Submitted:
10 October 2025
Posted:
13 October 2025
You are already at the latest version
Abstract
The etiology of inflammatory bowel diseases (IBD) is not precisely defined, however they display environmental factors, genetic predisposition, gut microbiota involvement, and abnormal immunity.Angiogenesis seems to be an integral part of IBD. Impaired intestinal barrier probably express an initiating or early feature in the disease. A disruption of epithelial barrier leads to penetration of microtiota and other antigens into the mucosa leading to an enhanced immune response, whereas the vascular barrier damage is related to endothelium activation and pathological angiogenesis which promote inflammation. Angiogenesis is a very complex phenomenon which including endothelial and immune cells, growth factors, cytokines, adhesion molecules, intestinal microbiota, and signal transduction. It seems that intestinal microvasculature hemostasis turns to prothrombotic state, and microthrombi formation enhance ischemia. The angiogenic process in IBD is in part regulated by intestinal microbiota. Antiangiogenic therapy is a novel and significant approach for IBD treatment. Biologic anti-inflammatory therapy of IBD simultaneously attenuates angiogenesis to a similar degree. However expression of VEGF and other grow factors may have dual and opposing effect probably related to stage of the disease. Thus anti- angiogenic treatment in IBD patients is still controversial and clinical trials should be performed using anti-angiogenic drugs.
Keywords:
inflammatory bowel disease
; IBD
; angiogenesis
; VEGF
; intestinal barrier
; microbiota
; coagulation
; bradykinin
1. Introduction
Crohn’s disease and ulcerative colitis (UC) are two entities of inflammatory bowel disease (IBD) with chronic local and systemic inflammatory changes and spontaneous relapsing course. The etiology and pathogenesis of IBD are still not fully known, but they show genetic and environmental factors and display immunologic alterations caused in part by intestinal microbiome[1].
IBD is classically considered as a disease of Western countries, but over the past few decades the incidence of the disease rapidly increased in newly industrialized Word regions. UC primarily impacts the large bowel, whereas Crohn’s disease may affect any part of the gastrointestinal tract, from the oral cavity to the rectum. About 20-30% of IBD cases are associated with extraintestinal complications [1].
Intestinal barrier is created by epithelial and vascular layers to potentially harmful components. When the intestinal barrier is disrupted, bacterial and other antigens may enter the usually sterile submucosa and change immunological response, thus may play a role in the pathogenesis of IBD. The normal immunologic reactions suppress inflammation, however in genetically susceptible subjects inflammatory response is altered and amplified with defective role and regulation of innate immune cells, faulty epithelial barrier function and pathological angiogenesis. Extensive tissue injury is characterized by expression of adhesive molecules in the vessel wall with inflammatory cell infiltration, release of cytokines and other inflammatory mediators. It also may perpetuate of the commensal microbiota composition contribute to dysbiosis [2].
In the last years many reports have presented a role of intestinal microbiota in intestinal homeostasis and intestinal barrier dysfunction in IBD. In light of the recent evidence, this review is mainly focused on a proangiogenic activators, contribution of microbiota to activation of intestinal microvasculature, relation between dysregulation of intestinal barriel and pathological angiogenesis, and summarizes the effectiveness of anti- angiogenic therapy in IBD.
2. Angiogenesis
Angiogenesis, the process of new vessel formation, physiologically takes place during embryonic development and menstrual cycle. However in pathology formation of new vessels may persists which can induce or augment several pathological conditions as cancer and chronic inflammation. Angiogenesis consist of multiple steps including vascular endothelial cells (ECs) stimulation and proliferation, maturation and lumen formation, degradation of basal membrane and extracellular matrix (ECM) and remodeling [3]. Pathological angiogenesis is component of inflammation and is perpetuated by the immune response. Chronic inflammation and angiogenesis show a significant role in the pathogenesis of various chronic diseases, such as rheumatoid arthritis, psoriasis, and metabolic syndrome [4]. Evidences have accumulated that pathological angiogenesis is pivotal in IBD (Table 1) [5].
2.1. Activators of Angiogenesis
There are multiple components in angiogenesis cascade, however vascular endothelial growth factor (VEGF) is the main angiogenic factor. Family of VEGF bind to three specific receptors (VEGFR1, VEGFR2, and VEGFR3). VEGF-A acts via four main isoforms; VEGF165 is the major activator of pathological angiogenesis and acts by binding to VEGFR2 [6].VEGF mediates angiogenesis by stimulating ECs to proliferation and migration, and in addition it restrains apoptosis of ECs. [7]. VEGF may also increases vascular permeability, and activates metalloproteinases (MPs) which participate in degradation of extracellular matrix (ECM) [8].
Angiogenesis during chronic inflammation is considered to be induced primarily by hypoxia [9,10] with hypoxia-inducible factor-1 and -2 (HIF), which transcriptionally activate the expression of VEGF-A [11]... VEGF activates nuclear factor κB (NFκB) and in turn stimulates to release pro-inflammatory cytokines such as IL-1, TNFα, growth factors and chemokines by leukocytes and ECs [12,13,14,15].
Growth factors have been implicated in activation and preservation of angiogenesis.
Among the growth factors fibroblast growth factor (FGF), transforming growth factor - β (TGF-β) and platelet-derived growth factor (PDGF) play especially important role. PDGF is released in reaction to hypoxia, cytokines, other growth factors and thrombin. It mediates enrollment of vascular muscle cells to the angiogenic milieu [16]. FGF supports angiogenesis via ECs proliferation, differentiation and migration, whereas (TGF-β) modulates survival and differentiation of ECs and regulates vascular homeostasis [17,18]. In addition, hepatocyte growth factor (HGF) mediates proliferation and differentiation of ECs [19], and placental growth factor (PGF) is thought to be a sensitive promotor of pathological angiogenesis [20].
Besides the classic angiogenic factors the angiopoietin/ tyrosine-kinase receptor (Ang/Tie) system plays a significant role in the late phase of angiogenesis via maturation and stabilization of blood vessels [21,22]. Angiopoietins may regulate angiogenesis via Tie-2 receptor. Activated ECs produce proangiogenic angiopoietin-2, which is not present in normal vessels, Angiopoietin-1 acts as a regulator of blood vessel maturation and has anti-inflammatory properties, whereas Angiopoetin-2 facilitates ECs activation in response to VEGF-A and other classic growth factors. It promotes angiogenesis by allowing ECs to be more sensitive to VEGF-An activated cells proliferation [23,24]. In fact the coordination of Ang-Tie-2 signaling and NFκB may provide to the vicious circle of inflammation and angiogenesis [25,26].
2.2. Antiangiogenic Factors
Thrombospondins (TSPs), calcium binding extracellular glycoproteins are well-known antiangiogenic factors. They inhibit angiogenesis due to stimulation ECs apoptosis and regulate inflammation [27,28]. TSP-1, the major antiangiogenic molecule is shown to be upregulated by HIF-1, and enhances the phagocytosis by macrophages and neutrophils in hypoxia [29].Other factors inhibiting angiogenesis are angiostatin and endostatin, which are 20kDa and 50 kDa components cleaved from plasminogen and collagen XVIII, respectively. These molecules may induce ECs apoptosis, and may inhibit proliferation and migration of ECs.
3. Intestinal Barrier
Epithelial cells, enterocytes and goblet cells which produced mucus, and the Paneth cells built the first layer that isolates the microorganisms in the gut milieu [30]. This intestinal epithelial barrier precludes the penetration of microbiota or microbial products into the tissue. Directly below the epithelial barrier the vascular barrier was identified which is consisted of ECs with smooth basal membrane and regular ECM. This is the second layer of preservation, which prevents microbial penetration into vessels [31,32]. At present, there is agreement that a disruption of intestinal barrier concomitant with bacteria dissemination probably determines an initiating or early feature of disease [33,34].
Mediators of inflammation , therein cytokines such as IFN-γ, IL-6 and TNF-α were found to increase intestinal permeability in experimental colitis models and IBD [35].On the other hand , recent reports in IBD patients indicated that an increase of epithelial permeability forego exacerbation of bowel inflammation suggesting a causal significance of epithelial barrier disruption in the intestinal inflammation [35]. Cytokine cascade also may activate ECs with induction of cell adhesion molecules (CAMs) such as E-selectin, intercellular adhesion molecule (ICAM)-1 or vascular cell adhesion molecule (VCAM)-1 and the synthesis of chemokines leading to leukocyte infiltration and mucosal damage [36].
VEGF which is synthesized by intestinal epithelium may provokes loosening of tight junctions (TJs) and adherents junctions (AJs) between enterocytes, and impair the intestinal barrier which cause the translocation of intestinal bacteria from the intestinal lumen leading to an enhanced immune response in genetically predisposed host [37]. Permeability route in intestinal microvascular vessels is controlled by TJs and AJs similarly as in epithelial barrier [38]. AJs are formed by vascular endothelial VC - cadherin and β-catenin [39], whereas TJs in intestinal endothelium are composed mainly of junctional adhesion molecule (JAM)-A, ocludin, and cingulin [31]. In addition claudin family molecules; claudin-3, -5, and -12 and enteric glial cells which are expressed in intestinal endothelia [40,41] develop the gut-vascular barrier which regulates soluble paracellular transport, compared to the brain-blood barrier (BBB) [42,43]. However in contrast to BBB, which has a size exclusion threshold of 500 Da, the gut – vascular barrier is permeable to molecules as large as 4 kDa [31]. Thus intestinal permeability is mediated by both the epithelial and endothelial barriers disturbances.
4. Intestinal Hemostasis in IBD
An ischemic inflammation in the intestinal microvasculature due to pro-coagulant activity and thrombi formation further enhance tissue damage and pathological angiogenesis. Long time ago Wakefield et al [44,45] observed granulomatous vasculitis in intestinal microvasculature during early stage of Crohn’s disease, which suggests its pathogenic significance. Capillary thrombi linked with fibrin, and an expression of the tissue factor were also observed in intestinal vasculature in Crohn’s disease and UC [46,47].
Activated platelets shown in intestinal microcirculation of IBD patients may activate endothelial cells via the expression of CD40. Danese et al [48] have shown higher platelet expression of surface CD40 ligand (CD40L), and an elevated concentration of platelets origin soluble CD40L in plasma of UC and Cronh’s disease patients as compared to healthy subjects. They also observed CD40L positive platelets adherents to ECs in intestinal circulation, which may initiate inflammatory response [49].
In addition, the interaction of platelets expressed CD40 with CD40 expressed vascular components and may increase inflammatory cell adhesion molecules ICAM-1 and VCAM-1, and white blood cells migration to extravascular space. Independently, platelet adhesion to inflamed intestinal endothelial cells may favor angiogenesis by release of VEGF and platelet-derived growth factor (PDGF), which is also a candidate for therapeutic target for IBD [50].
In the inflamed mucosa of IBD patients de Jong et al [51] presented a decrease of tissue plasminogen activator (t-PA) and increase of urokinase plasminogen activator (u-PA). U-PA, in contrast to t-PA, is less fibrin dependent; thus plasmin generated due to u-PA may act as proinflammatory protease. Cytokines IL-1 and TNF–α are able to cause procoagulant action by induction of tissue factor in ECs, platelets and monocytes; and suppress the anticoagulant potential of thrombomodulin by downregulation of endothelial protein C receptor, which is sustained normally by ECs [52].This disruption of the protein C system is also related to elevated adherence of ECs, thus support leukocyte enrolment. Recent investigations has indicated that the protein C pathway is expressed not only in ECs of the intestinal vessels, but also in epithelial cells and plays a significant role in enhancing the integrity of tight junctions [53]. In epithelium of patients with Crohn’s disease and UC expression of protein C is changed, which may amplify intestinal permeability [54].
5. Angiogenesis in IBD
Vessel quality are probably critical in pathogenesis of IBD (Table 2). In fact, newly formed vessels in IBD tissues are strongly disorganized and leaky as indicated by associated edema [55,56]. The induction and propagation of angiogenesis during IBD has been partly connected with hypoxia. In fact in IBD mucosa hypoxia-inducible factor-1 and -2 transcriptionally activate the expression of VEGF-A [11]. The VEGF gene may also be turn on by other factors, which are significant in IBD such as growth factor; mainly EGF and TGFβ and cytokines e.g. TNFα, IL6 and Il-1β. [8,57]. We and other were found VEGF higher expression in endothelial cells as well as epithelial cells in IBD- inflamed intestine [5,58]. Increased VEGF levels in both serum and plasma was observed in patients with IBD which may reflect VEGF overexpression in intestinal inflammatory tissue [56,58,59,60]. In addition increased VEGF has been found in mucosal extracts obtained from IBD patients as compared to controls [5].
Danese et al [5] first documented the significance of angiogenesis as a novel component both in UC and in Crohn’s disease pathogenesis (Figure 1). These authors demonstrated higher density of microvesels as well as VEGF expression within intestinal mucosa of active and inactive UC and Chron’s disease phase in comparison with the controls. Is seems that VEGF may activate two tyrosine kinase receptors; Flt-1 (fms-like tyrosine kinase-1 receptor) and KDR (kinase domain receptor) [61,62]. The Flt-1 receptor has closer propinquity to VEGF than KDR receptor. We observed significant increase amount of VEGF gene as well as Flt-1 gene, and higher levels of VEGF and Flt-1 protein in intestinal mucosa of active UC stage in comparison with control group and inactive UC stage, but we found only trace KDR gene expression in active UC phase [58]. Previously German investigators Griga et al. [63,64], have shown higher immunochemical reaction indicated VEGF protein in epithelium, and in lamina propria of colonic tissue in active UC stage than in inactive UC stage and normal colonic tissue, which is in agreement with our results (Figure 2 and Figure 3). In striking contrast Greek investigators Giatromanolaki et al [11] and Kapsoritakis et al [65] using immunohistochemical methods observed only weak specific reaction for VEGF in endothelial cells and enterocytes in UC inflamed colon. One could postulate that in different geographical regions i.e. northern Europe and Mediterranean area, varied environment and/or unknown genetic pattern may modulate VEGF and its receptors intestinal levels.
The endothelial damage may precede epithelial barrier dysfunction in IBD. The investigators at the La University using functional, morphologic, and molecular biologic studies in four animal models of UC indicated that endothelial permeability occurs earlier than epithelial barrier dysfunction that is followed by erosions, ulceration, and inflammation suggesting that endothelial disruption might be critical in disease pathogenesis [66]. In addition Lakatos et al [67] shown that serum antigen concentration of MMP-9 and tissue inhibitors of metalloproteinases TIMP-1 and TIMP-2 were higher in UC and Chron’s disease patients as compared to controls and that MMP-9 and TIMP levels significantly correlated with disease activity. Thus MMPs can contribute to remodeling processes in IBD and serum MMP-9 and TIMP might be used as biomarkers of disease activity.
Elevated levels of other angiogenic growth factors including basic fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF) have been found in the inflamed mucosa and in the blood of IBD patients [56,68,69,70]. Placental growth factor (PlGF), a specific regulator of pathological angiogenesis was found to be increased in the serum of IBD patients [20,71]. Recently Zhou et al presented proangiogenic effects of PlGF on human intestinal microvascular cells (HIMECs) via PI3K/Akt signaling pathway activation [20].
In relation to angiopoietin system, increased of Ang-2 and Tie-2 was observed in serum of patients with IBD [72]. In later studies it was shown immunologically higher levels of Ang-1 and Ang-2 in epithelia of crypt abscesses from patients with active UC as compared with UC patients with remission [73]. It suggests that angiogenic response is regulated by the Ang/Tie, and this pathway may play a role in the progression of UC. Another unique molecular pattern of ECs from angiogenic vessels represent the expression of adhesion molecules integrins ανβ3 and ανβ5 involved with recruitment of T-cells to intestinal mucosa. Importantly higher expression of ανβ3 has been shown in the mucosa of IBD patients [5,74].
In addition an effect of kinins in angiogenesis has been suggested. Kinins may mediate angiogenesis by upregulation of basic FGF via B1 bradykinin receptor and by activation of VEGF synthesis via both B1 and B2 bradykinin receptors [75]. Our data [76] demonstrated B1 and B2 receptors in intestinal tissues of human IBD, and indicated that B1 receptor upregulation is critical for kinin function. Kinins have an ability to stimulate proliferation, and interact with growth factors. Bradykinin as growth factor (also as a member autacoids family) is responsible for capillary permeability and edema. Thus, in all probability kinins may support angiogenesis in IBD although relationship between kinins and cytokines and growth factors in this aspect needs more research.
The chloride channels (ClCs) which are expressed in intestinal mast cells and epithelial cells support tight junction integrity, and regulate chloride ion transport, thus contribute to intestinal barrier function. Recent report indicate that dysregulation of these channels in mast cells may mediate mast cell activation and degranulation and immune cell recruitment in inflamed tissue leading to barrier dysregulation and aggravation some IBD symptoms mainly diarrhea [ 77] . Importantly the number of mast cells and mast cells tryptase expression (a marker of mast cell degranulation) were found to be increased in colonic mucosa and submucosa in experimental and human IBD [78]. Thus chloride channel modulation in mast cells may have a therapeutic potential in IBD.
5.1. Angiogenesis and Microbiome in IBD
Healthy intestinal bacteria consist mainly of Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria which Firmicutes and Bacteroidetes are dominant [79]. Intestinal tissue injury and break of the intestinal barrier lead to the dysregulation of the intestinal milieu and the reactions between the mucosal components and the commensal microbiota. This disequilibrium influence the function of the barrier and it also may alter of the commensal microbiota composition towards dysbiosis [2,80]. In fact, recent reports have indicated alterations in gut microbiome in IBD towards a less heterogeneous gut microbial composition due to a decrease in commensal anaerobic bacteria; Firmicutes and Bacteroidetes and an increase in the abundance of gram-negative Proteobacteria [81]. Dysbiosis in IBD is characterized by changes in microbiome profile and also by metabolic derangements, mainly decreased short-chain fatty acid (SCFA), and enhanced hydrogen sulfide (H2S) production, alteration of bile acid and tryptophan metabolism leading to the impairment of epithelial homeostasis and inflammation.
Interestingly, relationship of the gut – vascular barrier with the microbiota was shown to increase angiogenesis as observed in human IBD and murine models of colitis [40,82,83].
In fact, intestinal microbes are source of angiogenic activators in the form of ligands for Toll-like receptors (TLr) and protease – activated receptors (PARs) [82,83].
Ex vivo activated intestinal vessels as well as human intestinal microvascular cells (HIMECs) exposed to microbial products may sprout angiogenesis. These effects are mediated through Toll-like and NOD-like receptors (TLRs and NLRs) which represent innate immune responses [84]. Expression of TLRs on vascular endothelial cells is upregulated by vascular inflammation as well as by bacterial lipopolysaccharide (LPS).
Recently it has been shown that activation of TLRs and NLRs by specific bacterial ligands selectively upregulates the levels of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) produced by HIMECs, and simultaneously induces angiogenesis. These results suggest that a cooperation of endogenous and exogenous innate immune factors by reciprocal action of CEACAM1 and microbiota is essential to promote intestinal angiogenesis in IBD [85].
Microbial biomarkers are emerging as promising non-invasive tools to predict disease activity’ disease risk, and recurrence after surgery. The most cost-effective and less invasive method is the analysis of bacterial DNA in plasma. Importantly, bacterial genomic fragments (bactDNA) was observed in the blood for up to 50% of IBD patients [86], and bacterial DNA translocation can be regarded as risk factor of relapse at 6 months in patients with Crohn’s disease, and also independently it is connected with an increased risk of hospitalization and steroids induction [87]. Recent report indicate combination of sequencing techniques (methylation-based human cell-specific profiling together with shotgun metagenomics) to characterize the human and the microbial DNA content in feces. Combining neutrophil and other cell type fecal DNA fractions was demonstrated as non-invasive way to distinguish between inactive and active IBD and better disease monitoring [88]. Other approach which may indicate potential targets to aid diagnosis and direct therapeutic options in IBD refers to stool 16S rRNA gene sequencing and multi-system metabolomic phenotyping (using nuclear magnetic and mass spectroscopy), with subsequent integrative network analysis to delineate novel microbiota -metabolome interactions [89].
In addition some authors have reported endotoxemia and LPS-binding protein in systemic circulation of IBD patients [91,92]. Of note a link exist between the gut – vascular barrier and the blood–brain barrier (BBB) dysfunction in IBD. Depression and anxiety disorders have been demonstrated in up to 40% of patients with active IBD [93], and less frequently deterioration in cognitive functions [93,94]. Thus circulating bacterial products and pro-inflammatory mediators in IBD patients may enhance central nervous system disorders.
It is documented that in IBD microbiota and their products may modulate the function of immune cells as well as non –immune cells, mainly ECs, thus play a role in initiation or progression of inflammation. Importantly, in IBD microbial products via activating TLRs in ECs may trigger angiogenesis. Thus, it is fascinating to suggest that decrease of the gut microbial load might suppress angiogenesis.
5.2. Treatment of Angiogenesis in IBD
The TNF-α as central mediator to induce the inflammatory reactions has been documented in Crohn’s disease and in UC.Thus anti-TNF–α compounds are currently in the biological treatment of moderate and active Crohn’s disease and UC [95].Italian investigators in clinical study have presented the ability of infliximab to affect mucosal angiogenesis in patients with Crohn’s disease demonstrated that administration of infliximab downregulates mucosal angiogenesis suggesting that inflammation-driven angiogenesis in the gut mucosa may contributes to the therapeutic efficacy of TNF-α blockade of [96].Importantly Algaba et al. have shown that circulating VEGF and angiopoietin -1 levels decreased during anti – TNF therapy in UC and Crohn’s disease patients [97]. Whether these changes are a signs of improvement is a matter of debate. In addition, the treatment with infliximab downregulates the CD40/CD40L pathway in patients with Crohn’s disease [98], which may improve endothelial dysfunction reducing thrombi formation in intestinal vasculature.
Therapeutic strategies targeting VEGF represent an interesting way to reduce intestinal angiogenesis and mucosal inflammation. Upregulation of VEGF, a potent vascular permeability factor during early stages of UC may provide evidence to impaired endothelial barrier. Tolstanova et al [99] showing in experimental rat model of UC that anti-VEGF antibody diminished colonic vascular permeability and improved morphologic signs of colitis which indicate the pathogenic role of VEGF. Other pro-antigenic growth factor e.g. placental growth factor (PlGF) is upregulated in IBD patients via activation of tyrosine kinase -phosphoinositide 3-kinase / protein kinase B (PI3K/Akt) signaling pathway. Thus PlGF/PI3K/Akt signaling can be as an appropriate therapeutic target in IBD [20].
Bevacizumab a humanized IgG1 monoclonal antibody directed against VEGF-A is used as anti-angiogenic agent in various cancers including colorectal cancer. However up to now there are only case reports showing beneficial effect of bevacizumab (anti-VEGF-antibody) and sunitinib (multiple tyrosine kinase inhibitor of VEGF and PDGF) treatment in patients with Crohn’s disease [100,101]. Importantly, unwanted effects of bevacizumab may contain impaired wound healing, bleeding and even intestinal perforation, although such complications are rare [102,103]. Moreover in the light of case report of Loriot et al [104] anti – angiogenic treatment may exacerbate IBD. Recently safety of bevacizumab in association with chemotherapy in colonic cancers coexisting with IBD has been retrospectively evaluated. The authors have documented that bevacizumab is safe in cancer patients with IBD, and no clinical IBD exacerbation were found during bevacizumab treatment [105].
Other option for vessel-directed therapy of IBD are anti-adhesion molecules, such as vedolizumab and etrolizumab [106].Vedolizumab, the anti-integrin- α4β7-specific antibody may induce long-term remission in refractory patients with Crohn disease and UC [107,108]. This drug blocks reactions between α4-β7 and mucosal adhesion molecule (MAdCAM) involved in leukocyte recruitment, whereas etrolizumab, a monoclonal antibody against MAdCAM-1 acts as targets for the β7 subunit suppresses accumulation of T lymphocytes [109]. Of note Danese et al. in an experimental mouse model of ulcerative colitis have been shown inhibition angiogenesis employing the integrin αvβ3 inhibitor (ATN161) and at the same time decrease of intestinal inflammatory alterations [110]. However it should be emphasize that a beneficial effect of anti- angiogenic drugs as additional IBD treatment are still controversial. In fact up to now no clinical trial has been performed using anti anti-angiogenic treatment in IBD patients.
Among possible microbiota-targeted interventions, pro-biotics application provide limited effectiveness, in part because currently the limited microbial species available as probiotics. In contrast, fecal microbiota transplantation (FMT) replace intestinal milieu of a recipient by fecal solution from a donor [111]. Successful results from the use of FMT for recurrent Clostridium difficile infection indicated new approaches for IBD to replace existing microbiota and enhance protective bacterial numbers and metabolism. A number of clinical trials in patients with UC and Crohn’s disease showed the safety but variable efficacy of FMT in achieving clinical remission and maintaining remission after being achieved via drug therapy [112]. Involvement of angiogenesis in efficacy of FMT was indicated in murine irradiation model. In this model was found that application of FMT alleviated and protected against radiation induced intestinal injury via upregulation of intestinal VEGF expression [113]. Expanding insights into donor microbial composition, recipient factors, and post-transplant microbial profiles may define frequency of FMT administration and predict successful FMT outcomes.
5.3. Dual Role of Angiogenesis
The data of many studies indicate the significance of angiogenesis in IBD which may initiate and/or amplify chronic inflammation and fibrosis in the intestine. VEGF induces an abnormal angiogenesis, and other growth factors mainly placental growth factor (PlGF), basic epidermal growth factor (bFGF) and transforming growth factor-β (TGF-β) may play a critical role in pathogenesis of IBD. On the other hand these growth factors can be favorable in healing and reparation processes [114]. It has been shown in experimental studies that administration of PDGF or FGF significantly accelerated healing of UC [115]. In fact EGF enemas have been demonstrated to be useful in patients with UC [116].The researchers at the La University documented in rats models that the molecular mechanisms of bFGF in UC healing is connected with increased cell proliferation, especially angiogenesis and simultaneously decrease of cytokines and inflammatory cells [117]. Other authors also demonstrated that activation of Rac1 (a key factor of the Rho guanosine triphosphatase (GTPase) family) improved VEGF-induced angiogenesis in vivo as indicated by measurement of vascular density and diminished vessel leakiness in an angiogenic model [118]. Moreover anti-angiogenic factors are upregulated in inflamed intestine such as thrombospondin, endostatin—a cleaved fragment of collagen XVIII and angiostatin—a cleaved fragment of plasminogen [28,119]. Thus, therapeutic inhibition of angiogenesis may reduce wound healing in experimental colitis models and IBD.
6. Conclusions
It is indicated that in IBD intestinal vascular remodeling is disturbed by intestinal dysbiosis under conditions of intestinal inflammation. Defective immune response may promote intestinal angiogenesis through the selective induction of specific pro-angiogenic pathways. In turn angiogenesis promote recruitment of immune cells via intestinal barrier dysfunction and sustains inflammation. However an unresolved question in IBD pathogenesis is whether pathological angiogenesis is the cause or consequence of intestinal inflammation mediated by microbiota. Consequently current treatment of angiogenesis in IBD is not fully satisfactory, and its effect probably depends of the phase of the diseas.While intestinal microorganisms may switch on the angiogenic cascade in IBD, interference with gut microbiome and microbiota-targeted interventions would be a strategy for prevention or/and treatment of pathological angiogenesis.
Author Contributions
Antoni S., suggested the topic, writing— original draft preparation, review and editing, A.S., performed the literature, contributed to the writing Of the paper, W.P-W, critically reviewed the manuscript, providing critical comments. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
authors declare no conflict of interests.
References
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Benelli, R.; Lorusso, G.; Albini, A.; Noonan, D.M. Cytokines and Chemokines as Regulators of Angiogenesis in Health and Disease. Curr. Pharm. Des. 2006, 12, 3101–3115. [Google Scholar] [CrossRef]
- Sajib, S.; Zahra, F.T.; Lionakis, M.S.; German, N.A.; Mikelis, C.M. Mechanisms of angiogenesis in microbe-regulated inflammatory and neoplastic conditions. Angiogenesis 2017, 21, 1–14. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; De La Motte, C.; Graziani, C.; West, G.; Phillips, M.H.; Pola, R.; Rutella, S.; Willis, J.; Gasbarrini, A.; et al. Angiogenesis as a Novel Component of Inflammatory Bowel Disease Pathogenesis. Gastroenterology 2006, 130, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Cébe-Suarez, S.; Zehnder-Fjällman, A.; Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 2006, 63, 601–15. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Felmeden, D.C.; Blann, A.D.; Lip, G.Y. Angiogenesis: basic patho-physiology and implications for disease. Eur Heart J 2003, 24, 586–603. [Google Scholar] [CrossRef]
- Dachs, G.; Tozer, G. Hypoxia modulated gene expression: angiogenesis, metastasis and therapeutic exploitation. Eur. J. Cancer 2000, 36, 1649–1660. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Maltezos, E.; Papazoglou, D.; Simopoulos, C.; Gatter, K.C.; Harris, A.L.; et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol 2003, 56, 209–213. [Google Scholar] [CrossRef]
- Paleolog, E.M.; Young, S.; Stark, A.C.; McCloskey, R.V.; Feldmann, M.; Maini, R.N. Modulation of angiogenic vascular endothelial growth factor by tumor necrosis factor alpha? and interleukin-1 in rheumatoid arthritis. Arthritis Rheum. 1998, 41, 1258–1265. [Google Scholar] [CrossRef]
- Bottomley, M.J.; A Webb, N.J.; Watson, C.J.; Holt, P.J.L.; Freemont, A.J.; Brenchley, P.E.C. Peripheral blood mononuclear cells from patients with rheumatoid arthritis spontaneously secrete vascular endothelial growth factor (VEGF): specific up-regulation by tumour necrosis factor-alpha (TNF-α) in synovial fluid. Clin. Exp. Immunol. 1999, 117, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone, J.; Bandarra, D.; Rocha, S. The role of hypoxia in inflammatory disease (Review). Int. J. Mol. Med. 2015, 35, 859–869. [Google Scholar] [CrossRef]
- Bakirtzi, K.; West, G.; Fiocchi, C.; Law, I.K.M.; Iliopoulos, D.; Pothoulakis, C. The Neurotensin–HIF-1α–VEGFα Axis Orchestrates Hypoxia, Colonic Inflammation, and Intestinal Angiogenesis. Am. J. Pathol. 2014, 184, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. Biology of Platelet-Derived Growth Factor and Its Involvement in Disease. Mayo Clin. Proc. 2006, 81, 1241–1257. [Google Scholar] [CrossRef]
- Ierardi, E.; Giorgio, F.; Zotti, M.; Rosania, R.; Principi, M.; Marangi, S.; Della Valle, N.; De Francesco, V.; Di Leo, A.; Ingrosso, M.; et al. Infliximab therapy downregulation of basic fibroblast growth factor/syndecan 1 link: a possible molecular pathway of mucosal healing in ulcerative colitis. J. Clin. Pathol. 2011, 64, 968–972. [Google Scholar] [CrossRef]
- Kanazawa, S.; Tsunoda, T.; Onuma, E.; Majima, T.; Kagiyama, M.; Kikuchi, K. VEGF, basic-FGF, and TGF-β in Crohn’s disease and ulcerative colitis: a novel mechanism of chronić intestinal inflammation. Amer J. Gastroenterol. 2001, 96, 822–828. [Google Scholar]
- Itoh, H.; Kataoka, H. Roles of hepatocyte growth factor activator (HGFA) and its inhibitor HAI-1 in the regeneration of injured gastrointestinal mucosa. J. Gastroenterol. 2002, 37, 15–21. [Google Scholar] [CrossRef]
- Zhou, Y.; Tu, C.; Zhao, Y.; Liu, H.; Zhang, S. Placental growth factor enhances angiogenesis in human intestinal microvascular endothelial cells via PI3K/Akt pathway: Potential implications of inflammation bowel disease. Biochem. Biophys. Res. Commun. 2016, 470, 967–974. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Kim, I.; Kim, J.-H.; Moon, S.-O.; Kwak, H.J.; Kim, N.-G.; Koh, G.Y. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene 2000, 19, 4549–4552. [Google Scholar] [CrossRef]
- Lobov, I.B.; Brooks, P.C.; Lang, R.A. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11205–11210. [Google Scholar] [CrossRef]
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 sensitizes endothelial cells to TNF-α and has a crucial role in the induction of inflammation. Nat. Med. 2006, 12, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Linares, P.M.; Chaparro, M.; Gisbert, J.P. Angiopoietins in inflammation and their implication in the development of inflammatory bowel disease. A review. J. Crohn’s Colitis 2014, 8, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zak, S.; Treven, J.; Nash, N.; Gutierrez, L.S. Lack of thrombospondin-1 increases angiogenesis in a model of chronic inflammatory bowel disease. Int. J. Color. Dis. 2007, 23, 297–304. [Google Scholar] [CrossRef]
- Danese, S. Negative Regulators of Angiogenesis in Inflammatory Bowel Disease: Thrombospondin in the Spotlight. Pathobiology 2008, 75, 22–24. [Google Scholar] [CrossRef]
- Ortiz-Masià, D.; Díez, I.; Calatayud, S.; Hernández, C.; Cosín-Roger, J.; Hinojosa, J.; Esplugues, J.V.; Barrachina, M.D. Induction of CD36 and Thrombospondin-1 in Macrophages by Hypoxia-Inducible Factor 1 and Its Relevance in the Inflammatory Process. PLOS ONE 2012, 7, e48535. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, A.; Banan, A.; Fields, J.; Keshavarzian, A. Intestinal barrier: An interface between health and disease. J. Gastroenterol. Hepatol. 2003, 18, 479–497. [Google Scholar] [CrossRef]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Brescia, P.; Rescigno, M. The gut vascular barrier: A new player in the gut-liver-brain axis. Trends Mol. Med 2021, 27, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Mankertz, J.; Schulzke, J.-D. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Stürzl, M.; Kunz, M.; Krug, S.M.; Naschberger, E. Angiocrine Regulation of Epithelial Barrier Integrity in Inflammatory Bowel Disease. Front. Med. 2021, 8, 643607. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Himadri, R.; Shalini, B.; Seppo, Y.H. Biology of vascular endothelial growth factors. FEBS Letters 2006, 580, 2879–2887. [Google Scholar] [CrossRef]
- Spadoni, I.; Fornasa, G.; Rescigno, M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat. Rev. Immunol. 2017, 17, 761–773. [Google Scholar] [CrossRef]
- López-Posadas, R.; Stürzl, M.; Atreya, I.; Neurath, M.F.; Britzen-Laurent, N. Interplay of GTPases and Cytoskeleton in Cellular Barrier Defects during Gut Inflammation. Front. Immunol. 2017, 8, 1240. [Google Scholar] [CrossRef]
- Kabouridis, P.S.; Lasrado, R.; McCallum, S.; Chng, S.H.; Snippert, H.J.; Clevers, H.; Pettersson, S.; Pachnis, V. Microbiota Controls the Homeostasis of Glial Cells in the Gut Lamina Propria. Neuron 2015, 85, 289–295. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Claudins and the Modulation of Tight Junction Permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.-H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [CrossRef]
- Wakefield, A.; Dhillon, A.; Rowles, P.; Sawyerr, A.; Pittilo, R.; Lewis, A.; Pounder, R. Pathogenesis of Crohn’s disease: multifocal gastrointestinal infarction. Lancet 1989, 334, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, A.J.; Sankey, E.A.; Dhillon, A.P.; Sawyerr, A.; More, L.; Sim, R.; Pittilo, R.M.; Rowles, P.M.; Hudson, M.; Lewis, A.A.; et al. Granulomatous vasculitis in crohn's disease. Gastroenterology More, L.; Sim, R,; Hudson, M.; et al. Immunohistochemical study of tissue factor expression in normal intestine and idiopathic inflammatory bowel disease. J Clin Pathol 1993, 46, 703-708.. 1991, 100, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Granger, D.N. Inflammatory Bowel Diseases: a paradigm for the link between coagulation and inflammation. Inflamm Bowel Dis 2009, 15, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.; Anthony, A.; Sim, R.; Wakefield, A.; Sankey, E.; Hudson, M.; Allison, M.; Pounder, R. Mucosal capillary thrombi in rectal biopsies. Histopathology 1992, 21, 127–133. [Google Scholar] [CrossRef]
- Danese, S.; Katz, J.A.; Saibeni, S.; et al. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut 2003, 52, 1435–41. [Google Scholar] [CrossRef]
- Danese, S.; de la Motte, C.; Sturm, A.; Vogel, J.D.; A West, G.; A Strong, S.; A Katz, J.; Fiocchi, C. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology 2003, 124, 1249–1264. [Google Scholar] [CrossRef]
- Rutela, S.; Vetrano, S.; Correale, C.; Graziani, C.; Sturm, A.; Spinelli, A.; et al. Enhanced platelet adhesion induces angiogenesis in intestinal inflammation and inflammatory bowel disease microvasculature. J Cell Mol Med 2011, 15, 625–634. [Google Scholar] [CrossRef]
- de Jong, E.; Porte, R.J.; A Knot, E.; Verheijen, J.H.; Dees, J. Disturbed fibrinolysis in patients with inflammatory bowel disease. A study in blood plasma, colon mucosa, and faeces. Gut 1989, 30, 188–194. [Google Scholar] [CrossRef]
- Van de Wouwer, M.; Collen, D.; Conway, E.M. Thrombomodulin-Protein C-EPCR System: integrated to regulate coagulation and inflammation. Arter. Thromb. Vasc. Biol. 2004, 24, 1374–1383. [Google Scholar] [CrossRef]
- Vetrano, S.; Ploplis, V.A.; Sala, E.; Sandoval-Cooper, M.; Donahue, D.L.; Correale, C.; Arena, V.; Spinelli, A.; Repici, A.; Malesci, A.; et al. Unexpected role of anticoagulant protein C in controlling epithelial barrier integrity and intestinal inflammation. Proc. Natl. Acad. Sci. 2011, 108, 19830–19835. [Google Scholar] [CrossRef]
- Faioni, E.M.; Ferrero, S.; Fontana, G.; Gianelli, U.; Ciulla, M.M.; Vecchi, M.; Saibeni, S.; Biguzzi, E.; Cordani, N.; Franchi, F.; et al. Expression of endothelial protein C receptor and thrombomodulin in the intestinal tissue of patients with inflammatory bowel disease. Crit. Care Med. 2004, 32, S266–S270. [Google Scholar] [CrossRef] [PubMed]
- E Cromer, W.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–93. [Google Scholar] [CrossRef] [PubMed]
- Alkim, C.; Alkim, H.; Koksal, A.R.; Boga, S.; Sen, I. Angiogenesis in Inflammatory Bowel Disease. Int. J. Inflamm. 2015, 2015, 970890. [Google Scholar] [CrossRef] [PubMed]
- Stadnicki, A.; Machnik, G.; Klimacka-Nawrot, E.; Wolanska-Karut, A.; Labuzek, K. Transforming growth factor-β1 and its receptors in patients with ulcerative colitis. Int. Immunopharmacol. 2009, 9, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Frysz-Naglak, D.; Fryc, B.; Klimacka-Nawrot, E.; Mazurek, U.; Suchecka, W.; Kajor, M.; Kurek, J.; Stadnicki, A. Expression, localization and systemic concentration of vascular endothelial growth factor (VEGF) and its receptors in patients with ulcerative colitis. Int. Immunopharmacol. 2011, 11, 220–225. [Google Scholar] [CrossRef]
- Scaldaferri, F.; Vetrano, S.; Sans, M.; Arena, V.; Straface, G.; Stigliano, E.; Repici, A.; Sturm, A.; Malesci, A.; Panes, J.; et al. VEGF-A Links Angiogenesis and Inflammation in Inflammatory Bowel Disease Pathogenesis. Gastroenterology 2009, 136, 585–595.e5. [Google Scholar] [CrossRef]
- Bousvaros, A.; Leichtner, A.; Zurakowski, D.; Kwon, J.; Law, T.; Keough, K.; Fishman, S. Elevated Serum Vascular Endothelial Growth Factor in Children and Young Adults with Crohn's Disease. Dig. Dis. Sci. 1999, 44, 424–430. [Google Scholar] [CrossRef]
- Himadri, R.; Shalini, B.; Seppo, Y.H. Biology of vascular endothelial growth factors. FEBS Letters 2006, 580, 2879–87. [Google Scholar] [CrossRef]
- Proczka, R.M.; Polański, J.A.; Małecki, M.; Wikieł, K. The significance of vascular endothelial growth factor in the neoangiogenesis process. The role of hypoxia in the endothelial cells proliferation process and in the formation of collateral circulation. Acta Angiol 2003, 9, 143–149. [Google Scholar]
- Griga, T.; Gutzeit, A.; Sommerkamp, C.; May, B. Increased production of vascular endothelial growth factor by peripheral blood mononuclear cells in patients with inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 1999, 11, 175–180. [Google Scholar] [CrossRef]
- Griga, T.; May, B.; Pfisterer, O.; Müller, K.-M.; Brasch, F. Immunohistochemical localization of vascular endothelial growth factor in colonic mucosa of patients with inflammatory bowel disease. Hepato-Gastroenterology 2002, 49, 116–23. [Google Scholar]
- Kapsoritakis, A.; Sfiridaki, A.; Maltezos, E.; Simopoulos, K.; Giatromanolaki, A.; Sivridis, E.; Koukourakis, M.I. Vascular endothelial growth factor in inflammatory bowel disease. Int. J. Color. Dis. 2003, 18, 418–422. [Google Scholar] [CrossRef]
- Tolstanova, G.; Deng, X.; French, S.W.; Lungo, W.; Paunovic, B.; Khomenko, T.; Ahluwalia, A.; Kaplan, T.; Dacosta-Iyer, M.; Tarnawski, A.; et al. Early endothelial damage and increased colonic vascular permeability in the development of experimental ulcerative colitis in rats and mice. Mod. Pathol. 2012, 92, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, G.; Hritz, I.; Varga, M.Z.; Juhász, M.; Miheller, P.; Cierny, G.; Tulassay, Z.; Herszényi, L. The Impact of Matrix Metalloproteinases and Their Tissue Inhibitors in Inflammatory Bowel Diseases. Dig. Dis. 2012, 30, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Neubauer, K.; Matusiewicz, M. Platelet-derived growth factor-BB reflects clinical, inflammatory and angiogenic disease activity and oxidative stress in inflammatory bowel disease. Clin. Biochem. 2009, 42, 1602–1609. [Google Scholar] [CrossRef]
- Macé, V.; Ahluwalia, A.; Coron, E.; Le Rhun, M.; Boureille, A.; Bossard, C.; Mosnier, J.; Matysiak-Budnik, T.; Tarnawski, A.S. Confocal laser endomicroscopy: A new gold standard for the assessment of mucosal healing in ulcerative colitis. J. Gastroenterol. Hepatol. 2015, 30, 85–92. [Google Scholar] [CrossRef]
- Ippolito, C.; Colucci, R.; Segnani, C.; Errede, M.; Girolamo, F.; Virgintino, D.; Dolfi, A.; Tirotta, E.; Buccianti, P.; Di Candio, G.; et al. Fibrotic and Vascular Remodelling of Colonic Wall in Patients with Active Ulcerative Colitis. J. Crohn’s Colitis 2016, 10, 1194–1204. [Google Scholar] [CrossRef]
- Pousa, I.D.; Maté, J.; Salcedo-Mora, X.; Abreu, M.T.; Moreno-Otero, R.; Gisbert, J.P. Role of vascular endothelial growth factor and angiopoietin systems in serum of Crohnʼs disease patients. Inflamm. Bowel Dis. 2008, 14, 61–67. [Google Scholar] [CrossRef]
- Koutroubakis, I.E.; Xidakis, C.; Karmiris, K.; Sfiridaki, A.; Kandidaki, E.; Kouroumalis, E.A. Potential role of soluble angiopoietin-2 and Tie-2 in patients with inflammatory bowel disease. Eur. J. Clin. Investig. 2006, 36, 127–132. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Nakayama, T.; Naito, S.; Sekine, I. Expression Patterns of Angiopoietin-1, -2, and Tie-2 Receptor in Ulcerative Colitis Support Involvement of the Angiopoietin/Tie Pathway in the Progression of Ulcerative Colitis. Dig. Dis. Sci. 2008, 54, 2094–2099. [Google Scholar] [CrossRef]
- Dotan, I.; Allez, M.; Danese, S.; Keir, M.; Tole, S.; McBride, J. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.W. Regulation of Angiogenesis by the Kallikrein-Kinin System. Curr. Pharm. Des. 2006, 12, 2599–2607. [Google Scholar] [CrossRef]
- Stadnicki, A. Intestinal tissue kallikrein-kinin system in inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Aljameeli AM; Aswaid B; Bharati B; Gohri V; Mohite M, Singh S; Chidrawar V et al. Chloride channels and mast cell function: pioneering new frontiers in IBD therapy. Mol Cell Biochem 2025, 480, 3951–3969.
- He, S.-H. Key role of mast cells and their major secretory products in inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 309–318. [Google Scholar] [CrossRef]
- Khandagale, A.; Reinhardt, C. Gut microbiota ndash architects of small intestinal capillaries. Front. Biosci. 2018, 23, 752–766. [Google Scholar] [CrossRef]
- Mishima, Y.; Sartor, R.B. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J. Gastroenterol. 2019, 55, 4–14. [Google Scholar] [CrossRef]
- Alam, M.T.; Amos, G.C.A.; Murphy, A.R.J.; Murch, S.; Wellington, E.M.H.; Arasaradnam, R.P. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; Bergentall, M.; Greiner, T.U.; Schaffner, F.; Östergren-Lundén, G.; Petersen, L.C.; Ruf, W.; Bäckhed, F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 2012, 483, 627–631. [Google Scholar] [CrossRef]
- Schirbel, A.; Kessler, S.; Rieder, F.; West, G.; Rebert, N.; Asosingh, K.; McDonald, C.; Fiocchi, C. Pro-Angiogenic Activity of TLRs and NLRs: A Novel Link Between Gut Microbiota and Intestinal Angiogenesis. Gastroenterology 2013, 144, 613–623.e9. [Google Scholar] [CrossRef]
- Shin, H.-S.; Xu, F.; Bagchi, A.; Herrup, E.; Prakash, A.; Valentine, C.; Kulkarni, H.; Wilhelmsen, K.; Warren, S.; Hellman, J. Bacterial Lipoprotein TLR2 Agonists Broadly Modulate Endothelial Function and Coagulation Pathways In Vitro and In Vivo. J. Immunol. 2011, 186, 1119–1130. [Google Scholar] [CrossRef]
- Schirbel, A.; Rebert, N.; Sadler, T.; West, G.; Rieder, F.; Wagener, C.; Horst, A.; Sturm, A.; de la Motte, C.; Fiocchi, C. Mutual Regulation of TLR/NLR and CEACAM1 in the Intestinal Microvasculature: Implications for IBD Pathogenesis and Therapy. Inflamm. Bowel Dis. 2018, 25, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Francés, R.; Amorós, A.; Zapater, P.; Garmendia, M.; Ndongo, M.; Caño, R.; Jover, R.; Such, J.; Pérez-Mateo, M. Cytokine association with bacterial DNA in serum of patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Zapater, P.; Juanola, O.; Sempere, L.; García, M.; Laveda, R.; Martínez, A.; Scharl, M.; González-Navajas, J.M.; Such, J.; et al. Gut Bacterial DNA Translocation is an Independent Risk Factor of Flare at Short Term in Patients With Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni Ch; Ochana B; Focht G; Harpenas F; Quteineh A; Orlansky E et al. Human DNA levels in feces reflect gut inflammation and associate with presence of gut species in IBD patients across the age spectrum. Res Sq 2025 :rs.3.rs-6809327. [CrossRef]
- Radhakrishnan, S.T.; Mullish, B.H.; Olbei, M.L.; Danckert, N.P.; Valdivia-Garcia, M.A.; Serrano-Contreras, J.I.; Chrysostomou, D.; Balarajah, S.; Perry, R.W.; Thomas, J.P.; et al. Deciphering the microbiome–metabolome landscape of an inflammatory bowel disease inception cohort. Gut Microbes 2025, 17, 2527863. [Google Scholar] [CrossRef]
- Gardiner, K.R.; Halliday, M.I.; Barclay, G.R.; et al. Significance of systemic endotoxemia in inflammatory bowel disease. Gut 1995, 36, 897–901. [Google Scholar] [CrossRef]
- Rojo, Ó.P.; Román, A.L.S.; Arbizu, E.A.; Martínez, A.d.l.H.; Sevillano, E.R.; Martínez, A.A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef]
- Navabi, S.; Gorrepati, V.S.; Yadav, S.; Chintanaboina, J.; Maher, S.; Demuth, P.; Stern, B.; Stuart, A.; Tinsley, A.; Clarke, K.; et al. Influences and Impact of Anxiety and Depression in the Setting of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 2303–2308. [Google Scholar] [CrossRef]
- Byrne, G.; Rosenfeld, G.; Leung, Y.; Qian, H.; Raudzus, J.; Nunez, C.; Bressler, B. Prevalence of Anxiety and Depression in Patients with Inflammatory Bowel Disease. Can. J. Gastroenterol. Hepatol. 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Hadjina, I.T.; Zivkovic, P.M.; Matetic, A.; Rusic, D.; Vilovic, M.; Bajo, D.; Puljiz, Z.; Tonkic, A.; Bozic, J. Impaired neurocognitive and psychomotor performance in patients with inflammatory bowel disease. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Błoński W, Stadnicki A, Lichtenstein G., Burke, A. Infliximab use in ulcerative colitis. In: Ulcerative Colitis. The complete quid to medical management. Ed: GR Lichtenstein, EJ Scherl New York Slack Inc 2011, 237-254.
- Rutella, S.; Fiorino, G.; Vetrano, S.; Correale, C.; Spinelli, A.; Pagano, N.; Arena, V.; Maggiano, N.; Repici, A.; Malesci, A.; et al. Infliximab Therapy Inhibits Inflammation-Induced Angiogenesis in the Mucosa of Patients With Crohn's Disease. Am. J. Gastroenterol. 2011, 106, 762–770. [Google Scholar] [CrossRef]
- Algaba, A.; Linares, P.M.; Fernández-Contreras, M.; Figuerola, A.; Calvet, X.; Guerra, I.; et al. The effects of infliximab or adalimumab on vascular endothelial growth factor and angiopoietin 1 angiogenic factor levels in inflammatory bowel disease: serial observations in 37 patients. Inflamm Bowel Dis 2014, 20, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Sans, M.; Scaldaferri, F.; Sgambato, A.; Rutella, S.; Cittadini, A.; Piqué, J.M.; Panes, J.; Katz, J.A.; Gasbarrini, A.; et al. TNF-α Blockade Down-Regulates the CD40/CD40L Pathway in the Mucosal Microcirculation: A Novel Anti-Inflammatory Mechanism of Infliximab in Crohn’s Disease. J. Immunol. 2006, 176, 2617–2624. [Google Scholar] [CrossRef]
- Tolstanova, G.; Khomenko, T.; Deng, X.; Chen, L.; Tarnawski, A.; Ahluwalia, A.; Szabo, S.; Sandor, Z. Neutralizing Anti-Vascular Endothelial Growth Factor (VEGF) Antibody Reduces Severity of Experimental Ulcerative Colitis in Rats: Direct Evidence for the Pathogenic Role of VEGF. J. Pharmacol. Exp. Ther. 2009, 328, 749–757. [Google Scholar] [CrossRef]
- Magro, F.; Costa, C. Long-standing remission of Crohn’s disease under imatinib therapy in a patient with Crohn’s disease. Inflamm Bowel Dis 2006, 12, 1087–1089. [Google Scholar] [CrossRef]
- Coriat, R.; Mir, O.; Leblanc, S.; Ropert, S.; Brezault, C.; Chaussade, S.; Goldwasser, F. Feasibility of anti-VEGF agent bevacizumab in patients with Crohn’s disease. Inflamm Bowel Dis 2010, 17, 1632. [Google Scholar] [CrossRef]
- Hapani, S.; Chu, D.; Wu, S. Risk of gastrointestinal perforation in patients with cancer treated with bevacizumab: a meta-analysis. Lancet Oncol. 2009, 10, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Monk, B.J.; Walker, J.L.; Homesley, H.D.; Fowler, J.; Greer, B.E.; Boente, M.; Fleming, G.F.; et al. Risk Factors for GI Adverse Events in a Phase III Randomized Trial of Bevacizumab in First-Line Therapy of Advanced Ovarian Cancer: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2014, 32, 1210–1217. [Google Scholar] [CrossRef]
- Loriot, Y.; Boudou-Rouquette, P.; Billemont, B.; Ropert, S.; Goldwasser, F. Acute exacerbation of hemorrhagic rectocolitis during antiangiogenic therapy with sunitinib and sorafenib. Ann. Oncol. 2008, 19, 1975–1975. [Google Scholar] [CrossRef]
- Herrera-Gómez, R.G.; Grecea, M.; Gallois, C.; Boige, V.; Pautier, P.; Pistilli, B.; Planchard, D.; Malka, D.; Ducreux, M.; Mir, O. Safety and Efficacy of Bevacizumab in Cancer Patients with Inflammatory Bowel Disease. Cancers 2022, 14, 2914. [Google Scholar] [CrossRef]
- Löwenberg, M.; D’haens, G. Next-Generation Therapeutics for IBD. Curr. Gastroenterol. Rep. 2015, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.-F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.-J.; Danese, S.; et al. Vedolizumab as Induction and Maintenance Therapy for Ulcerative Colitis. New Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.-F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; Lopez-Posadas, R.; Watson, A.; Neufert, C.; et al. Blockade of alpha Ebeta7 integrin suppresses accumulation of CD8+ and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Sans, M.; Spencer, D.M.; Beck, I.; Doñate, F.; Plunkett, M.L.; de la Motte, C.; Redline, R.; E Shaw, D.; Levine, A.D.; et al. Angiogenesis blockade as a new therapeutic approach to experimental colitis. Gut 2007, 56, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Biazzo, M.; Deidda, G. Fecal Microbiota Transplantation as New Therapeutic Avenue for Human Diseases. J. Clin. Med. 2022, 11, 4119. [Google Scholar] [CrossRef]
- Sartor, B. Beyond random fecal microbial transplants: next generation personalized approaches to normalize dysbiotic microbiota for treating IBD. Gastrenterol Clin North Amer 2025, 54, 333–350. [Google Scholar] [CrossRef]
- Cui, M.; Xiao, H.; Li, Y.; Zhou, L.; Zhao, S.; Luo, D.; Zheng, Q.; et al. Faecal microbiota transplantation protects against radiation-induced toxicity. EMBO Mol Med 2017, 9, 448–461. [Google Scholar] [CrossRef]
- Dignass, A.U.; Podolsky, D.K. Epithelial restitution and intestinal repair. In Kirsner’s inflammatory bowel diseases; Sartor, R.B., Sandborn, W.J., Eds.; Saunders: Philadelphia, 2004; pp. 18–29. [Google Scholar]
- Deng, X.; Szabo, S.; Khomenko, T.; Tolstanova, G.; Paunovic, B.; French, S.W.; Sandor, Z. Novel Pharmacologic Approaches to the Prevention and Treatment of Ulcerative Colitis. Curr. Pharm. Des. 2012, 19, 17–28. [Google Scholar] [CrossRef]
- Sinha, A.; Nightingale, J.M.; West, K.P.; Berlanga-Acosta, J.; Playford, R.J. Epidermal Growth Factor Enemas with Oral Mesalamine for Mild-to-Moderate Left-Sided Ulcerative Colitis or Proctitis. New Engl. J. Med. 2003, 349, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Paunovic, B.; Deng, X.; Khomenko, T.; Ahluwalia, A.; Tolstanova, G.; Tarnawski, A.; Szabo, S.; Sandor, Z. Molecular Mechanisms of Basic Fibroblast Growth Factor Effect on Healing of Ulcerative Colitis in Rats. J. Pharmacol. Exp. Ther. 2011, 339, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.V.; Nagy, J.A.; Senger, D.R. Active Rac1 improves pathologic VEGF neovessel architecture and reduces vascular leak: mechanistic similarities with angiopoietin-1. Blood 2011, 117, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Singh, N.P.; Murphy, E.A.; Price, R.L.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 2016, 77, 44–49. [Google Scholar] [CrossRef]
Figure 1.
Main factors driving angiogenesis in inflammatory bowel disease. Insight into bowel lumen, intestinal inflamed mucosa/submucosa, and activated and altered endothelium. Infiltrating neutrophils (N), monocytes/macrophages (M), lymphocytes (L), and dendritic cells (DC)) secreted cytokines, growth factors, and other factors to stimulate EC (endothelial cells). Activated ECs produced vascular endothelial growth factor (VEGF), expressed of Toll – like receptor (TLR), the specific receptor for bacterial products. Activated ECs with expression of adhesion molecules cause leukocyte recruitment to mucosa/ submucosa and platelet adhesion, whereas activation of coagulation and platelets cause thrombi formation in microvasculature, and in turn ischemia. Vascular dysfunction leads to neovascularization and vascular remodeling which characterize pathological angiogenesis.
Figure 1.
Main factors driving angiogenesis in inflammatory bowel disease. Insight into bowel lumen, intestinal inflamed mucosa/submucosa, and activated and altered endothelium. Infiltrating neutrophils (N), monocytes/macrophages (M), lymphocytes (L), and dendritic cells (DC)) secreted cytokines, growth factors, and other factors to stimulate EC (endothelial cells). Activated ECs produced vascular endothelial growth factor (VEGF), expressed of Toll – like receptor (TLR), the specific receptor for bacterial products. Activated ECs with expression of adhesion molecules cause leukocyte recruitment to mucosa/ submucosa and platelet adhesion, whereas activation of coagulation and platelets cause thrombi formation in microvasculature, and in turn ischemia. Vascular dysfunction leads to neovascularization and vascular remodeling which characterize pathological angiogenesis.
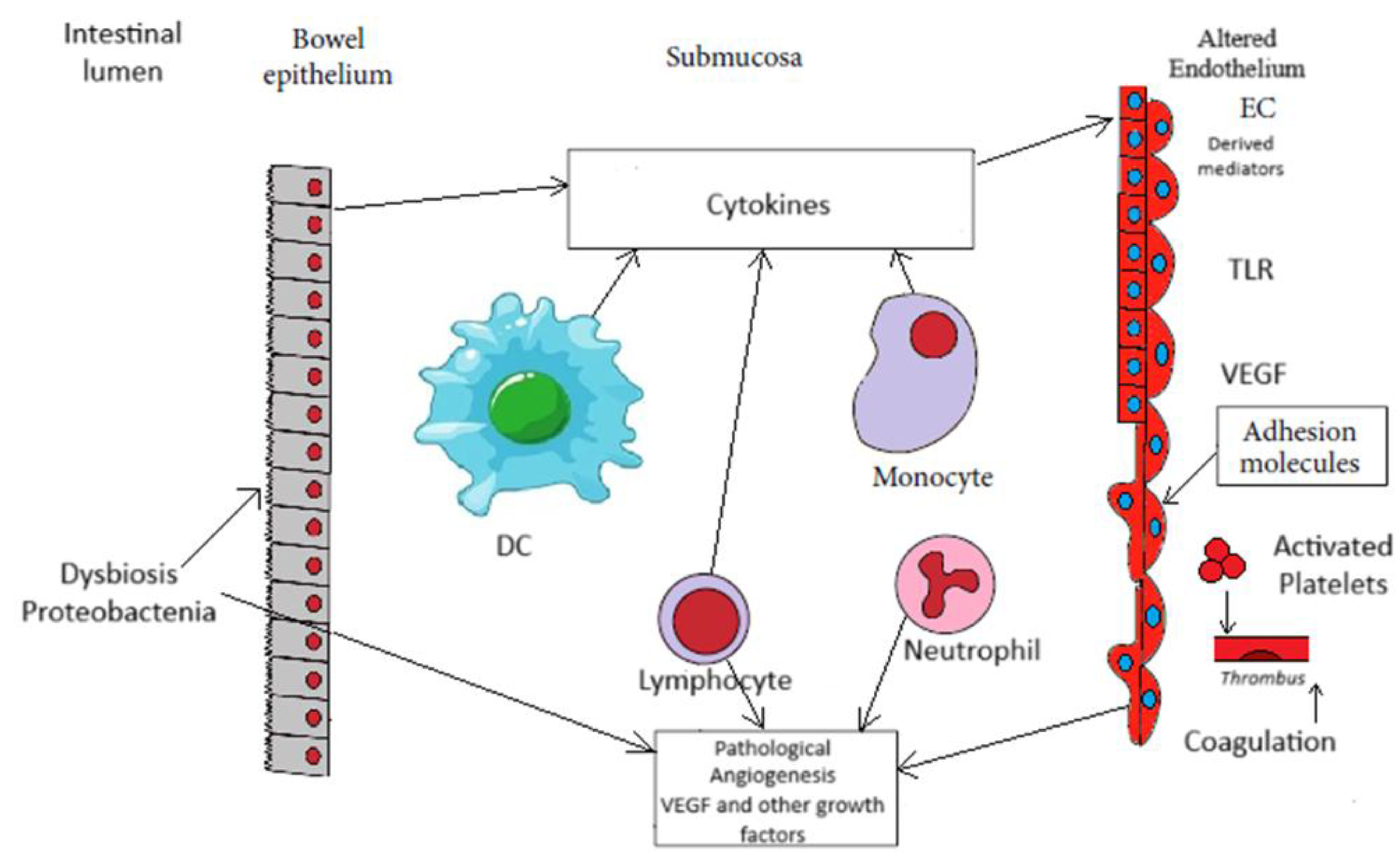
Figure 2.
VEGF immunohistochemical localization in UC - inflamed intestine and normal colon (100x). A: Colon in active UC phase, arrows mark specific staining reaction within enterocytes. B: Colon in active UC phase, yellow arrow - specific staining reaction in endothelium of vessel, black arrow - specific staining reaction in lumen vessel, red arrow - specific staining reaction within intestinal smooth mucles cells. C: Normal colon, arrow marks specific staining reaction in enterocytes (from Reference 58; Frysz-Naglak D. et al.,with permission of Elsevier publishing).
Figure 2.
VEGF immunohistochemical localization in UC - inflamed intestine and normal colon (100x). A: Colon in active UC phase, arrows mark specific staining reaction within enterocytes. B: Colon in active UC phase, yellow arrow - specific staining reaction in endothelium of vessel, black arrow - specific staining reaction in lumen vessel, red arrow - specific staining reaction within intestinal smooth mucles cells. C: Normal colon, arrow marks specific staining reaction in enterocytes (from Reference 58; Frysz-Naglak D. et al.,with permission of Elsevier publishing).
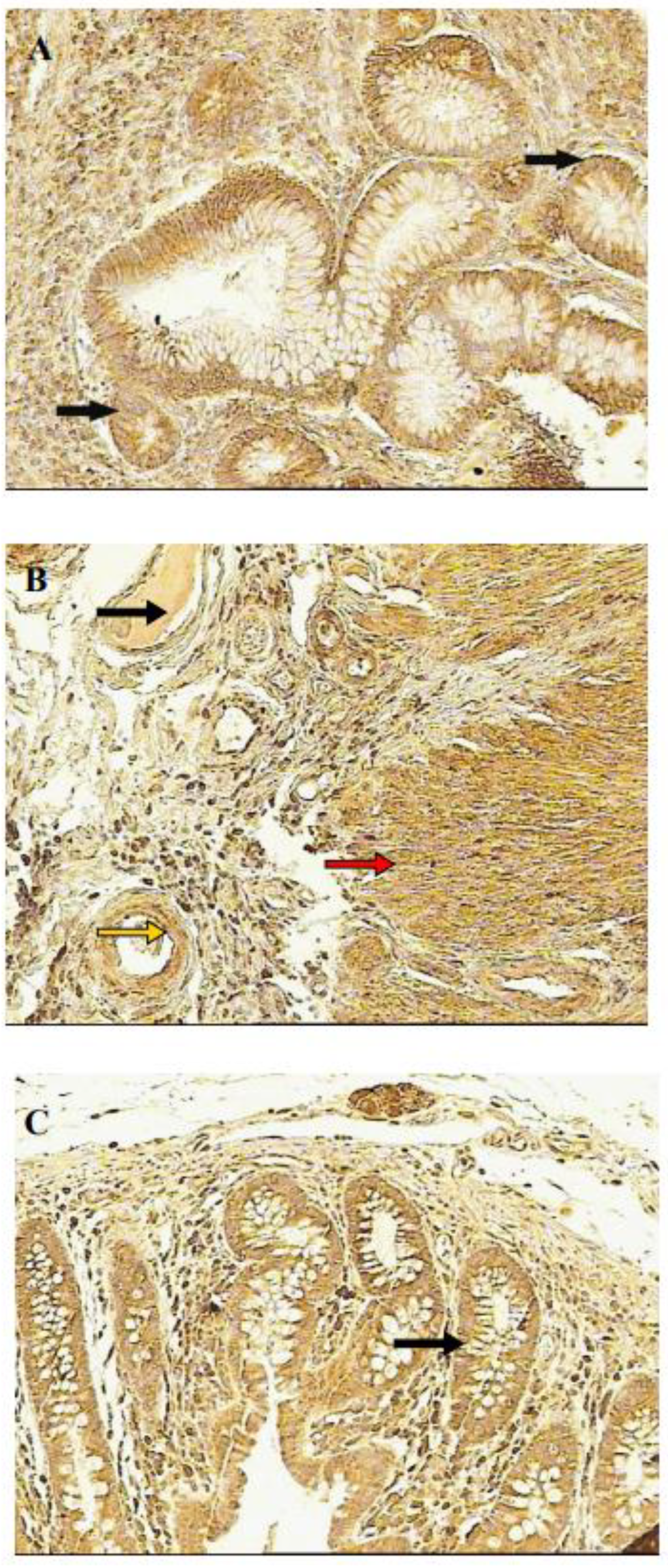
Figure 3.
Flt-1 receptor localization in UC - inflamed intestine and normal colon (100x).A: Colon in active UC phase, arrow marks specific staining reaction in enterocytes. B: Colon in active UC phase, arrow marks specific staining reaction in endothelium. C: Normal colon, arrows mark specific staining reaction in enterocytes (from Reference 58; Frysz-Naglak D. et al.,with permission of Elsevier publishing).
Figure 3.
Flt-1 receptor localization in UC - inflamed intestine and normal colon (100x).A: Colon in active UC phase, arrow marks specific staining reaction in enterocytes. B: Colon in active UC phase, arrow marks specific staining reaction in endothelium. C: Normal colon, arrows mark specific staining reaction in enterocytes (from Reference 58; Frysz-Naglak D. et al.,with permission of Elsevier publishing).
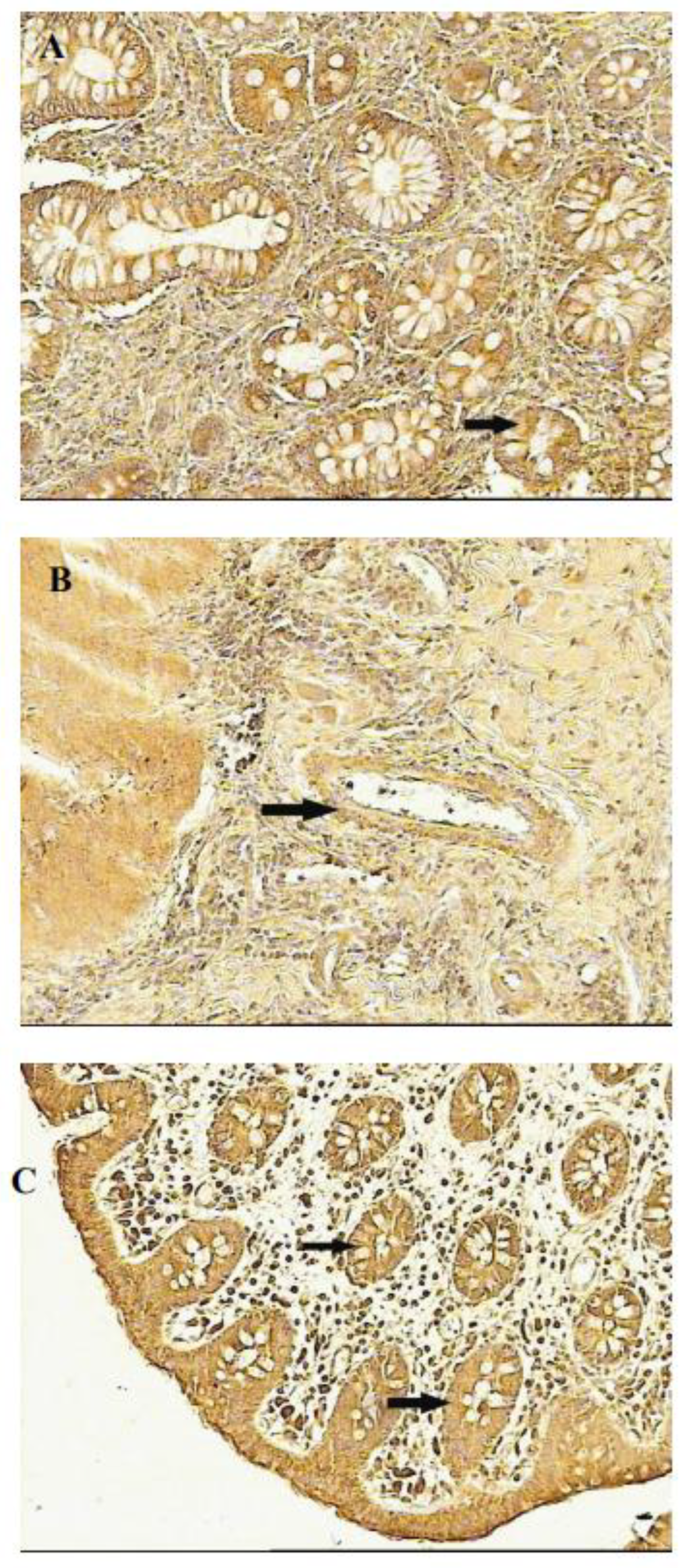

Table 1.
Main proangiogenic components in IBD.
| ■ Dysbiosis ■ Endothelial activation ■ Intestinal barrier dysfunction ■ Active inflammation ■ Hypercoagulability ■ Thrombi formation ■ Ischemia |
Table 2.
Manifestation of endothelial/vascular responses in IBD.
| Endothelial activation and expression of adhesion molecules | leucocyte recruitment platelet adhesion inflammation |
|---|---|
| VEGF expression | ECs proliferation and migration up-regulation of adhesion molecules immune cells recruitment vascular permeability sprouting of angiogenesis |
| b FGF expression | sprouting of angiogenesis |
| PDGF expression | sprouting of angiogenesis vascular coverage |
| Toll – like receptor expression by EC | regulation of endothelial barrier homeostasis specific receptor for bacterial products |
| Coagulation activation | platelet adhesion and activation impaired of protein C pathway thrombi formation ischemia |
| Microvascular dysfunction | granulomatous vasculitis ulceration |
| Angiogenesis | neovascularization remodeling of vasculature initiation /promotion of inflammation |
Abbreviations: ECs; endothelial cells, VEGF; vascular endothelial growth factor, b FGF ; basic fibroblast growth factor, PDGF; platelet-derived growth factor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



