Submitted:
22 September 2025
Posted:
23 September 2025
You are already at the latest version
Abstract
Background/Objectives: 3-Hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD) is an extremely rare autosomal recessive metabolic disorder caused by mutations in the HMGCL gene. HMGCLD disrupts ketogenesis and β-oxidation, leading to energy failure during fasting or stress, with clinical episodes characterized by hypoglycemia, hyperammonemia, lactic acidosis, and encephalopathy. Only 211 cases have been reported worldwide, with no prior reports on anesthetic management in these patients. Clinical features: We report a 14.5-year-old girl with known HMGCLD who was admitted with abdominal pain and nausea following a fatty meal. Imaging confirmed acute cholecystitis. Initial conservative management failed due to persistent vomiting and inability to tolerate feeding. Deviation from the metabolic protocol led to lactic acidosis and hypoglycemia, requiring intensive care with bicarbonate, carnitine, and glucose infusion. Once optimized, she underwent emergency laparoscopic cholecystectomy under sevoflurane-based anesthesia. Propofol was avoided given the patient’s compromised lipid metabolism. Intraoperative glucose and acid-base status were closely monitored, with balanced dextrose-based fluids. The patient remained hemodynamically stable throughout and was discharged three days postoperatively. Conclusions: This case highlights the anesthetic challenges of HMGCLD, where system-level miscommunication can trigger severe metabolic decompensation. A review of the literature emphasizes fasting avoidance, continuous glucose supplementation, careful drug and fluid selection, and multidisciplinary coordination. This report provides the first anesthetic roadmap for HMGCLD, underscoring the need for individualized care and meticulous perioperative metabolic control.
Keywords:
case reports
; 3-hydroxy-3-methylglutaryl CoA lyase deficiency
; fatty acid oxidation disorders
; anesthesia
; general
; perioperative care
; adolescent
; emergency surgical procedures
1. Introduction
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) lyase deficiency or HMGCLD is an autosomal recessive disorder first described by Faull et al. [1]. It results from mutations in the HMGCL gene [2,3], which affects the pathways of ketogenesis and leucine catabolism. It is considered a very rare disease with an estimated prevalence of less than 1 in 100,000 live births [4]; only approximately 200 cases have been reported in the literature [5]. Clinical manifestations include recurrent episodes of metabolic acidosis, hyperammonemia, and hypoglycemia [2,6]. During periods of prolonged starvation, glycogen depletion leads to lactic acidosis because of impaired β-oxidation and the inability to generate ketones from fatty acids.
To our knowledge, there are no published case reports regarding the anesthetic management of patients with HMGCLD. This report provides the first documented approach to perioperative anesthetic management in this population, offering practical guidance to avoid life-threatening decompensation in similar cases. We present a pediatric patient with HMGCLD who underwent emergency surgery and review the literature on perioperative care in fatty acid oxidation disorders. The objective of this article is to illustrate the anesthetic considerations in such patients and to underscore the critical importance of individualized anesthetic planning and strict adherence to metabolic protocols. Written informed consent for publication was obtained from the patient’s guardians.
2. Case Presentation
A 14.5-year-old girl (72 kg, 160 cm) with a known history of HMGCLD was admitted to the Pediatrics department of our hospital for investigation of abdominal pain and nausea that occurred after a fatty meal. An initial abdominal ultrasound suggested cholelithiasis. The patient was managed conservatively with intravenous fluids containing glucose and was able to tolerate a fat-free diet.
Despite this management, her condition worsened over three days, prompting a reassessment that confirmed the diagnosis of acute cholecystitis. Consequently, she was transferred to the General Surgery Department for further conservative treatment, which involved an NPO (nil per os) protocol, symptomatic therapy, and intravenous Maintelyte solution. During this period, she developed recurrent moderate to severe hypoglycemia requiring glucose boluses. After two days of marginal clinical improvement, a fluid diet was introduced, but she experienced recurrent vomiting with each feeding attempt over the following five days.
On day 10, her IV fluids were switched to Ringer’s Lactated solution, leading to clinical and laboratory deterioration within 12 hours. Ongoing vomiting prompted re-evaluation and the decision for surgical intervention, with consultation from anesthesiology and intensive care specialists. Because of persistent lactic acidosis and hypoglycemia, she was transferred to the Pediatric Intensive Care Unit (PICU) for preoperative stabilization. In the PICU, an arterial line was placed, and she received bicarbonate, L-carnitine, and 10% dextrose with electrolytes (NaCl, KCl, MgSO₄) at 150 mL/h, resulting in clinical improvement.
Following optimization, the patient underwent emergency laparoscopic cholecystectomy under standard ASA and invasive blood pressure monitoring. Anesthesia was induced with IV midazolam (5 mg), fentanyl (250 mcg), and cisatracurium (15 mg), with uncomplicated tracheal intubation using a size 7 cuffed endotracheal tube. Intraoperative anesthesia was maintained with sevoflurane at MAC 1.0. Ventilation was set at FiO₂ 35%, with decelerating flow and volume guarantee. Continuous monitoring of arterial blood gases and glucose ensured metabolic stability throughout the procedure. The patient’s glucose levels and arterial blood gas results are shown in Table 1.
The patient received maintenance fluids consisting of 0.9% normal saline at 200 mL/hour and the enriched glucose solution at 120 mL/hour. Antiemetic prophylaxis included 8 mg of dexamethasone and 4 mg of ondansetron. Postoperative analgesia was managed with a 30-minute intraoperative infusion of dexmedetomidine (25 μg), a 3 mg intravenous bolus of morphine, and preoperative administration of 1 g of IV paracetamol. The procedure lasted approximately 40 minutes and was uneventful, with the patient maintaining stable hemodynamic parameters. Neuromuscular blockade was reversed with 2.5 mg neostigmine and 1 mg atropine, and the patient emerged uneventfully. The patient remained in the PICU for two days, followed by transfer to the General Surgery Department for one additional day. Her postoperative course was uncomplicated, with minimal pain and a high level of patient satisfaction.
3. Discussion
The HMGCL gene expresses the 3-hydroxy-3-methylglutaryl-coenzyme A lyase (3-HMG-CoA lyase). 3-HMG-CoA lyase is the final mitochondrial enzyme in ketone-producing pathways: fatty acid oxidation and leucine catabolism. Without this enzyme, acetoacetate and acetyl-CoA cannot be produced from either pathway, depriving the body of key energy sources during starvation (Figure 1) [3]. This explains the profound hypoglycemia during fasting and why prolonged NPO status is especially dangerous in these patients.
In a healthy individual, during periods of starvation, the resulting hypoglycemia activates compensatory metabolic pathways. Specifically, increased glucagon production triggers hepatic glycogenolysis as well as hepatic and renal gluconeogenesis [9]. Glucogenic amino acids like alanine and aspartate serve as gluconeogenic substrates [10], if glucose remains unavailable. As prolonged fasting depletes glycogen stores, lipolysis is also utilized as an energy source.
In lipolysis, each triacylglycerol molecule yields glycerol and free fatty acids. While glycerol enters the gluconeogenic pathway, free fatty acids are beta-oxidized in the inner mitochondrial membrane to produce acetyl-CoA in the liver [11,12]. In addition to transporting fatty acids across mitochondrial membranes, carnitine helps clear toxic byproducts from the organelle [13]. Acetyl-CoA from fatty acid oxidation enters the citric acid cycle or is converted to ketone bodies. Ketones are essential for glucose-dependent tissues, such as the brain and the heart [11]. In glucose deprivation, these tissues possess the necessary enzymes to use ketones for energy production [12].
Lipolysis and gluconeogenesis are intricately interconnected. Specifically, beta-oxidation is a major supplier for molecules such as NADH+ and acetyl-CoA, which in turn stimulate pyruvate carboxylase (PC) and inhibit pyruvate dehydrogenase (PDH) enzymes [14]. PC is the enzyme responsible for the conversion of pyruvate to oxaloacetate, to serve as a substrate for gluconeogenesis. PDH, on the other hand, converts pyruvate into acetyl- CoA, thus promoting glycolysis, when fatty acid oxidation is impaired. Gluconeogenic substrates such as alanine and lactate are normally converted into pyruvate to enter the gluconeogenesis path. As a result, increased lipolysis and fatty acid oxidation promote diversion of pyruvate towards gluconeogenesis and inhibit glycolysis in the liver, preserving the available glucose for other tissues [14,15]. Through this delicate balance, there is an ongoing supply of glucose and ketone bodies to the tissues, as summarized in Figure 2.
In HMGCLD, as seen in our patient, key metabolic processes are disrupted. Due to impaired beta-oxidation, the liver cannot produce ketones or effectively use gluconeogenic substrates like lactate and alanine. Additionally, 3-HMG-CoA lyase plays a role in leucine catabolism into acetoacetate and acetyl-CoA (Figure- 1) [5]. During starvation, with no glucose intake and lipolysis insufficient for energy, the body relies on protein catabolism and gluconeogenesis. Although proteolysis occurs, leucine cannot be used for energy, as it cannot be converted into ketone bodies [2,3], and other ketogenic and glucogenic amino acids enter a compromised gluconeogenic cycle [14] (Figure 3).
This metabolic rigidity means that even brief catabolic stressors, such as prolonged fasting, vaccination, infection or surgical stress, can precipitate metabolic crisis. in patients with HMG-CoA lyase or synthase deficiency. As shown by Thompson et al. [17], even brief fasting may cause severe hypoketotic hypoglycemia, with a clinical spectrum ranging from vomiting, food refusal and altered mental status to encephalopathy, seizures, coma, or even death. This underscores the indispensable role of hepatic ketogenesis in preserving cerebral energy balance under catabolic stress. In our case, the patient was fasting for one week, with any feeding attempt resulting in vomiting. However, she did not yet present metabolic decompensation, because she was constantly receiving supplementary exogenous glucose through an IV infusion. A day after this solution was switched to Ringer’s Lactated, she was deprived of glucose supplementation, and deteriorated.
Laboratory findings during decompensation episodes, typically reveal profound hypoglycemia (without ketonemia), hyperammonemia, lactic acidosis, and abnormal liver function tests, all of which were present in our case. Usual complications are hepatomegaly, cardiomyopathy, arrhythmias, skeletal myopathy, rhabdomyolysis and pancreatitis due to lipotoxicity, and lack of energy [3,15].
Our patient was diagnosed with HMGCLD soon after birth. Management at home involved sufficient caloric intake, frequent meals and moderate protein and fat restriction as described in literature [18]. She was not on regular L-carnitine due to lack of consensus in the literature [8,19]. This was the second metabolic decompensation episode that required PICU, out of 25 total mild decompensation episodes during our patient’s lifetime. Metabolic decompensation is critical, as 1 out of 3 patients may suffer permanent, severe neurological deficit [3].
Clinical awareness and perioperative protocols must emphasize strict fasting avoidance, early glucose administration per os or IV, and close metabolic monitoring to prevent life-threatening complications. In case of severe metabolic acidosis (HCO3- <16 mEq/L), administration of intravenous bicarbonate at a dose of 1 mEq/kg is recommended [5,18]. In our case, the patient’s HCO3- levels upon admission to the PICU were found very low (15mEq/L), and she therefore received an IV infusion of 70 mEq NaHCO3. Some authors suggest L-carnitine intravenous administration at 30- 50 mg/kg/day, which was also administered during preoperative optimization, in order to avoid secondary L-carnitine deficiency, oxidative stress and intracellular depletion of free coenzyme A [3,8,18]. Carnitine molecules generally bind to the accumulating toxic products of the impaired fatty acid metabolism, to excrete them through the urine. The resulting secondary L-carnitine deficiency may lead to impaired neutralization of oxygen free radicals and accumulation of lipotoxic byproducts [20].
It is now apparent that patients with HMGCLD require very careful anesthetic planning. Given that there were no reported cases detailing anesthetic management, we relied on published case reports of patients with other beta-oxidation deficiencies [21,22,23] and general practice guidelines [5]. Bearing in mind the complex pathophysiological processes, we decided to avoid propofol, because it is prepared as a liquid lipid emulsion, thus increasing the lipid load of an already compromised fatty acid metabolism pathway [23]. Furthermore, propofol infusion syndrome seems to involve similar derangements in the oxidative processes of the inner mitochondrial membrane, as in HMGCLD. We also chose not to administer lidocaine or other local anesthetics, either intravenously or in regional anesthetic techniques as the main antidote in case of inadvertent local anesthetic toxicity is a lipid emulsion, contraindicated in our patient.
Regarding muscle relaxation, cisatracurium was preferred over rocuronium due to its lower hepatic metabolism, as it is primarily degraded by plasma esterases, advantageous given the patient's mildly elevated liver enzymes. Additionally, if rocuronium had been used, the reversal with sugammadex would have raised theoretical concerns, as sugammadex can bind steroid hormones such as cortisol in experimental settings, although clinical studies in humans have not demonstrated clinically significant adrenal supression [24]. In HMGCLD, disruptions in beta-oxidation may indirectly affect cholesterol synthesis and homeostasis by altering acetyl-CoA availability or cellular pathways regulating cholesterol metabolism [25]. This cautious approach avoided potential metabolic interference and ensured safe neuromuscular blockade.
Finally, we had to be diligent regarding the choice of intravenous fluids. We did not administer Ringer’s Lactated solution, as it contains lactate that could further deteriorate existing lactic metabolic acidosis. Instead, we administered Normal Saline 0.9% combined with Dextrose 10% with added electrolytes to correct hypoglycemia and electrolyte imbalances.
A major challenge was the necessity for coordinated collaboration among healthcare professionals from multiple departments. Due to case rarity, clinical decisions diverged from standard protocols, underscoring the critical importance of non-technical skills, particularly communication, situational awareness, and teamwork, vital for minimizing errors [26].
A key miscommunication involved IV glucose as a core element of HMGCLD management. On the fourth day of hospitalization, the glucose-rich solution (Maintelyte) was replaced with Ringer’s Lactated solution, which was consistent with standard practice in the surgical department but not appropriate for this patient’s fragile metabolic state. This substitution resulted in deterioration, highlighting the risks of applying routine protocols in complex metabolic disorders. This scenario exemplifies three recurrent issues in multidisciplinary collaborations: a lack of comprehensive situational awareness among team members, ineffective closed-loop communication, and confirmation bias, where clinicians default to familiar procedures based on previous experience rather than customized patient needs [26]. Recognizing these pitfalls is essential for optimizing interdisciplinary teamwork, especially in rare metabolic disorders requiring precise, collaborative decision-making.
4. Conclusions
HMGCLD is an exceptionally rare metabolic disturbance of fatty acid oxidation, with complex pathophysiology and only a few hundred cases reported. The absence of previous reports on anesthetic management makes this case particularly valuable, as it highlights the interplay between metabolic fragility, anesthetic drug choice, and system-level communication. Our experience underscores that individualized planning and multidisciplinary cooperation are essential to maintain stability and avoid life-threatening decompensation.
Key anesthetic lessons from this case include:
- Strict avoidance of prolonged fasting with early and continuous glucose supplementation.
- Avoidance of propofol and lipid-based therapies, due to impaired fatty acid metabolism.
- Use of lactate-free intravenous solutions to prevent worsening acidosis.
- Selection of cisatracurium over rocuronium, reducing hepatic load and avoiding sugammadex concerns.
- Close interdisciplinary communication to prevent errors such as inappropriate fluid substitutions.
Together, these principles provide practical guidance for the safe perioperative management of patients with HMGCLD and may inform future development of tailored anesthetic protocols for rare fatty acid oxidation disorders.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, V.N., F.C. and G.P.; formal analysis, V.N., G.P.; writing—original draft preparation, A.K., C.M.; writing—review and editing, V.N., I.G., S.I., A.P., G.P.; supervision, V.N.
Funding
This research received no external funding
Institutional Review Board Statement
This case report did not require ethical approval.
Informed Consent Statement
Written informed consent for publication was obtained from the patient’s guardians.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We would like to thank the patient and their guardians for providing consent to publish this case report. We also extend our gratitude to the operating room and Pediatric Intensive Care Unit teams for their professionalism, expertise, and collaborative care.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| HMGCLD | 3-hydroxy-3-methylglutaryl-CoA lyase deficiency |
| HMGCL | 3-hydroxy-3-methylglutaryl-CoA lyase gene |
| HMG-CoA | 3-hydroxy-3-methylglutaryl coenzyme A |
| PICU | Pediatric Intensive Care Unit |
| ASA | American Society of Anesthesiologists |
| IV | Intravenous |
| ABG(s) | Arterial Blood Gas(es) |
| PaO₂ | Partial pressure of oxygen in arterial blood |
| FiO₂ | Fraction of inspired oxygen |
| PaCO₂ | Partial pressure of carbon dioxide in arterial blood |
| BE | Base excess |
| HCO₃⁻ | Bicarbonate |
| NaCl | Sodium chloride |
| KCl | Potassium chloride |
| MgSO₄ | Magnesium sulfate |
| MAC | Minimum alveolar concentration |
| PC | Pyruvate carboxylase |
| PDH | Pyruvate dehydrogenase |
| LDH | Lactate dehydrogenase |
| ALT | Alanine aminotransferase |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| G6Pase | Glucose-6-phosphatase |
| G3PDH | Glyceraldehyde-3-phosphate dehydrogenase |
| NADH | Nicotinamide adenine dinucleotide (reduced form) |
| FADH₂ | Flavin adenine dinucleotide (reduced form) |
References
- Faull K, Bolton P, Halpern B, others. Patient with Defect in Leucine Metabolism. N Engl J Med. 1976;294:1013–5.
- Grünert SC, Schlatter SM, Schmitt RN, Gemperle-Britschgi C, Mrázová L, Balcı MC, et al. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Clinical presentation and outcome in a series of 37 patients. Mol Genet Metab. 2017 Jul;121(3):206–15.
- Grünert SC, Sass JO. 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: one disease - many faces. Orphanet J Rare Dis. 2020 Feb 14;15(1):48.
- Pié J, López-Viñas E, Puisac B, Menao S, Pié A, Casale C, et al. Molecular genetics of HMG-CoA lyase deficiency. Mol Genet Metab. 2007 Nov;92(3):198–209.
- Thompson S, Hertzog A, Selvanathan A, Batten K, Lewis K, Nisbet J, et al. Treatment of HMG-CoA Lyase Deficiency-Longitudinal Data on Clinical and Nutritional Management of 10 Australian Cases. Nutrients. 2023 Jan 19;15(3):531.
- Leipnitz G, Vargas CR, Wajner M. Disturbance of redox homeostasis as a contributing underlying pathomechanism of brain and liver alterations in 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. J Inherit Metab Dis. 2015 Nov;38(6):1021–8.
- Puisac B, Arnedo M, Concepcion M, Teresa E, Pie A, Bueno G, et al. HMG–CoA Lyase Deficiency. In: Ikehara K, editor. Advances in the Study of Genetic Disorders [Internet]. InTech; 2011 [cited 2025 Aug 3]. Available from: http://www.intechopen.
- Alfadhel M, Abadel B, Almaghthawi H, Umair M, Rahbeeni Z, Faqeih E, et al. HMG-CoA Lyase Deficiency: A Retrospective Study of 62 Saudi Patients. Front Genet. 2022;13:880464.
- Hatano R, Lee E, Sato H, Kiuchi M, Hirahara K, Nakagawa Y, et al. Hepatic ketone body regulation of renal gluconeogenesis. Mol Metab. 2024;84:101934.
- Holeček, M. Origin and Roles of Alanine and Glutamine in Gluconeogenesis in the Liver, Kidneys, and Small Intestine under Physiological and Pathological Conditions. Int J Mol Sci. 2024 Jun 27;25(13):7037.
- Kolb H, Kempf K, Röhling M, Lenzen-Schulte M, Schloot NC, Martin S. Ketone bodies: from enemy to friend and guardian angel. BMC Med. 2021 Dec 9;19(1):313.
- Barendregt K, Soeters P, Allison S, Sobotka L. Basics in clinical nutrition: Simple and stress starvation. Eur E-J Clin Nutr Metab. 2008 Dec 1;3(6):e267–71.
- Xiang F, Zhang Z, Xie J, Xiong S, Yang C, Liao D, et al. Comprehensive review of the expanding roles of the carnitine pool in metabolic physiology: beyond fatty acid oxidation. J Transl Med. 2025 Mar 14;23(1):324.
- Olpin, SE. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J Inherit Metab Dis. 2013;36(4):645–58.
- Houten SM, Violante S, Ventura FV, Wanders RJA. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu Rev Physiol. 2016 Feb 10;78(1):23–44.
- Suzuki T, Komatsu T, Shibata H, Inagaki T. Metabolic Responses to Energy-Depleted Conditions. In: Takada A, Himmerich H, editors. Psychology and Pathophysiological Outcomes of Eating [Internet]. Rijeka: IntechOpen; 2021. Available from. [CrossRef]
- Thompson GN, Hsu BYL, Pitt JJ, Treacy E, Stanley CA. Fasting Hypoketotic Coma in a Child with Deficiency of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase. N Engl J Med. 1997 Oct 23;337(17):1203–7.
- New England Consortium of Metabolic Programs [Internet]. [cited 2025 Apr 24]. 3-hydroxymethylglutaryl-CoA (3-HMG CoA) Lyase Deficiency. Available from: https://www.newenglandconsortium.
- Orphanet: 3-hydroxy-3-methylglutaric aciduria [Internet]. [cited 2025 Jun 15]. Available from: http://www.orpha.
- Delgado CA, Balbueno Guerreiro GB, Diaz Jacques CE, Moura Coelho D, Sitta A, Manfredini V, et al. Prevention by L-carnitine of DNA damage induced by 3-hydroxy-3-methylglutaric and 3-methylglutaric acids and experimental evidence of lipid and DNA damage in patients with 3-hydroxy-3-methylglutaric aciduria. Arch Biochem Biophys. 2019 Jun 15;668:16–22.
- Yu HK, Ok SH, Kim S, Sohn JT. Anesthetic management of patients with carnitine deficiency or a defect of the fatty acid β-oxidation pathway: A narrative review. Medicine (Baltimore). 2022 Feb 18;101(7):e28853.
- Martins, P. Glutaric acidaemia type 1. Anästh Intensiv. 2020;61(Suppl 6):S108–14.
- Robbins KA, León-ruiz EN. Anesthetic Management of a Patient with 3-Methylcrotonyl-CoA Carboxylase Deficiency. Anesth Analg. 2008 Aug;107(2):648.
- Gunduz Gul G, Ozer AB, Demirel I, Aksu A, Erhan OL. The effect of sugammadex on steroid hormones: A randomized clinical study. J Clin Anesth. 2016 Nov;34:62–7.
- obin KAR, Steineger HH, Alberti S, Spydevold Ø, Auwerx J, Gustafsson JÅ, et al. Cross-Talk between Fatty Acid and Cholesterol Metabolism Mediated by Liver X Receptor-α. Mol Endocrinol. 2000 May 1;14(5):741–52.
- Berthet, V. The Impact of Cognitive Biases on Professionals’ Decision-Making: A Review of Four Occupational Areas. Front Psychol. 2022 Jan 4;12:802439.
Figure 1.
Ketone body synthesis from fatty acid β-oxidation and leucine catabolism. HMG-CoA lyase deficiency (red) blocks conversion of HMG-CoA into acetoacetate, impairing ketogenesis. An HMG-CoA lyase deficiency (red) impairs ketogenesis [7,8]. Abbreviations: HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A.
Figure 1.
Ketone body synthesis from fatty acid β-oxidation and leucine catabolism. HMG-CoA lyase deficiency (red) blocks conversion of HMG-CoA into acetoacetate, impairing ketogenesis. An HMG-CoA lyase deficiency (red) impairs ketogenesis [7,8]. Abbreviations: HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A.
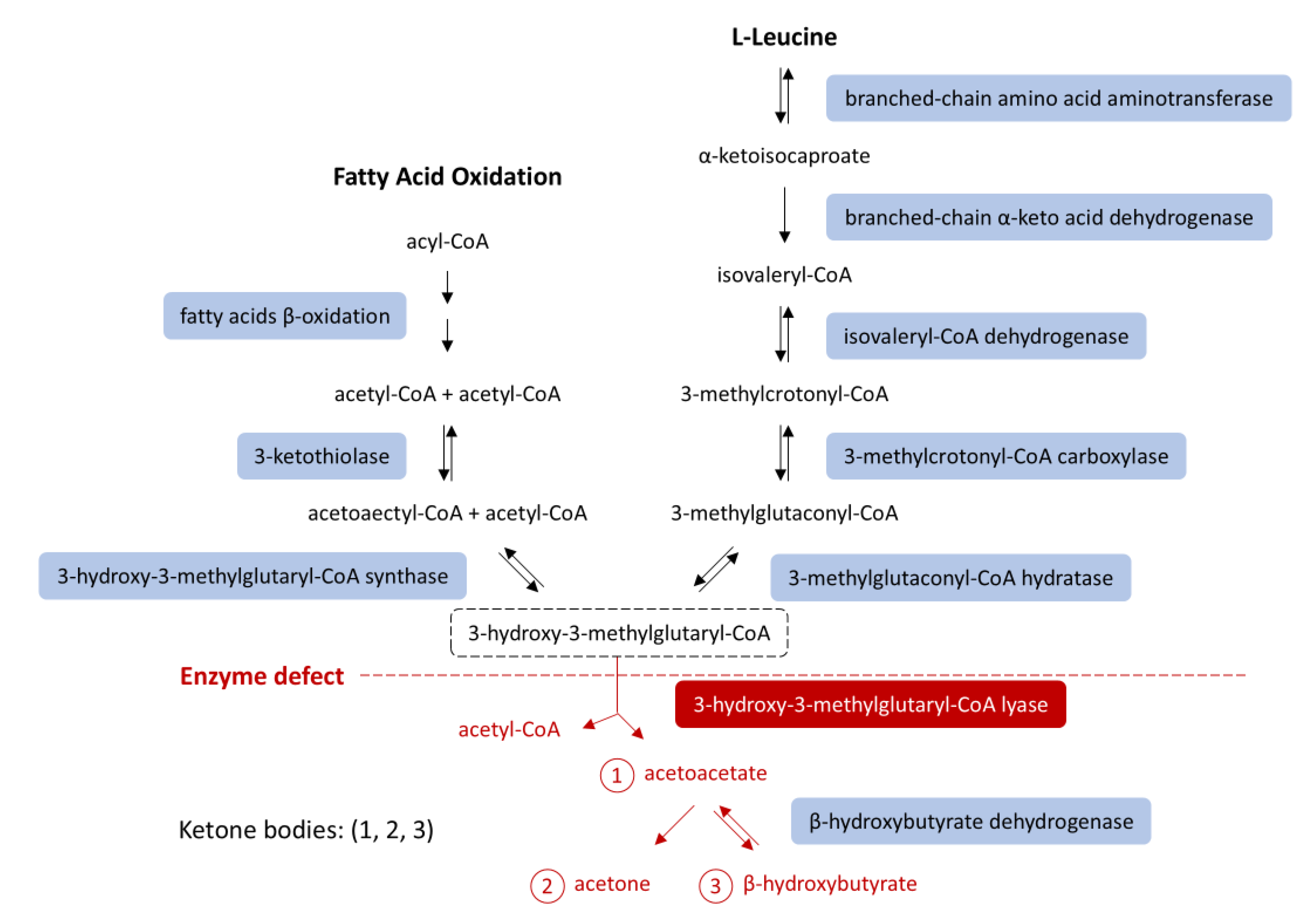
Figure 2.
Starvation metabolism in a healthy individual. Acetyl-CoA from β-oxidation and leucine catabolism enters the citric acid cycle, stimulating gluconeogenesis (via PC) and inhibiting glycolysis (via PDH), thereby diverting pyruvate - originating from lactate and glucogenic amino acids such as alanine - toward gluconeogenesis. Solid arrows = single-step processes; dashed arrows = simplified multi-step processes. A stylized mitochondrion highlights the intra-mitochondrial pathways. Abbreviations: LDH, lactate dehydrogenase; ALT, alanine aminotransferase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PEPCK, phosphoenolpyruvate carboxykinase; G6Pase, glucose-6-phosphatase; G3PDH, glyceraldehyde-3-phosphate dehydrogenase; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; NADH, nicotinamide adenine dinucleotide; FADH₂, flavin adenine dinucleotide [5,16].
Figure 2.
Starvation metabolism in a healthy individual. Acetyl-CoA from β-oxidation and leucine catabolism enters the citric acid cycle, stimulating gluconeogenesis (via PC) and inhibiting glycolysis (via PDH), thereby diverting pyruvate - originating from lactate and glucogenic amino acids such as alanine - toward gluconeogenesis. Solid arrows = single-step processes; dashed arrows = simplified multi-step processes. A stylized mitochondrion highlights the intra-mitochondrial pathways. Abbreviations: LDH, lactate dehydrogenase; ALT, alanine aminotransferase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PEPCK, phosphoenolpyruvate carboxykinase; G6Pase, glucose-6-phosphatase; G3PDH, glyceraldehyde-3-phosphate dehydrogenase; HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A; NADH, nicotinamide adenine dinucleotide; FADH₂, flavin adenine dinucleotide [5,16].
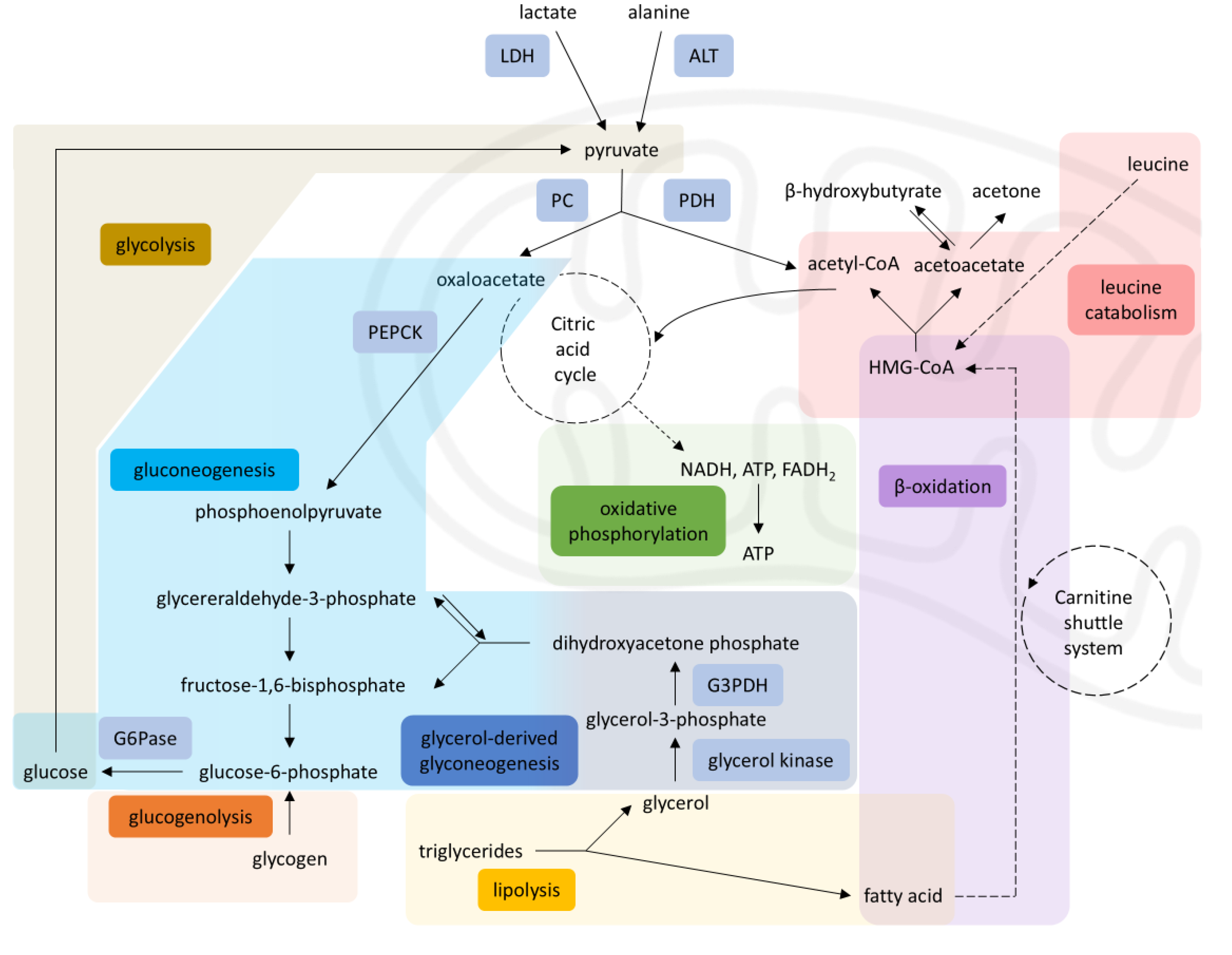
Figure 3.
Disrupted metabolism in HMG-CoA lyase deficiency. Impaired β-oxidation and leucine catabolism prevent acetyl-CoA production, inhibiting gluconeogenesis (via PC) and enhancing glycolysis (via PDH), leading to energy failure during starvation, diverting pyruvate toward the citric acid cycle and limiting its availability for gluconeogenesis. Solid arrows = single-step processes; dashed arrows = simplified multi-step processes; Red arrows = reduced activity; Red cross = blocked pathways; Bold arrows = increased activity. A stylized mitochondrion highlights the intra-mitochondrial pathways. Abbreviations: LDH: lactate dehydrogenase, ALT: alanine aminotransferase, PC: pyruvate carboxylase, PDH: pyruvate dehydrogenase, PEPCK: phosphoenolpyruvate carboxykinase, G6Pase: glucose-6-phosphatase, G3PDH: glyceraldehyde-3-phosphate dehydrogenase, HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A, NADH: nicotinamide-adenine dinucleotide, FADH₂: flavin adenine dinucleotide [5,7,8,16].
Figure 3.
Disrupted metabolism in HMG-CoA lyase deficiency. Impaired β-oxidation and leucine catabolism prevent acetyl-CoA production, inhibiting gluconeogenesis (via PC) and enhancing glycolysis (via PDH), leading to energy failure during starvation, diverting pyruvate toward the citric acid cycle and limiting its availability for gluconeogenesis. Solid arrows = single-step processes; dashed arrows = simplified multi-step processes; Red arrows = reduced activity; Red cross = blocked pathways; Bold arrows = increased activity. A stylized mitochondrion highlights the intra-mitochondrial pathways. Abbreviations: LDH: lactate dehydrogenase, ALT: alanine aminotransferase, PC: pyruvate carboxylase, PDH: pyruvate dehydrogenase, PEPCK: phosphoenolpyruvate carboxykinase, G6Pase: glucose-6-phosphatase, G3PDH: glyceraldehyde-3-phosphate dehydrogenase, HMG-CoA: 3-hydroxy-3-methylglutaryl coenzyme A, NADH: nicotinamide-adenine dinucleotide, FADH₂: flavin adenine dinucleotide [5,7,8,16].
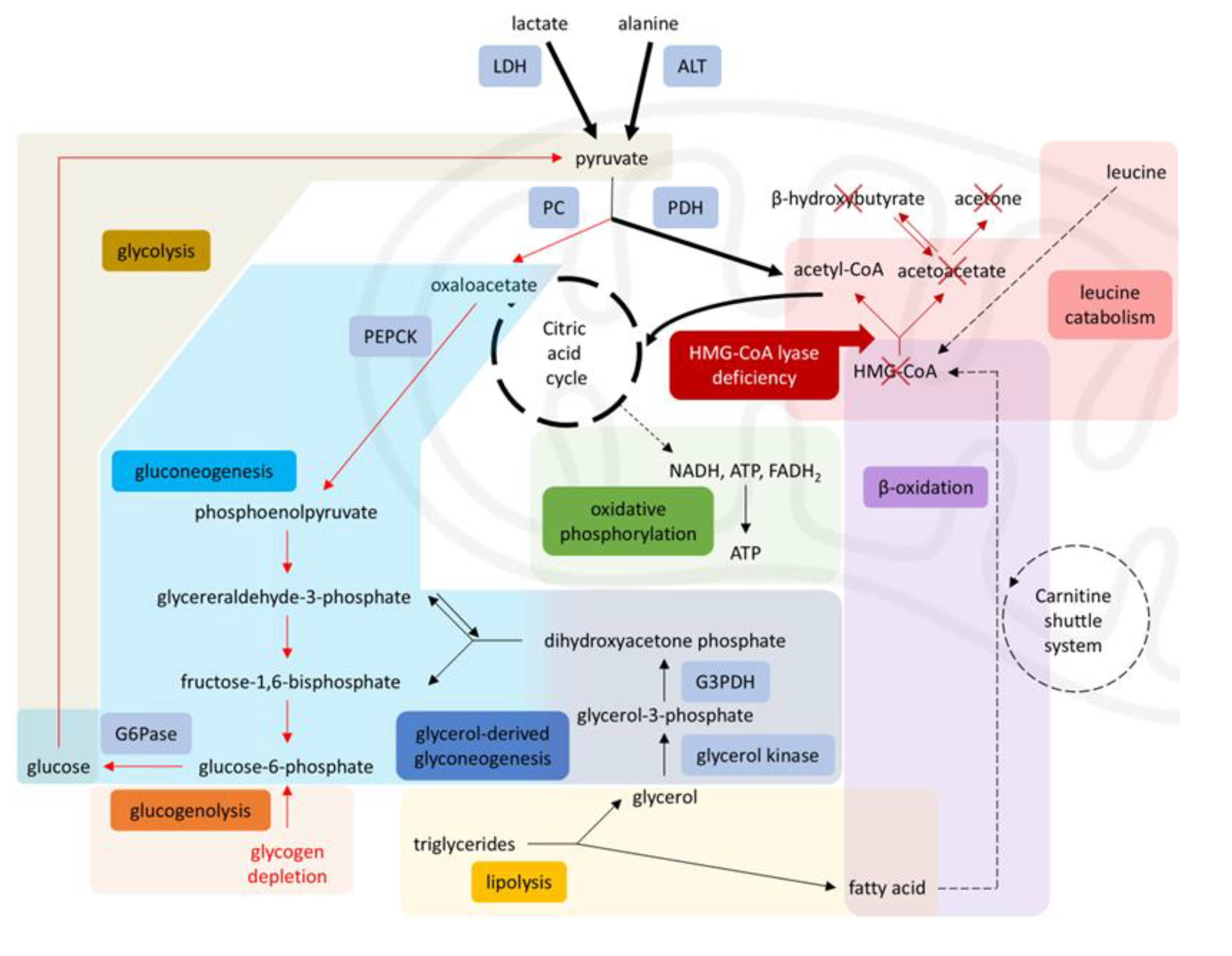

Table 1.
Perioperative values of arterial blood gases and glucose.
| ABGs | Pre-PICU | PICU prior to surgery |
Intraoperative 1 | Intraoperative 2 | Postoperative | |||
|---|---|---|---|---|---|---|---|---|
| pH | 7.577 | 7.472 | 7.406 | 7.368 | 7.414 | |||
|
PaO2 (mmHg) |
234 | 166 | 201 | 204 | 188.7 | |||
| FiO2 | 0.5 | ~ 0.28 | 0.35 | 0.35 | ~ 0.28 | |||
|
PaCO2 (mmHg) |
16.4 | 28.4 | 30,6 | 34 | 33 | |||
| BE | -4.8 | -2.5 | -5.1 | -5.4 | -1.3 | |||
| HCO3 | 15 | 20.3 | 18.8 | 19.3 | 20.4 | |||
|
Lactate (mmol/L) |
3.38 | 1.64 | 1.8 | 1.8 | 1.5 | |||
|
Glucose (mg/dL) |
74 | 173 | 144 | 177 | 204 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



