
Submitted:
08 December 2025
Posted:
12 December 2025
You are already at the latest version
Abstract
Background: Over the past century, numerous pharmacological regimens have been developed for multiple myeloma (MM), yet relapse remains inevitable. Although these regimens prolong overall survival (OS), repetitive treatment cycles and cumulative toxicity progressively impair quality of life (QoL). This study aimed to compare conventional stepwise therapies with CAR-T cell therapy in terms of QoL, toxicity, cost, and ethical value.Methods: Kaplan-Meier survival curves were analyzed to estimate overall (OS) and progression-free survival (PFS). Based on these durations, treatment costs were calculated. A simple and transparent utility-based model was developed to enable clinicians, researchers, and health policy authorities to easily estimate quality-adjusted life years (QALY) and incremental cost-effectiveness ratios (ICER).Results: Among heavily pretreated patients, CAR-T therapy approximately doubled OS and PFS compared with other late-line regimens and provided markedly better quality of life with lower overall treatment burden. While daratumumab-based combinations improved survival and patient well-being, they were associated with very high treatment costs (~USD 1 million per patient). Carfilzomib-based regimens remained essential for managing high-risk disease despite their expense. In contrast, VMP represented a practical and accessible option, especially for transplant-ineligible or resource-limited patients.Conclusion: CAR-T therapy provided significant improvements in survival and quality of life compared with conventional regimens among patients who had received three or more prior lines of therapy. Its earlier use appears promising. However, the limited availability of CAR-T across only a few countries raises ethical concerns regarding treatment accessibility. Contrary to common assumptions, CAR-T can be less expensive than many traditional therapies, though its single-payment structure poses barriers for patients with limited financial means. Further analyses are needed to refine toxicity management and optimize its broader clinical application in multiple myeloma.
Keywords:
multiple myeloma
; CAR‐T cell therapy
; drug toxicity
; quality‐adjusted life years (QALY)
; cost‐effectiveness
; eastern cooperative oncology group (ECOG) performance status
; Value‐based medicine
Dedication
This work is dedicated to all patients and families living with multiple myeloma, whose resilience continues to inspire. Our goal is not only to advance science, but to restore dignity, equity, and hope in treatment. May these findings serve as a reminder that true progress is measured not solely in survival curves, but in the quality of lives preserved.
1. Introduction
Multiple myeloma (MM) is a multisystemic malignancy characterized by the uncontrolled proliferation of plasma cells in the bone marrow, leading to disruption of bone, renal, and immune integrity. The monoclonal immunoglobulins produced by neoplastic plasma cells correlate with disease severity and alter the hematopoietic balance by disturbing the bone marrow microenvironment. Clinically, the disease defined by anemia, hypercalcemia, bone lesions, and renal impairment (CRAB criteria), presents with variable progression and therapeutic response due to its heterogeneous nature [1].
Over the past two decades, therapeutic strategies have evolved from conventional chemotherapy to proteasome inhibitors, immunomodulatory agents, monoclonal antibodies, and most recently, CAR-T cell therapies [2,3]. These advances have markedly extended survival, yet introduced new dimensions requiring assessment toxicity, quality of life, and cost. Today, therapeutic success is measured not only by survival outcomes but also by quality-of-life indicators, cost-effectiveness, and equitable access [4].
Despite numerous pharmacologic and technical interventions developed over the past two centuries, myeloma continues to relapse and develop resistance to therapy [5,6]. In recent years, CAR-T cell therapy has introduced a paradigm shift by re-engineering the patient’s immune system to target malignant plasma cells. Although applied in advanced disease stages, CAR-T therapy has demonstrated significant improvements in overall survival (OS) and progression-free survival (PFS), stimulating trials that explore its potential in earlier treatment lines. Current research focuses on minimizing toxicity, shortening manufacturing time, and enhancing antigen-targeting capacity [7,8,9,10,11,12,13]. This study provides a comprehensive overview of the clinical, economic, and ethical dimensions of conventional and CAR-T therapies. It also introduces a simple pharmacoeconomic model based on ECOG performance scores, designed to allow clinicians, non-economist researchers, policymakers, and health authorities to easily evaluate cumulative toxicity across treatment lines. This approach serves not only as a numerical tool for cost-effectiveness but also as a patient-centered, ethically grounded, and translational framework for evaluating therapeutic value.
2. Myeloma Therapies
The treatment of multiple myeloma has undergone a long evolution, progressing from the rudimentary practices of the 1800s such as rhubarb, opium, and corset applications to today’s sophisticated therapeutic approaches. Over time, the introduction of corticosteroids, alkylating agents, proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), and monoclonal antibodies (mAbs) has profoundly reshaped the therapeutic paradigm. In recent years, this transformation has accelerated with the advent of bispecific antibodies, antibody-drug conjugates (ADCs), and particularly, CAR-T cell therapies [3,14].
Contemporary myeloma therapy is individualized according to disease stage, cytogenetic risk profile, performance status, and comorbidities. Patients are typically stratified by transplant eligibility. In eligible candidates, high-dose melphalan followed by autologous stem cell transplantation (ASCT) remains the gold standard. Eligibility is determined not by chronological age but by biological fitness, performance status, and organ function. Patients with adequate cardiac, renal, hepatic, and pulmonary reserve (ECOG ≤2) are considered suitable for transplantation, whereas frail or organ-compromised individuals are treated with more tolerable regimens aimed at long-term disease control [15].
Standard therapeutic steps can be summarized as follows:
(1) Induction therapy: Combinations of proteasome inhibitors (bortezomib, carfilzomib), IMiDs (lenalidomide, thalidomide, pomalidomide), and dexamethasone. The addition of monoclonal antibodies such as daratumumab has enhanced efficacy. (2)Transplantation: In eligible patients, ASCT achieves durable remission. (3) Maintenance therapy: Typically involves lenalidomide to sustain disease suppression. (4) Relapsed/refractory stage: At this phase, next-generation PIs, IMiDs, monoclonal and bispecific antibodies, ADCs, and CAR-T cell therapies are employed [16] (see Figure 1).
2.1. Induction Therapies
Induction therapy represents the first step in the management of multiple myeloma. Its primary goals are to reduce tumor burden, achieve deep remission, and prepare eligible patients for autologous stem cell transplantation (ASCT) [2,3].
Currently, the VRd regimen (bortezomib, lenalidomide, dexamethasone) is regarded as the gold standard for transplant-eligible patients in both Europe and the United States, achieving high response rates (≥VGPR above 70%) and prolonged progression-free survival (PFS) [17]. The KRd regimen (carfilzomib, lenalidomide, dexamethasone) offers deeper responses in patients with high-risk cytogenetic profiles [18].
The addition of CD38-targeted monoclonal antibodies has further enhanced induction efficacy. In the GRIFFIN trial, Dara-VRd significantly improved minimal residual disease (MRD) negativity and response depth compared with conventional VRd [19]. Similarly, Dara-KRd has shown promising results in high-risk patients [20].
For transplant-ineligible, elderly, or highly comorbid patients, more tolerable regimens such as D-Rd (daratumumab, lenalidomide, dexamethasone) and VRd-lite are preferred [21]. The D-VMP regimen (daratumumab, bortezomib, melphalan, prednisone) became the standard of care in this population following the ALCYONE trial, in which daratumumab addition significantly improved PFS and OS [17,22]. The Rd regimen (lenalidomide, dexamethasone) remains the mainstay for frail or very elderly patients [23,24].
Common adverse events include peripheral neuropathy related to bortezomib, cardiotoxicity associated with carfilzomib, and hematologic toxicity from lenalidomide. In contrast, daratumumab is generally well tolerated, with only mild infusion-related reactions such as fever, chills, or nasal congestion [25]. The incorporation of anti-CD38 antibodies enhances treatment depth without increasing toxicity, markedly improving MRD negativity rates and extending PFS (see Table 1 and Appendix Table A1).
2.2. Autologous Stem Cell Transplantation
Autologous stem cell transplantation (ASCT) is a therapeutic procedure in which the patient’s own hematopoietic stem cells are reinfused following high-dose melphalan conditioning. For many years, ASCT has been regarded as the standard of care for transplant-eligible patients with multiple myeloma. The process includes several key steps: clinical preparation; stem-cell mobilization using cyclophosphamide and granulocyte colony-stimulating factor (G-CSF, e.g., filgrastim); CD34+ cell collection by apheresis; cryopreservation of the harvested cells in 10% dimethyl sulfoxide (DMSO); high-dose melphalan conditioning chemotherapy; and subsequent reinfusion of the stem cells. Once reinfused, the stem cells home to the bone-marrow microenvironment, initiate proliferation, and restore hematopoietic and immune functions within approximately 2-4 weeks [26] (see Figure 2).
When combined with modern induction regimens such as VRd or Dara-VRd, ASCT further reduces tumor burden, deepens response, and significantly prolongs progression-free survival (PFS) [27,28]. However, the procedure is associated with substantial biological stress and transient toxicities. During mobilization and conditioning, increases in cytokines, oxidative stress, and mucosal injury are commonly observed [29,30].
Administration of G-CSF can trigger transient inflammatory responses; some studies have reported p53-mediated DNA damage and reduced engraftment efficiency [31,32]. Melphalan induces DNA damage and apoptosis in rapidly dividing cells, contributing to mucositis, pancytopenia, and infection risk, and has also been linked to long-term bone fragility and secondary malignancies [33,34,35]. DMSO, used during cryopreservation, may cause transient cardiovascular or gastrointestinal side effects during reinfusion [36].
Patients are closely monitored for infections during neutropenic and immunosuppressive phases, with appropriate supportive therapy provided. Despite these challenges, long-term survival outcomes following ASCT remain remarkable. Transplantation enhances the depth of hematologic remission and nearly doubles median overall survival [17,22,23,37].
Although transplantation-related toxicities may create clinical uncertainty [38] and novel agents are showing increasing efficacy in transplant-ineligible populations [17,23]. ASCT continues to offer unmatched benefits in survival, quality of life, and durable disease control when applied to appropriately selected patients.
2.3. Maintenance Therapies
The primary goal of maintenance therapy is to sustain remission achieved after first-line treatment or transplantation and to suppress residual malignant plasma cells. Currently, lenalidomide remains the standard maintenance agent, while bortezomib or monoclonal antibodies such as daratumumab represent effective options for high-risk patients [39].
Despite prolonged maintenance, the emergence of drug-resistant clones and an immunosuppressive bone marrow microenvironment often set the stage for inevitable relapse [5,6].
Upon relapse, treatment selection depends on the patient’s overall condition, comorbidities, cytogenetic risk profile, and prior treatment response. In the relapsed/refractory (R/R) setting, second-line regimens are typically employed, which may involve introducing a new drug class or modifying the existing protocol [2].
However, with successive relapses, therapeutic efficacy diminishes due to clonal evolution, cumulative toxicity, and emerging resistance mechanisms. At this stage, less effective agents are discontinued or replaced with alternatives, while each subsequent line of therapy becomes progressively more complex and less tolerable [40,41].
2.4. Relapsed/Refractory Phase
In the relapsed/refractory (R/R) phase, proteasome inhibitors (bortezomib, carfilzomib, ixazomib), immunomodulatory agents (lenalidomide, pomalidomide), CD38 monoclonal antibodies (daratumumab, isatuximab), SLAMF7-targeted antibodies (elotuzumab), and newer modalities such as bispecific antibodies and antibody-drug conjugates (ADCs) are used sequentially [2].
Each treatment line provides temporary disease control, yet relapse remains almost inevitable. This resistance process is linked to (1) the persistence of refractory clones, (2) the immunosuppressive myeloma microenvironment, and (3) the survival of stem-like cell populations [5,42,43]. Over time, clonal evolution, cumulative toxicity, and declining organ reserve progressively reduce therapeutic efficacy. Ineffective agents are therefore discontinued or replaced with drugs of different classes. Minimal residual disease (MRD) positivity and high-risk cytogenetic profiles are key indicators for early treatment modification [44,45].
Prolonged therapy imposes biological, psychological, and economic burdens on patients [46]. Exhaustion of hematopoietic reserve leads to progenitor suppression and pancytopenia [47]. Immunomodulatory and antibody-based therapies suppress T/NK-cell function, increasing the risk of hypogammaglobulinemia and opportunistic infections [48,49]. Moreover, microenvironmental disruption reduces stromal support, enhances osteoclast activity, and weakens the hematopoietic niche. Consequently, each new line of therapy not only reduces tumor burden but also erodes biological resilience, immune defense, and overall quality of life [26,50].
Advanced treatment lines impose substantial toxicity not only on the hematopoietic system but also on the liver, kidneys, and central nervous system. Hepatic enzyme induction, impaired renal elimination, and neuro-inflammatory changes further compromise treatment tolerance and drug metabolism. This multi-organ load is one of the major factors limiting the clinical benefit of combination regimens in late-line therapy [51,52,53].The resulting cycle of cumulative exhaustion progressively diminishes efficacy and renders the patient increasingly fragile (see Figure 3).
Although multiple myeloma does not progress as aggressively as acute leukemias, its chronic and relentless nature gradually exhausts patients biologically, psychologically, and economically. Since the 19th century, therapeutic advances have dramatically extended survival, allowing many patients today to live five years or longer after diagnosis. Yet this progress comes at a cost: with each subsequent line of therapy, progression-free survival (PFS) shortens, cumulative toxicity increases, and survival curves narrow.
By occupying the bone marrow niche, myeloma not only disrupts hematopoiesis but also destabilizes systemic homeostasis. Anemia, immune deficiency, bone destruction, and organ dysfunction often reduce quality of life more than they contribute directly to mortality [46,54].
In this study, we comprehensively evaluated the clinical and economic impact of multiple myeloma treatment regimens by comparing estimated costs and mean overall survival (OS) values derived from published Kaplan-Meier curves. To quantitatively assess quality of life, quality-adjusted life years (QALYs) were calculated. These QALY values were not extracted directly from prior literature but were instead modeled using ECOG performance scores and survival data, in accordance with the study’s central hypothesis (see Table 2, Supplementary Table S1, Supplementary Table S2 and see section ‘’4.Quality-Adjusted Life Year (QALY) and ECOG-Based Modeling’’).
This approach allows comparison of treatment regimens not only in terms of clinical efficacy, but also through the lens of cost-effectiveness and ethical sustainability (see Supplementary Material for details). However, model-based QALY estimates presented here should not be directly compared with classical QALY values derived from prospective patient surveys. Although absolute QALY values are lower, the model provides internally consistent and reliable comparative insights across regimens (see Methods and Supplementary Methods).
3. CAR-T Therapies
A major milestone in multiple myeloma therapy was the identification of B-cell maturation antigen (BCMA) as a therapeutic target. To date, two autologous CAR-T cell products targeting BCMA have received regulatory approval.
Idecabtagene vicleucel (ide-cel, Abecma) was approved by the FDA in April 2021 following the KarMMa trials and demonstrated remarkable efficacy in heavily pretreated patients (≥3 prior lines including PI, IMiD, and anti-CD38). In this population, the overall response rate (ORR) reached 76% versus 32% with standard of care; ≥VGPR was 58% versus 14%; one-year progression-free survival (PFS) was 55% versus 30%; and minimal residual disease (MRD) negativity was approximately 20%. The most frequently reported toxicities included neutropenia, leukopenia, and cytokine release syndrome (CRS), with grade ≥3 CRS observed in roughly 14% of patients [13,72].
Ciltacabtagene autoleucel (cilta-cel, Carvykti), a second-generation BCMA-directed CAR-T construct with dual-epitope binding capacity, received FDA approval in February 2022. According to 2025 analyses, the median overall survival (OS) is approximately 56 months and median PFS about 18 months, with an MRD-negativity rate of 68%. Reported toxicities include low-grade CRS, immune effector cell-associated neurotoxicity syndrome (ICANS), cytopenias, and infections [70,73]
3.1. Treatment Process and Clinical Application
The CAR-T therapy process involves the collection of a patient’s T lymphocytes by apheresis, their genetic modification ex vivo, expansion in culture, and reinfusion as a single intravenous dose. Before infusion, lymphodepleting chemotherapy (fludarabine + cyclophosphamide) is administered to facilitate in vivo CAR-T expansion, reduce immune suppression within the tumor microenvironment, and enhance persistence. Manufacturing typically requires 2-4 weeks and includes the following main steps [74] (see Figure 4):
- Activation: stimulation using anti-CD3/CD28 microbeads and cytokines such as IL-2, IL-7, and IL-15.
- Genetic modification: transduction of the CAR construct, most commonly via lentiviral vectors.
- Expansion: large-scale proliferation in controlled culture systems to generate millions of CAR-T cells.
- Quality control and cryopreservation: verification of cell viability, microbial safety, and CAR expression, followed by freezing and storage until infusion.
The prepared cells are thawed and administered as a single intravenous infusion. The most common adverse events are cytokine release syndrome (CRS) and neurotoxicity, both of which require management in experienced centers with intensive-care support.
3.2. Clinical Efficacy, Innovations, and Limitations of CAR-T Cell Therapy
CAR-T cell therapy achieves high response rates (≥70%) and prolonged remissions in patients with relapsed/refractory multiple myeloma (RRMM). Clinical trials such as CARTITUDE-4 have demonstrated that CAR-T therapy can be used as early as the second line, aiming to improve both survival and quality of life with earlier intervention [12].
Its single-dose, potentially curative nature positions CAR-T as one of the most promising modalities in modern oncology. However, high cost, lengthy manufacturing, and the need for intensive toxicity management currently limit its widespread application. The comparative analysis in this study highlights that ciltacabtagene autoleucel (cilta-cel) offers unexpectedly favorable outcomes in terms of both economic sustainability and biological toxicity profile [74].
Cilta-cel’s single-infusion design reduces cumulative toxicity associated with multiple treatment lines and improves not only survival but also quality-of-life metrics (QALY) [59]. Although upfront costs are high, the long-term reduction in complications, lower hospitalization rates, and extended treatment-free intervals make CAR-T therapy a relatively cost-effective investment [74] (see Supplementary Material for details).
Recent advances have focused on moving CAR-T therapy into earlier lines of treatment, shortening manufacturing time, and enhancing antigen-binding efficiency (“docking efficiency”). Traditional autologous CAR-T production takes several weeks, during which many patients may experience disease progression before infusion. To address this, rapid manufacturing platforms such as FasT CAR-T have been developed. For instance, BCMA/CD19 dual-target GC012F CAR-T cells can be generated within 48-72 hours [7]. Similarly, the NEX-T process integrates non-viral gene-transfer systems (e.g., Sleeping Beauty transposons) and automated quality-control algorithms, potentially reducing production time to just a few days [8,9]. In addition, “off-the-shelf” allogeneic CAR-T products aim to eliminate wait times and improve accessibility [10].
At the molecular level, optimization of antigen-binding domains including linker length and conformational stability has improved docking efficiency. Dual-target constructs (e.g., BCMA + GPRC5D or BCMA + CD38) show promise in preventing antigen escape [11]. Early-line clinical testing (before third-line therapy) suggests these approaches may preserve immune-cell function and extend durable remission [12,13].However, as manufacturing time decreases, careful evaluation of cell quality, CRS incidence, and long-term efficacy remains critical.
Despite impressive response rates, CAR-T therapy faces major limitations due to severe toxicities and restricted T-cell persistence. The most frequent and potentially life-threatening adverse event is cytokine release syndrome (CRS), triggered by excessive release of pro-inflammatory cytokines such as IL-6, IFN-γ, and TNF-α, leading to systemic inflammation, fever, hypotension, hypoxia, and multi-organ dysfunction [75]. CRS typically occurs within the first few days post-infusion and is managed with tocilizumab or corticosteroids.
A second key toxicity is immune effector cell-associated neurotoxicity syndrome (ICANS), commonly observed with CD19- and BCMA-directed CAR-T therapies. Symptoms include confusion, aphasia, tremor, or seizures, linked to endothelial activation and disruption of the blood-brain barrier [76]. Prolonged neutropenia and hypogammaglobulinemia further increase the risk of secondary infections and delay immune reconstitution [75].
Despite these remarkable outcomes, limited accessibility remains a major challenge. Ide-cel is currently available only in the United States, France, Switzerland, Japan, and Germany, while cilta-cel is approved solely in the United States and Germany. Manufacturing delays, logistical barriers, and strict eligibility criteria continue to restrict broader use. The autologous nature of most CAR-T products limits functional capacity in older or heavily pretreated patients, reducing T-cell persistence [77,78]. Additional barriers include antigen loss, immunosuppressive microenvironmental effects (e.g., TGF-β, IL-10, accumulation of MDSCs), and high production costs. In allogeneic (“off-the-shelf”) CAR-T approaches, safety concerns such as graft-versus-host disease (GVHD) and immune rejection remain unresolved [79].
To overcome these challenges, next-generation strategies such as armored CAR-T, self-destruct switches, and regulated CAR systems are being developed to enhance persistence and minimize toxicity.
3.3. CAR-T Cell Trials
The first-generation BCMA-targeted CAR-T therapy, idecabtagene vicleucel (ide-cel; bb2121), represents the pioneering autologous CAR-T construct evaluated in the KarMMa trials. Its single-epitope binding design limited both antigen affinity and cellular persistence. In vivo proliferation peaked early, leading to rapid but short-lived responses [80,81]. Consequently, KarMMa-1 reported a median progression-free survival (PFS) of 8.8 months and a median overall survival (OS) of 19.4 months values substantially lower than those achieved by later dual-epitope designs [81].
By contrast, ciltacabtagene autoleucel (cilta-cel; LCAR-B38M), a second-generation BCMA-directed CAR-T product developed through the LEGEND-2 and CARTITUDE programs, demonstrated markedly improved outcomes [11,68]. Its dual-epitope binding configuration enhances antigen affinity, strengthens T-cell proliferation, and promotes durable immunologic memory [70].
Among clinical trials evaluating CAR-T efficacy, LEGEND-2 and CARTITUDE-1 provide the most comprehensive long-term data. CARTITUDE-1 confirmed that deep and durable responses can be achieved even in heavily pretreated patients. After 61 months of follow-up, median OS reached 60.7 months, while the mean OS (area-under-curve analysis) was estimated at ~85 months [70].
LEGEND-2 (NCT03090659), the first-in-human, open-label phase 1 study of the dual-epitope construct LCAR-B38M, enrolled 74 patients with RRMM previously treated with at least three therapy lines including proteasome inhibitors and immunomodulatory agents. Following lymphodepletion with fludarabine and cyclophosphamide, patients received 0.5-1.0 × 106 CAR+ T cells/kg as single or split doses. Response rates were high: overall response rate (ORR) 88-90%, complete response (CR) 74%, and MRD negativity ~68% [68]. Median PFS was 20 months and median OS 36 months. CRS occurred in most patients but was predominantly grade 1-2 and manageable; neurotoxicity was rare. This study provided the first clinical evidence that BCMA-directed CAR-T therapy could induce deep and durable remissions.
Following these successes, CARTITUDE-2, a multicenter phase 2 trial, explored CAR-T efficacy across diverse patient subgroups: Cohort A: triple-class-refractory patients (≥3 prior lines), Cohort B: lenalidomide-refractory patients with 1-3 prior lines [82], Cohort C: patients at first relapse [83], Cohort D: transplant-ineligible, lenalidomide-refractory patients [84], Cohort E: high-risk or MRD-positive early-stage disease [73].
In CARTITUDE-4, cilta-cel demonstrated statistical superiority over standard regimens in early-relapse settings in terms of progression-free survival [85]. The ongoing CARTITUDE-5 and CARTITUDE-6 studies [86,87] are now evaluating whether CAR-T therapy can replace the conventional ASCT + maintenance paradigm as a first-line treatment option, independent of transplant eligibility. Together, these consecutive trials establish CAR-T therapy not merely as a “last resort,” but as an emerging early-line, immune-rejuvenating strategy in multiple myeloma (see Table 3).
4. Quality-Adjusted Life Year (QALY) and ECOG-Based Modeling
The Quality-Adjusted Life Year (QALY) is an integrative measure that captures both the quantity and quality of life. One QALY represents one year lived in perfect health; for an individual experiencing 50% health quality, one year corresponds to 0.5 QALY. This concept allows treatment outcomes to be compared not only by survival duration but also by their impact on quality of life. In multiple myeloma, cumulative toxicity and treatment-related complications affect patients as profoundly as survival itself. Physical limitations, fatigue, immune suppression, and infection risk directly influence therapeutic success. Modern oncology therefore emphasizes combined metrics of longevity and quality most notably, the QALY as standard of value-based care. Quality of life is commonly assessed using validated questionnaires such as EORTC QLQ-C30, EQ-5D, or FACT-MM. The resulting utility scores, ranging from 0 to 1, are multiplied by the number of life-years gained to calculate QALY values [89]:
QALY=Utility×Life Years
In parallel, the ECOG Performance Status (0-4) provides a clinician-rated, objective measure of functional capacity and independence: 0 = fully active; 1 = mild restriction; 2 = capable of limited activity; 3 = restricted to self-care; 4 = completely disabled; 5 = death.
Multiple studies have demonstrated a strong correlation between ECOG scores and HRQoL or EQ-5D utility values [89,90,91,92,93]. However, this relationship has rarely been formalized into a simple, quantitative model.
In this study, based on [94] the following ECOG-to-utility conversion was applied: ECOG 0 = 0.85, 1 = 0.75, 2 = 0.65, 3 = 0.55, 4 = 0.45. Each one-point increase in ECOG corresponds to an average utility decrement of 0.05–0.10, representing a clinically meaningful change (MID).
This method directly links performance status to quality of life and enables standardized, transparent, and practical QALY estimation even in the absence of patient-reported data. Supplementary materials detail how this ECOG-based model was adapted to specific treatment arms and integrated with cost data. Furthermore, the model incorporates Incremental Cost-Effectiveness Ratio (ICER) analysis, which quantifies the additional cost required to gain one extra QALY compared with an alternative treatment:
- Low ICER → higher cost-effectiveness
- High ICER → lower value or higher cost per QALY [95]
This framework assesses not only the economic burden of therapy but also the value gained per unit of quality-adjusted survival. In our analysis, CAR-T therapies particularly those from CARTITUDE-1 showed lower ICER values than most conventional regimens. This finding suggests that CAR-T represents not a “high-cost innovation,” but a high-value therapy whose long-term gains in quality of life justify its initial expense. Ultimately, this study proposes a simple, mathematically consistent, and ethically grounded model that enables clinicians, policymakers, and researchers to evaluate myeloma therapies not only in clinical and economic terms but also through a humanistic and justice-oriented lens.
5. Methods (Summary)
This analysis was designed to evaluate multiple myeloma treatment regimens within a comparative, model-based framework from a healthcare system perspective. The methodological approach focused on estimating cost, overall survival (OS), and quality-adjusted life years (QALY) across sequential lines of therapy. All values were derived from published clinical trial data using Kaplan-Meier survival curves and corresponding cost-effectiveness parameters.
QALY estimates were calculated by weighting survival duration with utility values derived from ECOG performance scores. The utility coefficients were obtained according to the model described in the hypothesis section (ECOG 0 = 0.85; 1 = 0.75; 2 = 0.65; 3 = 0.55; 4 = 0.45). In studies where ECOG data were not reported, estimated utilities were derived by optimizing toxicity (TEAE) and mortality data. Since all regimens were analyzed within the same methodological framework, results are proportionally comparable across treatment lines (Supplementary Material Table S1).
Mean overall survival (OS) values were computed by calculating the area under the Kaplan-Meier curve (AUC). Graphical data were digitized using WebPlotDigitizer software, and geometric integration was used to estimate the area beneath survival curves. Similarly, progression-free survival (PFS) values were extracted using the same method. Missing curve segments were interpolated geometrically based on slope trajectory and directional trends.
Cost estimation was performed according to the primary endpoint reported in each trial, which was PFS in all cases. Drug costs were calculated based on dosage, administration frequency, and treatment duration, using unit prices adapted from recent literature and national reimbursement databases (see Supplementary Material, Table S2). A detailed description of the model structure, parameter sources, dose calculations, and sensitivity analyses is provided in the Supplementary Material - Methods section.
6. Discussion and Conclusion
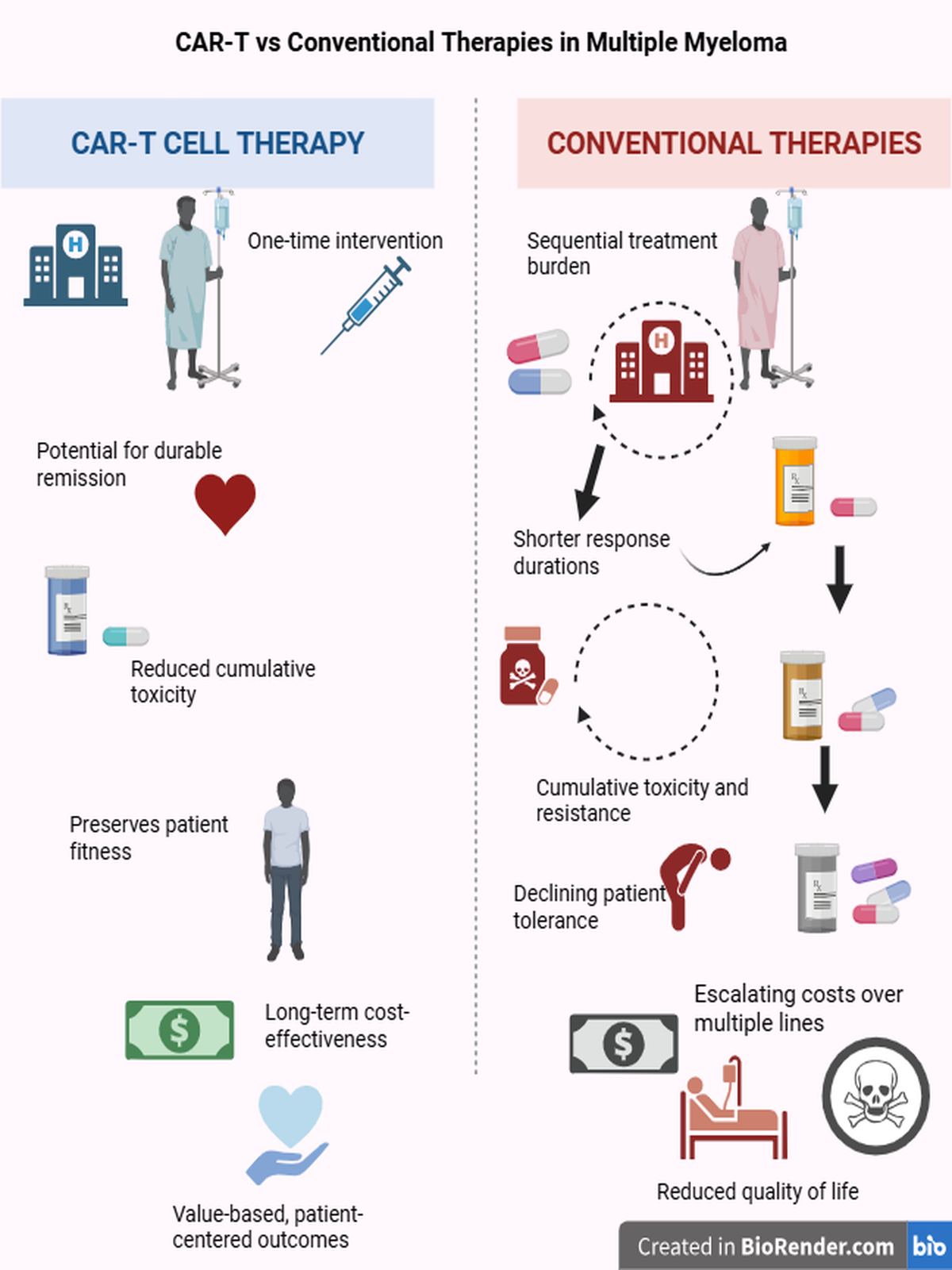
The primary aim of this study was to evaluate the advantages of CAR-T cell therapies over conventional regimens in terms of toxicity, progression-free survival (PFS), and overall survival (OS). Within this scope, QALY calculations and other pharmacoeconomic analyses were developed to interpret this major paradigm shift in hematology through an ethical and value-based framework. The study contrasts traditional approaches characterized by repeated chemotherapy cycles, high costs, and continuous hospital dependency with the transformative potential of CAR-T therapy: a single infusion that offers prolonged survival, reduced toxicity, and sustained remission. The goal, therefore, was not only to demonstrate clinical superiority but also to examine the ethical and human dimensions of modern therapeutic strategies. QALY-based cost analyses were used as a numerical representation of this ethical framework, seeking a clinical and philosophical answer to the question: “Is progress measured by the length of life, or by the quality of living?”
Most existing pharmacoeconomic studies in the literature rely on predefined QALY values and Markov-based cost-effectiveness models [59]. However, such models are often complex, making them difficult for clinicians and biomedical researchers to apply or interpret. In this context, our analysis provides a simpler but systematic framework that allows direct comparison of toxicity and survival outcomes without requiring complex modeling. Moreover, the cumulative toxicity observed with conventional therapies and the biological-economic transformation brought by CAR-T cells highlight an urgent need to reassess therapeutic priorities in hematology. This study responds to that need by evaluating treatment lines in terms of toxicity, survival, and quality of life, proposing an ethical, clinically meaningful, and practical framework for decision-making.
During the analysis, significant data gaps and transparency issues were encountered across clinical trials. Research reports should be written not only for clinicians but also for investigators from other disciplines. Yet, many pivotal phase studies compromise scientific clarity through excessive technical terminology, incomplete or unclear data presentation (particularly omission of death counts), and TEAE graphics that fail to reflect the patient’s actual clinical condition. These limitations blur the boundaries between methodological reliability and ethical accountability. Broadly defined treatment categories such as “2nd–10th line” often encompass heterogeneous patient populations that differ greatly in biological and functional capacity. A patient in the second line of therapy and one in the tenth line are clinically incomparable. Such grouping may distort both positive and negative interpretations of efficacy and safety. Therefore, publishers, reviewers, and researchers share an ethical obligation to report baseline and final patient status using functional indices (e.g., ECOG, quality-of-life scales) and to provide arm-specific mortality data transparently. Clear definition of heterogeneity, and, when necessary, separate analyses by treatment line, is essential not only for methodological accuracy but also for a patient-centered ethical evaluation.
In the transplant-ineligible group, the VMP regimen remains the most cost-effective option, offering a favorable balance between total cost and utility/QALY gains. Despite the abundance of new agents, the continued clinical efficacy of agents such as melphalan and prednisone drugs whose origins trace back to the post-World War II era demonstrates that progress in medicine is not always defined by innovation, but often by continuity and resilience [96]. Analysis of induction-phase regimens (VRd, D-VRd, D-VMP, VMP, D-Rd, and Rd) revealed the highest QALY and utility values at this early treatment stage (Table 2; Supplementary Table S2). This raises a broader question: should we favor well-known, lower-cost regimens with predictable toxicity and management profiles, or prioritize new, high-cost combinations whose long-term outcomes remain uncertain? Perhaps the time has come for a paradigm shift toward individualized treatments that strengthen the patient’s own immune system, integrating nutritional and lifestyle-based support with modern immunotherapies. Such a holistic approach may represent a more scientifically sound and ethically sustainable direction for the future [97,98].
In transplant-eligible patients, both survival and quality-of-life metrics were significantly superior. Adding daratumumab to VRd, which yielded an average QALY of 7.145, extended survival by more than 2.5 years though at approximately four times the cost. The most striking benefit of daratumumab was observed in progression-free survival (PFS): median PFS increased from 38 months with VRd to 84 months with D-VRd. Similar improvements were observed when daratumumab was added to VMP or Rd regimens, leading to sustained gains in both survival and quality of life [99].
Analysis of carfilzomib-based regimens (e.g., KRd, D-Kd) showed overall-survival advantages often exceeding 50 months, although progression-free survival (PFS) durations were shorter. This finding confirms that carfilzomib provides a mortality-reducing and quality-of-life-enhancing effect, particularly in high-risk patient groups [100]. Despite its short-term toxicity risks, carfilzomib continues to hold a valuable place within risk-adaptive therapeutic strategies.
Among late-line therapies, evaluated regimens include Elo-Rd, Elo-Pd, Elo-PVd, Isa-Pd, XVd, and CAR-T cell treatments such as those from the CARTITUDE-1 and LEGEND-2 trials (Appendix B, Figure A1). When daratumumab intolerance or resistance occurs, elotuzumab-based regimens become clinically relevant due to their low toxicity profile and unique SLAMF7-targeting mechanism [101]. In patients refractory to both lenalidomide and daratumumab, Elo-Pd remains a feasible alternative. Because most of these patients have previously received proteasome inhibitors, combinations such as Elo-PVd which include bortezomib are typically used in later treatment lines (Appendix B Figure A1).
Although late-line regimens such as PVd, Pd, and XVd achieve median overall survival (OS) approaching 40 months, the reliability of these data remains limited. The referenced studies involve highly heterogeneous patient populations, and the clinical characteristics of survivors are often underreported. In heavily pre-treated patients exposed to cumulative toxicity, the true clinical meaning of these survival durations is uncertain. The derived utility values were nearly ten-fold lower than those observed in the induction phase, with a mean progression-free survival (PFS) of only about 20 months. Treatment costs averaged roughly USD 800 000 per patient. Regimens such as Elo-Rd and Isa-Pd yielded QALY scores approaching 1.0, yet these modest gains came at an additional cost of about USD 100 000. Among them, Elo-Rd offered roughly one extra year of survival compared with other late-line options; however, this difference may partly reflect patient heterogeneity or residual lenalidomide sensitivity [102]. Within this landscape, the most remarkable finding was the CAR-T cell therapy results. Despite excluding patients from second- or third-line cohorts, cilta-cel demonstrated an estimated mean OS exceeding 85 months, PFS around 44 months, and a substantially lower annual cost (Table 2; Supplementary Table S2). In the ongoing CARTITUDE-1 [70], the mean OS exceeded 85 months, and the calculated QALY reached 2.744 approximately three times higher than conventional late-line regimens. These findings suggest that CAR-T therapy redefines the end-stage paradigm not only biologically, but also economically and ethically. To evaluate how much it costs to provide one additional year of quality-adjusted life, we performed an Incremental Cost-Effectiveness Ratio (ICER) analysis (Figure 5). This approach quantifies the comparative cost of achieving one year in which the patient perceives themselves as “healthy,” integrating clinical, economic, and ethical dimensions into a unified assessment of therapeutic value.
In the late-line analysis, the calculated ICER values indicate that, despite its seemingly high upfront cost, CAR-T therapy stands among the most value-based interventions in multiple myeloma. Its single-infusion design, minimal hospitalization time, and lack of cumulative toxicity management substantially reduce total healthcare expenditures. In contrast, conventional treatments administered “piece by piece” across multiple cycles create far greater long-term economic and biological burdens. Therefore, CAR-T should not be viewed as an “expensive innovation,” but rather as a high-value and sustainable solution. When cost optimization, insurance coverage, and public reimbursement mechanisms are implemented, its financial impact may become lighter than that of conventional regimens over time. Current evidence shows that CAR-T therapy provides remarkable improvements in survival and quality of life, even in heavily pretreated patients. Although results from early-line trials (such as the CARTITUDE and KarMMa series) are still maturing, preliminary findings suggest that CAR-T could be administered earlier, with lower toxicity and longer-lasting remission. This potential points toward a new therapeutic paradigm in myeloma one focused not on multiple sequential treatments, but on single-intervention, durable control. In this sense, CAR-T may represent not only an early intervention candidate, but also the first genuinely personalized therapy capable of achieving lasting remission with one infusion.
From an ethical standpoint, a therapy initially limited by high cost but ultimately capable of providing more life years, less toxicity, and lower overall expenses demands a re-examination of what constitutes justice and value in modern medicine. Today, CAR-T cell therapy remains accessible only in a few countries and primarily to patients who can afford its one-time infusion price. This disparity raises critical questions about how biotechnological innovation can be balanced with equity and accessibility. In a patient-centered and socially sustainable healthcare model, CAR-T should be recognized not only as a scientific breakthrough, but also as a transformative model redefining ethical, economic, and human values in medicine. Expanding its accessibility will not only improve clinical outcomes, but also strengthen equality, humanism, and the principle of “kill the tumor, not the human.
See Supplementary Material for details.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Gülüzar Gülnur İtez conceptualized the study, designed the QALY model, and drafted the manuscript. Asuman Sunguroğlu supervised the work and critically reviewed the final version. Both authors read and approved the final manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics Approval and Consent to Participate
This study did not involve any new studies with human participants or animals performed by any of the authors. All data were derived from previously published studies. Therefore, ethical approval and informed consent were not required.
Availability of Data and Materials
All data used in this study were obtained from publicly available clinical trial publications and databases, which are cited in the manuscript.
Acknowledgments
The authors thank Ankara University Stem Cell Institute for academic support.
Competing Interests
The authors declare no competing interests.
Consent for Publication
Not applicable.
Appendix A

Table A1.
Modern Therapies in Multiple Myeloma: Clinical Role.
| Therapy / Class | Year (Approval / Milestone) | Mechanism / Target | Current Use in MM | Key Toxicities |
| Autologous Stem Cell Transplant (ASCT) | 1983 (MM application) | Hematopoietic rescue | Standard in eligible patients | Myelosuppression, infection risk |
| Bortezomib (first PI) [103,104] | 2003 | Reversible 20S proteasome inhibitor | Backbone of induction (VRd etc.) | Peripheral neuropathy |
| Carfilzomib (2nd-gen PI) [104] | 2012 | Irreversible PI | Relapsed/refractory, combo regimens | Cardiac toxicity |
| Ixazomib (oral PI) [104] | 2015 | Oral PI | Maintenance, frail patients | GI, mild cytopenias |
| Lenalidomide (IMiD) [102] | 2005 | IMiD, cytokine modulation, T-cell activation | Frontline & maintenance backbone | Cytopenias, thrombosis |
| Pomalidomide (IMiD) [105] | 2013 | Next-gen IMiD | Relapsed/refractory settings | Cytopenias, infections |
| Daratumumab (anti-CD38 mAb) [1,107] | 2015 | ADCC, CDC, ADCP, direct apoptosis | Widely used frontline & RRMM | Neutropenia, infusion reactions |
| Isatuximab (anti-CD38 mAb) [1,106] | 2020 | Distinct CD38 epitope, direct apoptosis | Combo with Pd in RRMM | Neutropenia, infections |
| Selinexor (XPO1 inhibitor) [1,106] | 2019 | Nuclear export inhibition, p53 reactivation | Triple-class refractory | GI toxicity, cytopenias |
| Venetoclax (BCL-2 inhibitor) [1,106] | Investigation | BCL-2 inhibition | Targeted subgroup (t(11;14)) | Tumor lysis, cytopenias |
| Melflufen (peptide-drug conjugate) [1,106] | 2021 (revoked FDA approval) | Alkylating payload via peptide conjugate | EMA-approved, not FDA | Cytopenias, survival concern |
| HDAC inhibitors (e.g., panobinostat) [1,106] | 2015 (panobinostat FDA) | Histone deacetylase inhibition | Adjunct in refractory disease | GI, fatigue, cytopenias |
| Checkpoint inhibitors (PD-1/PD-L1) [1,106] | Trials (halted in MM) | Immune checkpoint blockade | Investigational only | Immune toxicities |
| Bispecific Abs / CAR-T / NK [1,106] | 2020s | Redirected T/NK cytotoxicity | Rapidly emerging | CRS, neurotoxicity, cytopenias |
Appendix B
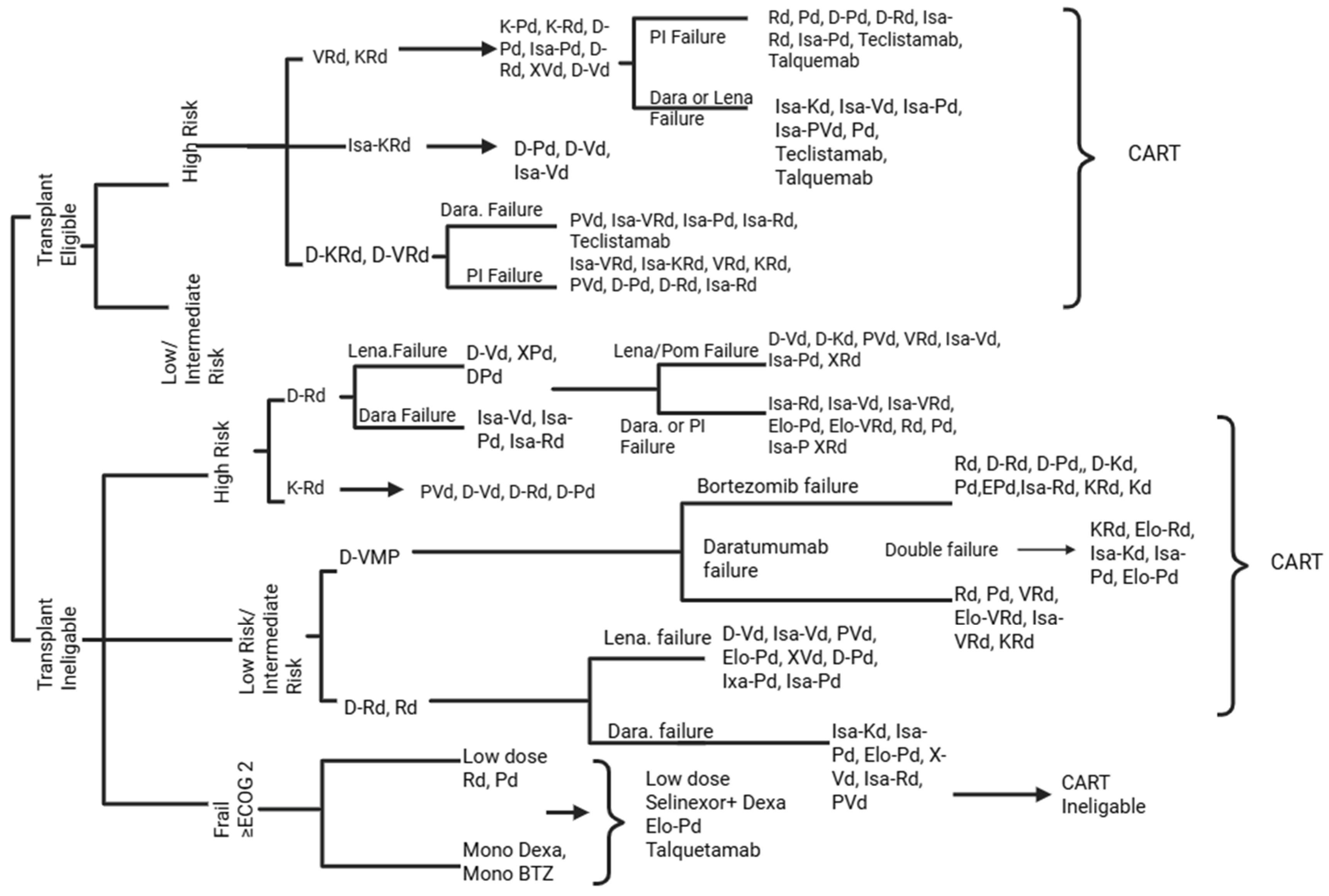
Figure A1.
Proposed treatment pathway for multiple myeloma based on the author’s synthesis of recent clinical studies (2018-2025), reflecting risk stratification, treatment eligibility, and therapeutic failure sequences. This algorithm was constructed by integrating data from the clinical trials and publications cited in the main text. It represents an interpretative synthesis rather than an adaptation of any single guideline. All references used to generate this figure are included in the manuscript’s reference list.
Figure A1.
Proposed treatment pathway for multiple myeloma based on the author’s synthesis of recent clinical studies (2018-2025), reflecting risk stratification, treatment eligibility, and therapeutic failure sequences. This algorithm was constructed by integrating data from the clinical trials and publications cited in the main text. It represents an interpretative synthesis rather than an adaptation of any single guideline. All references used to generate this figure are included in the manuscript’s reference list.
References
- Rajkumar SV. Multiple myeloma: 2024 update on diagnosis, risk-stratification, and management. Am J Hematol. 2024;99(9):1802–1824. [CrossRef]
- Mavrothalassitis E, Triantafyllakis K, Malandrakis P, et al. Current treatment strategies for multiple myeloma at first relapse. J Clin Med. 2025;14(5):1655. [CrossRef]
- Gay F, Marchetti E, Bertuglia G. Multiple myeloma unpacked. Hematol Oncol. 2025;43(Suppl 2):e70067. [CrossRef]
- Fonseca R, Hinkel J. Value and cost of myeloma therapy- we can afford it. Am Soc Clin Oncol Educ Book. 2018;38:647–655. [CrossRef]
- Lopes R, Caetano J, Ferreira B, et al. The immune microenvironment in multiple myeloma: friend or foe? Cancers (Basel). 2021;13(4):625. [CrossRef]
- Chroma K, Skrott Z, Gursky J, et al. A drug repurposing strategy for overcoming human multiple myeloma resistance to standard-of-care treatment. Cell Death Dis. 2022;13:203. [CrossRef]
- Du J, Fu W, Jiang H, et al. Updated results of a phase I, open-label study of BCMA/CD19 dual-targeting FASTCAR-T GC012F for patients with relapsed/refractory multiple myeloma (RRMM). Hemasphere. 2023;7(Suppl):e84060bf. [CrossRef]
- Swan D, Madduri D, Hocking J. CAR-T cell therapy in multiple myeloma: current status and future challenges. Blood Cancer J. 2024;14:206. [CrossRef]
- Prommersberger S, Reiser M, Beckmann J, et al. CARAMBA: a first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021;28:560–571. [CrossRef]
- Moradi V, Omidkhoda A, Ahmadbeigi N. The paths and challenges of “off-the-shelf” CAR-T cell therapy: an overview of clinical trials. Biomed Pharmacother. 2023;169:115888. [CrossRef]
- Steinhardt MJ, Schaefers C, Leypoldt LB, et al. Activity of CAR-T cells and bispecific antibodies in multiple myeloma with extramedullary involvement. Blood Cancer J. 2025;15:126. [CrossRef]
- San-Miguel J, Dhakal B, Yong K, et al. Cilta-cel or standard care in lenalidomide-refractory multiple myeloma. N Engl J Med. 2023;389(4):335–347. [CrossRef]
- Rodriguez-Otero P, Ailawadhi S, Arnulf B, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med. 2023;388(11):1002–1014. [CrossRef]
- Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008;111(6):2962–2972. [CrossRef]
- Mian O, Puts M, McCurdy A, et al. Decision-making factors for an autologous stem cell transplant for older adults with newly diagnosed multiple myeloma: a qualitative analysis. Front Oncol. 2023;12:974038. [CrossRef]
- Cornell RF, D’Souza A, Kassim AA, et al. Maintenance versus induction therapy choice on outcomes after autologous transplantation for multiple myeloma. Biol Blood Marrow Transplant. 2017;23(2):269–277. [CrossRef]
- Mateos MV, San-Miguel J, Cavo M, et al. Bortezomib, melphalan, and prednisone with or without daratumumab in transplant-ineligible patients with newly diagnosed multiple myeloma (ALCYONE): final analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2025;26(5):596–608. [CrossRef]
- Lee JH, Choi J, Min CK, et al. Superior outcomes and high-risk features with carfilzomib, lenalidomide, and dexamethasone combination therapy for relapsed/refractory multiple myeloma: results of the multicenter KMMWP2201 study. Haematologica. 2024;109(11):3681–3692. [CrossRef]
- Voorhees PM, Sborov DW, Laubach J, et al. Addition of daratumumab to lenalidomide, bortezomib, and dexamethasone for transplantation-eligible patients with newly diagnosed multiple myeloma (GRIFFIN): final analysis of an open-label, randomised, phase 2 trial. Lancet Haematol. 2023;10(10):e825–e837. [CrossRef]
- Landgren CO, Yeh JC, Hillengass J, et al. Randomized, multicenter study of carfilzomib, lenalidomide, and dexamethasone (KRd) with or without daratumumab in newly diagnosed multiple myeloma (ADVANCE trial). J Clin Oncol. 2025;43(16 Suppl):7503. [CrossRef]
- Ogura M, Ishida T, Nomura M, et al. Efficacy of modified VRd-lite for transplant-ineligible multiple myeloma. Blood. 2020;136(Suppl 1):4. [CrossRef]
- Fu W, Bang SM, Huang H, et al. Bortezomib, melphalan, and prednisone with or without daratumumab in transplant-ineligible Asian patients with newly diagnosed multiple myeloma: the phase 3 OCTANS study. Clin Lymphoma Myeloma Leuk. 2023;23(6):446–455.e4. [CrossRef]
- Facon T, Moreau P, Weisel K, et al. Daratumumab/lenalidomide/dexamethasone in transplant-ineligible newly diagnosed myeloma: MAIA long-term outcomes. Leukemia. 2025;39(4):942–950. [CrossRef]
- Grant SJ, Lipe B. Management of frail older adults with newly diagnosed multiple myeloma moving toward a personalized approach. Clin Lymphoma Myeloma Leuk. 2020;20(Suppl 1):S76–S80. [CrossRef]
- Geraldes C, Roque A, Sarmento-Ribeiro AB, et al. Practical management of disease-related manifestations and drug toxicities in patients with multiple myeloma. Front Oncol. 2024;14:1282300. [CrossRef]
- Wang YN, Zhang CW, Gao YX, Ge XL. The progress of autologous hematopoietic stem cell transplantation in the treatment of multiple myeloma (review). Technol Cancer Res Treat. 2025;24:15330338251321349. [CrossRef]
- Richardson PG, Jacobus SJ, Weller EA, et al. Triplet therapy, transplantation, and maintenance until progression in myeloma. N Engl J Med. 2022;387(2):132–147. [CrossRef]
- Garfall AL. Updated analysis of EMN02 demonstrated overall survival benefit to early ASCT for multiple myeloma. Blood Cancer J. 2025;15(1):1. [CrossRef]
- Lee SI, Kang KS. Function of capric acid in cyclophosphamide-induced intestinal inflammation, oxidative stress, and barrier function in pigs. Sci Rep. 2017;7:16530. [CrossRef]
- Le D, Deau P, Roche B, et al. Thiotepa, busulfan, cyclophosphamide: effective but toxic conditioning regimen prior to autologous hematopoietic stem cell transplantation in central nervous system lymphoma. Med Sci (Basel). 2023;11(1):14. [CrossRef]
- İlhan Ç, Suyanı E, Sucak GT, et al. Inflammatory markers, oxidative stress, and antioxidant capacity in healthy allo-HSCT donors during hematopoietic stem cell mobilization. J Clin Apher. 2015;30(4):197–203. [CrossRef]
- Araki D, Chen V, Redekar N, et al. Post-transplant G-CSF impedes engraftment of gene-edited human hematopoietic stem cells by exacerbating p53-mediated DNA damage response. Cell Stem Cell. 2025;32(1):53–70.e8. [CrossRef]
- Shah G, Giralt S, Dahi P. Optimizing high-dose melphalan. Blood Rev. 2024;64:101162. [CrossRef]
- Chai RC, McDonald MM, Terry RL, et al. Melphalan modifies the bone microenvironment by enhancing osteoclast formation. Oncotarget. 2017;8(40):68047–68058. [CrossRef]
- Jonsdottir G, Björkholm M, Turesson I, et al. Cumulative exposure to melphalan chemotherapy and subsequent risk of developing acute myeloid leukemia and myelodysplastic syndromes in multiple myeloma. Eur J Haematol. 2021;107(2):275–282. [CrossRef]
- Bekkem A, Selby G, Chakrabarty JH. Retrospective analysis of intravenous DMSO toxicity in transplant patients. Transplant Cell Ther. 2013;19(2 Suppl):S313. [CrossRef]
- Joseph NS, Kaufman JL, Gupta VA, et al. Quadruplet therapy for newly diagnosed myeloma: comparative analysis of sequential cohorts with triplet therapy lenalidomide, bortezomib and dexamethasone (RVd) versus daratumumab with RVd (DRVd) in transplant-eligible patients. Blood Cancer J. 2024;14(1):159. [CrossRef]
- Banerjee R, Williams L, Mikhael JR. Should I stay or should I go (to transplant)? Managing insufficient responses to induction in multiple myeloma. Blood Cancer J. 2023;13(1):89. [CrossRef]
- Zweegman S, van de Donk NWCJ. Maintain maintenance in multiple myeloma? Blood. 2023;142(18):1501–1502. [CrossRef]
- Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. [CrossRef]
- Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA–ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(3):309–322. Erratum in: Ann Oncol. 2022;33(1):117. doi:10.1016/j.annonc.2021.10.001. [CrossRef]
- Teoh PJ, Koh MY, Mitsiades C, Gooding S, Chng WJ. Resistance to immunomodulatory drugs in multiple myeloma: the cereblon pathway and beyond. Haematologica. 2025;110(5):1074–1091. [CrossRef]
- Franqui-Machin R, Wendlandt EB, Janz S, Zhan F, Tricot G. Cancer stem cells are the cause of drug resistance in multiple myeloma: fact or fiction? Oncotarget. 2015;6(38):40496–40506. [CrossRef]
- Vijjhalwar R, Kannan A, Fuentes-Lacouture C, Ramasamy K. Approaches to managing relapsed myeloma: switching drug class or retreatment with same drug class? Indian J Hematol Blood Transfus. 2025;41(3):478–493. [CrossRef]
- Medina-Herrera A, Sarasquete ME, Jiménez C, Puig N, García-Sanz R. Minimal residual disease in multiple myeloma: past, present, and future. Cancers (Basel). 2023;15(14):3687. [CrossRef]
- Banerjee R, Cowan AJ, Ortega M, et al. Financial toxicity, time toxicity, and quality of life in multiple myeloma. Clin Lymphoma Myeloma Leuk. 2024;24(7):446–454.e3. [CrossRef]
- Barberio J, Lash TL, Nooka AK, Naimi AI, Patzer RE, Kim C. Real-world risk of severe cytopenias in multiple myeloma patients sequentially treated with immunomodulatory drugs. Acta Haematol. 2025;148(2):135–147. [CrossRef]
- Teh BW, Harrison SJ, Worth LJ, et al. Risks, severity and timing of infections in patients with multiple myeloma: a longitudinal cohort study in the era of immunomodulatory drug therapy. Br J Haematol. 2015;171(1):100–108. [CrossRef]
- Hong JS, Zhou B, Yee AJ, Barmettler S. Hypogammaglobulinemia and daratumumab in multiple myeloma: risk factors, infections, immunoglobulin replacement, and mortality. Blood. 2024;144(Suppl 1):3353. [CrossRef]
- Reagan MR, Rosen CJ. Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. Nat Rev Rheumatol. 2016;12(3):154–168. [CrossRef]
- Dougherty JA, Elder CT. Managing multiple myeloma in the face of drug-induced adverse drug reaction. J Pharm Pract. 2022;35(3):500–504. [CrossRef]
- Karam K, Chebbo H, Saleh S, et al. Bortezomib-induced hepatotoxicity in a patient with multiple myeloma: a case report. Med Rep. 2024;6:100099. [CrossRef]
- Patel UH, Mir MA, Sivik JK, et al. Central neurotoxicity of immunomodulatory drugs in multiple myeloma. Hematol Rep. 2015;7(1):5704. [CrossRef]
- El-Cheikh J, Moukalled N, Malard F, Bazarbachi A, Mohty M. Cardiac toxicities in multiple myeloma: an updated and deeper look into the effect of different medications and novel therapies. Blood Cancer J. 2023;13(1):83. [CrossRef]
- Sonneveld P, Dimopoulos MA, Boccadoro M, et al. Daratumumab, bortezomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2024;390(4):301–313. [CrossRef]
- Jakubowiak AJ, et al. Carfilzomib, lenalidomide, and dexamethasone (KRd) as maintenance therapy after autologous stem-cell transplantation in newly diagnosed multiple myeloma. J Clin Oncol. 2025;43(16 Suppl):7535. [CrossRef]
- Richardson P, Beksaç M, Oriol A, et al. Pomalidomide, bortezomib, and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: final survival and subgroup analyses from the OPTIMISMM trial. Eur J Haematol. 2025;114(5):822–831. [CrossRef]
- Usmani SZ, Quach H, Mateos MV, et al. Final analysis of carfilzomib, dexamethasone, and daratumumab vs carfilzomib and dexamethasone in the CANDOR study. Blood Adv. 2023;7(14):3739–3748. [CrossRef]
- Wu W, Ding S, Zhang M, et al. Cost-effectiveness analysis of CAR-T cell therapy for patients with relapsed/refractory multiple myeloma in China. J Med Econ. 2023;26(1):701–709. [CrossRef]
- Sonneveld P, Chanan-Khan A, Weisel K, et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone alone in previously treated multiple myeloma: overall survival results from the phase 3 CASTOR trial. Hemasphere. 2022;6(Suppl):12. [CrossRef]
- Pourrahmat MM, Kim A, Kansal AR, et al. Health state utility values by cancer stage: a systematic literature review. Eur J Health Econ. 2021;22(8):1275–1288. [CrossRef]
- Zayad A, Magee G, Mansour R, et al. Efficacy and safety of daratumumab, pomalidomide and dexamethasone compared to daratumumab, carfilzomib and dexamethasone in relapsed multiple myeloma: multicenter real-world experience. Clin Lymphoma Myeloma Leuk. 2025 Aug 27. [CrossRef]
- Dimopoulos MA, Lonial S, White D, et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020;10(9):91. [CrossRef]
- Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for relapsed/refractory multiple myeloma: final overall survival analysis from the randomized phase II ELOQUENT-3 trial. J Clin Oncol. 2023;41(3):568–578. [CrossRef]
- Yee AJ, Laubach JP, Campagnaro EL, et al. Elotuzumab in combination with pomalidomide, bortezomib, and dexamethasone in relapsed and refractory multiple myeloma. Blood Adv. 2025;9(5):1163–1170. [CrossRef]
- Martin T, Dimopoulos MA, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma: updated results from IKEMA, a randomized phase 3 study. Blood Cancer J. 2023;13(1):72. Erratum in: Blood Cancer J. 2023;13(1):152. doi:10.1038/s41408-023-00923-6. [CrossRef]
- Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (ICARIA-MM): randomised, multicentre, open-label, phase 3 study. Lancet. 2019;394(10214):2096–2107. Erratum in: Lancet. 2019;394(10214):2072. doi:10.1016/S0140-6736(19)32944-7. [CrossRef]
- Xu J, Wang BY, Yu SH, et al. Long-term remission and survival in patients with relapsed or refractory multiple myeloma after treatment with LCAR-B38M CAR T cells: 5-year follow-up of the LEGEND-2 trial. J Hematol Oncol. 2024;17:23. [CrossRef]
- Canadian Agency for Drugs and Technologies in Health (CADTH). Ciltacabtagene autoleucel (Carvykti) indication: reimbursement recommendation [Internet]. Ottawa (ON): CADTH; 2024 Nov. Report No.: PG0361. PMID: 39693466.
- Jagannath S, Martin TG, Lin Y, et al. Long-term (≥5-year) remission and survival after treatment with ciltacabtagene autoleucel in CARTITUDE-1 patients with relapsed/refractory multiple myeloma. J Clin Oncol. 2025;43(25):e700–e710. [CrossRef]
- Richard S, Chari A, Delimpasi S, et al. Selinexor, bortezomib, and dexamethasone versus bortezomib and dexamethasone in previously treated multiple myeloma: outcomes by cytogenetic risk. Am J Hematol. 2021;96(9):1120–1130. [CrossRef]
- Sheykhhasan M, Ahmadieh-Yazdi A, Vicidomini R, et al. CAR T therapies in multiple myeloma: unleashing the future. Cancer Gene Ther. 2024;31(5):667–686. [CrossRef]
- Chekol Abebe E, Yibeltal Shiferaw M, Tadele Admasu F, Asmamaw Dejenie T. Ciltacabtagene autoleucel: the second anti-BCMA CAR T-cell therapeutic armamentarium of relapsed or refractory multiple myeloma. Front Immunol. 2022;13:991092. [CrossRef]
- Ayala Ceja M, Khericha M, Harris CM, Puig-Saus C, Chen YY. CAR-T cell manufacturing: major process parameters and next-generation strategies. J Exp Med. 2024;221(2):e20230903. [CrossRef]
- Afrough A, Abraham PR, Turer L, et al. Toxicity of CAR T-cell therapy for multiple myeloma. Acta Haematol. 2025;148(3):300–314. [CrossRef]
- Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. 2023;141(20):2430–2442. [CrossRef]
- Liu Q, Hu T, Wu H, et al. Prolonged haematologic toxicity in CAR-T-cell therapy: a review. J Cell Mol Med. 2023;27(23):3662–3671. [CrossRef]
- Beyar-Katz O, Rejeski K, Shouval R. Immune effector cell-associated hematotoxicity: mechanisms, clinical manifestations, and management strategies. Haematologica. 2025;110(6):1254–1268. [CrossRef]
- Chen S, van den Brink MRM. Allogeneic “off-the-shelf” CAR T cells: challenges and advances. Best Pract Res Clin Haematol. 2024;37(3):101566. [CrossRef]
- Anderson LD Jr. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol. 2022;18(3):277–289. [CrossRef]
- Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716. [CrossRef]
- Hillengass J, Cohen AD, Agha ME, et al. The phase 2 CARTITUDE-2 trial: updated efficacy and safety of ciltacabtagene autoleucel in patients with multiple myeloma and 1–3 prior lines of therapy (Cohort A) and with early relapse after first-line treatment (Cohort B). Blood. 2023;142(Suppl 1):1021. [CrossRef]
- Cohen AD, Mateos MV, Cohen YC, et al. Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood. 2023;141(3):219–230. [CrossRef]
- Arnulf B, Kerre T, Agha M, et al. MM-505: efficacy and safety of ciltacabtagene autoleucel ± lenalidomide maintenance in newly diagnosed multiple myeloma with suboptimal response to frontline autologous stem cell transplant: CARTITUDE-2 Cohort D. Lancet Haematol. 2024;[Epub ahead of print]. [CrossRef]
- Mina R, Mylin AK, Yokoyama H, et al. Patient-reported outcomes following ciltacabtagene autoleucel or standard of care in lenalidomide-refractory multiple myeloma (CARTITUDE-4): a randomised, open-label, phase 3 trial. Lancet Haematol. 2025;12(1):e45–e56. [CrossRef]
- Dytfeld D, Dhakal B, Agha M, et al. Bortezomib, lenalidomide and dexamethasone (VRd) followed by ciltacabtagene autoleucel versus VRd followed by lenalidomide and dexamethasone (Rd) maintenance in newly diagnosed multiple myeloma not intended for transplant: a randomized, phase 3 study (CARTITUDE-5). Blood. 2021;138(Suppl 1):1835. [CrossRef]
- Broijl A, San-Miguel J, Suzuki K, et al. EMAGINE/CARTITUDE-6: a randomized phase 3 study of DVRd followed by ciltacabtagene autoleucel versus DVRd followed by autologous stem cell transplant in transplant-eligible newly diagnosed multiple myeloma. Hemasphere. 2023;7(Suppl):22–23. [CrossRef]
- Rodriguez-Otero P, Ailawadhi S, Arnulf B, et al. Plain language summary of the KarMMa-3 study of ide-cel or standard-of-care regimens in relapsed or refractory multiple myeloma. Future Oncol. 2024;20(18):1221–1235. [CrossRef]
- Seyringer S, Pilz MJ, Al-Naesan I, et al. Validation of the cancer-specific utility measure EORTC QLU-C10D using evidence from four lung cancer trials covering six country value sets. Sci Rep. 2025;15:14907. [CrossRef]
- Osoba D, Zee B, Pater J, et al. Psychometric properties and responsiveness of the EORTC QLQ-C30 in patients with breast, ovarian and lung cancer. Qual Life Res. 1994;3(5):353–364. [CrossRef]
- Pickard AS, Ray S, Ganguli A, Cella D. Comparison of FACT- and EQ-5D-based utility scores in cancer. Value Health. 2012;15(2):305–311. [CrossRef]
- Yang SC, Kuo CW, Lai WW, et al. Dynamic changes of health utility in lung cancer patients receiving different treatments: a 7-year follow-up. J Thorac Oncol. 2019;14(11):1892–1900. [CrossRef]
- Harrison JP, Kim H. Eastern Cooperative Oncology Group performance status is an independent predictor of HRQoL in unresectable or metastatic melanoma. Value Health. 2015;18(7):A474. [CrossRef]
- Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. [CrossRef]
- Ronquest NA, Paret K, Gould IG, Barnett CL, Mladsi DM. The evolution of ICER’s review process for new medical interventions and a critical review of economic evaluations (2018–2019). J Manag Care Spec Pharm. 2021;27(11):1601–1612. [CrossRef]
- Bayraktar UD, Bashir Q, Qazilbash M, Champlin RE, Ciurea SO. Fifty years of melphalan use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19(3):344–356. [CrossRef]
- Konudula RD, Gorantla CSR, Athe R. Exploring holistic healing of cancer: German New Medicine (GNM) and homeopathic treatment beyond traditional therapies. J Appl Toxicol. 2025;45(9):1669–1674. [CrossRef]
- Pawlyn C, Davies FE. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood. 2019;133(7):660–675. [CrossRef]
- Souto Filho JTD, Cantadori LO, Crusoe EQ, Hungria V, Maiolino A. Daratumumab-based quadruplet versus triplet induction regimens in transplant-eligible newly diagnosed multiple myeloma: a systematic review and meta-analysis. Blood Cancer J. 2025;15(1):37. [CrossRef]
- Mateos MV, Martínez-López J, Rodriguez Otero P, et al. Curative strategy for high-risk smoldering myeloma: KRd followed by transplant, KRd consolidation, and Rd maintenance. J Clin Oncol. 2024;42(27):3247–3256. [CrossRef]
- Magen H, Muchtar E. Elotuzumab: the first approved monoclonal antibody for multiple myeloma treatment. Ther Adv Hematol. 2016;7(4):187–195. [CrossRef]
- Zhang CW, Wang YN, Ge XL. Lenalidomide use in multiple myeloma (review). Mol Clin Oncol. 2023;20(1):7. [CrossRef]
- Sogbein O, Paul P, Umar M, et al. Bortezomib in cancer therapy: mechanisms, side effects, and future proteasome inhibitors. Life Sci. 2024;358:123125. [CrossRef]
- Fricker LD. Proteasome inhibitor drugs. Annu Rev Pharmacol Toxicol. 2020;60:457–476. [CrossRef]
- Hoy SM. Pomalidomide: a review in relapsed and refractory multiple myeloma. Drugs. 2017;77(17):1897–1908. [CrossRef]
- El Khatib HH, Abdulla K, Nassar LK, Ellabban MG, Kakaruogkas A. Advancements in multiple myeloma therapies: a comprehensive review by disease stage. Lymphatics. 2025;3(1):2. [CrossRef]
Figure 1.
Simplified treatment algorithm for multiple myeloma according to transplant eligibility. Eligibility for ASCT is based on biological rather than chronological age. Transplant-ineligible patients receive non-transplant regimens aiming to sustain long-term disease control and quality of life (Created with BioRender.com).
Figure 1.
Simplified treatment algorithm for multiple myeloma according to transplant eligibility. Eligibility for ASCT is based on biological rather than chronological age. Transplant-ineligible patients receive non-transplant regimens aiming to sustain long-term disease control and quality of life (Created with BioRender.com).
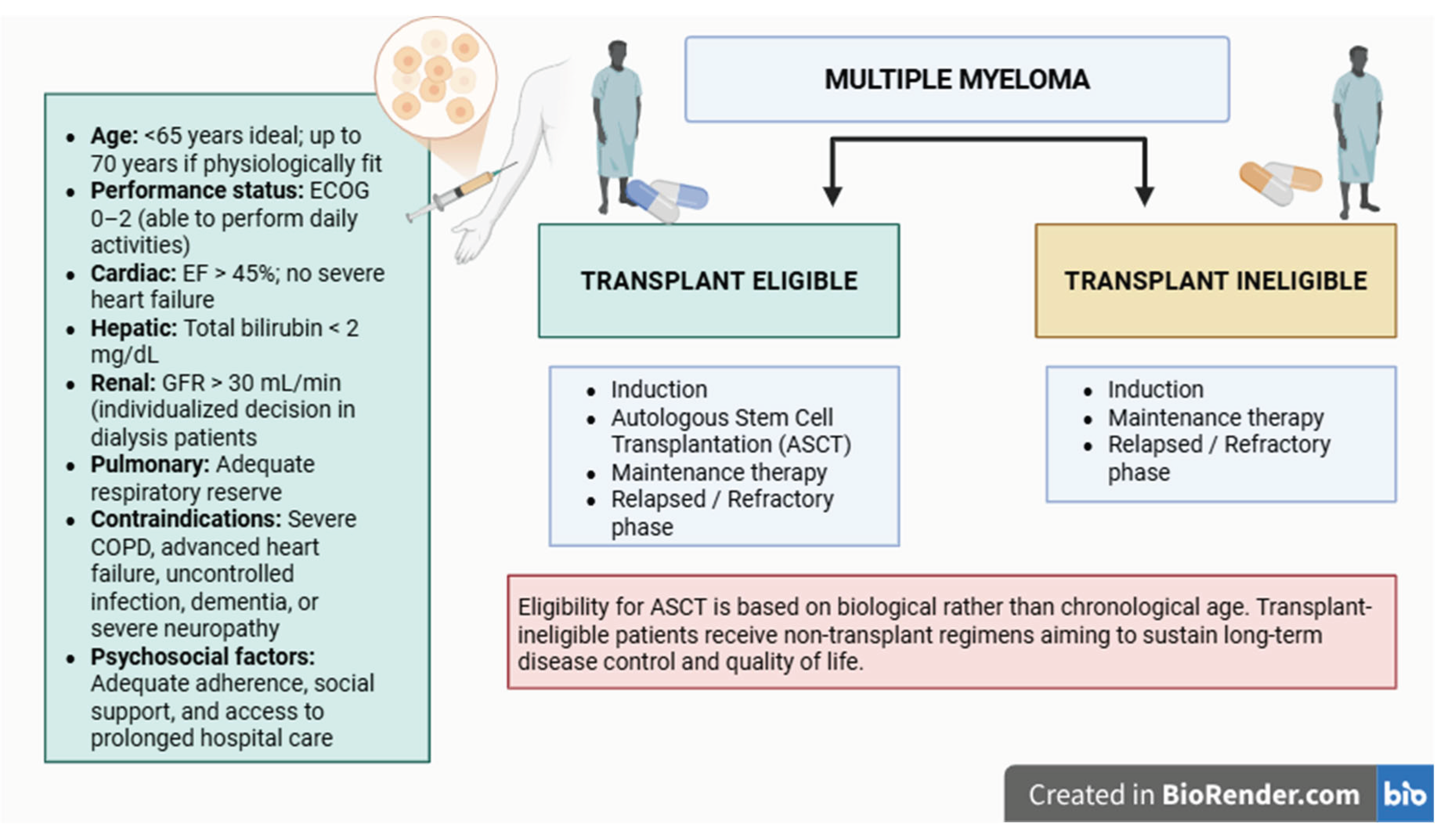
Figure 2.
Schematic representation of the autologous stem cell transplantation (ASCT) process. The procedure comprises sequential stages: patient preparation, hematopoietic stem cell mobilization using cyclophosphamide (Day 1) and granulocyte colony-stimulating factor (G-CSF, Days 4-9), peripheral stem cell collection through apheresis (Days 9-10), and cryopreservation of harvested stem cells in 10% DMSO. Following a resting interval, high-dose melphalan conditioning (Day 15) eradicates residual myeloma cells and prepares the bone marrow niche for reinfusion. Autologous CD34+ cells are then reinfused (Day 17). After transplantation, the patient is monitored in a protective environment to prevent infection during the neutropenic phase. Engraftment occurs as reinfused stem cells home to the marrow, proliferate, and reconstitute hematopoiesis and immune function within approximately 2-4 weeks (Created with BioRender.com).
Figure 2.
Schematic representation of the autologous stem cell transplantation (ASCT) process. The procedure comprises sequential stages: patient preparation, hematopoietic stem cell mobilization using cyclophosphamide (Day 1) and granulocyte colony-stimulating factor (G-CSF, Days 4-9), peripheral stem cell collection through apheresis (Days 9-10), and cryopreservation of harvested stem cells in 10% DMSO. Following a resting interval, high-dose melphalan conditioning (Day 15) eradicates residual myeloma cells and prepares the bone marrow niche for reinfusion. Autologous CD34+ cells are then reinfused (Day 17). After transplantation, the patient is monitored in a protective environment to prevent infection during the neutropenic phase. Engraftment occurs as reinfused stem cells home to the marrow, proliferate, and reconstitute hematopoiesis and immune function within approximately 2-4 weeks (Created with BioRender.com).
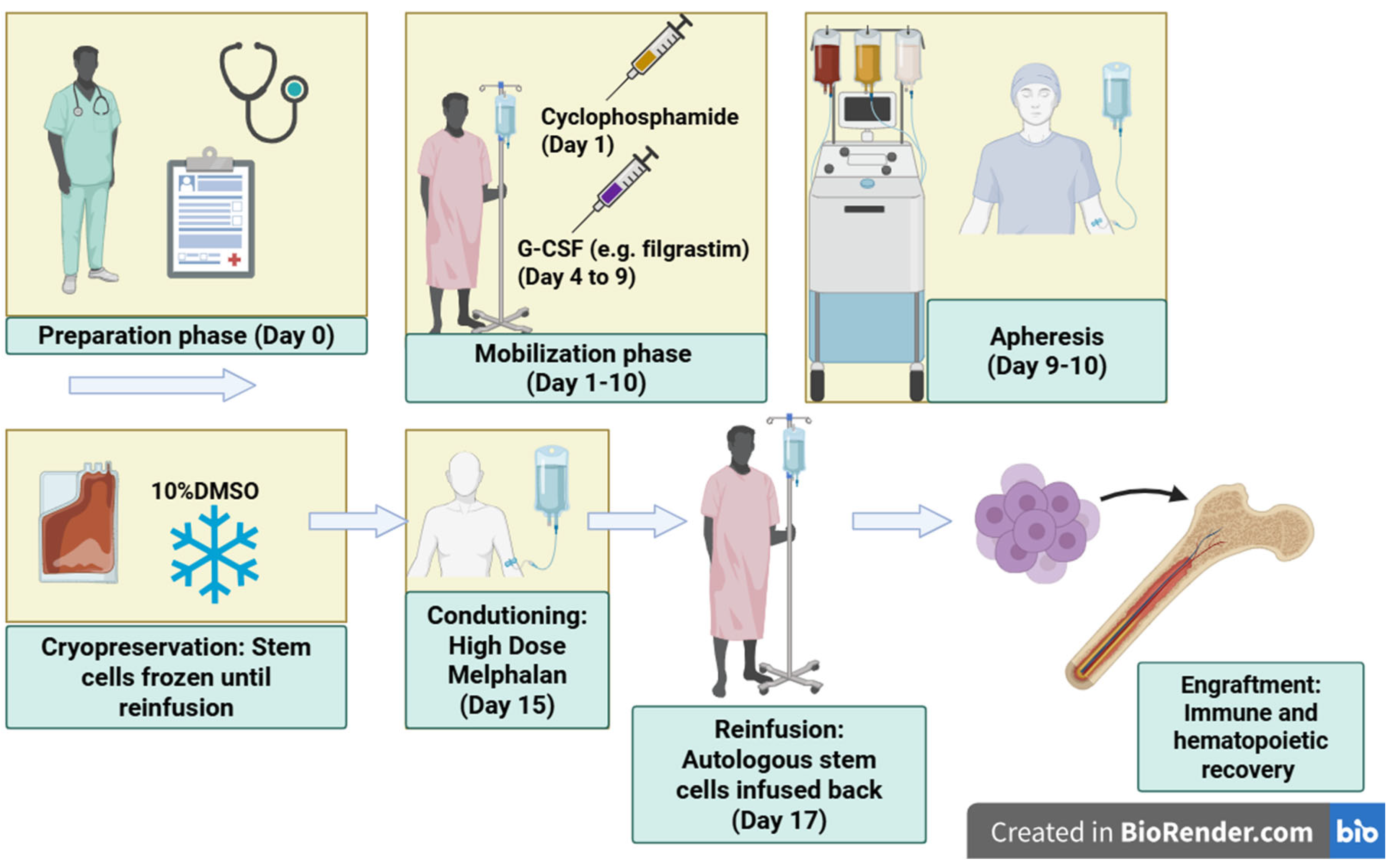
Figure 3.
Schematic representation of disease progression from premalignant stages (MGUS, SMM) to active multiple myeloma, and the cumulative impact of sequential therapy lines. Each treatment line contributes to cumulative toxicity, bone marrow damage, and declining patient tolerance, ultimately leading to poor outcomes (Created with BioRender.com).
Figure 3.
Schematic representation of disease progression from premalignant stages (MGUS, SMM) to active multiple myeloma, and the cumulative impact of sequential therapy lines. Each treatment line contributes to cumulative toxicity, bone marrow damage, and declining patient tolerance, ultimately leading to poor outcomes (Created with BioRender.com).
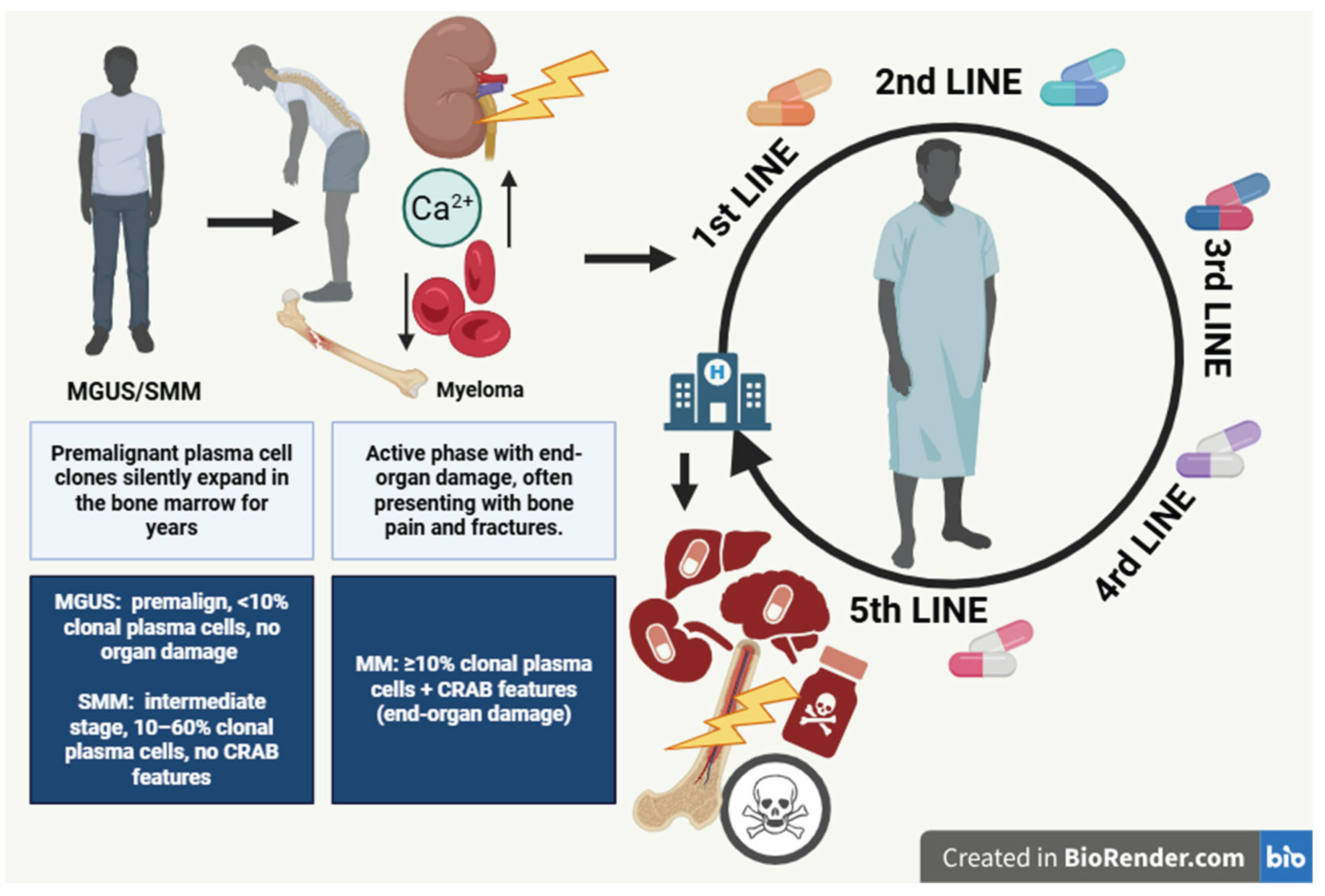
Figure 4.
Schematic representation of the CAR-T cell manufacturing process. Autologous T cells are harvested from the patient via leukapheresis, then enriched and activated through CD3/CD28 co-stimulation with cytokine support (IL-2, IL-7, IL-15). The CAR construct is introduced using lentiviral vectors, followed by large-scale expansion, quality control, and cryopreservation. After bridging therapy and lymphodepleting chemotherapy, the engineered CAR-T cells are thawed and administered intravenously (Created with BioRender.com).
Figure 4.
Schematic representation of the CAR-T cell manufacturing process. Autologous T cells are harvested from the patient via leukapheresis, then enriched and activated through CD3/CD28 co-stimulation with cytokine support (IL-2, IL-7, IL-15). The CAR construct is introduced using lentiviral vectors, followed by large-scale expansion, quality control, and cryopreservation. After bridging therapy and lymphodepleting chemotherapy, the engineered CAR-T cells are thawed and administered intravenously (Created with BioRender.com).
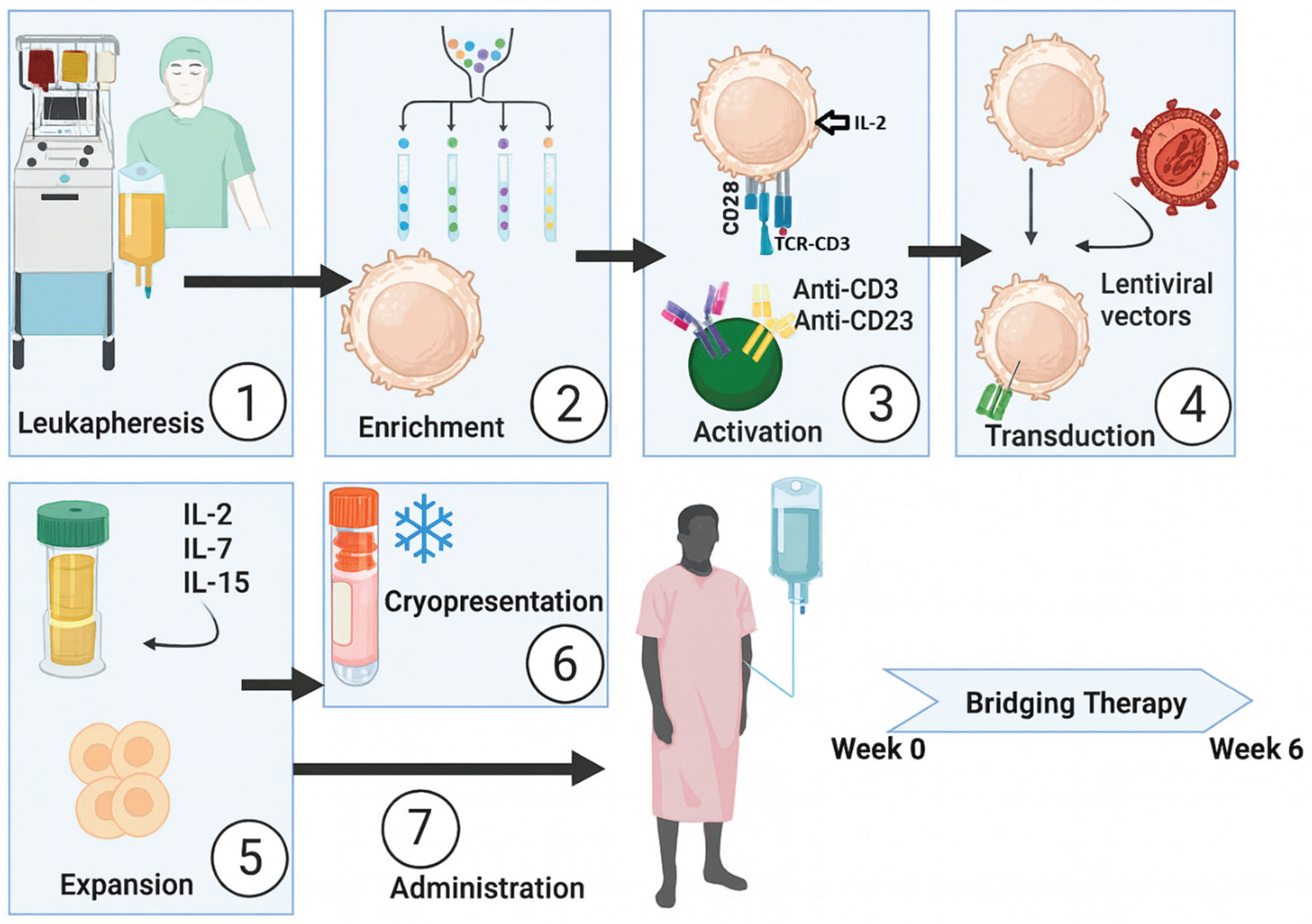
Figure 5.
ICER distribution across myeloma therapies based on ECOG-adjusted utility approach. Each bar represents the incremental cost-effectiveness ratio (ICER) expressed as cost per QALY gained. Green tones illustrate relative cost-effectiveness proximity among regimens, highlighting CAR-T (CARTITUDE-1) within the lowest ICER range, comparable to D-VRd and other advanced monoclonal antibody combinations.
Figure 5.
ICER distribution across myeloma therapies based on ECOG-adjusted utility approach. Each bar represents the incremental cost-effectiveness ratio (ICER) expressed as cost per QALY gained. Green tones illustrate relative cost-effectiveness proximity among regimens, highlighting CAR-T (CARTITUDE-1) within the lowest ICER range, comparable to D-VRd and other advanced monoclonal antibody combinations.
Table 1.
Current Induction Regimens and Their Main Adverse Effects.
| Regimen | Patient Group | Main Advantages | Main Toxicities |
| VRd | Transplant-eligible or ineligible (standard-risk) | High response rate, prolonged PFS | Peripheral neuropathy, hematologic toxicity |
| D-VRd | Transplant-eligible | Increased MRD negativity, well tolerated | Infusion reactions, hematologic toxicity |
| KRd | Younger, fit, high-risk cytogenetics | Deeper response, increased MRD negativity | Cardiotoxicity, hypertension |
| D-KRd | Transplant-eligible, high-risk | Deep and durable MRD negativity | Cardiac and hematologic toxicity |
| D-VMP | Transplant-ineligible, elderly | OS benefit, effective in elderly | Hematologic toxicity, infections |
| D-Rd | Transplant-ineligible, frail or elderly | Long OS, oral administration convenience | Immunosuppression, hematologic toxicity |
| Rd | Frail or relapsed patients | Oral administration, convenience | Myelosuppression, fatigue |
| VRd-Lite | Elderly, frail, or comorbid | Better tolerability | Potential lower efficacy |
V: Velcade(Bortezomib), R: Revlimid(Lenalidomide), D:Darzalex(Daratumumab), K:Kyprolis(Carfilzomib), M:Melphalan, P: Prednisone, d: Dexamethasone. MRD: Minimal Residual Disease. OS=Overall Survival, PFS: Progression Free Survival.
Table 2.
Estimated costs, mean overall survival (OS), and model-derived QALY values for major myeloma therapy regimens used in clinical practice.
Table 2.
Estimated costs, mean overall survival (OS), and model-derived QALY values for major myeloma therapy regimens used in clinical practice.
| Therapy | Setting/Lines | Mean OS*a | Mean PFS*a |
Cost*b USD |
QALY*c |
| Rd [23] | First Line/Frail | 57 months | 40 months | 995,924 USD | 3.6195 |
| D-Rd [23] | First Line/TIE | 70 months | 54.1 months | 1,924,803 USD | 4.434 |
| D-VRd [37,55] | First Line/TE | 159.12 months | 83.75 months | 2,184,110 USD | 9.9874 |
| VRd [37,55] | First Line/TE | 128.9 months | 37.98 months | 508,940 USD | 7.145 |
| KRd [56] | 2-5th Line | 56.9 months | 23 months | 795,518 USD | 2.608 |
| D-VMP [17,22] | First Line/TIE | 87 months | 66.7 months | 766,416 USD | 4.9778 |
| VMP [17,22] | First Line/TIE | 68 months | 42.4 months | 72,731 USD | 3.0866 |
| PVd [57] | 2-4th+ Lines | 43 months | 24 months | 633,028 USD | *d 0.7478 |
| Vd [57] | 2-4th+ Lines | 38 months | 18 months | 70,333 USD | *d 0.6727 |
| D-Kd [58,59] | 2-4th Lines | 61 months | 37 months | 2,153,071 USD | 2.103 |
| D-Vd [58,59,60,61] | 2-4+Lines | 66 months | 56 months | 611,243 USD | 1.9338 |
| D-Pd [58,59,62] | 2-4th Lines | 73 months | 32 months | 1,176,157 USD | 2.4164 |
| Kd [58,59] | 2-4th Lines | 55 months | 20 months | 935,796 USD | 1.7618 |
| Elo-Rd [63] | 2-4th Lines | 47.1 months | 31 months | 995,764 USD | 0.9655 |
| Elo-Pd [64] | 3-5th+ Lines | 34 months | 22 months | 835,752 USD | 0.8139 |
| Elo-PVd [65] | 2-10th Lines | 35 months | 18 months | 700,628 USD | 0.875 |
| Isa-Kd [58,66] | 2-4th Lines | 54 months | 34 months | 1,700,907 USD | 2.511 |
| Isa-Pd [67] | 3+ Lines | 38 months | 23.3 months | 907,989 USD | 0.95 |
| CAR-T Legend-2 [68,69] | 4+ Lines | 54 months+ | 30 months | 640,000 USD | 1.7433 |
| CARTITUDE-1 [69,70] | 4+ Lines | 85 months+ | 43.85 months | 640,000 USD | 2.7384 |
| X-Vd [71] | 2-4th Lines | 34.98 months | 18.5 | 839,679 USD | 1.575 |
*a: Mean OS values were calculated by digitalizing Kaplan–Meier survival curves and estimating the area under the survival percentage curve. Values represent fully observed cohorts only; extrapolation for censored data was performed using a simple geometric projection model. *b Cost estimation reflects total drug acquisition and administration expenses up to treatment discontinuation, based on list prices obtained from Drugs.com. Supportive care and concomitant therapy costs were included within the same time horizon. *c QALY values were derived from mean OS and ECOG-based utility adjustments within the hypothesis framework of this study (see Methods and Supplementary Material for details). d QALY could not be modeled due to insufficient follow-up data. V: Velcade(Bortezomib), R: Revlimid(Lenalidomide), D:Darzalex(Daratumumab), K:Kyprolis(Carfilzomib), M:Melphalan, P: Prednisone(VMP), d: Dexamethasone., P:Pomalyst (Pomalidomide), X:Xpovio (Selinexor), Elo:Empliciti(Elotuzumab), Isa:Sarclisa(Isatuximab). OS=Overall Survival. PFS: Progression Free Survival. USD: United States Dollar. QALY: The quality-adjusted life-year.
Table 3.
Clinical Summary of Cilta-cel and Ide-cel Based CAR-T Trials.
| Study Name | Phase / Design | Patient Profile | Treatment Line | Regimen / Comparator | Key Message |
| LEGEND-2 (NCT03090659) [68] | Phase 1, open-label, single-arm | ≥3 prior lines (PI + IMiD-treated RRMM) | 4+ line | LCAR-B38M single infusion | First-in-human BCMA-CAR-T; deep and durable responses |
| CARTITUDE-1 (NCT03548207) [70] | Phase 1b/2, multicenter | ≥3 prior lines (PI + IMiD + anti-CD38) | 4+ line | Cilta-cel single infusion | Global validation; high depth and long PFS |
| CARTITUDE-2 (NCT04133636) | Phase 2, multi-cohort | Cohorts A-E: RRMM, len-refractory, 1-3 prior lines | 2nd–3rd line | Cilta-cel single infusion | High efficacy maintained in early-line settings |
| CARTITUDE-4 (NCT04181827) [85] | Phase 3, global | 1–3 prior lines, len-refractory | 2nd–3rd line | Cilta-cel vs PVd / DPd | Phase 3 confirmation of superiority vs SOC |
| CARTITUDE-5 (NCT04923893) [86] | Phase 3 (ongoing) | Newly diagnosed, transplant ineligable | 1st line | VRd+Cilta-cel vs VRd+Rd maintenance | CAR-T as potential maintenance alternative in frontline |
| CARTITUDE-6 (NCT05257083) [87] | Phase 3, comparative | Newly diagnosed, transplant-eligible | 1st line | D-VRd+Cilta-cel + Lenalidomide vs D-VRd+ASCT+DVRd+Lenalidomide | Immune-regenerative CAR-T as ASCT replacement |
| KarMMa-1 (NCT03361748) [13] | Phase 2, single-arm | ≥3 prior lines RRMM | 4+ line | Ide-cel (bb2121) | First approved BCMA-CAR-T (US) |
| KarMMa-3 (NCT03651128) [88] | Phase 3, randomized | 2nd–4th line | - | Ide-cel vs SOC | Ide-cel effective but with shallower depth of response |
BCMA: B-cell maturation antigen; SOC: Standard of Care; NDMM: Newly Diagnosed Multiple Myeloma; RRMM: Relapsed Refractory Multiple Myeloma; CAR: Chimeric antigen receptor; PFS: Progression Free Survival.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



