
Submitted:
16 September 2025
Posted:
18 September 2025
You are already at the latest version
Abstract
PDAC remains one of the most lethal cancer malignancies, characterized by a highly desmoplastic ECM that promotes an immunosuppressive signaling network and dampens the effectiveness of traditional therapies. Among the several key contributors to its immune evasion pathways are chemokine signaling axes, which orchestrate the recruitment of regulatory immune cell populations, promote metastasis, and remodel the TME in favor of tumor progression. This review comprehensively examines the roles of major CCR-CCL signaling pathways—primarily focusing on the CCR2–CCL2, CCR5–CCL5, CCR4–CCL17/22, CCR6–CCL20, and CCR7–CCL19/21 axes—in PDAC development, detailing their expression patterns, immunologic impact, and downstream signaling mechanisms and outcomes. We further detail past and ongoing therapeutic efforts and trials addressing these axes in both PDAC and relevant non-PDAC settings via several small-molecule antagonists and monoclonal antibodies: BMS-813160, Maraviroc, Leronlimab, FLX475, PF-07054894, IDOR-1117-2520, and CAP-100. Despite continuous advances in the field, the current body of evidence still remains limited and presents significant research gaps in areas such as spatial profiling, stage-specific analyses, and general mechanistic validation in PDAC-specific settings. Addressing these shortcomings will be key to obtaining a more comprehensive knowledge of the field and improve future therapeutic strategies to overcome PDAC.
Keywords:
pancreatic ductal adenocarcinoma (PDAC)
; tumor microenvironment (TME)
; Extracellular matrix (ECM)
; immune evasion
; chemokine signaling
; CCR2–CCL2
; CCR5–CCL5
; CCR4–CCL17/22
; CCR6–CCL20
; CCR7–CCL19/21
; tumor-associated macrophages (TAMs)
; chemokine receptor antagonists
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers worldwide, ranking sixth among the leading causes of cancer-related mortalities [1]. In 2022 alone, PDAC was responsible for over 510,000 new cases and roughly 467,000 deaths worldwide with incidence rates projected to increase by 95.4% by 2050 [1]. The overall five-year survival rate for all stages of pancreatic cancer in 2011 was 4.2%, and only marginal improvements have come to fruition from the advances made in chemotherapy and immunotherapy in recent years [2,3]. While early-stage diagnoses—stages IA and IB—have seen modest survival gains from 47% to 75% and from 38% to 68% between 2004 and 2015, respectively, over 80% of patients still present with advanced or metastatic disease, where median survival drops to a few months [2,4]. Surgical resection remains the only potentially curative intervention, yet only around 20% of patients at diagnosis are eligible for resection due to the extent of local invasion or remote metastasis [5,6]. This aggressive progression is tightly linked to the immunosuppressive PDAC tumor microenvironment (TME), characterized by dense desmoplasia, regulatory cell infiltration, and a signaling network that collectively suppress anti-tumor responses [3,7,8].
Among these various signaling groups is the CC chemokine family, constituting one of the four principal chemokine subfamilies (CXC, CC, XC, CX3C) distinguished by the arrangement of conserved cysteine residues near the N-terminus [9,10,11]. Chemokines are small, secreted proteins that play essential roles in chemotaxis—the directed migration of immune cells such as monocytes/macrophages, T cells, and dendritic cells—through concentration gradients. CC chemokine receptor-ligand (CCR-CCL) interactions also play pivotal roles in immune cell polarization and the maintenance of immune homeostasis by directing leukocyte trafficking, although there have been implications of direct influence from chemokine signaling [9,10]. The CC chemokine family includes 27 known ligands (CCL1 to CCL28, with CCL9 and CCL10 representing the same gene product) and 10 known receptors (CCR1 to CCR10) [9,10]. Interactions within this predominantly non-exclusive ligand–receptor family (Figure 1)—with the exception of a few binding pairs—create an intricate signaling network leading to diverse downstream outcomes that extend beyond simple chemotaxis to contain immune cell activation, differentiation, and survival [9,10,12].
In the context of cancer, CC chemokines exert both anti-tumor and pro-tumor effects, though their influence is frequently skewed toward promoting tumor progression [13,14]. This pro-tumoral activity is mediated via several mechanisms, including the recruitment of immunosuppressive cell populations, support for cancer-associated fibroblasts (CAFs) and endothelial cell function, and the facilitation of angiogenesis, ECM remodeling and fibrosis, and metastasis [15,16,17,18,19]. However, it is important to note that factors like tumor stage or the immediate surrounding stromal context may cause the downstream outcomes of a specific CCR-CCL axis to differ significantly. Factors such as hypoxia, an inflammatory cytokine milieu, and/or abundant stromal signals, which are especially pronounced in PDAC, can influence the expression and activity of chemokines and their receptors, leading to context-dependent effects which will be discussed in more detail below [17,20,21]. Several CCR-CCL axes—including but not limited to CCR2–CCL2, CCR5–CCL5, CCR4–CCL17/22, CCR6–CCL20, and CCR7–CCL19/21—have been identified as drivers in the recruitment and functional modulation of several different cell populations in PDAC [15,19,22,23,24,25].
2. Upstream Sources and Induction of CCR-CCL Axes in PDAC
PDAC arises through a well-defined multistep progression involving the accumulation of genetic and epigenetic alterations in pancreatic ductal cells. The earliest and most common initiating event is an activating mutation in the KRAS oncogene, most frequently at codon 12 (G12D or G12V), which is present in over 90% of low-grade PanIN lesions—the principal noninvasive precursors to PDAC [26,27]. The near-universal presence of KRAS mutations creates a permissive landscape for downstream chemokine signaling, driving persistent activation of signaling cascades including MAPK/ERK and PI3K/AKT, which collectively enhance NF-B and HIF-1 transcriptional activity of chemokine genes [28,29,30,31]. Mutant KRAS can also activate NF-B via TAK1-mediated phosphorylation of IKK[32]. KRAS-induced positive feedback loops, such as IL-6 and TGF- secretion, activate JAK-STAT3 and SMAD pathways—also upstream to chemokine transcription and heavy stromal remodeling characteristic to PDAC [33,34,35]. It is important to note here that while the neoplastic pancreatic ductal cells themselves may not produce all the chemokine populations—though they do produce some—the cytokines whose downstream expression are driven by KRAS oncogene mutations are responsible for coordinating chemokine upregulation via the previously described cascades [36]. This genetic-epigenetic synergy between KRAS-mutant neoplastic cells, stromal cells, and dysregulated signaling pathways establishes a chemokine-rich milieu that drives PDAC progression, metastasis, and therapeutic resistance.

Table 1.
Upstream Pathways and Sources of CC Chemokines in the PDAC TME. Cell types and intracellular pathways associated with CCL expression. Asterisks denote cell types or signals for which CCL induction has been reported only in non-PDAC contexts, under ambiguous circumstances, or where findings were inconclusive.
Table 1.
Upstream Pathways and Sources of CC Chemokines in the PDAC TME. Cell types and intracellular pathways associated with CCL expression. Asterisks denote cell types or signals for which CCL induction has been reported only in non-PDAC contexts, under ambiguous circumstances, or where findings were inconclusive.
| Ligand | Cellular Sources | Upstream Pathways |
|---|---|---|
| CCL2 | Basal-like tumor cells, CAFs (iCAFs, CAFs*) | NF-B, STAT3, AP-1, HIF-1, PPAR |
| CCL5 | Tumor cells, Granulocytes, Dendritic cells, TAMs, CD T cells, CD T cells, NK cells, Monocytes, CAFs* | NF-B, STAT3, FOXP3, CD73, AP-1* |
| CCL17 | Dendritic cells, M2a TAMs* | TSLP, CD40/CD40L*, STAT6*, GM-CSF*, NF-B*, AP-1* |
| CCL22 | Dendritic cells, M2a TAMs* | TSLP, IL-1, CD40/CD40L*, PU.1*, IL-4/IL-13*, NF-B*, AP-1* |
| CCL20 | Senescent tumor cells, TAMs, Dendritic cells*, Eosinophils* | NF-B, IL-17B/IL-17RB, AP-1*, C/EBP*, SP1*, EGFR/Ras* |
| CCL19 | TLS fibroblasts (rCAFs*) | Noncanonical p52/RelB NF-B, LTR, AP-1* |
| CCL21 | TLS fibroblasts (rCAFs*) | Noncanonical p52/RelB NF-B, LTR* |
2.1. CCL2-Specific Expresssion Origins
One prominent example of a chemokine that drives PDAC progression is CCL2, also known as monocyte chemoattractant protein-1 (MCP-1). CCL2 is abundantly secreted by basal-like tumor cells and CAFs—influenced upstream by signals from mutant KRAS oncogene bearing tumor cells [37,38]. Among these CAF subtypes, iCAFs stand out in particular by being able secrete CCL2 at levels exceeding 5000 pg/mL compared to a baseline lack of CCL2 expression in normal pancreatic tissues, placing it significantly higher than the typical 1300–1800pg/mL and 500–700 pg/mL levels observed in breast or prostate cancer, respectively [39,40,41,42]. Another significant CAF subtype— CAFs—has also exhibited higher CCL2 activation in aggressive liver cancer models, and given their tightly shared desmoplastic traits, this mechanism is likely conserved in PDAC as well [35].
Intracellularly, several key transcription factors support CCL2 expression. The NF-B and STAT3 pathway—the latter triggered by IL-6 transignaling across diverse tumor settings—serves as central drivers, not only for CCL2 but for a broad range of other chemokine axes as well [30,34,43]. In basal-like PDAC tumor cells, BRD4-mediated cJun/AP-1 expression has also been implicated in sustaining CCL2 levels and its basal-like neoplastic state [38]. Likewise, HIF-1—a protein stabilised under the hypoxic conditions resulting from poorly developed vasculature and dense desmoplasia found in PDAC—further prompts CCL2 transcription [20,31]. The lipid-rich nature of the pancreas adds a unique layer, as sustained activation of PPARs induces their binding to PPREs within the CCL2 promoter, amplifying CCL2 expression [44].
2.2. CCL5-Specific Expresssion Origins
CCL5—also known as Regulated upon Activation, Normal T Cell Expressed and Secreted (RANTES)—is also a notably overactive chemokine in PDAC, and particularly pancreatic tumor cells, granulocytes, and dendritic cells (DCs) appear to be a major source of its production [36]. However, additional evidence shows that CCL5 is also produced by tumor-associated macrophages (TAMs), CD T cells, CD T cells, NK cells, and monocytes, though not nearly at the level of the tumor cells, granulocytes, and DCs [36]. Translation evidence from an ovarian cancer model also suggests CAFs can secrete CCL5 as well [45]. Thus, understanding what drives CCL5 production in PDAC requires considering a relatively greater range of tumor and stromal cell populations.
Many of the transcription factors that mediate CCL5 expression in PDAC overlap with those regulating CCL2. Mentioned previously, both NF-B and STAT3 are consistently observed as primary regulators of CCL5 in both general and pancreatic contexts [30,46,47]. On the other hand, while cJun/AP-1-mediated CCL5 promotion has not been explicitly examined in the context of PDAC, it has shown to be an upstream CCL5 activator in breast cancer settings, hinting at potential cross-tumor relevance [48]. FOXP3—a transcription factor expressed by both regulatory T cells (Tregs) and certain cancer cells—can directly bind to the CCL5 promoter as well, enhancing its transcription and promoting the recruitment of CCR Tregs into the tumor bed [16,36,49]. Uniquely, CD73 has demonstrated CCL5-mediated Treg recruitment via p38-STAT1 signaling [36]. These findings imply a positive feedback system whereby CCL5 enhances Treg recruitment and establishes a Treg-FOXP3-CCL5-Treg signaling loop that maintains immunosuppression in PDAC. More targeted research is warranted before any definitive claims can be drawn.
2.3. CCL17/22-Specific Expresssion Origins
CCL17 and CCL22—also known as thymus and activation-regulated chemokine (TARC) and macrophage-derived chemokine (MDC), respectively—are primarily secreted by DCs and macrophages in general settings [50,51]. In the context of PDAC however, literature evidence remains focused on DC derived CCL17/22 upregulation, particularly emphasizing DCs influenced by factors originating from tumor cells and fibroblasts [52,53]. On the other hand, macrophages/TAMs—while not explicitly mentioned—were implied via observations supporting CCL22 origins from immune cells of myeloid phenotype [52]. Among macrophage subtypes, M2a phenotypes—largely known for their key role in fibrosis, a major element PDAC desmoplasia, and characterized by their activation via Th2 cytokines IL-4 and IL-13—seem to be the most relevant in the context of CCL17 and CCL22 upregulation [54,55,56,57].
Several common intercellular signals drive the expression of CCL17 and CCL22, such as CD40L and TSLP via tumor cells and DCs, respectively [53,58]. However, while the CD40–CD40L pathway has been observed in follicular lymphoma, its relevance to PDAC remains hypothetical at best, warranting more tailored investigation [58]. Despite shared inducers, the transcriptional routes of CCL17 and CCL22 can diverge. CCL17 depends primarily on IL-4-mediated STAT6 activation and GM-CSF signaling—although IL-4/IL-13 has also shown simultaneous CCL22 upregulation [56,59,60]. In contrast, CCL22 is regulated by IL-1 and PU.1 [50,52]. This allows for context-dependent regulation, yet both chemokines are frequently co-expressed in tumor tissues, particularly within myeloid cell populations constitutively exhibiting both STAT6 and PU.1 [61,62]. NF-B and AP-1 are likely central mediators of CCL17/22 transcription in PDAC as well. Although clear PDAC-specific evidence is lacking, studies from other disease contexts show that NF-B and AP-1 driven expression of these chemokines is a common mechanism that likely stretches to PDAC [59,63,64].
2.4. CCL20-Specific Expresssion Origins
CCL20—also known as macrophage inflammatory protein-3 (MIP-3)—originates primarily from pancreatic tumor cells and M2 TAMs [65,66,67]. Among tumor cell subtypes however, senescent pancreatic tumor cells show the highest expression levels of CCL20, driven as part of the cytokine-rich senescence-associated secretory phenotype (SASP) [23]. On the other hand, M2 TAMs typically rely on IL-4 signaling to induce CCL20, while other cell types—such as DCs and eosinophils—have also displayed CCL20 secretion, however these observations have been restricted to non-PDAC settings, pointing out the need for pancreas-specific models [66,67].
Multiple intracellular and intercellular pathways regulate CCL20 expression. NF-B continues to be a key chemokine mediator in PDAC and plays a central role in promoting CCL20 levels [68]. Additionally, the IL-17B–IL-17RB signaling axis has also demonstrated CCL20 upregulation in a murine model, where subsequent IL-17RB inhibition resulted in reduced metastasis, highlighting roles of CCR6–CCL20 in PDAC invasion [21]. Separately, transcription factors AP-1, C/EBP, and SP1—likely upregulated via KRAS-driven p38 MAPK activity—have all also demonstrated binding to CCL20 promoter regions [21,69,70,71]. Across several different cancer types—breast, colon, head and neck, and melanoma—EGFR-Ras-induced CCL20 expression has also been observed in murine models, suggesting this pathway may stretch to pancreatic contexts as well [72].
2.5. CCL19/21-Specific Expresssion Origins
CCL19 and CCL21—also known as macrophage inflammatory protein-3 (MIP-3) and secondary lymphoid tissue chemokine (SLC), respectively—are primarily produced and active by fibroblast populations surrounding TME driven tertiary lymphoid structures (TLSs) and lymph vessels [73,74]. Among these different fibroblast populations, a scRNA-seq analysis on a stromal cell dataset in breast cancer models show CCL19 and CCL21 production particularly enriched in the reticular-like CAF (rCAF) subset, compared to the seven other CAF subtypes classified in the study [75]. These CAF subtypes, including rCAFs, have also been identified in PDAC as well [75].
The intracellular regulation of CCL19 and CCL21 expression is largely mediated by NF-B signaling, especially through the noncanonical p52/RelB NF-B pathway, which has been shown to constitutively drive CCL19 and CCL21 expression in pancreatic cancer [76]. Other signaling elements lymphotoxin- receptor (LTR) and AP-1—in coordination with NF-B—also enhanced CCR7, CCL19, and CCL21 expression, supported by explicit preliminary PDAC evidence and/or translational models [73,77,78,79].
3. CCR-CCL Axes in PDAC
3.1. Immune Cell Recruitment and Polarization
In the PDAC TME, chemokines orchestrate the selective recruitment of immune cell subsets that predominantly reinforce immunosuppression. Monocyte infiltration—a hallmark of the PDAC TME—is primarily mediated by the CCR2–CCL2 and CCR5–CCL5 axes. Currently, PDAC literature mainly emphasizes CCR2–CCL2-induced monocyte chemotaxis, whereas studies on CCR5–CCL5-mediated monocyte recruitment in PDAC are lacking, although a breast cancer study has confirmed this function, suggesting a likely supplementary role in the context of PDAC [15,80]. However, while the CCR2–CCL2 axis remains restricted to monocytes—although under radiotherapy conditions, CCL2 has also been shown to recruit Tregs in HNSCC—CCR5–CCL5 exerts slightly broader immunoregulatory influence, also facilitating a portion of Treg infiltration and potentially recruiting CAFs as shown in a PDAC and ESCC model, respectively [16,81,82]. General Treg accumulation, however, is most strongly associated with CCR4–CCL17/22 signaling, yet relevant studies examining PDAC-specific CCL17 remain absent, whereas the CCR4–CCL22 axis demonstrates a much clearer line of evidence—likely more focused on due to its binding superiority to CCR4 over CCL17 [52,83,84,85,86]. The CCR6–CCL20 axis may provide secondary support for Treg recruitment, while the CCR7–CCL19 pathway has been implicated in Treg chemotaxis in PDAC, based on findings translated from gastric cancer models [25,87]. Notably, although CCR4 is classically linked to Treg migration, recent patient-derived data in PDAC reveal an unconventional CCR4–CCL2 interaction that mediates monocytic myeloid derived suppressor cell (M-MDSC) recruitment [88].
Although limited in effect, several CCR-CCL axes also contribute to the recruitment of anti-tumor immune cells in the PDAC TME. The CCR7–CCL19/21 axis—particularly the CCR7–CCL21 interaction—is strongly associated with the infiltration of effector populations, including CD T cells, DCs, NK cells, and NKT cells, likely around lymphoid structures [74,89]. However, these cells are likely rendered dysfunctional by comparatively higher levels of immunoinhibitory signals in the primary TME characteristic to PDAC—including, but not limited to, PD-1, CTLA-4, IL-10, and TGF- [90,91]. Additional CCR-CCL interactions offer only minor contributions to anti-tumor infiltration: the CCR4–CCL17/22 axis can attract Th2 cells, though this is likely outweighed by its dominant role in Treg trafficking; similarly, the CCR6–CCL20 axis may recruit Th17 cells under inflammatory conditions, but this potential is suppressed by the immunologically ’cold’ nature of PDAC [87,92,93].
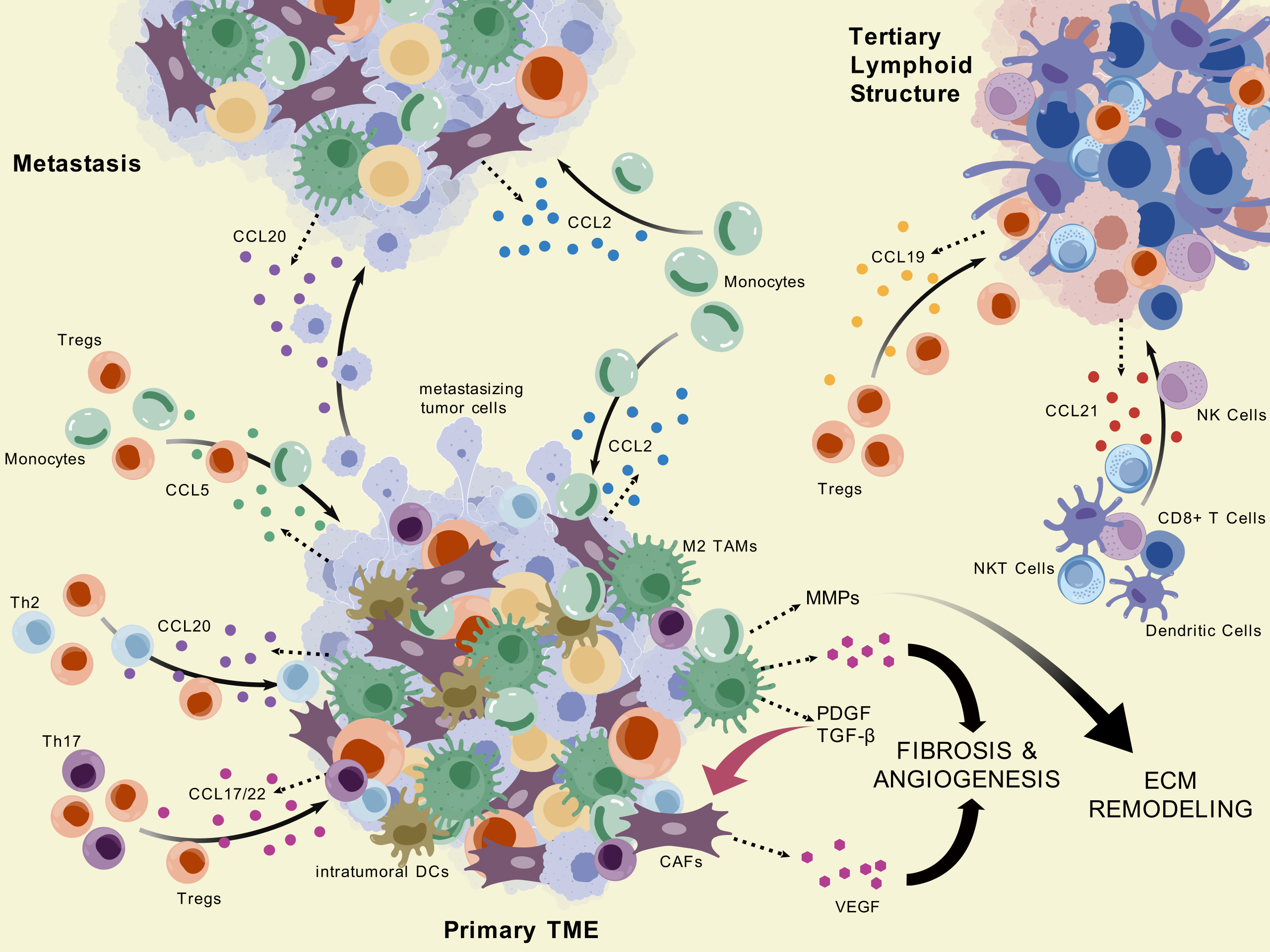
Figure 2.
CC Chemokine-Mediated Regulation of Immune and Stromal Interactions in PDAC. This diagram highlights the roles of CC chemokines across the primary tumor microenvironment, metastatic sites, and tertiary lymphoid structures in PDAC, illustrating their involvement in cell recruitment, tumor progression, and immune organization. Visualization generated with BioGDP.com [94].
Figure 2.
CC Chemokine-Mediated Regulation of Immune and Stromal Interactions in PDAC. This diagram highlights the roles of CC chemokines across the primary tumor microenvironment, metastatic sites, and tertiary lymphoid structures in PDAC, illustrating their involvement in cell recruitment, tumor progression, and immune organization. Visualization generated with BioGDP.com [94].
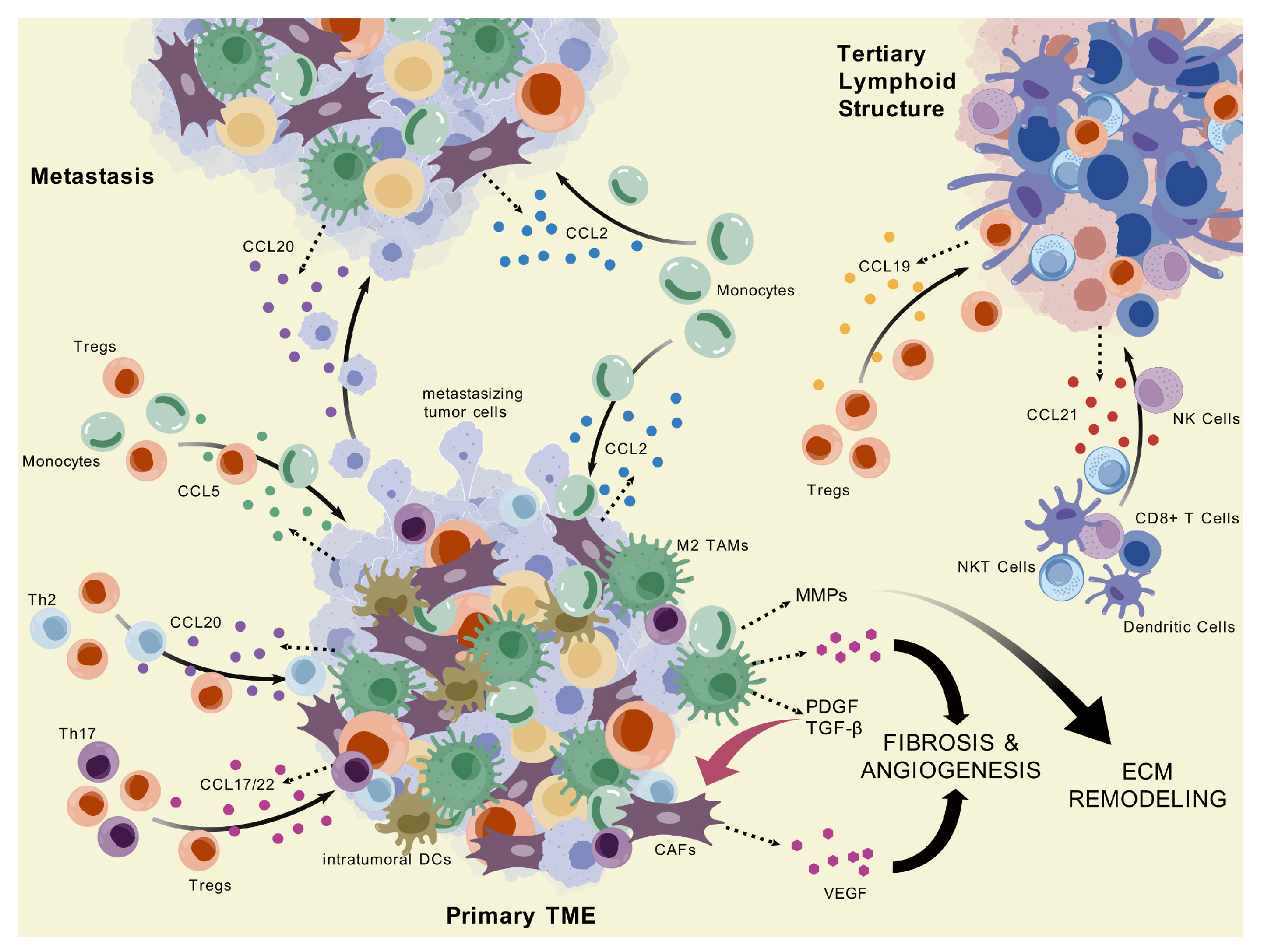
Beyond recruitment, CCR-CCL signaling axes play key roles in shaping the immunosuppressive characteristic of the PDAC TME by driving the polarization of both myeloid and lymphoid cells. M2 macrophage polarization is promoted directly by mutant KRAS-mediated downstream signals, such as elevated levels of GM-CSF with lactate and REG4 which converge onto AKT-mediated cascades [95,96]. MDSC polarization—which often produces overlapping results with TAMs—appears to rely mainly on GM-CSF signaling [97]. Among the previously covered chemokine axes, CCR2–CCL2 and CCR5–CCL5 are implicated in inducing both M-MDSC and M2 TAM phenotypes largely in part due to their function in monocyte recruitment—the shared progenitor of M-MDSCs and TAMs [98,99]. Signaling axes CCR6–CCL20 and CCR7–CCL19/21 have both also demonstrated M2-polarizing effects via either explicit observations in PDAC or translational evidence from other solid cancer studies [23,100]. The CCR6–CCL20 axis has also shown context-dependent effects on Treg plasticity. In the context of surrounding tissue inflammation, CCR6 expression in conjunction with RORγ transcriptional programming can facilitate the conversion of iTregs into Th17 cells, suggesting a dynamic immunomodulatory potential, although whether RORγ Tregs represent a population with reduced suppressive capacity remains unclear and appears to depend on additional contextual factors [91,101,102]. However, as the TME advances toward a more immunosuppressive cytokine profile—marked by high levels of IL-10 and TGF-—surrounding signals contribute to Treg stabilization, effectively suppressing Th17 differentiation and reinforcing the dominance of regulatory phenotypes [91].
3.2. TME Remodeling
3.2.1. Angiogenesis and Fibrosis
Angiogenesis and fibrosis are hallmarks of TME remodeling, and CCR-CCL signaling axes play a central supporting role in inducing VEGF-driven vascular growth. M2 TAMs—recruited and activated via CCL2—serve as major secretors of VEGF and other angiogenic factors like FGF as shown [15,103,104]. Under this logic, the other previously mentioned chemokine axes—CCR5–CCL5, CCR4–CCL17/22, CCR6–CCL20, and CCR7–CCL19/21—may further contribute to VEGF secretion through their variable influences on macrophage polarization and/or recruitment, though direct causal evidence remains limited [23,24,99,105]. In parallel, the CCR10–CCL28 axis mediates the migration of PSCs into the TME, where the CCL2-induced M2 TAMs also secrete signals like PDGF and TGF- which activate these recruited PSCs—which have been known to be the primary CAF precursor [17,106]. In recent years, however, the notion of PSC dominance among CAF precursor populations has been challenged as new studies demonstrate PSCs as a rather minor subset among other distinct precursor populations such as Gli cells [107,108]. Nevertheless, PSCs and CAFs have both demonstrated VEGF production—particularly in response to HIF-1 presence as a result of chronic hypoxia—and secretion of fibrotic components such as collagen, reflecting their significant role as key mediators of TME fibrosis [109,110].
Interestingly, CCR10–CCL28-mediated PSC migration to the PDAC TME was shown to be inflammation-driven, implying that PSCs tend to accumulate in peritumoral regions compared to central desmoplastic zones, which are characterized by a more anti-inflammatory cytokine milieu—an inference supported by spatial distribution analysis [17,111,112]. Peritumoral aggregation of PSCs and CAFs may represent a key driver of outward tumor expansion, as these cells prime the surrounding stroma for invasion by promoting fibrotic and angiogenic remodeling that facilitates tumor cell spread—an interconnected mechanism that warrants tailored investigation. Notably, MDSCs have also been linked to VEGF production in various tumor models, linking MDSC chemotaxis and polarization contributing axes—predominantly CCR2–CCL2—as upstream influencers of this observation although studies connecting CC chemokine axes to MDSC-mediated VEGF production in PDAC specifically remains unexplored [98,99,113]. The same literature pattern is seen in Tregs, suggesting that the CCR4–CCL17/22 axis, and to a lesser extent CCR5–CCL5 or CCR10–CCL28, may significantly influence angiogenesis by facilitating the recruitment of Tregs, although VEGF secretion has not been specified [16,84,114].
Beyond immune cell-driven mechanisms, tumor cells themselves have shown VEGF secretion in response to CCL5 via bone cancer models [115,116]. However, this pathway remains to be validated in the context of PDAC. In contrast, CCR7–CCL21 and VEGF-C expression has shown high correlation explicitly in PDAC and has been implicated in promoting both angiogenesis and lymphangiogenesis—supported by its association with elevated microvessel and lymphatic vessel densities, although concrete mechanistic studies have yet to happen [19,117]. Despite these insights, PDAC-specific evidence for CCR-CCL-driven angiogenesis and fibrosis remains preliminary, where much literature draws from data obtained in broader tumor studies and predominantly reveal correlative findings rather than mechanistic data, thus highlighting the need for more targeted research in PDAC.
3.2.2. Matrix Metalloproteinases
MMPs are key mediators of ECM degradation in PDAC, regulated in part by CC chemokine signaling. In one study, high incidence rates of MMP2, MMP7, and MMP9 expression (79.31%, 55.17%, 24.13%, respectively) among 29 patients have been observed in the PDAC stroma compared to normal tissue regions—all of which demonstrated a relative lack of these same signals (3.45%, 6.90%, 0%, respectively) [118]. A different investigation utilizing single cell RNA sequencing analysis revealed that CCR2/CCL macrophages directly facilitated MMP9 promotion in PDAC [119]. Likewise, CD40-mediated CCL2 and IFN- upregulation promoted the recruitment of Ly6 inflammatory monocytes, which subsequently secreted MMP10, MMP12, and MMP13 [120]. Given that IFN- is a well-characterized pro-inflammatory cytokine, this observation may be especially relevant to peritumoral regions as well, where the surrounding cytokine milieu is comparatively more inflammatory and may require ECM breakdown and subsequent immunosuppressive conditioning prior to outwards tumor invasion—similar to the potential peritumoral PSC/CAF aggregation pathway described previously. This proposed regional relationship remains to be validated. Separately, in vitro PANC-1 cell cultures stimulated by CCL20 and CCL21 have shown a specific upregulation of MMP9 [65,121]. Beyond these observations, additional PDAC-specific experiments investigating CCR-CCL-induced downstream MMP production remain scarce, however many translational liver cancer and HCC models have shown several cases of this general pathway—particularly highlighting MMP2 and MMP9 over other MMP types [122,123,124,125]. More broadly, CCR-CCL driven MMP induction has also been reported in lung, ovarian, prostate, breast, and colon cancer studies [126,127,128,129,130]. But based on current literature consensus, the CXCR4–CXCL12 axis seems more widely supported as the primary mediator of MMP induction in PDAC [131,132,133]. However, because this axis lies outside the scope of the present review, it will not be discussed in further detail here. Taken together, while there is a growing pool of preliminary evidence suggesting that CCR-CCL signaling causes MMP upregulation in PDAC, this field lacks mechanistic proof. Most studies remain associative, leaving a critical gap in our understanding of PDAC ECM remodeling which holds eventual implications for metastasis as well, a topic explored in more detail below.
3.2.3. Tertiary Lymphoid Structures
The formation of TLSs within the PDAC TME is increasingly recognized as a critical mediator of anti-tumor immunity. TLSs are immune cell aggregates that form through inflammatory signals that arise during diseases such as cancer [134]. In general, the presence of TLS has been highly associated with improved prognosis and better survival rates across several cancer models, particularly in concert with immune CPIs—which have been ineffective as PDAC monotherapies [3,135]. The CXCR5–CXCL13 and CCR7–CCL19/21 axes play key roles in TLS formation, serving as primary promoters of B and T lymphocyte infiltration in coordination with LTi and LTo cells triggered by IL-7 and LTR–LT12 signals during the early stages of TLS formation across GI cancers including PDAC [136,137]. The roles of CCL19 and CCL21 in particular appear to be concentrated in DC and T cell activation and migration [137,138,139]. In addition, the CCR6–CCL20 axis—although not as mechanistically investigated in literature—has shown potential in TLS development and maintenance due to its role in effector cell migration, including Th17 infiltration in inflammatory settings as well as B cells and DCs in other cancer models [87,140,141]. However, a clear association towards TLSs is never made in these studies, and the specific subset of the recruited immune cells should be carefully considered before making any hypotheses. Nevertheless, Pushpamali et al. showed upregulation of several signals in tumors containing TLSs, one of which was CCL20, suggesting a larger than previously understood role of the CCR6–CCL20 axis in TLS functioning [142]. TLS presence, however, remains generally uncommon in PDAC patients. A study analyzing the prognoses of PDAC TLSs reported that improved outcomes were mostly correlated with an intratumoral subset of TLSs, observed in only around 15-20% of patients. [143]. Together, these findings underscore roles of CC chemokines in TLS promotion, while highlighting the potential strategy of TLS induction as a therapy to overcome PDAC.
3.3. Metastasis and PMN Formation
In PDAC, TAMs contribute to metastatic progression through CCR-CCL axis-mediated recruitment and polarization, while MMPs, which often intersect with TAM-driven pathways, facilitate invasion by degrading the ECM. Sanford and colleagues, via an experiment dealing with the investigational drug PF-04136309, demonstrated that it is monocytes after differentiation into TAMs—rather than the initial CCR inflammatory monocytes—that primarily mediate immunosuppression [15]. In this study, PF-04136309 reduced TAM levels in the pre-metastatic liver, whereas—in a separate phase 1b trial using the same investigational drug PF-04136309 (NCT02732938)—TAM levels in the primary tumor body showed minimal and/or ambiguous change, a discrepancy likely attributable to the more pronounced desmoplastic and immunosuppressed stroma in the primary tumor compared to the PMN, which is only in the preparatory stages of tumor cultivation [15,144,145]. While this phase 1b trial did not report detailed efficacy endpoints on metastasis due to its early-stage nature, the available evidence suggests that CCR2–CCL2 contributes to metastatic progression primarily through TAM accumulation at the PMN rather than at the primary tumor site, a mechanism that warrants further investigation [15,145]. Supporting this idea, in vitro pancreatic models have demonstrated CCL20 dose-dependent invasion of collagen by PANC-1 cells, while ex vivo findings indicate that TAMs frequently upregulate CCL20 [65,67,146]. This may suggest that CCR2–CCL2 driven PMN TAM proliferation is a prerequisite for CCL20-mediated early metastasis, enabling both initial tumor cell recruitment and MMP9 upregulation, which helps establish a permissive environment for tumor dissemination beyond the primary tumor mass [65,67]. CCR macrophages have specifically been shown to induce MMP9 in pancreatic lymph node metastases, a pathway that likely stretches to other pancreatic metastatic sites [118]. Although direct indications of CCR-CCL driven MMP expression in PDAC remains rare, extensive data from other cancer models back this relationship. In nasopharyngeal carcinoma, CCR2–CCL2 has shown to upregulate MMP9 and MMP2 [147]. The CCR5–CCL5 signaling axis exhibited MMP9 promotion via pulmonary mesenchymal cells in lung cancer and through PLC, PKC, and NF-B in oral cancer [148,149]. And the CCR4–CCL17 axis has demonstrated MMP13 activation via ERK1/2 signaling in bladder cancer [150]. These cascades serve as potential therapeutic targets in PDAC metastasis, but their exact roles in the pancreatic setting remain unclear.
Currently, despite additional literature around CCR-CCL driven PDAC-specific organ metastasis remaining narrow, other correlative cancer models still underscore further research necessity in this field. For example, studies of CCR5–CCL5 in pancreatic tumor cell invasion are largely confined to in vitro correlative models [22,151]. In contrast, breast cancer—particularly TNBC, which shares many immunosuppressive and mesenchymal features with PDAC—offers more extensive insights [152]. TNBC cell-secreted IL-6 showed LEC priming in pre-metastatic organs to promote CCL5 levels in coordination with VEGF—via a pSTAT3/p-cJun/pATF-2 ternary complex—thereby recruiting CCR tumor cells to developing PMNs [153]. LncSNHG5 and IGF2BP2 further stabilise these niches by supporting ZNF281—a CCL2/5 transcription factor— and activating the p38-MAPK signaling axis in HUVECs, which upregulates angiogenesis and vascular permeability [154]. In a separate TNBC model, Chen et al. identified a CCR5-driven mechanism linking EMT and metastasis via HIF-1, YAP1, ZEB1/2, and -catenin [155]. Beyond CCR5, CCR Tregs showed breast cancer lung metastasis promotion via GBP-driven NK cell suppression [156]. Gastric cancer models showcasing peritoneal spread—which are particularly relevant given that peritoneal spread is the second most common metastatic site in PDAC after the liver—also show proof for CCR-CCL axis involvement in peritoneal invasion [157,158]. In vitro gastric cancer studies have shown upregulation of CCL5 among many other signals leads to peritoneal metastasis, and ex vivo human analyses confirm strong correlative patterns [159,160]. Another experiment uniquely reported CCR4 expression in gastric cancer peritoneal metastasis exhibiting preferential aggregation towards omental milky spots—an important consideration in potential future PDAC investigations [161]. Separately, CCL17 demonstrated significant upregulation as early as 10 weeks before micrometastatic formation in a melanoma model, suggesting that the CCR4–CCL17 axis carry a part in early PMN establishment [162]. These findings across several different cancer models may offer translatable frameworks when investigating CCR-CCL-driven metastasis in the context of PDAC.
Multiple CCR-CCL axes have been implicated in promoting PDAC LNM, acting via both tumor-intrinsic and immune-mediated mechanisms. LNM prevalence in PDAC stands at roughly 65-85% and serves as a strong prognostic factor that correlates with greatly reduced OS compared to node-free patients [163,164,165]. The CCR7 signaling axis has been identified as a primary driver of LNMs in PDAC. Elevated CCR7 levels in pancreatic tumor cells with nodal metastases relative to normal tissues have also been confirmed via ex vivo observational analyses [19,166]. In particular, cells expressing CD133—a commonly associated marker of CSCs—showed significantly increased CCR7 expression levels [166]. A different study examining CD133 and not CCR7 also separately verified CD133 as a key distinguishing element between metastatic and non-metastatic pancreatic cancer cell lines, heavily indicating a link between CD133 and metastasis [167]. This relationship between CD133 and CCR7 remains to be elucidated. Interestingly, CCR7 upregulation was found absent in PDAC liver or lung metastases, suggesting an exclusivity to lymphatic spread [168] However, this same observation was absent in a different model featuring liver metastases in colorectal cancer, indicating a context-dependent nuance [169]. Other CCR-CCL axes have shown signs of involvement in LNM as well. One study revealed high levels of CCL CCR macrophages in PDAC LNMs, which appeared to recruit Tregs and dysfunctional CD T cells, likely cultivating the same suppressive setting observed in the primary TME [118]. The CCR5–CCL5 axis showed strong correlation with LNM in a gastric cancer model and appeared to induce metastasis by skewing the Th1/Th2 immune ratio in favor of an immunosuppressive Th2 phenotype, although further mechanistic examination is warranted [170]. These observations highlight CC chemokine axes—particularly CCR7—as key drivers of nodal metastases in PDAC.
4. Chemokine Mediated Downstream Immune Evasion
Several immune evasion pathways are influenced by chemokine-mediated regulatory cell populations in the PDAC microenvironment. Among these immunosuppressive populations, Tregs are a key player in helping maintain an immunologically “cold” setting by suppressing effector immune responses via paracrine signaling and transcriptional modulation involving proteins like CTLA-4, PD-1, IL-10, TGF-, and FOXP3, among others [83,91,171]. In PDAC and colon cancer investigations, the CCR5–CCL5 signaling axis seems to have a large influence over these proteins. [99,172]. In another model examining various cancer types, upregulated glycolytic activity in tumor-infiltrating Tregs has been linked to the CCR6–CCL20 axis, which have also been correlated with reduced OS [173]. The CCR4–CCL22 axis has also demonstrated a role in Treg activation, distinct from Treg recruitment, via inducing DC-Treg interactions in nodal settings [174]. Interestingly, although CCL22-deficient mice showed elevated anti-tumor immune responses, Treg infiltration remained comparable to wild-type controls, suggesting that CCR4–CCL22 may perform a larger than known role in Treg activation versus recruitment, likely compensated by alternative cascades [174]. In parallel, M2 TAMs and MDSCs represent additional chemokine-driven immunosuppressive populations that synergize with Tregs. M2 TAMs produce inhibitory mediators like IL-10, TGF-, PD-L1, and IL-6—which suppress effector cell function and support Treg activity—and MDSCs exert many of these same roles also via ROS, NO, and ARG1 production [175,176,177,178]. M2 TAMs and MDSCs further back tumor progression by secreting growth factors VEGF and PDGF, working in concert with CAFs to support demoplasia [103,179,180]. By producing dense stroma components—primarily fibronectin and type I collagen—CAFs construct physical and biochemical barriers that impairs vascular perfusion, drug delivery, and nearby inflammatory signals [181,182,183]. Consequently, the PDAC TME develops into a highly integrated network between different interdependent cell populations. M2 TAMs and Tregs participate in extensive crosstalk—where autocrine signaling and positive feedback mechanisms foster mutual reinforcement—and MDSCs possess the capacity to differentiate into TAM-like cells under hypoxic conditions [184,185]. Separately, CAFs have also shown to assist in M2-like polarization of monocytes as well [186].
Th2 and Th17 cells also play important roles in the immune landscape of PDAC, albeit in a more nuanced and functionally plastic manner. Under inflammatory conditions, the CCR6–CCL20 axis has the ability to recruit Th17 cells, although to a lesser extent compared to other immune cell types [87]. The current body of literature represents a mix of both pro-tumorigenic and anti-tumorigenic functions of Th17 cells. In PDAC-specific models, Th17 cells exert pro-tumor development modulation via NETs and control of gut microbiota-tumor interactions via IL-17 [187,188]. A different subset of IL-17 producing cells, dubbed Tc17 cells, have also shown pro-tumor outcomes via CD T cell suppression and iCAF differentiation [189]. On the other hand, broader tumor models exhibit Th17-mediated anti-tumor immunity through DC induction and/or differentiation into Th1-like or Th17/Th1 hybrid phenotypes, strengthening cytotoxic immune responses [190,191]. Likewise, elevated levels of IL-6 in concert with TGF- leading to Th17 cell differentiation over Treg development resulted in delayed tumor growth and improved survival in a murine pancreatic model [91,192]. Taken together, the dual nature of Th17 responses underscores their plasticity in PDAC, while in contrast, Th2 cells—recruited via CCR4–CCL17/22 signaling—exhibit a functional bias toward tumor-promoting effects rather than a balance, one of which is M2 macrophage polarization through IL-4 and IL-13 expression [106,193]. IL-4 has also clearly demonstrated a dose-dependent promoting role in tumor proliferation and metastasis within pancreatic cancer [194,195]. On the contrary, findings by Jacenik and colleagues revealed that Th2 transduction resulted in eosinophil anti-tumor activity—partially driven by Th2 cell secreted IL-5—in pancreatic cancer lines [196]. This IL-5-secreting Th2 subset suggests a unique immunological profile that warrants more investigation to clarify its relevance in PDAC. Th2 and Th17 cells thus contribute to PDAC from several angles, where pro-tumorigenic outcomes remain dominant, although context-specific anti-tumor exceptions remain deserving of attention.
5. Therapeutic Targeting of CCR-CCL Axes and its Patient Outcomes
Despite several decades of research, the current PDAC standard of care remains largely limited. Upon diagnosis, treatment typically starts with surgical resection—-when feasible—and is paired with neoadjuvant and/or adjuvant chemotherapy or radiotherapy [197]. Depending on the patient’s condition, first-line or neoadjuvant/adjuvant chemotherapy generally entails either a gemcitabine and nab-paclitaxel combination or a 5-fluorouracil (5-FU) based regimen among other options [197]. Since its approval in 1995, gemcitabine has acted as the standard of care over 5-FU, and nab-paclitaxel was introduced in 2013 as a combinatorial partner to enhance efficacy [198,199,200]. FOLFIRINOX—a regimen comprising 5-FU, leucovorin, irinotecan, and oxaliplatin—entered clinical practice following a pivotal 2011 study as the first 5-FU-based regimen to demonstrate comparable outcomes, followed shortly thereafter by the 2015 approval of Onivyde [201,202]. NALIRIFOX, another 5-FU-based chemotherapy approved more recently in early 2024, has shown improved mOS (11.1 vs 9.2 months) with a virtually identical AE profile when placed head to head with a gemcitabine-nab-paclitaxel treatment arm, currently standing as a first-line alternative to FOLFIRINOX [203,204,205]. In addition to standard therapies, biomarker-driven treatments such as olaparib, pembrolizumab, zenocutuzumab, larotrectinib, and entrectinib target select PDAC patient subsets [197,206,207,208,209,210]. However, only a small fraction of the total population meet the given criteria, typically applicable to less than 5% or—in some cases—roughly 1% of the total patient population [205,206,207,208,209]. Conventional chemotherapy and radiotherapy also face challenges, such as consequent chemokine upregulation—particularly CCL2 among other chemokine types—which limit the treatments’ efficacy [81,210,211]. While 5-FU-based chemotherapies have not yet demonstrated similar outcomes in PDAC, preliminary evidence suggests that CCL upregulation via the STING pathway seems plausible and may lead to disease relapse [212,213,214]. Given these shortcomings, recent therapeutic strategies have begun to explore combinatorial approaches that integrate standard chemotherapy or radiotherapy with immune-based interventions.
Among these immune-based targets, CCR-CCL inhibition has emerged as a promising therapeutic avenue. While no CCR-CCL targeting drugs have been currently approved for PDAC yet, several mid-stage drug candidates are currently under trials to address this therapeutic gap. BMS-813160—a small-molecule dual antagonist of CCR2 and CCR5—remains the most explored in the context of PDAC. Although the efficacy results of two trials (NCT03184870, NCT03767582) investigating BMS-813160 in PDAC have yet to be released, a third study (NCT03496662) reported promising outcomes in BR (ORR 42%, mPFS 11.9 mo, mOS 18.2 mo) and LA PDAC (ORR 20%, mPFS 14.7 mo, mOS 17 mo), representing comparable to moderate improvements relative to historical benchmarks, particularly in LA PDAC [215,216,217]. Leronlimab—–a IgG4 monoclonal antibody for CCR5—also demonstrates translational potential in PDAC via activity in TNBC-centered trials [152]. A pooled analysis of three mTNBC studies (NCT03838367, NCT04313075, NCT04504942) showed that higher doses (525-700mg), compared to lower doses (350-525mg), resulted in >75% improvement in mPFS and mOS and outcomes reaching 6.1 months and over 12 months, respectively [218]. CytoDyn, the developers of leronlimab, has also announced through a press release that the high-dose patient arm showed subsequent PD-L1 upregulation—suggesting a potential combination therapy with CPIs—and that long-term follow-up observed >36 months OS in some cases [225,226]. Maraviroc—a small-molecule CCR5 antagonist currently only approved for HIV—has also emerged as a therapeutic candidate, supported by extensive preclinical PDAC models [22,151,219]. In the context of clinical trials, however, maraviroc has been primarily tested in CRC (NCT01736813, NCT03274804), although a separate trial (NCT04721301) named LUMINESCENCE has assessed it in PDAC [220,221]. Oddly, results for the LUMINESCENCE trial have yet to be released despite its completion in March 2023.
Table 2.
Clinical Stage Drug Candidates Targeting the CCR-CCL Signaling Network. Overview of therapeutic agents in clinical development as of September 2025 targeting the CCR-CCL signaling network. The table lists molecular targets, drug names, trial identifiers (NCT/ISRCTN), phases, conditions under investigation (including cancer and select nonmalignant diseases), and trial statuses, where applicable.
Table 2.
Clinical Stage Drug Candidates Targeting the CCR-CCL Signaling Network. Overview of therapeutic agents in clinical development as of September 2025 targeting the CCR-CCL signaling network. The table lists molecular targets, drug names, trial identifiers (NCT/ISRCTN), phases, conditions under investigation (including cancer and select nonmalignant diseases), and trial statuses, where applicable.
| Target | Drug Name | NCT/ISRCTN | Phase | Condition(s) | Status |
|---|---|---|---|---|---|
| CCR2 /CCR5 | BMS-813160 | NCT01049165 | 1 | Healthy | Completed Nov 2011 |
| NCT03184870 | 1b/2 | mCRC, mPDAC | Completed Jun 2023, Under QC Review | ||
| NCT03767582 | 1/2 | LA PDAC | Completed Sep 2024 | ||
| NCT03496662 | 1/2 | BR/LA PDAC | Completed Jul 2024 | ||
| NCT04123379 | 2 | NSCLC, HCC | Active | ||
| CCR5 | Maraviroc | NCT01736813 | 1 | mCRC | Completed Sep 2014 |
| NCT03274804 | 1 | MSS CRC | Completed Mar 2020 | ||
| NCT04721301 | 1 | mCRC, mPDAC | Completed Mar 2023 | ||
| Leronlimab | NCT03838367 | 1/2 | mTNBC | Active | |
| NCT04313075 | Expanded Access | mTNBC | N/A | ||
| NCT04504942 | 2 | LA/metastatic solid tumors | Unknown | ||
| NCT06699836 | 2 | MSS CRC | Active | ||
| CCR4 | FLX475 (Tivumecirnon) | NCT03674567 | 1b/2 | Advanced Cancer | Completed Dec 2024 |
| NCT04768686 | 2 | Advanced/metastatic GC | Completed Aug 2024 | ||
| CCR6 | PF-07054894 | NCT04388878 | 1 | Healthy | Completed Jun 2022 |
| NCT06327880 | 1 | Healthy | Completed Jul 2024 | ||
| NCT05549323 | 1b | Ulcerative Colitis | Active | ||
| NCT07009353 | 1 | Healthy | Active | ||
| IDOR-1117-2520 | ISRCTN28892128 | 1 | Healthy | Active | |
| CCR7 | CAP-100 | NCT04704323 | 1 | CLL, SLL | Active |
Shifting toward less focused on axes, FLX475—a small-molecule CCR4 antagonist also referred to as tivumecirnon—has been clinically tested in CPI-naive NSCLC, CPI-experienced HNSCC, and mGC (NCT03674567, NCT04768686). Across these studies, FLX475 exhibited greater efficacy in tumors characterized by high PD-L1 expression, HPV positivity, and/or EBV positivity, though the mid-stage nature of the trials and small sample sizes require the findings be assessed conservatively [222,223,224]. PDAC-specific studies will be necessary to support clinical translation of FLX475, given the pro-tumor roles of CCR4 in the pancreatic setting [24]. Clinical targeting of CCR6 and CCR7 remain even more scarce. CCR6 antagonists, PF-07054894 and IDOR-117-2520, are both under early-stage clinical investigation, primarily focused on evaluating safety and toxicity in healthy individuals or ulcerative colitis patients (NCT07009353, NCT05549323, ISRCTN28892128). Preclinical studies, however, have demonstrated that PF-07054894 and IDOR-117-2520-mediated blockade can reduce immune cell infiltration into tissues in autoimmune models, indicating a mechanism that may be viable in tumors but warrants more data [225,226]. CAP-100—an anti-CCR7 monoclonal antibody—is currently under phase 1 evaluation for CLL and SLL (NCT04704323), indications in which CCR7 plays a relatively well-defined role in disease progression [227]. Although CCR7 expression in PDAC is more variable, preliminary findings suggest that CCR7 blockade may exert anti-LNM effects in pancreatic cancer, though again, the approach should be pursued with caution [228]. These efforts highlight the growing interest in CCR-CCL axis targeting for drug development in PDAC.
6. Conclusions and Future Vision
Through this review, we have sought to provide a comprehensive look into our current understanding of CC chemokine functions in the context of PDAC. Generally, literature evidence has shown to point towards CCR-CCL axes as central drivers of immunosuppression in the PDAC TME—although there have been nuanced cases of inflammatory, anti-tumor outcomes—and enhance overall immune evasion not only via chemotaxis and polarization of regulatory cell types, but also via ECM remodeling and metastasis promotion through downstream cascades. While this review predominantly examines select signaling pairs, it is important to note that other CC chemokine pathways that were only briefly covered (CCR10–CCL28 and CCR4–CCL2) or were not mentioned at all (CCR8–CCL1, CCR1–CCL3/15) also play relevant functions yet in likely subtler ways [17,88,229,230,231]. Likewise, the CXC chemokine family—often functioning in parallel or in coordination with CC chemokines—introduces another added layer of intricacy and was not covered in detail here in order to prioritize greater depth and synthesis over breadth [232]. Insights based in this field should, if not already, take these other important elements into account as well.
Looking ahead, despite rising recognition and research, this particular field still possesses several knowledge gaps that may cause hesitancy in its therapeutic applicability, as portrayed by the apparent lack of clinical trials in explicitly PDAC among the already few number of relevant drugs covered. A recurring theme that showed potential for future investigation is spatial mapping analysis around peritumoral regions of PDAC in the context of inflammatory markers. In several instances, cross paper analysis suggested inflammatory signals promoting chemokine axes to recruit immune cells that condition peritumoral zones for subsequent invasion. Another area deserving of more attention is stage-specific studies in the context of inflammatory-signal-driven immune cell chemotaxis, specifically Th17 and Th2 cells under early PDAC development conditions. Yet an obstacle to this proposal is the current lack of diagnostic efficiency in the healthcare system to adequately detect early stage PDACs and analyze their development in sample sizes large enough to make any significant claims, likely limiting the current scope to murine models. Lastly, there exists a general absence of PDAC-specific models across the different aspects of cancer in the context of CC chemokines, particularly around mapping of different cellular and intracellular sources of CCLs (CCL17, CCL22, CCL20, CCL19, CCL21), MMP expression, and non-nodal metastases among others. Addressing these shortcomings will bring us closer to understanding the full complexity of PDAC, offer translational insights to other cancer types, and provide a more defined framework for other therapeutic strategies, if not directly identify therapeutic targets themselves.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Leiphrakpam, P.D.; Chowdhury, S.; Zhang, M.; Bajaj, V.; Dhir, M.; Are, C. Trends in the Global Incidence of Pancreatic Cancer and a Brief Review of its Histologic and Molecular Subtypes. 56, 71. [CrossRef]
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. 10, 16425. [CrossRef]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. 12, 173–181. [CrossRef]
- Perrotta, G.; Abboud, Y.; Osipov, A.; Mohamed, G.; El Helou, M.; Pandol, S.J.; Lo, S.K.; Gaddam, S. Trends in incidence and survival of early-stage pancreatic ductal adenocarcinoma in the United States. 42, 633–633. [CrossRef]
- Jo, J.H.; Cho, I.R.; Jung, J.H.; Lee, H.S.; Chung, M.J.; Bang, S.; Park, S.W.; Chung, J.B.; Song, S.Y.; Park, J.Y. Clinical characteristics of second primary pancreatic cancer. 12, e0179784. [CrossRef]
- Farnes, I.; Kleive, D.; Verbeke, C.S.; Aabakken, L.; Issa-Epe, A.; Småstuen, M.C.; Fosby, B.V.; Dueland, S.; Line, P.D.; Labori, K.J. Resection rates and intention-to-treat outcomes in borderline and locally advanced pancreatic cancer: real-world data from a population-based, prospective cohort study (NORPACT-2). 7, zrad137. [CrossRef]
- Li, Y.; Xiang, S.; Pan, W.; Wang, J.; Zhan, H.; Liu, S. Targeting tumor immunosuppressive microenvironment for pancreatic cancer immunotherapy: Current research and future perspective. 13, 1166860. [CrossRef]
- Guo, S.; Wang, Z. Unveiling the immunosuppressive landscape of pancreatic ductal adenocarcinoma: implications for innovative immunotherapy strategies. 14, 1349308. [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines. 12, 121–127. [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. 285, 2944–2971. [CrossRef]
- Miller, M.; Mayo, K. Chemokines from a Structural Perspective. 18, 2088. [CrossRef]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. 317, 575–589. [CrossRef]
- Korbecki, J.; Kojder, K.; Simińska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. 21, 8412. [CrossRef]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of Receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 Ligands. 21, 7619. [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. 19, 3404–3415. [CrossRef]
- Tan, M.C.B.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. 182, 1746–1755. [CrossRef]
- Roy, I.; Boyle, K.A.; Vonderhaar, E.P.; Zimmerman, N.P.; Gorse, E.; Mackinnon, A.C.; Hwang, R.F.; Franco-Barraza, J.; Cukierman, E.; Tsai, S.; et al. Cancer cell chemokines direct chemotaxis of activated stellate cells in pancreatic ductal adenocarcinoma. 97, 302–317. [CrossRef]
- Dixit, A.; Sarver, A.; Zettervall, J.; Huang, H.; Zheng, K.; Brekken, R.A.; Provenzano, P.P. Targeting TNF-α–producing macrophages activates antitumor immunity in pancreatic cancer via IL-33 signaling. 7, e153242. [CrossRef]
- Guo, J.; Lou, W.; Ji, Y.; Zhang, S. Effect of CCR7, CXCR4 and VEGF-C on the lymph node metastasis of human pancreatic ductal adenocarcinoma. 5, 1572–1578. [CrossRef]
- Li, N.; Li, Y.; Li, Z.; Huang, C.; Yang, Y.; Lang, M.; Cao, J.; Jiang, W.; Xu, Y.; Dong, J.; et al. Hypoxia Inducible Factor 1 (HIF-1) Recruits Macrophage to Activate Pancreatic Stellate Cells in Pancreatic Ductal Adenocarcinoma. 17, 799. [CrossRef]
- Wu, H.H.; Hwang-Verslues, W.W.; Lee, W.H.; Huang, C.K.; Wei, P.C.; Chen, C.L.; Shew, J.Y.; Lee, E.Y.H.; Jeng, Y.M.; Tien, Y.W.; et al. Targeting IL-17B–IL-17RB signaling with an anti–IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. 212, 333–349. [CrossRef]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.E.A.; Bae, S.; Lillard, J.W.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. 8, 1323. [CrossRef]
- Wu, M.; Han, J.; Wu, H.; Liu, Z. Proteasome-dependent senescent tumor cells mediate immunosuppression through CCL20 secretion and M2 polarization in pancreatic ductal adenocarcinoma. 14, 1216376. [CrossRef]
- Khabipov, A.; Trung, D.N.; Van Der Linde, J.; Miebach, L.; Lenz, M.; Erne, F.; Von Bernstorff, W.; Schulze, T.; Kersting, S.; Bekeschus, S.; et al. CCR4 Blockade Diminishes Intratumoral Macrophage Recruitment and Augments Survival of Syngeneic Pancreatic Cancer-Bearing Mice. 11, 1517. [CrossRef]
- Xu, D.; Liu, X.; Ke, S.; Guo, Y.; Zhu, C.; Cao, H. CCL19/CCR7 drives regulatory T cell migration and indicates poor prognosis in gastric cancer. 23, 464. [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. 142, 730–733.e9. [CrossRef]
- Zhang, X.; Mao, T.; Zhang, B.; Xu, H.; Cui, J.; Jiao, F.; Chen, D.; Wang, Y.; Hu, J.; Xia, Q.; et al. Characterization of the genomic landscape in large-scale Chinese patients with pancreatic cancer. 77, 103897. [CrossRef]
- Kang, R.; Hou, W.; Zhang, Q.; Chen, R.; Lee, Y.J.; Bartlett, D.L.; Lotze, M.T.; Tang, D.; Zeh, H.J. RAGE is essential for oncogenic KRAS-mediated hypoxic signaling in pancreatic cancer. 5, e1480–e1480. [CrossRef]
- Asano, T.; Yao, Y.; Zhu, J.; Li, D.; Abbruzzese, J.L.; Reddy, S.A.G. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-κB and c-Myc in pancreatic cancer cells. 23, 8571–8580. [CrossRef]
- Takaya, H.; Andoh, A.; Shimada, M.; Hata, K.; Fujiyama, Y.; Bamba, T. The Expression of Chemokine Genes Correlates with Nuclear Factor-κB Activation in Human Pancreatic Cancer Cell Lines. 21, 32.
- Mojsilovic-Petrovic, J.; Callaghan, D.; Cui, H.; Dean, C.; Stanimirovic, D.B.; Zhang, W. Hypoxia-inducible factor-1 (HIF-1) is involved in the regulation of hypoxia-stimulated expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in astrocytes. 4, 12. [CrossRef]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α Promotes Oncogenic KRAS Function in Pancreatic Cancer via TAK1–TAB Stabilization and Regulation of Noncanonical NF-κB. 3, 690–703. [CrossRef]
- D’Amico, S.; Kirillov, V.; Petrenko, O.; Reich, N.C. STAT3 is a genetic modifier of TGF-beta induced EMT in KRAS mutant pancreatic cancer. 13, RP92559. [CrossRef]
- Lesina, M.; Kurkowski, M.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. 19, 456–469. [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3–CCL2 Signaling. 76, 4124–4135. [CrossRef]
- Tang, T.; Huang, X.; Lu, M.; Zhang, G.; Han, X.; Liang, T. Transcriptional control of pancreatic cancer immunosuppression by metabolic enzyme CD73 in a tumor-autonomous and -autocrine manner. 14, 3364. [CrossRef]
- Pausch, T.M.; Aue, E.; Wirsik, N.M.; Freire Valls, A.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. 10, 5420. [CrossRef]
- Tu, M.; Klein, L.; Espinet, E.; Georgomanolis, T.; Wegwitz, F.; Li, X.; Urbach, L.; Danieli-Mackay, A.; Küffer, S.; Bojarczuk, K.; et al. TNF-α-producing macrophages determine subtype identity and prognosis via AP1 enhancer reprogramming in pancreatic cancer. 2, 1185–1203. [CrossRef]
- Boelaars, K.; Rodriguez, E.; Huinen, Z.R.; Liu, C.; Wang, D.; Springer, B.O.; Olesek, K.; Goossens-Kruijssen, L.; Van Ee, T.; Lindijer, D.; et al. Pancreatic cancer-associated fibroblasts modulate macrophage differentiation via sialic acid-Siglec interactions. 7, 430. [CrossRef]
- Lin, J.; Caress, A.; Ahmed, F.; Taylor, N.; Gong, Y.; Oh, W.K. Elevated circulating CCL2 in prostate cancer patients. 38, 201–201. Publisher: Wolters Kluwer, https://doi.org/10.1200/JCO.2020.38.6_suppl.201.
- Wang, J.; Zhuang, Z.G.; Xu, S.F.; He, Q.; Shao, Y.G.; Ji, M.; Yang, L.; Bao, W. Expression of CCL2 is significantly different in five breast cancer genotypes and predicts patient outcome. 8, 15684–15691.
- Saurer, L.; Reber, P.; Schaffner, T.; Büchler, M.W.; Buri, C.; Kappeler, A.; Walz, A.; Friess, H.; Mueller, C. Differential expression of chemokines in normal pancreas and in chronic pancreatitis. 118, 356–367. [CrossRef]
- Sun, K.; Zhang, X.; Shi, J.; Huang, J.; Wang, S.; Li, X.; Lin, H.; Zhao, D.; Ye, M.; Zhang, S.; et al. Elevated protein lactylation promotes immunosuppressive microenvironment and therapeutic resistance in pancreatic ductal adenocarcinoma. 135, e187024. [CrossRef]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid acceleration of KRAS-mutant pancreatic carcinogenesis via remodeling of tumor immune microenvironment by PPARδ. 13, 2665. [CrossRef]
- Zhou, B.; Sun, C.; Li, N.; Shan, W.; Lu, H.; Guo, L.; Guo, E.; Xia, M.; Weng, D.; Meng, L.; et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. 48, 2087–2097. [CrossRef]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. 21, 1396–1408. [CrossRef]
- Gong, J.; Li, X.; Feng, Z.; Lou, J.; Pu, K.; Sun, Y.; Hu, S.; Zhou, Y.; Song, T.; Shangguan, M.; et al. Sorcin can trigger pancreatic cancer-associated new-onset diabetes through the secretion of inflammatory cytokines such as serpin E1 and CCL5. 56, 2535–2547. [CrossRef]
- Jiao, X.; Katiyar, S.; Willmarth, N.E.; Liu, M.; Ma, X.; Flomenberg, N.; Lisanti, M.P.; Pestell, R.G. c-Jun Induces Mammary Epithelial Cellular Invasion and Breast Cancer Stem Cell Expansion. 285, 8218–8226. [CrossRef]
- Wang, X.; Lang, M.; Zhao, T.; Feng, X.; Zheng, C.; Huang, C.; Hao, J.; Dong, J.; Luo, L.; Li, X.; et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. 36, 3048–3058. [CrossRef]
- Yashiro, T.; Nakano, S.; Nomura, K.; Uchida, Y.; Kasakura, K.; Nishiyama, C. A transcription factor PU.1 is critical for Ccl22 gene expression in dendritic cells and macrophages. 9, 1161. [CrossRef]
- Vulcano, M.; Albanesi, C.; Stoppacciaro, A.; Bagnati, R.; D’Amico, G.; Struyf, S.; Transidico, P.; Bonecchi, R.; Del Prete, A.; Allavena, P.; et al. Dendritic cells as a major source of macrophage-derived chemokine/CCL22 in vitro and in vivo. 31, 812–822. [CrossRef]
- Wiedemann, G.M.; Knott, M.M.L.; Vetter, V.K.; Rapp, M.; Haubner, S.; Fesseler, J.; Kühnemuth, B.; Layritz, P.; Thaler, R.; Kruger, S.; et al. Cancer cell-derived IL-1α induces CCL22 and the recruitment of regulatory T cells. 5, e1175794. [CrossRef]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. 208, 469–478. [CrossRef]
- Hao, N.B.; Lü, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in Tumor Microenvironments and the Progression of Tumors. 2012, 1–11. [CrossRef]
- Wang, L.x.; Zhang, S.x.; Wu, H.j.; Rong, X.l.; Guo, J. M2b macrophage polarization and its roles in diseases. 106, 345–358. [CrossRef]
- Bonecchi, R.; Sozzani, S.; Stine, J.T.; Luini, W.; D’Amico, G.; Allavena, P.; Chantry, D.; Mantovani, A. Divergent Effects of Interleukin-4 and Interferon-γ on Macrophage-Derived Chemokine Production: An Amplification Circuit of Polarized T Helper 2 Responses. 92, 2668–2671. [CrossRef]
- Liddiard, K.; Welch, J.S.; Lozach, J.; Heinz, S.; Glass, C.K.; Greaves, D.R. Interleukin-4 induction of the CC chemokine TARC (CCL17) in murine macrophages is mediated by multiple STAT6 sites in the TARC gene promoter. 7, 45. [CrossRef]
- Rawal, S.; Chu, F.; Zhang, M.; Park, H.J.; Nattamai, D.; Kannan, S.; Sharma, R.; Delgado, D.; Chou, T.; Lin, H.Y.; et al. Cross Talk between Follicular Th Cells and Tumor Cells in Human Follicular Lymphoma Promotes Immune Evasion in the Tumor Microenvironment. 190, 6681–6693. [CrossRef]
- Wirnsberger, G.; Hebenstreit, D.; Posselt, G.; Horejs-Hoeck, J.; Duschl, A. IL-4 induces expression of TARC/CCL17 via two STAT6 binding sites. 36, 1882–1891. [CrossRef]
- Achuthan, A.; Cook, A.D.; Lee, M.C.; Saleh, R.; Khiew, H.W.; Chang, M.W.; Louis, C.; Fleetwood, A.J.; Lacey, D.C.; Christensen, A.D.; et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. 126, 3453–3466. [CrossRef]
- Nutt, S.L.; Metcalf, D.; D’Amico, A.; Polli, M.; Wu, L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. 201, 221–231. [CrossRef]
- Hsu, A.T.; Lupancu, T.J.; Lee, M.C.; Fleetwood, A.J.; Cook, A.D.; Hamilton, J.A.; Achuthan, A. Epigenetic and transcriptional regulation of IL4-induced CCL17 production in human monocytes and murine macrophages. 293, 11415–11423. [CrossRef]
- Nakayama, T.; Hieshima, K.; Nagakubo, D.; Sato, E.; Nakayama, M.; Kawa, K.; Yoshie, O. Selective Induction of Th2-Attracting Chemokines CCL17 and CCL22 in Human B Cells by Latent Membrane Protein 1 of Epstein-Barr Virus. 78, 1665–1674. [CrossRef]
- Kwon, D.J.; Bae, Y.S.; Ju, S.M.; Goh, A.R.; Youn, G.S.; Choi, S.Y.; Park, J. Casuarinin suppresses TARC/CCL17 and MDC/CCL22 production via blockade of NF-κB and STAT1 activation in HaCaT cells. 417, 1254–1259. [CrossRef]
- Campbell, A.S.; Albo, D.; Kimsey, T.F.; White, S.L.; Wang, T.N. Macrophage inflammatory protein-3α promotes pancreatic cancer cell invasion. 123, 96–101. [CrossRef]
- Rubie, C.; Frick, V.O.; Ghadjar, P.; Wagner, M.; Grimm, H.; Vicinus, B.; Justinger, C.; Graeber, S.; Schilling, M.K. CCL20/CCR6 expression profile in pancreatic cancer. 8, 45. [CrossRef]
- Liu, B.; Jia, Y.; Ma, J.; Wu, S.; Jiang, H.; Cao, Y.; Sun, X.; Yin, X.; Yan, S.; Shang, M.; et al. Tumor-associated macrophage-derived CCL20 enhances the growth and metastasis of pancreatic cancer. 48, 1067–1074. [CrossRef]
- Geismann, C.; Grohmann, F.; Dreher, A.; Häsler, R.; Rosenstiel, P.; Legler, K.; Hauser, C.; Egberts, J.H.; Sipos, B.; Schreiber, S.; et al. Role of CCL20 mediated immune cell recruitment in NF-κB mediated TRAIL resistance of pancreatic cancer. 1864, 782–796. [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.; Wang, H.; et al. KrasG12D-Induced IKK2/β/NF-κB Activation by IL-1α and p62 Feedforward Loops Is Required for Development of Pancreatic Ductal Adenocarcinoma. 21, 105–120. [CrossRef]
- Tjomsland, V.; Bojmar, L.; Sandström, P.; Bratthäll, C.; Messmer, D.; Spångeus, A.; Larsson, M. IL-1α Expression in Pancreatic Ductal Adenocarcinoma Affects the Tumor Cell Migration and Is Regulated by the p38MAPK Signaling Pathway. 8, e70874. [CrossRef]
- Li, Q.; Laumonnier, Y.; Syrovets, T.; Simmet, T. Recruitment of CCR6-expressing Th17 cells by CCL20 secreted from plasmin-stimulated macrophages. 45, 593–600. [CrossRef]
- Hippe, A.; Braun, S.A.; Oláh, P.; Gerber, P.A.; Schorr, A.; Seeliger, S.; Holtz, S.; Jannasch, K.; Pivarcsi, A.; Buhren, B.; et al. EGFR/Ras-induced CCL20 production modulates the tumour microenvironment. 123, 942–954. [CrossRef]
- Kirschstein, E.P.; Harder, O.; Khanal, S.; Gunderson, A.J.; Mack, M.; Ware, C.F.; Young, K.; Chen, W. 1007 Modulating CAF phenotypes in pancreatic cancer to enhance formation of tertiary lymphoid structures. In Proceedings of the Regular and Young Investigator Award Abstracts. BMJ Publishing Group Ltd, pp. A1128–A1128. [CrossRef]
- Sperveslage, J.; Frank, S.; Heneweer, C.; Egberts, J.; Schniewind, B.; Buchholz, M.; Bergmann, F.; Giese, N.; Munding, J.; Hahn, S.A.; et al. Lack of CCR7 expression is rate limiting for lymphatic spread of pancreatic ductal adenocarcinoma. 131. [CrossRef]
- Cords, L.; Tietscher, S.; Anzeneder, T.; Langwieder, C.; Rees, M.; De Souza, N.; Bodenmiller, B. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. 14, 4294. [CrossRef]
- Wharry, C.E.; Haines, K.M.; Carroll, R.G.; May, M.J. Constitutive noncanonical NFκB signaling in pancreatic cancer cells. 8, 1567–1576. [CrossRef]
- Lo, J.C.; Chin, R.K.; Lee, Y.; Kang, H.S.; Wang, Y.; Weinstock, J.V.; Banks, T.; Ware, C.F.; Franzoso, G.; Fu, Y.X. Differential regulation of CCL21 in lymphoid/nonlymphoid tissues for effectively attracting T cells to peripheral tissues. 112, 1495–1505. [CrossRef]
- Mathas, S. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappaB. 21, 4104–4113. [CrossRef]
- Mburu, Y.K.; Egloff, A.M.; Walker, W.H.; Wang, L.; Seethala, R.R.; Van Waes, C.; Ferris, R.L. Chemokine Receptor 7 (CCR7) Gene Expression Is Regulated by NF-κB and Activator Protein 1 (AP1) in Metastatic Squamous Cell Carcinoma of Head and Neck (SCCHN). 287, 3581–3590. [CrossRef]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. 25, 3873–3886. [CrossRef]
- Mondini, M.; Loyher, P.L.; Hamon, P.; Gerbé De Thoré, M.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. 7, 376–387. [CrossRef]
- Dunbar, K.J.; Karakasheva, T.A.; Tang, Q.; Efe, G.; Lin, E.W.; Harris, M.; Sahu, V.; Sachdeva, U.M.; Hu, J.; Klein-Szanto, A.J.; et al. Tumor-Derived CCL5 Recruits Cancer-Associated Fibroblasts and Promotes Tumor Cell Proliferation in Esophageal Squamous Cell Carcinoma. 21, 741–752. [CrossRef]
- Sivakumar, S.; Jainarayanan, A.; Arbe-Barnes, E.; Sharma, P.K.; Leathlobhair, M.N.; Amin, S.; Reiss, D.J.; Heij, L.; Hegde, S.; Magen, A.; et al. Distinct immune cell infiltration patterns in pancreatic ductal adenocarcinoma (PDAC) exhibit divergent immune cell selection and immunosuppressive mechanisms. 16, 1397. [CrossRef]
- Rapp, M.; Grassmann, S.; Chaloupka, M.; Layritz, P.; Kruger, S.; Ormanns, S.; Rataj, F.; Janssen, K.P.; Endres, S.; Anz, D.; et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. 5, e1105428. [CrossRef]
- D’Ambrosio, D.; Albanesi, C.; Lang, R.; Girolomoni, G.; Sinigaglia, F.; Laudanna, C. Quantitative Differences in Chemokine Receptor Engagement Generate Diversity in Integrin-Dependent Lymphocyte Adhesion. 169, 2303–2312. [CrossRef]
- Slack, R.; Hall, D. Development of operational models of receptor activation including constitutive receptor activity and their use to determine the efficacy of the chemokine CCL17 at the CC chemokine receptor CCR4. 166, 1774–1792. [CrossRef]
- Drouillard, D.; Volkman, B.; Dwinell, M. Roles for the chemokine receptor CCR6 in pancreatitis and pancreas cancer. 212, 0368_5421–0368_5421. [CrossRef]
- Sharma, V.; Sachdeva, N.; Gupta, V.; Nada, R.; Jacob, J.; Sahni, D.; Aggarwal, A. CCR4+ monocytic myeloid-derived suppressor cells are associated with the increased epithelial-mesenchymal transition in pancreatic adenocarcinoma patients. 227, 152210. [CrossRef]
- Turnquist, H.; Lin, X.; Ashour, A.; Hollingsworth, M.; Singh, R.; Talmadge, J.; Solheim, J. CCL21 induces extensive intratumoral immune cell infiltration and specific anti-tumor cellular immunity. [CrossRef]
- Wang, H.; Chen, L.; Qi, L.; Jiang, N.; Zhang, Z.; Guo, H.; Song, T.; Li, J.; Li, H.; Zhang, N.; et al. A Single-Cell Atlas of Tumor-Infiltrating Immune Cells in Pancreatic Ductal Adenocarcinoma. 21, 100258. [CrossRef]
- Wang, X.; Wang, L.; Mo, Q.; Dong, Y.; Wang, G.; Ji, A. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. 8, 5702–5708.
- Mariani, M.; Lang, R.; Binda, E.; Panina-Bordignon, P.; D’Ambrosio, D. Dominance of CCL22 over CCL17 in induction of chemokine receptor CCR4 desensitization and internalization on human Th2 cells. 34, 231–240. [CrossRef]
- Rodriguez, E.; Zwart, E.S.; Affandi, A.A.; Verhoeff, J.; De Kok, M.; Boyd, L.N.C.; Meijer, L.L.; Le Large, T.Y.S.; Olesek, K.; Giovannetti, E.; et al. In-depth immune profiling of peripheral blood mononuclear cells in patients with pancreatic ductal adenocarcinoma reveals discriminative immune subpopulations. 115, 2170–2183. [CrossRef]
- Jiang, S.; Li, H.; Zhang, L.; Mu, W.; Zhang, Y.; Chen, T.; Wu, J.; Tang, H.; Zheng, S.; Liu, Y.; et al. Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. 53, D1670–D1676. [CrossRef]
- Ma, X.; Wu, D.; Zhou, S.; Wan, F.; Liu, H.; Xu, X.; Xu, X.; Zhao, Y.; Tang, M. The pancreatic cancer secreted REG4 promotes macrophage polarization to M2 through EGFR/AKT/CREB pathway. 35, 189–196. [CrossRef]
- Boyer, S.; Lee, H.J.; Steele, N.; Zhang, L.; Sajjakulnukit, P.; Andren, A.; Ward, M.H.; Singh, R.; Basrur, V.; Zhang, Y.; et al. Multiomic characterization of pancreatic cancer-associated macrophage polarization reveals deregulated metabolic programs driven by the GM-CSF–PI3K pathway. 11, e73796. [CrossRef]
- Bayne, L.; Beatty, G.; Jhala, N.; Clark, C.; Rhim, A.; Stanger, B.; Vonderheide, R. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. 21, 822–835. [CrossRef]
- Gu, H.; Deng, W.; Zheng, Z.; Wu, K.; Sun, F. CCL2 produced by pancreatic ductal adenocarcinoma is essential for the accumulation and activation of monocytic myeloid-derived suppressor cells. 9, 1686–1695. [CrossRef]
- Wang, J.; Saung, M.T.; Li, K.; Fu, J.; Fujiwara, K.; Niu, N.; Muth, S.; Wang, J.; Xu, Y.; Rozich, N.; et al. CCR2/CCR5 inhibitor permits the radiation-induced effector T cell infiltration in pancreatic adenocarcinoma. 219, e20211631. [CrossRef]
- Zhou, W.H.; Wang, Y.; Yan, C.; Du, W.D.; Al-Aroomi, M.A.; Zheng, L.; Lin, S.F.; Gao, J.X.; Jiang, S.; Wang, Z.X.; et al. CC chemokine receptor 7 promotes macrophage recruitment and induces M2-polarization through CC chemokine ligand 19&21 in oral squamous cell carcinoma. 13, 67. [CrossRef]
- Kulkarni, N.; Meitei, H.T.; Sonar, S.A.; Sharma, P.K.; Mujeeb, V.R.; Srivastava, S.; Boppana, R.; Lal, G. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. 88, 121–130. [CrossRef]
- Chellappa, S.; Hugenschmidt, H.; Hagness, M.; Line, P.D.; Labori, K.J.; Wiedswang, G.; Taskén, K.; Aandahl, E.M. Regulatory T cells that co-express RORγt and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. 5, e1102828. [CrossRef]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. 5, e1191731. [CrossRef]
- Im, J.H.; Buzzelli, J.N.; Jones, K.; Franchini, F.; Gordon-Weeks, A.; Markelc, B.; Chen, J.; Kim, J.; Cao, Y.; Muschel, R.J. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. 11, 4064. [CrossRef]
- Geraldo, L.H.; Garcia, C.; Xu, Y.; Leser, F.S.; Grimaldi, I.; De Camargo Magalhães, E.S.; Dejaegher, J.; Solie, L.; Pereira, C.M.; Correia, A.H.; et al. CCL21-CCR7 signaling promotes microglia/macrophage recruitment and chemotherapy resistance in glioblastoma. 80, 179. [CrossRef]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. 6, 7158. [CrossRef]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. 12, 484–501. [CrossRef]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. 10, 581–599. [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. 295, G709–G717. [CrossRef]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. 48, 919–922. [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. 214, 579–596. [CrossRef]
- Croft, W.; Pearce, H.; Margielewska-Davies, S.; Lim, L.; Nicol, S.M.; Zayou, F.; Blakeway, D.; Marcon, F.; Powell-Brett, S.; Mahon, B.; et al. Spatial determination and prognostic impact of the fibroblast transcriptome in pancreatic ductal adenocarcinoma. 12, e86125. [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. 118, 3367–3377. [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. 475, 226–230. [CrossRef]
- Wang, S.W.; Liu, S.C.; Sun, H.L.; Huang, T.Y.; Chan, C.H.; Yang, C.Y.; Yeh, H.I.; Huang, Y.L.; Chou, W.Y.; Lin, Y.M.; et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. 36, 104–114. [CrossRef]
- Wang, L.H.; Lin, C.Y.; Liu, S.C.; Liu, G.T.; Chen, Y.L.; Chen, J.J.; Chan, C.H.; Lin, T.Y.; Chen, C.K.; Xu, G.H.; et al. CCL5 promotes VEGF-C production and induces lymphangiogenesis by suppressing miR-507 in human chondrosarcoma cells. 7, 36896–36908. [CrossRef]
- Zhao, B.; Cui, K.; Wang, C.; Wang, A.; Zhang, B.; Zhou, W.; Zhao, W.; Li, S. The chemotactic interaction between CCL21 and its receptor, CCR7, facilitates the progression of pancreatic cancer via induction of angiogenesis and lymphangiogenesis. 18, 821–828. [CrossRef]
- Wang, Y.; Bian, Z.; Xu, L.; Du, G.; Qi, Z.; Zhang, Y.; Long, J.; Li, W. The scRNA-sequencing landscape of pancreatic ductal adenocarcinoma revealed distinct cell populations associated with tumor initiation and progression. 12, 101323. [CrossRef]
- Jakubowska, K.; Pryczynicz, A.; Januszewska, J.; Sidorkiewicz, I.; Kemona, A.; Niewiński, A.; Lewczuk, .; Kędra, B.; Guzińska-Ustymowicz, K. Expressions of Matrix Metalloproteinases 2, 7, and 9 in Carcinogenesis of Pancreatic Ductal Adenocarcinoma. 2016, 1–7. [CrossRef]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNγ and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. 6, 400–413. [CrossRef]
- Cui, K.; Zou, H.; Shi, M.; Ou, Y.; Han, L.; Zhang, B.; Hu, D.; Li, S. Gene Expression Profiles in Chemokine (C-C Motif) Ligand 21-Overexpressing Pancreatic Cancer Cells. 26, 201–208. [CrossRef]
- Li, H.; Li, H.; Li, X.; Zou, H.; Liu, L.; Liu, W.; Duan, T. C-C chemokine receptor type 2 promotes epithelial-to-mesenchymal transition by upregulating matrix metalloproteinase-2 in human liver cancer. [CrossRef]
- Bai, H.; Weng, Y.; Bai, S.; Jiang, Y.; Li, B.; He, F.; Zhang, R.; Yan, S.; Deng, F.; Wang, J.; et al. CCL5 secreted from bone marrow stromal cells stimulates the migration and invasion of Huh7 hepatocellular carcinoma cells via the PI3K-Akt pathway. 45, 333–343. [CrossRef]
- Cheng, X.; Wu, H.; Jin, Z.J.; Ma, D.; Yuen, S.; Jing, X.Q.; Shi, M.M.; Shen, B.Y.; Peng, C.H.; Zhao, R.; et al. Up-regulation of chemokine receptor CCR4 is associated with Human Hepatocellular Carcinoma malignant behavior. 7, 12362. [CrossRef]
- Du, D.; Liu, Y.; Qian, H.; Zhang, B.; Tang, X.; Zhang, T.; Liu, W. The Effects of the CCR6/CCL20 Biological Axis on the Invasion and Metastasis of Hepatocellular Carcinoma. 15, 6441–6452. [CrossRef]
- Lu, J.; Zhong, H.; Chu, T.; Zhang, X.; Li, R.; Sun, J.; Zhong, R.; Yang, Y.; Alam, M.S.; Lou, Y.; et al. Role of anlotinib-induced CCL2 decrease in anti-angiogenesis and response prediction for nonsmall cell lung cancer therapy. 53, 1801562. [CrossRef]
- Long, H.; Xie, R.; Xiang, T.; Zhao, Z.; Lin, S.; Liang, Z.; Chen, Z.; Zhu, B. Autocrine CCL5 Signaling Promotes Invasion and Migration of CD133+ Ovarian Cancer Stem-Like Cells via NF-κB-Mediated MMP-9 Upregulation. 30, 2309–2319. [CrossRef]
- Kato, T.; Fujita, Y.; Nakane, K.; Mizutani, K.; Terazawa, R.; Ehara, H.; Kanimoto, Y.; Kojima, T.; Nozawa, Y.; Deguchi, T.; et al. CCR1/CCL5 interaction promotes invasion of taxane-resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. 64, 251–257. [CrossRef]
- Lee, S.K.; Park, K.K.; Kim, H.J.; Park, J.; Son, S.H.; Kim, K.R.; Chung, W.Y. Human antigen R-regulated CCL20 contributes to osteolytic breast cancer bone metastasis. 7, 9610. [CrossRef]
- Li, J.; Sun, R.; Tao, K.; Wang, G. The CCL21/CCR7 pathway plays a key role in human colon cancer metastasis through regulation of matrix metalloproteinase-9. 43, 40–47. [CrossRef]
- Shen, B.; Zheng, M.Q.; Lu, J.W.; Jiang, Q.; Wang, T.H.; Huang, X.E. CXCL12-CXCR4 Promotes Proliferation and Invasion of Pancreatic Cancer Cells. 14, 5403–5408. [CrossRef]
- Zhang, J.; Liu, C.; Mo, X.; Shi, H.; Li, S. Mechanisms by which CXCR4/CXCL12 cause metastatic behavior in pancreatic cancer. [CrossRef]
- Xu, Q.; Wang, Z.; Chen, X.; Duan, W.; Lei, J.; Zong, L.; Li, X.; Sheng, L.; Ma, J.; Han, L.; et al. Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. 6, 4717–4732. [CrossRef]
- Hiraoka, N.; Ino, Y.; Yamazaki-Itoh, R.; Kanai, Y.; Kosuge, T.; Shimada, K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. 112, 1782–1790. [CrossRef]
- Vanhersecke, L.; Brunet, M.; Guégan, J.P.; Rey, C.; Bougouin, A.; Cousin, S.; Le Moulec, S.; Besse, B.; Loriot, Y.; Larroquette, M.; et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. 2, 794–802. [CrossRef]
- Luther, S.A.; Ansel, K.M.; Cyster, J.G. Overlapping Roles of CXCL13, Interleukin 7 Receptor α, and CCR7 Ligands in Lymph Node Development. 197, 1191–1198. [CrossRef]
- Luther, S.A.; Bidgol, A.; Hargreaves, D.C.; Schmidt, A.; Xu, Y.; Paniyadi, J.; Matloubian, M.; Cyster, J.G. Differing Activities of Homeostatic Chemokines CCL19, CCL21, and CXCL12 in Lymphocyte and Dendritic Cell Recruitment and Lymphoid Neogenesis. 169, 424–433. [CrossRef]
- Loef, E.J.; Sheppard, H.M.; Birch, N.P.; Dunbar, P.R. Live-Cell Microscopy Reveals That Human T Cells Primarily Respond Chemokinetically Within a CCL19 Gradient That Induces Chemotaxis in Dendritic Cells. 12, 628090. [CrossRef]
- Wendland, M.; Willenzon, S.; Kocks, J.; Davalos-Misslitz, A.; Hammerschmidt, S.; Schumann, K.; Kremmer, E.; Sixt, M.; Hoffmeyer, A.; Pabst, O.; et al. Lymph Node T Cell Homeostasis Relies on Steady State Homing of Dendritic Cells. 35, 945–957. [CrossRef]
- He, H.; Wu, J.; Zang, M.; Wang, W.; Chang, X.; Chen, X.; Wang, R.; Wu, Z.; Wang, L.; Wang, D.; et al. CCR6+ B lymphocytes responding to tumor cell-derived CCL20 support hepatocellular carcinoma progression via enhancing angiogenesis. 7, 1151–1163.
- Bell, D.; Chomarat, P.; Broyles, D.; Netto, G.; Harb, G.M.; Lebecque, S.; Valladeau, J.; Davoust, J.; Palucka, K.A.; Banchereau, J. In Breast Carcinoma Tissue, Immature Dendritic Cells Reside within the Tumor, Whereas Mature Dendritic Cells Are Located in Peritumoral Areas. 190, 1417–1426. [CrossRef]
- De Silva, P.; Garaud, S.; Solinas, C.; De Wind, A.; Van Den Eyden, G.; Jose, V.; Gu-Trantien, C.; Migliori, E.; Boisson, A.; Naveaux, C.; et al. FOXP1 negatively regulates tumor infiltrating lymphocyte migration in human breast cancer. 39, 226–238. [CrossRef]
- Zou, X.; Lin, X.; Cheng, H.; Chen, Y.; Wang, R.; Ma, M.; Liu, Y.; Dai, Z.; Tasiheng, Y.; Yan, Y.; et al. Characterization of intratumoral tertiary lymphoid structures in pancreatic ductal adenocarcinoma: cellular properties and prognostic significance. 11, e006698. [CrossRef]
- Guo, J.; Wang, S.; Gao, Q. An integrated overview of the immunosuppression features in the tumor microenvironment of pancreatic cancer. 14, 1258538. [CrossRef]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. 38, 800–811. [CrossRef]
- Kimsey, T.F.; Campbell, A.S.; Albo, D.; Wang, T.N. Co-localization of Macrophage Inflammatory Protein-3α (Mip-3α) and Its Receptor, CCR6, Promotes Pancreatic Cancer Cell Invasion. 10, 374.
- Yang, J.; Lv, X.; Chen, J.; Xie, C.; Xia, W.; Jiang, C.; Zeng, T.; Ye, Y.; Ke, L.; Yu, Y.; et al. CCL2-CCR2 axis promotes metastasis of nasopharyngeal carcinoma by activating ERK1/2-MMP2/9 pathway. 7, 15632–15647. [CrossRef]
- Van Deventer, H.W.; Wu, Q.P.; Bergstralh, D.T.; Davis, B.K.; O’Connor, B.P.; Ting, J.P.Y.; Serody, J.S. C-C Chemokine Receptor 5 on Pulmonary Fibrocytes Facilitates Migration and Promotes Metastasis via Matrix Metalloproteinase 9. 173, 253–264. [CrossRef]
- Chuang, J.; Yang, W.; Chen, H.; Huang, C.; Tan, T.; Lin, Y.; Hsu, C.; Fong, Y.; Tang, C. CCL5/CCR5 axis promotes the motility of human oral cancer cells. 220, 418–426. [CrossRef]
- Zhao, H.; Bo, Q.; Wang, W.; Wang, R.; Li, Y.; Chen, S.; Xia, Y.; Wang, W.; Wang, Y.; Zhu, K.; et al. CCL17-CCR4 axis promotes metastasis via ERK/MMP13 pathway in bladder cancer. 120, 1979–1989. [CrossRef]
- Huang, H.; Zepp, M.; Georges, R.B.; Jarahian, M.; Kazemi, M.; Eyol, E.; Berger, M.R. The CCR5 antagonist maraviroc causes remission of pancreatic cancer liver metastasis in nude rats based on cell cycle inhibition and apoptosis induction. 474, 82–93. [CrossRef]
- Peng, J.; Madduri, S.; Clontz, A.D.; Stewart, D.A. Clinical trial-identified inflammatory biomarkers in breast and pancreatic cancers. 14, 1106520. [CrossRef]
- Lee, E.; Fertig, E.J.; Jin, K.; Sukumar, S.; Pandey, N.B.; Popel, A.S. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. 5, 4715. [CrossRef]
- Zeng, H.; Hou, Y.; Zhou, X.; Lang, L.; Luo, H.; Sun, Y.; Wan, X.; Yuan, T.; Wang, R.; Liu, Y.; et al. Cancer-associated fibroblasts facilitate premetastatic niche formation through lncRNA SNHG5-mediated angiogenesis and vascular permeability in breast cancer. 12, 7351–7370. [CrossRef]
- Chen, L.; Xu, G.; Song, X.; Zhang, L.; Chen, C.; Xiang, G.; Wang, S.; Zhang, Z.; Wu, F.; Yang, X.; et al. A novel antagonist of the CCL5/CCR5 axis suppresses the tumor growth and metastasis of triple-negative breast cancer by CCR5-YAP1 regulation. 583, 216635. [CrossRef]
- Olkhanud, P.B.; Baatar, D.; Bodogai, M.; Hakim, F.; Gress, R.; Anderson, R.L.; Deng, J.; Xu, M.; Briest, S.; Biragyn, A. Breast Cancer Lung Metastasis Requires Expression of Chemokine Receptor CCR4 and Regulatory T Cells. 69, 5996–6004. [CrossRef]
- del Castillo, C.F.; Warshaw, L. Peritoneal metastases in pancreatic carcinoma. 40, 430–432.
- Yachida, S.; Iacobuzio-Donahue, C.A. The Pathology and Genetics of Metastatic Pancreatic Cancer. 133, 413–422. [CrossRef]
- Fukui, R.; Nishimori, H.; Hata, F.; Yasoshima, T.; Ohno, K.; Nomura, H.; Yanai, Y.; Tanaka, H.; Kamiguchi, K.; Denno, R.; et al. Metastases-Related Genes in the Classification of Liver and Peritoneal Metastasis in Human Gastric Cancer. 129, 94–100. [CrossRef]
- Wang, T.; Wei, Y.; Tian, L.; Song, H.; Ma, Y.; Yao, Q.; Feng, M.; Wang, Y.; Gao, M.; Xue, Y. C-C motif chemokine ligand 5 (CCL5) levels in gastric cancer patient sera predict occult peritoneal metastasis and a poorer prognosis. 32, 136–142. [CrossRef]
- Cao, L.; Hu, X.; Zhang, J.; Huang, G.; Zhang, Y. The role of the CCL22-CCR4 axis in the metastasis of gastric cancer cells into omental milky spots. 12, 267. [CrossRef]
- Klein, A.; Sagi-Assif, O.; Meshel, T.; Telerman, A.; Izraely, S.; Ben-Menachem, S.; Bayry, J.; Marzese, D.M.; Ohe, S.; Hoon, D.S.; et al. CCR4 is a determinant of melanoma brain metastasis. 8, 31079–31091. [CrossRef]
- Yamamoto, Y.; Ikoma, H.; Morimura, R.; Konishi, H.; Murayama, Y.; Komatsu, S.; Shiozaki, A.; Kuriu, Y.; Kubota, T.; Nakanishi, M.; et al. The clinical impact of the lymph node ratio as a prognostic factor after resection of pancreatic cancer. 34, 2389–2394.
- Malleo, G.; Maggino, L.; Casciani, F.; Lionetto, G.; Nobile, S.; Lazzarin, G.; Paiella, S.; Esposito, A.; Capelli, P.; Luchini, C.; et al. Importance of Nodal Metastases Location in Pancreatoduodenectomy for Pancreatic Ductal Adenocarcinoma: Results from a Prospective, Lymphadenectomy Protocol. 29, 3477–3488. [CrossRef]
- Kanda, M.; Fujii, T.; Nagai, S.; Kodera, Y.; Kanzaki, A.; Sahin, T.T.; Hayashi, M.; Yamada, S.; Sugimoto, H.; Nomoto, S.; et al. Pattern of Lymph Node Metastasis Spread in Pancreatic Cancer. 40, 951–955. [CrossRef]
- Zhang, L.; Wang, D.; Li, Y.; Liu, Y.; Xie, X.; Wu, Y.; Zhou, Y.; Ren, J.; Zhang, J.; Zhu, H.; et al. CCL21/CCR7 Axis Contributed to CD133+ Pancreatic Cancer Stem-Like Cell Metastasis via EMT and Erk/NF-κB Pathway. 11, e0158529. [CrossRef]
- Lee, H.J.; You, D.D.; Choi, D.W.; Choi, Y.S.; Kim, S.J.; Won, Y.S.; Moon, H.J. Significance of CD133 as a cancer stem cell markers focusing on the tumorigenicity of pancreatic cancer cell lines. 81, 263. [CrossRef]
- Saur, D.; Seidler, B.; Schneider, G.; Algül, H.; Schmid, R. CXCR4 expression increases liver and lung metastasis in a mouse model of pancreatic cancer. 43, s–2005–920101. [CrossRef]
- Jiao, X.; Shu, G.; Liu, H.; Zhang, Q.; Ma, Z.; Ren, C.; Guo, H.; Shi, J.; Liu, J.; Zhang, C.; et al. The Diagnostic Value of Chemokine/Chemokine Receptor Pairs in Hepatocellular Carcinoma and Colorectal Liver Metastasis. 67, 299–308. [CrossRef]
- Cao, Z.; Xu, X.; Luo, X.; Li, L.; Huang, B.; Li, X.; Tao, D.; Hu, J.; Gong, J. Role of RANTES and its receptor in gastric cancer metastasis. 31, 342–347. [CrossRef]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. 20, 558–571. [CrossRef]
- Chang, L.Y.; Lin, Y.C.; Mahalingam, J.; Huang, C.T.; Chen, T.W.; Kang, C.W.; Peng, H.M.; Chu, Y.Y.; Chiang, J.M.; Dutta, A.; et al. Tumor-Derived Chemokine CCL5 Enhances TGF-β–Mediated Killing of CD8+ T Cells in Colon Cancer by T-Regulatory Cells. 72, 1092–1102. [CrossRef]
- Pant, A.; Jain, A.; Chen, Y.; Patel, K.; Saleh, L.; Tzeng, S.; Nitta, R.T.; Zhao, L.; Wu, C.Y.J.; Bederson, M.; et al. The CCR6–CCL20 Axis Promotes Regulatory T-cell Glycolysis and Immunosuppression in Tumors. 12, 1542–1558. [CrossRef]
- Rapp, M.; Wintergerst, M.W.; Kunz, W.G.; Vetter, V.K.; Knott, M.M.; Lisowski, D.; Haubner, S.; Moder, S.; Thaler, R.; Eiber, S.; et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. 216, 1170–1181. [CrossRef]
- Le, Y.; Gao, H.; Richards, W.; Zhao, L.; Bleday, R.; Clancy, T.; Zhu, Z. VentX expression in tumor-associated macrophages promotes phagocytosis and immunity against pancreatic cancers. 5, e137088. [CrossRef]
- He, Z.; Wang, J.; Zhu, C.; Xu, J.; Chen, P.; Jiang, X.; Chen, Y.; Jiang, J.; Sun, C. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. 548, 215751. [CrossRef]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. 7, 255. [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. 181, 5791–5802. [CrossRef]
- Hsu, Y.L.; Yen, M.C.; Chang, W.A.; Tsai, P.H.; Pan, Y.C.; Liao, S.H.; Kuo, P.L. CXCL17-derived CD11b+Gr-1+ myeloid-derived suppressor cells contribute to lung metastasis of breast cancer through platelet-derived growth factor-BB. 21, 23. [CrossRef]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K.; et al. Hyaluronan Synthase HAS2 Promotes Tumor Progression in Bone by Stimulating the Interaction of Breast Cancer Stem–Like Cells with Macrophages and Stromal Cells. 72, 537–547. [CrossRef]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J.; et al. Designing Liposomes To Suppress Extracellular Matrix Expression To Enhance Drug Penetration and Pancreatic Tumor Therapy. 11, 8668–8678. [CrossRef]
- Lee, H.O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. 11, 245. [CrossRef]
- Provenzano, P.; Cuevas, C.; Chang, A.; Goel, V.; Von Hoff, D.; Hingorani, S. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. 21, 418–429. [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. 207, 2439–2453. [CrossRef]
- Shi, X.; Liu, H.; Liang, Z. Cellular crosstalk of regulatory T cells in pancreatic ductal adenocarcinoma. 7, 131–140. [CrossRef]
- Kuen, J.; Darowski, D.; Kluge, T.; Majety, M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. 12, e0182039. [CrossRef]
- Chandra, V.; Li, L.; Le Roux, O.; Zhang, Y.; Howell, R.M.; Rupani, D.N.; Baydogan, S.; Miller, H.D.; Riquelme, E.; Petrosino, J.; et al. Gut epithelial Interleukin-17 receptor A signaling can modulate distant tumors growth through microbial regulation. 42, 85–100.e6. [CrossRef]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. 217, e20190354. [CrossRef]
- Picard, F.S.R.; Lutz, V.; Brichkina, A.; Neuhaus, F.; Ruckenbrod, T.; Hupfer, A.; Raifer, H.; Klein, M.; Bopp, T.; Pfefferle, P.I.; et al. IL-17A-producing CD8+ T cells promote PDAC via induction of inflammatory cancer-associated fibroblasts. 72, 1510–1522. [CrossRef]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. 31, 787–798. [CrossRef]
- Lei, X.; Xiao, R.; Chen, Z.; Ren, J.; Zhao, W.; Tang, W.; Wen, K.; Zhu, Y.; Li, X.; Ouyang, S.; et al. Augmenting antitumor efficacy of Th17-derived Th1 cells through IFN-γ-induced type I interferon response network via IRF7. 121, e2412120121. [CrossRef]
- Gnerlich, J.L.; Mitchem, J.B.; Weir, J.S.; Sankpal, N.V.; Kashiwagi, H.; Belt, B.A.; Porembka, M.R.; Herndon, J.M.; Eberlein, T.J.; Goedegebuure, P.; et al. Induction of Th17 Cells in the Tumor Microenvironment Improves Survival in a Murine Model of Pancreatic Cancer. 185, 4063–4071. [CrossRef]
- Little, A.C.; Pathanjeli, P.; Wu, Z.; Bao, L.; Goo, L.E.; Yates, J.A.; Oliver, C.R.; Soellner, M.B.; Merajver, S.D. IL-4/IL-13 Stimulated Macrophages Enhance Breast Cancer Invasion Via Rho-GTPase Regulation of Synergistic VEGF/CCL-18 Signaling. 9, 456. [CrossRef]
- Prokopchuk, O.; Liu, Y.; Henne-Bruns, D.; Kornmann, M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: evidence for autocrine and paracrine actions. 92, 921–928. [CrossRef]
- Traub, B.; Sun, L.; Ma, Y.; Xu, P.; Lemke, J.; Paschke, S.; Henne-Bruns, D.; Knippschild, U.; Kornmann, M. Endogenously Expressed IL-4Rα Promotes the Malignant Phenotype of Human Pancreatic Cancer In Vitro and In Vivo. 18, 716. [CrossRef]
- Jacenik, D.; Karagiannidis, I.; Beswick, E.J. Th2 cells inhibit growth of colon and pancreas cancers by promoting anti-tumorigenic responses from macrophages and eosinophils. 128, 387–397. [CrossRef]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. 15, 2403–2413. [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. 364, 1817–1825. [CrossRef]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Pazo Cid, R.; Chandana, S.R.; De La Fouchardière, C.; Dean, A.; Kiss, I.; Lee, W.J.; Goetze, T.O.; et al. NALIRIFOX versus nab-paclitaxel and gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (NAPOLI 3): a randomised, open-label, phase 3 trial. 402, 1272–1281. [CrossRef]
- Kindler, H.L.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Overall Survival Results From the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. 40, 3929–3939. [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. 38, 1–10. [CrossRef]
- Schram, A.M.; Goto, K.; Kim, D.W.; Macarulla, T.; Hollebecque, A.; O’Reilly, E.M.; Ou, S.H.I.; Rodon, J.; Rha, S.Y.; Nishino, K.; et al. Efficacy of Zenocutuzumab in NRG1 Fusion–Positive Cancer. 392, 566–576. [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; Van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. 21, 531–540. [CrossRef]
- Pishvaian, M.J.; Rolfo, C.D.; Liu, S.V.; Multani, P.S.; Chow Maneval, E.; Garrido-Laguna, I. Clinical benefit of entrectinib for patients with metastatic pancreatic cancer who harbor NTRK and ROS1 fusions. 36, 521–521. [CrossRef]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. 33, 3124–3129. [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. 70, 148–156. [CrossRef]
- Kim, D.W.; Schram, A.M.; Hollebecque, A.; Nishino, K.; Macarulla, T.; Rha, S.Y.; Duruisseaux, M.; Liu, S.V.; Al Hallak, M.N.; Umemoto, K.; et al. The phase I/II eNRGy trial: Zenocutuzumab in patients with cancers harboring NRG1 gene fusions. 20, 1057–1067. [CrossRef]
- Allen, M.J.; Zhang, A.; Bavi, P.; Kim, J.C.; Jang, G.H.; Kelly, D.; Perera, S.; Denroche, R.E.; Notta, F.; Wilson, J.M.; et al. Molecular characterisation of pancreatic ductal adenocarcinoma with NTRK fusions and review of the literature. 76, 158–165. [CrossRef]
- Knepper, T.C.; Kim, D.W.; Mauer, E.; Ronski, K.; Gulhati, P. Comparative analysis of the targetable landscape in KRAS -mutant and wild-type pancreatic adenocarcinoma. 40, 4155–4155. [CrossRef]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. 23, 137–148. [CrossRef]
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F.; et al. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. 80, 3101–3115. [CrossRef]
- Tian, J.; Zhang, D.; Kurbatov, V.; Wang, Q.; Wang, Y.; Fang, D.; Wu, L.; Bosenberg, M.; Muzumdar, M.D.; Khan, S.; et al. 5-Fluorouracil efficacy requires anti-tumor immunity triggered by cancer-cell-intrinsic STING. 40, e106065. [CrossRef]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. 8, 1736. [CrossRef]
- Wan, Z.; Huang, H.; West Iii, R.E.; Zhang, M.; Zhang, B.; Cai, X.; Zhang, Z.; Luo, Z.; Chen, Y.; Zhang, Y.; et al. Overcoming pancreatic cancer immune resistance by codelivery of CCR2 antagonist using a STING-activating gemcitabine-based nanocarrier. 62, 33–50. [CrossRef]
- Grierson, P.M.; Wolf, C.; Suresh, R.; Wang-Gillam, A.; Tan, B.R.; Ratner, L.; Oppelt, P.; Aranha, O.; Frith, A.; Pedersen, K.S.; et al. Neoadjuvant BMS-813160, Nivolumab, Gemcitabine, and Nab-Paclitaxel for Patients with Pancreatic Cancer. 31, 3644–3651. [CrossRef]
- Napolitano, F.; Formisano, L.; Giardino, A.; Girelli, R.; Servetto, A.; Santaniello, A.; Foschini, F.; Marciano, R.; Mozzillo, E.; Carratù, A.C.; et al. Neoadjuvant Treatment in Locally Advanced Pancreatic Cancer (LAPC) Patients with FOLFIRINOX or Gemcitabine NabPaclitaxel: A Single-Center Experience and a Literature Review. 11, 981. [CrossRef]
- Choi, Y.H.; Lee, S.H.; You, M.S.; Shin, B.S.; Paik, W.H.; Ryu, J.K.; Kim, Y.T.; Kwon, W.; Jang, J.Y.; Kim, S.W. Prognostic Factors for Patients with Borderline Resectable or Locally Advanced Pancreatic Cancer Receiving Neoadjuvant FOLFIRINOX. 15, 315–323. [CrossRef]
- Adams, D.L.; Raghavakaimal, A.; Tang, C.M.; Gardner, K.P.; Kelly, S.; Pourhassan, N.; Ray, N. Safety, efficacy, and clinical outcomes of the anti-CCR5 inhibitor (Leronlimab): A pooled analysis of three clinical trials in patients with mTNBC. 40, e13062–e13062. [CrossRef]
- Luo, H.; Ikenaga, N.; Nakata, K.; Higashijima, N.; Zhong, P.; Kubo, A.; Wu, C.; Tsutsumi, C.; Shimada, Y.; Hayashi, M.; et al. Tumor-associated neutrophils upregulate Nectin2 expression, creating the immunosuppressive microenvironment in pancreatic ductal adenocarcinoma. 43, 258. [CrossRef]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. 29, 587–601. [CrossRef]
- Haag, G.M.; Springfeld, C.; Grün, B.; Apostolidis, L.; Zschäbitz, S.; Dietrich, M.; Berger, A.K.; Weber, T.F.; Zoernig, I.; Schaaf, M.; et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer – The PICCASSO phase I trial. 167, 112–122. [CrossRef]
- Kim, T.M.; Ngamphaiboon, N.; Lee, K.H.; Desai, P.; Lin, C.C.; Muzaffar, J.; Feng, Y.H.; Wu, S.Y.; Brahmer, J.; Chisamore, M.; et al. 629-C Phase 2 safety and efficacy of oral CCR4 antagonist FLX475 (tivumecirnon) plus pembrolizumab in subjects with non-small cell lung cancer not previously treated with checkpoint inhibitor. In Proceedings of the Regular Abstracts – Part 2. BMJ Publishing Group Ltd, pp. A1808–A1808. [CrossRef]
- Muzaffar, J.; Kirtane, K.; Redman, R.; Yang, M.H.; Kim, T.M.; Liu, S.; Lynch, R.; Brahmer, J.; LoRusso, P.; Henick, B.; et al. Abstract CT226: Phase 2 study of oral CCR4 antagonist FLX475 (tivumecirnon) plus pembrolizumab in subjects with head and neck squamous cell carcinoma (HNSCC) previously treated with checkpoint inhibitor. 84, CT226–CT226. [CrossRef]
- Oh, D.Y.; Ryu, M.H.; Hwang, J.E.; Cho, J.; Zang, D.Y.; Oh, S.C.; Lee, J.; Lee, K.W.; Rha, S.Y.; Shim, B.Y.; et al. A phase 2 study to assess the safety and efficacy of FLX475 (tivumecirnon) combined with pembrolizumab in patients with advanced or metastatic gastric cancer. 43, 426–426. [CrossRef]
- Li, W.; Crouse, K.K.; Alley, J.; Frisbie, R.K.; Fish, S.C.; Andreyeva, T.A.; Reed, L.A.; Thorn, M.; DiMaggio, G.; Donovan, C.B.; et al. A Novel C-C Chemoattractant Cytokine (Chemokine) Receptor 6 (CCR6) Antagonist (PF-07054894) Distinguishes between Homologous Chemokine Receptors, Increases Basal Circulating CCR6+ T Cells, and Ameliorates Interleukin-23-Induced Skin Inflammation. 386, 80–92. [CrossRef]
- Meyer, E.A.; Croxford, A.L.; Gnerre, C.; Kulig, P.; Murphy, M.J.; Jacob, E.M.; Schäfer, G.; Richard-Bildstein, S.; Aissaoui, H.; Bouis, P.; et al. Discovery of the Clinical Candidate IDOR-1117-2520: A Potent and Selective Antagonist of CCR6 for Autoimmune Diseases. 67, 8077–8098. [CrossRef]
- Cuesta-Mateos, C.; Terrón, F.; Herling, M. CCR7 in Blood Cancers – Review of Its Pathophysiological Roles and the Potential as a Therapeutic Target. 11, 736758. [CrossRef]
- Nakata, B.; Fukunaga, S.; Noda, E.; Amano, R.; Yamada, N.; Hirakawa, K. Chemokine Receptor CCR7 Expression Correlates with Lymph Node Metastasis in Pancreatic Cancer. 74, 69–75. [CrossRef]
- Cadilha, B.L.; Benmebarek, M.R.; Dorman, K.; Oner, A.; Lorenzini, T.; Obeck, H.; Vänttinen, M.; Di Pilato, M.; Pruessmann, J.N.; Stoiber, S.; et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. 7, eabi5781. [CrossRef]
- Pietrobono, S.; Bertolini, M.; De Vita, V.; Sabbadini, F.; Fazzini, F.; Frusteri, C.; Scarlato, E.; Mangiameli, D.; Quinzii, A.; Casalino, S.; et al. CCL3 predicts exceptional response to TGFβ inhibition in basal-like pancreatic cancer enriched in LIF-producing macrophages. 8, 246. [CrossRef]
- Messex, J.; Adams, K.; Hawkins, W.; DeNardo, D.; Bardeesy, N.; Billadeau, D.; Liou, G.Y. Oncogenic Kras-Mediated Cytokine CCL15 Regulates Pancreatic Cancer Cell Migration and Invasion through ROS. 14, 2153. [CrossRef]
- Lekan, A.A.; Weiner, L.M. The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma. 16, 559. [CrossRef]
Figure 1.
Chord Diagram of Known CC Chemokine Ligand-Receptor Interactions in Humans. Binding relationships between human CC chemokines (CCL1-CCL28) and their corresponding receptors (CCR1-CCR10). Chemokines are shown on the left half of the circle, and their receptors on the right. Only human interactions are shown; mouse-specific ligands (e.g., CCL6, CCL9/10, and CCL12) are excluded. Adapted from Hughes et al. [10]. Visualization generated with pyCirclize v1.9.1 in Python.
Figure 1.
Chord Diagram of Known CC Chemokine Ligand-Receptor Interactions in Humans. Binding relationships between human CC chemokines (CCL1-CCL28) and their corresponding receptors (CCR1-CCR10). Chemokines are shown on the left half of the circle, and their receptors on the right. Only human interactions are shown; mouse-specific ligands (e.g., CCL6, CCL9/10, and CCL12) are excluded. Adapted from Hughes et al. [10]. Visualization generated with pyCirclize v1.9.1 in Python.
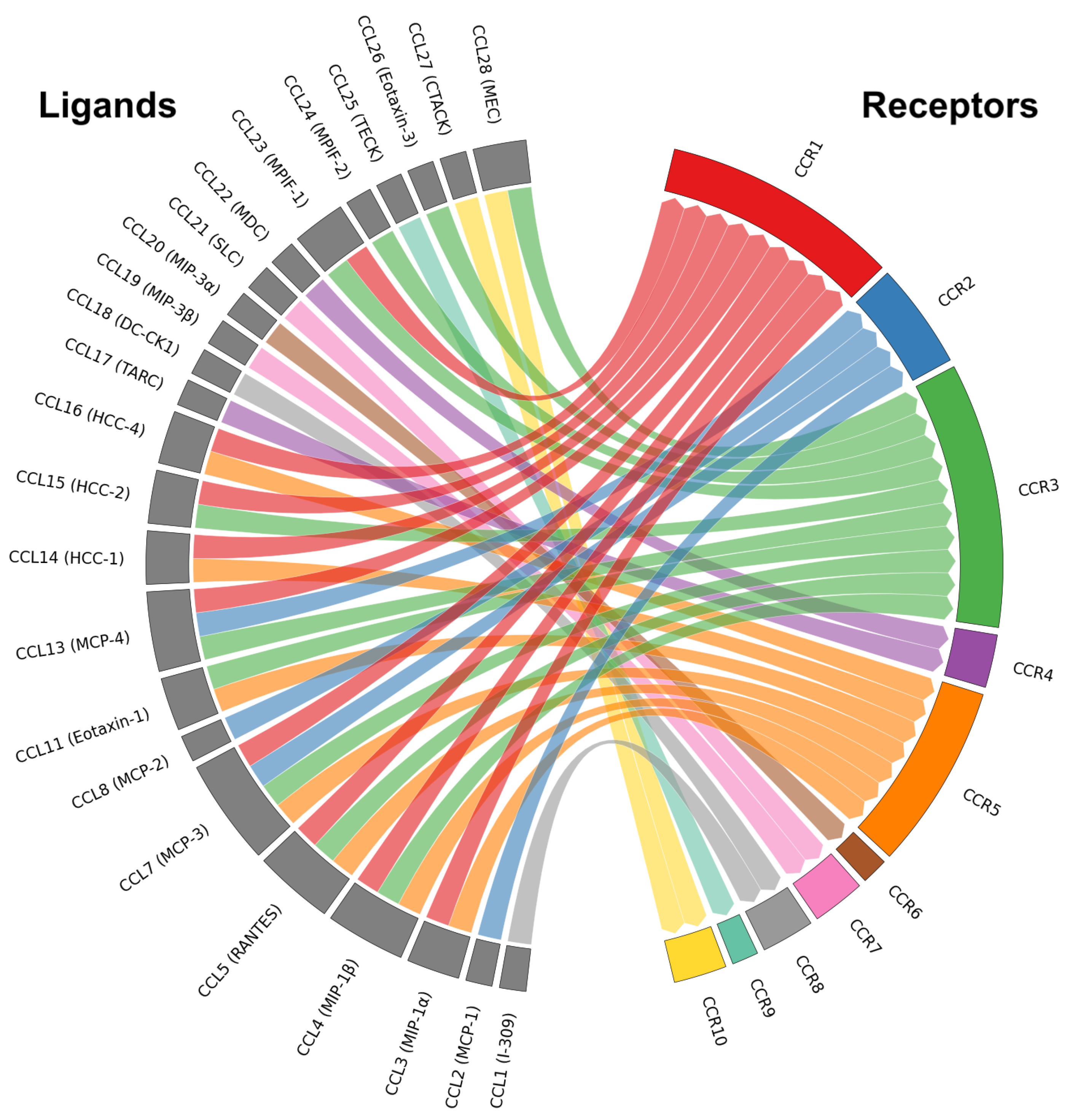
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



