Submitted:
17 September 2025
Posted:
18 September 2025
You are already at the latest version
Abstract
Bile acids are widely distributed in the human gastrointestinal tract. A literature review indicates a role for bile acids in initiating cancers in every organ of the digestive system. The estimated number of new digestive system cancers world-wide in 2022 was about 5 million. For esophageal adenocarcinoma, stomach cancer and small intestine cancer, bile reflux plays a significant role in carcinogenesis. For colon cancer, secondary bile acids produced in response to a high fat diet disrupt colonic epithelial cell mitochondrial membranes. This disruption leads to release of oxidative free radicals that damage DNA and can cause carcinogenic mutations. Cancers of the pancreas, liver, and biliary tract can be caused by constriction of the common bile duct leading to reflux of bile acids back into these organs. Gastroesophageal reflux involving bile acids may also contribute to hypopharyngeal squamous cell carcinogenesis. Thus, bile acids are a likely major contributory cause of cancer throughout the digestive tract.
Keywords:
1. Introduction
2. Background
2.1. Bile Acids in the Gastrointestinal System
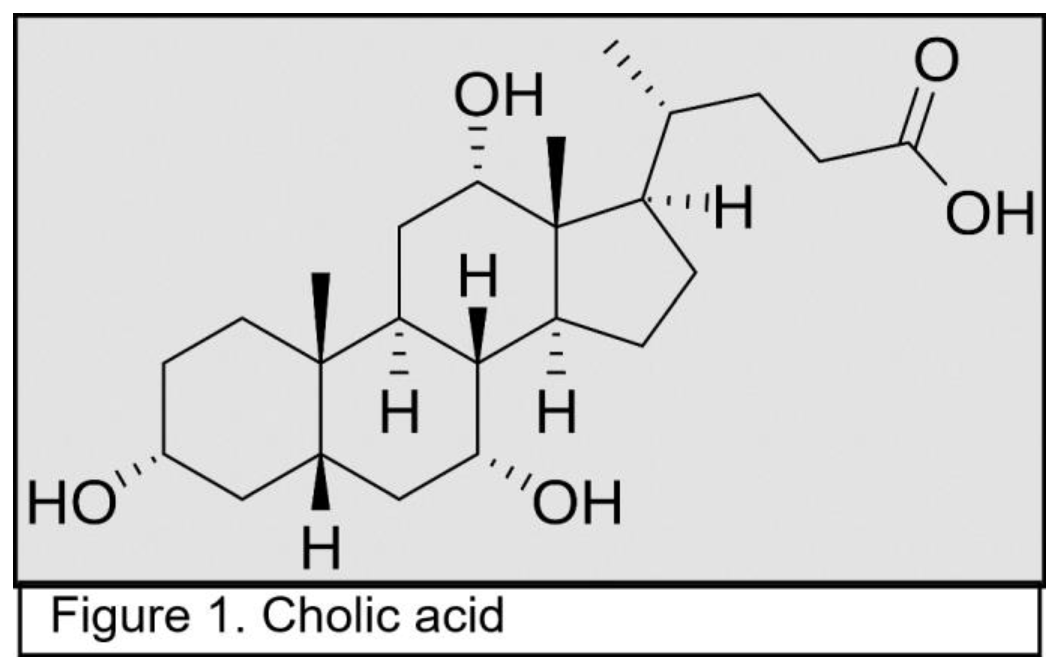
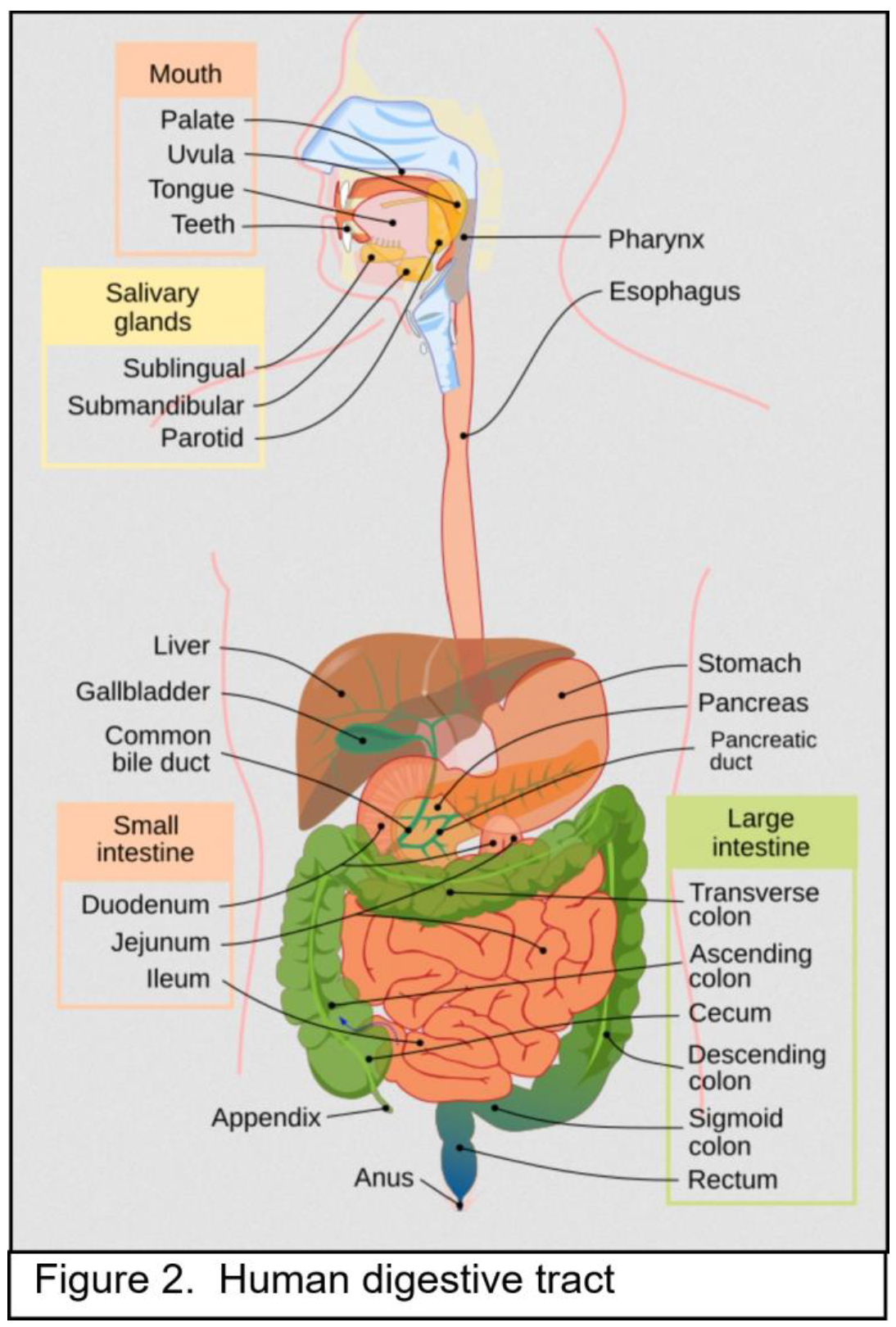
2.2. Elevated Bile Acids Cause Oxidative DNA Damages
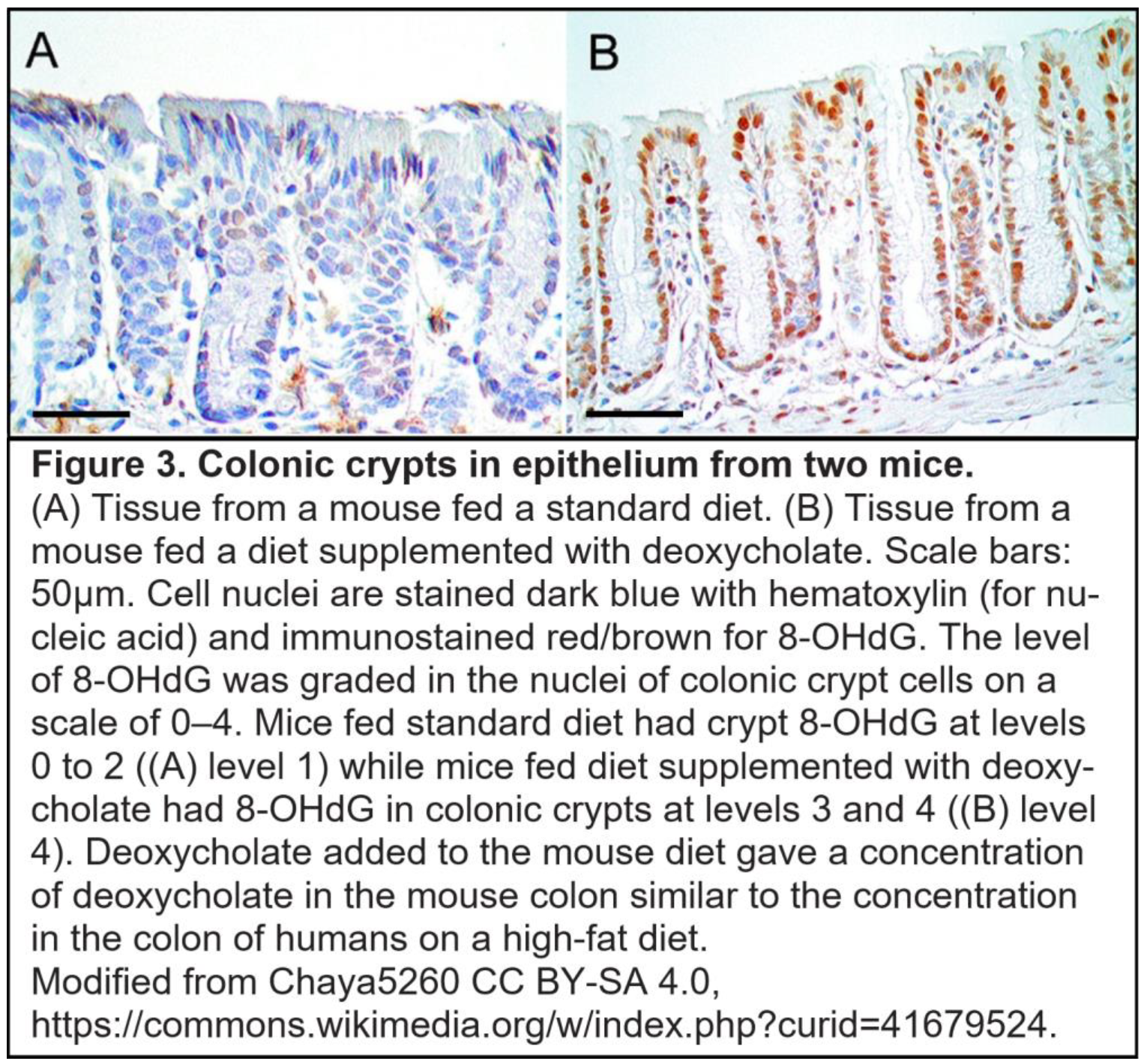
2.3. Frequencies of Incidence and Mortality Due to Gastrointestinal System Cancers
3. Roles of Elevated Bile Acids in Gastrointestinal System Cancers
3.1. Esophageal Cancer
3.2. Stomach Cancer
3.3. Small Intestine Cancer
3.4. Colon and Rectum Cancer
3.4.1. A Bile Acid Based Mouse Model of Colon Cancer
3.4.2. Epigenetic Alterations in Colon Cancer are More Frequent than Mutations
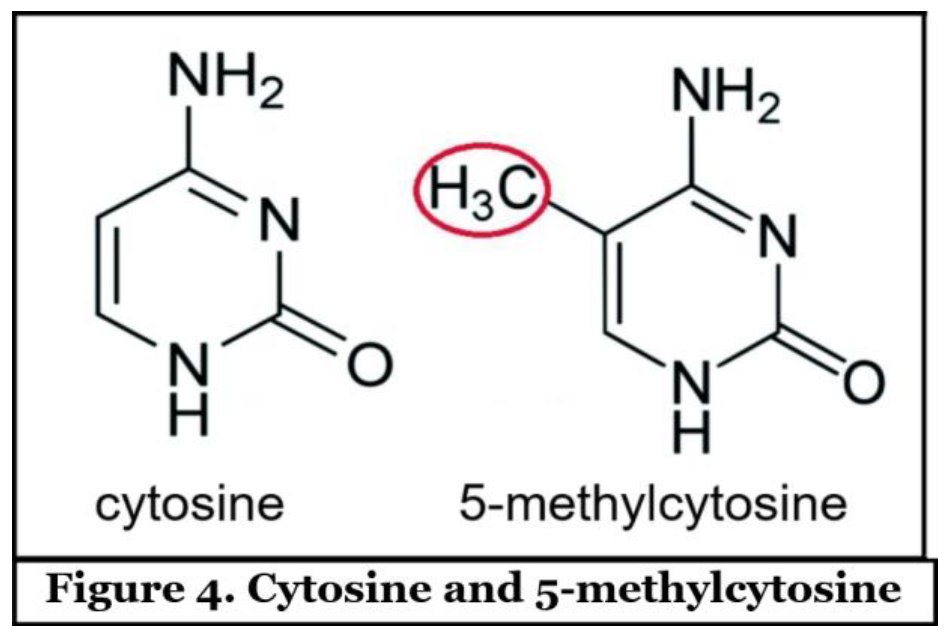
3.5. Liver Cancer (Hepatocellular Carcinoma)
3.6. Gallbladder Removal (Cholecystectomy) and Cancer
3.7. Gallbladder and Biliary Tract Cancer
3.8. Pancreatic Cancer
3.9. Oral Cavity and Pharynx
4. Bile Acids May Cause Field Defects Leading to Gastrointestinal Cancers
5. Discussion
References
- Xie, C.; Huang, W.; Young, R.L.; Jones, K.L.; Horowitz, M.; Rayner, C.K.; Wu, T. Role of Bile Acids in the Regulation of Food Intake, and Their Dysregulation in Metabolic Disease. Nutrients 2021, 13, 1104. [Google Scholar] [CrossRef]
- Sousa, T.; Castro, R.E.; Pinto, S.N.; Coutinho, A.; Lucas, S.D.; Moreira, R.; Rodrigues, C.M.P.; Prieto, M.; Fernandes, F. Deoxycholic acid modulates cell death signaling through changes in mitochondrial membrane properties. J. Lipid Res. 2015, 56, 2158–2171. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: an underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164–164. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.D.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Hundt M, Basit H, John S. Physiology, Bile Secretion. In: StatPearls. 2025. Available from: https://www.ncbi.nlm.nih. 13 September 4702.
- Chiang, J.Y. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Hofmann, A.F. Bile Acids: The Good, the Bad, and the Ugly. Physiology 1999, 14, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Shiffman ML, Sugerman HJ, Moore EW. Human gallbladder mucosal function. Effect of concentration and acidification of bile on cholesterol and calcium solubility. Gastroenterology. 1990;99(5):1452-9.
- Angelin B, Björkhem I, Einarsson K, Ewerth S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J Clin Invest. 1982;70(4):724-31.
- Hamilton, J.P.; Xie, G.; Raufman, J.-P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human cecal bile acids: concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef]
- Hepner GW, Hofmann AF, Malagelada JR, Szczepanik PA, Klein PD. Increased bacterial degradation of bile acids in cholecystectomized patients. Gastroenterology. 1974;66(4):556-64.
- Marciani, L.; Cox, E.F.; Hoad, C.L.; Totman, J.J.; Costigan, C.; Singh, G.; Shepherd, V.; Chalkley, L.; Robinson, M.; Ison, R.; et al. Effects of various food ingredients on gall bladder emptying. Eur. J. Clin. Nutr. 2013, 67, 1182–1187. [Google Scholar] [CrossRef]
- Patti, M.; Houten, S.M.; Bianco, A.C.; Bernier, R.; Larsen, P.R.; Holst, J.J.; Badman, M.K.; Maratos-Flier, E.; Mun, E.C.; Pihlajamaki, J.; et al. Serum Bile Acids Are Higher in Humans With Prior Gastric Bypass: Potential Contribution to Improved Glucose and Lipid Metabolism. Obesity 2009, 17, 1671–1677. [Google Scholar] [CrossRef]
- Payne, C.; Weber, C.; Crowley-Skillicorn, C.; Dvorak, K.; Bernstein, H.; Bernstein, C.; Holubec, H.; Dvorakova, B.; Garewal, H. Deoxycholate induces mitochondrial oxidative stress and activates NF- B through multiple mechanisms in HCT-116 colon epithelial cells. Carcinog. 2007, 28, 215–222. [Google Scholar] [CrossRef]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat. Res. Mutat. Res. 2005, 589, 47–65. [Google Scholar] [CrossRef]
- Prasad AR, Prasad S, Nguyen H, Facista A, Lewis C, Zaitlin B, et al. Novel diet-related mouse model of colon cancer parallels human colon cancer. World J Gastrointest Oncol. 2014;6(7):225-43.
- American Cancer Society. Cancer Facts & Figures 2024. Web site. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2024-cancer-facts-figures.html Accessed . 2025. 13 September.
- Bernstein, H.; Bernstein, C. Bile acids as carcinogens in the colon and at other sites in the gastrointestinal system. Exp. Biol. Med. 2022, 248, 79–89. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.J.S.; Cronin, J.; Alhamdani, A.; Rawat, N.; D'SOuza, F.; Thomas, T.; Eltahir, Z.; Griffiths, A.P.; Baxter, J.N. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF- B activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis 2008, 23, 399–405. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile Acid and Inflammation Activate Gastric Cardia Stem Cells in a Mouse Model of Barrett-Like Metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef]
- Cronin, J.; Williams, L.; McAdam, E.; Eltahir, Z.; Griffiths, P.; Baxter, J.; Jenkins, G. The role of secondary bile acids in neoplastic development in the oesophagus. Biochem. Soc. Trans. 2010, 38, 337–342. [Google Scholar] [CrossRef]
- Režen, T.; Rozman, D.; Kovács, T.; Kovács, P.; Sipos, A.; Bai, P.; Mikó, E. The role of bile acids in carcinogenesis. Cell. Mol. Life Sci. 2022, 79, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Yao, W.Z.; Zhang, D.L.; Feng, C.C.; He, Q.; Lv, H.H.; Cao, Y.P.; Wang, J.; Qi, Y.; et al. The relationship between gastric cancer, its precancerous lesions and bile reflux: A retrospective study. J. Dig. Dis. 2020, 21, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Shi, Y. Bile reflux and bile acids in the progression of gastric intestinal metaplasia. Chin. Med J. 2022, 135, 1664–1672. [Google Scholar] [CrossRef]
- Zhang, M.; Zhong, J.; Shen, Y.; Song, Z. Crosstalk between bile acids and gut microbiota: a potential target for precancerous lesions of gastric cancer. Front. Pharmacol. 2025, 16, 1533141. [Google Scholar] [CrossRef]
- Huang, G.; Wang, S.; Wang, J.; Tian, L.; Yu, Y.; Zuo, X.; Li, Y. Bile reflux alters the profile of the gastric mucosa microbiota. Front. Cell. Infect. Microbiol. 2022, 12, 940687. [Google Scholar] [CrossRef]
- Wang, S.; Kuang, J.; Zhang, H.; Chen, W.; Zheng, X.; Wang, J.; Huang, F.; Ge, K.; Li, M.; Zhao, M.; et al. Bile Acid–Microbiome Interaction Promotes Gastric Carcinogenesis. Adv. Sci. 2022, 9, e2200263. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Cui, Z.Y.; Huang, X.J. Exploration of gastric carcinogenesis from the relationship between bile acids and intestinal metaplasia and intragastric microorganisms (H. pylori and non-H. pylori). J. Cancer Res. Clin. Oncol. 2023, 149, 16947–16956. [Google Scholar] [CrossRef] [PubMed]
- Noto, J.M.; Piazuelo, M.B.; Shah, S.C.; Romero-Gallo, J.; Hart, J.L.; Di, C.; Carmichael, J.D.; Delgado, A.G.; Halvorson, A.E.; Greevy, R.A.; et al. Iron deficiency linked to altered bile acid metabolism promotes Helicobacter pylori–induced inflammation–driven gastric carcinogenesis. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.; Raufman, J.-P. Gastrointestinal neoplasia: carcinogenic interaction between bile acids and Helicobacter pylori in the stomach. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Canakis, A.; Lee, A.; Halvorson, A.E.; Noto, J.M.; Peek, R.M.; Wilson, O.; Hung, A.; Roumie, C.L.; Greevy, R.; Shah, S.C. Bile Acid Sequestrant Use and Gastric Cancer: A National Retrospective Cohort Analysis. Clin. Transl. Gastroenterol. 2023, 14, e00596. [Google Scholar] [CrossRef]
- Maguire, A.; Sheahan, K. Primary small bowel adenomas and adenocarcinomas-recent advances. Virchows Arch. 2018, 473, 265–273. [Google Scholar] [CrossRef]
- Ross, R.; Hartnett, N.; Bernstein, L.; Henderson, B. Epidemiology of adenocarcinomas of the small intestine: is bile a small bowel carcinogen? Br. J. Cancer 1991, 63, 143–145. [Google Scholar] [CrossRef]
- Cross, A.J.; Leitzmann, M.F.; Subar, A.F.; Thompson, F.E.; Hollenbeck, A.R.; Schatzkin, A. A Prospective Study of Meat and Fat Intake in Relation to Small Intestinal Cancer. Cancer Res. 2008, 68, 9274–9279. [Google Scholar] [CrossRef]
- Lagergren, J.; Ye, W.; Ekbom, A. Intestinal cancer after cholecystectomy: Is bile involved in carcinogenesis? Gastroenterology 2001, 121, 542–547. [Google Scholar] [CrossRef]
- IARC of World Health Organization. (2015, ). Cancer: Carcinogenicity of the consumption of red meat and processed meat (News release). https://www.who. 26 October 2025; 13.
- IARC Monographs evaluate consumption of red meat and processed meat (PDF). IARC. 26 October 2025.
- Chan, D.S.M.; Lau, R.; Aune, D.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Red and Processed Meat and Colorectal Cancer Incidence: Meta-Analysis of Prospective Studies. PLoS ONE 2011, 6, e20456. [Google Scholar] [CrossRef]
- Fogelson, K.A.; Dorrestein, P.C.; Zarrinpar, A.; Knight, R. The Gut Microbial Bile Acid Modulation and Its Relevance to Digestive Health and Diseases. Gastroenterology 2023, 164, 1069–1085. [Google Scholar] [CrossRef]
- Dalal, N.; Jalandra, R.; Bayal, N.; Yadav, A.K.; Harshulika; Sharma, M. ; Makharia, G.K.; Kumar, P.; Singh, R.; Solanki, P.R.; et al. Gut microbiota-derived metabolites in CRC progression and causation. J. Cancer Res. Clin. Oncol. 2021, 147, 3141–3155. [Google Scholar] [CrossRef]
- Yang, S.; Wang, Y.; Sheng, L.; Cui, W.; Ma, C. The effect of fecal bile acids on the incidence and risk-stratification of colorectal cancer: an updated systematic review and meta-analysis. Sci. Rep. 2025, 15, 1–13. [Google Scholar] [CrossRef]
- Loftfield, E.; Falk, R.T.; Sampson, J.N.; Huang, W.-Y.; Hullings, A.; Murphy, G.; Weinstein, S.J.; Albanes, D.; Freedman, N.D.; Sinha, R. Prospective Associations of Circulating Bile Acids and Short-Chain Fatty Acids With Incident Colorectal Cancer. JNCI Cancer Spectr. 2022, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dong, W.; Liu, L.; Xu, M.; Wang, Y.; Liu, T.; Zhang, Y.; Wang, B.; Cao, H. Interplay between bile acids and the gut microbiota promotes intestinal carcinogenesis. Mol. Carcinog. 2019, 58, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Kühn, T.; Stepien, M.; López-Nogueroles, M.; Damms-Machado, A.; Sookthai, D.; Johnson, T.; Roca, M.; Hüsing, A.; Maldonado, S.G.; Cross, A.J.; et al. Prediagnostic Plasma Bile Acid Levels and Colon Cancer Risk: A Prospective Study. JNCI J. Natl. Cancer Inst. 2020, 112, 516–524. [Google Scholar] [CrossRef]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 1–23. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Farsetti, A.; Illi, B.; Gaetano, C. How epigenetics impacts on human diseases. Eur. J. Intern. Med. 2023, 114, 15–22. [Google Scholar] [CrossRef]
- Beggs, A.D.; Jones, A.; El-Bahwary, M.; Abulafi, M.; Hodgson, S.V.; Tomlinson, I.P. Whole-genome methylation analysis of benign and malignant colorectal tumours. J. Pathol. 2012, 229, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, G.; Dang, S.; Zhou, Y.; Zeng, K.; Liu, M. Discovery and Validation of Hypermethylated Markers for Colorectal Cancer. Dis. Markers 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: a historical perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Gurtan, A.M.; Sharp, P.A. The Role of miRNAs in Regulating Gene Expression Networks. J. Mol. Biol. 2013, 425, 3582–3600. [Google Scholar] [CrossRef]
- Price, C.; Chen, J. MicroRNAs in cancer biology and therapy: Current status and perspectives. Genes Dis. 2014, 1, 53–63. [Google Scholar] [CrossRef]
- Schetter AJ, Okayama H, Harris CC. The role of microRNAs in colorectal cancer. Cancer J. 2012;18(3):244-52.
- Gulyaeva, L.F.; Kushlinskiy, N.E. Regulatory mechanisms of microRNA expression. J. Transl. Med. 2016, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Guo, S.; Zhou, Y.; Zhu, J.; Zhao, J.; Wang, M.; Sang, L.; Wang, B.; Chang, B. Hepatocellular carcinoma: Novel understandings and therapeutic strategies based on bile acids (Review). Int. J. Oncol. 2022, 61, 1–12. [Google Scholar] [CrossRef]
- Colosimo, S.; Tomlinson, J.W. Bile acids as drivers and biomarkers of hepatocellular carcinoma. World J. Hepatol. 2022, 14, 1730–1738. [Google Scholar] [CrossRef]
- Song, Y.; Lau, H.C.; Zhang, X.; Yu, J. Bile acids, gut microbiota, and therapeutic insights in hepatocellular carcinoma. Cancer Biol. Med. 2023, 21, 1–19. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Thrift, A.P.; Duong, H.; Ning, J.; Khaderi, S.; Singal, A.G.; Asrani, S.K.; Marrero, J.A.; Powell, H.; Rizwan, K.; et al. Serum levels of total bile acids are associated with an increased risk of HCC in patients with cirrhosis. Hepatol. Commun. 2024, 8. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Lopez-Nogueroles, M.; Lahoz, A.; Kühn, T.; Perlemuter, G.; Voican, C.; Ciocan, D.; Boutron-Ruault, M.; Jansen, E.; Viallon, V.; et al. Prediagnostic alterations in circulating bile acid profiles in the development of hepatocellular carcinoma. Int. J. Cancer 2021, 150, 1255–1268. [Google Scholar] [CrossRef]
- Wu, L.; Feng, J.; Li, J.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed. Pharmacother. 2021, 133, 111036. [Google Scholar] [CrossRef]
- Jia, W.; Rajani, C.; Xu, H.; Zheng, X. Gut microbiota alterations are distinct for primary colorectal cancer and hepatocellular carcinoma. Protein Cell 2020, 12, 374–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Q.; Ding, M.; Yao, J.; Guo, Y.; Yan, W.; Yu, S.; Shen, Q.; Huang, M.; Zheng, Y.; et al. Alcohol triggered bile acid disequilibrium by suppressing BSEP to sustain hepatocellular carcinoma progression. Chem. Interactions 2022, 356, 109847. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Turumin JL, Shanturov VA, Turumina HE. The role of the gallbladder in humans. Rev Gastroenterol Mex. 2013;78(3):177-87.
- Latta, R.; Fiander, H.; Ross, N.; Simpson, C.; Schneider, H. Toxicity of bile acids to colon cancer cell lines. Cancer Lett. 1993, 70, 167–173. [Google Scholar] [CrossRef]
- Wahlström, A.; Brumbaugh, A.; Sjöland, W.; Olsson, L.; Wu, H.; Henricsson, M.; Lundqvist, A.; Makki, K.; Hazen, S.L.; Bergström, G.; et al. Production of deoxycholic acid by low-abundant microbial species is associated with impaired glucose metabolism. Nat. Commun. 2024, 15, 1–11. [Google Scholar] [CrossRef]
- Roda, E.; Aldini, R.; Mazzella, G.; Roda, A.; Sama, C.; Festi, D.; Barbara, L. Enterohepatic circulation of bile acids after cholecystectomy. Gut 1978, 19, 640–649. [Google Scholar] [CrossRef]
- Mu, L.; Li, W.; Ren, W.; Hu, D.; Song, Y. The association between cholecystectomy and the risk of colorectal cancer: an updated systematic review and meta-analysis of cohort studies. Transl. Cancer Res. 2023, 12, 1452–1465. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y. Correlation Between Cholecystectomy and the Risk of Gastric Cancer. Am. J. Gastroenterol. 2024, 120, 1884–1884. [Google Scholar] [CrossRef]
- Johansen, C.; Chow, W.H.; Jorgensen, T.; Mellemkjaer, L.; Engholm, G.; Olsen, J.H. Risk of colorectal cancer and other cancers in patients with gall stones. Gut 1996, 39, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Jin, E.H.; Lim, J.H.; Shin, C.M.; Kim, N.; Han, K.; Lee, D.H. Increased Risk of Cancer after Cholecystectomy: A Nationwide Cohort Study in Korea including 123,295 Patients. Gut Liver 2022, 16, 465–473. [Google Scholar] [CrossRef]
- Wu N, Bayatpour S, Hylemon PB, Aseem SO, Brindley PJ, Zhou H. Gut Microbiome and Bile Acid Interactions: Mechanistic Implications for Cholangiocarcinoma Development, Immune Resistance, and Therapy. Am J Pathol. 2025;195(3):397-408.
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203-14.
- Sharma, B.; Twelker, K.; Nguyen, C.; Ellis, S.; Bhatia, N.D.; Kuschner, Z.; Agriantonis, A.; Agriantonis, G.; Arnold, M.; Dave, J.; et al. Bile Acids in Pancreatic Carcinogenesis. Metabolites 2024, 14, 348. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, H.; Zhang, X.; Ma, J.; Xue, S.; Shentu, D.; Mao, T.; Li, S.; Yue, M.; Cui, J.; et al. The Role of Bile Acids in Pancreatic Cancer. Curr. Cancer Drug Targets 2024, 24, 1005–1014. [Google Scholar] [CrossRef]
- Winter, J.M.; Maitra, A.; Yeo, C.J. Genetics and pathology of pancreatic cancer. HPB 2006, 8, 324–336. [Google Scholar] [CrossRef]
- Feng, H.-Y.; Chen, Y.-C. Role of bile acids in carcinogenesis of pancreatic cancer: An old topic with new perspective. World J. Gastroenterol. 2016, 22, 7463–7477. [Google Scholar] [CrossRef]
- Rees, D.O.; Crick, P.J.; Jenkins, G.J.; Wang, Y.; Griffiths, W.J.; Brown, T.H.; Al-Sarireh, B. Comparison of the composition of bile acids in bile of patients with adenocarcinoma of the pancreas and benign disease. J. Steroid Biochem. Mol. Biol. 2017, 174, 290–295. [Google Scholar] [CrossRef]
- Malhotra, P.; Palanisamy, R.; Caparros-Martin, J.A.; Falasca, M. Bile Acids and Microbiota Interplay in Pancreatic Cancer. Cancers 2023, 15, 3573. [Google Scholar] [CrossRef] [PubMed]
- Gál, E.; Veréb, Z.; Kemény, L.; Rakk, D.; Szekeres, A.; Becskeházi, E.; Tiszlavicz, L.; Takács, T.; Czakó, L.; Hegyi, P.; et al. Bile accelerates carcinogenic processes in pancreatic ductal adenocarcinoma cells through the overexpression of MUC4. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Tajima, Y.; Kuroki, T.; Mishima, T.; Kitasato, A.; Fukuda, K.; Tsutsumi, R.; Kanematsu, T. Bile-Reflux into the Pancreatic Ducts is Associated with the Development of Intraductal Papillary Carcinoma in Hamsters. J. Surg. Res. 2006, 136, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Available online:. Available online: https://seer.cancer.gov/statfacts/html/oralcav.html (accessed on 3 September 2021).
- Sasaki, C.T.; Doukas, S.G.; Costa, J.; Vageli, D.P. Biliary reflux as a causal factor in hypopharyngeal carcinoma: New clinical evidence and implications. Cancer 2019, 125, 3554–3565. [Google Scholar] [CrossRef]
- Vageli, D.P.; Doukas, S.G.; Doukas, P.G.; Judson, B.L. Bile reflux and hypopharyngeal cancer (Review). Oncol. Rep. 2021, 46, 1–14. [Google Scholar] [CrossRef]
- Vageli, D.P.; Doukas, P.G.; Siametis, A.; Judson, B.L. Targeting STAT3 prevents bile reflux-induced oncogenic molecular events linked to hypopharyngeal carcinogenesis. J. Cell. Mol. Med. 2021, 26, 75–87. [Google Scholar] [CrossRef]
- Bernstein, C.; Bernstein, H.; Payne, C.M.; Dvorak, K.; Garewal, H. Field defects in progression to gastrointestinal tract cancers. Cancer Lett. 2008, 260, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef]
- Williams, GC. Pleotropy, natural selection, and the evolution of senesence. 3: Evolution 1957;11(4), 1957. [Google Scholar]
- Li, Z.; Zhang, W.; Chen, Y.; Guo, W.; Zhang, J.; Tang, H.; Xu, Z.; Zhang, H.; Tao, Y.; Wang, F.; et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 2016, 23, 1765–1777. [Google Scholar] [CrossRef] [PubMed]

| Global | United States | |
| Whole digestive system | 4,957,675 | 353,820 |
| Colon and Rectum | 1,926,118 | 152,810 |
| Pancreas | 510,566 | 66,440 |
| Liver & intrahepatic bile duct | 865,269 | 41,630 |
| Stomach | 968,350 | 26,890 |
| Esophagus | 510,716 | 22,370 |
| Small intestine (duodenum) | 12,440 | |
| Gall bladder and other biliary | 122,462 | 12,350 |
| Anus, anal canal & anorectum | 54,194 | 10,540 |
| Other digestive organs | 8,350 |
| Bile acid | Pool size (μM per kilogram) | |
| Cholecystectomy | Healthy controls | |
| Cholic | 28.2 | 48.7 |
| Chenodeoxycholic | 33.0 | 31.1 |
| Deoxycholic | 42.5 | 21.4 |
| Lithocholic | 11.3 | 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).



