These formations have the ability to subsequently polymerize into linear mesoscale colloidal chains (Figure 52f).
By displaying a change in morphology when the corona volume is adjusted by altering the corona length NC or the corona block’s solvent quality, Gröschel et al. proved the concept’s universal applicability. What’s more, they demonstrated how the idea works with various ABC systems.[769] They used PS-b-PB-b-PVP, PS-b-PB-b-PtBMA, PtBMA-b-PVP-b-PB, pH-sensitive PtBMA-b-PCEMA-b-PDMAEMA to study the self-assembly of multicompartment structures and previously reported poly (tert-butoxy styrene)-block-poly( C6F13C2H4S- ethylethylene)-block-poly(tert-butyl methacrylate)[1005] and poly-(N-isopropyl acrylamide)-block-poly(n-butyl acrylate)-block-poly(ethylene oxide)polymers.[989] Based on this, the same group has introduced rational design of multiple domain morphologies on several micelle morphologies, thereby expanding the field of ABC multicompartment structures (Figure 53).[771] In acetone/isopropanol combinations, they employed the PS-b-PB-b-PtBMA model system, in which PB (B) is insoluble, PtBMA (C) is always soluble, and the composition of the solvent determines when PS (A) collapses. Regarding the earlier systems, PB and PS are not interchangeable. The corona length (rows in Figure 53) was used to adjust the overall micelle shape because:
N is the degree of polymerization; v is the monomer unit’s volume; l is the monomer unit’s length; q is the correction factor for the A block’s selective swelling; Surface tension at the A/solvent interface is measured as kBTγA/solvent. As with micelle geometry, one can adjust the aspect ratio between blocks B and C (columns in Figure 53) to modify domain morphology.
where surface tension at the B/solvent and B/A interfaces is taken into account by . These multicompartment structures could be used as models for nanoelectronics and nanooptics. Amphiphilic ABC polymers can be designed for use as smart drug delivery devices with controlled pharmacokinetic release patterns based on the proposed theory.[771]
9.2.4.2. Soluble A and C Blocks in ABC Polymers
Triblock copolymers of this kind are useful for creating self-assembling structures known as “patchy” structures, which have domains in their corona. Many polymers are now on the market, but I will concentrate on PEO-b-PCL-b-PMOXA polymers that have been well studied. PEO and PMOXA are water-soluble, while PCL is a semicrystalline hydrophobic block.[948,1006] Their aqueous self-assembly was examined in relation to the preparation technique and block lengths of PCL and PMOXA (PEO length was constant). Self-assembly of such ABC polymers was studied in dependence on the hydrophilic weight fraction f, just as amphiphilic AB diblock copolymers:
When AB polymers have a fixed A block length, the length of the B block can be changed to vary f. Polymersomes – rods – micelles with increasing f are a common self-assembly trend.[1007,1008,1009,1010] The accessible structures in the case of ABC polymers are significantly more diversified than those generated by AB polymers, and in the case of ABC polymers with fixed A block length, f can be modified through the change in B or C block length. When f is altered throughout the length of B (PCL) (Figure 54a, red and blue dashed lines), the morphology of PEO-b-PCL-b-PMOXA changes in the row of spherical particles (Figure 54b), irregularly shaped particles (Figure 54c), polymersomes (Figure 54d), and with an increase in f. The morphology of the assemblies changes in the row spherical particles (Figure 54b), polymersomes (Figure 54d), and cloud-like aggregates (Figure 54e) with an increase in f when f is altered along the length of the C (PMOXA) block for polymers with B of around 60 - 130 units. By employing various preparation techniques, the quantity of self-assembled structures made of ABC polymers can be raised even more. The packing geometry model provides an explanation for the formation of the observed self-assembled structures (Figure 54f - 54h). Packing geometry for AB diblock copolymers is determined by the packing parameter p:[1011]
where vB and lB represent the volume and critical length of the hydrophobic block B, respectively, and αA–B is the ideal area of the hydrophilic block A at the interface A-B. The packing geometry of AB polymers is shaped like a cone because they self-assemble into spherical particles (Figure 54f). The region αC–B at the C-B interface (Figure 54g) is likely introduced by adding the C block because PEO and PMOXA polymers are immiscible in aqueous solution.[1012,1013] In the above case, the packing geometry of ABC molecules corresponds to a cylinder that is somewhat truncated at the C side because the A block is longer than the C block. A longer A block forms the outside surface of the resulting polymersomes, while a shorter C block forms the inner surface. Stronger repulsive forces within the hydrophilic corona are produced by further lengthening the C block, and these forces compete with the hydrophobic forces that are attracted between the B chains. In this instance, the ABC molecules’ packing geometry approaches the double cone form, producing aggregates that resemble clouds (Figure 54h).
Making polymersomes with an asymmetric membrane is presumably the most popular use of ABC polymers containing A and C soluble components. Three possible compositions of polymersome membranes exist in principle: A patchy or mixed membrane is formed by A block outside, C block outside, or A and C blocks together (Figure 55).
Because the outer curvature of a polymersome has a wider radius than the inner curvature, a longer block is typically found on the outer surface. Asymmetric membrane polymersomes have been reported for PEO-b-PCL-b-PMOXA,[948] PEO-b-PDMS-b-PMOXA,[949,983] (PAA-b-PS-b-PVP) (PAA = poly(acrylic acid)),[1014] PEO-b-PCL-b-PAA,[1015] glycosylated PEO-b-PB,[1016] PEO-b-PCL-b-PDEAEMA,[1017] PMPC-b-PDPA-b-PDMEAEMA,[1018] and PEO-b-PMA-b-PAA (PMA = poly(methyl acrylate).[1019] Since (de)protonation affected the surface charge, asymmetry in the case of charged blocks, such as PAA, PDMAEMA, and PVP, was demonstrated by measuring the ζ-potential of the structures in solution at various pH values.[1014,1015,1019] This approach, however, cannot be used when both blocks are neutral and does not rule out the possibility of the presence of the second hydrophilic block on the outer polymersome surface. Raman spectroscopy was used to demonstrate the asymmetry of the polymersomes made of glycosylated PEO-b-PB through the interaction of gold nanoparticles with glucose. The lack of contacts between the glycosylated PB block and the PEO block, as detected by 2D-NOESY NMR, suggested the absence of PEO on the exterior. The presence of PEO-b-PDMS-b-PMOXA, which has a fluorescently tagged PMOXA block that is longer than PEO, was demonstrated externally by adding an external quencher to quench the associated dye’s fluorescence. Two distinct methods were employed to ascertain the asymmetry of PEO-b-PCL-b-PMOXA polymersomes. ABC polymersomes having terminal functional groups on either the A or C side were able to pair with a reactive fluorescent dye in the first method. The presence of the A block outside was demonstrated by the interaction that transpired between the A block and a dye. The lack of a reaction between C blocks and a dye indicated that there were no external C chain endings. The Bicinhoninic Acid Assay (BCA)[1020] demonstrated additional evidence of the asymmetry by its ability to react selectively with PMOXA. Schrage et al. demonstrated yet another method for producing polymersomes with an asymmetric membrane.[320] Two diblock copolymers were utilized by the authors: PS-b-P4VP (P4VP = poly(1-methyl-4-vinylpyridinium iodide)) and PB-b-PCsMA (PCsMA = poly(cesium methacrylate)), which self-assembled in THF as a result of electrostatic interactions between the PCsMA and P4VP blocks. Since the corona blocks in this instance, PS and PB, have distinct space needs and strong incompatibilities, it is likely that they separated on opposing sides of the polymersome membrane. Compared to traditional symmetric membranes, asymmetric membrane ABC polymersomes provide greater design flexibility for complex systems. The inner and outer surfaces of A and C blocks may have distinct chemical and physical characteristics that allow for selective catalysis, response to particular stimuli, immobilization of particular molecules, etc. An ABC membrane, for instance, can cause transmembrane proteins to insert directedly, as demonstrated by the insertion of Aquaporin 0 with a histidine tag into polymersomes composed of PEO-b-PDMS-b-PMOXA.[983] An insertion with 72% of His-tags on the outside was brought about by the creation of an ABC membrane, or PEO outside. On the other hand, a CBA membrane (PMOXA outside) caused 19% of the His-tags to be outside, causing the inverted orientation. For polymersomes derived from the symmetric PMOXA-b-PDMS-b-PMOXA block copolymer, no preferred orientation (47% of the His-tags outside) was observed. Polymersomes made of ABC polymers not only exhibit preferential orientation but also improve the delivery of some medications.[1017,1021,1022] Self-assembling ABC polymers containing A and C soluble blocks can form structures with patchy, mixed, and Janus corona.[893,989,1005,1023,1024,1025,1026,1027,1028,1029] with a wide range of possible uses, from healthcare to materials science.[1030,1031] Additionally, they can create novel structural forms[1006] that are not possible with traditional AB polymers.
9.2.5. Non-Linear Architectures
A great deal of work has gone into creating non-linear, well-defined amphiphilic block copolymers. Such synthetic techniques are motivated by the effects of architecture on the characteristics of double hydrophilic and amphiphilic block copolymers. Numerous non-linear block copolymers were created using this polymerization technique because anionic polymerization provides the most control over macromolecular architecture. These copolymers were subsequently converted to amphiphilic copolymers using post-polymerization chemical treatments. The reactivity and chemical stability of the branching points during the chemical modification reaction should also be considered when creating branched structures via chemical functionalization of premade block copolymer. This is one of the reasons why branching macromolecules have not been subjected to some of the functionalization procedures discussed previously. For instance, under the typical conditions used for PS sulfonation, it is not possible to sulfonate PS including star copolymers with chlororosilane linked arms because of the degradation of the central branching point (instability of the Si-C bonds). Although other graft copolymers have also been described, the majority of the study is concentrated on the synthesis of star polymers. Anionic polymerization can be used to create (PS)n(P2VP)n hetero-arm stars. First, divinylbenzene is used to link prefabricated PS arms. Next, 2VP monomer is polymerized using the anionic active sites in the DVB core of the produced star (a process known as the “in-out” approach). Due to the protonation/deprotonation of P2VP arms at low and high pH, respectively, these star copolymers exhibit amphiphilic character. They also exhibit intriguing micellization behavior and complexation capabilities towards inorganic salts.[1032,1033] By hydrolyzing the (PS)n(PtBA)n copolymer precursor, a hetero-arm star copolymer of type AnBn made up of polystyrene and poly(acrylic acid) arms was created. This copolymer was then neutralized to the (polystyrene)n(poly(sodium acetate)n, (PS)n(NaPAA)n form using a stoichiometric quantity of NaOH.[1034] A significant level of hydrolysis was attained. When the PAA and PS arms formed micelles with a PAA core and were symmetric, the PS/PAA star copolymer was soluble in dioxane. The hetero-arm star was insoluble in dioxane when it consisted of long PAA arms and short PS arms. Ampholytic (P2VP)n(PAA)n hetero-arm stars with basic and acidic arms were also produced in an identical chemical sequence.[1035] Anionic polymerization and acidic hydrolysis were used to create multiarm star-shaped copolymers with the design An(B-b-C)n, where A = PS, B = P2VP, and C = PAA. This process followed a similar “in-out” approach. Under various solution circumstances, the hetero-arm star terpolymers that are amphiphilic/polyampholytic exhibited intriguing solubility and gelation behavior.[1036] Amphiphilic star block copolymers have also been synthesized through the use of sulfonation and neutralization. By connecting PS-PtBS living chains with DVB, the precursor (PS-PtBS)n star block copolymers with PS outer arms were produced. Using SO3, the PS blocks of the stars were sulfonated selectively.[1037] Elements analysis has shown that the star block copolymers had a sulfonation degree of 85-95%. The salt form of the amphiphilic star block polyelectrolytes was obtained by neutralization with sodium methoxide in methanol (Figure 56). Additionally, amphiphilic star block copolymers with an inverted molecular topology were created. From the PCL-b-P4VP precursor graft copolymer, a unique amphiphilic graft copolymer with the structure poly(e-caprolactone)-b-quaternized poly(2-vinyl pyridine) (PCL-b-PQ4VP) was created by quaternization with a significant excess of iodomethane in DMF.[1038] In a similar manner, the graft copolymer PCL-b-PQDMA-EMA has been produced. Compared to their precursors, the quaternized graft copolymers exhibit better water solubility and more clearly characterized micellar aggregates.
10. Mechanisms of C3M Formation: Mechanism of Micelle Assembly and Disassembly
10.1. Mechanism of Aggregation: Factors Influencing and Impact of External Factors, Polymer Architecture, the Length of the Core-Forming Block and Charged Functionality’s Structure towards C3M Formation
When the cationic and anionic charges of the diblock copolymer and homopolymer are present in about equal amounts, complex coacervation core micelles are produced.[30,1039] This mixture is known as the preferred micellar mixture (PMC). No micelles form at compositions far from the PMC. Even if there is interaction, the aggregates most likely have a relatively loose structure.[1039] The aggregates’ extra charge prevents them from aggregating into denser, bigger objects. These kinds of aggregates will be referred to as soluble complex particles (SCPs). They often have low aggregate numbers, and the extra component’s charge sign determines their charge sign. A speciation diagram of a mixture of diblock copolymers with opposite charges and homopolymers is shown in Figure 56 as a function of composition. The titration of a solution containing anionic diblock copolymers with cationic homopolymers is depicted in this diagram, along with a schematic representation of the speciation as a function of f+, which is described as
where [PB] and [PA], described in terms of their chargeable, monomeric units, are the anionic component (diblock copolymer) and cationic component (homopolymer), respectively. Figure 57 shows the speciation as a function of f+ for each type of particle: micelles in (c), SCP+,- in (b), and free diblock copolymers (dbp) and homopolymers (hp) in (a).
As f+ increases, we observe that the concentration of diblock copolymers falls because the molecules are used up in the process of forming SCP-. The concentration of SCP- is at its greatest and the concentration of free diblock copolymers has decreased to zero at the critical excess anionic charge (CEAC). Now, as more homopolymers are added, the SCP- are used up in the process of creating CCCMs until the PMC is attained. When the homopolymers are overdosed, the micelles break down beyond the PMC and SCP+ is created. The concentration of micelles has once again dropped to zero at the critical excess cationic charge (CECC). As a result, micellar concentrations are highest at the PMC and are exclusively present between the CEAC and the CECC. The maximum and minimum SCP and CCCM concentrations are listed in Table 5.
If a system follows this relationship, it is precisely at the PMC.
where fi is the mole fraction of component i in the polyacid/polybase combination and R is the linear charge density of the polyacid (PA) or polybasic (PB) in the core. Because the charge densities in the core are different from those in the bulk solution, it is not possible to predict the PMC directly from bulk charge densities. Because the complex coacervate has a high concentration of cationic and anionic groups, its environment differs from that of the bulk solution, affecting how the charged groups dissociate. Both polyelectrolytes exhibit an increase in linear charge densities. Protons will be consumed by the polybasic chains and expelled by the polyacid chains. This change in charge could result in a net uptake or release of protons. Keep in mind that if the charges on the polymers are quenched, then these modifications won’t happen. By performing a light scattering titration experiment, in which the composition of the system is varied by titrating a concentrated solution of one component with a diluted solution of the other, and then monitoring the scattered intensity, the PMC can be experimentally determined. Light scattering theory can be used to generate a light scattering intensity (I) vs f+ using the concentration profiles in Figure 57. I is typically stated in terms of the excess Rayleigh ratio, or ∆R. The complete mathematical formula for light scattering is provided by
where C is the weight concentration of the scattering particles, Mw is their molar mass, P(q) is the particle form factor, S(q) is the interparticle structure factor, and K is the optical constant, which may be found by 4π2nsol2(∂n/∂c)2/ (NAVλ04). The following simplifications will allow equation 45 to be reduced to a scaling relationship: (a) K ∼ (∂n/∂c)2, (b) the micelle radii are around λ/20, thus P(q) → 1,[1040] and (c) considering the low concentrations, S(q) → 1. [1040] We obtain ∆R ∼ I by ignoring the solvent scattering. Now, equation 45 becomes
The components are either free in solution, form SCP, or form micelles, as Figure 56 illustrates. There should be a maximum in scattered intensity between the CEAC and CECC, which is at the PMC, because the molar masses of these distinct particles are predicted to be significantly different, namely, MCCCM > MSCP > Mhp,dbp. At this scale, the dispersion of HP and DBP is negligible. A schematic light scattering diagram is displayed in Figure 58.
The following arguments can be used to calculate the molar mass of the micelles based on this diagram. Because of their excess charge, the SCP-prefer to reduce the number of their aggregations. This indicates that when homopolymers are used as the minor component, the SCP-consists of a single homopolymer chain that is completely saturated with diblock copolymer blocks that have opposite charges. The length of the homopolymer Nhp, the block length of the core block from the diblock-copolymer Ncore, and fCEAC determine the number of diblock copolymers per SCP-, PSCP.
The molar mass of the SCP- follows naturally from equation 47 as MSCP = Mhp + PSCPMSCP. Observe that MSCP ∼ Mhp. Now that we have followed equation 46 and disregarded tiny variations in ∂n/∂c between the PMC and the CEAC, we can determine the molar mass of a CCCM from
The light scattering mass analysis (LSMA) is the term we give this technique. Keep in mind that the technique can also be used for systems in which the homopolymer and diblock copolymer have the opposite charge sign.A CCCM’s aggregation number (P), which is the number of diblock copolymers per micelle, may be computed from its molar mass. The core radius can be computed using the molar volumes of the constituent core components and an assumed polymer volume density.
where φ is the polymer volume fraction in the core and Vdry is the dry volume of the core’s constituent parts. The grafting density of corona chains at the core/corona interface can be determined using the core radius and aggregation number as follows:
Within Flory-Huggins theory, the corona thickness is determined by σ, Rcore, Ncorona, the corona chain length, and the excluded volume parameter υ. These parameters describe the binary interactions between polymer segments (υ = 1 - 2χ) The two equations that follow were extracted from Wijmans and Zhulina.[282] First, using a good solvent on a flat surface, the thickness of a polymeric brush is provided by
the monomer length is represented by l. The relative reciprocal curvature ω = Rcore/Hflat and Hcorona) determines the brush height for spherical particles.
We now have Rmicel = Rcore + Hcorona, which gives the theoretical micellar radius. We can also forecast possible pH changes during a titration experiment based on the speciation profiles displayed in Figure 57. The system contains free anionic groups when f+ is below the PMC, free groups are least concentrated near the PMC, and free cationic groups are present above the PMC. Free, annealed groups have the ability to function as a pH buffer. As we reasoned before, complexation will cause the pH of the solution to alter. The buffering effect of free annealed groups will reduce these changes when the system composition is (far) away from the PMC. This buffering ability reaches its minimum around the PMC, hence a maximum in ∂pH/∂f+ is anticipated.
Supramolecular nanostructures known as polymer micelles and polymersomes are typically created when amphiphilic block copolymers self-assemble.[1041] Traditionally, they consist of block copolymers with segments that are both hydrophilic and hydrophobic. Diblock, triblock, or multiblock copolymers can yield polymer micelles and polymersomes.[722,836] Depending on the polymer composition, the block copolymers assemble into nanostructures in aqueous conditions. The relative volumes filled by the hydrophilic(s) and hydrophobic(s) blocks produce the distinct assemblages. This regulates the geometry of the final assembly and specifies the geometric packing of block copolymers in the copolymer assemblies that are formed in aqueous solution. For instance, depending on how long the hydrophobic block is, a diblock copolymer made of hydrophilic and hydrophobic blocks can assemble into worm-like micelles, spherical micelles, or vesicles (or polymersomes).[755] The assembly of polymer micelles used to transport BTAs typically takes place in water, and the micelle core is formed when the hydrophobic portion of the block copolymer is kept out of the surrounding aqueous solution. Payloads, such as BTAs, are usually enclosed in this hydrophobic pocket located at the middle of the micelle. Next, using a brush-like topology, the hydrophilic portion of the block copolymer constructs the nanoparticle shell (Figure 59).
Diblock copolymer-based polymersomes self-assemble into a bilayer polymer membrane,[1043] which is similar to the liposome's phospholipidic double layer. To some extent, the synthetic mimics of phospholipid that are employed are the block copolymers.[1044] A semi-permeable hydrophobic membrane typically divides the aqueous core of the polymersomes employed to deliver BTAs from the surrounding aqueous environment (Figure 59). With polymersomes, there is greater flexibility to encapsulate a wider range of payloads because the payloads can be found inside the hydrophilic core or in the hydrophobic membrane.[1043]
Ionic block copolymers and proteins are typically mixed in an aqueous buffer solution to create C3Ms. A standard experiment involves gradually titrating the polymer into the protein solution; however, an alternative method could also be employed, but it might not yield identical outcomes. What polymer protein molar ratios are best for obtaining well-defined C3Ms remains the main question at this point. As mentioned earlier, van der Burgh et al. conducted a thorough investigation on the relationship between the quantity of charged species present and the development of PIC micelles.[30] The charge ratio f+ was established by the authors as , where [A] represents the concentration of anionic charges and [C] represents the concentration of cationic charges. When both charges are present in equimolar proportions, as for f+ = 0.5, the maximum scattering intensity shown in Figure 58 occurs; in contrast, species with an excess of one charge should be less densely packed.[30] As proteins’ surface charge may differ from polymers’ in terms of spatial distribution, more polymer may be needed to saturate the protein than would be the case with equimolar charged quantities. It should be mentioned here that the synthesis of C3Ms involving proteins differs from that occurring between two polymers. Proteins are rigid structures that do not adapt very much. Therefore, it is the polymer’s responsibility to offer enough flexibility to allow it to wrap around the protein’s structure and cross two charged regions. Stiff polymers hence have a lower binding strength.[1045] Entropically unfavorable chain stretching results from even extremely flexible polymers having to adapt to the often unequal distribution of charges on the protein surface.[1046] While one can visualize the shape of C3Ms in the presence of equimolar charges, alternative situations arising from excess negative or positive charges species are illustrated in Figure 60. Proteins will share the few available polymers at low polymer concentrations, most likely forming C3Ms with many proteins, resulting in huge particles. In a second situation, free protein stays in the solution and forms tiny C3Ms as they wait for additional polymer to be added. C3Ms with a low size dispersity will form when the polymer concentration is increased toward a charge-neutral point, but an excess of polymer should result in a bimodal size distribution of C3Ms and free polymer. As an alternative, C3Ms could enlarge to make room for additional polymer. Therefore, it is possible that the charged polymer repeating units and charged amino acids from the protein will be found in an equimolar ratio around the minimum size dispersity. It was definitely discovered by Harada et al. that the second situation shown in Figure 60.[281] Bimodal distributions were discovered at low polymer concentrations, in this case PEG-P (Asp), indicating free lysozyme, which served as the model protein. There was no change in the size related to the C3Ms. In contrast, in a study, a decrease in the measured particle size was observed where the authors combined poly(ethylene glycol) methyl ether acrylate (PEGMEA)-PAA with lysozyme, indicating that the first model applies in this system (Figure 60). The optimal size distribution was found at an equimolar ratio of charges, as was predicted. Equimolar ratios are not required, even if this can be a first approximation, and they must be found for each system. This point is easily ascertained since, as was previously noted, there is a correlation between the molar ratio of polymer to protein and the polydispersity obtained from scattering tests.[1047] Scattering techniques were employed in Harada et al.’s investigation[281] to probe deeper into the C3Ms. 36 lysozymes and 42 polymer chains with an equimolar ratio of both charges were estimated to be present in the C3Ms, which had a hydrodynamic diameter of Dh = 47 nm. With an increase in the quantity of polymer per particle and a slight decrease in the amount of protein, particle size rose as the amount of PEG-P(Asp) increased.[280] Remarkably, when referring to the charge concentration, it was discovered that the critical association concentration was independent of the mixing ratio. The scientists suggested, based on density estimates, that increased PEG stretching at high polymer–protein ratios explains why the PEG shell grows as the amount of polymer increases.[280] Since each system is based on a unique set of polymers and proteins with varying sizes, it is impossible to predict with certainty what situation would occur. For instance, at increased polymer content, C3Ms based on the much bigger glucose oxidase (GOx) resulted in significantly smaller sizes.[1047] Although C3Ms are dynamic structures, it is vital to keep in mind that, depending on the system, reaching equilibrium may take longer. The addition of the polymer to the protein or the other way around should be insignificant, theoretically. The equilibrium can really take several days to adjust, and it can take much longer if powerful polyelectrolytes are utilized. Therefore, the two methods-protein or polymer first-may produce somewhat different results.[94] A closer examination of the polyion complex generation process will help to clarify this. Random aggregates are formed by the mixing of negatively and positively charged polymers or proteins, which are decided by random collision events. Diffusion events are the only factors that control this quick process, which is why complexes form in milliseconds. These first structures lack ideal packing characteristics and are ill-defined. The polymers must then reorganize along the protein or exchange polymer with the solvent in order to reach equilibrium. However, the second stage may be a laborious procedure that takes many days to complete.[94] Typically, only electrostatic interactions are discussed in relation to C3Ms. These are the strongest forces, however there are additional attractive forces that can also be present and strong enough to attach a polymer to a protein, like H-bonding.[1048,1049]
Dynamic structures, polyion complex micelles can be assembled and destroyed in response to external stimuli. C3Ms have a critical aggregate concentration (cac), just like conventional micelles, at which point the formed structures begin to disintegrate into free polymers and proteins.[63,280] The block copolymer’s structure as well as outside factors like ionic strength and pH have an impact on the cac. The C3Ms always disassembles when salt is added. Stepwise disassembly may be necessary because the addition of salt may cause the core structure to relax or reorganize before complete disintegration is seen.[75] In general, in high ionic strength conditions, polymers with longer charged blocks exhibit greater durability against disintegration.[1050] The ionic strength variation and the corresponding assembly and disassembly were utilized to adjust the lysozyme’s activity against Micrococcus luteus cells. Because the shell inhibited cell lysis, the entire C3Ms was unable to exhibit any enzymatic activity on the cells. Protein release and the start of enzymatic activity were caused by the addition of NaCl. The C3Ms reassembled when the NaCl was removed.[284] As with higher ionic strength, surfactant addition may obstruct the production of C3Ms. While the use of surfactants like cetyltrimethylammonium bromide may not always result in disassembly, it can promote big particle development and precipitation thereafter.[1050] An electric field can have an impact on the binding process since the C3Ms production is dependent on electrostatic interactions. C3Ms micelles based on PEG-b-P (Asp) allowed for the control of the lysozyme activity by applying a threshold voltage to the solution.[1051] The length of the charged polymer, or more accurately, the quantity of charged functional groups, ought to have an impact on how the polymer and protein bond together. Many publications on this subject have used charged polymers-rather than proteins-for encapsulation. Nonetheless, proteins can benefit from the knowledge gained from attempting to capture polymers. With P(Lys) with 20-82 repeating units, the block copolymer PEG-P(Asp) with different P (Asp) block lengths was condensed. Remarkably, the hydrodynamic radius was discovered to be 24 nm regardless of the polymer blocks’ length. The amount of P(Lys) and the aggregation number N in the PIC micelles varied, despite their comparable exterior appearance. Longer charged block copolymers generally resulted in lower PEG chain densities in the shell as well as greater aggregation numbers N and P(Lys) in the core, which correlates with a larger core and smaller corona.[393] It was discovered that encasing the enzyme in polymers increased the enzymatic activity of the encapsulated protein-as long as the substrate was tiny enough to permeate through the polymer shell. For instance, it was discovered that the PEG-PAA C3Ms’ core lysozyme activity was higher than that of the free enzyme when compared to p-nitrophenyl-N-acetyl-b-chitooligosides. However, C3Ms based on smaller charged polymer blocks-in this case, poly(α,β-aspartic acid)-exhibited higher enzymatic activity. This was explained by the charged block copolymer’s location in respect to the binding pockets, which are made up of longer polymers that span many enzyme active sites.[1051] Utilizing trypsin (∼23 kDa) in place of lysozyme (∼14 kDa) produced differing outcomes. In general, larger charged polymer blocks led to higher enzymatic activity; however, the polymer to protein mixing ratio also affected the improvement of activity. As the molecular weight increased, the optimal ratio for mixing the polymer and protein was observed to decrease.[1052] It was looked into how long the copolymers with grafted PEG chains were and how it affected the way C3Ms with GOx (160 kDa) formed. PEG-graft-poly(allyl amine) had a molecular mass that ranged from 31 to 258 kDa, however the resulting C3Ms with GOx had a gyration radius of 12.6 nm instead of 10.8 nm. Extensive analyses demonstrated that C3Ms derived from longer polymers had a lower concentration of enzyme molecules. Since PEG acted as no barrier and the substrate glucose was able to penetrate quickly to the enzyme, the enzymatic activity was independent of the molecular mass.[1047] Since graft or comb polymers were also used to generate C3Ms containing proteins, the impact of the polymer architecture must be examined in terms of the molar ratio of neutral and charged blocks. To study the binding with bovine serum albumin (BSA) below its isoelectric point, that is, with BSA in a positively charged state, the negative neutral PDMAM content in the graft copolymers poly(sodium acrylate-co-sodium 2-acrylamido-2-methyl-1-propanesulfonate)-graft-poly(N,N-dimethy-lacrylamide) (P(NaA-coNaAMPS)-g-PDMAMx) was varied from 0 to 75%. The presence of big particle formation was indicated by the significant turbidity in the absence of stabilizing neutral PDMAM, whereas PDMAM was able to stabilize the system and minimize viscosity.[1053] The stability and production of C3Ms can be significantly impacted by the existence of a single hydrophobic endgroup. By adding phenyl, naphthyl, and pyrenyl terminal functionalities, the endgroups of PEG-P(Asp)-X were changed. These functionalities were then found in the center of the C3Ms loaded with lysozyme. The endgroup’s higher hydrophobicity was correlated with the size of the resultant C3Ms. Hydrophobic endgroups increased stability in the presence of NaCl and resulted in a greater aggregation number and more lysozyme in the core. Large aromatic group additions had even wider ramifications because light-scattering experiments indicated non-spherical particle formation. It was suggested that the stretching of the P(Asp) in the C3Ms was the reason behind the sphere-rod transition.[1054] Block length discussions cannot be limited to the charged block since the shell-forming block is also essential to the achievement of colloidal stability.[30] While very lengthy non-charged polymers might not cause any C3Ms production, too short non-charged polymers can cause nanoparticles to precipitate. Thus, longer, oppositely charged polymers in the core may contribute to the C3Ms’ stability.
When three block copolymers with comparable repeating units but distinct ionic block monomer structures were compared, it became clear that the pKa value-among other things besides polymer length-also matters when it comes to the immediate surrounds of the charged functionality. Trypsin was used to complex the three block copolymers PEG-block-poly(glutamic acid) (PEG-PGA), PEG-block-poly(α,β-aspartic acid) (PEG-P(Asp)), and PEG-block-poly(methacrylic acid) (PEG-PMA). It was discovered that trypsin activity was influenced by the solution’s pH. Since the imidazolium in the binding pocket-which is essential to amidase activity-is protonated at pH values below 6, free trypsin only exhibits minimal activity in these conditions. Negatively charged polymers have the ability to act as localized buffers, which can lead to increased activity at low pH levels. PEG-PMA and other high pKa polymers are more likely to protonate, making it difficult for them to stabilize the enzyme imidazolium and reducing the stabilizing effect.[1055] In other systems, the difference in protonation is also noticeable. PEG-PAsp(DET) has two pKa values, 9.9 and 6.1, which can have a significant buffering effect. In contrast, PEG-P(Lys) has a pKa value of 9.4. Similar in size, the C3Ms generated from the two polymers using Cu/Zn superoxide dismutase both caused a ∼50% reduction in enzymatic activity. PEG-PAsp(DET) has a benefit in this instance because of its reduced cytotoxicity, which makes it a desirable property for drug administration.[1056] The activity of the enzyme can be affected by structural changes in cationic polymers that revolve around the positive charge. PEG-PAMA, a cationic polymer, often reduced the α-amylase’s capacity to interact with the substrate p-nitrophenyl-α-D-maltoside (Figure 61). The catalytic process was restarted by adding salt and disassembling the C3Ms at the same time. The polymer’s polarity, however, affected how well it assembled and disassembled; the least polar polymer-which included a phenyl group-exhibited the strongest binding among the other polymers.[1057]
Because C3M formation is dependent on electrostatic interactions, the degree of ionization-which is determined by the pH of the surrounding environment and the pKa of the charged functionality-determines the stability of C3M. Polymers containing pendant sulfonate and quaternary amino groups are examples of permanently charged polymers that can be used to reduce the binding process’s sensitivity to the environment.[1058] As of right now, there doesn't seem to be much interest in trying to trap proteins using this method. Rather, the focus has been on developing systems that react rapidly to pH variations. The degree of polymer-protein binding can be somewhat tuned by varying the polymer’s pKa value. It does not, however, have a switch that, in response to slight pH changes, switches strong protein binding to rapid protein release. As Lee et al. showed, this can be accomplished by building a charge conversion system.[1059] Here, the charged species is supposed to be concealed by a labile functional group that carries the opposing charge. This group cleaves in mildly acidic situations, freeing the functional group underlying that is oppositely charged. The earliest application of this idea was seen in lysozyme, which was altered with citraconic anhydride to produce a negatively charged protein that, upon the breakdown of the labile amide group, could return to its original positively charged state.[1060] Numerous researchers then used this theory to stimulate the release of drugs. PEG-b-P(Lys) was changed by Chen et al. using four distinct anhydrides: dimethylmaleic anhydride (DMMA), succinic anhydride (SA), cis-cyclohexene-1,2-dicarboxylic anhydride (CDA), and cis-aconitic anhydride (CA).[1061] C3Ms containing positively charged doxorubicin (DOX) were subsequently formed by utilizing the resulting negatively charged block copolymers with pH-sensitive amide linkages. It was discovered that polymers containing CA and DMMA were more cytotoxic to HepG2 cells than DOX in its free form, but C3Ms with stable amide connections formed by SA and CDA were comparatively less lethal. The delivery of proteins was also considered in this context. PEG-b-poly[(N/-citraconyl-2-aminoethyl)aspartamide] (PEG-b-pAsp(EDA-CA)) was synthesized by reacting citraconic anhydride with the principal amine on PEG-b-poly[(2-aminoethyl)aspartamide] to produce C3Ms.[1059] An unresponsive SA system was set up as a control. At endosomal pH, the lysozyme carrying C3Ms underwent charge conversion, which resulted in the protein being repelled out (Figure 62).
10.2. Mechanism of Micelle Assembly and Disassembly
As mentioned earlier, micelles composed of micellar cores of ionic complexes and coronas of neutral polymers are known as polyelectrolyte complexes (PEC)/nanoscale self-assemblies. While positively charged polymers can condense large nucleic acids into small nanostructures and neutralize the negatively charged moieties on the nucleic acid chains, protecting them from potential enzymatic degradation and promoting successful transfection into various cell types, polyelectrolyte complex micelles perform well in gene and protein delivery.[271,1062,1063,1064] Because they are noncovalent assemblies, when they react to ionic or chemical gradients, they can dissociate and release the payload. The fundamental mechanism that controls the kinetics of C3M dissociation is still not well understood, despite its relevance. Over the past 20 years, the great bulk of research has concentrated on the static characteristics of C3M, including as their response to stimuli, their ability to comicellize with different biological macromolecules, and their potential uses in medicine.[27] In contrast, relatively little is known about the dynamic characteristics of C3M. It is equally important to comprehend C3M breakdown as it is to comprehend their production. An efficient release mechanism, for example, is required for therapeutic delivery. To induce such a change, pH, for instance, may be utilized as a trigger,[143] albeit the precise mechanism is still unknown. By sheltering charges, the addition of salt reduces polyelectrolyte interactions and may function as a trigger for C3M breakdown.[90] Similar to bulk PECs, adding salt to C3M first causes swelling due to water absorption and a weakening of the polyelectrolyte interaction.[1065,1066] Accordingly, Wu et al. puts forward that the core fragments into two charge-neutral micelle-like entities with overlaid corona segments, which subsequently split into smaller micelles.[90] The disassembly kinetics of PEO-b-PVBTMA/PAA C3Ms with stopped-flow light scattering were investigated by Wu et al. at various salt and temperature levels.[90] The fitting results are displayed in Table 6 at (i) 20, 37, and 57 0C, with a constant 500 mM NaCl salt jump, and at (ii) 300, 400, 500, and 600 mM NaCl, with a continuous 20 0C temperature jump. When the temperature was raised, τ dropped from 61.5 to 39.2 minutes, which accelerated the dissociation process. If β is set to 2, this implies second-order kinetics linked to a fission/fusion mechanism of micelle separation and fragmentation. Similarly, more salt led to faster relaxation kinetics by altering the salt-jump concentration and letting both τ and β to adjust, but disassembly took time following the salt-jump. Furthermore, the β shifted from around 1 to 2, indicating that the kinetics of C3M disassembly could not be fully explained by either the fission-fusion mechanism or single-chain expulsion/insertion. All things considered, this method gives researchers a quantitative approach to evaluate how resilient C3M s are to shifting solution conditions.
Additionally, C3Ms have been shown to be extremely dynamic structures, capable of ongoing dimer insertion/expulsion and fusion/fission events.[89] According to recent findings, the PEC core’s Coulombic forces in diluted systems do not extend past the micellar corona, and corona segments typically do not overlap with micelles that contact.[1067] Based on ζ-potential measurements of C3Ms solutions, it appears that the micelles collectively tend to enforce charge neutrality in all situations.[1067] In bulk PEC solutions, there is also a tendency to preserve charge neutrality.[1068] Nevertheless, no particular study comparing the performance of liquid-core versus solid-core micelles has been carried out. This includes examining the impact of the PEC phase and the interfacial tension with water on core fragmentation and/or expulsion/insertion events. Although there haven’t been many reports of direct investigations into the kinetics of dissociation (or creation) in C3Ms to date, work on micelles generated by nonionic block copolymers and low weight surfactants has advanced, and this work may offer some preliminary insights. Aniansson and Wall postulated two potential mechanisms in the 1970s for the kinetics of dissociation in surfactant micelles: (1) a collective micelle fission and fusion mechanism, and (2) a stepwise single chain ejection and insertion method.[1069] Subsequent light scattering experiments amply supported the idea that two distinct relaxation phases can characterize the kinetics of surfactant micelle dissociation-formation equilibrium. Halperin and Alexander used that information to create a fundamental theory for amphiphilic block copolymer (ABC) micelles.[1070] They asserted that the large free energy penalty micelle fission (or fusion) experiences as a result of the coronal interaction, which scales as
where P1 and P are the aggregation numbers of the fissionable aggregate and the starting micelle, respectively, and NB is the length of the core-forming block. It is evident that when P1 = 1, the fission activation energy minimizes, favoring the single chain expulsion mechanism. Notably, this result holds only for systems that experience negligible deviations from states of equilibrium. Some experiment results indicated that the phenomenon is not effectively explained by a single chain expulsion/insertion. For instance, Esselink et al. studied the evolution of mixed ABC micelles made up of two polymers with the same composition but differing coronal block lengths, and they found that micelle fusion is the primary mechanism via which polymer chains are redistributed among micelles.[1071] Micelle fission is a slow process at dynamic equilibrium states, but it becomes important when micelles re-equilibrate after a significant disturbance, as in temperature-jump tests, according to Dormidontova's theoretical prediction.[96] Micelle fission requires to overcome the free energy increase corresponding to the separation of micellar cores, in contrast to unimer ejection where the entropic cost originates from the exposure of the solvophobic chains in selected solvents. Ufiss, the fission activation energy, is expressed as
where Peq, P1, and P2 stand for the corresponding aggregation numbers of postfission micelle 1 and 2 and prefission micelle in equilibrium. According to this equation, micelle fission or fusion takes precedence when a micelle system deviates significantly from its equilibrium condition. Recently, there have been attempts to directly witness micelle fission through experimentation. Burke et al., for instance, noted that micelle breakup of block copolymer occurred when micelles morphologically changed from spherical micelles to rod-like aggregates.[420] According to Rharbi, micelle fission occurred in PEO-PPO-PEO micelles even at equilibrium, albeit 106 times more slowly than chain insertion and expulsion.[1072] Conversely, there is a dearth of experimental research on the kinetics of PEC micelle dissociation. C3Ms are frequently thought of in terms of ABC micelles, despite the clear differences in the forces that drive self-assembly. Regarding static micellar structures, which have received the most of attention thus far, this comparison might make sense. A recent paper showed that C3Ms in diluted fluids interact similarly to their uncharged counterparts, indicating that long-range electrostatic interactions do not significantly alter micelle-micelle correlation.[1067] On the other hand, differences appear regarding kinetic transitions between nonequilibrium states. First, C3Ms are typically a multicomponent system in which the interplay between positively charged blocks, negatively charged blocks, one or more coronal blocks, counterions, and solvent (typically water) complicates micellar dynamics. In contrast, ABC micelles can frequently be a single component system that contains only a single type of polymer with core-forming solvophobic block. Second, the core-forming chains of C3Ms are hydrophilic and maintain their complex state through ionic interactions, in contrast to ABC micelles. This indicates that no unfavorable chains are exposed when a chain is expelled from micelle cores; nonetheless, the Coulomb attraction between charged moieties may need to be overcome, depending on the ionic environment. The kinetics could be slowed down by the electrostatic connections breaking. Third, the cores of C3Ms can be fluidic and contain a significant quantity of water (around 30-90%), which is dependent on external parameters including system temperature and salt ion concentration. In contrast, the cores of ABC micelles are frequently solid or glassy. Studies on the mechanism of C3Ms production or dissociation are complicated by the combination of all these characteristics. Zhang et al. examined the kinetics of salt-induced disintegration of polyelectrolyte complex micelles made of poly(ethylene oxide)-b-poly(quaternized dimethyl amino methacrylate) and poly(ethlyene oxide)-b-poly(sodium styrenesulfonate) (PEO-b-PSS) (PEO-b- PQDMA).[1073] A double-exponential function is utilized to fit the relaxation curves. The slow relaxation is ascribed to a micelle fusion-fission mechanism, whereas the fast relaxation process is ascribed to the initial complex creation. When two strongly charged polymers were studied in the past for their formation and dissociation behaviors, the authors discovered that pairings of strong polyelectrolytes generated kinetically confined transient aggregates instead of spherical micelles. There were two steps involved in the breakdown of these linked aggregates when salt was added.[92] Using well-defined polyelectrolyte complex-based micelles, their goal in this research was to advance the understanding of the mechanism behind dissociation kinetics. Through the integration of the understanding of micelle scaling laws and polyelectrolyte complexation, they were able to: (1) explore the kinetic pathway of C3Ms dissociation during a salinity jump through experimental means; (2) create a theoretical model to account for this process; and (3) obtain a quantitative expression of fission relaxation kinetics in relation to temperature, salt concentration, and polyelectrolyte molecular weights. Poly(ethylene oxide)-block-poly(vinyl benzyl trimethylammonium chloride) (PEO225-b-PVBTMA100), a positively charged diblock polyelectrolyte, and poly(acrylic acid sodium) (PAA158), a negatively charged homopolymer, make up the experimental setup we use here.[92,1074] The numbers of repeat units are indicated by the subscripts. While the PAA block has been thoroughly investigated as a stand-in for weakly charged biomacromolecules, the PVBTMA block has been shown to be an efficient cationic vector for DNA encapsulation and delivery.[404,1075,1076] A mixture of methods, such as cryogenic transmission electron microscopy (cryo-TEM), small-angle X-ray scattering (SAXS), and dynamic light scattering (DLS), were used to evaluate the physical properties of the micelles in equilibrium.[90] Using time-resolved static light scattering, the evolution of PEC micelles in response to an abrupt salinity ascendance was studied in relation to salt. In order to explain the dissociation rate’s reliance on temperature, salt concentration, and ionic chain length, the authors created a mathematical model that may be further investigated and roughly matched their findings. To summarize, the mechanism of dissociation kinetics in complex micelles of polyelectrolyte is illustrated by a mix of theoretical modeling and light scattering experiments. Theories on polyelectrolyte complexes, kinetic theories for amphiphilic block copolymers, and the scaling law of PEC micelles form the foundation of the theoretical framework for PEC micelle dissociation. It is known that there are three stages involved in salt-induced micelle deconstruction. First, the addition of salt causes micelles to swell right away as the electrostatic contact weakens. The micelle cores tend to break into two charge-balanced aggregates during the second stage of the micelle chain rearrangement. As we see in the time-dependent light scattering tests, theoretical predictions indicate that splitting into two equal-sized aggregates is the most effective strategy to attain new equilibrium states, which results in a sharp decline of the scattering intensity. Third, the developing aggregates split apart. Furthermore, the authors derived an analytical equation for fission relaxation rates, taking into consideration the recent progress in understanding the link between interfacial tension and salt concentration/temperature in polyelectrolyte complexes. There is good agreement between the predictions and their experimental findings. The authors also talked about the scaling law that can be further investigated between the ionic block and the relaxation rate in the crew-cut and starlike scenarios. It can be expected that these results will stimulate more research on the dynamics of polyelectrolyte-based self-assemblies and serve as a great foundation for our understanding of micelle dissociation. Much less is known about the (dis)assembly pathways, structural rearrangements within the (transient) complexes, chain exchange between micelles, and morphological transitions than about the steady-state structure and characteristics of C3Ms, which have been the subject of extensive research. The few studies that concentrate on these processes provide an insight into the complexities involved and highlight the unrealized potential of using kinetic handles to construct structures that are not reachable by other means. As was previously indicated, a number of variables that have been researched for almost 20 years affect the development and morphology of C3Ms.[27,113] The static characteristics of C3Ms in relation to comicellization with biological molecules including proteins and nucleic acids,[113,825,1077,1078] stimuli responsiveness,[80,421,1079] and application in biological systems for therapeutic delivery[157,1077] are the main focus of our work. But research on the dynamics that lead to C3Ms production and ultimate disintegration due to altered solution conditions is relatively new. It seems sense to start by trying to apply notions from the much better understood systems of neutral micelles. The two most frequently suggested processes for the creation of neutral micelles are micelle fusion/fission and single-chain insertion/expulsion.[96,1080] Further investigation into the formation kinetics is necessary due to scenarios specific to charged polymers, such as the release of counterions following complexation, even if hydrophilicity and hydrophobicity can similarly influence the formation of C3Ms. Ion pairing between the oppositely charged polyelectrolytes is how micelle production begins.[88,1081] The process that comes after this is debatable, though. While it is widely accepted that early electrostatic interaction forms clusters of polymers, the size and charge distribution of these clusters are yet unknown. An unhomogenized complex core appears to be the cause of one set of results that suggests the production of bigger aggregates that might not be charge neutral; nevertheless, over time, chain redistribution causes a decrease in cluster size and the formation of charge-neutral micelles (Figure 63).[88]
According to a different study by Wu et al., merging clusters-referred to as cluster-micelle insertion-form smaller, potentially charge-neutral clusters that enlarge with time.[1081] Due to observational limitations, Wu et al. were unable to witness kinetics during the first 100 ms; nevertheless, Amann et al. propose that the initial cluster formation takes place within 2.6 ms of mixing. This almost instantaneous mechanism is believed to occur without enthalpic barriers and is preferred by electrostatic interactions.[88] The difference between the two ideas most likely results from the polyanion that was selected for each situation. Wu et al. utilize poly (acrylic acid) (pAA), while Amann et al. use poly (styrene sulfonate) (pSS). It has been demonstrated that pSS and poly (vinyl benzyl trimethylammonium chloride) combine to produce kinetically locked solid precipitate complexes that are initially bigger but eventually redistribute into smaller structures when salt is added.[92] Longer cationic blocks exhibit noticeably slower kinetics as well, according to Amann et al.,[88] who also mention the creation of a frozen nonequilibrium state. These findings imply that the formation and maybe even dissociation procedures of solid-core and liquid-core micelles, which are distinguished by the characteristics of the PEC core, may be different. Furthermore, both investigations were able to show that longer relaxation periods were associated with longer neutral and poly-cationic block block lengths, and they did not report any discernible changes in the formation kinetics in response to variations in solution polymer concentration. Note that simulation work by Bos et al. recently suggested the formation of neutral clusters but confirmed rapid ion pairing.[89] These investigations have all focused on C3Ms created with a BCP and an HP, but since ion pairing and cluster formation can happen in the same way in systems created with two BCPs, it is expected that the method of creation would not change. The relaxing process would be slowed by a BCP’s reduced rearrangement freedom as opposed to an HP’s. Although the kinetics of production have been covered thus far, a number of factors contribute to the final forms at thermodynamic equilibrium. Similar to neutral block copolymer self-assembly, the morphology of the produced structures is mostly determined by the ratio of the lengths of the charged and neutral blocks.[1082] It has been demonstrated by earlier experimental work on C3Ms that nanoscale stability was not achievable with shorter neutral block lengths than charged blocks.[30] Nonetheless, at lower polymer and salt concentrations, micelle production with shorter neutral blocks is feasible, according to simulation work using revised coacervation theory models.[1083] There is macrophase separation and the neutral blocks are absorbed into the bulk phase for charged to neutral block length ratios of five or higher. Nonetheless, there is microphase separation for the investigated charged to neutral block length ratios of 2 and 3. According to the phase diagram, lamellae form at even greater polymer fractions, while micelles form at lower polymer fractions and a hexagonal phase forms as the polymer fraction increases.[1083] These structures bear resemblance to mixes of triblock polyelectrolytes with opposite charges that have been discovered in experiments.[1084] It is also feasible to have different discrete nanoparticle structures. For instance, when the neutral block polymer content is less than 10%, polyion complex vesicles (PICsomes) develop, generating a bilayered PEC shell with an empty cavity.[545,1085] According to Kim et al.,[545] the bilayer is a PEC membrane with a pEG-shell layer on both the interior and outside of the vesicle. The membrane’s thickness is between 11 and 17 nm. Additionally, Lui et al. showed that selectively complexed domains in a variety of nanostructures were produced by in-situ polymerization of charged monomers.[1086] With increasing degrees of polymerization, the resulting structures ranged from micelles to nanosheets to nanocages, depending on the molecular weight of the resulting polycation.[1086] A solvent-immiscible polymeric block that was conjugated to the polyanion produced its own clusters inside the structures. This technique shows that multicompartment nanostructures may be created, which may make it easier to give a wide variety of treatments. Micellization pathways have been the main focus of the majority of kinetic investigations on C3Ms. As mentioned earlier, there are two primary mechanisms that allow initially created transitory (metastable) complexes to reorganize into steady-state (micellar) super-structures: fusion-fission and expulsion-insertion (Figure 63). The term “fusion-fission” describes the formation of structures that are thermodynamically preferred as a result of “embryonic” complexes breaking and merging. As an alternative, steady-state aggregates can form as a result of several expulsion and insertion events involving the constituent building blocks, which can occur as ion pairs, tiny soluble complexes, or unimeric form. We still don’t know which mechanism-fusion-fission or expulsion-insertion-dominates and under what circumstances. A transitory liquid-liquid phase separation is seen in a number of processes, such as protein crystallization,[1087] mineralization,[1088] and polymersome formation,[1089] before a new (condensed) phase emerges. Such transitory structures were originally observed during the production of C3M by Cohen Stuart et al. in a groundbreaking light scattering investigation. They show that brief, macroscopic phase separation occurs before micellization, which happens seconds after poly(N,N-dimethylamino ethyl methacrylate)35-co-poly(glyceryl methacrylate)105 (PDMAEMA-co-PGMA) and poly(acrylic acid)158 (PAA) are mixed.[145] Lund and colleagues have used cutting-edge methods to study the early phases of S-C3M production. They combined time-resolved small-angle X-ray scattering (trSAXS) studies with an amazing time-resolution of 5 ms with stopped-flow mixing with a 2.6 ms dead time.[88] Additionally, these trSAXS experiments revealed transient structures: large aggregates with an inhomogeneous charge distribution formed immediately following the mixing of poly(sodium 4-styrenesulfonate)19 (PSS) and poly(vinylbenzyl trimethylammonium chloride)y-block-poly(ethylene oxide)46 (PVBTA-b-PEO; y = 6, 8, 12, 19). These aggregates then rearranged into smaller C3Ms over a period of seconds (Figure 63). The observed transient complexes were bigger and reorganized more slowly with increasing PVBTA block length. The equilibration period increased by more than an order of magnitude, from around 100 ms to 30 s, when the PVBTA block length was doubled from 6 to 12 monomers. The observed nonequilibrium state became kinetically locked when the cationic block length was raised further from 12 to 19 (corresponding to the oppositely charged PSS block), as no relaxation took place over time. Fitting the scattering profile revealed pearl-necklace-like clusters in place of single micelles, and eventually the previously turbid solution phase-separated macroscopically. The authors connect multivalency to this impact. Since more noncovalent bonds need to be broken in order for a dbp to be expelled from the complex, dbps with longer ionic blocks have a lower dissociation probability than dbps with shorter ones. Consequently, as the degree of ionic block polymerization increases, the ionic-neutral dbp exchange rate falls off noticeably. The authors noticed two processes with distinct rate constants when they investigated the relaxation kinetics in more detail (Figure 63). Chain rearrangements in the large aggregates cause the quick process (k1 ∼ 10 s−1), which produces smaller clusters with a more uniform charge distribution. There is a connection between the rearrangement into equilibrium micelles and the sluggish process (k2 ∼ 0.3 s-1). Remarkably, this second, gradual relaxing process did not depend on concentration, indicating that polymer chain exchange, rather than the fusion and fission of (pre)micelles, is the primary mechanism driving equilibration. Time-resolved SAXS investigations on longer dbps (PVBTA53 -b-PEO112 and PVBTA100 -b-PEO225) mixed with the weak polyelectrolyte PAA158 were carried out by Tirrell and colleagues.[1081] The trSAXS measurements revealed charge-neutral clusters formed within the 100 ms of experimental dead time, which reassembled into small equilibrium micelles (RH = 10 nm) within a matter of seconds, in good accord with their molecular dynamics predictions. Additionally, the kinetic profiles indicated the low likelihood of micellar fusion-fission events. What is in contrast to Cohen Stuart and Lund’s previous findings, though, is the absence of bigger charged aggregates in the initial milliseconds following mixing. The Tirrell team explains this difference by pointing out that the polyanion is different (strong vs. weak), but other interactions might also be involved. For instance, in S-C3Ms made of PVBTA-b-PEO/PSS, π-π stacking interactions between the phenyl rings of the core-forming blocks may occur; in S-C3Ms made of PVBTA-b-PEO and PAA, similar interactions are not present. In comparison to C3Ms containing PAA, PSS-bearing C3Ms may have cohesive connections inside their micellar core that are further enhanced by their more hydrophobic nature. Zhang et al. used time-resolved static light scattering (trSLS) studies in conjunction with stopped-flow mixing to study the effects of salt on the relaxation from transient to stable structures because the ionic strength affects the balance of driving forces for the production of C3Ms.[1073] The two ionic-neutral copolymers with pH-independent charge density that made up the D-C3Ms were PSS47 and quaternized poly-(N,N-dimethylaminoethyl methacrylate)48 (PQDMAEMA), both of which were blockwise attached to PEO113. Around charge stoichiometry, the largest C3Ms with the lowest dispersity were discovered. A rise in scattering intensity was seen in the first few milliseconds following mixing at 0.47 < f+ < 0.52; this increase peaked in 0.4 s. This finding was explained as the tiny complexes forming and then equilibrating into stable C3Ms in the first second following mixing. Since it took up to twice as long to attain a plateau in scattering intensity, the addition of salt-up to 0.2 M NaCl-slowed the relaxation. Greater amounts of salt screened the electrostatic interactions to the point that fewer, looser complexes-those with more water and ions-formed. The rate constant of the first relaxation from tiny complexes to bigger micelles, which is interesting, rose linearly with rising dbp concentration at low ionic strength, indicating a considerable contribution from micellar fusion-fission processes. They draw the conclusion that, in their circumstances, expulsion-insertion events have to be unfavorable. These results are in opposition to those of Lund and colleagues, who observed the temporary creation of larger aggregates before PEO-b-PVBTA/PSS micellized, not smaller ones. This could be the result of other interactions that are missing from C3Ms made up of PQDMAEMA and PSS, like π−π stacking for PVBTA and PSS. Furthermore, Lund and associates looked at S-C3Ms (PVBTA19-b-PEO46 and PSS19), whereas Zhang et al. explored D-C3Ms (PQDMAEMA48-b-PEO113 and PSS47-b-PEO113). Next, Zhang et al.[1073] looked into the kinetics of salt-induced dissociation. The C3Ms disassemble when the salt concentration rises above the cs,cr. Remarkably, they found via kinetics analysis that dissociation involved two opposing processes: a concentration-independent, first-order process and a second-order, concentration-dependent process. On the other hand, micellization happened by a solitary, second-order mechanism, meaning that insertion-expulsion events were not advantageous. The dissociation process screens charges from the additional salt, which lowers the energy barrier for polyion chains to fully unbind and for unimers to be ejected, which explains the discrepancy. Therefore, both micellar fission (second-order) and chain insertion-expulsion (first-order) events comprise the dissociation process. When other parameters remain constant, it becomes clearer how the relaxation paths for S-C3Ms and D-C3Ms differ from one another. Shah and Leon discovered very slow relaxation in a fascinating investigation on remarkable changes in morphology and equilibration periods between (otherwise similar) D-C3Ms and S-C3Ms.[408] When poly(L-lysine)50 (PLL) complexed with anionic-neutral poly(acrylic acid)49-block-poly(N-isopropylacrylamide)70 dbp (PAA49-b-PNI-PAM70), wormlike S-C3Ms were created; in contrast, spherical D-C3Ms were generated when PLL50-b-PEO113 was used in place of PLL50. Furthermore, the authors note that because of their different micellar morphologies, the D-C3Ms relaxed in 6 min whereas the S-C3Ms required nearly 20 h to attain a steady-state. To determine if coassembled micelles are equilibrium structures or not, it is common practice to look at whether the steady-state structure and characteristics remain the same independent of the conditions during the assembly process and processing. In order to do this, Tirrell and colleagues examined the dimensions of D-C3Ms made using PSS100-b-PEO227 and PVBTA100-b-PEO227 using two different methods.[92] While the salt concentration is raised postmixing in the salt annealing method, polymer solutions with the desired salt concentration are mixed in their direct dissolving method. Comparing this method to other research, when salt is first supplied and then eliminated using techniques like dialysis, shows how uncommon it is.[148,149] Remarkably, the restricted distribution of tiny C3Ms (RH = 25 nm) produced by the direct dissolution widened over several months. On the other hand, following salt annealing, bigger complexes (RH = 100 nm) were first seen, which eventually equilibrated into smaller C3Ms (RH = 25–30 nm). These findings suggest that when the oppositely charged polymers are mixed without salt, there are strong attraction interactions that lead to the formation of kinetically trapped complexes where some of the PEO blocks may be buried. These complexes can be rearranged into smaller micelles with a coacervate core and PEO corona-a thermodynamically preferred state-by adding salt afterwards. Their data provide a useful illustration of how preparatory conditions affect the creation of transitory structures. While kinetically confined structures can be a challenge, they can also offer access to nanostructures that would not otherwise be possible. Thus, it is crucial to examine the coassembly process in considerable spatiotemporal depth. Tirrell and colleagues used theoretical modeling and complementing trSLS experiments to study the mechanisms underlying salt-induced C3M dissociation.[90] Within the measurement dead time of 1 s, the micellar radius rose as the salt concentration increased from no added salt to 0.5 M NaCl. Subsequently, there is a marked decrease in both the intensity and the radius, and at last, the intensity reaches a plateau. Their theoretical model separates the dissociation process into three steps, which are derived from complex coacervate scaling rules. First, the model assumes that a rise in water and ion absorption is the cause of the observed micellar radius increase. The micellar cores begin to fragment in a second stage, with fission being preferred over unimer ejection. This result is consistent with the abrupt drop in intensity, which is to be expected when micelles split in half as opposed to gradually losing trace amounts of polymer by ejection. The complete separation of the coronas of these freshly created complexes, which happens with a low relaxation time and a rise in entropy, is the final stage as stated in the model.
11. Kinetics of Micellization and Kinetics of Exchange
11.1. Kinetics of Micellization
There is still a dearth of research on the kinetics of association (and dissociation) in aqueous mixes of oppositely charged polymers. Primary complexes undergo structural rearrangements, addition reactions, and exchange reactions. Reactions that result in the redistribution of salt bonds are referred to as exchange reactions. Examples of these reactions include the substitution of one copolymer for another of the same type. When a copolymer is added to an already-existing oppositely charged complex, for example, the number of inter-polymeric salt bonds changes. This can lead to an entropic gain because a single macroion replaces many small counterions, albeit partially. The local complex geometry and the number of interpolymeric salt bonds can both be changed by structural rearrangements. When a diblock copolymer is mixed with a homopolymer that has the opposite charge, metastable or unstable complexes are first formed. These complexes then reorganize into micellar particles, most likely within a few milliseconds to a few hours.[71,93,145,271,1090] For more hydrophilic monomers (PAA versus PSS), in the presence of salt,[84,145,271,1091] and mixing of stock solutions of matched pH,[93] the transition to a stable state occurs more quickly. These mixing fractions correspond to shortage of copolymer and excess charge (i.e., non-stoichiometric conditions). If the produced particles are in a stable or metastable condition, a pH cycle may reveal this.[93] It is difficult to measure aggregation kinetics directly because most studies use probe molecules, like fluorescent markers, which can affect the observed kinetics. This is because labelled polymers have a higher binding affinity because the hydrophobic label makes them more hydrophobic. The kinetics of micellization, as generally observed in the micellization of amphiphilic polymers, involve two processes: “insertion and expulsion” of single chains and “merging and splitting” of micelles, with the latter process purportedly being faster, according to Holappa et al.’s conclusion from their experiments on C3Ms of PEO-b- PMAA + PMOTAC.[86] Comprehending the mechanisms underlying micellization, molecular exchange, and evolution is essential for managing cargo exposure in nanocarrier applications.[60,103,419] Greater control over the physical self-assembly process, nanocarrier stability over time, and encapsulation/release dynamics may be possible with a deeper comprehension of the PCM equilibration process. Using mostly scattering methods, we center our comments in this part on a number of recent advances in PCM dynamics. For a complete solution and in an exact time-resolved way, small-angle scattering is an effective instrument for simultaneously collecting numerous orders of magnitude of size information. A lab has recently published detailed methods for small-angle X-ray scattering (SAXS) to help with the planning of experiments and the analysis of SAXS data.[1092,1093] The model systems employed in these research are displayed in Figure 64A. PEO-b-PVBTMA, PSS, poly(ethylene oxide)-block-poly(sodium 4-styrenesulfonate) (PEO-b-PSS), and sodium poly(acrylate) (PAA) are examples of polyelectrolytes. The controlled production of these polyelectrolytes in water has been described experimentally before.[1074,1094] This allows for the preparation of exact neutral and charged block lengths with low dispersity in the molar mass distribution. The assemblies that develop can resemble spherical, core/shell PCMs, or polydisperse colloidal aggregates, depending on the block lengths and pairing of the PEO-b- PVBTMA polycation with PAA, PEO-b-PSS, or PSS polyanion. The specifics of both kinds of nanostructures, as well as some open-ended problems these findings bring up for the field of physical chemistry, will be covered in length below.
Time-resolved small-angle X-ray scattering (TR-SAXS) with millisecond temporal resolution can be used to observe in situ the ultrafast synthesis of PCMs by adhering to established stopped-flow techniques from amphiphilic block copolymer literature (Figure 64B). In order to perform scattering measurements, solutions of oppositely charged polymers are injected into individual syringes, pumped into a turbulent mixer, then dispensed into a capillary cell without additional flow. The behavior of ionic nanomaterials, such as the complex coacervate coalescence of poly(allylamine hydrochloride) and PAA in response to the addition of NaCl salt, has been better understood physically thanks to this approach.[1095] That being said, the first complexation of block polyelectrolytes has only recently been documented. The kinetic process of micellization appears to be significantly influenced by the chemical and electrostatic character of the polyelectrolyte pairing, highlighting the significance of careful polymer selection in the creation of PCM nanocarriers. Figure 65 displays two separate situations that demonstrate very different paths. Wu and colleagues examined the spatiotemporal formation kinetics of PEO-b-PVBTMA using PAA in the first instance.[1081] At the Stanford Synchrotron Radiation Lighthouse (SSRL, SLAC National Accelerator Laboratory),[1096] they used a stopped-flow apparatus with high-throughput data collection capabilities to directly observe the SAXS profiles and assembly kinetics of PEO-b-PVBTMA/PAA PCMs via TR-SAXS from 100 ms to 5 s. These profiles showed spherical particles (∼q0 power law dependence of intensity for q < 0.01 Å-1) that grew in size over time (Figure 65A). The apparent Guinier radius of gyration (Rg), which was used to assess the structural evolution of PCMs, revealed progressive micelle growth from Rg ∼ 10 to Rg ∼ 12 nm over 5 s due to the gradual insertion of either unimer chains or ion-paired clusters. In the second instance, Amann and associates looked at PCMs that included PSS and PEO-b-PVBTMA.[88] The formation of metastable aggregates for internal charge neutralization at 3 ms was observed by the researchers using an SFM-400 stopped-flow apparatus at the European Synchrotron Radiation Facility (ESRF). This was followed by a 30 s period of rearrangement and pinch-off into small micellar particles (Figure 65B). Rearranging unimer chains or ion-paired clusters becomes more unfavorable as block length increases, according to the relaxation processes that explained the equilibration data as a function of the degree of polymerization (N) of PVBTMA from N = 6 to N = 19. It has been demonstrated that the homopolymers PVBTMA + PAA create liquid-like coacervates, whereas PVBTMA + PSS form solid-like complexes, in an effort to explain the variations in kinetic paths.[1094] This finding prompts me to hypothesize that the formation kinetics may be significantly influenced by the chemical makeup of the polymer elements; nevertheless, more research is required to determine whether the complex cores’ makeup is similar to that of macroscopic complexes. Furthermore, the rate at which PCMs either fragment from larger colloidal structures upon complexation or expand incrementally may also be influenced by the block length of the block polyelectrolytes. Wu et al. previously demonstrated that by fitting the SAXS data, PEO-b-PVBTMA and PEO-b-PSS at N ≈ 50 for the charged blocks create nonequilibrium complexes that are far from well-defined spheres.[92] Work in progress is to investigate this system that matches the neutral and charged block lengths to PEO-b-PVBTMA/PAA for a direct comparison with the findings of Amann et al. Together, these examples highlight just two of the several formation routes that might result in micellization driven by charge. Expanding TR-SAXS to investigate more PCM systems and adjusting variables such as polyelectrolyte choice, block lengths, and molecular architecture can assist in advancing PCM design in the direction of more effective and predictable cargo encapsulation.
11.2. Kinetics of Exchange
The last ten years have seen a significant increase in interest in the field of micelleization kinetics in block copolymer solutions, as seen by several recent articles.[45,1070,1071,1097,1098,1099,1100,1101,1102,1103,1104,1105,1106,1107,1108,1109] To gather detailed information about the kinetics of micellar formation or reequilibration, a variety of experimental techniques have been used, including the stopped-flow method,[1097] temperature-jump experiments (T-jump),[1103,1106,1107,1108] nonradiative energy transfer and fluorescence-quenching techniques,[1098,1101,1102] time-resolved light scattering,[1103,1106,1107,1108] transmission electron microscopy,[1071,1109] and sedimentation velocity methods for comicellization experiments.[1099,1100] Even with the vast amount of experimental data gathered, a cohesive picture of micellar evolution has not yet been provided. Thus far, theoretical models explaining micellization kinetics in surfactant systems have been used to examine experimental results of relaxation kinetics in block copolymer solutions.[1069,1080,1110,1111,1112] It is reasonable to anticipate that the micellization kinetics in these polymer systems will differ from those of block copolymer micelles due to the structural and equilibrium differences between surfactant micelles. The evolution of surfactant micelles is divided into two primary mechanisms. The unimer exchange mechanism states that micelle expansion is achieved by the sequential inclusion of unimers, as schematically depicted in Figure 66, as expressed by the following equation:
where NQ is the number density of micelles with aggregation number Q.
The process of unimer exchange is quite quick, and its related relaxation time is τunim.
the average relative concentration deviation from the equilibrium among the micelles is Co, the relative fraction of micelles (compared to free unimers of concentration cun) is X (c-cun/cun), the average aggregation number of the micelles is Q, and the width of the micellar size distribution is σ. [1069,1080,1110] For modest departures from the equilibrium state, Aniansson and Wall[1069,1080,1110] presented this model of unimer exchange between surfactant micelles. In consecutive unimer association, the model suggests that the amount of micelles remains constant. However, surfactant micelles may periodically split into unimers, resulting in an increased quantity of free unimers and therefore promoting subsequent association. The second mechanism of surfactant association states that micellar fusion/fission, which is schematically depicted in Figure 67, is the evolutionary pathway followed by the following equation:
Micellar fusion/fission is thought to be the cause of the frequently seen slowly relaxing component.
where β is the mean dissociation rate constant measured. Similar to the unimer exchange model, the micellar fusion/fission[1111,1112] model operates under the supposition of minor departures from the equilibrium state, which is the state in which the processes of association and dissociation are in balance. Although surfactant and block copolymer micelles appear to be identical on the surface, the latter are significantly different from the former due to the presence of lengthy entangled polymer blocks in the micellar core structure, which replaces the relatively short alkyl chain of a surfactant. Block copolymer chains hence require different expulsion rate constants than surfactants. Halperin and Alexander have carried out an analogous analysis of the unimer expulsion rate constant for block copolymer micelles.[1070] Their paper’s[1070] principal finding states that the only mechanism for the growth of block copolymer micelles is unimer exchange. But it’s important to remember the following: (i) Only minor departures from the equilibrium state are valid for the results obtained.[1070] (ii) Because the unimer must spread throughout the entire corona region-which is significantly bigger than the size of a blob surrounding the core, or Rcore, as was previously thought by Halperin and Alexander[1070] – the typical time for unimer expulsion is underestimated. Furthermore, the time required for the disentanglement of intractable blocks has also not been considered. (iii) No calculations have been made on the association/dissociation rates for micellar fusion/fission. As a result, it is difficult to draw firm conclusions regarding the unique function of unimer exchange.[1070] Several experimental findings[1071,1100,1101,1103,1105,1107] appear to be unaccounted for under a model that solely takes into account unimer exchange. Computer simulations supported the hypothesis that micelle fusion/fission might occur in block copolymer solutions.[45,1101,1104,1113]
Understanding the exchange dynamics of C3M encapsulators is crucial for designing effective ones. The pace at which cargo in the core is exposed to the environment and, consequently, the degree of protection provided by the encapsulation vehicle, are determined by the exchange dynamics between C3Ms. Furthermore, the preparation method of C3Ms can occasionally influence their structure, indicating that kinetic effects can control the C3M structure and, consequently, the encapsulation properties.[91,92,93,94] The significance of exchange dynamics for amphiphilic diblock copolymer micelles is well known, and their exchange dynamics has been extensively researched.[55,94,96,97,98,102,1114,1115,1116,1117,1118] One macromolecular species with a soluble and an insoluble block makes up these micelles. Based on the theoretical framework provided by Dormidontova, two distinct mechanisms are frequently recognized to characterize the interchange of these micelles.[96] Unimer exchange appears to be the first mechanism. This is the point at which a single polymer, or several polymers, separate and enter another micelle. Fission and then fusion constitute the second mechanism. A micelle can then unite with another micelle after splitting into two sections of equal considerable size. The corona structure is present in both created sections of the fission. A micelle corona cannot form during the expulsion process because there are only one or two soluble blocks in the expelled component. Since the corona differs, the rate-limiting steps for each mechanism vary, and as a result, so do their dependencies on the system parameters. Thus, the dominating mechanism or whether both occur can be altered by altering one of the system characteristics. Therefore, system factors such as core block length,[98,1115,1116,1117,1118] corona block length,[102,1115,1118] polymer concentration,[98,1115,1116] chain flexibility,[1118] and interfacial tension between the core and solvent might have a complex dependence on micelle exchange rates.[55,1115,1116] While the theory formulated for amphiphilic diblock copolymer micelle exchange offers a useful foundation for explaining C3M exchange, it falls short of providing a comprehensive explanation for C3M exchange. The interactions that propel the core formation in the two forms of micelles are different. Whereas the core creation of C3Ms is caused by electrostatic attraction between the oppositely charged polyelectrolytes, which permits the release of counterions, the core formation of amphiphilic diblock copolymer micelles is often driven by hydrophobic attraction. The extensive research that has already been done on polyelectrolyte complexes helps to explain some of the variation in interaction. For instance, it has been demonstrated that the dynamics inside the complex coacervate phase are dependent upon both the concentration of salt and the length of the polyelectrolyte.[1119] This can aid in explaining the C3M core’s relaxation. Furthermore, measurements have been made of the strength of an ionic connection and the interfacial tension of specific complex coacervates, which can be used to explain the release of polyelectrolytes from the C3M core. [1120,1121] However, new tuning factors are introduced that are not present for amphiphilic diblock copolymer micelles since the core formation is based on the attraction between two different block types rather than just one. The block lengths of the positive and negative polymer blocks can be changed separately from one another, as opposed to just changing the length of one core block. Additionally, the micelle characteristics can change depending on whether the corona block is attached to both core blocks or only one of them.[25] Therefore, to comprehend the exchange of C3Ms, it is not adequate to concentrate solely on amphiphilic diblock copolymer micelles. Regretfully, there hasn’t been much research done on the interchange of C3Ms, and the few that have been done on the topic[86,87] have interpreted their findings in an oblique manner. The scientists combined C3Ms labeled with an acceptor fluorophore and a donor fluorophore, measuring the micelle exchange rates by measuring the rise in FRET (Förster resonance energy transfer). In this method, they discovered that the exchange rate is fast for proteins containing C3Ms and that it depends on the polyelectrolyte length and charge stoichiometry.[86,87] Observed exchange rates, rather than firsthand observations, served as the primary basis for later interpretations of the exchange mechanisms. These investigations are restricted in providing mechanistic descriptions, but they do provide highly helpful insights into the temporal scales at which micelle interchange might occur. A previous report (see below) provided an understanding of the molecular mechanisms of exchange in C3Ms.[89] Their approach involves utilizing simulations of coarse-grained dynamics. In investigations on amphiphilic diblock copolymer micelle exchange, this kind of simulation has previously proven to be useful.[1115,1116,1117,1118] The creation of a single C3M, complicated coacervation,[1122,1123,1124] and the static characteristics of several C3Ms have all been studied using coarse-grained dynamics simulations.[1125,1126,1127] The initial micellization kinetics of numerous C3Ms were tracked using the coarse-grained simulations. By doing this, the authors were able to increase the kinetic stability of C3Ms and gain fresh mechanistic insights into their interaction. The kinetic stability information can be used to supplement the static C3M stability previously discussed,[106,261,401,404] which is often defined as the critical salt concentration at which micelles disintegrate. Specifically, the authors demonstrated that adjusting the polyelectrolyte length ratio and nonelectrostatic interactions can further enhance the kinetic stability of C3Ms. Since the rate and mechanism by which sequestered medicinal molecules are exposed to the surrounding environment will limit their efficacy, an understanding of chain exchange in diluted micelle solutions is essential to their development as effective delivery vehicles. Generally speaking, there are two main ways that chain exchange between equilibrium polymeric micelles happens: micelle fusion/fission and chain expulsion/insertion.[96,1080,1128] Although amphiphilic polymer assemblies have been used to study chain exchange mechanisms, relatively few experimental research have, as far as we know, looked at chain exchange for C3Ms. One practical method for maybe looking into the C3Ms’ underlying exchange process is fluorescent imaging. Fluorescently tagged proteins in C3Ms have been used by Nolles and others to study the dynamics of exchange and formation.[87] An other tactic is synchrotron scattering. It has been previously demonstrated how interparticle effects manifest as a structure factor in SAXS profiles for concentrated micelle solutions, thereby preserving C3M stability over time.[1067] However, these experiments lack molecular contrast, making it impossible to uncover the intricate details of unimer chain exchange or fusion/fission. Unfortunately, the usefulness and accessibility of scattering methods with suitable contrast, like SANS, are limited by the need for extended time scales and deuterated/hydrogenated systems. On the other hand, improvements in molecular dynamic simulations have provided fresh perspectives on possible exchange pathways where electrostatics interact with other rival noncovalent interactions. In line with the findings of Bakeev et al., who discovered a significant rise in the rate of polyion exchange in complexes of long polyanions (also known as “hosts”) and short polycations (also known as “guests”) with increasing ionic strength,[1091] the rate of exchange kinetics in C3Ms is shown to be significantly salt-dependent. The majority of research in this area focuses on DNA-containing C3Ms and the issue of how well DNA can be incorporated into them, specifically whether it is possible to stop DNA release caused by exchange interactions with anionic macroions that are present in physiological settings.[84,1129] According to a number of studies, DNA incorporation is (at least partially) reversible since PSS can (at least partially) replace DNA from DNA/PHPMA-g-PLys C3Ms[1130] and PAsp can replace DNA in DNA/PEO-b-PLys C3Ms.[36] Using a model diblock-homopolymer C3M system, Bos et al. conducted coarse-grain dynamics simulations and described physical properties determining C3M stability and the manner of chain exchange.[89] Nonelectrostatic interactions, such as hydrogen bonding or hydrophobicity, tended to work against the chain expulsion mechanism in these Langevin dynamics simulations because they produce an enthalpic cost that balances the entropic gain that propels the expulsion of small neutral complex clusters from C3Ms (Figure 68A). It is noteworthy that these effects were contingent upon the nonelectrostatic contacts (indicated by the Lennard-Jones potential εLJ) being intermolecular, and were altered if the interactions were limited to intramolecular interactions alone. Chain expulsion and fusion/fission events increased in the scenario where one polyelectrolyte showed strong nonelectrostatic interactions with itself but not with the oppositely charged polyelectrolyte, indicating that the C3Ms became less stable. As εLJ was increased, Figure 68B illustrates how macromolecular design characteristics like block length had different effects on the two pathways. The system’s charged blocks’ relative lengths had an impact on chain expulsion, which showed a noticeable rise for matching chain lengths compared to unmatched chain lengths. Conversely, it seemed that fusion/fission was dependent on the homopolymer’s overall length rather than the ratio of the block lengths. When combined, experiment and simulation make a compelling justification for giving serious thought to nonelectrostatic interactions between polyelectrolytes and polyelectrolyte length.
More recently, Bos, Timmerman, and Sprakel used Fischer resonance energy transfer (FRET) to show the exchange dynamics of C3Ms.[103] The model systems employed in this work were fluorescently labeled poly(3-sulfopropyl methacrylate) (PSPMA) and poly(ethylene oxide)-b- poly(trimethylammonioethyl methacrylate chloride) (PEO-b- PTMAEMA). An analytical model that connected variations in salt concentration, polymer length, and micelle concentration to the broad dispersion of the detected exchange rate was created. The model linked the FRET efficiency between fluorophores and the exchange rates of polyelectrolyte chains. As far as we are aware, this is the first experimental proof that C3M equilibration mostly happens via expulsion/insertion pathways rather than fusion/fission. The possible relocation of nucleic acids over time upon immersion in various biological environments is a significant practical consequence of dynamic chain exchange across micellar assemblies for C3Ms containing nucleic acids. Molecular engineering techniques to increase stability in nanocarriers are nontrivial due to the complex delivery paths involved in overcoming different biological obstacles. Simplified fundamental experiments, on the other hand, may be able to provide structure-property connections for C3M stability, providing insights that advance our objective. Can foreign polyelectrolytes that have a high tendency to associate with C3M hosts, for instance, produce mixed micelles? If yes, what characteristics are essential for this macromolecular exchange feature? As far as we are aware, only a small number of published publications have looked into these kinds of questions. At physiological salt conditions, Dautzenberg et al. performed polyanion exchange processes using C3Ms containing model oligophosphates and competing higher MW polymers (PSS and DNA).[1130] Harada and Kataoka conducted a second study that used polymer architecture to demonstrate how diblock polyelectrolytes replace homopolyelectrolytes in C3Ms generated with an oppositely charged diblock polyelectrolyte. This finding suggests that there is more association in (AB + AC) C3M systems than in (AB + C) systems.[1131] These examples shed light on how molecular recognition based on polyelectrolyte compatibility and dynamics can lead to C3M chain exchange. This is an area where developments in noninvasive characterization techniques, like liquid-phase transmission electron microscopy, small-angle neutron scattering, and fluorescence microscopy, are well-suited to make significant advancements in our knowledge of polyelectrolyte complex micellization and chain exchange.[1132,1133,1134] Equilibration of the nanostructures and their characteristics requires a significant amount of material exchange between (pre)micelles. We may not see a transition on the experimental time scale when the exchange rates of the building blocks are very low because systems may become kinetically locked and unable to equilibrate. Neutral complexes, ion pairs, and unimers are among the species that can be traded. Sensitive component encapsulation by C3Ms is influenced by the species involved in the exchange as well as the pace of exchange. For instance, in situations where the primary exchange mechanism is the expulsion-insertion of individual constituents or small neutral complexes, (therapeutic) proteins and polynucleotides encapsulated with-in S-C3Ms (i.e., being C3Ms comprising dbp complexed with proteins, DNA, or RNA with or without auxiliary hp) are momentarily exposed during material exchange between these S-C3Ms. The water-soluble, biomacromolecular payload enclosed in C3Ms would be more susceptible to nuclease or protease destruction in vivo as a result of this brief solvent exposure than hydrophobic cargo enclosed in amphiphilic systems. Conversely, in the event that fusion-fission processes predominate in exchange, shielding might still be adequate to provide the necessary cargo protection. Comprehending the relationship between the relative rates of expulsion-insertion and fusion-fission events and their building block qualities is crucial for the development of encapsulation agents in nanomedicine, among other applications. Factors like (changes in) salt concentration will affect the type and rate of exchange because the exchange depends on the relative contact strength. Through the use of simulations, it was determined that the displacement of charged macromolecules from complexes by competitive species-that is, polyelectrolyte chains that are similar in nature but longer than those in the complex-occurs more quickly for larger competitive species and at higher salt concentrations.[1135] These aspects need to be taken into account while selecting polymeric components. Nolles et al. synthesized spherical C3Ms with a radius of 35 nm at pH 9 using fluorescent proteins (FPs) with a net negative charge of around −10 and PM2VP128-b- PEO477 in order to observe the dynamic dynamics of protein-polymer C3Ms.[87] The process of C3M production and material exchange between micelles involved the affiliation and dissociation of small, almost neutral protein-polymer complexes, or SCPs, made up of one dbp and approximately 10 FPs. The authors combined C3M solutions with either donor or acceptor FPs in order to monitor the exchange of FPs between the micelles via the technique of Fischer resonance energy transfer, or FRET. A high FRET signal is seen when the two types of proteins are combined and contained in the micellar core; in the case of smaller complexes, the signal is diminished. When these tiny complexes and C3Ms coexist, the protein cargo that is meant to be delivered in vivo is put at risk. Furthermore, the decrease in the FRET signal upon raising the salt concentration from 0 to 20 mM showed that about half of the C3Ms had fragmented, perhaps into tiny complexes. They are not suited for usage at physiological circumstances (i.e., an ionic strength of approximately 140 mM) due to their weak salt stability. By adding supplementary homopolymers,[159,1136] protein super-charging,[1137] tethering of charged polypeptides,[1138] complementary nonelectrostatic interactions,[1139] and cross-linking of the polymeric shell, the stability can be increased to get over this obstacle to biomedical applications.[1140,1141] More research is needed to determine how these tactics affect loading efficiency and exchange dynamics. Murmiliuk et al.[1142] used fluorescence quenching studies to study the ability of polyelectrolyte chains to alter their conformation in equilibrated S-C3Ms. Poly(methacrylic acid)1267 (PMA) homopolymers tagged at the chain ends with an umbelliferone fluorescent label and poly[3,5-bis(trimethylammonium methyl)-4-hydroxystyrene iodide]167-block-poly(ethylene oxide)320 copolymers (QNPHOS167-b-PEO320)were used to create C3Ms, because the QNPHOS will quench the umbelliferone's fluorescence, which can happen statically or dynamically, the system was built in this manner. A nonfluorescent compound arises between the dye and the cationic monomer in the event of static quenching. The QNPHOS and the excited umbelliferone collide to cause dynamic quenching, which necessitates the participating species’ freedom of motion. Since most fluorescently labeled chains were either free in solution or formed tiny complexes when sufficient PMA was present, the quenching efficiency was low. The equilibrium spherical C3Ms, on the other hand, show higher static and dynamic quenching when formed at stoichiometric mixing ratio. Given the near closeness of the species, this is to be expected in the case of static quenching. Even more unexpected is the ten-fold rise in dynamic quenching efficiency. After analyzing the data, the authors draw the conclusion that QNPHOS and the fluorophore can collide because the polyelectrolyte chains are still mobile inside the core. The main effects of hydrogen bond formation and salt concentration on the relaxation dynamics of viscoelastic, macroscopically phase-separated complex coacervates were shown using macrorheology.[1119,1143] By using salt-time superposition, the rheological data may be rescaled to access temporal scales that would not have been possible otherwise. Extending these research to C3Ms and directly comparing the relaxation dynamics of macroscopic coacervates with the exchange dynamics of micellar coacervates would be very interesting. Van der Kooij et al. concluded that the stability of macro- and mesoscopic coacervates is similar and significantly more dependent on the chain length of the ionic blocks than on the presence or absence of the solubilizing neutral blocks through a quantitative comparison of experimental results on micellar coacervates with theoretical descriptions established for macroscopic coacervates.[106] Li et al. compared the polyion exchange rates of S-C3Ms with similar ionic blocks that generated under the same conditions to those of tiny, off-stoichiometric, soluble complexes (f+ = 0.17) comprising two homopolymers.[1144] Their poly(N-ethyl-4-vinylpyridinium bromide)55 (PEVP55) reacted with PMA1560 to generate SCPs with a radius of 16 nm, and with PMA180-b- PEO170 to form C3Ms with a radius of 25 nm. To extract polyion exchange rates, the quenching of fluorescence caused by the coupling of cationic monomers to fluorescein dyes coupled to PMA monomers (dye/monomer ≈ 1:750) was observed. Remarkably, the C3Ms were discovered to have higher exchange rates than the SCPs. Because of the latter’s excess PMA chains, free HP chains in solution must get past the complex's repulsive barrier in order to be swapped for a chain in the complex. In this study, nonstoichiometric circumstances were used to generate SCPs rather than macroscopic complex coacervates. To clarify the parallels and discrepancies between the exchange mechanisms of stoichiometric SCPs and C3Ms, more research is required. Our understanding of the relationship between the C3M building blocks and their exchange dynamics is aided by experimental study insights. On the one hand, theory and simulations can support and expand on these discoveries; on the other hand, their predictions can direct synthesis efforts and experimental design. Bos and Sprakel studied chain exchange between S-C3Ms made up of anionic homopolymers with 20 charged monomers and cationic-neutral block copolymers using Langevin dynamics simulations. I looked into the two primary mechanisms: fusion-fission events and expulsion-insertion events. The evacuation of a single polyelectrolyte unimer is not preferred in amphiphilic micelles, because unimers are often transferred between micelles. Small (near-)neutral complexes are introduced and ejected in the case of C3Ms. The authors conclude that numerous expulsion and insertion processes take place when the polyion lengths are precisely matched, resulting in the smallest neutral complex consisting of only two chains. It is determined that the number of fusion and fission events is orders of magnitude smaller. Crucially, enhancing the nonelectrostatic interaction between the two polymers considerably reduces fission and expulsion-insertion, but it appears to have less of an impact on fusion events. Experiments frequently employ polymers with some dispersity and small (block) length inequalities, in contrast to simulations. Bos and Sprakel showed that the rates of insertion and expulsion sharply decreased in their simulations by altering the ratio between the cationic and anionic chain lengths.[89] The reason for this effect is because the smallest neutral complex now has more than two chains instead of just two. The tiny complex can only exit the micelle when more ionic connections are broken, which reduces the likelihood of ejection. This pattern is not followed by the number of fission events, which rises for longer hps but decreases for shorter anionic hps. The authors draw the conclusion that short HP chains allow for easier ionic bond rearrangement. Expulsion-to-insertion events were observed to have a greater overall influence on the exchange rate than fission-to-fusion events. Micelles with just a few hp chains in the core could occur in the extreme scenario where the homopolymer is extremely lengthy in comparison to the charged block of the dbp. Since the smallest near-neutral complexes that can be expelled are on the order of the C3M in size, such a scenario could result in a dominance of fusion− fission events. We expect that contrast variation in small-angle neutron scattering (SANS) will also be utilized in future studies on exchange and displacement dynamics. A multitude of insights into the exchange dynamics of micelles from polymeric amphiphiles have been made possible by this potent technique.[55,97,98,1145,1146] This is achieved by mixing solutions of hydrogenated and (partially) deuterated micelles, and then tracking the gradual loss of their distinct scattering characteristics as a result of exchange processes.[1132] By using this method, it is possible to derive the rates and mechanisms that control exchange in C3M equilibration.
11.3. Time-Resolved in situ Polyelectrolyte Complex Micelle Formation Kinetics Uncovered by Small-Angle X-ray Scattering
Macromolecules known as block polymers are made up of covalently bonded segments of chemically different repeat units. Depending on the relative length, chemical makeup, and molecular architecture of the hydrophilic and hydrophobic polymer blocks, amphiphilic block polymers can be engineered to self-assemble into a variety of unique morphological shapes in selected solution conditions.[775,1147] Two potential processes have been proposed by theoretical studies by Wall and Aniansson,[1069,1080,1110] Halperin and Alexander[1070] and Dormidontova[96,1071] for the kinetics of polymeric self-assemblies towards equilibrium: (i) single-chain insertion/expulsion and (ii) micelle fusion/fission. These model frameworks have been evaluated experimentally by varying the quality of the solvent to cause the creation of micelles. For example, Lund and colleagues used millisecond time-resolved small-angle X-ray scattering (TR-SAXS) under stopped-flow to provide nanoscale spatial resolution and millisecond temporal resolution. They found that the initial micellization kinetics of poly(ethylene-alt-propylene)-block-poly(ethylene oxide) (PEP-b-PEO) can be understood as a slow growth process with a fast nucleation (within 10 ms) and a slow growth process, where the elemental PEP-b-PEO aggregate growth adheres to a single-chain insertion mechanism similar to the Aniansson-Wall theory.[1128] By contrast, Kalkowski et al. used synchrotron X-ray scattering equipped with a microfluidic system to study the micellization kinetics of poly(ethylene glycol)-block-poly(caprolactone) (PEG-b-PCL).[1148] Their findings were supported by the temporal evolution of the radii of gyration (Rg), which showed three sequential steps of micellization in this system: nucleation, micelle fusion, and polymer insertion. As free unimers linked into nuclei, the Rg grew continuously from 0 to 250 ms. The Rg suddenly doubled in size at 250 ms, suggesting that emerging nuclei started to combine, and that free PEG-b-PCL chains thereafter continued to expand structurally. The formation kinetics are in good agreement with the particle-particle fusion model reported by Dormidontova,[96] although different from the Halperin-Alexander and Lund studies. Such collision events in solution have been caught by more sophisticated techniques such in silico simulations and in situ liquid-cell transmission electron microscopy.[1149] To put it briefly, the ability to comprehend kinetic formation in terms of tunable fundamental variables (polymer size/composition, concentration, and interfacial tension) has opened up design principles that make it easier to prepare nanoparticle assemblies for intended applications with more fidelity, control, reproducibility, and stability over time. But in contrast to the tremendous progress made in the last ten years in comprehending the assembly kinetics of micelles generated by uncharged amphiphilic polymers,[96,420,1070,1116,1150,1151,1152,1153,1154] not much is understood about the early stages of self-assembly of polyelectrolyte complexation-driven systems. In the case of block polyelectrolytes, the process is assumed to be more complex due to the interaction of several factors, such as the polycation/polyanion pairing (i.e., polymer polarity, charge density, sequence effect, chirality, asymmetric block length ratios, etc.), high water content and related excluded volume considerations, and the presence of salts in physiological settings. Polyelectrolyte complexes (PECs) are created through an associative phase separation (coacervation) process between oppositely charged polyelectrolytes in water.[27,1155] It is fair to assume that the formation kinetics of C3Ms are related to the thermodynamics of homopolyelectrolyte complexation as the formation of C3Ms is thought to be primarily driven by electrostatic interaction between oppositely charged moieties.[407,1156] Using TR-SAXS in conjunction with a stopped-flow device, Takahashi and colleagues examined the complexation kinetics of sodium poly(acrylate) and poly(allylamine hydrochloride), two oppositely charged homopolyelectrolytes, in aqueous NaCl solutions.[1095] Over a time span of 2.5 to 8733 ms, they detailed the changes in the complex droplet's size, structure, and molar mass. These changes occurred in three stages: (i) an instantaneous (less than 2.5 ms) pairing step, in which electrostatic interactions pair oppositely charged chains; (ii) a complexation step, in which the ion pairs further coalesce into nearly neutral aggregates due to van der Waals and hydrophobic effects; and (iii) a growth step, in which aggregates expanded in a manner reminiscent of the Brownian-coagulation kinetics of spherical colloidal particles. Later on, the same group also reported morphological changes in C3Ms with salt from cylindrical to spherical shapes using TR-SAXS,[1151] which were qualitatively comparable to amphiphilic micelles.[1146] To the best of our knowledge, there has never been a publication on the dynamics of C3Ms self-assembly at the millisecond time scale. In a letter,[1157] as a first step toward developing more precise predictions of formation kinetics, the authors experimentally studied the nonequilibrium self-assembly of a single set of well-defined C3Ms using in situ TR-SAXS with millisecond temporal precision. As seen in Figure 69(A), the components of this model C3M system are sodium poly(acrylate) (PAA) and poly(ethylene oxide)-block-poly(vinyl benzyl trimethylammonium chloride) (PEO-b-PVBTMA). In the past, the authors have shown how easy it is to create polyelectrolytes in parallel synthesis[1074] and have examined how this pairing behaves in terms of self-assembly and establishes clear equilibrium structures in the dilute regime.[1067] The Biological Small Angle Scattering Beamline BL4-2 at the Stanford Synchrotron Radiation Lighthouse (SSRL), SLAC National Accelerator Laboratory, was the platform for all TR-SAXS investigations.[1096,1157] Access to solution kinetics on the millisecond time scale and beyond is made possible by the specially designed Bio-Logic 4 syringe stopped-flow mixer (SFM 400) at the SSRL BL4-2 TR-SAXS facility. In summary, the steps of a typical TR-SAXS experiment are as follows, as schematically depicted in Figure 69(B). First, two different syringes are filled with aqueous solutions of positively and negatively charged polymers. Second, the motors pump equal amounts of polycation and polyanion solutions into the mixer, which correspond to the cationic and anionic monomer units’ equimolar concentrations. About 4 ms is the dead time. Third, 30 μL of the mixed solution is added to the capillary cell, which is the area through which the incident X-ray radiates. The capillary cell's two ends are sealed with the sample solution, which is buffered by solvent. Every measurement had an exposure time of 20 milliseconds. In between trials, automated capillary cleaning helped to check for possible radiation damage. All things considered, the authors have outlined[1081] the spatiotemporal evolution and kinetic pathways of a well-researched PEC micelle candidate using the in situ TR-SAXS approach with millisecond time resolution. It was demonstrated that in less than 100 ms, micelles with an average Rg of 10 nm were fully produced. These C3Ms grew steadily until they reached a size of about 12-14 nm. The authors arrived at the conclusion that neither the micelle fusion mechanism nor the standard single chain insertion holds up by using a quantitative physical model. Rather, a cluster-micelle insertion process is most likely to be used to carry out C3Ms synthesis. This discovery, the authors hypothesized, offered an unique starting point to gain a deeper understanding of comparable ionic soft matter systems that display enigmatic features arising from fast structural dynamics. These findings, for example, open up new avenues for investigating the kinetic rate dependency of molecular characteristics that influence coacervation, such as the lengths of the neutral and charged blocks, the nature and strength of the charged moieties, the valency of the counterion, the hydrophobicity of the polyelectrolyte chain backbone, and the ratio of the neutral to charged blocks for complexation.[1158] Using a molecular engineering approach to uncover the underlying polymer physics of complex creation may also yield new biological understandings.[1159] It is simple to study physiologically significant external characteristics in a systematic way, particularly when assessing micelle stability and disassembly. Examples of these parameters include solution pH, temperature, and salt salinity.[1081] TR-SAXS studies using C3Ms containing nucleic acids[404,1064] or bioactives[115,1139] can provide a basis for the translation of biopharmaceutical formulations into future pharmaceuticals that are more dependable in terms of efficacy, robustness, and safety.
12. Balance of Micellar Free Energy
Phase separation in complex coacervation is driven by an entropic and energetic component that increases the free energy contribution, or Fcomplex. The release of counterions from the polyelectrolyte double layers is linked to the entropic phase. There is an electrostatic component to the energetic portion. A compromise between minimizing electrostatic energy and maximizing counterion entropy is found in the distance between a polyelectrolyte charge and its counterions. The counterions’ entropy encourages a uniform distribution across the system. Close contact between the opposing charges is encouraged by the electrostatic energy. The counterions in a complex coacervate have been swapped out for polyions with the same charge sign. Once more, the distance between the polyions is a trade-off between minimising energy and maximising the polyion entropy. The average distance between the oppositely charged groups can be significantly lower since the entropy loss of polyions is quite minor in comparison to that of tiny ions. As a result, the complex coacervate will have substantially less electrostatic energy. Voorn and Overbeek[81] conducted an early theoretical investigation on complex coacervation for low charge density polyelectrolytes. Odijk[1160] conducted a further theoretical investigation in which they computed phase transitions resulting from the attraction between highly charged rod-like macromolecules and weakly charged polyelectrolyte chains. At charge stoichiometric conditions, a macroscopic phase separation is expected in these models. Water and tiny ions make up the majority of the second phase, while polyelectrolytes are abundant in the first. This phase separation limits the case of complicated coacervation core micelles to colloidal dimensions. The intricate interaction of several forces produces the equilibrium aggregation number (P). P is the number of diblock copolymers in a micelle, expressed in this way. In order to reduce the area of contact between the solvent and the coacervate cores, the surface free energy promotes micellar development. Fsurface is a negative contribution on its own. Because there is a decrease in surface area per unit core material as P increases, the contribution of Fsurface becomes less unfavorable. The stretching of core and corona blocks that prefer low values for P is the penalty underlying micellization. We shall refer to these contributions as Fcore and Fcorona. Fcomplex is the catalyst that drives the production of micelles. Thus, for each diblock copolymer, the micellar free energy is given by
Fmicelle = Fsurface + Fcorona + Fcore + Fcomplex
Two conditions yield the equilibrium aggregation number: ∂Fmicelle/∂P=0 and Fmicelle < 0. In the case of substantially asymmetric diblock copolymers where we neglect Fcore, P is only determined by Fsurface and Fcorona if we assume that Fcomplex is independent of P, which seems like a reasonable assumption for large P. By drawing comparisons with hydrophobically associating systems – for which scaling relationships have been derived – the effect of block length variation can be qualitatively investigated. Scaling theories for micelles with a neutral-hydrophobic architecture[726,1070] and substantially asymmetric diblock coppolymers say that, based on the chain lengths N of the participating blocks, two limiting situations can be identified: (i) star-shaped micelles in which the radius of the core is relatively small in relation to the micelles’ total dimensions, with Ncorona >> Ncore, and (ii) the so-called “crew-cut” micelles, in which the core Ncore >> Ncorona dominates the micellar radius. All that we shall think about is the good solvent limit. The equilibrium aggregation number for the star-like micelles is[726]
where Rcorona ∼ Ncorona3/5υ1/5P1/5 in the case of favorable solvent conditions. υ and γ, on the other hand, characterize the solvent quality for the corona block and the interfacial tension between the solvent and the core, respectively. We get the core radius using equation 61.
where φ is the polymer volume fraction in the core and Vdry is the dry volume of the core’s constituent parts.We can infer from equation 60 that when Rcorona /Rcore rises, P falls. A decreasing P is anticipated for growing Ncorona in a system with a constant Ncore and changing Ncorona when we use equation 61. Nevertheless, there is no direct power law dependence of P on Ncorona. When crew-cut micelles are involved, the equilibrium value for P is provided by[726]
Using this equation, we can observe once more that when Ncore remains constant, the aggregate number falls but Ncorona rises. Because of the strong interactions between the chains on the core-corona interface, P is extremely sensitive to changes in corona block length, given the scaling exponent for Ncorona in equation 13. Because of the lower P values that result in a drop in the grafting density of corona chains on the core surface, the interactions between the corona chains are substantially weaker in starlike micelles. This leads us to the conclusion that a decrease in P is anticipated with rising Ncorona for both limiting situations of block length ratios and a constant value for Ncore. Furthermore, it is evident how υ influences everything. The aggregate number will drop with an increase in υ. We expect that P will decrease with increasing Ncorona and that the qualitative link between Ncorona and P will hold even though formulae for intermediate block length ratios are not provided. Since Rcore ∼ P1/3, the drop in P with Ncorona will result in a decrease in core radius. On the other hand, when Ncorona increases, so will the corona’s thickness, H. Rmicel = Rcore + Hcorona increases with rising Ncorona, as was discovered by Willner et al.[876] in a SANS investigation on amphiphilic diblock copolymers with constant Ncore and changing Ncorona. This is because the increase in corona thickness overcompensated for the decrease in core radius. The micellar physics, including aggregation number and radius, are thus determined by the block lengths of the core blocks (Ncore) and corona blocks (Ncorona) in the dilute regime, when intermicellar interactions are insignificant. Eqs. 60 and 62 only take ∂Fmicelle/∂P=0 into account.and that there is no explicit discussion of the constraint Fmicelle < 0. The micellar system’s stability is also determined by the block lengths. It is evident that stable micelles are not expected for excessively asymmetric block length ratios. On the one hand, the entropy and energy gain from the complexation are inadequate to counteract the corona blocks’ stretching, preventing the formation of micelles, if Ncore << Ncorona. However, if Ncore >> Ncorona, we anticipate a macroscopic phase because there is insufficient core corona stop-mechanism due to the crowding of (short) corona chains on the core surface. A far more nuanced balance between stability and instability exists for intermediate block length ratios. The minimal necessary condition for the accumulation of osmotic pressure in the corona is that σ-1/2 < Rg, where Rg is the unperturbed dimension of the corona block and σ represents the grafting density of corona chains on the core surface.
13. Complex Coacervate Droplets and Micelles: DNA Dynamics
Positively charged macro-ions can form electrostatic complexes with negatively charged polyelectrolyte DNA. When DNA attaches to positively charged histone proteins in chromatin, a process known as DNA condensation occurs, which is explained by this theory.[1161,1162] Furthermore, the creation of membraneless organelles within cells frequently depends on the liquid-liquid phase separation of proteins that have opposing charges from nucleic acids.[1163,1164,1165] The liquid phase created by the polyelectrolyte complex is typically referred to as a complex coacervate. This liquid–liquid phase separation also happens for synthetic oppositely charged polyelectrolytes. In addition to their capacity to phase separate, other characteristics of membraneless organelles have recently been replicated in artificial systems through the use of natural and synthetic polyelectrolytes. These characteristics include their capacity to improve catalysis,[1166,1167] or to form multiple phases within a single complex coacervate droplet.[1168,1169] The formation of complex coacervate core micelles (C3Ms) rather than complex coacervate droplets can occur when DNA is mixed with a cationic-neutral diblock copolymer. This is because the neutral blocks’ repulsive interactions stop the complex coacervate from growing further, resulting in the formation of thermodynamically stable nanostructures rather than the macroscopic complex coacervates that eventually form when DNA is mixed with polycations without neutral blocks. The micelle corona is created by the neutral blocks, while the C3M core is generated by the cationic and DNA blocks. Micelle corona: a barrier that shields the micelle core and keeps micelle coalescence from happening.[1067] Thus, micelles are formedthat are able to shield their core components from outside components.[27,107,394] These micelles have specified polymer aggregation numbers and typically range in size from 10 to 100 nm. These DNA complex coacervate core micelles are intriguing DNA-based drug delivery methods due to their protective qualities and well-defined tiny size.[37,84,1170,1171] Researching liquid-like DNA polyelectrolyte complexes can provide basic understanding of polyelectrolyte complexes in general as well as aid in the development of DNA-based medical delivery devices and a better understanding of the genesis of membraneless organelles. Specifically, because unique DNA sequences can be synthesized, the effect of chain length and chain length polydispersity can be examined systemically using DNA. This results in monodisperse DNA whose length is systematically tunable by sequence modification. We currently don’t fully understand how chain length affects C3Ms and complicated coacervates. As I will discuss below, the chain length effect on the coacervate dynamics in particular is not well known. Complex coacervate dynamics dictate how complex coacervate materials react to deformation[1172,1173] and may also influence how quickly they react to dissociation triggers.[1174] The sticky Rouse model,[1175] in which the ionic bonds function as sticky points to slow down the dynamics, is typically used to explain the chain length influence on the dynamics in complex coacervates.[1172,1173] According to the sticky Rouse model, the relaxation time of a polymer increases with the square of the number of sticky bonds between molecules. This implies that for polyelectrolytes, the relaxation time should increase with the square of the polyelectrolyte length. This description appears to be effective for complex coacervates with matching chain lengths;[1172] however, the situation becomes more complicated for complexes where the cationic and anionic chain lengths differ. In certain instances, it appears that the dynamics are exclusively controlled by one of the two polyelectrolytes, with the second polyelectrolyte’s length having no bearing at all.[1172] Due to the small number of studies that have concentrated on polyion pairs with incommensurate lengths, a thorough explanation of coacervate dynamics and a comprehension of these asymmetry effects are absent. The degree of protection that the C3M provides to a cargo that it encapsulates is determined by the molecular exchange dynamics of complicated coacervate core micelles, which also dictates how frequently the core components are exposed to the environment. When a study measured the molecular exchange of C3Ms using Förster resonance energy transfer (FRET) in the past, the authors found a wide range of exchange rates,[103] which is consistent with what has been seen before for other C3Ms.[86] They postulated that chain polydispersity is the cause of this significant variation in exchange rates. Recently, small angle neutron scattering (SANS) was used to assess the molecular exchange of C3Ms. In this case, too, the existence of various exchange rates was explained by chain polydispersity.[1176] Since only the interchange of polydisperse polymers has been quantified thus far, the polydispersity hypothesis has not yet been verified. This theory may be fully tested by measuring the interchange of C3Ms with ssDNA since the DNA is monodisperse and can have its length routinely changed. In addition to the scant understanding of the influence of chain lengths on the dynamics in complex coacervates and C3Ms, little is understood about the relationship between the dynamics within bulk coacervates and the nanoscopically confined interior of C3Ms, as well as the degree to which the dynamics in bulk coacervates predicts the exchange dynamics in micelles. As a result, a study[1177] compared and examined in this work the dynamics of single-stranded DNA in complicated coacervate droplets and micelles (Figure 70).
The authors employed fluorescence recovery after photo-bleaching (FRAP) to determine the diffusion coefficient of fluorescently labelled single-stranded DNA for complex coacervates droplets. They tracked the exchange of the ssDNA and the diblock copolymers for the C3Ms using FRET-based exchange measurements, which they supplemented with simulations of Langevin dynamics. In this way, the authors demonstrated how the length of the cationic and DNA chains affects both the DNA exchange of C3Ms and the DNA diffusion in complex coacervates. The authors found that, contrary to what the sticky Rouse model anticipated, the DNA diffusion coefficient has a greater reliance on DNA length, which is only partially explained by the effect of chain length on coacervate density. Furthermore, they deduced from a comparison of the exchange of the polydisperse diblock copolymer and the monodisperse ssDNA that the previously noted large variety of exchange times is, in fact, mostly due to chain length polydispersity. They postulated that variations in the interactions between the donor and acceptor fluorophore labels account for the majority of the observed exchange rates for the monodisperse DNA, and their data support the notion that fluorophores can significantly impact C3M exchange. In conclusion, the report[1177] go over a revised explanation of the C3M exchange that takes into consideration the impact of the oppositely charged core species’ chain length on the exchange rate, among other factors. The findings collectively can contribute to a better understanding of complex coacervates droplets and micelles in general, as well as DNA-specific systems. As previously mentioned, the C3M exchange rate (Figure 71) is determined by both activation and relaxation processes in the core, much as the interchange of amphiphilic diblock copolymer micelles is determined by these activities.[98] In contrast to the amphiphilic diblock copolymer micelles, the C3M exchange relies on the length of the oppositely charged core species to determine the exchange rate of a core species. In the case of the amphiphilic diblock copolymer, on the other hand, the exchange rate is solely dependent on the length of the copolymer, as there is only one core species. Thus, the charge ratio may be more significant than the absolute polyelectrolyte length in C3M exchange, and the energy barrier of the activated process may not always rise with increasing polyelectrolyte length. The inability to use the amphiphilic diblock copolymer model to describe the exchange of PSPMA/ PEG-PTMAEMA micelles[103] and the requirement for a lower polydispersity than measured in a recent SANS study of C3M exchange to obtain a good fit between the exchange data and the amphiphilic diblock copolymer model can be attributed to these differences between C3Ms and amphiphilic diblock copolymer micelles.[1176] These variations also highlight how crucial it is to investigate C3Ms in particular in order to fully understand how chain lengths and other parameters affect the C3M exchange rate.
14. C3M in Dilute Solutions: Interparticle Interactions
Researchers have made an effort to comprehend C3Ms by drawing comparisons to the micelles created by the neutral and amphiphilic block copolymers, despite the clear differences in the underlying driving factors. It’s not immediately clear that this is necessary. The relevant characterisation methods, including using the Guinier approximation to determine the radius of gyration (Rg), are typically arbitrarily borrowed and applied to C3Ms. A group has reported on the polymer concentration-dependent phase diagram of C3Ms, which exhibit certain similarities with uncharged, amphiphilic block copolymer micelles. For instance, the phase-separated complex domains' structural evolution changed from disorder arrays to body center cubic lattices and finally to hexagonally packed cylindrical morphologies when the polymer concentration of the C3M solutions increased from moderate to high (roughly 5-40 wt%).[140,1156,1178] A recent study conducted by a group, however, discovered that the phenomenological consistency deviates at lower concentrations (0.5−2.0 wt%). This is demonstrated by the difference between the formation of interconnected micellar networks in complexation-driven self-assemblies and the individual micelles driven by the solvophobicity−solvophilicity balance.[1179] Others have also reported findings that are comparable.[69] The Tirrell’s team reported an in-depth analysis of C3Ms in diluted fluids (polymer concentration < 0.5 wt%), encompassing an assessment of their internal compositions and an estimation of the effective interparticle interactions.[1067] Over the past twenty years, diluted solutions of C3Ms have generated a great deal of attention and a surge of articles, most of which focus on micelle stimuli-responsiveness, comicellization with different biological compounds, and other medicinal applications.[26,283,393] Nevertheless, there haven't been many reports of a thorough analysis of their physical characteristics, and certain significant behaviors are still poorly understood. For instance, (i) less is known about the response of the coronal layers to micelle contacts and the ways in which electrostatic interactions affect micelle dynamics; (ii) less is known about whether the electrostatic force of the core is present outside of the coronas. The spatial filling limits that C3Ms face in diluted fluids are revealed by the micelle-micelle interaction, and this information is further connected to the micelle stability, diffusivity, and repeatability over the long term. Because of its implications for dosage optimization and durability under physiological settings, deepening our understanding of the fundamental behaviors of polymeric nanocarriers in diluted fluids is crucial for the advancement of biomedical applications. A PEC system that forms uniform core/shell spherical micelles has been studied by a group previously.[1074,1096] It was made up of a negatively charged homopolyelectrolyte (poly(sodium acrylate), PAA158) and a positively charged block polyelectrolyte (poly(ethylene oxide)-block-poly(vinyl benzyl trimethylammonium chloride, PEO225-b-PVBTMA100). Each block’s level of polymerization was indicated by a subscript. Multi-angle dynamic light scattering, SAXS characterisation, and cryogenic electron microscopy were used to confirm that this polyelectrolyte pairing produced assemblies with variable nanoparticle sizes, contingent upon the constituent block lengths. Moreover, PEO-b- PVBTMA controllably binds with DNA oligonucleotides form spheroidal or cylindrical morphologies, contingent on block lengths and DNA hybridization, as recently shown by Marras and coworkers.[92] Hence, the PEO-b-PVBTMA/PAA model system serves as a useful artificial framework for examining the dynamics and assembly creation of C3Ms in the direction of biologically meaningful nanocarriers. A previous study[1067] presented the identification of repulsive intermicellar interactions in C3Ms at low polymer concentrations (< 0.5 wt%), when two-body interactions should predominate, if not be the only mode of contact. Using synchrotron small-angle X-ray scattering (SAXS), structural features of C3Ms in diluted concentrations were explored multiparametrically, including the diameters of the core, corona, radius of gyration, aggregation number, volume fraction, and pair distribution correlations. Every SAXS experiment was conducted at the Stanford Synchrotron Radiation Lighthouse (SSRL), SLAC National Accelerator Laboratory, using the Biological Small Angle Scattering Beamline BL4−2.[404] SAXS curves were fitted using a combinatorial form factor that included a Gaussian coil function to account for the blob scattering from the coronal polymer chains and a polydisperse core-shell sphere model to account for micelle shape. Structure factor contributions become visible when there is an increasing divergence in the low-q range between the form factor and the experimental data. The authors used both the Debye-Huckel screening Coulomb potential and the hard sphere potential to represent the electrostatic repulsive interaction. The increasing discrepancy between the real-space Rg derived from the pair distribution functions calculated by excluding the low q region and the apparent Rg of the micelle obtained through a Guinier approximation to the low q data further supports the effective interparticle repulsive interaction. Overall, the authors presented the measurement and identification of the repulsive micelle-micelle interaction in C3Ms at polymer concentrations between 1.0 and 5.0 mg mL−1. The interparticle interactions between PEC micelles are not as negligible as is usually believed at such a dilute regime, as was reported. The structure of C3Ms was demonstrated to be characterized by hard spherical cores connected by compressible Gaussian coronal chains. Furthermore, the electrostatic force was observed to be limited to the micelle coronas and there was no long-range screened Coulomb potential. Differences between the radii of gyration obtained by pair distribution functions and Guinier approximation further supported the repulsive intermicellar contact. The authors also highlighted the fact that the correctness of the widely used Guinier approximation is dependent on micelle concentrations due to the nontrivial nature of the interparticle interaction among C3Ms in diluted fluids. These results offer essential understandings of C3Ms dynamics, including micelle fission/fusion kinetics.[96] They may also clarify the behavior of micelles when administered intravenously into the bloodstream, a process that greatly dilutes the micelle concentration. These self-assembling nanoparticles’ structure-property correlations have been thoroughly investigated and used as delivery vehicles for enzymes,[1051] proteins.[1059] DNA,[1180,1181,1182] RNA,[1183,1184,1185] and oligonucleotides,[107,1186] Relying on these subtle yet crucial design concerns, such nanoparticle formulations with control and accuracy over functions will be advanced as nanomedicine.
15. Methods
General features observed in experiments (i) probing micellar formation (viscosimetry, conductometry, ζ-potential measurements, static light scattering), (ii) probing micellar structure (small angle neutron and X-ray scattering, static and dynamic light scattering, cryogenic electron microscopy, atomic force microscopy), (iii) probing micellar dynamics, and (iv) probing micellar function are described in this section.
15.1. ζ- Potential and Viscosimetry
The ζ- potential, which is determined by light scattering detection in a Zetasizer, typically passes zero for f+ = 0.5 (if α+ = α−, where α denotes the degree of dissociation of the cationic (α+) or anionic (α−) polyelectrolyte) as f+ increases from 0 to 1 for C3Ms made up of synthetic and/or biopolymers.[72,1187] There are significant differences between systems in how these findings should be interpreted in terms of the kind of nanoparticles that are generated at different f+. According to some groups, for every f+, a single type of particle is generated, carrying excess positive charge after the charge neutralization condition and excess negative charge before it.[72] Some groups contend that for any f+, there are two sorts of particles in solution with constant composition. According to symmetry arguments, the cationic equivalents for low f+ are referred to as cationic soluble complexes and excess cationic homopolymer, whereas for large f+, they are known as anionic soluble complexes and excess anionic homopolymer. Anionic or cationic soluble complexes are assumed to exist in equilibrium with charge-neutral C3Ms for f+ close to charge stoichiometry.[25,30] Therefore, in the latter molecular image, the excess charge is never included in the micelle but is always included in either single polymers or soluble complexes. Assuming that the measured ζ-potential is an average of the ζ-potential of all species present in solution, the experimentally observed fluctuation of ζ-potential with increasing f+ is compatible with both explanations. One may anticipate variations in the slope of the ζ-potential versus f+ for the second interpretation and light scattering detection (discriminating towards the larger particles; i.e. stronger scatterers) where the kinks should coincide with the CEAC, CECC, and PMC.[30] Since the species’ charge-neutral percentage is the biggest scatterer, it will be mostly identified in the charge stoichiometric area, where a relatively flat slope with absolute values close to zero may be expected. Additionally, for C3Ms made up of an oppositely charged surfactant and a neutral-ionic copolymer, the ζ-potential passes zero for f+ = 0.5 as f+ increases from 0 to 1.[1188,1189,1190,1191,1192,1193,1194,1195,1196] As the quantity of surfactant/copolymer complexes (that coexist with excess polyion) rises, binding of surfactant to the polyion causes the ζ-potential to increase or fall towards zero for values of f+ where the polyion is in excess. When surfactant concentration is increased for values of f+ where surfactant is excess, the absolute ζ-potential is typically increased as well.[1188,1189,1190,1192,1193,1194,1195] This is interpreted as excess surfactant incorporated into the surfactant/copolymer complexes because of hydrophobic interactions,[1189,1190,1192,1195] surfactant interaction with the neutral corona blocks,[1191] and/or formation of surfactant micelles in solution.[1191]
The ratio of the mixture’s experimentally observed reduced viscosity value at a given f+, ηrsample to the mixture's reduced viscosity value in the event of ideal mixing at the same f+, ηrideal is known as the reduced viscosity ratio, or rrη. The latter represents the mass average of the two polymers' decreased viscosities, ηr+ and ηr−, combined at a specific mass fraction, m+ and m−.
As complexation lowers solution viscosity, one typically sees a parabolic dependence of rrη on f+ with a minimum at the PMC.[26,72,1131,1197] Because rrη is (mostly) influenced by the degree of complexation and Ncorona/Ncore does not affect the position of the PMC, it was shown that the fluctuation of rrη with f+ is nearly independent of Ncorona/Ncore. Moreover, it was discovered that as the strength of complexation diminishes, rrη increases as ionic strength increases (for values below the critical ionic strength).[72] When comparing the experimental data with the theoretical predictions made by Colby et al. regarding the rheological properties of aqueous polyelectrolyte solutions with oppositely charged surfactants, one can represent the viscosity data for neutral-ionic copolymer/surfactant systems as the specific viscosity against the surfactant concentration. It has been demonstrated that theory and experiment agree rather frequently in a number of instances.[1188]
15.2. Conductometry and Static Light Scattering (SLS)
When two oppositely charged macromolecules (a neutral-ionic copolymer and an oppositely charged homopolymer) combine to form C3Ms, solution conductivity (κ) typically reaches a maximum at the preferred micellar composition as a function of f+.[72,1197,1198] This phenomenon may be related to the release of counterions during the complexation process. Such an extremum might not exist for C3Ms made up of an oppositely charged surfactant and a neutral-ionic copolymer, even when the PMC is within the composition range under investigation.[1189] The linear dependence of κ on f+ typically exhibits a break point at the CAC.[1189,1190] This kind of solution has a higher conductivity than can be determined only by looking at the elements (surfactant, polymer, and counterions).[1189,1190] Counter ion hopping by polymer-surfactant nanoparticles has been proposed to require consideration.[1199,1200] Numerous investigations[72,1189,1190] document a rise in the absolute value of κ in tandem with a reduction in the mass of polyions per charge (i.e., an increase in the total degree of dissociation of polyions), suggesting that polyions indeed play a role in the specific conductivity.[72]
In a static light scattering experiment, C3M production is easily observed.[25,30,36,70,72,91,159,278,393,401,421,546,663,724,1129,1187,1197,1201,1202,1203,1204,1205,1206,1207,1208,1209,1210,1211,1212,1213,1214,1215,1216,1217,1218,1219,1220,1221,1222,1223,1224,1225,1226,1227,1228,1229,1230,1231,1232,1233,1234,1235,1236,1237] When f+ increases from 0 to 1, one usually observes a rise in total scattered intensity followed by a fall, peaking at the desired micellar composition. This is readily explained by differences in the complexes’ molar mass and aggregation number.[25,30,72] To connect the experimentally determined excess Rayleigh ratio, R(θ,C), to the micellar mass, one must calculate or determine the specific refractive index increase of the micelles, dn=dc. The micellar dn=dc is often calculated assuming additivity. In 10 mM sodium phosphate buffer, Harada and colleagues showed good agreement between estimated and experimentally measured values for C3Ms consisting of PLys(-b-PEO) and PAsp-b-PEO.[393] The SLS experiments on C3Ms as a function of mixing fraction, f+, are presented in a less straightforward manner than one might think because the simple interpretation of I900 versus f+ in terms of excess scattering relative to the scattering of the single components is precluded by the known[1236] or unknown, but potential difference in dn/dc for the single components. Therefore, in an I900 versus f+ plot, a significant non-zero slope baseline may be masked, and I900 at f+ of 0 and 1 are not always equal.[72,1236] Furthermore, one must account for dilution effects throughout the measuring process when performing so-called mole fraction light scattering titrations, or f+ LS-T. Many techniques have been employed to get around the issues mentioned above without carrying out a complete Zimm-analysis for each f+, or extrapolation to zero concentration and scattering vector. Some writers plot the hypothetical scattering of the ideal, or non-interacting mixture, in addition to the sample scattering (solvent scattering subtracted).[72,1235,1236] Note that if one divides the sample scattering with the hypothetical ideal mixture scattering, one should exercise extreme caution when attempting to obtain absolute values for micellar mass and aggregation numbers by this method. This is because the excess scattering obtained in this manner may contain large errors if the single components scatter little. This could apply to S- C3Ms, D- C3Ms, and C3Ms that are created through complexation with low-MW surfactants in diluted solutions. Therefore, there may be (very significant) differences between the values from this method and those from a thorough Zimm analysis. A different approach that is less likely to produce significant mistakes has been proposed recently,[27] in which the excess sample scattering is basically calculated by calibration with a toluene standard following the subtraction of solvent and single component scattering. Because of a decrease in particle mass and aggregation number upon micellar dissociation, the critical ionic strength can be found by analyzing the inclination point in a plot of the (initially decreasing) static light scattering intensity upon increasing ionic strength. This process can be repeated until the critical ionic strength is reached and no more C3Ms can be observed.[159,1201,1238,1239] A plot of the static light scattering intensity against pH can also be used to assess the critical pH for C3M production in systems containing at least one weak polyelectrolyte.[1201,1240]
15.3. Dynamic Light Scattering (DLS) and Other Methods
In the C3M literature, dynamic light scattering (DLS) is mostly used for particle size. The measured Rh represents a weighted distribution over all objects present in solution, weighted with their relative scattering power. This is because aqueous mixtures of two oppositely charged components can contain several types of scattering objects, such as C3Ms and one excess component or unimerically dissolved non-interaction species. Therefore, the method of cumulants, which assumes a single type of scattering object (with a specified polydispersity), should be avoided for DLS data processing. Instead, inverse Laplace transform (ILT) programs, like CONTIN, should be used. According to a number of publications, charge neutralization causes a minimum in particle size under isoelectric conditions; in other words, excess charge causes particle swelling.[68,72,280,1195,1197,1241,1242,1243,1244,1245] But there are also reports of the opposite. For instance, it was discovered that Rh passes through a maximum for C3Ms of PTEA-b-PAAm and CeO2 or γ-Fe2O3 nanoparticles at (near) isoelectric conditions.[1236] The combination of an automated titration and (static and dynamic) light scattering setup has proven to be very beneficial for a group’s laboratory research on the effects of the mixing fraction, pH, and ionic strength on micellar formation and characteristics.[25,27,159,401,421,1201] A wide range of other spectroscopic, scattering, and imaging techniques can offer helpful information on C3M production, structure, dynamics, and function in addition to the conventional experimental methods mentioned above. The secondary structure of encapsulated (or attached) DNA and protein/peptide molecules has been studied by applying fluorescence resonance energy transfer (FRET)[1181,1219,1246] and circular dichroism (CD) [36,68,71,79,93,129,163,278,283,546,600,663,1129,1187,1196,1197,1204,1205,1206,1207,1208,1209,1210,1211,1212,1213,1214,1215,1216,1217,1218,1219,1220,1221,1222,1223,1224,1225,1226,1227,1228,1229,1230,1231,1232,1233,1234,1235,1236,1247,1248,1249,1250,1251,1252,1253,1254,1255,1256,1257,1258,1259,1260,1261,1262,1263,1264,1265,1266,1267] to DNA and protein/peptide-containing C3Ms. Micellar dynamics has also been studied using FRET.[86] Studies on pyrene solubilization have produced CMCs (and CACs)[63,1188,1194,1268,1269] as well as details on the micellar core’s micropolarity and microstructure.[1188,1189,1190,1196,1241] Since the co-assembly process may result in a decrease in the mobility and solvation of protons of core-forming blocks, which normally results in a decrease in NMR peak intensity, 1H nuclear magnetic resonance (NMR) has been used to analyze C3M formation.[548] Moreover, 2D 1H NOESY NMR investigations may be used to examine the distribution of chemically distinct segments inside the C3M core and corona.[318,421,724,1201,1270,1271,1272] Surfactant- and nanoparticle-containing C3Ms have been successfully subjected to small angle neutron scattering (SANS) [91,146,724,1201,1202,1203,1271,1273,1274,1275,1276,1277,1278,1279] and small angle X-ray scattering (SAXS)[1205] to study a number of intriguing micellar characteristics, including mass, size, shape, aggregation number, and microstructure. Utilizing anomalous short angle X-ray scattering (ASAXS), size distributions of platinum nanoparticles inside the micellar core have been obtained.[1234] The distribution of γ-Fe2O3-containing C3Ms’ core size was obtained using magnetic sedimentation,[1280] and the size of the gold nanoparticles inside the C3Ms was determined using UV-VIS spectroscopy.[1232] Using attenuated total reflectance infrared spectroscopy (ATR-FTIR), the formation of coordination bonds between polyelectrolyte blocks and metal ions in the C3M core has been investigated.[1281] The visualization of (the core of) C3Ms has been achieved through the use of cryogenic transmission electron microscopy (cryo-TEM),[145,401,1252,1253,1276,1277,1278] transmission electron microscopy (TEM),[548,1191,1192,1195,1204,1244,1251,1258,1282] and atomic force microscopy (AFM).[273,546,1204,1205,1206,1237,1248,1266,1269] To investigate the binding equilibrium between the oppositely charged species, potentiometric titrations have been carried out.[1192] The examination of whether the complexation is endothermic or exothermic has been done using isothermal titration calorimetry (ITC),[25,1227] DSC,[1266] and microcalorimetry.[1195] Chromatography has been employed to investigate recognition phenomena and the aggregation mechanism.[283,1131]
16. Applications
Our understanding of C3Ms at the molecular level and the fundamental research on physical scaling and kinetics continue to spark interest in scientists. The ultimate goal, in my opinion, is to create tunable polymeric nanoparticles for the delivery of biomolecules. Research changes when medicinal biomolecules are replaced with model polymers and C3Ms are placed in challenging conditions. A strong reciprocal relationship exists in this sector between application-based science and basic research, as seen by the many developments that are advancing the possibility of using C3Ms for biomolecule delivery. C3Ms have a broad range of uses, including drug delivery devices, diffusional nanoprobes, anti-biofouling coatings, and nanoreactors.[165,602,1283,1284]
16.1. Biomedical Applications
16.1.1. Control of Enzymatic Activity: Optimizing Enzyme Encapsulation Stability and Efficiency in Complicated Coacervate Core Micelles
Enzyme-encapsulated C3Ms are used to produce nanoparticles that have the ability to regulate catalytic activity and stabilize enzymes. It is believed that the enzyme is enclosed and shielded from the environment within the nanoparticle's core. On the other hand, if the polymer causes the protein to unfold or if the substrate is too large to fit through the polymer shell, this may decrease enzymatic activity. As long as the substrate is tiny, C3Ms generally appear to increase, or at least not inhibit, the catalytic activity of enzymes including GOx,[1047] lysozyme,[1051] and trypsin.[1052] On the other hand, big substrates can totally impair enzymatic action.[284] In this situation, a switch that can dismantle the C3Ms and restore the enzyme is needed, such as an electric field[1051] or the injection of salt.[1057] The structure of the protein after it has complexed with the charged polymer has not received much attention up to this point. In order to determine when lysozyme folds and unfolds, thermal and circular dichroism analysis has been used. The results show that the C3Ms enzyme exhibits distinct secondary and tertiary structures, a reduced thermostability, and distinct unfolding and refolding behaviors.[1285] The solubility of C3Ms in aqueous solution, their ability to encapsulate many protein molecules in a single micelle,[1286] and their potential for controlled release are advantages as a protein packing technology.[1287] Enzyme-containing C3Ms can be employed as a microreactor to overcome incompatibilities between polar enzymes and nonpolar substrates, and encapsulation can shield proteins from harmful environmental impacts and protease activity.[1288] The total charge of the protein and the distribution of charges across its surface are critical factors for protein encapsulation in C3Ms and are influenced by the pH, structure, and amino acid composition of the surrounding solution.[126] Furthermore, due to the hydrophobic properties of different amino acid residues, hydrophobic interactions may play a substantial role in the synthesis of protein-containing C3Ms in addition to electrostatic interactions and entropy gain from counterion release.[8,1289] The fact that C3Ms readily dissolve, frequently as a result of proteins’ low charge density, presents one of the biggest obstacles to their usage as protein packing devices. Using dynamic light scattering (DLS), Lindhoud et al. demonstrated that the most stable enzyme-containing C3Ms could be generated by supplementing the two-component system with an excess of homopolymer with the same charge sign as the protein above the concentration of protein.[159] Black et al.’s research shown that coacervate droplets of two oppositely charged polypeptides could encapsulate bovine serum albumin (BSA). By employing a Bradford colorimetric test, it was observed that there was a less than linear increase in BSA in the coacervate when the ratio of BSA to the total amount of polypeptides in the system was increased.[220] This highlights the necessity of conducting additional research on three-component coacervate systems for protein encapsulation, as previously recommended by Blocher and Perry.[1290] In 2010, Gapinski and colleagues demonstrated that micellar sizes and shapes may be measured using the relatively new method of fluorescence correlation spectroscopy (FCS).[1291] Nolles et al.’s research revealed that when it came to the hydrodynamic radius and preferred micellar composition (PMC) of two-component C3Ms containing green fluorescent protein (GFP), DLS and FCS yielded results that were comparable.[1292] Furthermore, they were able to determine the quantity of protein molecules absorbed into each micelle as well as the distribution of protein throughout the solution and micelles thanks to FCS.[1286] Because of the fluorescence emission's Stokes shift, FCS has a comparatively low background noise as compared to DLS, and measurements can be made at extremely low (nanomolar) concentrations. Moreover, FCS's selectivity makes it possible to analyze particular fluorescent compounds within systems.[1293] However, compared to DLS measurements, FCS experiments and analysis take more labor, and fluorescent tagging of biomolecules is frequently necessary. The model enzyme used in an investigation is the spore coat protein A (CotA) laccase. The outer coat layer of the Bacillus subtilis endospore is where CotA was first discovered.[1294] CotA has a molar weight of 65 kDa and an isoelectric point (pI) of 5.84 at pH. Four copper ions are present, indicating that it is a multicopper oxidase (MCO) (Figure 72).[1295]
Based on the ultraviolet/visible (UV/vis) and electronic paramagnetic resonance (EPR) spectra, these four copper ions are categorized into three types, designated as type 1 (T1), type 2 (T2), and type 3 (T3).[1298] The enzyme has a deep blue color due to the absorption of the T1 Cu ion, which is visible at a wavelength of approximately 600 nm. The trinuclear center (TNC) in the protein structure is made up of two T3 and T2 Cu ions.[1299] CotA can use dioxygen as an electron acceptor to catalyze the oxidation of a broad range of substrates. After the T1 binding pocket experiences substrate oxidation, electrons are moved to the TNC, where dioxygen reduction takes place.[1296,1300] A area of positive charge, comprising 10 lysine and 5 arginine amino acid residues, is present on the surface of CotA near the interface between domains 1, 2, and 3.[1301] This positively charged patch’s role in the assembly of CotA into the spore outer coat layer is its biological function.[1283] Enzyme immobilization has previously been studied using models based on specific laccase types. For instance, Bryjak et al.[1302] immobilized fungal laccase by covalently bonding it to a copolymer of butyl acrylate and ethylene glycol dimethacrylate, whereas Pich et al. employed fungal laccase in their investigation of composite magnetic particles as enzyme carriers.[1303] However, encapsulation in intricate C3M has not yet been studied using laccases. In a recent study,[1136] the encapsulation effectiveness and stability of C3M-containing CotA was looked on. The authors demonstrated that the best compromise between micelle stability and encapsulation efficiency requires the use of both DLS and FCS data. It was demonstrated via DLS and FCS that the enzyme CotA can be encapsulated with the diblock copolymer PM2PV-b-PEO forming complex coacervate core micelles at a high pH (10.8). Micelle formation is inhibited at neutral pH, most likely due to the enzyme’s low net negative charge and the existence of a positively charged patch on its surface. The negatively charged homopolymer PSS was added to the micelles to increase their salt stability, resulting in the formation of three-component C3Ms. FCS measurements revealed that less CotA is encapsulated per micelle in the three-component C3Ms (hydro-dynamic radius of 24 versus 32 nm for the two-component system), but the fraction of encapsulated enzyme is still substantial (80 versus 84%). The three-component C3Ms are, in fact, more salt-resistant than the two-component C3Ms, according to DLS measurements. Nevertheless, FCS analysis showed that even at comparatively low salt concentrations, CotA is already ejected from the three-component C3Ms. Their findings demonstrate the importance of FCS studies in gaining understanding of the salt stability and composition of the three-component C3Ms, as they allow for the distinction between free and encapsulated CotA. Based on these results, it is not recommended to add a homopolymer that has the same charge sign as the protein in order to increase the salt stability of C3Ms that contain protein. The authors proposed that the bioconjugation approach plays a crucial role in increasing the enzyme’s charge density, which in turn makes C3Ms containing enzymes more resistant to salt.
16.1.2. C3M-Based Biomolecule Delivery
The delivery of various other bioactive compounds, including proteins, enzymes, peptides, genes, bioactive nanoparticles, diagnostic agents, etc., in addition to conventional hydrophobic drug delivery, is unquestionably one of the most significant and widely applied fields for BCP self-assembly, and as such, it has been thoroughly studied and reviewed.[37,1304,1305,1306,1307,1308,1309] Aiming for selective and controlled drug delivery, liposomes, polymer-drug conjugates, nanoparticles, polymeric micelles, and vesicles are used in nanomedicine. This maximizes biodistribution and bioavailability, improves pharmacokinetics and cellular uptake, and minimizes unfavorable side effects. Hybrid protein/peptide-polymer conjugates have also been successfully used in pharmaceutics and drugs delivery.[1310,1311,1312,1313] Because of the ability to effectively regulate the size, shape, morphology, surface chemistry, and even the responsiveness of generated nanostructures to environmental changes, recent developments in BCP self-assembly have led to the widespread use of BCP micelles and vesicles in drug delivery applications. Their broad range of applications stems from several aspects, the first of which is their capacity to entrap and transfer hydrophobic medications within their core, even at quantities higher than their inherent water solubility. Furthermore, because they can prevent opsonization and do not obstruct blood vessels, their tunable size in the nanometer range (often less than 100 nm) and narrow size distribution make them perfect for extended blood circulation. This characteristic is further improved by selecting biocompatible polymers, such as the well-known PEO (or PEG), for the micellar corona's construction. These polymers resist cellular adhesion and protein adsorption, shielding the hydrophobic drug from hydrolysis and enzymatic degradation while also preventing recognition by the reticuloendothelial system and macrophage phagocytosis. BCP micelles are utilized as a passive targeting technique because of their extended circulation capacity and tiny size, which increases accumulation at tumor locations due to the enhanced permeability and retention (EPR) effect. BCP micelles also have the benefit of having great static and dynamic structural stability due to their low CMC, which prevents them from dissociating under extremely diluted circumstances found in bodily fluids during in vivo applications.[37,1304,1306] Naturally, core- or corona cross-linking, which solidifies the structures and permits their dispersion in a broad range of media,[1314] can be used to further improve micelle structural integrity if needed. Alternatively, two or more amphiphilic copolymers can be combined to create mixed micelles, which also show improved stability and drug loading capacity.[1315] In an attempt to accomplish active targeting, tailored surface functionalization may be able to significantly improve their chances of successful delivery. To put this into practice, the outside of the nanocarriers must be coated with suitable ligands (such as functional organic compounds like folic acid, peptides and proteins, sugars, antibodies, aptamers, etc.) that can bind and recognize specific locations in the human body through matching receptors.[1316] Moreover, it has occasionally been demonstrated that adding fluorinated moieties to polymeric nanoparticles increases the absorption of such particles by live cells.[1317,1318,1319,1320] The enhancement of intracellular penetration and uptake, which improves therapeutic efficacy without harming healthy tissues, is the primary advantage of active targeting. Upon arrival at the intended location, the nanocarrier must then be optimized for the delivery of the bioactive payload. This is typically accomplished by creating carriers that release their contents in response to specific stimuli. These methods capitalize on the physiology of tumor tissues, which is very different from normal tissues in that it exhibits greater temperatures, lower pH values, and higher concentrations of certain enzymes, among other differences. In order to achieve the controlled release of the drug, these characteristics may therefore act as catalysts for the structural modification or direct breakdown of the loaded nanocarriers. In addition to these internal stimulus sources, responsive delivery can also be achieved by applying external stimulus sources such heat, magnetic fields, ultrasound, and radiation. [37,1304,1306] The need for controlled or triggered release has resulted in the creation of "smart" nanocarrier systems, which can react to external stimuli originating from chemical, physical, or biological sources.[1321,1322,1323,1324,1325,1326,1327,1328,1329,1330] Since the extracellular pH of the majority of tumor tissues is strongly acidic (pH 6.5-6.8), slightly lower than that of normal tissues (pH 7.2-7.4), pH is one often used trigger. Protonation of ionizable groups or the breakdown of pH-sensitive linkages are the main causes of the pH sensitivity of polymers; poly(L-histidine) (PHis), PAsp, PGlu, PDMAEMA, poly(2-[diisopropylamino]ethyl methacrylate) (PDPA), P2VP and P4VP, poly (methacrylic acid) (PMAA), etc. are some common examples. The redox potential is another distinctive stimulus that is used to differentiate between the external and intracellular environments, as well as between normal tissues and tumors, because of the variation in glutathione (GSH) content. Thus far, a number of redox-sensitive nanocarriers have primarily been designed using disulfide linkages, which are easily broken down by GSH. Comparably, oxidation-responsive systems take advantage of the elevated local concentrations of reactive oxygen species (ROS) such as H2O2, which are linked to a range of oxidative stress-related illnesses, such as various malignancies, Parkinson’s, and Alzheimer’s diseases. In a similar vein, because of their remarkable catalytic qualities and remarkable selectivity, enzymes are essential to numerous biological and metabolic processes within the body. In addition, the uncontrolled expression of some enzymes, such as lipases, peptidases, and proteases, in neoplastic settings, may serve as a trigger for the delivery of drugs that are responsive to enzymes. Temperature is a final extensively studied internal stimulus that has the ability to function as an external one. As was already established, tumor areas have a temperature that is around 40 0C higher than that of healthy tissue, and this can result in a particular thermal response. An external heating source or gadget can be used as an alternative to achieve this. Since PNIPAM has a lower critical solution temperature (LCST) of about 32 0C, which is near to the physiological temperature of the human body, it is typically the basis for temperature-sensitive BCP-based nanocarriers. PNIPAM is the most extensively used thermo-responsive polymer.[1321,1322,1324,1325] Ultrasound, light, and magnetic fields are other commonly used external sources. In addition to being used for tumor targeting in the presence of an external magnetic field, magnetic field-responsive carriers can also be used for rapid drug release brought on by the elevated temperature brought on by an alternating magnetic field (magnetic hyperthermia). These carriers normally include both therapeutic payloads and a magnetically active component. The most widely employed iron oxide nanoparticles as magnetic agents are those that are smaller than 10 nm, such as magnetite (Fe3O4) and maghemite (Fe2O3) nanoparticles. Because of their superparamagnetic properties, these nanoparticles are also known as superparamagnetic iron oxide nanoparticles (SPIONs). Ultrasound can be used for imaging at low frequencies (less than 20 kHz) or for destroying nanocarriers to release cargo at specified areas at high frequencies (more than 20 kHz). Additionally, by creating cavitation bubbles and producing heat, ultrasound can increase the permeability of biological barriers, including cell membranes and the blood-brain barrier, which will improve the drugs’ absorption by cells. Ultimately, light-which includes UV, visible, and infrared/NIR-represents an alluring external stimulus among the several triggers employed in biomedical applications because of its noninvasiveness, potential for remote control, and high spatiotemporal resolution. Furthermore, light-induced reactions can be controlled for a range of uses by varying exposure times, wavelengths, and intensities. In actuality, chromophores such azobenzene, pyrene, cinnamoyl, spirobenzopyran, or nitrobenzyl groups are usually included and/or conjugated to the polymeric structure to produce light-responsive polymeric carriers. Without a doubt, combining two or more distinct trigger types in a single delivery system can only be beneficial since it allows for precisely regulated drug administration and release, which enhances its anticancer effects in vitro and/or in vivo. As a result, platforms for multi-responsive therapy are beginning to emerge, allowing for the simultaneous or sequential occurrence of coupled sensitivities to various stimuli. The recent reviews by Zhou et al.[1322] and Mi[1324] provide detailed details of representative cases of stimuli-responsive nanocarriers based on trigger type and carrier morphology. Table 7 summarizes some common examples.
All of the aforementioned statements are unquestionably true for all of the many morphological types of BCP micelles that are now accessible, with polymeric vesicles providing more options for compartmentalization because of their inherent hollow nanostructure. Furthermore, polymersomes are appealing options for drug administration, diagnostic imaging, nanoreactor vessels, and artificial organelles due to their enhanced robustness and stability, as well as their chemical diversity that permits variable membrane characteristics and surface functionalization. One step further, stimuli-responsive polymersomes have been produced that may conduct “on demand” release in dose-, spatial-, and temporal-controlled ways in response to a variety of external physical or internal biological environmental stimuli. It should come as no surprise that polymersomes have been the subject of extensive scientific investigation in recent years as prospective biomimetic platforms appropriate for a broad spectrum of applications connected to biomedicine. Table 7 provides some typical instances of these systems; additional details can be found in a number of recent, in-depth reviews.[748,749,750,751,752,753,754,755,756,757,758] Utilizing electrostatic interactions to produce C3Ms with electrostatically binned payloads at their core is an alternate method of inducing self-assembly relevant to biomedical applications, aside from the standard hydrophobically formed nanostructures.[61,824,1362,1363] Numerous biologically significant macromolecules, including as genes, polysaccharides, proteins, enzymes, antibodies, nucleic acids, and so forth, are regulated by charge-charge interactions because they possess multiple charges. As a result, charged medicinal compounds can be simply added to the C3Ms core, and because of the micelle's sensitivity to pH and salt, its subsequent release can be trigger-controlled. Genes and proteins are the two main classes of charged bioactive entities meant to be delivered intracellularly for therapeutic purposes. Cell adhesion, immunological responses, cell signaling, and the cell cycle are all significantly influenced by proteins. Over the past few decades, there has been a noticeable growth in the use of protein therapeutics, including monoclonal antibodies, antibody fragments, peptides, replacement factors, enzymes, and vaccinations.[116,1364,1365] As a result, significant progress has been achieved in the development of protein delivery methods, such as PIC formulations, which effectively transport proteins into tumor locations and cells while shielding cargo proteins from unfavorable physiological environments. When it comes to gene delivery,[1366,1367] cationic dihydrogen bond bridges (DHBCs) are typically used for direct ionic complexation with negatively charged nucleic acids. In this scenario, the complex formed by the nucleic acid and the cationic block forms the core. On the other hand, nucleic acids can interact with polymeric micelles that contain a cationic corona to generate micelleplexes. The hydrophilic/cationic portion of the copolymer is often made up of PEI, PLL, or PDMAEMA blocks, whereas the hydrophobic core of amphiphilic BCPs used to build micelleplexes is composed of polyesters like PLGA, PLA, and PCL.[1368] A few noteworthy examples are also provided in Table 7, and the thorough explanations of PIC formulations pertinent to biomedical applications that have been recently reviewed by Magana et al.[61] and Jundi et al.[824] are also worthy of note. It is clear that there are countless ways to create functional, adequate, complex, and custom pharmaceutical formulations using BCPs. These drug delivery vehicles must meet the following requirements in order to be used: they must be nontoxic, have good biocompatibility, have a long half-life in circulation, have regulated release and biodegradability, and target specific cells. In order to be licensed for commercial use, these items also need to meet quality, safety, and efficacy standards set by regulatory authorities and organizations, as well being clinically and financially efficient. Thankfully, a large number of polymeric nanoformulations have previously received approval, moved forward to become commercialized products, or are presently undergoing clinical development and trials with encouraging outcomes for potential commercial use in the future, demonstrating the systems’ boundless potential.[1369,1370,1371,1372] C3Ms are well-suited as delivery vehicles for nanoscopic cargo because of their adjustable nanoscale size and morphology, unique capacity to partition hydrophilic material, and dynamic responsivity to environmental changes and stimuli. A significant obstacle for nanomedicine is the administration of treatments nonvirally, a field that has been developing for many years. Among medicinal nanoparticles, C3Ms are distinct since they are made entirely of hydrophilic components and are therefore extremely hydrated.[1373,1374,1375,1376] C3Ms do not have some of the severe limits on biodistribution that hydrophobically driven assemblies do, like accumulating in the liver.[1377] They also possess the rare capacity to sequester hydrophilic cargo, however only charged or modifiablely charged cargo is capable to doing so. By delivering therapeutic nucleic acids, proteins, and other materials, recent in vivo investigations employing C3Ms have set the stage for future applications in nanomedicine. Here are some highlights of promising engineering approaches that have been used to distribute and sequester different kinds of therapeutic biomacromolecules.[1377]
- 16.1.2.1. Nucleic Acid Delivery
Diseases can be treated using nucleic acids because of their high selectivity in either promoting or suppressing gene expression. Nucleic acids such as DNA, plasmid DNA (pDNA), messenger RNA (mRNA), small interfering RNA (siRNA), micro RNA (miRNA), and antisense oligonucleotides are typically introduced to achieve this.[825,1378,1379] Due to their large negative charge and vulnerability to physiological breakdown due to the presence of nucleases, nucleic acids are difficult to transport.[1380] The need for these treatments to pass cellular and occasionally nuclear membranes presents an additional difficulty for the delivery of nucleic acids.[1381] Nanoscale carriers composed of lipids, polymers, or even metals enable this transport; numerous platforms and delivery methods have been examined elsewhere.[155] Since C3Ms are easily formed by complexation between nucleic acids and diblock copolymers with cationic blocks, they are uniquely positioned to be ideal carriers for nucleic acids, even though features like size, composition, and surface properties can be tuned and engineered to provide effective biocompatible delivery platforms. Kataoka’s research[36] was among the first to focus on C3Ms nucleic acid carriers. Despite the fact that the main focus of that work was on the stability and formation of C3Ms with DNA, important advancements in in vitro and in vivo research have been made, demonstrating encouraging outcomes for site-specific targeting, effective cellular transfection, and the treatment of diseases and cancer.[37,113,1382,1383] Nucleic acids are arguably the easiest cargos for C3Ms to handle. As previously noted, molecular features can have a significant impact, but in their single-stranded form, DNA and RNA behave similarly to the linear charged polymers that are used to encapsulate them.[396,404,417] C3Ms have demonstrated potential in the delivery of antisense oligonucleotides,[285,1384] microRNA inhibitors,[1077,1385] small interfering RNA,[395,396,1386,1387] messenger RNA,[547,1345,1388] plasmid DNA,[547,1389,1390] and other biomolecules by means of cationic polyelectrolytes that sequester the intrinsically anionic nucleic acids in the micelle’s core, shielding the cargo from enzymatic threats and harsh environments. These investigations, which involve cellular delivery and animal models, are advancing C3Ms research toward practical uses in vaccination and gene therapy. When employing C3Ms as vectors for the transport of nucleic acids, the usual obstacles are their low bioavailability and potential loss of therapeutic efficacy due to polyion exchange interactions with physiologically occurring anionic biomacromolecules. This mostly necessitates the creation of stable micelles. Notably, the stability of polycations in nucleic acids is largely dependent on the charge-related stoichiometry of their mixing. The N/P ratio-the ratio of the amines of the polycations to the phosphates of the nucleic acids-is the definition of the charge ratio for carriers that particularly contain nucleic acids. According to recent research, C3Ms that have been overcharged and have larger N/P ratios (>1) create more stable micelles.[1346,1391,1392,1393] The higher N/P ratio micelles provide increased colloidal stability, especially with more stiff nucleic acids like mRNA, where packing them into noticeably smaller PEC cores is challenging.[1346] This is probably due to the increased quantity of binding sites that are accessible. Charge density and the cationic block's chain flexibility have both been demonstrated to have an impact on micelle stability.[1346,1391] Nucleic acid can be packed more tightly when there is a higher charge density. Fischer resonance energy transfer (FRET) was used in a recent study to compare the mRNA delivery potential of a pEG-pLK system with modified pLK side chains. This allowed for the identification of more compact packaging of mRNA with polycations that had higher charge densities and greater binding potential due to higher electrostatic interaction.[1391] The effectiveness of the previously employed method of disulfide crosslinking the PEC core for extra micelle stability was also compared in this study.[600] These incredibly long molecules are restricted to cellular nuclei by the packing of nucleic acids when complexed with histone to form chromatin; this mechanism has been duplicated to encapsulate nucleic acids in C3Ms cores.[1393,1394] It has been investigated how the lengths of the cationic and neutral blocks utilized affect the packing of DNA in C3Ms. Briefly put, the block lengths determine the shape of the resulting particles, which can be toroidal, rod-like, or globular.[1394] Micelles change from being globular to being rod-like as the length of the cationic block decreases.[1394] Due to DNA’s long persistence duration, this would not always be feasible.[1395] Jiang et al.[1393] demonstrated a hybrid system that is an alternative to packaging DNA. It is made up of three blocks: a hydrophobic, a cationic, and a neutral hydrophilic copolymer. The rigid pDNA that is employed, known as “micelleplexes” (Figure 73), connects several micelles to create a supramolecular assembly that resembles “beads on a chain”.[1393] It’s interesting to note that due of the increased distance between micelles, it was found that increasing the corona-forming block reduced the number of micelles per micelleplex. Two pDNA chains per complex, however, remained the same.[1393] According to this experiment, the length of the corona-forming block determines the carriers’ colloidal stability.
Solid-core micelles are expected to have irregular shapes because, as was previously indicated, complexes of polycations with more stiff chains (dsDNA) create precipitate-like complexes.[404] Rather than trying to modify the nucleic acid, a recent study examined the impact of changed chain flexibility of the cationic block on the encapsulation ability and micelle stability.[1346] Micelle stability was enhanced by flexible nucleic acids by increasing conformational entropy. According to Miyazaki et al., the polycation may also contribute to conformational entropy.[1346] According to that theory, the micelles that were created with flexible cationic blocks had better bioavailability and cellular absorption because they were more resilient to attack by anionic biomolecules like heparin.[1346] The significance of the PEC phase has been emphasized multiple times in this text. The cationic block’s longer chain flexibility is thought to enhance mRNA binding, albeit at the expense of hydration. This could be an additional instance where C3Ms that are kinetically confined are more resilient to adverse physiological conditions. In addition to variations in flexibility, ssDNA and dsDNA show variations in therapeutic efficiency. Nevertheless, during cell transfection, the adeno-associated virus has demonstrated ssDNA to active dsDNA conversion, indicating a potential mechanism for ssDNA’s therapeutic effectiveness.[1396] To increase the efficacy of dsDNA, several methods exist. Recent research, for instance, has shown that heating dsDNA to linearize it improved encapsulation stability and allowed for the production of smaller spherical micelles, which in turn allowed extravasation through the dense stroma of pancreatic cancer tumors.[1392] This work demonstrated gene transfection, the generation of anticancer effects, and the much larger amount of cargo that C3Ms can contain following linearization. This was demonstrated by contrasting the micellar system’s (11,000 base pair) capability with a viral vector’s (4800 base pair).[1392] Regretfully, the precise process of gene transfection was not disclosed by the authors of this paper.
The ability to blend many polymer segments into a single polymer chain facilitates the creation of customized designs and multipurpose micellar delivery systems. The creation of micelles made of branching polyethylenimine modified with PEG is one intriguing method. The micelles were employed to introduce small interfering RNA (siRNA) targeting transcription factor-1 into the lung (Figure 74).[1397] The goal of this type of therapy is to prevent lung-resident mesenchymal stem cells from differentiating into myofibroblasts in order to treat pulmonary fibrosis. The siRNA was successfully encapsulated in the copolymers of cationic PEI grafted with maleimide-PEG (PEI-g-PEG-Mal). Their surface was then further modified with a stem cell antigen 1 antibody (anti-Sca1 Fab/) to target lung mesenchymal stem cells (Figure 74).
Because they ablated the expression of an RUNX1 gene, the siRNA-loaded micelles modified with anti-Sca1 Fab/ aggregated preferentially in the lung and were able to decrease the myofibroblast development of lung-resident mesenchymal stem cells. Using a similar method, copolymers of PEG and cationic poly[N/-[N-(2-aminoethyl)-2 aminoethtyl]aspartamide] were able to further self-assemble into micelles by electrostatically binding mRNA.[1398] Studies on this micellar system in animals for Alzheimer’s therapy have moved forward. An imbalance between amyloid-beta peptide creation and breakdown in brain cells is the hallmark of Alzheimer’s disease. Protein neprilysin has the ability to regulate the peptides’ breakdown. The introduction of mRNA encoding neprilysin may make it possible to stop the pathogenic alterations in the brain and reduce the amount of amyloid-beta accumulation. According to research on animals, administering this micellar system containing the mRNA raised neprilysin levels, which in turn decreased amyloid-beta peptide concentrations. The micellar formulation’s effectiveness can be attributed to its ability to impede the digestion of mRNA by nucleases and the immune system’s detection of the biomacromolecule, both of which boosted the efficacy of mRNA distribution to the cells. Preclinical testing has begun on polymersome DDSs intended for BTA delivery. Polymersomes that were prepared from a poly(ethylene glycol)-b-poly(dithiolane trimethylene carbonate-co-trimethylene carbonate)-b-polyethylenimine triblock copolymers, for example, were functionalized with peptides derived from apolipoprotein E, demonstrating an exceptional capacity to cross the blood–brain barrier and intended for the treatment of glioblastoma, an incurable primary brain tumor.[1399] In animal tests, the polymersomes containing the therapeutic protein saporin demonstrated a strong and highly specific anticancer response against orthotopic human glioblastoma xenografts. Polymersomes made of 1,2-dioleoyl-3-trimethylammonium-propane block copolymers and polybutadiene-b-polyethylene glycol were created in a different recent work to co-deliver the SARS-CoV-2 spike protein with an immune-stimulatory adjuvant based on DNA nucleotides as a SARS-CoV-2 vaccine.[1400] The polymersome formulation produced neutralizing antibody titers in mice for as long as 40 days when given in two doses, according to the animal study, and the adjuvant's co-administration decreased the amount of SARS-CoV-2 spike protein needed.Smart membranes can be constructed in polymersomes by including functional polymer segments into the polymer chain. The membrane can then be functionalized with molecules that adjust its permeability and increase the mass transport of the active molecules, so controlling the release of the encapsulated molecules. It is particularly important to boost the membrane’s permeability while working with macromolecular therapeutic agents.[1401] Numerous methods have been devised to achieve this, including the incorporation of channel-forming transmembrane proteins, smart gates, or DNA nanopores into the polymersome membrane.[1401] For example, membrane proteins like aquaporin ionomycin were introduced into the membrane shell of polymersomes with selective membrane permeability, modeling them after biological cell membranes where different proteins facilitate ion transport.[1402,1403] To do this, the block copolymers can be combined with aquaporin, a naturally occurring membrane protein that forms pores as a result of their assembly as trimers, before the self-assembly process. A selective release of molecules is made possible by the proteins integrated into the membrane during the development of the polymersome.[1404] An additional option for controlled transport across polymersome membranes is offered by DNA nanopores.[1405] They can be created by oligonucleotides self-assembling, and they can create precisely sized holes in the polymer layer.[1406] After polymersomes were synthesized, DNA fragments were added and the polymersomes were then incubated with the fragments. Depending on the amount of oligonucleotides present, the DNA pores may tether to the membrane before completely inserting themselves. The resultant nanopores function as gates, regulating the release of payloads in a size-dependent manner.[1406] The release of medications and tiny molecules under control is made possible by the intricate engineering of polymersome membranes; nonetheless, one of the present problems is the release of biomacromolecules through polymersome pores. One way to address this is through the construction of smart membranes, which can include heterogeneous membrane portions with responsive and finely adjustable behavior.[1401] It is possible to regulate the membrane’s permeability by adding polymers that are responsive to external stimuli. The polymers employed in these situations have the ability to modify their characteristics in reaction to external stimuli, which may cause the membrane to inflate and the payload to release under regulated conditions. A prime example of a system that forms smart gates in polymersome membranes makes use of the responsive polymers poly(diethylamino)ethyl methacrylate (polyDEA) and polyNIPAM.[1401] Following the self-assembly in a tetrahydrofuran and water mixture, the pH-responsive polyDEA functionality is deprotonated and the thermoresponsive polyNIPAM dehydrates. Polymersomes with closed membrane gates are the outcome of this. The polyNIPAM-based section permits the membrane to be designed with a “boarding gate” that opens below the PNIPAM (20-30 0C) LCST. Biomacromolecules can be loaded under these circumstances, and the payload can be locked in place by closing the gate at physiological temperature (37 0C) (Figure 75a). The pH-sensitive polyDEA segments found in the polymersome membrane were intended to act as a “release gate,” allowing the encapsulated plasmid DNA to be released into an acidic environment (like endosomes) when the polyDEA is protonated. The polymersomes swell as a result of protonation, growing from 450 nm at pH 6.8 (Figure 75b) to 610 nm at pH 5.4. The successful release of the payload both in vitro and in vivo has been achieved through the employment of such polymersomes in the transfection of cells with plasmid DNA encoding GFP (Figure 75c).
A highly controlled platform is made available for the administration of BTAs via polymer micelles and polymersomes, which can accommodate a variety of therapeutic agents or a mix of them. One effective way to increase the half-life and get past physiological barriers without sacrificing the molecules' activity is to encapsulate BTAs in polymersomes. It is now possible to build sophisticated micelle structures with a variety of morphologies and compositions thanks to modern methods for synthesizing well-defined and regulated block copolymers. The resulting micelles have distinct interactions with biological systems, mostly because of their distinctive form, which allows for cellular uptake and targeting. Moreover, the dense polymer shell shields the therapeutic agents from denaturation and eliminates the need for harsh chemical changes for the encapsulation of the macromolecular therapeutic agents. Regretfully, because of the possibility of disruption in the non-covalent contacts that control the assembly, micelles, like other self-assembled systems, may experience intrinsically low stability in vivo.[1407]
16.1.2.2. Brain Delivery
- 16.1.2.2.1. Passing the Brain-Blood Barrier
The most common type of dementia and one of the most expensive neurological conditions is Alzheimer's disease (AD). There are no treatment approaches available at this time that can stop the AD’s ongoing neuronal deterioration.[1408,1409,1410,1411,1412,1413,1414,1415,1416,1417] Antibodies and their fragments hold great promise as therapeutic possibilities as they can reduce the toxic activity and aggregation of amyloid beta (Aβ) peptide,[1418,1419,1420,1421] which is the principal pathogenic mechanism responsible for dementia. Nevertheless, antibody therapies have not been very successful in the clinic, and their advantages in terms of neurodegeneration and cognitive decline have been negligible. The blood-brain barrier (BBB), a highly restrictive physiological barrier primarily made up of endothelial cells lining the brain microvasculature, is one of the main causes of the lack of therapeutic effect. Free antibodies’ ability to enter the brain is severely limited. Therefore, in order to effectively treat AD, methods for getting across the BBB and delivering therapeutic antibody compounds into the brain are desperately needed. The blood-brain barrier (BBB), a very restrictive physiological barrier primarily made up of endothelial cells lining the brain microvasculature,[1422] severely limits the ability of free antibodies to enter the brain, which is one of the main causes of the lack of therapeutic effect. Therefore, in order to effectively treat AD, methods for getting across the BBB and delivering therapeutic antibody compounds into the brain are desperately needed. Antibodies can be delivered across the blood-brain barrier (BBB) with nanocarrier systems, which have the advantage of a lengthy blood circulation period, high tissue penetration, the ability to install ligands, sensitivity to stimuli, and controlled toxicity.[37,154] Nevertheless, creating nanocarriers that can effectively bridge the blood-brain barrier and release antibodies into the brain parenchyma while retaining most of their bioactivity is still a difficult task. The safe passage of treatments and/or vehicles beyond the brain-blood barrier (BBB), a very selectively permeable endothelial cell membrane, has proven to be one of the more difficult problems in drug delivery. Promising findings from recent research indicate that PEC micelles can be transported across the blood-brain barrier (BBB) by conjugating a glucose transporter protein (Glut-1) with the micelle corona, as Figure 76 illustrates.[1423] Xie et al.’s work, which claims the highest brain accumulation of antibody agents reported to date, created antibody fragment (Fab) carrying micelles labeled with glucose transporter molecules to treat Alzheimer's disease by preventing the aggregation of amyloid beta (Aβ) peptides in brain parenchyma.[1423] Both in vitro and in vivo evaluations of the pEG-pLK system complexed with charge-modified Fab and partial disulfide crosslinking for stability were conducted.[1423] Micellar breakdown and 90% Fab release were achieved by using a reductive and acidic media, which simulated the environment in brain parenchyma and intracellular endosomes, respectively. Using a mouse model, the effectiveness of this system was investigated against a variety of controls. Results showed a 56% reduction in Aβ aggregation, a seven-fold increase in circulation time compared with free Fab, and approximately a 42-fold increase in Fab accumulation in the brain with almost no accumulation in peripheral organs. While targeting brain parenchyma, a different approach developed by Min et al. showed how to encapsulate and transport synthetic nucleic acid molecules called antisense oligonucleotides, which are intended to target and regulate RNA and protein.[285] Here, the Glut-1 transporter molecule was used in the translocation, which happened via the same route as the earlier investigation.
- 16.1.2.2.2. Potential Applications of C3M in Glioblastoma Treatment.
- 16.1.2.2.2.1. The Importance of Micelle-based Glioblastoma Multiforme (GBM) Therapy Implementation Hindrances
Astrocytomas, oligodendrogliomas, and ependymomas are the three types of malignant gliomas that make up the majority of primary brain tumors in adult patients.[179] The most common and lethal of these tumor types is GBM, a grade IV astrocytoma. According to Stupp et al.,[1424] following adjuvant temozolomide radiation, patients with GBM have a median survival of 14.6 months and a less than 10% 5-year survival rate.[180] Over the past ten years, not much has changed in terms of the standard of treatment for these individuals. A few other therapies, including oncolytic viruses, liposomal doxorubicin, and anti-angiogenic drugs like bevacizumab, have also been used with varying degrees of clinical success.[181,182,183,184] A growing number of clinically implemented nanoparticle systems are being made available to cancer patients. For instance, liposomal doxorubicin formulations (e.g., Doxil®, Caelyx®) are being tested for efficacy in patients with GBM and brain metastases from solid tumors[189,190] and are currently being used for patients with a variety of cancers.[191,192,193,194] To boost the dispersion of a drug injected intratumorally, patients have also been treated using novel delivery techniques such convection-enhanced delivery (CED).[185,186] However, there is currently no known “cure” for this illness, highlighting the need for both innovative drug delivery approaches and an improved knowledge of the underlying disease process in GBM. Another class of nanoparticles entering the clinical domain are polymeric micelles.[1425] As mentioned earlier, these amphiphilic nanoparticles have proven to be capable of delivering a variety of therapeutic agents, such as proteins, siRNA, DNA, and chemotherapy drugs.[369,1426,1427,1428,1429,1430,1431,1432,1433,1434] It is concerning that these micelle formulations have not yet been used to treat GBM, despite the fact that they have been effectively used to treat a wide variety of solid tumors in preclinical and clinical settings. Nonetheless, there are a few noteworthy obstacles that might be impeding their use in the context of GBM treatment:
- Due to the CNS’s location of these cells, systemically delivered therapeutic medicines are unable to reach the target malignant tissue without first passing through the BBB. Many therapeutic drugs still do not reach significantly hazardous levels within tumors, despite the existence of a weakened vasculature that may exacerbate the intratumoral EPR impact.[188]
- Necrotic and hypoxic tissue can be found in certain regions of GBM tumors, while neovascularization can be found in other locations. Hypovascularization, fibrosis, and necrotic pockets are the main reasons for reduced intratumoral drug distribution, whereas hypervascularized regions promote accumulation in the surrounding tissue. It's critical to comprehend the distribution of nanoparticles throughout tumors since certain cell populations, such self-renewing GBM cancer stem cells that sustain a tumor, may be restricted to particular vascular habitats.[1435,1436]
- Depending on how it is administered, therapeutic delivery has inherent flaws. Many systemically administered therapeutic drugs produce non-specific organ toxicity and are quickly cleared from circulation by reticuloendothelial cells. Thus, enhancements in the duration of drug circulation and the precision of targeting represent significant advancements for this mode of delivery. The delivery of various dose regimens to a patient and high interstitial pressures that result in poor molecular dispersion limit intratumoral administration of therapeutic medicines.[1437]
- While these clinical micelle formulations improve the potency of the medicine in several solid tumor types, they do not yet have any targeting moieties that could enable increased accumulation in brain tumors or the central nervous system. To increase the effectiveness of currently available formulations, it could be necessary to target alternative receptors expressed on glioma cells molecularly.
- Systemic toxicity may unavoidably be a problem if high dosages of given micelles are required to guarantee sufficient intratumoral accumulation because they lack a controlled-release capability. In order to minimize non-specific release prior to reaching the tumor location, it would be ideal to include stimulus-triggered releasing mechanisms of encapsulated compounds, which would enable release only inside the tumor area.
- 16.1.2.2.2.2. Glioma-Specific Targeting Moieties
As was previously established, one of the main (perhaps the biggest) drawbacks of systemically delivered unmodified micelles is their poor BBB penetration. Despite the possibility of some particle buildup at the site of a brain tumor due to the EPR effect, the administered medication concentrations might still be subtherapeutic. Improving localization to a tumor by covalently attaching targeting molecules to the surface of drug-loaded micelles is one way to address this problem. Table 8 provides an overview of the targeted moieties listed below as well as others that were not covered.
Using the Arg-Gly-Asp (RGD) tripeptide is one such example of a technique for targeting gliomas. High affinity binding of the RGD peptide has been observed for αvβ3 integrin, a receptor that is overexpressed on tumor cells and the tumor vasculature.[1438,1439] The cyclic RGD peptide (cRGD) has been used in a number of studies’ micellar systems to target GBM. A cRGD-PEG-PEI polymeric micelle was created by Zhan et al.[1430] to transport the gene for tumor necrosis factor-related apoptosis-inducing ligand (pORF-hTRAIL). With the application of this targeting moiety, targeted gene transfer with a greater gene transfer efficiency in an intracranial U87 mouse model could be accomplished than with unmodified particles. These mice’s life times were extended (23.5 vs. 19 days; p < 0.05) and their levels of TRAIL expression were elevated as a result of this targeted approach.[1430] Subsequently, mice with intracranial GBM tumors were given cRGD-PEG-PEI/pORG-hTRAIL particles together with PEG-PLA micelles modified with candoxin-derived peptide and loaded with paclitaxel.[1431] Since it has been demonstrated to bind nicotinic acetylcholine receptors expressed on the BBB, a candoxin derivative was selected.[1460] Combining the two drugs was found to boost the apoptotic impact because paclitaxel increased the transfection of the TRAIL gene into U87 cells.[1431] In particular, Jiang et al.[1471] investigated, in an intracranial U87MG mouse model, the penetrating depth of cRGD-modified poly(trimethylene carbonate)-based micelles delivering paclitaxel into glioma tissue as well as the dispersion of systemic particles following intravenous injection. It was discovered that cRGD alteration improved micellar penetration into cerebral tumors in vivo and into U87MG glioma spheroids in culture. Additionally, compared to non-modified micelles containing paclitaxel (27 days, p = 0.012) and Taxol (23 days, p < 0.001), these particles increased the median survival of U87MG glioma-bearing mice (32 days).[1471] In order to facilitate [64]Cu labeling and PET imaging, Xiao et al.[1444] created cRGD modified micelles coupled to doxorubicin (via a pH-sensitive hydrazone link) and 1,4,7-triazacyclononane-N,N′,N′′-triaceticacid (NOTA), an amacrocyclic chelator. With the use of a front flank U87MG mouse model, the in vivo particle distribution could be quantitatively measured thanks to this technique. 4 h after the cyclic-RGD modified micelles were injected, the tumor showed 5.7% ID/g, which was much greater than that of the non-modified micelles. In addition to the tumor site, the liver, kidney, lungs, and intestines showed the highest levels of particle accumulation.[1444] The targeting kinetics of cRGD-modified micelles containing ultra-sensitive SPIO nanoparticles were also investigated by Kessinger et al.[1442] Using a one-compartment pharmacokinetic model, αvβ3-specific accumulation of these particles was seen in subcutaneous U87 tumors during the first five minutes of treatment, with an accumulation rate of 0.24 min−1.[1442] Fibrin deposits in the stroma and vasculature of tumors present another intriguing target for nanomedicines. According to Bardos et al.,[1447] these deposits are present throughout primary and metastatic brain cancers. Lately, cysteine-arginine-glutamic acid-lysine-alanine (CREKA) peptide-modified micelles have targeted these deposits. When compared to micelles without the CREKA alteration, Chung et al.[1448] showed that Cy7-labeled CREKA-micelles may accumulate more in GL261 glioma bearing mice at 3 and 24 hours after treatment. Using CREKA-micelles, future research may try to deliver therapeutic medicines in GBM animal models in an effort to see if this increases survival. Adding transferrin (Tf) to the structure of micelles is yet another targeted tactic.[1445,1456,1457] Normally, endothelial cells use a Tf receptor-mediated pathway to deliver Tf into the central nervous system.[1453] Tf-modified polyphosphoester hybrid micelles containing paclitaxel were created by Zhang et al.[1457] to treat mice with intracranial U87MG tumors. Compared to mice treated with Taxol (33.6 days, p < 0.01), mice treated with Tf-modified micelles exhibited significantly longer survival (39.5 days). Greater %ID/g reaching brain tissue with transferrin functionalization was seen in biodistribution experiments. But compared to other human organs including the liver (∼15%ID/g), spleen (∼5%ID/g), lungs (∼4%ID/g), and kidney (∼5%ID/g), this amount (∼2.5 X 10−4 %ID/g) was minuscule. Thus, penetration into the central nervous system remained low even after functionalization. To capitalize on a tumor-specific targeting mechanism as well as a BBB crossing pathway, the same group also created Tf-modified micelles loaded with cRGD-paclitaxel conjugates.[1445] In mice with intracranial U87MG tumors, this method resulted in a significantly longer mean survival time (42.8 days) than Tf-modified paclitaxel-loaded micelles (39.5 days, p < 0.05), paclitaxel-loaded micelles (34.8 days), and Taxol (33.6 days). Once more, biodistribution tests demonstrated enhanced intratumoral accumulation of particles (∼0.7%ID/g at 4 h) when both targeting components were utilized; however, these levels were still insignificant compared to the amounts that reached the lungs (∼4% ID/g), spleen (∼4% ID/g), and liver (∼10%ID/g). Aptamers have also been utilized in addition to peptide-based moieties to help target glioma cells. Gao et al.[1432] functionalized micelles using GMT8 aptamers, which were demonstrated to bind to U87 cells alone. The aptamers were chosen using the cell-based systematic evolution of ligands by exponential enrichment (SELEX) approach.[1472] When loaded with docetaxel, aptamer-modified micelles showed an improved mean survival time of 40 days, which was significantly longer than that of unmodified micelles carrying docetaxel (35 days, p < 0.05) and free docetaxel (30 days, p < 0.05).[1432] These modified micelles were also more effective at penetrating U87 tumor spheroids.
- 16.1.2.2.2.3. Therapeutic Micelle Delivery to Brain Tumors
Micelles can be administered locally or systemically at the location of a brain tumor. Each approach has benefits and disadvantages of its own. Because systemically delivered drug-loaded micelles are relatively non-invasive, they are an appealing delivery strategy. Figure 77 shows how systemic distribution of particles can lead to their entry into the brain tumor and the central nervous system (CNS) through both endothelial cell trafficking into the parenchyma and the EPR effect. However, surface modification is necessary for this mode of administration since, following intravenous injection, unaltered micelles may be quickly removed from the circulation by antibody opsonization. The reticuloendothelial system’s macrophages ingest these opsonized particles, trapping them in the liver or spleen and reducing their therapeutic efficiency.[1473] Micelles have been designed with a smaller size (usually less than 100 nm), hydrophilic blocks like PEG,[1474,1475] or an extra coating surfactant[1476] to prevent such absorption. The hydrophilic surface of this material greatly reduces macrophage identification and complement activation. As a result, their circulatory circulation time increases significantly, and these particles are better delivered to the tumor burden.[187] Even though modified micelles have been shown to successfully cross the blood-brain barrier, accumulate within a tumor site, and improve animal survival following intravenous administration, they still have significant drawbacks, including uneven intratumoral distribution because of variations in the vasculature within a GBM tumor and widespread particle deposition in non-cancerous areas like the liver, spleen, and lungs. Alternative delivery methods seek to get around these restrictions. Kanazawa et al.[1477] showed that, following intraperitoneal administration, coumarin 6 was delivered to the brains of rats developing C6 gliomas using micelles modified with the cell penetration peptide Tat. In either hemisphere of the brain, 1.6% ID/g was seen around one hour after intranasal delivery.[1477] Notably, intranasal distribution of micelles did not result in preferential accumulation within the brain’s tumor side, indicating that this delivery mechanism was unrelated to the EPR effect. A “nose-to-brain” micellar delivery method for neuroprotective peptides targeted against Alzheimer's diseases was created by Liu et al.[1454] PEG-PCL micelles were coupled with lactoferrin. After being administered intranasally, this group observed that lactoferrin-modified micelles loaded with coumarin-6 localized to the olfactory bulb, olfactory tract, hippocampus, cerebellum, and cerebrum. In a Morris water maze experiment, this approach increased choline acetyltransferase activity and decreased acetylcholinesterase. It also improved memory performance. This approach implies that a comparable setup including the distribution of glioma toxic ants may be a potential path for investigation, even though it was not utilized in a brain tumor model. Local delivery of drug-loaded micelles can produce substantially larger quantities of nanoparticles at the tumor site than intravenous or intranasal delivery. CED, a technique that uses a pressure gradient to supplement local diffusion in order to achieve an efficient intratumoral distribution of the injected compound, can solve the frequently observed issues of injection backflow and limited intratumoral diffusion using this method.[1478,1479] Consequently, compared to local diffusion alone, CED can treat broader brain areas.[1480] Despite being intrusive, this treatment method has been effective in treating patients with gliomas in several clinical trials.[1481,1482] In glioma xenograft models, preclinical findings using CED for paclitaxel-loaded nanoparticle administration have shown improved animal survival.[1483] However, there are a few significant drawbacks to this strategy. First, large volume infusions almost always result in elevated intracranial pressure. Secondly, even while CED promotes a more uniform intratumoral spread, medication distribution remains erratic. Finally, because prolonged intracranial infusion exposes more brain tissue to the outside world, it may result in local infection. Loading these therapeutic compounds into neural or mesenchymal stem cell carriers provides an alternative to use CED. According to Thaci et al.[1484] and Huang et al.,[1485] these vehicles have intrinsic immunosuppressive and tumor-tropic qualities that can result in intratumoral dispersion and targeted administration to infiltrative tumor locations without being harmful to non-neoplastic tissues. Stem cells have been exploited as carriers for several nanoparticle-drug conjugate systems, even though they haven't been particularly utilized for the delivery of micelles.[1486,1487,1488]
- 16.1.2.3. Drug Delivery
The biological actions of the majority of systemically delivered medications are felt both at the target and non-target locations, which frequently leads to unwanted side effects and reduces the therapeutic potential of the treatments. This highlights how crucial drug delivery systems (DDS) are for delivering biologically active substances to the diseased location in a targeted manner. Since the side effects of cytostatic and other medications can be extremely harmful, targeted drug delivery is especially important for the treatment of life-threatening disorders like cancer.[422,423,424] Thus, the creation of drug carriers that permit selective and tissue-specific therapeutic targeting is one of the primary research problems in DDS. Drug carriers that serve this purpose should ideally have a high drug loading capacity, sufficient circulatory stability, extended circulation, selective accumulation at the site of action, a suitable drug release profile, and acceptable biocompatibility. A wide range of nanoscopic drug carriers have been developed as a result of intensive research conducted in the last 25 years to find the perfect, all-purpose drug carrier system.[1490,1491] As their name suggests, nanoscale dimensions (usually ranging from 10 to 200 nm) characterize nanosized drug carriers, which are classified into two groups: particulate systems and water-soluble macromolecular systems. Lipid-based systems like liposomes,[1492,1493] surfactant-based systems like emulsions,[1494,1495] and synthetic polymer-based systems like nanoparticles,[1496,1497] polymeric micelles,[43,430,637] and polymeric vesicles[1498,1499] are all included in the first group. Polymer-drug conjugates[1500,1501] and dendrimers[1502,1503] are included in the second group of nanoscopic drug carrier systems. Numerous clinical evaluations are being conducted to show these systems’potential for patient treatment, and some of them are currently available on the market.[455,1504]
- 16.1.2.3.1. Polymeric Micelles and Vesicles: Their Characteristics
As mentioned earlier, polymeric micelles, which consist of amphiphilic block copolymers, are self-assembling core-shell nanostructures that are created in an aqueous solution (Figure 77).[270,1505] The core of the micelles is usually formed via hydrophobic interaction between the hydrophobic blocks of the copolymers, however other interactions, such electrostatic interaction[283] and stereocomplex formation,[574] can also be used as the driving force for the core formation. The micelle shell is formed by the hydrophilic blocks of the copolymers, which also serve to stabilize the micellar structure. Amphiphilic block copolymers can also form vesicular assemblies, depending on preparation circumstances and polymer content (Figure 78). These are referred to as “polymersomes” because they resemble liposomes in that they have an aqueous interior surrounded by a bilayer structure. Polymersomes are thought to be more robust, resilient, and adaptable than lipid vesicles.[1498] Both hydrophilic and hydrophobic medicines can be transported by polymersomes (in the interior) and in the bilayer.
The use of polymeric micelles as a drug delivery mechanism was first demonstrated in 1984 by the H. Ringsdorf group.[1506] Later, in the early 1990s, Kataoka employed this technique by creating doxorubicin-conjugated block copolymer micelles.[436] The first description of polymeric micelles containing drugs non-covalently integrated was made by Kabanov.[1507] Due to their attractive properties that meet the requirements for selective drug delivery, polymeric micelles are currently being researched intensively as a prospective nanoscopic drug carrier.[425,426,427,431,434] Most remarkably, hydrophobic medications can be accommodated in high quantities by the hydrophobic core. Although polymeric micelles were once thought to be most suited for intravenous (i.v.) administration, they are now also being studied as an oral drug delivery strategy.[1284,1287] The submicron size of i.v. administered polymeric micelles (usually ranging from 10 to 100 nm) allows them to bypass biological barriers in the human body that arise upon oral administration, such as limited gastrointestinal absorption and high hepatic first-pass effect, and allows them to potentially reach the pathological sites. When the concentration of the block copolymer rises above a particular value, known as the critical micelle concentration (CMC) or critical aggregation concentration (CAC), micelles or vesicles form in aqueous solution. A vesicular or core-shell micellar structure is formed when hydrophobic segments of block copolymers join at the CAC or CMC to reduce interaction with water molecules. Polymeric micelles usually have a CMC of approximately 10− 6 to 10− 7 M, whereas low molecular weight surfactant micelles have a CMC of approximately 10− 3 to 10− 4 M.[425] This suggests that at low concentrations, C3Ms are less prone to dissociate than surfactant micelles. Given that C3Ms are diluted when administered intravenously, having a low CMC is beneficial for preserving the micellar structure, which enables extended bloodstream circulation.
Polymeric micelles should preferentially deliver their payloads to the target areas following intravenous delivery. It is well recognized that this kind of targeting, for example, to tumor tissues, is significantly aided by the so-called increased permeability and retention (EPR) effect.[458,459] Maeda et al. first hypothesized the EPR effect in the 1980s, and it is caused by two things. First off, because of its discontinuous endothelium, the blood vessels in angiogenic tumors and other diseased tissues have a higher permeability than those in normal tissues. Second, research has demonstrated that tumors lack completely established lymphatic drainage. These characteristics cause colloidal particles, or polymeric micelles, to enter tumors and other inflammatory tissues through the “leaky” epithelial layer and stay there. It should be noted that the polymeric micelles must circulate in the bloodstream for a substantial amount of time in order to accomplish this “passive” targeting. Since they dictate their biological destiny, the surface characteristics of polymeric micelles are significant in this regard. Reducing the rate of non-specific identification and uptake by the reticuloendothelial system (RES) is crucial for extended circulation. Grafting hydrophilic polymers (polyethylene glycol, poloxamer, etc.) onto particle surfaces has been demonstrated to be an efficient means of preventing opsonization and the consequent uptake by the RES cells found in the liver, spleen, and bone marrow.[464,506] Nevertheless, a number of variables, including particle size, surface layer properties, and rigidity, affect the precise biodistribution and pharmacokinetics of polymeric micelles.[467,486,1508,1509] Although polymeric micelles are typically removed from experimental animals’ systemic circulation within the first 8 to 10 hours following intravenous administration,[433,434] poly(ethylene glycol)-b-poly(D,L-lactide) micelles demonstrated appropriate circulation times, with 25% of the injected dose still being detected in the blood after 24 hours.[472] The polymeric micelles’ virus-like size allows them to enter the targeted tissue and internalize into the cells through processes including fluid-state endocytosis, even in the absence of a targeting ligand on their surface.[358,442,1510] But “active” targeting is achieved by affixing a particular ligand-such as antibodies-to polymeric micelles that are filled with drugs. The drugs are delivered directly into the cells through accelerated internalization of the loaded vehicles, which occurs through ligand–receptor contact with the target cells.[1511] Moreover, adding stimulus-sensitive sections to the block copolymers produces nanoparticles with controlled release mechanisms in the environment (see infra). It is important to remember that polymeric micelles eventually split into single block copolymer chains in the body, raising concerns about their toxicity. Because block copolymers for micellar applications typically have a molecular weight of less than 50,000 g/mol, the body cannot accumulate them over time because of renal clearance.[431,621] Moreover, polymeric micelles, such as poly(lactic acid) or poly(ε-caprolactone), that aid in their removal can be created using biodegradable building blocks. Finally, the ease with which polymeric micelles can be sterilised using 0.2 μm filtering provides a benefit for pharmaceutical development.
- 16.1.2.3.2. The Building Blocks
Polymeric micelles and vesicles are often designed using amphiphilic block copolymers of the A-B type, where A denotes a hydrophilic block and B denotes a hydrophobic block. A-B-A triblock copolymers[1512] and graft copolymers[84,1513] are two more examples. Because it is a non-toxic polymer with FDA approval for use in a variety of pharmaceutical formulations and because of its special physicochemical properties (high water solubility, high flexibility, and large exclusion volume) that provide good “stealth” properties, poly(ethylene glycol) (PEG) is most frequently used as the hydrophilic segment of copolymers.[465,481,1514] Other polymers, such as poly(N-vinyl-2-pyrrolidone) (PVP)[513] and poly(acrylic acid),[527] can also be utilized as the shell-forming segment. On the other hand, a greater range of polymers have been investigated as the hydrophobic segment in polymeric micelles. These include poly(propylene glycol) (PPO, Pluronics®),[488] poly(aspartic acid) with chemically conjugated doxorubicin (PAsp(DOX)),[498] poly(β-benzyl-L-aspartate) (PBLA),[1515] and poly(ester)s like poly(lactic acid) (PLA),[336,469] poly(ε-caprolactone) (PCL),[549,570] and poly(trimethylene carbonate) (PTMC).[550] This explains why so many core-forming hydrophobic polymers have been used in the development of polymeric micelles: the choice of the core-forming segment is the major determinant for important properties of polymeric micelles, such as stability, drug loading capacity, and drug release profile (described in more detail in the next section).
- 16.1.2.3.3. Polymeric Micelles: Loading, Retention, and Release of Drugs
Numerous drugs, including doxorubicin (DOX),[436,437,439,440,441,442,1516] paclitaxel (PTX),[336,358,443,444,445,446,447,448,1517] cisplatin,[497,1518] amphotericin B,[449] indomethacin,[1519,1520] photosensitizers,[451,452,515] and novel cytotoxic drug candidates, such as KRN 5500,[1521,1522] have been loaded into polymeric micelles. Drugs can be chemically conjugated or physically trapped into polymeric micelles. However, there are a number of drawbacks to a drug’s chemical conjugation to the core-forming block: i) the drug must contain reactive groups; ii) the coupling reaction may be harmful to the drug's activity; iii) the drug should be released as close to the target site as possible, such as through enzymatic or chemical hydrolysis; iv) the type of conjugated drug determines the final polymer properties and micellar characteristics; and it is preferable to have a platform technology where the polymer backbone alone determines the micellar behavior, independent of the drug to be encapsulated. Drugs can be physically loaded into the core of polymeric micelles using a variety of techniques. Organic solvent loading approaches, such as o/w emulsion,[427] dialysis,[1523] and solid dispersion,[1517,1524] are typically employed in loading operations. The efficacy of a polymeric micelle release mechanism that is unique to the target site is contingent upon the reduction of drug release from the micelles prior to their arrival at the target site, as seen in Figure 79.
Drugs that are physically entrapped in polymeric micelles can be released through diffusion through the micellar core and partition coefficient over the micellar core and aqueous phase, as long as the micelles don’t break. Very tiny diffusion constants of 10− 18 to 10− 16 cm2/s[1525] indicate a sluggish release of pyrene from polymeric micelles with a glassy core, such as poly(styrene) (Tg = 100 °C) and poly(tert-butyl acrylate) (Tg = 40-43 °C). However, the rate at which pyrene was released from poly(2-vinylpyridine)-b-PEG, whose core in the experimental conditions resembles a liquid, was too quick to evaluate.[1525] Therefore, if the goal of polymeric micelles is targeted drug administration, they should ideally have a glassy, solid core at body temperature. The length of the polymer segment that forms the core, the affinity of the drug for the core (i.e., the partition coefficient between the aqueous phase and the core), and the amount of loaded drug are other factors that impact drug release. Research shown that when the molecular weight of the PCL block and the amount of indomethacin entrapped increased, the pace at which indomethacin was released from PCL-b-PEG micelles reduced.[559] A strong affinity between the drug and the block copolymer’s core-forming segment-measured, for example, by the Flory-Huggins interaction parameter[426] – allows for the most stable drug loading. Pyrene partition coefficients, for instance, range from 102 for Pluronics®,[1526] 104 for PBLA-b-PEG,[1527] and 105 for PS-b-PEG.[564,1528] A suitable choice of block copolymer can maximize a drug’s affinity for the hydrophobic core. According to research, PTX-loaded polymeric micelles with a PLA core did not exhibit improved PTX target distribution in vivo when compared to Taxol.[371,450] In contrast, PTX-loaded polymeric micelles with a polyaspartate core modified with 4-phenyl-1-butanol significantly increased PTX's plasma AUC (90-fold vs. Taxol) and tumor AUC (25-fold vs. Taxol).[444] Based on these findings, it appears that a micellar core with more hydrophobic and PTX-compatible groups (such phenyl groups) is more suited for a robust interaction with PTX and its long-term retention in vivo. Poly(N-(6-hexylstearate)-L-aspartamide)-b-PEG (PHSA-b-PEG) for amphotericin B,[561,573] poly(aspartic acid-co-phenylalanine)-b-PEG for diminazene aceturate,[563] and poly(C16-benzyl-L-aspartate)-b-PEG for KRN 5000[1521] are further instances where the drug and polymer compatibility has been optimized. Covalently attaching a medication to the core-forming block opens up an additional way to enhance the drug’s physical entrapment. Clinical study of this formulation (NK911) is currently underway. This was demonstrated for PAsp(DOX)-b-PEG, which stably incorporated DOX by π–π stacking between conjugated and non-conjugated molecules.[498,1529] A fascinating method for creating a polymer with a core-forming block that has a strong affinity for the medication to be entrapped was recently disclosed by Park et al. The most efficient hydrotropic agent for PTX was discovered to be N,N-diethylnicotinamide (NNDENA), after a significant number of hydrotropic agents were evaluated to determine their capacity to increase the solubility of PTX in water.[562] Afterwards, block copolymers of NNDENA, 2- (4-(vinylbenzyloxyl)-N,N-diethylnicotinamide (DENA monomer), and their monomers were produced using PEG. As a result, PTX was up to 37% (w/w) soluble in the resultant pDENA-b-PEG block copolymer, forming 100 nm-sized polymeric micelles that remained stable for months without leaking or precipitating PTX.[445]
- 16.1.2.4. Delivery of Therapeutic Proteins
C3Ms have also been used to encapsulate and deliver proteins, in a manner similar to that of nucleic acid encapsulation and delivery. Proteins are very specialized and effective in treating illnesses, but, similar to nucleic acids, they are easily broken down by enzymes. Proteins differ greatly in terms of charge density, charge distribution, and hydrophobicity since they are made up of different amino acid sequences. The implication is that encapsulation in C3Ms cores may be mediated by interactions other than electrostatic ones, and that they are not just susceptible to external factors like pH and salt content. Furthermore, as is extensively covered in other studies,[115,134] proteins can act as polyion scaffolding for C3Ms. A recent study examined the impact of hydrophobicity on protein encapsulation and salt stability by modifying the monomer branches of the anionic block of a block copolymer with different lengths of hydrocarbon spacers.[1530] This led to an effective increase in chain flexibility as well as hydrophobicity. More hydrophobicity and enhanced stability against salt have recently been shown by a group, but in bulk complexes.[1139] This was also noted for the C3Ms that had been hydrophobically changed; however, the charge distribution produced opposing effects that led to insufficient binding. It was discovered that there was an initial decrease in salt stability when spacer length increased, i.e., from pAA to poly (acryloylaminooctanoic acid) (pAAOA), because shorter spacers could not stretch between the surface charges of the protein. However, this was followed by an increase in salt stability with pAAOA because the spacers were long enough to allow for better binding. The stability against acidic pH, on the other hand, was the opposite, allowing for better release in cancer cell spheroids. Another recent work examined nonelectrostatic interactions, including hydrophobic contacts, which were shown by a divergence from the charge stoichiometric ratio of polyanion to polycation upon the inclusion of a third component having hydrophobic branches.[1136] The goal of this work was to introduce a second polyelectrolyte, pSS, with the same charge as the protein in order to increase the stability of C3Ms as protein carriers. When pSS increased, the amount of protein encapsulated reduced and the protein was preferentially rejected from the complex with additional salt,[1136] despite the fact that the stability of the micelles against salt did improve. Additionally, encapsulation through electrostatic mediation has improved. While adjusting the pH can change the charge distribution of the protein,[1136] this could have unfavorable impacts on protein folding and, consequently, the molecule’s therapeutic efficacy. Increasing the charged block of the block copolymer’s charge density is one way to increase electrostatic interactions.[1531] Short poly (L-glutamic acid) chains were grafted along the charged block to accomplish this. When kept at 4 0C, the micelle system demonstrated protein loading stability, maintained bioactivity for at least a month, and allowed for delayed release kinetics due to enhanced binding.[1531] Nevertheless, interactions with the oppositely charged polymers may negate therapeutic activity even though no deliberate modifications are performed to the protein, as observed with melittin and poly (methacrylic acid).[1532] The design parameters of protein delivery platforms should take this impact into account as it might be unique to certain combinations of polymer-proteins.
Therapeutic protein delivery has in fact been demonstrated via C3Ms. In a particular instance, the protein Sprouty 1, which has the ability to suppress angiogenesis naturally, was introduced into a C3Ms consisting of albumin as the charged component and a pegylated polymer as the neutralizing stabilizing block. The goal was to develop a breast cancer therapy system that would be effective.[1533] Reduced levels of brain-derived neurotrophic factor (BDNF) are hypothesized to be the cause of poor neurological recovery, depression, and cognitive impairment following strokes. This protein was delivered by condensing the block copolymer PEG-PGA with BDNF.[1048] Before the polymer was incubated with the proteins, the authors conducted molecular simulation simulations to determine the electrostatic interaction and H-bonding nature of the contact points between the polymer and the protein. The mixture was injected intravenously and had the potential to build up in the brain.[1048] After a stroke, mice’s brain tissue loss was considerably lessened by the C3Ms that delivered BDNF as compared to free BDNF.[1534] Based on catalase, C3Ms have been directed towards Parkinson’s disease. C3Ms were formed with catalase using PEI-PEG. The end product, which the scientists called nanozymes, were non-toxic C3Ms that ranged in size from 60 to 100 nm and shielded the enzyme from hydrolytic breakdown. To take advantage of the receptors expressed on the surface of these cells, the resultant nanoparticle was loaded into bone marrow macrophages as a cell-based drug carrier. The enzyme was gradually released from the cell after a few days of being fully absorbed by the cells in just one hour.[1535] These carriers may be used to treat Parkinson’s disease because nanozyme loading had no effect on the macrophages’ levels of α4-integrin.[1536] When compared to free catalase, it was demonstrated that the nanozymes were more readily absorbed by rat astrocytes, neurons, and murine brain microvascular endothelial cells (BMVEC).[1537] Then, employing a non-degradable (bi(sulfosuccinimidyl)suberate sodium salt) and a degradable (3,3/-dithiobis(sulfosuccinimidyl propionate) linker, this catalyse-loaded C3Ms was stabilized by crosslinking. In mice, the crosslinked nanozyme administered via macrophages decreased inflammation of the nervous system.[1538] The key to treating disorders affecting the central nervous system and reducing damage to brain tissue is reducing oxidative stress. Angiotensin II (AngII)-induced illnesses like hypertension and heart failure have been found to be less affected by the enzyme copper/zinc superoxide dismutase (CuZn-SOD), which scavenges superoxide. The delivery of the enzymes in PEI-PEG micelles was found to be more effective than the enzyme alone in reducing the generation of peroxide in neurons.[1539] A rat model of middle cerebral artery occlusion (MCAO) was used to evaluate the same system, but with the enzyme now crosslinked to stabilize it. The amount of oxidative damage was decreased as a result of the enzyme delivery. In rat models, the C3Ms loaded with crosslinked CuZnSOD was able to decrease the size of infarcts and enhance motor performance.[1540] Further in-depth analysis of the rat brain was conducted to comprehend how the crosslinked PIC micelle was able to exhibit this increased therapeutic impact. The blood vessel lumen contained the nanoparticles, but they were unable to get through the blood–brain barrier. The buildup of the nanoparticle in the infarct zone was thought to be responsible for its therapeutic action.[1541] After it was discovered that the polymer being employed was very cytotoxic, PEG-PAsp(DET) was utilized instead, as it was not only less toxic but also showed less accumulation in the liver and spleen.[1056]
The precise interactions between polyelectrolytes and biological entities, such as cells and tissue, are not as well understood, despite the numerous successful demonstrations of medicinal administration employing PEC micelles that have been made, as mentioned in this article and elsewhere. While it would be great to be able to detect these events in living biological systems, such animal models, there does not appear to be a noninvasive method at this time. On the other hand, a novel use of light-sheet microscopy on 3D multicellular spheroids provides a way to achieve deeper penetration with less light intensity than previous techniques (fluorescent lifetime imaging microscopy and confocal laser scanning microscopy).[1542] A fluorescently labeled cationic protein (hen egg white lysozyme) was complexed with two variants of an anionic diblock copolymer, one of which contained a crosslinked PEC core and the other did not. Confocal laser scanning microscopy, when combined with the conventional method of incubating the micelles in two-dimensionally cultivated human breast cancer cells, revealed that the cells eliminated the protein while retaining the polymer. The discovery made with light-sheet microscopy on a 3D cancer spheroid (400 mm diameter) was strikingly different from this outcome. The absence of crosslinking in the micelles indicated that the polymer was retained at the spheroid surface and that the protein was distributed uniformly throughout the spheroid. This suggests that the micelles completely disassembled, most likely as a result of acidic environments and other competing charged species. On the other hand, the crosslinked micelle revealed some protein and complete micelles inside the spheroid, with notably less polymer on the surface.[1542] It is also evident from this work that there are no clear similarities between 2D cell culture studies and physiological applications, despite the elegant demonstration of a new method for examining 3D cellular environments. This method has also been used by a few other studies covered in this article to look into comparable therapeutic distributions.[1530,1532] This technique could provide insight into the destiny of many PEC micelles in various applications, as there is limited knowledge on the internal trafficking of exogenous therapeutics and carriers in thick tissue environments.
16.1.3. Diagnostics, Imaging and Theranostics: Combination of Diagnosis and Treatment
C3Ms have other applications beyond drug transport, such as disease monitoring and detection. C3Ms’ capacity to encapsulate coordination metal complexes was demonstrated in work by Wang et al.,[1543] which supported the use of C3Ms as MRI contrast agents. After complexing with a cationic block copolymer, the metal ion and an anionic ligand created a coordination structure. A cationic block copolymer that lacked stability when salt was added was combined with a smaller, more flexible anionic ligand with twofold symmetry in their earlier work.[1544] Because a ligand with a higher metal coordination complexity is employed in this instance, the degree of crosslinking per supramolecular assembly is increased along with its rigidity and availability of binding sites. Coordination of ligands with Mn, Fe, Zn, and Ni demonstrated the system's adaptability. Two hours following a tail vein injection, strong MRI contrast was observed in vivo using a mouse model, with the liver and kidney exhibiting the most brightness.[1543] Nguyen et al. provide another illustration of the encapsulation of inorganic species within PEC micelles. In this instance, magnetite nanoparticles (Fe3O4) are templated using polystyrene-graft-poly (2-vinylpyridine) and subsequently complexed with poly (acrylic acid)-block-poly (2-hydroxyethyl acrylate) to form C3Ms with Fe3O4- nanoparticles in the core.[1545] It was demonstrated that these micelles exhibited up to 80% cellular internalization and produced cytotoxicity with a somewhat low level of 91% cell viability. The objective of this research was to induce and control localized hyperthermia using radiofrequency magnetic fields in order to induce apoptosis in sick tissue, such as cancerous tumors.[1545] Following internalization and rinsing, exposure to the magnetic field resulted in hyperthermia, where cell viability dropped from roughly 75% to roughly 30% with increased exposure and increasing micellar concentration.[1545] This approach not only caused the targeted killing of cancer cells while preventing any major macroscopic heating, but it also demonstrated high stability over a pH range of 3-9.5. These days, the focus of breakthroughs in nanomedicine is on building multifunctional nanocarriers that may be used for both therapeutic and diagnostic purposes. This has made it possible to create what are known as theranostic systems.[1546,1547,1548] When it comes to implementing such complex systems, polymeric nanocarriers can be of great assistance because of their ability to encapsulate drugs and bio-imaging agents on the same platform. This allows for the simultaneous monitoring and treatment of diseased tissues while preserving the pharmacological result. Clinical applications have made extensive use of a variety of imaging techniques, including optical imaging, nuclear imaging, single-photon computed tomography (SPECT), positron emission tomography (PET), X-ray computed tomography (CT), ultrasound, photoacoustic imaging (PA), etc. A direct, easy, and real-time method of obtaining anatomical or molecular information on organs and tissues is offered by molecular imaging, which has numerous uses in illness screening, organ positioning, and therapeutic impact monitoring. To visualize the target area, contrast agents are the foundation of most traditional bioimaging techniques. The majority of clinically accessible imaging probes, however, are currently insufficiently sensitive for a precise diagnosis and lack selectivity and targeting to certain malignant areas. When it comes to delivering probes and bioactive compounds to tumor sites for molecular imaging and therapy, BCP nanoassemblies have a lot to offer (high stability, biocompatibility, simple formulation, co-loading multiple payloads, controllable size and morphology, etc.).[1546] Among the available molecular imaging modalities, magnetic resonance imaging (MRI) and optical imaging are the two most commonly used that have greatly profited from the application of polymeric nanocarriers (see Table 7 for pertinent examples).[1369,1546,1548] Because optical imaging is noninvasive and robust near-infrared fluorescence imaging devices have been developed, it has seen exponential growth recently. In an effort to get the best possible imaging within the human body, a number of widely accessible fluorescent probes and dyes have been encapsulated within various polymer nanoconstructs. With its excellent spatial resolution that permits tissue investigation without the risk of ionizing radiation, magnetic resonance imaging (MRI) is another popular nonintrusive method utilized in a range of therapeutic imaging procedures. Hydrogen atoms are excited by an appropriate resonant frequency magnetic field, which is then applied. The excited hydrogen atoms return to equilibrium and generate a radio-frequency signal, which can be recognized and translated into an image. Different tissues exhibit varying rates of relaxation, making it easier to distinguish between healthy and injured tissues. Greater MRI sensitivity is necessary for an accurate diagnosis and prognosis of early-stage malignancies. SPIONs and/or paramagnetic gadolinium (Gd)-based nanoparticles can be used as contrast agents to increase the sensitivity. As a result, BCP-based carriers can offer an anticancer medication and magnetic nanoparticles together as diagnostic instruments for MRI cancer detection.[1548] Of course, combining the appropriate contrast agents can always result in multi-modal imaging. Creating a single, multipurpose platform for an all-encompassing approach to the clinical care of cancer and other comparable, life-threatening disorders is the ultimate goal of pharmaceutical nanotechnology. For these reasons, theranostic devices offer a way to image the degree of the illness, administer treatment, and track the effectiveness of real-time therapies. This allows for the proper drug dosage to be given to the right place at the right time.[1369] Naturally, this has led to the proposal of ever-more-complex nanocarrier systems that incorporate several interventions against a tumor. For instance, Xiao et al.[1549] recently described the production of a PLGA-PEI-mPEG copolymer functionalized with the cRGD peptide (cRGD-PLGA, for short), which is used to encapsulate superparamagnetic iron oxide (SPIO) and doxorubicin (DOX) for MRI-guided cancer therapy (Figure 80).
The resultant multicompartment micelles demonstrated good integrin-targeting and pH-responsive drug release characteristics under physiological settings. Furthermore, when compared to DOX medication treatment, the cRGD-PLGA-SPIO@DOX micelles showed enhanced anticancer activity and decreased toxicity in vivo. These micelles can function as a theranostic agent for real-time treatment monitoring, according to the study’s findings. In a similar vein, Deng et al.[1550] created and synthesized a triBCP, or poly(fluorene)-b-PNIPAAM-b-poly(oligo(ethylene glycol) monomethyl ether methacrylate) (PF-b-PNIPAAM-b-POEGMA) that is capable of self-assembling into micelles. Its structure is comprised of an inner, hydrophobic PF moiety for drug encapsulation and fluorescent tracking, a thermosensitive middle shell of PNIPAAm block for thermo-modulated drug loading and release, and an outer corona of the POEGMA segment that is hydrophilic, which helps stabilize the micelle. It's interesting to note that the DOX-loaded micelles made at 25 0C had a higher drug loading capacity than their 37 0C counterparts due to the former formulation's improved stability, which increased the drug’s in vitro cytotoxicity against HeLa cells. This formulation has considerable potential for cancer theranostics, as demonstrated by the integration of a localized hyperthermia-triggered drug release profile and effective intracellular trafficking of the nanocarriers by measuring the fluorescence of the PF moiety. The case study by Ke et al.[1551] is particularly noteworthy as it showcases a nanoreactor that employs a PEG-b-poly[(thioketal-linked camptothecin methacry- late)-co-(2-[pentamethyleneimino] ethyl methacrylate)] (PEG-b-P[CPTKMA-co-PEMA]) BCP polymersome. The polymersome is loaded with glucose oxidase (GOD) in the inner aqueous cavity and membrane, respectively. In order to initiate cascade events for coordinated cooperative cancer treatment, the nanoreactor demonstrates tumor acidity-responsive membrane permeability (Figure 81). Tumor acidity-responsive membrane permeability can specifically activate the cascade reactions, which include fast iron ion release, radical-triggered rapid release of parent drugs, Fenton reaction between H2O2 and iron ion to produce hydroxyl radicals, and glucose consumption to generate H2O2. Through in situ consumption and production of chemicals, high-efficiency tumor suppression is achieved through the coordinated use of chemotherapy, chemodynamic therapy, and starvation therapy in this process of coordinated cooperative cancer therapy. An innovative paradigm for accurate combinatorial cancer therapy is represented by this nanoreactor, which is intended for tumor-activable cascade reactions. Table 7 provides other instances of multimodal/multifunctional systems that integrate diagnostic and therapeutic modes. Each of these examples exemplifies the countless opportunities for building extremely complex theranostic systems.
16.2. Nanofabrication
Directed synthesis of inorganic nanoparticles (NPs) is an intriguing use of BCP self-assembly techniques.[1552,1553,1554] Because of their distinct qualities that set them apart from their bulk counterparts, inorganic NPs have recently become the subject of intense study interest across a wide range of disciplines. A crucial component of nanoparticle creation is controlling the size and form of the particles. One of the most dependable methods for precisely adjusting the size and form of nanoparticles is to utilize BCP templates during the synthesis of inorganic NPs.[1554] NPs are often created via a top-down or bottom-up approach. The first explains how the application of powerful external forces breaks down bulk materials, leading to the formation of nanoparticles. Top-down approaches typically call for harsh circumstances, which can be expensive. In addition, it might be challenging to further functionalize or regulate the size and form of the resultant nanoparticles. Bottom-up approaches involve nucleating and growing particles, which allows for precise control over the size and morphology of the resultant nanoparticles (NPs) by manipulation of the crystallization process’s direction and duration. Controlling the dispersity of NPs in solution and their size and shape are the two most important issues associated with their synthesis and application. This is because NPs have a strong tendency to congregate in solution due to their high surface energy and huge interfacial area. The inability to manage the size and shape remains a problem, despite the emergence of “grafting-to” and “grafting-from” technologies to passivate NPs surfaces and so inhibit aggregation. Thus, it has been proposed to use BCPs as nanoreactors for NP production. NPs are prevented from aggregating by the capping ligand of the other polymer block, while the functional groups in one block of polymers provide a guided location for particle nucleation and growth in the approaches based on BCP templates. Furthermore, it is feasible to modify the size and form of the generated NPs by varying the lengths of each block.[1554] By adhering to these guidelines, the Schmidt group demonstrated the synthesis of hybrid organic-inorganic core–shell Au NPs in a single step or phase. This process was made possible by amino-functionalized amphiphilic BCPs, which serve as both a reductant and a stabilizer. To be more precise, they employed a pH-responsive triblock terpolymer called PEO-b-poly(2,3-dihydroxypropyl methacrylate)-b-poly[2-(diisopropylamino)ethyl methacrylate] (PEO-b-PDHPMA-b-PDPAEMA) that enables direct chemical cross-linking of the formed micellar structures. The aqueous dissociation equilibrium involving tertiary amino groups, the Au(III) speciation, and electrochemical redox potentials was discovered to be reliant on the production of Au NP.[1555] Similarly, ZnS nanoparticles with a PS-b-P2VP reverse micelle-templated substructure have been reported by Podhorska et al. They consist of a larger (∼20 nm) amorphous organic–inorganic hybrid matrix surrounded by tiny crystallites (∼4 nm). The creation of this structure was explained by the fact that, as opposed to the amount of accessible metal ions, the radius of the metal-loaded core mostly determines the NP size.[1556] An additional instance akin to this one involves the utilization of core-cross-linked PS-b-P4VP reverse micelles in the Zhang et al. study to produce Au and Pd thermally stable nanoparticles.[1557] There have also been reports of more complicated morphologies than core-shell spherical micelles. For instance, the Kim group saw Au NP-Pluronic BCPs complexes produce spherical, cylindrical, and vesicular self-assembled structures in aqueous solution.[1558] In a similar vein, Sohn et al. described a nanoscale morphology-regulation method using square planar Pt compounds to create self-assembled double hydrophilic PAA-PEG BCP.[1559] Normal core-shell and crew-cut spheres gave way to anisotropic pearl-string formations when PAA was varied block-length with constant PEG. Conversely, significant hydrogen bonding caused by Pt adsorption on PEG blocks might further alter the molecular geometry of metal-chelated unimers by reducing the volume of hydrophilic segments, which will ultimately cause a shape shift to vesicular structures. The architecture of the BCP is equally significant, as demonstrated by the ABA and BAB triBCPs with PEGMA and 2-(diethylamino)ethyl methacrylate (DEAEMA) units, which were utilized as templates to fabricate Au NPs and produced micelles with Au NPs in their core that were either core-shell or “flower-like.”[1560] Apart from linear, easy in situ synthesis of Au NPs has also been achieved using miktoarm “star-like” β-cyclodextrin-based copolymer unimolecular micelles.[1561] It appears that the ability of BCPs to self-assemble offers a versatile approach for the controlled creation of a wide variety of useful materials with various ordered structures. Because of their capacity to absorb, store, and interact with guest species on their exterior/interior surfaces as well as in the pore space, a variety of mesoporous materials that are appropriate for energy storage and conversion have also been prepared using this soft-templating technique in recent years.[1562] The creation of nano-catalysts with the use of BCP templates is very intriguing, since it has a wide range of potential uses.[1563]
16.3. BCP Self-Assembly Applications in Ionic Liquids (ILs)
16.3.1. Soft Actuators
Electronic and ionic electroactive polymers (EAPs) are two types of EAPs that can alter their size and form in response to voltage variations.[1564,1565] The coulombic pull between the electrodes causes electronic EAPs to react. These actuators provide lengthy room temperature endurance, quick reactions, and comparatively high actuation forces. The enormous voltage (41 kV) needed to activate electronic EAP actuators is a significant barrier, though. On the other hand, because of the driving force that is regulated by ion migration and diffusion processes, ionic EAP actuators can function at low voltage ranges (1-2 V). The issue of employing volatile solvents, which hinder long-term operation in an open atmosphere, may be mitigated by the use of ILs with low vapor pressure as a medium for ion transport. High ionic conductivity, mechanical integrity, and solution processability are among benefits of using ion gel-based BCPs as materials for ionic EAP actuators. At a low voltage (2 V), the rectangular-shaped bending movements of the manufactured ionic EAP actuator utilizing PS-b-PMMA-b-PS/[EMIM] [NTf2] ion gel were demonstrated (Figure 82A).[1566] A dielectric double layer forms on the electrode surface as a result of the ion transfer between cationic and anionic electrodes via ILs under voltage changes, which ultimately results in a bending motion.[1567] The behavior of the swelling anionic and zwitterionic ABA triblock copolymers in ILs and their use as a medium in ionic EAP actuators were investigated by Long et al. following this finding.[1568,1569] Park et al. reported actuators made of zwitterionic molecules and single-ion conducting diblock copolymers that could bend huge motions and respond quickly (tens of milliseconds) (Figure 82B).[1570] Under the influence of an electric field, the applied sulfonated polystyrene-nonionic polymethylbutylene (PSS-b-PMB) diblock copolymer was able to construct microphase-separated assemblies with only cations moving. Furthermore, the inclusion of imidazolium-type zwitterion (ZImS) improved the ionic conductivity. The latter may make the solution more polar and make it easier for cation ions to migrate and dissociate (Figure 82C).
16.3.2. Electrochemical Applications and Devices
Such a system is a promising candidate as an electrolyte in next-generation electrochemical devices,[1571] such as lithium-ion batteries,[1572] solar cells,[1573] electro-mechanical actuators,[1574,1575] electrolyte-gated transistors, and light-emitting electrochemical cells[1576,1577] due to the ionic conductivity and electrochemical stability of the self-assembled ABC triblock in ILs. Because pyridine, ether, and acrylate have the ability to solvate a variety of ions, the most often utilized block copolymers are those that contain a PEO, PMMA, or P2VP block.[1578] Examples of the use of BCPs/ILs systems in various applications are shown in Table 9.
16.3.3. Lithium-Ion Batteries
Lithium-ion batteries (LIBs) typically use liquid electrolytes made of polar organic solvents and dissolved lithium salts. Although organic solvents have low interfacial resistance and great ionic conductivity, their flammability has raised significant safety issues. Because of their high ionic conductivity and lack of flammability, ILs may be desirable alternatives to organic solvents.[1580,1581] Another problem with liquid/electrolyte LIBs is electrolyte leakage, which may be resolved by using polymers like poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP). Lithium salts have been demonstrated to gel when PS-b-PMMA-b-PS triblock copolymer is combined with common ILs (Figure 83).[1582] Other block copolymers, including PS-b-PEO-b-PS and PS-b-poly(n-butyl acrylate)-b-PS, have been used in addition to PS-b-PMMA-b-PS triblock copolymer to produce gel-electrolyte-based LIBs.[1583] The results obtained from the use of triblock copolymers have demonstrated that the B middle block rule is critical and has a significant impact on battery performance.[1583] Furthermore, by creating robust physical crosslinks, substituting poly(lithium acrylate-r-acrylic acid) ionomer blocks for the PS end-block improves the mechanical strength and electrochemical characteristics.[1584]
16.3.4. The Electrolyte-Gated Transistors
PS-b-PEO-b-PS triblock copolymer was employed in the first documented electrolyte-gated transistors based on self-assembly of BCPs/ILs systems in [BMIM]PF6 and [EMIM][NTf2].[1576,1585] The self-assembled BCPs/ILs systems demonstrated a notable improvement over the traditional solid polymer-electrolytes, as evidenced by the improved gate capacitance at higher frequencies and the rapid polarization response (Figure 84A, B). Furthermore, BCPs/ILs systems can create thin-film transistors using transfer printing,[1586] spin coating,[1587] and aerosol jet printing[1576] because BCPs are physically cross-linked. Only semiconductor materials that are resistant to organic solvents, which are employed as cosolvents for ion gel printing, can be used to overcome the challenges of fabricating thin ion gel films since the ion gels must be built directly over the semiconductor. Iwasa et al. reported PS-b-PMMA-b-PS copolymer-based flat ion gel films for this purpose by adjusting the spin-coating conditions and organic solvents (Figure 84C).[1588] In one instance, they deposited chemical vapor deposition-grown molybdenum disulfide (MoS2) onto a flexible polyimide substrate, and then they poured ethyl propionate-dissolved PS-b-PMMA-b-PS/[EMIM] [NTf2] ion gels over the substrate. Under bending conditions, the resulting film showed remarkable flexibility without electrochemical deterioration (Figure 84D).
Weinheim. (C) Atomic force microscopy image of the thin, flat PS-b-PMMA-b-PS/[EMIM][NTf2] ion gel sheet and chemical structures of the SMS triblock copolymer and ILs. Adapted with permission from ref 1588. Copyright 2012, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (D) The ion-gel-gated flexible MoS2 EDLT is shown, along with the transfer properties prior to (black), during (red), and following bending (blue). Adapted with permission from ref 1589. Copyright 2012, American Chemical Society.
16.4. Other Applications
Apart from the aforementioned medicinal uses, PEC micelles have also been investigated in other settings, including biosensing. As was previously mentioned, a variety of external factors can affect the structure of C3Ms, and stimuli-responsive characteristics, like ligands on the corona, can be produced. These characteristics make them highly versatile biosensors.[1590,1591,1592] An alkaline phosphatase enzyme was recently encapsulated in a C3Ms with a functionalized corona that would undergo crosslinking when exposed to UV light.[1593] When exposed to UV light, the micelles, which were initially colloidally stable, precipitate and form a thin layer on a substrate. The utilized enzyme exhibits fluorescence upon reconstitution with Zn2+, thereby serving as a highly selective heavy metal sensor for Zn2+, with a sensitivity as low as 50 ppb.[1593] Catalysis is another field in which C3Ms find application. The reactants’ ability to reach wide surface areas is one of the more important characteristics of catalysts. C3Ms are excellent choices for nanoreactors to enhance catalytic activity because of their great colloidal stability and capacity to produce a very large surface area per mass of material. Recently, electrostatic interactions were used to enclose gold (Au) nanoparticles in C3Ms. Water contaminant 4-nitrophenol is known to be efficiently reduced by Au nanoparticles acting as catalysts. 8 minutes were required for the full conversion of 4-nitrophenol to 4-aminophenol at a molar ratio of 0.00125 of Au in Au-containing C3Ms. Lately, C3Ms have also been employed in templating to control the silica mesostructure by adjusting the pH to modify the silica condensation rate, the pEG-silanol interactions, and the polyelectrolyte interactions.[1594] Different pH values changed the aforementioned interactions and resulted in the production of a variety of mesoporous structures, ranging from cylindrical pore morphologies at higher pH levels to long-range organized 2D hexagonal structures at lower pH levels. These structures have the potential to be used in adsorption, separation, and catalysis due to their high-order porosity.
17. Micellar Formulations in Clinical Trials
Polymeric micelles have been employed as carriers of strong chemotherapeutic drugs to target various types of solid tumors in several clinical trials conducted in the past ten years. We provide an overview of these clinical studies’ findings in the section that follows, focusing on the effectiveness of different micellar formulations.
17.1. Genexol-PM and NK105
According to Gonge et al.,[1425] Genexol-PM is a micellar formulation consisting of a monomethoxy-PEG-block-poly(D,L-lactide) copolymer with paclitaxel embedded in its hydrophobic core. The absence of Cremophor EL, a hazardous surfactant typically employed in the therapeutic formulation of Taxol to solubilize paclitaxel in circulation, is a significant benefit of this formulation. Numerous Phase I and II clinical studies have looked into the application of Genexol-PM in the treatment of solid tumors. Kim et al.[446] treated patients with lung, colorectal, renal cell, breast, ovarian, and esophageal malignancies with Genexol-PM in a phase I clinical research. 3 patients (14.3%) out of 21 had partial responses, while 6 patients (28.6%) had stable disease. Myalgia, sensory neuropathy, and neutropenia were among the toxicities that were dose-limiting.[446] Lim et al.[1595] treated patients with breast, head & neck, lung, and nasopharyngeal cancer with a modified dose regimen (weekly delivery rather than once every three weeks) of Genexol-PM. Of the 21 patients, 9 (42.9%) had stable illness and 5 (23.8%) had partial responses.[1595] According to Lee et al.,[1596] among 41 patients included in a phase II clinical trial evaluating Genexol-PM’s effectiveness in treating metastatic breast cancer, 5 (12.2%) saw complete responses, 19 (46.3%) had partial responses, and 13 (31.7%) had stable illness. Even with a median follow-up of 17 months, the study’s median overall survival was not met. In the setting of this illness, it would have been intriguing to observe how this formulation affected brain metastasis; however, CNS metastases were one of the study’s exclusion criteria.[1596] Kim et al.[1597] employed cisplatin and Genexol-PM in combination to treat advanced non-small cell lung cancer in another phase II clinical research. 20 patients (29.0%) had stable illness, and 26 patients (37.7%) had a partial response. The study found that a median of 21.7 months was the overall survival span. Kim et al.[1597] noted that the absence of Cremophor EL in this formulation allows for the possibility of administering greater dosages of paclitaxel without experiencing an increase in toxicity. Additionally, in a phase II clinical trial, Genexol-PM was evaluated against advanced pancreatic cancer.[1598] The mean duration of survival for patients receiving 300 or 350 mg/m2 was 6.5 months. Of the 45 patients receiving this dosage, 1 (2.2%) had a full response, 2 (4.4%) had a partial response, and 24 (53.3%) had stable disease. There are other therapeutic micelle formulations containing paclitaxel besides Genexol-PM. According to Gong et al.,[1425] NK105 is a core-shell micelle made of PEG and poly(aspartic acid) that has been treated with 4-phenyl-1-butanol to make it more hydrophobic. Hamaguchi et al.[444] examined the effectiveness of treatment in a colorectal cancer xenograft mouse model in a preclinical investigation. In nude mice transplanted with HT-29 colon cancer cells, NK105 had better anti-tumor efficacy than free paclitaxel; also, NK105's tumor area under the curve (AUC) was 25 times larger than that of free paclitaxel.[444] Hamaguchi et al.[711] examined the use of NK105 as a treatment for pancreatic, bile duct, and colon cancer in a phase I clinical research. 6 (31.6%) of the 19 patients had stable disease, and 1 patient each had a partial response (mean of 10.5%). 2 patients had metastatic stomach cancer and pancreatic cancer. According to Hamaguchi et al.,[711] the AUC and total clearance rate of NK105 at 150 mg/m2 were approximately 32 times higher and 72 times lower, respectively, than those of Genexol-PM at a dose of 300 mg/m2. This suggests that NK105 is more stable in circulation. In a phase II clinical trial, NK105 was also administered to patients with advanced or recurring gastric cancer.[1599] 2 (3.6%) of the 56 patients who were eligible for an effectiveness evaluation had full responses, 12 (21.4%) had partial responses, and 17 (30.4%) had stable disease. The average duration of survival was 14.4 months. July 2012 saw the start of a phase III clinical trial using NK105 to treat breast cancer; however, no updates have been provided to date (NCT01644890).
17.2. SP1049C AND NK911
Furthermore, doxorubicin has been encapsulated into micelles for usage with solid tumor patients. For this objective, a polymeric micelle called SP1049C was created, which is composed of a poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) block copolymer.[1425] Several solid tumors, including colorectal, esophageal, lung, ovarian, kidney, and hepatic malignancies, as well as soft-tissue sarcoma, mesothelioma, neuroblastoma, cholangiocarcinoma, and Ewing’s sarcoma, were treated with SP1049C in a phase I clinical trial.[1600] 8 patients (31%) had stable disease; about 21 patients were evaluable for response, but none showed complete or partial responses. Patients with advanced esophageal and gastroesophageal junction adenocarcinomas were included in a phase II clinical trial with SP1049C.[1601] 9 (47.3%) of the 19 patients who were eligible for evaluation had partial responses, while 8 (42.1%) had mild responses or stable disease. The compound's primary toxicity was determined to be neutropenia, with a median overall survival of 10 months. NK911, a different micelle formulation including PEG-poly(Asp) block copolymers linked to doxorubicin, is another example of a microcapsule encapsulating the drug.[1425] NK911 was utilized to treat a number of different solid tumors, including leiomyosarcoma, pancreatic, colonic, esophageal, gall bladder, and stomach cancer, in a phase I clinical study.[454] Of the 23 patients, 8 (34.7%) had stable disease, while 1 (4.3%) showed some degree of response. Similar to SP1049C, the main hematologic hazard was neutropenia.
17.3. NC-6004 AND NC-4016
For therapeutic uses, different medications have been added to micelles in addition to paclitaxel. Following systemic injection, NC-6004 (NanoplatinTM) is a polymeric micelle consisting of PEG and poly(glutamic acid) with integrated cisplatin. Normally, this micelle is quickly excreted by the kidneys, resulting in nephrotoxicity.[1425] In a mouse model of colon adenocarcinoma 26, human gastric cancer (MKN-45) harboring mice, and HT29 oxaliplatin-resistant bearing mice, NC-6004 demonstrated strong anti-tumor activities.[497,1602,1603] NC-6004 was utilized in a phase I clinical trial to treat a number of solid tumors, including melanoma, esophageal cancer, lung, colon, hepatic, pancreatic, and renal cancer.[1604] 7 (41.2%) of the 17 patients had stable disease. High dosages of NC-6004 still resulted in renal impairment, but overall, toxicities were less severe and less common than those caused by cisplatin. NC-4016, a formulation including PEG and a coordination combination of poly amino acid and 1,2-diaminocyclohexane platinum (II), is another micelle platform for the administration of a platinum-based drug.[1425] March 2009 saw the beginning of a phase I clinical trial,[1425] but no updates have been released to date.
17.4. NC-6300 and NK102
Just on the horizon could be another micelle system intended for clinical application. According to Takahashi et al.,[1605] mice with human hepatocellular carcinoma xenograft tumors were administered NC-6300, an epirubicin-incorporated micelle. When NC-6300 was administered at doses of 10 and 15 mg/kg, mice’s survival rates significantly increased as compared to control and epirubicin-treated mice. According to Takahashi et al.,[1605] the micellar formulation also seems to lessen the cardiotoxicity that epirubicin often causes. Unfortunately, neither clinical trials nor extensive preclinical testing in suitable glioma animal models have been conducted using these previously stated micelle formulations in patients with GBM. On the other hand, NK012, a PEG-poly(Glu) block copolymer micelle containing covalently attached SN-38, has demonstrated potential in the management of malignant gliomas. As a topoisomerase I inhibitor, CPT-11 (Irinotecan) is the active metabolite of SN-38.[1425,1606,1607] Kuroda et al.[1608] used a U87MG xenograft mouse model to assess the effects of NK012 against CPT-11 therapy. NK012 was 34-44 times more powerful than CPT-11 in vitro, according to research using five distinct human glioma cell lines. According to in vivo investigations, mice treated with NK012 (30 mg/kg/day) had a noticeably longer lifespan than mice treated with CPT-11 (66.7 mg/kg/day; p = 0.0014) or control (p = 0.001). Kuroda et al.[1609] conducted a follow-up trial to investigate the effectiveness of NK012 ± bevacizumab. When NK012 monotherapy (30 mg/kg/day) was administered to mice with orthotopic intracranial tumors (U87MG) instead of any combination of CPT-11 and bevacizumab (40 or 66.7 mg/kg/day CPT-11 + 5 mg/kg/day bevacizumab; p < 0.05), the mice showed larger gains in survival. NK012 mice and those given NK012 with bevacizumab did not vary from one another.[1609] These experiments’ in vivo bioluminescence investigations are shown in Figure 85.
Regarding clinical trials, a phase I clinical research of NK012 was carried out for the treatment of lung malignancies (small cell, carcinoid, and non-small cell) as well as colorectal, pancreatic, and esophageal cancers.[1610] Two patients (8.7%) had partial responses and five patients (21.7%) had stable disease out of the 23 patients that were evaluable for response. Out of 16 evaluable patients treated with NK012, 2 (12.5%) had partial responses, and 10 (62.5%) had stable disease, according to Burris et al.[1611] in another phase I trial. Nevertheless, no micelle-drug combinations have been employed in clinical trials to target GBM or other types of brain cancer, similar to the other micelle formulations.
Conclusion and Perspective
I have discussed current progress toward treatments as well as breakthroughs in customized C3Ms with an emphasis on their physical and dynamic properties. However, C3Ms’ remarkable potential to advance nanomedicine through improved biodistribution, targeting, and controlled release for uses in gene therapy, gene editing, and protein- or peptide-based drug delivery is far greater than their current level of success. Building on these and other medical research endeavors, C3Ms for nanomedicine will play a significant part in meeting the growing need for nanotherapeutics in the future. Versatile biomolecule carriers are becoming increasingly important, as highlighted by the COVID-19 pandemic. Nanoparticle delivery holds promise not only for vaccinations but also for gene silencing, monoclonal antibody treatment, and small molecule immunotherapy therapies. Although many obstacles still need to be overcome, C3Ms research has shown that these tasks can be completed with entirely hydrophilic components for enhanced distribution, customized particle size, shape, stability, release, and surface alterations. Although C3Ms have demonstrated efficacy in cellular delivery, more research is necessary to fully understand the processes involved. Therapeutic C3M efficaciousness will be further enhanced by further advancements in cell targeting. Delivery to cells that overexpress the folate receptor,[1612,1613] such as breast cancer cells, can be mediated by attaching folate to the corona. It has been shown that sequence-defined peptides can be targeted to inflammatory vascular endothelial cells.[1077] Similarly, RGD peptides have been shown to biodistribute, target cancer cells that upregulate integrins, and enhance cell adhesion.[1614,1615,1616] It is also necessary to define C3M function and stability in the endosome's mildly acidic environment. Because it is susceptible to acidic environments, RNA in particular has to be preserved during this phase. The cargo must ultimately exit the endosome and be liberated from the C3M into the cytoplasm, which presents a significant risk for C3Ms. However, initial research indicates that endosomal escape and nucleic acid release can be programmed with promising results employing endosomolytic peptides[1617] and membrane rupture caused by deprotecting cations.[252,1618] Amidst the extensive C3M design space that we have covered here, the primary tool is the selection of physical and chemical polymer properties. When creating a delivery system that must safeguard and discharge cargo in intricate situations, these efforts are essential. Numerous studies have demonstrated that strengthening a complex through increased charge density or polyelectrolyte length will enhance salt resistance. The McCormick group has demonstrated that longer polyelectrolytes, albeit with a time delay due to greater binding constants, boost C3M effectiveness when utilizing siRNA for gene knockdown and silencing applications.[395] Furthermore, improving siRNA release through lowering polyelectrolyte binding strength by decreasing charge density increases C3M susceptibility to enzymatic degradation, which in turn reduces cell transfection effectiveness.[396] Moreover, it is widely known that cytotoxicity increases with increasing polycation molecular weight or charge density.[1619,1620,1621] While designing therapeutic micelles, polymer selection is crucial for striking an appropriate balance between managing cytotoxic effects, transfection efficiency, and release kinetics. We have evaluated the effects of polymer choice on C3M size, stability, and efficacy in core-forming blocks; however, polymer choice for neutral blocks is rarely investigated, probably because the existing standards are effective. Better alternatives are rarely investigated. PEO is the most frequently used neutral hydrophilic polymer that forms nanoparticle coronas. It is readily soluble in aqueous solutions, commercially available, and exhibits little concern in vivo. Zwitterionic polymers have been added to C3Ms as net-neutral blocks recently due to their strong biocompatibility, hydrated lubricating capabilities, and superior antiprotein resistance.[1094,1622] Since protein adsorption to the corona is a major mechanism influencing nanoparticle expulsion from the bloodstream, protein-resistant corona materials provide a pathway to improved nanoparticle stealth.[1623] A deeper comprehension of the functioning of the zwitterionic corona and how it affects the transport of biomolecules would be beneficial to this research. C3Ms have already proven to be an effective platform for the delivery of nucleic acids, a range of proteins, and several small molecule drugs,[1624] and novel approaches to targeted delivery are constantly being discovered. When planning for delivery, it will be essential to comprehend the variables that affect C3M stability, disassembly, and physical characteristics. Driven by advancements in structural design, tailored C3Ms that protect, distribute, and release therapeutic cargo with control over targeting and transport can improve precision medicine. I have examined recent advancements in the theory of flexible diblock copolymer self-assembly in a diluted solution, which is both solvophobic and solvophilic. We believe that there is now enough development in the analytical theory of non-ionic diblock copolymer micelles to enable a thorough quantitative comparison with experiment. There is less development in the theory of ionic micelles with PE coronae. Here, a basic description of self-assembled aggregates is primarily obtained at the numerical modeling or asymptotic power law dependency levels. The analytical developments are restricted to linear elasticity of PE coronal chains and simplified models of coronal charge and ion distributions. Triblock terpolymer self-assembly theory is a difficult group of questions that is mainly undeveloped. It is reasonable to predict that in the years to come, BCP solution self-assembly will continue to pique scientific curiosity and open up new avenues for investigation. The synthesis of specially created copolymers with a range of compositions and topologies has been made possible by advancements in polymer chemistry and polymerization techniques. Because of their amphiphilic nature, BCPs’ thermodynamically driven self-organization can produce a variety of nanostructures, giving them easy control over their size, stability, and surface chemistry. Numerous potential nanoassembled morphologies have already been identified, ranging in complexity from straightforward spherical micelles to multicompartment segregated superstructures with fascinating shapes. There’s little doubt that the discoveries to come will continue to wow us. Self-assembled morphologies never seen before, such as those caused by the incorporation of more and more polymeric blocks into a single macromolecular chain, the development of new methods to control self-assembly, such as gradient control of block junctions or frustration at interfaces, and the increase in architectural complexity with new types of hyperbranched copolymers.
The growing popularity of BCPs is explicable not just from the perspective of basic comprehension but also from the range of real-world applications that bridge various scientific domains. The use of BCPs has shown beneficial in a variety of applied scientific fields connected to materials and nanotechnology, such as nanomedicine, formulations for treatment and diagnosis, and the creation of functionalized nanomaterials. These days, we may take use of BCPs’ ability to self-assemble to create “smart” multifunctional systems that can recognize and interact with their surroundings and even carry out desired behaviors in response to particular inputs. Block copolymer micelles show potential for gene and medication delivery. Block copolymer micelles’ creation of a distinctive core-shell architecture is their most pertinent feature for this aim. The shell-forming segments can be any flexible hydrophilic polymer, such as PEG, that assembles into dense palisades of tethered chains with effective steric stabilizing propensities. The primary driving force behind micellization is core segregation from the aqueous milieu, which is achieved by a variety of intermolecular processes such as hydrophobic contact, electrostatic interaction, metal complexation, and hydrogen bonding of the component block copolymers. Different medications with different properties are stored in the segregated core buried in the hydrophilic palisade. Moreover, pilot molecules can be positioned on the micelle's periphery to enhance the micelle’s absorption into the specific cells that express the desired receptors. Polymer strands having thermosensitive characteristics, like poly(N-isopropylacrylamide), can even be used to target polymeric micelles led by physical stimuli, such as local heating.[558] Therefore, it is anticipated that polymeric micelles will be widely used in the field of drug delivery, especially for the regulated distribution of cytotoxic agents and genes. Whenever the achievable sophistication of these systems rises, new and interesting implementation paths will inevitably appear.
In recent decades, the electrostatic coassembly of (diblock) copolymers containing oppositely charged species has drawn a lot of attention as a flexible method for producing a novel type of polymer micelles with intriguing characteristics. The main focus has been on comprehending and fine-tuning steady-state structure-function interactions through chemical and physical components under carefully regulated settings. This has significantly improved our understanding of C3M behavior and laid the groundwork for a number of exciting new research avenues that are gaining traction quickly. Macroscopic materials such hydrogels, thin films, adhesives, and saloplastics that are made from C3Ms and coacervates are expected to attract more attention in the future. Their steady-state characteristics, such as their reliance on the selection of one or two block copolymers, polymer length (ratios), and component composition, have all been well investigated. The precise way in which these factors affect the dynamic properties of C3Ms remains unclear. Different mechanisms are said to predominate in the relaxation from transitory structures to (steady-state) micelles, according to contradicting studies. Further systematic research will clarify when precisely the fusion-fission or expulsion-insertion mechanism governs the kinetics of C3M production. Salt concentration modifies the micellar dynamics and stability by altering the balance of driving factors for complexation. Hydrophobic effects and hydrogen bonding are examples of additional core interactions that influence the pathways and time scale of relaxation from transient to stable structures. Specifically, focusing solely on stability and steady-state characteristics is not sufficient to fully understand the application of C3Ms as biomacromolecule carriers. The delicate payload of C3Ms made of polypeptides (such as proteins) or polynucleotides paired with ionic-neutral copolymers needs to be shielded from degradation in vivo. Studies on exchange kinetics will provide additional insight into the process by which these building blocks are transferred across micelles as well as the duration of their exposure. The design of C3Ms with improved delivery efficiency will benefit from this knowledge. Although it was once thought of as a bottleneck, the generation of kinetically trapped states is today recognized as a useful technique for producing a wide range of nanostructures with previously unheard-of complexity using the same basic components. The change in emphasis from equilibrium to out-of-equilibrium conditions is another significant development. These mixed micelles are being thoroughly investigated for their (dis)assembly kinetics and exchange dynamics through the use of fluorescence spectroscopy and time-resolved scattering techniques. These findings reveal transitory (nano)-structures that could expand the range of possible architectures in the future by being annealed or vitrified. Reaction-assembly networks are particularly interesting in this regard, as they reveal the importance of compositional changes and kinetic parameters in the assembly paths. Because of the high concentrations that may be used, they also show tremendous potential as an effective platform for the creation of colloidally stable nanostructures of varied forms and dimensions in high yield. Developments in characterization and the application of single-molecule instruments, like single-molecule localization microscopy, will clarify the significance of minor species in groups and the destiny of C3Ms in non-stoichiometric compositions,[110,1625] out-of-equilibrium settings, and within cellular contexts.[1626] The design of C3Ms for targeted therapeutic delivery will progress with the application of such strategies. Lastly, I anticipate a growing endeavor within the polymer chemistry community to synthesize and integrate specially designed materials and replace PEO with alternative stabilizers, like zwitterionic/antifouling blocks,[1094] in order to optimize the functionality, responsiveness, stability, and dynamicity of C3Ms both in vitro and in vivo for enhanced performance in intricate (biological) settings.
Author Information
Corresponding Author: Present Address: §P.S.R.: Department of Pharmaceutical Chemistry, School of Pharmacy, Sister Nivedita University, Newtown, Kolkata, West Bengal, 700156, India; Email: royps2005@yahoo.com; parthasarathi.r@snuniv.ac.in Notes: The author disclaims any financial conflicts of interest.
Acknowledgments
P.S.R gratefully acknowledges the postdoctoral research assistantships provided by University of the Pacific (UoP)-Stockton, California, and University of Missouri-Kansas City (UMKC), U.S.A. Additional support from the University Grants Commission (UGC), Government of India, New Delhi, India, for funding doctoral work with a Junior Research Fellowship (JRF) in Engineering and Technology [Fellowship 10-01/2008 (SA-I)] and a postdoctoral fellowship through the RUSA 2.0 scheme (Ref. No. R-11/214/19) are also gratefully acknowledged.
References
- Mitragotri, S.; Burke, P.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G. F.; Adams, D. J.; Alewood, P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Kulkarni, J. A.; Witzigmann, D.; Thomson, S. B.; Chen, S.; Leavitt, B. R.; Cullis, P. R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Evans, B. C.; Fletcher, R. B.; Kilchrist, K. V.; Dailing, E. A.; Mukalel, A. J.; Colazo, J. M.; Oliver, M.; Cheung-flynn, J.; Brophy, C. M.; Tierney, J. W.; Isenberg, J. S.; Hankenson, K. D.; Ghimire, K.; Lander, C.; Gersbach, C. A.; Duvall, C. L. An Anionic, Endosome-Escaping Polymer to Potentiate Intracellular Delivery of Cationic Peptides, Biomacromolecules, and Nanoparticles. Nat. Commun. 2019, 10, 5012. [Google Scholar] [CrossRef]
- Walsh, G. Therapeutic insulins and their large-scale manufacture. Appl. Microbiol. Biotechnol. 2005, 67, 151–159. [Google Scholar] [CrossRef]
- Sheridan, C. First generic biologics finally approved. Nat. Rev. Drug Discov. 2006, 5, 445. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A. L.; Dhimolea, E.; Reichert, J. M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Wolfram Julie, A.; Donahue, J. K. J. Am. J . Am. Heart Assoc. 2013, 2, e000119. [Google Scholar]
- Lostalé-Seijo, I.; Montenegro, J. Synthetic materials at the forefront of gene delivery. Nat. Rev. Chem. 2018, 2, 258–277. [Google Scholar] [CrossRef]
- Rainov, N. G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef]
- Marks, W. J. Jr.; Ostrem, J. L.; Verhagen, L.; Starr, P. A.; Larson, P. S.; Bakay, R. A.; Taylor, R.; Cahn-Weiner, D. A.; Stoessl, A. J.; Olanow, C. W.; Bartus, R. T. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Bartus, R. T.; Baumann, T. L.; Siffert, J.; Herzog, C. D.; Alterman, R.; Boulis, N.; Turner, D. A.; Stacy, M.; Lang, A. E.; Lozano, A. M.; Olanow, C. W. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 2013, 80, 1698–1701. [Google Scholar] [CrossRef]
- Polack, F, P. ; Thomas, S. J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J. L.; P ́erez Marc, G.; Moreira, E. D.; Zerbini, C.; Bailey, R.; Swanson, K. A.; Roychoudhury, S.; Koury, K.; Li, P.; Kalina, W. V.; Cooper, D.; Frenck, R.W.; Hammitt, L. L.; Türeci, ̈ O.; Nell, H.; Schaefer, A.; Ünal, S.; Tresnan, D. B.; Mather, S.; Dormitzer, P. R.; -Sahin, U.; Jansen, K. U.; Gruber, W. C. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Kisby, T.; Yilmazer, A.; Kostarelos, K. Reasons for success and lessons learnt from nanoscale vaccines against COVID-19. Nat. Nanotechnol. 2021, 16, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D. J. A.; Anchordoquy, T. J.; Volkin, D. B.; Jiskoot, W.; Mastrobattista, E. Addressing the Cold Reality of mRNA Vaccine Stability. J Pharm Sci. 2021, 110, 997–1001. [Google Scholar] [CrossRef]
- Vahedifard, F,; Chakravarthy, K. Nanomedicine for COVID-19: the role of nanotechnology in the treatment and diagnosis of COVID-19. Emergent Mater. 2021, 4, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A. C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef]
- Fenton, O. S.; Olafson, K. N.; Pillai, P. S.; Mitchell. M. J.; Langer, R. Advances in Biomaterials for Drug Delivery. Adv Mater. 2018, 30, e1705328. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K. J.; Islam, J.; Thurecht, K. J.; Corrie, S. R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D. A Comprehensive Map of FDA-Approved Pharmaceutical Products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef]
- Sims, K. R.; Maceren, J. P.; Strand, A. I.; He, B.; Overby, C.; Benoit, D. S. W. RSC Adv. 2020, 10, 2513–2518. 10.
- Bungenberg de Jong, H. G.; Kruyt, H. R. Coacervation (Partial Miscibility in Colloid Systems). Proc. K. Ned. Akad. Wet. 1929, 32, 849–856. [Google Scholar]
- Hofs, B.; Voets, I. K.; De Keizer, A.; Cohen Stuart, M. A. Comparison of Complex Coacervate Core Micelles from Two Diblock Copolymers or a Single Diblock Copolymer with a Polyelectrolyte. Phys. Chem. Chem. Phys. 2006, 8, 4242–4251. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. Formation of Polyion Complex Micelles in an Aqueous Milieu from a Pair of Oppositely-Charged Block Copolymers with Poly(Ethylene Glycol) Segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar] [CrossRef]
- Voets, I. K.; de Keizer, A.; Cohen Stuart, M. A. Complex Coacervate Core Micelles. Adv. Colloid Interface Sci. 2009, 147–148, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Pergushov, D. V.; Müller, A. H. E.; Schacher, F. H. Micellar Interpolyelectrolyte Complexes. Chem. Soc. Rev. 2012, 41, 6888–6901. [Google Scholar] [CrossRef] [PubMed]
- Cingil, H. E.; Boz, E. B.; Wang, J.; Cohen Stuart, M. A.; Sprakel, J. Probing Nanoscale Coassembly with Dual Mechanochromic Sensors. Adv. Funct. Mater. 2016, 26, 1420–1427. [Google Scholar] [CrossRef]
- van der Burgh, S.; de Keizer, A.; Cohen Stuart, M. A. Complex Coacervation Core Micelles. Colloidal Stability and Aggregation Mechanism. Langmuir 2004, 20, 1073–1084. [Google Scholar] [CrossRef]
- Zhang, Q.; Ko, N. R.; Oh, J. K. Recent Advances in Stimuli-Responsive Degradable Block Copolymer Micelles: Synthesis and Controlled Drug Delivery Applications. Chem. Commun. (Camb) 2012, 48, 7542–7552. [Google Scholar] [CrossRef]
- Baek, C.; Ha, T.-L.; Kim, E.; Jeong, S.W.; Lee, S.G.; Lee, S.J.; Kim, H.-C. Bioreducible Micelles Self-Assembled from Poly(ethylene glycol)-Cholesteryl Conjugate As a Drug Delivery Platform. Polymers 2015, 7, 2245–2258. [Google Scholar] [CrossRef]
- Ding, J.; Chen, L.; Xiao, C.; Chen, L.; Zhuang, X.; Chen, X. Noncovalent interaction-assisted polymeric micelles for controlled drug delivery. Chem. Commun. 2014, 50, 11274–11290. [Google Scholar] [CrossRef]
- Biswas, S.; Kumari, P.; Lakhani, P. M.; Ghosh, B. Recent advances in polymeric micelles for anti-cancer drug delivery. Eur. J. Pharm. Sci. 2016, 83, 184–202. [Google Scholar] [CrossRef]
- Bae, Y.; Fukushima, S.; Harada, A.; Kataoka, K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew. Chem. Int. Ed. Engl. 2003, 42, 4640–4643. [Google Scholar] [CrossRef]
- Katayose, S.; Kataoka, K. Water-soluble polyion complex associates of DNA and poly(ethylene glycol)-poly(L-lysine) block copolymer. Bioconjug. Chem. 1997, 8, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed]
- Mukerabigwi, J. F.; Ge, Z.; Kataoka, K. Therapeutic nanoreactors as in vivo nanoplatforms for cancer therapy. Chem.-Eur. J. 2018, 24, 15706–15724. [Google Scholar] [CrossRef]
- Facciotti, C.; Saggiomo, V.; Bunschoten, A.; ten Hove, J. B.; Rood, M. T. M.; van Leeuwen, F. W. B.; Velders, A. H. Assembly, Disassembly and Reassembly of Complex Coacervate Core Micelles with Redox-Responsive Supramolecular Cross-Linkers. ChemSystemsChem. 2020, 2, e1900032. [Google Scholar] [CrossRef]
- Orilall, M. C.; Wiesner, U. Block copolymer based composition and morphology control in nanostructured hybrid materials for energy conversion and storage: solar cells, batteries, and fuel cells. Chem. Soc. Rev. 2011, 40, 520–535. [Google Scholar] [CrossRef]
- Dwars, T.; Paetzold, E.; Oehme, G. Reactions in micellar systems. Angew Chem Int Ed Engl. 2005, 44, 7174–7199. [Google Scholar] [CrossRef]
- Mortensen, K. PEO-related block copolymer surfactants oids Surf. A Physicochem. Eng. Asp. 2001, 183-185, 277-292.
- Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, M.; Prytkova, T.; Tonzani, S.; Winter, N. D.; Schatz, G. C. Modeling Self-Assembly Processes Driven by Nonbonded Interactions in Soft Materials. J. Phys. Chem. B 2008, 112, 10388–10398. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I.; Erman, B.; Mattice, W. L. Mechanisms of the Exchange of Diblock Copolymers between Micelles at Dynamic Equilibrium. Macromolecules 1996, 29, 4764–4771. [Google Scholar] [CrossRef]
- Wang, Y.; Mattice, W. L.; Napper, D. H. Simulation of the Formation of Micelles by Diblock Copolymers under Weak Segregation. Langmuir 1993, 9, 66–70. [Google Scholar] [CrossRef]
- De Gennes, P. G. Scaling theory of polymer adsorption. J. Phys. (Paris) 1976, 37, 1445–1452. [Google Scholar] [CrossRef]
- De Gennes, P. G. Conformation of Polymers Attached to an Interface. Macromolecules 1980, 13, 1069–1075. [Google Scholar] [CrossRef]
- Halperin, A. Polymeric Micelles: A Star Model. Macromolecules 1987, 20, 2943–2946. [Google Scholar] [CrossRef]
- Noolandi, J.; Hong, K. M. Theory of Block Copolymer Micelles in Solution. Macromolecules 1983, 16, 1443–1448. [Google Scholar] [CrossRef]
- Leibler, L.; Orland, H.; Wheeler, J. C. Theory of critical micelle concentration for solutions of block copolymers. J. Chem. Phys. 1983, 79, 3550–3557. [Google Scholar] [CrossRef]
- Pépin, M. P.; Whitmore, M. D. Monte Carlo and Mean Field Study of Diblock Copolymer Micelles. Macromolecules 2000, 33, 8644–8653. [Google Scholar] [CrossRef]
- Cantu, L.; Corti, M.; Salina, P. Direct measurement of the formation time of mixed micelles. J. Phys. Chem. 1991, 95, 5981–5983. [Google Scholar] [CrossRef]
- Won, Y.-Y.; Davis, H. T.; Bates, F. S. Molecular Exchange in PEO-PB Micelles in Water. Macromolecules 2003, 36, 953–955. [Google Scholar] [CrossRef]
- Lund, R.; Willner, L.; Richter, D.; Dormidontova, E. E. Equilibrium Chain Exchange Kinetics of Diblock Copolymer Micelles: Tuning and Logarithmic Relaxation. Macromolecules 2006, 39, 4566–4575. [Google Scholar] [CrossRef]
- Sing, C. E.; Perry, S. L. Recent progress in the science of complex coacervation. Soft Matter 2020, 16, 2885–2914. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Schlenoff, J. B. Driving Forces for Oppositely Charged Polyion Association in Aqueous Solutions: Enthalpic, Entropic, but Not Electrostatic. J. Am. Chem. Soc. 2016, 138, 980–990. [Google Scholar] [CrossRef]
- Marras, A. E.; Ting, J. M.; Stevens, K. C.; Tirrell, M. V. Advances in the Structural Design of Polyelectrolyte Complex Micelles. J. Phys. Chem. B. 2021, 125, 7076–7089. [Google Scholar] [CrossRef]
- Rumyantsev, A. M.; Zhulina, E. B.; Borisov, O.V. ; Zhulina, E. B.; Borisov O.V. Scaling Theory of Complex Coacervate Core Micelles. ACS Macro Lett. 2018, 7, 7–811. [Google Scholar] [CrossRef]
- Sproncken, C. C. M.; Magana, J. R.; Voets, I. K. 100th Anniversary of Macromolecular Science Viewpoint: Attractive Soft Matter: Association Kinetics, Dynamics, and Pathway Complexity in Electrostatically Coassembled Micelles. ACS Macro Lett. 2021, 10, 167–179. [Google Scholar] [CrossRef]
- Magana, J. R.; Sproncken, C. C. M.; Voets, I. K. On Complex Coacervate Core Micelles: Structure-Function Perspectives. Polymers (Basel) 2020, 12, 1953. [Google Scholar] [CrossRef] [PubMed]
- Riggleman, R. A.; Kumar, R.; Fredrickson, G. H. Investigation of the interfacial tension of complex coacervates using field-theoretic simulations. J. Chem. Phys. 2012, 136, 024903. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A. V.; Bronich, T. K.; Kabanov, V. A.; Yu, K.; Eisenberg, A. Soluble stoichiometric complexes from poly(N-ethyl-4- vinylpyridinium) cations and poly(ethylene oxide)-block-polymetha- crylate anions. Macromolecules 1996, 29, 6797–6802. [Google Scholar] [CrossRef]
- Yan, Y.; de Keizer, A.; Cohen Stuart, M. A.; Drechsler, M.; Besseling, N. A. M. Stability of complex coacervate core micelles containing metal coordination polymer. J. Phys. Chem. B 2008, 112, 10908–10914. [Google Scholar] [CrossRef]
- Wang, J.; de Keizer, A.; Fokkink, R.; Yan, Y.; Cohen Stuart, M. A.; van der Gucht, J. Complex coacervate core micelles from iron-based coordination polymers. J. Phys. Chem. B 2010, 114, 8313–8319. [Google Scholar] [CrossRef]
- Gaucher, G.; Dufresne, M.-H.; Sant, V. P.; Kang, N.; Maysinger, D.; Leroux, J.-C. Block copolymer micelles: preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef]
- Kramarenko, E.Y.; Khokhlov, A.R.; Reineker, P. Stoichiometric polyelectrolyte complexes of ionic block copolymers and oppositely charged polyions. J. Chem. Phys. 2006, 125, 194902. [Google Scholar] [CrossRef]
- Ma, R.; Wang, B.; Liu, X.; An, Y.; Li, Y.; He, Z.; Shi, L. Pyranine- induced micellization of poly(ethylene glycol)-block-poly(4-vinyl- pyridine) and pH-triggered release of pyranine from the complex micelles. Langmuir 2007, 23, 7498–7504. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, M.; Voets, I. K.; Cohen Stuart, M. A.; van der Gucht, J. Transient network topology of interconnected polyelectrolyte complex micelles. Soft Matter 2011, 7, 1378–1389. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of stable and monodispersive polyion complex micelles in aqueous medium from poly(L-lysine) and poly(ethylene glycol)-poly(aspartic acid) block copolymer. J. Macromol. Sci. Part A. 1997, 34, 2119–2133. [Google Scholar] [CrossRef]
- Dautzenberg, H.; Rother, G. Response of polyelectrolyte complexes to subsequent addition of sodium chloride: time-dependent static light scattering studies. Macromol. Chem. Phys. 2004, 205, 114–121. [Google Scholar] [CrossRef]
- Matralis, A.; Sotiropoulou, M.; Bokias, G.; Staikos, G. Water- soluble stoichiometric polyelectrolyte complexes based on cationic comb-type copolymers. Macromol. Chem. Phys. 2006, 207, 1018–1025. [Google Scholar] [CrossRef]
- Park, J.-S.; Akiyama, Y.; Yamasaki, Y.; Kataoka, K. Preparation and characterization of polyion complex micelles with a novel thermosensitive poly(2-isopropyl-2-oxazoline) shell via the complexation of oppositely charged block ionomers. Langmuir 2007, 23, 138–146. [Google Scholar] [CrossRef]
- Pispas, S. Complexes of polyelectrolyte-neutral double hydrophilic block copolymers with oppositely charged surfactant and polyelectrolyte. J. Phys. Chem. B 2007, 111, 8351–8359. [Google Scholar] [CrossRef] [PubMed]
- Lindhoud, S.; Voorhaar, L.; de Vries, R.; Schweins, R.; Cohen Stuart, M. A.; Norde, W. Salt-induced disintegration of lysozyme- containing polyelectrolyte complex micelles. Langmuir 2009, 25, 11425–11430. [Google Scholar] [CrossRef] [PubMed]
- Soliman, G. M. Polysaccharide-Based Polyion Complex Micelles as New Delivery Systems for Hydrophilic Cationic Drugs. Ph.D. thesis, Faculty of Pharmacy, University of Montreal, 09; pp 1-258. 20 August.
- Brzozowska, A.; Zhang, Q.; de Keizer, A.; Norde, W.; Cohen Stuart, M. Reduction of protein adsorption on silica and polysulfone surfaces coated with complex coacervate core micelles with poly(vinyl alcohol) as a neutral brush forming block. Colloids Surf. A 2010, 368, 96–104. [Google Scholar] [CrossRef]
- Brzozowska, A.; de Keizer, A.; Christophe, D.; Cohen Stuart, M. A.; Norde, W. Grafted ionomer complexes and their effect on protein adsorption on silica and polysulfone surfaces. Colloid Polym. Sci. 2010, 288, 1621–1632. [Google Scholar] [CrossRef]
- Danial, M.; Klok, H.-A.; Norde, W.; Cohen Stuart, M. A. Complex coacervate core micelles with a lysozyme-modified corona. Langmuir 2007, 23, 8003–8009. [Google Scholar] [CrossRef]
- Lindhoud, S.; de Vries, R.; Schweins, R.; Cohen Stuart, M. A.; Norde, W. Salt-induced release of lipase from polyelectrolyte complex micelles. Soft Matter 2009, 5, 242–250. [Google Scholar] [CrossRef]
- Overbeek, J. T.; Voorn, M. J. Phase separation in polyelectrolyte solutions; theory of complex coacervation. J. Cell Physiol. Suppl. 1957, 49 (Suppl. 1), 7–22; discussion, 22-26. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J. N.; Feldman, K. E.; Lynd, N. A.; Deek, J.; Campos, L. M.; Spruell, J. M.; Hernandez, B. M.; Kramer, E. J.; Hawker, C. J. Tunable, high modulus hydrogels driven by ionic coacervation. Adv. Mater. 2011, 23, 2327–2331. [Google Scholar] [CrossRef]
- Capito, R. M.; Azevedo, H. S.; Velichko, Y. S.; Mata, A.; Stupp, S. I. Self-assembly of large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. [Google Scholar] [CrossRef]
- Kakizawa, Y.; Kataoka, K. Block copolymer micelles for delivery of gene and related compounds. Adv. Drug Delivery Rev. 2002, 54, 203–222. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Supramolecular assemblies of block copolymers in aqueous media as nanocontainers relevant to biological applications. Prog. Polym. Sci. 2006, 31, 949–982. [Google Scholar] [CrossRef]
- Holappa, S.; Kantonen, L.; Andersson, T.; Winnik, F.; Tenhu, H. Overcharging of polyelectrolyte complexes by the guest polyelectrolyte studied by fluorescence spectroscopy. Langmuir 2005, 21, 11431–11438. [Google Scholar] [CrossRef] [PubMed]
- Nolles, A.; Hooiveld, E.; Westphal, A. H.; van Berkel, W. J.; Kleijn, J. M.; Borst, J. W. FRET Reveals the Formation and Exchange Dynamics of Protein-Containing Complex Coacervate Core Micelles. Langmuir 2018, 34, 12083–12092. [Google Scholar] [CrossRef]
- Amann, M.; Diget, J. S.; Lyngsø, J.; Pedersen, J. S.; Narayanan, T.; Lund, R. Kinetic Pathways for Polyelectrolyte Coacervate Micelle Formation Revealed by Time-Resolved Synchrotron SAXS. Macromolecules 2019, 52, 8227–8237. [Google Scholar] [CrossRef]
- Bos, I.; Sprakel, J. Langevin Dynamics Simulations of the Exchange of Complex Coacervate Core Micelles: The Role of Nonelectrostatic Attraction and Polyelectrolyte Length. Macromolecules 2019, 52, 8923–8931. [Google Scholar] [CrossRef]
- Wu, H.; Ting, J. M.; Tirrell, M. V. Mechanism of Dissociation Kinetics in Polyelectrolyte Complex Micelles. Macromolecules 2020, 53, 102–111. [Google Scholar] [CrossRef]
- Berret, J.-F.; Cristobal, G.; Herve,́ P. ; Oberdisse, J.; Grillo, I. Structure of colloidal complexes obtained from neutral/polyelectrolyte copolymers and oppositely charged surfactants. Eur. Phys. J. E Soft Matter 2002, 9, 301–311. [Google Scholar] [CrossRef]
- Wu, H.; Ting, J. M.; Werba, O.; Meng, S.; Tirrell, M. V. Non- equilibrium phenomena and kinetic pathways in self-assembled polyelectrolyte complexes. J. Chem. Phys. 2018, 149, 163330. [Google Scholar] [CrossRef]
- Hofs, B.; De Keizer, A.; Cohen Stuart, M. A. On the stability of (highly aggregated) polyelectrolyte complexes containing a charged- block-neutral diblock copolymer. J. Phys. Chem. B 2007, 111, 5621–5627. [Google Scholar] [CrossRef] [PubMed]
- Lindhoud, S.; Norde, W.; Cohen Stuart, M. A. Reversibility and relaxation behavior of polyelectrolyte complex micelle formation. J. Phys. Chem. B 2009, 113, 5431–5439. [Google Scholar] [CrossRef]
- Bos, I.; Terenzi, C.; Sprakel, J. Chemical Feedback in Templated Reaction-Assembly Networks. Macromolecules 2020, 53, 10675–10685. [Google Scholar] [CrossRef] [PubMed]
- Dormidontova, E. E. Micellization kinetics in block copolymer solutions: Scaling model. Macromolecules 1999, 32, 7630–7644. [Google Scholar] [CrossRef]
- Lund, R.; Willner, L.; Stellbrink, J.; Lindner, P.; Richter, D. Logarithmic chain-exchange kinetics of diblock copolymer micelles. Phys. Rev. Lett. 2006, 96, 068302. [Google Scholar] [CrossRef]
- Choi, S.-H.; Lodge, T. P.; Bates, F. S. Mechanism of molecular exchange in diblock copolymer micelles: hypersensitivity to core chain length. Phys. Rev. Lett. 2010, 104, 047802. [Google Scholar] [CrossRef]
- Zinn, T.; Willner, L.; Lund, R.; Pipich, V.; Richter, D. Equilibrium exchange kinetics in n-alkyl-PEO polymeric micelles: single exponential relaxation and chain length dependence. Soft Matter 2012, 8, 623–626. [Google Scholar] [CrossRef]
- Lu, J.; Choi, S.; Bates, F.; Lodge, T. Molecular exchange in diblock copolymer micelles: bimodal distribution in core-block molecular weights. ACS Macro Lett. 2012, 1, 982–985. [Google Scholar] [CrossRef]
- Lu, J.; Bates, F.; Lodge, T. Chain exchange in binary copolymer micelles at equilibrium: confirmation of the independent chain hypothesis. ACS Macro Lett. 2013, 2, 451–455. [Google Scholar] [CrossRef]
- Wang, E.; Lu, J.; Bates, F. S.; Lodge, T. P. Effect of corona block length on the structure and chain exchange kinetics of block copolymer micelles. Macromolecules 2018, 51, 3563–3571. [Google Scholar] [CrossRef]
- Bos, I.; Timmerman, M.; Sprakel, J. FRET-Based Determination of the Exchange Dynamics of Complex Coacervate Core Micelles. Macromolecules 2021, 54, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Semenov. A. N. Contribution to the theory of microphase layering in block-copolymer melts. Sov. Phys. JETP 1985, 61, 733–742. [Google Scholar]
- Borisov, O. V.; Zhulina, E. B.; Leermakers, F. A. M.; Müller, A. H. E. Self-Assembled Structures of Amphiphilic Ionic Block Copolymers: Theory, Self-Consistent Field Modeling and Experiment. Adv. Polym. Sci. 2011, 241, 57–129. [Google Scholar]
- van der Kooij, H. M.; Spruijt, E.; Voets, I. K.; Fokkink, R.; Cohen Stuart, M. A.; van der Gucht, J. . On the Stability and Morphology of Complex Coacervate Core Micelles: From Spherical to Wormlike Micelles Langmuir 2012, 28 (40), 14180-14191.
- Lueckheide, M. , Vieregg, J. R.; Bologna, A. J.; Leon, L.; Tirrell, M. V. Structure-property relationships of oligonucleotide polyelectrolyte complex micelles. Nano Lett. 2018, 18, 7111–7117. [Google Scholar] [CrossRef] [PubMed]
- Nakata, M.; Zanchetta, G.; Chapman, B. D.; Jones, C. D.; Cross, J. O.; Pindak, R.; Bellini, T.; Clark, N. A. End-to-end stacking and liquid crystal condensation of 6 to 20 base pair DNA duplexes. Science 2007, 318, 1276–1279. [Google Scholar] [CrossRef]
- Rumyantsev A., M. , de Pablo J. J., Liquid crystalline and isotropic coacervates of semiflexible polyanions and flexible polycations. Macromolecules 2019, 52, 5140–5156. [Google Scholar] [CrossRef]
- Aloi, A.; Guibert, C.; Olijve, L. L. C.; Voets, I. K. Morphological Evolution of Complex Coacervate Core Micelles Revealed by iPAINT Microscopy. Polymer 2016, 107, 450–455. [Google Scholar] [CrossRef]
- Shakya, A.; Girard, M.; King, J. T.; De La Cruz, M. O. Role of Chain Flexibility in Asymmetric Polyelectrolyte Complexation in Salt Solutions. Macromolecules 2020, 53, 1258–1269. [Google Scholar] [CrossRef]
- Blocher McTigue, W. C.; Perry, S. L. Protein Encapsulation Using Complex Coacervates: What Nature Has to Teach Us. Small 2020, 16, e1907671. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Polyion Complex Micelle Formation from Double-Hydrophilic Block Copolymers Composed of Charged and Non-Charged Segments in Aqueous Media. Polym. J. 2018, 50, 95–100. [Google Scholar] [CrossRef]
- Roy, P.S. Complex Coacervate-Based Materials for Biomedicine: Recent Advancements and Future Prospects. Ind. Eng. Chem. Res. 2024, 63, 5414–5487. [Google Scholar] [CrossRef]
- Horn, J. M.; Kapelner, R. A.; Obermeyer, A. C. Macro- and Microphase Separated Protein-Polyelectrolyte Complexes: Design Parameters and Current Progress. Polymers (Basel, Switz.) 2019, 11, 578. [Google Scholar] [CrossRef]
- Chen, F.; Stenzel, M. H. Polyion Complex Micelles for Protein Delivery. Aust. J. Chem. 2018, 71, 768–780. [Google Scholar] [CrossRef]
- Zhang, Y.; Batys, P.; O’Neal, J. T.; Li, F.; Sammalkorpi, M.; Lutkenhaus, J. L. Molecular Origin of the Glass Transition in Polyelectrolyte Assemblies. ACS Cent. Sci. 2018, 4, 638–644. [Google Scholar] [CrossRef]
- Gineste, S.; Di Cola, E.; Amouroux, B.; Till, U.; Marty, J.-D.; Mingotaud, A.-F.; Mingotaud, C.; Violleau, F.; Berti, D.; Parigi, G.; Luchinat, C.; Balor, S.; Sztucki, M.; Lonetti, B. Mechanistic Insights into Polyion Complex Associations. Macromolecules 2018, 51, 1427–1440. [Google Scholar] [CrossRef]
- Rathee, V. S.; Sidky, H.; Sikora, B. J.; Whitmer, J. K. Role of Associative Charging in the Entropy-Energy Balance of Polyelectrolyte Complexes. J. Am. Chem. Soc. 2018, 140, 15319–15328. [Google Scholar] [CrossRef] [PubMed]
- Roy, P. S.; Samanta, A.; Mukherjee, M.; Roy, B.; Mukherjee, A. Designing Novel pH-Induced Chitosan-Gum Odina Complex Coacervates for Colon Targeting. Ind. Eng. Chem. Res. 2013, 52, 15728–15745. [Google Scholar] [CrossRef]
- Takemoto, H.; Miyata, K.; Hattori, S.; Ishii, T.; Suma, T.; Uchida, S.; Nishiyama, N.; Kataoka, K. Acidic PH-Responsive SiRNA Conjugate for Reversible Carrier Stability and Accelerated Endosomal Escape with Reduced IFNα-Associated Immune Response. Angew. Chem. Int. Ed. 2013, 52, 6218–6221. [Google Scholar] [CrossRef]
- van der Gucht, J.; Spruijt, E.; Lemmers, M.; Cohen Stuart, M. A. Polyelectrolyte Complexes: Bulk Phases and Colloidal Systems. J. Colloid Interface Sci. 2011, 361, 407–422. [Google Scholar] [CrossRef]
- Stapert, H. R.; Nishiyama, N.; Jiang, D. L.; Aida, T.; Kataoka, K. Polyion Complex Micelles Encapsulating Light-Harvesting Ionic Dendrimer Zinc Porphyrins. Langmuir 2000, 16, 8182–8188. [Google Scholar] [CrossRef]
- Sadman, K.; Wang, Q.; Chen, Y.; Keshavarz, B.; Jiang, Z.; Shull, K. R. Influence of Hydrophobicity on Polyelectrolyte Complexation. Macromolecules 2017, 50, 9417–9426. [Google Scholar] [CrossRef]
- De Kruif, C. G.; Weinbreck, F.; De Vries, R. Complex Coacervation of Proteins and Anionic Polysaccharides. Curr. Opin. Colloid Interface Sci. 2004, 9, 340–349. [Google Scholar] [CrossRef]
- Mills, C. E.; Obermeyer, A.; Dong, X.; Walker, J.; Olsen, B. D. Complex Coacervate Core Micelles for the Dispersion and Stabilization of Organophosphate Hydrolase in Organic Solvents. Langmuir 2016, 32, 13367–13376. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Zhou, J.; Pan, W.; Li, Z.; Liang, D. Assembly and Reassembly of Polyelectrolyte Complex Formed by Poly(Ethylene Glycol)-Block-Poly(Glutamate Sodium) and S5R4 Peptide. Macromolecules 2016, 49, 4627–4633. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Chang, H.; Cheng, Y. Surface-Engineered Dendrimers in Gene Delivery. Chem. Rev. 2015, 115, 5274–5300. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Shi, L.; Zhang, W.; An, Y.; Zhu, X.-X.; Zhang, X.; Li, Z. Formation of Hybrid Micelles between Poly(Ethylene Glycol)-Block- Poly(4-Vinylpyridinium) Cations and Sulfate Anions in an Aqueous Milieu. Soft Matter 2005, 1, 455–459. [Google Scholar] [CrossRef]
- Sanson, N.; Bouyer, F.; Destarac, M.; In, M.; Gérardin, C. Hybrid Polyion Complex Micelles Formed from Double Hydrophilic Block Copolymers and Multivalent Metal Ions: Size Control and Nanostructure. Langmuir 2012, 28, 3773–3782. [Google Scholar] [CrossRef]
- Carl, N.; Prévost, S.; Schweins, R.; Houston, J. E.; Morfin, I.; Huber, K. Invertible Micelles Based on Ion-Specific Interactions of Sr2+ and Ba2+ with Double Anionic Block Copolyelectrolytes. Macromolecules 2019, 52, 8759–8770. [Google Scholar] [CrossRef]
- Facciotti, C.; Saggiomo, V.; van Hurne, S.; Bunschoten, A.; Kaup, R.; Velders, A. H. Oxidant-Responsive Ferrocene-Based Cyclodextrin Complex Coacervate Core Micelles. Supramol. Chem. 2020, 32, 30–38. [Google Scholar] [CrossRef]
- Yoon, H.; Dell, E. J.; Freyer, J. L.; Campos, L. M.; Jang, W. D. Polymeric Supramolecular Assemblies Based on Multivalent Ionic Interactions for Biomedical Applications. Polymer 2014, 55, 453–464. [Google Scholar] [CrossRef]
- Gao, S.; Holkar, A.; Srivastava, S. Protein-Polyelectrolyte Complexes and Micellar Assemblies. Polymers (Basel, Switz.) 2019, 11, 1097. [Google Scholar] [CrossRef]
- Smith, A. E.; Sizovs, A.; Grandinetti, G.; Xue, L.; Reineke, T. M. Diblock Glycopolymers Promote Colloidal Stability of Polyplexes and Effective PDNA and siRNA Delivery under Physiological Salt and Serum Conditions. Biomacromolecules 2011, 12, 3015–3022. [Google Scholar] [CrossRef]
- Qian, Y.; Zha, Y.; Feng, B.; Pang, Z.; Zhang, B.; Sun, X.; Ren, J.; Zhang, C.; Shao, X.; Zhang, Q.; Jiang, X. PEGylated Poly(2- (Dimethylamino) Ethyl Methacrylate)/DNA Polyplex Micelles Decorated with Phage-Displayed TGN Peptide for Brain-Targeted Gene Delivery. Biomaterials 2013, 34, 2117–2129. [Google Scholar] [CrossRef]
- Tan, Z.; Jiang, Y.; Zhang, W.; Karls, L.; Lodge, T. P.; Reineke, T. M. Polycation Architecture and Assembly Direct Successful Gene Delivery: Micelleplexes Outperform Polyplexes via Optimal DNA Packaging. J. Am. Chem. Soc. 2019, 141, 15804–15817. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Diociaiuti, M.; Cametti, C.; Masci, G. Hyaluronic Acid and Alginate Covalent Nanogels by Template Cross-Linking in Polyion Complex Micelle Nanoreactors. Carbohydr. Polym. 2014, 101, 96–103. [Google Scholar] [CrossRef]
- Seo, E.; Lee, S. H.; Lee, S.; Choi, S. H.; Hawker, C. J.; Kim, B. S. Highly Stable Au Nanoparticles with Double Hydrophilic Block Copolymer Templates: Correlation Between Structure and Stability. Polym. Chem. 2017, 8, 4528–4537. [Google Scholar] [CrossRef]
- Krogstad, D. V.; Choi, S. H.; Lynd, N. A.; Audus, D. J.; Perry, S. L.; Gopez, J. D.; Hawker, C. J.; Kramer, E. J.; Tirrell, M. V. Small Angle Neutron Scattering Study of Complex Coacervate Micelles and Hydrogels Formed from Ionic Diblock and Triblock Copolymers. J. Phys. Chem. B 2014, 118, 13011–13018. [Google Scholar] [CrossRef] [PubMed]
- Ortony, J. H.; Choi, S. H.; Spruell, J. M.; Hunt, J. N.; Lynd, N. A.; Krogstad, D. V.; Urban, V. S.; Hawker, C. J.; Kramer, E. J.; Han, S. Fluidity and Water in Nanoscale Domains Define Coacervate Hydrogels. Chem. Sci. 2014, 5, 58–67. [Google Scholar] [CrossRef]
- Fu, J.; Fares, H. M.; Schlenoff, J. B. Ion-Pairing Strength in Polyelectrolyte Complexes. Macromolecules 2017, 50, 1066–1074. [Google Scholar] [CrossRef]
- Cingil, H. E.; Meertens, N. C. H.; Voets, I. K. Temporally Programmed Disassembly and Reassembly of C3Ms. Small 2018, 14, e1802089. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, Y.; Zhang, J.; Gao, H.; Liu, G.; Ma, R.; An, Y.; Kong, D.; Shi, L. pH/Sugar Dual Responsive Core-Cross-Linked PIC Micelles for Enhanced Intracellular Protein Delivery. Biomacromolecules 2013, 14, 3434–3443. [Google Scholar] [CrossRef]
- Cohen Stuart, M. A.; Besseling, N. A. M.; Fokkink, R. G. Formation of Micelles with Complex Coacervate Cores. Langmuir 1998, 14, 6846–6849. [Google Scholar] [CrossRef]
- Voets, I. K.; De Vries, R.; Fokkink, R.; Sprakel, J.; May, R. P.; De Keizer, A.; Cohen Stuart, M. A. Towards a Structural Characterization of Charge-Driven Polymer Micelles. Eur. Phys. J. E: Soft Matter Biol. Phys. 2009, 30, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Fresnais, J.; Lavelle, C.; Berret, J. F. Nanoparticle Aggregation Controlled by Desalting Kinetics. J. Phys. Chem. C 2009, 113, 16371–16379. [Google Scholar] [CrossRef]
- Qi, L.; Fresnais, J.; Berret, J. F.; Castaing, J. C.; Destremaut, F.; Salmon, J. B.; Cousin, F.; Chapel, J. P. Influence of the Formulation Process in Electrostatic Assembly of Nanoparticles and Macromolecules in Aqueous Solution: The Interaction Pathway. J. Phys. Chem. C 2010, 114, 16373–16381. [Google Scholar] [CrossRef]
- Qi, L.; Fresnais, J.; Berret, J. F.; Castaing, J. C.; Grillo, I.; Chapel, J. P. Influence of the Formulation Process in Electrostatic Assembly of Nanoparticles and Macromolecules in Aqueous Solution: The Mixing Pathway. J. Phys. Chem. C 2010, 114, 12870–12877. [Google Scholar] [CrossRef]
- Blocher McTigue, W. C.; Voke, E.; Chang, L.-W.; Perry, S. L. The Benefit of Poor Mixing: Kinetics of Coacervation. Phys. Chem. Chem. Phys. 2020, 22, 20643–20657. [Google Scholar] [CrossRef]
- Voets, I. K. Electrostatically Driven Assembly of Polyelectrolytes. In: Fluorescence Studies of Polymer Containing Systems; Procházka, K., Ed.; Springer Series on Fluorescence, Springer International Publishing, Cham, 2016; Vol 16, pp 65-89. [CrossRef]
- Shi, B.; Zheng, M.; Tao, W.; Chung, R.; Jin, D.; Ghaffari, D.; Farokhzad, O. C. Challenges in DNA Delivery and Recent Advances in Multifunctional Polymeric DNA Delivery Systems. Biomacromolecules 2017, 18, 2231–2246. [Google Scholar] [CrossRef]
- Uchida, S.; Kataoka, K. Design concepts of polyplex micelles for in vivo therapeutic delivery of plasmid DNA and messenger RNA. J. Biomed. Mater. Res. A. 2019, 107, 978–990. [Google Scholar] [CrossRef]
- Kataoka K, Harada A, Nagasaki Y: Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. [CrossRef]
- Benjamin, C.; Brohlin, O.; Shahrivarkevishahi, A.; Gassensmith, J. J. Virus like Particles: Fundamental Concepts, Biological Interactions, and Clinical Applications. Chung E. J., Leon L., Rinaldi C., Eds.; Nanoparticles for Biomedical Applications; Elsevier Publishing: Amsterdam, The Netherlands, 2020; Chapter 11, pp. 153-174. [Google Scholar] [CrossRef]
- Tockary, T. A.; Osada, K.; Chen, Q.; Machitani, K.; Dirisala, A.; Uchida, S.; Nomoto, T.; Toh, K.; Matsumoto, Y.; Itaka, K.; Nitta, K.; Nagayama, K.; Kataoka, K. Tethered Peg Crowdedness Determining Shape and Blood Circulation Profile of Polyplex Micelle Gene Carriers. Macromolecules 2013, 46, 6585–6592. [Google Scholar] [CrossRef]
- Kanazawa, T.; Sugawara, K.; Tanaka, K.; Horiuchi, S.; Takashima, Y.; Okada, H. Suppression of tumor growth by systemic delivery of anti-VEGF siRNA with cell-penetrating peptide-modified MPEG-PCL nanomicelles. Eur. J. Pharm. Biopharm. 2012, 81, 470–477. [Google Scholar] [CrossRef]
- Jeong, J. H.; Kim, S. W.; Park, T. G. A new antisense oligonucleotide delivery system based on self-assembled ODN-PEG hybrid conjugate micelles. J. Control Release 2003, 93, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lindhoud, S.; de Vries, R.; Norde, W.; Cohen Stuart, M. A. Structure and stability of complex coacervate core micelles with lysozyme. Biomacromolecules 2007, 8, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K. Lactosylated poly(ethylene glycol)-siRNA conjugate through acid- labile β-thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J. Am. Chem. Soc. 2005, 127, 1624–1625. [Google Scholar] [CrossRef]
- Lee, Y.; Ishii, T.; Kim, H. J.; Nishiyama, N.; Hayakawa, Y.; Itaka, K.; Kataoka, K. Efficient delivery of bioactive antibodies into the cytoplasm of living cells by charge-conversional polyion complex micelles. Angew. Chem. Int. Ed. Engl. 2010, 49, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.-D.; Nakagishi, Y.; Nishiyama, N.; Kawauchi, S.; Morimoto, Y.; Kikuchi, M.; Kataoka, K. Polyion complex micelles for photodynamic therapy: incorporation of dendritic photosensitizer excitable at long wavelength relevant to improved tissue-penetrating property. J. Controlled Release 2006, 113, 73–79. [Google Scholar] [CrossRef]
- Yan, Y.; Besseling, N. A. M.; de Keizer, A.; Marcelis, A.; Drechsler, M.; Cohen Stuart, M. A. Hierarchical self-assembly in solutions containing metal ions, ligand, and diblock copolymer. Angew. Chem. Int. Ed. Engl. 2007, 46, 1807–1809. [Google Scholar] [CrossRef]
- Schwarz, S.; Dragan, S. Nonstoichiometric interpolyelectrolyte complexes as colloidal dispersions based on NaPAMPS and their interaction with colloidal silica particles. Macromol. Symp. 2004, 210, 185–192. [Google Scholar] [CrossRef]
- van der Burgh, S.; Fokkink, R.; de Keizer, A.; Cohen Stuart, M. A. Complex coacervation core micelles as anti-fouling agents on silica and polystyrene surfaces. Colloids Surf. A 2004, 242, 167–174. [Google Scholar] [CrossRef]
- Brzozowska, A.; Hofs, B.; de Keizer, A.; Fokkink, R.; Cohen Stuart, M.; Norde, W. Reduction of protein adsorption on silica and polystyrene surfaces due to coating with complex coacervate core micelles. Colloids Surf. A 2009, 347, 146–155. [Google Scholar] [CrossRef]
- Tinkle, S.; McNeil, S. E.; Mühlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y. C.; Tamarkin, L.; Desai, N. Nanomedicines: addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef]
- Sarmento, B. Have nanomedicines progressed as much as we'd hoped for in drug discovery and development? Expert Opin. Drug Discov. 2019, 14, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Krukemeyer, M. G.; Krenn, V.; Huebner, F.; Wagner, W.; Resch, R. History and Possible Uses of Nanomedicine Based on Nanoparticles and Nanotechnological Progress. J. Nanomed. Nanotechnol. 2015, 6, 1000336. [Google Scholar]
- Decuzzi, P.; Peer, D.; Mascolo, D. D.; Palange, A. L.; Manghnani, P. N.; Moghimi, S. M.; Farhangrazi, Z. S.; Howard, K. A.; Rosenblum, D.; Liang, T.; Chen, Z.; Wang, Z.; Zhu, J. J.; Gu, Z.; Korin, N.; Letourneur, D.; Chauvierre, C.; van der Meel, R.; Kiessling, F.; Lammers, T. Roadmap on nanomedicine. Nanotechnology 2021, 32, 012001. [Google Scholar] [CrossRef] [PubMed]
- Cicha, I.; Chauvierre, C.; Texier, I.; Cabella, C.; Metselaar, J. M.; Szebeni, J.; Dézsi, L.; Alexiou, C.; Rouzet, F.; Storm, G.; Stroes, E.; Bruce, D.; MacRitchie, N.; Maffia, P.; Letourneur, D. From design to the clinic: practical guidelines for translating cardiovascular nanomedicine. Cardiovasc Res. 2018, 114, 1714–1727. [Google Scholar] [CrossRef]
- Fu, X.; Shi, Y.; Qi, T.; Qiu, S.; Huang, Y.; Zhao, X.; Sun, Q.; Lin, G. Precise design strategies of nanomedicine for improving cancer therapeutic efficacy using subcellular targeting. Signal Transduct Target Ther. 2020, 5, 262. [Google Scholar] [CrossRef]
- Bhise, K.; Sau, S.; Alsaab, H.; Kashaw, S. K.; Tekade, R. K.; Iyer, A. K. Nanomedicine for cancer diagnosis and therapy: advancement, success and structure-activity relationship. Ther. Deliv. 2017, 8, 1003–1018. [Google Scholar] [CrossRef]
- Macchione, M. A.; Aristizabal Bedoya, D.; Figueroa, F. N.; Munoz-Fernandez, M. A.; Strumia, M. C. Nanosystems applied to HIV infection: Prevention and treatments. Int. J. Mol. Sci. 2020, 21, 8647. [Google Scholar] [CrossRef] [PubMed]
- Binda, A.; Murano, C.; Rivolta, I. Innovative Therapies and Nanomedicine Applications for the Treatment of Alzheimer's Disease: A State-of-the-Art (2017-2020). Int. J. Nanomedicine 2020, 15, 6113–6135. [Google Scholar] [CrossRef]
- Kim, K.; Khang, D. Past, Present, and Future of Anticancer Nanomedicine. Int. J. Nanomedicine. 2020, 15, 5719–5743. [Google Scholar] [CrossRef]
- Desland, F. A.; Hormigo, A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int. J. Mol. Sci. 2020, 21, 7358. [Google Scholar] [CrossRef]
- Sharma, H. S.; Muresanu, D. F.; Castellani, R. J.; Nozari, A.; Lafuente, J. V.; Tian, Z. R.; Sahib, S.; Bryukhovetskiy, I.; Bryukhovetskiy, A.; Buzoianu, A. D.; Patnaik, R; Wiklund, L. ; Sharma, A. Pathophysiology of blood-brain barrier in brain tumor. Novel therapeutic advances using nanomedicine. Int. Rev. Neurobiol. 2020, 151, 1–66. [Google Scholar]
- Maher, E. A.; Furnari, F. B.; Bachoo, R. M.; Rowitch, D. H.; Louis, D. N.; Cavenee, W. K.; DePinho, R. A. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Stupp, R. , Hegi, M. E., Mason, W. P., van den Bent, M. J., Taphoorn, M. JB., Janzer, R. C., Ludwin, S. K., Allgeier, A., Fisher, B., Belanger, K., Hau, P., Brandes, A. A., Gijtenbeek, J., Marosi, C., Vecht, C. J., Mokhtari, K., Wesseling, P., Villa, S., Eisenhauer, E., Gorlia, T.; Weller, M.; Lacombe, D.; Gregory Cairncross, J.; Mirimanoff, R. O. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar]
- Hau, P.; Fabel, K.; Baumgart, U.; Rümmele, P.; Grauer, O.; Bock, A.; Dietmaier, C.; Dietmaier, W.; Dietrich, J.; Dudel, C.; Hübner, F.; Jauch, T.; Drechsel, E.; Kleiter, I.; Wismeth, C.; Zellner, A.; Brawanski, A.; Steinbrecher, A.; Marienhagen, J.; Bogdahn, U. Pegylated liposomal doxorubicin-efficacy in patients with recurrent high-grade glioma. Cancer 2004, 100, 1199–1207. [Google Scholar] [CrossRef]
- Friedman, H. S.; Prados, M. D.; Wen, P. Y.; Mikkelsen, T.; Schiff, D.; Abrey, L. E.; Yung, W. K.; Paleologos, N.; Nicholas, M. K.; Jensen, R.; Vredenburgh, J.; Huang, J.; Zheng, M.; Cloughesy, T. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Westphal, M.; Ylä-Herttuala, S.; Martin, J.; Warnke, P.; Menei, P.; Eckland, D.; Kinley, J.; Kay, R.; Ram, Z.; ASPECT Study Group. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Zustovich, F.; Landi, L.; Lombardi, G.; Porta, C.; Galli, L.; Fontana, A.; Amoroso, D.; Galli, C.; Andreuccetti, M.; Falcone, A.; Zagonel, V. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: a phase II study. Anticancer Res. 2013, 33, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; Croteau, D.; Pedain, C.; Leland, P.; Husain, S. R.; Joshi, B. H.; Puri, R. K.; PRECISE Study Group. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Bienemann, A.; Taylor, H.; Hopkins, K.; Cameron, A.; Gill, S. A phase I trial of carboplatin administered by convection-enhanced delivery to patients with recurrent/progressive glioblastoma multiforme. Contemp. Clin. Trials 2012, 33, 320–331. [Google Scholar] [CrossRef]
- Tiwari, S. B.; Amiji, M. M. A review of nanocarrier-based CNS delivery systems. Curr. Drug Deliv. 2006, 3, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Caraglia, M.; Addeo, R.; Costanzo, R.; Montella, L.; Faiola, V.; Marra, M.; Abbruzzese, A.; Palmieri, G.; Budillon, A.; Grillone, F.; Venuta, S.; Tagliaferri, P.; Del Prete, S. Phase II study of temozolomide plus pegylated liposomal doxorubicin in the treatment of brain metastases from solid tumours. Cancer Chemother. Pharmacol. 2006, 57, 34–39. [Google Scholar] [CrossRef]
- Beier, C. P.; Schmid, C.; Gorlia, T.; Kleinletzenberger, C.; Beier, D.; Grauer, O.; Steinbrecher, A.; Hirschmann, B.; Brawanski, A.; Dietmaier, C.; Jauch-Worley, T.; Kölbl, O.; Pietsch, T.; Proescholdt, M.; Rümmele, P.; Muigg, A.; Stockhammer, G.; Hegi, M.; Bogdahn, U.; Hau, P. RNOP-09: pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma-a phase II study. BMC Cancer 2009, 9, 308. [Google Scholar] [CrossRef]
- Northfelt, D. W.; Dezube, B. J.; Thommes, J. A.; Miller, B. J.; Fischl, M. A.; Friedman-Kien, A.; Kaplan, L. D.; Du Mond, C.; Mamelok, R. D.; Henry, D. H. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J. Clin. Oncol. 1998, 16, 2445–2451. [Google Scholar] [CrossRef]
- Gordon, A. N.; Tonda, M.; Sun, S.; Rackoff, W.; Doxil Study 30-49 Investigators. Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory epithelial ovarian cancer. Gynecol. Oncol. 2004, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, M. E. R.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D. G.; Tomczak, P.; Ackland, S. P.; Orlandi, F.; Mellars, L.; Alland, L.; Tendler, C.; CAELYX Breast Cancer Study Group. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [CrossRef]
- Orlowski, R. Z.; Nagler, A.; Sonneveld, P.; Bladé, J.; Hajek, R.; Spencer, A.; San Miguel, J.; Robak, T.; Dmoszynska, A.; Horvath, N.; Spicka, I.; Sutherland, H. J.; Suvorov, A. N.; Zhuang, S. H.; Parekh, T.; Xiu, L.; Yuan, Z.; Rackoff, W.; Harousseau, J. L. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J. Clin. Oncol. 2007, 25, 3892–3901. [Google Scholar] [CrossRef]
- Khaitan, D.; Reddy, P. L.; Ningaraj, N. Targeting Brain Tumors with Nanomedicines: Overcoming Blood Brain Barrier Challenges. Curr. Clin. Pharmacol. 2018, 13, 110–119. [Google Scholar] [CrossRef]
- Dunton, C. J. Management of treatment-related toxicity in advanced ovarian cancer. Oncologist 2002, 7, 11–19. [Google Scholar] [CrossRef]
- Yallapu, M. M.; Jaggi, M.; Chauhan, S. C. Scope of nanotechnology in ovarian cancer therapeutics. J. Ovarian Res. 2010, 3, 19. [Google Scholar] [CrossRef]
- Waite, C. L.; Roth, C. M. Nanoscale drug delivery systems for enhanced drug penetration into solid tumors: current progress and opportunities. Crit. Rev. Biomed. Eng. 2012, 40, 21–41. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J. M.; Hong, S.; Farokhzad, O. C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Petros, R. A.; DeSimone, J. M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Tian, L. R.; Lin, M. Z.; Zhong, H. H.; Cai, Y. J.; Li, B.; Xiao, Z. C.; Shuai, X. T. Nanodrug regulates lactic acid metabolism to reprogram the immunosuppressive tumor microenvironment for enhanced cancer immunotherapy. Biomater. Sci. 2022, 10, 3892–3900. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Zhang, B.; Xu, Z. P. MnO2-shelled Doxorubicin/Curcumin nanoformulation for enhanced colorectal cancer chemo-immunotherapy. J. Colloid Interface Sci. 2022, 617, 315–325. [Google Scholar] [CrossRef]
- Han Y, Wen P, Li J, Kataoka K. Targeted nanomedicine in cisplatin-based cancer therapeutics. J. Control Release 2022, 345, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Nabi, B.; Javed, A.; Khan, T.; Iqubal, A.; Ansari, M. J.; Baboota, S.; Ali, J. Unraveling enhanced brain delivery of paliperidone-loaded lipid nanoconstructs: pharmacokinetic, behavioral, biochemical, and histological aspects. Drug Deliv. 2022, 29, 1409–1422. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, T.; Qin, S.; Huang, Z.; Zhou, L.; Shi, J.; Nice, E. C.; Xie, N.; Huang, C.; Shen, Z. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. J. Hematol. Oncol. 2022, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, Z.; Huang, C.; Qiu, M.; Song, D.; Li, Y.; Xu, Q. Lipid nanoparticle-mediated lymph node-targeting delivery of mRNA cancer vaccine elicits robust CD8+ T cell response. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2207841119. [Google Scholar] [CrossRef] [PubMed]
- Singh, V. K.; Chau, E.; Mishra, A.; DeAnda, A.; Hegde, V. L.; Sastry, J. K.; Haviland, D.; Jagannath, C.; Godin, B.; Khan, A. CD44 receptor targeted nanoparticles augment immunity against tuberculosis in mice. J. Control Release. 2022, 349, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Yan, H.; Zhang, J.; Thomas, R. K. Binding of sodium dodecyl sulfate with linear and branched polyethyleneimines in aqueous solution at different pH values. Langmuir 2006, 22, 1526–1533. [Google Scholar] [CrossRef]
- Wang, Y.; Kimura, K.; Dubin, P. L.; Jaeger, W. Polyelectrolyte−Micelle Coacervation: Effects of Micelle Surface Charge Density, Polymer Molecular Weight, and Polymer/Surfactant Ratio. Macromolecules 2000, 33, 3324–3331. [Google Scholar] [CrossRef]
- Wang, Y.; Kimura, K.; Huang, Q.; Dubin P., L.; Jaeger, W. Effects of Salt on Polyelectrolyte−Micelle Coacervation. Macromolecules 1999, 32, 7128–7134. [Google Scholar] [CrossRef]
- Kim, B. S.; Park, S. W.; Hammond, P. T. Hydrogen-bonding layer-by-layer-assembled biodegradable polymeric micelles as drug delivery vehicles from surfaces. ACS Nano 2008, 2, 386–392. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, G.; Ma, Y.; Cui, Z.; Binks, B. P. Smart worm-like micelles responsive to CO₂/N₂ and light dual stimuli. Soft Matter 2017, 13, 2727–2732. [Google Scholar] [CrossRef]
- Son, S.; Shin, E.; Kim, B. S. Light-responsive micelles of spiropyran initiated hyperbranched polyglycerol for smart drug delivery. Biomacromolecules 2014, 15, 628–634. [Google Scholar] [CrossRef]
- Oh, H.; Ketner A., M.; Heymann, R.; Kesselman, E.; Danino, D.; Falvey, D. E.; Raghavan, S. R. A Simple Route to Fluids with Photo-Switchable Viscosities Based on a Reversible Transition between Vesicles and Wormlike Micelles. Soft Matter 2013, 9, 5025. [Google Scholar] [CrossRef]
- Davies, T. S.; Ketner, A. M.; Raghavan, S. R. Self-assembly of surfactant vesicles that transform into viscoelastic wormlike micelles upon heating. J. Am. Chem. Soc. 2006, 128, 6669–6675. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, Y.; Chu, Z.; He, S.; Zhang, J.; Feng, Y. Thermally induced structural transitions from fluids to hydrogels with pH-switchable anionic wormlike micelles. J. Colloid Interface Sci. 2013, 394, 319–328. [Google Scholar] [CrossRef]
- Mezei, A.; Meszaros, R.; Varga, I.; Gilanyi, T. Effect of mixing on the formation of complexes of hyperbranched cationic polyelectrolytes and anionic surfactants. Langmuir 2007, 23, 4237–4247. [Google Scholar] [CrossRef]
- Chiappisi, L.; Leach, S. D.; Gradzielski, M. Precipitating polyelectrolyte–surfactant systems by admixing a nonionic surfactant – a case of cononsurfactancy. Soft Matter 2017, 13, 4988–4996. [Google Scholar] [CrossRef]
- Zhao, M.; Eghtesadi, S. A.; Dawadi, M. B.; Wang, C.; Huang, S; Seymore, A. E.; Vogt, B. D.; Modarelli, D. A.; Liu, T.; Zacharia, N. S. Partitioning of Small Molecules in Hydrogen-Bonding Complex Coacervates of Poly(acrylic acid) and Poly(ethylene glycol) or Pluronic Block Copolymer. Macromolecules 2017, 50, 3818–3830. [Google Scholar] [CrossRef]
- Black, K. A.; Priftis, D.; Perry, S. L.; Yip, J.; Byun, W. Y.; Tirrell, M. Protein Encapsulation via Polypeptide Complex Coacervation. ACS Macro Lett. 2014, 3, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zacharia, N. S. Sequestration of Methylene Blue into Polyelectrolyte Complex Coacervates. Macromol Rapid Commun. 2016, 37, 1249–1255. [Google Scholar] [CrossRef]
- Lv, K.; Perriman, A. W.; Mann, S. Photocatalytic multiphase micro-droplet reactors based on complex coacervation. Chemical Communications 2015, 51, 8600–8602. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liu, X. K.; Yuan, W. C.; Brown, L. J.; Wang, D. Y. Confined flocculation of ionic pollutants by Poly(l-dopa)-based polyelectrolyte complexes in hydrogel beads for three-dimensional, quantitative, efficient water decontamination. Langmuir 2015, 31, 6351–6366. [Google Scholar] [CrossRef]
- Xu, Y.; Mazzawi, M.; Chen, K.; Sun, L.; Dubin, P. L. Protein purification by polyelectrolyte coacervation: influence of protein charge anisotropy on selectivity. Biomacromolecules 2011, 12, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Dora Tang, T. Y.; Rohaida Che Hak, C.; Thompson, A. J.; Kuimova, M. K.; Williams, D. S.; Perriman, A. W.; Mann, S. Fatty acid membrane assembly on coacervate microdroplets as a step towards a hybrid protocell model. Nat. Chem. 2014, 6, 527–533. [Google Scholar] [CrossRef]
- Stano, P.; Luisi, P. L. Achievements and open questions in the self-reproduction of vesicles and synthetic minimal cells. Chem. Commun. (Camb) 2010, 46, 3639–3653. [Google Scholar] [CrossRef]
- Kurihara, K.; Tamura, M.; Shohda, K.; Toyota, T.; Suzuki, K.; Sugawara, T. Self-reproduction of supramolecular giant vesicles combined with the amplification of encapsulated DNA. Nat Chem. 2011, 3, 775–781. [Google Scholar] [CrossRef]
- Mansy, S. S.; Schrum, J. P.; Krishnamurthy, M.; Tobé, S.; Treco, D. A.; Szostak, J. W. Template-directed synthesis of a genetic polymer in a model protocell. Nature 2008, 454, 122–125. [Google Scholar] [CrossRef]
- Stadler, B.; Price, A. D.; Chandrawati, R.; Hosta-Rigau, L.; Zelikin, A. N.; Caruso, F. Polymer hydrogel capsules: En route toward synthetic cellular systems. Nanoscale 2009, 1, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Bai, S.; Ansorge-Schumacher, M. B.; Wang, D. Nanoparticle cages for enzyme catalysis in organic media. Adv. Mater. 2011, 23, 5694–5699. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A. B.; Wan, J.; Gopinath, A.; Stone, H. A. Semi-permeable vesicles composed of natural clay. Soft Matter 2011, 7, 2600–2612. [Google Scholar] [CrossRef]
- Luisi, P. L.; Ferri, F.; Stano, P. Approaches to semi-synthetic minimal cells: a review. Naturwissenschaften 2006, 93, 1–13. [Google Scholar] [CrossRef]
- Dzieciol, A. J.; Mann, S. Designs for life: protocell models in the laboratory. Chem. Soc. Rev. 2012, 41, 79–85. [Google Scholar] [CrossRef]
- Priftis, D.; Farina, R.; Tirrell, M. Interfacial energy of polypeptide complex coacervates measured via capillary adhesion. Langmuir 2012, 28, 8721–8729. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, M.; Dawadi, M. B.; Cai, Y.; Lapitsky, Y.; Modarelli, D. A.; Zacharia, N. S. Effect of small molecules on the phase behavior and coacervation of aqueous solutions of poly(diallyldimethylammonium chloride) and poly(sodium 4-styrene sulfonate). J. Colloid Interface Sci. 2018, 518, 216–224. [Google Scholar] [CrossRef]
- Lapitsky, Y.; Kaler, E. W. Surfactant and polyelectrolyte gel particles for encapsulation and release of aromatic oils. Soft Matter 2006, 2, 779–784. [Google Scholar] [CrossRef]
- Tehrani-Bagha, A. R.; Holmberg, K. Solubilization of Hydrophobic Dyes in Surfactant Solutions. Materials (Basel) 2013, 6, 580–608. [Google Scholar] [CrossRef]
- Tehrani-Bagha, A. R.; Singh, R. G.; Holmberg, K. Solubilization of two organic water-insoluble dyes by anionic, cationic and nonionic surfactants. Colloid Surf. Physicochem. Eng. Asp. 2013, 417, 133–139. [Google Scholar] [CrossRef]
- Thalberg, K.; Lindman, B.; Karlstroem, G. Phase-behavior of systems of cationic surfactant and anionic polyelectrolyte: influence of surfactant chain-length and polyelectrolyte molecular-weight. J. Phys. Chem. 1991, 95, 3370–3376. [Google Scholar] [CrossRef]
- Jain, N.; Trabelsi, S.; Guillot, S.; McLoughlin, D.; Langevin, D.; Letellier, A. P.; Turmine, M. Critical Aggregation Concentration in Mixed Solutions of Anionic Polyelectrolytes and Cationic Surfactants. Langmuir 2004, 20, 8496–8503. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. J.; Xia, J. L.; Dubin, P. L. Complex formation between polyelectrolyte and oppositely charged mixed micelles: Static and dynamic light scattering study of the effect of polyelectrolyte molecular weight and concentration. Macromolecules 1994, 27, 7049–7055. [Google Scholar] [CrossRef]
- Dubin P., L.; Rigsbee, D. R.; Gan, L.; Fallon, M. A. Equilibrium Binding of Mixed Micelles to Oppositely Charged Polyelectrolytes. Macromolecules 1988, 21, 2555–2559. [Google Scholar] [CrossRef]
- Comert, F.; Nguyen, D.; Rushanan, M.; Milas, P.; Xu, A. Y.; Dubin, P. L. Precipitate–Coacervate Transformation in Polyelectrolyte–Mixed Micelle Systems. J. Phys. Chem. B. 2017, 121, 4466–4473. [Google Scholar] [CrossRef]
- Dubin, P. L.; Oteri, R. Association of polyelectrolytes with oppositely charged micelles. J. Colloid Interface Sci. 1983, 95, 453–461. [Google Scholar] [CrossRef]
- Xia, J.; Zhang, H.; Rigsbee, D. R.; Dubin, P. L.; Shaikh, T. Structural elucidation of soluble polyelectrolyte-micelle complexes: Intra- vs interpolymer association. Macromolecules 1993, 26, 2759–2766. [Google Scholar] [CrossRef]
- Wang, Y.; Kimura, K.; Huang, Q.; Dubin, P.L.; Jaeger, W. Effects of Salt on Polyelectrolyte−Micelle Coacervation. Macromolecules 1999, 32, 7128–7134. [Google Scholar] [CrossRef]
- McQuigg, D. W.; Kaplan, J. I.; Dubin, P. L. Critical conditions for the binding of polyelectrolytes to small oppositely charged micelles. J. Chem. Phy. 1992, 96, 1973–1978. [Google Scholar] [CrossRef]
- Mészáros, R.; Thompson, L.; Bos, M.; Varga, I.; Gilányi, T. Interaction of Sodium Dodecyl Sulfate with Polyethyleneimine: Surfactant-Induced Polymer Solution Colloid Dispersion Transition. Langmuir 2003, 19, 609–615. [Google Scholar] [CrossRef]
- Mezei, A.; Ábrahám, Á.; Pojják, K.; Mészáros, R. The impact of electrolyte on the aggregation of the complexes of hyperbranched poly(ethyleneimine) and sodium dodecylsulfate. Langmuir 2009, 25, 7304–7312. [Google Scholar] [CrossRef]
- Petzold, G.; Schwarz, S. Dye removal from solutions and sludges by using polyelectrolytes and polyelectrolyte-surfactant complexes. Sep. Purif. Technol. 2006, 51, 318–324. [Google Scholar] [CrossRef]
- Chatterjee, S.; Lee, M. W.; Woo, S. H. Adsorption of congo red by chitosan hydrogel beads impregnated with carbon nanotubes. Bioresour. Technol. 2010, 101, 1800–1806. [Google Scholar] [CrossRef]
- Takae, S.; Miyata, K.; Oba, M.; Ishii, T.; Nishiyama, N.; Itaka, K.; Yamasaki, Y.; Koyama, H.; Kataoka, K. PEG-detachable polyplex micelles based on disulfide-linked block catiomers as bioresponsive nonviral gene vectors. J. Am. Chem. Soc. 2008, 130, 6001–6009. [Google Scholar] [CrossRef]
- Ge, Z.; Liu, S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bu, Y.; Zhang, L.; Wang, X.; Yang, Y.; Zhuang, Y.; Yang, F.; Shen, H.; Wu, D. Facile construction of pH- and redox-responsive micelles from a biodegradable poly(β-hydroxyl amine) for drug delivery. Biomacromolecules 2016, 17, 291–300. [Google Scholar] [CrossRef]
- Zhu, C.; Zheng, M.; Meng, F.; Mickler, F. M.; Ruthardt, N.; Zhu, X.; Zhong, Z. Reversibly shielded DNA polyplexes based on bioreducible PDMAEMA-SS-PEG-SS-PDMAEMA triblock copolymers mediate markedly enhanced nonviral gene transfection. Biomacromolecules 2012, 13, 769–778. [Google Scholar] [CrossRef]
- Wang, J. Y.; Velders, A. H.; Gianolio, E.; Aime, S.; Vergeldt, F. J.; Van As, H.; Yan, Y.; Drechsler, M.; de Keizer, A.; Stuart, M. A. C.; van der Gucht, J. Controlled Mixing of Lanthanide(III) Ions in Coacervate Core Micelles. Chem. Commun. 2013, 49, 3736–3738. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Groeneveld, A.; Oikonomou, M.; Prusova, A.; Van As, H.; van Lent, J.W.; Velders, A.H. Revealing and Tuning the Core, Structure, Properties and Function of Polymer Micelles with Lanthanide-coordination Complexes. Soft Matter 2016, 12, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ding, Y.; Yang, Y.; Yan, Y.; Huang, J.; de Keizer, A.; Cohen Stuart, M. A. Fluorescence enhancement by microphase separation-induced chain extension of Eu3+ coordination polymers: phenomenon and analysis. Soft Matter 2011, 7, 2720–2724. [Google Scholar] [CrossRef]
- Wang. J.; Cohen Stuart, M. A.; Marcelis, A. T. M., Colomb-Delsuc, M.; Otto, S.; van der Gucht, J. Stable polymer micelles formed by metal coordination. Macromolecules 2012, 45, 7179–7185. [Google Scholar] [CrossRef]
- Ten Hove, J. B.; Wang, J.; van Leeuwen, F. W. B.; Velders, A. H. Dendrimer-encapsulated Nanoparticle-core Micelles as a Modular Strategy for Particle-in-a-box-in-a-box Nanostructures. Nanoscale 2017, 9, 18619–18623. [Google Scholar] [CrossRef]
- Wang, J.; Voets, I. K.; Fokkink, R.; van der Gucht, J.; Velders, A. H. Controlling the Number of Dendrimers in Dendrimicelle Nanoconjugates from 1 to More than 100. Soft Matter 2014, 10, 7337–7345. [Google Scholar] [CrossRef]
- Ten Hove, J. B.; van Oosterom, M. N.; van Leeuwen, F. W. B.; Velders, A. H. Nanoparticles Reveal Extreme Size-Sorting and Morphologies in Complex Coacervate Superstructures. Sci. Rep. 2018, 8, 13820–13827. [Google Scholar] [CrossRef]
- Facciotti, C.; Saggiomo, V.; Bunschoten, A.; Fokkink, R.; Hove, J. B. T.; Wang, J.; Velders, A. H. Cyclodextrin-based Complex Coacervate Core Micelles with Tuneable Supramolecular Host-guest, Metal-to-ligand and Charge Interactions. Soft Matter 2018, 14, 9542–9549. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Leon, L. Structural dynamics, phase behavior, and applications of polyelectrolyte complex micelles. Curr. Opin. Colloid Interface Sci. 2021, 53, 101424. [Google Scholar] [CrossRef]
- Cohen Stuart, M. A.; Hofs, B.; Voets, I. K.; de Keizer, A. Assembly of polyelectrolyte-containing block copolymers in aqueous media. Curr. Opin. Colloid Interface Sci. 2005, 10 (1–2), 30-36.
- Hales, K.; Pochan, D. J. Using polyelectrolyte block copolymers to tune nanostructure assembly. Curr. Opin. Colloid Interface Sci. 2007, 11, 330–336. [Google Scholar] [CrossRef]
- Colfen, H. Double-hydrophilic block copolymers: synthesis and application as novel surfactants and crystal growth modifiers. Macromol. Rapid Commun. 2001, 22, 219–252. [Google Scholar] [CrossRef]
- Dou, H. J.; Kang, S. Double-Hydrophilic Block Copolymers and Their Self-Assembly. Prog. Chem. 2005, 17, 854–859. [Google Scholar]
- Gohy, J. F. Block Copolymer Micelles. In Block Copolymers II; Abetz, V., Ed.; Advances in Polymer Science; Springer: Berlin, Heidelberg, 2005; Vol. 190, pp 65-136. [Google Scholar] [CrossRef]
- Riess, G. Micellization of block copolymers. Prog. Polym. Sci. 2003, 28, 1107–1170. [Google Scholar] [CrossRef]
- Kabanov, V. A.; Kabanov, A. V. Interpolyelectrolyte and block ionomer complexes for gene delivery: physico-chemical aspects. Adv. Drug Deliv. Rev. 1998, 30, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Bronich, T. K.; Kabanov, A. V.; Eisenberg, A.; Kabanov, V. A. , Block ionomer complexes: Implications for drug delivery. Abstr. Pap. Am. Chem. Soc. 2002, 223, U438–U438. [Google Scholar]
- Harada, A.; Kataoka, K. Polyion complex micelles with core-shell structure: their physicochemical properties and utilities as functional materials. Macromol. Symp. 2001, 172, 1–9. [Google Scholar] [CrossRef]
- Gebhart, C. L.; Kabanov, A. V. Perspectives on Polymeric Gene Delivery. J. Bioact. Compat. Polym. 2003, 18, 147–166. [Google Scholar] [CrossRef]
- Gillies, E. R.; Frechet, J. M. J. Development of acid-sensitive copolymer micelles for drug delivery. Pure Appl. Chem. 2004, 76, 1295–1307. [Google Scholar] [CrossRef]
- Nishiyama, N.; Jang, W-D. ; Kataoka, K. Supramolecular nanocarriers integrated with dendrimers encapsulating photosensitizers for effective photodynamic therapy and photochemical gene delivery. New J. Chem. 2007, 31, 1074–1082. [Google Scholar] [CrossRef]
- Yang, L.; Alexandridis, P. Physicochemical aspects of drug delivery andrelease from polymer-based colloids. Curr. Opin. Colloid Interface Sci. 2000, 5, 132–143. [Google Scholar] [CrossRef]
- Kataoka, K.; Togawa, H.; Harada, A.; Yagusi, K.; Matsumoto, T.; Katayose, S. Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline. Macromolecules 1996, 29, 8556–8557. [Google Scholar] [CrossRef]
- Cohen Stuart, M. A.; Besseling, N. A. M.; Fokkink, R. G. Formation of Micelles with Complex Coacervate Cores. Langmuir 1998, 14, 6846–6849. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Novel Polyion Complex Micelles Entrapping Enzyme Molecules in the Core. 2. Characterization of the Micelles Prepared at Nonstoichiometric Mixing Ratios. Langmuir 1999, 15, 4208–4212. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Novel Polyion Complex Micelles Entrapping Enzyme Molecules in the Core: Preparation of Narrowly-Distributed Micelles from Lysozyme and Poly(ethylene glycol)−Poly(aspartic acid) Block Copolymer in Aqueous Medium. Macromolecules 1998, 31, 288–294. [Google Scholar] [CrossRef]
- Wijmans, C. M.; Zhulina, E. B. Polymer brushes at curved surfaces. Macromolecules 1993, 26, 7214–7224. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Chain length recognition: core-shell supramolecular assembly from oppositely charged block copolymers. Science (Washington, DC, U. S.) 1999, 283, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. On-off Control of Enzymatic Activity Synchronizing With Reversible Formation of Supramolecular Assembly from Enzyme and Charged Block Copolymers. J. Am. Chem. Soc. 1999, 121, 9241–9242. [Google Scholar] [CrossRef]
- Min, H. S.; Kim, H. J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B. S.; Fukushima, S.; Anraku, Y.; Miyata, K.; Kataoka, K. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. Int. Ed. Engl. 2020, 59, 8173–8180. [Google Scholar] [CrossRef]
- Cammas, S.; Kataoka, K. Site specific drug-carriers: polymeric micelles as high potential vehicles for biologically active molecules. In Solvents and Self-organization of Polymers; Webber, S. E., Ed.; Kluwer Academic Publishers: Dordrecht, 1996. [Google Scholar]
- Quémener, D.; Deratani, A.; Lecommandoux, S. Dynamic Assembly of Block-Copolymers; Barboiu, M., Ed.; Constitutional Dynamic Chemistry. Topics in Current Chemistry; Springer: Berlin, Heidelberg, 2011; Vol.322, pp 165-192. [Google Scholar] [CrossRef]
- Meier, D. J. Theory of block copolymers. I. Domain formation in A-B block copolymers. J. Polym. Sci. Part C 1969, 26, 81–98. [Google Scholar] [CrossRef]
- Helfand, E.; Tagami, Y. Theory of the Interface between Immiscible Polymers. II. J. Chem. Phys. 1972, 56, 3592–3601. [Google Scholar] [CrossRef]
- Helfand, E.; Tagami, Y. Theory of the Interface Between Immiscible Polymers. J. Chem. Phys. 1972, 57, 1812–1813. [Google Scholar] [CrossRef]
- Helfand, E.; Wasserman, Z. R. Block Copolymer Theory. 4. Narrow Interphase Approximation. Macromolecules 1976, 9, 879–888. [Google Scholar]
- Helfand, E.; Wasserman, Z. R. Block Copolymer Theory. 5. Spherical Domains. Macromolecules 1978, 11, 960–966. [Google Scholar] [CrossRef]
- Helfand, E.; Wasserman, Z. R. Block Copolymer Theory. 6. Cylindrical Domains. Macromolecules 1980, 13, 994–998. [Google Scholar] [CrossRef]
- Leermakers, F. A. M.; & Scheutjens, J. M. H. M.; Scheutjens, J. M. H. M. Statistical thermodynamics of associated colloids. 1. Lipid bilayer membranes. Journal of Chemical Physics 1988, 89, 3264–3274. [Google Scholar] [CrossRef]
- Yuan, X. F.; Masters, A. J.; Price, C. Self-consistent field theory of micelle formation by block copolymers. Macromolecules 1992, 25, 6876–6884. [Google Scholar] [CrossRef]
- Fleer, G.; Stuart, M. C.; Scheutjens, J. M.; Cosgrove, T.; Vincent, B. Polymers at interfaces. Chapman and Hall: London, 1993.
- Leermakers, F. A. M.; Eriksson, J. C.; Lyklema, J. Association colloids and their equilibrium modelling. In Fundamentals of Interface and Colloid Science; Lyklema, J., Elsevier Publishing: Amsterdam, The Netherlands, 2005; Vol. V: Soft Colloids, pp 4.1-4.123. [Google Scholar]
- Edwards, S. F. The statistical mechanics of polymers with excluded volume. Proc. Phys. Soc. 1965, 85, 613–624. [Google Scholar] [CrossRef]
- Ulrich, R.D.; Flory, P. J. In: Macromolecular Science. Contemporary Topics in Polymer Science; Ulrich, R. D., Ed.; Springer: Boston, MA, 1978; Vol. 1, pp 69-98. [Google Scholar] [CrossRef]
- Kik, R. A.; Leermakers, F. A.; Kleijn, J. M. Molecular modeling of lipid bilayers and the effect of protein-like inclusions. Phys. Chem. Chem. Phys. 2005, 7, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- Kik, R. A.; Lermakers, F. A. M.; Kleijn, J. M. Molecular modeling of protein like inclusions in lipid bilayers: Lipid-mediated interactions. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2010, 81 (2 Pt 1)), 021915/1-12. [Google Scholar] [CrossRef] [PubMed]
- Shi, A. C.; Noolandi, J. Theory of inhomogeneous weakly charged polyelectrolytes. Macromol. Theory Simul. 1999, 8, 214–229. [Google Scholar] [CrossRef]
- Wang, Q.; Taniguchi, T.; Fredrickson, G. H. Self-Consistent Field Theory of Polyelectrolyte Systems. J. Phys. Chem. B 2004, 108, 6733–6744. [Google Scholar] [CrossRef]
- De Gennes, P. G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, AZ, USA; London, UK, 1979. [Google Scholar]
- Birshtein, T. M.; Gridnev, V. N.; Gotlib, Y.Y.; Skvortsov, A. M Simulating the dynamic behaviour of polymer chains using the Monte Carlo method. Polymer Sci. USSR. 1977, 19, 1612–1621. [Google Scholar] [CrossRef]
- Schaefer, D. W.; Joanny, J.-F.; Pincus, P. Dynamics of Semiflexible Polymers in Solution. Macromolecules 1980, 13, 1280–1289. [Google Scholar] [CrossRef]
- de Gennes, P.-G. In Solid State Physics; Liebert, L. (Eds.) ; Academic: New York, 1978; Suppl. 14.
- Zhulina, Y. B.; Birshtein, T. M. Conformations of block-copolymer molecules in selective solvents (micellar structures). Polym. Sci. USSR. 1985, 27, 570–578. [Google Scholar] [CrossRef]
- Birshtein, T. M.; Zhulina, E. B. Scaling theory of supermolecular structures in block copolymer-solvent systems: 1. Model of micellar structures. Polymer 1989, 30, 170–177. [Google Scholar] [CrossRef]
- Pergushov, D. V.; Remizova, E. V.; Gradzielski, M.; Lindner, P.; Feldthusen, J.; Zezin, A.B.; Müller, A. H. E.; Kabanov, V. A. Micelles of polyisobutylene-block-poly(methacrylicacid) diblock copolymers and their water-soluble interpolyelectrolyte complexes formed with quaternized poly(4 vinylpyridine). Polymer 2004, 45, 367–378. [Google Scholar] [CrossRef]
- Lysenko, E. A.; Chelushkin, P. S.; Bronich, T. K.; Eisenberg, A.; Kabanov, V. A.; Kabanov, A. V. Formation of Multilayer Polyelectrolyte Complexes by Using Block Ionomer Micelles as Nucleating Particles. J. Phys. Chem. B 2004, 108, 12352–12359. [Google Scholar] [CrossRef]
- Zhang, W. Q.; Shi, L. Q.; Gao, L. C.; An, Y. L.; Wu, K. Formation of Core-Shell-Corona Micellar Complexes through Adsorption of Double Hydrophilic Diblock Copolymers into Core-Shell Micelles. Macromol. Rapid Commun. 2005, 26, 1341–1345. [Google Scholar] [CrossRef]
- Zhu, R. P.; Wang, Y. M.; He, W. D. Multiple morphological micelles formed from the self-assembly of poly(styrene)-b-poly( 4-vinylpyridine) containing cobalt dodecyl benzene sulfonate. Eur. Polym. J. 2005, 41, 2088–2096. [Google Scholar] [CrossRef]
- Hu, Z. J.; Jonas, A. M.; Varshney, S. K.; Gohy, J. F. Dilution-induced spheres-to-vesicles morphological transition in micelles from block copolymer/surfactant complexes. J. Am. Chem. Soc. 2005, 127, 6526–6527. [Google Scholar] [CrossRef]
- Zhang, W. Q.; Shi, L. Q.; Gao, L. C.; An, Y. L.; Li, G. Y.; Wu, K.; Liu, Z. Comicellization of Poly(ethylene glycol)-block-poly(acrylic acid) and Poly(4-vinylpyridine) in Ethanol. Macromolecules 2005, 38, 899–903. [Google Scholar] [CrossRef]
- Duan, H. W.; Kuang, M.; Wang. ; Chen, D. Y.; Jiang, M. Self-assembly of rigid and coil polymersinto hollow spheres in their common solvent. J. Phys. Chem. B 2004, 108, 550–555. [Google Scholar] [CrossRef]
- Kuang, M.; Duan, H. W.; Wang, J.; Jiang, M. Structural factors of rigid-coil polymer pairs influencing their self-assembly in common solvent. J. Phys. Chem. B. 2004, 108, 16023–16029. [Google Scholar] [CrossRef]
- Guo, Y.; Moffitt, M. G. Semiconductor quantum dots with environmentally responsive mixed polystyrene/poly (methyl methacrylate) brush layers. Macromolecules 2007, 40, 5868–5878. [Google Scholar] [CrossRef]
- Moffitt, M.; Eisenberg, A. Scaling Relations and Size Control of Block Ionomer Microreactors Containing Different Metal Ions. Macromolecules 1997, 30, 4363–4373. [Google Scholar] [CrossRef]
- Schrage, S.; Sigel, R.; Schlaad, H. : Formation of amphiphilic polyion complex vesicles from mixtures of oppositely charged block ionomers. Macromolecules 2003, 36, 1417–1420. [Google Scholar] [CrossRef]
- Topouza, D.; Orfanou, K.; Pispas, S. Thermosensitive Non-covalently Bonded Block Copolymer-like Micelles from Interpolymer Complexes. J. Polym. Sci. Part A: Polym. Chem. 2004, 42, 6230–6237. [Google Scholar] [CrossRef]
- Gohy, J. F.; Khousakoun, E.; Willet, N.; Varshney, S. K.; Jerome, R. Segregation of coronal chains in micelles formed by supramolecular interactions. Macromol. Rapid Commun. 2004, 25, 1536–1539. [Google Scholar] [CrossRef]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol. 2008, 26, 57–64. [Google Scholar] [CrossRef]
- Quan, C. Y.; Wei, H.; Sun, Y. X.; Cheng, S. X.; Shen, K.; Gu, Z. W.; Zhang, X. Z.; Zhuo, R. X. Polyethyleneimine modified biocompatible poly(N-isopropylacrylamide)- based nanogels for drug delivery. J. Nanosci. Nanotechnol. 2008, 8, 2377–2384. [Google Scholar] [CrossRef]
- Diez, S.; Navarro, G.; de Ilarduya, C. T. In vivo targeted gene delivery by cationic nanoparticles for treatment of hepatocellular carcinoma. J. Gene Med. 2008, 11, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Sortino, S.; Mazzaglia, A.; Monsu Scolaro, L.; Marino Merlo, F.; Valveri, V.; Sciortino, M. T. Nanoparticles of cationic amphiphilic cyclodextrins entangling anionic porphyrins as carrier-sensitizer system in photodynamic cancer therapy. Biomaterials 2006, 27, 4256–4265. [Google Scholar] [CrossRef]
- Xiong, M. P.; Bae, Y.; Fukushima, S.; Forrest, M. L.; Nishiyama, N.; Kataoka, K.; Kwon, G. S. pH-responsive Multi-PEGylated dual cationic nanoparticles enable charge modulations for safe gene delivery. ChemMedChem. 2007, 2, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Breunig, M.; Bauer, S.; Goepferich, A. Polymers and nanoparticles: intelligent tools for intracellular targeting? Eur. J. Pharm. Biopharm. 2008, 68, 112–128. [Google Scholar] [CrossRef]
- Lim, Y. T.; Cho, M. Y.; Lee, J. M.; Chung, S. J.; Chung, B. H. Simultaneous intracellular delivery of targeting antibodies and functional nanoparticles with engineered protein G system. Biomaterials 2009, 30, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Ishida, E.; Hashimoto, K.; Kobayashi, H.; Aoki, T.; Yasuda, J.; Obata, K.; Kikuchi, H.; Ishida, T.; Kiwada, H.; Harashima, H. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int. J. Pharm. 2007, 342, 194–200. [Google Scholar] [CrossRef]
- Kirpotin, D. B.; Drummond, D. C.; Shao, Y.; Shalaby, M. R.; Hong, K.; Nielsen, U. B.; Marks, J. D.; Benz, C. C.; Park, J. W. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006, 66, 6732–6740. [Google Scholar] [CrossRef]
- Liu, P.; Wang, B.; Weili Qiao, J. L. Multi-anticancer drugs encapsulated in the micelle: a novel chemotherapy to cancer. Med. Hypotheses 2008, 71, 379–381. [Google Scholar] [CrossRef]
- Matsumura, Y. Basic Aspects and Clinical Trials of Micelle Carrier System. MEMS, NANO and Smart Systems, ICMENS 2004: 25-27 Aug. 2004. p 610.
- Hwang, M. J.; Suh, J. M.; Bae, Y. H.; Kim, S. W. ; Jeong B: Caprolactonic poloxamer analog: PEG-PCL-PEG. Biomacromolecules 2005, 6, 885–890. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Y.; Allen, C. Polymer-drug compatibility: a guide to the development of delivery systems for the anticancer agent, ellipticine. J. Pharm. Sci. 2004, 93, 132–143. [Google Scholar] [CrossRef]
- Liggins, R. T.; Burt, H. M. Polyether-polyester diblock copolymers for the preparation of paclitaxel loaded polymeric micelle formulations. Adv. Drug Deliv. Rev. 2002, 54, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M. M.; Jaggi, M.; Chauhan, S. C. beta-Cyclodextrin-curcumin self- assembly enhances curcumin delivery in prostate cancer cells. Colloids Surf. B Biointerf. 2010, 79, 113–125. [Google Scholar] [CrossRef]
- Yallapu, M. M.; Jaggi, M.; Chauhan, S. C. Poly(β-cyclodextrin)/curcumin self-assembly: a novel approach to improve curcumin delivery and its therapeutic efficacy in prostate cancer cells. Macromol. Biosci. 2010, 10, 1141–1151. [Google Scholar] [CrossRef]
- Yallapu, M. M.; Gupta, B. K.; Jaggi, M.; Chauhan, S. C. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J. Colloid Interface Sci. 2010, 351, 19–29. [Google Scholar] [CrossRef]
- Yasugi, K.; Nagasaki, Y.; Kato, M.; Kataoka, K. Preparation and characterization of polymer micelles from poly(ethylene glycol)-poly(D,L-lactide) block copolymers as potential drug carrier. J. Control Release 1999, 62, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Solomon, C. H.; Pho, L. N.; Burt, R. W. Current status of genetic testing for colorectal cancer susceptibility. Oncology 2002, 16, 161–171. [Google Scholar] [PubMed]
- Yoo, H. S.; Park, T. G. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J. Control Release 2001, 70, 63–70. [Google Scholar] [CrossRef]
- Jette, K. K.; Law, D.; Schmitt, E. A.; Kwon, G. S. Preparation and drug loading of poly(ethylene glycol)-block-poly(epsilon-caprolactone) micelles through the evaporation of a cosolvent azeotrope. Pharm. Res. 2004, 21, 1184–1191. [Google Scholar] [CrossRef]
- Chen, C.; Yu, C. H.; Cheng, Y. C.; Yu, P. H.; Cheung, M. K. Biodegradable nanoparticles of amphiphilic triblock copolymers based on poly(3- hydroxybutyrate) and poly(ethylene glycol) as drug carriers. Biomaterials 2006, 27, 4804–4814. [Google Scholar] [CrossRef]
- La, S. B.; Okano, T.; Kataoka, K. Preparation and characterization of the micelle-forming polymeric drug indomethacin-incorporated poly (ethylene oxide)-poly(beta-benzyl L-aspartate) block copolymer micelles. J. Pharm. Sci. 1996, 85, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yu, C. H.; Cheng, Y. C.; Yu, P. H.; Cheung, M. K. Micelle formation and sol- gel transition behavior of comb-like amphiphilic poly((PLGA-b-PEG)MA) copolymers. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 1954–1963. [Google Scholar]
- Park, I.; Lee, H.; Chang, T.; Kim, S. W.; Lee, D. S. Poly(D,L-lactic acid-co-glycolic acid)-b-poly(ethylene glycol)-b-poly (D,L-lactic acid-co-glycolic acid) triblock copolymer and thermoreversible phase transition in water. J. Biomed. Mater. Res. 2002, 61, 188–196. [Google Scholar]
- Lee, S. J.; Han, B. R.; Park, S. Y.; Han, D. K.; Kim, S. C. Sol-gel transition behavior of biodegradable three-arm and four-arm star-shaped PLGA-PEG block copolymer aqueous solution. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 888–899. [Google Scholar] [CrossRef]
- Kim, H. K.; Park, T. G. Surface Stabilization of Diblock PEG-PLGA Micelles by Polymerization of N-Vinyl-2-pyrrolidone. Macromol. Rapid Commun. 2002, 23, 26–31. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, C. H.; Park, T. G. Poly[lactic-co-(glycolic acid)]-Grafted Hyaluronic Acid Copolymer Micelle Nanoparticles for Target-Specific Delivery of Doxorubicin. Macromol. Biosci. 2009, 9, 336–342. [Google Scholar] [CrossRef]
- Kim, S. H.; Jeong, J. H.; Chun, K. W.; Park, T. G. Target-Specific Cellular Uptake of PLGA Nanoparticles Coated with Poly(l-lysine)-Poly(ethylene glycol)- Folate Conjugate. Langmuir 2005, 21, 8852–8857. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S. V.; Zeman, A. D.; Batrakova, E. V.; Kabanov, A. V. Polyplex Nanogel formulations for drug delivery of cytotoxic nucleoside analogs. J. Control Release 2005, 107, 143–157. [Google Scholar] [CrossRef]
- Vinogradov, S. V.; Bronich, T. K.; Kabanov, A. V. Nanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cells. Adv. Drug Deliv. Rev. 2002, 54, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C. M.; Warren, G.; Kohli, E.; Zeman, A.; Mitin, A.; Vinogradov, S. V. Polymeric nanogels containing the triphosphate form of cytotoxic nucleoside analogues show antitumor activity against breast and colorectal cancer cell lines. Mol. Cancer Ther. 2008, 7, 3373–3380. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Kasoju, N.; Bora, U. Fluorescence Study of the Curcumin-Casein Micelle Complexation and Its Application as a Drug Nanocarrier to Cancer Cells. Biomacromolecules 2008, 9, 2905–2912. [Google Scholar] [CrossRef]
- Kim, S. C.; Kim, D. W.; Shim, Y. H.; Bang, J. S.; Oh, H. S.; Wan Kim, S.; Seo, M. H. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J. Control Release 2001, 72, 191–202. [Google Scholar] [CrossRef]
- Jubo, L.; Yuehua, X.; Christine, A. Polymer-drug compatibility: A guide to the development of delivery systems for the anticancer agent, ellipticine. J. Pharm. Sci. 2004, 93, 132–143. [Google Scholar]
- Shuai, X.; Merdan, T.; Schaper, A. K.; Xi, F.; Kissel, T. Core-Cross-Linked Polymeric Micelles as Paclitaxel Carriers. Bioconj. Chem. 2004, 15, 441–448. [Google Scholar] [CrossRef]
- Shuai, X.; Ai, H.; Nasongkla, N.; Kim, S.; Gao, J. Micellar carriers based on block copolymers of poly(epsilon-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J. Control Release 2004, 98, 415–426. [Google Scholar] [CrossRef]
- Wang, Y-C. ; Tang, L-Y.; Sun, T-M.; Li, C-H.; Xiong, M-H.; Wang, J. Self-Assembled Micelles of Biodegradable Triblock Copolymers Based on Poly(ethyl ethylene phosphate) and Poly(ε-caprolactone) as Drug Carriers. Biomacromolecules 2008, 9, 388–395. [Google Scholar] [CrossRef]
- Wang, J.; Woodcock, S. E.; Buck, S. M.; Chen, C.; Chen, Z. Different Surface- Restructuring Behaviors of Poly(methacrylate)s Detected by SFG in Water. J. Am. Chem. Soc. 2001, 123, 9470–9471. [Google Scholar] [CrossRef]
- Licciardi, M.; Craparo E., F.; Giammona, G.; Armes, S. P.; Tang, Y.; Lewis, A. L. in vitro biological evaluation of folate-functionalized block copolymer micelles for selective anti-cancer drug delivery. Macromol. Biosci. 2008, 8, 615–626. [Google Scholar] [CrossRef]
- Kim, S. H.; Jeong, J. H.; Lee, S. H.; Kim, S. W.; Park, T. G. LHRH Receptor-Mediated Delivery of siRNA Using Polyelectrolyte Complex Micelles Self- Assembled from siRNA-PEG-LHRH Conjugate and PEI. Bioconj. Chem. 2008, 19, 2156–2162. [Google Scholar] [CrossRef]
- Lee, H.; Hu, M.; Reilly, R. M. ; Allen, C: Apoptotic Epidermal Growth Factor (EGF)-Conjugated Block Copolymer Micelles as a Nanotechnology Platform for Targeted Combination Therapy. Mol. Pharm. 2007, 4, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Nishiyama, N.; Kataoka, K. In vivo antitumor activity of the folate- conjugated pH-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjug. Chem. 2007, 18, 1131–1139. [Google Scholar] [CrossRef]
- Kim, D.; Lee, E. S.; Park, K.; Kwon, I. C.; Bae, Y. H. Doxorubicin loaded pH-sensitive micelle: antitumoral efficacy against ovarian A2780/DOXR tumor. Pharm. Res. 2008, 25, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Kyung Taek Oh, E. S. L. Cancer-associated pH-responsive tetracopolymeric micelles composed of poly(ethylene glycol)-b-poly(L-histidine)-b-poly(L- lactic acid)-b-poly(ethylene glycol). Polym. Adv. Technol. 2008, 19, 1907–1913. [Google Scholar]
- Lee, E. S.; Gao, Z.; Kim, D.; Park, D.; Kwon, I. C.; Bae, Y. H. Super pH-sensitive multifunctional polymeric micelle for tumor pHe specific TAT exposure and multidrug resistance. J. Control Release 2008, 129, 228–236. [Google Scholar] [CrossRef]
- Yoo, H. S.; Lee, E. A.; Park, T. G. Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J. Control Release 2002, 82, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Park, S. Y.; Mok, H.; Park, T. G. Synthesis, Characterization, Antitumor Activity of Pluronic Mimicking Copolymer Micelles Conjugated with Doxorubicin via Acid-Cleavable Linkage. Bioconj. Chem. 2008, 19, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Burt, H. M.; Zhang, X.; Toleikis, P.; Embree, L.; Hunter, W. L. Development of copolymers of poly(D,L-lactide) and methoxypolyethylene glycol as micellar carriers of paclitaxel. Colloids Surf. B Biointer. 1999, 16, 161–171. [Google Scholar] [CrossRef]
- Chen, H.; Kim, S.; He, W.; Wang, H.; Low, P. S.; Park, K.; Cheng, J. X. Fast Release of Lipophilic Agents from Circulating PEG-PDLLA Micelles Revealed by in Vivo Förster Resonance Energy Transfer Imaging. Langmuir 2008, 24, 5213–5217. [Google Scholar] [CrossRef]
- O’Reilly, R. K.; Hawker, C. J.; Wooley, K. L. Cross-linked block copolymer micelles: functional nanostructures of great potential and versatility. Chem. Soc. Rev. 2006, 35, 1068–1083. [Google Scholar] [CrossRef]
- Bontha, S.; Kabanov, A. V.; Bronich, T. K. Polymer micelles with cross-linked ionic cores for delivery of anticancer drugs. J Control Release 2006, 114, 163–174. [Google Scholar] [CrossRef]
- Chan, Y.; Wong, T.; Byrne, F.; Kavallaris, M.; Bulmus, V. Acid-Labile Core Cross- Linked Micelles for pH-Triggered Release of Antitumor Drugs. Biomacromolecules 2008, 9, 1826–1836. [Google Scholar] [CrossRef]
- Nishiyama, N.; Kataoka, K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol. Ther. 2006, 112, 630–648. [Google Scholar] [CrossRef]
- Aoyagi, T.; Sugi, K.; Sakurai, Y.; Okano, T.; Kataoka, K. Peptide drug carrier: studies on incorporation of vasopressin into nano-associates comprising poly(ethylene glycol)-poly(-aspartic acid) block copolymer. Colloids Surf. B Biointerf. 1999, 16, 237–242. [Google Scholar] [CrossRef]
- Yokoyama, M.; Miyauchi, M.; Yamada, N.; Okano, T.; Sakurai, Y.; Kataoka, K. Characterization and anticancer activity of the micelle-forming polymeric anticancer drug adriamycin-conjugated poly(ethylene glycol)- poly(aspartic acid) block copolymer. Cancer Res. 1990, 50, 1693–1700. [Google Scholar]
- Gao, Z.; Fain, H. D.; Rapoport, N. Ultrasound-enhanced tumor targeting of polymeric micellar drug carriers. Mol. Pharm. 2004, 1, 317–330. [Google Scholar] [CrossRef]
- Marin, A.; Sun, H.; Husseini, G. A.; Pitt, W. G.; Christensen, D. A.; Rapoport, N. Y. Drug delivery in pluronic micelles: effect of high-frequency ultrasound on drug release from micelles and intracellular uptake. J. Control Release 2002, 84, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Namdeo, M.; Sutanjay, S.; Tankhiwale, R.; Bajpai, M.; Yallapu, M. M.; Bajpai, S. K. Magnetic Nanoparticles for Drug Delivery Applications. J. Nanosci. Nanotech. 2008, 8, 3247–3271. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J. R.; Weissleder, R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv. Drug Deliv. Rev. 2008, 60, 1241–1251. [Google Scholar] [CrossRef]
- Bertorelle, F.; Wilhelm, C.; Roger, J.; Gazeau, F.; Menager, C.; Cabuil, V. Fluorescence-modified superparamagnetic nanoparticles: intracellular uptake and use in cellular imaging. Langmuir 2006, 22, 5385–5391. [Google Scholar] [CrossRef]
- Cinteza, L. O.; Ohulchanskyy, T. Y.; Sahoo, Y.; Bergey, E. J.; Pandey, R. K.; Prasad, P. N. Diacyllipid micelle-based nanocarrier for magnetically guided delivery of drugs in photodynamic therapy. Mol. Pharm. 2006, 3, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Zhang, L.; Qian, H.; Li, R.; Jiang, X.; Liu, B. Multifunctional nanocarriers for cell imaging, drug delivery, and near-IR photothermal therapy. Langmuir 2010, 26, 5428–5434. [Google Scholar] [CrossRef]
- Guthi, J. S.; Yang, S. G.; Huang, G.; Li, S.; Khemtong, C.; Kessinger, C. W.; Peyton, M.; Minna, J. D.; Brown, K. C.; Gao, J. MRI-visible micellar nanomedicine for targeted drug delivery to lung cancer cells. Mol. Pharm. 2010, 7, 32–40. [Google Scholar] [CrossRef]
- Pradhan, P.; Giri, J.; Rieken, F.; Koch, C.; Mykhaylyk, O.; Doblinger, M.; Banerjee, R.; Bahadur, D.; Plank, C. Targeted temperature sensitive magnetic liposomes for thermo-chemotherapy. J. Control Release 2010, 142, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Retama, J.; Zafeiropoulos, N. E.; Serafinelli, C.; Rojas-Reyna, R.; Voit, B.; Cabarcos, E. L.; Stamm, M. Synthesis and characterization of thermosensitive PNIPAM microgels covered with superparamagnetic gamma-Fe2O3 nanoparticles. Langmuir 2007, 23, 10280–10285. [Google Scholar] [CrossRef] [PubMed]
- Shubayev, V. I.; Pisanic, T. R.; Jin, S. Magnetic nanoparticles for theragnostics. Adv. Drug Deliv. Rev. 2009, 61, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Z.; Zhang, Y.; Wang, L. Biofunctional nanoparticles with magnetization and luminescence. J. Phys. Chem. C 2009, 113, 3955–3959. [Google Scholar] [CrossRef]
- Jain, T. K.; Foy, S. P.; Erokwu, B.; Dimitrijevic, S.; Flask, C. A.; Labhasetwar, V. Magnetic resonance imaging of multifunctional pluronic stabilized iron-oxide nanoparticles in tumor-bearing mice. Biomaterials 2009, 30, 6748–6756. [Google Scholar] [CrossRef]
- Ridolfo, R.; Tavakoli, S.; Junnuthula, V.; Williams, D. S.; Urtti, A.; van Hest, J. C. Exploring the impact of morphology on the properties of biodegradable nanoparticles and their diffusion in complex biological medium. Biomacromolecules 2021, 22, 126–133. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Effect of Charged Segment Length on Physicochemical Properties of Core–Shell Type Polyion Complex Micelles from Block Ionomers. Macromolecules 2003, 36, 4995–5001. [Google Scholar] [CrossRef]
- Marras, A. E.; Campagna, T. R.; Vieregg, J. R.; Tirrell, M. V. Physical Property Scaling Relationships for Polyelectrolyte Complex Micelles. Macromolecules 2021, 54, 6585–6594. [Google Scholar] [CrossRef]
- Holley, A. C.; Parsons, K. H.; Wan, W. M.; Lyons, D. F.; Bishop, G. R.; Correia, J. J.; Huang, F. Q.; McCormick, C. L. Block ionomer complexes consisting of siRNA and aRAFT-synthesized hydrophilic-block-cationic copolymers: the influence of cationic block length on gene suppression. Polym. Chem. 2014, 5, 6967–6976. [Google Scholar] [CrossRef]
- Parsons, K. H.; Holley, A. C.; Munn, G. A.; Flynt, A. S.; McCormick, C. L. Block ionomer complexes consisting of siRNA and aRAFT-synthesized hydrophilic-block-cationic copolymers II: The influence of cationic block charge density on gene suppression. Polym. Chem. 2016, 7, 6044–6054. [Google Scholar] [CrossRef]
- Heo, T. Y.; Kim, I.; Chen, L.; Lee, E.; Lee, S.; Choi, S. H. Effect of Ionic Group on the Complex Coacervate Core Micelle Structure. Polymers (Basel) 2019, 11, 455. [Google Scholar] [CrossRef]
- Koide, A.; Kishimura, A.; Osada, K.; Jang, W. D.; Yamasaki, Y.; Kataoka, K. Semipermeable polymer vesicle (PICsome) self-assembled in aqueous medium from a pair of oppositely charged block copolymers: physiologically stable micro-/nanocontainers of water-soluble macromolecules. J. Am. Chem. Soc. 2006, 128, 5988–5989. [Google Scholar] [CrossRef] [PubMed]
- Chuanoi, S.; Anraku, Y.; Hori, M.; Kishimura, A.; Kataoka, K. Fabrication of polyion complex vesicles with enhanced salt and temperature resistance and their potential applications as enzymatic nanoreactors. Biomacromolecules 2014, 15, 2389–2397. [Google Scholar] [CrossRef]
- Sakamoto, S.; Sanada, Y.; Sakashita, M.; Nishina, K.; Nakai, K.; Yusa, S. I.; Sakurai, K. Chain-Length Dependence of Polyion Complex Architecture Bearing Phosphobetaine Block Explored Using SAXS and FFF-MALS. Polym. J. 2014, 46, 617–622. [Google Scholar] [CrossRef]
- Voets, I. K.; De Keizer, A.; Cohen Stuart, M. A.; Justynska, J.; Schlaad, H. Irreversible Structural Transitions in Mixed Micelles of Oppositely Charged Diblock Copolymers in Aqueous Solution. Macromolecules 2007, 40, 2158–2164. [Google Scholar] [CrossRef]
- Anraku, Y.; Kishimura, A.; Oba, M.; Yamasaki, Y.; Kataoka, K. Spontaneous formation of nanosized unilamellar polyion complex vesicles with tunable size and properties. J. Am. Chem. Soc. 2010, 132, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Osada, K.; Matsuda, H.; Anraku, Y.; Hirose, H.; Kishimura, A.; Kataoka, K. Morphology Control in Water of Polyion Complex Nanoarchitectures of Double-Hydrophilic Charged Block Copolymers through Composition Tuning and Thermal Treatment. Macromolecules 2014, 47, 3086–3092. [Google Scholar] [CrossRef]
- Marras, A. E.; Vieregg, J. R.; Ting, J. M.; Rubien, J. D.; Tirrell, M. V. Polyelectrolyte Complexation of Oligonucleotides by Charged Hydrophobic-Neutral Hydrophilic Block Copolymers. Polymers 2019, 11, 83. [Google Scholar] [CrossRef]
- Takahashi, R.; Sato, T.; Terao, K.; Yusa, S. Intermolecular Interactions and Self-Assembly in Aqueous Solution of a Mixture of Anionic-Neutral and Cationic-Neutral Block Copolymers. Macromolecules 2015, 48, 7222–7229. [Google Scholar] [CrossRef]
- Takahashi, R.; Sato, T.; Terao, K.; Yusa, S. Reversible Vesicle- Spherical Micelle Transition in a Polyion Complex Micellar System Induced by Changing the Mixing Ratio of Copolymer Components. Macromolecules 2016, 49, 3091–3099. [Google Scholar] [CrossRef]
- Perry, S. L.; Leon, L.; Hoffmann, K. Q.; Kade, M. J.; Priftis, D.; Black, K. A.; Wong, D.; Klein, R. A.; Pierce, C. F., 3rd; Margossian, K. O.; Whitmer, J. K.; Qin, J.; de Pablo, J. J.; Tirrell, M. Chirality-selected phase behaviour in ionic polypeptide complexes. Nat. Commun. 2015, 6, 6052. [Google Scholar] [CrossRef]
- Shah, S.; Leon, L. Structural transitions and encapsulation selectivity of thermoresponsive polyelectrolyte complex micelles. J. Mater. Chem. B 2019, 7, 6438–6448. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, J.; Lecommandoux, S. Reversible inside- out micellization of pH-responsive and water-soluble vesicles based on polypeptide diblock copolymers. J. Am. Chem. Soc. 2005, 127, 2026–2027. [Google Scholar] [CrossRef]
- Holder, S. J.; Sommerdijk, N. A. J. M. New micellar morphologies from amphiphilic block copolymers: disks, toroids and bicontinuous micelles. Polym. Chem. 2011, 2, 1018–1028. [Google Scholar] [CrossRef]
- Srivastava, S.; Tirrell, M. V. Polyelectrolyte Complexation. In Advances in Chemical Physics; Rice, S. A., Dinner, A. R., Eds.; Advances in Chemical Physics; Wiley-Blackwell: Malden, 2016; Vol. 161, pp 499-544. [Google Scholar]
- Burke, P. A.; Pun, S. H.; Reineke, T. M. Advancing polymeric delivery systems amidst a nucleic acid therapy renaissance. ACS Macro Lett. 2013, 2, 928–934. [Google Scholar] [CrossRef]
- Hays, J. B.; Magar, M. E.; Zimm, B. H. Persistence Length of DNA. Biopolymers 1969, 8, 531–536. [Google Scholar] [CrossRef]
- Tinland, B.; Pluen, A.; Sturm, J.; Weill, G. Persistence length of single-stranded DNA. Macromolecules 1997, 30, 5763–5765. [Google Scholar] [CrossRef]
- Vieregg, J. R.; Lueckheide, M.; Marciel, A. B.; Leon, L.; Bologna, A. J.; Rivera, J. R.; Tirrell, M. V. Oligonucleotide-Peptide Complexes: Phase Control by Hybridization. J. Am. Chem. Soc. 2018, 140, 1632–1638. [Google Scholar] [CrossRef]
- Shakya, A.; King, J. T. DNA Local-Flexibility-Dependent Assembly of Phase-Separated Liquid Droplets. Biophys. J. 2018, 115, 1840–1847. [Google Scholar] [CrossRef]
- Hayashi, K.; Chaya, H.; Fukushima, S.; Watanabe, S.; Takemoto, H.; Osada, K.; Nishiyama, N.; Miyata, K.; Kataoka, K. Influence of RNA Strand Rigidity on Polyion Complex Formation with Block Catiomers. Macromol. Rapid Commun. 2016, 37, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K. M.; Osada, K.; Tockary, T. A.; Dirisala, A.; Chen, Q.; Kataoka, K. Poly(ethylene glycol) Crowding as Critical Factor To Determine pDNA Packaging Scheme into Polyplex Micelles for Enhanced Gene Expression. Biomacromolecules 2017, 18, 36–43. [Google Scholar] [CrossRef]
- Takahashi, R.; Narayanan, T.; Yusa, S. I.; Sato, T. Kinetics of Morphological Transition between Cylindrical and Spherical Micelles in a Mixture of Anionic-Neutral and Cationic-Neutral Block Copolymers Studied by Time-Resolved SAXS and USAXS. Macromolecules 2018, 51, 3654–3662. [Google Scholar] [CrossRef]
- Burke, S. E.; Eisenberg, A. Kinetics and Mechanisms of the Sphere-to-Rod and Rod-to-Sphere Transitions in the Ternary System PS310-b-PAA52/Dioxane/Water. Langmuir 2001, 17, 6705–6714. [Google Scholar] [CrossRef]
- Voets, I. K.; Moll, P. M.; Aqil, A.; Jeŕôme, C.; Detrembleur, C.; De Waard, P.; De Keizer, A.; Cohen Stuart, M. A. Temperature Responsive Complex Coacervate Core Micelles with a PEO and PNIPAAm Corona. J. Phys. Chem. B 2008, 112, 10833–10840. [Google Scholar] [CrossRef]
- Crommelin, D. J. A.; Scherphof, G.; Storm, G. Active targeting with particulate carrier systems in the blood compartment. Adv. Drug Deliv. Rev. 1995, 17, 49–60. [Google Scholar] [CrossRef]
- Gros, L.; Ringsdorf, H.; Schupp, H. Polymeric antitumor agents on a molecular and on a cellular level? Angew. Chem. Int. Ed. Engl. 1981, 20, 305–325. [Google Scholar] [CrossRef]
- Moses, M. A.; Brem, H.; Langer, R. Advancing the field of drug delivery: taking aim at cancer. Cancer Cell 2003, 4, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Adams, M. L.; Lavasanifar, A.; Kwon, G. S. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [Google Scholar] [CrossRef]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf. B Biointerfaces. 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Jones, M.; Leroux, J. Polymeric micelles - a new generation of colloidal drug carriers. Eur. J. Pharm. Biopharm. 1999, 48, 101–111. [Google Scholar] [CrossRef]
- Kabanov, A. V.; Batrakova, E. V.; Melik-Nubarov, N. S.; Fedoseev, N. A.; Dorodnich, T. Y.; Alakhov, V. Y.; Chekhonin, V. P.; Nazarova, I. R.; Kabanov, V. A. A new class of drug carriers: Micelles of poly (oxyethylene)-poly (oxypropylene) block copolymers as microcontainers for drug targeting from blood in brain. J. Control. Release 1992, 22, 141–157. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticles--a historical perspective. Int. J. Pharm. 2006, 2006 331, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kwon, G. S. Polymeric micelles for delivery of poorly water-soluble compounds. Crit. Rev. Ther. Drug Carrier Syst. 2003, 20, 357–403. [Google Scholar] [CrossRef] [PubMed]
- Lavasanifar, A.; Samuel, J.; Kwon, G. S. Poly(ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv. Drug. Deliv. Rev. 2002, 54, 169–190. [Google Scholar] [CrossRef]
- Liu, J.; Zeng, F.; Allen, C. In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur. J. Pharm. Biopharm. 2007, 65, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S. M.; Hunter, A. C.; Murray, J. C. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar] [CrossRef]
- Torchilin, V. P. Structure and design of polymeric surfactant-based drug delivery systems. J. Control Release 2001, 73, 137–172. [Google Scholar] [CrossRef]
- Torchilin, V. P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef]
- Yokoyama, M.; Kwon, G. S.; Okano, T.; Sakurai, Y.; Seto, T.; Kataoka, K. Preparation of micelle-forming polymer-drug conjugates. Bioconjug. Chem. 1992, 3, 295–301. [Google Scholar] [CrossRef]
- Gillies, E. R.; Fréchet, J. M. pH-Responsive copolymer assemblies for controlled release of doxorubicin. Bioconjug. Chem. 2005, 16, 361–368. [Google Scholar] [CrossRef]
- Hrubý, M.; Konák, C.; Ulbrich, K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J. Control Release 2005, 103, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A. V.; Batrakova, E. V.; Alakhov, V. Y. Pluronic block copolymers for overcoming drug resistance in cancer. Adv. Drug Deliv. Rev. 2002, 54, 759–779. [Google Scholar] [CrossRef]
- Lee, E. S.; Na, K.; Bae, Y. H. Super pH-sensitive multifunctional polymeric micelle. Nano Lett. 2005, 5, 325–329. [Google Scholar] [CrossRef]
- Nakanishi, T.; Fukushima, S.; Okamoto, K.; Suzuki, M.; Matsumura, Y.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Development of the polymer micelle carrier system for doxorubicin. J. Control. Release 2001, 74, 295–302. [Google Scholar] [CrossRef]
- Rapoport, N. Combined cancer therapy by micellar-encapsulated drug and ultrasound. Int. J. Pharm. 2004, 277, 155–162. [Google Scholar] [CrossRef]
- Cavallaro, G.; Licciardi, M.; Giammona, G.; Caliceti, P.; Semenzato, A.; Salmaso, S. Poly(hydroxyethylaspartamide) derivatives as colloidal drug carrier systems. J. Control. Release 2003, 89, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Matsumura, Y.; Suzuki, M.; Shimizu, K.; Goda, R.; Nakamura, I.; Nakatomi, I.; Yokoyama, M.; Kataoka, K.; Kakizoe, T. NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br. J. Cancer 2005, 92, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Huh, K. M.; Lee, S. C.; Cho, Y. W.; Lee, J.; Jeong, J. H.; Park, K. Hydrotropic polymer micelle system for delivery of paclitaxel. J. Control. Release 2005, 101, 59–68. [Google Scholar] [CrossRef]
- Kim, T-Y. ; Kim, D-W.; Chung, J-Y.; Shin, S. G.; Kim, S-C.; Heo, D. S.; Kim, N. K.; Bang, Y-J. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin. Cancer Res. 2004, 10, 3708–3716. [Google Scholar] [CrossRef]
- Krishnadas, A.; Rubinstein, I.; Onyüksel, H. Sterically stabilized phospholipid mixed micelles: in vitro evaluation as a novel carrier for water-insoluble drugs. Pharm. Res. 2003, 20, 297–302. [Google Scholar] [CrossRef]
- Torchilin, V. P.; Lukyanov, A. N.; Gao, Z.; Papahadjopoulos-Sternberg, B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6039–6044. [Google Scholar] [CrossRef] [PubMed]
- Lavasanifar, A.; Samuel, J.; Sattari, S.; Kwon, G. S. Block copolymer micelles for the encapsulation and delivery of amphotericin B. Pharm. Res. 2002, 19, 418–422. [Google Scholar] [CrossRef]
- Le Garrec, D.; Gori, S.; Luo, L.; Lessard, D.; Smith, D. C.; Yessine, M-A; Ranger, M. ; Leroux, J. C. Poly(N-vinylpyrrolidone)-block-poly(D,L-lactide) as a new polymeric solubilizer for hydrophobic anticancer drugs: in vitro and in vivo evaluation. J. Control. Release 2004, 99, 83–101. [Google Scholar] [CrossRef]
- van Nostrum, C. F. Polymeric micelles to deliver photosensitizers for photodynamic therapy. Adv. Drug Deliv. Rev. 2004, 56, 9–16. [Google Scholar] [CrossRef]
- Zhang, G. D.; Harada, A.; Nishiyama, N.; Jiang, D. L.; Koyama, H.; Aida, T.; Kataoka, K. Polyion complex micelles entrapping cationic dendrimer porphyrin: effective photosensitizer for photodynamic therapy of cancer. J. Control. Release 2003, 93, 141–150. [Google Scholar] [CrossRef]
- Barratt, G. M. Therapeutic applications of colloidal drug carriers. Pharm. Sci. Technol. Today 2000, 3, 163–1171. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Hamaguchi, T.; Ura, T.; Muro, K.; Yamada, Y.; Shimada, Y.; Shirao, K.; Okusaka, T.; Ueno, H.; Ikeda, M.; Watanabe, N. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br. J. Cancer. 2004, 91, 1775–1781. [Google Scholar] [CrossRef]
- Torchilin, V. P. Micellar nanocarriers: pharmaceutical perspectives. Pharm. Res. 2007, 24, 1–16. [Google Scholar] [CrossRef]
- Torchilin, V. P. PEG-based micelles as carriers of contrast agents for different imaging modalities. Adv. Drug Deliv. Rev. 2002, 54, 235–252. [Google Scholar] [CrossRef]
- Maeda, H.; Fang, J.; Inutsuka, T.; Kitamoto, Y. Vascular permeability enhancement in solid tumor: various factors, mechanisms involved and its implications. Int. Immunopharmacol. 2003, 3, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Illum, L.; Jacobsen, L. O.; Müller, R. H.; Mak, E.; Davis, S. S. Surface characteristics and the interaction of colloidal particles with mouse peritoneal macrophages. Biomaterials 1987, 8, 113–117. [Google Scholar] [CrossRef]
- Leroux, J. C.; De Jaeghere, F.; Anner, B.; Doelker, E.; Gurny, R. An investigation on the role of plasma and serum opsonins on the internalization of biodegradable poly(D,L-lactic acid) nanoparticles by human monocytes. Life Sci. 1995, 57, 695–703. [Google Scholar] [CrossRef]
- Moghimi, S. M.; Porter, C. J. H.; Illum, L.; Davis, S. S. The effect of poloxamer-407 on liposome stability and targeting to bone marrow: comparison with polystyrene microspheres. Int. J. Pharm. 1991, 68, 121–126. [Google Scholar] [CrossRef]
- Owens, D. E. , 3rd.; Peppas, N. A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Stolnik, S.; Illum, L.; Davis, S. S. Long circulating microparticulate drug carriers. Adv. Drug Deliv. Rev. 1995, 16, 195–214. [Google Scholar] [CrossRef]
- Woodle, M. C.; Lasic, D. D. Sterically stabilized liposomes. Biochim. Biophys. Acta. 1992, 1113, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J. P. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Rossin, R.; Turner, J. L.; Becker, M. L.; Joralemon, M. J.; Welch, M. J.; Wooley, K. L. An assessment of the effects of shell cross-linked nanoparticle size, core composition, and surface PEGylation on in vivo biodistribution. Biomacromolecules 2005, 6, 2541–2554. [Google Scholar] [CrossRef]
- Carstens, M. G.; Bevernage, J. J. L.; van Nostrum, C. F.; van Steenbergen, M. J.; Flesch, F. M.; Verrijk, R.; de Leede, L. G. J.; Crommelin, D. J. A.; Hennink, W. E. Small Oligomeric Micelles Based on End Group Modified mPEG–Oligocaprolactone with Monodisperse Hydrophobic Blocks. Macromolecules 2007, 40, 116–122. [Google Scholar] [CrossRef]
- Hagan, S. A.; Coombes, A. G. A.; Garnett, M. C.; Dunn, S. E.; Davies, M. C.; Illum, L.; Davis, S. S.; Harding, S. E.; Purkiss, S.; Gellert, P. R. Polylactide-poly(ethylene glycol) copolymers as drug delivery systems. 1. Characterization of water dispersible micelle-forming systems. Langmuir 1996, 12, 2153–2161. [Google Scholar] [CrossRef]
- Luo, L.; Tam, J.; Maysinger, D.; Eisenberg, A. Cellular internalization of poly(ethylene oxide)-b-poly(epsilon-caprolactone) diblock copolymer micelles. Bioconjug. Chem. 2002, 13, 1259–1265. [Google Scholar] [CrossRef]
- Shi, B.; Fang, C.; You, M. X.; Zhang, Y.; Fu, S.; Pei, Y. Stealth MePEG-PCL micelles: effects of polymer composition on micelle physicochemical characteristics, in vitro drug release, in vivo pharmacokinetics in rats and biodistribution in S180 tumor bearing mice. Colloid Polym. Sci. 2005, 283, 954–967. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nagasaki, Y.; Kato, Y.; Sugiyama, Y.; Kataoka, K. Long-circulating poly(ethylene glycol)-poly(D,L-lactide) block copolymer micelles with modulated surface charge. J. Control. Release 2001, 77, 27–38. [Google Scholar] [CrossRef]
- Lukyanov, A. N.; Torchilin, V. P. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Deliv. Rev. 2004, 56, 1273–1289. [Google Scholar] [CrossRef]
- Weissig, V.; Whiteman, K. R.; Torchilin, V. P. Accumulation of protein-loaded long-circulating micelles and liposomes in subcutaneous Lewis lung carcinoma in mice. Pharm. Res. 1998, 15, 1552–1556. [Google Scholar] [CrossRef] [PubMed]
- Neradovic, D.; Soga, O.; Van Nostrum, C. F.; Hennink, W. E. The effect of the processing and formulation parameters on the size of nanoparticles based on block copolymers of poly(ethylene glycol) and poly(N-isopropylacrylamide) with and without hydrolytically sensitive groups. Biomaterials 2004, 25, 2409–2418. [Google Scholar] [CrossRef] [PubMed]
- Rijcken, C. J. F.; Veldhuis, T. F. J.; Ramzi, A.; Meeldijk, J. D.; van Nostrum, C. F.; Hennink, W. E. Novel fast degradable thermosensitive polymeric micelles based on PEG-block-poly(N-(2-hydroxyethyl)methacrylamide-oligolactates). Biomacromolecules 2005, 6, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Soga, O.; van Nostrum, C. F.; Ramzi, A.; Visser, T.; Soulimani, F.; Frederik, P. M.; Bomans, P. H.; Hennink, W. E. Physicochemical characterization of degradable thermosensitive polymeric micelles. Langmuir 2004, 20, 9388–9395. [Google Scholar] [CrossRef]
- Topp, M. D. C, Dijkstra P. J.; Talsma, H.; Feijen, J. Thermosensitive micelle-forming block copolymers of poly(ethylene glycol) and poly(N-isopropylacrylamide). Macromolecules 1997, 30, 8518–8520. [Google Scholar] [CrossRef]
- Allen, T. M.; Hansen, C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim. Biophys. Acta 1991, 1068, 133–141. [Google Scholar] [CrossRef]
- Lee, K. Y.; Ha, W. S.; Park, W. H. Blood compatibility and biodegradability of partially N-acylated chitosan derivatives. Biomaterials 1995, 16, 1211–1216. [Google Scholar] [CrossRef]
- Molineux, G. Pegylation: engineering improved pharmaceuticals for enhanced therapy. Cancer Treat Rev. 2002, 28 (Suppl. A), 13–16. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. P.; Trubetskoy, V. S. Which polymers can make nanoparticulate drug carriers long-circulating? Adv. Drug Deliver. Rev. 1995, 16, 141–155. [Google Scholar] [CrossRef]
- Klibanov, A. L.; Maruyama, K.; Torchilin, V. P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Soppimath, K. S.; Aminabhavi, T. M.; Kulkarni, A. R.; Rudzinski, W. E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Michel, R.; Pasche, S.; Textor, M.; Castner, D. G. Influence of PEG architecture on protein adsorption and conformation. Langmuir 2005, 21, 12327–12332. [Google Scholar] [CrossRef]
- Kwon, G. S.; Suwa, S.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Enhanced Tumor Accumulation and Prolonged Circulation Times of Micelle-Forming Poly (Ethylene Oxide-Aspartate) Block Copolymer-Adriamycin Conjugates. J. Control. Release 1994, 29, 17–23. [Google Scholar] [CrossRef]
- Kwon, G. S.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Biodistribution of micelle-forming polymer-drug conjugates. Pharm. Res. 1993, 10, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A. V.; Batrakova, E. V.; Alakhov, V. Y. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Simard, P.; Leroux, J. C.; Benoit, J. P. Evaluation of pegylated lipid nanocapsules versus complement system activation and macrophage uptake. J. Biomed. Mater. Res. A 2006, 78, 620–628. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, J.; Tong, Y. W.; Wang, C. H. Effect of PEG conformation and particle size on the cellular uptake efficiency of nanoparticles with the HepG2 cells. J. Control. Release 2007, 118, 7–17. [Google Scholar] [CrossRef]
- Peracchia, M. T.; Vauthier, C.; Passirani, C.; Couvreur, P.; Labarre, D. Complement consumption by poly(ethylene glycol) in different conformations chemically coupled to poly(isobutyl 2-cyanoacrylate) nanoparticles. Life Sci. 1997, 61, 749–761. [Google Scholar] [CrossRef]
- Moghimi, S. M.; Szebeni, J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Carstens, M. G.; Romberg, B.; Laverman, P.; Boerman, O. C.; Oussoren, C.; Storm. G. Observations on the disappearance of the stealth property of PEGylated liposomes. Effects of lipid dose and dosing frequency. In Liposome Technology, 3rd ed.; Gregoriadis, G. Ed.; CRC Press: London, 2006; Vol III, pp 79-93.
- Dams, E. T.; Laverman, P.; Oyen, W. J.; Storm, G.; Scherphof, G. L.; van Der Meer, J. W.; Corstens, F. H.; Boerman, O. C. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Harashima, H.; Kiwada, H. Liposome clearance. Biosci. Rep. 2002, 22, 197–224. [Google Scholar] [CrossRef]
- Laverman, P.; Brouwers, A. H.; Dams, E. T.; Oyen, W. J.; Storm, G.; van Rooijen, N.; Corstens, F. H.; Boerman, O. C. Preclinical and clinical evidence for disappearance of long-circulating characteristics of polyethylene glycol liposomes at low lipid dose. J. Pharmacol. Exp. Ther. 2000, 293, 996–1001. [Google Scholar] [CrossRef]
- Nishiyama, N.; Okazaki, S.; Cabral, H.; Miyamoto, M.; Kato, Y.; Sugiyama, Y.; Nishio, K.; Matsumura, Y.; Kataoka, K. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003, 63, 8977–8983. [Google Scholar]
- Yokoyama, M.; Fukushima, S.; Uehara, R.; Okamoto, K.; Kataoka, K.; Sakurai. ; Y, Okano, T. Characterization of physical entrapment and chemical conjugation of adriamycin in polymeric micelles and their design for in vivo delivery to a solid tumor. J. Control. Release 1998, 50, 79–92. [Google Scholar] [CrossRef]
- Yokoyama, M.; Okano, T.; Sakurai, Y; Fukushima, S. ; Okamoto, K.; Kataoka, K. Selective delivery of adriamycin to a solid tumor using a polymeric micelle carrier system. J. Drug Target. 1999, 7, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M. C.; Engbers, C. M.; Zalipsky, S. New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug. Chem. 1994, 5, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Okuizumi, S.; Ishida, O.; Yamauchi, H.; Kikuchi, H.; Iwatsuru, M. Phosphatidyl polyglycerols prolong liposome circulation in vivo. Int. J. Pharm. 1994, 111, 103–107. [CrossRef]
- Torchilin, V. P.; Levchenko, T. S.; Whiteman, K. R.; Yaroslavov, A. A.; Tsatsakis, A. M.; Rizos, A. K.; Michailova, E. V.; Shtilman, M. I. Amphiphilic poly-N-vinylpyrrolidones: synthesis, properties and liposome surface modification. Biomaterials 2001, 22, 3035–3044.
- Torchilin, V. P.; Shtilman, M. I.; Trubetskoy, V. S.; Whiteman, K.; Milstein, A. M. Amphiphilic vinyl polymers effectively prolong liposome circulation time in vivo. Biochim. Biophys. Acta (Bba) Biomembr. 1994, 1195, 181–184. [CrossRef]
- Takeuchi, H.; Kojima, H.; Yamamoto, H.; Kawashima, Y. Evaluation of circulation profiles of liposomes coated with hydrophilic polymers having different molecular weights in rats. J. Control Release 2001, 75, 83–91. [CrossRef]
- Whiteman, K. R.; Subr, V.; Ulbrich, K.; Torchilin, V. P. Poly(Hpma)-coated liposomes demonstrate prolonged circulation in mice. J. Liposome Res. 2001, 11, 153–164. [CrossRef] [PubMed]
- Metselaar, J. M.; Bruin, P.; de Boer, L. W.; de Vringer, T.; Snel, C.; Oussoren, C.; Wauben, M. H.; Crommelin, D. J.; Storm, G.; Hennink, W. E. A novel family of L-amino acid-based biodegradable polymer-lipid conjugates for the development of long-circulating liposomes with effective drug-targeting capacity. Bioconjug. Chem. 2003, 14, 1156–1164.
- Romberg, B.; Oussoren, C.; Snel. C. J.; Carstens, M. G.; Hennink, W. E.; Storm, G. Pharmacokinetics of poly(hydroxyethyl-l-asparagine)-coated liposomes is superior over that of PEG-coated liposomes at low lipid dose and upon repeated administration. Biochim. Biophys. Acta. 2007, 1768, 737–743. [CrossRef] [PubMed]
- Hsiue, G. H.; Wang, C. H.; Lo, C. L.; Wang, C. H.; Li, J. P.; Yang, J. L. Environmental-sensitive micelles based on poly(2-ethyl-2-oxazoline)-b-poly(L-lactide) diblock copolymer for application in drug delivery. Int. J. Pharm. 2006, 317, 69–75. [CrossRef]
- Kim, C.; Lee, S. C.; Shin, J. H.; Kwon, I. C.; Jeong, S. Y. Amphiphilic diblock copolymers based on poly(2-ethyl-2-oxazoline) and poly(1,3-trimethylene carbonate): Synthesis and micellar characteristics. Macromolecules 2000, 33, 7448–7452. [CrossRef]
- Lee, S.C.; Chang, Y. K.; Yoon, J. S.; Kim, C. H.; Kwon, I. C.; Kim, Y. H.; Jeong, S. Y. Synthesis and micellar characterization of amphiphilic diblock copolymers based on poly(2-ethyl-2-oxazoline) and aliphatic polyesters. Macromolecules 1999, 32, 1847–1852. [CrossRef]
- Lee, S. C.; Lee, H. J. pH-controlled, polymer-mediated assembly of polymer micelle nanoparticles. Langmuir 2007, 23, 488–495. [CrossRef] [PubMed]
- Volet, G.; Chanthavong, V.; Wintgens, V.; Amiel, C. Synthesis of Monoalkyl End-Capped Poly(2-Methyl-2-Oxazoline) and Its Micelle Formation in Aqueous Solution. Macromolecules 2005, 38, 5190–5197. [CrossRef]
- Benahmed, A.; Ranger, M.; Leroux, J. C. Novel polymeric micelles based on the amphiphilic diblock copolymer poly(N-vinyl-2-pyrrolidone)-block-poly(D,L-lactide). Pharm. Res. 2001, 18, 323–328. [CrossRef] [PubMed]
- Chung, T. W.; Cho, K. Y.; Lee, H. C.; Nah, J. W.; Yeo, J. H.; Akaike, T.; Cho, C. S. Novel micelle-forming block copolymer composed of poly(e-caprolactone) and poly(vinyl pyrrolidone). Polymer 2004, 45, 1591–1597. [CrossRef]
- Le Garrec, D.; Taillefer, J.; Van Lier, J. E.; Lenaerts, V.; Leroux J. C. Optimizing pH-responsive polymeric micelles for drug delivery in a cancer photodynamic therapy model. J. Drug Target. 2002, 10, 429–437. [CrossRef]
- Lele, B. S.; Lereux, J. C. Synthesis and Micellar Characterization of Novel Amphiphilic A-B-A Tricopolymers of N-(2-Hydroxypropyl) Methacrylamide or N-Vinyl-2-pyrrolidone with Poly (caprolactone). Macromolecules 2002, 35, 6714–6723. [CrossRef]
- Luo, L. B.; Ranger, M.; Lessard, D. G.; Le Garrec, D.; Gori, S.; Leroux, J. C.; Rimmer, S.; Smith, D. Novel amphiphilic diblock copolymer of low molecular weight poly(N-vinylpyrrolidone)-block-poly(D,L-lactide): Synthesis, characterization, and micellization. Macromolecules 2004, 37, 4008–4013.
- Lemarchand, C.; Gref, R.; Lesieur, S.; Hommel, H.; Vacher, B.; Besheer, A.; Maeder, K.; Couvreur, P. Physico-chemical characterization of polysaccharide-coated nanoparticles. J. Control. Release 2005, 108, 97–111. [CrossRef]
- Rouzes, C.; Gref, R.; Leonard, M.; De Sousa Delgado, A.; Dellacherie, E. Surface modification of poly(lactic acid) nanospheres using hydrophobically modified dextrans as stabilizers in an o/w emulsion/evaporation technique. J. Biomed. Mater. Res. 2000, 50, 557–565. [CrossRef]
- Lemarchand, C.; Gref, R.; Couvreur, P. Polysaccharide-decorated nanoparticles. Eur. J. Pharm. Biopharm. 2004, 58, 327–341. [CrossRef] [PubMed]
- Passirani, C.; Barratt, G.; Devissaguet, J. P.; Labarre, D. Long-circulating nanoparticles bearing heparin or dextran covalently bound to poly(methyl methacrylate). Pharm. Res. 1998, 15, 1046–1050. [CrossRef]
- Besheer, A.; Hause, G.; Kressler, J.; Mäder, K. Hydrophobically modified hydroxyethyl starch: synthesis, characterization, and aqueous self-assembly into nano-sized polymeric micelles and vesicles. Biomacromolecules 2007, 8, 359–367. [CrossRef]
- Lemarchand, C.; Gref, R.; Passirani, C.; Garcion, E.; Petri, B.; Müller, R.; Costantini, D.; Couvreur, P. Influence of polysaccharide coating on the interactions of nanoparticles with biological systems. Biomaterials 2006, 27, 108–118. [CrossRef]
- Maruyama, A.; Ishihara, T.; Adachi, N.; Akaike, T. Preparation of nanoparticles bearing high density carbohydrate chains using carbohydrate-carrying polymers as emulsifier. Biomaterials 1994, 15, 1035–1042. [CrossRef]
- Bougard, F.; Jeusette, M.; Mespouille, L.; Dubois, P.; Lazzaroni, R. Synthesis and supramolecular organization of amphiphilic diblock copolymers combining poly(N,N-dimethylamino-2-ethyl methacrylate) and poly(epsilon-caprolactone). Langmuir 2007, 23, 2339–2345. [CrossRef]
- Nam, Y. S.; Kang, H. S.; Park, J. Y.; Park, T. G.; Han, S. H.; Chang, I. S. New micelle-like polymer aggregates made from PEI-PLGA diblock copolymers: micellar characteristics and cellular uptake. Biomaterials 2003, 24, 2053–2059. [CrossRef] [PubMed]
- Inoue, T.; Chen, G.; Nakamae, K.; Hoffman, A. S. An AB block copolymer of oligo(methyl methacrylate) and poly(acrylic acid) for micellar delivery of hydrophobic drugs. J. Control. Release 1998, 51, 221–229. [CrossRef]
- Jeong, J. H.; Kang, H. S.; Yang, S. R.; Park, K.; Kim, J-D. Biodegradable poly(asparagine) grafted with poly(caprolactone) and the effect of substitution on self-aggregation. Colloids Surf. A: Physicochem. Eng. Asp. 2005, 264, 187–194. [CrossRef]
- Kohori, F.; Sakai, K.; Aoyagi, T.; Yokoyama, M.; Yamato, M.; Sakurai, Y.; Okano, T. Control of adriamycin cytotoxic activity using thermally responsive polymeric micelles composed of poly (N-isopropylacrylamide-co-N,N-dimethylacrylamide)-β-poly (D,L-lactide). Colloid Surf B: Biointerfaces 1999, 16, 195–205.
- Zuccari, G.; Carosio, R.; Fini, A.; Montaldo, P. G.; Orienti, I. Modified polyvinylalcohol for encapsulation of all-trans-retinoic acid in polymeric micelles. J. Control. Release 2005, 103, 369–380. [CrossRef] [PubMed]
- Moghimi, S. M.; Porter, C. J.; Muir, I. S.; Illum, L.; Davis, S. S. Non-phagocytic uptake of intravenously injected microspheres in rat spleen: influence of particle size and hydrophilic coating. Biochem. Biophys. Res. Commun. 1991, 177, 861–866. [CrossRef]
- Hobbs, S. K.; Monsky, W. L.; Yuan, F.; Roberts, W. G.; Griffith, L.; Torchilin, V. P.; Jain, R. K. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 4607–4612. [CrossRef] [PubMed]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D. A.; Torchilin, V. P.; Jain, R. K. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756.
- Bae, Y.; Nishiyama, N.; Fukushima, S.; Koyama, H.; Yasuhiro, M.; Kataoka, K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug. Chem. 2005, 16, 122–130.
- Oku, N.; Namba, Y. Long-circulating liposomes. Crit. Rev. Ther. Drug Carrier Syst. 1994, 11, 231–270.
- Senior, J.; Gregoriadis, G. Stability of small unilamellar liposomes in serum and clearance from the circulation: the effect of the phospholipid and cholesterol components. Life Sci. 1982, 30, 2123–2136. [CrossRef]
- Abra, R. M.; Bosworth, M. E.; Hunt, C. A. Liposome disposition in vivo: effects of pre-dosing with lipsomes. Res. Commun. Chem. Pathol. Pharmacol. 1980, 29, 349–360.
- Gao, Z.; Lukyanov, A. N.; Chakilam, A. R.; Torchilin, V. P. PEG-PE/phosphatidylcholine mixed immunomicelles specifically deliver encapsulated taxol to tumor cells of different origin and promote their efficient killing. J. Drug Target. 2003, 11, 87–92. [CrossRef]
- Jeong, Y. I.; Seo, S. J.; Park, I. K.; Lee, H. C.; Kang, I. C.; Akaike, T.; Cho, C. S. Cellular recognition of paclitaxel-loaded polymeric nanoparticles composed of poly(gamma-benzyl L-glutamate) and poly(ethylene glycol) diblock copolymer endcapped with galactose moiety. Int. J. Pharm. 2005, 296, 151–161.
- Park, E. K.; Kim, S. Y.; Lee, S. B.; Lee, Y. M. Folate-conjugated methoxy poly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric micelles for tumor-targeted drug delivery. J. Control. Release 2005, 109, 158–168. [CrossRef]
- Xiong, X. B.; Mahmud, A.; Uludağ, H.; Lavasanifar, A. Conjugation of arginine-glycine-aspartic acid peptides to poly(ethylene oxide)-b-poly(epsilon-caprolactone) micelles for enhanced intracellular drug delivery to metastatic tumor cells. Biomacromolecules 2007, 8, 874–884. [CrossRef]
- Sethuraman, V. A.; Bae, Y. H. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Control. Release 2007, 118, 216–224. [CrossRef]
- Fernandez-Villamarin, M.; Sousa-Herves, A.; Porto, S.; Guldris, N.; Martinez-Costas, J.; Riguera, R.; Fernandez-Megia, E. A dendrimer- hydrophobic interaction synergy improves the stability of polyion complex micelles. Polym. Chem. 2017, 8, 2528–2537. [CrossRef]
- Izumrudov, V. A.; Zhiryakova, M. V.; Kudaibergenov, S. E. Controllable stability of DNA-containing polyelectrolyte complexes in water-salt solutions. Biopolymers 1999, 52, 94–108. [CrossRef]
- Kim, B. S.; Chuanoi, S.; Suma, T.; Anraku, Y.; Hayashi, K.; Naito, M.; Kim, H. J.; Kwon, I. C.; Miyata, K.; Kishimura, A.; Kataoka K. Self- assembly of siRNA/PEG-b-catiomer at integer molar ratio into 100 nm- sized vesicular polyion complexes (siRNAsomes) for RNAi and codelivery of cargo macromolecules. J. Am. Chem. Soc. 2019, 141, 3699–3709.
- Miyata, K.; Kakizawa, Y.; Nishiyama, N.; Harada, A.; Yamasaki, Y.; Koyama, H.; Kataoka, K. Block catiomer polyplexes with regulated densities of charge and disulfide cross-linking directed to enhance gene expression. J. Am. Chem. Soc. 2004, 126, 2355–2361. [CrossRef] [PubMed]
- Wang, Y.; Ma, B.; Abdeen, A. A.; Chen, G.; Xie, R.; Saha, K.; Gong, S. Versatile Redox-Responsive Polyplexes for the Delivery of Plasmid DNA, Messenger RNA, and CRISPR-Cas9 Genome-Editing Machinery. ACS Appl. Mater. Interfaces 2018, 10, 31915–31927. [CrossRef]
- Bronich, T. K.; Keifer, P. A.; Shlyakhtenko, L. S.; Kabanov, A. V. Polymer micelle with cross-linked ionic core. J. Am. Chem. Soc. 2005, 127, 8236–8237. [CrossRef] [PubMed]
- Allen, C.; Han, J.; Yu, Y.; Maysinger, D.; Eisenberg, A. Polycaprolactone-b-poly(ethylene oxide) copolymer micelles as a delivery vehicle for dihydrotestosterone. J. Control. Release 2000, 63, 275–286. [CrossRef]
- Zhang, Z.; Grijpma, D. W.; Feijen, J. Thermo-sensitive transition of monomethoxy poly(ethylene glycol)-block-poly(trimethylene carbonate) films to micellar-like nanoparticles. J. Control. Release 2006, 112, 57–63. [CrossRef]
- Kwon, G. S.; Naito, M.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Micelles based on AB block copolymers of poly(ethylene oxide) and poly(b-benzyl l-aspartate). Langmuir 1993, 9, 945–949. [CrossRef]
- Watanabe, M.; Kawano, K.; Yokoyama, M.; Opanasopit, P.; Okano, T.; Maitani, Y. Preparation of camptothecin-loaded polymeric micelles and evaluation of their incorporation and circulation stability. Int. J. Pharm. 2006, 308, 183–189. [CrossRef]
- Yokoyama, M.; Opanasopit, P.; Okano, T.; Kawano, K.; Maitani, Y. Polymer design and incorporation methods for polymeric micelle carrier system containing water-insoluble anti-cancer agent camptothecin. J. Drug Target. 2004, 12, 373–384. [CrossRef]
- Neradovic, D.; Van Nostrum, C. F.; Hennink, W. E. Thermoresponsive Polymeric Micelles with Controlled Instability Based on Hydrolytically Sensitive N-Isopropylacrylamide Copolymers. Macromolecules 2001, 34, 7589–7591. [CrossRef]
- Rapoport, N.; Marin, A.; Luo, Y.; Prestwich, G. D.; Muniruzzaman, M. D. Intracellular uptake and trafficking of Pluronic micelles in drug-sensitive and MDR cells: effect on the intracellular drug localization. J. Pharm. Sci. 2002, 91, 157–170. [CrossRef]
- Dong, Y.; Feng, S. S. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials 2004, 25, 2843–2849. [CrossRef] [PubMed]
- Attwood, D.; Florence, A. T. Surfactant Systems. Their Chemistry, Pharmacy and Biology. Chapman and Hall: London, 1983.
- Kwon, G. S.; Okano, T. Polymeric micelles as new drug carriers. Adv. Drug Deliv. Rev. 1996, 21, 107–116. [CrossRef]
- Kim, S. Y.; Shin, I. G.; Lee, Y. M.; Cho, C. S.; Sung, Y. K. Methoxy poly(ethylene glycol) and epsilon-caprolactone amphiphilic block copolymeric micelle containing indomethacin. II. Micelle formation and drug release behaviours. J Control. Release 1998, 51, 13–22. [CrossRef]
- Forrest, M. L.; Zhao, A.; Won, C. Y.; Malick, A. W.; Kwon, G. S. Lipophilic prodrugs of Hsp90 inhibitor geldanamycin for nanoencapsulation in poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J. Control. Release 2006, 116, 139–149. [CrossRef]
- Lavasanifar, A.; Samuel, J.; Kwon, G. S. The effect of fatty acid substitution on the in vitro release of amphotericin B from micelles composed of poly(ethylene oxide)-block-poly(N-hexyl stearate-L-aspartamide). J. Control. Release 2002, 79, 165–172. [CrossRef] [PubMed]
- Lee, J.; Lee, S. C.; Acharya, G.; Chang, C. J.; Park, K. Hydrotropic solubilization of paclitaxel: analysis of chemical structures for hydrotropic property. Pharm Res. 2003, 20, 1022–1030. [CrossRef]
- Prompruk, K.; Govender, T.; Zhang, S.; Xiong, C. D.; Stolnik, S. Synthesis of a novel PEG-block-poly(aspartic acid-stat-phenylalanine) copolymer shows potential for formation of a micellar drug carrier. Int. J. Pharm. 2005, 297, 242–253. [CrossRef]
- Wilhelm, M.; Zhao, C.; Wang, Y.; Xu, R.; Winnik, M. A.; Mura, J. L; Riess, G.; Croucher, M. D. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: a fluorescence probe study. Macromolecules 1991, 24, 1033–1040. [CrossRef]
- Konan-Kouakou, Y. N.; Boch, R..; Gurny, R.; Allemann, E. In vitro and in vivo activities of verteporfin-loaded nanoparticles. J. Control Release 2005, 103, 83–91. [CrossRef]
- Liu, J.; Zeng, F.; Allen, C. Influence of serum protein on polycarbonate-based copolymer micelles as a delivery system for a hydrophobic anti-cancer agent. J. Control. Release 2005, 103, 481–497. [CrossRef]
- Lo, C. L.; Huang, C. K.; Lin, K. M.; Hsiue, G. H. Mixed micelles formed from graft and diblock copolymers for application in intracellular drug delivery. Biomaterials 2007, 28, 1225–1235. [CrossRef]
- Opanasopit, P.; Yokoyama, M.; Watanabe, M.; Kawano, K.; Maitani, Y.; Okano, T. Influence of serum and albumins from different species on stability of camptothecin-loaded micelles. J. Control. Release 2005, 104, 313–321. [CrossRef]
- Savić, R.; Azzam, T.; Eisenberg, A.; Maysinger, D. Assessment of the integrity of poly(caprolactone)-b-poly(ethylene oxide) micelles under biological conditions: a fluorogenic-based approach. Langmuir 2006, 22, 3570–3578. [CrossRef] [PubMed]
- Letchford, K.; Zastre, J.; Liggins, R.; Burt H. Synthesis and micellar characterization of short block length methoxy poly(ethylene glycol)-block-poly(caprolactone) diblock copolymers. Colloids Surf. B Biointerfaces 2004, 35, 81–91. [CrossRef] [PubMed]
- Mahmud, A.; Xiong, X.B.; Lavasanifar, A. Novel self-associating poly(ethylene oxide)-block-poly(ε-caprolactone) block copolymers with functional side groups on the polyester block for drug delivery. Macromolecules 2006, 39, 9419–9428. [CrossRef]
- Opanasopit, P.; Yokoyama, M.; Watanabe, M.; Kawano, K.; Maitani, Y.; Okano, T. Block copolymer design for camptothecin incorporation into polymeric micelles for passive tumor targeting. Pharm Res. 2004, 21, 2001–2008. [CrossRef]
- Lavasanifar, A.; Samuel, J.; Kwon, G. S. The effect of alkyl core structure on micellar properties of poly(ethylene oxide)-block-poly(L-aspartamide) derivatives. Colloids Surf. B Biointerfaces 2001, 22, 115–126. [CrossRef]
- Kang, N.; Perron, M. E.; Prud'homme, R. E.; Zhang, Y.; Gaucher, G.; Leroux, J. C. Stereocomplex block copolymer micelles: core-shell nanostructures with enhanced stability. Nano Lett. 2005, 5, 315–319. [CrossRef] [PubMed]
- Zhang, J.; Wang, L. Q.; Wang, H.; Tu, K. Micellization phenomena of amphiphilic block copolymers based on methoxy poly(ethylene glycol) and either crystalline or amorphous poly(caprolactone-b-lactide). Biomacromolecules 2006, 7, 2492–2500. [CrossRef] [PubMed]
- Yoshida, E.; Kunugi, S. Micelle formation of poly(vinyl phenol)-block-polystyrene by alfa,omega-diamines. J. Polym. Sci. Part A Pol. Chem. 2002, 40, 3063–3067. [CrossRef]
- 577. Rijcken, C. Tuneable & degradable polymeric micelles for drug delivery: from synthesis to feasibility in vivo. Ph.D Thesis, Utrecht University, Sep 17, 2007.
- Rodriguez-Hernandez, J.; Checot, F.; Gnanou, Y.; Lecommandoux, S. Toward ‘smart’ nano- objects by self-assembly of block copolymers in solution. Prog. Polym. Sci. 2005, 30, 691–724. [CrossRef]
- Rösler, A.; Vandermeulen, G. W.; Klok, H. A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2001, 53, 95–108. [CrossRef]
- Iijima, M.; Nagasaki, Y.; Okada, T.; Kato, M.; Kataoka, K. Core-polymerized reactive micelles from heterotelechelic amphiphilic block copolymers. Macromolecules 1999, 32, 1140–1146. [CrossRef]
- Kim, J.-H.; Emoto, K.; Lijima, M.; Nagasaki, Y.; Aoyagi, T.; Okano, T.; Sakurai, Y.; Kataoka, K. Core-stabilized polymeric micelle as potential drug carrier: Increased solubilization of taxol. Polym. Adv. Technol. 1999, 10, 647–654. [CrossRef]
- Bae, K. H.; Choi, S. H.; Park, S. Y.; Lee, Y.; Park, T. G. Thermosensitive pluronic micelles stabilized by shell cross-linking with gold nanoparticles. Langmuir 2006, 22, 6380–6384. [CrossRef]
- Liu, S.; Weaver, J. V. M.; Tang, Y.; Billingham, C.; Armes, S. P.; Tribe, K. Synthesis of shell cross-linked micelles with pH-responsive cores using ABC triblock copolymers. Macromolecules 2002, 35, 6121–6131. [CrossRef]
- Jiang, J.; Qi, B.; Lepage, M.; Zhao, Y. Polymer micelles stabilization on demand through reversible photo-cross-linking. Macromolecules 2007, 40, 790–792. [CrossRef]
- Jiang, X.; Luo, S.; Armes, S. P.; Shi, W.; Liu, S. UV Irradiation-induced shell cross-linked micelles with pH-responsive cores using ABC triblock copolymers. Macromolecules 2006, 39, 5987–5994. [CrossRef]
- Joralemon, M. J.; O'Reilly, R. K.; Hawker, C. J.; Wooley, K. L. Shell click-crosslinked (SCC) nanoparticles: a new methodology for synthesis and orthogonal functionalization. J. Am. Chem. Soc. 2005, 127, 16892–16899. [CrossRef] [PubMed]
- Li, X.; Ji, J.; Shen, J. Synthesis of hydroxyl-capped comb-like poly(ethylene glycol) to develop shell cross-linkable micelles. Polymer 2006, 47, 1987–1994. [CrossRef]
- Li, Y.; Lokitz, B. S.; Armes, S. P.; McCormick, C. L. Synthesis of reversible shell cross-linked micelles for controlled release of bioactive agents. Macromolecules 2006, 39, 2726–2728. [CrossRef]
- Rodríguez-Hernández J, Babin J, Zappone B, Lecommandoux S. Preparation of shell cross-linked nano-objects from hybrid-peptide block copolymers. Biomacromolecules 2005, 6, 2213–2220. [CrossRef]
- 590. Pilon, L. N.; Armes, S. P.; Findlay, P.; Rannard, S. P. Synthesis and characterization of shell cross-linked micelles with hydroxy-functional coronas: a pragmatic alternative to dendrimers? Langmuir 2005, 21, 3808–3813.
- Thurmond, K. B., II.; Huang H, Clark, C. G., Jr.; Kowalewski, T.; Wooley, K. L. Shell cross-linked polymer micelles: stabilized assemblies with great versatility and potential. Colloid Surf. B - Biointerfaces 1999, 16, 45–54. [CrossRef]
- Hu, F. Q.; Ren, G. F.; Yuan, H.; Du, Y. Z.; Zeng, S. Shell cross-linked stearic acid grafted chitosan oligosaccharide self-aggregated micelles for controlled release of paclitaxel. Colloids Surf. B - Biointerfaces 2006, 50, 97–103. [CrossRef] [PubMed]
- Liu, S.; Weaver, J. V. M.; Save, M.; Armes, S. P. Synthesis of pH-responsive shell crosslinked micelles and their use as nanoreactors for the preparation of gold nanoparticles. Langmuir 2002, 18, 8350–8357. [CrossRef]
- Zhang, L; Katapodi, K; Davis, T. P; Stenzel, M; Barner-Kowollik, C. W. Using the Reversible Addition-Fragmentation Chain Transfer Process to Synthesize Core-Crosslinked Micelles. J. Polym. Sci., Part A: Polym. Chem. 2006, 44, 2177–2194. [CrossRef]
- Li, Z.; Ding, J.; Day, M.; Tao, Y. Molecularly imprinted polymeric nanospheres by diblock copolymer self-assembly. Macromolecules 2006, 39, 2629–2636. [CrossRef]
- 596. Rheingans, O.; Hugenberg, N.; Harris, J. R.; Fischer, K.; Maskos, M. Nanoparticles built of cross- linked heterotechelic amphiphilic poly(dimethylsiloxane)-b-poly(ethylene oxide) diblock copolymers, Macromolecules 2000, 33, 4780–4790.
- Lee, B. H.; West, B.; McLemore, R.; Pauken, C.; Vernon, B. L. In-situ injectable physically and chemically gelling NIPAAm-based copolymer system for embolization. Biomacromolecules 2006, 7, 2059–2064. [CrossRef]
- Petrov, P.; Bozukov, M.; Tsvetanov, C. B. Innovative approach for stabilizing poly(ethylen oxide)-b-poly(propylene oxide)-b-poly(ethylen oxide) micelles by forming nano-sized networks in the micelle. J. Mater. Chem. 2005, 15, 1481–1486. [CrossRef]
- Jaturanpinyo, M.; Harada, A.; Yuan, X.; Kataoka, K. Preparation of bionanoreactor based on core-shell structured polyion complex micelles entrapping trypsin in the core cross-linked with glutaraldehyde. Bioconjug. Chem. 2004, 15, 344–348. [CrossRef] [PubMed]
- Kakizawa, Y.; Harada, A.; Kataoka, K. Environment-sensitive stabilization of core-shell structured polyion complex micelle by reversible cross-linking of the core through disulfide bond. J. Am. Chem. Soc. 1999, 121, 11247–11248. [CrossRef]
- Rijcken, C. J.; Snel, C. J.; Schiffelers, R. M.; van Nostrum, C. F.; Hennink, W. E. Hydrolysable core-crosslinked thermosensitive polymeric micelles: synthesis, characterisation and in vivo studies. Biomaterials 2007, 28, 5581–5593. [CrossRef] [PubMed]
- Bourouina, N.; Cohen Stuart, M. A.; Mieke Kleijn, J. Complex coacervate core micelles as diffusional nanoprobes. Soft Matter 2014, 10, 320–331. [CrossRef]
- Shao, Y.; Huang, W.; Shi, C.; Atkinson, S. T.; Luo, J. Reversibly crosslinked nanocarriers for on-demand drug delivery in cancer treatment. Ther. Deliv. 2012, 3, 1409–1427. [CrossRef]
- 604. Hebels, E. R.; van Steenbergen, M. J.; Haegebaert, R.; Cornelis W. Seinen, C. W.; Mesquita, B. S.; van den Dikkenberg, A.; Remaut, K.; Rijcken, C. J. F.; van Ravensteijn, B. G. P.; Hennink, W. E.; Vermonden, T. Mechanistic Study on the Degradation of Hydrolysable Core-Crosslinked Polymeric Micelles. Langmuir 2023, 39, 12132–12143.
- Talelli, M.; Barz, M.; Rijcken, C. J.; Kiessling, F.; Hennink, W. E.; Lammers, T. Core-Crosslinked Polymeric Micelles: Principles, Preparation, Biomedical Applications and Clinical Translation. Nano Today 2015, 10, 93–117. [CrossRef] [PubMed]
- Shim, M. S.; Kwon. Y. J. Acid-transforming polypeptide micelles for targeted nonviral gene delivery. Biomaterials 2010, 31, 3404–3413. [CrossRef]
- Krishnakumar, G. S.; Sampath, S.; Muthusamy, S.; John, M. A. Importance of crosslinking strategies in designing smart biomaterials for bone tissue engineering: A systematic review. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 96, 941–954. [CrossRef]
- Hennink, W. E.; van Nostrum, C. F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 13–36. [CrossRef] [PubMed]
- Van Nostrum, C. F. Covalently cross-linked amphiphilic block copolymer micelles. Soft Matter 2011, 7, 3246–3259. [CrossRef]
- Tillet, G.; Boutevin, B.; Ameduri. B. Chemical Reactions of Polymer Crosslinking and Post-Crosslinking at Room and Medium Temperature. Prog. Polym. Sci. 2011, 36, 191–217. [CrossRef]
- Ayyub, O. B.; Ibrahim, M. B.; Kofinas, P. Synthesis and characterization of microphase separated primary amine functionalized polystyrene-b-poly(2-vinylpyridine). Polymer 2014, 55, 6227–6231. [CrossRef]
- Kembaren, R.; Mieke Kleijn, J.; Borst, J. W.; Kamperman, M.; Hofman, A. H. Enhanced stability of complex coacervate core micelles following different core-crosslinking strategies. Soft Matter 2022, 18, 3052–3062. [CrossRef]
- Mantovani, G.; Lecolley, F.; Tao, L.; Haddleton, D. M.; Clerx, J.; Cornelissen, J. J.; Velonia, K. Design and synthesis of N-maleimido-functionalized hydrophilic polymers via copper-mediated living radical polymerization: a suitable alternative to PEGylation chemistry. J. Am. Chem. Soc. 2005, 127, 2966–2973.
- Ju, M.; Gong, F.; Cheng, S.; Gao, Y. Fast and Convenient Synthesis of Amine-Terminated Polylactide as a Macroinitiator for ω-Benzyloxycarbonyl-L-Lysine-N-Carboxyanhydrides. Int. J. Polym. Sci. 2011, 2011, 1–8.
- McMaster, P. D.; Byrnes, E. W.; Block, A. J.; Paul A. Tenthorey, P. A. New antiarrhythmic agents. 5..alpha.-Aminoaceto-2,6-xylidides with functionalized amide alkyl substituents. J. Med. Chem. 1981, 24, 53–58. [CrossRef]
- Jiang, J.; Tong, X.; Morris, D.; Zhao, Y. Toward photocontrolled release using light-dissociable block copolymer micelles. Macromolecules 2006, 39, 4633–4640. [CrossRef]
- 617. Liu, D-Z.; Hsieh, J-H.; Fan, X-C.; Yang, J-D.; Chung, T-W. Synthesis, characterization and drug delivery behaviors of new PCP polymeric micelles, Carbohydr. Polym. 2007, 68, 544–554.
- Tian, L.; Yam, L.; Wang, J.; Tat, H.; Uhrich, K. Core crosslinkable polymeric micelles from PEG- lipid amphiphiles as drug carriers. J. Mater. Chem. 2004, 14, 2317–2324. [CrossRef]
- Yokoyama, M.; Miyauchi, M.; Yamada, N.; Okano, T.; Sakurai, Y.; Shohei, I.; Kataoka, K. Polymeric Micelles as Novel Drug Carrier: Adriamycin-Conjugated Poly(Ethylene Glycol)–Poly(Aspartic Acid) Block Copolymer. J. Control. Release 1990, 11, 269–278. [CrossRef]
- Kataoka, K.; Matsumoto, T.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Fukushima, S.; Okamoto, K.; Kwon, G. S. Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-L-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J. Control. Release 2000, 64, 143–153.
- Seymour, L. W.; Duncan, R.; Strohalm, J.; Kopecek, J. Effect of molecular weight (Mw) of N-(2-hydroxypropyl)methacrylamide copolymers on body distribution and rate of excretion after subcutaneous, intraperitoneal, and intravenous administration to rats. J. Biomed. Mater. Res. 1987, 21, 1341–1358. [CrossRef]
- Mart, R. J.; Osborne, R. D.; Stevens, M. M.; Ulijn, R. V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. [CrossRef]
- Rijcken, C. J.; Soga, O.; Hennink, W. E.; van Nostrum, C. F. Triggered destabilisation of polymeric micelles and vesicles by changing polymers polarity: an attractive tool for drug delivery. J. Control. Release 2007, 120, 131–148. [CrossRef]
- Chilkoti, A.; Dreher, M. R.; Meyer, D. E.; Raucher, D. Targeted drug delivery by thermally responsive polymers. Adv. Drug Deliv. Rev. 2002, 54, 613–630. [CrossRef]
- Hoffman, A. S.; Stayton, P. S.; Volga, B.; Chen, G.; Chen, J.; Cheung, C.; Chilkoti, A.; Ding, Z.; Dong, L.; Fong, R.; Lackey, C. A.; Long, C. J.; Miura, M.; Morris, J. E.; Murthy, N.; Nabeshima, Y.; Park, T. G.; Press, O. W.; Shimoboji, T.; Shoemaker, S.; Yang, H. J.; Monji, N.; Nowinski, R. C.; Cole, C. A.; Priest, J. H.; Harris, J. M.; Nakamae, K.; Nishino, T.; Miyata, T. Really smart bioconjugates of smart polymers and receptor proteins. J. Biomed. Mater. Res. 2000, 52, 577–586.
- Jeong, B.; Bae, Y. H.; Lee, D. S.; Kim, S. W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860–862. [CrossRef]
- Heskins, M.; Guillet, J. E. Solution properties of poly (N-isopropylacrylamide). J. Macromol. Sci. A Chem. 1968, 2, 1441–1455. [CrossRef]
- Pelton, R. Temperature-sensitive aqueous microgels. Adv. Colloid Interface Sci. 2000, 85, 1–33. [CrossRef]
- Schild, H. G. Poly (N-isopropylacrylamide) experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [CrossRef]
- Feil, H.; Bae, Y. H.; Feijen J.; Kim, S. W. Effect of comonomer hydrophilicity and ionization on the lower critical solution temperature of N-isopropylacrylamide copolymers. Macromolecules 1993, 26, 2496–2500. [CrossRef]
- Park, J.-S.; Kataoka, K. Precise control of lower critical solution temperature of thermosensitive poly(2-isopropyl-2-oxazoline) via gradient copolymerization with 2-ethyl-2-oxazoline as a hydrophilic comonomer. Macromolecules 2006, 39, 6622–6630. [CrossRef]
- Shibayama, M.; Mizutani, S.; Nomura, S. Thermal properties of copolymer gels containing N- isopropylacrylamide. Macromolecules 1996, 29, 2019–2024. [CrossRef]
- Soga, O.; van Nostrum, C. F.; Hennink, W. E. Poly(N-(2-hydroxypropyl) methacrylamide mono/di lactate): a new class of biodegradable polymers with tuneable thermosensitivity. Biomacromolecules 2004, 5, 818–821. [CrossRef] [PubMed]
- Qiu, X. P.; Wu, C. Study of the core-shell nanoparticle formed through the "coil-to-globule" transition of poly(N-isopropylacrylamide) grafted with poly(ethylene oxide). Macromolecules 1997, 30, 7921–7926. [CrossRef]
- Zhu, P. W.; Napper, D. H. Effect of heating rate on nanoparticle formation of poly(N- isopropylacrylamide)-poly(ethylene glycol) block copolymer microgels. Langmuir 2000, 16, 8543–8545. [CrossRef]
- Engin, K.; Leeper, D. B.; Cater, J. R.; Thistlethwaite, A. J.; Tupchong, L.; McFarlane, J. D. Extracellular pH distribution in human tumours. Int. J. Hyperthermia 1995, 11, 211–216.
- Haag, R. Supramolecular drug-delivery systems based on polymeric core-shell architectures. Angew. Chem. Int. Ed. Engl. 2004, 43, 278–282. [CrossRef]
- Lee, E. S.; Na, K.; Bae, Y. H. Polymeric micelle for tumor pH and folate-mediated targeting. J Control. Release 2003, 91, 103–113. [CrossRef]
- Lee, E. S.; Shin, H. J.; Na, K.; Bae, Y. H. Poly(L-histidine)-PEG block copolymer micelles and pH-induced destabilization. J. Control. Release 2003, 90, 363–374. [CrossRef]
- Martin, T. J.; Procházka, K.; Munk, P.; Webber, S. E. pH-Dependent Micellization of Poly(2-vinylpyridine)-block-poly(ethylene oxide). Macromolecules 1996, 29, 6071–6073. [CrossRef]
- Lee, A. S.; Gast, A. P.; Bütün, V.; Armes, S. P. Characterizing the structure of pH dependent polyelectrolyte block copolymer micelles. Macromolecules 1999, 32, 4302–4310. [CrossRef]
- Tang, Y.; Liu, S. Y.; Armes, S. P.; Billingham, N. C. Solubilization and controlled release of a hydrophobic drug using novel micelle-forming ABC triblock copolymers. Biomacromolecules 2003, 4, 1636–1645. [CrossRef]
- Na, K.; Bae, Y. H. Self-assembled hydrogel nanoparticles responsive to tumor extracellular pH from pullulan derivative/sulfonamide conjugate: characterization, aggregation, and adriamycin release in vitro. Pharm. Res. 2002, 19, 681–688. [CrossRef]
- Na, K.; Lee, K. H.; Bae, Y. H. pH-sensitivity and pH-dependent interior structural change of self-assembled hydrogel nanoparticles of pullulan acetate/oligo-sulfonamide conjugate. J. Control. Release 2004, 97, 513–525. [CrossRef]
- Giacomelli, C.; Le Men, L.; Borsali, R.; Lai-Kee-Him, J.; Brisson, A.; Armes, S. P.; Lewis, A. L. Phosphorylcholine-based pH-responsive diblock copolymer micelles as drug delivery vehicles: light scattering, electron microscopy, and fluorescence experiments. Biomacromolecules 2006, 7, 817–828.
- Licciardi, M.; Giammona, G.; Du, J.; Armes, S. P.; Tang, Y.; Lewis, A. L. New folate-functionalized biocompatible block copolymer micelles as potential anti-cancer drug delivery systems. Polymer 2006, 47, 2946–2955. [CrossRef]
- Kumar, N.; Ravikumar, M. N.; Domb, A. J. Biodegradable block copolymers. Adv. Drug Deliv. Rev. 2001, 53, 23–44. [CrossRef]
- Zweers, M. L.; Engbers, G. H.; Grijpma, D. W.; Feijen, J. In vitro degradation of nanoparticles prepared from polymers based on DL-lactide, glycolide and poly(ethylene oxide). J Control. Release 2004, 100, 347–356. [CrossRef] [PubMed]
- Hu, Y.; Zhang, L.; Cao, Y.; Ge, H.; Jiang, X.; Yang, C. Degradation behavior of poly(epsilon-caprolactone)-b-poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles in aqueous solution. Biomacromolecules 2004, 5, 1756–1762. [CrossRef] [PubMed]
- Carstens, M. G.; van Nostrum, C. F.; Verrijk, R.; de Leede, L. G.; Crommelin, D. J.; Hennink, W. E. A mechanistic study on the chemical and enzymatic degradation of PEG-Oligo(epsilon-caprolactone) micelles. J. Pharm. Sci. 2008, 97, 506–518. [CrossRef] [PubMed]
- Geng, Y.; Discher, D. E. Hydrolytic degradation of poly(ethylene oxide)-block-polycaprolactone worm micelles. J. Am. Chem. Soc. 2005, 127, 12780–12781. [CrossRef]
- De Jong, S. J.; Arias, E. R.; Rijkers, D. T. S., van Nostrum, C. F.; Kettenes-van den Bosch, J. J.; Hennink, W. E. New insights into the hydrolytic degradation of poly (lactic acid): Participation of the alcohol terminus. Polymer 2001, 42, 2795–2802. [CrossRef]
- Heller, J.; Barr, J. Poly(ortho esters)--from concept to reality. Biomacromolecules 2004, 5, 1625–1632. [CrossRef]
- Heller J.; Barr, J.; Ng, S. Y, Abdellauoi, K. S, Gurny R. Poly(ortho esters): synthesis, characterization, properties and uses. Adv. Drug Deliv. Rev. 2002, 54, 1015–1039. [CrossRef]
- Geng, Y.; Discher, D. E. Visualization of degradable worm micelle breakdown in relation to drug release. Polymer 2006, 47, 2519–2525. [CrossRef]
- Gillies, E. R.; Frechet, J. M. A new approach towards acid sensitive copolymer micelles for drug delivery. Chem. Comm. 2003, 0, 1640–1641.
- Hrubý, M.; Konák, C.; Ulbrich, K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J. Control. Release 2005, 103, 137–148. [CrossRef] [PubMed]
- Gillies, E. R.; Jonsson, T. B.; Fréchet, J. M. Stimuli-responsive supramolecular assemblies of linear-dendritic copolymers. J. Am. Chem. Soc. 2004, 126, 11936–11943. [CrossRef] [PubMed]
- Xie, Z.; Guan, H,; Chen, X.; Lu, C.; Chen, L.; Hu, X.; Shi, Q.; Jing, X. A novel polymer-paclitaxel conjugate based on amphiphilic triblock copolymer. J. Control. Release 2007, 117, 210–216. [CrossRef]
- Gan, Z.; Jim, T. F.; Li, M.; Yuer, Z.; Wang, S.; Wu, C. Enzymatic Biodegradation of Poly(ethylene oxide-b-ε-caprolactone) Diblock Copolymer and Its Potential Biomedical Applications. Macromolecules 1999, 32, 590–594. [CrossRef]
- Akagi, T.; Higashi, M.; Kaneko, T.; Kida, T.; Akashi, M. Hydrolytic and enzymatic degradation of nanoparticles based on amphiphilic poly(gamma-glutamic acid)-graft-L-phenylalanine copolymers. Biomacromolecules 2006, 7, 297–303. [CrossRef]
- Ghosh, S.; Basu, S.; Thayumanavan, S. Simultaneous and reversible functionalization of copolymers for biological applications. Macromolecules 2006, 39, 5595–5597. [CrossRef]
- Kakizawa, Y.; Harada, A.; Kataoka, K. Glutathione-sensitive stabilization of block copolymer micelles composed of antisense DNA and thiolated poly(ethylene glycol)-block-poly(L-lysine): a potential carrier for systemic delivery of antisense DNA. Biomacromolecules 2001, 2, 491–497. [CrossRef]
- Anton, P.; Heinze, J.; Laschewsky, A. Redox-active monomeric and polymeric surfactants. Langmuir 1993, 9, 77–85. [CrossRef]
- Takeoka, Y.; Aoki, T.; Sanui, K.; Ogata, N.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Watanabe, M. Electrochemical control of drug release from redox-active micelles. J. Control. Release 1995, 33, 79–87. [CrossRef]
- Jiang, J.; Tong, X.; Zhao, Y. A new design for light-breakable polymer micelles. J. Am. Chem. Soc. 2005, 127, 8290–8291. [CrossRef]
- Goodwin, A. P.; Mynar, J. L.; Ma, Y.; Fleming, G. R.; Fréchet, J. M. Synthetic micelle sensitive to IR light via a two-photon process. J. Am. Chem. Soc. 2005, 127, 9952–9953. [CrossRef] [PubMed]
- Tong, X.; Wang, G.; Soldera, A.; Zhao, Y. How can azobenzene block copolymer vesicles be dissociated and reformed by light? J. Phys. Chem. B. 2005, 109, 20281–20287. [CrossRef] [PubMed]
- Konak, C.; Rathi, R. C.; Kopeckova, P.; Kopecek, J. Photoassociation of water-soluble copolymers containing photochromic spirobenzopyran moieties. Polym. Adv. Technol. 1998, 9, 641–648. [CrossRef]
- Kono, K.; Nishihara, Y.; Takagishi, T. Photoresponsive permeability of polyelectrolyte complex capsule membrane containing triphenylmethane leucohydroxide residues. J. Appl. Polym. Sci. 1995, 56, 707–713. [CrossRef]
- Gao, Z. G.; Fain, H. D.; Rapoport, N. Controlled and targeted tumor chemotherapy by micellar-encapsulated drug and ultrasound. J. Control. Release 2005, 102, 203–222. [CrossRef]
- Rapoport, N.; Pitt, W. G.; Sun, H.; Nelson, J. L. Drug delivery in polymeric micelles: from in vitro to in vivo. J. Control. Release 2003, 91, 85–95. [CrossRef]
- Rapoport, N. Y.; Christensen, D. A.; Fain, H. D.; Barrows, L.; Gao, Z. Ultrasound-triggered drug targeting of tumors in vitro and in vivo. Ultrasonics 2004, 42, 943–950. [CrossRef] [PubMed]
- Lecommandoux, S.; Sandre, O.; Checot, F.; Perzynski, R. Smart hybrid magnetic self-assembled micelles and hollow capsules. Prog. Solid State Chem. 2006, 34, 171–179. [CrossRef]
- Lecommandoux, S.; Sandre, O.; Checot, F.; Rodriguez-Hernandez, J.; Perzynski, R. Self- assemblies of magnetic nanoparticles and di-block copolymers: Magnetic micelles and vesicles. J. Magn. Magn. Mater. 2006, 300, 71–74. [CrossRef]
- Liu, S. Q.; Wiradharma, N.; Gao, S. J.; Tong, Y. W.; Yang, Y. Y. Bio-functional micelles self-assembled from a folate-conjugated block copolymer for targeted intracellular delivery of anticancer drugs. Biomaterials 2007, 28, 1423–1433. [CrossRef]
- Soppimath, K. S.; Tan, DC-W.; Yang, Y-Y. pH-triggered thermally responsive polymer core-shell nanoparticles for drug delivery. Adv. Mater. 2005, 17, 318–323. [CrossRef]
- Liu, X-M.; Wang, L-S.; Wang, L.; Huang, J.; He, C. The effect of salt and pH on the phase-transition behaviors of temperature-sensitive copolymers based on N-isopropylacrylamide. Biomaterials 2004, 25, 5659–5666. [CrossRef]
- Mertoglu, M.; Garnier, S.; Laschewsky, A.; Skrabania, K.; Storsberg, J. Stimuli responsive amphiphilic block copolymers for aqueous media synthesised via reversible addition fragmentation chain transfer polymerisation (RAFT). Polymer 2005, 46, 7726–7740. [CrossRef]
- Pişkin E. Molecularly designed water soluble, intelligent, nanosize polymeric carriers. Int. J. Pharm. 2004, 277, 105–118. [CrossRef]
- Salgado-Rodriguez, R.; Licea-Claverie, A.; Arndt, K. F. Random copolymers of N- isopropylacrylamide and methacrylic acid monomers with hydrophobic spacers: pH- tunable temperature sensitive materials. Eur. Polym. J. 2004, 40, 1931–1946. [CrossRef]
- Yin, X.; Hoffman, A. S.; Stayton, P. S. Poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers that respond sharply to temperature and pH. Biomacromolecules 2006, 7, 1381–1385. [CrossRef] [PubMed]
- Lee, B. H.; Vernon, B. Copolymers of N-isopropylacrylamide HEMA-lactate and acrylic acid with time-dependent lower critical solution temperature as a bioresorbable carrier. Polym. Int. 2005, 54, 418–422. [CrossRef]
- Neradovic, D.; Hinrichs, W. L. J.; Kettenes-van den Bosch, J. J.; Hennink, W. E. Poly(N-isopropylacrylamide) with hydrolyzable lactic acid ester side groups: a new type of thermosensitive polymer. Macromol. Rapid Commun. 1999, 20, 577–581. [CrossRef]
- Shah, S. S.; Wertheim, J.; Wang, C. T.; Pitt, C. G. Polymer-drug conjugates: manipulating drug delivery kinetics using model LCST systems. J. Control. Release 1997, 45, 95–101. [CrossRef]
- Soga, O.; van Nostrum, C. F.; Fens, M.; Rijcken, C. J.; Schiffelers, R. M.; Storm, G.; Hennink, W. E. Thermosensitive and biodegradable polymeric micelles for paclitaxel delivery. J. Control. Release 2005, 103, 341–353.
- Cui, Z.; Lee, B. H.; Vernon, B. L. New hydrolysis-dependent thermosensitive polymer for an injectable degradable system. Biomacromolecules 2007, 8, 1280–1286. [CrossRef] [PubMed]
- Hruby, M.; Kueka, J.; Lebeda, O.; Mackova, H.; Babie, M.; Kooak, E.; Studenovsky, M.; Sikora, A.; Kozempel, J.; Ulbrich, K. New bioerodable thermoresponsive polymers for possible radiotherapeutic applications. J. Control. Release 2007, 119, 25–33.
- Huang, X.; Rong, F. D.; Li, J. Z. Novel acid-labile, thermoresponsive poly(methacrylamide)s with pendent ortho ester moieties. Macromol. Rapid Commun. 2007, 28, 597–603. [CrossRef]
- Ivanov, A. E.; Eremeev, N. L.; Wahlund, P-O.; Galaev, I. Y.; Mattiasson, B. Photosensitive copolymer of N-isopropylacrylamide and methacryloyl derivative of spyrobenzopyran. Polymer 2002, 43, 3819–3823. [CrossRef]
- Laschewsky, A.; Rekai, E. D. Photochemical modification of the lower critical solution temperature of cinnamoylated poly(N-2-hydropropylmethacrylamide) in water. Macromol. Rapid Commun. 2000, 21, 937–940. [CrossRef]
- Sugiyama, K.; Sono, K. Characterization of photo- and thermoresponsible amphiphilic copolymers having azobenzene moieties as side groups. J. Appl. Polym. Sci. 2000, 81, 3056–3063.
- Ravi, P.; Sin, S. L.; Gan, L. H.; Gan, Y. Y.; Tam, K. C.; Xia, X. L.; Hu, X. New water soluble azobenzene- containing diblock copolymers: synthesis and aggregation behavior. Polymer 2005, 46, 137–146. [CrossRef]
- 694. Desponds, A.; Freitag, R. Synthesis and characterization of photoresponsive N- isopropylacrylamide cotelomers. Langmuir 2003, 19, 6261–6270.
- 695. Szczubialka, K.; Moczek, L.; Blaskiewicz, S.; Nowakowska, M. Photocrosslinkable smart terpolymers responding to pH, temperature and ionic strength. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 3879–3886.
- Bütün, V.; Billingham, N. C.; Armes, S. P. Unusual Aggregation Behavior of a Novel Tertiary Amine Methacrylate-Based Diblock Copolymer: Formation of Micelles and Reverse Micelles in Aqueous Solution. J. Am. Chem. Soc. 1998, 120, 11818–11819. [CrossRef]
- Bütün, V.; Liu, S.; Weaver, J. V. M; Bories-Azeau, X.; Cai, Y.; Armes, S. P. A Brief Review of “Schizophrenic” Block Copolymers React. Funct. Polym. 2006, 66, 157–165.
- Katayama, Y.; Sonoda, T.; Maeda, M. A polymer micelle responding to the protein kinase A signal. Macromolecules 2001, 34, 8569–8573. [CrossRef]
- Wickline, S. A.; Lanza, G. M. Nanotechnology for molecular imaging and targeted therapy. Circulation 2003, 107, 1092–1095. [CrossRef]
- Trubetskoy, V. S.; Frank-Kamenetsky, M. D.; Whiteman, K. R.; Wolf, G. L.; Torchilin, V. P. Stable polymeric micelles: lymphangiographic contrast media for gamma scintigraphy and magnetic resonance imaging. Acad. Radiol. 1996, 3, 232–238. [CrossRef]
- Otsuka, H.; Nagasaki, Y.; Kataoka, K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev. 2003, 55, 403–419. [CrossRef]
- Ai, H.; Flask, C.; Weinberg, B.; Shuai, X.; Pagel, M. D.; Farrell, D.; Duerk, J.; Gao, J. Magnetite-loaded polymeric micelles as ultrasensitive magnetic-resonance probes. Adv. Mater. 2005, 17, 1949–1952. [CrossRef]
- Nakamura, E.; Makino, K.; Okano, T.; Yamamoto, T.; Yokoyama, M. A polymeric micelle MRI contrast agent with changeable relaxivity. J. Control. Release 2006, 114, 325–333. [CrossRef]
- Yoon, T. J.; Kim, J. S.; Kim, B. G.; Yu, K. N.; Cho, M. H.; Lee, J. K. Multifunctional nanoparticles possessing a "magnetic motor effect" for drug or gene delivery. Angew. Chem. Int. Ed. Engl. 2005, 44, 1068–1071. [CrossRef]
- Kopelman, R.; Lee Koo, Y. E. L.; Philbert, M.; Moffat, B. A.; Ramachandra Reddy, G.; McConville, P.; Hall, D. E.; Chenevert, T. L.; Bhojani, M. S.; Buck, S. M.; Rehemtulla, A.; Ross, B. D. Multifunctional nanoparticle platforms for in vivo MRI enhancement and photodynamic therapy of a rat brain cancer. J. Magn. Magn. Mater. 2005, 293, 404–410.
- Reddy, G. R.; Bhojani, M. S.; McConville, P.; Moody, J.; Moffat, B. A.; Hall, D. E.; Kim, G.; Koo, Y. E.; Woolliscroft, M. J.; Sugai, J. V.; Johnson, T. D.; Philbert, M. A.; Kopelman, R.; Rehemtulla, A.; Ross, B. D. Vascular targeted nanoparticles for imaging and treatment of brain tumors. Clin. Cancer Res. 2006, 12, 6677–6686.
- Sahoo, S. K.; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov. Today 2003, 8, 1112–1120. [CrossRef] [PubMed]
- 708. Maysinger, D.; Lovrić, J.; Eisenberg, A.; Savić, R. Fate of micelles and quantum dots in cells. Eur. J. Pharm. Biopharm. 2007, 65, 270–281.
- Dubertret, B.; Skourides, P.; Norris, D. J.; Noireaux, V.; Brivanlou, A. H.; Libchaber, A. In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science 2002, 298, 1759–1762. [CrossRef] [PubMed]
- Gao, X.; Cui, Y.; Levenson, R. M.; Chung, L. W.; Nie, S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat. Biotechnol. 2004, 22, 969–976. [CrossRef] [PubMed]
- Hamaguchi, T.; Kato, K.; Yasui, H.; Morizane, C.; Ikeda, M.; Ueno, H.; Muro, K.; Yamada, Y.; Okusaka, T.; Shirao, K.; Shimada, Y.; Nakahama, H.; Matsumura, Y. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br. J. Cancer 2007, 97, 170–176.
- Doncom, K. E. B.; Blackman, L. D.; Wright, D. B.; Gibson, M. I.; O'Reilly, R. K. Dispersity effects in polymer self-assemblies:a matter of hierarchical control. Chem. Soc. Rev. 2017, 46, 4119–4134. [CrossRef]
- Israelachvili, J. N.; Mitchell, D. J.; Ninham, B. W. Theory of self-assembly of lipid bilayers and vesicles. Biochim. Biophys. Acta 1977, 470, 185–201. [CrossRef] [PubMed]
- Tritschler, U.; Pearce, S.; Gwyther, J.; Whittell, G. R.; Manners, I. 50th Anniversary Perspective: Functional Nanoparticles from the Solution Self-Assembly of Block Copolymers. Macromolecules 2017, 50, 3439–3463. [CrossRef]
- 715. Wyman, I. W.; Liu, G. Micellar structures of linear triblock terpolymers: Three blocks but many possibilities. Polymer 2013, 54, 54–1950.
- 716. Gröschel, A. H.; Müller, A. H. Self-assembly concepts for multicompartment nanostructures. Nanoscale 2015, 7, 7–11841.
- Mai, Y.; Eisenberg, A. Self-Assembly of Block Copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [CrossRef]
- Lin, P. C.; Lin, S.; Wang, P. C.; Sridhar, R. Techniques for physicochemical characterization of nanomaterials. Biotechnol. Adv. 2014, 32, 711–726. [CrossRef] [PubMed]
- Patterson, J. P.; Robin, M. P.; Chassenieux, C.; Colombani, O.; O'Reilly, R. K. The analysis of solution self-assembled polymeric nanomaterials. Chem. Soc. Rev. 2014, 43, 2412–2425. [CrossRef]
- Epps, T. H., III; O’Reilly, R. K. Block copolymers: controlling nanostructure to generate functional materials – synthesis, characterization, and engineering. Chem. Sci. 2016, 7, 1674–1689. [CrossRef]
- 721. Agrahari, V.; Agrahari, V. Advances and applications of block-copolymer-based nanoformulations. Drug. Discov. Today. 2018, 23, 1139–1151.
- Zhulina, E. B.; Borisov, O. V. Theory of Block Polymer Micelles: Recent Advances and Current Challenges. Macromolecules 2012, 45, 4429–4440. [CrossRef]
- Zhang, Q.; Lin, J. P.; Wang, L. Q.; Xu, Z. W. Theoretical modeling and simulations of self-assembly of copolymers in solution. Prog. Polym. Sci. 2017, 75, 1–30. [CrossRef]
- 724. Voets, I. K.; van der Burgh, S.; Farago, B.; Fokkink, R.; Kovacevic, D.; Hellweg, T.; de Keizer, A.; Cohen Stuart, M. A. Electrostatically driven coassembly of a diblock copolymer and an oppositely charged homopolymer in aqueous solutions. Macromolecules 2007, 40, 8476–8482.
- Zhulina, E. B.; Adam, M.; LaRue, I.; Sheiko, S. S.; Rubinstein, M. Diblock copolymer micelles in a dilute solution. Macromolecules 2005, 38, 5330–5351. [CrossRef]
- Borisov, O.; Zhulina, E. Effect of salt on self-assembly in charged block copolymer micelles. Macromolecules 2002, 35, 4472–4480. [CrossRef]
- Deng, Z.; Liu, S. Emerging trends in solution self-assembly of block copolymers. Polymer 2020, 207, 122914. [CrossRef]
- Jiao, W.; Yang, H.; Wu, Z.; Liu, J.; Zhang, W. Self-assembled block polymer aggregates in selective solution: controllable morphology transitions and their applications in drug delivery. Expert Opin. Drug Deliv. 2020, 17, 947–961. [CrossRef] [PubMed]
- Rangelov, S.; Pispas, S. Polymer and Polymer-Hybrid Nanoparticles: From Synthesis to Biomedical Applications, Ed.; CRC Press: Taylor and Francis Group, 2014. [Google Scholar]
- 730. Crassous, J. J.; Schurtenberger, P.; Ballauff, M.; Mihut, A. M. Design of Block. Copolymer Micelles via Crystallization. Polymer 2015, 62, 62–A1.
- Foster, J. C.; Varlas, S.; Couturaud, B.; Coe, Z.; O'Reilly, R. K. Getting into Shape: Reflections on a New Generation of Cylindrical Nanostructures’ Self-Assembly Using Polymer Building Blocks. J. Am. Chem. Soc. 2019, 141, 2742–2753. [CrossRef] [PubMed]
- 732. Ganda, S.; Stenzel, M.H. Concepts, fabrication methods and applications of living crystallization-driven self-assembly of block copolymers. Prog. Polym. Sci. 2020, 101, 101195.
- 733. MacFarlane, L.; Zhao, C.; Cai, J.; Qiu, H.; Manners, I. Emerging applications for living crystallization-driven self-assembly. Chem. Sci. 2021, 12, 4661–4682.
- Qiu, H.; Gao, Y.; Du, V. A.; Harniman, R.; Winnik. M. A.; Manners, I. Branched Micelles by Living Crystallization-Driven Block Copolymer Self-Assembly under Kinetic Control. J. Am. Chem. Soc. 2015, 137, 2375–2385. [CrossRef]
- 735. Hayward D. W. Gilroy J. B. Rupar P. A. Chabanne L. Pizzey C. Winnik M. A. Whittell G. R. Manners I. Richardson R. M. Tailored assemblies of block copolymers in solution: it is all about the process. Macromolecules, 2015; 48 (5), 1579-1591.
- Song, S.; Liu, X.; Nikbin, E.; Howe, J. Y.; Yu, Q.; Manners, I.; Winnik, M. A. Uniform 1D Micelles and Patchy & Block Comicelles via Scalable, One-Step Crystallization-Driven Block Copolymer Self-Assembly. J. Am. Chem. Soc. 2021, 143, 6266–6280.
- Gwyther, J.; Gilroy, J. B.; Rupar, P. A.; Lunn, D. J.; Kynaston, E.; Patra, S. K.; Whittell, G. R.; Winnik, M. A.; Manners, I. Dimensional Control of Block Copolymer Nanofibers with a π-Conjugated Core: Crystallization-Driven Solution Self-Assembly of Amphiphilic Poly(3-hexylthiophene)-b-poly(2-vinylpyridine). Chem. Eur. J. 2013, 19, 9186–9197.
- Qian, J.; Li, X.; Lunn, D. J.; Gwyther, J.; Hudson, Z. M.; Kynaston, E.; Rupar, P. A.; Winnik, M. A.; Manners, I. Uniform, High Aspect Ratio Fiber-like Micelles and Block Co-micelles with a Crystalline π-Conjugated Polythiophene Core by Self-Seeding. J. Am. Chem. Soc. 2014, 136, 4121–4124. [CrossRef]
- Li, X.; Wolanin, P. J.; MacFarlane, L. R.; Harniman, R. L.; Qian,J.; Gould, O. E. C.; Dane, T. G.; Rudin, J.; Cryan, M. J.; Schmaltz, T.;Frauenrath, H.; Winnik, M. A.; Faul, C. F. J.; Manners, I. Uniform Electroactive Fibre-like Micelle Nanowires for Organic Electronics. Nat. Commun. 2017, 8, 15909/1-8 (Erratum 1).
- Karayianni M.; Pispas S. Block copolymer solution self-assembly: Recent advances, emerging trends, and applications. J. Polym. Sci. 2021, 59, 1874–1898. [CrossRef]
- Sun, L.; Pitto-Barry, A.; Kirby, N.; Schiller, T. L.; Sanchez, A. M.; Dyson, M. A.; Sloan, J.; Wilson, N. R.; O'Reilly, R. K.; Dove, A. P. Structural reorganization of cylindrical nanoparticles triggered by polylactide stereocomplexation. Nat. Commun. 2014, 5, 5746.
- Pitto-Barry, A.; Kirby, N.; Dove, A. P.; O'Reilly, R. K. Expanding the scope of the crystallization-driven self-assembly of polylactide-containing polymers. Polym. Chem. 2014, 5, 1427–1436. [CrossRef]
- Yu, W.; Inam, M.; Jones, J. R.; Dove, A.P.; O’Reilly, R. K. Understanding the CDSA of poly(lactide) containing triblock copolymers. Polym. Chem. 2017, 8, 5504–5512. [CrossRef]
- 744. Coe, Z.; Weems, A.; Dove, A. P.; O’Reilly, R. K. Synthesis of Monodisperse Cylindrical Nanoparticles via Crystallization-driven Self-assembly of Biodegradable Block Copolymers. J. Vis. Exp. 2019, 20, e59772.
- Yu, W.; Foster, J. C.; Dove, A. P.; O’Reilly, R. K. Length Control of Biodegradable Fiber-Like Micelles via Tuning Solubility: A Self-Seeding Crystallization-Driven Self-Assembly of Poly(ε-caprolactone)-Containing Triblock Copolymers. Macromolecules 2020, 53, 1514–1521. [CrossRef]
- Pearce, A. K.; Wilks, T. R.; Arno, M. C.; O'Reilly, R. K. Synthesis and applications of anisotropic nanoparticles with precisely defined dimensions. Nat. Rev. Chem. 2021, 5, 21–45.
- Lefley, J.; Waldron, C.; Becer, C.R. Macromolecular design and preparation of polymersomes. Polym. Chem. 2020, 11, 7124–7136. [CrossRef]
- Palivan, C. G.; Goers, R.; Najer, A.; Zhang, X.; Car, A.; Meier, W. Bioinspired polymer vesicles and membranes for biological and medical applications. Chem. Soc. Rev. 2016, 45, 377–411. [CrossRef]
- Che, H.; van Hest, J. C. M. Stimuli-responsive polymersomes and nanoreactors. J. Mater. Chem. B 2016, 4, 4632–4647. [CrossRef]
- Hu, X.; Zhang, Y.; Xie, Z.; Jing, X.; Bellotti, A.; Gu, Z. Stimuli-Responsive Polymersomes for Biomedical Applications. Biomacromolecules 2017, 18, 649–673. [CrossRef]
- Zhu, Y. Q.; Yang, B.; Chen, S.; Du, J. Z. Polymer vesicles: Mechanism, preparation, application, and responsive behavior. Prog. Polym. Sci. 2017, 64, 1–22. [CrossRef]
- Leong, J.; Teo, J. Y.; Aakalu, V. K.; Yang, Y. Y.; Kong, H. Engineering Polymersomes for Diagnostics and Therapy. Adv. Healthc. Mater. 2018, 7, e1701276.
- Klermund, L.; Castiglione, K. Polymersomes as nanoreactors for preparative biocatalytic applications: current challenges and future perspectives. Bioprocess Biosyst Eng. 2018, 41, 1233–1246. [CrossRef]
- Wang, F.; Xiao, J.; Chen, S.; Sun, H.; Yang, B.; Jiang, J.; Zhou, X.; Du, J. Polymer Vesicles: Modular Platforms for Cancer Theranostics. Adv. Mater. 2018, 30, e1705674. [CrossRef] [PubMed]
- Yorulmaz Avsar, S.; Kyropoulou, M.; Di Leone, S.; Schoenenberger, C.-A.; Meier, W. P.; Palivan, C. G. Biomolecules turn self-assembling amphiphilic block co-polymer platforms into biomimetic interfaces. Front. Chem. 2019, 6, 645. [CrossRef]
- Iqbal, S.; Blenner, M.; Alexander-Bryant, A.; Larsen, J. Polymersomes for Therapeutic Delivery of Protein and Nucleic Acid Macromolecules: From Design to Therapeutic Applications. Biomacromolecules 2020, 21, 1327–1350. [CrossRef]
- Liu, D.; Sun, H.; Xiao, Y.; Chen, S.; Cornel, E. J.; Zhu, Y.; Du, J. Design principles, synthesis and biomedical applications of polymer vesicles with inhomogeneous membranes. J. Control. Release 2020, 326, 365–386. [CrossRef] [PubMed]
- Moreno, S.; Voit, B.; Gaitzsch, J. The chemistry of cross-linked polymeric vesicles and their functionalisation towards biocatalytic nanoreactors. Colloid Polym. Sci. 2021, 299, 309–324. [CrossRef]
- 759. Liarou, E.; Varlas, S.; Skoulas, D.; Tsimblouli, C.; Sereti, E.; Dimas, K.; Iatrou, H. Smart polymersomes and hydrogels from polypeptide-based polymer systems through α-amino acid N-carboxyanhydride ring-opening polymerization. From chemistry to biomedical applications. Prog. Polym. Sci. 2018, 83, 28–78.
- De Oliveira, H.; Thevenot, J.; Lecommandoux, S. Smart polymersomes for therapy and diagnosis: fast progress toward multifunctional biomimetic nanomedicines. WIREs Nanomed. Nanobiotechnol. 2012, 4, 525–546. [CrossRef] [PubMed]
- Konishcheva E. Daubian D. Gaitzsch J. Meier W. Synthesis of Linear ABC Triblock Copolymers and Their Self-Assembly in Solution. Helv. Chim. Acta. 2018, 101, e1700287. [CrossRef]
- Brendel, J. C.; Schacher, F. H. Block Copolymer Self-Assemblyin Solution-Quo Vadis? Chem. - Asian J. 2018, 13, 230–239. [CrossRef]
- Lunn, D. J.; Finnegan, J. R.; Manners, I. Self-assembly of “patchy” nanoparticles: a versatile approach to functional hierarchical materials. Chem Sci. 2015, 6, 3663–3673. [CrossRef]
- Huang J.; Guo, Y.; Gu, S.; Han, G.; Duan, W.; Gao, C.; Zhang, W. Multicompartment block copolymer nanoparticles: recent advances and future perspectives. Polym. Chem. 2019, 10, 3426–3435. [CrossRef]
- Nghiem, T.-L.; Steinhaus, A., Chakroun, R., Tjaberings, S., Qiang, X., Chen, C., Tjaberings, A., Quintieri, G., Gröschel, A. H. In Comprehensive Nanoscience and Nanotechnology, Eds.; Amsterdam: Elsevier, 2019; Vol. 1-5, p 217. [Google Scholar]
- Hils, C.; Manners, I.; Schöbel, J.; Schmalz, H. Patchy Micelles with a Crystalline Core: Self-Assembly Concepts, Properties, and Applications. Polymers 2021, 13, 1481. [CrossRef]
- Nayanathara, U.; Kermaniyan, S. S.; Such, G. K. Multicompartment Polymeric Nanocarriers for Biomedical Applications. Macromol. Rapid Commun. 2020, 41, e2000298. [CrossRef]
- Lu, Y.; Lin, J.; Wang, L.; Zhang, L.; Cai, C. Self-Assembly of Copolymer Micelles: Higher-Level Assembly for Constructing Hierarchical Structure. Chem. Rev. 2020, 120, 4111–4140. [CrossRef]
- Gröschel, A. H.; Schacher, F. H.; Schmalz, H.; Borisov, O. V.; Zhulina, E. B.; Walther, A.; Müller, A. H. Precise hierarchical self-assembly of multicompartment micelles. Nat. Commun. 2012, 3, 710/1-10.
- Gröschel, A. H.; Walther, A.; Löbling, T. I.; Schacher, F. H.; Schmalz, H.; Müller, A. H. Guided hierarchical co-assembly of soft patchy nanoparticles. Nature 2013, 503, 247–251. [CrossRef] [PubMed]
- Löbling, T. I.; Borisov, O. V.; Haataja, J. S.; Ikkala, O.; Gröschel, A. H.; Müller, A. H. E. Rational design of ABC triblock terpolymer solution nanostructures with controlled patch morphology. Nat Commun 2016, 7, 12097. [CrossRef] [PubMed]
- Löbling, T. I.; Ikkala, O.; Gröschel, A. H.; Müller, A. H. E. Controlling Multicompartment Morphologies Using Solvent Conditions and Chemical Modification. ACS Macro Lett. 2016, 5, 1044–1048. [CrossRef] [PubMed]
- Nghiem, T.-L.; Löbling, T. I.; Gröschel. A. H. Supracolloidal chains of patchy micelles in water. Polym. Chem. 2018, 9, 1583–1592. [CrossRef]
- Moreno, N.; Sutisna B.; Fried, E. Entropic factors and structural motifs of triblock-terpolymer-based patchy nanoparticles. Nanoscale 2020, 12, 22059–22069. [CrossRef]
- Moughton, A. O.; Hillmyer, M. A.; Lodge. T. P. Multicompartment Block Polymer Micelles. Macromolecules 2012, 45, 2–19. [CrossRef]
- Moughton, A. O.; Sagawa, T.; Yin, L.; Lodge, T. P.; Hillmyer, M. A. Multicompartment Micelles by Aqueous Self-Assembly of μ-A(BC) nMiktobrush Terpolymers. ACS Omega 2016, 1, 1027–1033. [CrossRef]
- Hanisch A.; Gröschel A. H.; Förtsch M.; Drechsler M.; Jinnai H.; Ruhland T. M.; Schacher F. H.; Müller A. H. E. Counterion-Mediated Hierarchical Self-Assembly of an ABC Miktoarm Star Terpolymer. ACS Nano 2013, 7, 4030–4041. [CrossRef]
- Xu, F.; Wu, D.; Huang, Y.; Wei, H.; Gao, Y.; Feng, X.; Yan, D.; Mai, Y. Multi-Dimensional Self-Assembly of a Dual-Responsive ABC Miktoarm Star Terpolymer. ACS Macro Lett. 2017, 6, 426–430. [CrossRef] [PubMed]
- Huo, H.; Liu, J.; Kannan, S.; Chen, L.; Zhao, Y.; Zhang, L.; Chang, G.; Zhang, Q.; Liu, F. Multicompartment nanoparticles bearing hydrophilic/hydrophobic subdomains by self-assembly of star polymers in aqueous solution. Macromolecules 2020, 54, 35–43.
- Liu, R.; Rong, Z.; Han, G.; Yang, X.; Zhang, W. Synthesis and self-assembly of star multiple block copolymer of poly (4-vinylpyridine)-block-polystyrene. Polymer 2021, 215, 123431. [CrossRef]
- Gädt, T.; Ieong, N. S.; Cambridge, G.; Winnik, M. A.; Manners, I. Complex and hierarchical micelle architectures from diblock copolymers using living, crystallization-driven polymerizations. Nat. Mater. 2009, 8, 144–150. [CrossRef]
- Schmelz, J.; Schmalz, H.. Corona structure on demand: Tailor-made surface compartmentalization in worm-like micelles via random cocrystallization. Polymer 2012, 53, 4333–4337. [CrossRef]
- Hudson, Z. M.; Boott, C. E.; Robinson, M. E.; Rupar, P. A.; Winnik, M. A.; Manners, I. Tailored hierarchical micelle architectures using living crystallization-driven self-assembly in two dimensions. Nat. Chem. 2014, 6, 893–898. [CrossRef] [PubMed]
- Qiu, H.; Cambridge, G.; Winnik, M. A.; Manners I. Multi-armed micelles and block co-micelles via crystallization-driven self-assembly with homopolymer nanocrystals as initiators. J. Am. Chem. Soc. 2013, 135, 12180–12183. [CrossRef] [PubMed]
- Li, X.; Gao, Y.; Boott, C. E.; Winnik, M. A.; Manners, I. Non-covalent synthesis of supermicelles with complex architectures using spatially confined hydrogen-bonding interactions. Nat. Commun. 2015, 6, 8127. [CrossRef]
- Gegenhuber, T.; Gröschel, A. H.; Löbling, T. I.; Drechsler, M.; Ehlert, S.; Förster, S.; Schmalz, H. Noncovalent Grafting of Carbon Nanotubes with Triblock Terpolymers: Toward Patchy 1D Hybrids. Macromolecules 2015, 48, 1767–1776. [CrossRef]
- Schöbel, J.; Hils, C.; Weckwerth, A.; Schlenk, M.; Bojer, C.; Stuart, M. C. A.; Breu, J.; Förster, S.; Greiner, A.; Karg, M.; Schmalz, H. Strategies for the selective loading of patchy worm-like micelles with functional nanoparticles. Nanoscale 2018, 10, 18257–18268. [CrossRef]
- Oliver, A. M.; Gwyther, J.; Winnik, M. A.; Manners, I. Cylindrical Micelles with “patchy” Coronas from the Crystallization-Driven Self-Assembly of ABC Triblock Terpolymers with a Crystallizable Central Polyferrocenyldimethylsilane Segment. Macromolecules 2018, 51, 222–231. [CrossRef]
- Oliver, A. M.; Spontak, R. J.; Manners, I. Solution self-assembly of ABC triblock terpolymers with a central crystallizable poly(ferrocenyldimethylsilane) core-forming segment. Polymer Chemistry 2019, 10, 2559–2569. [CrossRef]
- Finnegan, J. R.; Lunn, D. J.; Gould, O. E.; Hudson, Z. M.; Whittell, G. R.; Winnik, M. A.; Manners I. Gradient crystallization-driven self-assembly: cylindrical micelles with “patchy” segmented coronas via the coassembly of linear and brush block copolymers. J. Am. Chem. Soc. 2014, 136, 13835–13844.
- Cruz, M.; Xu, J.; Yu, Q.; Guerin, G.; Manners, I.; Winnik, M. A. Visualizing Nanoscale Coronal Segregation In Rod-Like Micelles Formed By Co-Assembly Of Binary Block Copolymer Blends. Macromol. Rapid Commun. 2018, 39, 1800397. [CrossRef] [PubMed]
- Xu, J.; Zhou, H.; Yu, Q.; Manners, I.; Winnik, M. A. Competitive Self-Assembly Kinetics as a Route to Controlling the Morphology of Core-Crystalline Cylindrical Micelles. J. Am. Chem. Soc. 2018, 140, 2619–2638. [CrossRef]
- Atanase, L. I.; Riess, G. Self-Assembly of Block and Graft Copolymers in Organic Solvents: An Overview of Recent Advances. Polymers (Basel) 2018, 10, 62. [CrossRef]
- Daubian, D.; Gaitzsch, J.; Meier, W. Synthesis and complex self-assembly of amphiphilic block copolymers with a branched hydrophobic poly(2-oxazoline) into multicompartment micelles, pseudo-vesicles and yolk/shell nanoparticles. Polym. Chem. 2020, 11, 1237–1248. [CrossRef]
- Wang, W.-L.; Jin, R.-H. Synthesis and self-assembly of amphiphilic comb-copolymers possessing polyethyleneimine and its derivatives: Site-selective formation of loop-cluster covered vesicles and flower micelles. Polymer 2021, 212, 123289. [CrossRef]
- Qiang, X.; Chakroun, R.; Janoszka, N.; Gröschel, A. H. Self-assembly of Multiblock Copolymers. Isr. J. Chem. 2019, 59, 945–958. [CrossRef]
- Haque, F. M.; Grayson, S. M. The synthesis, properties and potential applications of cyclic polymers. Nat Chem. 2020, 12, 433–444. [CrossRef] [PubMed]
- Varlas, S.; Lawrenson, S. B.; Arkinstall, L. A.; O’Reilly, R. K.; Foster, J. C. Self-assembled nanostructures from amphiphilic block copolymers prepared via ring-opening metathesis polymerization (ROMP). Prog. Polym. Sci. 2020, 107, 101278. [CrossRef]
- Nam, J.; Kim, Y. J.; Kim, J. G.; Seo, M. Self-Assembly of Monolayer Vesicles via Backbone-Shiftable Synthesis of Janus Core-Shell Bottlebrush Polymer. Macromolecules 2019, 52, 9484–9494. [CrossRef]
- 800. Shao, F.; Wang, Y.; Tonge, C. M.; Sauvé, E. R.; Hudson, Z. M. Self-assembly of luminescent triblock bottlebrush copolymers in solution Polym. Chem. 2020, 11, 1062–1071.
- Ma, H.; Kim, K.T.. Self-assembly of bottlebrush block copolymers into triply periodic nanostructures in a dilute solution. Macromolecules 2020, 53, 711–718. [CrossRef]
- Ahmed, E.; Tyler Womble, C.; & Weck, M. Synthesis and aqueous self-assembly of ABCD bottlebrush block copolymers. Macromolecules 2020, 53, 9018–9025.
- Kim, S.; Lee, S.; Choi, S.-H.; Char, K. Chain Exchange Kinetics of Bottlebrush Block Copolymer Micelles. Macromolecules 2021, 54, 4739–4746. [CrossRef]
- Kepola, E. J.; Loizou, E.; Patrickios, C. S.; Leontidis, E.; Voutouri, C.; Stylianopoulos, T.; Schweins, R.; Gradzielski, M.; Krumm, C.; Tiller, J. C.; Kushnir, M.; Wesdemiotis, C. Amphiphilic Polymer Conetworks Based on End-linked “Core-first” Star Block Copolymers: Structure Formation with Long-range Order. ACS Macro Lett. 2015, 4, 1163–1168.
- Varnava, C. K.; Patrickios, C. S. Model Amphiphilic Polymer Conetworks in Water: Prediction of Their Ability for Oil Solubilization. ACS Omega 2019, 4, 4721–4738. [CrossRef]
- Gröschel, A. H.; Walther, A. Block copolymer micelles with inverted morphologies. Angew. Chem. Int. Ed. 2017, 56, 10992–10994. [CrossRef]
- Ha, S.; La, Y.; Kim, K. T. Polymer Cubosomes: Infinite Cubic Mazes and Possibilities. Acc. Chem. Res. 2020, 53, 620–631. [CrossRef]
- Kim, J.; Wang, V.; Kim, K. T. Block Copolymers Composed of Main-Chain Cyclic Polymers: Morphology Transition and Covalent Stabilization of Self-Assembled Nanostructures via Intra- and Interchain Cyclization of Styrene-co-isoprene Blocks. Macromolecules 2020, 53, 10725–10733. [CrossRef]
- Pei, Y.; Lowe, A. B.; Roth, P. J. Stimulus-Responsive Nanoparticles and Associated (Reversible) Polymorphism via Polymerization Induced Self-assembly (PISA). Macromol. Rapid Commun. 2017, 38, 1600528. [CrossRef]
- Penfold, N. J. W.; Yeow, J.; Boyer, C.; Armes, S. P. Emerging Trends in Polymerization-Induced Self-Assembly. ACS Macro Lett. 2019, 8, 1029–1054. [CrossRef]
- Canning, S. L.; Smith, G. N.; Armes, S. P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. [CrossRef] [PubMed]
- D’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed. Engl. 2020, 59, 8368–8392. [CrossRef]
- Varlas, S.; Foster, J. C.; O'Reilly, R. K. Ring-opening metathesis polymerization-induced self-assembly (ROMPISA). Chem. Commun. 2019, 55, 9066–9071. [CrossRef] [PubMed]
- Liu, C.; Hong, C. Y.; Pan, C. Y. Polymerization techniques in polymerization-induced self-assembly (PISA). Polym. Chem. 2020, 11, 3673–3689. [CrossRef]
- Boott, C. E.; Gwyther, J.; Harniman, R. L.; Hayward, D. W.; Manners, I. Scalable and uniform 1D nanoparticles by synchronous polymerization, crystallization and self-assembly. Nat. Chem. 2017, 9, 785–792. [CrossRef]
- Oliver, A. M.; Gwyther, J.; Boott, C. E.; Davis, S.; Pearce, S.; Manners, I. Scalable Fiber-like Micelles and Block Co-micelles by Polymerization-Induced Crystallization-Driven Self-Assembly. J. Am. Chem. Soc. 2018, 140, 18104–18114. [CrossRef]
- Sha, Y.; Rahman, M. A.; Zhu, T.; Cha, Y.; McAlister, C. W.; Tang, C. ROMPI-CDSA: ring-opening metathesis polymerization-induced crystallization-driven self-assembly of metallo-block copolymers. Chem. Sci. 2019, 10, 9782–9787. [CrossRef]
- Hurst P. J.; Rakowski A. M.; Patterson J. P. Ring-Opening Polymerization-Induced Crystallization-Driven Self-Assembly of Poly-L-Lactide-Block-Polyethylene Glycol Block Copolymers (ROPI-CDSA. Nat. Commun. 2020, 11, 4690. [CrossRef]
- Yeow, J.; Boyer, C. Photoinitiated Polymerization-Induced Self-Assembly (Photo-PISA): New Insights and Opportunities. Adv. Sci. (Weinh) 2017, 4, 1700137. [CrossRef]
- Zhang, W.-J.; Hong, C.-Y.; Pan, C.-Y. Polymerization-Induced Self-Assembly of Functionalized Block Copolymer Nanoparticles and Their Application in Drug Delivery. Macromol. Rapid Commun. 2019, 40, 1800279. [CrossRef] [PubMed]
- Pearce S.; Perez-Mercader J. PISA: Construction of Self-Organized and Self-Assembled Functional Vesicular Structures. Polym. Chem. 2021, 12, 29–49. [CrossRef]
- 822. Phan, H.; Taresco, V.; Penelle, J.; Couturaud, B. Polymerisation-induced self-assembly (PISA) as a straightforward formulation strategy for stimuli-responsive drug delivery systems and biomaterials: Recent advances. Biomater. Sci. 2021, 9, 38–50.
- Zhang, W. J.; Kadirkhanov, J.; Wang, C. H.; Ding, S. G.; Hong, C. Y.; Wang, F.; You, Y. Z. Polymerization-induced self-assembly for the fabrication of polymeric nano-objects with enhanced structural stability by cross-linking. Polym. Chem. 2020, 11, 3654–3672. [CrossRef]
- El Jundi, A.; Buwalda, S. J.; Bakkour, Y.; Garric, X.; Nottelet, B. Double hydrophilic block copolymers self-assemblies in biomedical applications. Adv. Colloid Interface Sci. 2020, 283, 102213. [CrossRef]
- Shah, S.; Eyler, A., Tabandeh, S., Leon, L. Electrostatically driven self-assembled nanoparticles and coatings. In Nanoparticles for Biomedical Applications, Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 349–370. [Google Scholar]
- Ku, K. H. Responsive Nanostructured Polymer Particles. Polymers 2021, 13, 273. [CrossRef]
- Dai, X.; Qiang, X.; Hils, C.; Schmalz, H.; Gröschel, A. H. Frustrated Microparticle Morphologies of a Semicrystalline Triblock Terpolymer in 3D Soft Confinement. ACS Nano 2021, 15, 1111–1120. [CrossRef]
- Qiang, X.; Steinhaus, A.; Chen, C.; Chakroun, R.; Gröschel, A. H. Angew. Chemie Int. Ed. 2019, 58, 7122–7126.
- Steinhaus, A.; Chakroun, R.; Müllner, M.; Nghiem, T. L.; Hildebrandt, M.; Gröschel, A. H. Confinement Assembly of ABC Triblock Terpolymers for the High-Yield Synthesis of Janus Nanorings. ACS Nano 2019, 13, 6269–6278. [CrossRef]
- Tamate, R.; Hashimoto, K.; Ueki, T.; Watanabe, M. Block copolymer self-assembly in ionic liquids. Phys. Chem. Chem. Phys. 2018, 20, 25123–25139. [CrossRef]
- Ghorbanizamani, F.; Moulahoum, H.; Zihnioglu, F.; Timur, S. Self-assembled block copolymers in ionic liquids: Recent advances and practical applications. J. Mol. Liq. 2021, 323, 115076. [CrossRef]
- He, Y.; Li, Z.; Simone, P.; Lodge, T. P. Self-assembly of block copolymer micelles in an ionic liquid. J. Am. Chem. Soc. 2006, 128, 2745–2750. [CrossRef]
- Zhao, D.; Ma, Y.; Lodge, T. P. Exchange Kinetics for a Single Block Copolymer in Micelles of Two Different Sizes. Macromolecules 2018, 51, 2312–2320. [CrossRef]
- Bhushan, V.; Heitz, M. P.; Baker, G. A.; Pandey, S. Ionic Liquid-Controlled Shape Transformation of Spherical to Nonspherical Polymersomes via Hierarchical Self-Assembly of a Diblock Copolymer. Langmuir 2021, 37, 5081–5088. [CrossRef] [PubMed]
- He, Z.; Ma, Y.; Alexandridis, P. Comparison of ionic liquid and salt effects on the thermodynamics of amphiphile micellization in water. Colloids Surf. A Physicochem. Eng. Asp. 2018, 559, 159–168. [CrossRef]
- Rideau, E.; Dimova, R.; Schwille, P.; Wurm F. R.; Landfester, K. Liposomes and polymersomes: A comparative review towards cell mimicking. Chem. Soc. Rev. 2018, 47, 8572–8610. [CrossRef]
- Kita-Tokarczyk, K.; Grumelard, J.; Haefele, T.; Meier, W. Block copolymer vesicles-using concepts from polymer chemistry to mimic biomembranes. Polymer 2005, 46, 3540–3563. [CrossRef]
- Chen, Q.; Zhao, H.; Ming, T.; Wang, J.; Wu, C. Nanopore extrusion-induced transition from spherical to cylindrical block copolymer micelles. J. Am. Chem. Soc. 2009, 131, 16650–16651. [CrossRef]
- Caire da Silva, L.; Cao, S.; Landfester, K. Bursting and Reassembly of Giant Double Emulsion Drops Form Polymer Vesicles. ACS Macro Lett. 2021, 10, 401–405. [CrossRef] [PubMed]
- Chariou, P. L.; Ortega-Rivera, O. A.; Steinmetz, N. F. Nanocarriers for the Delivery of Medical, Veterinary, and Agricultural Active Ingredients. ACS Nano 2020, 14, 2678–2701. [CrossRef] [PubMed]
- Rameez, S.; Alosta, H.; Palmer, A. F. Biocompatible and biodegradable polymersome encapsulated hemoglobin: a potential oxygen carrier. Bioconjug. Chem. 2008, 19, 1025–1032. [CrossRef] [PubMed]
- Kishimura, A.; Koide, A.; Osada, K.; Yamasaki, Y.; Kataoka, K. Encapsulation of myoglobin in PEGylated polyion complex vesicles made from a pair of oppositely charged block ionomers: a physiologically available oxygen carrier. Angew. Chem. Int. Ed. Engl. 2007, 46, 6085–6088. [CrossRef]
- Forster, S.; Antonietti, M. Amphiphilic block copolymers in structure- controlled nanomaterials hybrids. Adv. Mater. 1998, 10, 195–217. [CrossRef]
- Hadjichristidis, N.; Pispas, S., Ed.; Floudas, G. Block copolymers: synthetic strategies, physical properties, and applications. John Wiley & Sons, Inc.: Hoboken, New Jersey, 2003. [Google Scholar]
- Gohy, J. F.; Antoun, S.; Jerome, R. pH-dependent micellization of poly(2- vinylpyridine)-block-((dimethylamino)ethyl methacrylate) diblock copolymers. Macromolecules 2001, 34, 7435–7440. [CrossRef]
- Antoun, S.; Gohy, J. F.; Jerome, R. Micellization of quaternized poly(2-(dimethylamino)ethyl methacrylate)-block-poly(methyl methacrylate) copolymers in water. Polymer 2001, 42, 3641–3648. [CrossRef]
- Wang, X.; Winnik, A.; Manners, I. Synthesis and self assembly of poly(ferrocenyldimethylsilane-b-dimethylaminoethyl methacrylate): toward water-soluble cylinders with an organometallic core. Macromolecules 2005, 38, 1928–1935. [CrossRef]
- Valint Jr, P. L.; Bock, J. Synthesis and characterization of hydrophobically associating block copolymers. Macromolecules 1988, 21, 175–179. [CrossRef]
- Guenoun, P.; Davis, H. T.; Tirrell, M.; Mays, J. W. Aqueous micellar solutions of hydrophobically modified polyelectrolytes. Macromolecules 1996, 29, 3965–3969. [CrossRef]
- Yang, J. C.; Jablonsky, M. J.; Mays, J. W. NMR and FT-IR studies of sulfonated styrene-based homopolymers and copolymers. Polymer 2002, 43, 5125–5132. [CrossRef]
- Muller, F.; Delsanti, M.; Auvray, L.; Yang, J.; Chen, Y. J.; Mays, J. W.; Demé, Tirrell, M.; Guenoun, P. Ordering of urchin-like charged copolymer micelles: electrostatic, packing and polyelectrolyte correlations. Eur. Phys. J. E 2000, 3, 45–53. [CrossRef]
- Muller, F.; Fontaine, P.; Delsanti, M.; Belloni, L.; Yang, J.; Chen, Y. J.; Mays, J. W.; Lesieur, P.; Tirrell, M.; Guenoun, P. Counterion distribution in a spherical charged sparse brush. Eur. Phys. J. E 2001, 6, 109–115.
- Muller, F.; Guenoun, P.; Delsanti, M.; Demé, B.; Auvray, L.; Yang, J.; Mays, J. W. Spherical polyelectrolyte block copolymer micelles: structural change in presence of monovalent salt. Eur. Phys. J. E Soft Matter 2004, 15, 465–472. [CrossRef]
- Szczubialka, K.; Ishikawa, K.; Morishima, Y. Micelle formationof diblock copolymers of styrene and sulfonated isoprene inaqueous solution. Langmuir 1999, 15, 454–462. [CrossRef]
- Tauer, K.; Zimmermann, A.; Schlaad, H. New reactive block copolymers as stabilizers in emulsion polymerization. Macromol. Chem. Phys. 2002, 203, 319–327. [CrossRef]
- Justynska, J.; Hordyjewucz, Z.; Schlaad, H. Toward a toolbox of functional block copolymers via free-radical addition of mercaptans. Polymer 2005, 46, 12057–12064. [CrossRef]
- Geng, Y.; Discher, D.; Justynska, J.; Schladd, H. Grafting short peptides onto polybutadiene-blockpoly(ethylene oxide): a platform for self- assembling hybrid amphiphiles. Angew. Chem. Int. Ed. 2006, 118, 7578–7581.
- Uchman, M.; Procházka, K.; Stepánek, M.; Mountrichas, G.; Pispas, S.; Spírková, M.; Walther, A. pH-dependent self-assembly of polystyrene-block-poly((sulfamate-carboxylate)isoprene) copolymer in aqueous media. Langmuir 2008, 24, 12017–12025. [CrossRef]
- Pispas, S.; Siakali-Kioulafa, E.; Hadjichristidis, N.; Mavromoustakos, T. Block copolymers with crystalline/amorphous, crystalline/ polyelectrolyte and amorphous/polyelectrolyte blocks. Macromol. Chem. Phys. 2002, 203, 1317–1327. [CrossRef]
- Prochazka, K.; Kiserow, D.; Ramireddy, C.; Tuzar, Z.; Munk, P.; Webber, S. E. Time-resolved fluorescence studies of the chain dynamics of naphthalene-labeled polystyrene-blocks-poly(methacrylic acid) micelles in aqueous media. Macromolecules 1992, 25, 454–460. [CrossRef]
- André X.; Zhang M.; Müller A. H. E. Thermo- and pH-Responsive Micelles of Poly(acrylic acid)-block-Poly(N,N-diethylacrylamide). Macromol. Rapid Commun. 2005, 26, 558–563. [CrossRef]
- Perrin, R.; Elomaa, M.; Jannasch, P. Nanostructured proton conducting polystyrene-poly(vinylphosphonic acid)block copolymers prepared via sequential anionic polymerizations. Macromolecules 2009, 42, 5146–5154. [CrossRef]
- Kuo, S. W.; Tung, P.; Chang, F. Syntheses and the study of strongly hydrogen-bonded poly(vinyphenol-b-vinylpyridine)diblock copolymer through anionic polymerization. Macromolecules 2006, 39, 9388–9395. [CrossRef]
- Kaditi, E.; Pispas, S. β-Lactam functionalized poly(isoprene-b- ethylene oxide) amphiphilic block copolymers. J. Polym. Sci. Part A: Polym. Chem. 2010, 48, 24–33. [CrossRef]
- Hrubý, M.; Koňák, Č.; Ulbrich, K. Poly(allyl glycidyl ether)-Block-Poly(ethylene oxide): A Novel Promising Polymeric Intermediate for the Preparation of Micellar Drug Delivery Systems. J. Appl. Polym. Sci. 2005, 95, 201–211. [CrossRef]
- Mahltig, B.; Gohy, J. F.; Antoun, S.; Jerome, R.; Stamm, M. Adsorption and structure formation of the weak polyelectrolytic diblock copolymer, PVP-b-PDMAEMA. Colloid Polym. Sci. 2002, 280, 495–502. [CrossRef]
- Fragouli, P. G.; Iatrou, H.; Hadjichristidis, N. Synthesis and characterization of linear diblock and triblock copolymers of 2-vinyl pyridine and ethylene oxide. Polymer 2002, 43, 7141–7144. [CrossRef]
- Pispas, S. Double hydrophilic block copolymer of sodium(2- sulfamate-3-carboxylate)isoprene and ethylene oxide. J. Polym. Sci Part A: Polym. Chem. 2006, 44, 606–613. [CrossRef]
- Mountrichas, G.; Pispas, S. Synthesis and pH responsive self-assembly of new double hydrophilic block copolymers. Macromolecules 2006, 39, 4767–4774. [CrossRef]
- Mountrichas, G.; Pispas, S. Novel double hydrophilic block copolymers based on poly(p-hydroxystyrene) derivatives and poly(ethylene oxide). J. Polym. Sci. Part A: Polym. Chem. 2007, 45, 5790–5799. [CrossRef]
- Kaluzynski, K., Pretula, J., Lapienis, G., Basko, M., Bartczak, Z., Dworak, A., Penczek, S. Dihydrophilic block copolymers with ionic and nonionic blocks. I. poly(ethylene oxide)-b-polyglycidol with OP(O)(OH)2, COOH, OR SO3H functions: synthesis and influence for CaCO3 crystallization. J. Polym. Sci. Part A: Polym. Chem. 2001, 39, 955–963.
- Kaditi, E.; Mountrichas, G.; Pispas, S. Amphiphilic block copolymers by a combination of anionic polymerization and selective post-polymerization functionalization. Eur. Polym. J. 2011, 47, 415–434. [CrossRef]
- 873. Alexander, S. Adsorption of chain molecules with a polar head - a scaling description. J. Phys. (Paris) 1977, 38, 38–983.
- Daoud, M.; Cotton. J. P. Star shaped polymers : a model for the conformation and its concentration dependence. J. Phys. (Paris) 1982, 43, 531–538. [CrossRef]
- Birshtein, T. M.; Zhulina, E. B. Conformations of star-branched macromolecules. Polymer 1984, 25, 1453–1461. [CrossRef]
- Willner, L.; Poppe, A.; Allgaier, J.; Monkenbusch, M.; Lindner, P.; Richter, D. Micellization of amphiphilic diblock copolymers: Corona shape and mean-field to scaling crossover. Europhys. Lett. 2000, 51, 628–634. [CrossRef]
- Jensen, G. V.; Shi, Q.; Hernansanz, M. J.; Oliveira, C. L. P.; Deen, G. R.; Almdal, K.; Pederson, J. S. Structure of PEP-PEO block copolymer micelles: Exploiting the complementarity of small-angle X-ray scattering and static light scattering. J. Appl. Crystallogr. 2011, 44, 473–482. [CrossRef]
- Förster, S.; Zisenis, M.; Wenz, E.; Antonietti, M. Micellization of strongly segregated block copolymers. J. Chem. Phys. 1996, 104, 9956–9970. [CrossRef]
- Zhulina, E. B.; Birshtein, T. M.; Borisov, O. V. Curved polymer and polyelectrolyte brushes beyond the Daoud-Cotton model. Eur. Phys. J. E Soft Matter 2006, 20, 243–256. [CrossRef]
- Birshtein, T.; Lyatskaya, Yu. Theory of the Collapse-Stretching Transition of a Polymer Brush in a Mixed Solvent. Macromolecules 1994, 27, 1256–1266. [CrossRef]
- Lai, P.-Y.; Halperin, A. Polymer brushes in mixed solvents: chromatography and collapse. Macromolecules 1992, 25, 6693–6695. [CrossRef]
- Birshtein, T. M.; Zhulina, E. B.; Merkurieva, A. A. Amphiphilic polymer brush in a mixture of incompatible liquids. Macromol. Theory Simul. 2000, 9, 47–55. [CrossRef]
- Merkurieva, A. A.; Leermakers, F. A. M.; Birshtein, T. M.; Fleer, G. L.; Zhulina, E. B. Amphiphilic Polymer Brush in a Mixture of Incompatible Liquids. Numerical Self-Consistent-Field Calculations. Macromolecules 2000, 33, 1072–1081. [CrossRef]
- Nagarajan, R.; Ganesh, K. Block copolymer self-assembly in selective solvents: theory of solubilization in spherical micelles. Macromolecules 1989, 22, 4312–4325. [CrossRef]
- Cogan, K. A.; Leermakers, F. A. M.; Gast, A. P. Predictions of copolymer micelle behavior in immiscible solvents. Langmuir 1992, 8, 429–436. [CrossRef]
- MacEwan, S. R.; Chilkoti, A. Elastin-like polypeptides: biomedical applications of tunable biopolymers. Biopolymers 2010, 94, 60–77. [CrossRef]
- Dreher, M. R.; Simnick, A. J.; Fischer, K.; Smith, R. J.; Patel, A.; Schmidt, M.; Chilkoti, A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. J. Am. Chem. Soc. 2008, 130, 687–694. [CrossRef] [PubMed]
- Trent, A.; Marullo, R.; Lin, B.; Black, M.; Tirrell, M. Structural properties of soluble peptide amphiphile micelles. Soft Matter 2011, 7, 9572–9582. [CrossRef]
- 889. Kastantin, M.; Tirrell, M. Helix Formation in the Polymer Brush. Macromolecules 2011, 44, 4977–4987.
- Gervais, M.; Gallot, B. Phase diagram and structural study of polystyrene-poly (ethylene oxide) block copolymers, 1. Systems polystyrene/poly (ethylene oxide)/diethyl phthalate. Makromol. Chem. 1973, 171, 157–178. [CrossRef]
- Lin, E. K.; Gast, A. P. Semicrystalline Diblock Copolymer Platelets in Dilute Solution. Macromolecules 1996, 29, 4432–4441. [CrossRef]
- Yin, L.; Hillmyer, M. A. Disklike Micelles in Water from Polyethylene-Containing Diblock Copolymers. Macromolecules 2011, 44, 3021–3028. [CrossRef]
- Schmalz, H.; Schmelz, J.; Drechsler, M.; Yuan, J.; Walther, A.; Schweimer, K.; Mihut, A. M. Thermo-Reversible Formation of Wormlike Micelles with a Microphase-Separated Corona from a Semicrystalline Triblock Terpolymer. Macromolecules 2008, 41, 3235–3242. [CrossRef]
- Schmelz, J.; Karg, M.; Hellweg, T.; Schmalz, H. General pathway toward crystalline-core micelles with tunable morphology and corona segregation. ACS Nano 2011, 5, 9523–9534. [CrossRef]
- Yu-Su, S. Y.; Sheiko, S. S.; Lee, H.-I.; Jakubowski, W.; Nese, A.; Matyjaszewski, K.; Anokhin, D.; Ivanov, D. A. Crystallization of molecular brushes with block copolymer side chains. Macromolecules 2009, 42, 9008–9017.
- DiMarzio, E. A.; Guttman, C. M.; Hoffman, J. D. Calculation of lamellar thickness in a diblock copolymer, one of whose components is crystalline. Macromolecules 1980, 13, 1194–1198. [CrossRef]
- Birshtein, T. M.; Zhulina, E. B. Scaling theory of supermolecular structures in block copolymer-solvent systems: 2. Supercrystalline structures. Polymer 1990, 31, 1312–1320. [CrossRef]
- Zhang, L.; Eisenberg, A. Multiple Morphologies of “Crew-Cut” Aggregates of Polystyrene-b-poly(acrylic acid) Block Copolymers. Science 1995, 268, 1728–1731. [CrossRef]
- Zhang, L.; Eisenberg, A. Multiple morphologies and characteristics of “crew-cut” micelle-like aggregates of polystyrene-b-poly (acrylic acid) diblock copolymers in aqueous solutions. J. Am. Chem. Soc. 1996, 118, 3168–3181. [CrossRef]
- Zhang, L.; Barlow, R. J.; Eisenberg, A. Scaling relations and coronal dimensions in aqueous block polyelectrolyte micelles. Macromolecules 1995, 28, 6055–6066. [CrossRef]
- Manning, G. J. Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. J. Chem. Phys. 1969, 51, 924–933. [CrossRef]
- 902. Zhulina, E. B.; Borisov, O. V. Structure and interaction of weakly charged polyelectrolyte brushes: Self-consistent field theory. J. Chem. Phys. 1997, 107, 5952–5967.
- Zhulina, E. B.; Borisov, O. V. Poisson-Boltzmann theory of pH-sensitive (annealing) polyelectrolyte brush. Langmuir 2011, 27, 10615–10633. [CrossRef]
- 904. Tamashiro, M. N.; Hernández-Zapata, E.; Schorr, P. A.; Balastre, M.; Tirrell, M.; Pincus, P. Salt dependence of compression normal forces of quenched polyelectrolyte brushes, J. Chem. Phys. 2001, 115, 1960–1969.
- Lauw, Y.; Leermakers, F. A. M.; Cohen Stuart, M. A.; Borisov, O. V.; Zhulina, E. B. Coexistence of crew-cut and starlike spherical micelles composed of copolymers with an annealed polyelectrolyte block. Macromolecules 2006, 39, 3628–3641. [CrossRef]
- Marko, J. F.; Rabin, Y. Microphase separation of charged diblock copolymers: melts and solutions. Macromolecules 1992, 25, 1503–1509. [CrossRef]
- Wittmer, J.; Joanny, J.-F. Charged diblock copolymers at interfaces. Macromolecules 1993, 26, 2691–2697. [CrossRef]
- Pincus, P. Colloid stabilization with grafted polyelectrolytes. Macromolecules 1991, 24, 2912–2919. [CrossRef]
- 909. Borisov, O. V.; Birshtein, T. M.; Zhulina, E. B. Collapse of Grafted Polyelectrolyte Layer. J. Phys. II 1991, 1, 521–526.
- Borisov, O. V.; Zhulina, E. B.; Birshtein, T. M. Diagram of the states of a grafted polyelectrolyte layer. Macromolecules 1994, 27, 4795–4803. [CrossRef]
- Shusharina, N. P.; Nyrkova, I. A.; Khokhlov, A. R. Diblock copolymers with a charged block in a selective solvent: micellar structure. Macromolecules 1996, 29, 3167–3174. [CrossRef]
- Dan, N.; Tirrell, M. Self-assembly of block copolymers with a strongly charged and a hydrophobic block in a selective, polar solvent. Micelles and adsorbed layers. Macromolecules 1993, 26, 4310–4315. [CrossRef]
- Zhulina, E. B.; Borisov, O. V. Self-assembly in solution of block copolymers with annealing polyelectrolyte blocks. Macromolecules 2002, 35, 9191–9203. [CrossRef]
- Borisov, O. V.; Zhulina, E. B. Reentrant morphological transitions in copolymer micelles with pH-sensitive corona. Langmuir 2005, 21, 3229–3231. [CrossRef]
- Zhulina, E. B.; Borisov, O. V. Theory of morphological transitions in weakly dissociating diblock polyelectrolyte micelles. Macromolecules 2005, 38, 6726–6741. [CrossRef]
- Förster, S.; Hermsdorf, N.; Böttcher, C.; Lindner, P. Structure of polyelectrolyte block copolymer micelles. Macromolecules 2002, 35, 4096–4105. [CrossRef]
- Burkhardt, M.; Martinez-Castro, N.; Tea, S.; Drechsler, M.; Babin, I.; Grishagin, I.; Schweins, R.; Pergushov, D. V.; Gradzielski, M.; Zezin, A. B.; Müller, A. H. E. Polyisobutylene-Block-Poly(Methacrylic Acid) Diblock Copolymers: Self-Assembly in Aqueous Media. Langmuir 2007, 23, 12864–12874.
- Xu, L.; Zhu, Z.; Borisov, O. V.; Zhulina, E. B.; Sukhishvili, S. A. pH-Triggered Block Copolymer Micelle-to-Micelle Phase Transition. Phys. Rev. Lett. 2009, 103, 118301. [CrossRef]
- Borisov, O. V.; Zhulina, E. B. Morphology of micelles formed by diblock copolymer with a polyelectrolyte block. Macromolecules 2003, 36, 10029–10036. [CrossRef]
- Victorov, A. I.; Plotnikov, N. V.; Hong, P.-D. Molecular thermodynamic modeling of the morphology transitions in a solution of a diblock copolymer containing a weak polyelectrolyte chain. J. Phys. Chem. B 2010, 114, 8846–8860. [CrossRef] [PubMed]
- Netz, R. R. Micellar morphologies of charged diblock-copolymers. Europhys. Lett. 1999, 47, 391–397. [CrossRef]
- Venev, S. V.; Reineker, P.; Potemkin, I. I. Direct and inverse micelles of diblock copolymers with a polyelectrolyte block: Effect of equilibrium distribution of counterions. Macromolecules 2010, 43, 10735–10742. [CrossRef]
- Nyrkova, I.; Semenov, A. N. On the theory of aggregation and micellization: PEO–PVP copolymer in water. Faraday Discuss 2005, 128, 113–127. [CrossRef]
- Munk, P.; Ramieddi, C.; Tian, M.; Webber, S. E.; Prochazka, K.; Tuzar, Z. Block copolymer micelles in aqueous media. Macromol. Chem., Macromol. Symp. 1992, 58, 195–199. [CrossRef]
- Rager T.; Meyer W. H.; Wegner G. Micelle Formation of Poly(acrylic acid)-block-Poly(methyl methacrylate) Block Copolymers in Mixtures of Water with Organic Solvents. Macromol. Chem. Phys. 1999, 200, 1672–1680. [CrossRef]
- Deniau-Lejeune, E.; Drechsler, M.; Jestin, J.; Müller, A. H.; Chassenieux, C.; Colombani, O. Amphiphilic diblock copolymers with a moderately hydrophobic block: Toward dynamic micelles. Macromolecules 2010, 43, 2667–2671. [CrossRef]
- Charbonneau, C.; Chassenieux, C.; Colombani, O.; Nicolai, T. Controlling the Dynamics of Self-Assembled Triblock Copolymer Networks via the pH. Macromolecules 2011, 44, 4487–4495. [CrossRef]
- Borisova, O. V.; Billon, L.; Zaremski, M.; Grassl, B.; Bakaeva, Z.; Lapp, A.; Stepanek, P.; Borisov, O. V. pH-triggered reversible sol–gel transition in aqueous solutions of amphiphilic gradient copolymers. Soft Matter 2011, 7, 10824–10833. [CrossRef]
- Borisova, O.; Billon, L.; Zaremski, M.; Gassl, B.; Bakaeva, Z.; Lapp, A.; Stepanekc, P.; Borisov, O. Synthesis and pH-and salinity-controlled self-assembly of novel amphiphilic block-gradient copolymers of styrene and acrylic acid. Soft Matter 2012, 8, 7649–7659. [CrossRef]
- Carlsen, A.; Lecommandoux, S. Self-assembly of polypeptide-based block copolymer amphiphiles. Curr. Opin. Colloid Interface Sci. 2009, 14, 329–339. [CrossRef]
- Checot, F.; Rodriguez-Hernandez, J.; Gnanou, Y.; Lecommandoux, S. pH-responsive micelles and vesicles nanocapsules based on polypeptide diblock copolymers. Biomed. Eng. 2007, 24, 81–85.
- Checot, F.; Brulet, A.; Oberdisse, J.; Gnanou, Y.; Mondain- Monval, O.; Lecommandoux, S. Structure of polypeptide-based diblock copolymers in solution: Stimuli-responsive vesicles and micelles. Langmuir 2005, 21, 4308–4315. [CrossRef]
- Tirrell, M. Modular materials by self-assembly. AIChE J. 2005, 51, 2386–2390. [CrossRef]
- Teixeira, F. Jr.; Rigler, P.; Vebert-Nardin, C. Nucleo-copolymers: oligonucleotide-based amphiphilic diblock copolymers. Chem. Commun. 2007, 11, 1130–1132.
- Halperin, A.; Buhot, A.; Zhulina, E. B. On the hybridization isotherms of DNA microarrays: the Langmuir model and its extensions. J. Phys.: Condens. Matter 2006, 18, S463–S490.
- Konradi, R.; Rühe, J. Interaction of poly(methacrylic acid) brushes with metal ions: swelling properties. Macromolecules 2005, 38, 4345–4354. [CrossRef]
- 937. Li, F. Polyelectrolyte Brushes at Interfaces: Properties, Structure and Interactions. Ph.D Thesis, University of California, Santa Barbara, 2005.
- Birshtein, T. M.; Zhulina, E. B. The effect of polyvalent salt ions on the properties of annealed polyelectrolyte brushes. Ber. Bunsen-Ges. Phys. Chem. 1996, 100, 929–935. [CrossRef]
- Zhulina, E. B.; Borisov, O. V.; Birshtein, T. M. Polyelectrolyte Brush Interaction with Multivalent Ions. Macromolecules 1999, 32, 8189–8196. [CrossRef]
- Shusharina, N. A.; Rubinstein, M. Concentration Regimes in Solutions of Polyelectrolyte Stars. Macromolecules 2008, 41, 203–217. [CrossRef]
- Skandalis, A.; Pispas, S. PDMAEMA-b-PLMA-b-POEGMA Tri- block Terpolymers via RAFT Polymerization and Their Self- Assembly in Aqueous Solutions. Polym. Chem. 2017, 8, 4538–4547. [CrossRef]
- Jin, J.; Tian, J.; Lian, X.; Sun, P.; Zhao, H. Reactive Triblock Copolymer Micelles Induced by Click Reaction: A Platform for RAFT Polymerization. Soft Matter 2011, 7, 11194–11202. [CrossRef]
- Löbling, T. I.; Hiekkataipale, P.; Hanisch, A.; Bennet, F.; Schmalz, H.; Ikkala, O.; Gröschel, A.; Müller, A. H. Bulk morphologies of polystyrene-block-polybutadiene-block-poly(tert-butyl methacrylate) triblock terpolymers. Polymer 2015, 72, 479–489. [CrossRef]
- Barthel, M. J.; Mansfeld, U.; Hoeppener, S.; Czaplewska, J. A.; Schacher, F. H.; Schubert, U. S. Understanding and tuning the self-assembly of polyether-basedtriblock terpolymers in aqueous solution. Soft Matter 2013, 9, 3509–3520. [CrossRef]
- Kempe, K.; Baumgaertel, A.; Hoogenboom, R.; Schubert, U. S. Design of New Amphiphilic Triblock Copoly(2-oxazoline)s Containing a Fluorinated Segment. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5100–5108. [CrossRef]
- Wang, H.; Yao, Y.; Han, L.; Che, S. A. Amphiphilic ABC Triblock Terpolymer Templating for Mesoporous Silica. Chem. Res. Chin. Univ. 2014, 30, 863–867. [CrossRef]
- Petrova, S.; Venturini, C. G.; Jäger, A.; Jäger, E.; Černoch, P.; Kereïche, S.; Kovácik, L.; Raška, I.; Štěpánek, P. Novel Thermo- Responsive Double-Hydrophilic and Hydrophobic MPEO-b- PEtOx-b-PCL Triblock Terpolymers: Synthesis, Characterization and Self-Assembly Studies. Polymer 2015, 59, 215–225.
- Konishcheva, E. V.; Zhumaev, U. E.; Meier, W. P. PEO-b-PCL- b-PMOXA Triblock Copolymers: From Synthesis to Micro- scale Polymersomes with Asymmetric Membrane. Macromolecules 2017, 50, 1512–1520. [CrossRef]
- Stoenescu, R.; Meier W. Vesicles with Asymmetric Membranes from Amphiphilic ABC Triblock Copolymers. Chem Commun (Camb). 2002, (24), 3016–3017.
- Bian, J.; Hao, Y.; He, J.; Zhang, W.; Zhang, M.; Ni, P. Synthesis and Characterization of a Biodegradable ABC Triblock Terpolymer as Co-Delivery Carrier of Doxorubicin and DNA. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 3005–3016. [CrossRef]
- Matter, Y.; Enea, R.; Casse, O.; Lee, C. C.; Baryza, J.; Meier, W. Amphiphilic PEG-b-PMCL-b-PDMAEMA Triblock Copolymers : from Synthesis to Physico-Chemistry of Self-Assembled Structures. Macromol. Chem. Phys. 2011, 212, 937–949.
- He, X.; Liang, L.; Xie, M.; Zhang, Y.; Lin, S.; Yan, D. Synthesis of Novel Linear PEO-b-PS-b-PCL Triblock Copolymers by the Combination of ATRP, ROP, and a Click Reaction. Macromol. Chem. Phys. 2007, 208, 1797–1802. [CrossRef]
- Tsitsilianis, C.; Iliopoulos, I.; Ducouret, G. An associative polyelectrolyte end-capped with short polystyrene chains. Synthesis and reological behavior. Macromolecules 2000, 33, 2936–2943. [CrossRef]
- Tsitsilianis, C.; Iliopoulos, I. Viscoelastic properties of physical gels formed by associative telechelic polyelectrolytes in aqueous media. Macromolecules 2002, 35, 3662–3667. [CrossRef]
- Sfika, V.; Tsitsilianis, C. Association phenomena of poly(acrylic acid)- b-poly(2-vinylpyrine)-b-poly(acrylic acid) triblock polyampholyte in aqueous solution from transient network to compact micelles. Macromolecules 2003, 36, 4983–4988. [CrossRef]
- Sfika, V.; Tsitsilianis, C.; Bossard, F.; Yannopoulos, S.; Petekidis. G. A novel thermothickening phenomenon exhibited by a triblock polyampholyte in aqueous salt-free solutions. Macromolecules 2005, 38, 2883–2888. [CrossRef]
- Stavrouli, N.; Tsitsilianis, C.; Aubry, T. Rheological properties of ABA telechelic polyelectrolyte and ABA polyampholyte reversible hydrogels: a comparative study. Polymer 2008, 49, 1249–1256. [CrossRef]
- Karanikolas, A.; Tsolakis, P.; Bokias, G.; Tsitsilianis, C. Stimuli-responsive poly(ethylene oxide)-b-(2-vinylpyridine)-b-poly(ethylene oxide) triblock copolymers and complexation with poly(acrylic acid) at low pH. Eur. Phys. J. E 2008, 27, 335–343. [CrossRef]
- Angelopoulos, S.; Tsistilianis, C. Thermo-reversible hydrogels based on poly(N,N-diethylamine)-block-poly(acrylicacid)-block-poly(N,N- diethylamine double hydrophilic triblock copolymer. Macromol. Chem. Phys. 2006, 207, 2188–2194. [CrossRef]
- Uchman, M.; Stepanek, M.; Prochazka, K.; Mountrichas, G.; Pispas, S.; Voets, I. K.; Walther, A. Multicompartment Nanoparticles Formed by a Heparin-Mimicking Block Terpolymer in Aqueous Solutions. Macromolecules 2009, 42, 5605–5613. [CrossRef]
- Katsampas, I.; Roiter, Y.; Minko, S.; Tsitsilianis, C. Multifunctional stimuli responsive ABC terpolymers: From 3-compartment micelles to 3-dimentional Network. Macromol. Rapid Commun. 2005, 26, 1371–1376. [CrossRef]
- Koutalas, G.; Pispas, S.; Hadjichristidis, N. Micelles of poly(isoprene-b-2-vinylpyridine-b-ethylene oxide) terpolymers in aqueous media and their interaction with surfactants. Eur. Phys. J. E Soft Matter 2004, 15, 457–464. [CrossRef] [PubMed]
- Park, J.; Liu, M.; Mays, J.; Dadmum, M.; Advincula, R. Nano-donuts from pH dependent block restructuring in amphiphilic ABA triblock copolymer vesicles at the air–water interface. Soft Matter 2009, 5, 747–749. [CrossRef]
- Sfika, V.; Tsitsilianis, C.; Kiriy, A.; Gorodyska, G.; Stamm, M. pH responsive heteroarm starlike micelles from double hydrophilic ABC terpolymers with ampholitic A and C blocks. Macromolecules 2004, 37, 9551–9560. [CrossRef]
- 965. Tsitsilianis, C.; Stavrouli, N.; Bocharova, V.; Angelopoulos, S.; Kiriy, A.; Katsampas, I.; Stamm, M. Stimuli responsive associative polyampholytes based on ABCBA pentablock terpolymer architecture. Polymer 2008, 49, 2996–3006.
- Stavrouli, N.; Tsitsilianis, C.; Angelopoulos, A.; Katsampas, I. pH/ thermosensitive hydrogels formed at low pH by a PMMA-PAA- P2VP-PAA-PMMA pentablock terpolymer. Macromol. Rapid Commun. 2008, 29, 130–135. [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S. H. RAFT polymerization and some of its applications. Chem. Asian J. 2013, 8, 1634–1644. [CrossRef] [PubMed]
- Davaran, S.; Ghamkhari, A.; Alizadeh, E.; Massoumi, B.; Jaymand, M. Novel dual stimuli-responsive ABC triblock copolymer: RAFT synthesis, “schizophrenic” micellization, and its performance as an anticancer drug delivery nanosystem. J. Colloid Interface Sci. 2017, 488, 282–293. [CrossRef]
- Wang, W.; Lin, L.; Lu, J.; Liang, H.; Feng, H.; Hu, L., Synthesis, self-assembly, and formation of photo-crosslinking-stabilized fluorescent micelles covalently containing zinc(II)-bis(8-hydroxyquinoline) for ABC triblock copolymer bearing cinnamoyl and 8-hydroxyquinoline side groups. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1056–1064.
- Hadasha, W.; Klumperman, B. Atom Transfer Radical Polymerization as a Powerful Tool in the Synthesis of Molecular Brushes. Polym. Int. 2014, 63, 824–834. [CrossRef]
- Siegwart, D. J.; Oh, J. K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [CrossRef]
- Gaynor, S.; Qiu, J., Matyjaszewski, K. Using atom transfer radical polymerization (ATRP) in environmentally benign processes. In Advancing Sustainability Through Green Chemistry and Engineering, Lankey, R. L., Anastas, P. T., Eds.; American Chemistry Society (ACS) Symposium Series, Washington; DC: American Chemical Society, 2002; Vol. 823, Chapter 9, pp. 113-126. [Google Scholar]
- Lomas, H.; Canton, I.; MacNeil, S.; Du, J.; Armes, S. P.; Ryan, A. J.; Lewis, A. L.; Battaglia, G. Biomimetic pH Sensitive Polymersomes for Efficient DNA Encapsulation and Delivery. Adv. Mater. 2007, 19, 4238–4246. [CrossRef]
- Baskaran, D.; Müller, A. H. E. Anionic Vinyl Polymerization. In Controlled and Living Polymerizations; Müller, A. H. E., Matyjaszewski, K., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010; pp 1-56.
- Penczek, S.; Cypryk, M.; Duda, A.; Kubisa, P.; Slomkowski, S. Living Ring-Opening Polymerizations of Heterocyclic Monomers. Progr. Polym. Sci. 2007, 32, 247–282. [CrossRef]
- Lecomte, P.; Jérôme, C. Recent developments in ring-opening polymerization of lactones. In Synthetic biodegradable polymers, Rieger, B., Künkel, A., Coates, G., Reichardt, R., Dinjus, E., Zevaco, T., Eds.; Advances in polymer science; Springer: Berlin, Heidelberg, 2011; Vol 245, pp 173-217. [Google Scholar] [CrossRef]
- Verbraeken, B.; Monnery, B. D.; Lava, K.; Hoogenboom, R. The Chemistry of Poly(2-oxazoline)s. Eur. Polym. J. 2017, 88, 451–469.
- Hoogenboom, R.; Wiesbrock, F.; Huang, H.; Leenen, M. A.; Thijs, H. M.; van Nispen, S. F.; van der Loop, M.; Fustin, C. A.; Jonas, A. M.; Gohy, J. F.; Schubert, U. S. Microwave-assisted cationic ring-opening polymerization of 2-oxazolines: A powerful method for the synthesis of amphiphilic triblock copolymers. Macromolecules 2006, 39, 4719–4725.
- Kempe, K.; Hoogenboom, R.; Hoeppener, S.; Fustin, C.-A.; Gohy, J.-F.; Schubert, U. S. Discovering New Block Terpolymer Micellar Morphologies. Chem. Commun. 2010, 46, 6455–6457. [CrossRef]
- Fetsch, C.; Luxenhofer, R. Highly defined multiblock copolypeptoids: pushing the limits of living nucleophilic ring-opening polymerization. Macromol. Rapid Commun. 2012, 33, 1708–1713. [CrossRef] [PubMed]
- Barthel, M. J.; Schacher, F. H.; Schubert, U. S. Poly(ethylene oxide) (PEO)-Based ABC Triblock Terpolymers – Synthetic Complexity vs. Application Benefits. Polym. Chem. 2014, 5, 2647–2662. [CrossRef]
- 982. Sun, J.; Černoch, P.; Völkel, A.; Wei, Y.; Ruokolainen, J.; Schlaad, H. Aqueous self-assembly of a protein-mimetic ampholytic block copolypeptide. Macromolecules 2016, 49, 5494–5501.
- Stoenescu, R.; Graff, A.; Meier, W. Asymmetric ABC-triblock copolymer membranes induce a directed insertion of membrane proteins. Macromol. Biosci. 2004, 4, 930–935. [CrossRef] [PubMed]
- Konishcheva, E.; Häussinger, D.; Lörcher, S.; Meier, W. Key aspects to yield low dispersity of PEO-b-PCL diblock copolymers and their mesoscale self-assembly. Eur. Polym. J. 2016, 83, 300–310. [CrossRef]
- Semenov, A. M.; Joanny, J.-F.; Khokhlov, A. R. Associating Polymers: Equilibrium and Linear Viscoelasticity. Macromolecules 1995, 28, 1066–1075, pp 156-164. [CrossRef]
- Halperin, A. 986. Halperin, A., Zhulina, E. B. Triblock copolymers, mesogels and deformation behavior in poor solvents. In Physics of Polymer Networks; Progress in Colloid & Polymer Science; Steinkopff; 1992; Vol. 90. [CrossRef]
- Erhardt, R.; Zhang, M.; Böker, A.; Zettl, H.; Abetz, C.; Frederik, P.; Krausch, G.; Abetz, V.; Müller, A. H. E. Amphiphilic Janus micelles with polystyrene and poly(methacrylic acid) hemispheres. J. Am. Chem. Soc. 2003, 125, 3260–3267. [CrossRef]
- Walther, A.; Müller, A. H. E. Janus particles. Soft Matter 2008, 4, 663–668.
- Walther, A.; Barner-Kowollik, C.; Müller A. H. E. Mixed, multicompartment, or Janus micelles? A systematic study of thermoresponsive bis-hydrophilic block terpolymers. Langmuir 2010, 26, 12237–12246. [CrossRef]
- Charlaganov, M. I.; Borisov, O. V.; Leermakers, F. A. M. Modeling of Triblock Terpolymer Micelles with a Segregated Corona. Macromolecules 2008, 41, 3668–3677. [CrossRef]
- Bütün, V.; Wang, X.-S.; de Paz Báñez, M. V.; Robinson, K. L.; Billingham, N. C.; Armes, S. P.; Tuzar, Z. Synthesis of shell cross-linked micelles at high solids in aqueous media. Macromolecules 2000, 33, 1–3. [CrossRef]
- Laschewsky, A. Polymerized micelles with compartments. Curr. Opin. Colloid Interface Sci. 2003, 8, 274–281. [CrossRef]
- Liu, Y.; Abetz, V.; Müller, A. H. E. Janus Cylinders. Macromolecules 2003, 36, 7894–7898.
- Stewart, S.; Liu, G. Block Copolymer Nanotubes. Angew. Chem. Int. Ed. 2000, 39, 340–344. [CrossRef]
- Holmes, R. M.; Williams, D. R. M. Single Chain Asymmetric Block Copolymers in Poor Solvents. Candidates for Patchy Colloids. Macromolecules 2011, 44, 6172–6181. [CrossRef]
- Li, Z.; Hillmyer, M. A.; Lodge, T. P. Morphologies of multicompartment micelles formed by ABC miktoarm star terpolymers. Langmuir 2006, 22, 9409–9417. [CrossRef]
- Saito, N.; Liu, C.; Lodge, T. P.; Hillmyer, M. A. Multicompartment micelle morphology evolution in degradable miktoarm star terpolymers. ACS Nano 2010, 4, 1907–1912. [CrossRef]
- Li, Z.; Kesselman, E.; Talmon, Y.; Hillmyer, M. A.; Lodge, T. P. Multicompartment micelles from ABC miktoarm stars in water. Science 2004, 306, 98–101. [CrossRef] [PubMed]
- He, X.; Huang, L.; Liang, H.; Pan, C. Self-assembly of star block copolymers by dynamic density functional theory. J. Chem. Phys. 2002, 116, 10508–10513. [CrossRef]
- He, X. H.; Huang, L.; Liong, H. J.; Pan, C. Y. Localizations of junction points of ABC 3-miktoarm star terpolymers. J. Chem. Phys. 2003, 118, 9861–9863. [CrossRef]
- Birshtein, T. M.; Polotsky, A.; Abetz, V. Theory of the Lamellar Superstructure of an ABC 3-Miktoarm Star-Terpolymer. Macromol. Theory Simul. 2004, 13, 512–519. [CrossRef]
- Gemma, T.; Hatono, A.; Dotera, T. Monte Carlo Simulations of the Morphology of ABC Star Polymers Using the Diagonal Bond Method. Macromolecules 2002, 35, 3225–3237. [CrossRef]
- Zhu, Y.; Li, R. K. Y.; Jiang, W. A Monte Carlo simulation for the micellization of ABC 3-miktoarm star terpolymers in a selective solvent. Chem. Phys. 2006, 327, 137–143. [CrossRef]
- Zhulina, E. B.; Borisov, O. V. (). Scaling theory of 3-miktoarm ABC copolymer micelles in selective solvent. Macromolecules 2008, 41, 5934–5944. [CrossRef]
- Fang, B.; Walther, A.; Wolf, A.; Xu, Y.; Yuan, J.; Müller, A. H. Undulated multicompartment cylinders by the controlled and directed stacking of polymer micelles with a compartmentalized corona. Angew. Chem. Int. Ed. Engl. 2009, 48, 2877–2880. [CrossRef]
- Konishcheva, E. V.; Zhumaev, U. E.; Kratt, M.; Oehri, V.; Meier, W. Complex Self-Assembly Behavior of Bis-hydrophilic PEO-b-PCL-b-PMOXA Triblock Copolymers in Aqueous Solution. Macromolecules 2017, 50, 7155–7168. [CrossRef]
- Jain, S.; Bates, F. S. On the origins of morphological complexity in block copolymer surfactants. Science 2003, 300, 460–464. [CrossRef] [PubMed]
- Zupancich, J. A.; Bates, F. S.; Hillmyer, M. A. Aqueous dispersions of poly(ethylene oxide)-b-poly(γ-methyl-ε-caprolactone) block copolymers. Macromolecules 2006, 39, 4286–4288. [CrossRef]
- Adams, D. J.; Butler, M. F.; Weaver, A. C. Effect of block length, polydispersity, and salt concentration on PEO-PDEAMA block copolymer structures in dilute solution. Langmuir 2006, 22, 4534–4540. [CrossRef]
- Wu, D.; Spulber, M.; Itel, F.; Chami, M.; Pfohl, T.; Palivan, C. G.; Meier, W. Effect of Molecular Parameters on the Architec- ture and Membrane Properties of 3D Assemblies of Amphiphilic Copolymers. Macromolecules 2014, 47, 5060–5069. [CrossRef]
- Israelachvili, J. N. Intermolecular and Surface Forces; Academic Press: New York, 2010; 3rd edition.
- Taubert, A.; Furrer, E.; Meier, W. Water-in-water mesophases for templating inorganics. Chem. Commun. (Camb). 2004, (19), 2170–2171.
- Casse, O.; Shkilnyy, A.; Linders, J.; Mayer, C.; Haussinger, D.; Volkel, A.; Thunemann, A. F.; Dimova, R.; Colfen, H.; Meier, W.; Schlaad, H.; Taubert, A. Solution Behavior of Double-Hydrophilic Block Copolymers in Dilute Aqueous Solution. Macromolecules 2012, 45, 4772–4777.
- Liu, F.; Eisenberg, A. Preparation and pH triggered inversion of vesicles from poly(acrylic acid)-block-polystyrene-block-poly(4-vinyl pyridine). J. Am. Chem. Soc. 2003, 125, 15059–15064. [CrossRef]
- Wittemann, A.; Azzam, T.; Eisenberg, A. Biocompatible polymer vesicles from biamphiphilic triblock copolymers and their interaction with bovine serum albumin. Langmuir 2007, 23, 2224–2230. [CrossRef] [PubMed]
- Schlaad, H.; You, L.; Sigel, R.; Smarsly, B.; Heydenreich, M.; Mantion, A.; Masić, A. Glycopolymer vesicles with an asymmetric membrane. Chem. Commun. (Camb). 2009, (12), 1478–1480.
- Liu, G.; Ma, S.; Li, S.; Cheng, R.; Meng, F.; Liu, H.; Zhong, Z. The highly efficient delivery of exogenous proteins into cells mediated by biodegradable chimaeric polymersomes. Biomaterials 2010, 31, 7575–7585. [CrossRef]
- Du, J.; Fan, L.; Liu, Q. pH-Sensitive Block Copolymer Vesicles with Variable Trigger Points for Drug Delivery. Macromolecules 2012, 45, 8275–8283. [CrossRef]
- Mason, A. F.; Thordarson, P. Polymersomes with Asymmetric Membranes Based on Readily Accessible Di- and Triblock Copolymers Synthesized via SET-LRP. ACS Macro Lett. 2016, 5, 1172–1175. [CrossRef]
- Smith, P. K.; Krohn, R. I.; Hermanson, G. T.; Mallia, A. K.; Gartner, F. H.; Provenzano, M. D.; Fujimoto, E. K.; Goeke, N. M.; Olson, B. J.; Klenk, D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85.
- Blanazs, A.; Massignani, M.; Battaglia, G.; Armes, S. P.; Ryan, A. J. Tailoring macromolecular expression at polymersome surfaces. Adv. Funct. Mater. 2009, 19, 2906–2914. [CrossRef]
- Liu, Q.; Chen, J.; Du, J. Asymmetrical polymer vesicles with a “stealthy” outer corona and an endosomal-escape-accelerating inner corona for efficient intracellular anticancer drug delivery. Biomacromolecules 2014, 15, 3072–3082. [CrossRef]
- Njikang, G.; Han, D.; Wang, J.; Liu, G. ABC Triblock Copolymer Micelle-Like Aggregates in Selective Solvents for A and C. Macromolecules 2008, 41, 9727–9735. [CrossRef]
- Du, J.; Armes, S. P. Patchy multi-compartment micelles are formed by direct dissolution of an ABC triblock copolymer in water. Soft Matter 2010, 6, 4851–4857. [CrossRef]
- Walther, A.; Millard, P.-E.; Goldmann, A. S.; Lovestead, T. M.; Schacher, F.; Barner-Kowollik, C.; Mueller, A. H. E. Bis-Hydrophilic Block Terpolymers via RAFT Polymerization: Toward Dynamic Micelles with Tunable Corona Properties. Macromolecules 2008, 41, 8608–8619. [CrossRef]
- Erhardt, R.; Böker, A.; Zettl, H.; Kaya, H.; Pyckhout-Hintzen, W.; Krausch, G.; Abetz, V.; Müller, A. H. E. Macromolecules 2001, 34, 1069–1075.
- Saito, R.; Fujita, A.; Ichimura, A.; Ishizu, K. Synthesis of microspheres with microphase-separated shells. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 2091–2097. [CrossRef]
- Hoppenbrouwers, E.; Li, Z.; Liu G. Triblock nanospheres with amphiphilic coronal chains. Macromolecules 2003, 36, 876–881. [CrossRef]
- Dag, A.; Zhao, J.; Stenzel, M. H. Origami with ABC Triblock Terpolymers Based on Glycopolymers: Creation of Virus-Like Morphologies. ACS Macro Lett. 2015, 4, 579–583. [CrossRef]
- Walther, A.; Müller, A. H. Janus particles: synthesis, self-assembly, physical properties, and applications. Chem. Rev. 2013, 113, 5194–5261. [CrossRef]
- Du, J.; O’Reilly, R. K. Anisotropic particles with patchy, multicompartment and Janus architectures: preparation and application. Chem Soc Rev. 2011, 40, 2402–2416. [CrossRef]
- Tsitsilianis, C.; Voulgaris, D.; Stepanek, M.; Podhajecka, K.; Prochazka, K.; Tuzar, Z.; Brown, W. Polystyrene/poly(2-vinylpyridine) heteroarm star copolymer micelles in aqueous media and onion type micelles stabilized by diblock copolymers. Langmuir 2000, 16, 6868–6876. [CrossRef]
- Gorodyska, G.; Kiriy, A.; Minko, S.; Tsitsilianis, C.; Stamm, M. Reconformation and Metallization of Unimolecular Micelles in Controlled Environment. Nano Lett. 2003, 3, 365–368. [CrossRef]
- Voulgaris, D.; Tsitsilianis, C. Aggregation behavior of polystyrene/poly(acrylic acid) heteroarm star copolymers in 1,4-dioxane and aqueous media. Macromol. Chem. Phys. 2001, 202, 3284–3292. [CrossRef]
- 1035. Hammond, M.R.; Li, C.; Tsitsilianis, C.; Mezzenga, R. Hierarchical self-organization in polyelectrolyte-surfactant complexes based on heteroarm star block copolyampholytes. Soft Matter 2009, 5, 2371–2377.
- 1036. Stavrouli, N.; Kyriazis, A.; Tsitsilianis, C. Reversible hydrogels from an ampholytic An(B-b-C)n heteroarm star block terpolymer. Macromol. Chem. Phys. 2008, 209, 2241–2247.
- Yang, J. C.; Mays J. W. Synthesis and Characterization of Neutral/Ionic Block Copolymers of Various Architectures. Macromolecules 2002, 35, 3433–3438. [CrossRef]
- Nottelet, B.; Vert, M.; Coudane, J. Novel Amphiphilic Degradable Poly(-caprolactone)-graft-poly(4-vinyl pyridine), Poly(-caprolactone)-graft-poly(dimethylaminoethyl methacrylate) and Water-Soluble Derivatives. Macromol. Rapid Commun. 2008, 29, 743–750. [CrossRef]
- Cohen Stuart, M. A.; Besseling, N. A. M.; Fokkink, R. G. Formation of Micelles with Complex Coacervate Cores. Langmuir 1998, 14, 6846–6849. [CrossRef]
- Higgins, J. S.; Benoit, H. C. Polymers and Neutron Scattering, Ed.; Oxford University Press: New York, 1994. [Google Scholar]
- Jhaveri, A. M.; Torchilin, V. P. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front. Pharmacol. 2014, 5, 77.
- Machtakova, M.; Thérien-Aubin, H.; Landfester, K. Polymer nano-systems for the encapsulation and delivery of active biomacromolecular therapeutic agents. Chem. Soc. Rev. 2022, 51, 128–152. [CrossRef] [PubMed]
- Lee, J. S.; Feijen, J. Polymersomes for drug delivery: design, formation and characterization. J. Control. Release 2012, 161, 473–483. [CrossRef] [PubMed]
- LoPresti, C.; Lomas, H.; Massignani, M.; Smart, T.; Battaglia, G. Polymersomes: nature inspired nanometer sized compartments. J. Mater. Chem. 2009, 19, 3576–3590. [CrossRef]
- Jonsson, M.; Linse, P. Polyelectrolyte–macroion complexation. II. Effect of chain flexibility. J.Chem.Phys. 2001, 115, 10975–10985. [CrossRef]
- Park, J. M.; Muhoberac, B. B.; Dubin, P. L.; Xia, J. Effects of protein charge heterogeneity in protein-polyelectrolyte complexation. Macromolecules 1992, 25, 290–295. [CrossRef]
- Kawamura, A.; Kojima, C.; Iijima, M.; Harada, A.; Kono. K. Polyion complex micelles formed from glucose oxidase and comb-type polyelectrolyte with poly(ethylene glycol) grafts. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 3842–3852. [CrossRef]
- Jiang, Y.; Fay, J. M.; Poon, C. D.; Vinod, N.; Zhao, Y.; Bullock, K.; Qin, S.; Manickam, D. S.; Yi, X.; Banks, W. A.; Kabanov, A. V. Nanoformulation of Brain-Derived Neurotrophic Factor with Target Receptor-Triggered-Release in the Central Nervous System. Adv. Funct. Mater. 2018, 28, 1703982. [CrossRef]
- De Luca, S.; Chen, F.; Seal, P.; Stenzel, M. H.; & Smith, S. C. Binding and Release between Polymeric Carrier and Protein Drug: pH-Mediated Interplay of Coulomb Forces, Hydrogen Bonding, van der Waals Interactions, and Entropy. Biomacromolecules 2017, 18, 3665–3677.
- Kaczmarek, D.; Diget, J. S.; Nyström, B.; Gyulai, G.; Mészáros, R.; Gilányi, T.; Imre Varga, T. Response of block copolyelectrolyte complexes to addition of ionic surfactants. Colloids Surf. A 2017, 532, 290–296. [CrossRef]
- Harada, A.; Kataoka, K. Switching by Pulse Electric Field of the Elevated Enzymatic Reaction in the Core of Polyion Complex Micelles. J.Am.Chem.Soc. 2003, 125, 15306–15307. [CrossRef] [PubMed]
- Kawamura, A.; Yoshioka, Y.; Harada, A.; Kono, K. Acceleration of enzymatic reaction of trypsin through the formation of water-soluble complexes with poly(ethylene glycol)-block-poly(α,β-aspartic acid). Biomacromolecules 2005, 6, 627–631. [CrossRef]
- Sotiropoulou, M.; Bokias, G.; Staikos, G. Water-soluble complexes through coulombic interactions between bovine serum albumin and anionic polyelectrolytes grafted with hydrophilic nonionic side chains. Biomacromolecules 2005, 6, 1835–1838. [CrossRef]
- Yuan, X.; Harada, A.; Yamasaki, Y.; Kataoka, K. Stabilization of lysozyme-incorporated polyion complex micelles by the ω-end derivatization of poly(ethylene glycol)-poly(α,β-aspartic acid) block copolymers with hydrophobic groups. Langmuir 2005, 21, 2668–2674. [CrossRef]
- Harada, A.; Yoshioka, Y.; Kawamura, A.; Kojima, C.; Kono, K. Effect of polycarboxylate blocks on the amidase activity of trypsin through complexation with PEG/polycarboxylate block ionomers. Macromol. Biosci. 2007, 7, 339–343. [CrossRef] [PubMed]
- Jiang, Y.; Arounleut, P.; Rheiner, S.; Bae, Y.; Kabanov, A. V.; Milligan, C.; Manickam, D. S. SOD1 nanozyme with reduced toxicity and MPS accumulation. J. Control. Release 2016, 231, 38–49. [CrossRef] [PubMed]
- Kuwada K., Kurinomaru T., Tomita S., Shiraki K. Noncovalent PEGylation-based enzyme switch in physiological saline conditions using quaternized polyamines. Colloid Polym. Sci. 2016, 294, 1551–1556. [CrossRef]
- Nakai, K.; Nishiuchi, M.; Inoue, M.; Ishihara, K.; Sanada, Y.; Sakurai, K.; Yusa, S. Preparation and characterization of polyion complex micelles with phosphobetaine shells. Langmuir 2013, 29, 9651–9661. [CrossRef] [PubMed]
- Lee, Y.; Fukushima, S.; Bae, Y.; Hiki, S.; Ishii, T.; Kataoka, K. A protein nanocarrier from charge-conversion polymer in response to endosomal pH. J. Am. Chem. Soc. 2007, 129, 5362–5363. [CrossRef]
- Shetty, J. K.; Kinsella, J. E. Ready separation of proteins from nucleoprotein complexes by reversible modification of lysine residues. Biochem. J. 1980, 191, 269–272. [CrossRef]
- Chen, J.; Ding, J.; Zhang, Y.; Xiao, C.; Zhuang, X.; Chen, X. Polyion complex micelles with gradient pH-sensitivity for adjustable intracellular drug delivery. Polym. Chem. 2015, 6, 397–405. [CrossRef]
- Park, T. G.; Jeong, J. H.; Kim, S. W. Current status of polymeric gene delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 467–486. [CrossRef]
- Mundra, V.; Mahato, R. I. Design of nanocarriers for efficient cellular uptake and endosomal release of small molecule and nucleic acid drugs: learning from virus. Front. Chem. Sci. Eng. 2014, 8, 387–404. [CrossRef]
- Van Bruggen, C.; Hexum, J. K.; Tan, Z.; Dalal, R. J.; Reineke, T. M. Nonviral Gene Delivery with Cationic Glycopolymers. Acc. Chem. Res. 2019, 52, 1347–1358. [CrossRef]
- Wang, Q.; Schlenoff, J. B. The polyelectrolyte complex/coacervate continuum. Macromolecules 2014, 47, 3108–3116. [CrossRef]
- Schlenoff, J. B.; Yang, M.; Digby, Z. A.; Wang, Q. Ion Content of Polyelectrolyte Complex Coacervates and the Donnan Equilibrium. Macromolecules 2019, 52, 9149–9159. [CrossRef]
- Wu, H.; Ting, J. M.; Weiss, T. M.; Tirrell, M. V. Interparticle Interactions in Dilute Solutions of Polyelectrolyte Complex Micelles. ACS Macro Lett. 2019, 8, 819–825. [CrossRef]
- Liu, X.; Haddou, M.; Grillo, I.; Mana, Z.; Chapel, J. P.; Schatz, C. Early stage kinetics of polyelectrolyte complex coacervation monitored through stopped-flow light scattering. Soft Matter 2016, 12, 9030–9038. [CrossRef]
- Aniansson E. A. G.; Wall S. N. Kinetics of step-wise micelle association. Correction and improvement. J. Phys. Chem. 1975, 79, 857–858. [CrossRef]
- Halperin A.; Alexander S. Polymeric micelles: their relaxation kinetics. Macromolecules 1989, 22, 2403–2412. [CrossRef]
- Esselink F. J., Dormidontova E., Hadziioannou G. Evolution of Block Copolymer Micellar Size and Structure Evidenced with Cryo Electron Microscopy. Macromolecules 1998, 31, 2925–2932. [CrossRef]
- Rharbi, Y. Fusion and Fragmentation Dynamics at Equilibrium in Triblock Copolymer Micelles. Macromolecules 2012, 45, 9823–9826. [CrossRef]
- Zhang J.; Chen S.; Zhu Z.; Liu S. Stopped-Flow Kinetic Studies of the Formation and Disintegration of Polyion Complex Micelles in Aqueous Solution. Phys. Chem. Chem. Phys. 2014, 16, 117–127. [CrossRef]
- Ting, J. M.; Wu, H.; Herzog-Arbeitman, A.; Srivastava, S.; Tirrell, M. V. Synthesis and Assembly of Designer Styrenic Diblock Polyelectrolytes. ACS Macro Lett. 2018, 7, 726–733. [CrossRef]
- 1075. Haladjova, E.; Mountrichas, G.; Pispas, S.; Rangelov, S. Poly(vinyl benzyl trimethylammonium chloride) Homo and Block Copolymers Complexation with DNA. J. Phys. Chem. B. 2016, 120, 2586–2595.
- Lou, B.; Beztsinna, N.; van den Dikkenberg, J. B.; Pispas, S.; Hennink, W. E. Small nanosized poly(vinyl benzyl trimethylammonium chloride) based polyplexes for siRNA delivery. Int. J. Pharm. 2017, 525, 388–396. [CrossRef]
- Kuo, C. H.; Leon, L.; Chung, E. J.; Huang, R. T.; Sontag, T. J, Reardon, C. A,; Getz, G. S.; Tirrell, M.; Fang, Y. Inhibition of atherosclerosis-promoting microRNAs via targeted polyelectrolyte complex micelles. J. Mater. Chem. B 2014, 2, 8142–8153. [CrossRef] [PubMed]
- Marciel, A. B.; Chung, E. J.; Brettmann, B. K.; Leon, L. Bulk and nanoscale polypeptide based polyelectrolyte complexes. Adv. Colloid Interface Sci. 2017, 239, 187–198. [CrossRef] [PubMed]
- Ramasamy, T.; Kim, H.; Choi, Y.; Tran, H. pH sensitive polyelectrolyte complex micelles for highly effective combination chemotherapy. J. Mater. Chem. B 2014, 2, 6324–6333. [CrossRef]
- Aniansson, E. A. G.; Wall, S. N. On the kinetics of step-wise micelle association. J. Phys. Chem. 1974, 78, 1024–1030. [CrossRef]
- Wu, H.; Ting, J.; Yu, B.; Jackson, N.; Meng, S.; De Pablo, J.; Tirrell, M. Spatiotemporal formation and growth kinetics of polyelectrolyte complex micelles with millisecond resolution. ACS Macro Lett. 2020, 9, 1674–1680. [CrossRef]
- Lynd, N. A.; Meuler, A. J.; Hillmyer, M. A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 2008, 33, 875–893. [CrossRef]
- Ong, G. M. C.; Sing, C. E. Mapping the phase behavior of coacervate-driven self-assembly in diblock copolyelectrolytes. Soft Matter 2019, 15, 5116–5127. [CrossRef]
- Srivastava, S.; Levi, A. E.; Goldfeld, D. J.; Tirrell, M. V. Structure, morphology, and rrheology of polyelectrolyte complex hydrogels formed by self-assembly of oppositely charged triblock polyelectrolytes. Macromolecules 2020, 53, 5763–5774. [CrossRef]
- Ohara, Y.; Nakai, K.; Ahmed, S.; Matsumura, K.; Ishihara, K.; Yusa, S. I. pH-Responsive Polyion Complex Vesicle with Polyphosphobetaine Shells. Langmuir 2019, 35, 1249–1256. [CrossRef] [PubMed]
- Liu, Q.; Wang, X.; Ma, L.; Yu, K.; Xiong, W.; Lu, X.; Cai, Y. Polymerization-Induced Hierarchical Electrostatic Self-Assembly: Scalable Synthesis of Multicompartment Polyion Complex Micelles and Their Monolayer Colloidal Nanosheets and Nanocages. ACS Macro Lett. 2020, 9, 454–458. [CrossRef] [PubMed]
- Dumetz, A. C.; Chockla, A. M.; Kaler, E. W.; Lenhoff, A. M. Protein Phase Behavior in Aqueous Solutions: Crystallization, Liquid- Liquid Phase Separation, Gels, and Aggregates. Biophys. J. 2008, 94, 570–583. [CrossRef]
- Wallace, A. F.; Hedges, L. O.; Fernandez-Martinez, A.; Raiteri, P.; Gale, J. D.; Waychunas, G. A.; Whitelam, S.; Banfield, J. F.; De Yoreo, J. J. Microscopic Evidence for Liquid-Liquid Separation in Supersaturated CaCO3 Solutions. Science (Washington, DC, U. S.) 2013, 341, 885–889.
- Ianiro, A.; Wu, H.; van Rijt, M. M. J.; Vena, M. P.; Keizer, A. D. A.; Esteves, A. C. C.; Tuinier, R.; Friedrich, H.; Sommerdijk, N. A. J. M.; Patterson, J. P. Liquid−Liquid Phase Separation during Amphiphilic Self-Assembly. Nat. Chem. 2019, 11, 320–328.
- Kiriy, A.; Yu, J.; Stamm, M. Interpolyelectrolyte complexes: a single-molecule insight. Langmuir 2006, 22, 1800–1803. [CrossRef]
- Bakeev, K. N.; Izumrudov, V. A.; Kuchanov, S. I.; Zezin, A. B.; Kabanov, V. A. Kinetics and mechanism of interpolyelectrolyte exchange and addition reactions. Macromolecules 1992, 25, 4249–4254. [CrossRef]
- Marras, A. E.; Vieregg, J. R.; Tirrell, M. V. Assembly and Characterization of Polyelectrolyte Complex Micelles. J. Vis. Exp. 2020, 157, e60894.
- Marciel, A. B.; Srivastava, S.; Ting, J. M.; Tirrell, M. V. SAXS methods for investigating macromolecular and self-assembled polyelectrolyte complexes. Methods Enzymol. 2021, 646, 223–259.
- Ting, J. M.; Marras, A. E.; Mitchell, J. D.; Campagna, T. R.; Tirrell, M. V. Comparing Zwitterionic and PEG Exteriors of Polyelectrolyte Complex Micelles. Molecules 2020, 25, 2553. [CrossRef]
- Takahashi, R.; Narayanan, T.; Sato, T. Growth Kinetics of Polyelectrolyte Complexes Formed from Oppositely-Charged Homo- polymers Studied by Time-Resolved Ultra-Small-Angle X-ray Scattering. J. Phys. Chem. Lett. 2017, 8, 737–741. [CrossRef]
- Smolsky, I. L.; Liu, P.; Niebuhr, M.; Ito, K.; Weiss, T. M.; Tsuruta, H. Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 2007, 40, s453–s458. [CrossRef]
- Bednar, B.; Edwards, K.; Almgren, M.; Tormod, S.; Tuzar, Z. Rates of association and dissociation of block copolymer micelles: Light-scattering stopped-flow measurements. Makromol. Chem.-Rapid Commun. 1988, 9, 785–790. [CrossRef]
- Prochazka K.; Bednar B.; Mukhtar E.; Svoboda P.; Trnena J.; Almgren M. Nonradiative Energy Transfer in Block Copolymer Micelles. J. Phys. Chem. 1991, 95, 4563–4568. [CrossRef]
- Pacovska, M.; Prochazka, K., Tuzar, Z., Eds.; Munk, P. 1993: Formation of block copolymer micelles : a sedimentation study. Polymer (Guildford) 34, 4585-4588. [Google Scholar]
- Tian, M.; Qin, A.; Ramireddy, C.; Webber, S. E.; Munk, P.; Tuzar, Z.; Prochazka, K. Langmuir 1993, 9, 1741–1748.
- Wang, Y.; Kausch, C. M.; Chun, M.; Quirk, R. P.; Mattice, W. L. Exchange of chains between micelles of labeled polystyrene-block-poly (oxyethylene) as monitored by nonradiative singlet energy transfer. Macromolecules 1995, 28, 904–911. [CrossRef]
- Bednar, B.; Karasek, L.; Pokorny, J. Nonradiative energy transfer studies of block copolymers in selective solvents. Polymer 1996, 37, 5261–5268.
- 1103. Honda, C.; Abe, Y.; Nose, T. Relaxation Kinetics of Micellization in Micelle-Forming Block Copolymer in Selective Solvent. Macromolecules 1996, 29, 6778–6785.
- Haliloglu, T.; Mattice, W. L. In Solvent and Self-Assembly of Polymers, Webber, S. E., Eds.; Kluwer: Dortrecht, 1996. [Google Scholar]
- Z. In Solvent and Self-Assembly of Polymers; Webber, S. E., Ed.; Kluwer: Dortrecht, 1996. [Google Scholar]
- Honda, C.; Hasegawa, Y.; Hirunuma, R.; Nose, T. Micellization Kinetics of Block Copolymers in Selective Solvent. Macromolecules 1994, 27, 7660–7668. [CrossRef]
- Goldmints, I.; Holzwarth, J. F.; Smith, K. A.; Hatton, T. A. Micellar Dynamics in Aqueous Solutions of PEO-PPO-PEO Block Copolymers. Langmuir 1997, 13, 6130–6134. [CrossRef]
- Michels, B.; Waton, G.; Zana, R. Dynamics of micelles of poly(ethyleneoxide)-poly(propyleneoxide)-poly(ethylene oxide) block copolymers in aqueous solutions. Langmuir 1997, 13, 3111–3118. [CrossRef]
- Esselink F. J.; Dormidontova E. E.; Hadziioannou G. Redistribution of Block Copolymer Chains between Mixed Micelles in Solution. Macromolecules 1998, 31, 4873–4878. [CrossRef]
- Aniansson E. A. G.; Wall S. N.; Almgren M.; Hoffmann H.; Kielmann I.; Ulbricht W.; Zana R.; Lang J.; Tondre C. Theory of the kinetics of micellar equilibria and quantitative interpretation of chemical relaxation studies of micellar solutions of ionic surfactants. J. Phys. Chem. 1976, 80, 905–922.
- Lessner, E.; Teubner, M.; Kahlweit, M. Relaxation Experiments in Aqueous Solutions of Ionic Micelles. 1. Theory and Experiments on the System Water-Sodium Tetradecyl Sulfate-Sodium Perchlorate. J. Phys. Chem. 1981, 85, 1529–1536. [CrossRef]
- Kahlweit, M. Kinetics of formation of association colloids. J. Colloid Interface Sci. 1982, 90, 92–99. [CrossRef]
- Shusharina, N. P.; Saphonov, M. V.; Nyrkova, I. A; Khalatur, P. G.; Khokhlov. A. R. The Critical Micelle Concentration for the Solution of Polyelectrolyte/Neutral Block-Copolymers. Ber. Bunsenges. Phys. Chem. 1996, 100, 857–862. [CrossRef]
- Denkova, A. G.; Mendes, E.; Coppens, M.-O. Non-equilibrium dynamics of block copolymer micelles in solution: recent insights and open questions. Soft Matter 2010, 6, 2351–2357. [CrossRef]
- Li, Z.; Dormidontova, E. E. Kinetics of diblock copolymer micellization by dissipative particle dynamics. Macromolecules 2010, 43, 3521–3531. [CrossRef]
- Li, Z.; Dormidontova, E. E. Equilibrium chain exchange kinetics in block copolymer micelle solutions by dissipative particle dynamics simulations. Soft Matter 2011, 7, 4179–4188. [CrossRef]
- Sheng, Y.-J.; Wang, T.-Y.; Chen, W. M.; Tsao, H.-K. A- B diblock copolymer micelles: Effects of soluble-block length and component compatibility. J. Phys. Chem. B 2007, 111, 10938–10945. [CrossRef]
- Prhashanna, A.; Khan, S. A.; Chen, S. B. Kinetics of chain exchange between diblock copolymer micelles. Macromol. Theory Simul. 2016, 25, 383–391. [CrossRef]
- Spruijt, E.; Sprakel, J.; Lemmers, M.; Cohen Stuart, M. A.; Van Der Gucht, J. Relaxation dynamics at different time scales in electrostatic complexes: time-salt superposition. Phys. Rev. Lett. 2010, 105, 208301. [CrossRef] [PubMed]
- Spruijt, E.; Sprakel, J.; Cohen Stuart, M. A.; van der Gucht, J. Interfacial tension between a complex coacervate phase and its coexisting aqueous phase. Soft Matter 2010, 6, 172–178. [CrossRef]
- Spruijt, E.; van den Berg, S. A.; Cohen Stuart, M. A.; van der Gucht, J. Direct measurement of the strength of single ionic bonds between hydrated charges. ACS Nano 2012, 6, 5297–5303. [CrossRef] [PubMed]
- Li, L.; Srivastava, S.; Andreev, M.; Marciel, A. B.; de Pablo, J. J.; Tirrell, M. V. Phase behavior and salt partitioning in polyelectrolyte complex coacervates. Macromolecules 2018, 51, 2988–2995. [CrossRef]
- Ou, Z.; Muthukumar, M. Entropy and enthalpy of polyelectrolyte complexation: Langevin dynamics simulations. J. Chem. Phys. 2006, 124, 154902. [CrossRef] [PubMed]
- Ziebarth, J.; Wang, Y. Coarse-grained molecular dynamics simulations of DNA condensation by block copolymer and formation of core- corona structures. J. Phys. Chem. B 2010, 114, 6225–6232. [CrossRef]
- Šindelka, K.; Limpouchova,́ Z.;Lísal, M.; Prochaźka, K. Dissipative particle dynamics study of electrostatic self-assembly in aqueous mixtures of copolymers containing one neutral water-soluble block and one either positively or negatively charged polyelectrolyte block. Macromolecules 2014, 47, 6121–6134.
- Šindelka, K.; Limpouchova,́ Z.; Lísal, M.; Prochaźka, K. The electrostatic co-assembly in non-stoichiometric aqueous mixtures of copolymers composed of one neutral water-soluble and one polyelectrolyte (either positively or negatively charged) block: a dissipative particle dynamics study. Phys. Chem. Chem. Phys. 2016, 18, 16137–16151.
- Šindelka, K.; Limpouchova,́ Z.; Prochaźka, K. Computer study of the solubilization of polymer chains in polyelectrolyte complex cores of polymeric nanoparticles in aqueous media. Phys. Chem. Chem. Phys. 2018, 20, 29876–29888. [CrossRef]
- Lund, R.; Willner, L.; Monkenbusch, M.; Panine, P.; Narayanan, T.; Colmenero, J.; Richter, D. Structural observation and kinetic pathway in the formation of polymeric micelles. Phys. Rev. Lett. 2009, 102, 188301. [CrossRef]
- Katayose S, Kataoka K. Remarkable increase in nuclease resistance of plasmid DNA through supramolecular assembly with poly(ethylene glycol)-poly(L-lysine) block copolymer. J. Pharm. Sci. 1998, 87, 160–163. [CrossRef]
- Dautzenberg H.; Konak C.; Reschel T.; Zintchenko A.; Ulbrich K. Cationic graft copolymers as carriers for delivery of antisense-oligonucleotides. Macromol. Biosci. 2003, 3, 425–435. [CrossRef]
- Harada, A.; Kataoka, K. Selection between block- and homo- polyelectrolytes through polyion complex formation in aqueous medium. Soft Matter 2008, 4, 162–167. [CrossRef]
- Carl, N.; Prévost, S.; Schweins, R.; Huber, K. Contrast variation of micelles composed of Ca2+ and block copolymers of two negatively charged polyelectrolytes. Colloid Polym. Sci. 2020, 298, 663–679. [CrossRef]
- Qiang, Z.; Wang, M. 100th Anniversary of Macromolecular Science Viewpoint: Enabling Advances in Fluorescence Microscopy Techniques. ACS Macro Lett. 2020, 9, 1342–1356. [CrossRef] [PubMed]
- Parent, L. R.; Gnanasekaran, K.; Korpanty, J.; Gianneschi, N. C. 100th Anniversary of Macromolecular Science Viewpoint: Polymeric Materials by In Situ Liquid-Phase Transmission Electron Microscopy. ACS Macro Lett. 2021, 10, 14–38. [CrossRef] [PubMed]
- Peng, B.; Muthukumar, M. Modeling Competitive Substitution in a Polyelectrolyte Complex. J. Chem. Phys. 2015, 143, 243133. [CrossRef]
- Kembaren, R.; Fokkink, R.; Westphal, A. H.; Kamperman, M.; Kleijn, J. M.; Borst, J. W. Balancing Enzyme Encapsulation Efficiency and Stability in Complex Coacervate Core Micelles. Langmuir 2020, 36, 8494–8502. [CrossRef]
- Obermeyer, A. C.; Mills, C. E.; Dong, X. H.; Flores, R. J.; Olsen, B. D. Complex Coacervation of Supercharged Proteins with Polyelectrolytes. Soft Matter 2016, 12, 3570–3581. [CrossRef]
- Kapelner, R. A.; Obermeyer, A. C. Ionic Polypeptide Tags for Protein Phase Separation. Chem. Sci. 2019, 10, 2700–2707. [CrossRef]
- 1139. Tabandeh, S.; Leon, L. Engineering Peptide-Based Polyelectrolyte Complexes with Increased Hydrophobicity. Molecules 2019, 24, 868.
- Yang, Y.; Zhu, H.; Wang, J.; Fang, Q.; Peng, Z. Enzymatically Disulfide-Crosslinked Chitosan/Hyaluronic Acid Layer-by-Layer Self-Assembled Microcapsules for Redox-Responsive Controlled Release of Protein. ACS Appl. Mater. Interfaces 2018, 10, 33493–33506. [CrossRef]
- Wang, Y.; Cheng, Y. T.; Cao, C.; Oliver, J. D.; Stenzel, M. H.; Chapman, R. Polyion Complex-Templated Synthesis of Cross-Linked Single-Enzyme Nanoparticles. Macromolecules 2020, 53, 5487–5496. [CrossRef]
- Murmiliuk,̌ A.; Matějíček, P.; Filippov, S. K.; Janata, M.; Slouf, M.; Pispas, S.; Štěpánek, M. Formation of Core/CoronaNano- particles with Interpolyelectrolyte Complex Cores in Aqueous Solution: Insight into Chain Dynamics in the Complex from Fluorescence Quenching. Soft Matter 2018, 14, 7578–7585.
- Marciel, A. B.; Srivastava, S.; Tirrell, M. V. Structure and Rheology of Polyelectrolyte Complex Coacervates. Soft Matter 2018, 14, 2454–2464. [CrossRef]
- Li, Y.; Bronich, T. K.; Chelushkin, P. S.; Kabanov, A. V. Dynamic Properties of Block Ionomer Complexes with Polyion Complex Cores. Macromolecules 2008, 41, 5863–5868. [CrossRef]
- Pedersen, J. S.; Hamley, I. W.; Ryu, C. Y.; Lodge, T. P. Contrast Variation Small-Angle Neutron Scattering Study of the Structure of Block Copolymer Micelles in a Slightly Selective Solvent at Semidilute Concentrations. Macromolecules 2000, 33, 542–550. [CrossRef]
- Lund, R.; Willner, L.; Richter, D.; Lindner, P.; Narayanan, T. Kinetic Pathway of the Cylinder-to-Sphere Transition in Block Copolymer Micelles Observed in Situ by Time-Resolved Neutron and Synchrotron Scattering. ACS Macro Lett. 2013, 2, 1082–1087. [CrossRef]
- Cameron, N. S.; Corbierre, M. K.; Eisenberg, A. 1998 E.W.R. Steacie Award Lecture Asymmetric Amphiphilic Block Copolymers in Solution: A Morphological Wonderland. Can. J. Chem. 1999, 77, 1311–1326. [CrossRef]
- Kalkowski, J.; Liu, C.; Leon-Plata, P.; Szymusiak, M.; Zhang, P.; Irving, T.; Shang, W.; Bilsel, O.; Liu, Y. In Situ Measurements of Polymer Micellization Kinetics with Millisecond Temporal Resolution. ACS Macro Lett. 2019, 52, 3151–3157.
- Parent, L. R.; Bakalis, E.; Ramírez-Hernández, A.; Kammeyer, J. K.; Park, C.; de Pablo, J.; Zerbetto, F.; Patterson, J. P.; Gianneschi, N. C. Directly Observing Micelle Fusion and Growth in Solution by Liquid-Cell Transmission Electron Microscopy. J. Am. Chem. Soc. 2017, 139, 17140–17151. [CrossRef]
- Geng, Y.; Ahmed, F.; Bhasin, N.; Discher, D. E. Visualizing Worm Micelle Dynamics and Phase Transitions of a Charged Diblock Copolymer in Water. J. Phys. Chem. B 2005, 109, 3772–3779. [CrossRef]
- Loverde, S. M.; Ortiz, V.; Kamien, R. D.; Klein, M. L.; Discher, D. E. Curvature-Driven Molecular Demixing in the Budding and Breakup of Mixed Component Worm-Like Micelles. Soft Matter 2010, 6, 1419–1425. [CrossRef]
- Zhang, L.; Yu, K.; Eisenberg, A. Ion-Induced Morphological Changes in “Crew-Cut” Aggregates of Amphiphilic Block Copolymers. Science 1996, 272, 1777–1779. [CrossRef]
- Cui, H.; Chen, Z.; Zhong, S.; Wooley, K. L.; Pochan, D. J. Block Copolymer Assembly via Kinetic Control. Science 2007, 317, 647–650. [CrossRef]
- Meli, L.; Santiago, J. M.; Lodge, T. P. Path-Dependent Morphology and Relaxation Kinetics of Highly Amphiphilic Diblock Copolymer Micelles in Ionic Liquids. Macromolecules 2010, 43, 2018–2027. [CrossRef]
- Muthukumar, M. 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions. Macromolecules 2017, 50, 9528–9560. [CrossRef]
- Krogstad, D. V.; Lynd, N. A.; Miyajima, D.; Gopez, J.; Hawker, C. J.; Kramer, E. J.; Tirrell, M. V. Structural Evolution of Polyelectrolyte Complex Core Micelles and Ordered-Phase Bulk Materials. Macromolecules 2014, 47, 8026–8032. [CrossRef]
- Martel, A.; Liu, P.; Weiss, T. M.; Niebuhr, M.; Tsuruta, H. An Integrated High-Throughput Data Acquisition System for Biological Solution X-Ray Scattering Studies. J. Synchrotron Radiat. 2012, 19 Pt 3, 431–434. [CrossRef]
- de Pablo, J. J.; Jones, B.; Kovacs, C. L.; Ozolins, V.; Ramirez, A. P. The Materials Genome Initiative, the Interplay of Experiment, Theory and Computation. Curr. Opin. Solid. State Mater. Sci. 2014, 18, 99–117. [CrossRef]
- Acar, H.; Ting, J. M.; Srivastava, S.; LaBelle, J. L.; Tirrell, M. V. Molecular Engineering Solutions for Therapeutic Peptide Delivery. Chem. Soc. Rev. 2017, 46, 6553–6569. [CrossRef]
- Odijk, T. Binding of long flexible chains to a rodlike macromolecule. Macromolecules 1980, 13, 13–1542.
- Perico, A.; Penna, G. L; Arcesi, L. Electrostatic interactions with histone tails may bend linker DNA in chromatin. Biopolymers 2006, 81, 20–28. [CrossRef]
- 1162. Arcesi, L.; Penna, G. L.; Perico, A. Generalized electrostatic model of the wrapping of DNA around oppositely charged proteins. Biopolymers, 2007, 86, 127–135.
- Feric, M.; Vaidya, N.; Harmon, T. S.; Mitrea, D. M.; Zhu, L.; Richardson, T. M.; Kriwacki, R. W.; Pappu, R. V.; Brangwynne, C. P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697.
- Banani, S. F.; Lee, H. O.; Hyman, A. A.; Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [CrossRef]
- Yewdall, N. A.; André, A. A. M.; Lu, T.; Spruijt, E. Curr. Opin. Colloid Interface Sci. 2020, 52, 101416.
- Drobot, B.; Iglesias-Artola, J. M.; Le Vay, K.; Mayr, V.; Kar, M.; Kreysing, M.; Mutschler, H.; Tang, T. D. Compartmentalised RNA catalysis in membrane-free coacervate protocells. Nat. Commun. 2018, 9, 3643.
- Poudyal, R. R., Guth-Metzler, R. M., Veenis, A. J., Frankel, E. A., Keating, C. D., & Bevilacqua, P. C. Template-directed RNA polymerization and enhanced ribozyme catalysis inside membraneless compartments formed by coacervates. Nat. Commun. 2019, 10, 490.
- Mountain, G. A.; Keating, C. D. Formation of Multiphase Complex Coacervates and Partitioning of Biomolecules within them. Biomacromolecules 2020, 21, 630–640. [CrossRef]
- Lu, T.; Spruijt, E. Multiphase Complex Coacervate Droplets. J. Am. Chem. Soc. 2020, 142, 2905–2914. [CrossRef]
- Lee, Y.; Kataoka, K. Biosignal-sensitive polyion complex micelles for the delivery of biopharmaceuticals. Soft Matter 2009, 5, 3810–3817. [CrossRef]
- Kim, Y. H., Lee, K., Li, S. Nucleic acids based polyelectrolyte complexes: Their complexation mechanism, morphology, and stability. Chem. Mater. 2021, 33, 7923–7943. [CrossRef]
- Spruijt, E.; Cohen Stuart, M. A.; van der Gucht, J. Linear viscoelasticity of polyelectrolyte complex coacervates. Macromolecules 2013, 46, 1633–1641. [CrossRef]
- Liu, Y.; Winter, H. H.; Perry, S. L. Linear viscoelasticity of complex coacervates. Adv. Colloid Interface Sci. 2017, 239, 46–60. [CrossRef]
- Späth, F.; Donau, C.; Bergmann, A. M.; Kränzlein, M.; Synatschke, C. V.; Rieger, B.; Boekhoven, J. J. Am. Chem. Soc. 2021, 143, 4782–4789.
- Rubinstein, M.; Semenov, A. N. 1175. Rubinstein, M.; Semenov, A. N. Dynamics of entangled solutions of associating polymers. Macromolecules, 1058. [Google Scholar]
- Heo, T-Y; Kim, S; Chen, L; Sokolova, A; Lee, S and Choi, S-H. Molecular Exchange Kinetics in Complex Coacervate Core Micelles: Role of Associative Interaction. ACS Macro Lett. 2021, 10, 1138–1144. [CrossRef]
- Bos, I.; Brink, E.; Michels, L.; Sprakel, J. DNA dynamics in complex coacervate droplets and micelles. Soft Matter 2022, 18, 2012–2027. [CrossRef]
- Krogstad, D. V.; Lynd, N. A.; Choi, S.-H.; Spruell, J. M.; Hawker, C. J.; Kramer, E. J.; Tirrell, M. V. Effects of Polymer and Salt Concentration on the Structure and Properties of Triblock Copolymer Coacervate Hydrogels. Macromolecules 2013, 46, 1512–1518. [CrossRef]
- Srivastava, S.; Andreev, M.; Levi, A. E.; Goldfeld, D. J.; Mao, J.; Heller, W. T.; Prabhu, V. M.; De Pablo, J. J.; Tirrell, M. V. Gel Phase Formation in Dilute Triblock Copolyelectrolyte Complexes. Nat. Commun. 2017, 8, 14131.
- Kim, W. J.; Sato, Y.; Akaike, T.; Maruyama, A. Cationic Comb-Type Copolymers For DNA Analysis. Nat. Mater. 2003, 2, 815–820. [CrossRef]
- Kim, W. J.; Akaike, T.; Maruyama, A. DNA Strand Exchange Stimulated by Spontaneous Complex Formation with Cationic Comb- Type Copolymer. J. Am. Chem. Soc. 2002, 124, 12676–12677. [CrossRef]
- Bronich, T. K.; Nguyen, H. K.; Eisenberg, A.; Kabanov, A. V. Recognition of DNA Topology in Reactions between Plasmid DNA and Cationic Copolymers. J. Am. Chem. Soc. 2000, 122, 8339–8343. [CrossRef]
- Kanasty, R.; Dorkin, J. R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [CrossRef]
- Matsui, A.; Uchida, S.; Ishii, T.; Itaka, K.; Kataoka, K. Messenger RNA-Based Therapeutics for the Treatment of Apoptosis- Associated Diseases. Sci. Rep. 2015, 5, 15810. [CrossRef]
- Kaczmarek, J. C.; Kowalski, P. S.; Anderson, D. G. Advances in the Delivery of RNA Therapeutics: From Concept to Clinical Reality. Genome Med. 2017, 9, 60. [CrossRef]
- Kole, R.; Krainer, A. R.; Altman, S. RNA Therapeutics: Beyond RNA Interference and Antisense Oligonucleotides. Nat. Rev. Drug Discovery 2012, 11, 125–140. [CrossRef]
- Bromberg, L.; Deshmukh, S.; Temchenko, M.; Iourtchenko, L.; Alakhov, V.; Alvarez-Lorenzo, C.; Barreiro-Iglesias, R.; Concheiro, A.; Hatton, T. A. Polycationic Block Copolymers of Poly(ethylene oxide) and Poly(propylene oxide) for Cell Transfection. Bioconjugate Chem. 2005, 16, 626–633. [CrossRef]
- Balomenou, I.; Bokias. G. Water-Soluble Complexes between Cationic Surfactants and Comb-Type Copolymers Consisting of an Anionic Backbone and Hydrophilic Nonionic Poly(N,N-dimethylacrylamide) Side Chains. Langmuir 2005, 21, 9038–9043. [CrossRef]
- Kizhakkedathu, J. N.; Nisha, C. K.; Manorama, S. V.; Maiti, S. Water-Soluble Nanoparticles from Random Copolymer and Oppositely Charged Surfactant, 3. Macromolecular Bioscience 2005, 5, 549–558. [CrossRef]
- Nisha, C. K.; Manorama, S. V.; Kizhakkedathu, J. N.; Maiti, S. Water-soluble complexes from random copolymer and oppositely charged surfactant. 2. Complexes of poly (ethylene glycol)-based cationic random copolymer and bile salts. Langmuir 2004, 20, 8468–8475. [CrossRef]
- Chen, W.; Chen, H. R.; Hu, J. H.; Yang, W. L.; Wang, C. C. Synthesis and characterization of polyion complex micelles between poly (ethylene glycol)-grafted poly (aspartic acid) and cetyltrimethyl ammonium bromide. Colloid Surf A: Physicochem Eng Asp. 2006, 278, 60–66. [CrossRef]
- Bronich, T. K.; Cherry, T.; Vinogradov, S. V.; Eisenberg, A.; Kabanov, V. A., Kabanov, A. V. Self-Assembly in Mixtures of Poly (ethylene oxide)-graft-Poly (ethyleneimine) and Alkyl Sulfates. Langmuir 1998, 14, 6101–6106.
- Bronich, T. K.; Nehls, A.; Eisenberg, A.; Kabanov, V. A., & Kabanov, A. V. Novel drug delivery systems based on the complexes of block ionomers and surfactants of opposite charge. Colloids and Surfaces B: Biointerfaces 1999, 16, 243–251.
- Solomatin, S. V.; Bronich, T. K.; Bargar, T. W.; Eisenberg, A.; Kabanov, V. A.; Kabanov, A. V. Environmentally responsive nanoparticles from block ionomer complexes: effects of pH and ionic strength. Langmuir 2003, 19, 8069–8076. [CrossRef]
- Bronich, T. K.; Kabanov, A. V.; Kabanov, V. A.; Yu, K.; Eisenberg, A. Soluble complexes from poly(ethylene oxide)-block-polymethacrylate anions and n-alkylpyridinium cations. Macromolecules 1997, 30, 3519–3525. [CrossRef]
- Nisha, C. K.; Basak, P.; Manorama, S. V.; Maiti, S.; Jayachandran, K. N. Water-soluble complexes from random copolymer and oppositely charged surfactant. 1. Complexes of poly(ethylene glycol)-based cationic random copolymer and sodium dodecyl sulfate. Langmuir 2003, 19, 2947–2955. [CrossRef]
- Dubruel, P.; De Strycker, J.; Westbroek, P.; Bracke, K.; Temmerman, E.; Vandervoort, J.; Ludwig, A.; Schacht, E. Synthetic polyamines as vectors for gene delivery. Polym. Int. 2002, 51, 948–957.
- Holappa, S.; Andersson, T.; Kantonen, L.; Plattner, P.; Tenhu, H. Soluble polyelectrolyte complexes composed of poly(ethylene oxide)-block-poly(sodium methacrylate) and poly(methacryloyloxyethyl trimethylammonium chloride). Polymer 2003, 44, 7907–7916. [CrossRef]
- Winnik, M. A.; Bystryak, S. M.; Chassenieux, C.; Strashko, V.; Macdonald, P. M.; Siddiqui, J. Study of Interaction of Poly(ethylene imine) with Sodium Dodecyl Sulfate in Aqueous Solution by Light Scattering. Langmuir 2000, 16, 4495–4510. [CrossRef]
- Bystryak, S. M.; Winnik, M. A.; Siddiqui, J. Unusual conductivity changes for sodium dodecyl sulfate solutions in the presence of polyethyleneimine and polyvinylamine. Langmuir 1999, 15, 3748–3751. [CrossRef]
- Voets, I. K.; Fokkink, R.; Hellweg, T.; King, S. M.; de Waard, P.; de Keizer, A.; Stuart, M. A. C. Spontaneous symmetry breaking: formation of Janus micelles. Soft Matter 2009, 5, 999–1005.
- Annaka, M.; Morishita, K.; Okabe, S. Electrostatic Self-Assembly of Neutral and Polyelectrolyte Block Copolymers and Oppositely Charged Surfactant. J. Phys. Chem. B 2007, 111, 11700–11707. [CrossRef]
- Berret J.-F.; Vigolo, B.; Eng, R.; Hervé, P.; Grillo, I.; Yang, L. Electrostatic Self-assembly of Oppositely Charged Copolymers and Surfactants: A Light, Neutron, and X-ray Scattering Study. Macromolecules 2004, 37, 4922–4930. [CrossRef]
- Gohy, J. F.; Mores, S.; Varshney, S. K.; Jérôme, R. Self-Organization of WaterSoluble Complexes of a Poly (2-vinylpyridinium)-block-poly (ethylene oxide) Diblock with Fluorinated Anionic Surfactants. Macromolecules 2003, 36, 2579–2581.
- Laschewsky, A.; Mertoglu, M.; Kubowicz, S.; Thunemann, A. F. Lamellar Structured Nanoparticles Formed by Complexes of a Cationic Block Copolymer and Perfluorodecanoic Acid. Macromolecules 2006, 39, 9337–9345. [CrossRef]
- Elsabahy, M.; Zhang, M.; Gan, S. M.; Waldron, K. C.; Leroux, J. C. Synthesis and enzymatic stability of PEGylated oligonucleotide duplexes and their self-assemblies with polyamidoamine dendrimers. Soft Matter 2008, 4, 294–302. [CrossRef]
- Harada, A.; Togawa, H.; Kataoka, K.; Physicochemical properties and nuclease resistance of antisense-oligodeoxynucleotides entrapped in the core of polyion complex micelles composed of poly(ethylene glycol)-poly(L-Lysine) block copolymers. Eur. J. Pharm. Sci. 2001, 13, 35–42. [CrossRef]
- Kabanov, A. V.; Vinogradov, S. V.; Suzdaltseva, Y. G.; Alakhov, V. Y. Water-Soluble Block Polycations as Carriers for Oligonucleotide Delivery. Bioconjug. Chem. 1995, 6, 639–643. [CrossRef]
- Vinogradov, S. V.; Bronich, T. K.; Kabanov, A. V. Self-assembly of polyamine-poly(ethylene glycol) copolymers with phosphorothioate oligonucleotides. Bioconjug. Chem. 1998, 9, 805–812. [CrossRef]
- Dufresne, M. H.; Gauthier, M. A.; Leroux, J. C. Thiol-functionalized polymeric micelles: from molecular recognition to improved mucoadhesion. Bioconjug. Chem. 2005, 16, 1027–1033. [CrossRef]
- Jo, J. I.; Nagaya, N.; Miyahara, Y.; Kataoka, M.; Harada-Shiba, M.; Kangawa, K.; Tabata, Y. Transplantation of genetically engineered mesenchymal stem cells improves cardiac function in rats with myocardial infarction: benefit of a novel nonviral vector, cationized dextran. Tissue Eng. 2007, 13, 313–322.
- Alvarez-Lorenzo, C.; Barreiro-Iglesias, R.; Concheiro, A; Iourtchenko, L.; Alakhov, V.; Bromberg, L.; Temchenko, M.; Deshmukh, S.; Hatton, T. A. Biophysical characterization of complexation of DNA with block copolymers of poly(2-dimethylaminoethyl) methacrylate, poly(ethylene oxide), and poly(propylene oxide). Langmuir 2005, 21, 5142–5148.
- Wakebayashi, D.; Nishiyama, N.; Yamasaki, Y.; Itaka, K.; Kanayama, N.; Harada, A.; Nagasaki, Y.; Kataoka, K. Lactose-conjugated polyion complex micelles incorporating plasmid DNA as a targetable gene vector system: their preparation and gene transfecting efficiency against cultured HepG2 cells. J. Control. Release 2004, 95, 653–664.
- Wakebayashi, D.; Nishiyama, N.; Itaka, K.; Miyata, K.; Yamasaki, Y.; Harada, A.; Koyama, H.; Nagasaki, Y.; Kataoka, K., Polyion Complex Micelles of pDNA with Acetalpoly(ethylene glycol)-poly(2-(dimethylamino)ethyl methacrylate) Block Copolymer as the Gene Carrier System: Physicochemical Properties of Micelles Relevant to Gene Transfection Efficacy. Biomacromolecules 2004, 5, 2128–2136.
- Kataoka, K.; Harada, A.; Wakebayashi, D.; Nagasaki, Y. Polyion complex micelles with reactive aldehyde groups on their surface from plasmid DNA and end-functionalized charged block copolymers. Macromolecules 1999, 32, 6892–6894. [CrossRef]
- Harada-Shiba, M.; Yamauchi, K.; Harada, A.; Takamisawa, I.; Shimokado, K.; Kataoka, K. Polyion complex micelles as vectors in gene therapy--pharmacokinetics and in vivo gene transfer. Gene Ther. 2002, 9, 407–414. [CrossRef]
- Wolfert, M. A.; Schacht, E. H.; Toncheva, V.; Ulbrich, K.; Nazarova, O.; Seymour, L. W. Characterization of vectors for gene therapy formed by self-assembly of DNA with synthetic block co-polymers. Hum. Gene Ther. 1996, 7, 2123–2133. [CrossRef]
- Itaka, K.; Yamauchi, K.; Harada, A.; Nakamura, K.; Kawaguchi, H.; Kataoka, K. Polyion complex micelles from plasmid DNA and poly (ethylene glycol)–poly (l-lysine) block copolymer as serum-tolerable polyplex system: physicochemical properties of micelles relevant to gene transfection efficiency. Biomaterials 2003, 24, 4495–4506.
- Itaka, K.; Harada, A.; Nakamura, K.; Kawaguchi, H.; Kataoka K. Evaluation by fluorescence resonance energy transfer of the stability of nonviral gene delivery vectors under physiological conditions. Biomacromolecules 2002, 3, 841–845. [CrossRef]
- Choi, J. S., Lee, E. J., Choi, Y. H., Jeong, Y. J., Park, J. S. Poly (ethylene glycol)-block-poly (L-lysine) dendrimer: novel linear polymer/dendrimer block copolymer forming a spherical water-soluble polyionic complex with DNA. Bioconjug. Chem. 1999, 10, 62–65. [CrossRef]
- Oupický, D., Koňák, Č., Dash, P. R., Seymour, L. W., Ulbrich, K. (). Effect of albumin and polyanion on the structure of DNA complexes with polycation containing hydrophilic nonionic block. Bioconjug. Chem. 1999, 10, 764–772. [CrossRef]
- Oupický, D.; Koňák, Č.; Ulbrich, K. Preparation of DNA Complexes with Diblock Copolymers of Poly[N(2-Hydroxypropyl)Methacrylamide] and Polycations. Mater. Sci. Eng. C 1999, 7, 59–65. [CrossRef]
- Oupicky, D.; Konak, C.; Ulbrich, K. DNA complexes with block and graft copolymers of N-(2-hydroxypropyl)methacrylamide and 2-(trimethylammonio)ethyl methacrylate. J. Biomater. Sci. Polym. Ed. 1999, 10, 573–590. [CrossRef]
- Lee, H.; Jeong, J. H.; Park, T. G. A new gene delivery formulation of polyethylenimine/DNA complexes coated with PEG conjugated fusogenic peptide. J. Control. Release 2001, 76, 183–192. [CrossRef]
- Caputo, A.; Betti, M.; Altavilla, G.; Bonaccorsi, A.; Boarini, C.; Marchisio, M.; Buttò, S.; Sparnacci, K.; Laus, M.; Tondelli, L.; Ensoli, B. Micellar-type complexes of tailor-made synthetic block copolymers containing the HIV-1 tat DNA for vaccine application. Vaccine 2002, 20, 2303–2317.
- Laus, M.; Sparnacci, K.; Ensoli, B.; Buttò, S. O; Caputo, A.; Mantovani, I.; Zuccheri, G.; Samori, B.; Tondelli, L. Complex associates of plasmid DNA and a novel class of block copolymers with PEG and cationic segments as new vectors for gene delivery. J. Biomater. Sci. Polym. Ed. 2001, 12, 209–228. [CrossRef]
- Tan, J. F.; Too, H. P.; Hatton, T. A.; Tam, K. C. Aggregation behavior and thermodynamics of binding between poly (ethylene oxide)-block-poly (2-(diethylamino) ethyl methacrylate) and plasmid DNA. Langmuir 2006, 22, 3744–3750. [CrossRef]
- Liu, B.; Bazan, G. C. Homogeneous fluorescence-based DNA detection with water-soluble conjugated polymers. Chem. Mater. 2004, 16, 4467–4476. [CrossRef]
- Liu, B.; Bazan, G. C. Interpolyelectrolyte complexes of conjugated copolymers and DNA: Platforms for multicolor biosensors. J. Am. Chem. Soc. 2004, 126, 1942–1943. [CrossRef]
- Scales, C. W.; Huang, F. Q.; Li, N.; Vasilieva, Y. A, Ray, J.; Convertine, A. J.; McCormick, C. L. Corona-stabilized interpolyelectrolyte complexes of SiRNA with nonimmunogenic, hydrophilic/cationic block copolymers prepared by aqueous RAFT polymerization. Macromolecules 2006, 39, 6871–6881. [CrossRef]
- Itaka, K.; Kanayama, N.; Nishiyama, N.; Jang, W. D.; Yamasaki, Y.; Nakamura, K.; Kawaguchi, H.; Kataoka, K. Supramolecular nanocarrier of siRNA from PEG-based block catiomer carrying diamine side chain with distinctive pKa directed to enhance intracellular gene silencing. J. Am. Chem. Soc. 2004, 126, 13612–13613.
- Bronstein, L. H.; Sidorov, S. N.; Valetsky, P. M.; Hartmann, J.; Colfen, H.; Antonietti, M. Induced micellization by interaction of poly(2-vinylpyridine)-block-poly(ethylene oxide) with metal compounds. Micelle characteristics and metal nanoparticle formation. Langmuir 1999, 15, 6256–6262.
- Vamvakaki, M.; Papoutsakis, L.; Katsamanis, V.; Afchoudia, T.; Fragouli, P. G.; Iatrou, H.; Hadjichristidis, N.; Armes, S. P.; Sidorov, S.; Zhirov, D.; Zhirov, V.; Kostylev, M.; Bronstein, L. M.; Anastasiadis, S. H. Micellization in pH-sensitive amphiphilic block copolymers in aqueous media and the formation of metal nanoparticles. Faraday Discuss. 2005, 128, 129–147.
- Bronstein, L. M.; Sidorov, S. N.; Zhirov, V.; Zhirov, D.; Kabachii, Y. A.; Kochev, S. Y.; Valetsky, P. M.; Stein, B.; Kiseleva, O. I.; Polyakov, S. N.; Shtykova, E. V.; Nikulina, E. V.; Svergun, D. I.; Khokhlov, A. R. Metalated Diblock and Triblock Poly(ethylene oxide)-block-poly(4-vinylpyridine) Copolymers: Understanding of Micelle and Bulk Structure. J. Phys. Chem. B 2005, 109, 18786–18798.
- Berret, J. F.; Schonbeck, N.; Gazeau, F.; El Kharrat, D.; Sandre, O.; Vacher, A.; Airiau, M. Controlled clustering of superparamagnetic nanoparticles using block copolymers: design of new contrast agents for magnetic resonance imaging. J. Am. Chem. Soc. 2006, 128, 1755–1761. [CrossRef]
- Berret J-F. Stoichiometry of electrostatic complexes determined by light scattering. Macromolecules 2007, 40, 4260–4266. [CrossRef]
- Dufresne, M. H.; Leroux, J. C. Study of the Micellization Behavior of Different Order Amino Block Copolymers with Heparin. Pharm. Res. 2004, 21, 160–169. [CrossRef]
- Gohy, J. F.; Varshney, S. K.; Antoun, S.; Jerome, R. Water-soluble complexes formed by sodium poly(4-styrenesulfonate) and a poly(2-vinylpyridinium)-block-poly(ethyleneoxide) copolymer. Macromolecules 2000, 33, 9298–9305. [CrossRef]
- Gohy, J. F.; Varshney, S. K.; Jerome, R. Water-Soluble Complexes Formed by Poly (2-vinylpyridinium)-block-poly (ethylene oxide) and Poly (sodium methacrylate)-block-poly (ethylene oxide) Copolymers. Macromolecules 2001, 34, 3361–3366. [CrossRef]
- van der Burgh, S. 1240. van der Burgh, S. Complex Coacervate Core Micelles in Solution and at Interfaces. PhD thesis, Wageningen Universiteit, Wageningen, The Netherlands, ; pp 1-134. https://edepot.wur. 26 April 1935. [Google Scholar]
- Li, Y.; Nakashima, K. Fluorescence Studies on the Properties of the Nanoaggregates of Poly(ethylene oxide)-b-polymethacrylate Copolymer Formed by Binding of Cationic Surfactants to Polymethacrylate Block. Langmuir 2003, 19, 548–553. [CrossRef]
- Bronich, T. K.; Ming, O. Y.; Kabanov, V. A.; Eisenberg, A.; Szoka, F. C.; Kabanov, A. V. Synthesis of vesicles on polymer template. J. Am. Chem. Soc. 2002, 124, 11872–11873.
- Bronich, T. K., Popov, A. M., Eisenberg, A., Kabanov, V. A., Kabanov, A. V. Effects of block length and structure of surfactant on self-assembly and solution behavior of block ionomer complexes. Langmuir 2000, 16, 481–489. [CrossRef]
- Kabanov, A. V.; Bronich, T. K.; Kabanov, V. A.; Yu, K.; Eisenberg, A. Spontaneous formation of vesicles from complexes of block ionomers and surfactants. J. Am. Chem. Soc. 1998, 120, 9941–9942. [CrossRef]
- Solomatin, S. V.; Bronich, T. K.; Eisenberg, A.; Kabanov, V. A.; Kabanov, A. V., Nanomaterials from Ionic Block Copolymers and Single-, Double-, and Triple-Tail Surfactants. Langmuir 2007, 23, 2838–2842.
- Nagata, I.; Morawetz, H. Study of the association of anionic and cationic polymers by nonradiative energy transfer. Macromolecules 1981, 14, 87–91. [CrossRef]
- Maruyama, A.; Katoh, M.; Ishihara, T.; Akaike, T. Comb-type polycations effectively stabilize DNA triplex. Bioconjug. Chem. 1997, 8, 3–6. [CrossRef]
- Choi, Y. H.; Liu, F.; Choi, J. S.; Kim, S. W.; Park, J. S. Characterization of a targeted gene carrier, lactose-polyethylene glycol-grafted poly-L-lysine and its complex with plasmid DNA. Hum. Gene Ther. 1999, 10, 2657–2665. [CrossRef]
- Harada, A.; Kataoka K. Pronounced activity of enzymes through the incorporation into the core of polyion complex micelles made from charged block copolymers. J. Control. Release. 2001, 72, 85–91.
- Weaver, J. V. M.; Armes, S. P.; Liu, S. Y. A “holy trinity” of micellar aggregates in aqueous solution at ambient temperature: unprecedented self-assembly behavior from a binary mixture of a neutral-cationic diblock copolymer and an anionic polyelectrolyte. Macromolecules 2004, 36, 9994–9998.
- Gohy, J. F.; Varshney, S. K.; Jerome, R. Morphology of water-soluble interpolyelectrolyte complexes formed by poly (2-vinylpyridinium) -block-poly (ethylene oxide) diblocks and poly (4-styrenesulfonate) polyanions. Macromolecules 2001, 34, 2745–2747. [CrossRef]
- Yan, Y.; Besseling, N. A. M.; de Keizer, A.; Drechsler, M.; Fokkink, R.; Cohen Stuart, M. A. Wormlike aggregates from a supramolecular coordination polymer and a diblock copolymer. J. Phys. Chem. B 2007, 111, 11662–11669. [CrossRef]
- Yan, Y.; Besseling, N. A. M.; de Keizer, A.; Cohen Stuart, M. A. Characteristic differences in the formation of complex coacervate core micelles from neodymium and zinc-based coordination polymers. J. Phys. Chem. B 2007, 111, 5811–5818. [CrossRef]
- Berret, J.-F.; Sehgal, A.; Morvan, M.; Sandre, O.; Vacher, A.; Airiau, M. Stable oxide nanoparticle clusters obtained by complexation. J. Colloid Interface Sci. 2006, 303, 315–318. [CrossRef]
- Qi, L.; Chapel, J.-P.; Castaing, J.-P.; Fresnais, J.; Berret, J.-F. Stability and Adsorption Properties of Electrostatic Complexes: Design of Hybrid Nanostructures for Coating Applications. Langmuir 2007, 23, 11996–11998. [CrossRef]
- Wu, K.; Shi, L. Q.; Zhang, W. Q.; An, Y. L.; Zhang, X.; Li, Z. Y.; Zhu, X. X. Thermoresponsiveness of hybrid micelles from poly(ethylene glycol)-block-poly(4-vinylpyridium) cations annul SO42− anions in aqueous solutions. Langmuir 2006, 22, 1474–1477. [CrossRef]
- Jang, W. D.; Nishiyama, N.; Zhang, G. D.; Harada, A.; Jiang, D. L.; Kawauchi, S.; Morimoto, Y.; Kikuchi, M.; Koyama, H.; Aida, T.; Kataoka, K. Supramolecular Nanocarrier of Anionic Dendrimer Porphyrins with Cationic Block Copolymers Modified with Polyethylene Glycol to Enhance Intracellular Photodynamic Efficacy. Angew. Chem. Int. Ed. Engl. 2005, 44, 419–423.
- Li, Y.; Jang, W-D.; Nishiyama, N.; Kishimura, A.; Kawauchi, S.; Morimoto, Y.; Miake, S.; Yamashita, T.; Kikuchi, M.; Aida, T.; Kataoka, K. Dendrimer generation effects on photodynamic efficacy of dendrimer porphyrins and dendrimer-loaded supramolecular nanocarriers. Chem. Mater. 2007, 19, 5557–5562.
- Ideta, R.; Tasaka, F.; Jang, W. D.; Nishiyama, N.; Zhang, G. D.; Harada, A. Nanotechnology-based photodynamic therapy for neovascular disease using a supramolecular nanocarrier loaded with a dendritic photosensitizer. Nano Lett. 2005, 5, 2426–2431. [CrossRef]
- Karanikolopoulos, N.; Pitsikalis, M.; Hadjichristidis, N.; Georgikopoulou, K.; Calogeropoulou, T.; Dunlap, J. R. pH-Responsive aggregates from double hydrophilic block copolymers carrying zwitterionic groups. Encapsulation of antiparasitic compounds for the treatment of leishmaniasis. Langmuir 2007, 23, 4214–4224.
- Katakura, H., Harada, A., Kataoka, K., Furusho, M., Tanaka, F., Wada, H., & Ikenaka, K. Improvement of retroviral vectors by coating with poly (ethylene glycol)-poly (L-lysine) block copolymer (PEG-PLL). J. Gene Med. A cross-disciplinary journal for research on the science of gene transfer and its clinical applications. 2004, 6, 471–477.
- Dautzenberg, H. Polyelectrolyte complex formation in highly aggregating systems. 1. Effect of salt: polyelectrolyte complex formation in the presence of NaCl. Macromolecules 1997, 30, 7810–7815. [CrossRef]
- Dautzenberg, H.; Karibyants, N. Polyelectrolyte complex formation in highly aggregating systems. Effect of salt: response to subsequent addition of NaCl. Macromol. Chem. Phys. 1999, 200, 118–125. [CrossRef]
- Dautzenberg, H.; Kriz, J. Response of polyelectrolyte complexes to subsequent addition of salts with different cations. Langmuir 2003, 19, 5204–5211. [CrossRef]
- Ganguli, M.; Jayachandran, K. N.; Maiti, S. Nanoparticles from cationic copolymer and DNA that are soluble and stable in common organic solvents. J. Am. Chem. Soc. 2004, 126, 26–27. [CrossRef]
- Nisha, C. K.; Manorama, S. V.; Ganguli, M.; Maiti, S.; Kizhakkedathu, J. N. Complexes of poly (ethylene glycol)-based cationic random copolymer and calf thymus DNA: a complete biophysical characterization. Langmuir 2004, 20, 2386–2396. [CrossRef]
- Asayama, S.; Maruyama, A.; Cho, C. S.; Akaike, T. Design of comb-type polyamine copolymers for a novel pH-sensitive DNA carrier. Bioconjug. Chem. 1997, 8, 833–838. [CrossRef]
- Tsolakis, P.; Bokias, G. Temperature-sensitive water-soluble polyelectrolyte/surfactant complexes formed between dodecyltrimethylammonium bromide and a comb-type copolymer consisting of an anionic backbone and poly(N-isopropylacrylamide) side chains. Macromolecules 2006, 39, 393–398. [CrossRef]
- Khanal, A.; Nakashima, Y.; Li, Y.; Nakashima, K.; Kawasaki, N.; Oishi, Y. Fabrication of nanoaggregates of poly(ethylene oxide)-b-polymethacrylate by complex formation with chitosan or methylglycolchitosan. Colloids and Surfaces A: Physicochem. Eng. Aspects 2005, 260, 129–135. [CrossRef]
- Voets, I. K.; de Keizer, A.; Cohen Stuart, M. A.; de Waard, P. Core and corona structure of mixed polymeric micelles. Macromolecules 2006, 39, 5952–5955. [CrossRef]
- Voets, I. K.; de Keizer, A.; de Waard, P.; Frederik, P. M.; Bomans, P. H. H.; Schmalz, H.; Walther, A.; King, S. M.; Leermakers, F. A. M.; Cohen Stuart, M. A. Angew. Chem. Int. Ed. 2006, 45, 6673–6676.
- Voets, I. K.; Fokkink R.; De Keizer A.; May R. P.; De Waard P.; Cohen Stuart M. A. On the Transition between a Heterogeneous and Homogeneous Corona in Mixed Polymeric Micelles. Langmuir 2008, 24, 12221–12227. [CrossRef]
- Serefoglou, E.; Oberdisse, J.; Staikos, G. Characterization of the soluble nanoparticles formed through coulombic interaction of bovine serum albumin with anionic graft copolymers at low pH. Biomacromolecules 2007, 8, 1195–1199. [CrossRef]
- Bastardo, L. A.; Iruthayaraj, J.; Lundin, M.; Dedinaite, A.; Vareikis, A.; Makuška, R.; van der Wal, A.; Furó, I.; Garamus, V. M.; Claesson, P. M. Soluble Complexes in Aqueous Mixtures of Low Charge Density Comb Polyelectrolyte and Oppositely Charged Surfactant Probed by Scattering and NMR. J. Colloid Interface Sci. 2007, 312, 21–33.
- Berret, J-F. Evidence of overcharging in the complexation between oppositely charged polymers and surfactants. J. Chem. Phys. 2005, 123, 164703–164712. [CrossRef]
- Sanson, N.; Bouyer, F.; Gerardin, C.; In, M. Nanoassemblies formed from hydrophilic block copolymers and multivalent ions. Phys. Chem. Chem. Phys. 2004, 6, 1463–1466. [CrossRef]
- Sanson, N.; Putaux, J. L.; Destarac, M.; Gerardin, C.; Fajula, F. Hybrid Organic-Inorganic Colloids with a Core-Corona Structure: A Transmission Electron Microscopy Investigation. Macromol. Symp. 2005, 226, 279–288. [CrossRef]
- Gerardin, C.; Sanson, N.; Bouyer, F.; Fajula, F.; Putaux, J. L.; Joanicot, M.; Chopin, T. Highly Stable Metal Hydrous Oxide Colloids by Inorganic Polycondensation in Suspension. Angew. Chem. Int. Ed. 2003, 42, 3681–3685. [CrossRef]
- Bouyer, F.; Gérardin, C.; Fajula, F.; Putaux, J. L.; Chopin, T. Role of doublehydrophilic block copolymers in the synthesis of lanthanum-based nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2003, 217, 179–184. [CrossRef]
- Berret, J-F.; Sandre, O.; Mauger, A. Size distribution of superparamagnetic particles determined by magnetic sedimentation. Langmuir 2007, 23, 2993–2999. [CrossRef]
- Bouyer, F.; Sanson, N.; Destarac, M.; Gerardin, C. Hydrophilic block copolymer-directed growth of lanthanum hydroxidenanoparticles. New J. Chem. 2006, 30, 399–408. [CrossRef]
- Gu, C. F.; Chen, D. Y.; Jiang, M. Short-Life Core-Shell Structured Nanoaggregates Formed by the Self-Assembly of PEO-b-PAA/ETC (1-(3-Dimethylamino- propyl)-3-ethylcarbodiimide Methiodide) and Their Stabilization. Macromolecules 2004, 37, 1666–1669. [CrossRef]
- Khullar, P.; Singh, V.; Mahal, A.; Kumar, H.; Kaur, G.; Bakshi, M. S. Block Copolymer Micelles as Nanoreactors for Self-assembled Morphologies of Gold Nanoparticles. J. Phys. Chem. B 2013, 117, 3028–3039. [CrossRef]
- Mathot, F.; van Beijsterveldt, L.; Preat, V.; Brewster, M.; Arien, A. Intestinal Uptake and Biodistribution of Novel Polymeric Micelles after Oral Administration. J. Control. Release 2006, 111, 47–55. [CrossRef]
- Wu, F.-G.; Jiang, Y.-W.; Chen, Z.; Yu, Z.-W. Folding Behaviors of Protein (Lysozyme) Confined in Polyelectrolyte Complex Micelle. Langmuir 2016, 32, 3655–3664. [CrossRef]
- Nolles, A.; Westphal, A. H.; de Hoop, J. A.; Fokkink, R. G.; Kleijn, J. M.; van Berkel, W. J. H.; Borst, J. W. Encapsulation of GFP in Complex Coacervate Core Micelles. Biomacromolecules 2015, 16, 1542–1549.
- Sant, V. P.; Smith, D.; Leroux, J.-C. Enhancement of Oral Bioavailability of Poorly Water-Soluble Drugs by Poly(Ethylene Glycol)-block-Poly(Alkyl Acrylate-co-Methacrylic Acid) Self-Assemblies. J. Control. Release 2005, 104, 289–300. [CrossRef]
- Kotz, J.; Kosmella, S.; Beitz, T. Self-assembled Polyelectrolyte Systems. Prog. Polym. Sci. 2001, 26, 1199–1232. [CrossRef]
- Cooper, C. L.; Dubin, P. L.; Kayitmazer, A. B.; Turksen, S. Polyelectrolyte-Protein Complexes. Curr. Opin. Colloid Interface Sci. 2005, 10, 52–78.
- Blocher, W. C.; Perry, S. L. Complex Coacervate-Based Materials for Biomedicine. WIREs Nanomed. Nanobiotechnol. 2017, 9 (4), No. e1442.
- Gapinśki, J.; Szymanśki, J.; Wilk, A.; Kohlbrecher, J.; Patkowski, A.; Hołyst, R. Size and Shape of Micelles Studied by Means of SANS, PCS, and FCS. Langmuir 2010, 26, 9304–9314. [CrossRef]
- Nolles, A.; Westphal, A. H.; Kleijn, J. M.; van Berkel, W. J. H.; Borst, J. W. Colorful Packages: Encapsulation of Fluorescent Proteins in Complex Coacervate Core Micelles. Int. J. Mol. Sci. 2017, 18, 1557–1576. [CrossRef]
- Elson, E. L. Fluorescence Correlation Spectroscopy: Past, Present, Future. Biophys. J. 2011, 101, 2855–2870. [CrossRef]
- Martins, L. O.; Soares, C. M.; Pereira, M. M.; Teixeira, M.; Costa, T.; Jones, G. H.; Henriques, A. O. Molecular and Biochemical Characterization of a Highly Stable Bacterial Laccase that Occurs as a Structural Component of the Bacillus subtilis Endospore Coat. J. Biol. Chem. 2002, 277, 18849–18859.
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszynska, D. Immobilization as Strategy for Improving Enzyme Properties-Application to Oxidoreductases. Molecules 2014, 19, 8995–9018. [CrossRef]
- Enguita, F. J.; Martins, L. O.; Henriques, A. O.; Carrondo, M. A. Crystal Structure of a Bacterial Endospore Coat Component. A Laccase with Enhanced Thermostability Properties. J. Biol. Chem. 2003, 278, 19416–19425.
- Christopher, L. P.; Yao, B.; Ji, Y. Lignin Biodegradation with Laccase-Mediator Systems. Front. Energy Res. 2014, 2, 1–13.
- Shraddha; Shekher, R.; Sehgal, R.; Kamthania, M.; Kumar, A. Laccase: Microbial Sources, Production, Purification, and Potential Biotechnological Applications. Enzyme Res. 2011, 2011, 217861.
- Fernandes, A. T.; Lopes, C.; Martins, L. O.; Melo, E. P. Unfolding Pathway of CotA-Laccase and the Role of Copper on the Prevention of Refolding through Aggregation of the Unfolded State. Biochem. Biophys. Res. Commun. 2012, 422, 442–446. [CrossRef]
- Beneyton, T.; Beyl, Y.; Guschin, D. A.; Griffiths, A. D.; Taly, V.; Schuhmann, W. The Thermophilic CotA Laccase from Bacillus subtilis: Bioelectrocatalytic Evaluation of O2 Reduction in the Direct and Mediated Electron Transfer Regime. Electroanalysis 2011, 23, 1781–1789. [CrossRef]
- Liu, Z.; Xie, T.; Zhong, Q.; Wang, G. Crystal Structure of CotA Laccase Complexed with 2,2-azinobis-(3-ethylbenzothiazoline-6-sulfonate) at a Novel Binding Site. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 328–335. [CrossRef]
- Bryjak, J.; Kruczkiewicz, P.; Rekuc, A.; Peczynska-Czoch, W. Laccase Immobilization on Copolymer of Butyl Acrylate and Ethylene Glycol Dimethacrylate. Biochem. Eng. J. 2007, 35, 325–332. [CrossRef]
- Pich, A.; Bhattacharya, S.; Adler, H.-J. P.; Wage, T.; Taubenberger, A.; Li, Z.; van Pee, K.-H.; Böhmer, U.; Bley, T. Composite Magnetic Particles as Carriers for Laccase from Trametes versicolor. Macromol. Biosci. 2006, 6, 301–310. [CrossRef]
- Movassaghian, S.; Merkel, O. M.; Torchilin, V. P. Applications of polymer micelles for imaging and drug delivery. WIREs Nanomed. Nanobiotechnol. 2015, 7, 691–707. [CrossRef]
- Zheng, C. X.; Zhao, Y.; Liu, Y. Recent advances in self-assembled nano-therapeutics. Chin. J. Polym. Sci. 2018, 36, 322–346. [CrossRef]
- Martinelli, C.; Pucci, C.; Ciofani, G. Nanostructured carriers as innovative tools for cancer diagnosis and therapy. APL Bioeng. 2019, 3, 011502. [CrossRef]
- Majumder, N.; Das, N. G.; Das, S. K. Polymeric micelles for anticancer drug delivery. Ther. Deliv. 2020, 11, 613–635. [CrossRef]
- Perin, F.; Motta, A.; Maniglio, D. Amphiphilic copolymers in biomedical applications: Synthesis routes and property control. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 123, 111952. [CrossRef]
- Ghosh, B.; Biswas, S. Polymeric micelles in cancer therapy: State of the art. J. Control. Release 2021, 332, 127–147. [CrossRef]
- Shu, J. Y.; Panganiban, B.; Xu, T. Peptide-polymer conjugates: from fundamental science to application. Annu. Rev. Phys. Chem. 2013, 64, 631–657. [CrossRef]
- Wilson, P. Synthesis and Applications of Protein/Peptide-Polymer Conjugates. Macromol. Chem. Phys. 2017, 218, 1600595. [CrossRef]
- Paik, B. A.; Mane, S. R.; Jia, X.; Kiick, K. L. Responsive Hybrid (Poly)peptide-Polymer Conjugates. J. Mater. Chem. B. 2017, 5, 8274–8288. [CrossRef]
- Stevens, C.A.; Kaur, K.; Klok, H.-A. Self-Assembly of Protein-Polymer Conjugates for Drug Delivery. Adv. Drug Deliv. Rev. 2021, 174, 447–460. [CrossRef] [PubMed]
- Lu, Y.; Zhang, E.; Yang, J.; Cao, Z. Strategies to improve micelle stability for drug delivery. Nano Res. 2018, 11, 4985–4998. [CrossRef] [PubMed]
- Cagel, M.; Tesan, F. C.; Bernabeu, E.; Salgueiro, M. J.; Zubillaga, M. B.; Moretton, M. A.; Chiappetta, D. A. Polymeric mixed micelles as nanomedicines: Achievements and perspectives. Eur. J. Pharm. Biopharm. 2017, 113, 211–228. [CrossRef]
- Mi, P.; Cabral, H.; Kataoka, K. Ligand-Installed Nanocarriers toward Precision Therapy. Adv. Mater. 2020, 32, 1902604. [CrossRef]
- Wu, T.; Wang, L. H.; Ding, S. G.; You, Y. Z. Fluorinated PEG-polypeptide polyplex micelles have good serum-resistance and low cytotoxicity for gene delivery. Macromol. Biosci. 2017, 17, 1700114.
- Chen, G.; Wang, K.; Wang, Y.; Wu, P.; Sun, M.; Oupický, D. Fluorination Enhances Serum Stability of Bioreducible Poly(amido amine) Polyplexes and Enables Efficient Intravenous siRNA Delivery. Adv. Healthcare Mater. 2018, 7, 1700978. [CrossRef]
- Zhang, C.; Liu, T.; Wang, W.; Bell, C. A.; Han, Y.; Fu, C.; Peng, H.; Tan, X.; Král, P.; Gaus, K.; Gooding, J. J.; Whittaker, A. K. Tuning of the Aggregation Behavior of Fluorinated Polymeric Nanoparticles for Improved Therapeutic Efficacy. ACS Nano 2020, 14, 7425–7434. [CrossRef]
- 1320. Zerrillo, L.; Gupta, K.B.S.S.; Lefeber, F.A.W.M.; Da Silva, C.G.; Galli, F.; Chan, A.; Veltien, A.; Dou, W.; Censi, R.; Di Martino, P.; Srinivas, M.; Cruz, L. Pharmaceutics 2021, 13, 235.
- Yin, J.; Chen, Y.; Zhang, Z.-H.; Han, X. Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications. Polymers 2016, 8, 268. [CrossRef]
- Zhou, Q.; Zhang, L.; Yang, T.; Wu, H. Stimuli-responsive polymeric micelles for drug delivery and cancer therapy. Int J Nanomedicine. 2018, 13, 2921–2942. [CrossRef]
- Li, L.; Wang, J.; Kong, H.; Zeng, Y.; Liu, G. Functional biomimetic nanoparticles for drug delivery and theranostic applications in cancer treatment. Sci. Technol. Adv. Mater. 2018, 19, 771–790. [CrossRef]
- Mi, P. Stimuli-responsive nanocarriers for drug delivery, tumor imaging, therapy and theranostics. Theranostics 2020, 10, 4557–4588. [CrossRef]
- Dhara (Ganguly), M. Smart polymeric nanostructures for targeted delivery of therapeutics. J. Macromol. Sci. Part A 2020, 58, 269–284.
- Das, S. S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G. Z. Stimuli-Responsive Polymeric Nanocarriers for Drug Delivery, Imaging, and Theragnosis. Polymers 2020, 12, 1397. [CrossRef]
- Shahriari, M.; Torchilin, V. P.; Taghdisi, S. M.; Abnous, K.; Ramezani, M.; Alibolandi, M. “Smart” self-assembled structures: Toward intelligent dual responsive drug delivery systems. Biomater. Sci. 2020, 8, 5787–5803. [CrossRef] [PubMed]
- Mane, S. R.; Sathyan, A.; Shunmugam, R. Biomedical applications of pH-responsive amphiphilic polymer nanoassemblies. ACS Appl. Nano Mater. 2020, 3, 2104–2117. [CrossRef]
- Grimm, O.; Wendler, F.; Schacher, F. H. Micellization of Photo-Responsive Block Copolymers. Polymers 2017, 9, 396. [CrossRef]
- Tian, Q., Fei C., Yin H., Feng Y. Stimuli-responsive polymer wormlike micelles. Prog. Polym. Sci. 2019, 89, 108–132. [CrossRef]
- Yin, W.; Qiang, M.; Ke, W.; Han, Y.; Mukerabigwi, J. F.; Ge, Z. Hypoxia-responsive block copolymer radiosensitizers as anticancer drug nanocarriers for enhanced chemoradiotherapy of bulky solid tumors. Biomaterials 2018, 181, 360–371. [CrossRef]
- Quader, S.; Cabral, H.; Liu, X.; Paraiso, W. K. D.; Kinoh, H.; Kataoka, K. Engineered Nanomedicine Targets Intractable Cancers. Mater. Proc. 2021, 4, 84.
- Cao, Y.; Cao, J.; Zhang, J.; Zhang, D.; Li, M.; Xu, Y.; He, K.; Chen, G.; Yuan, C.; Dai, L. The research on multi-responsive azobenzene block copolymer and its self-assembly behavior. Polym. Adv. Technol. 2020, 31, 759–771. [CrossRef]
- Razavi, B.; Abdollahi, A.; Roghani-Mamaqani, H.; Salami-Kalajahi M. Light-, temperature-, and pH-responsive micellar assemblies of spiropyran-initiated amphiphilic block copolymers: Kinetics of photochromism, responsiveness, and smart drug delivery. Mater. Sci. Eng. C. 2020, 109, 110524. [CrossRef]
- Ji, C.; Deng, Y.; Yuan, H.; Yuan, W.; Song, Y.; Li, Z. Hypoxia/Temperature/pH Triple Stimuli–Responsive Block Copolymers: Synthesis, Self-Assembly, and Controlled Drug Release. Macromol. Mater. Eng. 2021, 306, 2100073. [CrossRef]
- Kim, J.; Shamul, J. G.; Shah, S. R.; Shin, A.; Lee, B. J.; Quinones-Hinojosa, A.; Green, J. J. Verteporfin-Loaded Poly(ethylene glycol)-Poly(beta-amino ester)-Poly(ethylene glycol) Triblock Micelles for Cancer Therapy. Biomacromolecules 2018, 19, 3361–3370. [CrossRef]
- Zhu, Q.; Zhang, B.; Wang, Y.; Liu, X.; Li, W.; Su, F.; Li, S. Self-assembled micelles prepared from poly(D, L -lactide-co-glycolide)-poly(ethylene glycol) block copolymers for sustained release of valsartan. Polym. Adv. Technol. 2021, 32, 1262–1271. [CrossRef]
- Zou, Y.; Meng, F.; Deng, C.; Zhong, Z. Robust, Tumor-Homing and Redox-Sensitive Polymersomal Doxorubicin: A Superior Alternative to Doxil and Caelyx? J. Control. Release 2016, 239, 149–158. [CrossRef] [PubMed]
- Chen, M.; Li, J. W.; Zhang, W. J.; Hong, C. Y.; Pan, C. Y. pH- and reductant-responsive polymeric vesicles with robust membrane-cross-linked structures: In situ cross-linking in polymerization-induced self-assembly. Macromolecules 2019, 52, 1140–1149. [CrossRef]
- Albuquerque, L. J. C.; Sincari, V.; Jäger, A.; Kucka, J.; Humajova, J.; Pankrac, J.; Paral, P.; Heizer, T.; Janouškova, O.; Davidovich, I.; Talmon, Y.; Pouckova, P.; Štěpánek, P.; Sefc, L.; Hruby, M.; Giacomelli, F. C.; Jäger, E. pH-responsive polymersome-mediated delivery of doxorubicin into tumor sites enhances the therapeutic efficacy and reduces cardiotoxic effects. J. Control. Release 2021, 332, 529–538.
- Nishida, H., Matsumoto, Y.; Kawana, K.; Christie, R. J.; Naito, M.; Kim, B. S.; Toh, K.; Min, H. S.; Yi, Y.; Matsumoto, Y.; Kim, H. J.; Miyata, K.; Taguchi, A.; Tomio, K.; Yamashita, A.; Inoue, T.; Nakamura, H.; Fujimoto, A.; Sato, M.; Yoshida, M.; Adachi, K.; Arimoto, T.; Wada-Hiraike, O.; Oda, K.; Nagamatsu, T.; Nishiyama, N.; Kataoka, K.; Osuga, Y.; Fujii, T. Systemic delivery of siRNA by actively targeted polyion complex micelles for silencing the E6 and E7 human papillomavirus oncogenes. J. Control. Release 2016, 231, 29–37.
- Feng, G.; Chen, H.; Li, J.; Huang, Q.; Gupte, M. J.; Liu, H.; Song, Y.; Ge, Z. Gene therapy for nucleus pulposus regeneration by heme oxygenase-1 plasmid DNA carried by mixed polyplex micelles with thermo-responsive heterogeneous coronas. Biomaterials 2015, 52, 1–13. [CrossRef]
- Yu, H.; Guo, C.; Feng, B.; Liu, J.; Chen, X.; Wang, D.; Teng, L.; Li, Y.; Yin, Q.; Zhang, Z.; Li, Y. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics 2016, 6, 14–27. [CrossRef]
- Tang, X.; Rao, J.; Yin, S.; Wei, J.; Xia, C.; Li, M.; Mei, L.; Zhang, Z.; He, Q. Eur. J. Pharm. Sci. 2019, 127, 161–174.
- Koji, K.; Yoshinaga, N.; Mochida, Y.; Hong, T.; Miyazaki, T.; Kataoka, K.; Osada, K.; Cabral, H.; Uchida, S. Bundling of mRNA strands inside polyion complexes improves mRNA delivery efficiency in vitro and in vivo. Biomaterials 2020, 261, 120332.
- Miyazaki, T.; Uchida, S.; Nagatoishi, S.; Koji, K.; Hong, T.; Fukushima, S.; Tsumoto, K.; Ishihara, K.; Kataoka, K.; Cabral, H. Polymeric nanocarriers with controlled chain flexibility boost mRNA delivery in vivo through enhanced structural fastening. Adv. Healthcare Mater. 2020, 9, 2000538. [CrossRef]
- Kanto R., Yonenuma R., Yamamoto M., Furusawa H., Yano S., Haruki M., Mori H. Mixed Polyplex Micelles with Thermoresponsive and Lysine-Based Zwitterionic Shells Derived from Two Poly(Vinyl Amine)-Based Block Copolymers. Langmuir 2021, 37, 3001–3014. [CrossRef]
- Hu, X.; Yu, J.; Qian, C.; Lu, Y.; Kahkoska, A. R.; Xie, Z.; Jing, X.; Buse, J. B.; Gu, Z. H2O2-Responsive Vesicles Integrated with Transcutaneous Patches for Glucose-Mediated Insulin Delivery. ACS Nano 2017, 11, 613–620. [CrossRef]
- Kamenova, K.; Haladjova, E.; Grancharov, G.; Kyulavska, M.; Tzankova, V.; Aluani, D.; Yoncheva, K.; Pispas, S.; Petrov, P. Co-assembly of block copolymers as a tool for developing novel micellar carriers of insulin for controlled drug delivery. Eur. Polym. J. 2018, 104, 1–9. [CrossRef]
- Kim, D. H.; Seo, Y. K.; Thambi, T.; Moon, G. J.; Son, J. P.; Li, G.; Park, J H.; Lee, J. H.; Kim, H. H.; Lee, D. S.; Bang, O. Y. Enhancing neurogenesis and angiogenesis with target delivery of stromal cell derived factor-1 alpha using a dual ionic pH-sensitive copolymer. Biomaterials. 2015, 61, 115–125.
- Kim, A.; Miura, Y.; Ishii, T.; Mutaf, O. F.; Nishiyama, N.; Cabral, H.; Kataoka, K. Intracellular Delivery of Charge-Converted Monoclonal Antibodies by Combinatorial Design of Block/Homo Polyion Complex Micelles. Biomacromolecules 2016, 17, 446–453. [CrossRef]
- Xie, J.; Gonzalez-Carter, D.; Tockary, T. A.; Nakamura, N.; Xue, Y.; Nakakido, M.; Akiba, H.; Dirisala, A.; Liu, X.; Toh, K.; Yang, T.; Wang, Z.; Fukushima, S.; Li, J.; Quader, S.; Tsumoto, K.; Yokota, T.; Anraku, Y.; Kataoka, K. Dual-sensitive nanomicelles enhancing systemic delivery of therapeutically active antibodies specifically into the brain. ACS Nano 2020, 14, 6729–6742.
- Tao, A.; Huang, G. L.; Igarashi, K.; Hong, T.; Liao, S.; Stellacci, F.; Matsumoto, Y.; Yamasoba, T.; Kataoka, K.; Cabral, H. Polymeric micelles loading proteins through concurrent ion complexation and pH-cleavable covalent bonding for in vivo delivery. Macromol. Biosci. 2020, 20, 1900161.
- Bai, T.; Shao, D.; Chen, J.; Li, Y.; Xu, B. B, Kong, J. pH-responsive dithiomaleimide-amphiphilic block copolymer for drug delivery and cellular imaging. J. Colloid Interface Sci. 2019, 552, 439–447. [CrossRef]
- Weng, J.; Huang, Z.; Pu, X.; Chen, X.; Yin, G.; Tian, Y.; Song, Y. Preparation of polyethylene glycol-polyacrylic acid block copolymer micelles with pH/hypoxic dual-responsive for tumor chemoradiotherapy. Colloids Surf. B Biointerfaces 2020, 191, 110943.
- Xiao, H.; Li, X.; Zheng, C.; Liu, Q.; Sun, C.; Huang, J.; Wang, Y.; Yuan, Y. Intracellular pH-responsive polymeric micelle for simultaneous chemotherapy and MR imaging of hepatocellular carcinoma. J Nanopart Res 2020, 22, 105. [CrossRef]
- Xiong, D.; Zhang, X.; Peng, S.; Gu, H.; Zhang, L. Smart pH-sensitive micelles based on redox degradable polymers as DOX/GNPs carriers for controlled drug release and CT imaging. Colloids Surf. B Biointerfaces 2017, 163, 29–40.
- Yan, K.; Zhang, S.; Zhang, K.; Miao, Y.; Qiu, Y.; Zhang, P.; Jia, X.; Zhao, X. Enzyme-responsive polymeric micelles with fluorescence fabricated through aggregation-induced copolymer self-assembly for anticancer drug delivery. Polym. Chem. 2020, 11, 7704–7713. [CrossRef]
- Lee, S-Y.; Yang, C-Y.; Peng, C-L.; Wei, M-F.; Chen, K-C.; Yao, C-J.; Shieh, M-J. A theranostic micelleplex co-delivering SN-38 and VEGF siRNA for colorectal cancer therapy. Biomaterials 2016, 86, 92–105. [CrossRef]
- Liu, Q.; Chen, S.; Chen, J.; Du, J. An asymmetrical polymer vesicle strategy for significantly improving T1 MRI sensitivity and cancer-targeted drug delivery. Macromolecules 2015, 48, 739–749. [CrossRef]
- Wang, F.; Gao, J.; Xiao, J.; Du, J. Dually gated polymersomes for gene delivery. Nano Lett. 2018, 18, 5562–5568. [CrossRef]
- Insua, I.; Wilkinson, A.; Fernandez-Trillo, F. Polyion complex (PIC) particles: Preparation and biomedical applications. Eur. Polym. J. 2016, 81, 198–215. [CrossRef]
- Abolmaali, S. S.; Tamaddon, A. M.; Salmanpour, M.; Mohammadi, S.; Dinarvand, R. Block ionomer micellar nanoparticles from double hydrophilic copolymers, classifications and promises for delivery of cancer chemotherapeutics. Eur. J. Pharm. Sci. 2017, 104, 393–405. [CrossRef]
- Pachioni-Vasconcelos, J. D. A.; Lopes, A. M.; Apolinário, A. C.; Valenzuela-Oses, J. K.; Costa, J. S. R.; Nascimento, L. D. O.; Pessoa, A.; Barbosa, L. R. S.; Rangel-Yagui, C. O. Nanostructures for protein drug delivery. Biomater. Sci. 2016, 4, 205–218.
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent Advances in Anti-cancer Protein/Peptide Delivery. Bioconjugate Chem. 2019, 30, 305–324. [CrossRef]
- Yousefpour Marzbali, M.; Yari Khosroushahi, A. Polymeric micelles as mighty nanocarriers for cancer gene therapy: a review. Cancer Chemother. Pharmacol. 2017, 79, 637–649. [CrossRef]
- Amjad, M.W.; Kesharwani, P.; Mohd Amin, M. C. I.; Iyer, A. K. Recent advances in the design, development, and targeting mechanisms of polymeric micelles for delivery of siRNA in cancer therapy. Prog. Polym. Sci. 2017, 64, 154–181. [CrossRef]
- Pereira-Silva, M.; Jarak, I.; Alvarez-Lorenzo, C.; Concheiro, A.; Santos, A. C.; Veiga, F.; Figueiras, A. Micelleplexes as nucleic acid delivery systems for cancer-targeted therapies. J. Control. Release. 2020, 323, 442–462. [CrossRef]
- Banik, B. L.; Fattahi, P.; Brown, J. L. Polymeric Nanoparticles: The Future of Nanomedicine: Polymeric Nanoparticles. WIREs Nanomed Nanobiotechnol. 2016, 8, 271–299. [CrossRef] [PubMed]
- Deshmukh, A. S.; Chauhan, P. N.; Noolvi, M. N.; Chaturvedi, K.; Ganguly, K.; Shukla, S. S.; Nadagouda, M. N.; Aminabhavi, T. M. Polymeric micelles: Basic research to clinical practice. Int. J. Pharm. 2017, 532, 249–268.
- Hwang, D.; Ramsey, J. D.; Kabanov, A. V. Polymeric micelles for the delivery of poorly soluble drugs: From nanoformulation to clinical approval. Adv. Drug Deliv. Rev. 2020, 156, 80–118. [CrossRef] [PubMed]
- Osorno, L. L.; Brandley, A. N.; Maldonado, D. E.; Yiantsos, A.; Mosley, R. J.; Byrne, M. E. Review of Contemporary Self-Assembled Systems for the Controlled Delivery of Therapeutics in Medicine. Nanomaterials 2021, 11, 278. [CrossRef] [PubMed]
- Park, K. Controlled drug delivery systems: Past forward and future back. J. Control. Release 2014, 190, 3–8. [CrossRef]
- Leroux J.-C. Editorial: Drug Delivery: Too Much Complexity, Not Enough Reproducibility? Angew. Chemie Int. Ed. 2017, 56, 15170–15171. [CrossRef]
- Lee, Y. W.; Luther, D. C.; Kretzmann, J. A.; Burden, A.; Jeon, T.; Zhai, S.; Rotello, V. M. Protein Delivery into the Cell Cytosol using Non-Viral Nanocarriers. Theranostics 2019, 9, 3280–3292. [CrossRef]
- Acar, H.; Srivastava, S.; Chung, E. J.; Schnorenberg, M. R.; Barrett, J. C.; LaBelle, J. L.; Tirrell, M. Self-assembling peptide-based building blocks in medical applications. Adv. Drug Delivery Rev.
- Lorenzer, C.; Dirin, M.; Winkler, A. M.; Baumann, V.; Winkler, J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [CrossRef]
- Mukalel, A. J.; Riley, R. S.; Zhang, R.; Mitchell, M. J. Nanoparticles for nucleic acid delivery: applications in cancer immunotherapy. Canc. Lett. 2019, 458, 102–112. [CrossRef]
- Peng, L.; Wagner, E. Polymeric Carriers for Nucleic Acid Delivery: Current Designs and Future Directions. Biomacromolecules 2019, 20, 3613–3626. [CrossRef]
- Kauffman, K. J.; Webber, M. J.; Anderson, D. G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [CrossRef]
- Stewart, M. P.; Sharei, A.; Ding, X. Y.; Sahay, G.; Langer, R.; Jensen, K. F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192.
- Kim, S. H.; Jeong, J. H.; Mok, H.; Lee, S. H.; Kim, S. W.; Park, T. G. Folate receptor targeted delivery of polyelectrolyte complex micelles prepared from ODN-PEG-folate conjugate and cationic lipids. Biotechnol. Prog. 2007, 23, 232–237. [CrossRef] [PubMed]
- Kim, J. H.; Ramasamy, T.; Tran, T. H.; Choi, J. Y.; Cho, H. J.; Yong, C. S.; Kim, J. O. Polyelectrolyte complex micelles by self-assembly of polypeptide-based triblock copolymer for doxorubicin delivery. Asian J. Pharm. Sci. 2014, 9, 191–198.
- Oishi, M.; Nagatsugi, F.; Sasaki, S.; Nagasaki, Y.; Kataoka, K. Smart polyion complex micelles for targeted intracellular delivery of PEGylated antisense oligonucleotides containing acid-labile linkages. ChemBioChem 2005, 6, 718–725. [CrossRef]
- Wang, X.; Xiao, X.; Zhang, B.; Li, J.; Zhang, Y. A self-assembled peptide nucleic acid-microRNA nanocomplex for dual modulation of cancer-related microRNAs. Chem. Commun. (Cambridge, U. K.) 2019, 55, 2106–2109. [CrossRef]
- Konate, K.; Dussot, M.; Aldrian, G.; Vaissière, A.; Viguier, V.; Neira, I. F.; Couillaud, F.; Vivès, E.; Boisguerin, P.; Deshayes, S. Peptide-based nanoparticles to rapidly and efficiently “Wrap’n Roll” siRNA into cells. Bioconjugate Chem. 2019, 30, 592–603.
- Christie, R. J.; Matsumoto, Y.; Miyata, K.; Nomoto, T.; Fukushima, S.; Osada, K.; Halnaut, J.; Pittella, F.; Kim, H. J.; Nishiyama, N.; Kataoka, K. Targeted polymeric micelles for siRNA treatment of experimental cancer by intravenous injection. ACS Nano 2012, 6, 5174–5189. [CrossRef]
- Uchida, S.; Kinoh, H.; Ishii, T.; Matsui, A.; Tockary, T. A.; Takeda, K. M.; Uchida, H.; Osada, K.; Itaka, K.; Kataoka, K. Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 2016, 82, 221–228. [CrossRef] [PubMed]
- Sprouse, D.; Reineke, T. M. Investigating the Effects of Block versus Statistical Glycopolycations Containing Primary and Tertiary Amines for Plasmid DNA Delivery. Biomacromolecules 2014, 15, 2616–2628. [CrossRef] [PubMed]
- Fukushima, S.; Miyata, K.; Nishiyama, N.; Kanayama, N.; Yamasaki, Y.; Kataoka, K. PEGylated polyplex micelles from triblock catiomers with spatially ordered layering of condensed pDNA and buffering units for enhanced intracellular gene delivery. J. Am. Chem. Soc. 2005, 127, 2810–2811. [CrossRef] [PubMed]
- Dirisala, A.; Uchida, S.; Tockary, T. A.; Yoshinaga, N.; Li, J.; Osawa, S.; Gorantla, L.; Fukushima, S.; Osada, K.; Kataoka, K. Precise tuning of disulphide crosslinking in mRNA polyplex micelles for optimising extracellular and intracellular nuclease tolerability. J. Drug Target. 2019, 27, 670–680.
- Tockary, T. A.; Foo, W.; Dirisala, A.; Chen, Q.; Uchida, S.; Osawa, S.; Mochida, Y.; Liu, X.; Kinoh, H.; Cabral, H.; Osada, K.; Kataoka, K. Single-stranded DNA-packaged polyplex micelle as adeno- associated-virus-iinspired compact vector to ssystemically target stroma-rich pancreatic cancer. ACS Nano 2019, 13, 12732–12742.
- Jiang, Y.; Lodge, T. P.; Reineke, T. M. Packaging pDNA by polymeric ABC micelles simultaneously achieves colloidal stability and structural control. J. Am. Chem. Soc. 2018, 140, 11101–11111. [CrossRef] [PubMed]
- Osada, K. Structural ppolymorphism of single pDNA ccondensates elicited by cationic block polyelectrolytes. Polymers 2020, 12, 1603. [CrossRef]
- Hagerman, P. J. Flexibility of DNA. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 265–286. [CrossRef]
- Balakrishnan, B.; Jayandharan, G. Basic biology of adeno- associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014, 14, 86–100. [CrossRef]
- Ji, Q.; Hou, J.; Yong, X.; Gong, G.; Muddassir, M.; Tang, T.; Xie, J.; Fan, W.; Chen, X. Targeted Dual Small Interfering Ribonucleic Acid Delivery via Non-Viral Polymeric Vectors for Pulmonary Fibrosis Therapy. Adv. Mater. 2021, 33, e2007798. [CrossRef]
- Lin, C.-Y.; Perche, F.; Ikegami, M.; Uchida, S.; Kataoka, K.; Itaka, K. Messenger RNA-based therapeutics for brain diseases: An animal study for augmenting clearance of beta-amyloid by intracerebral administration of neprilysin mRNA loaded in polyplex nanomicelles. J. Control. Release 2016, 235, 268–275.
- Jiang, Y.; Zhang, J.; Meng, F.; Zhong, Z. Apolipoprotein E Peptide-Directed Chimeric Polymersomes Mediate an Ultrahigh-Efficiency Targeted Protein Therapy for Glioblastoma. ACS Nano 2018, 12, 11070–11079. [CrossRef]
- Lam, J. H.; Khan, A. K.; Cornell, T. A.; Chia, T. W.; Dress, R. J.; Yeow, W. W. W.; Mohd-Ismail, N. K.; Venkataraman, S.; Ng, K. T.; Tan, Y. J.; Anderson, D. E.; Ginhoux, F.; Nallani, M. Polymersomes as Stable Nanocarriers for a Highly Immunogenic and Durable SARS-CoV-2 Spike Protein Subunit Vaccine. ACS Nano 2021, 15, 15754–15770.
- Wang, F., Gao, J., Xiao, J., & Du, J. Dually gated polymersomes for gene delivery. Nano Lett. 2018, 18, 5562–5568.
- Lomora, M.; Itel, F.; Dinu, I.A.; Palivan, C.G. Selective ion-permeable membranes by insertion of biopores into polymersomes. Phys. Chem. Chem. Phys. 2015, 17, 15538–15546. [CrossRef]
- Kumar, M.; Grzelakowski, M.; Zilles, J.; Clark, M.; Meier, W. Highly permeable polymeric membranes based on the incorporation of the functional water channel protein Aquaporin Z. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20719–20724. [CrossRef]
- Einfalt, T.; Goers, R.; Dinu, I. A.; Najer, A.; Spulber, M.; Onaca-Fischer, O.; Palivan, C. G. Stimuli-triggered activity of nanoreactors by biomimetic engineering polymer membranes. Nano Lett. 2015, 15, 7596–7603. [CrossRef]
- Burns, J. R.; Stulz, E.; Howorka, S. Self-assembled DNA nanopores that span lipid bilayers. Nano Lett. 2013, 13, 2351–2356. [CrossRef] [PubMed]
- Messager, L.; Burns, J. R.; Kim, J.; Cecchin, D.; Hindley, J.; Pyne, A. L. B.; Gaitzsch, J.; Battaglia, G.; Howorka, S. Biomimetic Hybrid Nanocontainers with Selective Permeability. Angew. Chem. Int. Ed. 2016, 55, 11106–11109.
- Xiao, F.-X.; Pagliaro, M.; Xu, Y.-J.; Liu, B. Layer-by-layer Assembly of Versatile Nanoarchitectures with Diverse Dimensionality: A New Perspective for Rational Construction of Multilayer Assemblies. Chem. Soc. Rev. 2016, 45, 3088–3121. [CrossRef]
- Bacskai, B.J.; Kajdasz, S.T.; McLellan, M.E.; Games, D.; Seubert, P.; Schenk, D.; Hyman, B.T. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J. Neurosci. 2002, 22, 7873–7878. [CrossRef] [PubMed]
- Sehlin, D.; Fang, X. T.; Cato, L.; Antoni, G.; Lannfelt, L.; and Syvanen, S. Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer’s disease. Nat. Commun. 2016, 7, 10759. [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P. H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; O’Gorman, J.; Qian, F.; Arastu, M.; Li, M.; Chollate, S.; Brennan, M. S.; Quintero-Monzon, O.; Scannevin, R. H.; Arnold, H. M.; Engber, T.; Rhodes, K.; Ferrero, J.; Hang, Y.; Mikulskis, A.; Grimm, J.; Hock, C.; Nitsch, R. M.; Sandrock, A. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56.
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R. L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; Kholodenko, D.; Lee, M.; Lieberburg, I.; Motter, R.; Nguyen, M.; Soriano, F.; Vasquez, N.; Weiss, K.; Welch, B.; Seubert, P.; Schenk, D.; Yednock, T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919.
- Habicht, G.; Haupt, C.; Friedrich, R. P.; Hortschansky, P.; Sachse, C.; Meinhardt, J.; Wieligmann, K.; Gellermann, G. P.; Brodhun, M.; Gotz, J.; Halbhuber, K.; Rocken, C.; Horn, U.; Fandrich, M. Directed Selection of a Conformational Antibody Domain That Prevents Mature Amyloid Fibril Formation by Stabilizing Aβ Protofibrils. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19232–19237.
- Perche, F.; Uchida, S.; Akiba, H.; Lin, C.; Ikegami, M.; Dirisala, A.; Nakashima, T.; Itaka, K.; Tsumoto, K.; Kataoka, K. Improved Brain Expression of Anti-Amyloid β scFv by Complexation of mRNA Including a Secretion Sequence With PEG-based Block Catiomer. Curr. Alzheimer Res. 2017, 14, 295–302. [CrossRef] [PubMed]
- Pul, R.; Dodel, R.; Stangel, M. Antibody-Based Therapy in Alzheimer’s Disease. Expert Opin. Biol. Ther. 2011, 11, 343–357. [CrossRef]
- Montoliu-Gaya, L.; Mulder, S. D.; Veerhuis, R.; Villegas, S.Effects of an Aβ Antibody Fragment on Aβ Aggregation and Astrocytic Uptake Are Modulated by Apolipoprotein E and J MimeticPeptides. PLoS One 2017, 12, e0188191. [CrossRef]
- Marín-Argany, M.; Rivera-Hernández, G.; Martí, J.; Villegas, S. An Anti-aβ (Amyloid β) Single-Chain Variable Fragment Prevents Amyloid Fibril Formation and Cytotoxicity by Withdrawing Aβ Oligomers from the Amyloid Pathway. Biochem. J. 2011, 437, 25–34. [CrossRef]
- Nelson, A. L.; Reichert, J. M. Development Trends for Therapeutic Antibody Fragments. Nat. Biotechnol. 2009, 27, 331–337. [CrossRef]
- Abbott, N. J.; Patabendige, A. A.; Dolman, D. E.; Yusof, S. R.; Begley, D. J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25.
- Lemere, C. A.; Masliah, E. Can Alzheimer Disease Be Prevented by Amyloid-Beta Immunotherapy? Nat. Rev. Neurol. 2010, 6, 108–119. [CrossRef]
- Arbel, M.; Solomon, B. Immunotherapy for Alzheimer’s Disease: Attacking Amyloid-β From the Inside. Trends Immunol. 2007, 28, 511–513. [CrossRef] [PubMed]
- van Dyck, C. H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [CrossRef]
- Neves, V.; Aires-Da-Silva, F.; Corte-Real, S.; Castanho, M. A. R. B. Antibody Approaches to Treat Brain Diseases. Trends Biotechnol. 2016, 34, 36–48. [CrossRef]
- Xie, J.; Gonzalez-Carter, D.; Tockary, T. A.; Nakamura, N.; Xue, Y.; Nakakido, M.; Akiba, H.; Dirisala, A.; Liu, X.; Toh, K.; Yang, T.; Wang, Z.; Fukushima, S.; Li, J.; Quader, S.; Tsumoto, K.; Yokota, T.; Anraku, Y.; Kataoka, K. Dual-Sensitive Nanomicelles Enhancing Systemic Delivery of Therapeutically Active Antibodies Specifically into the Brain. ACS Nano 2020, 14, 6729–6742.
- Stupp, R.; Mason, W. P.; van den Bent, M. J.; Weller, M.; Fisher, B.; Taphoorn, M. J.; Belanger, K.; Brandes, A. A.; Marosi, C.; Bogdahn, U.; Curschmann, J.; Janzer, R. C.; Ludwin, S. K.; Gorlia, T.; Allgeier, A.; Lacombe, D.; Cairncross, J. G.; Eisenhauer, E.; Mirimanoff, R. O. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Gong, J.; Chen, M.; Zheng, Y.; Wang, S.; Wang, Y. Polymeric micelles drug delivery system in oncology. J. Control Release 2012, 159, 312–323. [CrossRef]
- Oba, M.; Fukushima, S.; Kanayama, N.; Aoyagi, K.; Nishiyama, N.; Koyama, H.; Kataoka, K. Cyclic RGD peptide-conjugated polyplex micelles as a targetable gene delivery system directed to cells possessing αvβ3 and αvβ5 integrins. Bioconjug. Chem. 2007, 18, 1415–1423. [CrossRef]
- Heffernan, M. J., Murthy, N. Disulfide-crosslinked polyion micelles for delivery of protein therapeutics. Ann. Biomed. Eng. 2009, 37, 1993–2002. [CrossRef] [PubMed]
- Tong, Y-C.; Chang, S-F.; Kao, W. W-Y.; Liu, C-Y.; Liaw, J. Polymeric micelle gene delivery of bcl-xL via eye drop reduced corneal apoptosis following epithelial debridement. J. Control. Release 2010, 147, 76–83. [CrossRef]
- Zhan, C.; Gu, B.; Xie, C.; Li, J.; Liu, Y.; Lu, W. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J. Control. Release 2010, 143, 136–142. [CrossRef] [PubMed]
- Zhan, C.; Meng, Q.; Li, Q.; Feng, L.; Zhu, J.; Lu, W. Cyclic RGD- polyethylene glycol-polyethylenimine for intracranial glioblastoma-targeted gene delivery. Chem. Asian J. 2012, 7, 91–96. [CrossRef] [PubMed]
- Zhan, C.; Wei, X.; Qian, J.; Feng, L.; Zhu, J.; Lu, W. Co-delivery of TRAIL gene enhances the anti-glioblastoma effect of paclitaxel in vitro and in vivo. J. Control. Release 2012, 160, 630–636. [CrossRef]
- Gao, H.; Qian, J.; Yang, Z.; Pang, Z.; Xi, Z.; Cao, S.; Wang, Y.; Pan, S.; Zhang, S.; Wang, W.; Jiang, X.; Zhang, Q. Whole-cell SELEX aptamer-functionalised poly(ethyleneglycol)-poly(ε-caprolactone) nanoparticles for enhanced targeted glioblastoma therapy. Biomaterials 2012, 33, 6264–6272. [CrossRef]
- Yin, T.; Wang, P.; Li, J.; Zheng, R.; Zheng, B.; Cheng, D.; Li, R.; Lai, J.; Shuai, X. Ultrasound- sensitive siRNA-loaded nanobubbles formed by hetero-assembly of polymeric micelles and liposomes and their therapeutic effect in gliomas. Biomaterials 2013, 34, 4532–4543. [CrossRef] [PubMed]
- Zheng, C.; Zheng, M.; Gong, P.; Deng, J.; Yi, H.; Zhang, P., Zhang, Y.; Liu, P.; Ma, Y.; Cai, L. Polypeptide cationic micelles mediated co-delivery of docetaxel and siRNA for synergistic tumor therapy. Biomaterials 2013, 34, 3431–3438. [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T. L.; Fuller, C.; Hamner, B.; Oh, E. Y.; Gaber, M. W.; Finklestein, D.; Allen, M.; Frank, A.; Bayazitov, I. T.; Zakharenko, S. S.; Gajjar, A.; Davidoff, A.; Gilbertson, R. J. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82.
- Gilbertson, R. J.; Rich, J. N. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [CrossRef]
- Jain, R. K. Delivery of novel therapeutic agents in tumors: physiological barriers and strategies. J. Natl. Cancer Inst. 1989, 81, 570–576.
- Bello, L.; Francolini, M.; Marthyn, P.; Zhang, J.; Carroll, R.S.; Nikas, D.C.; Strasser, J. F.; Villani, R.; Cheresh, D. A.; Black P. M. αvβ3 and αvβ5 integrin expression in glioma periphery. Neurosurgery 2001, 49, 380–389.
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [CrossRef]
- Nasongkla, N.; Bey, E.; Ren, J.; Ai, H.; Khemtong, C.; Guthi, J. S.; Chin, S-F.; Sherry, A. D.; Boothman, D. A.; Gao, J. Multifunctional polymeric micelles as cancer-targeted, MRI-ultrasensitive drug delivery systems. Nano Lett. 2006, 6, 2427–2430. [CrossRef]
- Hu, Z.; Luo, F.; Pan, Y.; Hou, C.; Ren, L.; Chen, J.; Wang, J.; Zhang, Y. Arg-Gly-Asp (RGD) peptide conjugated poly(lactic acid)–poly(ethylene oxide) micelle for targeted drug delivery. J. Biomed. Mater. Res. A 2008, 85A, 797–807. [CrossRef]
- Kessinger, C. W.; Togao, O.; Khemtong, C.; Huang, G.; Takahashi, M.; and Gao, J. Investigation of in vivo targeting kinetics of αvβ3-specific super- paramagnetic nanoprobes by time-resolved MRI. Theranostics 2011, 1, 263–273. [CrossRef]
- Liu, X.; Cui, W.; Li, B.; Hong, Z. Targeted therapy for glioma using cyclic RGD-entrapped polyionic complex nanomicelles. Int. J. Nanomed. 2012, 7, 2853–2862.
- Xiao, Y.; Hong, H.; Javadi, A.; Engle, J. W.; Xu, W.; Yang, Y.; Zhang, Y.; Barnhart, T. E.; Cai, W.; Gong, S. Multifunctional unimolecular micelles for cancer-targeted drug delivery and positron emission tomography imaging. Biomaterials 2012, 33, 3071–3082. [CrossRef]
- Zhang, P.; Hu, L.; Yin, Q.; Feng, L.; Li, Y. Transferrin-modified c[RGDfK]-paclitaxel loaded hybrid micelle for sequential blood-brain barrier penetration and glioma targeting therapy. Mol. Pharm. 2012, 9, 1590–1598. [CrossRef]
- Simberg, D.; Duza, T.; Park, J. H.; Essler, M.; Pilch, J.; Zhang, L.; Derfus, A. M.; Yang, M.; Hoffman, R. M.; Bhatia, S.; Sailor, M. J.; Ruoslahti, E. Biomimetic Amplification of Nanoparticle Homing to Tumors. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 932–936.
- Bárdos, H., Molnár, P., Csécsei, G., and Adány, R. Fibrin deposition in primary and metastatic human brain tumours. Blood Coagul. Fibrinolysis 1996, 7, 536–548. [CrossRef]
- Chung, E. J.; Cheng, Y.; Morshed, R.; Nord, K.; Han, Y.; Wegscheid, M. L.; Auffinger, B.; Wainwright, D. A.; Lesniak, M. S.; Tirrel, M. V. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials 2013, 35, 1249–1256.
- Peters, D.; Kastantin, M.; Kotamraju, V. R.; Karmali, P. P.; Gujraty, K.; Tirrell, M.; Ruoslahti, E. Targeting atherosclerosis by using modular, multifunctional micelles. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 9815–9819. [CrossRef]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R. A.; Shapiro, L. H.; Arap, W.; Ruoslahti, E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727.
- Ellerby, H. M.; Arap, W.; Ellerby, L. M.; Kain, R.; Andrusiak, R.; Rio, G. D.; Krajewski, S.; Lombardo, C. R.; Rao, R.; Ruoslahti, E.; Bredesen, D. E. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038.
- Zhao, B-J.; Ke, X-Y.; Huang, Y.; Chen, X-M.; Zhao, X.; Zhao, B-X.; Lu, W-L.; Lou, J-N.; Zhang, X.; Zhang, Q. The antiangiogenic efficacy of NGR-modified PEG–DSPE micelles containing paclitaxel (NGR-M-PTX) for the treatment of glioma in rats. J. Drug Target. 2011, 19, 382–390. [CrossRef]
- Fishman, J. B.; Rubin, J. B.; Handrahan, J. V.; Connor, J. R.; Fine, R. E. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res. 1987, 18, 299–304. [CrossRef]
- Liu, Z.; Jiang, M.; Kang, T.; Miao, D.; Gu, G.; Song, Q.; Yao, L.; Hu, Q.; Tu, Y.; Pang, Z.; Chen, H.; Jiang, X.; Gao, X.; Chen, J. Lactoferrin-modified PEG-co-PCL nanoparticles for enhanced brain delivery of NAP peptide following intranasal administration. Biomaterials 2013, 34, 3870–3881. [CrossRef]
- Mu, C.; Dave N.; Hu, J.; Desai, P.; Pauletti, G.; Bai, S.; Hao, J. Solubilization of flurbiprofen into aptamer-modified PEG-PLA micelles for targeted delivery to brain-derived endothelial cells In Vitro. J. Microencapsul. 2013, 30, 701–708. [CrossRef]
- Ren, W.-h.; Chang, J.; Yan, C.-h.; Qian, X.-m.; Long, L.-x.; He, B.; Yuan, X.-b.; Kang, C.-s.; Betbeder, D.; Sheng, J.; Pu, P.-y Development of transferrin functionalized poly (ethylene glycol)/poly (lactic acid) amphiphilic block copolymeric micelles as a potential delivery system targeting brain glioma. J. Mater. Sci. Mater. Med. 2010, 21, 2673–2681.
- Zhang, P.; Hu, L.; Yin, Q.; Zhang, Z.; Feng, L.; Li, Y. Transferrin-conjugated polyphosphoester hybrid micelle loading paclitaxel for brain-targeting delivery: Synthesis, preparation and in vivo evaluation. J. Control. Rel. 2012, 159, 429–434. [CrossRef] [PubMed]
- Kalaria, R. N.; Homayoun, P.; Whitehouse, P. J. Nicotinic choliner- gic receptors associated with mammalian cerebral vessels. J. Auton. Nerv. Syst. 1994, 49, S3–S7. [CrossRef] [PubMed]
- MacKlin, K. D., Maus, A. D., Pereira, E. F., Albuquerque, E. X., and Conti-Fine, B. M. Human vascular endothelial cells express functional nicotinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 1998, 287, 435–439. [CrossRef]
- Zhan, C.; Li, B.; Hu, L.; Wei, X.; Feng, L.; Fu, W.; Lu, W. Micelle-based brain-targeted drug delivery enabled by a nicotine acetylcholine receptor ligand. Angew. Chem. Int. Ed. Engl. 2011, 50, 5482–5485. [CrossRef]
- Wong, A. J.; Bigner, S. H.; Bigner, D. D.; Kinzler, K. W.; Hamilton, S. R.; Vogelstein, B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplifi- cation. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 6899–6903. [CrossRef] [PubMed]
- Kuo, Y-C.; Liang, C-T. Inhibition of human brain malignant glioblas toma cells using carmustine-loaded catanionic solid lipid nanoparticles with surface anti-epithelial growth factor receptor. Biomaterials 2011, 32, 3340–3350. [CrossRef] [PubMed]
- Oliveira, S.; Schiffelers, R. M.; van der Veeken, J.; van der Meel, R.; Vongpromek, R.; van Bergen en Henegouwen, P. M. P.; Storm, G.; Roovers, R.C. Downregulation of EGFR by a novel multivalent nanobody-liposome platform. J. Control. Release 2010, 145, 165–175.
- Talelli, M.; Rijcken, C. J. F.; Oliveira, S.; van der Meel, R.; van Bergen en Henegouwen, P. M. P.; Lammers, T.; van Nostrum, C. F.; Storm, G.; Hennink, W. E. Nanobody-shell functionalized thermosensitive core-crosslinked polymeric micelles for active drug targeting. J. Control. Release 2011, 151, 183–192.
- Bu, G.; Maksymovitch, E. A.; Geuze, H.; Schwartz, A. L. Subcellular localization and endocytic function of low density lipoprotein receptor-related protein in human glioblastoma cells. J. Biol. Chem. 1994, 269, 29874–29882. [CrossRef]
- Yamamoto, M.; Ikeda, K.; Ohshima, K.; Tsugu, H.; Kimura, H.; Tomonaga, M. Increased expression of low density lipoprotein receptor-related protein/α2-macroglobulin receptor in human malignant astrocytomas. Cancer Res. 1997, 57, 2799–2805.
- Herz, J.; Bock, H. H. Lipoprotein receptors in the nervous system. Annu. Rev. Biochem. 2002, 71, 405–434. [CrossRef]
- Demeule, M.; Currie, J. C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [CrossRef]
- Shen, J.; Zhan, C.; Xie, C.; Meng, Q.; Gu, B.; Li, C.; Zhang, Y.; Lu, W. Poly(ethylene glycol)-block-poly(D,L-lactide acid) micelles anchored with angiopep-2 for brain-targeting delivery. J. Drug Target. 2011, 19, 197–203. [CrossRef]
- Bayrac, A. T.; Sefah, K.; Parekh, P.; Bayrac, C.; Gulbakan, B.; Oktem, H. A.; Tan, W. In vitro selection of DNA aptamers to glioblastoma multiforme. ACS Chem. Neurosci. 2011, 2, 175–181. [CrossRef] [PubMed]
- Jiang, X.; Sha, X.; Xin, H.; Xu, X.; Gu, J.; Xia, W.; Chen, S.; Xie, Y.; Chen, L.; Chen, Y.; Fang, X. Integrin-facilitated transcytosis for enhanced penetration of advanced gliomas by poly(trimethylene carbonate)-based nanoparticles encapsulating paclitaxel. Biomaterials 2013, 34, 2969–2979.
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z. C.; Chen, H. W.; Mallikaratchy, P.; Sefah, K.; Yang, C. J.; Tan, W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11838–11843. [CrossRef] [PubMed]
- Pardridge, W. M. Recent developments in peptide drug delivery to the brain. Pharmacol. Toxicol. 1992, 71, 3–10. [CrossRef]
- Calvo, P.; Gouritin, B.; Chacun, H.; Desmaele, D.; D’Angelo, J.; Noel, J.; Georgin, D.; Fattal, E.; Andreux, J.; Couvreur, P. Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. Pharm. Res. 2001, 18, 1157–1166. [CrossRef] [PubMed]
- Brigger, I.; Morizet, J.; Aubert, G.; Chacun, H.; Terrier-Lacombe, M. J.; Couvreur, P.; Vassal G. Poly(ethylene glycol)-coated hexadecylcyanoacrylate nanospheres display a combined effect for brain tumor targeting. J. Pharmacol. Exp. Ther. 2002, 303, 928–936. [CrossRef] [PubMed]
- Gelperina, S.; Maksimenko, O.; Khalansky, A.; Vanchugova, L.; Shipulo, E.; Abbasova, K.; Berdiev, R.; Wohlfart, S.; Chepurnova, N.; Kreuter, J. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: Influence of the formulation parameters. Eur. J. Pharm. Biopharm. 2010, 74, 157–163.
- Kanazawa, T.; Taki, H.; Tanaka, K.; Takashima, Y.; Okada, H. Cell-Penetrating Peptide-Modified Block Copolymer Micelles Promote Direct Brain Delivery via Intranasal Administration. Pharm. Res. 2011, 28, 2130–2139. [CrossRef]
- Bobo, R. H.; Laske, D. W.; Akbasak, A.; Morrison, P. F.; Dedrick, R. L.; and Oldfield, E. H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 2076–2080.
- Allard, E.; Passirani, C.; Benoit, J. P. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials 2009, 30, 2302–2318. [CrossRef]
- Lesniak, M. S. Novel advances in drug delivery to brain cancer. Technol. Cancer Res. Treat. 2005, 4, 417–428. [CrossRef]
- Sampson, J. H., Akabani, G., Archer, G. E., Bigner, D. D., Berger, M. S., Friedman, A. H., Friedman, H. S.; Herndon II, J. E.; Kunwar, S.; Marcus, S.; Mclendon, R. E.; Paolino, A.; Penne, K.; Provenzale, J.; Quinn, J.; Reardon, D. A.; Rich, J.; Stenzel, T.; Tourt-Uhlig, S.; Wikstrand, C.; Wong, T.; Williams, R.; Yuan, F.; Zalutsky, M. R.; Pastan, I. Progress report of a phase I study of the intracerebral microinfu- sion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. 2003, 65, 27–35.
- Weaver, M.; Laske, D. W. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003, 65, 3–14. [CrossRef]
- Vinchon-Petit, S.; Jarnet, D.; Paillard, A.; Benoit, J. P.; Garcion, E.; Menei, P. In vivo evaluation of intracellular drug-nanocarriers infused into intracranial tumours by convection-enhanced delivery: distribution and radiosensitisation efficacy. J. Neurooncol. 2010, 97, 195–205. [CrossRef]
- Thaci, B.; Ahmed, A. U.; Ulasov, I. V.; Tobias, A. L.; Han, Y.; Aboody, K. S.; Lesniak, M. S. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012, 19, 431–442.
- Huang, X.; Zhang, F.; Wang, H.; Niu, G.; Choi, K. Y.; Swierczewska, M.; Zhang, G.; Gao, H.; Wang, Z.; Zhu, L. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials 2013, 34, 1772–1780. [CrossRef] [PubMed]
- Roger, M.; Clavreul, A.; Venier-Julienne, M.-C.; Passirani, C.; Sindji, L.; Schiller, P.; Montero-Menei, C.; Menei, P. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials 2010, 31, 8393–8401. [CrossRef] [PubMed]
- Li, L.; Guan, Y.; Liu, H.; Hao, N.; Liu, T.; Meng, X.; Fu, C.; Li, Y.; Qu, Q.; Zhang, Y.; Ji, S.; Chen, L.; Chen, D.; Tang, F. Silica Nanorattle-Doxorubicin-Anchored Mesenchymal Stem Cells for Tumor-Tropic Therapy. ACS Nano 2011, 5, 7462–7470. [CrossRef]
- Cheng, Y.; Morshed, R.; Cheng, S-H.; Tobias, A.; Auffinger, B.; Wainwright, D. A.; Zhang, L.; Yunis, C.; Han, Y.; Chen, C-T.; Lo, L-W.; Aboody, K. S.; Ahmed, A. U.; Lesniak, M. S. Nanoparticle-programmed self-destructive neural stem cells for glioblastoma targeting and therapy. Small 2013, 9, 4123–4129.
- Morshed, R. A.; Cheng, Y.; Auffinger, B.; Wegscheid, M. L.; Lesniak, M. S. The potential of polymeric micelles in the context of glioblastoma therapy. Front. Pharmacol. 2013, 4, 157.
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discovery 2003, 2, 347–360. [CrossRef]
- Crommelin, D. J. A.; Storm, G.; Jiskoot, W.; Stenekes, R.; Mastrobattista, E.; Hennink, W. E. Nanotechnological approaches for the delivery of macromolecules. J. Control. Release 2003, 87, 81–88.
- Crommelin, D.J.A.; Storm, G. Liposomes: from the bench to the bed. J. Liposome Res. 2003, 13, 33–36. [CrossRef]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin. D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 1999, 51, 691–743. [CrossRef]
- Yamaguchi, T.; Mizushima, Y. Lipid microspheres for drug delivery from the pharmaceutical viewpoint. Crit. Rev. Ther. Drug Carr. Syst. 1994, 11, 215–229.
- Tamilvanan, S. Oil-in-water lipid emulsions: implications for parenteral and ocular delivering systems. Prog. Lipid Res. 2004, 43, 489–533. [CrossRef]
- Bala, I.; Hariharan, S.; Ravi Kumar, M. N. V. PLGA nanoparticles in drug delivery: the state of the art. Crit. Rev. Ther. Drug Carr. Syst. 2004, 21, 387–422. [CrossRef] [PubMed]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [CrossRef]
- Discher, D. E.; Eisenberg, A. Polymer vesicles. Science 2002, 297, 967–973. [CrossRef] [PubMed]
- Ahmed, F.; Discher, D. E. Self-porating polymersomes of PEG-PLA and PEG-PCL: hydrolysis-triggered controlled release vesicles. J. Control. Release 2004, 96, 37–53. [CrossRef]
- Nori, A.; Kopecek, J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv. Drug Deliv. Rev. 2005, 57, 609–636. [CrossRef]
- Satchi-Fainaro, R.; Puder, M.; Davies, J. W.; Tran, H. T.; Sampson, D. A.; Greene, A. K.; Corfas, G.; Folkman, J. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat. Med. 2004, 10, 255–261. [CrossRef] [PubMed]
- Gillies, E. R.; Fréchet, J. M. J. Dendrimers and dendritic polymers in drug delivery. Drug Discov. Today 2005, 10, 35–43. [CrossRef] [PubMed]
- Aulenta, F.; Hayes, W.; Rannard, S. Dendrimers: a new class of nanoscopic containers and delivery devices. Eur. Polym. J. 2003, 39, 1741–1771. [CrossRef]
- Zamboni, W. C. Liposomal, nanoparticle and conjugated formulations of anticancer agents. Clin. Cancer Res. 2005, 11, 8230–8234. [CrossRef]
- Forster, S.; Plantenberg, T. From self-organizing polymers to nanohybrid and biomaterials. Angew. Chem. Int. Ed. Engl. 2002, 41, 689–714.
- 1506. Bader, H.; Ringsdorf, H.; Schmidt, B. Water soluble polymers in medicine. Angew. Makromol. Chem. 1984, 123, 457–485.
- Kabanov, A. V.; Chekhonin, V. P.; Alakhov VYu; Batrakova, E. V.; Lebedev, A. S.; Melik-Nubarov, N. S.; Arzhakov, S. A.; Levashov, A. V.; Morozov, G. V.; Severin, E. S.; Kabanov, V. A. The neuroleptic activity of haloperidol increases after its solubilization in surfactant micelles. Micelles as microcontainers for drug targeting. FEBS Lett. 1989, 258, 343–345.
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Livaniou, E.; Evangelatos, G.; Ithakissios, D. S. Effect of Copolymer Composition on the Physicochemical Characteristics, in Vitro Stability, and Biodistribution of PLGA-mPEG Nanoparticles. Int. J. Pharm. 2003, 259, 115–127. [CrossRef]
- Neradovic, D.; van Nostrum, C. F.; Hennink, W. E. Thermoresponsive polymeric micelles with controlled instability based on hydrolytically sensitive N-Isopropylacrylamide copolymers. Macromolecules 2001, 34, 7589–7591. [CrossRef]
- Savic, R.; Luo, L.; Eisenberg, A.; Maysinger, D. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science 2003, 300, 615–618. [CrossRef]
- Allen, T. M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2002, 2, 750–763. [CrossRef]
- Kabanov, A. V.; Lemieux, P.; Vinogradov, S.; Alakhov, V. Pluronic block copolymers: novel functional molecules for gene therapy. Adv. Drug Deliv. Rev. 2002, 54, 223–233. [CrossRef]
- Francis, M. F.; Lavoie, L.; Winnik, F. M.; Leroux, J. C. Solubilization of cyclosporin A in dextran-g-polyethyleneglycolalkyl ether polymeric micelles. Eur. J. Pharm. Biopharm. 2003, 56, 337–346. [CrossRef] [PubMed]
- Lee, J. H.; Lee, H. B.; Andrade, J. D. Blood compatibility of polyethylene oxide surfaces. Prog. Polym. Sci. 1995, 20, 1043–1079. [CrossRef]
- Kwon, G. S.; Naito, M.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Physical entrapment of Adriamycin in AB block copolymer micelles. Pharm. Res. 1995, 12, 192–195. [CrossRef] [PubMed]
- Hrubý, M.; Konák, C.; Ulbrich, K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J. Control. Release 2005, 103, 137–148. [CrossRef]
- Kim, S. C.; Kim, D. W.; Shim, Y. H.; Bang, J. S.; Oh, H. S.; Wan Kim, S.; Seo, M. H. In Vivo Evaluation of Polymeric Micellar Paclitaxel Formulation: Toxicity and Efficacy. J. Control. Release 2001, 72, 191–202. [CrossRef]
- Nishiyama, N.; Kato, Y.; Sugiyama, Y.; Kataoka, K. Cisplatin-loaded polymer–metal complex micelle with time-modulated decaying property as a novel drug delivery system. Pharm. Res. 2001, 18, 1035–1041. [CrossRef]
- Lin, W-J.; Juang, L-W.; Lin, C-C. Stability and release performance of a series of pegylated copolymeric micelles. Pharm Res 2003, 20, 668–673. [CrossRef]
- Djordjevic, J.; Barch, M.; Uhrich, K. E. Polymeric micelles based on amphiphilic scorpion-like macromolecules: novel carriers for water- insoluble drugs. Pharm. Res. 2005, 22, 24–32. [CrossRef] [PubMed]
- Yokoyama, M.; Satoh, A.; Sakurai, Y.; Okano, T.; Matsumura, Y.; Kakizoe, T.; Kataoka, K. Incorporation of water-insoluble anticancer drug into polymeric micelles and control of their particle size. J. Control. Release 1998, 55, 219–229. [CrossRef]
- Matsumura, Y.; Yokoyama, M.; Kataoka, K.; Okano, T.; Sakurai, Y.; Kawaguchi, T.; Kakizoe, T. Reduction of the side effects of an antitumor agent, KRN5500, by incorporation of the drug into polymeric micelles. Jpn. J. Cancer Res. 1999, 90, 122–128. [CrossRef] [PubMed]
- Taillefer, J.; Jones, M. C.; Brasseur, N.; van Lier, J. E.; Leroux, J. C. Preparation and characterization of pH-responsive polymeric micelles for the delivery of photosensitizing anticancer drugs. J. Pharm. Sci. 2000, 89, 52–62. [CrossRef]
- Zhang, X. C.; Jackson, J. K.; Burt, H. M. Development of amphiphilic diblock copolymers as micellar carriers of taxol. Int. J. Pharm. 1996, 132, 195–206. [CrossRef]
- Teng, Y.; Morrison, M. E.; Munk, P.; Webber, S. E.; Procházka, K. Release Kinetics Studies of Aromatic Molecules into Water from Block Polymer Micelles. Macromolecules 1998, 31, 3578–3587. [CrossRef]
- Kabanov, A. V.; Nazarova, I. R.; Astafieva, I. V.; Batrakova, E. V.; Alakhov, V. Y.; Yaroslavov, A. A.; Kabanov, V. A. Micelle Formation and Solubilization of Fluorescent Probes in Poly(oxyethylene-b-oxypropylene-b-oxyethylene) Solutions. Macromolecules 1995, 28, 2303–2314. [CrossRef]
- Kwon, G. S.; Naito, M.; Kataoka, K.; Yokoyama, M.; Sakurai, Y.; Okano, T. Block copolymer micelles as vehicles for hydrophobic drugs. Colloids Surf. B Biointerface 1994, 2, 429–434. [CrossRef]
- Zhao, J. X.; Allen, C.; Eisenberg, A. Partitioning of pyrene between “crew cut” block copolymer micelles and H2O/DMF solvent mixtures. Macromolecules 1997, 30, 7143–7150. [CrossRef]
- Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Improved synthesis of adriamycin-conjugated poly (ethylene oxide)-poly (aspartic acid) block copolymer and formation of unimodal micellar structure with controlled amount of physically entrapped adriamycin. J. Control. Release 1994, 32, 269–277. [CrossRef]
- Li, K.; Chen, F.; Wang, Y.; Stenzel, M. H.; Chapman, R. Polyion complex micelles for protein delivery benefit from flexible hydrophobic spacers in the binding group. Macromol. Rapid Commun. 2020, 41, e2000208. [CrossRef] [PubMed]
- Li, X.; Lu, C.; Xia, W.; Quan, G.; Huang, Y.; Bai, X.; Yu, F.; Xu, Q.; Qin, W.; Liu, D.; Pan, X. Poly (L-glutamic acid)-based brush co- polymers : fabrication, self-assembly, and evaluation as efficient nnanocarriers for cationic protein drug delivery. AAPS PharmSciTech 2020, 21, 78. [CrossRef]
- Raveendran, R.; Chen, F.; Kent, B.; Stenzel, M. H. Estrone-decorated polyion complex micelles for targeted melittin delivery to hormone-responsive breast cancer cells. Biomacromolecules 2020, 21, 1222–1233. [CrossRef] [PubMed]
- Jiang, Y.; Lu, H.; Chen, F.; Callari, M.; Pourgholami, M.; Morris, D. L.; Stenzel, M. H. PEGylated albumin-based polyion complex micelles for protein delivery. Biomacromolecules 2016, 17, 808–817. [CrossRef] [PubMed]
- Harris, N. M.; Ritzel, R.; Mancini, N. S.; Jiang, Y.; Yi, X.; Manickam, D. S.; Banks, W. A.; Kabanov, A. V.; McCullough, L. D.; Verma, R. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol. Biochem. Behav. 2016, 150–151, 48–56.
- Batrakova, E. V.; Li, S.; Reynolds, A. D.; Mosley, R. L.; Bronich, T. K.; Kabanov, A. V.; Gendelman, H. E. A macrophage-nanozyme delivery system for Parkinson’s disease. Bioconjugate Chemistry 2007, 18, 1498–1506.
- Brynskikh, A. M.; Zhao, Y.; Mosley, R. L.; Li, S.; Boska, M. D.; Klyachko, N. L.; Kabanov, A. V.; Gendelman, H. E.; Batrakova, E. V. Macrophage Delivery of Therapeutic Nanozymes in A Murine Model of Parkinson’s Disease. Nanomedicine 2010, 5, 379–396.
- Haney, M. J.; Zhao, Y.; Li, S.; Higginbotham, S. M.; Booth, S. L.; Han, H. Y.; Vetro, J. A.; Mosley, R. L.; Kabanov, A. V.; Gendelman, H. E.; Batrakova, E. V. Cell-mediated transfer of catalase nanoparticles from macrophages to brain endothelial, glial and neuronal cells. Nanomedicine 2011, 6, 1215–1230.
- Klyachko, N. L.; Haney, M. J.; Zhao, Y.; Manickam, D. S.; Mahajan, V.; Suresh, P.; Hingtgen, S. D.; Mosley, R. L.; Gendelman, H. E.; Kabanov, A. V.; Batrakova, E. V. Macrophages offer a paradigm switch for CNS delivery of therapeutic proteins. Nanomedicine 2014, 9, 1403–1422.
- Rosenbaugh, E. G.; Roat, J. W.; Gao, L.; Yang, R.-F.; Manickam, D. S.; Yin, J.-X.; Schultz, H.D.; Bronich, T. K.; Batrakova, E. V.; Kabanov, A. V.; Zucker, I. H.; Zimmerman, M. C. The attenuation of central angiotensin II-dependent pressor response and intra-neuronal signaling by intracarotid injection of nanoformulated copper/zinc superoxide dismutase. Biomaterials 2010, 31, 5218–5226.
- 1540. Manickam, D. S.; Brynskikh, A. M.; Kopanic, J. L.; Sorgen, P. L.; Klyachko, N. L.; Batrakova, E. V.; Bronich, T. K.; Kabanov, A. V. Well-defined cross-linked antioxidant nanozymes for treatment of ischemic brain injury. J. Control. Release 2012, 162, 636–645.
- Jiang, Y.; Brynskikh, A. M.; Devika, S.; Kabanov, A. V. SOD1 nanozyme salvages ischemic brain by locally protecting cerebral vasculature. J. Control. Release 2015, 213, 36–44. [CrossRef] [PubMed]
- Chen, F.; Li, K.; Hart-Smith, G.; Xu, Y. D.; Jiang, Y.; Lu, H.; Fok, S.; Macmillian, A.; Pandzic, E.; Stenzel M. Light-sheet microscopy as a tool to understanding the behaviour of Polyion complex micelles for drug delivery. Chem. Commun. 2018, 54, 12618–12621. [CrossRef]
- Wang, J.; Wang, J.; Ding, P.; Zhou, W.; Li, Y.; Drechsler, M.; Guo, X.; Cohen Stuart, M. A. A supramolecular crosslinker to give salt- resistant polyion complex micelles and improved MRI contrast agents. Angew. Chem. Int. Ed Engl. 2018, 57, 12680–12684. [CrossRef]
- Yan, Y.; Besseling, N. A. M.; de Keizer, A.; Marcelis, A. T. M.; Drechsler, M.; Cohen Stuart, M. A. Hierarchical self-assembly in solutions containing metal ions, ligand, and diblock copolymer. Angew. Chem. Int. Ed. Engl. 2007, 46, 1807–1809. [CrossRef]
- Nguyen, V. T. A.; De Pauw-Gillet M. C.; Gauthier, M.; Sandre, O. Magnetic polyion complex micelles for cell toxicity induced by radiofrequency magnetic field hyperthermia. Nanomaterials 2018, 8, 1014. [CrossRef]
- Mi, P.; Wang, F.; Nishiyama, N.; Cabral, H. Molecular Cancer Imaging with Polymeric Nanoassemblies: From Tumor Detection to Theranostics. Macromol. Biosci. 2017, 17, 1600305.
- Silva, C. O.; Pinho, J. O.; Lopes, J. M.; Almeida, A. J.; Gaspar, M. M.; Reis, C. Current Trends in Cancer Nanotheranostics: Metallic, Polymeric, and Lipid-Based Systems. Pharmaceutics 2019, 11, 22. [CrossRef] [PubMed]
- Indoria, S.; Singh, V.; Hsieh, M. F. Recent advances in theranostic polymeric nanoparticles for cancer treatment: A review. Int. J. Pharm. 2020, 582, 119314. [CrossRef] [PubMed]
- Xiao, Z.; Chan, L.; Zhang, D.; Huang, C.; Mei, C.; Gao, P.; Huang, Y.; Liang, J.; He, L.; Shi, C.; Chen, T.; Luo, L. Precise delivery of a multifunctional nanosystem for MRI-guided cancer therapy and monitoring of tumor response by functional diffusion-weighted MRI. J. Mater. Chem. B 2019, 7, 2926–2937.
- Deng, K.; Zhao, X.; Liu, F.; Peng, J.; Meng, C.; Huang, Y.; Ma, L.; Chang, C.; Wei, H. Synthesis of Thermosensitive Conjugated Triblock Copolymers by Sequential Click Couplings for Drug Delivery and Cell Imaging. ACS Biomater. Sci. Eng. 2019, 5, 3419–3428. [CrossRef]
- Ke, W.; Li, J.; Mohammed, F.; Wang, Y.; Tou, K.; Liu, X.; Wen, P.; Kinoh, H.; Anraku, Y.; Chen, H.; Kataoka, K.; Ge, Z. Therapeutic Polymersome Nanoreactors with Tumor-Specific Activable Cascade Reactions for Cooperative Cancer Therapy. ACS Nano 2019, 13, 2357–2369.
- Hood, M. A.; Mari, M.; Muñoz-Espí, R. Synthetic Strategies in the Preparation of Polymer/Inorganic Hybrid Nanoparticles. Materials (Basel) 2014, 7, 4057–4087. [CrossRef]
- 1553. Tharmavaram, M.; Rawtani, D.; Pandey, G. Fabrication routes for one-dimensional nanostructures via block copolymers. Nano Converg. 2017, 4, 12.
- Li, X.; Iocozzia, J.; Chen, Y.; Zhao, S.; Cui, X.; Wang, W.; Yu, H.; Lin, S.; Lin, Z. From Precision Synthesis of Block Copolymers to Properties and Applications of Nanoparticles. Angew. Chem. Int. Ed. Engl. 2018, 57, 2046–2070. [CrossRef]
- Scaravelli, R. C.; Dazzi, R. L.; Giacomelli, F. C.; Machado, G.; Giacomelli, C.; Schmidt, V. Direct synthesis of coated gold nanoparticles mediated by polymers with amino groups. J. Colloid Interface Sci. 2013, 397, 114–121. [CrossRef] [PubMed]
- 1556. Podhorska, L.; Delcassian, D.; Goode, A. E.; Agyei, M.; McComb, D. W.; Ryan, M. P.; Dunlop, I. E. Mechanisms of Polymer-Templated Nanoparticle Synthesis: Contrasting ZnS and Au. Langmuir 2016, 32, 9216–9222.
- Zhang, G.; Tang, S.; Li, A. Thermally Stable Metallic Nanoparticles Prepared via Core-Cross-linked Block Copolymer Micellar Nanoreactors. Langmuir 2017, 33, 6353–6362. [CrossRef]
- Jang, J. D.; Jeon, S.-W.; Yoon, Y.-J.; Bang, J.; Han, Y. S.; Kim, T.-H. Self-assembly of gold nanoparticles in a block copolymer aggregate template driven by hydrophobic interactions. Polym. Chem. 2019, 10, 6269–6277. [CrossRef]
- Sohn, H.; Shin, H. W.; Lee, S. M. Metal-Mediated Morphology Regulation of Self-Assembled Double-Hydrophilic Block Copolymers. ACS Macro Lett. 2020, 9, 600–605. [CrossRef]
- Constantinou, A. P.; Marie-Sainte, U.; Peng, L.; Carroll, D. R.; McGilvery, C. M.; Dunlop, I. E.; Georgiou, T. K. Effect of block copolymer architecture and composition on gold nanoparticle fabrication. Polymer Chemistry 2019, 10, 4637–4642. [CrossRef]
- Yao, N.; Lin, W.; Zhang, X.; Gu, H.; Zhang, L. Amphiphilic β-cyclodextrin-based star-like block copolymer unimolecular micelles for facile in situ preparation of gold nanoparticles. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 186–196. [CrossRef]
- 1562. Li, C.; Li, Q.; Kaneti Y. V.; Hou, D.; Yamauchi, Y.; Mai, Y. Self-assembly of block copolymers towards mesoporous materials for energy storage and conversion systems. Chem. Soc. Rev. 2020, 49, 4681–4736.
- 1563. Kumar, L.; Singh, S.; Horechyy, A.; Fery, A.; Nandan B. Block copolymer template-directed catalytic systems: Recent progress and perspectives. Membranes 2021, 11, 318.
- Hara, S.; Zama, T.; Takashima, W.; Kaneto, K. Artificial muscles based on polypyrrole actuators with large strain and stress induced electrically. Polym. J. 2004, 36, 151–161. [CrossRef]
- O’Halloran, A.; O’Malley, F.; McHugh, P. A review on dielectric elastomer actuators, technology, applications, and challenges. J. Appl. Phys. 2008, 104, 071101. [CrossRef]
- Imaizumi, S.; Kokubo, H.; Watanabe M. Polymer actuators using ion-gel electrolytes prepared by self-assembly of ABA-triblock copolymers. Macromolecules 2012, 45, 401–409. [CrossRef]
- Imaizumi, S.; Kato, Y.; Kokubo, H.; Watanabe, M. Driving mechanisms of ionic polymer actuators having electric double layer capacitor structures. J. Phys. Chem. B 2012, 116, 5080–5089. [CrossRef]
- Margaretta, E.; Fahs, G. B.; Inglefield Jr., D. L.; Jangu, C.; Wang, D.; Heflin, J. R.; Moore, R. B.; Long, T. E. Imidazolium-containing ABA Triblock copolymers as Electroactive devices. ACS Appl. Mater. Interfaces 2016, 8, 1280–1288. [CrossRef] [PubMed]
- Wu, T.; Wang, D.; Zhang, M.; Heflin, J. R.; Moore, R. B.; Long, T. E. RAFT synthesis of ABA triblock copolymers as ionic liquid-containing electroactive membranes. ACS Appl. Mater. Interfaces 2012, 4, 6552–6559. [CrossRef] [PubMed]
- Kim, O.; Kim, H.; Choi, U. H.; Park, M. J. One-volt-driven superfast polymer actuators based on single-ion conductors. Nat. Commun. 2016, 7, 13576. [CrossRef]
- Park, M. J.; Choi, I.; Hong, J.; Kim, O. Polymer electrolytes integrated with ionic liquids for future electrochemical devices. J. Appl. Polym. Sci. 2013, 129, 2363–2376. [CrossRef]
- Seki, S.; Kobayashi, Y.; Miyashiro, H.; Ohno, Y.; Usami, A.; Mita, Y.; Kihira, N.; Watanabe, M.; Terada, N. Lithium secondary batteries using modified-imidazolium room-temperature ionic liquid. J. Phys. Chem. B 2006, 110, 10228–10230. [CrossRef]
- Zakeeruddin S. M.; Gratzel, M. Solvent-free ionic liquid electrolytes for Mesoscopic dye-sensitized solar cells. Adv. Funct. Mater. 2009, 19, 2187–2202. [CrossRef]
- Lu, W.; Fadeev, A. G.; Qi, B.; Smela, E.; Mattes, B. R.; Ding, J.; Spinks, G. M.; Mazurkiewicz, J.; Zhou, D.; Wallace, G. G.; MacFarlane, D. R.; Forsyth, S. A.; Forsyth, M. Use of ionic liquids for pi-conjugated polymer electrochemical devices. Science 2002, 297, 983–987.
- Ding, J.; Zhou, D.; Spinks, G.; Wallace, G.; Forsyth, S.; Forsyth, M.; MacFarlane, D. Use of Ionic Liquids as Electrolytes in Electromechanical Actuator Systems Based on Inherently Conducting Polymers. Chem. Mater. 2003, 15, 2392–2398. [CrossRef]
- Cho, J. H.; Lee, J.; Xia, Y.; Kim, B.; He, Y.; Renn, M. J.; Lodge, T. P.; Frisbie, C. D. Printable ion- gel gate dielectrics for low-voltage polymer thin-film transistors on plastic. Nat. Mater. 2008, 7, 900–906. [CrossRef]
- Cho, J. H.; Lee, J.; He, Y.; Kim, B.; Lodge, T. P.; Frisbie, C. D. High-capacitance ion gel gate dielectrics with faster polarization response times for organic thin film transistors. Adv. Mater. 2008, 20, 686–690. [CrossRef]
- Virgili, J. M.; Hexemer, A.; Pople, J. A.; Balsara, N. P.; Segalman, R. A. Phase behavior of polystyrene-block-poly(2-vinylpyridine) copolymers in a selective ionic liquid solvent. Macromolecules 2009, 42, 4604–4613. [CrossRef]
- 1579. Xie, R.; López-Barrón, C. R.; Wagner, N. J. Self-assembly of block copolymers in ionic liquids. ACS Symp. Ser. 2017, 1250, 83–142.
- Yoshida, K.; Nakamura, M.; Kazue, Y.; Tachikawa, N.; Tsuzuki, S.; Seki, S.; Dokko, K.; Watanabe, M. Oxidative-stability enhancement and charge transport mechanism in glyme-lithium salt equimolar complexes. J. Am. Chem. Soc. 2011, 133, 13121–13129. [CrossRef]
- Ueno, K.; Yoshida, K.; Tsuchiya, M.; Tachikawa, N.; Dokko, K.; Watanabe, M. Glyme-Lithium Salt Equimolar Molten Mixtures: Concentrated Solutions or Solvate Ionic Liquids? J. Phys. Chem. B 2012, 116, 11323–11331. [CrossRef] [PubMed]
- Kitazawa, Y.; Iwata, K.; Imaizumi, S.; Ahn, H.; Kim, S.Y.; Ueno, K.; Park, M.J.; Watanabe, M. Gelation of solvate ionic liquid by self-assembly of block copolymer and characterization as polymer electrolyte. Macromolecules 2014, 47, 6009–6016. [CrossRef]
- Kitazawa, Y.; Iwata, K.; Kido, R.; Imaizumi, S.; Tsuzuki, S.; Shinoda, W.; Ueno, K.; Mandai, T.; Kokubo, H.; Dokko, K.; Watanabe, M. Polymer electrolytes containing solvate ionic liquids: a new approach to achieve high ionic conductivity, thermal stability, and a wide potential window. Chem. Mater. 2017, 30, 252–261.
- 1584. Kawazoe, T.; Hashimoto, K.; Kitazawa, Y.; Kokubo, H.; Watanabe, M. A polymer electrolyte containing solvate ionic liquid with increased mechanical strength formed by self-assembly of ABA-type Ionomer triblock copolymer, Electrochim. Acta 2017, 235, 287–294.
- Lee, J.; Panzer, M. J.; He, Y. Y.; Lodge, T. P.; Frisbie, C. D. Ion gel gated polymer thin-film transistors. J. Am. Chem. Soc. 2007, 129, 4532–4533. [CrossRef]
- Lee, K. H.; Zhang, S.; Gu, Y.; Lodge, T. P.; Frisbie, C. D. Transfer printing of thermoreversible ion gels for flexible electronics. ACS Appl. Mater. Interfaces 2013, 5, 9522–9527. [CrossRef]
- Lee, K. H.; Zhang, S.; Lodge, T. P.; Frisbie, C. D. Electrical impedance of spin-coatable ion gel films. J. Phys. Chem. B 2011, 115, 3315–3321. [CrossRef]
- Yomogida, Y.; Pu, J.; Shimotani, H.; Ono, S.; Hotta, S.; Iwasa, Y.; Takenobu, T. Ambipolar organic single-crystal transistors based on ion gels. Adv. Mater. 2012, 24, 4392–4397. [CrossRef]
- Pu, J.; Yomogida, Y.; Liu, K. K.; Li, L. J.; Iwasa, Y.; Takenobu, T. Highly flexible MoS2 thin-film transistors with ion gel dielectrics. Nano Lett. 2012, 12, 4013–4017. [CrossRef]
- Zhang, M.; Yu, M.; Li, F.; Zhu, M.; Li, M.; Gao, Y.; Li, L.; Liu, Z.; Zhang, J.; Zhang, D.; Yi, T.; Huang, C. A highly selective fluorescence turn- on sensor for cysteine/homocysteine and its application in bioimaging. J. Am. Chem. Soc. 2007, 129, 10322–10323. [CrossRef]
- Li, Y.; Hu, X.; Tian, S.; Li, Y.; Zhang, G.; Zhang, G.; Liu, S. Polyion complex micellar nanoparticles for integrated fluorometric detection and bacteria inhibition in aqueous media. Biomaterials 2014, 35, 1618–1626. [CrossRef]
- Tian, S.; Liu. G.; Wang, X.; Wu, T.; Yang, J.; Ye, X.; Zhang, G.; Hu, J.; Liu, S. pH-Regulated Reversible Transition Between Polyion Complexes (PIC) and Hydrogen-Bonding Complexes (HBC) with Tunable Aggregation-Induced Emission. ACS Appl. Mater. Interfaces 2016, 8, 3693–3702. [CrossRef] [PubMed]
- Sureka, H.V.; Obermeyer, A.C.; Flores, R.J.; Olsen, B.D. Catalytic Biosensors from Complex Coacervate Core Micelles (C3M) Thin Films. ACS Appl. Mater. Interfaces 2019, 11, 32354–32365. [CrossRef]
- Molina, E.; Mathonnat, M.; Richard, J.; Lacroix-Desmazes, P.; In, M.; Dieudonné, P.; Cacciaguerra, T.; Gérardin, C.; Marcotte, N. pH-Mediated Control over the Mesostructure of Ordered Mesoporous Materials Templated by Polyion Complex Micelles. Beilstein J. Nanotechnol. 2019, 10, 144–156. [CrossRef]
- Lim, W. T.; Tan, E. H.; Toh, C. K.; Hee, S. W.; Leong, S. S.; Ang, P. C. S.; Wong, N. S.; Chowbay, B. Phase I pharmacokinetic study of a weekly liposomal paclitaxel formulation (Genexol-PM) in patients with solid tumors. Ann. Oncol. 2010, 21, 382–388. [CrossRef]
- Lee, K. S.; Chung, H. C.; Im, S. A.; Park, Y. H.; Kim, C. S.; Kim, S.-B.; Rha, S. Y.; Lee, M. Y.; Ro, J. Multicenter phase II trial of genexol-PM, a cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res. Treat. 2008, 108, 241–250. [CrossRef]
- Kim, D.- W.; Kim, S.- Y.; Kim, H.- K.; Kim, S.- W., Shin, S. W.; Kim, J. S.; Park, K.; Lee, M. Y.; Heo, D. S. Multicenter phase II trial of genexol-PM, a novel cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2007, 18, 2009–2014.
- Saif, M. W.; Podoltsev, N. A.; Rubin, M. S.; Figueroa, J. A.; Lee, M. Y.; Kwon, J.; Rowen, E.; Yu, J.; Kerr, R. O. Phase II clinical trial of paclitaxel loaded polymeric micelle in patients with advanced pancreatic cancer. Cancer Invest. 2010, 28, 186–194.
- Kato, K.; Chin, K.; Yoshikawa, T.; Yamaguchi, K.; Tsuji, Y.; Esaki, T.; Sakai, K.; Kimura, M.; Hamaguchi, T.; Shimada, Y.; Matsumura, Y.; Ikeda, R. Phase II study of NK105, a paclitaxel-incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer. Investig. New Drugs 2012, 30, 1621–1627.
- Danson, S.; Ferry, D.; Alakhov, V.; Margison, J.; Kerr, D.; Jowle, D.; Brampton, M.; Halbert, G.; Ranson, M. Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br. J. Cancer 2004, 90, 2085–2091. [CrossRef]
- 1601. Valle, J. W.; Armstrong, A.; Newman, C.; Alakhov, V.; Pietrzynski, G.; Brewer, J.; Campbell, S.; Corrie, P.; Rowinsky, E. K.; Ranson, M. A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Investig. New Drugs 2010, 29, 1029–1037.
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br. J. Cancer 2005, 93, 678–687. [CrossRef] [PubMed]
- Alami, N.; Banerjee, K.; Juste, S.; Page, V.; Brossard, M.; Hayashi, T.; Igarashi, E.; Jones, B. L. NC-6004, a novel cisplatin-incorporated polymeric micelle, is highly effective against oxaliplatin-resistant tumor models. AACR Meet. Abstr. 2006, 133.
- Plummer, R.; Wilson, R. H.; Calvert, H.; Boddy, A. V.; Griffin, M.; Sludden, J.; Tilby, M. J.; Eatock, M.; Pearson, D. G.; Ottley, C. J.; Matsumura, Y.; Kataoka, K.; Nishiya, T. A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. Br. J. Cancer 2011, 104, 593–598.
- 1605. Takahashi, A.; Yamamoto, Y.; Yasunaga, M.; Koga, Y.; Kuroda, J.; Takigahira, M.; Harada, M.; Saito, H.; Hayashi, T.; Kato, Y.; Kinoshita, T.; Ohkohchi, N.; Hyodo, I.; Matsumura, Y. NC-6300, an epirubicin-incorporating micelle, extends the antitumor effect and reduces the cardiotoxicity of epirubicin. Cancer Sci. 2013, 104, 920–925.
- Hsiang, Y. H.; Liu, L. F. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48, 1722–1726.
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991, 51, 4187–4191.
- Kuroda, J.; Kuratsu, J.; Yasunaga, M.; Koga, Y.; Saito, Y.; Matsumura, Y. Potent antitumor effect of SN-38-incorporating polymeric micelle, NK012, against malignant glioma. Int. J. Cancer. 2009, 124, 2505–2511. [CrossRef]
- Kuroda, J.; Kuratsu, J.; Yasunaga, M.; Koga, Y.; Kenmotsu, H.; Sugino, T.; Matsumura, Y. Antitumor effect of NK012, a 7-ethyl-10-hydroxycamptothecin-incorporating polymeric micelle, on U87MG orthotopic glioblastoma in mice compared with irinotecan hydrochloride in combination with bevacizumab. Clin Cancer Res. 2010, 16, 521–529.
- Hamaguchi, T.; Doi, T.; Eguchi-Nakajima, T.; Kato, K.; Yamada, Y.; Shimada, Y.; Fuse, N.; Ohtsu, A.; Matsumoto, S.; Takanashi, M.; Matsumura, Y. Phase I study of NK012, a novel SN-38-incorporating micellar nanoparticle, in adult patients with solid tumors. Clin Cancer Res. 2010, 16, 5058–5066. [CrossRef]
- Burris, H. A., III.; Infante, J. R.; Spigel, D. R.; Greco, F. A.; Thompson, D. S.; Matsumoto, S.; Kawamura, S.; Jones, S. F. A phase I dose-escalation study of NK012. Proc. Am. Soc. Clin. Oncol. 2008, Abstract No. 2538.
- Sudimack, J.; Lee, R. J. Targeted drug delivery via the folate receptor. Adv. Drug Delivery Rev. 2000, 41, 147–162. [CrossRef] [PubMed]
- Low, P. S.; Henne, W. A.; Doorneweerd, D. D. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc. Chem. Res. 2008, 41, 120–129. [CrossRef] [PubMed]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [CrossRef] [PubMed]
- Ge, Z.; Chen, Q.; Osada, K.; Liu, X.; Tockary, T. A.; Uchida, S.; Dirisala, A.; Ishii, T.; Nomoto, T.; Toh, K.; Matsumoto, Y.; Oba, M.; Kano, M. R.; Itaka, K.; Kataoka, K. Targeted gene delivery by polyplex micelles with crowded PEG palisade and cRGD moiety for systemic treatment of pancreatic tumors. Biomaterials 2014, 35, 3416–3426.
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [CrossRef]
- Varkouhi, A. K.; Scholte, M.; Storm, G.; Haisma, H. J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [CrossRef]
- Pittella, F.; Zhang, M.; Lee, Y.; Kim, H. J.; Tockary, T.; Osada, K.; Ishii, T.; Miyata, K.; Nishiyama, N.; Kataoka, K. Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity. Biomaterials 2011, 32, 3106–3114.
- Monnery, B. D.; Wright, M.; Cavill, R.; Hoogenboom, R.; Shaunak, S.; Steinke, J. H. G.; Thanou, M. Cytotoxicity of polycations: Relationship of molecular weight and the hydrolytic theory of the mechanism of toxicity. Int. J. Pharm. 2017, 521, 249–258. [CrossRef]
- Breunig, M.; Lungwitz, U.; Liebl, R.; Goepferich, A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14454–14459. [CrossRef]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [CrossRef]
- Ishihara, K. Revolutionary advances in 2-methacryloyloxyethyl phosphorylcholine polymers as biomaterials. J. Biomed. Mater. Res., Part A 2019, 107, 933–943. [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [CrossRef] [PubMed]
- Zhang, Y. A.; Ni, C. H.; Shi, G.; Wang, J.; Zhang, M.; Li, W. The polyion complex nano-prodrug of doxorubicin (DOX) with poly(lactic acid-co-malic acid)-block-polyethylene glycol: preparation and drug controlled release. Med. Chem. Res. 2015, 24, 1189–1195. [CrossRef]
- Feiner-Gracia, N.; Olea, R. A.; Fitzner, R.; El Boujnouni, N.; van Asbeck, A. H.; Brock, R.; Albertazzi, L. Super-Resolution Imaging of Structure, Molecular Composition, and Stability of Single Oligonucleotide Polyplexes. Nano Lett. 2019, 19, 2784–2792. [CrossRef] [PubMed]
- Riera, R.; Feiner-Gracia, N.; Fornaguera, C.; Cascante, A.; Borroś, S.; Albertazzi, L. Tracking the DNA Complexation State of PBAE Polyplexes in Cells with Super Resolution Microscopy. Nanoscale 2019, 11, 17869–17877. [CrossRef]
Figure 1.
An anionic HP and a cationic-neutral dbp combined to generate complex coacervate core micelles. A single hp binds several dbps at f+ ∼ 0.3, and condensed C3Ms develop when f+ increases near 0.5. Adapted with permission from ref [29]. Copyright 2016, Wiley-VCH Verlag GmbH & Co.
Figure 1.
An anionic HP and a cationic-neutral dbp combined to generate complex coacervate core micelles. A single hp binds several dbps at f+ ∼ 0.3, and condensed C3Ms develop when f+ increases near 0.5. Adapted with permission from ref [29]. Copyright 2016, Wiley-VCH Verlag GmbH & Co.
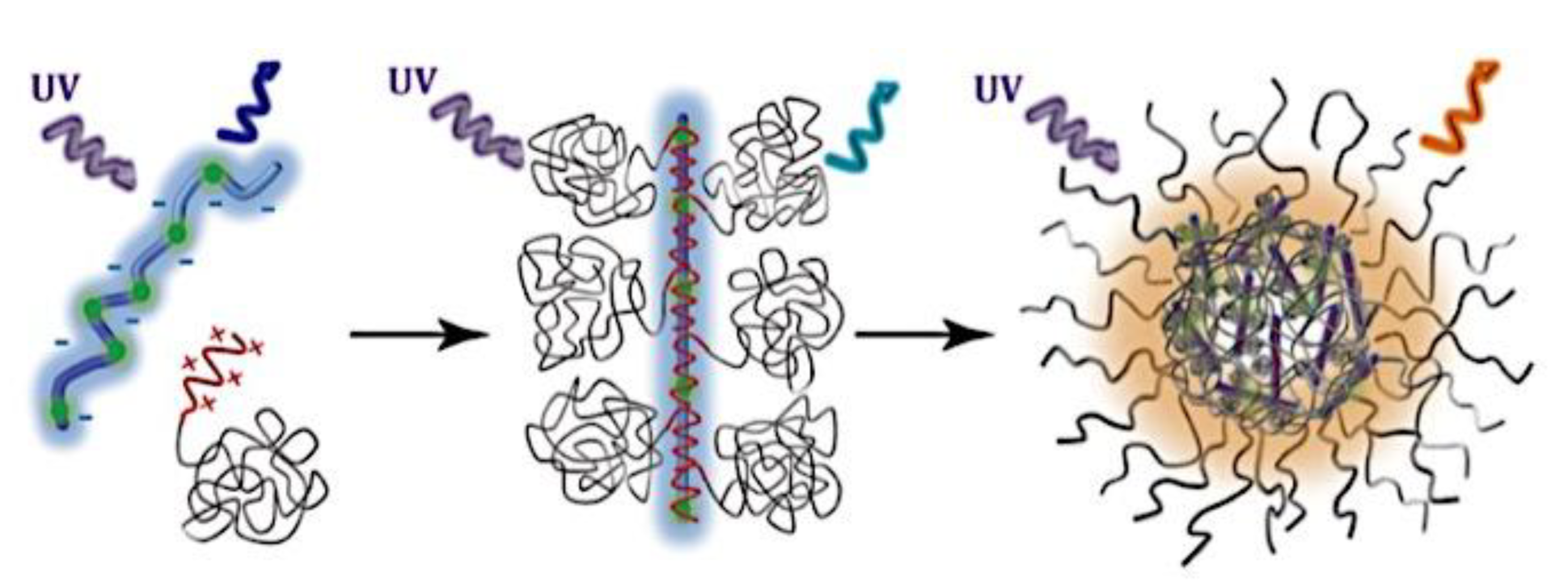
Figure 2.
A micelle with a complicated coacervation core represented schematically. The cationic block of the diblock copolymer and the anionic homopolymer complex together to produce the core. The diblock copolymer’s noncharged hydrophilic blocks make up the corona. The square root of the reciprocal grafting density σ is used to calculate the distance between the corona blocks. Hcorona is a representation of the corona's thickness or the corona blocks’ extension into the bulk solution. Rcore is the representation of the radius of the core. The equation Rmicelle = Rcore + Hcorona represents the entire micellar radius. Adapted with permission from ref [30]. Copyright 2004, American Chemical Society.
Figure 2.
A micelle with a complicated coacervation core represented schematically. The cationic block of the diblock copolymer and the anionic homopolymer complex together to produce the core. The diblock copolymer’s noncharged hydrophilic blocks make up the corona. The square root of the reciprocal grafting density σ is used to calculate the distance between the corona blocks. Hcorona is a representation of the corona's thickness or the corona blocks’ extension into the bulk solution. Rcore is the representation of the radius of the core. The equation Rmicelle = Rcore + Hcorona represents the entire micellar radius. Adapted with permission from ref [30]. Copyright 2004, American Chemical Society.
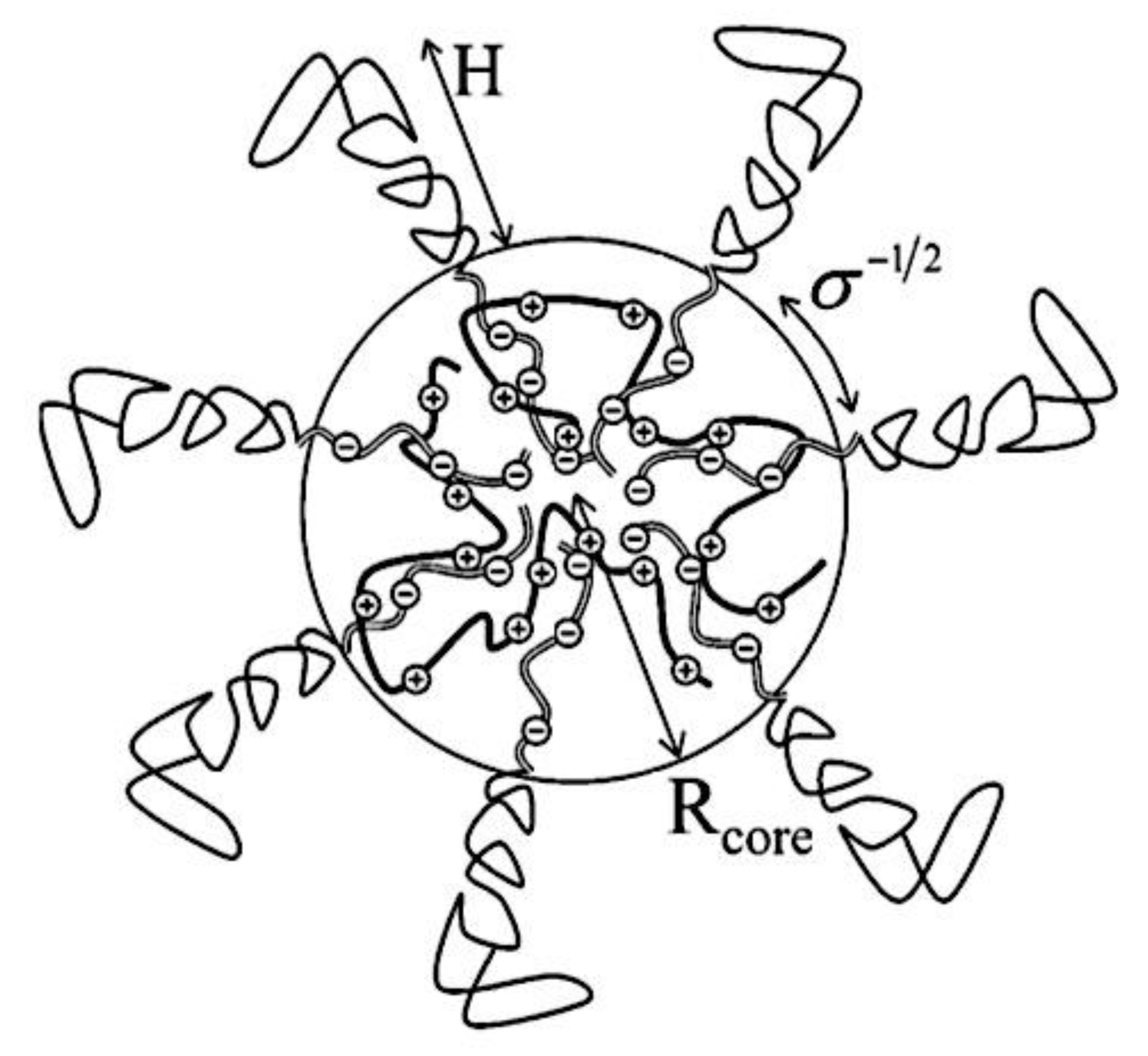
Figure 3.
The molecular structures and schematic representation of the synthesis of complex coacervate core micelles (C4Ms) based on cyclodextrin and a supramolecular redox-responsive cross-linker (Ad-SS-Ad). Four orthogonal interactions combine to generate Ad-SS-Ad-based C4Ms. The coordination complex is formed by metal-to-ligand coordination between one europium ion and three cyclodextrin-modified dipicolinic acids (βCD DPA) molecules. Additionally there are host-guest interactions between the βCD and the Ad-SS-Ad bislinker that result in oligomeric core-units along with reversible covalent disulfide crosslinks and coacervate interactions by adding the block copolymer (PMVP128-PEO477) with a positively charged part that neutralizes the oligomeric core-unit’s overall negative charge. Adapted with permission from ref [39]. Copyright 2020, The Authors, Published by Wiley-VCH Verlag GmbH & Co. KGaA.
Figure 3.
The molecular structures and schematic representation of the synthesis of complex coacervate core micelles (C4Ms) based on cyclodextrin and a supramolecular redox-responsive cross-linker (Ad-SS-Ad). Four orthogonal interactions combine to generate Ad-SS-Ad-based C4Ms. The coordination complex is formed by metal-to-ligand coordination between one europium ion and three cyclodextrin-modified dipicolinic acids (βCD DPA) molecules. Additionally there are host-guest interactions between the βCD and the Ad-SS-Ad bislinker that result in oligomeric core-units along with reversible covalent disulfide crosslinks and coacervate interactions by adding the block copolymer (PMVP128-PEO477) with a positively charged part that neutralizes the oligomeric core-unit’s overall negative charge. Adapted with permission from ref [39]. Copyright 2020, The Authors, Published by Wiley-VCH Verlag GmbH & Co. KGaA.
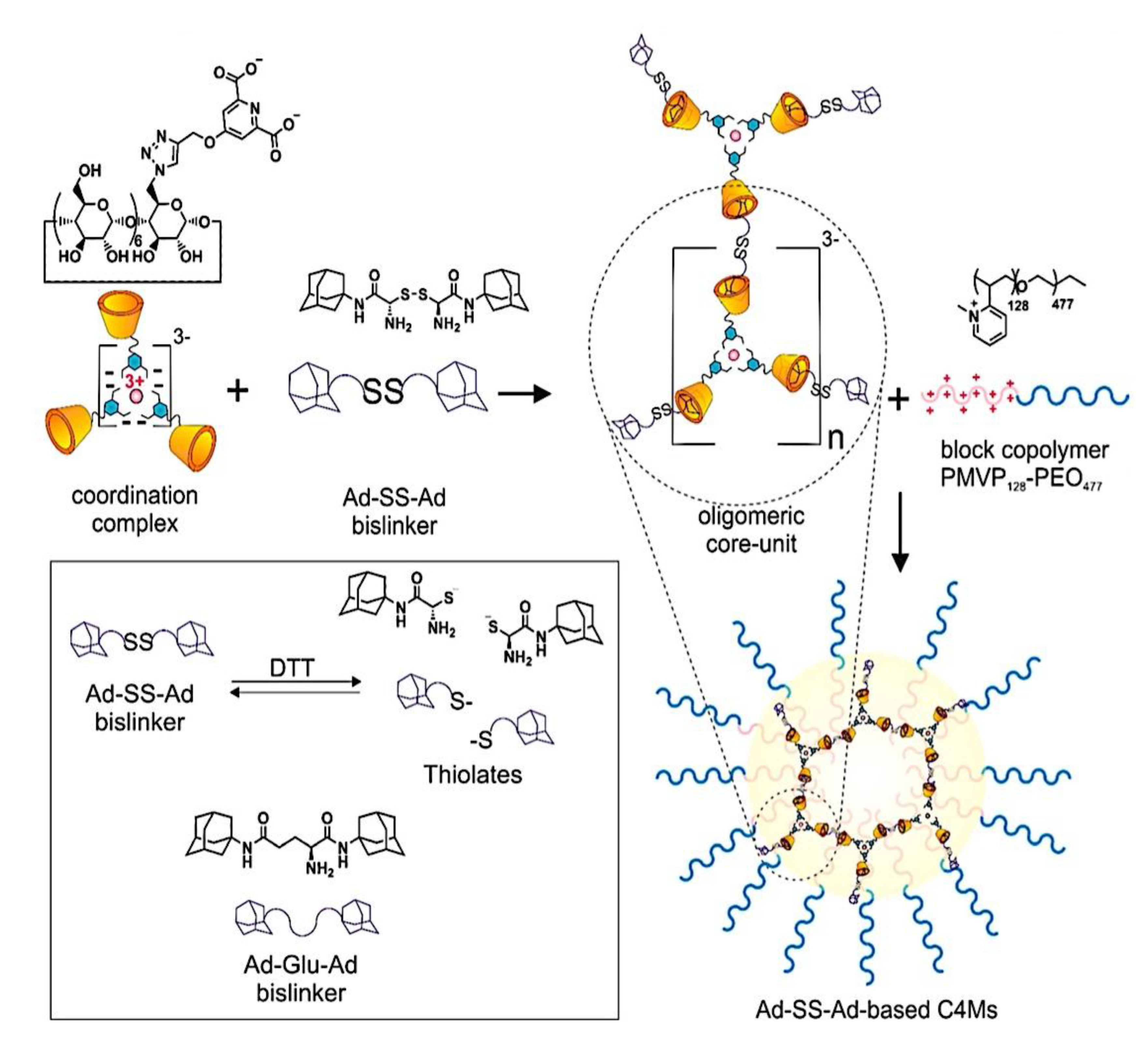
Figure 4.
C3Ms: constituent parts and microphase separation mechanism. In terms of nomenclature, a neutral, hydrophilic block is denoted by A, and oppositely charged polyelectrolyte blocks are represented by B/C. An AB diblock polycation plus either an AC diblock polyanion or a C homopolyanion make up a typical C3M. Adapted with permission from ref [58]. Copyright 2021, The Authors, Published by American Chemical Society.
Figure 4.
C3Ms: constituent parts and microphase separation mechanism. In terms of nomenclature, a neutral, hydrophilic block is denoted by A, and oppositely charged polyelectrolyte blocks are represented by B/C. An AB diblock polycation plus either an AC diblock polyanion or a C homopolyanion make up a typical C3M. Adapted with permission from ref [58]. Copyright 2021, The Authors, Published by American Chemical Society.
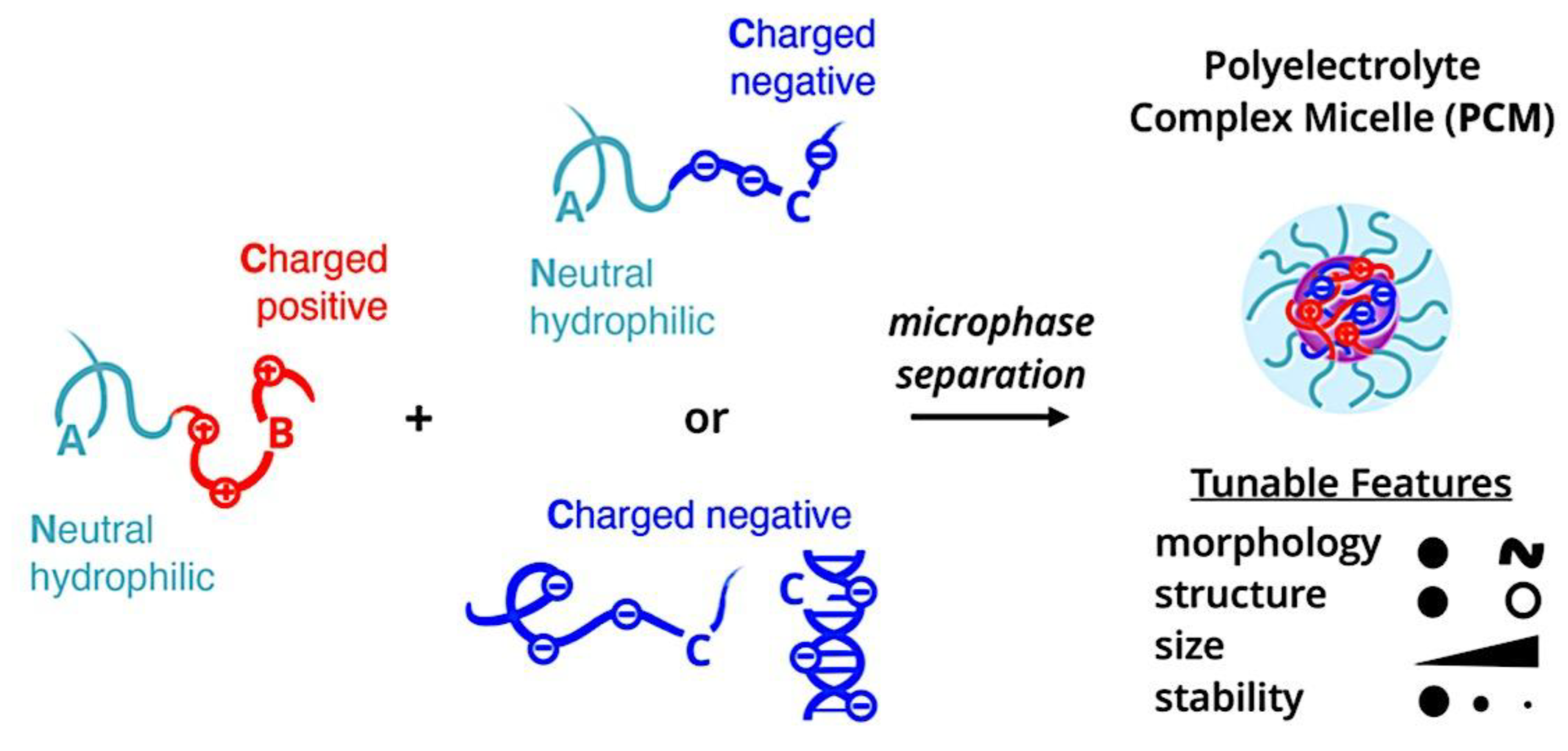
Figure 5.
Diagram illustrating the micelle exchange experiment based on FRET. Adapted from ref [103]. Copyright 2020, Published Open Access by American Chemical Society under the terms of the CC-BY-NC-ND license.
Figure 5.
Diagram illustrating the micelle exchange experiment based on FRET. Adapted from ref [103]. Copyright 2020, Published Open Access by American Chemical Society under the terms of the CC-BY-NC-ND license.
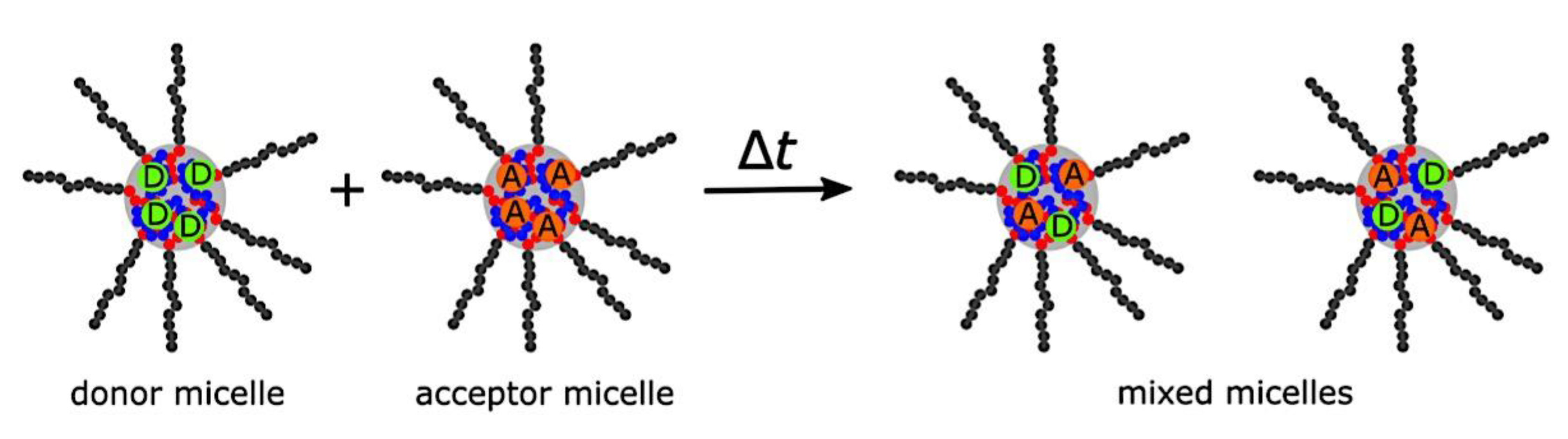
Figure 6.
Therapy plan for ovarian cancer over the long term that combines different anti-cancer medications. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 6.
Therapy plan for ovarian cancer over the long term that combines different anti-cancer medications. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
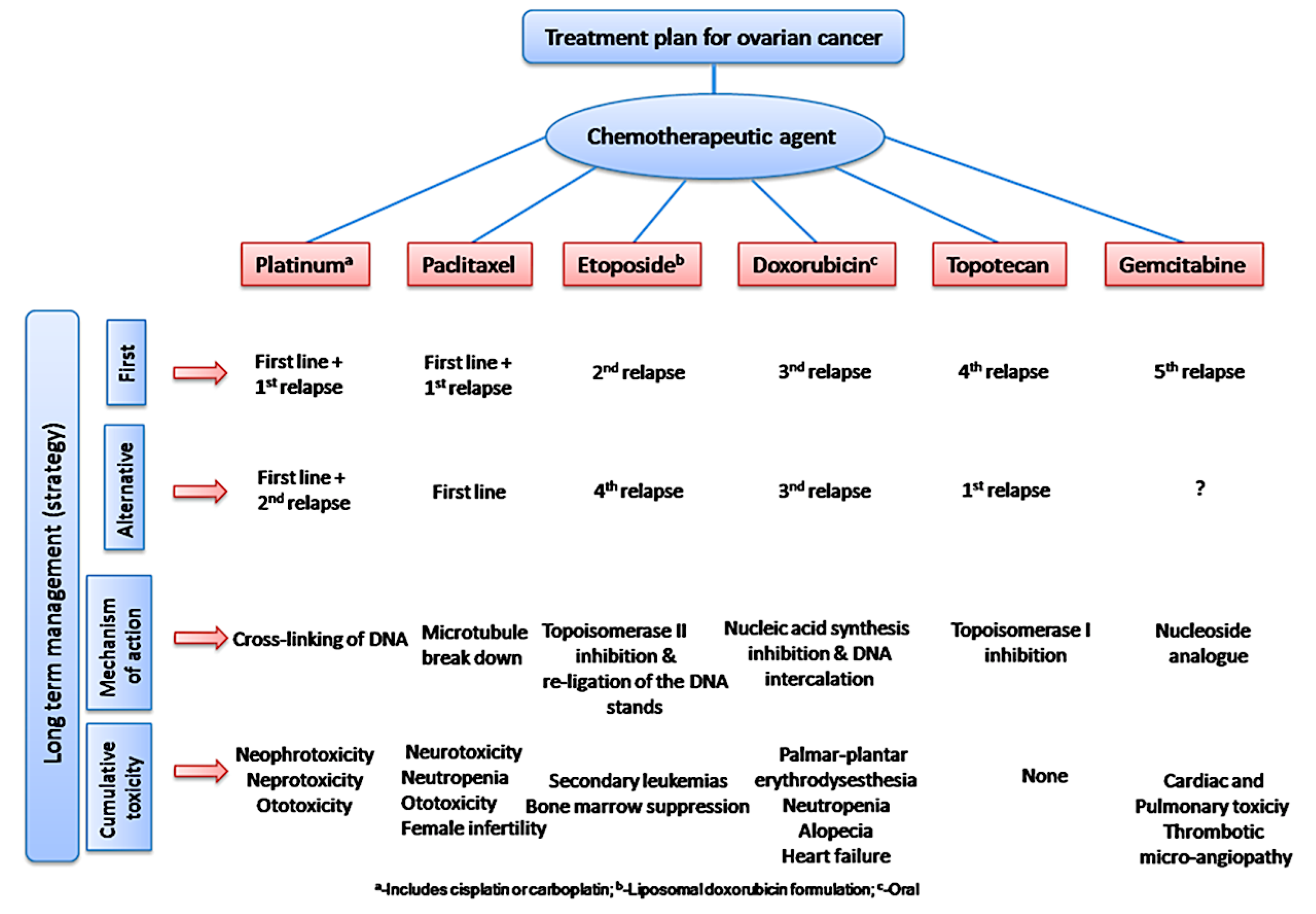
Figure 7.
When two oppositely charged polyions are combined in solution and either one or both of the polyions are conjugated to a neutral hydrophilic polymeric block, C3Ms are created. Adapted with permission from ref [264]. Copyright 2021, Elsevier Ltd. .
Figure 7.
When two oppositely charged polyions are combined in solution and either one or both of the polyions are conjugated to a neutral hydrophilic polymeric block, C3Ms are created. Adapted with permission from ref [264]. Copyright 2021, Elsevier Ltd. .
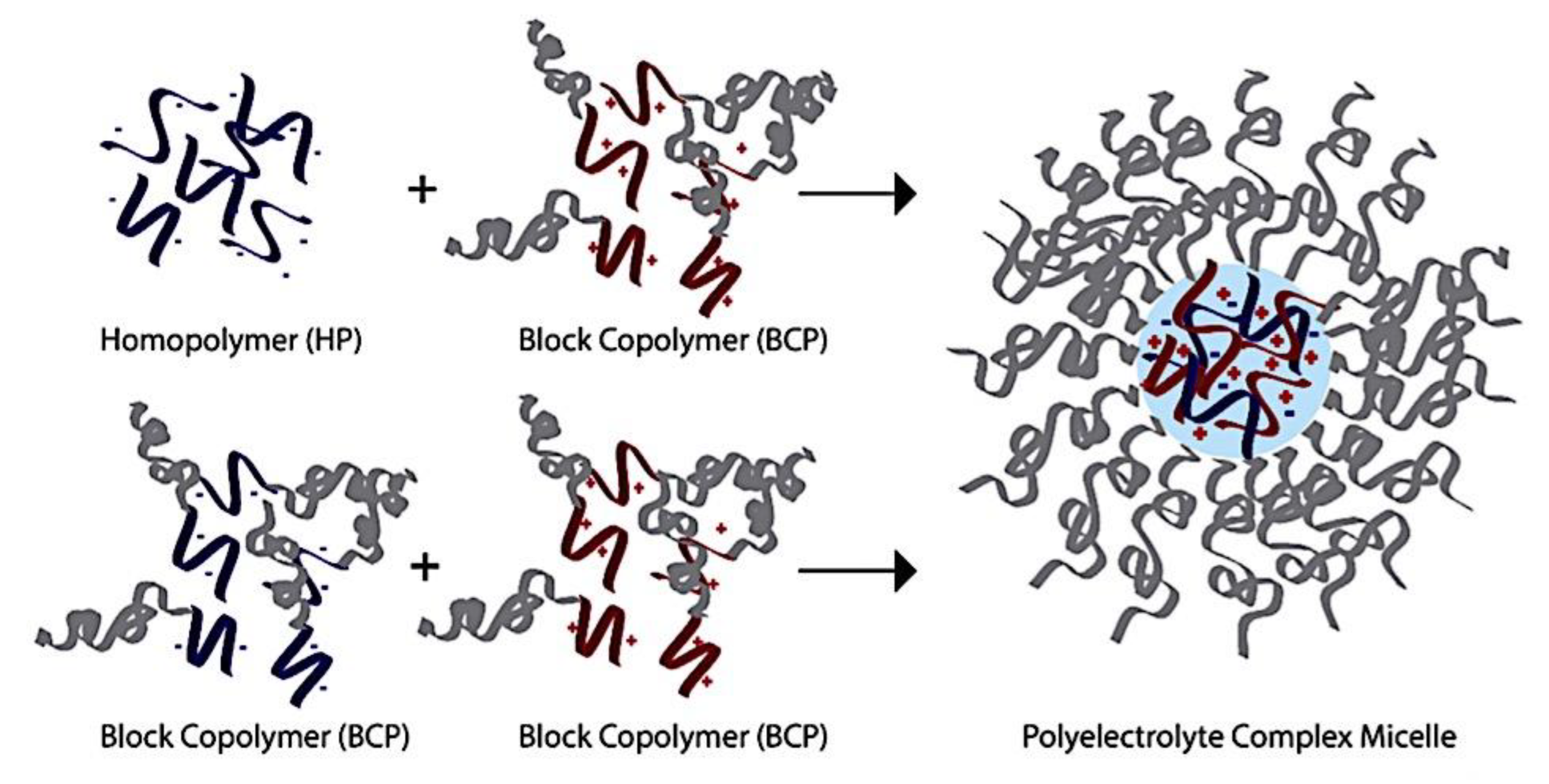
Figure 8.
Diagram showing the two diblock copolymers that make up a spherical complicated coacervate core micelle. The gray core of the neutral-ionic copolymers is made up of the polyelectrolyte blocks, while the micellar corona is home to the neutral blocks (blue and green). Adapted with permission from ref [27]. Copyright 2008, Elsevier B.V.
Figure 8.
Diagram showing the two diblock copolymers that make up a spherical complicated coacervate core micelle. The gray core of the neutral-ionic copolymers is made up of the polyelectrolyte blocks, while the micellar corona is home to the neutral blocks (blue and green). Adapted with permission from ref [27]. Copyright 2008, Elsevier B.V.
Figure 9.
Enhanced and prolonged therapeutic impact of nanotechnology-based chemotherapuetic drugs. (A) Delivery of conventional formulations and nanocarriers via oral or intravenous routes. (B) Illustrations of the pictorial structures of different drug delivery systems, including lipid nanoparticles/capsules, dendrimers, polymer micelles, and polymer-drug conjugates. (C) Using immunoconjugate nanosystems to increase the effectiveness of treatment. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 9.
Enhanced and prolonged therapeutic impact of nanotechnology-based chemotherapuetic drugs. (A) Delivery of conventional formulations and nanocarriers via oral or intravenous routes. (B) Illustrations of the pictorial structures of different drug delivery systems, including lipid nanoparticles/capsules, dendrimers, polymer micelles, and polymer-drug conjugates. (C) Using immunoconjugate nanosystems to increase the effectiveness of treatment. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
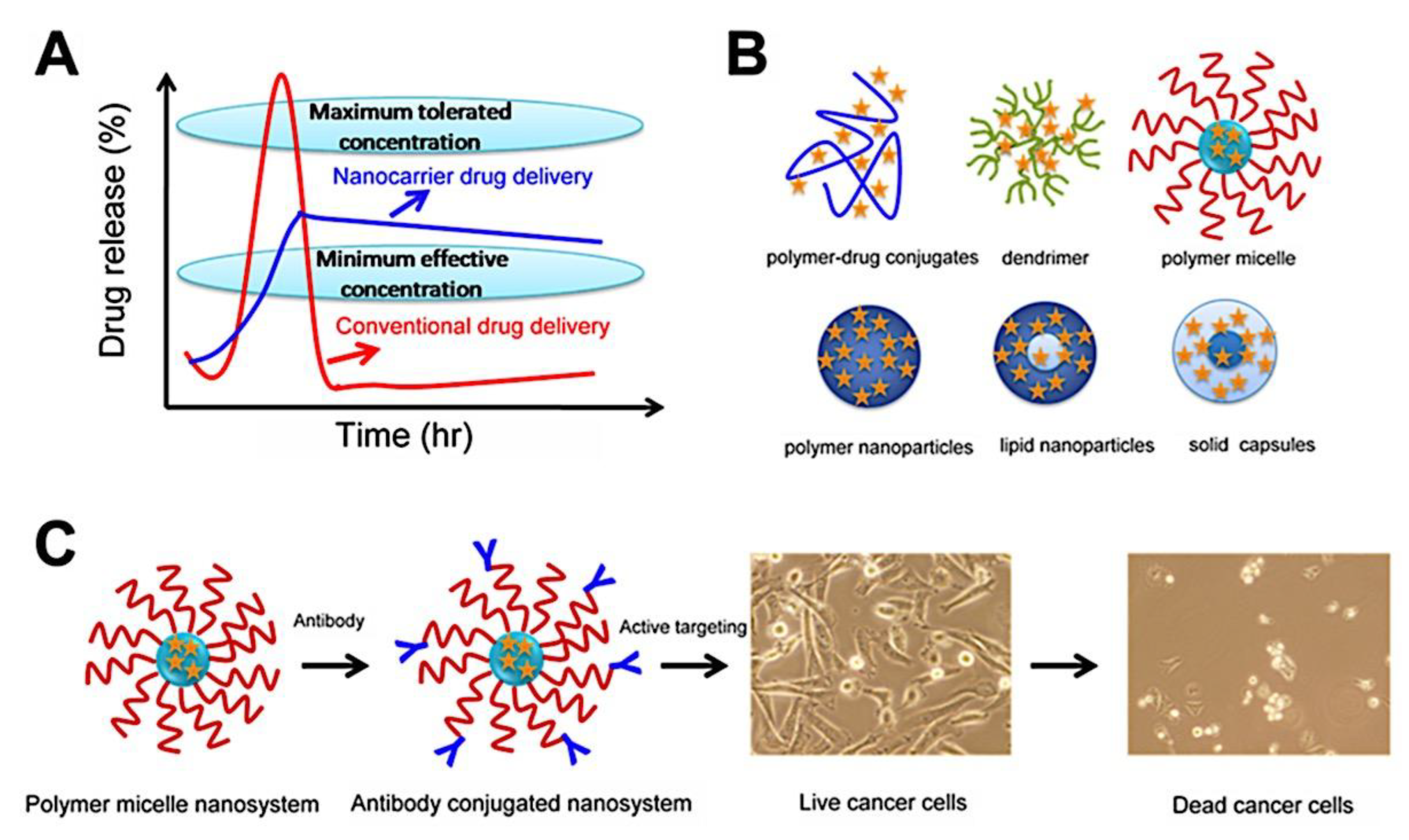
Figure 10.
An illustration of the method for employing immunoconjugated and nanoparticles to target cancer cells. Drug-loaded nanoparticles are used for passive targeting, while antibody-conjugated nanoparticles are used for active targeting. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 10.
An illustration of the method for employing immunoconjugated and nanoparticles to target cancer cells. Drug-loaded nanoparticles are used for passive targeting, while antibody-conjugated nanoparticles are used for active targeting. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
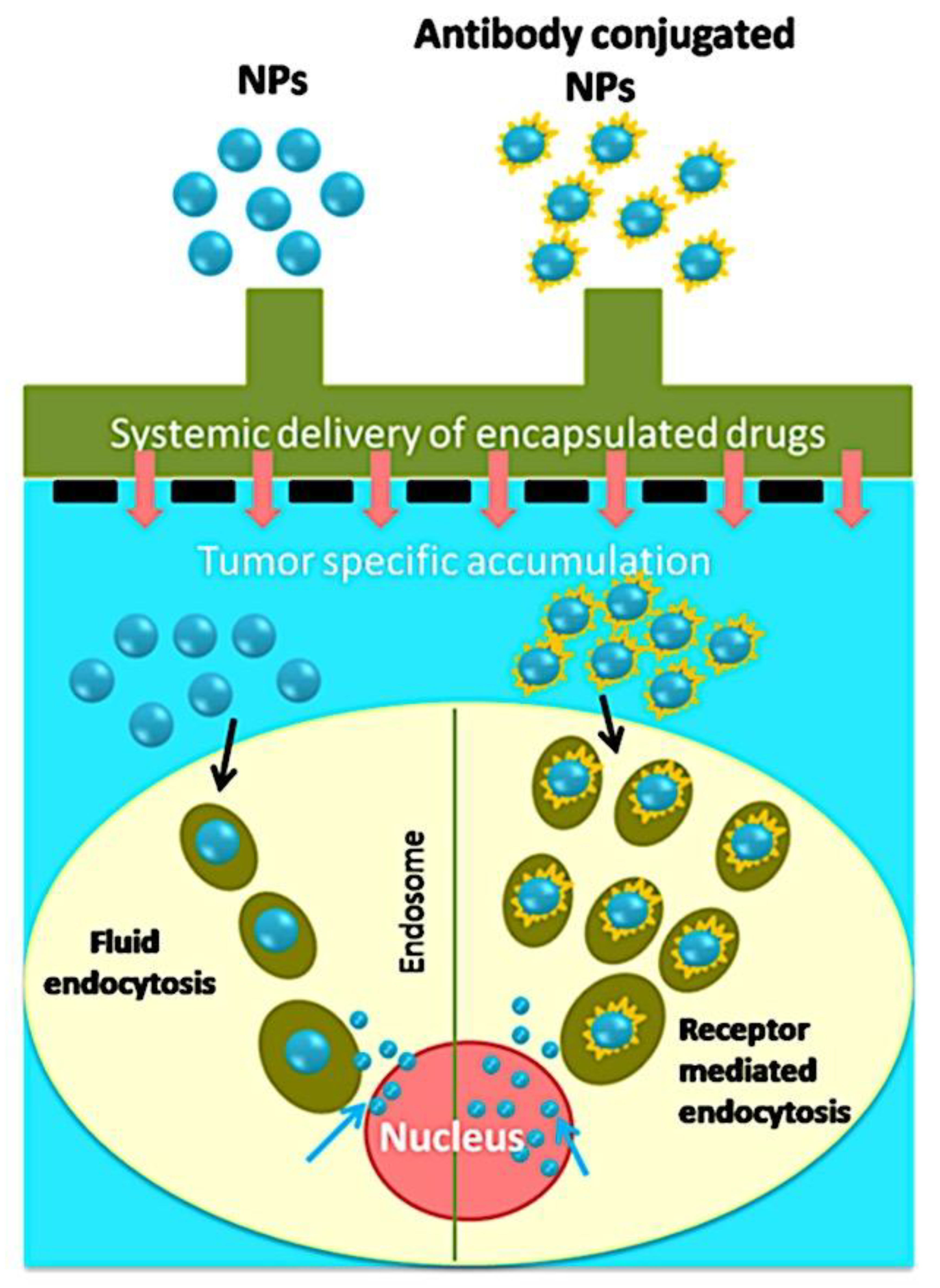
Figure 11.
Various polymer micelle forms achieved by the process of self-assembly. Within the block copolymers, hydrophobic-hydrophobic interactions always encourage this reaction. Since the core is totally hydrophobic, anti-cancer medications can be loaded into it. Antibody conjugations can make use of reactive functional groups. Their chemical structures serve as the basis for illustrations. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 11.
Various polymer micelle forms achieved by the process of self-assembly. Within the block copolymers, hydrophobic-hydrophobic interactions always encourage this reaction. Since the core is totally hydrophobic, anti-cancer medications can be loaded into it. Antibody conjugations can make use of reactive functional groups. Their chemical structures serve as the basis for illustrations. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
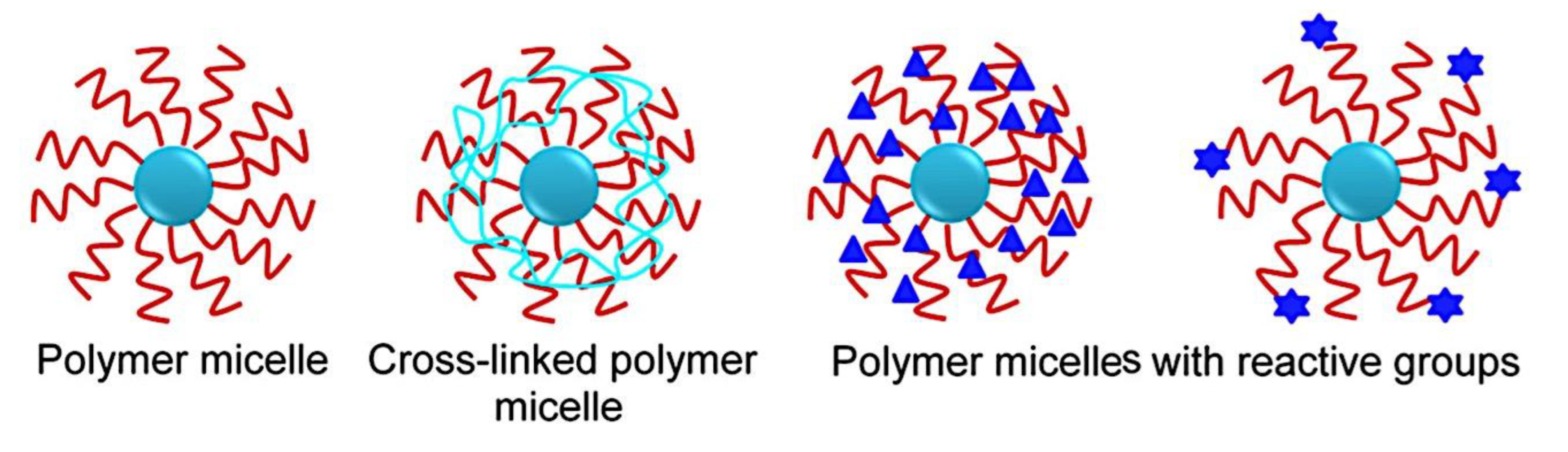
Figure 12.
Nanoparticle uptake by cancerous cells. (A-B) Fluorescence pictures of PLGA NPs, FITC in solution, and FITC-loaded PLGA NPs treated on A2780CP and MDA-MB-231 cells. DAPI is used to dye nuclei blue. (C-D) The fluorescence levels in A2780CP and MDA-MB-231 were measured using a flow cytometer. The control cells were shown as black, the nanoparticles in cells as blue, the FITC in cells as red, and the FITC-nanoparticles in cells as yellow lines. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 12.
Nanoparticle uptake by cancerous cells. (A-B) Fluorescence pictures of PLGA NPs, FITC in solution, and FITC-loaded PLGA NPs treated on A2780CP and MDA-MB-231 cells. DAPI is used to dye nuclei blue. (C-D) The fluorescence levels in A2780CP and MDA-MB-231 were measured using a flow cytometer. The control cells were shown as black, the nanoparticles in cells as blue, the FITC in cells as red, and the FITC-nanoparticles in cells as yellow lines. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
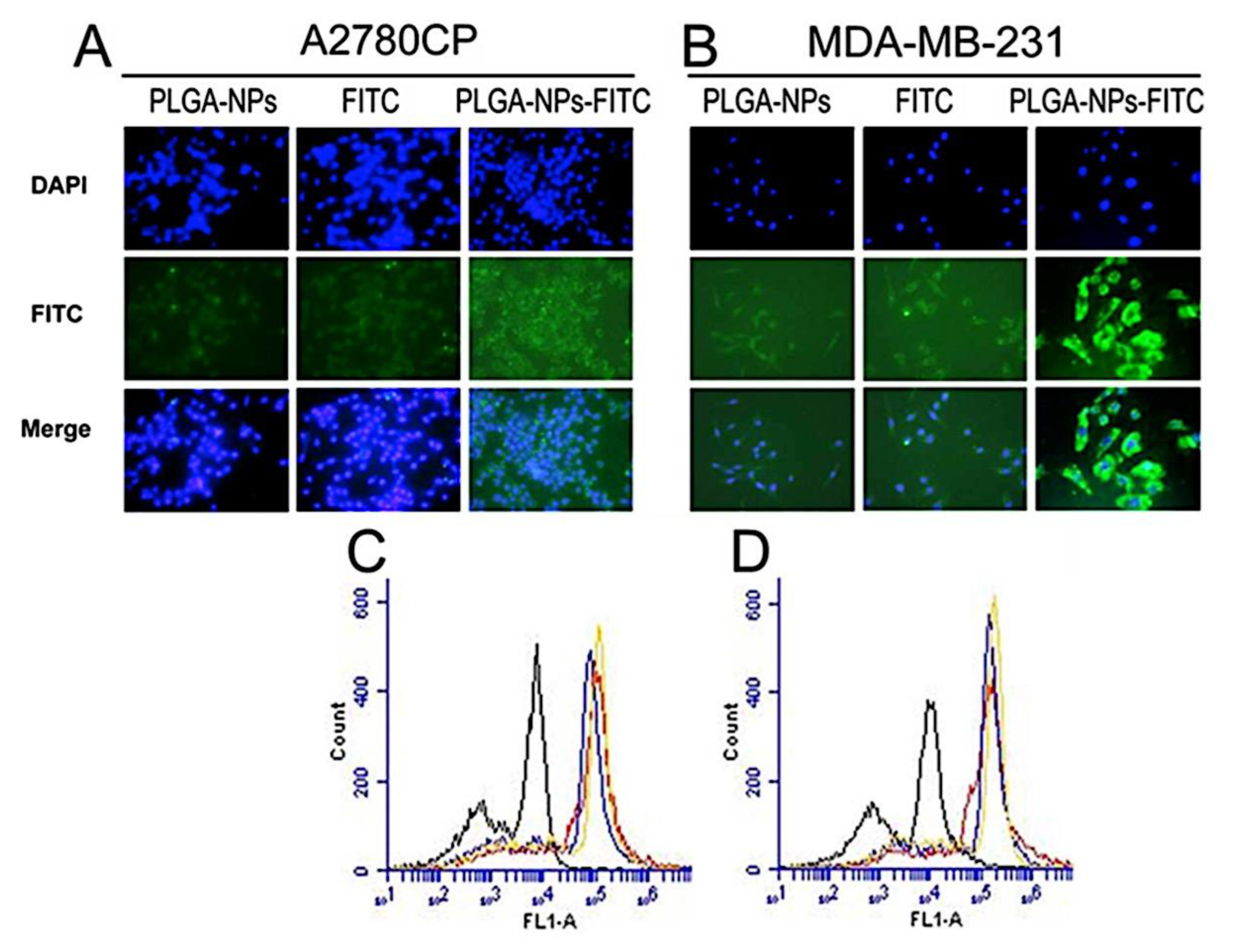
Figure 13.
Curcumin/photoactivator loaded, double-layered, antibody-conjugated magnetic nanoformulation having multiple uses in medicine. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 13.
Curcumin/photoactivator loaded, double-layered, antibody-conjugated magnetic nanoformulation having multiple uses in medicine. Adapted from ref [197]. Copyright 2010, The Authors, Published Open Access by BioMed Central Ltd. under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0).
Figure 14.
(A) An anionic HP and a cationic-neutral dbp combined to generate complex coacervate core micelles. A single hp binds several dbps at f+ ∼ 0.3, and condensed C3Ms develop when f+ increases near 0.5. (B) The hydrodynamic radii and intensity obtained from light scattering, adjusted for dilution, both rise as f+ increases. (C) As f+ increases (as indicated by the following arrows), photoluminescence (PL) spectra show a shift in vibronic bands (from I to II) due to micelle formation. Adapted with permission from ref [29]. Copyright 2016, Wiley-VCH Verlag GmbH & Co.
Figure 14.
(A) An anionic HP and a cationic-neutral dbp combined to generate complex coacervate core micelles. A single hp binds several dbps at f+ ∼ 0.3, and condensed C3Ms develop when f+ increases near 0.5. (B) The hydrodynamic radii and intensity obtained from light scattering, adjusted for dilution, both rise as f+ increases. (C) As f+ increases (as indicated by the following arrows), photoluminescence (PL) spectra show a shift in vibronic bands (from I to II) due to micelle formation. Adapted with permission from ref [29]. Copyright 2016, Wiley-VCH Verlag GmbH & Co.
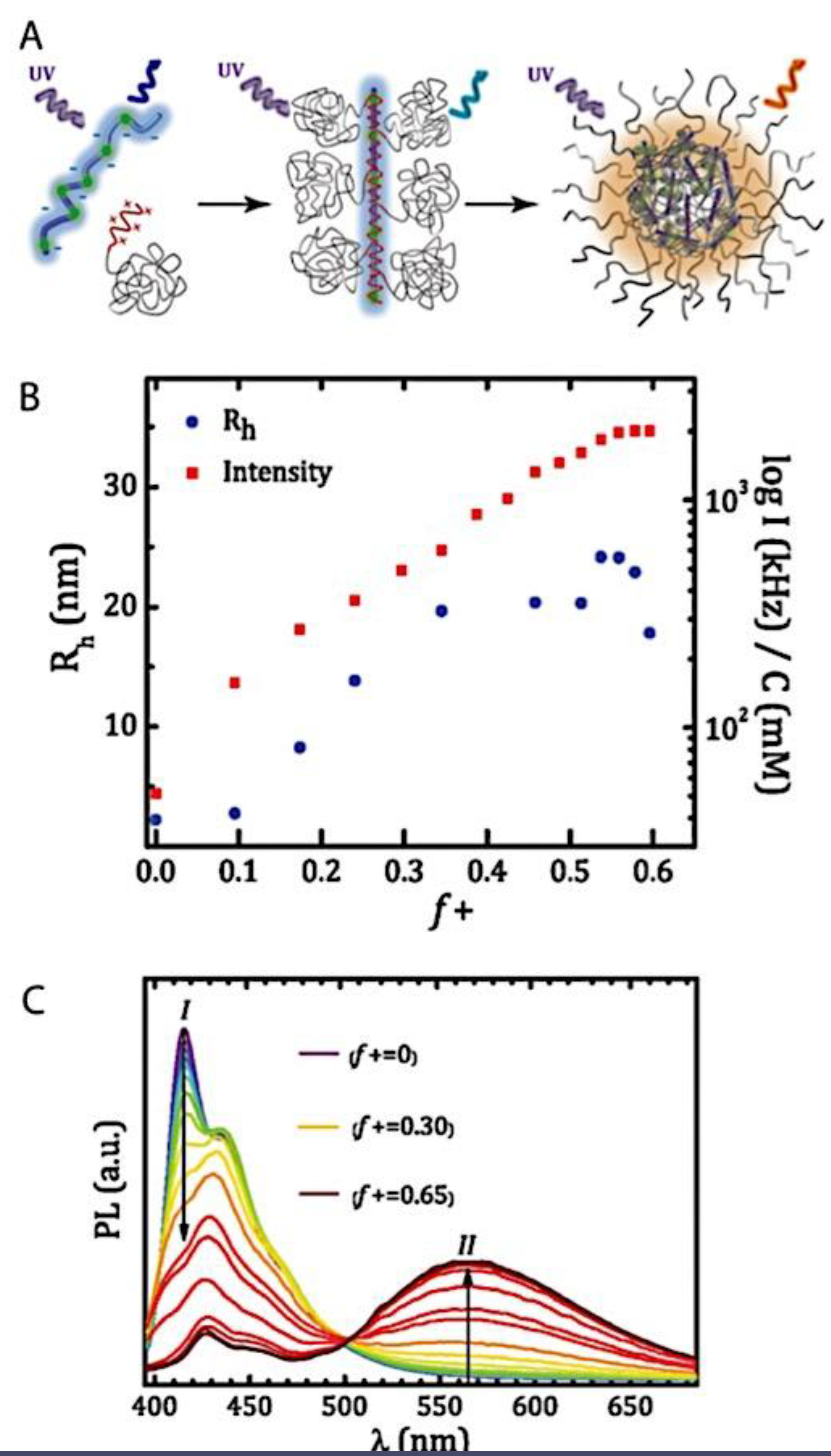
Figure 15.
C3Ms: constituent parts and microphase separation mechanism. In terms of nomenclature, a neutral, hydrophilic block is denoted by A, and oppositely charged polyelectrolyte blocks are represented by B/C. An AB diblock polycation plus either an AC diblock polyanion or a C homopolyanion make up a typical C3M. Adapted with permission from ref [58]. Copyright 2021, The Authors, published by American Chemical Society.
Figure 15.
C3Ms: constituent parts and microphase separation mechanism. In terms of nomenclature, a neutral, hydrophilic block is denoted by A, and oppositely charged polyelectrolyte blocks are represented by B/C. An AB diblock polycation plus either an AC diblock polyanion or a C homopolyanion make up a typical C3M. Adapted with permission from ref [58]. Copyright 2021, The Authors, published by American Chemical Society.
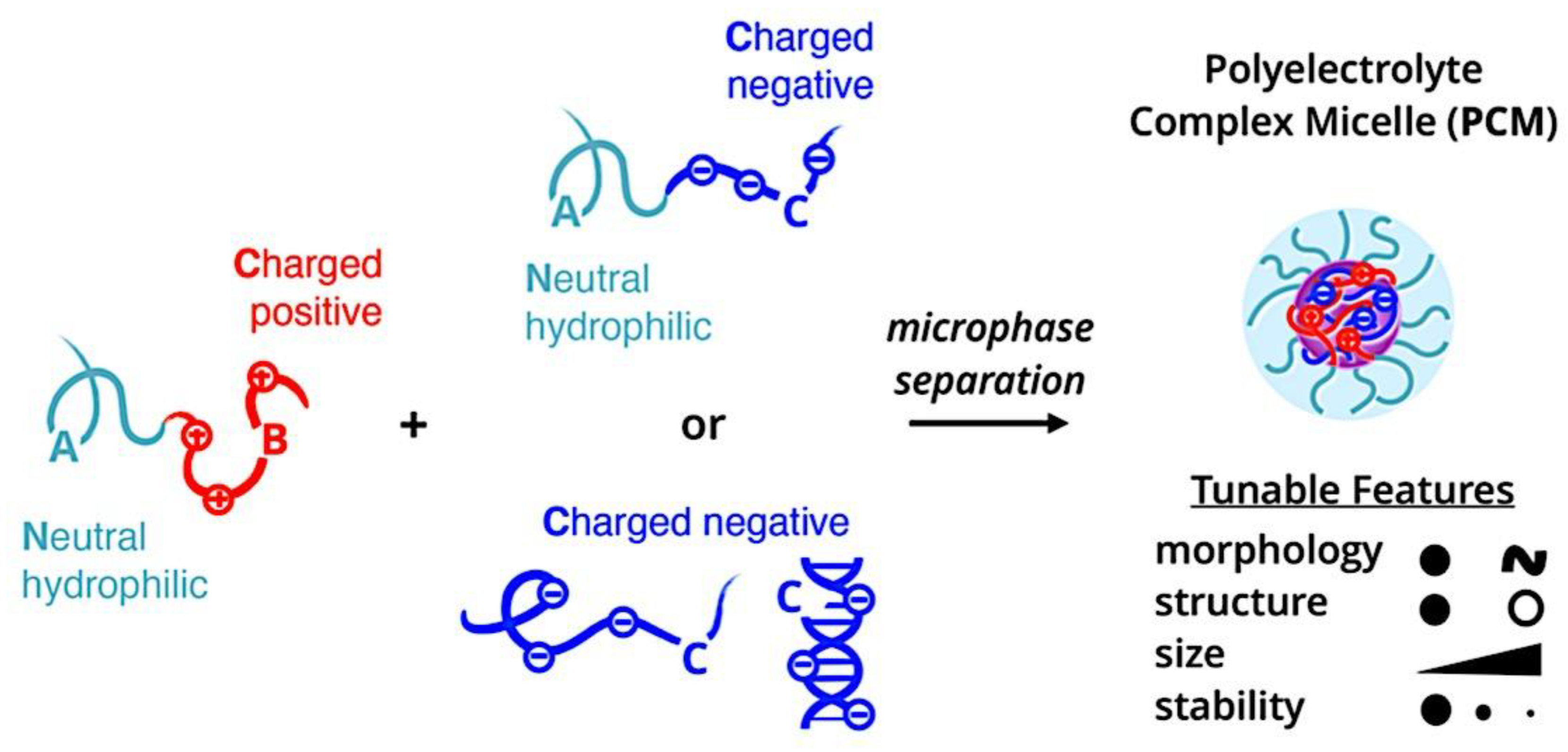
Figure 16.
The repulsion of opsonic proteins (P) is affected by the conformation (B) and chain density (A, large (a) and small (b) proteins). Adapted with permission from ref [466]. Copyright 2006, Elsevier Ltd. .
Figure 16.
The repulsion of opsonic proteins (P) is affected by the conformation (B) and chain density (A, large (a) and small (b) proteins). Adapted with permission from ref [466]. Copyright 2006, Elsevier Ltd. .
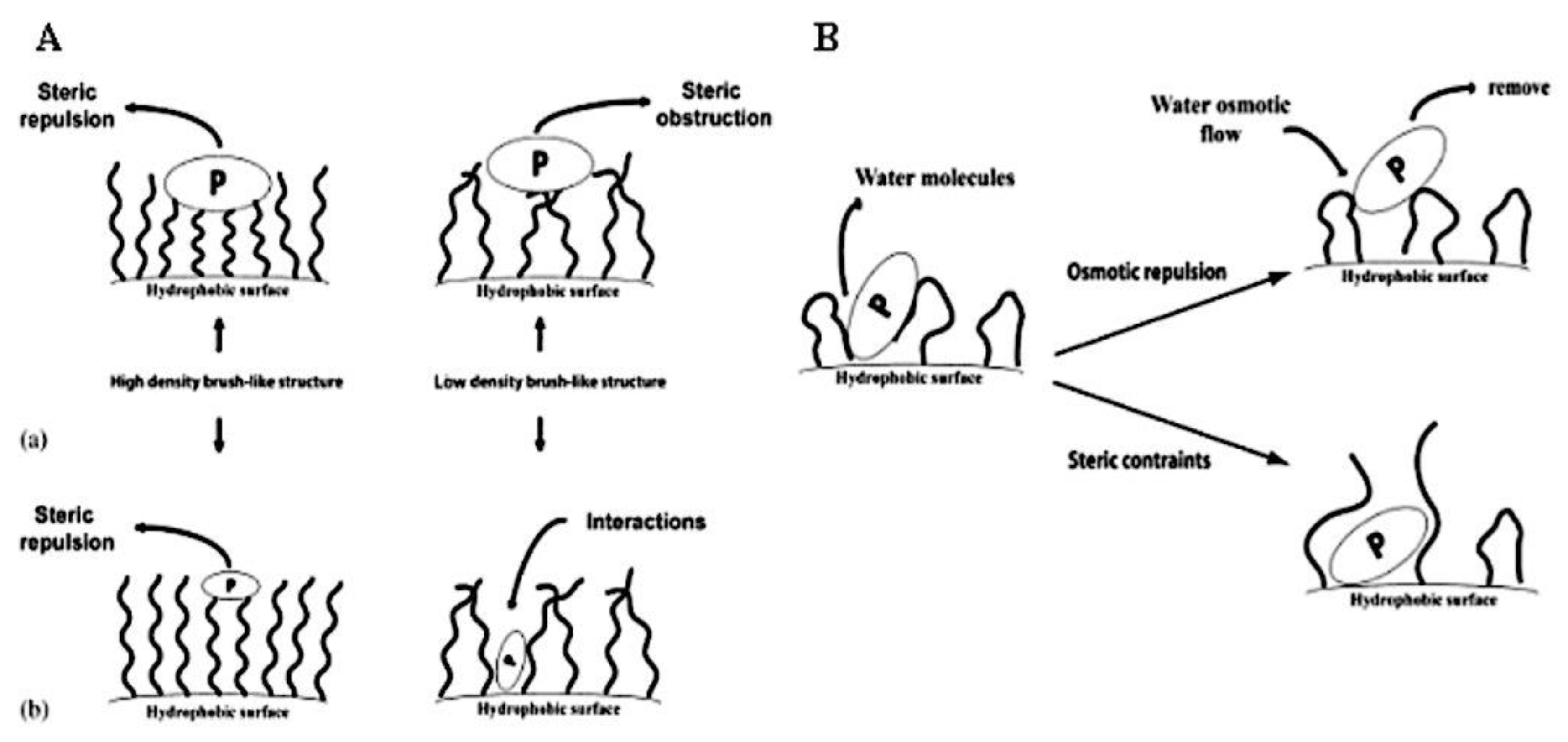
Figure 17.
Interactions that improve the kinetic stability of polymeric micelles within the micellar core. Adapted from ref [577]. Copyright 2007, Ricken.
Figure 17.
Interactions that improve the kinetic stability of polymeric micelles within the micellar core. Adapted from ref [577]. Copyright 2007, Ricken.
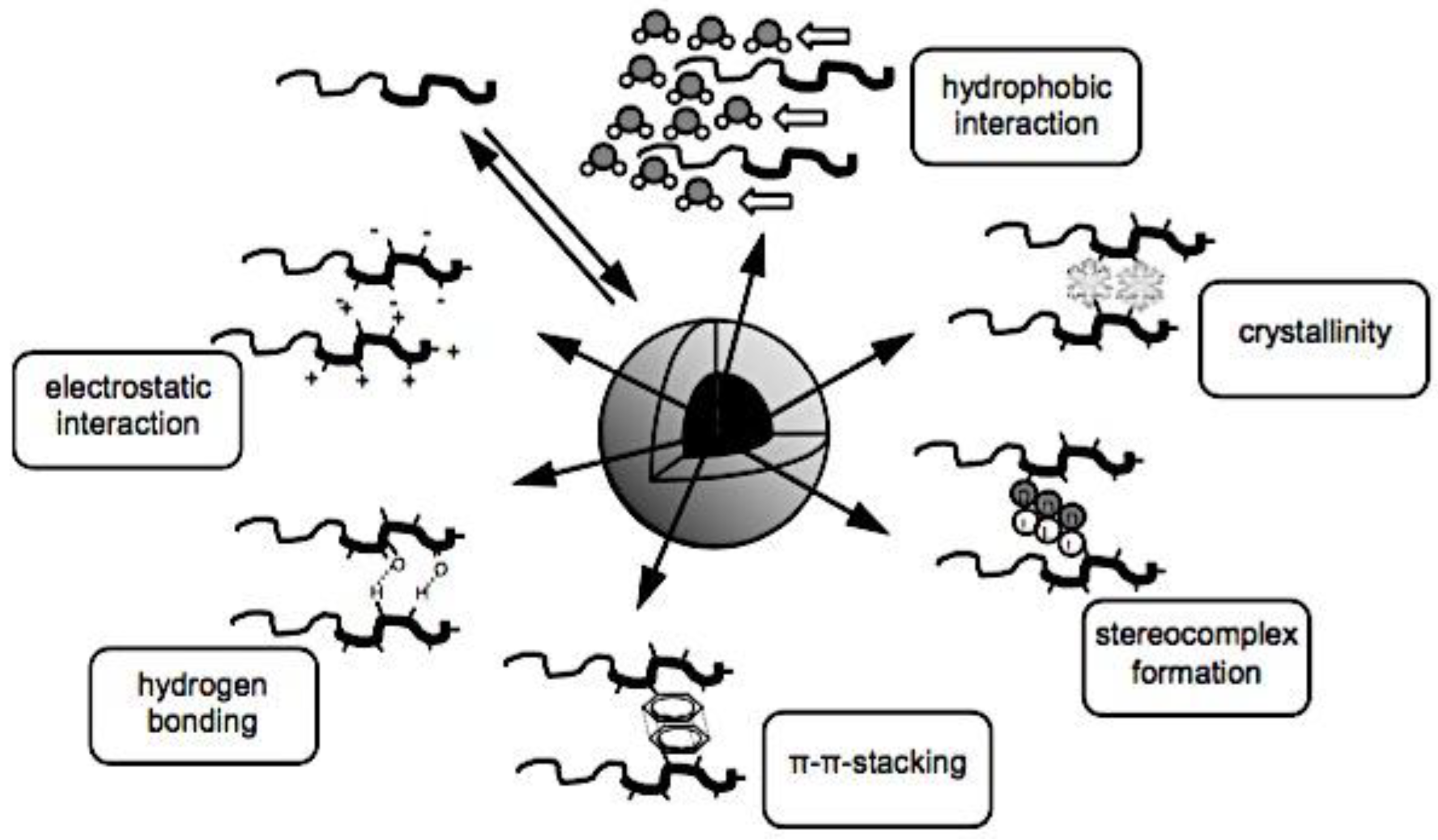
Figure 18.
C3M micelles are formed with a cationic poly(L-lysine) (p(Lys)) and anionic ionomer poly(aspartic acid) (p(Asp) in the core, and a thermosensitive poly(2-isopropyl-2-oxazoline) (PiPrOx) shell on the outside. Adapted with permission from ref [73]. Copyright 2007, American Chemical Society.
Figure 18.
C3M micelles are formed with a cationic poly(L-lysine) (p(Lys)) and anionic ionomer poly(aspartic acid) (p(Asp) in the core, and a thermosensitive poly(2-isopropyl-2-oxazoline) (PiPrOx) shell on the outside. Adapted with permission from ref [73]. Copyright 2007, American Chemical Society.
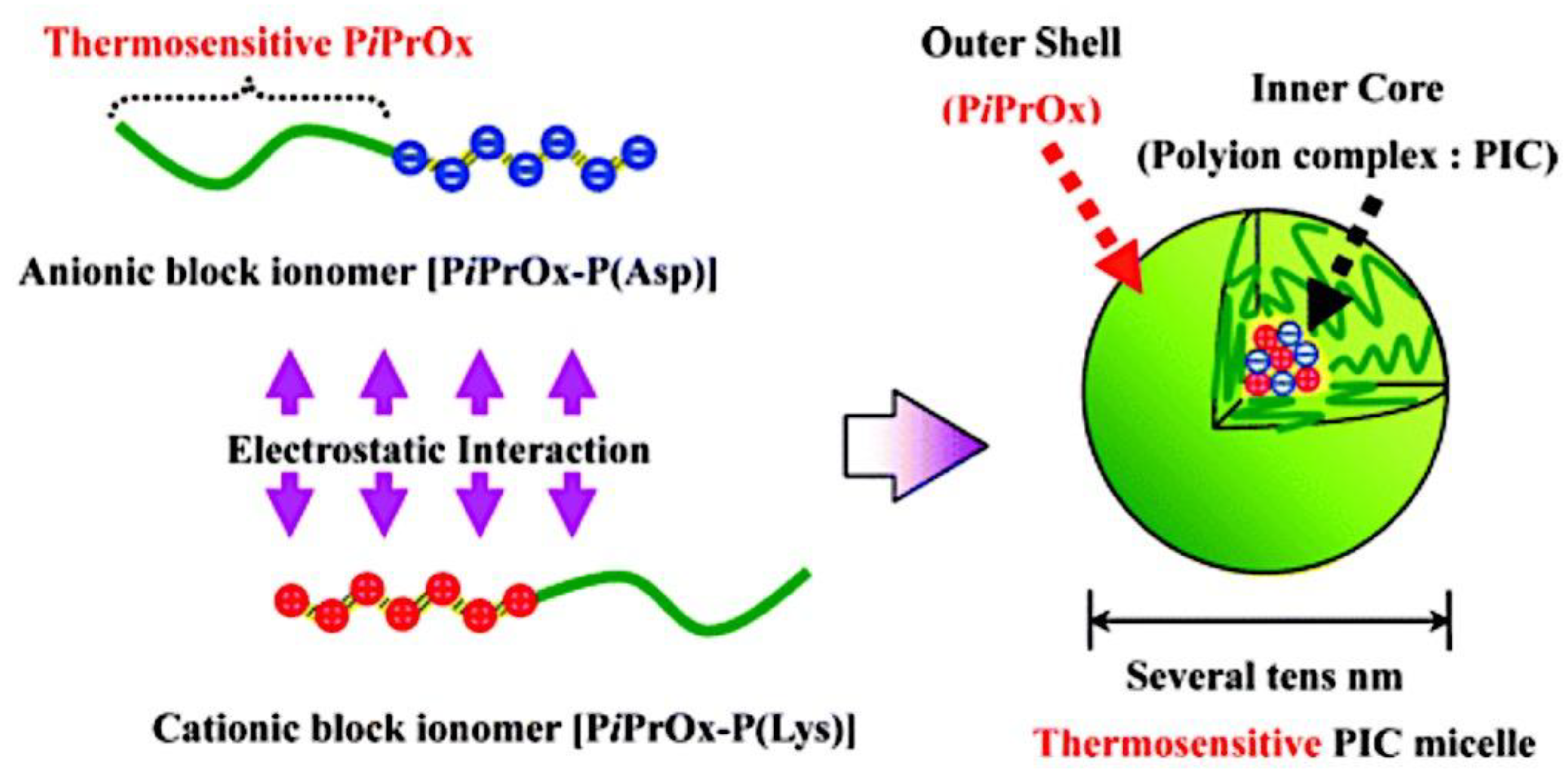
Figure 19.
Illustration of the covalent crosslinking of the micellar core, interfacial layer, and shell. Adapted from ref [577]. Copyright 2007, Ricken.
Figure 19.
Illustration of the covalent crosslinking of the micellar core, interfacial layer, and shell. Adapted from ref [577]. Copyright 2007, Ricken.
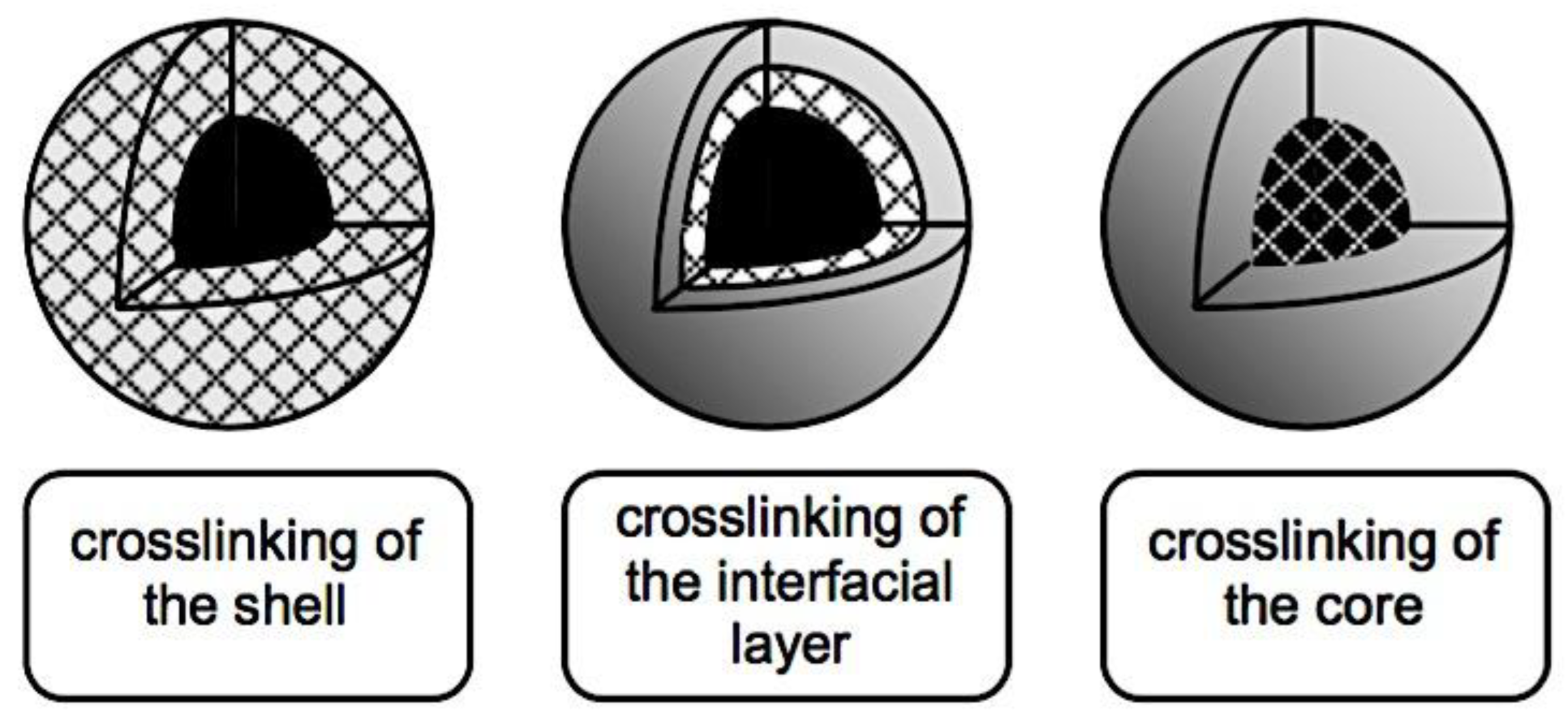
Figure 20.
Diagram showing the C3Ms’ core-crosslinking tactics employing the crosslinkers DTBP and EDC. Whereas DTBP creates crosslinks that can be broken by reducing agents such as 1,4-dithiothreitol (DTT), EDC creates crosslinks that are permanent. Red-blue: diblock copolymer of charges and neutrals. Black: homopolymer with opposing charges. Crosslinks created by DTBP and EDC are shown in yellow and green, respectively. Adapted from ref [612]. Copyright 2022, The Royal Society of Chemistry. Published by The Royal Society of Chemistry under the terms of the Creative Commons Attribution-NonCommercial 3.0 Unported Licence (CC BY-NC 4.0).
Figure 20.
Diagram showing the C3Ms’ core-crosslinking tactics employing the crosslinkers DTBP and EDC. Whereas DTBP creates crosslinks that can be broken by reducing agents such as 1,4-dithiothreitol (DTT), EDC creates crosslinks that are permanent. Red-blue: diblock copolymer of charges and neutrals. Black: homopolymer with opposing charges. Crosslinks created by DTBP and EDC are shown in yellow and green, respectively. Adapted from ref [612]. Copyright 2022, The Royal Society of Chemistry. Published by The Royal Society of Chemistry under the terms of the Creative Commons Attribution-NonCommercial 3.0 Unported Licence (CC BY-NC 4.0).
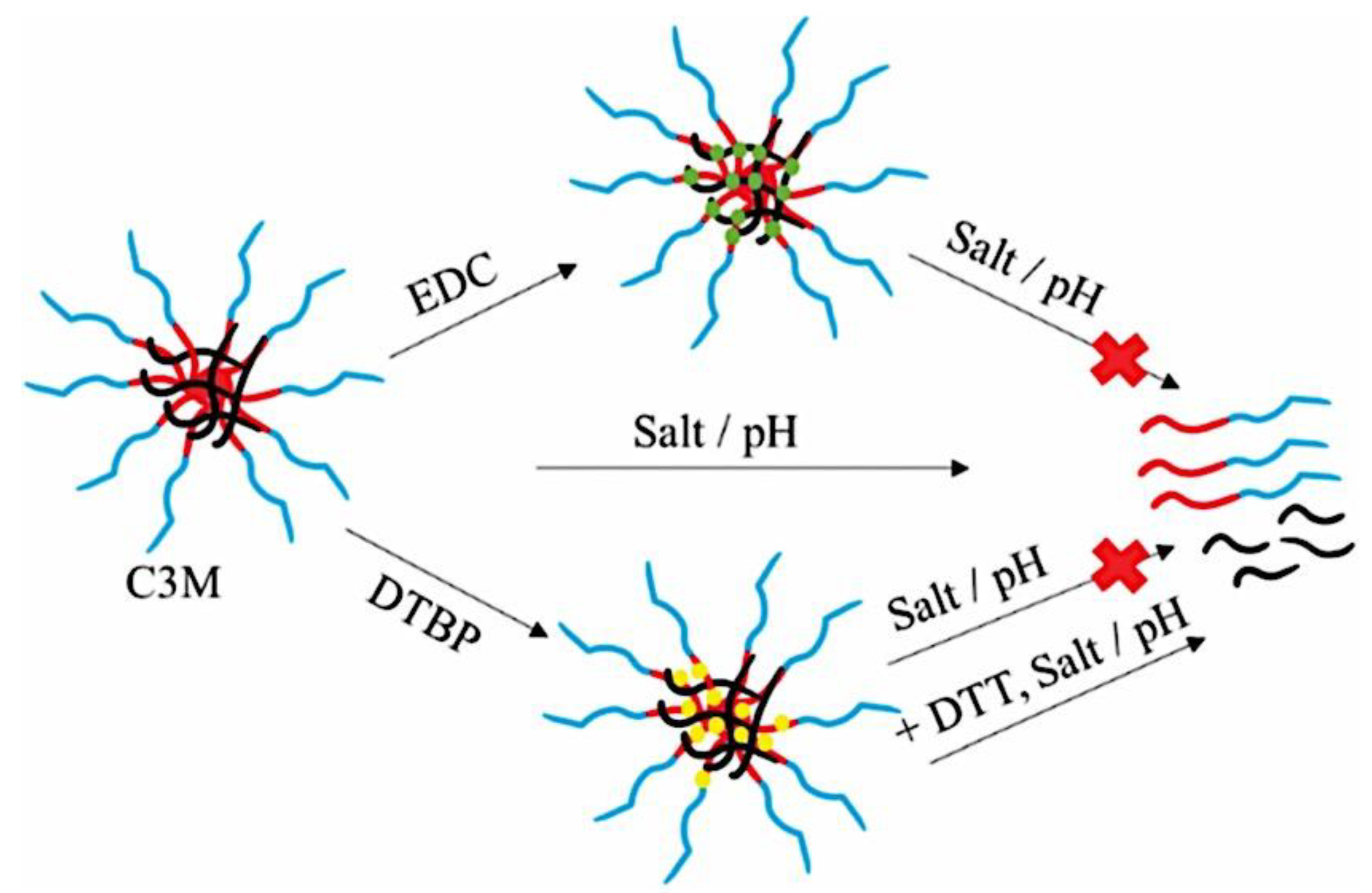
Figure 21.
Doxorubicin (DOX), also known as NK911, is physically and covalently encapsulated in PEG-b-poly(aspartic acid) micelles. Adapted with permission from ref [454]. Copyright 2004, The Author(s) (Cancer Research UK). Published by Springer Nature Limited under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License (Creative Commons CC-BY-NC-SA).
Figure 21.
Doxorubicin (DOX), also known as NK911, is physically and covalently encapsulated in PEG-b-poly(aspartic acid) micelles. Adapted with permission from ref [454]. Copyright 2004, The Author(s) (Cancer Research UK). Published by Springer Nature Limited under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License (Creative Commons CC-BY-NC-SA).
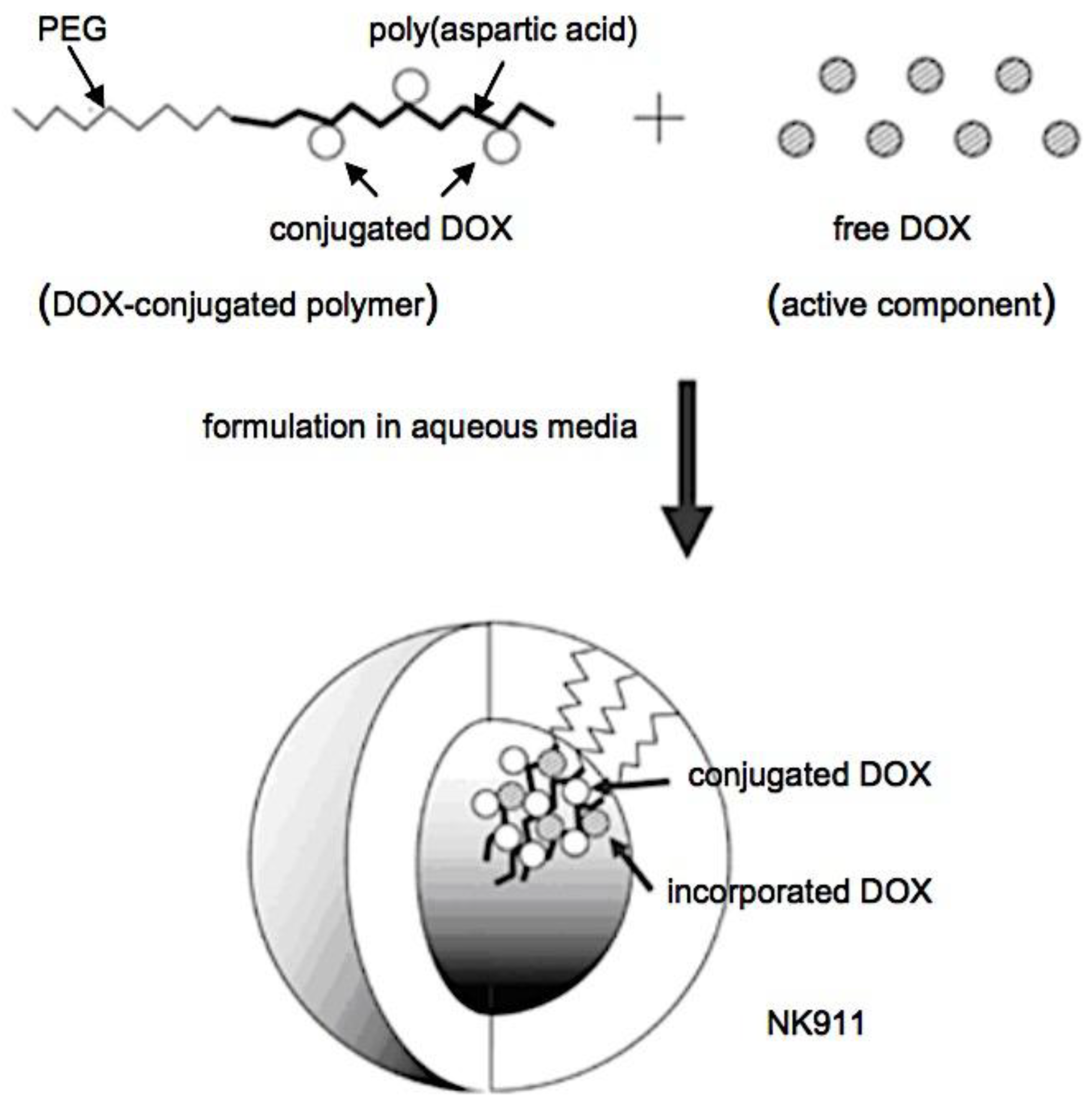
Figure 22.
Polymeric micelle destabilization processes that are sensitive to stimuli. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V. .
Figure 22.
Polymeric micelle destabilization processes that are sensitive to stimuli. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V. .
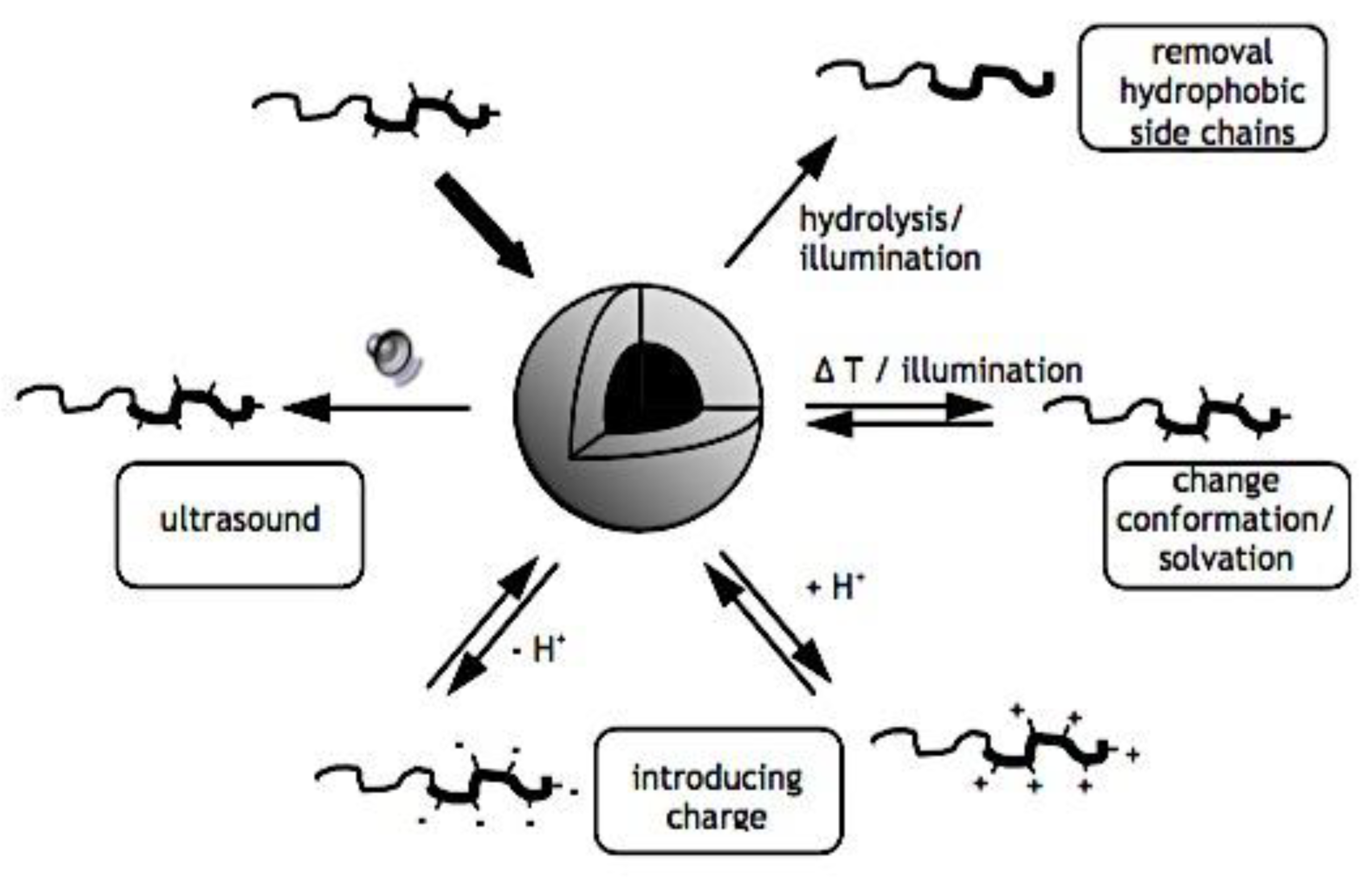
Figure 23.
Drug-loaded block copolymer micelles with a thermosensitive block that can be either the hydrophobic core above the CP (bottom) or the hydrophilic shell below the CP (top). The micellar structures can be distorted by heating or cooling, which will also cause the loaded medication to release simultaneously. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V. .
Figure 23.
Drug-loaded block copolymer micelles with a thermosensitive block that can be either the hydrophobic core above the CP (bottom) or the hydrophilic shell below the CP (top). The micellar structures can be distorted by heating or cooling, which will also cause the loaded medication to release simultaneously. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V. .
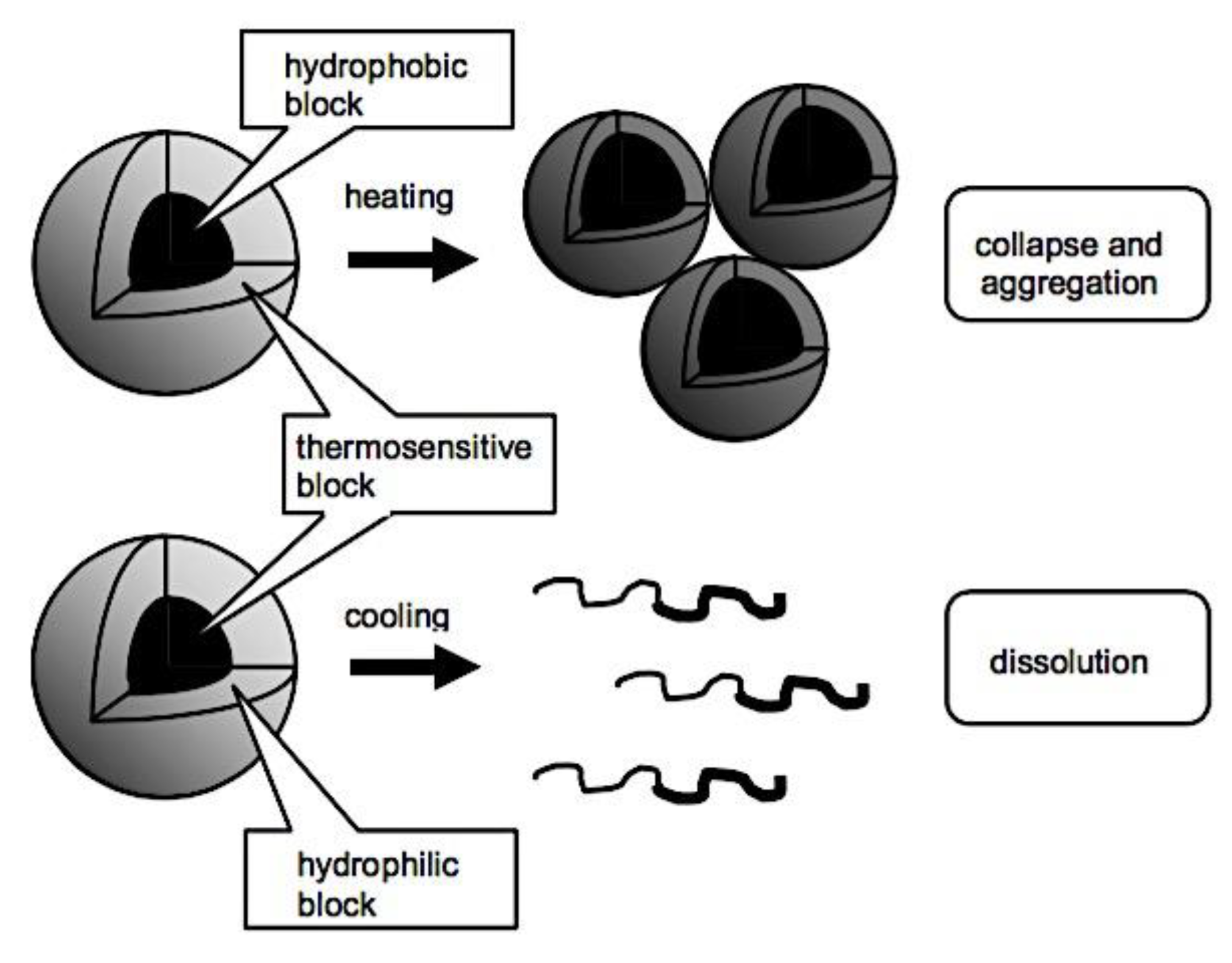
Figure 24.
2-Diazo-1,2-naphthoquinone derivatives’ solubility changes to 3-indenecarboxylate in buffered water following the Wolff rearrangement. Adapted from ref [577]. Copyright 2007, Ricken.
Figure 24.
2-Diazo-1,2-naphthoquinone derivatives’ solubility changes to 3-indenecarboxylate in buffered water following the Wolff rearrangement. Adapted from ref [577]. Copyright 2007, Ricken.
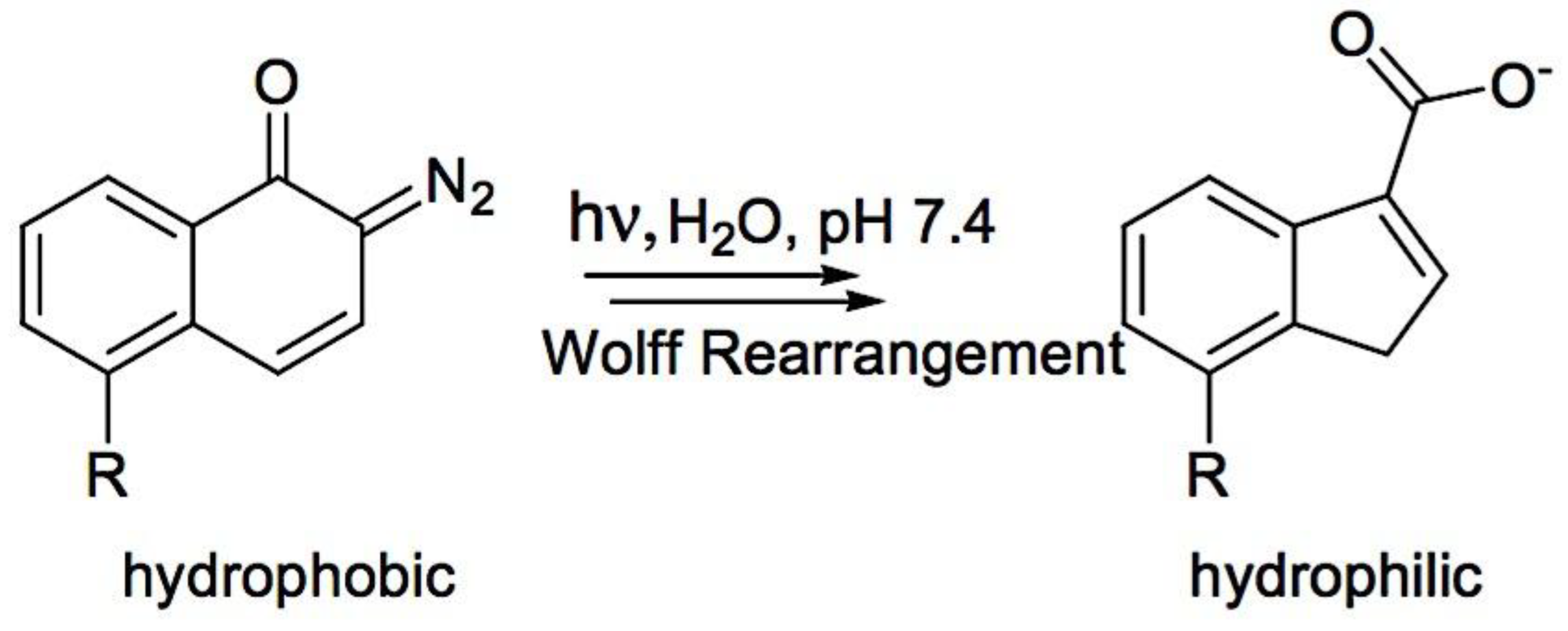
Figure 25.
Reversible photodimerization (bottom) and trans-to-cis isomerization (top) of the cinnamonoyl photoreactive group in response to UV radiation. Adapted from ref [577]. Copyright 2007, Ricken.
Figure 25.
Reversible photodimerization (bottom) and trans-to-cis isomerization (top) of the cinnamonoyl photoreactive group in response to UV radiation. Adapted from ref [577]. Copyright 2007, Ricken.
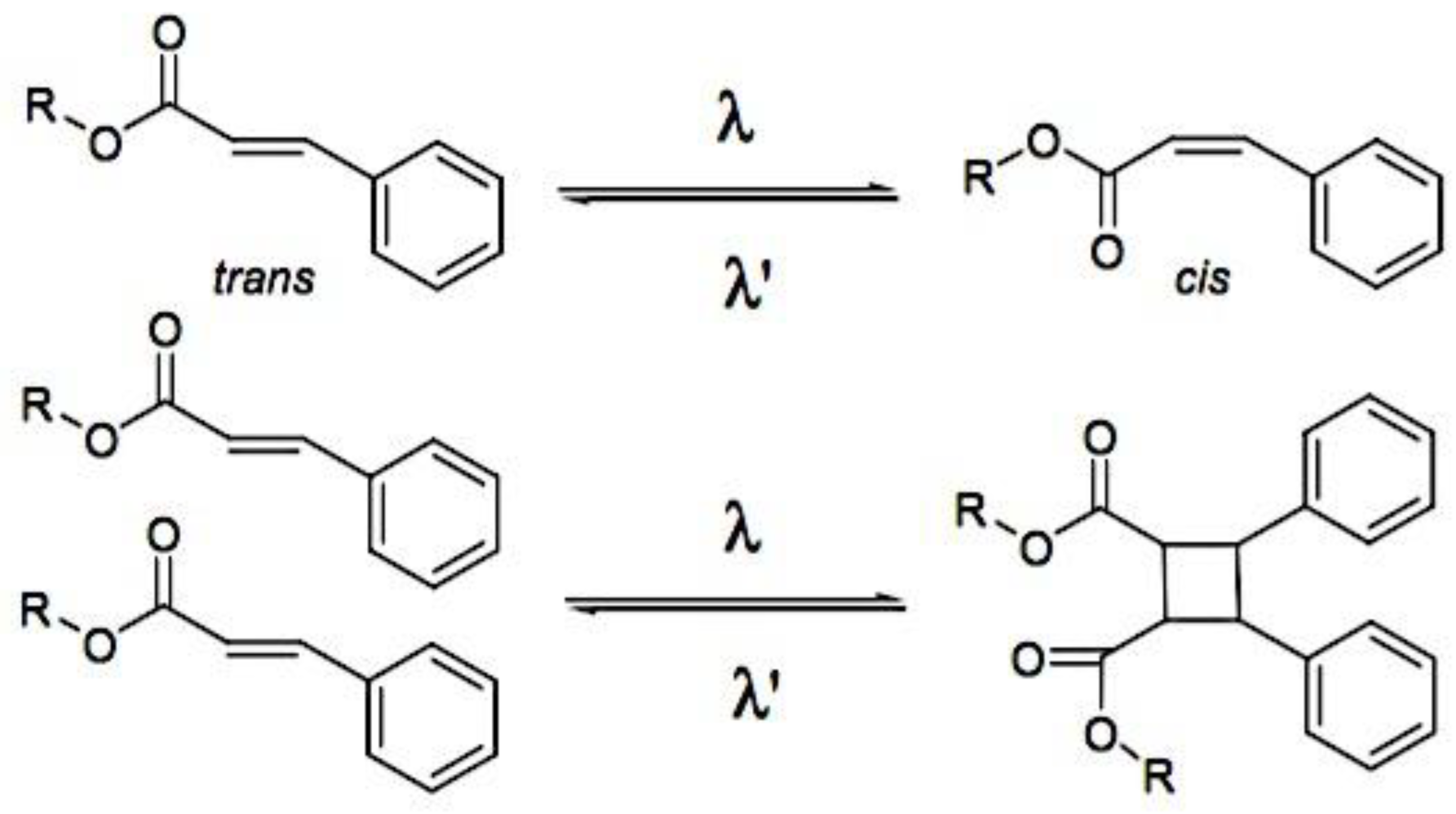
Figure 26.
Stabilized micelles are produced when cisplatin is complexed with carboxylate-containing polymers (PEG-b-p(glutamic acid)) (see above); cisplatin is released gradually by ligand exchange reactions in saline (see below). Adapted with permission from ref [497]. Copyright 2003, American Association for Cancer Research. .
Figure 26.
Stabilized micelles are produced when cisplatin is complexed with carboxylate-containing polymers (PEG-b-p(glutamic acid)) (see above); cisplatin is released gradually by ligand exchange reactions in saline (see below). Adapted with permission from ref [497]. Copyright 2003, American Association for Cancer Research. .
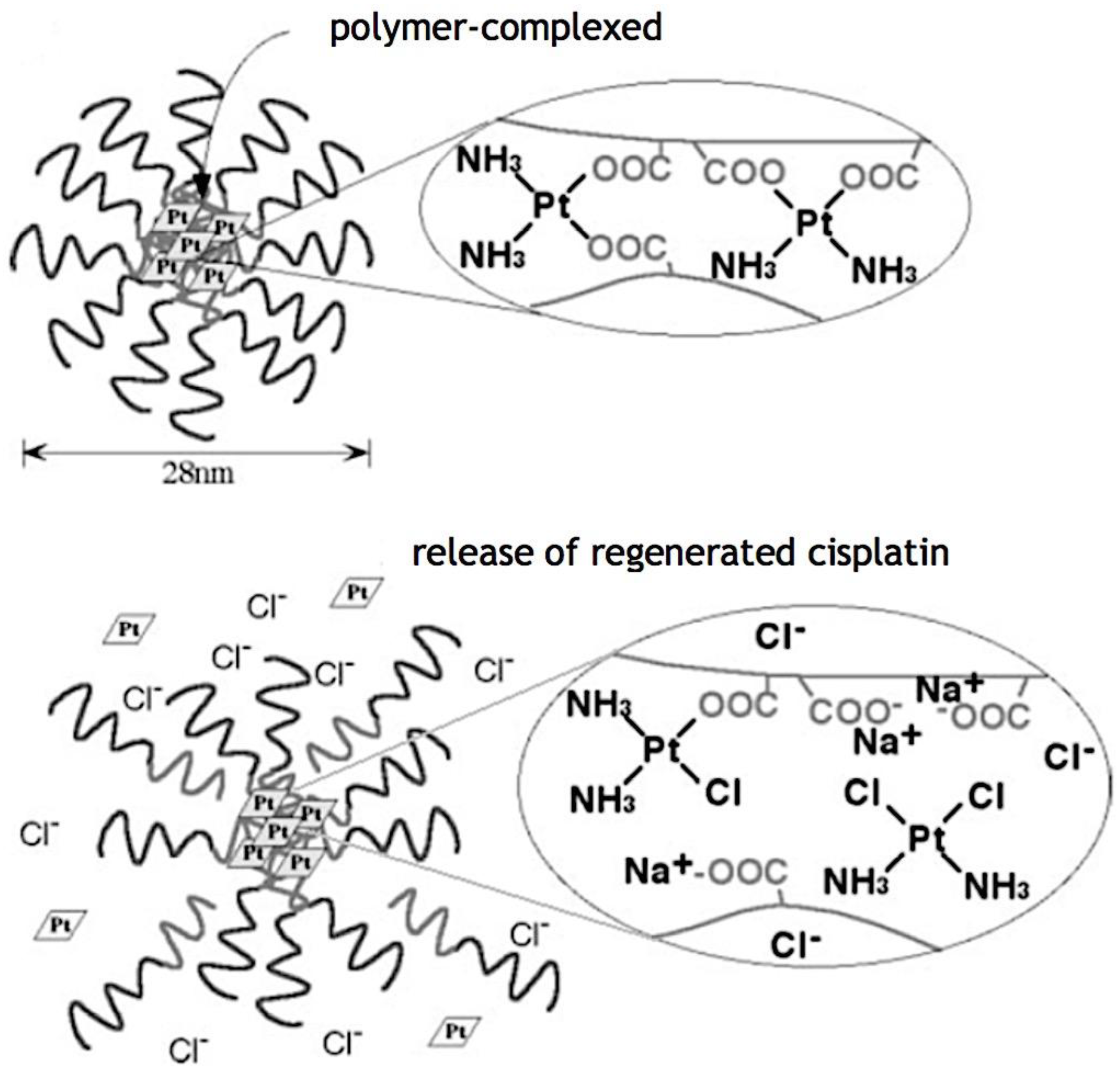
Figure 27.
The hydrolysis of mPEG-b-p(HPMAm-Lac2) (left) results in the elimination of the hydrophobic lactic acid groups (black dots; right), raising the block copolymer’s critical micelle temperature. Therefore, as the CMT rises over the incubation temperature, micelles that were produced above the cloud point of mPEG-b-p(HPMAm-Lac2) destabilize. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V. .
Figure 27.
The hydrolysis of mPEG-b-p(HPMAm-Lac2) (left) results in the elimination of the hydrophobic lactic acid groups (black dots; right), raising the block copolymer’s critical micelle temperature. Therefore, as the CMT rises over the incubation temperature, micelles that were produced above the cloud point of mPEG-b-p(HPMAm-Lac2) destabilize. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V. .
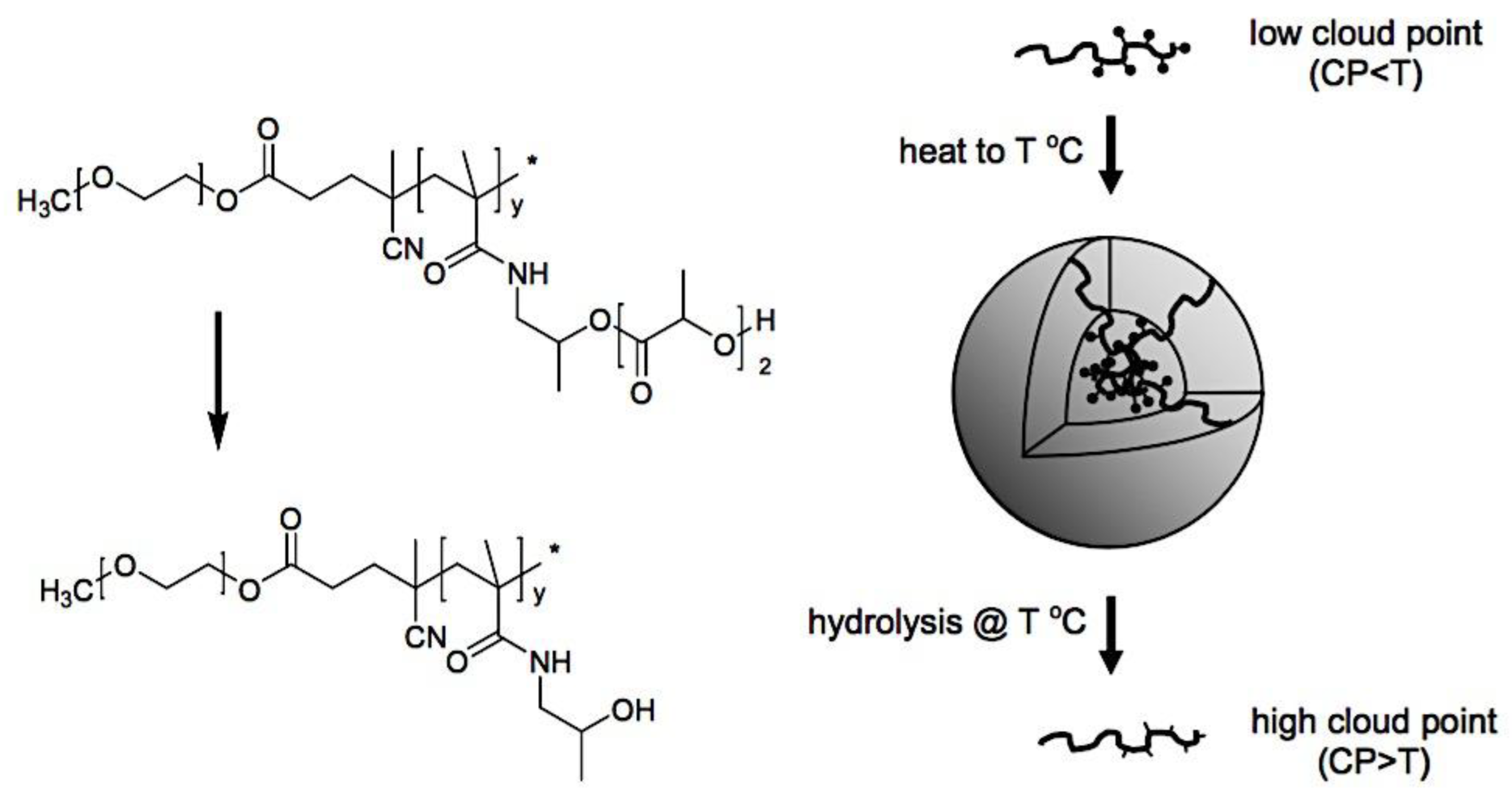
Figure 28.
Multipurpose quantum dots with micelle content. Adapted with permission from ref [708]. Copyright 2006, Elsevier B.V. .
Figure 28.
Multipurpose quantum dots with micelle content. Adapted with permission from ref [708]. Copyright 2006, Elsevier B.V. .
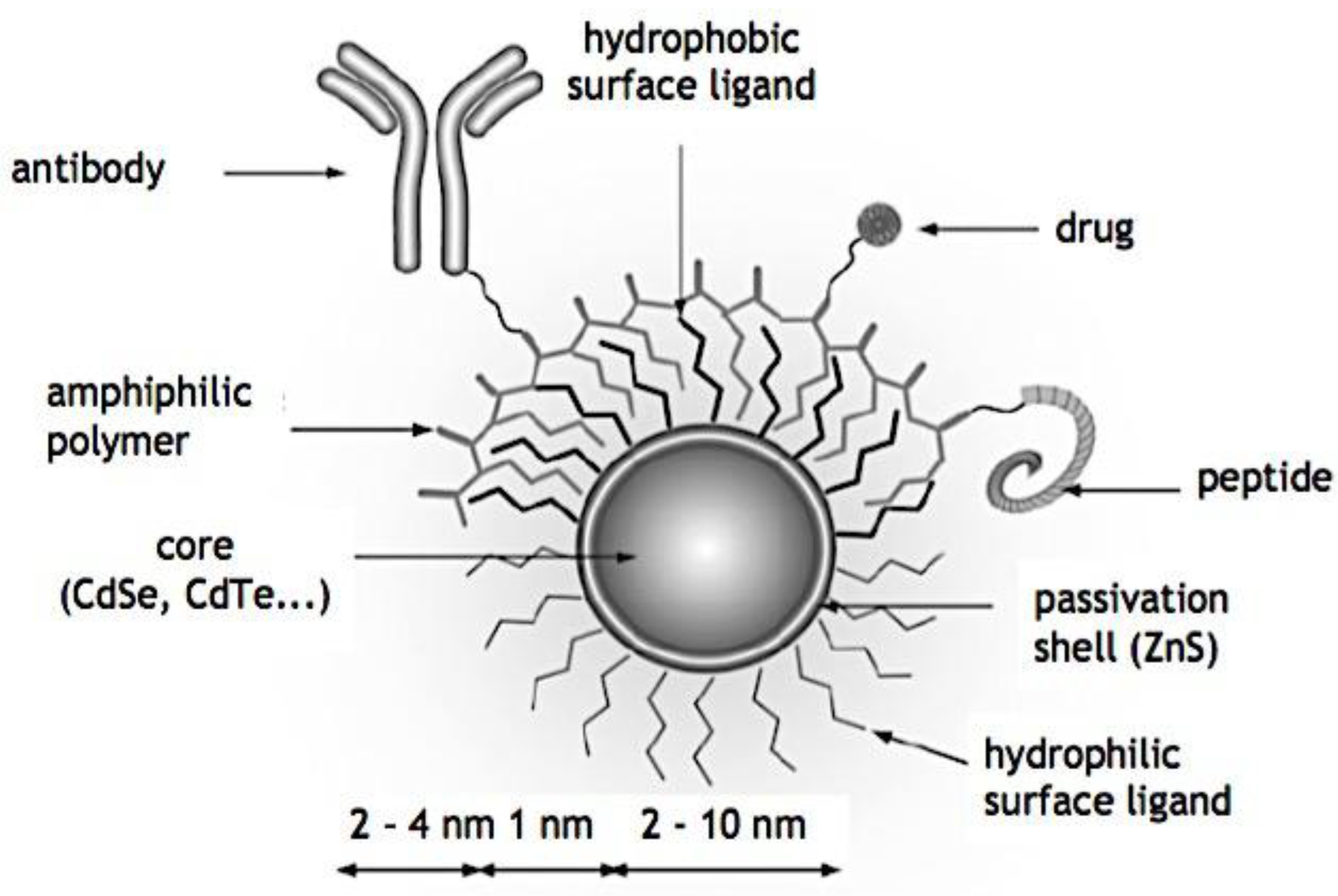
Figure 29.
Targeting distinct packing parameters, p., yielded varying morphologies. Adapted from ref [712]. Copyright 2017, The Royal Society of Chemistry. Published by The Royal Society of Chemistry under a Creative Commons Attribution 3.0 Unported Licence.
Figure 29.
Targeting distinct packing parameters, p., yielded varying morphologies. Adapted from ref [712]. Copyright 2017, The Royal Society of Chemistry. Published by The Royal Society of Chemistry under a Creative Commons Attribution 3.0 Unported Licence.
Figure 30.
Experimental scaling rules are displayed as black lines on top of aggregated data from published (AB + C) C3M experimental research utilizing strong polyelectrolytes at stoichiometric charge ratios. After applying scaling rules for two block lengths to standardize the data, the data were plotted against the third block length, collapsing to display scaling for the relevant block length. Aggregated data for core size (A-C), hydrodynamic size (D-E), and aggregation number (F) are available in the literature. The data uses a variety of synthetic and biological polymers to depict C3Ms from six papers.[30,106,107,146,394,404] The scaling laws are experimental and are in line with theoretical predictions[59,404] for C3Ms between the star-like and crew-cut regimes. Adapted with permission from ref [394]. Copyright 2021, American Chemical Society. .
Figure 30.
Experimental scaling rules are displayed as black lines on top of aggregated data from published (AB + C) C3M experimental research utilizing strong polyelectrolytes at stoichiometric charge ratios. After applying scaling rules for two block lengths to standardize the data, the data were plotted against the third block length, collapsing to display scaling for the relevant block length. Aggregated data for core size (A-C), hydrodynamic size (D-E), and aggregation number (F) are available in the literature. The data uses a variety of synthetic and biological polymers to depict C3Ms from six papers.[30,106,107,146,394,404] The scaling laws are experimental and are in line with theoretical predictions[59,404] for C3Ms between the star-like and crew-cut regimes. Adapted with permission from ref [394]. Copyright 2021, American Chemical Society. .
Figure 31.
Several spherical micelle varieties based on their shape. Micelles that self-assembled from an AB diBCP with an insoluble B block and varying relative block lengths were (A) core-corona symmetric, (B) “Star-like,” and (C) “Crew-cut.” Core-corona (D) and “Flower-like” micelles (E) made of insoluble B blocks and ABA and BAB triBCPs. Core-corona micelles that self-assembled from ABC triBCPs with an insoluble middle B block had a (F) Mixed or (G) Compartmentalized corona (Janus micelles). Core-shell-corona micelles derived from ABC terpolymers with insoluble just A or A and B blocks, respectively, have (H) soluble or (I) insoluble shells. Adapted with permission from ref [740]. Copyright 2021, Wiley Periodicals LLC.
Figure 31.
Several spherical micelle varieties based on their shape. Micelles that self-assembled from an AB diBCP with an insoluble B block and varying relative block lengths were (A) core-corona symmetric, (B) “Star-like,” and (C) “Crew-cut.” Core-corona (D) and “Flower-like” micelles (E) made of insoluble B blocks and ABA and BAB triBCPs. Core-corona micelles that self-assembled from ABC triBCPs with an insoluble middle B block had a (F) Mixed or (G) Compartmentalized corona (Janus micelles). Core-shell-corona micelles derived from ABC terpolymers with insoluble just A or A and B blocks, respectively, have (H) soluble or (I) insoluble shells. Adapted with permission from ref [740]. Copyright 2021, Wiley Periodicals LLC.
Figure 32.
Techniques to create monodisperse cylindrical micelles in solution with crystalline cores using “living” CDSA and “seeded growth” (bottom route) or “self-seeding” (top route). Adapted with permission from ref [734]. Copyright 2015, American Chemical Society.
Figure 32.
Techniques to create monodisperse cylindrical micelles in solution with crystalline cores using “living” CDSA and “seeded growth” (bottom route) or “self-seeding” (top route). Adapted with permission from ref [734]. Copyright 2015, American Chemical Society.
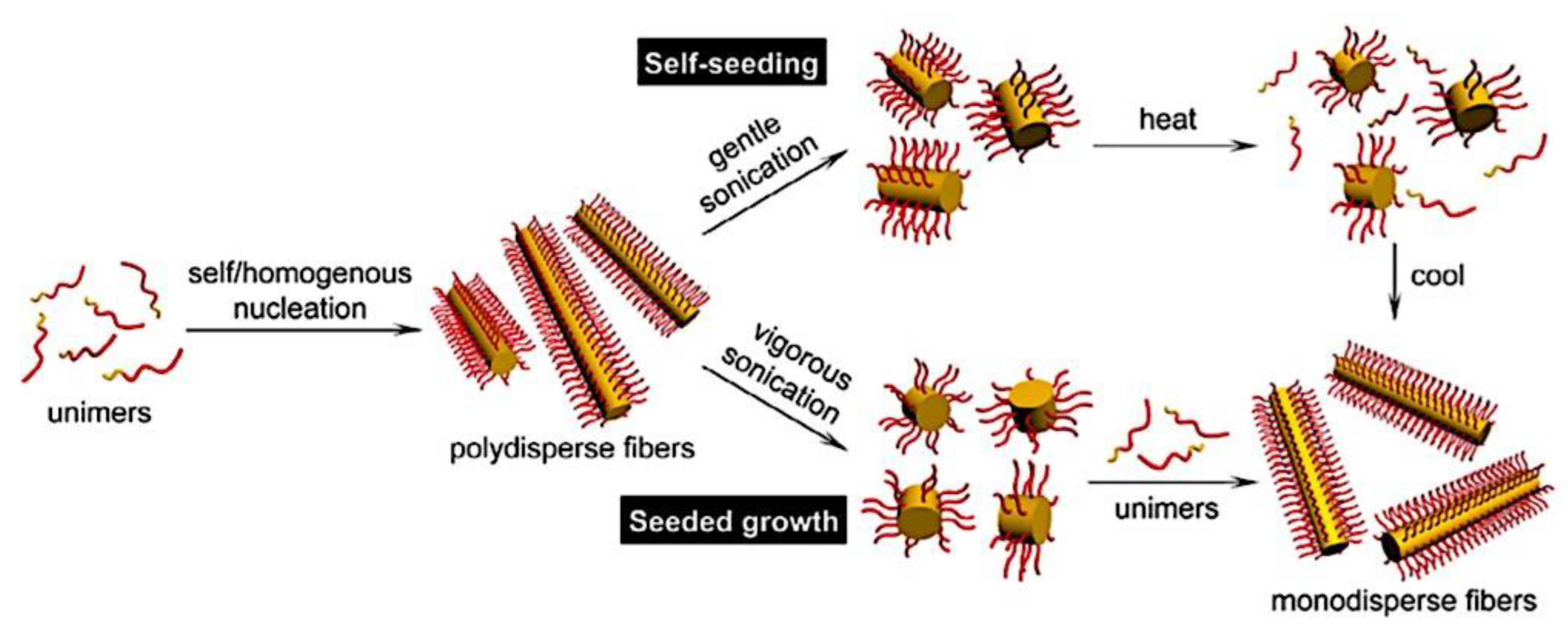
Figure 33.
PEO-PS-P2VP triblock terpolymer and the proposed route for self-assembly leading to patchy micelles and subsequent assembly into 1D supracolloidal chains. Adapted with permission from ref [773]. Copyright 2018, The Royal Society of Chemistry.
Figure 33.
PEO-PS-P2VP triblock terpolymer and the proposed route for self-assembly leading to patchy micelles and subsequent assembly into 1D supracolloidal chains. Adapted with permission from ref [773]. Copyright 2018, The Royal Society of Chemistry.
Figure 34.
Multicompartment micelles, vesicles, and strings are formed when PEG(PAA-PS)20 star polymer self-assembles. The PS, PAA, and PEG chains are shown as orange, blue, and green, respectively. Adapted with permission from Ref [779]. Copyright 2021, American Chemical Society.
Figure 34.
Multicompartment micelles, vesicles, and strings are formed when PEG(PAA-PS)20 star polymer self-assembles. The PS, PAA, and PEG chains are shown as orange, blue, and green, respectively. Adapted with permission from Ref [779]. Copyright 2021, American Chemical Society.
Figure 35.
Diagram showing a cylindrical “Janus” micelle (A), “Block” co-micelle (B), and “Patchy” micelle (C). Adapted with permission from Ref [766]. Copyright 2021, The Authors. Published by MDPI, Basel, Switzerland, under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/). .
Figure 35.
Diagram showing a cylindrical “Janus” micelle (A), “Block” co-micelle (B), and “Patchy” micelle (C). Adapted with permission from Ref [766]. Copyright 2021, The Authors. Published by MDPI, Basel, Switzerland, under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/). .
Figure 36.
A summary of the PISA procedure showing the values of each morphology’s packing parameter, p. Adapted with permission from ref [809]. Copyright 2016, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figure 36.
A summary of the PISA procedure showing the values of each morphology’s packing parameter, p. Adapted with permission from ref [809]. Copyright 2016, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
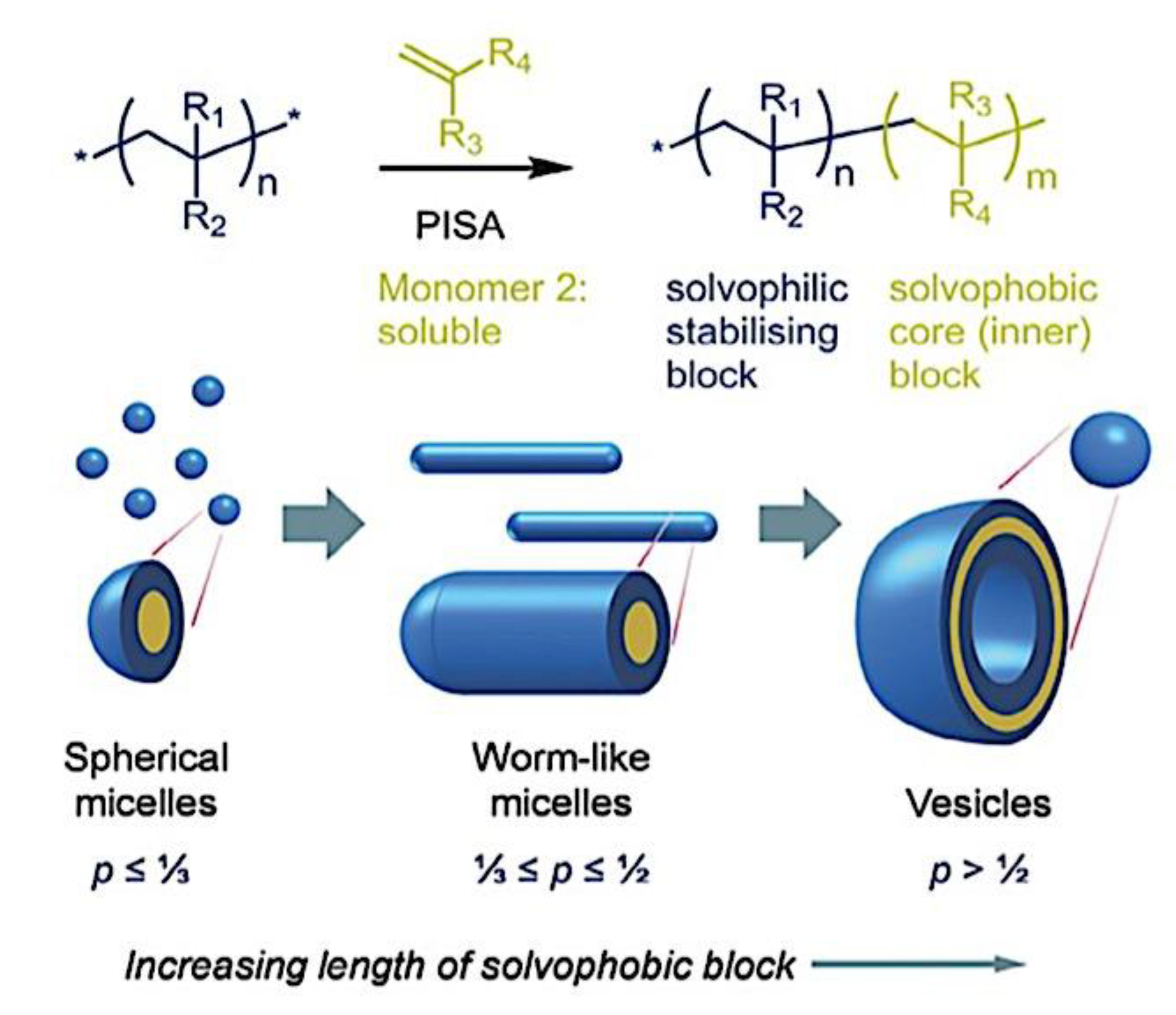
Figure 37.
The chemical structures of different kinds of steric stabilizer blocks used in (A) RAFT-mediated aqueous dispersion polymerization and (B) RAFT-mediated aqueous emulsion polymerization. Adapted from ref [811]. Copyright 2016, American Chemical Society. Published by American Chemical Society under ACS AuthorChoice/Editors’ Choice via Creative Commons CC-BY agreement (CC-BY).
Figure 37.
The chemical structures of different kinds of steric stabilizer blocks used in (A) RAFT-mediated aqueous dispersion polymerization and (B) RAFT-mediated aqueous emulsion polymerization. Adapted from ref [811]. Copyright 2016, American Chemical Society. Published by American Chemical Society under ACS AuthorChoice/Editors’ Choice via Creative Commons CC-BY agreement (CC-BY).
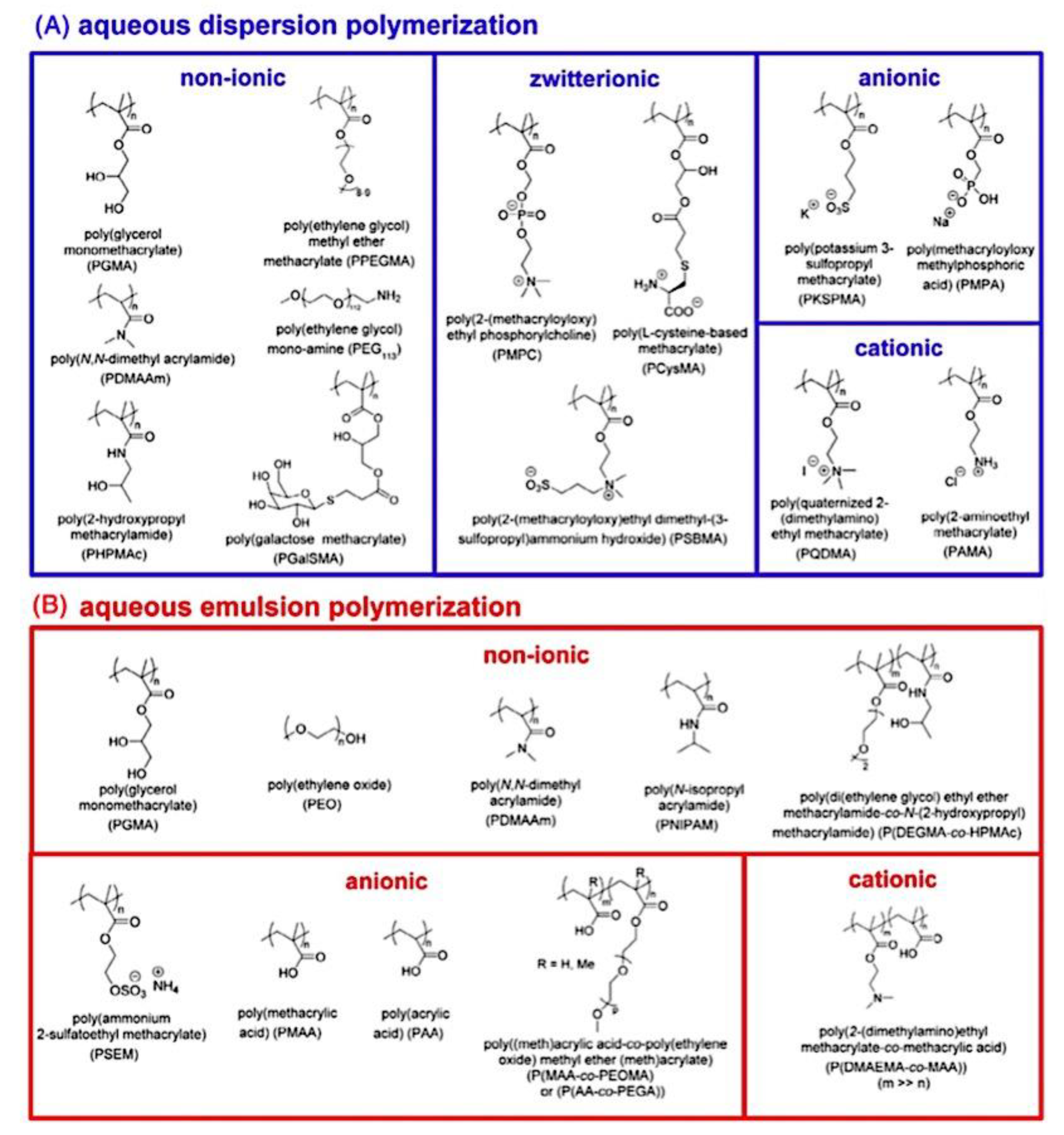
Figure 38.
Synthesis of polyion complex (PIC) micelles by the use of high- or low-MW active pharmaceutical ingredients (APIs) and degradable double hydrophilic block copolymers (DHBCs), followed by their stimuli-responsive release and disintegration. Adapted with permission from Ref [824]. Copyright 2020, Elsevier B.V.
Figure 38.
Synthesis of polyion complex (PIC) micelles by the use of high- or low-MW active pharmaceutical ingredients (APIs) and degradable double hydrophilic block copolymers (DHBCs), followed by their stimuli-responsive release and disintegration. Adapted with permission from Ref [824]. Copyright 2020, Elsevier B.V.
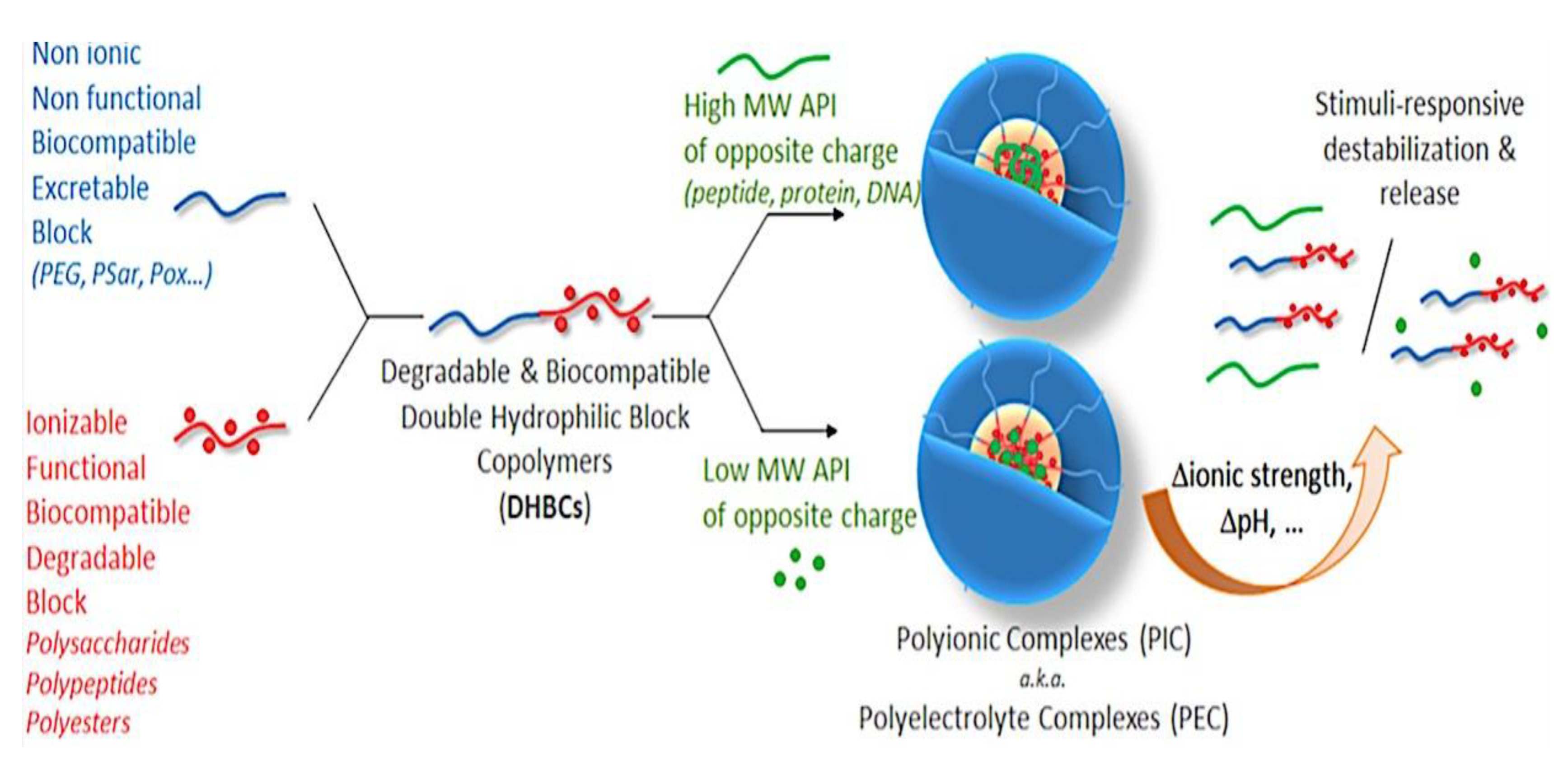
Figure 39.
Diagrammatic representation of two distinct paths for the PS-PE-PMMA triBCP’s oil-in-water emulsion confinement self-assembly at various temperatures, along with matching transmission electron microscopy (TEM) pictures. Adapted with permission from Ref [827]. Copyright 2020, American Chemical Society.
Figure 39.
Diagrammatic representation of two distinct paths for the PS-PE-PMMA triBCP’s oil-in-water emulsion confinement self-assembly at various temperatures, along with matching transmission electron microscopy (TEM) pictures. Adapted with permission from Ref [827]. Copyright 2020, American Chemical Society.
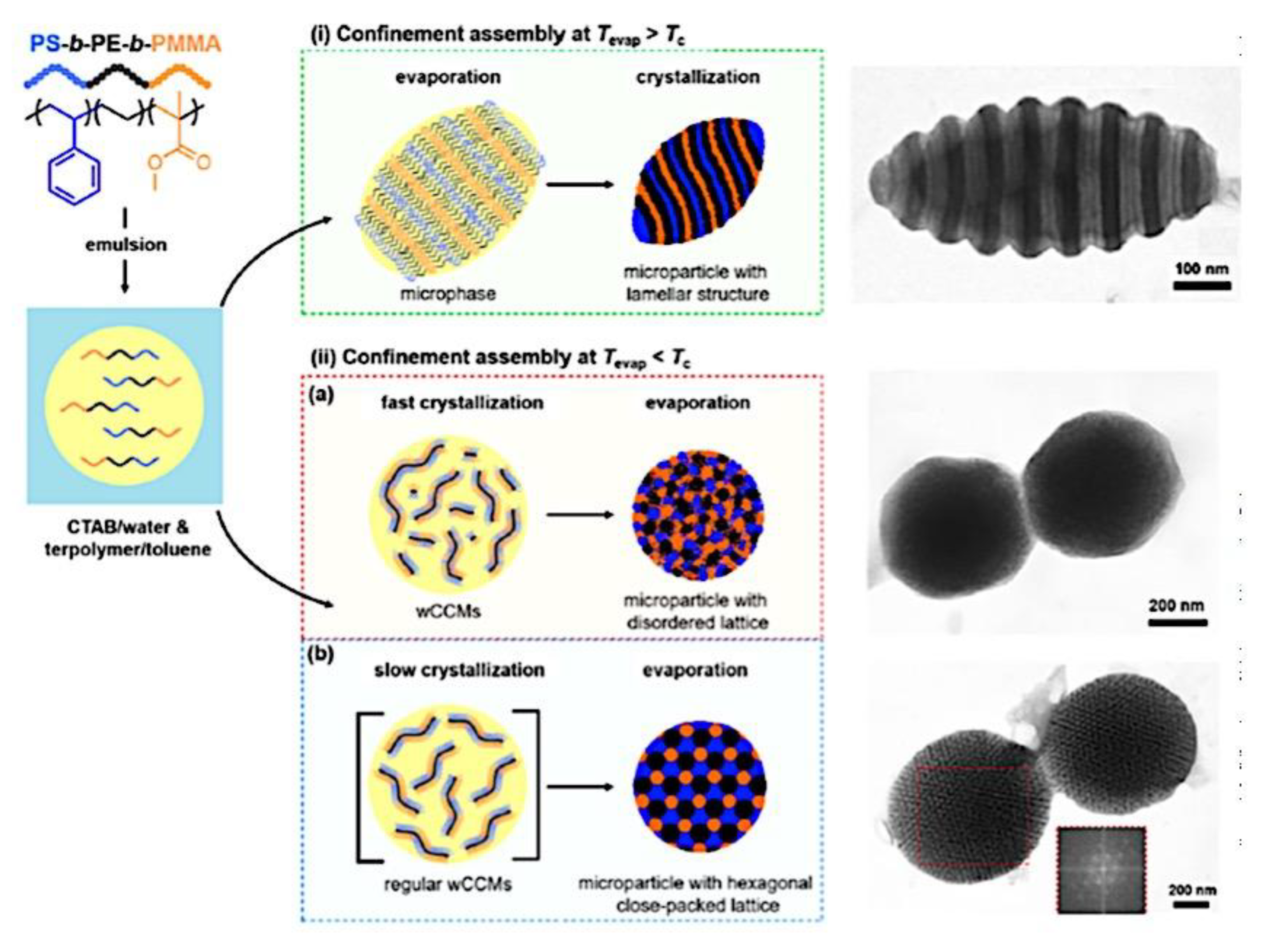
Figure 40.
Poly(2-dimethylamino)ethyl methacrylate)-b-poly(methyl methacrylate) diblock copolymer can be quaternized using alkyl halides according to a general synthesis approach. (40.1) PS-b-PI diblock copolymer reaction strategy for the sulfonation of the polyisopene block. (40.2) Up: radical insertion of ω-functional mercaptans into polybutadiene double bonds. Down: certain ω-functional mercaptans’ chemical structures are used. (40.3) Synthesis of a dipeptide-grafted PB-b-PEO diblock. (40.4) The functionalization reaction between chlorosulfonylisocyanate and poly(isoprene-b-ethylene oxide) diblock copolymers is described. (40.5) PtBOS-b-PMAA precursors are used to create PHOS-b-PMAA double hydrophilic block copolymers. (40.6) QNPHOS-b-PEO diblock copolymer synthesised from PtBOS-b-PEO precursor. (40.7) Adapted from Ref [872]. Copyright 2010, Elsevier Ltd. Published open access under CC BY-NC-ND license.
Figure 40.
Poly(2-dimethylamino)ethyl methacrylate)-b-poly(methyl methacrylate) diblock copolymer can be quaternized using alkyl halides according to a general synthesis approach. (40.1) PS-b-PI diblock copolymer reaction strategy for the sulfonation of the polyisopene block. (40.2) Up: radical insertion of ω-functional mercaptans into polybutadiene double bonds. Down: certain ω-functional mercaptans’ chemical structures are used. (40.3) Synthesis of a dipeptide-grafted PB-b-PEO diblock. (40.4) The functionalization reaction between chlorosulfonylisocyanate and poly(isoprene-b-ethylene oxide) diblock copolymers is described. (40.5) PtBOS-b-PMAA precursors are used to create PHOS-b-PMAA double hydrophilic block copolymers. (40.6) QNPHOS-b-PEO diblock copolymer synthesised from PtBOS-b-PEO precursor. (40.7) Adapted from Ref [872]. Copyright 2010, Elsevier Ltd. Published open access under CC BY-NC-ND license.
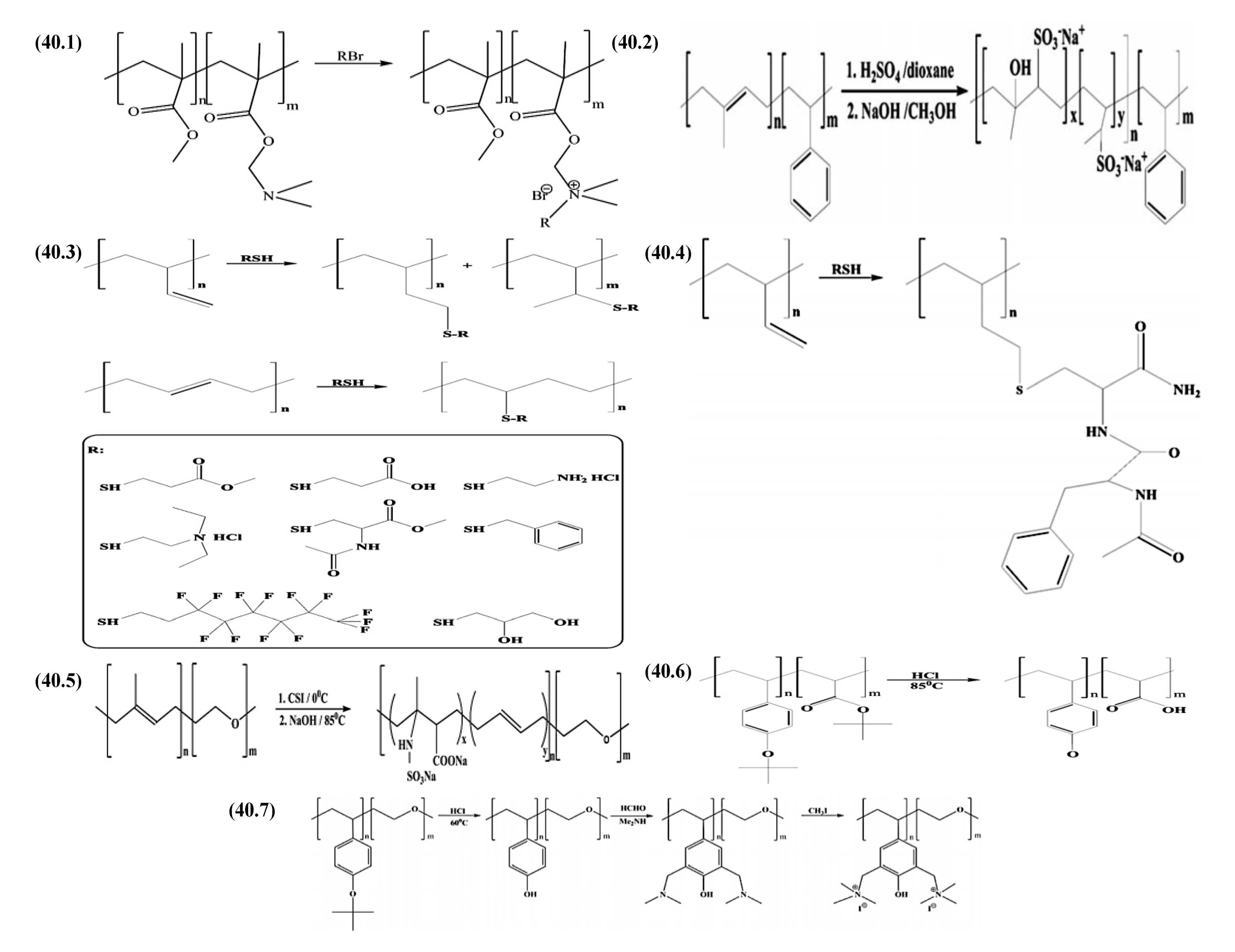
Figure 42.
Synthesis of an amphiphilic triblock terpolymer, poly((sulfamate-carboxylate)isoprene)-b-polystyrene-b-poly(ethyleneoxide)). Adapted from Ref [872]. Copyright 2010, Elsevier Ltd. Published open access under CC BY-NC-ND license.
Figure 42.
Synthesis of an amphiphilic triblock terpolymer, poly((sulfamate-carboxylate)isoprene)-b-polystyrene-b-poly(ethyleneoxide)). Adapted from Ref [872]. Copyright 2010, Elsevier Ltd. Published open access under CC BY-NC-ND license.
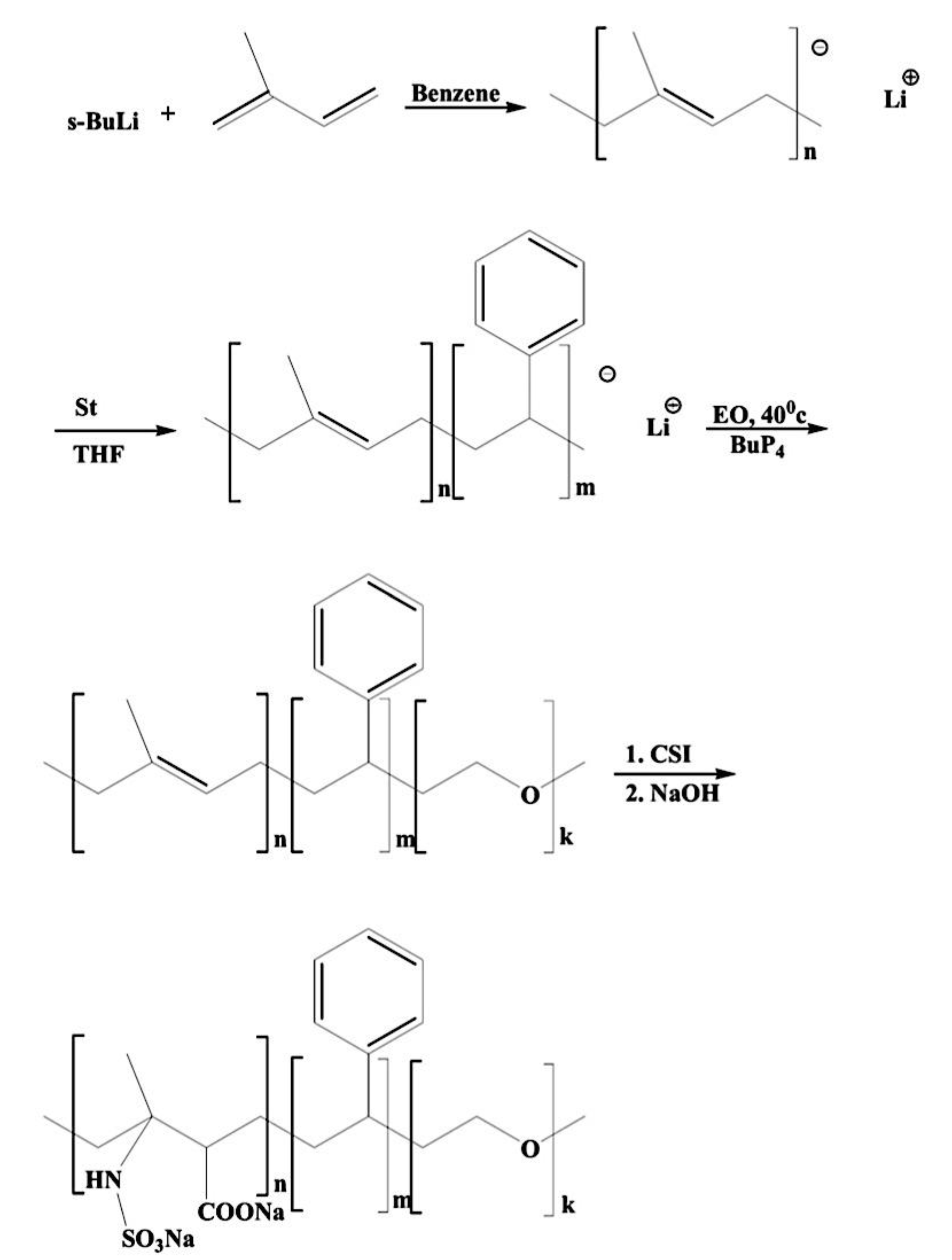
Figure 43.
Diagram showing the sequential RAFT and GPC traces used to synthesize PDMAEMA-b-PLMA-b-POEGMA. (Red: PDMAEMA, Blue: PDMAEMA-b-PLMA-b-POEGMA, Red: PDMAEMA-b-PLMA). GPC trace taken from ref [941]. Adapted with permission from ref [941]. Copyright 2017, The Royal Society of Chemistry.
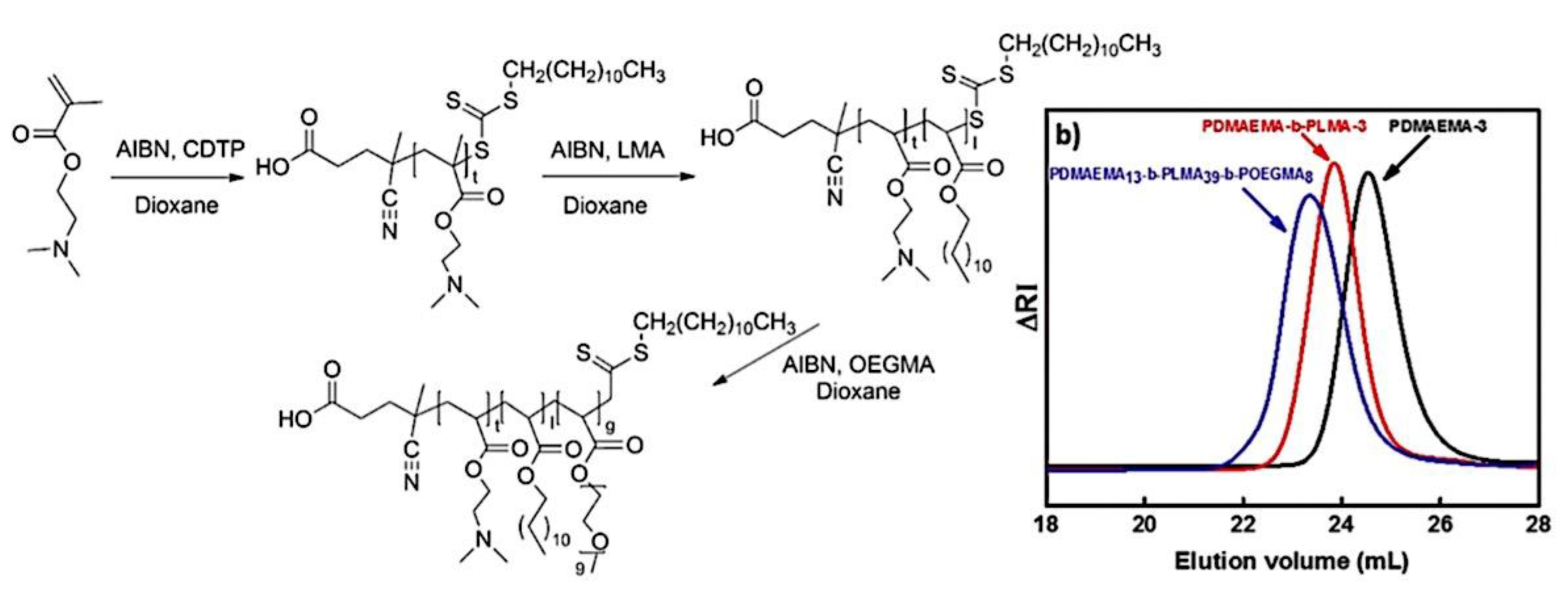
Figure 44.
Diagram illustrating the PEO-b-PHEMA-b-PtBA synthesis. Adapted with permission from ref [942]. Copyright 2011, The Royal Society of Chemistry. Published by DOUGLAS AIRCRAFT CO INC SANTA MONICA CA; ROYAL SOCIETY OF CHEMISTRY; US Dept of the Air Force.
Figure 44.
Diagram illustrating the PEO-b-PHEMA-b-PtBA synthesis. Adapted with permission from ref [942]. Copyright 2011, The Royal Society of Chemistry. Published by DOUGLAS AIRCRAFT CO INC SANTA MONICA CA; ROYAL SOCIETY OF CHEMISTRY; US Dept of the Air Force.
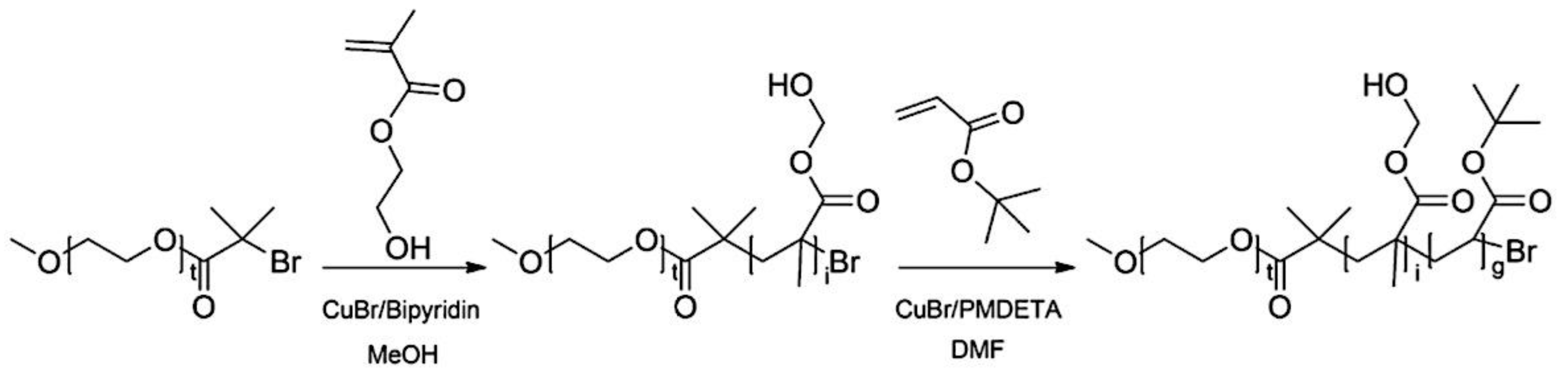
Figure 45.
Diagrammatic illustration of the sequential AP and GPC trace (Gray – PS, Black – PS-b-PB, Green – PS-b-PB-PtBMA) used in the synthesis of PS-b-PB-b-PtBMA. GPC trace taken from ref [943]. Adapted with permission from ref [943]. Copyright 2015, Elsevier Publishing Group.
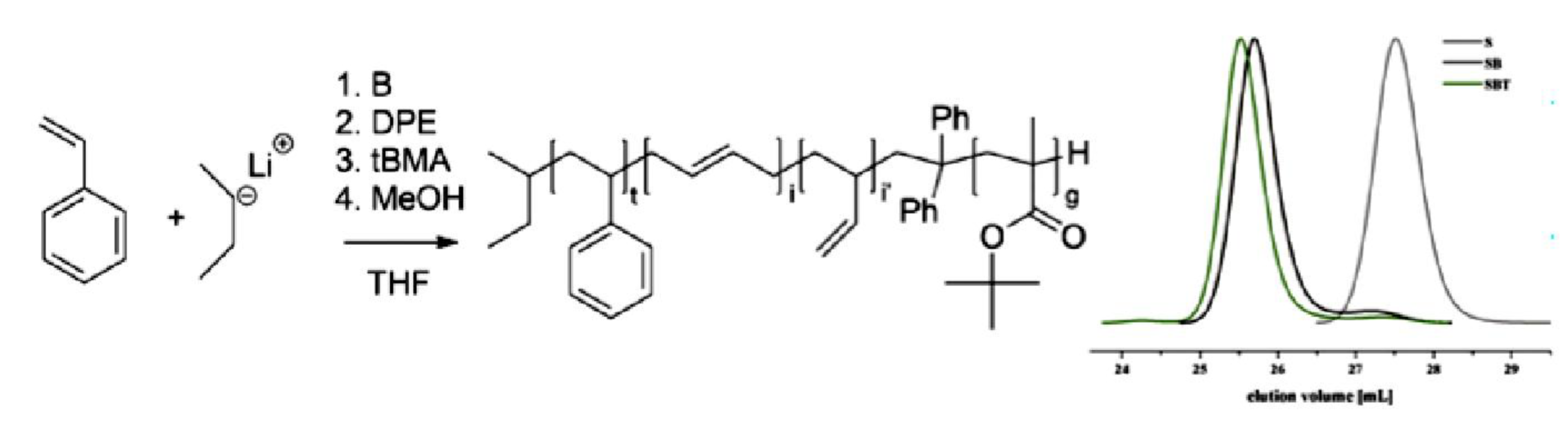
Figure 46.
Diagrammatic illustration of the sequential AROP and GPC tracing method used to synthesize PEO-b-PAGE-b-PtBGE. GPC trace taken from ref [944]. Adapted with permission from ref [944]. Copyright 2013, The Royal Society of Chemistry. Published by DOUGLAS AIRCRAFT CO INC SANTA MONICA CA; ROYAL SOCIETY OF CHEMISTRY; US Dept of the Air Force.
Figure 46.
Diagrammatic illustration of the sequential AROP and GPC tracing method used to synthesize PEO-b-PAGE-b-PtBGE. GPC trace taken from ref [944]. Adapted with permission from ref [944]. Copyright 2013, The Royal Society of Chemistry. Published by DOUGLAS AIRCRAFT CO INC SANTA MONICA CA; ROYAL SOCIETY OF CHEMISTRY; US Dept of the Air Force.
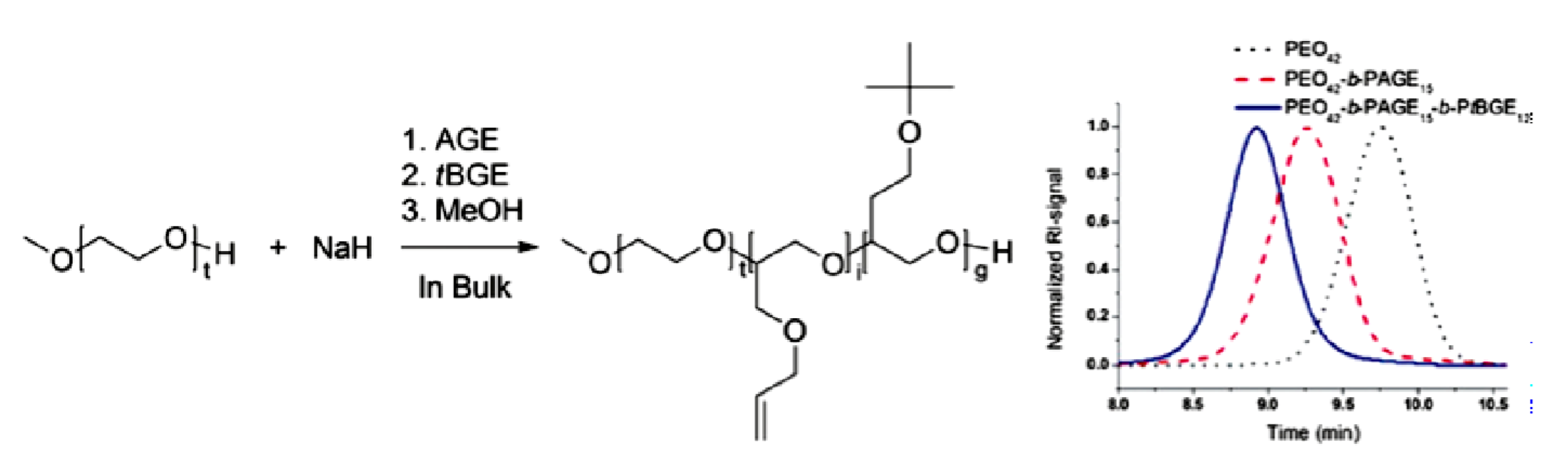
Figure 47.
Diagram showing the sequential CROP and GPC trace synthesis of PODFOx-b-PEPOx-b-PEtOx. GPC trace modified using ref [945]. Adapted with permission from ref [945]. Copyright 2017, John Wiley and Sons Publishing Group.
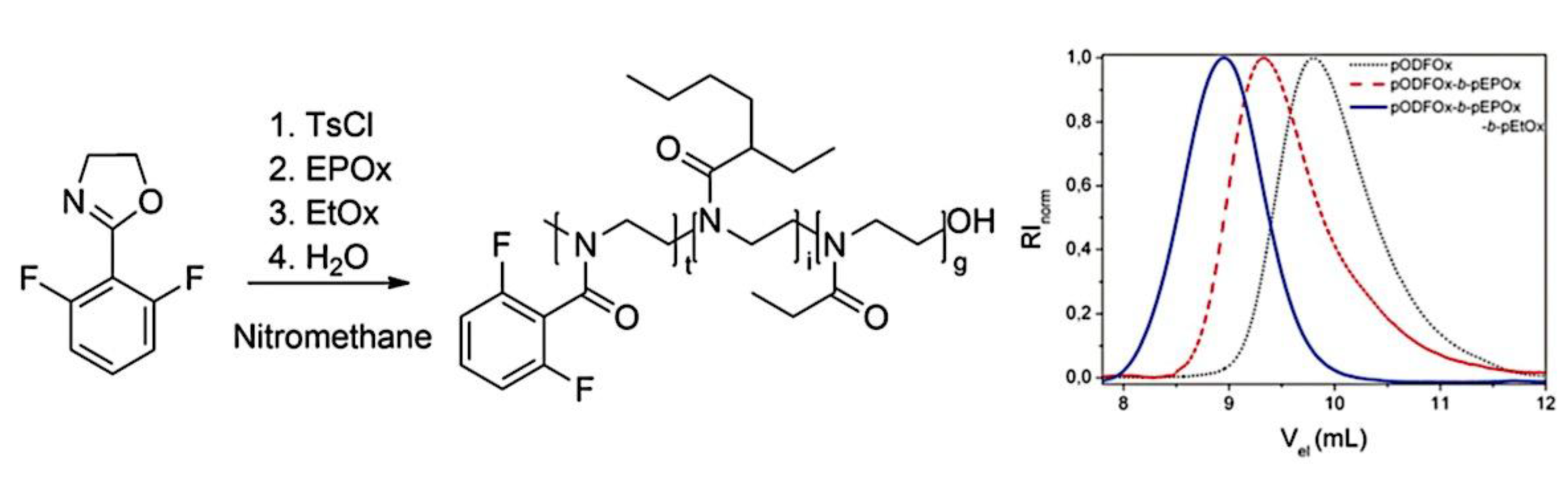
Figure 48.
Diagram showing PEO-b-PCL-b-PMOXA and GPC trace synthesis (blue for PEO, green for PEO-b-PCL, black for PEO-b-PCL-OTs, and red for PEO-b-PCL-b-PMOXA) taken from ref [948]. Adapted with permission from ref [948]. Copyright 2017, American Chemical Society.
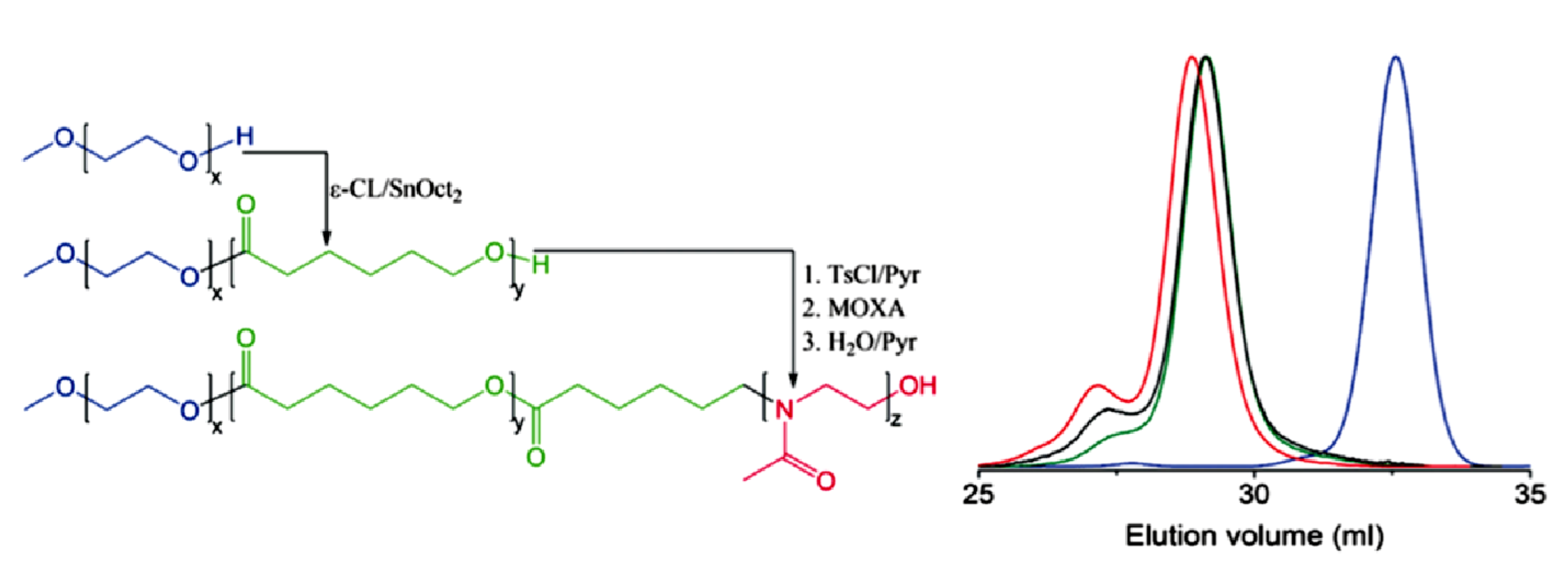
Figure 49.
Diagrammatic illustration of the PEO-b-PMCL-b-PDMAEMA synthesis and GPC trace of PEO45 (a), PEO45-b-PMCL47 (b), PEO45-b-PMCL47-b-PDMAEMA15 (c), PEO45-b-PMCL47-b-PDMAEMA31 (d), and PEO45-b-PMCL47-b-PDMAEMA42 (e). Adapted with permission from ref [951]. Copyright 2011, John Wiley and Sons Publishing Group.
Figure 49.
Diagrammatic illustration of the PEO-b-PMCL-b-PDMAEMA synthesis and GPC trace of PEO45 (a), PEO45-b-PMCL47 (b), PEO45-b-PMCL47-b-PDMAEMA15 (c), PEO45-b-PMCL47-b-PDMAEMA31 (d), and PEO45-b-PMCL47-b-PDMAEMA42 (e). Adapted with permission from ref [951]. Copyright 2011, John Wiley and Sons Publishing Group.
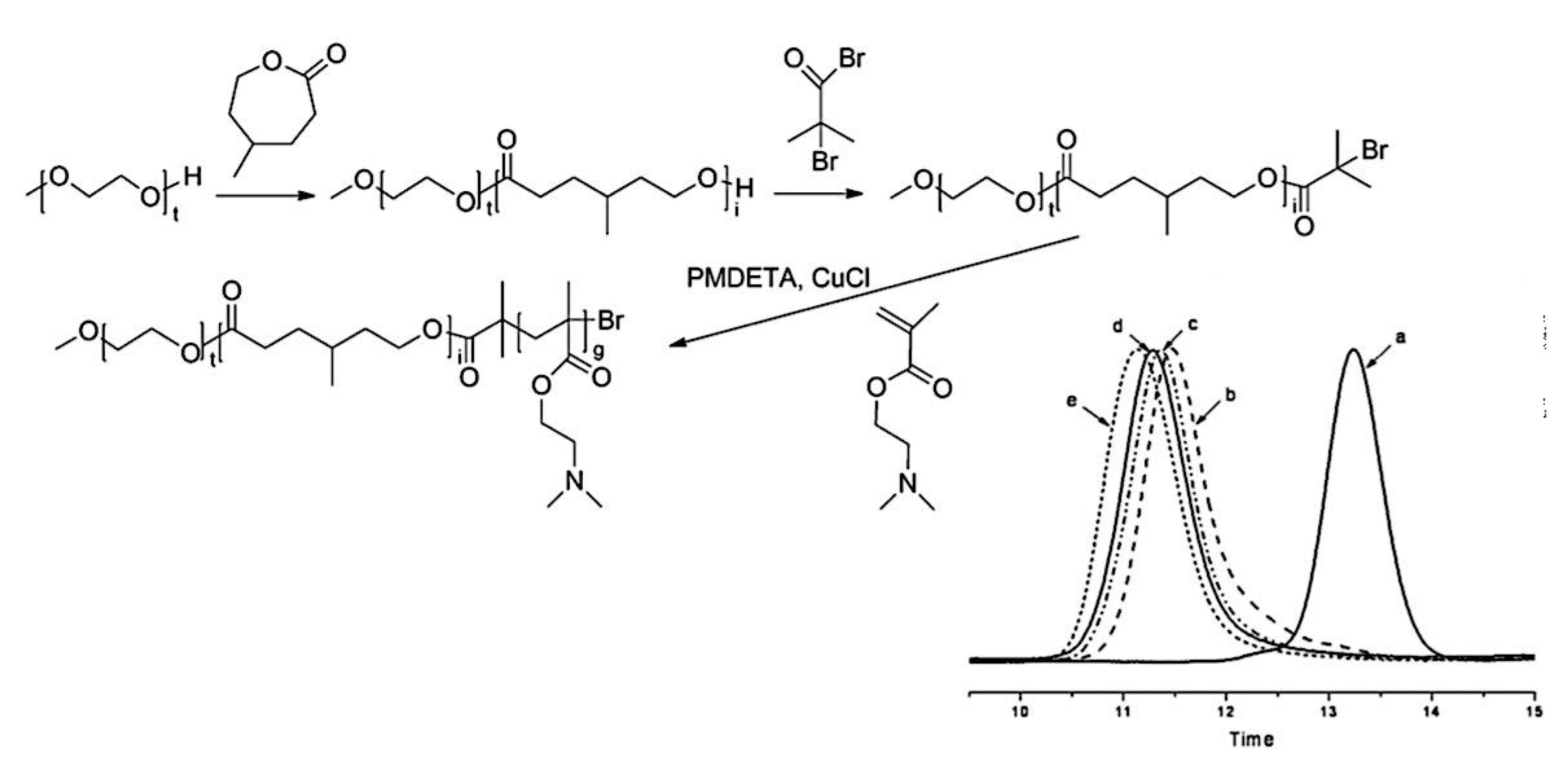
Figure 50.
Diagrammatic illustration of the PEO-b-PS-b-PCL synthesis and GPC trace. Adapted with permission from ref [952]. Copyright 2007, John Wiley and Sons Publishing Group.
Figure 50.
Diagrammatic illustration of the PEO-b-PS-b-PCL synthesis and GPC trace. Adapted with permission from ref [952]. Copyright 2007, John Wiley and Sons Publishing Group.
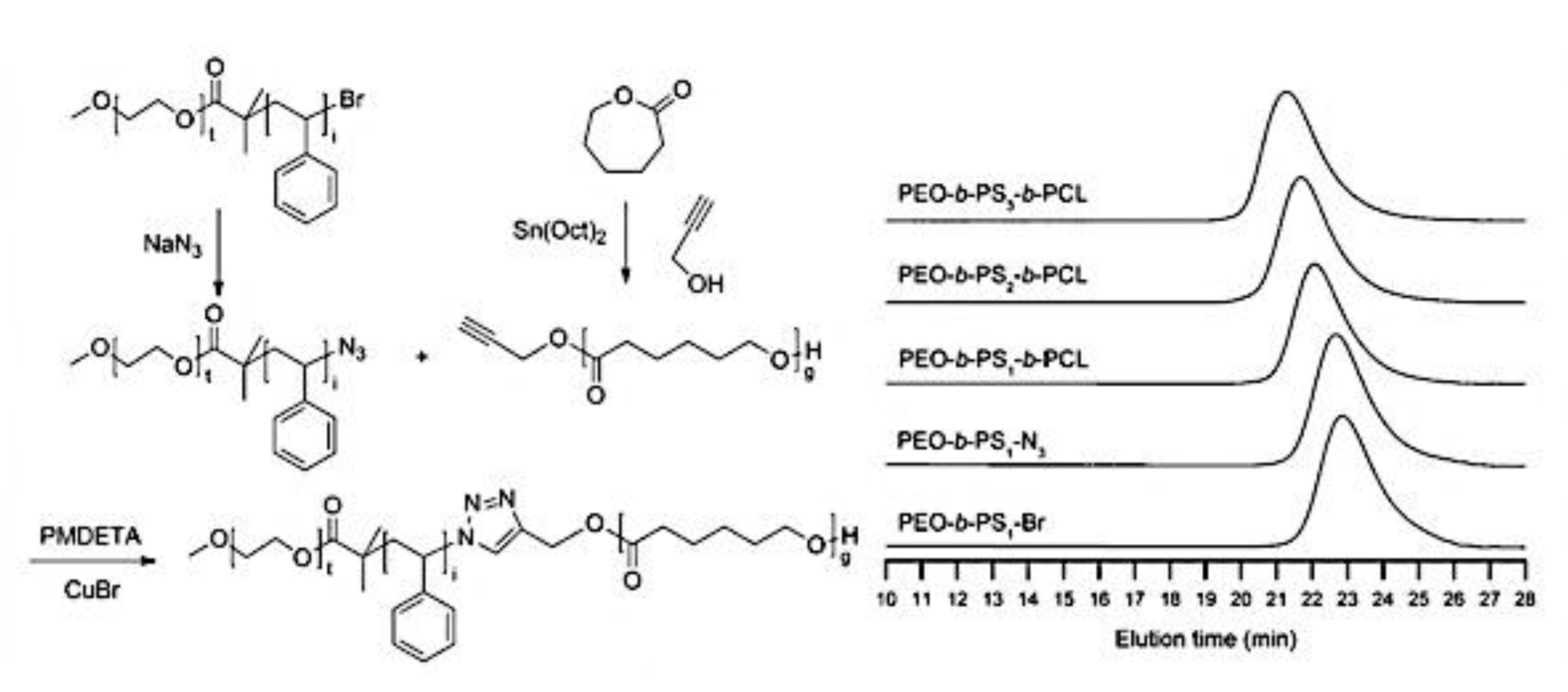
Figure 51.
3-miktoarm terpolymer micelle schematic with dumble-like (a) and segmented cylindrical core (b). Adapted with permission from ref [722]. Copyright 2012, American Chemical Society.
Figure 51.
3-miktoarm terpolymer micelle schematic with dumble-like (a) and segmented cylindrical core (b). Adapted with permission from ref [722]. Copyright 2012, American Chemical Society.
Figure 52.
Micelle geometry is classified according to compartment morphology created by PS-b-PB-b-PtBMA polymers in mixtures of acetone and isopropanol: spheres-on-spheres, spheres-on-cylinders, spheres-on-bilayer sheets, and polymersomes; cylinders-on-cylinders (double-helix compartment), cylinders-on-bilayer sheets, and polymersomes; sheets and polymersomes with bicontinuous membrane morphology; core–shell micelles, core-shell cylinders, lamellar sheets, and vesicles. TEM pictures: OsO4 staining (scale bars are 200 nm; PS is grey, PB is black, and PtBMA is not visible). Adapted with permission from ref [769]. Copyright 2012, The Authors. Published by Springer Nature under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. (Creative Commons CC-BY-NC-ND license, http://creativecommons.org/licenses/by-nc-nd/3.0/). .
Figure 52.
Micelle geometry is classified according to compartment morphology created by PS-b-PB-b-PtBMA polymers in mixtures of acetone and isopropanol: spheres-on-spheres, spheres-on-cylinders, spheres-on-bilayer sheets, and polymersomes; cylinders-on-cylinders (double-helix compartment), cylinders-on-bilayer sheets, and polymersomes; sheets and polymersomes with bicontinuous membrane morphology; core–shell micelles, core-shell cylinders, lamellar sheets, and vesicles. TEM pictures: OsO4 staining (scale bars are 200 nm; PS is grey, PB is black, and PtBMA is not visible). Adapted with permission from ref [769]. Copyright 2012, The Authors. Published by Springer Nature under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. (Creative Commons CC-BY-NC-ND license, http://creativecommons.org/licenses/by-nc-nd/3.0/). .
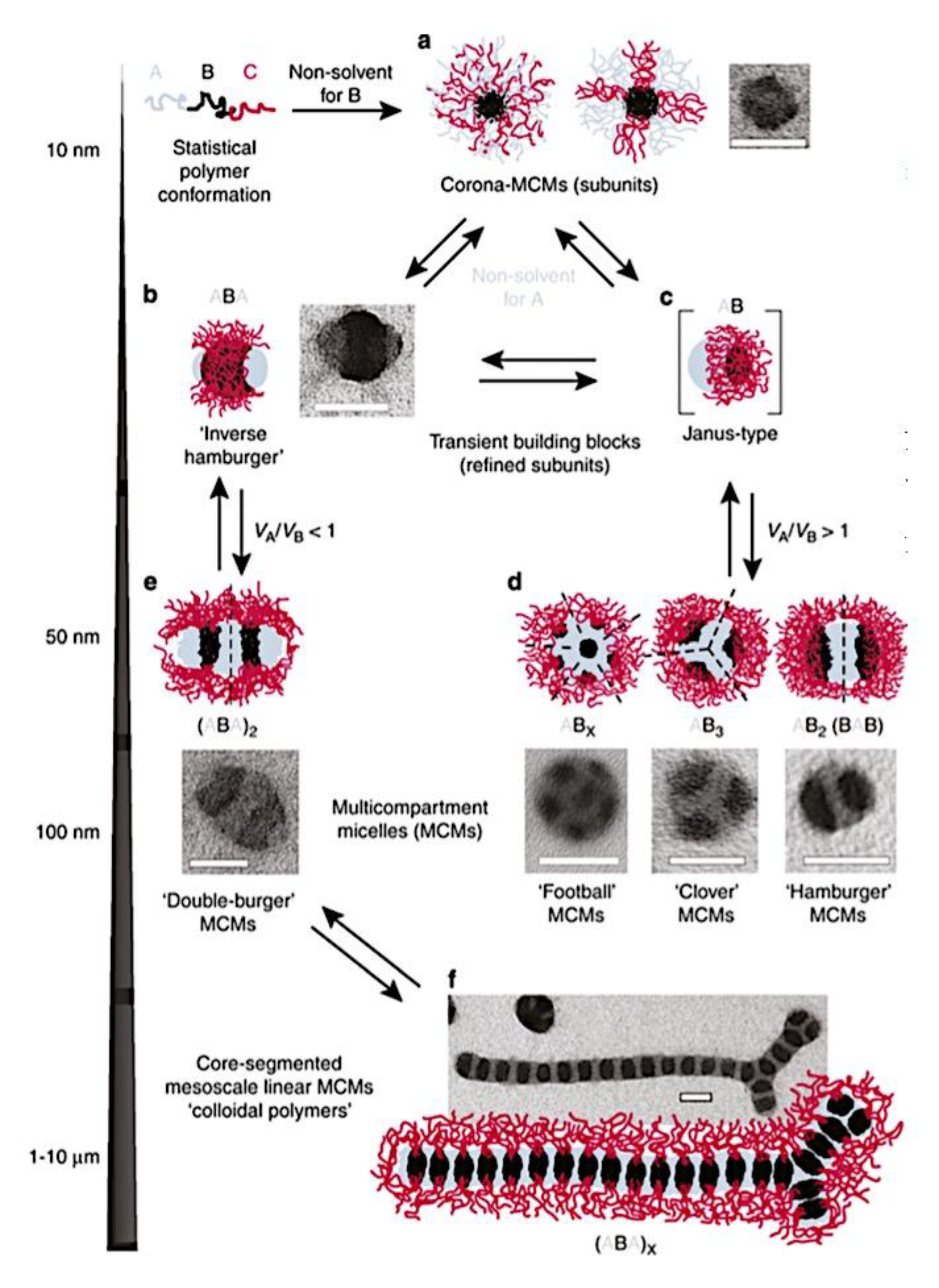
Figure 53.
Micelle geometry is classified according to compartment morphology created by PS-b-PB-b-PtBMA polymers in mixtures of acetone and isopropanol: spheres-on-spheres, spheres-on-cylinders, spheres-on-bilayer sheets, and polymersomes; cylinders-on-cylinders (double-helix compartment), cylinders-on-bilayer sheets, and polymersomes; sheets and polymersomes with bicontinuous membrane morphology; core-shell micelles, core-shell cylinders, lamellar sheets, and vesicles. TEM pictures: OsO4 staining (scale bars are 200 nm; PS is grey, PB is black, and PtBMA is not visible). Adapted from ref [771]. Copyright 2016, Löbling, T. I. et al. Published by Springer Nature under the terms of the Creative Commons CC BY (http://creativecommons.org/licenses/by/4.0/) license. .
Figure 53.
Micelle geometry is classified according to compartment morphology created by PS-b-PB-b-PtBMA polymers in mixtures of acetone and isopropanol: spheres-on-spheres, spheres-on-cylinders, spheres-on-bilayer sheets, and polymersomes; cylinders-on-cylinders (double-helix compartment), cylinders-on-bilayer sheets, and polymersomes; sheets and polymersomes with bicontinuous membrane morphology; core-shell micelles, core-shell cylinders, lamellar sheets, and vesicles. TEM pictures: OsO4 staining (scale bars are 200 nm; PS is grey, PB is black, and PtBMA is not visible). Adapted from ref [771]. Copyright 2016, Löbling, T. I. et al. Published by Springer Nature under the terms of the Creative Commons CC BY (http://creativecommons.org/licenses/by/4.0/) license. .
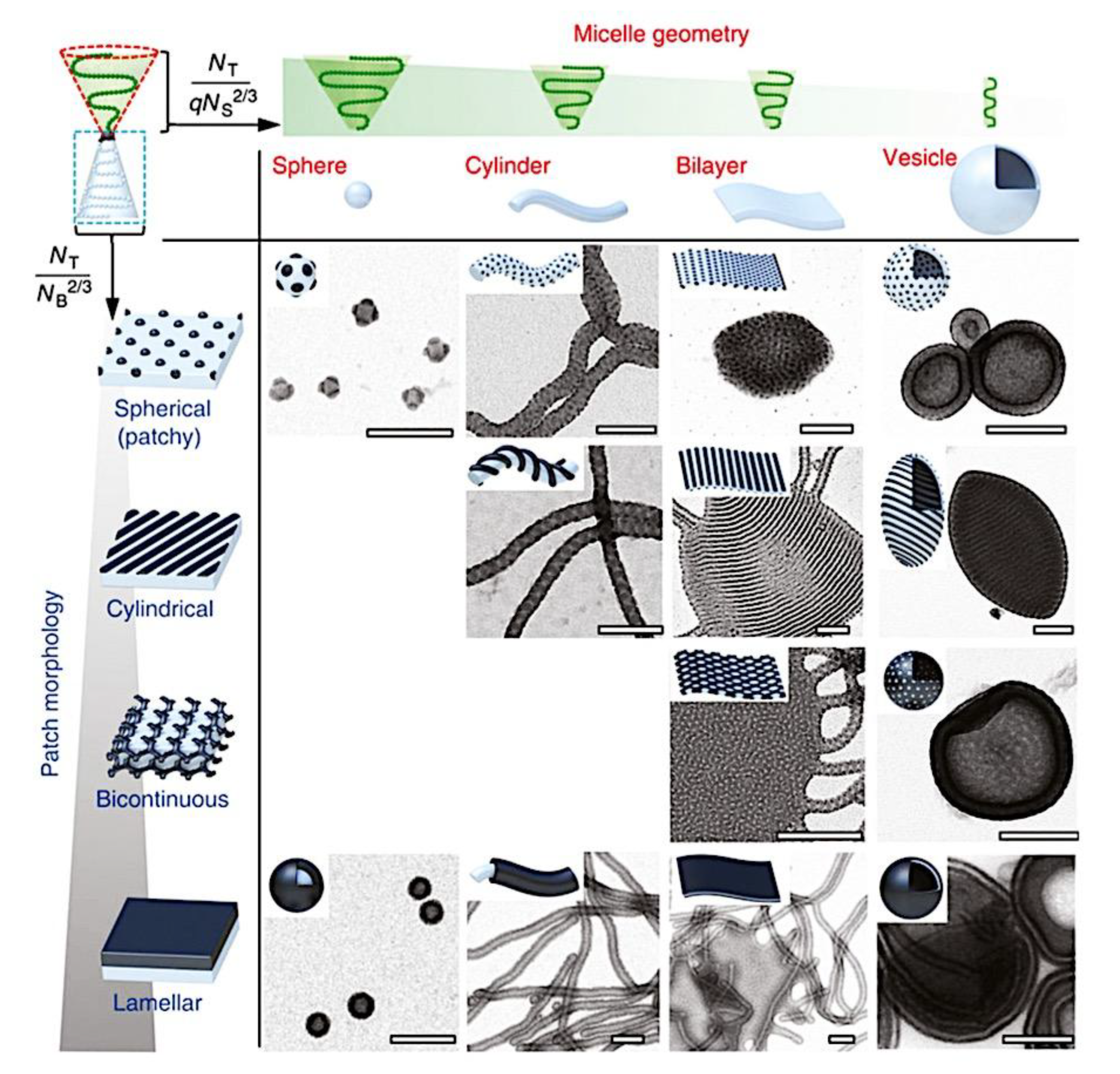
Figure 54.
(a) A morphology diagram that illustrates the structures created in an aqueous solution by PEO-b-PCL and PEO-b-PCL-b-PMOXA in relation to their molecular composition. Each color’s points represent polymers with a specific PMOXA length. Points of each shape represent a particular morphology: polymersomes (circles), irregularly shaped particles (diamonds), spherical particles (squares), and aggregates that resemble clouds (triangles). The regions with the same morphology are indicated by the gray areas. Exemplary light scattering (LSM) pictures of (b) round particles, (c) asymmetrical particles, (d) polymersomes, and (e) aggregates resembling clouds. Stains used on the structures were Bodipy 630/650. There are 5 μm scale bars. A sample TEM image of negatively stained spherical particles is shown in B inset; the scale bar is 200 nm. The packing geometry of the following polymers is illustrated: (f) AB, (g) ABC with short C block, and (h) ABC with long C block. The polymers have fixed A and B (around 60-130 units) but variable C block lengths. Adapted with permission from ref [1006]. Copyright 2017, American Chemical Society.
Figure 54.
(a) A morphology diagram that illustrates the structures created in an aqueous solution by PEO-b-PCL and PEO-b-PCL-b-PMOXA in relation to their molecular composition. Each color’s points represent polymers with a specific PMOXA length. Points of each shape represent a particular morphology: polymersomes (circles), irregularly shaped particles (diamonds), spherical particles (squares), and aggregates that resemble clouds (triangles). The regions with the same morphology are indicated by the gray areas. Exemplary light scattering (LSM) pictures of (b) round particles, (c) asymmetrical particles, (d) polymersomes, and (e) aggregates resembling clouds. Stains used on the structures were Bodipy 630/650. There are 5 μm scale bars. A sample TEM image of negatively stained spherical particles is shown in B inset; the scale bar is 200 nm. The packing geometry of the following polymers is illustrated: (f) AB, (g) ABC with short C block, and (h) ABC with long C block. The polymers have fixed A and B (around 60-130 units) but variable C block lengths. Adapted with permission from ref [1006]. Copyright 2017, American Chemical Society.
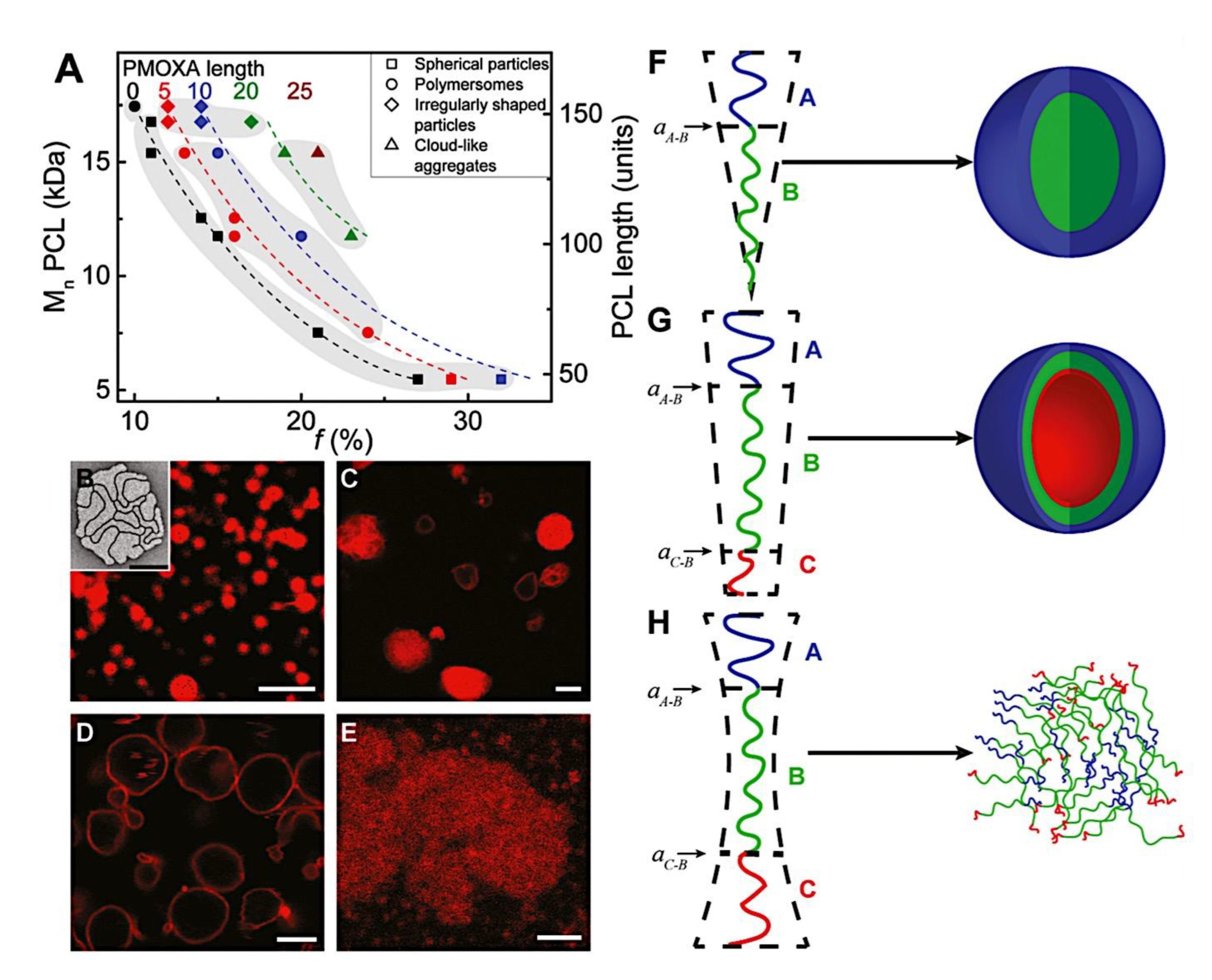
Figure 55.
Three possible membrane compositions for polymersomes made of ABC polymers and soluble blocks of A and C are as follows: a) A outside; b) C outside; and c) A and C create a mixed membrane. Adapted with permission from ref [948]. Copyright 2017, American Chemical Society.
Figure 55.
Three possible membrane compositions for polymersomes made of ABC polymers and soluble blocks of A and C are as follows: a) A outside; b) C outside; and c) A and C create a mixed membrane. Adapted with permission from ref [948]. Copyright 2017, American Chemical Society.
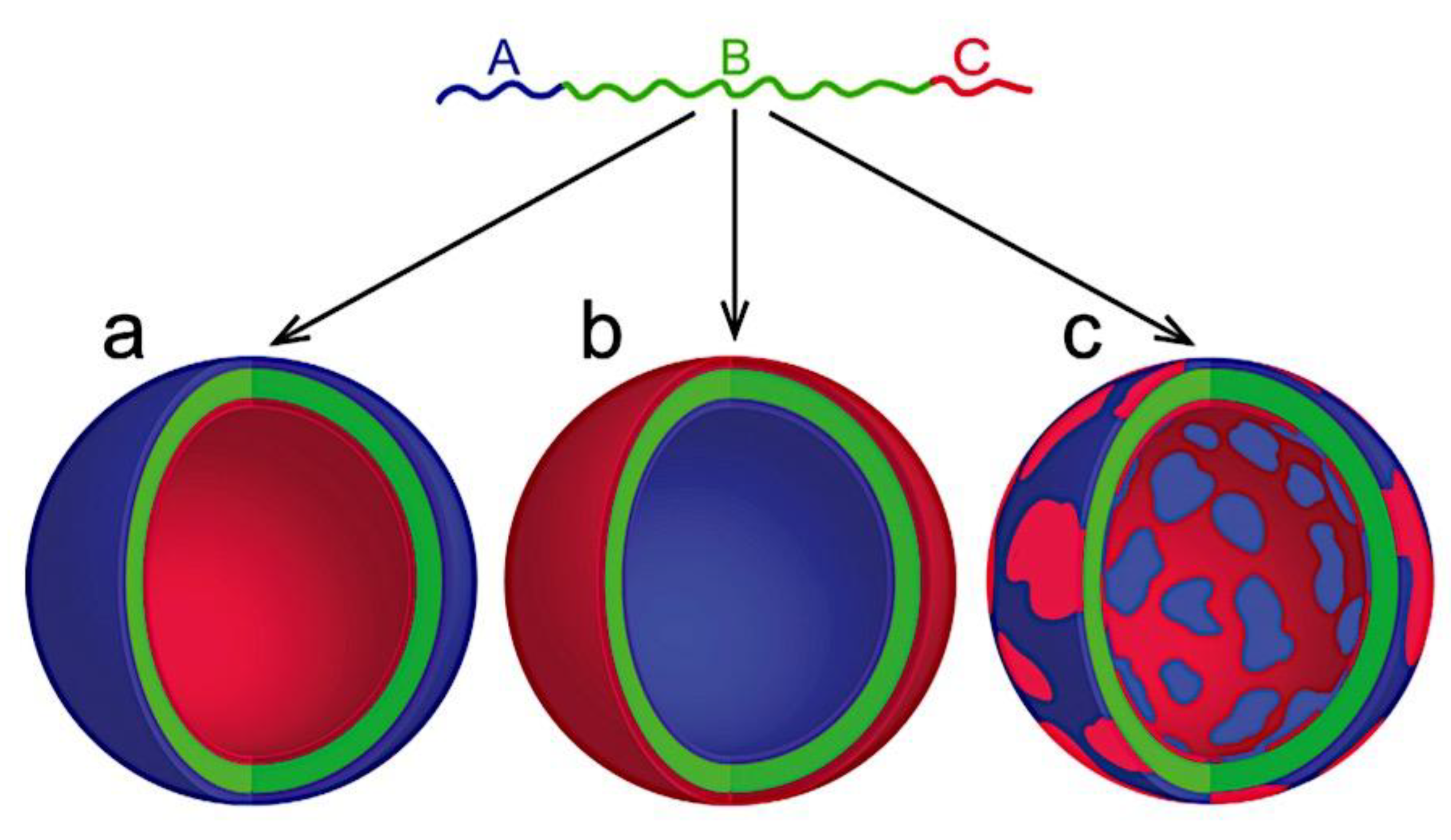
Figure 56.
Amphiphilic star block copolymer synthesis utilizing SPS-b-PtBS diblock arms. Adapted from Ref [872]. Copyright 2010, Elsevier Ltd. Published open access under CC BY-NC-ND license.
Figure 56.
Amphiphilic star block copolymer synthesis utilizing SPS-b-PtBS diblock arms. Adapted from Ref [872]. Copyright 2010, Elsevier Ltd. Published open access under CC BY-NC-ND license.
Figure 57.
As a function of composition, a combination of homopolymer (hp) and diblock coppolymer (dbp) with opposite charges is specifed into free molecules (a), soluble complex particles (SCP+,-), (b), and CCCMs (c) at constant concentration overall. Dotted vertical lines represent the PMC, the critical excess anionic charge (CEAC), and the critical excess cationic charge (CECC). Adapted with permission ref [30]. Copyright 2004, American Chemical Society.
Figure 57.
As a function of composition, a combination of homopolymer (hp) and diblock coppolymer (dbp) with opposite charges is specifed into free molecules (a), soluble complex particles (SCP+,-), (b), and CCCMs (c) at constant concentration overall. Dotted vertical lines represent the PMC, the critical excess anionic charge (CEAC), and the critical excess cationic charge (CECC). Adapted with permission ref [30]. Copyright 2004, American Chemical Society.
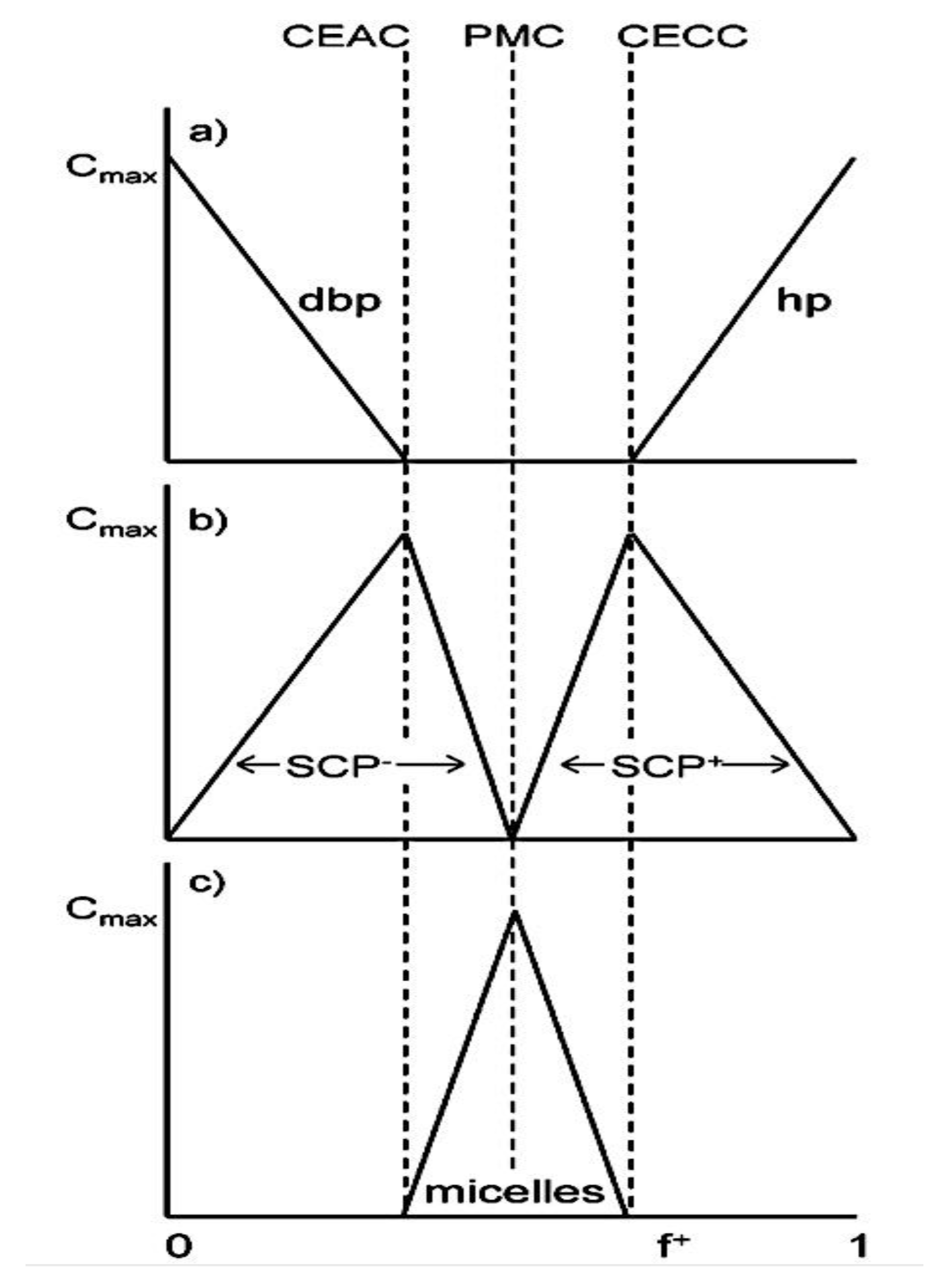
Figure 58.
Diagram showing the CCCMs’ light dispersion. The light scattering intensity is represented on the vertical axis. f+ is shown on the horizontal axis. Adapted with permission ref [30]. Copyright 2004, American Chemical Society.
Figure 58.
Diagram showing the CCCMs’ light dispersion. The light scattering intensity is represented on the vertical axis. f+ is shown on the horizontal axis. Adapted with permission ref [30]. Copyright 2004, American Chemical Society.
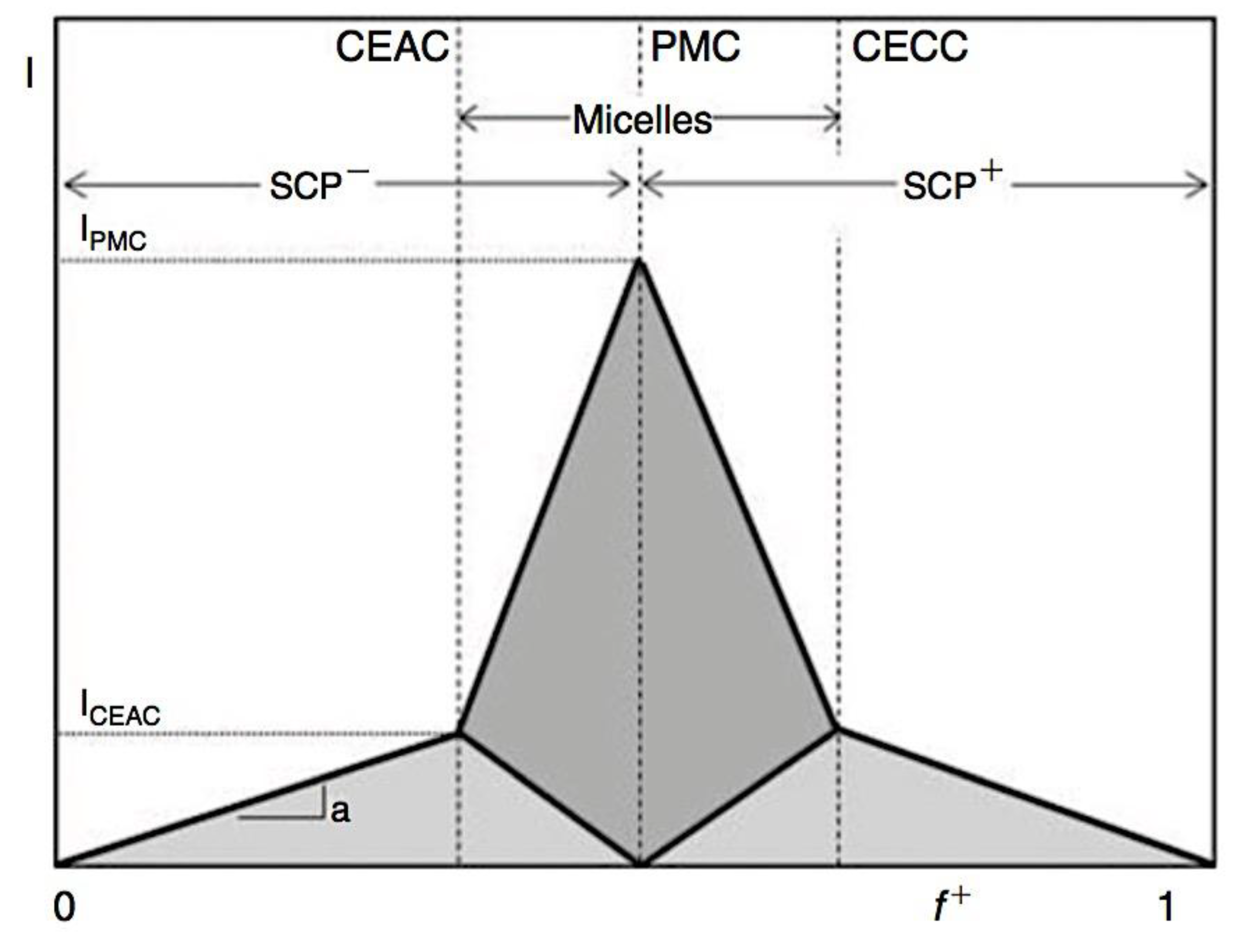
Figure 59.
Illustration of polymeric micelles and polymersomes schematically. Adapted from ref [1042]. Copyright 2022, The Royal Society of Chemistry. Published by The Royal Society of Chemistry under a Creative Commons Attribution 3.0 Unported Licence.
Figure 59.
Illustration of polymeric micelles and polymersomes schematically. Adapted from ref [1042]. Copyright 2022, The Royal Society of Chemistry. Published by The Royal Society of Chemistry under a Creative Commons Attribution 3.0 Unported Licence.
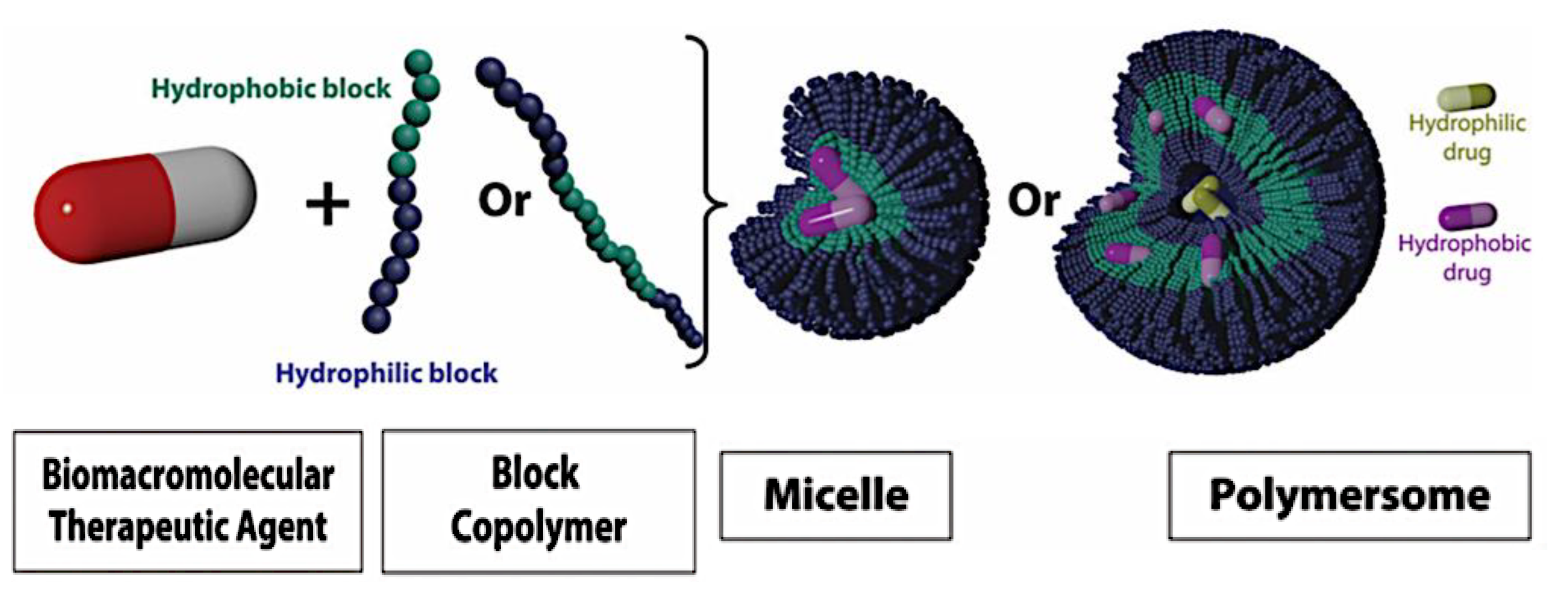
Figure 60.
Possible situations in which C3Ms develop. While Scenarios I and IV were seen by Stenzel’s group by employing PEGMEA-PAA with lysozyme, Scenarios II and III are described by Harada et al.[281] Orange is a charged polymer, blue is a water-soluble neutral polymer, and green is protein. Adapted with permission from ref [116]. Copyright 2018, CSIRO Publishing.
Figure 60.
Possible situations in which C3Ms develop. While Scenarios I and IV were seen by Stenzel’s group by employing PEGMEA-PAA with lysozyme, Scenarios II and III are described by Harada et al.[281] Orange is a charged polymer, blue is a water-soluble neutral polymer, and green is protein. Adapted with permission from ref [116]. Copyright 2018, CSIRO Publishing.
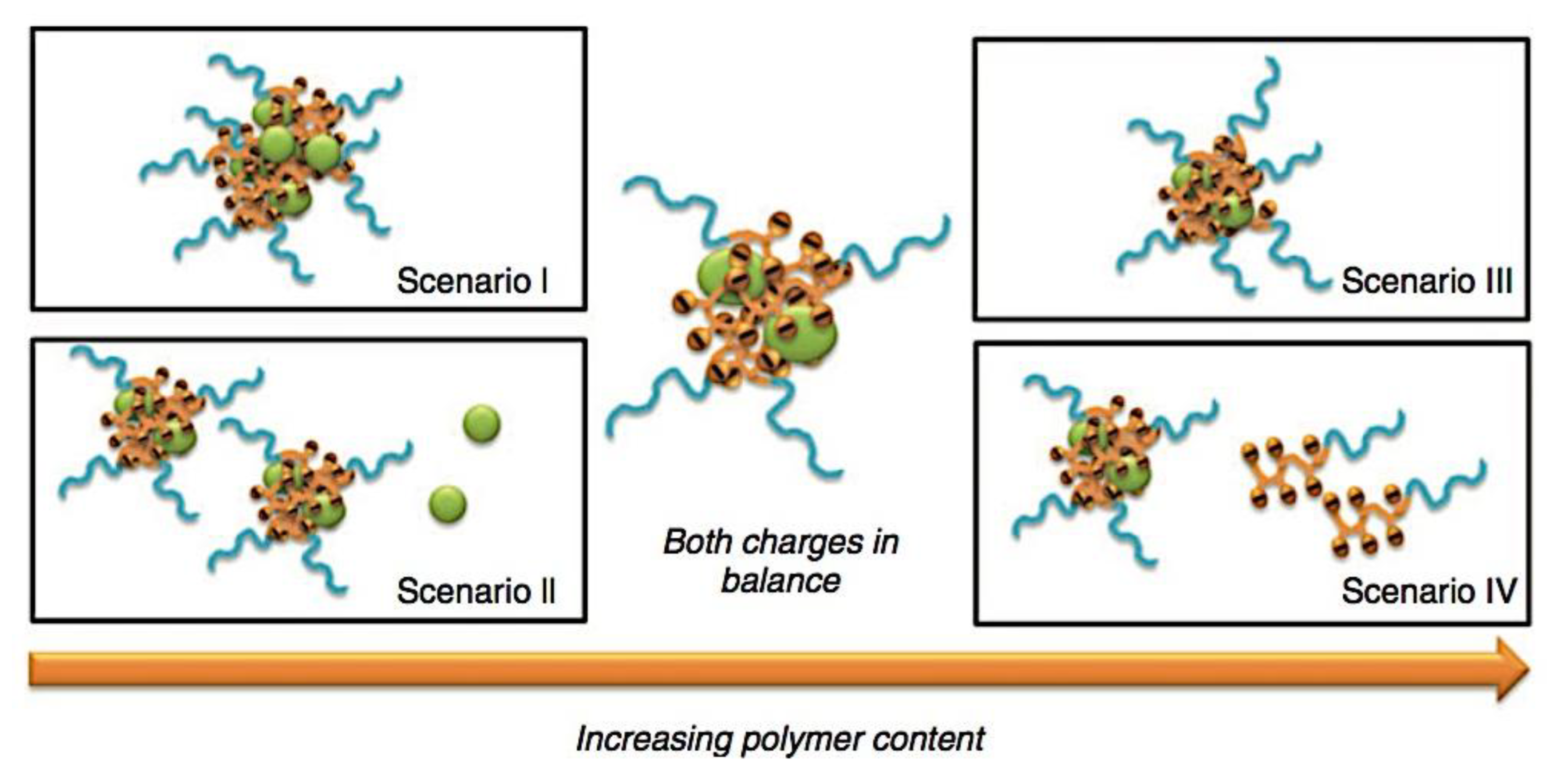
Figure 61.
Charged polymers for the production of PIC micelles containing proteins. Adapted with permission from ref [116]. Copyright 2018, CSIRO Publishing.
Figure 61.
Charged polymers for the production of PIC micelles containing proteins. Adapted with permission from ref [116]. Copyright 2018, CSIRO Publishing.
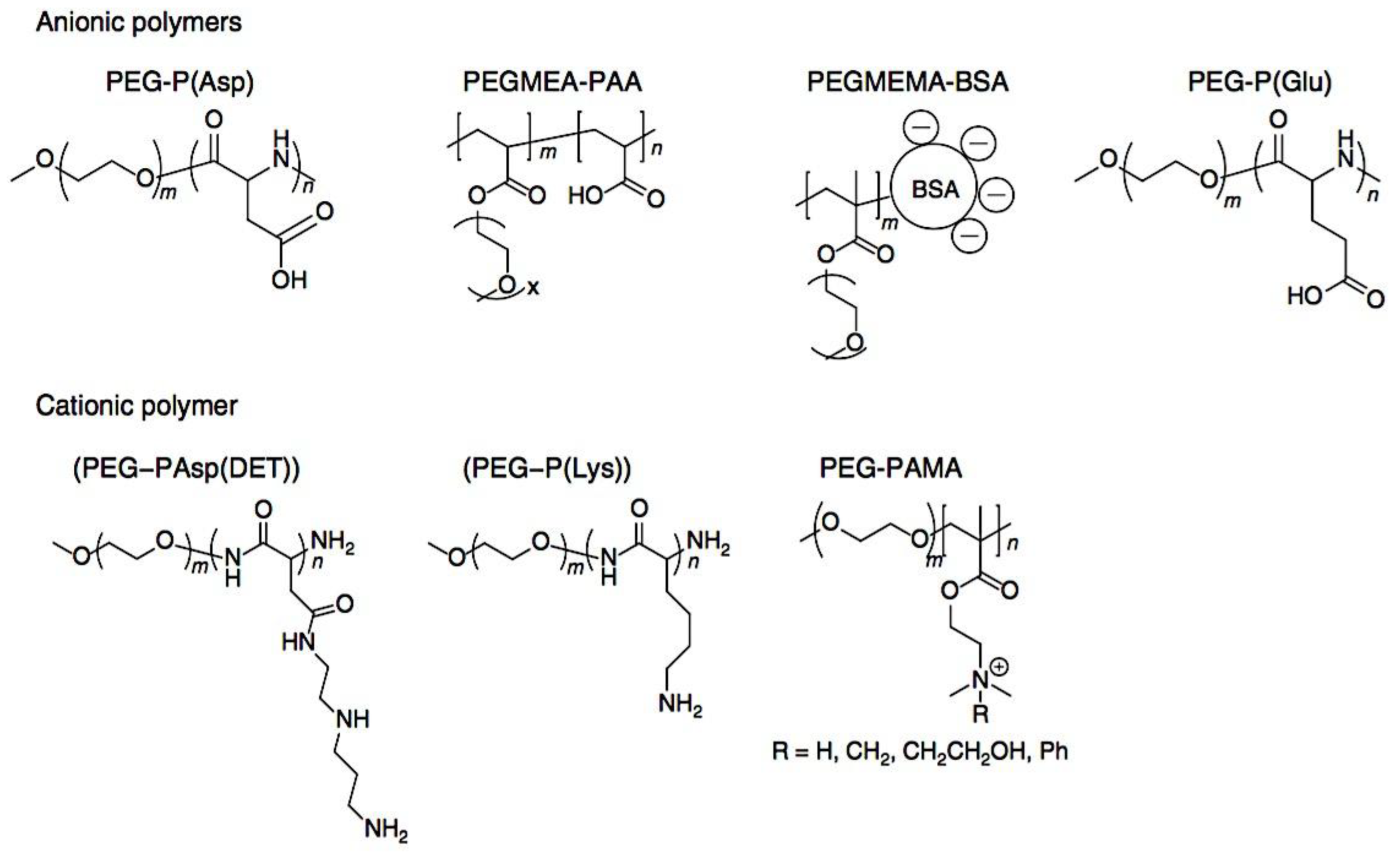
Figure 62.
(a) PIC micelle formation and dissociation upon charge conversion. Hydrodynamic diameter changes for (b) PEG-pAsp(EDA-CA) (labile group) and (c) PEG-pAsp(EDA-SA) (stable group) at pH 5.5 (filled symbol) and 7.4 (open symbols). Adapted with permission from ref [1059]. Copyright 2007, American Chemical Society. .
Figure 62.
(a) PIC micelle formation and dissociation upon charge conversion. Hydrodynamic diameter changes for (b) PEG-pAsp(EDA-CA) (labile group) and (c) PEG-pAsp(EDA-SA) (stable group) at pH 5.5 (filled symbol) and 7.4 (open symbols). Adapted with permission from ref [1059]. Copyright 2007, American Chemical Society. .
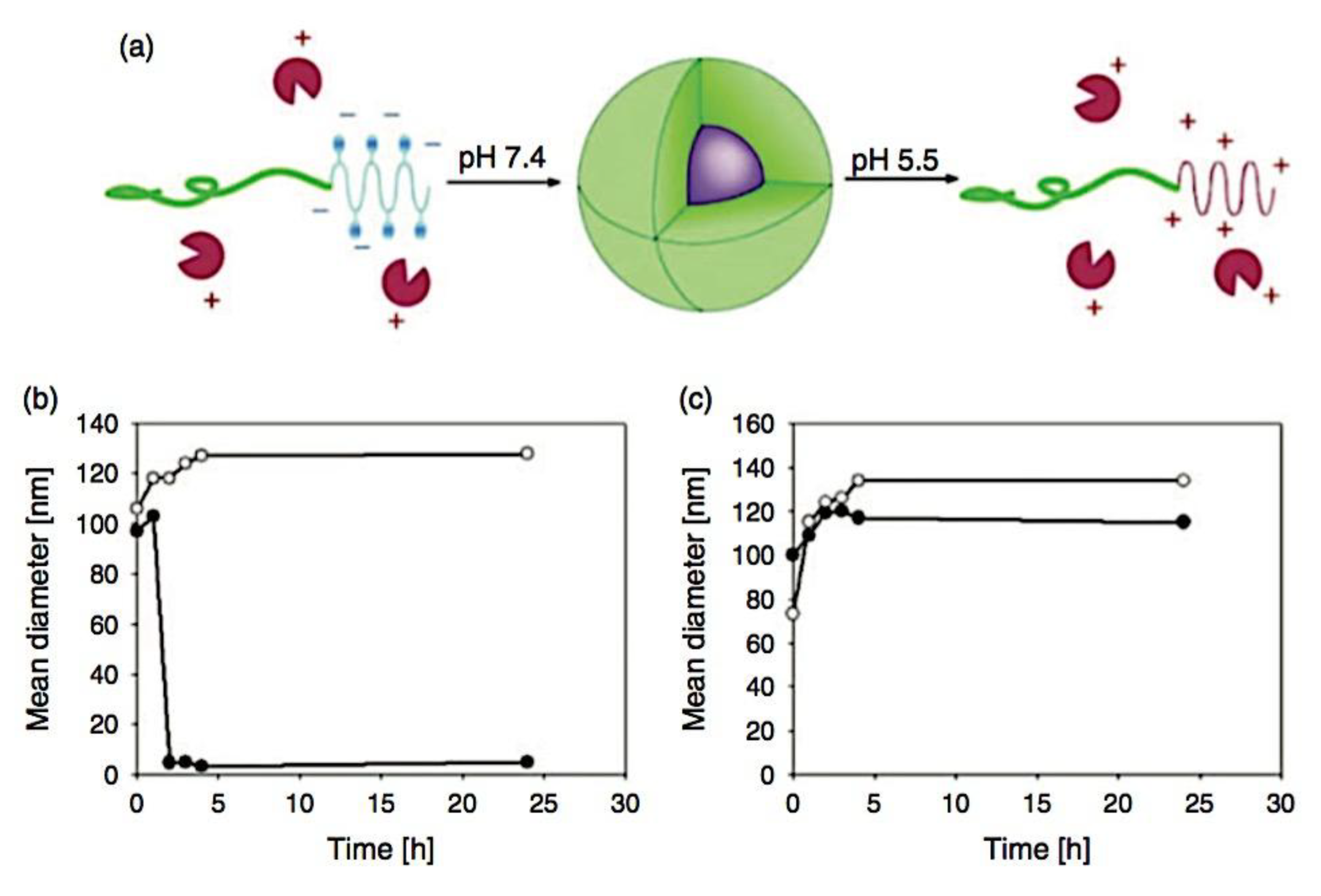
Figure 63.
Amann et al.’s suggested kinetic mechanism for PEC micelle assembly is most likely for solid-core micelles. The evolution of spherical micelles is caused by change exchange that occurs after the production of bigger irregular aggregates. Adapted with permission from ref [88]. Copyright 2019, American Chemical Society.
Figure 63.
Amann et al.’s suggested kinetic mechanism for PEC micelle assembly is most likely for solid-core micelles. The evolution of spherical micelles is caused by change exchange that occurs after the production of bigger irregular aggregates. Adapted with permission from ref [88]. Copyright 2019, American Chemical Society.
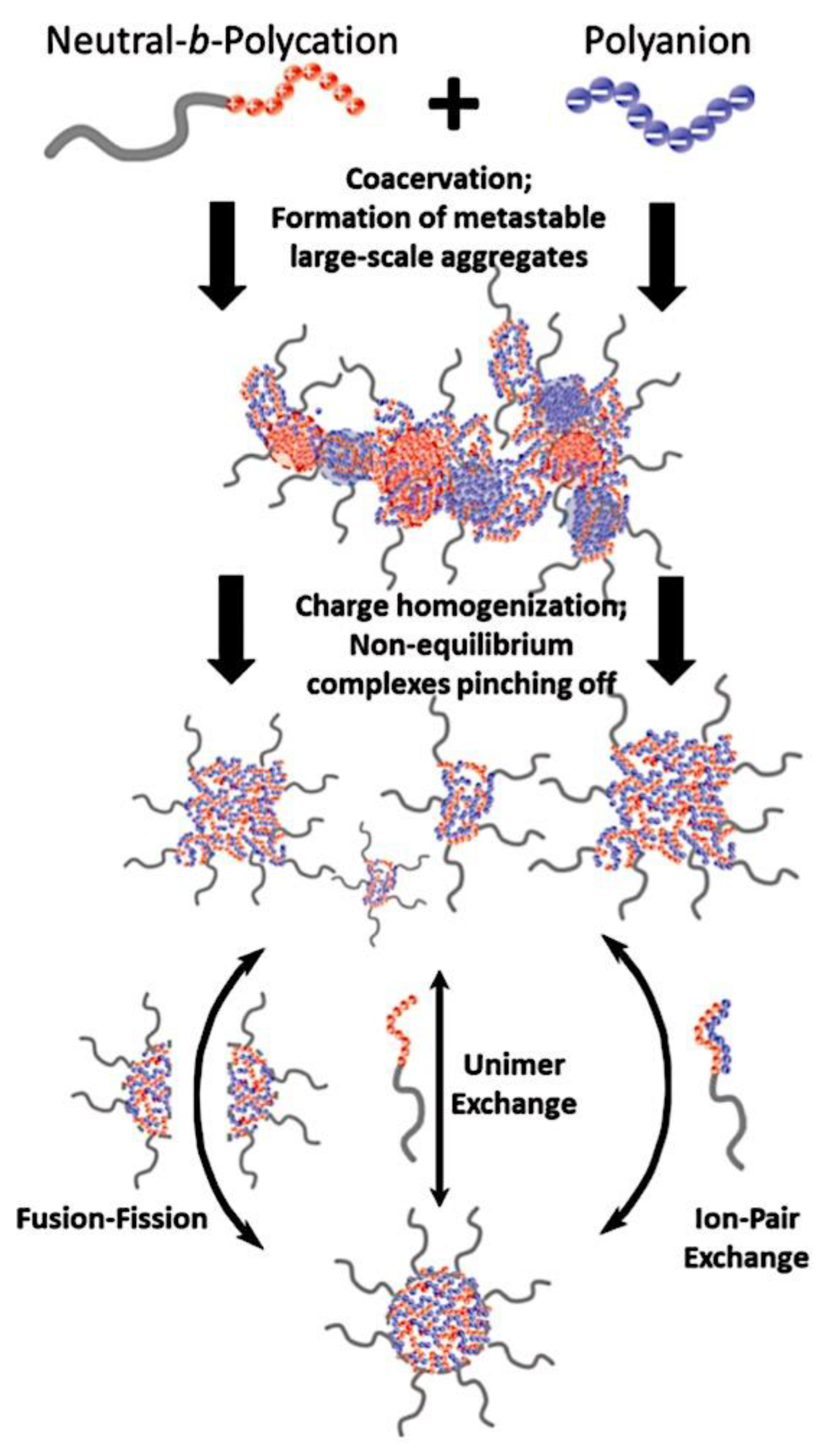
Figure 64.
Polyelectrolyte complex micelles (PCMs) dynamics. The chemical structures of sodium poly(acrylate) (PAA, boxed in blue), poly(ethylene oxide)-block-poly(sodium 4-styrenesulfonate) (PEO-b-PSS, boxed in blue), and poly(sodium 4-styrenesulfonate) (PSS, boxed in blue) are displayed in (A). (B) An illustration utilizing small-angle X-ray scattering, cryogenic electron microscopy, and dynamic light scattering of the pertinent time and length scales studied in PCM creation (purple), chain exchange (green), and disassembly (orange), extending from milliseconds to minutes. Adaped from ref [58]. Copyright 2021, The Authors. Published by American Chemical Society. This publication is licensed under CC-BY-NC-ND 4.0.
Figure 64.
Polyelectrolyte complex micelles (PCMs) dynamics. The chemical structures of sodium poly(acrylate) (PAA, boxed in blue), poly(ethylene oxide)-block-poly(sodium 4-styrenesulfonate) (PEO-b-PSS, boxed in blue), and poly(sodium 4-styrenesulfonate) (PSS, boxed in blue) are displayed in (A). (B) An illustration utilizing small-angle X-ray scattering, cryogenic electron microscopy, and dynamic light scattering of the pertinent time and length scales studied in PCM creation (purple), chain exchange (green), and disassembly (orange), extending from milliseconds to minutes. Adaped from ref [58]. Copyright 2021, The Authors. Published by American Chemical Society. This publication is licensed under CC-BY-NC-ND 4.0.
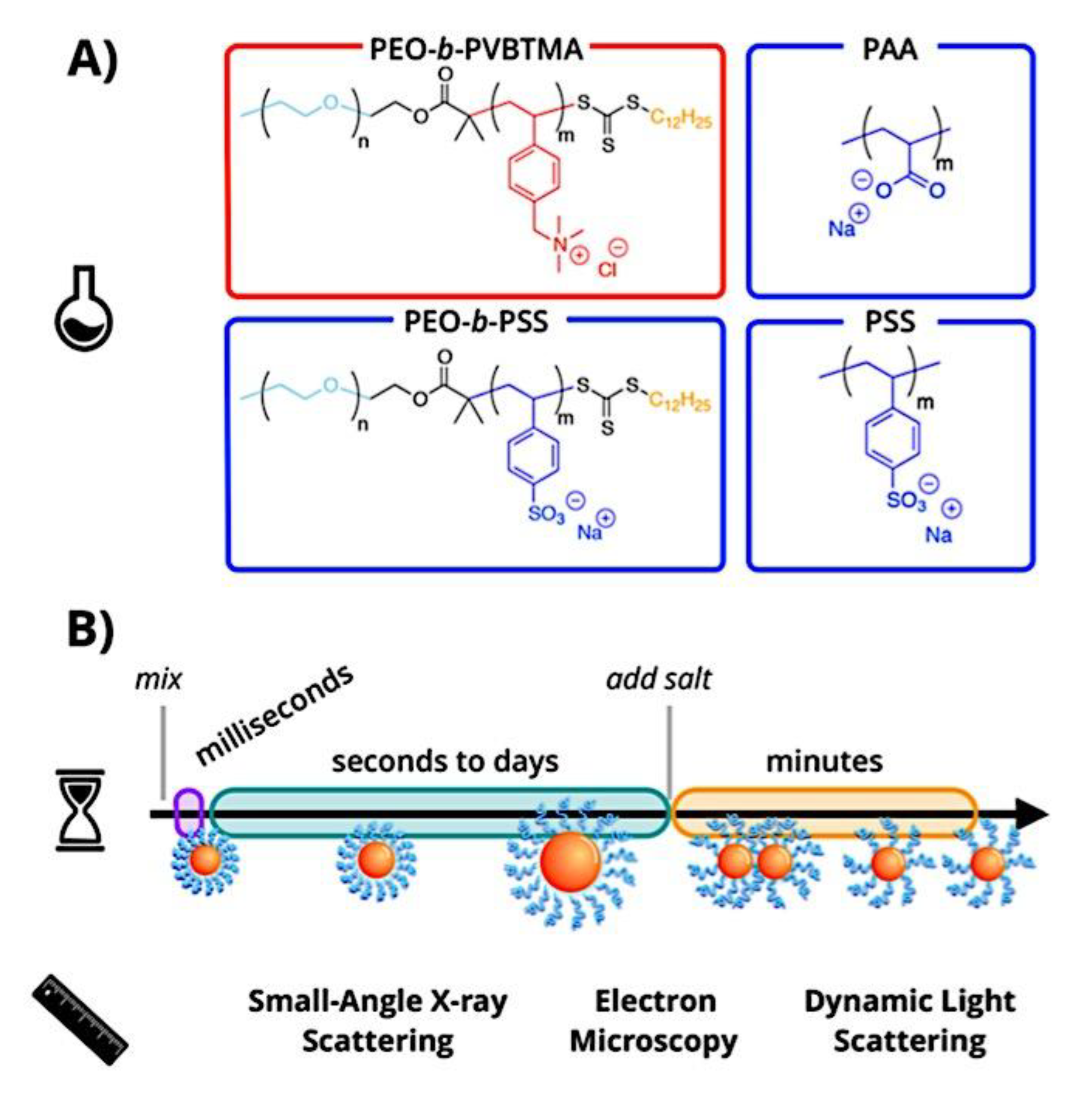
Figure 65.
Different production paths of polyelectrolyte complex micelles (PCMs) are revealed by time-resolved small-angle X-ray scattering (TR-SAXS). (A) The black arrow indicates the progressive growth of well-defined spherical micelles into bigger micellar entities within 100 ms for PEO-b-PVBTMA/PAA systems. Adapted with permission from ref [1081]. Copyright 2020, American Chemical Society. (B) The black arrow indicates that, for PEO-b-PVBTMA/PSS systems, aggregates fragment into smaller micellar entities in less than 3 ms. Adapted with permission from ref [88]. Copyright 2019, American Chemical Society. .
Figure 65.
Different production paths of polyelectrolyte complex micelles (PCMs) are revealed by time-resolved small-angle X-ray scattering (TR-SAXS). (A) The black arrow indicates the progressive growth of well-defined spherical micelles into bigger micellar entities within 100 ms for PEO-b-PVBTMA/PAA systems. Adapted with permission from ref [1081]. Copyright 2020, American Chemical Society. (B) The black arrow indicates that, for PEO-b-PVBTMA/PSS systems, aggregates fragment into smaller micellar entities in less than 3 ms. Adapted with permission from ref [88]. Copyright 2019, American Chemical Society. .
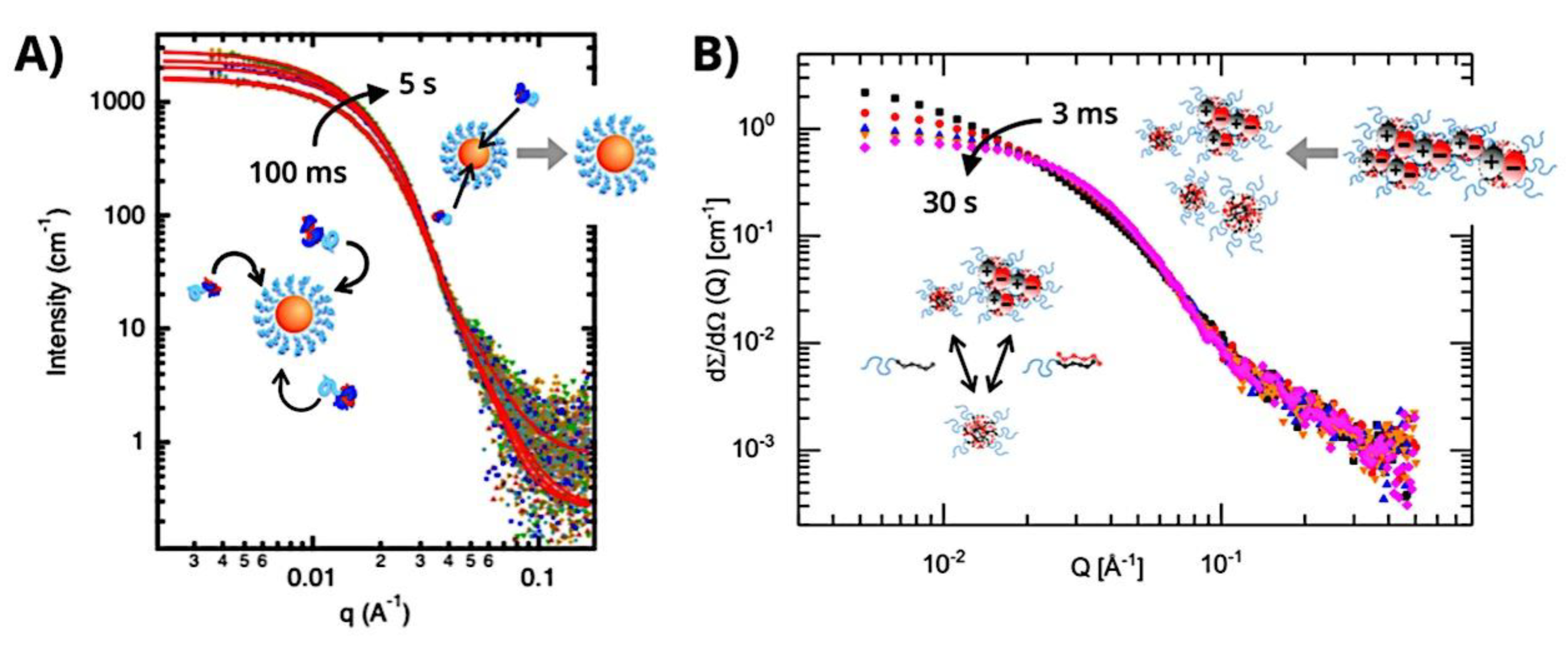
Figure 66.
Diagram showing the unimer exchange process. Adapted with permission from ref 96. Copyright 1999, American Chemical Society.
Figure 66.
Diagram showing the unimer exchange process. Adapted with permission from ref 96. Copyright 1999, American Chemical Society.
Figure 67.
Diagrammatic representation of micelle fission/fusion. Adapted with permission from ref [96]. Copyright 1999, American Chemical Society.
Figure 67.
Diagrammatic representation of micelle fission/fusion. Adapted with permission from ref [96]. Copyright 1999, American Chemical Society.
Figure 68.
During creation, chain exchange of C3Ms as a function of electrostatic interactions, nonelectrostatic interactions, and polyelectrolyte length is studied using Langevin dynamics simulations. (A) C3M size distribution histograms with three different nonelectrostatic attraction strength values between polyelectrolytes: εLJ = 0.05kBT (blue), εLJ = 0.15kBT (red), and εLJ = 0.25kBT (gray); snapshots of the simulated C3Ms with Nnegative = Npositive = 20 and Nnetural = 50 are displayed in the insets. (B) As nonelectrostatic attraction strengths increase, the number of micelle fission/fusion and chain expulsion/insertion events for C3Ms are compared as a function of the polyelectrolyte length ratio (Nnegative / Npositive). Adapted from ref [89]. Copyright 2019, American Chemical Society. This publication is licensed under CC-BY-NC-ND.
Figure 68.
During creation, chain exchange of C3Ms as a function of electrostatic interactions, nonelectrostatic interactions, and polyelectrolyte length is studied using Langevin dynamics simulations. (A) C3M size distribution histograms with three different nonelectrostatic attraction strength values between polyelectrolytes: εLJ = 0.05kBT (blue), εLJ = 0.15kBT (red), and εLJ = 0.25kBT (gray); snapshots of the simulated C3Ms with Nnegative = Npositive = 20 and Nnetural = 50 are displayed in the insets. (B) As nonelectrostatic attraction strengths increase, the number of micelle fission/fusion and chain expulsion/insertion events for C3Ms are compared as a function of the polyelectrolyte length ratio (Nnegative / Npositive). Adapted from ref [89]. Copyright 2019, American Chemical Society. This publication is licensed under CC-BY-NC-ND.
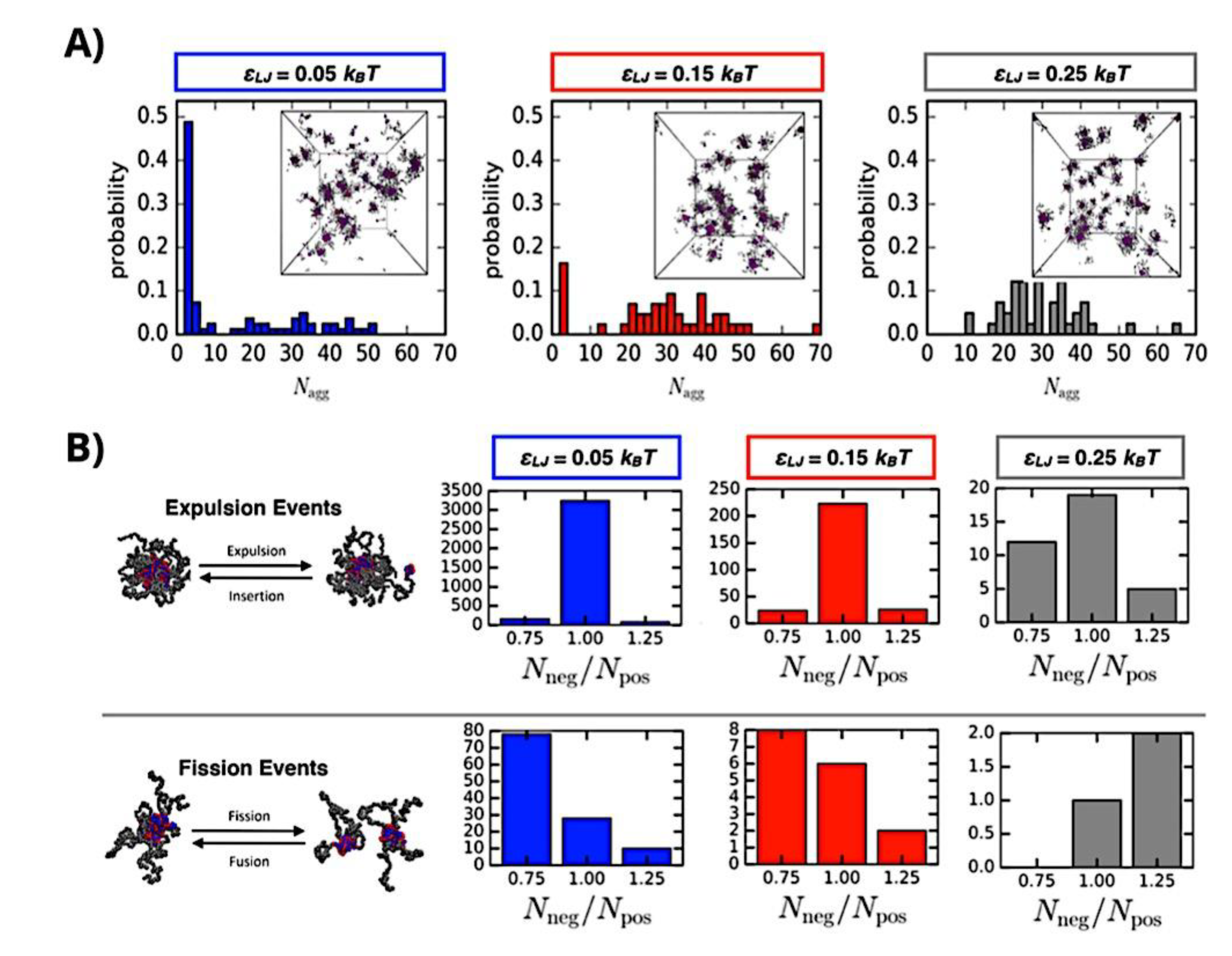
Figure 69.
(A) The time-resolved small-angle X-ray scattering experiments merged with a stopped-flow setup are schematically represented. (B) Forming polyelectrolyte complex micelles as a result of combining the polyanion PAA with the polycation PEO-b-PVBTMA. Adapted with permission from ref [1081]. Copyright 2020, American Chemical Society.
Figure 69.
(A) The time-resolved small-angle X-ray scattering experiments merged with a stopped-flow setup are schematically represented. (B) Forming polyelectrolyte complex micelles as a result of combining the polyanion PAA with the polycation PEO-b-PVBTMA. Adapted with permission from ref [1081]. Copyright 2020, American Chemical Society.
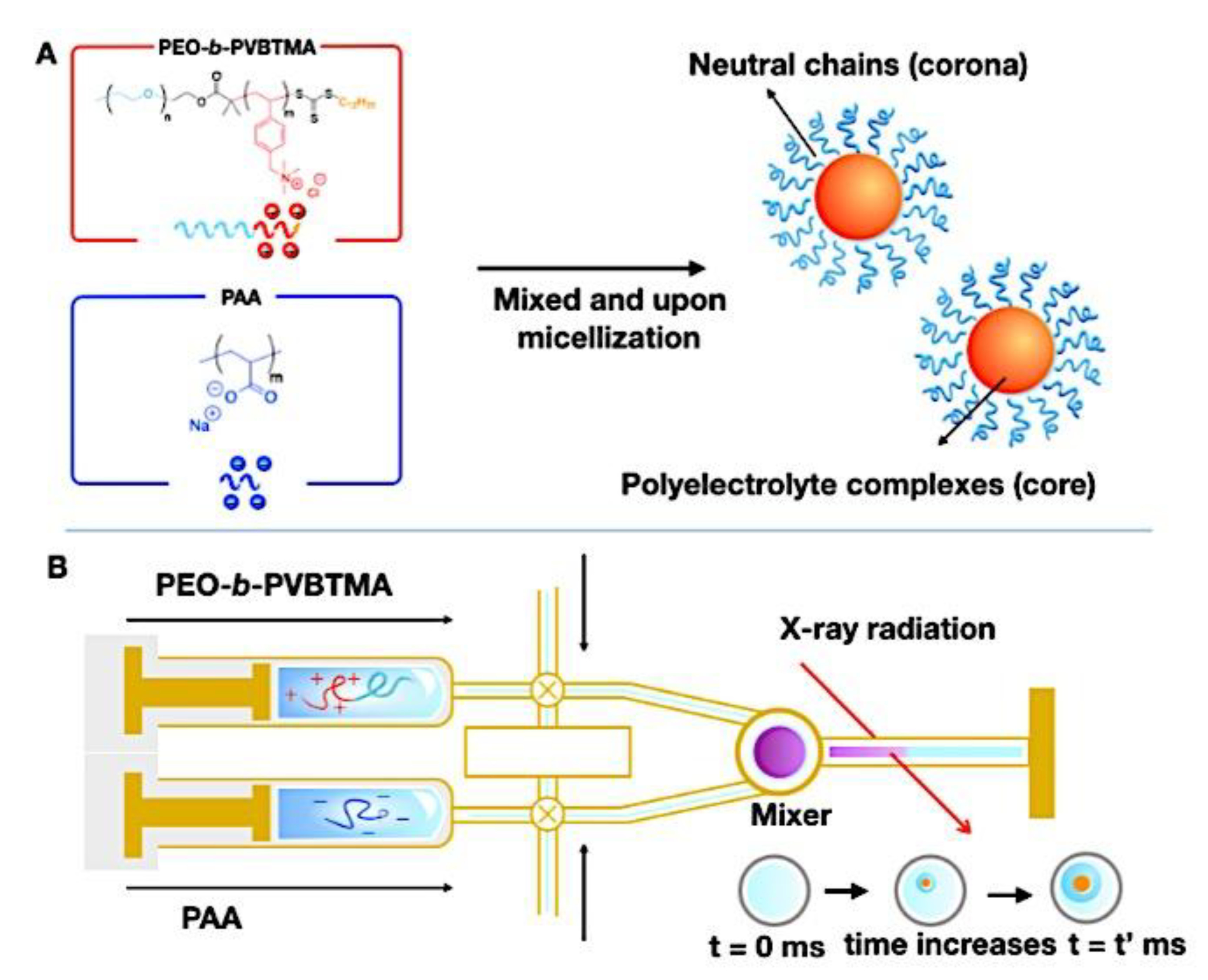
Figure 70.
Diagram illustrating the intricate development of micelles and coacervate droplets. (a) The structures of poly(L-lysine) (pLL), poly(ethyleneglycol)–poly(L-lysine) (PEG–pLL), and single-stranded DNA. The nucleobase (adenine, guanine, cytosine, or thymine) is indicated by the letter R in the ssDNA chemical structure. The degree of polymerization of ssDNA, pLL, the pLL block, and the PEG block is indicated by the subscripts a, b, b/, and c, respectively. (b) A schematic illustration of the polymer lengths required to generate complicated coacervate droplets and how these droplets develop. (c) A schematic overview of the lengths of polymers required to construct complex coacervate core micelles and their creation process. Adapted with ref [1177]. Copyright 2022, The Royal Society of Chemistry. This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
Figure 70.
Diagram illustrating the intricate development of micelles and coacervate droplets. (a) The structures of poly(L-lysine) (pLL), poly(ethyleneglycol)–poly(L-lysine) (PEG–pLL), and single-stranded DNA. The nucleobase (adenine, guanine, cytosine, or thymine) is indicated by the letter R in the ssDNA chemical structure. The degree of polymerization of ssDNA, pLL, the pLL block, and the PEG block is indicated by the subscripts a, b, b/, and c, respectively. (b) A schematic illustration of the polymer lengths required to generate complicated coacervate droplets and how these droplets develop. (c) A schematic overview of the lengths of polymers required to construct complex coacervate core micelles and their creation process. Adapted with ref [1177]. Copyright 2022, The Royal Society of Chemistry. This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
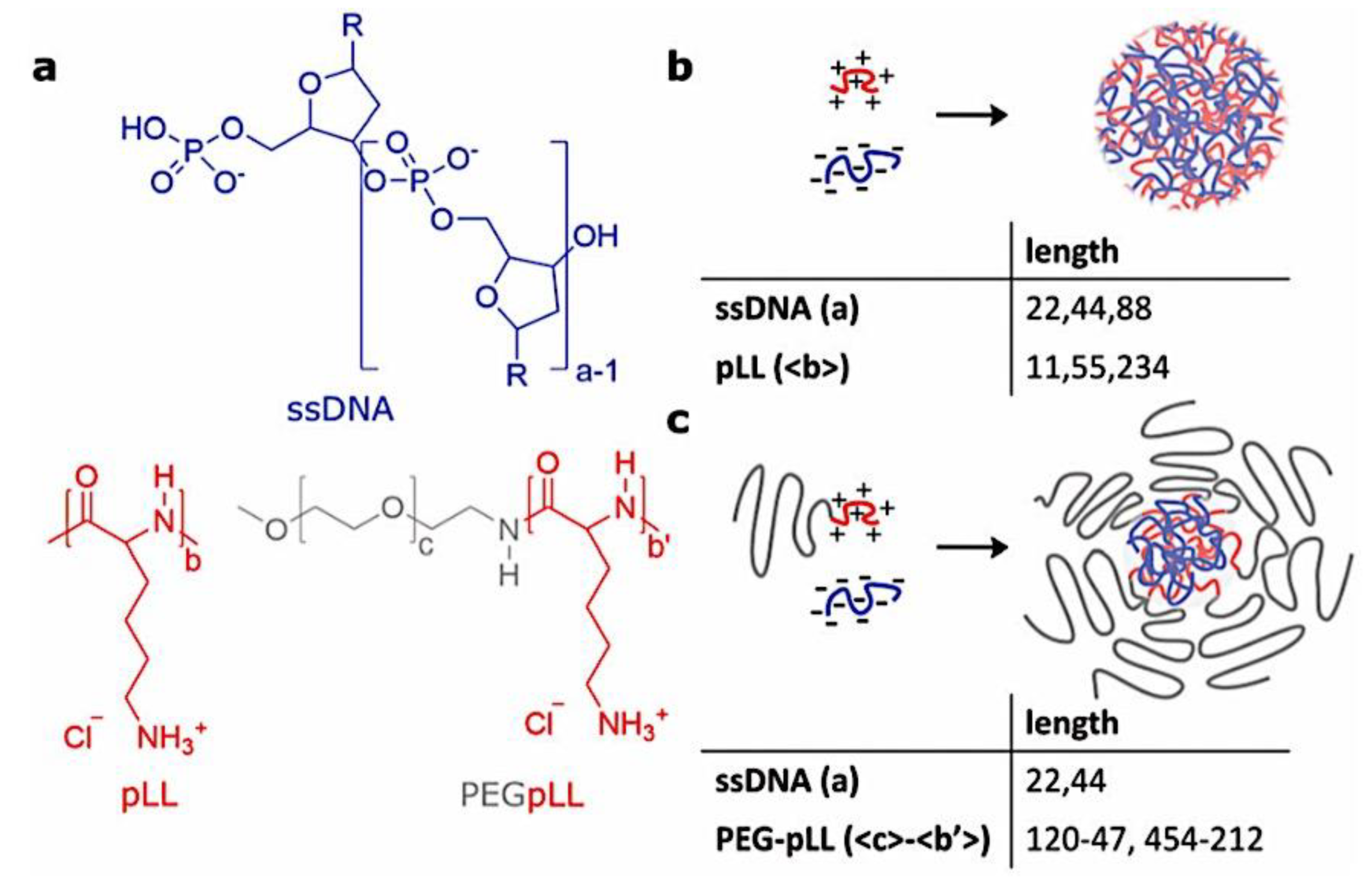
Figure 71.
A schematic representation of the C3M exchange mechanism (1) A distinct tiny polyelectrolyte complex inside the C3M core can only arise through relaxation processes. (2) Then, this distinct little complex can be released from the core. This is an active process; if the complex has a big net charge (2b), the expulsion probability is minimal; otherwise, it is large when the complex is neutrally charged (2a). Adapted from ref [1177]. Copyright 2022, The Royal Society of Chemistry. This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
Figure 71.
A schematic representation of the C3M exchange mechanism (1) A distinct tiny polyelectrolyte complex inside the C3M core can only arise through relaxation processes. (2) Then, this distinct little complex can be released from the core. This is an active process; if the complex has a big net charge (2b), the expulsion probability is minimal; otherwise, it is large when the complex is neutrally charged (2a). Adapted from ref [1177]. Copyright 2022, The Royal Society of Chemistry. This article is licensed under a Creative Commons Attribution 3.0 Unported Licence.
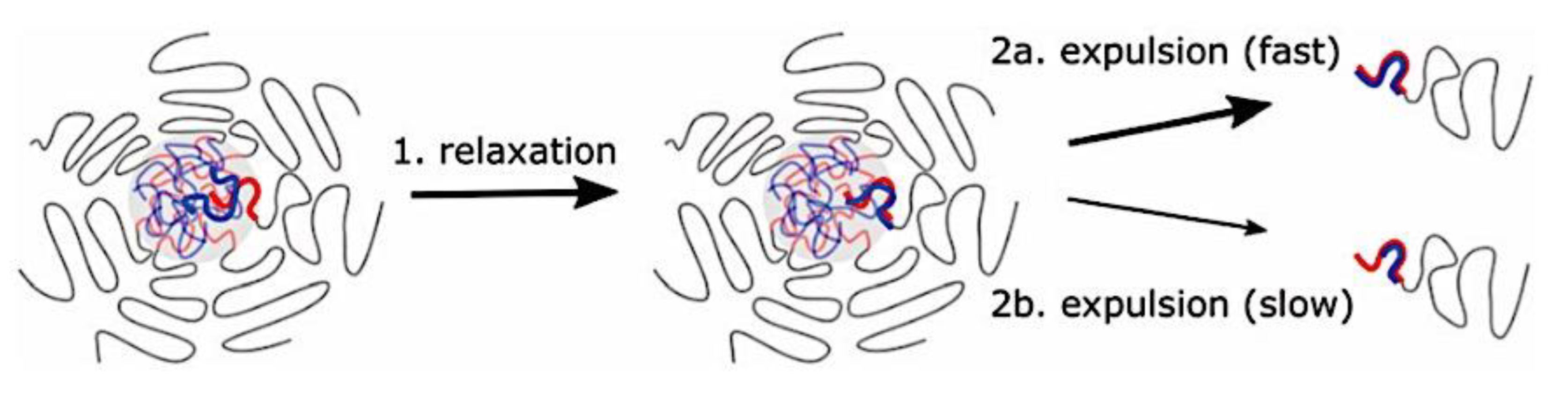
Figure 72.
(A) The wild-type structure of CotA. Cu atoms are shown as brown-colored spheres. A red line indicates the location of the single disulfide bridge found in the protein. Adapted from ref [1296]. Copyright 2003, The American Society for Biochemistry and Molecular Biology, Inc. Currently published by Elsevier Inc; originally published by American Society for Biochemistry and Molecular Biology. This is an open access article distributed under the terms of the Creative Commons CC-BY license. (B) The CotA laccase structure’s copper coordination. Adapted from ref [1297]. Copyright 2014, Christopher, Yao and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY).
Figure 72.
(A) The wild-type structure of CotA. Cu atoms are shown as brown-colored spheres. A red line indicates the location of the single disulfide bridge found in the protein. Adapted from ref [1296]. Copyright 2003, The American Society for Biochemistry and Molecular Biology, Inc. Currently published by Elsevier Inc; originally published by American Society for Biochemistry and Molecular Biology. This is an open access article distributed under the terms of the Creative Commons CC-BY license. (B) The CotA laccase structure’s copper coordination. Adapted from ref [1297]. Copyright 2014, Christopher, Yao and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY).
Figure 73.
As the length of the pEG increases, the hydrodynamic radius of the micelles decreases. A decrease in the quantity of micelles per micelleplex is also consistent with this. This work shows how to encapsulate rigid charged molecules like pDNA in a novel way. Adapted with permission from ref [1393]. Copyright 2018, American Chemical Society.
Figure 73.
As the length of the pEG increases, the hydrodynamic radius of the micelles decreases. A decrease in the quantity of micelles per micelleplex is also consistent with this. This work shows how to encapsulate rigid charged molecules like pDNA in a novel way. Adapted with permission from ref [1393]. Copyright 2018, American Chemical Society.
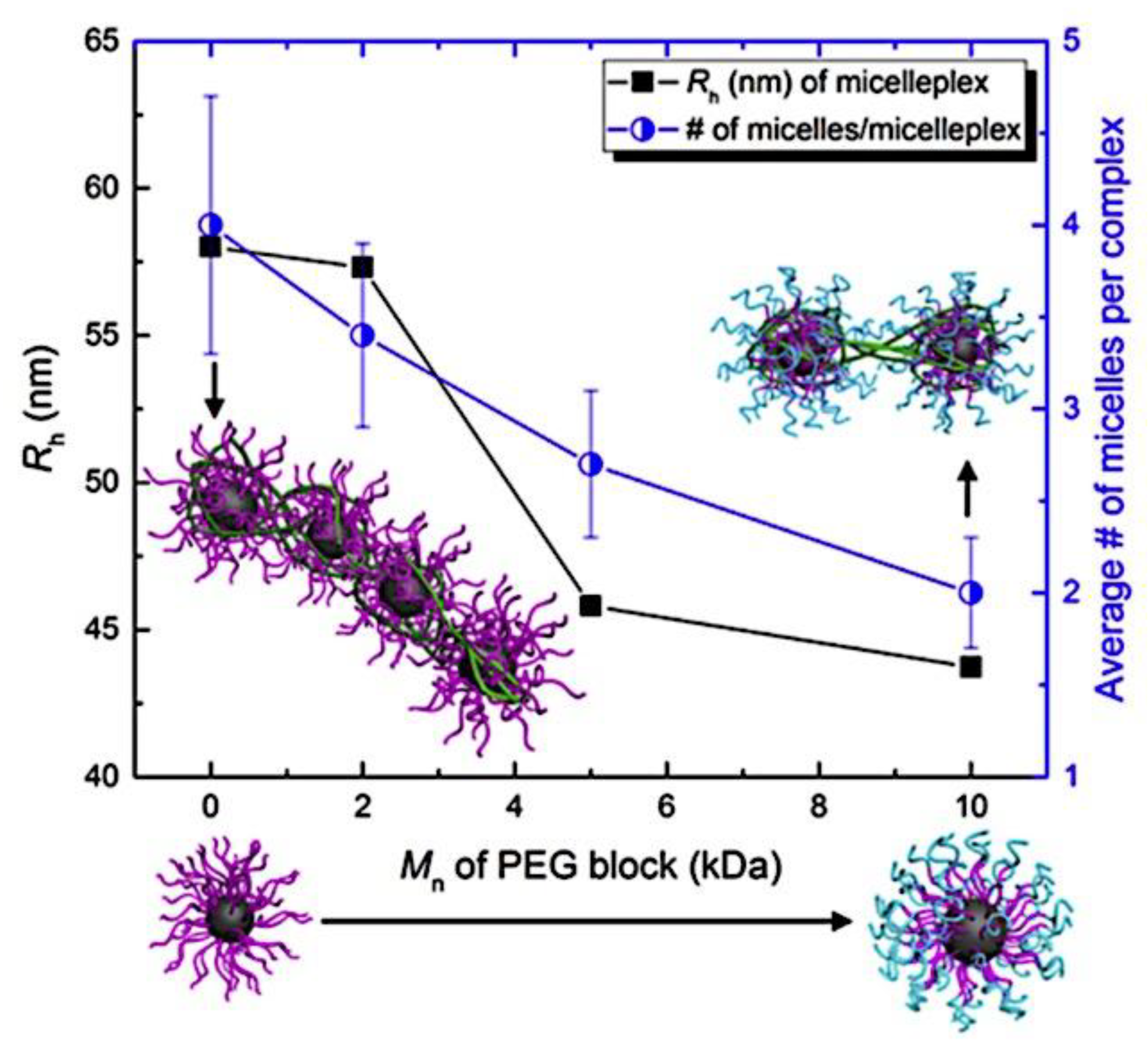
Figure 74.
The process of creating siRNA-loaded micelles involves joining negatively charged siRNA with positively charged PEI-g(20)-PEG-MAL block copolymers. Next, a stem cell antigen 1 antibody (Anti-sca 1 Fab/) is conjugated to target lung mesenchymal stem cells. The organs of a mouse administered with Fab/-conjugated Micelle are visualized using Alexa 647 fluorescence images. Adapted with permission from ref [1397]. Copyright 2021, Wiley-VCH GmbH.
Figure 74.
The process of creating siRNA-loaded micelles involves joining negatively charged siRNA with positively charged PEI-g(20)-PEG-MAL block copolymers. Next, a stem cell antigen 1 antibody (Anti-sca 1 Fab/) is conjugated to target lung mesenchymal stem cells. The organs of a mouse administered with Fab/-conjugated Micelle are visualized using Alexa 647 fluorescence images. Adapted with permission from ref [1397]. Copyright 2021, Wiley-VCH GmbH.
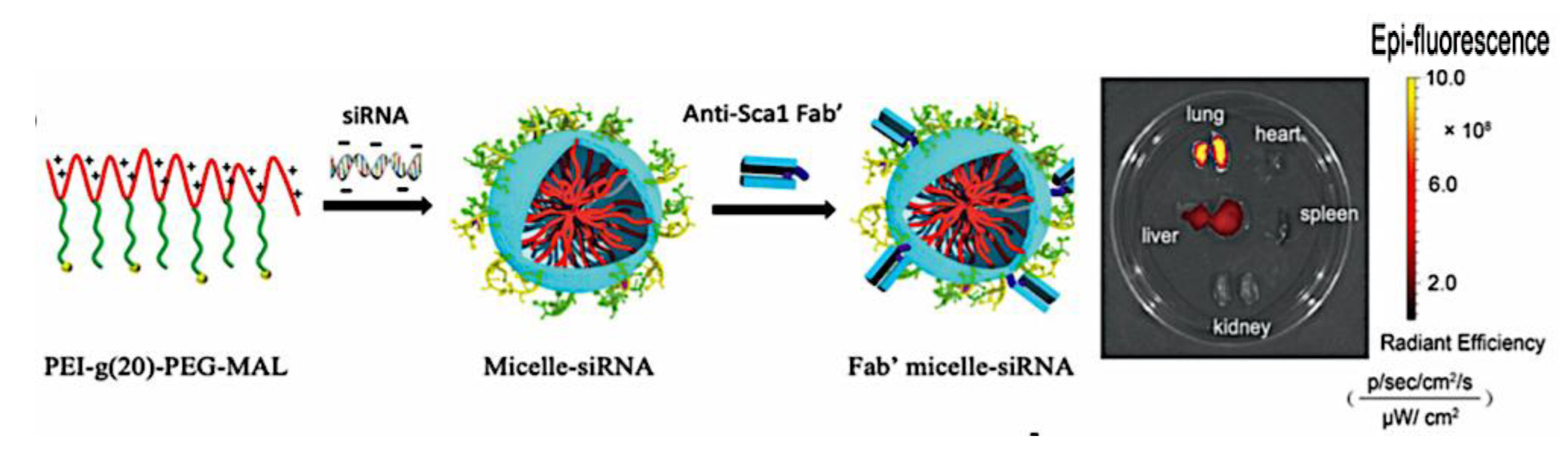
Figure 75.
(a) polyNIPAM-polyDEA block copolymers self-assembling into a polymersome featuring a “boarding gate” and a “release gate”; (b) DLS measurements of crosslinked polymersomes at varying temperatures; (c) in vivo transfection and expression of plasmid DNA distributed by polymersomes and controls (triplicate experiments). Adapted with permission from ref [1401]. Copyright 2018, The American Chemical Society.
Figure 75.
(a) polyNIPAM-polyDEA block copolymers self-assembling into a polymersome featuring a “boarding gate” and a “release gate”; (b) DLS measurements of crosslinked polymersomes at varying temperatures; (c) in vivo transfection and expression of plasmid DNA distributed by polymersomes and controls (triplicate experiments). Adapted with permission from ref [1401]. Copyright 2018, The American Chemical Society.
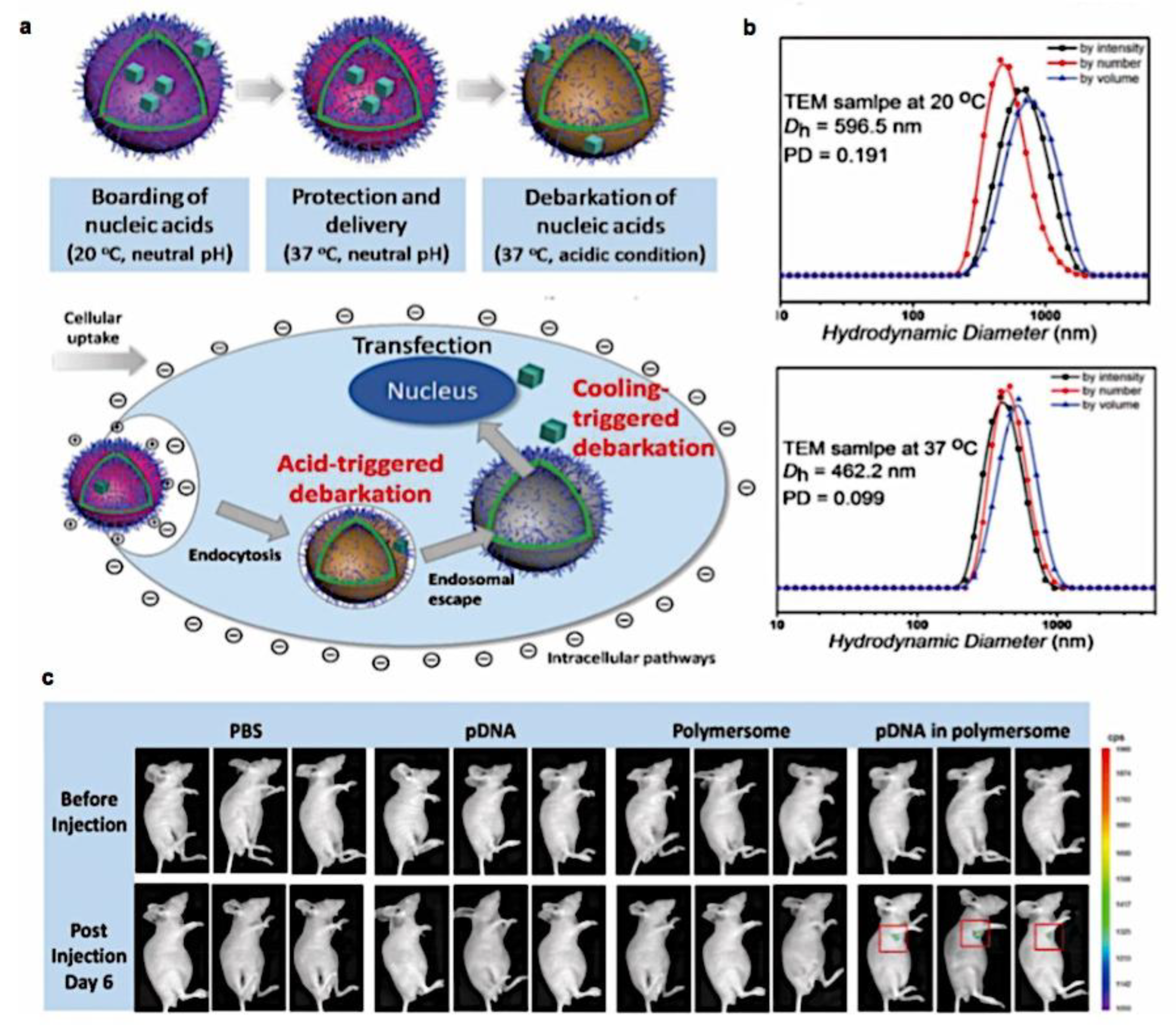
Figure 76.
A diagram illustrating the movement of PEC micelles coated with Glut-1 through the brain-blood barrier's endothelial cells (BBB). Adapted with permission from ref [1423]. Copyright 2020, American Chemical Society.
Figure 76.
A diagram illustrating the movement of PEC micelles coated with Glut-1 through the brain-blood barrier's endothelial cells (BBB). Adapted with permission from ref [1423]. Copyright 2020, American Chemical Society.
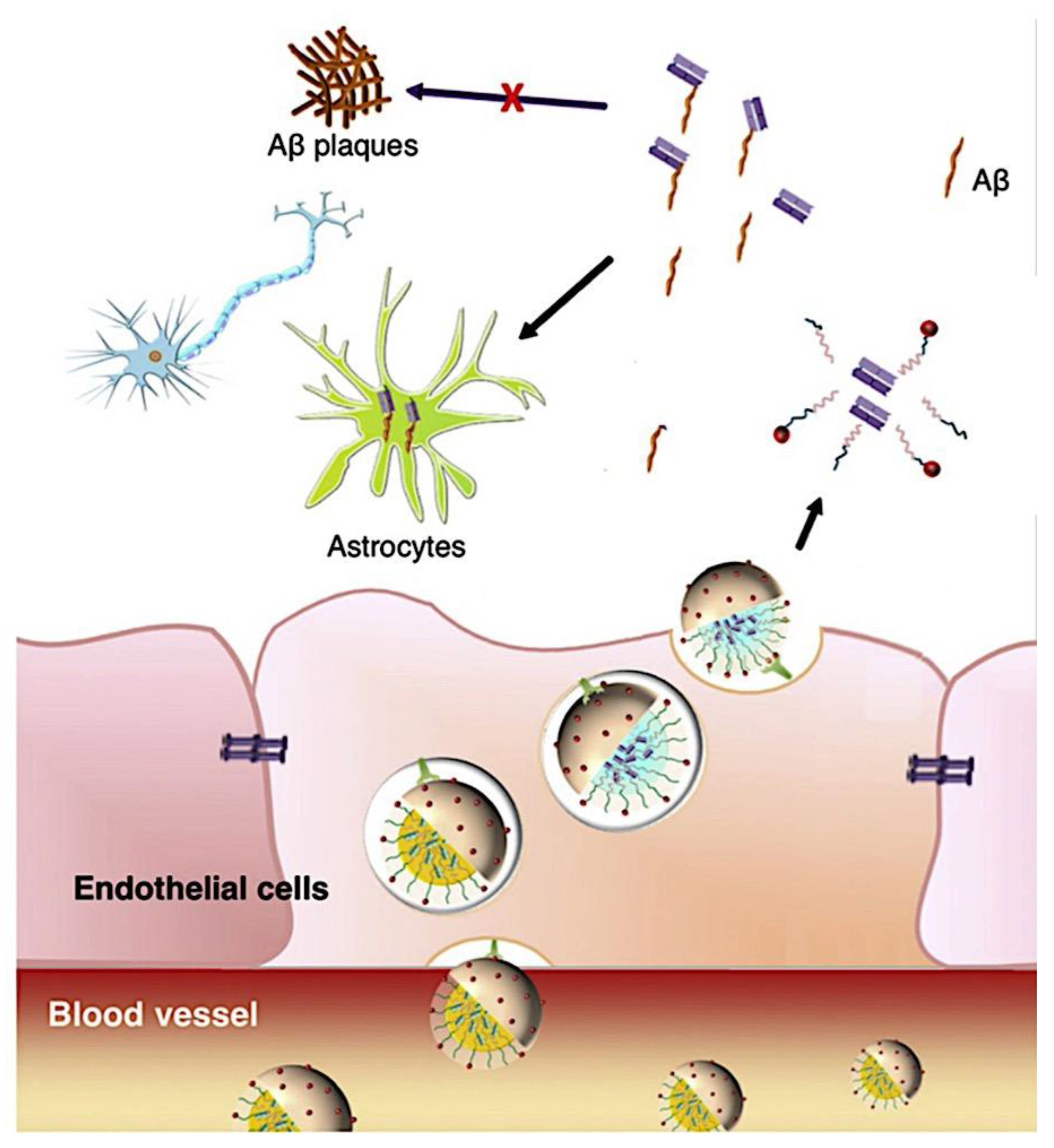
Figure 77.
Summary of micelle nanoparticles’ GBM-targeting mechanisms. (A) GBM tumor and normal brain parenchyma in the same section of the brain. (B) An enlargement of the glioma cells encircling the tumor vasculature. Tight connections between endothelial cells are disrupted in the vasculature here. Two primary methods exist for micelles to target tumor areas: (1) through the EPR effect, in which they passively diffuse past the compromised blood-brain barrier to glioma cells, or (2) through interactions with endothelial cells and transcytosis to the tumor parenchyma. Aminopeptidase N and αvβ3 integrin are two examples of receptors that are more particular to the tumor vasculature and glioma cells. (C) Normal brain parenchyma surrounds a close-up of the vasculature. Here, micelles can interact with the intact BBB to facilitate particle transcytosis. The Tf receptor is one type of receptor that mediates this pathway. Although these images show receptor-mediated endocytosis, these spots may also be the site of alternative endothelial cell uptake mechanisms, such as adsorptive-mediated endocytosis. Adapted from ref [1489]. Copyright 2013, The Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY).
Figure 77.
Summary of micelle nanoparticles’ GBM-targeting mechanisms. (A) GBM tumor and normal brain parenchyma in the same section of the brain. (B) An enlargement of the glioma cells encircling the tumor vasculature. Tight connections between endothelial cells are disrupted in the vasculature here. Two primary methods exist for micelles to target tumor areas: (1) through the EPR effect, in which they passively diffuse past the compromised blood-brain barrier to glioma cells, or (2) through interactions with endothelial cells and transcytosis to the tumor parenchyma. Aminopeptidase N and αvβ3 integrin are two examples of receptors that are more particular to the tumor vasculature and glioma cells. (C) Normal brain parenchyma surrounds a close-up of the vasculature. Here, micelles can interact with the intact BBB to facilitate particle transcytosis. The Tf receptor is one type of receptor that mediates this pathway. Although these images show receptor-mediated endocytosis, these spots may also be the site of alternative endothelial cell uptake mechanisms, such as adsorptive-mediated endocytosis. Adapted from ref [1489]. Copyright 2013, The Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY).
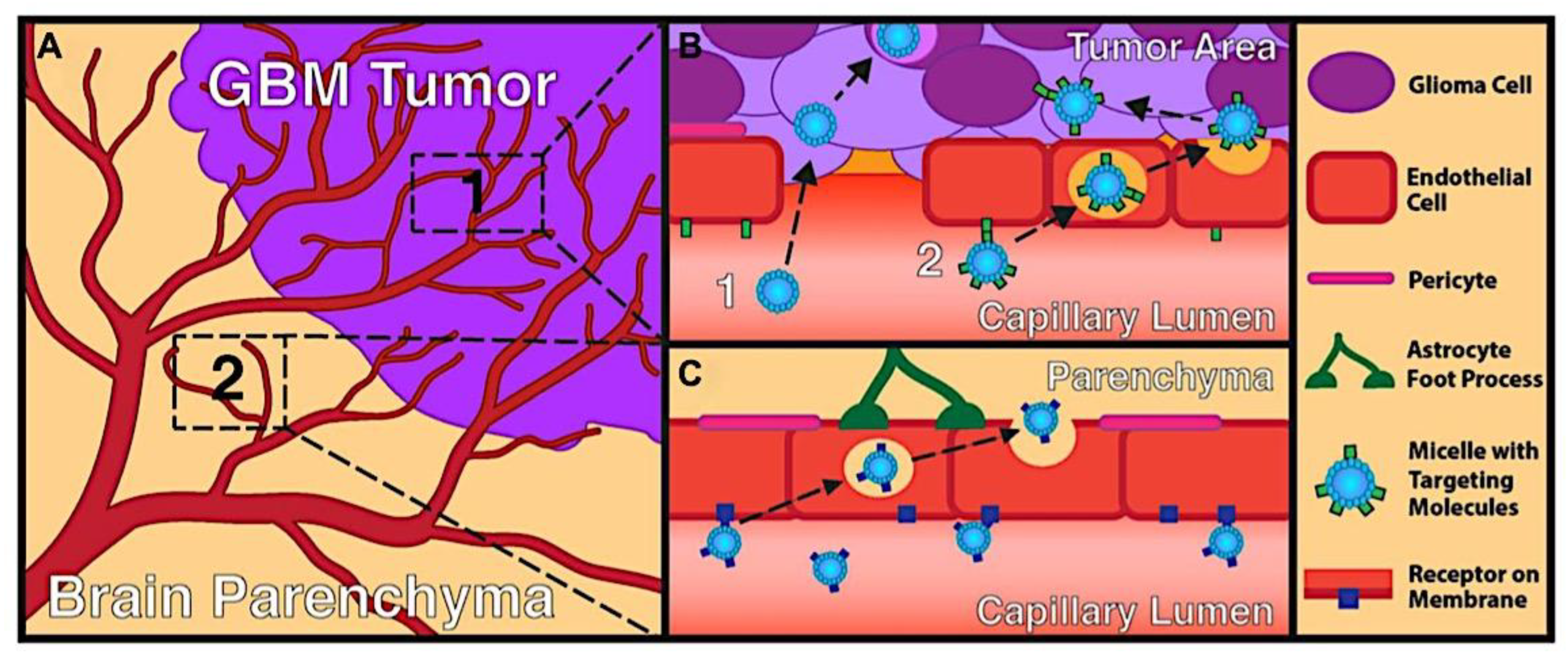
Figure 78.
Creation of bilayer vesicles (bottom) or core-shell micelles (top) from amphiphilic block copolymers. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V.
Figure 78.
Creation of bilayer vesicles (bottom) or core-shell micelles (top) from amphiphilic block copolymers. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V.
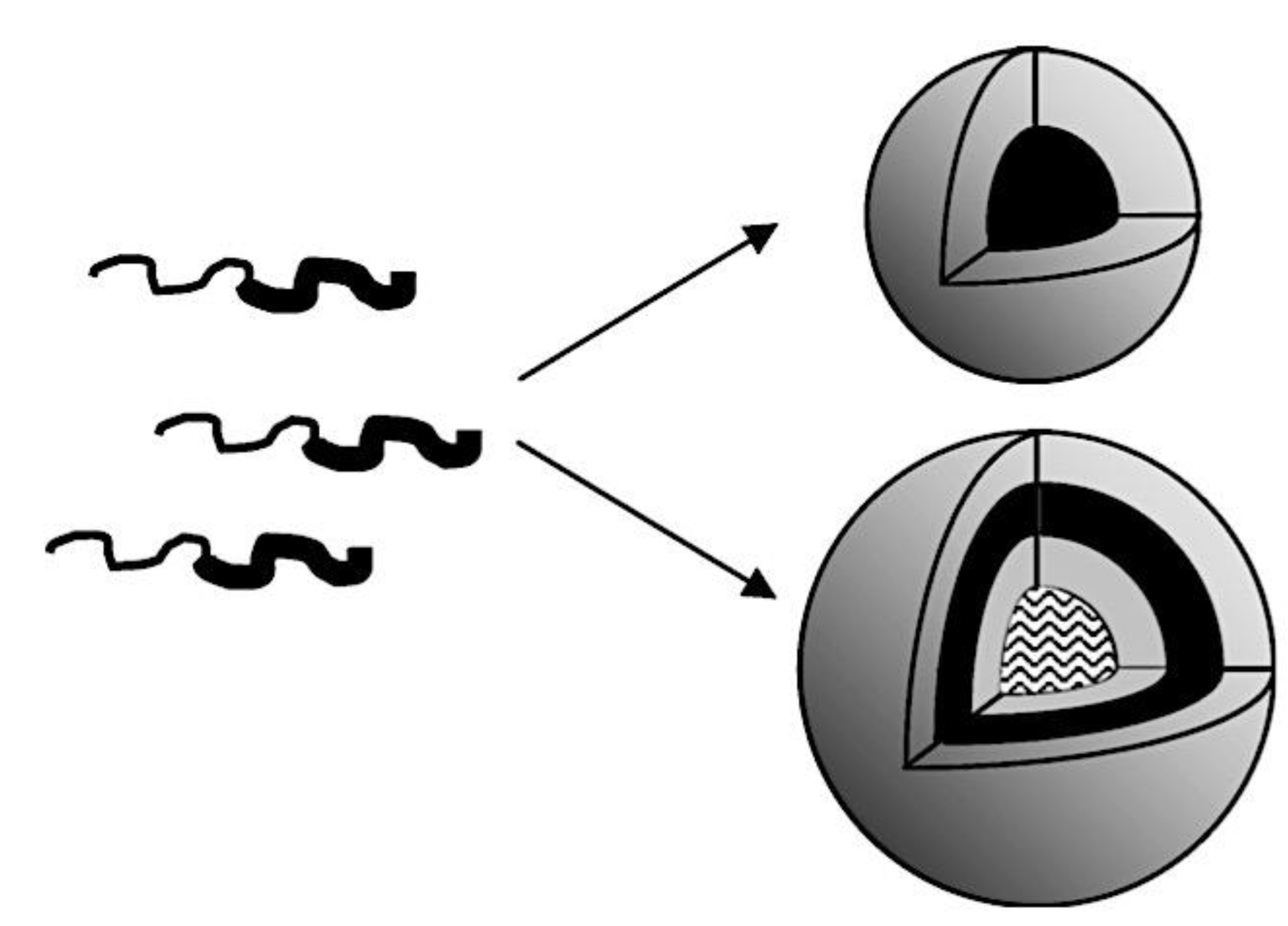
Figure 79.
An overview of the characteristics that promote drug release (right) and retention (left) in polymeric micelles. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V.
Figure 79.
An overview of the characteristics that promote drug release (right) and retention (left) in polymeric micelles. Adapted with permission from ref [623]. Copyright 2007, Elsevier B.V.
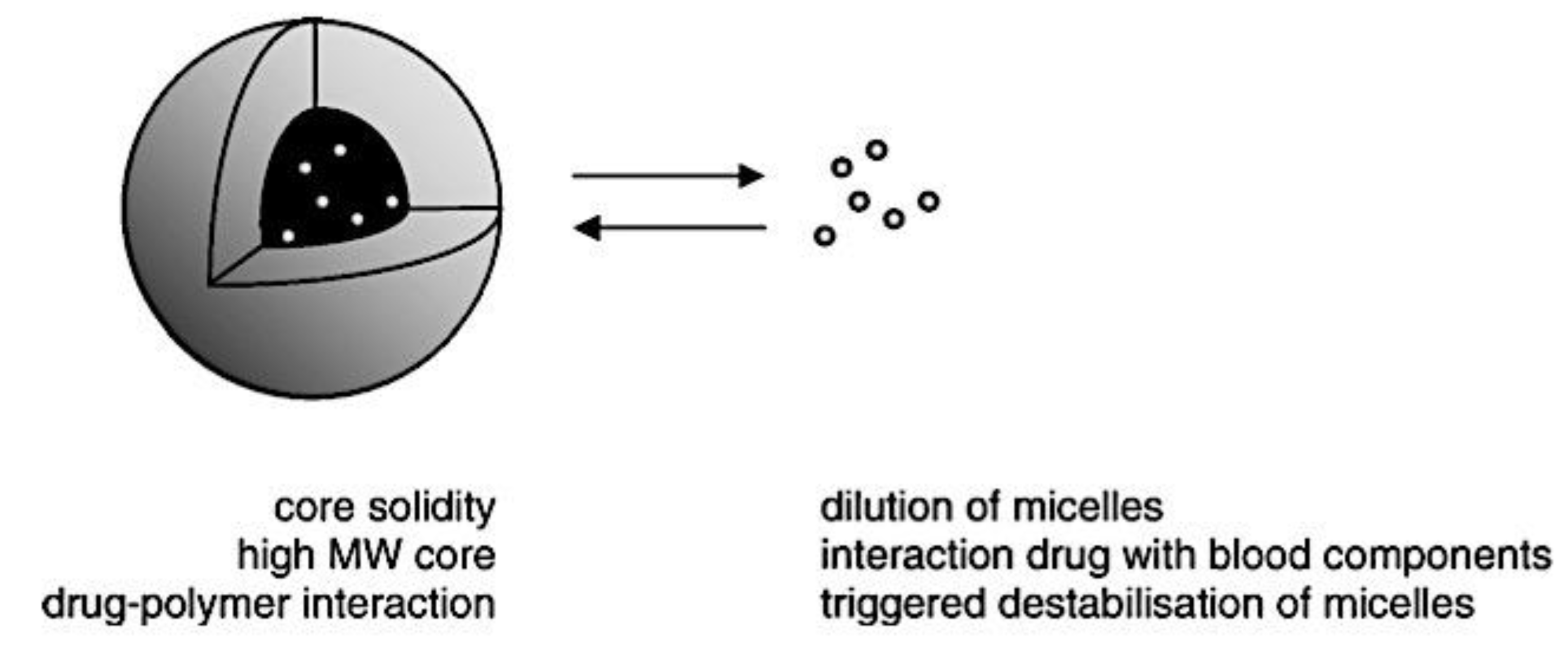
Figure 80.
Diagrams of the multifunctional cRGD-PLGA-SPIO@DOX nanoparticles for targeted tumor treatment and magnetic resonance imaging. To assess the therapeutic effect of the nanosystem dynamically from an anatomical and functional perspective, MRI was employed. Adapted with permission from ref [1549]. Copyright 2019. The Royal Society of Chemistry.
Figure 80.
Diagrams of the multifunctional cRGD-PLGA-SPIO@DOX nanoparticles for targeted tumor treatment and magnetic resonance imaging. To assess the therapeutic effect of the nanosystem dynamically from an anatomical and functional perspective, MRI was employed. Adapted with permission from ref [1549]. Copyright 2019. The Royal Society of Chemistry.
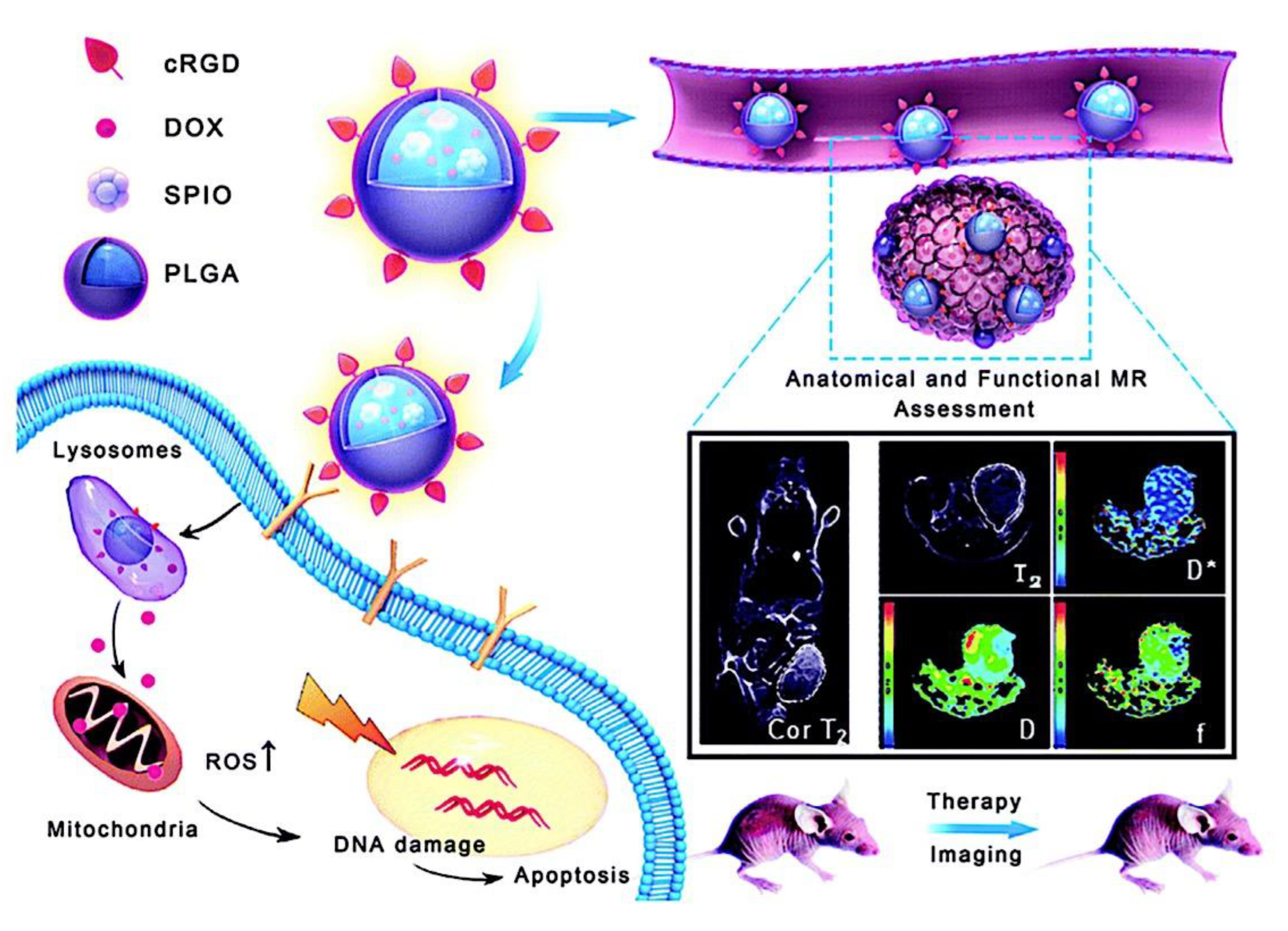
Figure 81.
Diagram illustrating the following: (A) the creation of polymersome nanoreactors for systemic administration-based cooperative cancer therapy; (B) tumor acidity-responsive membrane permeability at tumor sites can activate the nanoreactors’ cascade reactions; (C) the chemical structure of the pH-and ROS-responsive block copolymer polyprodrug, PEG-b-P(CPTKMA-co-PEMA); and (D) the cascade reactions that take place in the nanoreactors. Adapted with permission from ref [1551]. Copyright 2019, American Chemical Society.
Figure 81.
Diagram illustrating the following: (A) the creation of polymersome nanoreactors for systemic administration-based cooperative cancer therapy; (B) tumor acidity-responsive membrane permeability at tumor sites can activate the nanoreactors’ cascade reactions; (C) the chemical structure of the pH-and ROS-responsive block copolymer polyprodrug, PEG-b-P(CPTKMA-co-PEMA); and (D) the cascade reactions that take place in the nanoreactors. Adapted with permission from ref [1551]. Copyright 2019, American Chemical Society.
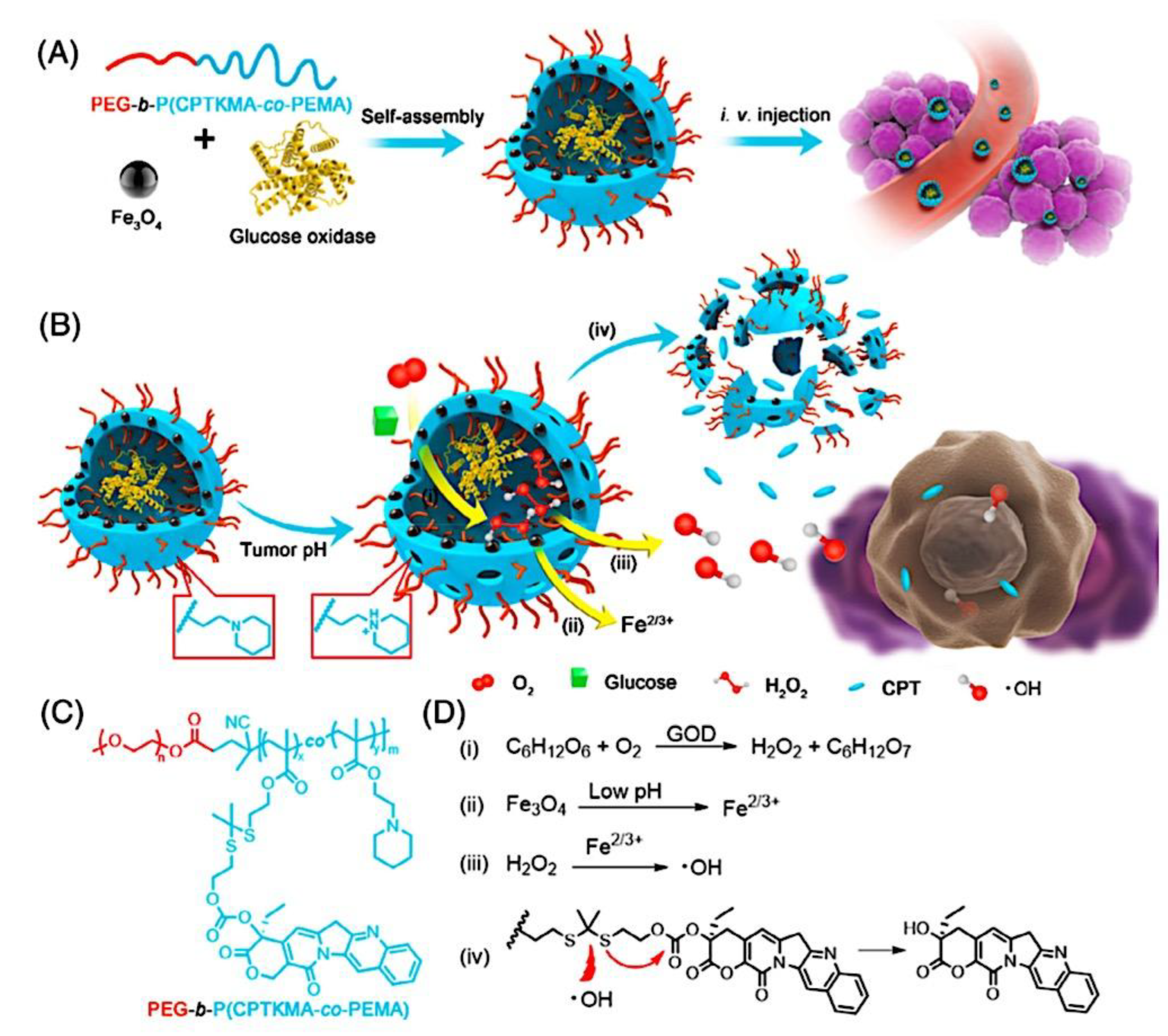
Figure 82.
(A) A diagram showing the bending motion under voltage and the SMS/[EMIM][NTf2] ion gel actuator. (B) Zwitterions and the PSS-b-PMB diblock copolymer’s chemical structures. (C) PSS-b-PMB-based actuator displacement and bending strain. Adapted from ref [1570]. Copyright 2016, The Authors. Published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Figure 82.
(A) A diagram showing the bending motion under voltage and the SMS/[EMIM][NTf2] ion gel actuator. (B) Zwitterions and the PSS-b-PMB diblock copolymer’s chemical structures. (C) PSS-b-PMB-based actuator displacement and bending strain. Adapted from ref [1570]. Copyright 2016, The Authors. Published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Figure 83.
The PS-b-PMMA-b-PS/lithium/tetraglyme bis(trifluoromethylsulfonyl)imide [Li(G4)][NTf2] ion gel’s chemical composition and appearance. Adapted with permission from ref [1582]. Copyright 2014, American Chemical Society.
Figure 83.
The PS-b-PMMA-b-PS/lithium/tetraglyme bis(trifluoromethylsulfonyl)imide [Li(G4)][NTf2] ion gel’s chemical composition and appearance. Adapted with permission from ref [1582]. Copyright 2014, American Chemical Society.
Figure 84.
(A) The SOS/[BMIM]PF6 ion-gel-gated EDLT’s transient response. Adapted with permission from ref 1585. Copyright 2007, American Chemical Society. (B) Capacitance frequency dependency for PS-b-PMMA-b-PS ion gels with various ILs. Adapted with permission from ref [1577]. Copyright 2008, WILEY-VCH Verlag GmbH & Co. KGaA,.
Figure 84.
(A) The SOS/[BMIM]PF6 ion-gel-gated EDLT’s transient response. Adapted with permission from ref 1585. Copyright 2007, American Chemical Society. (B) Capacitance frequency dependency for PS-b-PMMA-b-PS ion gels with various ILs. Adapted with permission from ref [1577]. Copyright 2008, WILEY-VCH Verlag GmbH & Co. KGaA,.
Figure 85.
A comparison between bevacizumab and CPT-11 therapy and the formulation of NK012 micelles. Eight days after tumor cell implantation, athymic mice were given cerebral injections of Luciferase-labeled U87MG and began receiving treatment. Three times every four days, an intravenous dose of 30 mg/kg of NK012 was administered. In addition to bevacizumab, which was administered intraperitoneally six times each four days at a dose of 5 mg/kg, CPT-11 was given at 67 or 40 mg/kg three times every four days. Adapted with permission from ref [1609]. Copyright 2010, American Association for Cancer Research.
Figure 85.
A comparison between bevacizumab and CPT-11 therapy and the formulation of NK012 micelles. Eight days after tumor cell implantation, athymic mice were given cerebral injections of Luciferase-labeled U87MG and began receiving treatment. Three times every four days, an intravenous dose of 30 mg/kg of NK012 was administered. In addition to bevacizumab, which was administered intraperitoneally six times each four days at a dose of 5 mg/kg, CPT-11 was given at 67 or 40 mg/kg three times every four days. Adapted with permission from ref [1609]. Copyright 2010, American Association for Cancer Research.
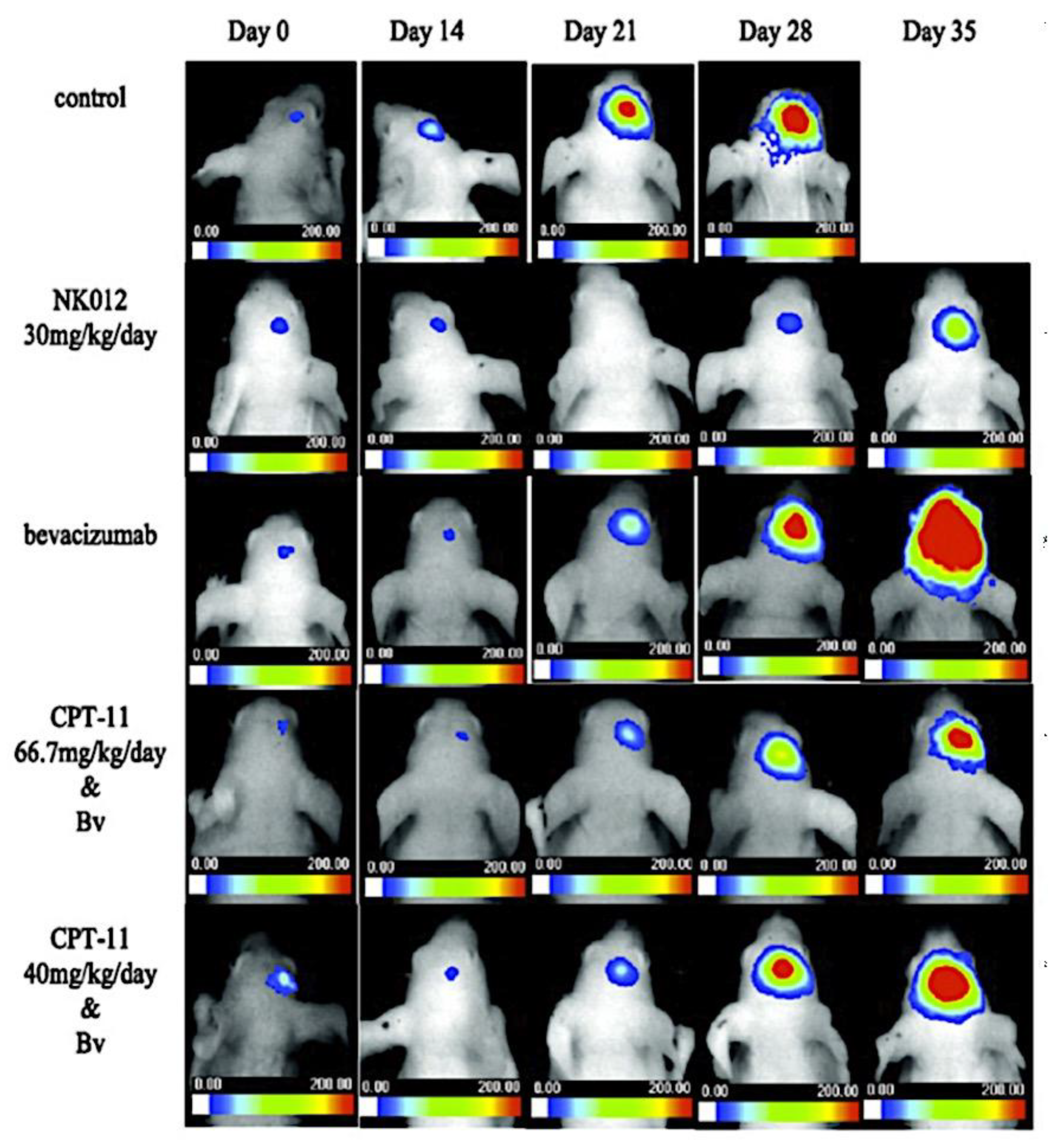

Table 1.
Different Hydrophilic Polymers that are Employed to Stabilize Micelles Sterically.
| Polymer | Abbreviation | Chemical Structure | References |
| Poly(vinyl alcohol) | PVA | 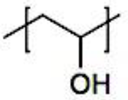 |
[508] |
| Poly(ethylene) glycol | PEG | 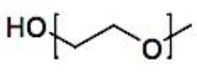 |
[428,468,469,470,471,472,473,474,475,476,477,478,487,498] |
| Poly(2-ethyl-2- oxazoline) | PEOx |  |
[508,509,510,511,512] |
| Poly(N- vinylpyrrolidone) | PVP |  |
[450,511,513,514,515,516,517] |
| Poly(acrylamide) | pAAm |  |
[426,482] |
| Poly(N-(2- hydroxypropyl) methacrylamide) | pHPMAm |  |
[516] |
| Dextran | Dex |  |
[518,519] |
Table 2.
Polymeric Micelles Employ Hydrophobic Polymers as Core-Forming Blocks.
| Polymer | Abbreviation | Chemical Structure | Reference |
| poly(methacryl- amide- oligolactates) | p(HEMAm- Lacn)or p(HPMAm- Lacn) | 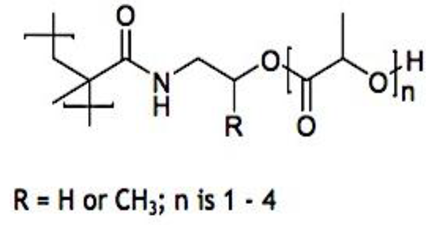 |
[476,477] |
| poly( -benzyl L- glutamate) or poly( -benzyl L- aspartate) | PBLG or PBLA | 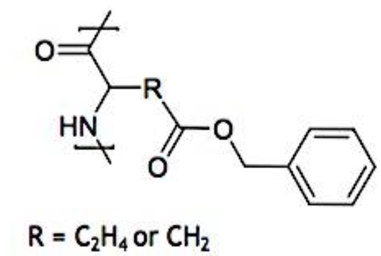 |
[539,551,552,553] |
| poly(N- isopropylacryl- amide) | pNIPAAm | 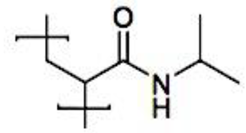 |
[478,554] |
| poly(propylene oxide) | PPO | 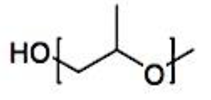 |
[428,439,555] |
| poly(lactic acid) | PLA |  |
[450,556] |
| poly( - caprolactone) | PCL |  |
[417,468,517] |
Table 3.
Encapsulated Biodegradable Components within Polymeric Micelles.
| Degradable group | Structure | Degradation products | Reference |
| Acetal |  |
 |
[656] |
| Ester | 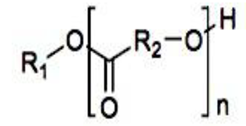 |
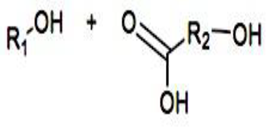 |
[476,650] |
| Hydrazone |  |
 |
[35,534,657] |
| Orthoester |  |
 |
[653] |
Table 4.
Examples and Characterisation of ABC Triblock Copolymers.
| Example Triblock | Mn [g mol-1] | ĐM [a] | Synthetic Route | Refs |
| PDMAEMA8-b-PLMA39-b-POEGMA8 | 15,600 [b] | 1.19 | Sequential RAFT | [941] |
| PEO120-b-PHEMA11-b-PtBA46 | 17600 [b] | 1.3 | Sequential ATRP | [942] |
| PS51-b-PB28-b-PtBMA21 | 104,000 [b] | 1.07 | Sequential AP | [943] |
| PEO42-b-PAGE15-b-PtBGE12 | 3400 [b] | 1.07 | Sequential AROP | [944] |
| PODFOx20-b-PEPOx20-b-PEtOx40 | 7900 [c] | 1.12 | Sequential CROP | [945] |
| PE20-b-PEO20-b-PCL10 | 2540 [b] | 1.23 | ROP | [946] |
| PEO44-b-PEtOx263-b-PCL175 | 46,600 [b] | 1.31 | CROP + ROP | [947] |
| PEO45-b-PCL103-b-PMOXA4 | 14,000 [b] | 1.14 | ROP + CROP | [948] |
| PEO45-b-PDMS40-b-PMOXA67 | 13,070 [b] | Not recorded | AROP + CROP | [949] |
| PEEP135-b-PCL50-b-PDMAEMA118 | 44,900 [b] | 1.31 | AROP + ROP + ATRP | [950] |
| PEO45-b-PMCL47-b-PDMAEMA31 | 12,890 [b] | 1.25 | ROP + ATRP | [951] |
| PEO30-b-PS90-b-PCL62 | 19,600 [b] | 1.09 | ATRP + ROP + Click | [952] |
[a] ĐM acquired via GPC. [b] Mn determined using 1H-NMR. [c] GPC-obtained Mn calculated using PS standards.
Table 5.
SCP-,+, and CCCM Concentrations at the CECC, PMC, and CEAC (Adapted with permission from ref [30]. Copyright 2004, American Chemical Society).
Table 5.
SCP-,+, and CCCM Concentrations at the CECC, PMC, and CEAC (Adapted with permission from ref [30]. Copyright 2004, American Chemical Society).
| Particle | CEAC | PMC | CECC |
| SCP- | maximum | 0 | 0 |
| CCCM | 0 | maximum | 0 |
| SCP+ | 0 | 0 | maximum |
Table 6.
Relaxation Rate and Exponential Micelle Dissociation Fits at Increasing Temperatures and Salt Concentrations (Adapted with permission from ref [90]. Copyright 2020, American Chemical Society).
Table 6.
Relaxation Rate and Exponential Micelle Dissociation Fits at Increasing Temperatures and Salt Concentrations (Adapted with permission from ref [90]. Copyright 2020, American Chemical Society).
| temp. (°C) | [salt] (mM) | τ (min) | β |
| 20 | 500 | 61.5 | 2.00 |
| 37 | 500 | 52.2 | 2.00 |
| 57 | 500 | 39.2 | 2.00 |
| 20 | 300 | 51.9 | 0.82 |
| 20 | 400 | 35.2 | 1.43 |
| 20 | 500 | 27.4 | 2.03 |
| 20 | 600 | 10.9 | 1.94 |
Table 7.
Typical Instances of BCP Self-Assembled Nanoformulations with a Focus on Biomedicine.
| Formulation | Stimulas/Ligand | Bioactive Compound | Block Copolymer | Intended Target/Function | Refs |
|---|---|---|---|---|---|
| Drug Delivery | |||||
| Micelle | Hypoxia | DOX | PEG-b-P(LG-g-MN) | Tumor chemoradiotherapy | [1331] |
| Micelle | pH | DAVBNH | Functionalized PEG-PAA | Glioblastoma | [1332] |
| Micelle | pH, light, and redox | Nile red | PAA-b-P (AzoMA-co- PEGMA) | Multi-responsiveness | [1333] |
| Micelle | Light, temperature, and pH | DOX | PDMAEMA-PMMA (with spiropyran chain end groups) | Multi-responsiveness | [1334] |
| Micelle | Hypoxia, temperature, and pH | DOX | P(MAA-co-NIMA)-b- PDMAEMA | Multi-responsiveness | [1335] |
| Spherical and filamentous micelles | pH | Verteporfin | PEG-PBAE-PEG | Breast and lung cancer | [1336] |
| Spherical and worm-like micelles | Valsartan | PLGA-PEG | Hypertension | [1337] | |
| Polymersome | Redox/ cNGQGEQc peptide | DOX | PEG-P(TMC-DTC) | Lung cancer | [1338] |
| Polymersome | pH and redox | Rhodamine B | PEG-P(DIPEMA-co-CBMA) | Drug delivery | [1339] |
| Polymersome | pH | DOX | PHPMA-b-PDPA | Lymphoma | [1340] |
| Gene Delivery | |||||
| PIC micelle | cRGD peptide | siRNA | PEG-PLL | Cervical cancer | [1341] |
| Mixed polyplex micelle | Temperature | pDNA | PEG- and PNIPAM-b-PAsp (DET) | Disc degeneration-associated diseases | [1342] |
| Core-shell- corona micelle | pH | siRNA and cisplatin | PEG-b-PAGA-b-PDPA | Breast cancer | [1343] |
| Mixed micelleplex | PD-L1 | siRNA and PTX | PCL-PEG and PCL-PEI | Melanoma | [1344] |
| Polyplex micelle | bundled mRNA | PEG-PLys | Improved mRNA delivery | [1345] | |
| PIC micelle | mRNA | PEG-PGBA or PEG-PLL | Improved mRNA delivery | [1346] | |
| Mixed polyplex micelles | pH and temperature | DNA | PVAm-b-PALysOH and PVAm-b-PNIPAM | DNA delivery | [1347] |
| Protein/Enzyme Delivery | |||||
| Vesicle | H2O2 and glucose | Glucose oxidase and insulin | mPEG-b-P(Ser-PBE) | Glucose regulation | [1348] |
| Mixed micelle | Insulin | PEO-b-PCL-b-PEO and PDMAEMA-b-PCL-b- PDMAEMA | Insulin delivery | [1349] | |
| PIC micelle | pH | SDF-1α | PEG-PUASM | Neuro-restoration | [1350] |
| PIC micelle | IgG (charge-converted | PEG-PAsp(DET) | Antibody delivery | [1351] | |
| PIC micelle | BDNF | PEG-PLE | Brain delivery and neuroprotection | [1048] | |
| PIC micelle | pH and redox | 3D6-Fab antibody | PEG-PLL | Brain delivery and amyloid b peptide inhibition | [1352] |
| PIC micelle | pH | Myoglobin | PEG-P(Lys-CDM) | Protein delivery | [1353] |
| Therapy and Diagnosis | |||||
| Micelle | pH | DOX | PEG-b-P(DPA/DBA-co- DTM) | Tumor chemotherapy and fluorescence imaging |
[1354] |
| Micelle | pH and hypoxia | DOX | PEG-PAA linked with metronidazole | Tumor chemoradiotherapy and fluorescence imaging |
[1355] |
| Micelle | pH | PTX and SPIONs | mPEG-b-PAsp(DIP)-co- PLLeu | Hepatocellular carcinoma treatment and MRI | [1356] |
| Micelle | pH and GSH | DOX and Au NPs | PCL-SS-PDMAEMA (SS: disulfide bond) | Cancer chemotherapy and CT imaging | [1357] |
| Micelle | Enzyme | DOX | mPEG-b-P(AA-g-TPE) | Tumor chemotherapy and fluorescence imaging | [1358] |
| Mixed micelleplex | SN-38, USPIO and VEGF siRNA | PDMA-b-PCL | Colorectal cancer gene silencing, chemotherapy and MRI | [1359] | |
| Vesicle | Folic acid | Gd and DOX | FA/DTPA-PGA-PCL | Cancer chemotherapy and MRI | [1360] |
| Polymersome | Temperature and pH | siRNA and pDNA | PEO-b-P(NIPAM-stat-CMA-stat-DEA) | Gene delivery and fluorescence imaging | [1361] |
Table 8.
Enhancing Micelle Selectivity Towards Brain Cancers by the use of Targets and Targeting Moieties.
Table 8.
Enhancing Micelle Selectivity Towards Brain Cancers by the use of Targets and Targeting Moieties.
| Target | Target location | Targeting molecule | Examples of incorporation onto micelles |
|---|---|---|---|
| αv β3 integrin | Tumor vasculature[1438] Glioma cells[1438] | RGD peptide[1439] | [1426,1429,1430,1431,1440,1441,1442,1443,1444,1445] |
| Fibrin deposits | Tumor vasculature [1446] Tumor stroma[1446,1447] | CREKA Peptide[1446] | [1448,1449] |
| Aminopeptidase N | Tumor vasculature[1450] | NGR peptide [1450,1451] | [1452] |
| Transferrin receptor | CNS vasculature[1453] | Transferrin[1453] Lactoferrin[1454] Aptamer[1455] | [1445,1454,1455,1456,1457] |
| nAchR | CNS vasculature[1458,1459] | Candoxin-derived peptide[1460] | [1431,1460] |
| EGFR | Glioma cells[1461] | Anti-EGFR Antibody[1462] EGa1[1463] | [1462,1464] |
| LRP1 | Glioma cells,[1465,1466] Neurons[1467] | Angiopep2[1468] | [1469] |
| Unknown | Glioma cells[1470] | GMT8 aptamer[1432,1470] | [1432] |
Table 9.
Applications and Operation of BCPs/ILs Systems. Adapted with permission from ref [1579]. Copyright 2017, American Chemical Society.
Table 9.
Applications and Operation of BCPs/ILs Systems. Adapted with permission from ref [1579]. Copyright 2017, American Chemical Society.
| Representative BCPs | Representative IL Cations | Applications | Working principle |
|---|---|---|---|
| PEO-based polymer (PS-PEO, PEO-PMMA) | AILs with cations and anions of alkyl pyrrolidinium, alkyl imidazolium, and alkyl sulfonium can differ. | Lithium batteries | By acting as a plasticizer, ILs can quicken the relaxation of polymer chains, which lowers the polymers' glass transition temperature (Tg). |
| PVDF-PHFP | |||
| NafionTM | AILs are made up of short alkyl chains of heterocyclic diazonium or alkyl imidazolium. Different cations and anions exist. | Fuel cell | One easy way to achieve high conductivity at high temperatures and without water is to incorporate nonvolatile, highly conductive ILs into polymer matrices. |
| PBI | |||
| PVDF-HFP | |||
| PMMA (and PMMA based copolymers | |||
| P2VP (and P2VP based copolymers) | |||
| PSS (and PSS based copolymers) | |||
| PVDF-PHFP | Electroactive Actuators | By lowering Young's modulus, promoting ion transport, and strengthening electrochemical stability, adding IL to the actuator's ionic polymer layer can enhance performance. | |
| NafionTM | |||
| Bi or triblock ABCs that can form micelles or vesicles in ILs | PILs as well as AILs. | Nanoreactor | In ILs, BCPs have the ability to self-assemble into vesicles or micelles, which can be employed as nanodelivery vehicles. |
| Triblock copolymers (PS-PEO-PS,PPO-PEO-PPO, PEO-PPO-PEO) | AILs containing cations and anions of alkyl imidazolium can differ. EAN is the PIL reported. | Wearable electronics | Wearable electronics applications can benefit from the ultra-stretchability and high ion conductivity of iono-elastomers made of self-assembled BCPs in ILs. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



