Submitted:
08 October 2025
Posted:
09 October 2025
You are already at the latest version
Abstract
Hypoxia, or lack of adequate oxygen saturation, activates a vast repertoire of vascular responses to increase cell survival and proliferation, driven primarily by activation of oxygen-sensing hypoxia-inducible factors (HIFs). Key hypoxia mediator HIF-1 alpha is capable of driving vascular restructuring in response to low oxygen tension and oxygen-independent signaling pathways, and thus serves as a promising therapeutic modulator for ischemic cardiovascular diseases such as peripheral artery disease and coronary artery disease. In this review, we discuss oxygen-dependent and oxygen-independent mechanisms of HIF-1 alpha regulation, the HIF protein family’s role in vessel collateralization, and translational efforts seeking to exploit HIF-1 alpha’s key role in hypoxia signaling for the purpose of therapeutic development of clinical treatments for ischemic cardiovascular disease.
Keywords:
HIF-1a
; oxygen
; hypoxia
; ischemia
; cardiovascular diseases
1. Introduction
Oxygen homeostasis, the balance of oxygen supply and demand, is crucial to the normal functioning of molecular and cellular processes involved in cell metabolism, differentiation, proliferation, and survival, as well as organ function and human survival [1,2]. Humans have adapted cellular and biochemical responses to combat hypoxic insult key to many disease processes. The most well studied are the hypoxia-inducible factors (HIFs) [3]. HIF-1 was identified in 1992 as a transcription factor that upregulates erythropoietin (EPO) production in response to hypoxia by binding to the EPO enhancer and increases its transcription [4,5,6]. EPO is a glycoprotein hormone produced by specialized interstitial peritubular fibroblast-like cells of the kidney which acts to promote erythropoiesis in the bone marrow in response to hypoxia and/or anemia, thereby increasing circulating red blood cell (RBC) number and increasing oxygen delivery to tissues [7].
Further studies have since uncovered the hypoxic regulatory mechanisms of HIF-1α and the more than 100 genes that it regulates [8]. HIF-1α plays a crucial role in promoting the formation of new blood vessels (angiogenesis) through upregulation of growth factors such as VEGF [9,10,11], facilitating an energy-conserving metabolic switch from aerobic to anaerobic metabolism via the upregulation of key glycolytic enzymes, increase in glucose transporters in the cell membrane, and repression of mitochondrial TCA cycle enzymes – all serving to effectively increase the intracellular oxygen tension [12,13]. Additionally, HIF-1α induction has been shown to increase EPO production to increase circulating RBC volume [7], dampen the inflammatory response via extracellular adenosine signaling [14], and promote cell proliferation and survival in hypoxic environments, including solid tumors [15,16].
These HIF-driven cellular mechanisms are central to tissue survival in the response to ischemic events, such as the growth of new collateral blood vessels in occluded cerebral, peripheral, and coronary arteries to restore local circulation [17]. The purpose of this review paper is to explore the biological regulation of the HIF proteins, the role of HIFs and its downstream targets in promoting angiogenesis, and the clinical implications of therapeutic angiogenesis in ischemic coronary and peripheral artery disease.
2. Oxygen-Dependent Regulation of the HIFs
HIF-1α and HIF-2α are heterodimeric transcription factors belonging to the basic helix-loop-helix PER-ARNT-SIM family (bHLH-PAS) consisting of an oxygen-sensitive alpha subunit and a constitutively expressed beta subunit (HIF-1β) [18]. The beta subunit is also known as the aryl hydrocarbon receptor nuclear translocator (ARNT) and is encoded by ARNT1 and ARNT2 [19]. HIF-1β forms a heterodimer with both HIF-1α and HIF-2α [18]. Three isoforms of the alpha subunit exist and are termed HIF-1α, HIF-2α, and HIF-3α, respectively [20]. The alpha subunits of HIF-1 and HIF-2 exhibit stable transcription; however, they are tightly regulated at the protein level [21]. The alpha subunit contains an oxygen-dependent degradation (ODD) domain which contains two specific proline residues which are subject to hydroxylation by several prolyl hydroxylase domain proteins (PHD1-4) under normal oxygen tension, or “normoxic” conditions [22]. Hydroxylation of the HIF alpha subunits occurs in the cytoplasm of the cell, which subsequently leads to the binding of the alpha subunit to Von Hippel Lindau protein (VHL) [23]. A complex formed with the E3 ubiquitin ligase results in polyubiquitylation and subsequent degradation via the ubiquitin-proteasome pathway [24]. The half-life of the HIF alpha subunits in the cytosol is approximately 5 minutes, resulting in rapid protein degradation in normoxic conditions [25] (Left, Figure 1).
The PHD-mediated hydroxylation of the HIF alpha subunit is dependent on the presence of molecular oxygen, α-ketoglutarate, ascorbate, as well as iron as a catalyst [26]. The interference with the iron catalyst through iron chelation with deferoxamine (DFO) or through competing with the PHD iron binding site with cobalt chloride (CoCl2) prevents PHD mediated hydroxylation of HIF alpha subunits and allows for chemical stabilization of HIF alpha subunits in in vitro experiments [27,28]. Under hypoxic conditions, PHD proteins cannot hydroxylate the HIF alpha subunits, and the HIF alpha subunit translocates to the nucleus where it forms a heterodimer with the HIF beta subunit [29]. The HIF heterodimer complex binds to specific core DNA sequences most often located near the promoters of HIF target genes termed hypoxic response elements (HREs) [30]. The bHLH sequence is crucial for DNA-binding and the three PAS regions, PAS-A, PAS-B, and PAS-associated C-terminal domain, are involved in heterodimerization [18]. HIF-1α and HIF-2α contain N-terminal and C-terminal transactivation domains (N-TAD and C-TAD, respectively) that are involved in the activation of HIF target genes [31]. These domains associate with additional transcriptional co-activators, most notably CBP and p300, which contain lysine acetyl-transferase activity [32]. The CTAD region of the HIF alpha subunit polypeptide contains an additional level of oxygen-dependent regulation via the Factor Inhibiting HIF (FIH). FIH hydroxylates an asparagine residue within the CTAD domains at even lower oxygen tensions than PHD proteins due to its lower Km for oxygen and thus exhibits negative regulation for the HIF alpha subunits even under hypoxic conditions [33].
HIF-1α and HIF-2α show strong sequence conservation between their bHLH and PAS regions which demonstrates their capacity to bind identical regions of DNA [18]. However, their NTAD regions confer target gene selectivity to the two proteins, likely secondary to distinct interactions with various transcriptional co-activators [34]. Interestingly, the CTAD region exhibits the least sequence conservation between the two proteins, however, they act to transactivate genes common to both HIF-1α and HIF-2α [35]. Several splice variants of HIF-3α exist which lack a functional CTAD region and may or may not contain an NTAD region [20]. The most well studied variant of HIF-3α, HIF-3AF, lacks both transactivation domains, and functions to negatively regulate HIF-1α in an oxygen-independent manner [36]. HIF-3α and its therapeutic potential for neovascularization are less characterized in the literature. Therefore, the remainder of this review paper will focus on HIF-1α and HIF-2α.
HIF-1α and HIF-2α demonstrate temporal differences in their gene expression. HIF-1α responds to acute hypoxia within minutes and quickly induces the expression of its downstream target genes [37,38]. At around 8 hours, HIF-1α levels peak and begin to decrease and HIF-2α levels begin to rise. By 24–48 hours, HIF-2α levels become the more active responder to chronic hypoxia [34,39]. The fall in HIF-1α protein levels is, in part, attributable to hypoxia-associated factor (HAF) mediated ubiquitination which targets HIF-1α for VHL-mediated protein degradation in proliferating cells regardless of oxygen tension [40]. This oxygen-independent regulation does not occur to HIF-2α [41].
3. Oxygen-Independent Regulation of the HIFs
While the HIF alpha subunits are canonically regulated at the post-translational level via oxygen-dependent hydroxylation, the subunits are also regulated by oxygen-independent crosstalk with other cell signaling pathways [42]. One area of signaling crosstalk comes from the NF-κB pathway [43]. It should come to no intuitive surprise that crosstalk exists between hypoxia and inflammatory cell signaling. Indeed, several studies have identified an NF-κB binding site within the promoter of HIF-1α [44]. One such study demonstrated that HIF-1α mRNA and protein levels increased in response to exogenous reactive oxygen species (ROS) administration, specifically H₂O₂, to cultured pulmonary artery smooth muscle cells (PASMCs) in normoxia [44,45]. This result implies direct HIF-1α transcriptional upregulation by NF-κB in an oxygen-independent manner [46].
Interestingly, TNF-α, a potent cell surface activator of NF-κB, has been demonstrated to promote HIF-1α protein activity, but the mechanism remains controversial given that there are conflicting results for increased HIF-1α DNA binding, increased HIF-1α mRNA levels, and post-translational protein stabilization that may vary based on cell types and experimental conditions [47,48,49]. For example, TNF-α has been shown to upregulate HIF-1α mRNA and protein levels via NF-κB in human pterygium fibroblasts in normoxic conditions [50] while TNF-α interferes with transcription of HIF target genes in cultured smooth muscle cells in hypoxia [51]. IL-1β can also upregulate HIF-1α in an NF-κB-dependent manner [52]. Additionally, NF-κB can upregulate HIF-1α expression in hypoxic environments, particularly in the central regions of the solid tumor microenvironment [53]. Hypoxia has been shown to upregulate HIF-1α mRNA via NF-κB through a PI3K/AKT pathway dependent mechanism in PASMCs [54].
The PI3K/AKT/mTOR and PI3K/AKT/FRAP pathway can also induce HIF-1α expression independently of NF-κB, as well as in both normoxia and hypoxia [55]. Various cell surface ligands and receptors can activate the PI3K/AKT pathway such as EGFR, PDGF, TNF-α, IL-1β, and insulin [9,56]. Interestingly, recent studies have identified growth hormone-releasing hormone (GHRH) as an upstream, oxygen-independent activator of HIF-1α in iPSC-derived cardiomyocytes via GHRH/GHRH-R/cAMP signaling, acting as a mediator of cardiomyocyte proliferation and oxidative phosphorylation [57,58].
Additionally, many post-translational modifications of the HIF proteins can occur, such as phosphorylation and acetylation of HIF-1α protein [59]. Positive or negative regulation of HIF-1α by these post-translational modifications can be exhibited depending on where the modified amino acid is located within the protein [60]. For example, phosphorylation of serine residues by ERK1/2 of the MAPK pathway promotes HIF-1α transcriptional activity and cell survival after hypoxic injury in cardiomyocytes [61]. Phosphorylation events occurring in the PAS or ODD regions inhibit HIF-1α protein activity [62].
Moreover, post-transcriptional modification of HIF-1α mRNA by microRNAs (miRNAs) adds an additional layer of regulation [63]. Active HIF-1α directly upregulates several small ~22 bp miRNAs which in turn regulate HIF-1α mRNA or protein stability in a positive or negative manner [64]. The most well-studied miRNA involved in regulating the activity of HIF-1α is miR-210 [65,66,67]. The expression of miR-210 is directly upregulated by HIF-1α in hypoxic conditions, which then binds to its target protein glycerol-3-phosphate dehydrogenase 1-like (GPD1L) [68]. GPD1L normally increases the activity of the PHD enzymes which subsequently promotes HIF-1α protein hydroxylation and degradation [68]. Increasing miR-210 by active HIF-1α in hypoxia results in a positive feedback loop with downregulation of GPD1L, resulting in less active PHD enzymes, and stable HIF-1α protein [65,69]. Other miRNAs, such as miR-155, can bind to the 3′ UTR region of HIF-1α mRNA transcripts and interfere with translation [70] (Right, Figure 1).
4. The HIF Proteins and Neovascularization
The term “neovascularization” refers to the various processes that generate new blood vessels which include vasculogenesis, arteriogenesis, and angiogenesis [71]. Vasculogenesis occurs during embryonic development and involves de novo formation of blood vessels from vascular progenitor cells [72]. The HIF proteins play a pivotal role in vasculogenesis, however, given the confinement of vasculogenesis to embryonic development, this is beyond the scope of this review.
Arteriogenesis refers to collateral formation from preexisting collateral vessels that occurs as a result from shifts in hemodynamic pressure from distal arterial occlusion [10]. These collaterals can be visible with iodinated contrast beyond the level of arterial occlusion during angiogram procedures. As the radius of the arterial lumen narrows with progressive atherosclerotic stenosis, increases in fluid shear stress remodel the pre-existing artery-arteriolar connections to allow blood flow down the path of least resistance [73]. The increase in fluid shear stress promotes the activity of endothelial nitric oxide synthase (eNOS), releasing nitric oxide, and promoting smooth muscle cell (SMC) relaxation and vasodilation [74].
VEGF is released along with monocyte chemotactic protein-1 (MCP-1) which promotes the upregulation of cell adhesion molecules (CAMs) on the endothelial cell surface and recruitment of monocytes, respectively [75]. Monocytes and platelets localize to the CAMs where they secrete various growth factors and cytokines to stimulate endothelial cell proliferation, a switch of SMCs from the contractile to proliferative phenotype, and ultimately, proliferation of collateral arterioles [74]. The process concludes with collateral vessel pruning, whereby, many smaller arterioles occlude in favor of fewer, larger arterioles, which favors flow and distal perfusion [74,76]. Arteriogenesis is often not enough to restore adequate distal perfusion, such as in the case of PAD. Collaterals formed via arteriogenesis are often present in patients undergoing surgical intervention with chronic limb-threatening ischemia (CLTI) [77].
Whereas the inciting event for the initiation of arteriogenesis is increases in fluid shear stress, angiogenesis is initiated by tissue ischemia itself [71,78]. Angiogenesis refers to the process by which new capillaries are formed in response to ischemia to increase the delivery of oxygen and nutrients to the tissue [72,79]. The HIF proteins play a crucial role in angiogenesis as the presence of hypoxia stabilizes the alpha subunits, promoting translocation to the nucleus, heterodimerization with the beta subunit and DNA binding, followed by the upregulation of many potent pro-angiogenic genes [10,80]. Angiogenesis can occur via two different mechanisms, sprouting and non-sprouting, or intussusceptive angiogenesis [79,81].
HIF expression can be upregulated in many cell types in the presence of ischemia, including fibroblasts, cardiomyocytes, skeletal muscle cells, immune cells, and solid tumor cells [80,82]. VEGF is the most well studied and potent stimulator of angiogenesis in the ischemic microenvironment and its expression is directly upregulated by HIF [10,83]. In sprouting angiogenesis, VEGF binds its receptor VEGFR-2 on endothelial cells which induces the formation of endothelial tip cells [76,84]. The tip cells are responsible for directing the growing vessel towards its chemotactic source by its tip projections, rather than elongation of the blood vessel.
Close interplay between VEGF and anti-angiogenic Notch signaling facilitates coordinated formation of the new vessel [77,78]. The tip cell exhibits high VEGF/VEGFR-2 and high delta like ligand 4 (Dll4) expression with low Notch sig[10,80naling [78]. The increased Dll4 increases Notch signaling in neighboring endothelial cells which inhibits their migration. These endothelial cells with higher Notch signaling and lower Dll4 expression comprise the stalk cells, which exhibit a proliferative phenotype that facilitates the elongation of the new vessel [79,80].
HIF signaling in stalk cells maintains a sustained glycolytic metabolism which promotes cellular proliferation in low oxygen tension [83,85]. Additionally, HIF-1α promotes the secretion of matrix metalloproteases (MMPs), urokinase plasminogen activator (uPA), and plasminogen activator inhibitor-1 (PAI-1), which function to degrade the basement membrane and surrounding extracellular matrix (ECM) components to generate room for new blood vessels to form [82]. As the lumen of the new vessel is formed via a process called tubulogenesis, HIF-2α upregulates the expression of VE-cadherin to form new endothelial cell junctions, promoting vascular integrity and preventing luminal collapse [80,86].
Additionally, HIF-1α recruits pericytes to surround the endothelial cells, adding structural integrity to the vessel and preventing leakage [87]. Furthermore, the delayed onset of HIF-2α relative to HIF-1α explains their complementary role in angiogenesis. HIF-1α quickly upregulates VEGF expression to initiate the process of angiogenesis, and HIF-2α sustains the pro-angiogenic response in chronic hypoxia to promote vascular remodeling and integrity [72,83].
HIF-1α also plays a key role in recruiting hematopoietic and endothelial progenitor cells (EPCs) from the bone marrow to sites of ischemic tissue via its direct upregulation of its downstream target stromal-derived factor 1-alpha (SDF-1α) [88,89]. SDF-1α is a secreted cytokine from cells of ischemic tissue that enters the peripheral circulation and mobilizes to the bone marrow, where it binds with its receptor, CXCR4, on the cell surface of EPCs [90]. SDF-1α works synergistically with other pro-angiogenic mobilizing factors such as VEGF, hepatocyte growth factor (HGF), and eNOS to mobilize EPCs from the bone marrow into the peripheral circulation [88,91].
A SDF-1α concentration gradient is established between the sites of ischemic insult and the peripheral circulation, resulting in homing of EPCs to sites of ischemia [92]. EPCs then proliferate and differentiate into mature endothelial cells that contribute to the formation of new blood vessels [89,90]. EPCs also secrete a variety of growth factors such as VEGF and SDF-1α that promote angiogenesis and further recruitment of EPCs to sites of ischemia [89,91]. Studies have shown that SDF-1 levels are increased after ischemic events, and cleavage-resistant gene delivery platforms of SDF-1 offer therapeutic potential in rodent models of myocardial infarction [93,94,95].
The HIF proteins upregulate a broad range of known pro-angiogenic genes. A full list can be seen in Table 1.
5. HIFs and Ischemic Cardiovascular Disease
Ischemic cardiovascular disease is the leading cause of death in the United States [96]. Arterial stenosis and subsequent occlusion due to the development of atherosclerotic plaque burden over time results in downstream tissue ischemia and hypoxia characterized by a reduction in blood flow and decreased oxygen supply that is insufficient for oxygen demand [97]. Atherosclerotic stenosis and occlusion are the pathological basis for many cardiovascular diseases including coronary artery disease (CAD), cerebral ischemia and stroke, mesenteric and renal ischemia, and PAD of the extremities [3,97]. Prolonged and worsening tissue hypoxia from severe atherosclerotic disease ultimately leads to end organ dysfunction such as ischemic cardiomyopathy in CAD and tissue loss in CLTI, the most severe form of PAD [3]. Additionally, acute plaque rupture and vessel thrombosis in the coronary, peripheral, or cerebral circulation results in acute severe hypoxia and infarction of tissue in myocardial infarction, acute limb ischemia, and stroke, respectively [98]. Moreover, myocardial conditions such as atrial fibrillation and left ventricular aneurysm, along with atherosclerotic aortic or carotid artery disease, can predispose patients to embolic events which result in acute tissue ischemia and infarction [96,97]. Abrupt onset of tissue ischemia is frequently more catastrophic given the lack of vessel collateralization that can be seen with chronic stenosis and occlusion [3].This section will focus on the role of HIF-1α in CAD and PAD followed by therapeutic implications for promoting angiogenesis and vessel collateralization.
As discussed previously, HIF-1α is the major driver of hypoxia-induced angiogenesis and vessel collateralization to ischemic cardiomyocytes due to coronary artery atherosclerosis [3,99]. Many patients with CAD present with vessel collateralization bypassing obstructive plaque, while others lack collaterals. Increased collateralization correlates with reduced infarct size, lower heart failure risk, and decreased mortality [100,101]. In a porcine model of acute myocardial infarction, overexpression of HIF-1α resulted in increased myocardial perfusion post-injury [102]. HIF-1α expression also supports cardioprotection, reduced infarct size, and ischemic preconditioning [103]. In the acute phase of ischemic insult, this HIF-1α–mediated response serves as a protective mechanism to rescue injured tissue and restore perfusion. However, when hypoxic and ischemic insults are prolonged or overwhelming, the compensatory capacity of HIF-1α becomes maladaptive, tipping the balance toward pathological remodeling, chronic inflammation, and disease progression (Figure 2).
Single nucleotide polymorphisms (SNPs) in the HIF-1 gene, specifically SNPs that lead to a Pro582Ser substitution, are associated with reduced collateral formation in coronary artery disease (CAD) and is linked a clinical presentation of stable exertional angina rather than acute myocardial infarction, indicating a potential role in earlier disease presentation [104]. In a Mexican population, the SNP rs2057482 is associated with decreased risk of developing premature CAD [105]. While out of scope of this review, the same SNP is associated with increased risk of various cancers and predictive of clinical outcomes, and has reduced binding to microRNA-199a, a negative regulator of HIF-1 levels that binds to the 3’-UTR [106,107]. This implies that increased HIF-1 protein levels confer protection against coronary ischemic events but may predispose patients to cancer progression that may be mediate by microRNA-199a. Indeed, the genetic diversity of HIF1A and the varying risks of cancer risk versus CAD protection is interesting and requires further study.
In contrast, a recent systemic review and meta-analysis by Chaar and colleagues have found no association between SNPs of HIF-1 and risk of peripheral artery disease. [108]. These risk factors are linked to decreased HIF-1α expression, reducing VEGF levels and endothelial progenitor cell recruitment. HIF-1α transcriptional activity drives endothelial cell sprouting, migration, and proliferation under hypoxia [3]. Vascular smooth muscle cells also promote vascular integrity during peripheral arterial perfusion [109]. Borton et al. showed that smooth muscle-specific deletion of ARNT (HIF-1β) increased vascular permeability and tissue damage in mice after femoral artery ligation, resembling acute limb ischemia [109]. These findings complicate the route to developing effective HIF-based therapies for PAD.
Acute limb ischemia, often from emboli, differs from chronic PAD but can occur in PAD patients as acute on chronic limb ischemia. Tuomisto et al. found higher HIF-1α, HIF-2α, VEGF, VEGFR-2, and TNF-α expression in acute on chronic limb ischemia compared to chronic limb ischemia [110]. Heterogeneity in PAD patient populations, including socioeconomic factors, may affect HIF-1α expression and collateralization [111].
6. HIF-1α Modulation for Therapeutic Angiogenesis and Ischemic Cardiovascular Diseases
The standard of care for ischemic cardiovascular disease is restoring arterial perfusion to alleviate hypoxia. In CAD and MI, this is typically achieved through percutaneous coronary intervention (PCI) using balloons and drug-eluting stents [112]. Some patients with multivessel disease or unfavorable anatomy are better suited for coronary artery bypass grafting (CABG), traditionally requiring sternotomy and cardiopulmonary bypass, though less invasive options are emerging [113]. Ischemic stroke is treated with tissue plasminogen activator (tPA) or mechanical thrombectomy to reestablish perfusion [114].
Chronic limb-threatening ischemia (CLTI) is marked by ischemic pain or tissue loss, most often in the distal lower extremities. Without intervention, these patients face a 22% annual risk of major limb amputation [115]. As with CAD, treatment involves endovascular or surgical revascularization to improve distal blood flow, oxygen delivery, pain relief, wound healing, and limb salvage. However, many patients are not candidates for revascularization due to comorbidities, previous failed interventions, or lack of suitable outflow targets.
Diabetes frequently coexists with CLTI and contributes to both macrovascular and microvascular disease [116]. Occlusions often occur in the tibial and foot arteries, making surgical bypass challenging and less durable due to the distal location. Even when large vessels are successfully treated, microvascular disease in the diabetic foot remains a barrier to healing. Patients who cannot undergo revascularization are deemed to have “no-option” CLTI. In these cases, therapies that enhance HIF-1α expression and angiogenesis may offer new options for improving tissue oxygenation [117].
7. Prolyl Hydroxylase Domain Inhibition
HIF-1α and HIF-2α are regulated by oxygen-dependent prolyl hydroxylase domain (PHD) enzymes, which target them for degradation. Inhibiting PHD enzymes stabilize HIF proteins and may promote angiogenesis [118]. Several preclinical studies have demonstrated the promise of this approach. In murine hindlimb ischemia models, PHD knockout or knockdown improved perfusion, motor function, and capillary density [119]. Studies using short hairpin RNA (shRNA) targeting PHD2 delivered via a minicircle vector (MC-shPHD2) achieved greater transfection efficiency, higher skeletal muscle HIF-1α levels, and up to 50% blood flow recovery compared to conventional vectors [120,121]. These results highlight the importance of delivery methods in gene-based therapies.
In myocardial infarction models, PHD2 knockout led to markedly increased HIF-1α and VEGF levels in peri-infarct tissue, resulting in enhanced neovascularization, reduced fibrosis, and improved cardiac function [122,123,124]. Dual knockdown of PHD and FIH further augmented angiogenesis, progenitor cell recruitment, and reduced apoptosis, with upregulation of downstream genes such as VEGF, FGF2, and KDR [125]. Similar cardioprotective effects have been shown in various mouse and human tissue models using pharmacologic or genetic silencing of PHD proteins [126]. However, not all findings have been favorable. In vitro treatment of human endothelial cells with dimethyloxalylglycine (DMOG), a chemical PHD inhibitor, reduced endothelial proliferation, migration, and tube formation, despite increased HIF levels [127]. This suggests that the method of HIF stabilization, cell type, and experimental context significantly influence the angiogenic response.
Clinically, translation has been limited for PAD patients. One randomized trial using an oral PHD inhibitor GSK1278863 in PAD patients failed to improve walking performance or increase expression HIF-1 target genes [128]. Limitations included short treatment duration, oral administration, and lack of angiographic assessment. While oral PHD inhibitors like Roxadustat, Daprodustat, and Vadadustat have been approved to stimulate erythropoiesis in chronic kidney disease, their role in promoting angiogenesis for PAD or CLTI remains unproven. Safety concerns, including risks of thromboembolism and pulmonary hypertension, further complicate their use.
8. HIF-1α Gene Overexpression
While inhibiting the inhibitor of HIF-1α is a strategy to promote HIF-1α protein stabilization, inducing HIF-1α overexpression is an alternative to promote neovascularization [117]. Gene therapy for therapeutic angiogenesis uses plasmids or viral vectors to deliver target genes to ischemic tissue. Viral vectors include adenovirus, adeno-associated virus, and retroviruses [129]. Early preclinical studies using downstream targets of HIF-1α like VEGF, HGF, and FGF showed promise, but clinical trials with these growth factors yielded inconsistent results in PAD and CAD [130,131]. A trial in diabetic patients with no-option CLTI using VEGF/HGF bicistronic plasmid therapy reported increased serum VEGF, ABIs, and vessel collateralization, along with improved rest pain [132]. However, the trial was limited by a small cohort. These results suggest that coordinated signaling from multiple factors, as induced by HIF-1α, may be necessary for robust angiogenesis.
Xue and colleagues used a transgenic diabetes mouse model to show that cardiomyocyte-specific HIF-1α overexpression increases myocardial capillary density and prevented diabetes-mediated cardiac hypertrophy and glycolytic metabolism remodeling [133]. In a mouse model of myocardial infarction, constitutive expression of HIF-1α attenuated infarct size, increased capillary density, and improved heart function 4 weeks after myocardial infarction [134]. This supports a rationale for targeting HIF-1α directly instead of its downstream factors. Preclinical studies have reinforced this. Intramyocardial injection of HIF-1α/VP16 hybrid increased capillary density and blood flow in rats post-LAD occlusion, similar to VEGF treatment [135]. Combined HIF-1α and VEGF therapy further increased vessel density but did not reduce infarct size. Remote quadriceps injection of HIF-1α promoted coronary vessel growth, reduced infarct size, and improved ventricular function, suggesting a role in ischemic preconditioning [136]. Sarkar et al. demonstrated in a mouse diabetic model of critical limb ischemia that adenoviral HIF-1α (AdCA5) increased arterial remodeling and perfusion, promoting both angiogenesis and arteriogenesis [137]. In diabetic mice, AdCA5 improved perfusion, tissue viability, and motor function and increased circulating angiogenic cells (CACs), which are typically diminished in diabetes [137].
Despite promising preclinical data, clinical trials with intramuscular HIF-1α gene therapy for PAD have been disappointing. A Phase 1 trial showed safety without tumorigenesis or ocular neovascularization, with some patients experiencing pain resolution and ulcer healing [131]. However, a larger double-blinded, randomized control trial in patients with intermittent claudication showed no improvement in walking time, ABIs, or biomarkers [138]. Low transfection efficiency may explain these results. Newer vectors like AAV2 and AAV9 may improve outcomes, though they have not been tested in humans. Inadequate preclinical models and differences in patient pathophysiology further complicate translation [117].
9. Cell-Based HIF-1α Therapies
Stem cell-based therapies show potential for ischemic cardiovascular disease. MSCs, ADSCs, EPCs, and iPSCs can differentiate into various cell types and secrete angiogenic factors [139]. MSCs are particularly attractive due to ease of harvest and low immunogenicity. Extracellular vesicles (EVs) from stem cells, such as exosomes, deliver pro-angiogenic molecules and influence target cells through paracrine signaling [139]. Stem cells also promote EPC homing via SDF-1α and can differentiate into relevant vascular and cardiac cells [139]. Nonetheless, clinical application of unmodified stem cells is limited by poor viability, retention, and homing. Hypoxic/ischemic environments, especially in diabetics, impair stem cell survival. Strategies to overcome this include genetic modification, chemical and physical surface modifications, and hydrogel encapsulation. HIF-1α is central to many of these enhancements [117].
Hypoxia preconditioning activates HIF-1α and improves stem cell survival, proliferation, and pro-angiogenic activity [140]. A systematic review of hypoxia-conditioned ADSCs showed consistent upregulation of pro-angiogenic markers and viability [141]. Studies using hypoxia-mimicking agents or reduced oxygen tension confirmed these findings in vitro and in vivo [139]. One murine study showed that hi-MSCs enhanced perfusion, vessel density, and HIF-1α/VEGF expression versus normoxic MSCs [139]. Direct HIF-1α overexpression in stem cells using plasmids or viral vectors also enhances pro-angiogenic function [142]. CSCs may outperform MSCs in this regard. A study using HIF-1α-transfected CSCs embedded in fibrin gel (HIF-CSC-Gel) improved limb perfusion more than CSCs without the gel [142]. Combined therapy using HIF-1α gene delivery and MSCs in a myocardial infarction model enhanced angiogenesis and cardiac function compared to monotherapies, possibly due to improved MSC engraftment [142]. Future studies should explore combined therapies to optimize outcomes.
Stem cell-derived EVs can also deliver miRNAs like miR-31 and miR-20b to ischemic tissues, promoting angiogenesis and reducing apoptosis in models of myocardial ischemia and reperfusion injury [143]. miR-31 targets FIH, reducing its expression and thereby enhancing HIF-1α activity [144]. Engineering stem cells or EVs with these miRNAs offers another avenue to boost HIF-1α-dependent neovascularization.
The role of HIF-2α in therapeutic angiogenesis has received less attention. While better studied in cancer, HIF-2α contributes to vascular remodeling and integrity during chronic hypoxia [117]. Combining HIF-1α and HIF-2α gene therapies may offer a more durable and functional angiogenic response in atherosclerotic cardiovascular disease [117].
In summary, ischemic cardiovascular diseases including CAD and PAD comprise a massive healthcare burden with regards to morbidity, mortality, and healthcare economic burden [96]. The mainstay of treatment for both conditions is via traditional lifestyle modifications and medical therapy aimed at cardiovascular risk reduction (statins, anti-platelet agents, glycemic control, smoking cessation, etc.) and endovascular or surgical revascularization in the cases of AMI and CLTI [112,115]. Surgically or anatomically unfit patients exist in both CAD and PAD, leading to the basis for gene and cell-based therapy to improve neovascularization and improve functional outcomes. Despite early preclinical promise for both mechanisms of therapy, clinical trials have demonstrated serious limitations to gene and cell-based therapy resulting in failed treatment outcomes [131,132]. Improved functional outcomes in cardiac function, maximal walking distance, amputation-free survival, quality of life, morbidity, and mortality is the goal of therapeutic angiogenesis with these treatment modalities.
5. Conclusions
HIF-1α is the master regulator of a myriad of pro-angiogenic growth factors and cell survival responses and thus, HIF-1α overexpression is a key strategy to improve gene and cell-based therapy outcomes in treating ischemic cardiovascular diseases. Future gene therapy-based strategies should focus on optimal vector delivery and transfection efficiency as well as duration of action and optimal dosing. Hypoxic preconditioning of stem cells has demonstrated improved pro-angiogenic phenotypes in multiple stem cell types and is mediated by induction of HIF-1α expression. Future clinical studies of cell-based treatment of ischemic cardiovascular disease should incorporate hypoxic preconditioning into their approach for optimized pro-angiogenic and cell retention efficacy. Combination treatments with gene and cell-based therapy may have additive effects in promoting stem cell engraftment, neovascularization, and functional outcomes. The addition of stem-cell derived exosomes for the delivery of miRNAs known to promote HIF-1α expression is also an active area of ongoing preclinical research and be additive to the above therapeutic strategies. The potential of HIF-2α for therapeutic angiogenesis in ischemic cardiovascular is not well studied in the literature and further investigation, either as a monotherapy or in combination with HIF-1α gene therapy, is warranted. Novel strategies to increase neovascularization of ischemic tissues which are not necessarily directly related to HIF-1α are under investigation in our research laboratory and involve the membrane-bound adhesion molecule, E-Selectin [145,146,147,148,149,150,151] To-date, no mechanistic intercept has been discovered that links the angiogenic signaling pathways of HIF-1α and membrane-bound E-Selectin. However, the potential for synergism in combination therapies may deserve additional study.
Author Contributions
Conceptualization, P.J.B., A-I.S.R., Y.Y.O., D.A.R, K.G; writing—original draft preparation, E.C., P.J.B., A-I.S.R.; writing—review and editing, B-N.N., A.B., Z-J.L., O.C.V.; visualization, A-I.S.A., P.J.B.; supervision, Z-J.L., O.C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health—NIH/NHLBI Catalyze R33 HL156141; NIH T32GM145462 Medical Scientist Training Program; Philanthropy: Eloise & David Kimmelman Foundation.
Acknowledgments
The authors thank all members in Surgical Vascular Research Labs at DeWitt Daughtry Family Department of Surgery, University of Miami Miller School of Medicine for helpful discussion and suggestion.
Conflicts of Interest
The authors Ethan Carmichael, Patrick J. Bosco, Anne-Isabelle S. Reme, Yulexi Y. Ortiz, Daniela Alexandra Ramos, Katherine Gomez, Bao-Ngoc Nguyen, and Arash Bornak declare no conflicts of interest. Authors: Zhao-Jun Liu (ZJL) and Dr. Omaida C. Velazquez (OCV) declare the following potential conflicts of interest with respect to the research, authorship, and/or presentation and/or publication of some aspects that are indirectly related to this work: E-selectin gene modification technologies aimed as pro neovascularization technologies were developed in our research laboratory and patented/licensed by the University of Miami. These E-Selectin technologies are currently under pre-clinical development by Ambulero Inc., a new start-up company out of the University of Miami that focuses on developing new vascular treatments for ischemic tissue conditions and limb salvage. ZJL and OCV serve as Ambulero Inc. consultants and chief scientific and medical advisory officers, respectively and are co-inventors of the E-Selectin technologies, and are minority shareholders in Ambulero Inc. ZJL and OCV are also funded by the NIH/NHLBI and philanthropy in preclinical investigations of E-Selectin technologies and other pro neovascularization technologies.
Abbreviations
The following abbreviations are used in this manuscript:
| HIFs | hypoxia inducible factors |
| EPO | erythropoietin |
| RBC | red blood cell |
| VEGF | Vascular Endothelial Growth Factor |
| TCA | tricarboxylic acid cycle |
| ARNT | aryl hydrocarbon receptor nuclear translocator |
| ODD | oxygen-dependent degradation |
| PHD | prolyl hydroxylase domain |
| VHL | Von Hippel Lindau |
| DFO | deferoxamine |
| CoCl2 | cobalt chloride |
| HREs | hypoxic response elements |
| N-TAD | N-terminal activation domains |
| C-TAD | C-terminal activation domains |
| FIH | Factor Inhibiting HIF |
| HAF | hypoxia-associated factor |
| ROS | reactive oxygen species |
| PASMCs | pulmonary artery smooth muscle cells |
| EGFR | Epidermal Growth Factor Receptor |
| PDGF | Platelet-Derived Growth Factor |
| TNF-α | Tumor Necrosis Factor-alpha |
| IL-1β | Interleukin-1 beta |
| GHRH | growth hormone-releasing hormone |
| miRNAs | microRNAs |
| GPD1L | glycerol-3-phosphate dehydrogenase 1-like |
| eNOS | endothelial nitric oxide synthase |
| SMC | smooth muscle cell |
| MCP-1 | monocyte chemotactic protein-1 |
| CAMs | cell adhesion molecules |
| PAD | Peripheral Artery Disease |
| CLTI | chronic limb-threatening ischemia |
| Dll4 | delta like ligand 4 |
| MMPs | matrix metalloproteases |
| uPA | urokinase plasminogen activator |
| PAI-1 | plasminogen activator inhibitor-1 |
| ECM | extracellular matrix |
| EPCs | endothelial progenitor cells |
| HGF | hepatocyte growth factor |
| SDF-1α | Stromal Cell-Derived Factor-1 alpha |
| CAD | coronary artery disease |
| SNPs | Single nucleotide polymorphisms |
| MI | Myocardial Infarction |
| PCI | percutaneous coronary intervention |
| CABG | coronary artery bypass grafting |
| tPA | tissue plasminogen activator |
| shRNA | short hairpin RNA |
| MC-shPHD2 | minicircle vector |
| FGF | Fibroblast Growth Factor |
| KDR | Kinase Insert Domain Receptor |
| DMOG | Dimethyloxalylglycine |
| ABI | Ankle-Brachial Index |
| AdCA5 | adenoviral HIF-1α |
| CACs | circulating angiogenic cells |
| AAV | Adeno-Associated Virus |
| MSCs | Mesenchymal Stem Cells |
| ADSCs | Adipose-Derived Stem Cells |
| EPCs | Endothelial Progenitor Cells |
| iPSCs | Induced Pluripotent Stem Cells |
| EVs | Extracellular vesicles |
| CSCs | Cardiac stem cells |
| HIF-CSC-Gel | HIF-1α-transfected CSCs embedded in fibrin gel |
| AMI | acute myocardial infarction |
References
- Semenza, G.L. , Regulation of Mammalian O2 Homeostasis by Hypoxia-Inducible Factor 1. Annu Rev Cell Dev Biol, 1999. 15: p. 551-578.
- Wenger, R.H. , Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J, 2002. 16(10): p. 1151-1162. [CrossRef]
- Semenza, G.L. , Hypoxia-inducible factors in physiology and medicine. Cell, 2012. 148(3): p. 399-408.
- Wang, G.L. , et al., Hypoxia-inducible factor 1 is a basic-helix–loop–helix–PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A, 1995. 92(12): p. 5510-5514.
- Semenza, G.L. , Wang G.L., A Nuclear Factor Induced by Hypoxia via De Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Molecular and Cellular Biology, 1992. 12: p. 5447-5454.
- Gregg, L. Semenza, M.K.N., Suzie M. Chi, Stylianos E. Antonarakis, Hypoxia-inducible nuclear factors bind to an enhancer element located 3' to the human erythropoietin gene. PNAS, 1991. 88: p. 5680-5684.
- Haase, V.H. , Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev, 2013. 27(1): p. 41-53. [CrossRef]
- Wei Liu, S.-M.S. , Xu-Yun Zhao, Guo-Qiang Chen, Targeted genes and interacting proteins of hypoxia inducible factor-1 Int J Biochem Mol Biol, 2012. 3: p. 165-178.
- Forsythe, J.A. , et al., Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol, 1996. 16(9): p. 4604-4613.
- Shweiki, D. , Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature, 1992. 359(6398): p. 843-845.
- Yuxiang Liu, S.R.C. , Toshisuke Morita, and Stella Kourembanas, Hypoxia Regulates Vascular Endothelial Growth Factor Gene Expression in Endothelial Cells. Circulation Research, 1995. 77(3): p. 638-643.
- Papandreou, I. , et al., HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab, 2006. 3(3): p. 187-197. [CrossRef]
- Semenza, G.L. , et al., Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem, 1994. 269(38): p. 23757-23763.
- Eltzschig, H.K. and P. Carmeliet, Hypoxia and inflammation. N Engl J Med, 2011. 364(7): p. 656-665. [CrossRef]
- P. H. Maxwell, G.U.D., J. M. Gleadle, L. G. Nicholls, A. L. Harris, I. J. Stratford, O. Hankinson, C. W. Pugh, and P. J. Ratcliffe, Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. PNAS, 1997. 94: p. 8104-8109.
- M.E. Hubbi, G.L.S., Regulation of cell proliferation by hypoxia-inducible factors. Am J Physiol Cell Physiol., 2015. 309. [CrossRef]
- S. Rey, G.L.S., Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res, 2010. 86: p. 236–242. [CrossRef]
- D. Wu et al., Structural characterization of mammalian bHLH-PAS transcription factors Curr Opin Struct Biol., 2016. 43: p. 1-9. [CrossRef]
- K. Hirose et al., cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol Cell Biol., 1996. 16(4): p. 1706-1713.
- M. A. Maynard et al., Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma FASEB J, 2005. 19(11): p. 1396-1406. [CrossRef]
- Maxwell, P.H. , et al., The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature, 1999. 399(6733): p. 271-275.
- Epstein, A.C. , et al., C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell, 2001. 107(1): p. 43-54. [CrossRef]
- W. G. Kaelin Jr et al., The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma Clin Cancer Res., 2007. 13: p. 680s-684s. [CrossRef]
- Ivan, M. , et al., HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science, 2001. 292(5516): p. 464-468. [CrossRef]
- Huang, L.E. , et al., Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J Biol Chem, 1996. 271(50): p. 32253-32259.
- Schofield, C.J. and P.J. Ratcliffe, Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol, 2004. 5(5): p. 343-354. [CrossRef]
- Goldberg, M.A., S. P. Dunning, and H.F. Bunn, Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science, 1988. 242(4884): p. 1412-1415.
- Yuan, Y. , et al., Cobalt inhibits the interaction between hypoxia-inducible factor-α and von Hippel–Lindau protein by direct binding to hypoxia-inducible factor-α. J Biol Chem, 2003. 278(18): p. 15911-15916. [CrossRef]
- Jaakkola, P. , et al., Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science, 2001. 292(5516): p. 468-472. [CrossRef]
- Semenza, G.L. , HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol, 2000. 88(4): p. 1474-1480. [CrossRef]
- Mahon, P.C., K. Hirota, and G.L. Semenza, FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev, 2001. 15(20): p. 2675-2686. [CrossRef]
- Arany, Z. , et al., An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A, 1996. 93(23): p. 12969-12973. [CrossRef]
- Lando, D. , et al., FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev, 2002. 16(12): p. 1466-1476. [CrossRef]
- Hu, C.J. , et al., Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol Cell Biol, 2003. 23(24): p. 9361-9374.
- Tian, H., S. L. McKnight, and D.W. Russell, Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev, 1997. 11(1): p. 72-82. [CrossRef]
- Makino, Y. , et al., Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3α locus. J Biol Chem, 2002. 277(36): p. 32405-32408. [CrossRef]
- Wenger, R.H. and M. Gassmann, Oxygen(es) and the hypoxia-inducible factor-1 (HIF-1): Approaching the next level of oxygen sensing. Biol Chem, 1997. 378(7): p. 609-616.
- Kietzmann, T. and A. Gorlach, Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol, 2005. 16(4-5): p. 474-486. [CrossRef]
- Koh, M.Y. and G. Powis, Passing the baton: the HIF switch. Trends Biochem Sci, 2012. 37(9): p. 364-372. [CrossRef]
- Koh, M.Y. , et al., The hypoxia-associated factor switches cells from HIF-1α to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res, 2011. 71(11): p. 4015-4027.
- Rocha, S. , Gene regulation under low oxygen: holding your breath for transcription. Trends Biochem Sci, 2007. 32(8): p. 389-397. [CrossRef]
- Semenza, G.L. , Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol, 2002. 64(5-6): p. 993-998. [CrossRef]
- Rius, J. , et al., NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature, 2008. 453(7196): p. 807-811. [CrossRef]
- Bonello, S. , et al., Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler Thromb Vasc Biol, 2007. 27(4): p. 755-761. [CrossRef]
- Diebold, I. , et al., The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Molecular Biology of the Cell, 2010. 21(12): p. 2087-2096. [CrossRef]
- Oliver, K.M., C. T. Taylor, and E.P. Cummins, Hypoxia. Regulation of NFκB signaling during inflammation: the role of hydroxylases. Arthritis Res Ther, 2009. 11(1): p. 215-215. [CrossRef]
- Richard, D.E., E. Berra, and J. Pouysségur, Angiogenesis: how a tumor adapts to hypoxia. Biochem Biophys Res Commun, 1999. 266(3): p. 718-722. [CrossRef]
- Taylor, C.T. and E.P. Cummins, The role of NF-κB in hypoxia-induced gene expression. Ann N Y Acad Sci, 2009. 1177: p. 178-184. [CrossRef]
- Ghosh et al., Tumor Necrosis Factor Alpha-Induced Hypoxia-Inducible Factor 1α–β-Catenin Axis Regulates Major Histocompatibility Complex Class I Gene Activation through Chromatin Remodeling. 2013: Mol Cell Biol. p. 2718–2731. [CrossRef]
- K. W. Kim et al., TNF-α upregulates HIF-1α expression in pterygium fibroblasts and enhances their susceptibility to VEGF independent of hypoxia. 2017: Experimental Eye Research, 164, 74–81. [CrossRef]
- S. Tsapournioti et al., TNFα induces expression of HIF-1α mRNA and protein but inhibits hypoxic stimulation of HIF-1 transcriptional activity in airway smooth muscle cells. J Cell Physiol., 2013. 228(8): p. 1745-1753. [CrossRef]
- Y.-J. Jung et al., IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis FASEB J., 2003. 17(14): p. 2115-2117. [CrossRef]
- Zhong, H., K. Chiles, and D. Feldser, Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells. Cancer Res, 2000. 60(6): p. 1541-1545.
- Görlach, A. and S. Bonello, The cross-talk between NF-κB and HIF-1: further evidence for a significant liaison. Biochem J, 2008. 412(3): p. e17-e19. [CrossRef]
- E. Laughner et al., HER2 (neu) Signaling Increases the Rate of Hypoxia-Inducible Factor 1α (HIF-1α) Synthesis: Novel Mechanism for HIF-1-Mediated Vascular Endothelial Growth Factor Expression Mol Cell Biol., 2001. 21(12): p. 3995-4004. [CrossRef]
- Treins, C. and et al., Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem, 2002. 277(31): p. 27975-27981. [CrossRef]
- R. Kanashiro-Takeuchi et al., Cardiomyocyte-specific expression of HIF-1α mediates the cardioprotective effects of Growth Hormone Releasing Hormone (GHRH). bioRxiv, 2025. [CrossRef]
- Wanschel et al., Oxygen-independent, hormonal control of HIF-1α regulates the developmental and regenerative growth of cardiomyocytes. bioRxiv, 2023. [CrossRef]
- A. Albanese and et al., The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity Int. J. Mol. Sci., 2021. 22(1). [CrossRef]
- J. Bárdos and et al., Negative and positive regulation of HIF-1: A complex network. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 2005. 1755(2): p. 107-120. [CrossRef]
- Y. Mizukami et al., ERK1/2 regulates intracellular ATP levels through alpha-enolase expression in cardiomyocytes exposed to ischemic hypoxia and reoxygenation J Biol Chem., 2004. 279(48): p. 50120-50131. [CrossRef]
- Sang, N. and et al., MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem, 2003. 278(16): p. 14013-14019. [CrossRef]
- Chan, S.Y. and J. Loscalzo, MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle, 2010. 9(6): p. 1072-1083. [CrossRef]
- C. Devlin and et al., miR-210: More than a silent player in hypoxia. IUBMB Life, 2011. 63(2): p. 94-100. [CrossRef]
- Huang, X. , Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol Cell, 2009. 35(6): p. 856-867. [CrossRef]
- G. Cao and et al., MiR-210 regulates lung adenocarcinoma by targeting HIF-1α. Heliyon, 2023. 9(5). [CrossRef]
- H. Wang and et al., Negative regulation of Hif1a expression and T H 17 differentiation by the hypoxia-regulated microRNA miR-210 Nature Immunology, 2014. 15: p. 393-401. [CrossRef]
- T. Kelly and et al., A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like Mol Cell Biol, 2011. 31(13): p. 2696-2706. [CrossRef]
- Fasanaro, P. , MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem, 2008. 283(23): p. 15878-15883. [CrossRef]
- Bruning, U. , MicroRNA-155 promotes resolution of hypoxia-inducible factor 1α activity during prolonged hypoxia. Mol Cell Biol, 2011. 31(19): p. 4087-4096. [CrossRef]
- Risau, W. , Mechanisms of angiogenesis. Nature, 1997. 386(6626): p. 671-674. [CrossRef]
- Carmeliet, P. , Angiogenesis in life, disease and medicine. Nature, 2005. 438(7070): p. 932-936. [CrossRef]
- F. Pipp et al., Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb Aterioscler Thromb Vascular Biol., 2004. 24(9): p. 1664-1668. [CrossRef]
- Helisch, A. and W. Schaper, Arteriogenesis: the development and growth of collateral arteries. Microcirculation, 2003. 10(1): p. 83-97. [CrossRef]
- Z. Peng et al., CCL2 promotes proliferation, migration and angiogenesis through the MAPK/ERK1/2/MMP9, PI3K/AKT, Wnt/β-catenin signaling pathways in HUVECs Exp Ther Med., 2022. 25(2). [CrossRef]
- Gerhardt, H. , et al., VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. Journal of Cell Biology, 2003. 161(6): p. 1163-1177. [CrossRef]
- Phng, L.K. and H. Gerhardt, Angiogenesis: a team effort coordinated by Notch. Developmental Cell, 2009. 16(2): p. 196-208. [CrossRef]
- Lobov, I.B. , et al., Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proceedings of the National Academy of Sciences U S A, 2007. 104(9): p. 3219-3224. [CrossRef]
- Adams, R.H. and K. Alitalo, Molecular regulation of angiogenesis and lymphangiogenesis. Nature Reviews Molecular Cell Biology, 2007. 8(6): p. 464-478. [CrossRef]
- Eilken, H.M. and R.H. Adams, Dynamics of endothelial cell behavior in sprouting angiogenesis. Current Opinion in Cell Biology, 2010. 22(5): p. 617-625. [CrossRef]
- S. J. Mentzer, M.A.K., Intussusceptive Angiogenesis: Expansion and Remodeling of Microvascular Networks Angiogenesis, 2014. 17: p. 499-509. [CrossRef]
- N. Tang et al., Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis Cancer Cell, 2004. 6(5): p. 485-495. [CrossRef]
- Semenza, G.L. , HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. Journal of Clinical Investigation, 2013. 123(9): p. 3664-3681. [CrossRef]
- Ferrara, N., H. P. Gerber, and J. LeCouter, The biology of VEGF and its receptors. Nat Med, 2003. 9(6): p. 669-676. [CrossRef]
- De Bock, K. , et al., Role of PFKFB3-driven glycolysis in vessel sprouting. Cell, 2013. 154(3): p. 651-663. [CrossRef]
- Yancopoulos, G.D. , et al., Vascular-specific growth factors and blood vessel formation. Nature, 2000. 407(6801): p. 242-248. [CrossRef]
- R. Carlsson et al., Molecular Regulation of the Response of Brain Pericytes to Hypoxia Int. J. Mol. Sci, 2023. 24(6). [CrossRef]
- Carmeliet, P. and R.K. Jain, Molecular mechanisms and clinical applications of angiogenesis. Nature, 2011. 473(7347): p. 298-307. [CrossRef]
- Urbich, C. and S. Dimmeler, Endothelial progenitor cells: characterization and role in vascular biology. Circ Res, 2004. 95(4): p. 343-353. [CrossRef]
- Ceradini, D.J. and G.C. Gurtner, Homing to hypoxia: HIF-1 as a mediator of progenitor cell recruitment to injured tissue. Trends Cardiovasc Med, 2005. 15(2): p. 57-63. [CrossRef]
- Y. Yin et al., SDF-1alpha involved in mobilization and recruitment of endothelial progenitor cells after arterial injury in mice Cardiovasc Pathol., 2010. 19(4): p. 218-227. [CrossRef]
- Askari, A.T. , et al., Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischemic cardiomyopathy. Lancet, 2003. 362(9385): p. 697-703. [CrossRef]
- S. Kanki et al., Stromal Cell-Derived Factor-1 Retention and Cardioprotection for Ischemic Myocardium 2011, Circulation: Heart Failure. [CrossRef]
- D. Rath et al., Platelet surface expression of SDF-1 is associated with clinical outcomes in the patients with cardiovascular disease Platelets, 2017. 28(1): p. 34-39. [CrossRef]
- J. Tang et al., Adenovirus-mediated stromal cell-derived factor-1 alpha gene transfer improves cardiac structure and function after experimental myocardial infarction through angiogenic and antifibrotic actions Mol Biol Rep., 2010. 37(4): p. 1957-1969. [CrossRef]
- Benjamin, E.J. , et al., Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation, 2019. 139(10): p. e56-e528. [CrossRef]
- Libby, P. , Inflammation in atherosclerosis. Nature, 2002. 420(6917): p. 868-874. [CrossRef]
- Eelen, G. , et al., Endothelial cell metabolism in normal and diseased vasculature. Circulation Research, 2015. 116(7): p. 1231-1244. [CrossRef]
- Lee, S.H. , et al., Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med, 2000. 342(9): p. 626-633. [CrossRef]
- G. B. Habib et al., Influence of coronary collateral vessels on myocardial infarct size in humans. Results of phase I thrombolysis in myocardial infarction (TIMI) trial. The TIMI Investigators Circulation, 1991. 83(3): p. 739-746. [CrossRef]
- J. Elias et al., Impact of Collateral Circulation on Survival in ST-Segment Elevation Myocardial Infarction Patients Undergoing Primary Percutaneous Coronary Intervention With a Concomitant Chronic Total Occlusion. JACC: Cardiovascular Interventions, 2017. 10(9). [CrossRef]
- A. Heinl-Green et al., The efficacy of a ‘master switch gene’ HIF-1α in a porcine model of chronic myocardial ischaemia European Heart Journal, 2005. 26(13) p. 1327-1332. [CrossRef]
- Milkiewicz, M., C. W. Pugh, and S. Egginton, Inhibition of endogenous HIF inactivation induces angiogenesis in ischaemic skeletal muscles of mice. J Physiol, 2004. 560(Pt 1): p. 21-26. [CrossRef]
- M. A. Hlatky et al., Polymorphisms in hypoxia inducible factor 1 and the initial clinical presentation of coronary disease Am Heart J., 2007. 154(6): p. 1035-1042. [CrossRef]
- A. López-Reyes et al., The HIF1A rs2057482 polymorphism is associated with risk of developing premature coronary artery disease and with some metabolic and cardiovascular risk factors. The Genetics of Atherosclerotic Disease (GEA) Mexican Study. Experimental and Molecular Pathology, 2014. 96(3). [CrossRef]
- L.-F. Wu et al., The association between hypoxia inducible factor 1 subunit alpha gene rs2057482 polymorphism and cancer risk: a meta-analysis BMC Cancer, 2019. 19. [CrossRef]
- X. Guo et al., SNP rs2057482 in HIF1A gene predicts clinical outcome of aggressive hepatocellular carcinoma patients after surgery Scientific Reports, 2015. 5. [CrossRef]
- C. Chaar et al., Systematic review and meta-analysis of the genetics of peripheral arterial disease JVS Vasc Sci., 2023. 5. [CrossRef]
- A. H. Borton et al., Aryl Hydrocarbon Receptor Nuclear Translocator in Vascular Smooth Muscle Cells Is Required for Optimal Peripheral Perfusion Recovery J Am Heart Assoc., 2018. 7(11). [CrossRef]
- Tuomisto, T.T. , HIF-VEGF-VEGFR-2, TNF-α and IGF pathways are upregulated in critical human skeletal muscle ischemia as studied with DNA array. Atherosclerosis, 2004. 174(1): p. 111-120. [CrossRef]
- Annex, B.H. , Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol, 2013. 10(7): p. 387-396. [CrossRef]
- O'Gara, P.T. , et al., 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction. Circulation, 2013. 127(4): p. e362-e425. [CrossRef]
- Neumann, F.J. , et al., 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J, 2019. 40(2): p. 87-165. [CrossRef]
- Powers, W.J. , et al., 2018 Guidelines for the early management of patients with acute ischemic stroke. Stroke, 2018. 49(3): p. e46-e110. [CrossRef]
- Conte, M.S. , et al., Global vascular guidelines on the management of chronic limb-threatening ischemia. J Vasc Surg, 2019. 69(6S): p. 3S-125S.e40.
- Jude, E.B., I. Eleftheriadou, and N. Tentolouris, Peripheral arterial disease in diabetes—a review. Diabet Med, 2010. 27(1): p. 4-14. [CrossRef]
- Semenza, G.L. , Targeting HIF-1 for cancer therapy. Nat Rev Cancer, 2003. 3(10): p. 721-732. [CrossRef]
- Takeda, K., A. Cowan, and G.H. Fong, Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Mol Cell Biol, 2007. 27(21): p. 7179-7185.
- M. T. Rishi et al., Deletion of prolyl hydroxylase domain proteins (PHD1, PHD3) stabilizes hypoxia inducible factor-1 alpha, promotes neovascularization, and improves perfusion in a murine model of hind-limb ischemia. Microvascular Research, 2015. 97: p. 181-188. [CrossRef]
- L. Zhang et al., Localized Delivery of shRNA against PHD2 Protects the Heart from Acute Myocardial Infarction through Ultrasound-Targeted Cationic Microbubble Destruction Theranostics, 2017. 7(1): p. 51-66. [CrossRef]
- M. Huang et al., Short hairpin RNA interference therapy for ischemic heart disease. Circulation, 2008. 118: p. S226-S233. [CrossRef]
- Milkiewicz, M., C. W. Pugh, and S. Egginton, Inhibition of endogenous HIF inactivation induces angiogenesis in ischaemic skeletal muscles of mice. Journal of Physiology, 2004. 560(Pt 1): p. 21-26. [CrossRef]
- S. R. Pradeep et al., Protective Effect of Cardiomyocyte-Specific Prolyl-4-Hydroxylase 2 Inhibition on Ischemic Injury in a Mouse MI Model J Am Coll Surg., 2022. 235(2): p. 240-254. [CrossRef]
- M. Thirunavukkarasu et al., Stabilization of Transcription Factor, HIF-1α by Prolylhydroxylase 1 Knockout Reduces Cardiac Injury After Myocardial Infarction in Mice Cells, 2025. 14(6). [CrossRef]
- M. Huang et al., Double Knockdown of Prolyyl Hydroxylase and Factor Inhibiting HIF with Non-Viral Minicircle Gene Therapy Enhances Stem Cell Mobilization and Angiogenesis After Myocardial Infarction Circulation, 2011. 124: p. S46-S54. [CrossRef]
- L. Xie et al., Depletion of PHD3 Protects Heart from Ischemia/Reperfusion Injury by Inhibiting Cardiomyocyte Apoptosis J Mol Cell Cardiol., 2015. 80: p. 156-165. [CrossRef]
- R. Tiwari et al., Chemical inhibition of oxygen-sensing prolyl hydroxylases impairs angiogenic competence of human vascular endothelium through metabolic reprogramming. iScience, 2022. 25(10). [CrossRef]
- E. Olson et al., Short-term treatment with a novel HIF-prolyl hydroxylase inhibitor (GSK1278863) failed to improve measures of performance in subjects with claudication-limited peripheral artery disease Vasc Med., 2014. 19(6): p. 473-482. [CrossRef]
- Y. Liu et al., Advances in the study of exosomes derived from mesenchymal stem cells and cardiac cells for the treatment of myocardial infarction Cell Commun Signal, 2023. 21. [CrossRef]
- H. Shigematsu et al., Randomized, double-blind, placebo-controlled clinical trial of hepatocyte growth factor plasmid for critical limb ischemia Gene Ther., 2010. 17(9): p. 1152-1161. [CrossRef]
- S. Rajagopalan et al., Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication Circulation, 2003. 108(16): p. 1933-1938. [CrossRef]
- P. Barć et al., Double VEGF/HGF Gene Therapy in Critical Limb Ischemia Complicated by Diabetes Mellitus. J Cardiovasc Transl Res, 2021. 14(3). [CrossRef]
- W. Xue et al., Cardiac-Specific Overexpression of HIF-1α Prevents Deterioration of Glycolytic Pathway and Cardiac Remodeling in Streptozotocin-Induced Diabetic Mice Am J Pathol, 2010. 177(1): p. 97-105. [CrossRef]
- M. Kido et al., Hypoxia-Inducible Factor 1-Alpha Reduces Infarction and Attenuates Progression of Cardiac Dysfunction After Myocardial Infarction in the Mouse. Journal of the American College of Cardiology, 2005. 46(11): p. 2116-2124. [CrossRef]
- K. Shyu et al., Intramyocardial injection of naked DNA encoding HIF-1alpha/VP16 hybrid to enhance angiogenesis in an acute myocardial infarction model in the rat Cardiovascular Res, 2002. 54(3): p. 576-583. [CrossRef]
- Czibik et al., In vivo Remote Delivery of DNA Encoding for Hypoxia-inducible Factor 1 Alpha Reduces Myocardial Infarct Size. American Society for Clinical Pharmacology & Therapeutics, 2009. 2(1): p. 33-40. [CrossRef]
- K. Sarkar et al., Adenoviral transfer of HIF-1α enhances vascular responses to critical limb ischemia in diabetic mice PNAS, 2009: p. 18769-18774. [CrossRef]
- M. Creager et al., Effect of Hypoxia-Inducible Factor-1α Gene Therapy on Walking Performance in Patients With Intermittent Claudication Circulation, 2011. 124. [CrossRef]
- Hu, X. , et al., Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J Thorac Cardiovasc Surg, 2008. 135(4): p. 799-808. [CrossRef]
- I. Rosová et al., Hypoxic Preconditioning Results in Increased Motility and Improved Therapeutic Potential of Human Mesenchymal Stem Cells Stem Cells, 2008. 26(8): p. 2173-2182. [CrossRef]
- J. Garcia et al., Hypoxia-preconditioning of human adipose-derived stem cells enhances cellular proliferation and angiogenesis: A systematic review J Clin Transl Res, 2022. 8(1): p. 61-70. [CrossRef]
- X. Pei et al., Local delivery of cardiac stem cells overexpressing HIF-1α promotes angiogenesis and muscular tissue repair in a hind limb ischemia model. Journal of Controlled Release, 2020. 322: p. 610-621. [CrossRef]
- R. C. Lai et al., Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury Stem Cell Res, 2010. 4(3): p. 214-222.
- T. Chen et al., MicroRNA-31 contributes to colorectal cancer development by targeting factor inhibiting HIF-1α (FIH-1) Cancer Biol Ther, 2014. 15(5): p. 516-523. [CrossRef]
- P. P. Parikh, R.M.L.-S., H. Shao et al.,, Intramuscular E-selectin/adeno-associated virus gene therapy promotes wound healing in an ischemic mouse model J Surg Res, 2018: p. 68-76. [CrossRef]
- HJ, Q., P. Parikh, RM. Lassance-Soares et al.,, Gangrene, revascularization, and limb function improved with E-selectin/adeno-associated virus gene therapy JVS Vasc Sci, 2020. 2: p. 20-32. [CrossRef]
- HJ. Quiroz, F.S., H. Shao et al.,, E-Selectin-Overexpressing Mesenchymal Stem Cell Therapy Confers Improved Reperfusion, Repair, and Regeneration in a Murine Critical Limb Ischemia Model Front Cardiovasc Med, 2022. 8:826687. [CrossRef]
- A. Ribieras, Y.Y.O., Y. Li, et al.,, E-Selectin/AAV2/2 Gene Therapy Alters Angiogenesis and Inflammatory Gene Profiles in Mouse Gangrene Model Frontiers in Cardiovascular Medicine, 2022. 9. [CrossRef]
- A. Ribieras, Y.O., Y. Li et al.,, E-Selectin/AAV Gene Therapy Promotes Myogenesis and Skeletal Muscle Recovery in a Mouse Hindlimb Ischemia Model Cardiovasc Ther, 2023: p. 1-10. [CrossRef]
- CT. Huerta, Y.O., Li Y, et al.,, Novel Gene-Modified Mesenchymal Stem Cell Therapy Reverses Impaired Wound Healing in Ischemic Limbs Ann Surg, 2023. 278: p. 383-395. [CrossRef]
- F. Voza, Y.O., Y. Li, et al.,, Codon-Optimized and de novo-Synthesized E-Selectin/AAV2 Dose-Response Study for Vascular Regeneration Gene Therapy Ann Surg, 2024. 280(4): p. 570-583. [CrossRef]
- Le Jan, S. , et al., Angiopoietin-Like 4 Is a Proangiogenic Factor Produced during Ischemia and in Conventional Renal Cell Carcinoma. Am J Pathol, 162, 1521-1528 (2003).
- A. Mammoti, et al., Endothelial Twist1-PDGFB signaling mediates hypoxia- induced proliferation and migration of αSMA-positive cells. Scientific Reports, 10, 7563 (2020). [CrossRef]
- G. Seghezzi, et al., Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol, 141, 1659-1673 (1998). [CrossRef]
- A. Luttun, et al., Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nature Medicine, 8, 831-840 (2002). [CrossRef]
- Y. Liu, et al., MMP-2 and MMP-9 contribute to the angiogenic effect produced by hypoxia/15-HETE in pulmonary endothelial cells. J Molecular and Cellular Cardiology, 121, 36-50 (2018). [CrossRef]
- E. Revuelta-López, et al., Hypoxia Induces Metalloproteinase-9 Activation and Human Vascular Smooth Muscle Cell Migration Through Low-Density Lipoprotein Receptor–Related Protein 1–Mediated Pyk2 Phosphorylation. Arterioscler Thromb Vasc Bio, 33, 2877-2887 (2013). [CrossRef]
- Y. S. Park et al., Expression of angiopoietin-1 in hypoxic pericytes: Regulation by hypoxia-inducible factor-2α and participation in endothelial cell migration and tube formation. Biochemical and Biophysical Research Communications, 2, 263-269 (2016). [CrossRef]
Figure 1.
Oxygen-dependent and oxygen-independent regulation of HIF-1α signaling. Under normoxic conditions (left), HIF-1α undergoes prolyl hydroxylation by prolyl hydroxylase domain proteins (PHDs) and factor inhibiting HIF (FIH), enabling recognition by von Hippel–Lindau protein (pVHL). This leads to ubiquitination and proteasomal degradation of HIF-1α, preventing transcriptional activity. Under hypoxic conditions (right), reduced hydroxylation stabilizes HIF-1α, allowing its accumulation and dimerization with HIF-1β in the nucleus. The HIF-1α/β complex, together with transcriptional co-activators CBP and p300, binds to hypoxia response elements (HREs) to activate transcription of target genes. In addition to this oxygen-dependent regulation, HIF-1α can also be stabilized through oxygen-independent mechanisms, largely mediated by growth factor/receptor signaling pathways. These include PI3K–Akt/mTOR, NF-κB, and EGFR signaling, which enhance HIF-1α synthesis and transcriptional activity even under normoxia. Growth factors such as PDGF, TNF-α, IL-1β, and GHRH further potentiate these effects, amplifying the hypoxic response. Through both oxygen-dependent and oxygen-independent mechanisms, HIF-1α drives the expression of target genes that promote adaptive responses including angiogenesis (VEGF), glycolysis (GLUT1), erythropoiesis (EPO), and cell proliferation (TGF-α). Created in BioRender. Reme, A. (2025) https://BioRender.com/wytkie.
Figure 1.
Oxygen-dependent and oxygen-independent regulation of HIF-1α signaling. Under normoxic conditions (left), HIF-1α undergoes prolyl hydroxylation by prolyl hydroxylase domain proteins (PHDs) and factor inhibiting HIF (FIH), enabling recognition by von Hippel–Lindau protein (pVHL). This leads to ubiquitination and proteasomal degradation of HIF-1α, preventing transcriptional activity. Under hypoxic conditions (right), reduced hydroxylation stabilizes HIF-1α, allowing its accumulation and dimerization with HIF-1β in the nucleus. The HIF-1α/β complex, together with transcriptional co-activators CBP and p300, binds to hypoxia response elements (HREs) to activate transcription of target genes. In addition to this oxygen-dependent regulation, HIF-1α can also be stabilized through oxygen-independent mechanisms, largely mediated by growth factor/receptor signaling pathways. These include PI3K–Akt/mTOR, NF-κB, and EGFR signaling, which enhance HIF-1α synthesis and transcriptional activity even under normoxia. Growth factors such as PDGF, TNF-α, IL-1β, and GHRH further potentiate these effects, amplifying the hypoxic response. Through both oxygen-dependent and oxygen-independent mechanisms, HIF-1α drives the expression of target genes that promote adaptive responses including angiogenesis (VEGF), glycolysis (GLUT1), erythropoiesis (EPO), and cell proliferation (TGF-α). Created in BioRender. Reme, A. (2025) https://BioRender.com/wytkie.
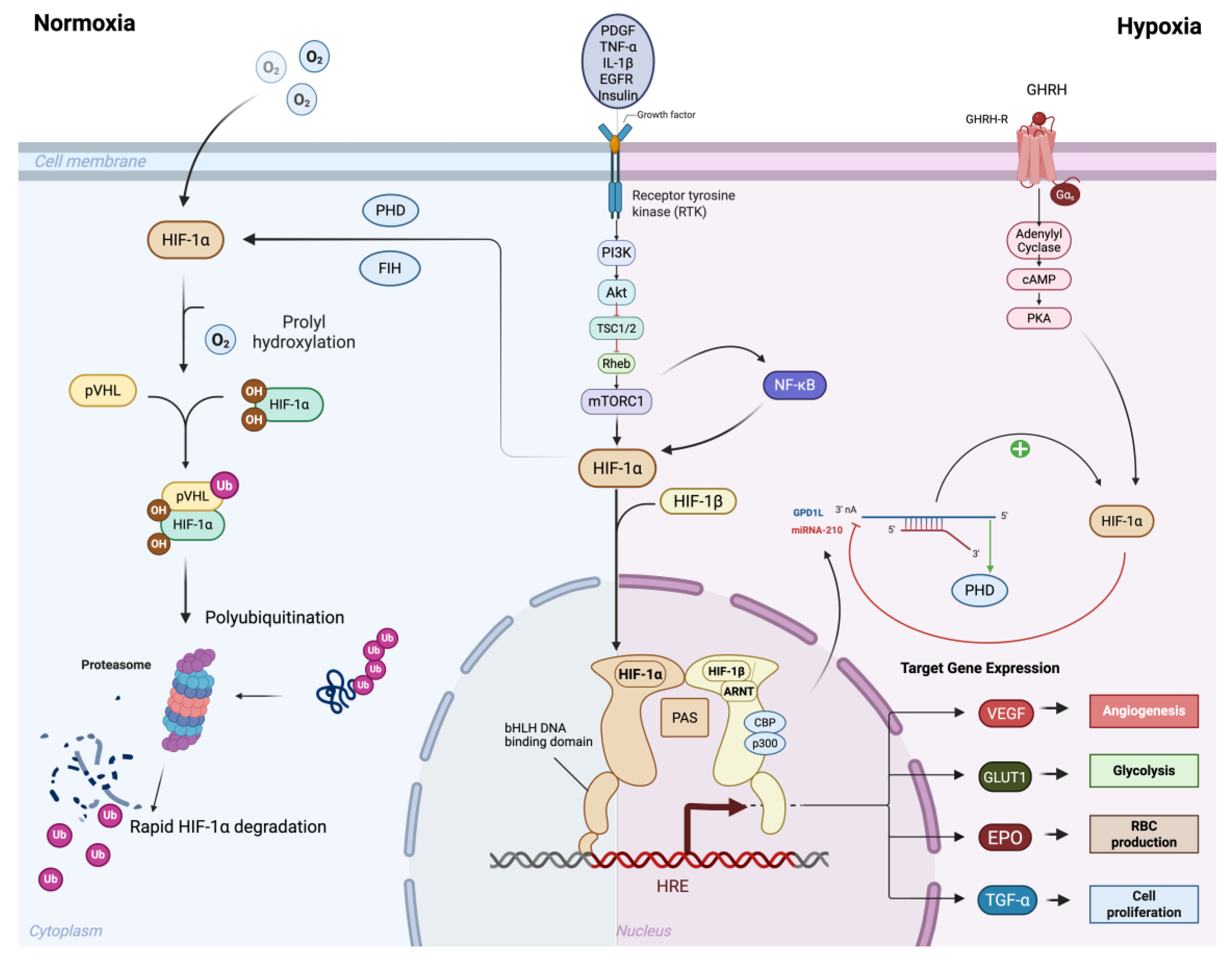
Figure 2.
HIF regulation and Ischemic Cardiovascular Diseases Atherosclerotic plaque–induced arterial stenosis causes tissue hypoxia from cardiovascular and peripheral ischemia. Hypoxia directly upregulates HIF-1α, which activates transcription of angiogenic targets (e.g., VEGF, SDF-1), promoting cell survival, angiogenesis, vessel collateralization, and improved cardiac/limb function. In the acute phase of ischemic insult, this HIF-1α–mediated response serves as a protective mechanism to rescue injured tissue and restore perfusion. However, when hypoxic and ischemic insults are prolonged or overwhelming, the compensatory capacity of HIF-1α becomes maladaptive, tipping the balance toward pathological remodeling, chronic inflammation, and disease progression. Created in BioRender. Reme, A. (2025) https://BioRender.com/wytkieo.
Figure 2.
HIF regulation and Ischemic Cardiovascular Diseases Atherosclerotic plaque–induced arterial stenosis causes tissue hypoxia from cardiovascular and peripheral ischemia. Hypoxia directly upregulates HIF-1α, which activates transcription of angiogenic targets (e.g., VEGF, SDF-1), promoting cell survival, angiogenesis, vessel collateralization, and improved cardiac/limb function. In the acute phase of ischemic insult, this HIF-1α–mediated response serves as a protective mechanism to rescue injured tissue and restore perfusion. However, when hypoxic and ischemic insults are prolonged or overwhelming, the compensatory capacity of HIF-1α becomes maladaptive, tipping the balance toward pathological remodeling, chronic inflammation, and disease progression. Created in BioRender. Reme, A. (2025) https://BioRender.com/wytkieo.
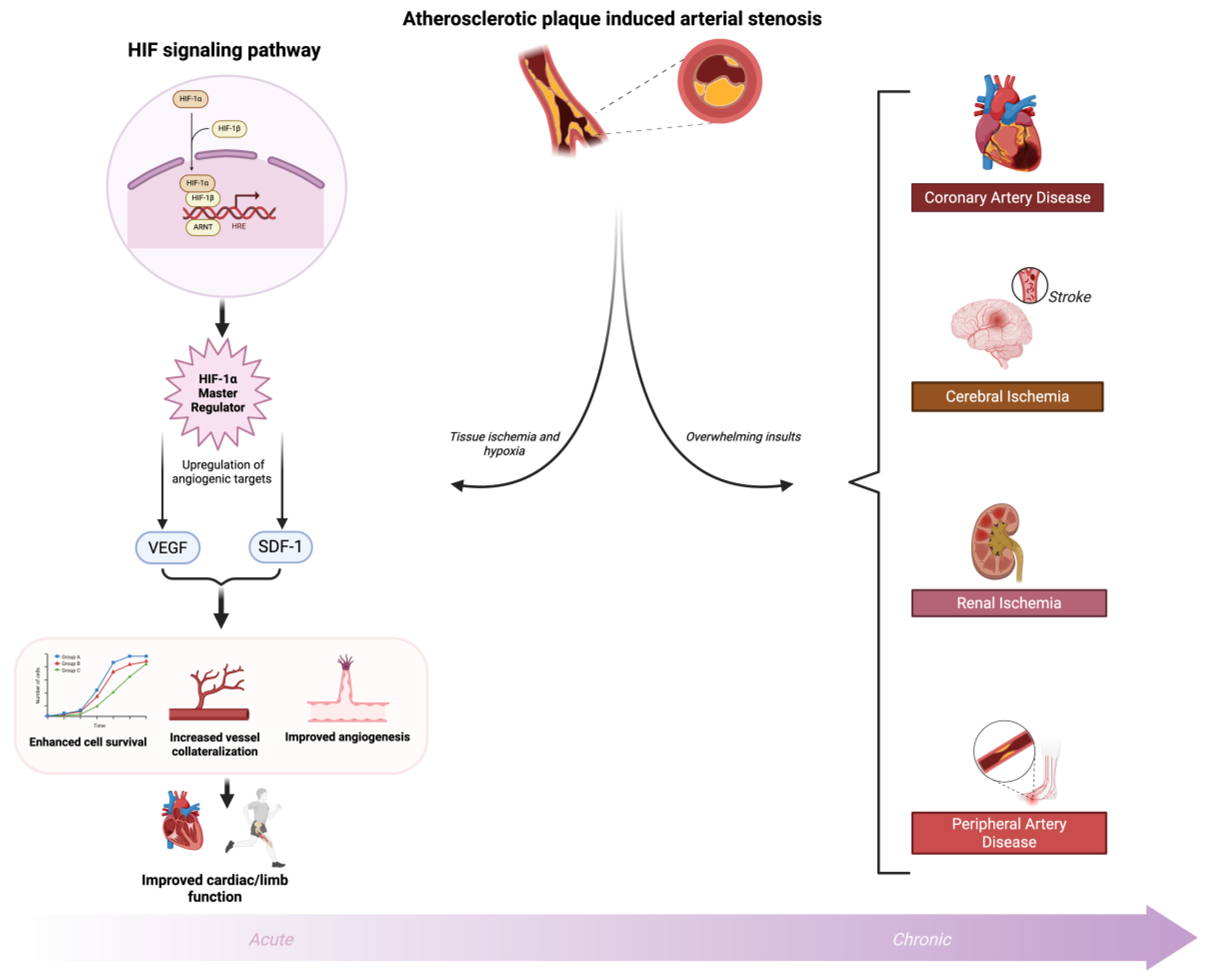

Table 1.
Pro-angiogenic targets of HIF proteins.
| Target Gene | Function | Citation |
|---|---|---|
| VEGF (VEGFA) | Stimulates endothelial cell proliferation, migration, and new blood vessel formation | [9] |
| ANGPT1 (Angiopoietin-1) | Stabilizes blood vessels and promotes maturation via Tie2 receptor | [158] |
| ANGPTL4 (angiopoietin-related protein 4) | Regulates vascular permeability and enhances endothelial cell survival | [152] |
| PDGFB (platelet-derived growth factor B) | Recruits pericytes and smooth muscle cells for vessel stabilization | [153] |
| FGF2 (Basic fibroblast growth factor) | Promotes proliferation and differentiation of endothelial cells | [154] |
| SDF-1 (CXCL12) | Attracts endothelial progenitor cells to ischemic tissue | [91] |
| PIG (Placental growth factor) | Enhances VEGF-driven angiogenesis and inflammatory cell recruitment | [155] |
| EPO (Erythropoietin) | Indirectly promotes angiogenesis by enhancing red blood cell mass and oxygen delivery | [3] |
| MMP2 (Matrix metalloproteinase-2) | Degrades extracellular matrix for endothelial migration and angiogenic sprouting | [156] |
| MMP9 (Matrix metalloproteinase-9) | Facilitates basement membrane remodeling during angiogenesis | [157] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



