
Submitted:
17 September 2025
Posted:
18 September 2025
You are already at the latest version
Abstract
Type-P5 ATPases are the least characterized among the P-type ATPases and this is especially true in the case of the malaria parasite. In this study, Spf1, a subtype-P5A ATPase of yeast, and ATP13A2, a subtype-P5B ATPase of humans, were used as templates to extensively characterize the sequences and structural features of haemosporidian type-P5 ATPases. Malaria parasites have both subtype-P5A and subtype-P5B ATPase genes and the structural features of the proteins recapitulate the known structures of subtype-P5A and subtype-P5B ATPases respectively. Detailed structural analysis detected an addition-al alpha-helix in the P-domain of subtype-P5A ATPases which is not found in subtype-P5B ATPases. This feature may be an additional signature to distinguish subtype-P5A and subtype-P5B ATPases in addition to the differences in the membrane loops of the N-terminal domain, the arm in the P-domain of subtype-P5A, and substrate differences. A notable difference in the type-P5 ATPases from the malaria parasite, as compared to the templates, is the insertion of multiple regions of variable and low-complexity regions that form intrinsically disorganized loops. These loops may form a shroud-like structure that protects the core ATPase structure and/or participates in low-affinity interprotein interactions. Homology modeling did not provide definitive answers about the substrate specificity of the haemosporidian type-P5 ATPases. However, the haemosporidian sub-type-P5A ATPase is likely an ER transmembrane dislocase as are the other subtype-P5A ATPases. In contrast, the subtype-P5B ATPases of the malaria parasite are not likely to be polyamine transporters in lysosomes, as have been described in fungi and metazoans. This suggests that subtype-P5B ATPases have undergone lineage specific divergence in regard to their function(s).
Keywords:
P‐type ATPase
; type‐P5 ATPase
; malaria parasite
; Plasmodium
; Haemosporida
; homology modeling
; intrinsically disordered proteins
1. Introduction
P-type ATPases move substances -- especially
cations and lipids -- across membranes through the hydrolysis of ATP and the
transient phosphorylation of a highly conserved aspartate (D) residue [1]. Despite their crucial importance in cellular
physiology, relatively little research has been done on the potential of P-type
ATPases as drug targets against the malaria parasite. Among the potential
twelve P-type ATPases that have been identified in the Plasmodium falciparum
genome [2] , most research has been done on the
SERCA orthologue and potential drugs that target this enzyme have been
identified [3,4]. The ability to generate
accurate 3-dimensional models of proteins using known structures as templates,
often called homology modeling, will certainly expedite the identification and
characterization of potential drug targets [5].
This may be especially true of P-type ATPases since their 3-dimensional
structures are relatively well characterized. Homology modeling is a powerful
and essential tool in pathogen research and drug discovery that will provide
insight into protein structure, function, and interactions.
1.1. P-Type ATPases
P-type ATPases comprise a large and ubiquitous gene
family that is defined by canonical structural features. The first and foremost
is the highly conserved phosphorylation motif DKTGT which defines P-type
ATPases. During substrate transport the aspartate (D) is transiently
phosphorylated via ATP hydrolysis and the resulting protein conformation
changes associated with the phosphorylation-dephosphorylation cycles facilitate
movement of substances across membranes. All P-type ATPases are comprised of
four highly conserved domains called the actuator (A) domain, the
phosphorylation (P) domain, the nucleotide-binding (N) domain, and membrane (M)
domain [6]. The A-domain has phosphatase
activity to remove the phosphate. The N-domain binds ATP so that the γ -phosphate is adjacent to the phosphorylated
aspartate residue of the DKTGT motif in the P-domain. These three domains are
located on the cytoplasmic face of the membrane. The M-domain is formed by six
core transmembrane helices (cTM) and a variable number of supporting
transmembrane helices (sTM). A substrate-binding groove that opens to the
extracytoplasmic (i.e., extracellular or luminal) side of the membrane is
formed by the cTM. There is a highly conserved proline in the fourth
transmembrane helix (cTM4) that forms a ‘kink’ which forms the base of the
substrate binding groove.
There are five major types (numbered 1-5) of P-type
ATPases based on sequence homology, additional structural domains, and
substrate specificity [1,7]. Subtypes have
also been defined and are designated with capital letters. Type-P5 ATPases, the
least characterized among the P-type ATPases, are defined through sequence
homology and structural features. For example, the signature motif is an
expanded FDKTGTLT, there is an N-terminal domain (NTD), and the M-domain
contains four sTM helices (Figure 1). The
two subtypes (A and B) of type-P5 ATPases are quite similar and differ
primarily in the NTD, the P-domain, and substrate specificity [1,8]. For example, the NTD of subtype-P5A has two
N-terminal transmembrane helices (nTM), whereas subtype-P5B has a triangular
N-terminal membrane loop (nML). Another distinguishing feature is a helical
projection from the P-domain called the ‘arm’ in subtype-P5A that is not found
in subtype-P5B. And subtype-P5A ATPase has been described as a transmembrane
helix dislocase of the ER [9] and subtype-P5B
ATPase has been described as a polyamine transporter of the lysosome [10,11].
1.2. Type-P5 ATPases of Plasmodium
Type-P5 ATPases are only found in eukaryotes and
most eukaryotes have a single copy of the subtype-P5A and multiple paralogues
of the subtype-P5B, especially in multicellular organisms [8,12]. A single gene for a P5A-subtype has been
identified in Plasmodium [12]. The
protein has not been characterized other than its presence in genomic databases
and the P. falciparum gene is described as a putative
cation-transporting P-type ATPase that is located on chromosome seven [2]. Two subtype-P5B genes designated as ATPase1 and
ATPase3 have been identified in P. falciparum. ATPase1 arose from a
duplication of ATPase3 early in the evolution of the malaria parasite and is
only found in P. falciparum and related parasites of the great apes
(i.e., Laverania), avian malaria parasites, and Haemoproteus [13]. Orthologues of ATPase3 can be positively
identified throughout the Apicomplexa, but no clear orthologues outside of the
Apicomplexa could be identified. ATPase1 and ATPase3 from all the apicomplexan
species exhibited a high level of sequence homology except for four rather
large variable regions composed of low-complexity sequence. Both ATPase1 and
ATPase3 genes also have a single intron located in the same position. The only
quasi-substantial difference between these two paralogues is an extended N-terminus
of variable sequence in ATPase3.
Homology
modeling with Phyre® was carried out in the previous study
[13]
to search
for structural homologues. Numerous P-type ATPases were identified as potential
templates and templates from type-P5 ATPases tended to produce robust
structural models with the expected canonical structural features. However, in
some of the structural models, conserved sequences were sometimes excluded and
replaced with sequences from variable regions to form the secondary elements of
the ATPase domains. This suggests that the variable regions may interfere with
the ability of the templates to generate accurate and robust models. If true,
this may be an important limitation in the analysis of Plasmodium proteins
which often contain large regions of low complexity sequence
[14,15]
.
In addition, detailed analyses of the
polyamine-binding site in the modeled structures of ATPase1 and ATPase3 were
carried out. The predicted structures using a P5B-subtype as the template are
quite similar to the experimentally determined polyamine-binding site and most
of the key amino acids are conserved, especially in ATPase1. The differences
between ATPase1 and ATPase3 opens the possibility of different substrate
specificities. However, as discussed in detail [13]
neither ATPase1 nor ATPase3 are likely to be polyamine transporters and neither
ATPase1 nor ATPase3 appear to be located in the lysosome, which is called the
digestive vacuole in the malaria parasite [16].
Immunofluorescence studies using antibodies against ATPase1 or ATPase3 reveal a
diffuse vesicle-like cytoplasmic staining [17–19]. As previously discussed [20] , this immunofluorescence pattern is consistent
with the known ultrastructure of the ER in malaria parasites [21,22]. The possible ER-localization of ATPase1 and
ATPase3 indicates that these Plasmodium subtype-P5B ATPases may have a
different function than the subtype-P5B ATPases of fungi or metazoans.
To address these potential limitations with
3-dimensional modeling and to gain insight into the structure and function of
type-P5 ATPases from the malaria parasite, Spf1, a rather well-characterized
subtype-P5A ATPase [9] , and ATP13A2, a rather
well-characterized subtype-P5B ATPase [11] ,
were used as templates to investigate sequence and structural homologies of Plasmodium
subtype-P5A ATPase (PlP5A), ATPase1, and ATPase3 from P.
falciparum and P. relictum. These two templates were particularly
robust in the previous study at predicting structures that recapitulated the
experimentally determined structures [13]. The
two species not only serve as replicate analyses but also may provide insight
into the effects of the variable regions since the P. relictum proteins
tend to have smaller variable regions.
2. Materials and Methods
2.1. Identification of Subtype-P5A ATPases from Haemosporidians and the SAR Supergroup
The P. falciparum subtype-P5A ATPase (Gene
ID: PF3D7_0727800) was used as a query in BLASTP searches [23] of non-redundant protein sequences at NCBI and
PlasmoDB [24]. Alignments associated with the
BLAST results were used to eliminate partial sequences and sequences with
obvious errors. Subsequently, the newly identified sequences were used as
queries in BLAST searches to ensure that no subtype-P5A sequences were missed.
In cases where complete sequences were available from multiple strains of a
single species, the reference strain for that species was chosen. For the
haemosporidians a chromosome number was recorded when available. A partial
genome sequence of Haemoproteus tartakovskyi which is not part of the
non-redundant database is available [25]. A
TBLASTN search of those contigs was performed using the subtype-P5A ATPase from
P. relictum as a query. A complete gene sequence was identified on a
single contig (HtScaffold0006) and corrected by the insertion of a single A
residue. A summary of all the sequences is found in Table S1.
A conserved-domain (CD) analysis was also carried
out [26] and the e-values associated with the
subtype-P5A (accession number cd07543) and subtype-P5B (accession number
TIGR01657) ATPases were recorded.
2.2. Sequence Alignments
The haemosporidian subtype-P5A sequences were
aligned with ClustalW within Mega XI [27].
Alignments were adjusted manually to accommodate variable regions and core
regions of P-type ATPases. The adjusted alignments were used to generate
phylogenetic trees (Figure S1).
Subtype-P5A and subtype-P5B ATPases from P.
falciparum and P. relictum were subjected to a detailed analysis of
their sequences and predicted 3-dimensional structures, which were compared to
Spf1 and ATP13A2 (Table 1). Spf1 was used
as a prototype of subtype-P5A ATPases and ATP13A2 was used as a prototype of
subtype-P5B ATPases. The P. falciparum subtype-P5A ATPase was originally
described in a survey of transporter genes with Gene ID PF07_0115 [28] , which was later changed to PF3D7_0727800. This
sequence was also used in the phylogenetic analysis of type-P5 ATPases [12]. The P. relictum orthologue of
subtype-P5A was identified in PlasmoDB [24].
ATPase3 is a subtype-P5B ATPase found in apicomplexans and ATPase1 arose from a
gene duplication early in the evolution of the malaria parasite and is only
found in some malaria species [13].
The initial alignment of these eight ATPases was
carried out using ClustalW within Mega 11 [27].
This alignment was adjusted manually using 3-dimensional structures generated
by Swiss Model [29] as a guide to determine
the boundaries between the various domains. The transmembrane helices of the
M-domain were determined from the membrane annotation of Swiss Model
predictions. The first residue after the β -sheet
in the NTD was used as the boundary between the NTD and the A-domain. The first
residue of β -strand-1 and the last
residue of β -strand-6 of the modified
Rossmann fold were used as the boundaries between the P-domain and N-domain.
Furthermore, aligning the β -strand-2
from the subtype-P5A and subtype-P5B ATPases greatly improved the alignment.
Boxshade (https://junli.netlify.app/apps/boxshade/) was used to denote
identical (shaded in black) or similar (shaded in gray) residues.
Pairwise distances of the conserved regions of the
ATPases were also determined in Mega 11 after removing the variable regions of
low-complexity sequence from the alignment as previously recommended [7,30].
2.3. Homology Modeling
Homology modeling of the eight type-P5 sequences
was carried out with Swiss Model® [29]
using two templates each from Spf1 (PDB ID 6xmq and 6xmu) and ATP13A2 (PDB ID
7m5v and 7m5x). Templates 6xmq and 7m5v were bound with a non-hydrolysable ATP
analog (AMP-PCP) and templates 6xmu and 7m5x were bound with BeF3 and
their respective substrates of a transmembrane helix or spermine. These
templates were previously identified as reliable templates in Phyre®
analyses using the ATPase1 and ATPase3 sequences to search structural databases
[13]. Homology modeling was also carried out
on the Plasmodium ATPases after removal of the large regions of low
complexity and variable sequence.
Images were generated with PyMOL 3.1.3
(Schrodinger, LLC). The various domains were demarcated in different colors,
and the color names refer to the names given by PyMOL. Color scheme: nMH
(limon), cTM (yelloworange), sTM (light orange), NTD (aquamarine), A-domain
(tv_green), N-domain (skyblue), P-domain (violetpurple), cTM4 kink and
signature motif (magenta), phosphorylated asp (yellow), P-domain arm
(purpleblue), and IDL (deepsalmon).
Four criteria were used to assess the quality of
the models: the Global Model Quality Estimation (GMQE), the QMEANDisCo, the
percentage of Ramachandran favorable and Θ angles, and the retention of ligands. GMQE
predicts the expected accuracy of a protein model. QMEANDisCo is a composite
scoring function that provides both global and local quality estimates for a
protein model. Non-covalently bound ligands are retained in models if there are
at least three coordinating residues in the protein and those residues are
conserved in the target–template alignment, and if the resulting atomic
interactions in the model are within the expected range for van der Waals interactions
and water-mediated contacts.
3. Results
3.1. Subtype-P5A ATPase of Haemosporida
The P. falciparum subtype-P5A ATPase (PfP5A)
was originally described in a survey of transporter genes as a presumptive
cation transporter [28] and subsequently
included in a phylogenetic analysis of type-P5 ATPases [12]. Searches of PlasmoDB and NCBI identified
orthologues of subtype-P5A ATPase from other haemosporidians (Table S1). All the haemosporidian genes have a
single exon and their chromosomal locations correspond to the known gene
synteny of Plasmodium [31]. A search of
conserved domains [26] identified cd07543
(subtype-P5A) and TIGR01657 (subtype-P5B) as the two highest scoring domains
based on E-values. In both cases the E-values were extremely low and as
expected the cd07543 tended to be lower than TIGR01657. This confirms that the
sequences are subtype-P5A ATPases but also indicates that subtype-P5A and
subtype-P5B are very similar.
Alignment of the haemosporidian subtype-P5A
orthologues revealed regions of very high sequence homology interspersed with
variable regions of low sequence homology (Figure S1a). These haemosporidian subtype-P5A ATPases exhibit a similar
phylogeny as reported for ATPase3 [13] and
separate into eight clades (Figure S1b-c)
that are consistent with the current views on the phylogeny of malaria
parasites [32,33]. The eight clades are Haemoproteus,
avian parasites, Laverania, rodent parasites, Hepatocystis,
malariae, ovale, and vivax-like. As previously observed with ATPase3 [13] , Haemoproteus, avian parasites, and Laverania
form a clade and the other mammalian parasites including Hepatocystis
form a second clade (Figure S1b).
The Plasmodium subtype-P5A ATPases (PlP5A)
have N-terminal extensions (NTE) not found in either Spf1 or ATP13A2. ATPase3
also has an NTE but ATPase1 does not [13]. The
NTE of PlP5A is longer than the NTE of ATPase3 and there is no shared
sequence homology (Figure S2). In the
case of ATPase3, there is substantial sequence homology of the NTE within the
major apicomplexan clades (i.e., haemosporidians, piroplasmids, and
coccidians), whereas there is little homology between these clades [13]. The first half of the NTE of subtype-P5A
exhibits a high degree of sequence homology across all haemosporidians and the
second half of the NTE exhibits little sequence homology (Figure S1). This creates an insertion of a
variable region between the NTE and the NTD designated as variable region 1
(VR1). Six additional variable regions (VR2-VR7) are found in the
haemosporidian orthologues of subtype-P5A ATPase. These inserts tend to be low
complexity sequence with a high preponderance of asparagine and lysine residues
and tandem repeats are sometimes observed (Figure S1a).
3.2. Sequence Homology between Subtype-P5A and Subtype-P5B ATPases of Malaria Parasites
ATPase1, ATPase3, and PlP5A from Plasmodium
falciparum and P. relictum were aligned with Spf1 (subtype-P5A) and
ATP13A2 (subtype-P5B). The alignment exhibited regions of high sequence
homology between all eight proteins that were interspersed with regions of
little sequence homology (Figure S2). The
region including the NTD and the N-terminal membrane loops (nML) exhibit a
moderate level of homology. The regions of highest sequence homology correspond
to the canonical domains of P-type ATPases (A, N, P, and M). Other than the
P-domain (discussed below), there were no substantial regions in which the
sequence homology clearly segregated into either subtype-P5A or subtype-P5B.
This is consistent with the paucity of specific signature sequences to
distinguish subtype-P5A and subtype-P5B [8,12].
Large stretches of low complexity sequence are
inserted between or within the highly conserved canonical domains of the Plasmodium
type-P5 ATPases. This was previously described in ATPase1 and ATPase3 as
four large variable regions inserted into the NTD, A-domain, N-domain, and
P-domain referred to as variable regions (VR) 1-4 respectively [13]. The positions of these variable regions are
the same in ATPase1 and ATPase3. Subtype-P5A ATPase of Plasmodium has
seven substantial inserts of low complexity sequence that exhibit sequence
variation (Figure S1a). The locations of
some of the inserts are exclusive for either subtype-P5A or subtype-P5B,
whereas three inserts have a shared location between the two subtypes (Table 2). Inserts with shared locations the
inserts tend to be substantially larger in one of the two subtypes. In general,
the inserts of the subtype-P5A ATPases tend to be smaller in size than the
subtype-P5B ATPases. The total percentage of the ATPases that are associated
with variable regions is approximately the same between subtype-P5A and
subtype-P5B. If present, the insert sizes within Spf1 and ATP13A2 are
substantially smaller.
An alignment with the low homology inserts removed
was used to generate a pairwise distance matrix of the eight type-P5 ATPases (Table 3). Comparisons of ATPases of the same
subtype were designated as concordant and comparisons between subtype-P5A and
subtype-P5B ATPases were designated as discordant. The distance between Spf1
(subtype P5A) and ATP12A2 (subtype P5B) is 1.39 and represents the divergence
subtype-P5A and subtype-P5B from early in the evolution of eukaryotes. The
distances between other discordant sequences exhibit similar values
(1.36-1.59). The concordant subtype-P5B sequences also exhibit similar values
(1.24-1.56), whereas the values from concordant subtype-P5A sequences were a
little lower (1.14-1.16). Even though ATPase1 and ATPase3 are subtype-P5B
ATPases, they are also essentially equal distance from subtype-P5A (1.41-1.59)
and subtype-P5B (1.24-1.56) templates. As expected, the P. falciparum
and P. relictum orthologues are the most closely related (0.13-0.42) and
reflect a substantially more recent divergence which is on the order of 10
million years ago [34].
3.3. Homology Modeling and Structure Comparisons
The experimentally determined structures of Spf1
and ATP13A2 were used as templates to carry out homology modeling of the Plasmodium
type-P5 ATPases. As controls, the Spf1 and ATP13A2 sequences were modeled with
the same template (i.e., concordant) or the other template (i.e., discordant).
Modeling with either the concordant or discordant templates produced structures
that were similar to each other and to the experimentally determined
structures. This is especially true for the predicted structures of the M-, A-,
N-, and P-domains. Neither template provided much insight into the structures
of the N-terminal and C-terminal extensions since these sequences were usually
excluded from the models.
As expected, differences between subtype-P5A and
subtype-P5B were primarily in the NTD, including the N-terminal membrane loops,
and the arm in the P-domain. Concordant templates produced more complete
structures than discordant templates. For example, modeling Spf1 with the Spf1
template generated two N-terminal transmembrane helices as is found in
subtype-P5A (Figure 2), and the
discordant template generated a single N-terminal transmembrane helix (Figure 3). Similarly, modeling ATP13A2 with the
concordant template generated the expected triangular membrane loop that is
found in subtype-P5B ATPases (Figure 3)
and triangular loop generated by the discordant template was somewhat
incomplete (Figure 2). Another feature
specific to subtype-P5A ATPases is a α -helical
‘arm’ that projects from the P-domain. The arm is evident in the experimentally
determined structure and a shorter version is predicted in the concordant model
while no arm is generated with the discordant template.
In general, the predicted structures of the Plasmodium ATPases are similar to the templates and the major domains of P-type ATPases are readily discernable. However, neither template did an extremely good job at generating the NTD including the associated membrane loops. No NTD was generated with the Spf1 template in four of the Plasmodium ATPases, and the first transmembrane helix was lacking in the other two ATPases (Figure 2). Similarly, the NTD was lacking in five of the Plasmodium ATPase models generated with the ATP13A2 template (Figure 3). However, in PrATPase1 a complete triangular membrane loop was generated.
3.4. Quality Assessment of Modeling
The quality of the structural models produced from concordant and discordant templates was assessed with two templates representing different conformational states of both Spf1 and ATP13A2 by four criteria as described in the Methods (Table 4). Models of Spf1 or ATP13A2 using concordant templates represent the maximum possible quality scores and serve as positive controls, whereas scores from discordant sequence and template pairs represent the magnitude of the difference between subtype-P5A and subtype-P5B. The PlP5A sequences modeled with the Spf1 templates (i.e., concordant) had notably higher GMQE scores and slightly higher QMEANDisCo scores than those modeled with the discordant templates (7m5x and 7m5v). This was not observed with the Plasmodium subtype-P5B ATPases which exhibited essentially the same quality scores with either the concordant or discordant templates. The P. falciparum sequences have lower GMQE scores than the P. relictum sequences, which is likely due to the smaller variable regions in the P. relictum proteins. Magnesium was the most often retained ligand and was only excluded in a few instances and only with templates bound with non-hydrolysable ATP analogs. The substrates and non-hydrolysable ATP analogs were not retained in any models. There were a few instances of models retaining the BeF3 using the ATP13A2 template.
The quality assessments indicate that the models of the Plasmodium type-P5 ATPases are relatively accurate, and the quality assessments did not substantially favor one template over the other in subtype-P5B ATPases. In contrast, the concordant template (i.e., Spf1) was favored in the subtype-P5A ATPases.
3.5. A-Domain
The A-domain is formed by a segment between the NTD and cTM1 plus the loop between cTM2 and cTM3. The loop between cTM2 and cTM3 is composed of eight primarily anti-parallel β-strands which form a structure called a distorted jelly roll [6]. The order of the β-strands in the β-sheet according to linear amino acid sequence is b2-b1-b3-b8-b4-b7-b5-b6, and b4 and b7 are parallel while the other β-strands are anti-parallel. Two α-helices connected to cTM1 and a third α-helix connected to cTM3 are stacked against this β-sheet. These basic features of the distorted jelly roll are found in the experimentally determined structures of Spf1 and ATP13A2 except for some minor differences (Figure 4a). For example, Spf1 appears to be missing β-strand-6 and both Spf1 and ATP13A2 have a short β-strand of two amino acids (designated b6’) that is found between β-strand-4 and β-strand-7 which is not found in the standard distorted jelly roll. This short β-strand (b6’) is not observed in any of the models generated with either concordant or discordant templates, and β-strand-6 is present in all the models generated with both templates (Figure S3). Except for the perplexing case of the experimentally determined structure of Spf1, the distorted jelly roll is the same as the known structure of the A-domain of type-P5 ATPases.
The predicted A-domain structures of the Plasmodium type-P5 ATPases are similar to the experimentally determined structures, and the same structures were produced with either concordant or discordant templates (Figure S3). Differences in the modeled Plasmodium proteins were minor, inconsistent, or of dubious importance. All three subtype-P5A ATPases modeled with the ATP13A2 template (discordant) had an extra helix in a short segment of low sequence homology between β-strand-7 and β-strand-8 (Figure 4b). However, this helix (h’) was not observed in the experimentally determined structure of Spf1 nor in the predicted structures with the concordant template (6xmu). An additional short β-strand (b’) that is not part of the β-sheet was observed in PfATPase1 and PrP5A and an additional short helix (h”) is seen in PrP5A. This additional β-strand is also seen in the experimentally determined ATP13A2 structure but not found in the modeled structures produced with either template. These additional elements are found in regions of low sequence homology (Figure 4b). A large portion of N-terminal sequence, including part of the A-domain, was excluded from the P. falciparum subtype-P5A structure modeled with either 6xmu or 7m5x templates and from the P. relictum subtype-P5A structure modeled with 7m5x. In summary, there are no major differences in the A-domain between subtype-P5A and subtype-P5B ATPases including the Plasmodium type-P5 ATPases.
3.6. N-Domain
N-domains have been described as either a six-stranded twisted antiparallel β-sheet [35] or a seven-stranded twisted antiparallel β-sheet [6,36] flanked by four [6,35] or five [36] α-helices. The basic structure of the N-domain of both Spf1 and ATP13A2 consists of a twisted six-stranded anti-parallel β-sheet flanked by four α-helices (Figure 5), thus, both are similar to the Cu-transporting P-type ATPase [35]. Additional β-strands are observed in both Spf1 and ATP13A2 (Figure 5). Two additional β-strands of two amino acids each are observed in Spf1 between α-helix-1 and α-helix-2 and are not likely to be incorporated into the twisted β-sheet. These short β-strands are not found in any of the modeled structures (Figure S4a). In ATP13A2 the additional β-strands are located between α-helix-2 and β-strand-2 and are potentially in close enough proximity to be part of the β-sheet. These two β-strands are found in the model structures of ATP13A2 and Spf1 using the ATP13A2 template but are not found in any of the modeled Plasmodium ATPases (Figure S4b). The sequence encoding these additional β-strands is in a short non-conserved region that tends to form IDL (Figure 5b), thus raising questions about their validity or significance. Except for the extra β-strands, the N-domain structures of Spf1 and ATP13A2 are nearly identical.
The predicted structures of the N-domains from the Plasmodium type-5 ATPases are very similar to the structures of Spf1 and ATP13A2 except for the lack of the extra β-strands (Figure S5). No substantial differences between subtype-P5A and subtype-P5B ATPases are seen in the N-domain and no unique features of the Plasmodium type-5 ATPases were observed. The N-domain of the type-P5 ATPases is highly conserved in regard to its secondary structural features.
3.7. P-Domain
The P-domain of SERCA is described as a modified Rossmann fold with a six-stranded parallel β-sheet and six associated α-helices [6]. The Rossmann fold is a common tertiary structure found in proteins that bind nucleotides [37]. This modified Rossmann fold is formed from alternating β-strands and α-helices with the resulting β-sheet being somewhat sandwiched between two rows of α-helices. The fourth helix has a kink resulting in two closely associated helices designated as helix-4a and helix-4b. For the most part, the β-strands and α-helices making up this modified Rossmann fold are readily identified in the experimentally determined structure of Spf1 and ATP13A2 (Figure 6 and Figure 7). In addition, two additional parallel β-strands (b7 and b8), two additional anti-parallel β-strands (b’ and b”), and an additional α-helix (h’) are observed.
The experimentally determined structures of Spf1 and ATP13A2 are quite similar to each other except that ATP13A2 is missing the α-helix-3 (Figure 7). However, homology modeling of the ATP13A2 sequence with the Spf1 template results in random-coil sequence in the shape of a helix at this position (Figure 6), and modeling of the Spf1 sequence with the ATP13A2 template results in the loss of this third helix (Figure 7). In other words, the structures generated with concordant templates better recapitulated the actual experimentally determined structures. Another unique feature of the subtype-P5A P-domain is the presence of a α-helical projection called the arm. Following the arm is an intrinsically disordered region that may interact with the membrane [9]. The arm is readily identified in the experimentally determined structure of Spf1 and in predicted structure of Spf1 using Spf1 as a template (Figure 6). As expected, no arm is seen in either the experimentally determined structure of ATP13A2 nor the predicted structure using the Spf1 template. These results confirm that subtype-P5A and subtype-P5B differ in regard to the presence of the arm structure and imply that there may be a difference between the Rossmann folds of subtype-P5A and subtype-P5B ATPases, especially in the region of the third helix.
Homology modeling of the Plasmodium subtype-P5A ATPases with the Spf1 (i.e., concordant) generates structures similar to the experimentally determined Spf1 with all the expected secondary structures of the Rossmann fold (Figure 6). However, a shorter than expected arm is predicted in the PrP5A structure, and no arm is predicted in the PfP5A structure. As expected, no arms are predicted in ATPase1 or ATPase3. In contrast, there were several missing secondary structural elements in ATPase1 and ATPase3 (i.e., subtype-P5B Plasmodium sequences) modeled with Spf1 (i.e., discordant), as well as β-strand-b” of PrATPase1 being generated from variable region-4 sequence. The ATP13A2 template performed approximately equally well with Plasmodium types-P5A (i.e., discordant) and subtype-P5B ATPases (i.e., concordant) (Figure 7). For example, the ATP13A2 template also generated several instances of variable sequence being incorporated into the secondary structures of the P-domain in both concordant and discordant structures. There were also several instances of missing secondary structural elements in both concordant and discordant structures. These anomalies are restricted to the central part of the P-domain in a region that does not exhibit a high level of sequence homology and that includes VR4 of ATPase1/3 and VR6 of PlP5A (Figure 8). In particular, β-strand-3, helix-3, β-strand-4, and helix-4a are in a region of low sequence homology.
3.8. Variable Regions Effects
Overall, the predicted structures of the Plasmodium type-P5 ATPases are quite similar to the experimentally determined structures, and this is especially true for the A- and N-domains. The relatively minor discrepancies in the P-domain of the predicted structures are often associated with the variable regions composed of IDL. The potential effects of the variable regions were analyzed by modeling the Plasmodium subtype-P5B ATPase sequences with or without variable regions using the Spf1 and ATP13A2 templates. In general, removing the variable regions more than doubled the GMQE scores, increased the QMEANDisCo scores by 20% or more, and increased the percentage of Ramachandran favorable and Θ angles by 10% or more (Table 4). The retention of ligands did not appear to be affected by the presence or absence of variable regions.
Removing the variable regions for the most part either had no effect on the predicted structures or improved the homology modeling by generating structures that better recapitulated the experimentally determined structures (Figure S5 and Table 5). For example, most of the predicted structures of ATPase1 and ATPase3 were missing part of the NTD and removing the variable regions partially or completely restored the membrane associated helices of the NTD. Similarly, for the most part, the minor discrepancies in the P-domain were corrected by removing the variable regions. When there were no discrepancies, removing the variable regions did not introduce discrepancies except for a loss of β-strands in the P-domain of PrATPase3. There was no obvious correlation between the size of the variable regions and whether their removal had a positive, neutral, or negative impact on the predicted 3-dimensional structures.
3.9. Substrate-Binding Site
The substrate-binding sites of P-type ATPases are formed from the core transmembrane helices, and cTM2, cTM4, and cTM6 often interact with the substrate [1]. These helices wrap around to form an incomplete cylindrical structure that makes up the substrate-binding groove. This groove typically opens to the extra-cytoplasmic face of the membrane and the kink in cTM4 forms the base. Subtype-P5A ATPase has a much wider substrate groove than subtype-P5B to accommodate the transmembrane-helix substrate, and the groove opens laterally within the lipid bilayer. This wider substrate groove is due to cTM5 and cTM6 rotating away from the other cTM [8]. For example, the distance between cTM2 and cTM6 is approximately 19 Å in Spf1 and 7 Å in ATP13A2 (Figure 9). Important residues in Spf1 are six non-polar amino acids on cTM2 and cTM4 that interact with the α-helical substrate [12]. These are F223, M227 and M231 of cTM2, P445 of the kink in cTM4, and M449 and M453 of cTM4b. The substrate-binding groove of ATP13A2 is stabilized by interactions between Y259 in cTM2, Y940 and Q944 in cTM5, and D967 in cTM6 called the tetrad [11]. In addition, aspartates that provide negative charges to interact with the positive charges of polyamines have also been identified.
Homology modeling of the Plasmodium subtype-P5A ATPases with the Spf1 template yields structures that are essentially identical to the experimentally determined Spf1 structures and the six residues implicated in substrate binding are all non-polar (Figure 9). Somewhat unexpectant though, modeling Plasmodium subtype-P5A ATPases with the discordant template produces a substrate-binding groove essentially identical to the experimentally determined ATP13A2 structure (i.e., subtype-P5B). However, none of the stabilizing tetrad amino acids are conserved and only one of the aspartates is conserved and the other is replaced with a positively charged histidine. Thus, even though the discordant structure recapitulates the subtype-P5B ATPase, key residues are missing, and therefore the Plasmodium subtype-P5A ATPases are not likely to be polyamine transporters.
Likewise, modeling of ATPase1 and ATPase3 with the concordant template produced models essentially identical to ATP13A2 and the discordant template produced models essentially identical to Spf1. As previously noted [13], the residues making up the tetrad and the aspartate residues of ATPase1 are identical to ATP13A2, whereas these residues are only partially conserved in ATPase3. In regard to the discordant template, the six non-polar residues implicated in substrate binding are all non-polar in ATPase3, whereas 5/6 of the residues are non-polar in PrATPase1 and 4/6 residues are non-polar in PfATPase1. However, these three polar residues are next to non-polar residues. The conservation of the non-polar residues that interact with the substrate suggests that Plasmodium subtype-P5B ATPases may be able to conform to α-helix dislocase activity.
4. Discussion
Type-P5 ATPases are not well characterized, and this is especially true for the type-P5 ATPases of the malaria parasite. Malaria parasites, like most other eukaryotes, have two type-P5 ATPases designated as subtype-P5A and subtype-P5B. The subtype-P5A gene and protein of Plasmodium have not yet been characterized other than to be listed as part of genomic surveys [28] or phylogenetic studies [12]. Two subtype-P5B ATPases have been identified in the major human pathogen Plasmodium falciparum and designated as ATPase1 and ATPase3. ATPase1 is not found in all species of malaria parasites and is limited to the parasites of great apes (i.e., Laverania), avian malaria parasites, and Haemoproteus [13]. ATPase1 arose from a duplication of the ATPase3 gene early in the evolution of the malaria parasite. Further characterization of the type-P5 ATPases from the malaria parasite using Spf1, a subtype-P5A ATPase of yeast, and ATP13A2, a subtype-P5B ATPase of humans, as templates provided insight into the structures of the Plasmodium type-P5 ATPases, revealed a possible role for the IDL associated with these proteins, allowed for speculation about the possible functions of type-P5 ATPases in the malaria parasite, and possibly contributed to a better understanding of the evolution of the type-P5 ATPases.
4.1. Homology Modeling and Predicted Structures
Sequence alignments and homology modeling of the Plasmodium type-P5 ATPases identified all the expected canonical domains found in P-type ATPases (i.e., A, N, P, and M). The predicted secondary structures of these canonical domains recapitulated experimentally determined structures with relatively minor discrepancies whether concordant (i.e., same subtype) or discordant (i.e., different subtype) templates were used in homology modeling. Many of these discrepancies disappeared if the regions corresponding to the IDL were removed from the sequences before modeling. This suggests that large intrinsically disorganized regions may interfere with homology modeling. Even though this effect is relatively minor, it may be prudent to carry out modeling on both complete protein sequences and sequences with large regions of low-complexity sequence removed. This may be especially important for proteins of the malaria parasite since large regions of low-complexity sequence are often found in Plasmodium proteins [15]. In contrast to the canonical P-type ATPase domains, the NTD and CTE were not always accurately modeled, and often the N-terminal and C-terminal regions were excluded from the predicted three-dimensional structures.
The secondary structural features of the A- and N-domains were nearly identical between the subtype-P5A and subtype-P5B ATPases whether concordant or discordant templates were used. This emphasizes the similarity between the two subtypes of the type-P5 ATPases. Overall, homology modeling predicted structures that recapitulated the known structures of type-P5 ATPases and it is likely that the subtype-P5A proteins from Plasmodium have a similar structure as other subtype-P5A ATPases and ATPase1 and ATPase3 have similar structures as other subtype-P5B ATPases. The good performance of homology modeling also indicates that homology modeling will be a useful tool in future studies of these proteins and may be useful in preliminary studies to search for possible drugs targeting P-type ATPases.
Differences between subtype-P5A and subtype-P5B ATPases are primarily in the membrane associated helices of the NTD, the arm of the P-domain, and the substrate specificity [8]. For example, the subtype-P5A NTD has two additional transmembrane helices and the subtype-P5B NTD has a triangular membrane loop that does not traverse the lipid bilayer. Homology modeling of the Plasmodium type-P5 ATPases did correctly generate these structures when concordant templates were used and especially if the variable regions were removed from the protein sequences. Another difference between subtype-P5A and subtype-P5B is a α-helical projection in the P-domain called the arm that is not found in subtype-P5B. As expected, no arm structures are found in ATPase1 or ATPase3 and this arm was predicted in the PrP5A sequence with the Spf1 (i.e., concordant) template and not the ATP13A2 (i.e., discordant) template. It is not clear why the arm was not predicted in PfP5A. An extra α-helix was observed in the Rossmann fold of the P-domain in the subtype-P5A ATPases which was not seen in the subtype-P5B ATPases. This extra helix may possibly be another structural feature to distinguish subtype-P5A and subtype-P5B ATPases and merits further investigation. To accommodate their distinct substrates, the substrate binding groove of Spf1 is much wider than the narrow groove of ATP13A2.
4.2. Limitations of Homology Modeling
Discordant templates revealed a potential limitation of homology modeling since the predicted 3-dimensional structures had a strong tendency to recapitulate the template, especially in the NTD and substrate-binding grove. For example, homology modeling of the Plasmodium subtype-P5A ATPase with the subtype-P5B template (i.e., discordant) generated the triangular membrane loop, whereas modeling with the concordant template produced the expected transmembrane helices. This same effect of the predicted structures reflecting the templates was also observed in the Plasmodium subtype-P5B ATPases. However, in the case of the NTD the concordant templates did produce structures of slightly higher quality in regard to recapitulating the experimentally determined structures than the discordant templates. This was not true with the substrate-binding groove. The homology models were essentially identical to the experimentally determined structures of the templates and both concordant and discordant templates appeared to generate structures of equal quality. Thus, it will be important to use the correct template when carrying out homology modeling and to have a means to choose the correct template among multiple possible templates. For example, the Spf1 template ranked equally well to subtype-P5B templates in an analysis of subtype-P5B ATPases using Phyre® [13].
Another limitation is that most experimental work on eukaryotes has been carried out in humans and other mammals or yeasts. In terms of eukaryotic diversity, fungi and metazoans are relatively closely related as members of the opisthokonts in a major eukaryotic supergroup, or clade, that includes some amoeba called Amorphea [38]. Little experimental work has been carried out in the other major eukaryotic supergroups other than perhaps plants and a smattering of pathogenic eukaryotes. It is not inconceivable that even highly conserved proteins may exhibit structural or functional differences between the major eukaryotic supergroups. In regard to this study, experimentally determined structures from other apicomplexans or members of SAR supergroup, which represents half of all eukaryote diversity [39], may be informative and useful as templates. Thus, more experimental data reflecting the extreme diversity of eukaryotes is clearly needed.
4.3. Possible Functions of IDL
An obvious unique feature of the predicted structures of the Plasmodium type-P5 ATPases is the presence of large loops of intrinsically disorganized sequence. Intrinsically disorganized regions in proteins are random coil secondary structures that lack bulky hydrophobic amino acids and do not adopt a tertiary structure with a hydrophobic core [40,41]. Traditionally, IDL were viewed as passive linkers between structured domains, but are now known to have functional roles [42]. For example, IDL can be involved in the formation of protein complexes and higher-order supramolecular structures [43]. The flexibility of the IDL allows them to take on many conformations and may allow for an induced fit as they bind ligands including other proteins. Thus, the IDL may allow for low-affinity interactions with other proteins. At the same time, the physical mass of the IDL, which ranges from one-third to one-half in the type-P5 ATPases of Plasmodium, could also provide spacing between proteins.
The flexibility of the IDL could also form a shroud-like structure that surrounds the core ATPase structure. Such a shroud could have a protective role and prevent damage to the primary domains. Similarly, such a shroud may provide a buffer zone for these ATPase molecules so that other proteins on the membrane do not inadvertently interact with them and thus possibly interfere with their function. A protective role of the shroud and a role in protein-protein interactions are not necessarily mutually exclusive.
The IDL are correlated with inserts of variable regions which intuitively would argue against a specific function. However, these variable regions only exhibit variation between the eight haemosporidian clades and are conserved within the individual haemosporidian clades for both subtype-P5A (Figure S1) and subtype-P5B [13]. These variable regions are composed of low-complexity sequences that are highly enriched in polar amino acids, as are IDL in general [40,41]. Thus, despite the lack of sequence conservation, the flexibility and similar amino acid composition of the IDL could preserve a function such as low affinity binding to other proteins. Indeed, low-complexity sequence has been demonstrated to be subject to natural selection [44]. The observation that tandem repeats are often observed in these low-complexity regions suggests a mechanism to explain the variability and preservation of the amino acid composition. A periodic replacement and expansion of tandem repeats through slipped-strand mispairing [45] or unequal crossing-over [46] could occur on a similar evolutionary timeframe as the formation of the major haemosporidian clades.
4.4. Substrate Specificity
The substrate specificity of P-type ATPases and their subcellular locations are key elements regarding their functions [8]. Subtype-P5A ATPases are localized to the ER [47] and have been previously hypothesized to transport calcium [48], manganese [49], or lipids [50]. Recent studies indicate that Spf1 is likely a transmembrane helix dislocase [8,11]. Presumably this dislocase activity serves as a quality control mechanism to remove mistargeted transmembrane helices from the ER membrane. Consistent with this function, a subtype-P5A ATPase from Caenorhabditis elegans plays a role in correctly targeting membrane and secretory proteins [51]. Other than some early speculation that subtype-P5B might be heavy metal transporters [52], most studies have characterized subtype-P5B ATPases as polyamine transporters of the late endosomes or lysosomes [10,53,54,55,56].
Homology modeling by itself cannot resolve the substrate specificity of the Plasmodium type-P5 ATPases since the substrate-binding sites conform to the template. Nonetheless, it is quite likely that Plasmodium subtype-P5A ATPases are helix dislocases. This assertion is supported by the observation that the key residues for binding to the α-helix substrate are conserved, whereas as key residues involved in polyamine binding are less conserved (Figure 9). Helix dislocase activity is likely a necessary quality control mechanism in the ER and disruption of the gene has adverse pleiotropic effects [8,57]. The subtype-P5A gene is found in all eukaryotes and with few exceptions it is a single copy gene [58], and it is believed that subtype-P5A may have a general and highly conserved function [47]. The very high level of sequence conservation, except for the variable region inserts, within the haemosporidians (Figure S1), and identification of clear orthologues throughout the SAR (Table S1) further support a conserved function of the subtype-P5A ATPases in all eukaryotes including the malaria parasite.
In contrast, as previously discussed [13], it is unlikely that the haemosporidian subtype-P5B ATPases are lysosomal polyamine transporters. Previously published immunofluorescence data [17,18,19] are more reminiscent of ER staining, and proteomic analysis did not detect any P-type ATPases in the lysosomal compartment (i.e., digestive vacuole) of the malaria parasite [59]. Furthermore, malaria parasites are capable of synthesizing polyamines [60] and have a plasma membrane associated polyamine transporter to take up polyamines from the host cell cytoplasm [61]. In addition, clear orthologues of ATPase3 can only be identified in the apicomplexans and ATPase1 is limited to only some of the malaria parasites [13]. If the apicomplexan and opisthokont subtype-P5B ATPases had the same function one might expect to be able to identify clear orthologues throughout the eukaryotes. Determining the substrate specificity of the haemosporidian or apicomplexan subtype-P5B ATPases will likely require experimental verification.
4.5. Divergent Evolution of Subtype-P5B ATPases
Type-P5 ATPases likely originated during the early evolution of eukaryotes, and its origin was likely coincident with the formation of the ER and other internal membrane systems [12,58]. The duplication and divergence into subtype-P5A and subtype-P5B also likely occurred in a primordial eukaryote before the formation of the major eukaryote supergroups. A limited number of subsequent duplications of subtype-P5A have also been noted in the archaeplastids and stramenopiles [58]. In contrast, subtype-P5B has been duplicated many times in several different eukaryotic lineages [8,12,13]. In addition, subtype-P5B is not found in all eukaryotes, and this is usually attributed to lineage specific losses of subtype-P5B [12]. For example, gene loss certainly explains the lack of subtype-P5B in Entamoeba since these organisms are amorpheans, as are fungi and metazoans which have subtype-P5B. Therefore, it is likely a loss of the subtype-P5B gene occurred after the amoebozoans split from the opisthokonts.
Duplicated genes can undergo divergent evolution, and this allows for an expansion and diversification of gene families and provides opportunities for innovation and adaptation to new environments or physiological milieus [62]. Following the split into subtype-P5A and subtype-P5B it is probable that subtype-P5A maintained its function as an ER helix dislocase in all eukaryotes. Subtype-P5B, on the other hand, may have evolved new functions in a lineage-specific manner. For example, subtype-P5B in the opisthokonts diverged into a polyamine transporter located in the lysosome. In contrast to the opisthokonts, the subtype-P5B ATPases of the malaria parasite appear to have maintained their location in the ER and did not evolve to transport polyamines. Therefore, it is unlikely that subtype-P5B ATPases have a common function in all eukaryotes and there could be lineage specific functions in the various eukaryotic supergroups.
The retention of subtype-P5B ATPase in the ER of malaria parasites implies a possible retention and localization of subtype-P5B ATPases to the ER throughout the Apicomplexa and even perhaps the SAR supergroup. This could represent redundancy or perhaps a divergence to a more specialized function. Furthermore, homology modeling with the discordant subtype-P5A template indicates that ATPase1 and ATPase3 can at least in theory might be capable of helix dislocase activity. In addition, the observation that the Plasmodium subtype-P5B ATPases are approximately equal distance from Spf1 and ATP13A2 in phylogenetic pairwise analyses (Table 3) is also consistent with Plasmodium subtype-P5B ATPases having a similar function as subtype-P5A ATPases. Similarly, the quality assessment scores of Plasmodium subtype-P5B ATPases are approximately the same with concordant or discordant templates suggesting no strong preference for either template. In contrast, the subtype-P5A ATPase shows a preference for concordant templates over discordant templates. Therefore, it is not inconceivable that the subtype-P5B ATPases of Plasmodium, other apicomplexans and members of SAR have a function similar to the subtype-P5A ATPases.
Maintenance of two genes with similar functions allows for divergence in regard to the precise substrate specificity. For example, subtype-P5A ATPases have also been implicated in the removal of signal peptides from the ER membrane [8]. Interestingly, the malaria parasite and other apicomplexans may have two distinct pathways targeting proteins for export [63]. One of these pathways involves the traditional signal peptide and the other involves a signal called PEXEL [64]. PEXEL signaling is also found in some stramenopiles [65]. As highly hypothetical speculation, one could propose the need for different helix dislocases to remove these rather different peptide signals from the ER membrane.
5. Conclusions
The subtype-P5A ATPase of malaria parasites is likely a helix dislocase of the ER as are other subtype-P5A ATPases. In contrast, the subtype-P5B ATPases of the malaria parasite have likely diverged from other subtype-P5B ATPases and are not polyamine transporters of the lysosome. Experimental verification of cellular locations and substrate specificities of the type-P5 ATPases of the malaria are needed. A notable difference between the type-P5 ATPases of the malaria parasite and other subtype-P5A ATPases are the insertion of variable regions composed of low complexity sequence. This low complexity sequence may form a shroud that surrounds the core of the ATPase which may function in low-affinity protein-protein interactions or protection of the core ATPase.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Identification of subtype-P5A ATPase genes in SAR (Stramenopiles, Alveolata, and Rhizaria); Figure S1: Alignment and phylogeny of subtype-P5A ATPases from haemosporidians; Figure S2: Alignment of Plasmodium type-P5 ATPases with Spf1 (type-P5A) and 13A2 (type-P5B); Figure S3: A-domain structure of type-P5 ATPases; Figure S4: N-domain structure of type-P5 ATPases; Figure S5. Effects of low complexity variable regions on homology modeling.
Author Contributions
Not applicable.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Contact author to receive data generated by this study.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| cTM | core transmembrane helix |
| CTE | C-terminal extension |
| IDL | intrinsically disorganized loops |
| PEXEL | Plasmodium export element |
| PlP5A | Plasmodium subtype-P5A ATPase |
| SAR | stramenopiles-alveolates-rhizarians |
| SERCA | sarcoplasmic-endoplasmic reticulum ATPase |
References
- Palmgren, M. P-Type ATPases: Many More Enigmas Left to Solve. J Biol Chem 2023, 299, 105352. [Google Scholar] [CrossRef]
- Martin, R.E.; Henry, R.I.; Abbey, J.L.; Clements, J.D.; Kirk, K. The “permeome” of the Malaria Parasite: An Overview of the Membrane Transport Proteins of Plasmodium Falciparum. Genome Biol 2005, 6, R26. [Google Scholar] [CrossRef] [PubMed]
- Arnou, B.; Montigny, C.; Morth, J.P.; Nissen, P.; Jaxel, C.; Møller, J.V.; Maire, M. le The Plasmodium Falciparum Ca2+-ATPase PfATP6: Insensitive to Artemisinin, but a Potential Drug Target. Biochem Soc Trans 2011, 39, 823–831. [Google Scholar] [CrossRef]
- Krishna, S.; Pulcini, S.; Fatih, F.; Staines, H. Artemisinins and the Biological Basis for the PfATP6/SERCA Hypothesis. Trends Parasitol 2010, 26, 517–523. [Google Scholar] [CrossRef]
- Fiser, A. Template-Based Protein Structure Modeling. Methods Mol Biol 2010, 673, 73–94. [Google Scholar] [CrossRef]
- Stokes, D.L.; Green, N.M. Structure and Function of the Calcium Pump. Annu Rev Biophys 2003, 32, 445–468. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, K.B.; Palmgren, M.G. Evolution of Substrate Specificities in the P-Type ATPase Superfamily. J Mol Evol 1998, 46, 84–101. [Google Scholar] [CrossRef]
- Sim, S.I.; Park, E. P5-ATPases: Structure, Substrate Specificities, and Transport Mechanisms. Curr Opin Struct Biol 2023, 79, 102531. [Google Scholar] [CrossRef]
- McKenna, M.J.; Sim, S.I.; Ordureau, A.; Wei, L.; Harper, J.W.; Shao, S.; Park, E. The Endoplasmic Reticulum P5A-ATPase Is a Transmembrane Helix Dislocase. Science 2020, 369. [Google Scholar] [CrossRef]
- Li, P.; Wang, K.; Salustros, N.; Grønberg, C.; Gourdon, P. Structure and Transport Mechanism of P5B-ATPases. Nat Commun 2021, 12, 3973. [Google Scholar] [CrossRef] [PubMed]
- Tillinghast, J.; Drury, S.; Bowser, D.; Benn, A.; Lee, K.P.K. Structural Mechanisms for Gating and Ion Selectivity of the Human Polyamine Transporter ATP13A2. Mol Cell 2021, 81, 4650–4662.e4. [Google Scholar] [CrossRef] [PubMed]
- Møller, A.B.; Asp, T.; Holm, P.B.; Palmgren, M.G. Phylogenetic Analysis of P5 P-Type ATPases, a Eukaryotic Lineage of Secretory Pathway Pumps. Mol Phylogenet Evol 2008, 46, 619–634. [Google Scholar] [CrossRef]
- Wiser, M.F. Duplication of a Type-P5B-ATPase in Laverania and Avian Malaria Parasites and Implications about the Evolution of Plasmodium. Parasitologia 2025, 5, 6. [Google Scholar] [CrossRef]
- Cortés, G.T.; Beltran, M.M.G.; Gómez-Alegría, C.J.; Wiser, M.F. Identification of a Protein Unique to the Genus Plasmodium That Contains a WD40 Repeat Domain and Extensive Low-Complexity Sequence. Parasitol Res 2021, 120, 2617–2629. [Google Scholar] [CrossRef]
- DePristo, M.A.; Zilversmit, M.M.; Hartl, D.L. On the Abundance, Amino Acid Composition, and Evolutionary Dynamics of Low-Complexity Regions in Proteins. Gene 2006, 378, 19–30. [Google Scholar] [CrossRef]
- Wiser, M.F. The Digestive Vacuole of the Malaria Parasite: A Specialized Lysosome. Pathogens 2024, 13, 182. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Cowan, G.; Meade, J.C.; Wells, R.A.; Stringer, J.R.; Robson, K.J. A Family of Cation ATPase-like Molecules from Plasmodium Falciparum. Journal of Cell Biology 1993, 120, 385–398. [Google Scholar] [CrossRef]
- Rozmajzl, P.J.; Kimura, M.; Woodrow, C.J.; Krishna, S.; Meade, J.C. Characterization of P-Type ATPase 3 in Plasmodium Falciparum. Mol Biochem Parasitol 2001, 116, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Tanabe, K.; Krishna, S.; Tsuboi, T.; Saito-Ito, A.; Otani, S.; Ogura, H. Gametocyte-Dominant Expression of a Novel P-Type ATPase in Plasmodium Yoelii. Mol Biochem Parasitol 1999, 104, 331–336. [Google Scholar] [CrossRef]
- Wiser, M.F.; Lanners, H.N.; Bafford, R.A.; Favaloro, J.M. A Novel Alternate Secretory Pathway for the Export of Plasmodium Proteins into the Host Erythrocyte. Proc Natl Acad Sci U S A 1997, 94, 9108–9113. [Google Scholar] [CrossRef]
- Aikawa, M. Parasitological Review. Plasmodium: The Fine Structure of Malarial Parasites. Exp Parasitol 1971, 30, 284–320. [Google Scholar] [CrossRef]
- Langreth, S.G.; Jensen, J.B.; Reese, R.T.; Trager, W. Fine Structure of Human Malaria in Vitro. J Protozool 1978, 25, 443–452. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A Better Web Interface. Nucleic Acids Res 2008, 36, W5–9. [Google Scholar] [CrossRef] [PubMed]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A Functional Genomic Database for Malaria Parasites. Nucleic Acids Res 2009, 37, 539–543. [Google Scholar] [CrossRef]
- Bensch, S.; Canbäck, B.; DeBarry, J.D.; Johansson, T.; Hellgren, O.; Kissinger, J.C.; Palinauskas, V.; Videvall, E.; Valkiūnas, G. The Genome of Haemoproteus Tartakovskyi and Its Relationship to Human Malaria Parasites. Genome Biol Evol 2016, 8, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The Conserved Domain Database in 2023. Nucleic Acids Res 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martin, R.E.; Henry, R.I.; Abbey, J.L.; Clements, J.D.; Kirk, K. The “permeome” of the Malaria Parasite: An Overview of the Membrane Transport Proteins of Plasmodium Falciparum. Genome Biol 2005, 6, R26. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Naylor, G.J.; Brown, W.M. Amphioxus Mitochondrial DNA, Chordate Phylogeny, and the Limits of Inference Based on Comparisons of Sequences. Systematic Bology 1998, 47, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, A.S.; Mello, B.; Pacheco, M.A.; Luo, Z.; Sullivan, S.A.; Carlton, J.M.; Escalante, A.A. The Genome of Plasmodium Gonderi: Insights into the Evolution of Human Malaria Parasites. Genome Biology and Evolutioniology and Evolution 2024, 16, evae027. [Google Scholar] [CrossRef]
- Loy, D.E.; Liu, W.; Li, Y.; Learn, G.H.; Plenderleith, L.J.; Sundararaman, S.A.; Sharp, P.M.; Hahn, B.H. Out of Africa: Origins and Evolution of the Human Malaria Parasites Plasmodium Falciparum and Plasmodium Vivax. Int J Parasitol 2017, 47, 87–97. [Google Scholar] [CrossRef]
- Pacheco, M.A.; Escalante, A.A. Origin and Diversity of Malaria Parasites and Other Haemosporida. Trends Parasitol 2023, 39, 501–516. [Google Scholar] [CrossRef] [PubMed]
- Böhme, U.; Otto, T.D.; Cotton, J.A.; Steinbiss, S.; Sanders, M.; Oyola, S.O.; Nicot, A.; Gandon, S.; Patra, K.P.; Herd, C.; et al. Complete Avian Malaria Parasite Genomes Reveal Features Associated with Lineage-Specific Evolution in Birds and Mammals. Genome Res 2018, 28, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Inagaki, S.; Migliardi, M.; Rosato, A. The Binding Mode of ATP Revealed by the Solution Structure of the N-Domain of Human ATP7A. J Biol Chem 2010, 285, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, K.O. The Crystallographic Structure of Na,K-ATPase N-Domain at 2.6Å Resolution. J Mol Biol 2003, 332, 1175–1182. [Google Scholar] [CrossRef]
- Shin, W.-H.; Kihara, D. 55 Years of the Rossmann Fold. Methods in Molecular Biology 2019, 1958, 1–13. [Google Scholar] [CrossRef]
- Wiser, M.F. Protozoa. In Encyclopedia of Biodiversity (Volume 2); Scheiner, S.M.B.T., Ed.; Academic Press: Oxford, 2024; ISBN 978-0-323-98434-8. [Google Scholar]
- Grattepanche, J.-D.; Walker, L.M.; Ott, B.M.; Paim Pinto, D.L.; Delwiche, C.F.; Lane, C.E.; Katz, L.A. Microbial Diversity in the Eukaryotic SAR Clade: Illuminating the Darkness between Morphology and Molecular Data. BioEssays 2018, 40, 1700198. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Chakravarty, D. Intrinsically Disordered Proteins/Regions and Insight into Their Biomolecular Interactions. Biophys Chem 2022, 283, 106769. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically Unstructured Proteins and Their Functions. Nat Rev Mol Cell Biol 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Uversky, V.N.; Kulkarni, P. Intrinsically Disordered Proteins: Chronology of a Discovery. Biophys Chem 2021, 279, 106694. [Google Scholar] [CrossRef]
- Wu, H.; Fuxreiter, M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165, 1055–1066. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Lwin, N.; Phelan, D.; Escalante, A.A.; Battistuzzi, F.U. Comparative Analysis of Low Complexity Regions in Plasmodia. Sci Rep 2018, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Viguera, E.; Canceill, D.; Ehrlich, S.D. Replication Slippage Involves DNA Polymerase Pausing and Dissociation. EMBO J 2001, 20, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.; Nofal, S.D.; McLaughlin, E.J.; Osborne, A.R. Repetitive Sequences in Malaria Parasite Proteins. FEMS Microbiol Rev 2017, 41, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, D.M.; Holemans, T.; van Veen, S.; Martin, S.; Arslan, T.; Haagendahl, I.W.; Holen, H.W.; Hamouda, N.N.; Eggermont, J.; Palmgren, M. Parkinson Disease Related ATP13A2 Evolved Early in Animal Evolution. PLoS One 2018, 13, e0193228. [Google Scholar] [CrossRef]
- Cronin, S.R.; Rao, R.; Hampton, R.Y. Cod1p/Spf1p Is a P-Type ATPase Involved in ER Function and Ca2+ Homeostasis. J Cell Biol 2002, 157, 1017–1028. [Google Scholar] [CrossRef]
- Cohen, Y.; Megyeri, M.; Chen, O.C.W.; Condomitti, G.; Riezman, I.; Loizides-Mangold, U.; Abdul-Sada, A.; Rimon, N.; Riezman, H.; Platt, F.M. The Yeast P5 Type ATPase, Spf1, Regulates Manganese Transport into the Endoplasmic Reticulum. PLoS One 2013, 8, e85519. [Google Scholar] [CrossRef]
- Sørensen, D.M.; Holen, H.W.; Pedersen, J.T.; Martens, H.J.; Silvestro, D.; Stanchev, L.D.; Costa, S.R.; Günther Pomorski, T.; López-Marqués, R.L.; Palmgren, M. The P5A ATPase Spf1p Is Stimulated by Phosphatidylinositol 4-Phosphate and Influences Cellular Sterol Homeostasis. Mol Biol Cell 2019, 30, 1069–1084. [Google Scholar] [CrossRef]
- Feng, Z.; Zhao, Y.; Li, T.; Nie, W.; Yang, X.; Wang, X.; Wu, J.; Liao, J.; Zou, Y. CATP-8/P5A ATPase Regulates ER Processing of the DMA-1 Receptor for Dendritic Branching. Cell Rep 2020, 32. [Google Scholar] [CrossRef]
- van Veen, S.; Sørensen, D.M.; Holemans, T.; Holen, H.W.; Palmgren, M.G.; Vangheluwe, P. Cellular Function and Pathological Role of ATP13A2 and Related P-Type Transport ATPases in Parkinson’s Disease and Other Neurological Disorders. Front Mol Neurosci 2014, 7. [Google Scholar] [CrossRef]
- Sim, S.I.; von Bülow, S.; Hummer, G.; Park, E. Structural Basis of Polyamine Transport by Human ATP13A2 (PARK9). Mol Cell 2021, 81, 4635–4649.e8. [Google Scholar] [CrossRef]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.-P.; Gelders, G.; Lambie, E. ATP13A2 Deficiency Disrupts Lysosomal Polyamine Export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Azfar, M.; van Veen, S.; Houdou, M.; Hamouda, N.N.; Eggermont, J.; Vangheluwe, P. P5B-ATPases in the Mammalian Polyamine Transport System and Their Role in Disease. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2022, 1869, 119354. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, M.; Zhang, S.; Yin, J.; Zhang, P.; Xuan, X.; Wang, P.; Liu, Z.; Zhou, B.; Yang, M. Cryo-EM Structures and Transport Mechanism of Human P5B Type ATPase ATP13A2. Cell Discov 2021, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, D.M.; Holen, H.W.; Holemans, T.; Vangheluwe, P.; Palmgren, M.G. Towards Defining the Substrate of Orphan P5A-ATPases. Biochimica et Biophysica Acta (BBA) - General Subjects 2015, 1850, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, M.; Sørensen, D.M.; Hallström, B.M.; Säll, T.; Broberg, K. Evolution of P2A and P5A ATPases: Ancient Gene Duplications and the Red Algal Connection to Green Plants Revisited. Physiol Plant 2020, 168, 630–647. [Google Scholar] [CrossRef]
- Lamarque, M.; Tastet, C.; Poncet, J.; Demettre, E.; Jouin, P.; Vial, H.; Dubremetz, J.-F. Food Vacuole Proteome of the Malarial Parasite Plasmodium Falciparum. Proteomics Clin Appl 2008, 2, 1361–1374. [Google Scholar] [CrossRef]
- Phillips, M.A. Polyamines in Protozoan Pathogens. J Biol Chem 2018, 293, 18746–18756. [Google Scholar] [CrossRef]
- Niemand, J.; Louw, A.I.; Birkholtz, L.; Kirk, K. Polyamine Uptake by the Intraerythrocytic Malaria Parasite, Plasmodium Falciparum. Int J Parasitol 2012, 42, 921–929. [Google Scholar] [CrossRef]
- Liao, X.; Zhu, W.; Zhou, J.; Li, H.; Xu, X.; Zhang, B.; Gao, X. Repetitive DNA Sequence Detection and Its Role in the Human Genome. Commun Biol 2023, 6, 954. [Google Scholar] [CrossRef]
- Wiser, M.F. Unique Endomembrane Systems and Virulence in Pathogenic Protozoa. Life (Basel) 2021, 11, 822. [Google Scholar] [CrossRef] [PubMed]
- Bullen, H.E.; Crabb, B.S.; Gilson, P.R. Recent Insights into the Export of PEXEL/HTS-Motif Containing Proteins in Plasmodium Parasites. Curr Opin Microbiol 2012, 15, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Hiller, N.L.; Liolios, K.; Win, J.; Kanneganti, T.-D.; Young, C.; Kamoun, S.; Haldar, K. The Malarial Host-Targeting Signal Is Conserved in the Irish Potato Famine Pathogen. PLoS Pathog 2006, 2, e50. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of type-P5 ATPases. The canonical A-domain, N-domain, and P-domain are located on the cytoplasmic face of the membrane along with an N-terminal domain (NTD) and a C-terminal extension (CTE) of variable length. The M-domain (gray background) consists of six core transmembrane (cTM) helices, four supporting transmembrane (sTM) helices, and membrane helices associated with the NTD. Subtype-P5A ATPases have two N-terminal transmembrane helices (nTM) and subtype-P5B has an N-terminal membrane loop (nML) formed by three short helices. Subtype-P5A ATPases have a helical projection from the P-domain that is not found in subtype-P5B ATPases called the “arm”. ATP binding occurs in a crevice between the N-domain and P-domain called the hinge. The transmembrane helix of cTM4 is disrupted by conserved prolines that results in a ‘kink’ in the helix (between 4a and 4b) which plays an essential role in the formation of the substrate-binding groove. Similar nomenclature and color schemes are used in all subsequent figures. A single subtype-P5A ATPase (PlP5A) and two subtype-P5B ATPases (ATPase1 and ATPase3) have been identified in genomes of malaria parasites. PlP5A has a single exon and ATPase1 and ATPase3 have two exons with the position of the intron denoted by a blue arrow near sTM9. PlP5A and ATPase3 have N-terminal extensions that are not found in other type-P5 ATPases. Alignment of PlP5A sequences reveals seven variable regions (VR1-7 green) composed of low-complexity sequence which are denoted with green arrows and lettering. ATPase1 and ATPase3 exhibit four variable regions (VR1-4), three of which are shared with PlP5A, composed of low-complexity sequence and are denoted with blue arrows and lettering.
Figure 1.
Schematic representation of type-P5 ATPases. The canonical A-domain, N-domain, and P-domain are located on the cytoplasmic face of the membrane along with an N-terminal domain (NTD) and a C-terminal extension (CTE) of variable length. The M-domain (gray background) consists of six core transmembrane (cTM) helices, four supporting transmembrane (sTM) helices, and membrane helices associated with the NTD. Subtype-P5A ATPases have two N-terminal transmembrane helices (nTM) and subtype-P5B has an N-terminal membrane loop (nML) formed by three short helices. Subtype-P5A ATPases have a helical projection from the P-domain that is not found in subtype-P5B ATPases called the “arm”. ATP binding occurs in a crevice between the N-domain and P-domain called the hinge. The transmembrane helix of cTM4 is disrupted by conserved prolines that results in a ‘kink’ in the helix (between 4a and 4b) which plays an essential role in the formation of the substrate-binding groove. Similar nomenclature and color schemes are used in all subsequent figures. A single subtype-P5A ATPase (PlP5A) and two subtype-P5B ATPases (ATPase1 and ATPase3) have been identified in genomes of malaria parasites. PlP5A has a single exon and ATPase1 and ATPase3 have two exons with the position of the intron denoted by a blue arrow near sTM9. PlP5A and ATPase3 have N-terminal extensions that are not found in other type-P5 ATPases. Alignment of PlP5A sequences reveals seven variable regions (VR1-7 green) composed of low-complexity sequence which are denoted with green arrows and lettering. ATPase1 and ATPase3 exhibit four variable regions (VR1-4), three of which are shared with PlP5A, composed of low-complexity sequence and are denoted with blue arrows and lettering.
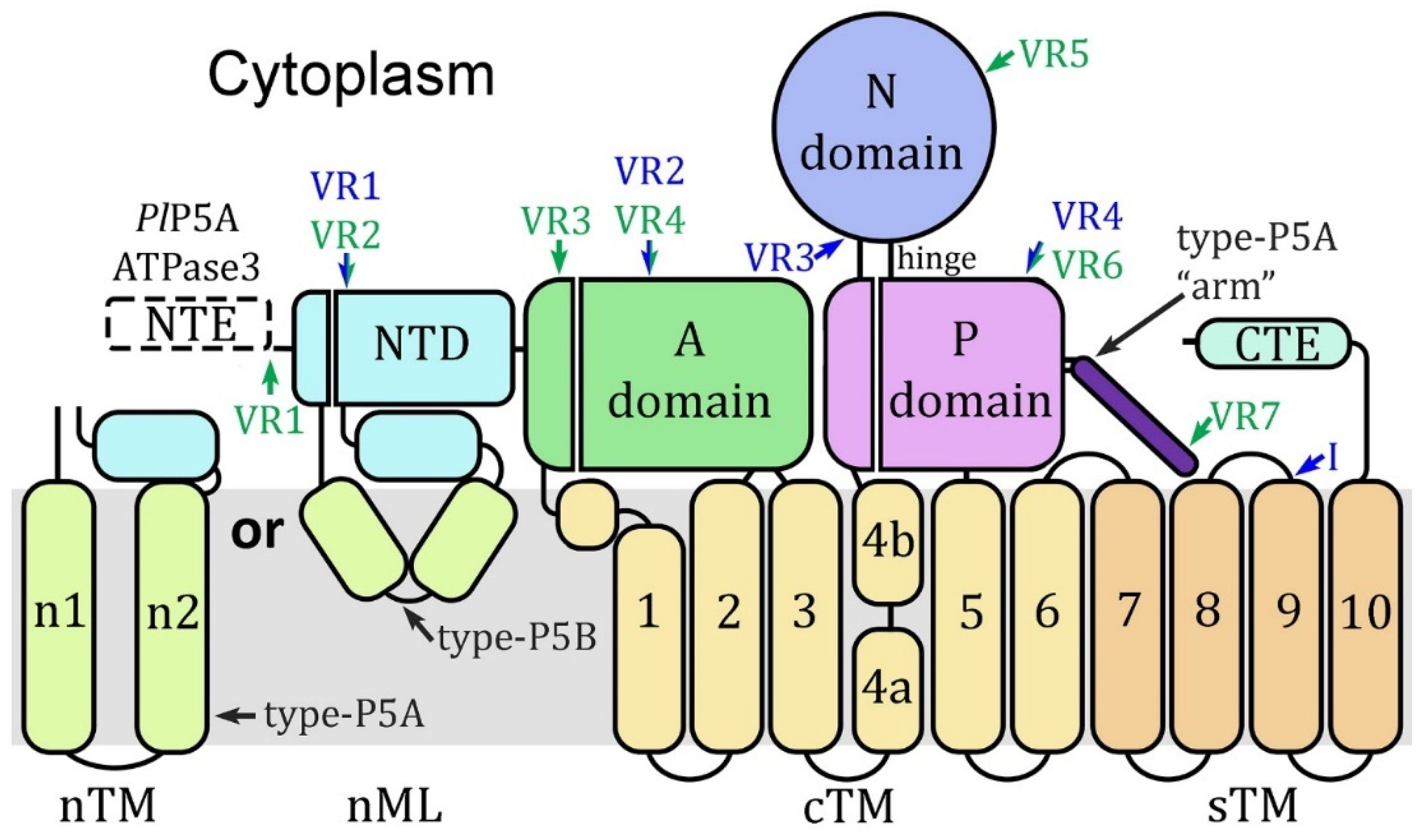
Figure 2.
Homology modeling with the Spf1 template. Plasmodium subtype-P5A, ATPase1 (A1), and ATPase3 (A3) sequences from P. falciparum (Pf) and P. relictum (Pr) were modeled by Swiss Model® with the Spf1 (PDB Acc. No. 6xmu) template. Spf1* is the experimentally determined structure and concordant (c) and discordant (d) models are noted. Dashed boxes denote missing elements in the modeled structures. The ‘arm’ (arrow, dark purple) is an α-helix that is specific to subtype-P5A. The A-domain is colored green, the N-domain blue, the P-domain purple, the transmembrane helices yellow, and the NTD aqua with the membrane associated loops light green. The intrinsically disordered loops (IDL) produced by the variable regions are salmon. The white asterisk (*) denotes the α-helix substrate (dark green) in the substrate-binding groove.
Figure 2.
Homology modeling with the Spf1 template. Plasmodium subtype-P5A, ATPase1 (A1), and ATPase3 (A3) sequences from P. falciparum (Pf) and P. relictum (Pr) were modeled by Swiss Model® with the Spf1 (PDB Acc. No. 6xmu) template. Spf1* is the experimentally determined structure and concordant (c) and discordant (d) models are noted. Dashed boxes denote missing elements in the modeled structures. The ‘arm’ (arrow, dark purple) is an α-helix that is specific to subtype-P5A. The A-domain is colored green, the N-domain blue, the P-domain purple, the transmembrane helices yellow, and the NTD aqua with the membrane associated loops light green. The intrinsically disordered loops (IDL) produced by the variable regions are salmon. The white asterisk (*) denotes the α-helix substrate (dark green) in the substrate-binding groove.
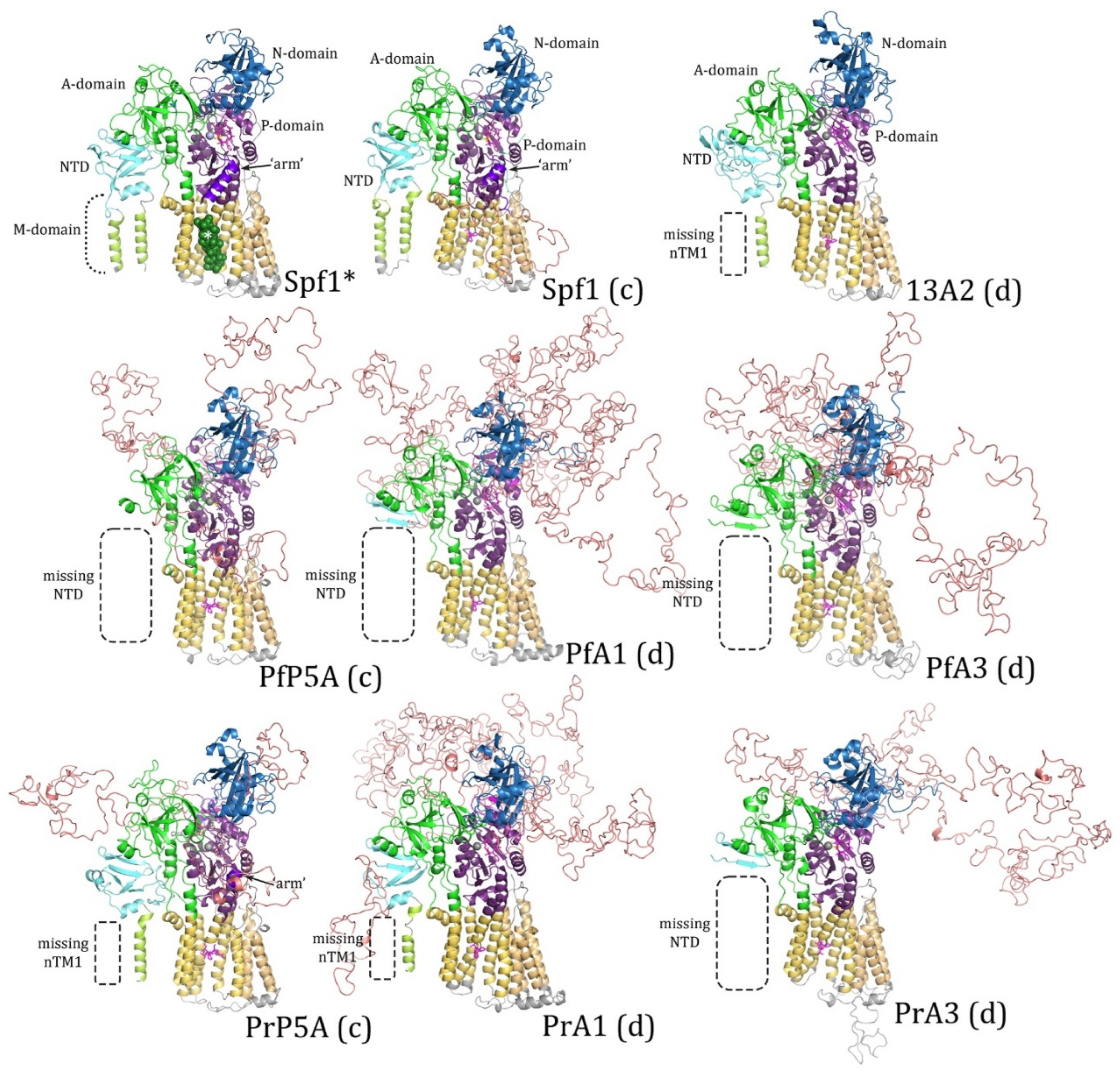
Figure 3.
Homology modeling with the ATP13A2 template. Plasmodium subtype-P5A, ATPase1 (A1), and ATPase3 (A3) sequences from P. falciparum (Pf) and P. relictum (Pr) were modeled by Swiss Model® with the ATP13A2 (PDB Acc. No. 7m5x) template. 13A2* is an experimentally determined structure and concordant (c) and discordant (d) models are noted. Dashed boxes denote missing elements in the modeled structures. The A-domain is colored green, the N-domain blue, the P-domain purple, the transmembrane helices yellow, and the NTD aqua with the membrane associated loops light green. IDL produced by the variable regions are colored salmon. The arrow denotes the spermine substrate in the substrate-binding groove.
Figure 3.
Homology modeling with the ATP13A2 template. Plasmodium subtype-P5A, ATPase1 (A1), and ATPase3 (A3) sequences from P. falciparum (Pf) and P. relictum (Pr) were modeled by Swiss Model® with the ATP13A2 (PDB Acc. No. 7m5x) template. 13A2* is an experimentally determined structure and concordant (c) and discordant (d) models are noted. Dashed boxes denote missing elements in the modeled structures. The A-domain is colored green, the N-domain blue, the P-domain purple, the transmembrane helices yellow, and the NTD aqua with the membrane associated loops light green. IDL produced by the variable regions are colored salmon. The arrow denotes the spermine substrate in the substrate-binding groove.
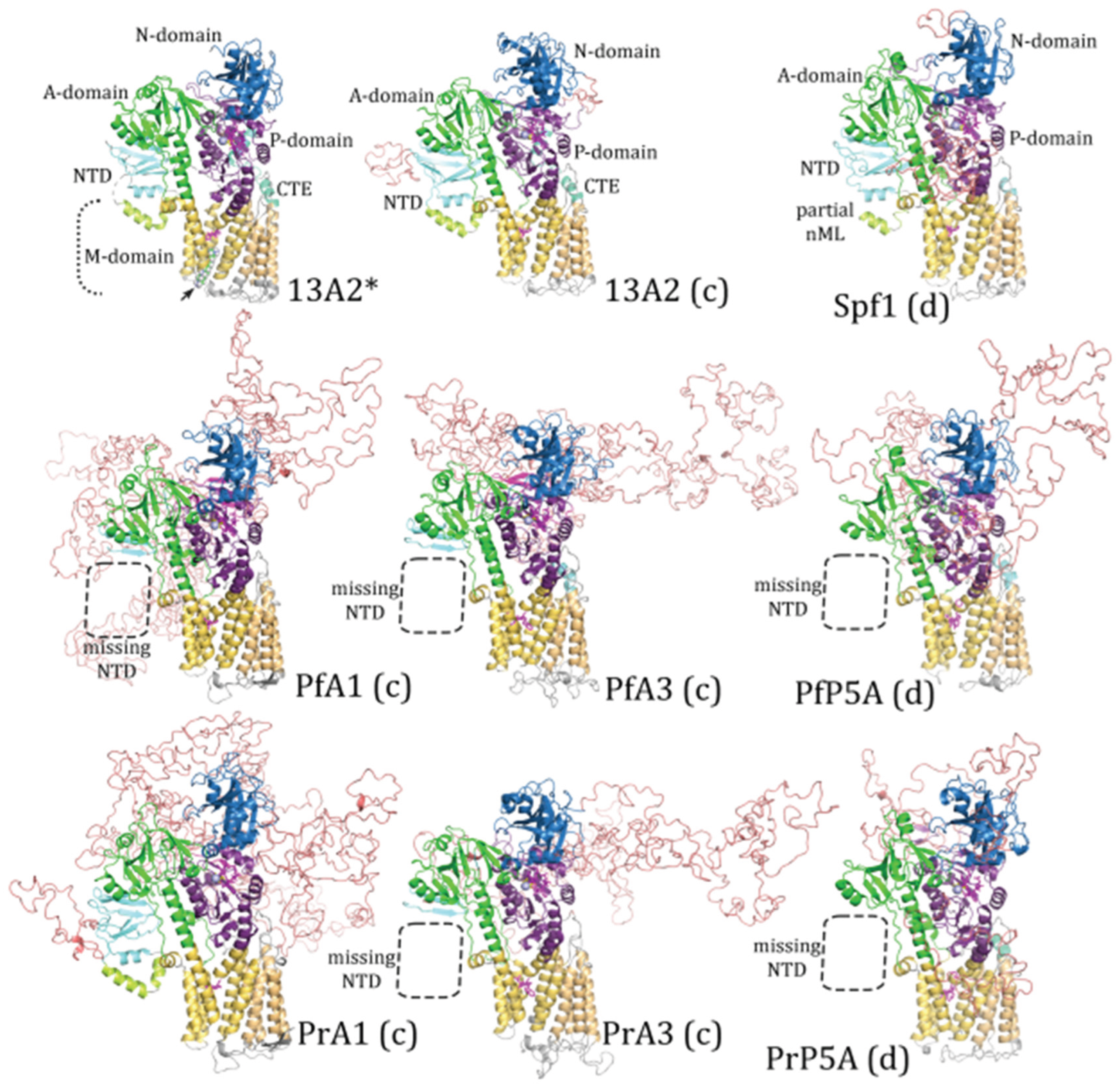
Figure 4.
A-domain of type-P5 ATPases. (a) Shown are the experimentally determined structures of the A-domain with the eight β-strands (b1-8) of the distorted jelly roll and the three associated α-helices (h1-3) denoted. Beta-strand-6 in ATP13A2 is colored differently as well as the corresponding segment in Spf1 (circled). The circled short β-strand (b6’) in teal is found in experimentally determined structures but none of the modeled structures (Figure S3). The blue annotations refer to differences in the Plasmodium type-P5 ATPases modeled with either template. (b) The sequence alignment corresponding to the distorted jelly roll with the β-strands boxed. Additional β-strands (b’) and helices (h’ and h”) are denoted. The green segment denotes a region of intrinsically disordered sequence that corresponds to variable region 2 of ATPase1/3 or variable region 4 of PlP5A. Black or gray shading indicates identical or similar residues, respectively..
Figure 4.
A-domain of type-P5 ATPases. (a) Shown are the experimentally determined structures of the A-domain with the eight β-strands (b1-8) of the distorted jelly roll and the three associated α-helices (h1-3) denoted. Beta-strand-6 in ATP13A2 is colored differently as well as the corresponding segment in Spf1 (circled). The circled short β-strand (b6’) in teal is found in experimentally determined structures but none of the modeled structures (Figure S3). The blue annotations refer to differences in the Plasmodium type-P5 ATPases modeled with either template. (b) The sequence alignment corresponding to the distorted jelly roll with the β-strands boxed. Additional β-strands (b’) and helices (h’ and h”) are denoted. The green segment denotes a region of intrinsically disordered sequence that corresponds to variable region 2 of ATPase1/3 or variable region 4 of PlP5A. Black or gray shading indicates identical or similar residues, respectively..

Figure 5.
N-domain of Spf1 and ATP13A2. (a) Shown are the experimentally determined structures of the N-domain with the six β-strands (b1-6) and four associated α-helices (h1-4) denoted. Two extra short β-strands (b*) are observed in Spf1 which are not modeled in any of the other ATPases (Figure S4). Similarly, two extra β-strands (b’ and b”) are observed in ATP13A2 which are not modeled in the Plasmodium ATPases. (b) The sequence alignment corresponding to the region of the extra β-strands in relation to helix-1, helix-2, and β-strand-2 (boxed). Black or gray shading indicates identical or similar residues, respectively.
Figure 5.
N-domain of Spf1 and ATP13A2. (a) Shown are the experimentally determined structures of the N-domain with the six β-strands (b1-6) and four associated α-helices (h1-4) denoted. Two extra short β-strands (b*) are observed in Spf1 which are not modeled in any of the other ATPases (Figure S4). Similarly, two extra β-strands (b’ and b”) are observed in ATP13A2 which are not modeled in the Plasmodium ATPases. (b) The sequence alignment corresponding to the region of the extra β-strands in relation to helix-1, helix-2, and β-strand-2 (boxed). Black or gray shading indicates identical or similar residues, respectively.
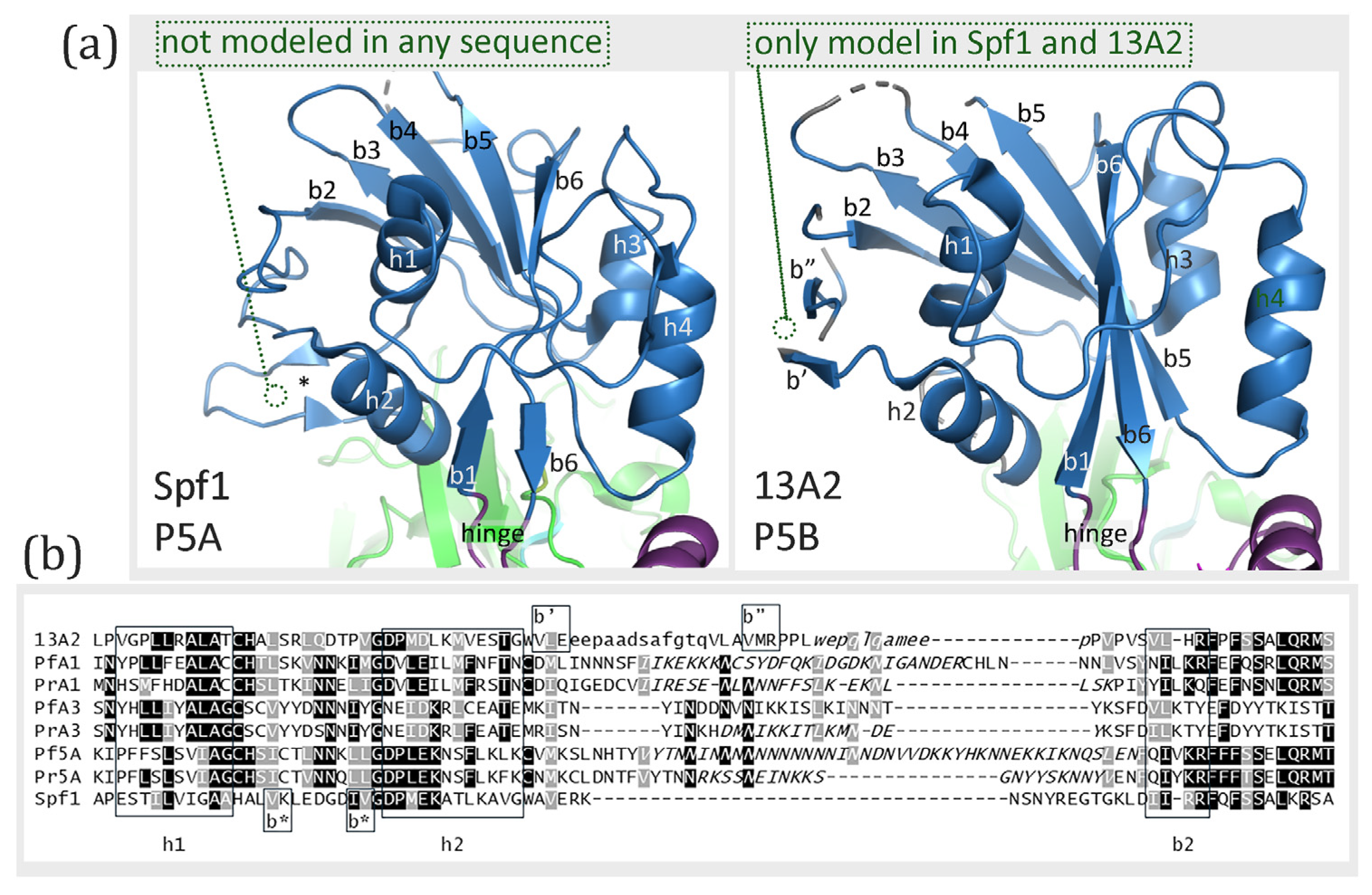
Figure 6.
Homology modeling of the P-domain with the Spf1 template. 3-dimensional structures of the P-domain from Spf1, ATP13A2, and the six Plasmodium type-P5 ATPases were generated with the Spf1 template (6xmu) and compared to the experimentally determined structure of Spf1 (*). Subtype-P5A sequences are designated as concordant (c) and subtype-P5B are designated as discordant (d). Eight parallel β-strands (b1-8) form a wavy β-sheet that is flanked by two anti-parallel β-strands (b’ and b”). The α-helices include two helices that continue from cTM4 (S4) and cTM5 (S5) and form stalks (S), six helices of the modified Rossmann fold (h1-6), and an additional helix (h’). Missing secondary structural elements are denoted with dashed circles and secondary elements derived from variable regions are magenta. The “h3” (arrow) denotes a region of random coil that has a helical shape (yellow) in ATP13A2.
Figure 6.
Homology modeling of the P-domain with the Spf1 template. 3-dimensional structures of the P-domain from Spf1, ATP13A2, and the six Plasmodium type-P5 ATPases were generated with the Spf1 template (6xmu) and compared to the experimentally determined structure of Spf1 (*). Subtype-P5A sequences are designated as concordant (c) and subtype-P5B are designated as discordant (d). Eight parallel β-strands (b1-8) form a wavy β-sheet that is flanked by two anti-parallel β-strands (b’ and b”). The α-helices include two helices that continue from cTM4 (S4) and cTM5 (S5) and form stalks (S), six helices of the modified Rossmann fold (h1-6), and an additional helix (h’). Missing secondary structural elements are denoted with dashed circles and secondary elements derived from variable regions are magenta. The “h3” (arrow) denotes a region of random coil that has a helical shape (yellow) in ATP13A2.
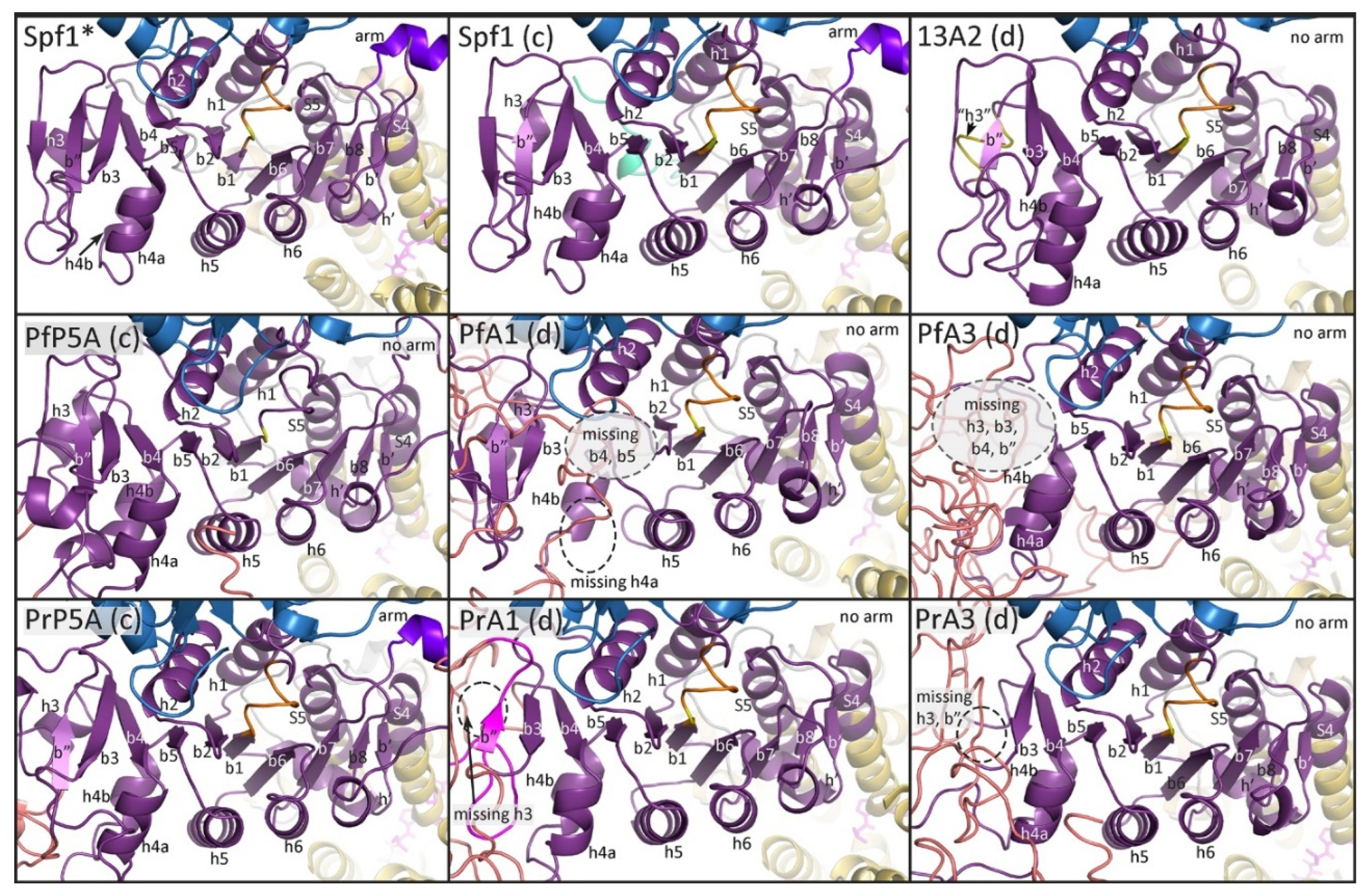
Figure 7.
Homology modeling of the P-domain with the ATP13A2 template. 3-dimensional structures of the P-domain from Spf1, ATP13A2, and the six Plasmodium type-P5 ATPases were generated with the ATP13A2 template (7m5x) and compared to the experimentally determined structure of ATP13A2 (*). Subtype-P5B sequences are designated as concordant (c) and subtype-P5A are designated as discordant (d). Eight parallel β-strands (b1-8) form a wavy β-sheet that is flanked by two anti-parallel β-strands (b’ and b”). The α-helices include two helices that continue from cTM4 (S4) and cTM5 (S5) and form stalks (S), five helices of the modified Rossmann fold (h1-2, h4-6), and an additional helix (h’). Missing secondary structural elements are denoted with dashed circles and secondary elements derived from variable regions are magenta. The no h3 box highlights that α-helix-3 is not found in ATP13A2, nor in any of the models generated with ATP13A2 as the template.
Figure 7.
Homology modeling of the P-domain with the ATP13A2 template. 3-dimensional structures of the P-domain from Spf1, ATP13A2, and the six Plasmodium type-P5 ATPases were generated with the ATP13A2 template (7m5x) and compared to the experimentally determined structure of ATP13A2 (*). Subtype-P5B sequences are designated as concordant (c) and subtype-P5A are designated as discordant (d). Eight parallel β-strands (b1-8) form a wavy β-sheet that is flanked by two anti-parallel β-strands (b’ and b”). The α-helices include two helices that continue from cTM4 (S4) and cTM5 (S5) and form stalks (S), five helices of the modified Rossmann fold (h1-2, h4-6), and an additional helix (h’). Missing secondary structural elements are denoted with dashed circles and secondary elements derived from variable regions are magenta. The no h3 box highlights that α-helix-3 is not found in ATP13A2, nor in any of the models generated with ATP13A2 as the template.
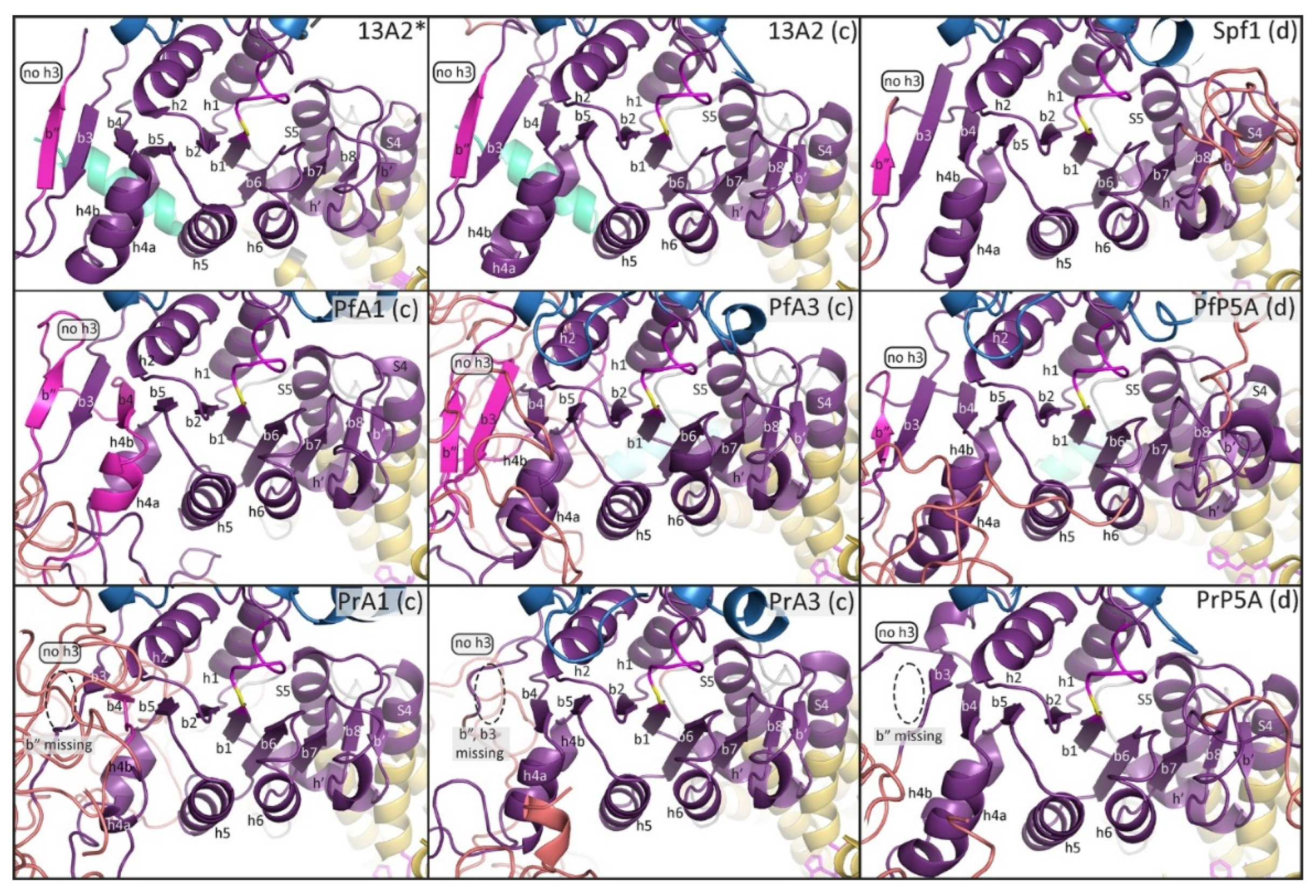
Figure 8.
P-domain alignment. The secondary structural elements of the P-domain (Figure 6 and Figure 7) are boxed (green dashes) and labeled as β-strands (b#), α-helices (h#), or stalks (s#). In the low homology region between h2 and h4b only the experimentally determined secondary structures are boxed. The yellow shading represents cTM4 and cTM5 which flank the P-domain; the blue shading represents the N-domain; the red shading represents VR6 in Pl5A or VR4 in ATPase1/3; and the dark blue shading between b7 and b8 represents the arm and associated IDL (VR7) of the subtype-P5A ATPases. Sequences in lower case are excluded from the experimentally-determined 3-dimensional structures and italicized sequence are IDL..
Figure 8.
P-domain alignment. The secondary structural elements of the P-domain (Figure 6 and Figure 7) are boxed (green dashes) and labeled as β-strands (b#), α-helices (h#), or stalks (s#). In the low homology region between h2 and h4b only the experimentally determined secondary structures are boxed. The yellow shading represents cTM4 and cTM5 which flank the P-domain; the blue shading represents the N-domain; the red shading represents VR6 in Pl5A or VR4 in ATPase1/3; and the dark blue shading between b7 and b8 represents the arm and associated IDL (VR7) of the subtype-P5A ATPases. Sequences in lower case are excluded from the experimentally-determined 3-dimensional structures and italicized sequence are IDL..

Figure 9.
Substrate-binding sites of Plasmodium type-P5 ATPases. (a) Predicted structures of the substrate binding sites of the Plasmodium type-P5 ATPases modeled with the Spf1 (6xmu) or ATP13A2 (7m5x) templates. The top row shows the experimentally determined structures of Spf1 or ATP13A2 with (+) or without their respective alpha-helix (α) or spermine (SPM) substrates. Other rows show ATPase1 (A1), ATPase3 (A3), or subtype-P5A from P. falciparum (Pf) and P. relictum (Pr) modeled with concordant (c) or discordant (d) templates. The dashed yellow line is the distance between cTM2 and cTM6 measured in angstroms (Å) demonstrating the width of the substrate groove. Sidechains of the amino acids making up the kink in cTM4 are shown and colored magenta. Sidechains of amino acids that may interact with the α-helical substrate of subtype-P5A are shown and colored light blue for nonpolar residues and light green for polar residues, except for the proline in the kink. Sidechains making up the tetrad that stabilizes the substrate groove of subtype-P5B are shown and colored orange if conserved with ATP13A2 or yellow if not conserved. The sidechains of the two aspartate residues that may interact with the polyamine substrate are shown and colored red. Nearby negatively-charged sidechains are also colored red and nearby positively charged residues are colored blue. (b) Alignment of sequences making up the substrate-binding groove. Carets (^) below the alignment denote residues that bind to substrate (b) in subtype-P5A, make up the stabilizing tetrad (t) in subtype-P5B, or provide negative charges (-) for polyamine binding in subtype-P5B. Shading corresponds to the colors used in the homology models. The |-> <-| denotes the residues used to determine the width of the substrate-binding groove. Three dashes (---) denote removed sequence.
Figure 9.
Substrate-binding sites of Plasmodium type-P5 ATPases. (a) Predicted structures of the substrate binding sites of the Plasmodium type-P5 ATPases modeled with the Spf1 (6xmu) or ATP13A2 (7m5x) templates. The top row shows the experimentally determined structures of Spf1 or ATP13A2 with (+) or without their respective alpha-helix (α) or spermine (SPM) substrates. Other rows show ATPase1 (A1), ATPase3 (A3), or subtype-P5A from P. falciparum (Pf) and P. relictum (Pr) modeled with concordant (c) or discordant (d) templates. The dashed yellow line is the distance between cTM2 and cTM6 measured in angstroms (Å) demonstrating the width of the substrate groove. Sidechains of the amino acids making up the kink in cTM4 are shown and colored magenta. Sidechains of amino acids that may interact with the α-helical substrate of subtype-P5A are shown and colored light blue for nonpolar residues and light green for polar residues, except for the proline in the kink. Sidechains making up the tetrad that stabilizes the substrate groove of subtype-P5B are shown and colored orange if conserved with ATP13A2 or yellow if not conserved. The sidechains of the two aspartate residues that may interact with the polyamine substrate are shown and colored red. Nearby negatively-charged sidechains are also colored red and nearby positively charged residues are colored blue. (b) Alignment of sequences making up the substrate-binding groove. Carets (^) below the alignment denote residues that bind to substrate (b) in subtype-P5A, make up the stabilizing tetrad (t) in subtype-P5B, or provide negative charges (-) for polyamine binding in subtype-P5B. Shading corresponds to the colors used in the homology models. The |-> <-| denotes the residues used to determine the width of the substrate-binding groove. Three dashes (---) denote removed sequence.
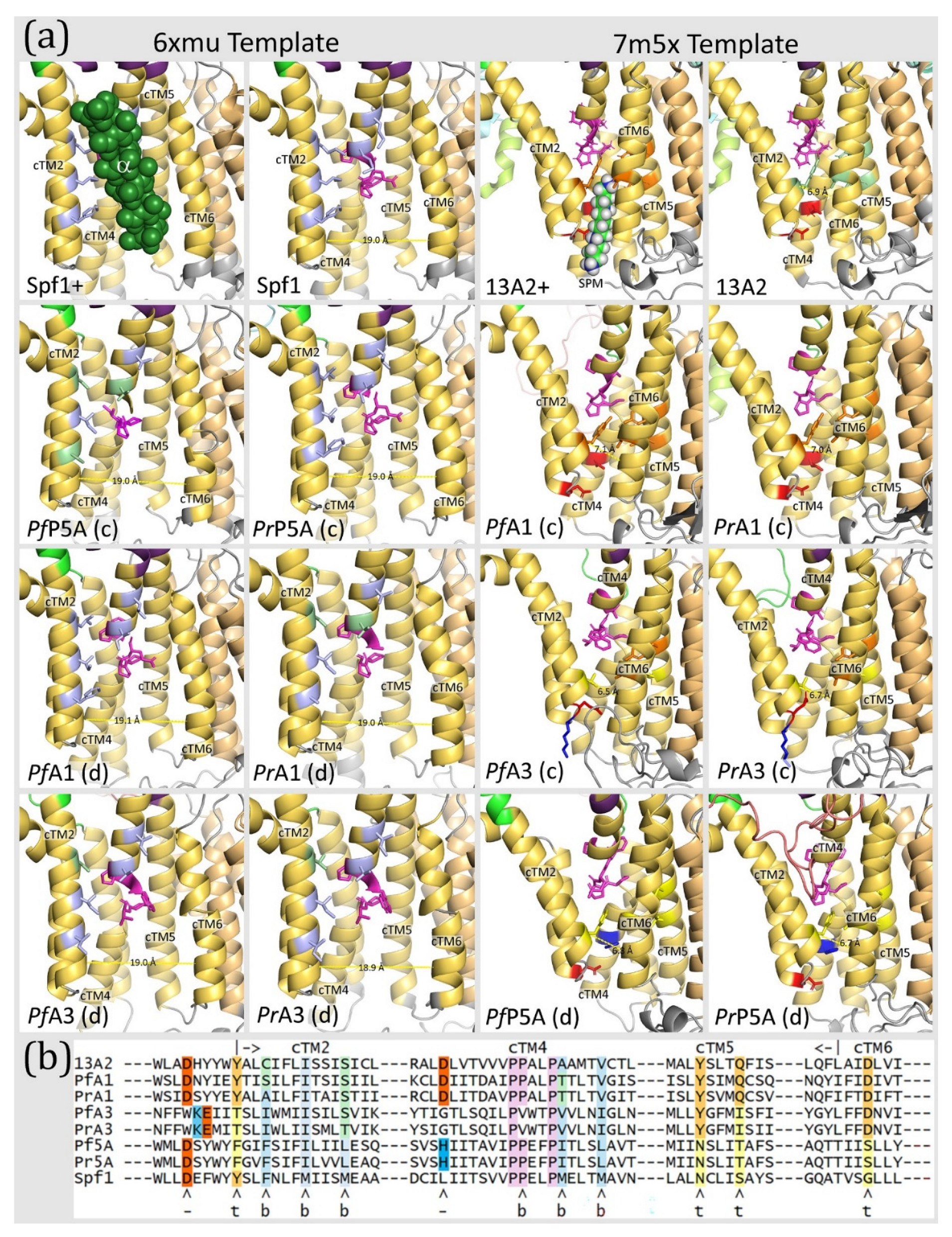

Table 1.
Sequences used in detailed sequence and structure analysis.
| Protein | Species | Abbr | Gene ID | Activity/Description | Ref |
|---|---|---|---|---|---|
| Spf1 | Saccharomyces cerevisiae | Spf1 | AAB64508.1 | Transmembrane helix dislocase | [9] |
| Uncharacterized subtype-P5A | Plasmodium falciparum | PfP5A | PF3D7_0727800 | Only identified in sequence databases | [12,28] |
| Uncharacterized subtype-P5A | Plasmodium relictum | PrP5A | PRELSG_0216200 | ||
| ATP13A2 | Homo sapiens | 13A2 | NP_071372.1 | Polyamine transporter | [11] |
| ATPase3 | Plasmodium falciparum | PfA3 | PF3D7_0504000 | Apicomplexan subtype-P5B of unknown substrate specificity | [13] |
| ATPase3 | Plasmodium relictum | PrA3 | PRELSG_1028500 | ||
| ATPase1 | Plasmodium falciparum | PfA1 | PF3D7_0516100 | Paralogue of ATPase3 only found in Laverania and avian Haemosporida | |
| ATPase1 | Plasmodium relictum | PrA1 | PRELSG_1015800 |
Table 2.
Sizes and locations of low complexity inserts in type-P5 ATPases1.
| Domain | NTE | NTD | A1 | A2 | N1 | N2 | P1 | P2 | Total |
|---|---|---|---|---|---|---|---|---|---|
| P5B-VR | VR1 | VR2 | VR3 | VR4 | |||||
| 13A2 | n.a. | 23 | 0 | 0 | 0 | 0 | 56 | 0 | 79 (7%) |
| PfA1 | n.a. | 126 | 0 | 168 | 281 | 37 | 664 | 0 | 1276 (53%) |
| PrA1 | n.a. | 45 | 0 | 136 | 145 | 0 | 681 | 0 | 1007 (48%) |
| PfA3 | 0 | 288 | 0 | 127 | 478 | 0 | 327 | 0 | 1220 (51%) |
| PrA3 | 0 | 89 | 0 | 52 | 332 | 0 | 280 | 0 | 753 (39%) |
| P5A-VR | VR1 | VR2 | VR3 | VR4 | VR5 | VR6 | VR7 | ||
| PfP5A | 79 | 25 | 180 | 107 | 2 | 147 | 158 | 103 | 801 (42%) |
| PrP5A | 79 | 24 | 119 | 15 | 2 | 43 | 152 | 91 | 525 (32%) |
| Spf1 | n.a. | 0 | 0 | 0 | 0 | 0 | 65 | 119 | 184 (15%) |
1The variable regions of subtype P5A and subtype P5B ATPases are colored green and blue respectively to match Figure 1. Between the NTE and NTD there is in insert (VR1) only found in subtype-P5A. The insert in the NTD is in the middle of an anti-parallel β-sheet and corresponds to variable region 1 (VR1) of subtype-P5B and VR2 of subtype-P5A. The first insert in the A-domain (A1) is located near the NTD/A-domain junction and is specific for subtype-P5A. The second A-domain insert (A2) is located between β-strand-5 and β-strand-6 of the distorted jelly roll. The first insert in the N-domain (N1) is VR3 of subtype-P5B and is located near the first P-domain/N-domain junction. Another N-domain insert is found between β-strand-3 and β-strand-4 of the twisted β-sheet of the N-domain (N2). The first insert in the P-domain (P1) is located between β-strand-3 and helix-3 of the Rossmann fold. The second P-domain insert (P2) is located after the ‘arm’ that is exclusive to subtype-P5A. Shown are the sizes of the variable regions expressed as number of amino acids and the percentage of the total amino acids found in the variable regions.
Table 3.
Pairwise distances between Plasmodium type-P5 ATPases, Spf1 (subtype-P5A), and ATP13A2 (subtype-P5B)1.
Table 3.
Pairwise distances between Plasmodium type-P5 ATPases, Spf1 (subtype-P5A), and ATP13A2 (subtype-P5B)1.
| 13A2 | PfA1 | PrA1 | PfA3 | PrA3 | Pf5A | Pr5A | |
|---|---|---|---|---|---|---|---|
| PfA1 | 1.32 | ||||||
| PrA1 | 1.24 | 0.42 | |||||
| PfA3 | 1.49 | 1.39 | 1.41 | ||||
| PrA3 | 1.56 | 1.40 | 1.40 | 0.13 | |||
| Pf5A | 1.43 | 1.37 | 1.38 | 1.51 | 1.47 | ||
| Pr5A | 1.45 | 1.36 | 1.40 | 1.48 | 1.44 | 0.25 | |
| Spf1 | 1.39 | 1.44 | 1.41 | 1.59 | 1.58 | 1.14 | 1.16 |
1Pairwise distances were generated with Mega XI from a ClustalW alignment with the variable regions removed. Concordant alignments are shaded in green and discordant alignments are shaded in red. The comparison of orthologues from P. falciparum and P. relictum are boxed. The distance between the Spf1 and ATP13A2 templates is underlined and the distances between the Plasmodium subtype-P5A ATPases and Spf1 are in bold..
Table 4.
Quality assessment of the predicted 3-dimensional structures of Plasmodium type-P5 ATPases1.
Table 4.
Quality assessment of the predicted 3-dimensional structures of Plasmodium type-P5 ATPases1.
| 6xmu (aHS, BeF, Mg) | 6xmq (ACP, Mg) | 7m5x (spm, BeF, Mg) | 7m5v (ANP, Mg) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATPase | GM | QM | RF | Ligand | GM | QM | RF | Ligand | GM | QM | RF | Ligand | GM | QM | RF | Ligand |
| Spf1 | 0.72 | 0.78 | 92.4 | Mg | 0.78 | 0.82 | 95.5 | ACP, Mg | 0.47 | 0.55 | 86.4 | Mg | 0.50 | 0.59 | 86.6 | Mg |
| ATP13A2 | 0.54 | 0.59 | 88.9 | Mg | 0.54 | 0.60 | 89.4 | Mg | 0.67 | 0.71 | 91.9 | BeF, Mg | 0.69 | 0.74 | 91.8 | ANP, Mg |
| PfP5A | 0.27 | 0.50 | 81.6 | BeF, Mg | 0.28 | 0.50 | 80.2 | Mg | 0.19 | 0.46 | 82.9 | BeF, Mg | 0.20 | 0.46 | 80.6 | Mg |
| PrP5A | 0.39 | 0.51 | 83.5 | Mg | 0.39 | 0.50 | 84.1 | Mg | 0.27 | 0.48 | 82.9 | Mg | 0.29 | 0.51 | 86.9 | Mg |
| PfA1 | 0.12 | 0.37 | 73.8 | Mg | 0.13 | 0.38 | 73.7 | Mg | 0.11 | 0.38 | 69.4 | BeF, Mg | 0.12 | 0.39 | 72.2 | - |
| PrA1 | 0.20 | 0.38 | 76.0 | Mg | 0.18 | 0.38 | 76.7 | - | 0.18 | 0.38 | 72.7 | Mg | 0.18 | 0.38 | 73.0 | Mg |
| PfA3 | 0.15 | 0.41 | 75.5 | Mg | 0.15 | 0.41 | 74.0 | Mg | 0.13 | 0.41 | 72.9 | Mg | 0.14 | 0.43 | 75.4 | Mg |
| PrA3 | 0.24 | 0.41 | 79.3 | Mg | 0.24 | 0.41 | 78.3 | Mg | 0.21 | 0.41 | 77.2 | Mg | 0.21 | 0.42 | 78.0 | Mg |
| PfP5Avrr | 0.54 | 0.58 | 91.4 | Mg | 0.54 | 0.58 | 91.6 | Mg | not analyzed | |||||||
| PrP5Avrr | 0.54 | 0.59 | 91.2 | Mg | 0.54 | 0.59 | 91.2 | Mg | ||||||||
| PfA1vrr | not analyzed | 0.49 | 0.53 | 89.6 | BeF, Mg | 0.51 | 0.55 | 91.0 | - | |||||||
| PrA1vrr | 0.52 | 0.56 | 87.1 | BeF, Mg | 0.54 | 0.56 | 90.4 | - | ||||||||
| PfA3vrr | 0.46 | 0.52 | 86.7 | Mg | 0.47 | 0.53 | 89.3 | Mg | ||||||||
| PrA3vrr | 0.49 | 0.52 | 88.0 | Mg | 0.49 | 0.53 | 89.7 | Mg | ||||||||
1The top row shows the PDB files used as templates to model the sequences in the first column. The features in the parentheses () are the ligands associated with the crystallized protein. Ligands include an α-helix substrate (aHS), spermine (spm), non-hydrolysable ATP analogs (either adenosine 5′-[β,γ-methylene]triphosphate, ACP, or β,γ-imidoadenosine 5′-triphosphate, ANP), BeF3 (mimics the phosphorylated state), or Mg2+. The second row are the quality assessments and include GMQE (GM), QMEANDisCo (QM), percentage of Ramachandran favorable and Θ angles (RF), and the ligands retained in the model. Dashes (-) indicate no ligand was retained in the model. Concordance or discordance with the prototype ATPases are denoted with light green or red shading, respectively. For the Plasmodium ATPases concordance and discordance are denoted with blue and gray shading respectively. Analysis of the Plasmodium type-P5 ATPases with the variable regions removed (vrr) was also carried out with concordant templates.
Table 5.
The effect of removing variable regions on discrepancies in the modeling of Plasmodium subtype-P5B ATPases1.
Table 5.
The effect of removing variable regions on discrepancies in the modeling of Plasmodium subtype-P5B ATPases1.
| 6xmu Template (discordant) | 7m5x Template (concordant) | |||||||
|---|---|---|---|---|---|---|---|---|
| Domain | ATPase | VR | Discrepancy | VRR Effect | Discrepancy | VRR Effect | ||
| NTD (VR1) | PfA1 | 126 | missing NTD | partial restoration of nTM2 | + | missing NTD | restoration of nML | + |
| PrA1 | 45 | missing nTM1 | none | 0 | none | none | 0 | |
| PfA3 | 288 | missing NTD | missing NTD | 0 | missing NTD | partial restoration of nML | + | |
| PrA3 | 89 | missing NTD | partial restoration of nTM2 | + | missing NTD | partial restoration of nML | + | |
| A (VR2) |
PfA1 | 168 | none | none | 0 | extra -strand | loss of extra -strand | + |
| PrA1 | 136 | none | none | 0 | none | none | 0 | |
| PfA3 | 127 | none | none | 0 | none | none | 0 | |
| PrA3 | 52 | none | none | 0 | none | none | 0 | |
| N (VR3) |
PfA1 | 278 | none | none | 0 | none | none | 0 |
| PrA1 | 153 | none | none | 0 | none | none | 0 | |
| PfA3 | 469 | none | none | 0 | none | none | 0 | |
| PrA3 | 323 | none | none | 0 | none | none | 0 | |
| P (VR4) |
PfA1 | 558 | b4 and b5 missing, extra helix derived from VR4 | b4 and b5 restored, loss of extra helix | + | b4 and h3 generated from VR4, extra b-strand derived from VR4 | b4 and h3 generated from expected sequence, loss of extra b-strand | + |
| PrA1 | 568 | extra b-strand derived from VR4 | loss of extra b-strand | + | none | none | 0 | |
| PfA3 | 217 | b3 and b4 missing | b3 and b4 restored | + | b3 generated from VR4, extra b-strand derived from VR4 | b3 generated from expected sequence, loss of extra b-strand | + | |
| PrA3 | 172 | none | b3 missing | - | b3 missing | additional loss of b4 and b5 | - | |
1Based on Figure S5 and Figure 3, Figure 4, Figure 5, Figure 6, Figure 7 and Figure 8. The b# and h# refer to specific β-strands and α-helices respectively found in the domains. VR = sizes of variable regions in that domain. The effects of variable regions are rated as positive (+) if removing the variable regions improved the predicted structure in regard to recapitulating the experimentally determined structures, neutral (0) if removing the variable regions have no effect, and negative (-) if removing the variable regions led to a loss of expected secondary structures.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



