Submitted:
15 September 2025
Posted:
15 September 2025
You are already at the latest version
Abstract
Polycystic ovary syndrome (PCOS) is a systemic metabolic and endocrine disorder that significantly disrupts reproductive physiology and endometrial function. In this narrative review, we examine the molecular impact of metabolic and hormonal imbalances on the endometrium of women with PCOS. We investigate the specific mechanisms that delineate how hyperinsulinemia and insulin resistance, chronic low-grade inflammation, and estrogen/progesterone/androgen imbalance, contribute to altered epigenetic, transcriptomic, metabolomic and signalling profiles in a wide array of different cell types within endometrial tissues. The synergistic interplay between upregulated inflammatory cytokines (e.g., IL-1,2,6,8,17,18, TNF-α), along with key changes in critical molecular pathways associated with hyperinsulinemia and insulin resistance (e.g., PI3K/AKT/MAPK, Wnt/β-catenin), in addition to aberrant sex steroid hormone signalling (e.g., CYP19A1, COX-2, PGE2, HOXA10, 11βHSD2), promotes deleterious changes within the endometrial microenvironment. These anomalies underpin a spectrum of clinical manifestations observed in women with PCOS at each stage of the life course, including abnormal uterine bleeding in reproductive-age women, impaired decidualization in pregnancy, and altered postmenopausal endometrial physiology. Clinically, these alterations are associated with abnormal uterine bleeding, subfertility, implantation failure, miscarriage, pregnancy complications, and postmenopausal endometrial hyperplasia and cancer. Overall, our review provides novel insights into the molecular mechanisms linking systemic metabolic and endocrine dysfunction with endometrial pathology in PCOS and has broader implications that apply to all women.
Keywords:
Polycystic ovary syndrome
; endometrium
; metabolic
; hormonal
; endocrine
; dysfunctional uterine bleeding
; pregnancy
; decidualization
; postmenopausal
; cancer
1. Introduction
Polycystic ovary syndrome (PCOS) is increasingly being viewed as an evolutionary mismatch disorder that becomes apparent after exposure to contemporary lifestyle, nutritional and environmental factors.(1–6). Having a metabolically thrifty metabolism was an evolutionary advantage in times of famine, as it enhanced the capacity to store energy (e.g., as triglycerides) for later use in times of food scarcity (1). However, in today’s environment, characterized by diminished diet quality, calorie surplus, sedentary behavior, and pervasive lifestyle shifts, these previously advantageous phenotypes have become maladaptive, exerting detrimental effects on both metabolic and reproductive health. Consistent with the evolutionary models, the international guidelines on PCOS provide evidence that many of the symptoms, biochemical and endocrine changes are reversible following diet, exercise, and other lifestyle and medical interventions (7). It is anticipated that characterizing PCOS in an evolutionary perspective may help create a framework conducive to promoting healthy lifestyle and preventative interventions (8).
PCOS affects 8-13% of women globally and encompasses a constellation of metabolic and endocrine disturbances that define it as a true systemic disorder (9,10). This is reflected in the current international consensus process to change the name to reflect the contribution of metabolic and endocrine factors (7,11). Analysis of the Global Burden of Disease Study data found that the global prevalence of PCOS and related infertility is increasing (12). The escalating prevalence of PCOS in lower socio-demographic regions is particularly concerning. The authors identified environmental and behavioural risks—such as high fat/high sugar diets, sedentary behavior, stress, circadian disruption, and exposure to endocrine disrupting chemicals (EDC)—as the fundamental drivers (12).
Key features of PCOS, including insulin resistance (IR), hyperinsulinemia, low-grade chronic systemic inflammation (CSI), and hyperandrogenism (HA), act synergistically to impair endometrial function, leading to menstrual disturbance, reduced fertility and adverse obstetric outcomes (13–15). Although PCOS is now appreciated to be a multisystem disorder, the precise molecular pathways that induce endometrial dysfunction are still under investigation (14,16,17). The convergence of high throughput profiling (e.g., transcriptomics), advanced bioinformatics (Gene Set Enrichment Analysis, Connectivity Map), and novel in-vitro models (e.g., organoids), are providing insights into how molecular networks regulate endometrial function (18–20).
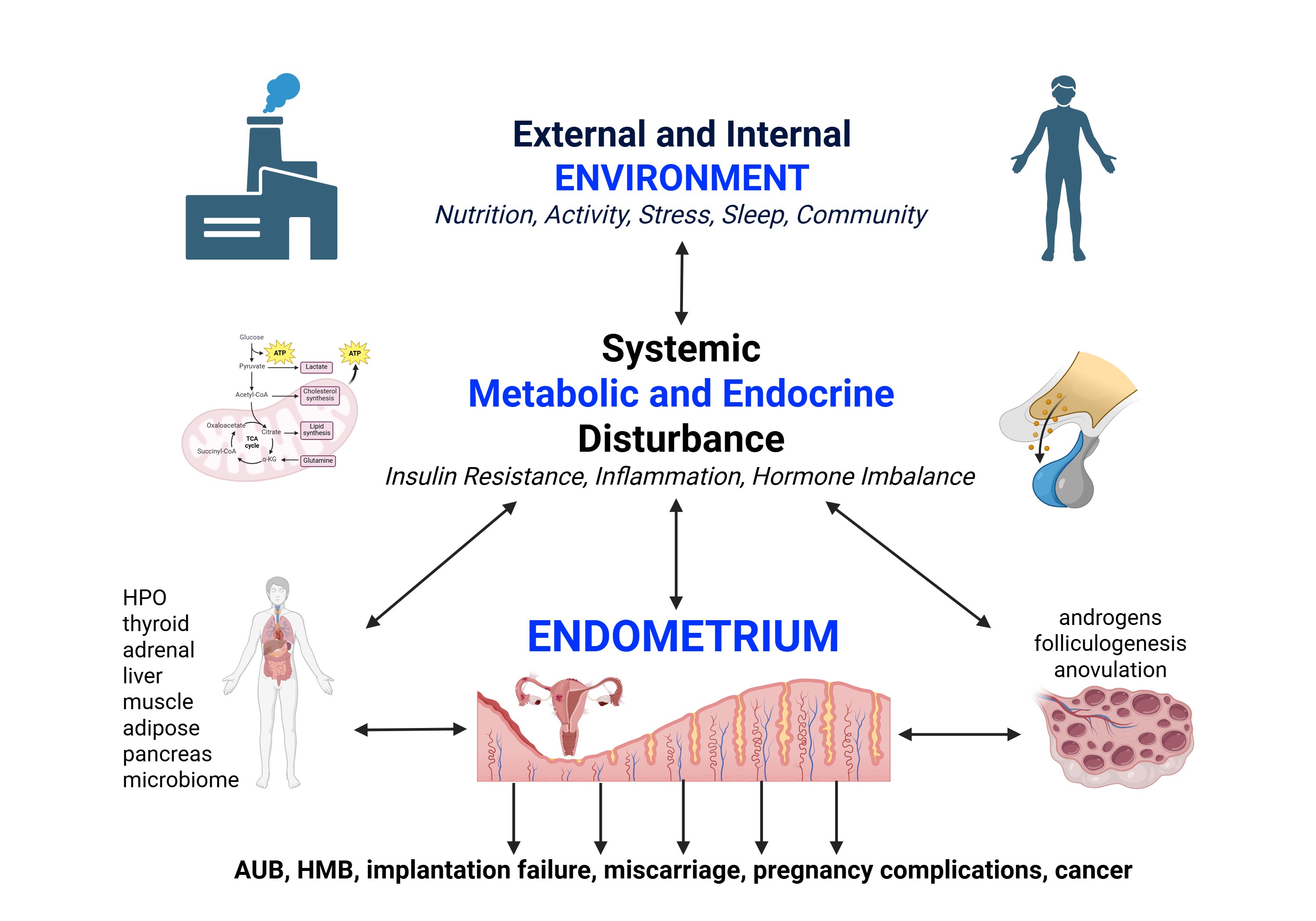
Emerging evidence implicates altered insulin receptor signalling (phosphoinositide 3-kinase/serine-threonine specific kinase-mitogen activated protein kinase (PI3K/AKT/MAPK) (21), androgen-mediated shifts in Wilms Tumour-1 (WT1)—a transcription factor important for cell development and survival (22)—and Wingless-related integration site (Wnt)/β-catenin activity (23), in disrupting precisely coordinated hormone-induced changes in the PCOS endometrium (24,25). At the same time, elevated inflammatory cytokines, such as tumour necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), disrupt local hormone networks by interfering with estrogen (E2), progesterone (P4), and insulin receptor signalling (14,26). Together, the resulting signalling defects alter epigenetic regulatory pathways (27), change the endometrial transcriptome (the set of all RNA transcripts) (28) and metabolome (the collection of small molecules produced by a cell) (29), and impair cell-to-cell communication by disrupting gap junction and paracrine signalling networks (17). Ultimately, the disruption of finely tuned proliferative and secretory phase hormonal and metabolic signalling networks play a pivotal role in the pathophysiology of endometrial dysfunction (Figure 1).
Clinically, these molecular events have unique effects at different life stages. In reproductive-age women they may present as altered bleeding patterns, excessive endometrial proliferation, impaired fertility, and reduced implantation potential (Figure 1) (30). In pregnancy, altered bidirectional communication leads to impaired stromal decidualization and trophoblast invasion increasing the risk of miscarriage, preterm birth, preeclampsia, and other pregnancy complications (31,32). After the menopausal transition, residual metabolic, inflammatory, and hormonal dysregulation may promote endometrial overgrowth and enhance neoplastic potential (33).
This narrative review integrates advances in our understanding of how IR, CSI, and E2/P4/androgen imbalance remodel the endometrial environment at a molecular level. Our aim is to highlight the role of the core pathophysiological components of PCOS in endometrial dysfunction. This should inform future research and guide precision-based approaches to the prevention and management of endometrial health across the lifespan.
2. Scope and Methodology
Section 3—Endometrial Function in Reproductive-Age Women—is a summary of normal endometrial function in women of reproductive age and provides the background for subsequent sections on the effect of IR, hyperinsulinemia, CSI, and hormonal imbalance on normal physiology. The normal role of the mucosal immune system and related microbiome (MB) are discussed. Readers are directed to the corresponding discussion in section 4 to link normal endometrial physiology with the detailed pathophysiology.
Section 4—Pathophysiology of Dysfunctional Endometrium—provides a review of endometrial changes related to IR, CSI, and E2/P4/androgen imbalance, particularly in women with PCOS. This section also includes a review of the molecular alterations in abnormal uterine bleeding (AUB) and heavy menstrual bleeding (HMB). This section provides an overview of the place of PCOS in the classification of AUB, followed by a detailed molecular description of endometrial events in AUB and PCOS. Section 4 also includes a summary of emerging research from endometrial organoid models.
The list of bibliographic references is based on MEDLINE, PubMed, Scopus, Google Scholar and Cochrane databases. Databases were searched from inception to September 2025. Papers reviewed include primary research, narrative reviews, and systematic reviews. Additional articles were obtained from the bibliography list of retrieved publications. To optimize our literature search, we also employed Microsoft Copilot (GPT-4; Microsoft 2025) to locate relevant articles using keywords and phrases. The resulting narrative synthesis aims to provide a contemporary summary of molecular research on endometrial dysfunction in PCOS and women with AUB. We synthesized the data qualitatively and no attempt was made to perform a systematic review due to the broad range of topics included and heterogeneity of studies identified.
3. Endometrial Function in Reproductive-Age Women
The human endometrium is the functional tissue that interacts with the embryo from the next generation to ensure implantation, appropriate feto-placental development and growth, and species survival (13,34,35). Female metabolism and endocrine physiology are intimately connected by reciprocal feedback mechanisms that ensure reproduction is coordinated with optimal metabolic health (1,3). Cyclical stimulation of endometrial epithelial, stromal, glandular, and muscular tissues by ovarian-derived sex steroids and locally produced paracrine hormones, cytokines, chemokines, exosomes, and other communication molecules, operate best when female physiology and metabolism are optimized. Lifestyle factors such as healthy diet and exercise are recommended for first-line treatment of PCOS in the international guidelines to optimize physiology, metabolism, and ultimately, molecular function in the endometrium (see Figure 2).
Altered physiology in the hypothalamic-pituitary-ovarian (HPO) tissues, can downregulate and limit reproduction in times of metabolic and systemic stress (1,2). Pathophysiological changes such as IR, CSI, HA, and other hormonal disturbances, can impact the endometrium indirectly via effects in distant tissues (ovary, adipose, liver, pancreas, brain), or cause direct negative effects in endometrial cells and tissues (24). This can result in endometrial changes that are reflected in multiple symptoms and pathologies that vary across the lifespan (see section 4) (36).
3.1. Normal endometrial physiology
Cyclical menstruation is a sign of both reproductive and metabolic health. The American College of Obstetricians and Gynecologists and American Academy of Pediatrics have recommended using the menstrual cycle as a vital sign (37,38). Abnormalities in cycle length or volume can both signal, and exacerbate, chronic conditions, making cycle tracking a valuable assessment tool from adolescence through menopause (39–41). Furthermore, elevated insulin levels, low-grade inflammatory mediators, and altered hormones, appear to crosstalk with HPO signals and endometrial responses, undermining normal cyclical menstrual function (see section 4.3).
3.2. Regulation of hypothalamic-pituitary gonadotrophin hormones
Ovarian function is driven by three-way regulatory feedback circuits linking the hypothalamus, pituitary and ovaries (42,43). Studies in cell cultures, tissue cultures and animal models show that hypothalamic neurons release a 10-amino-acid hormone, gonadotropin releasing hormone (GnRH), under the control of arcuate-nucleus neurons co-expressing kisspeptin, neurokinin B and dynorphin (the KNDy network) (42,44). Those KNDy cells generate GnRH in discrete bursts into the hypophyseal portal blood vessels, prompting the anterior pituitary to discharge follicle stimulating hormone (FSH) and luteinizing hormone (LH) (44). Slow GnRH oscillations (about one burst every 2–3 hours) bias the pituitary toward FSH output, while accelerated rhythms (one burst every 30–60 minutes), as is often observed in PCOS, drive up LH release and increase the LH:FSH ratio (45).
The pulsatile activity of GnRH is mainly regulated by indirect homeostatic feedback from gonadal steroid hormones to the neuronal network upstream of GnRH neurons (45). Specifically, GnRH neurons themselves carry estrogen receptor β (ERβ), while ERα, progesterone receptor (PR), and androgen receptor (AR) are localized to upstream KNDy and gamma-aminobutyric acid (GABA) circuits (43). When ovarian steroid signals are perturbed—whether by metabolic derangements, inflammation, or shifts in hormone levels—those neural circuits lose their ability to synchronize GnRH release, undermining normal HPO-axis regulation of ovarian hormone production and folliculogenesis.
3.3. Hormonal control of ovarian hormones—indirect control of the endometrium
3.3.1. Sex steroid regulation of the endometrium—systemic endocrine control
Ovarian granulosa and theca cells cooperate to produce E2 and androgens (46). LH binds to G-protein-coupled LH receptors on the theca interna—a highly vascularized layer of the follicle adjacent to the basement membrane of the granulosa cells—triggering adenylate cyclase and elevating cyclic adenosine 3’5’-monophosphate protein kinase A (cAMP-PKA) signalling. This cascade enhances transcription of key steroidogenic enzymes, such as CYP11A1 and CYP17A1, leading to increased synthesis of androstenedione and testosterone. Meanwhile, FSH binds to its receptors on granulosa cells, also raising cAMP-PKA levels. The resulting signal upregulates aromatase (CYP19A1) and 17β-hydroxysteroid dehydrogenase (HSD) activity. Androstenedione and testosterone from theca cells diffuse into granulosa cells, where aromatase converts them into estrone (E1) (low affinity for ER) and 17β-estradiol (E2) (high affinity for ER). Following ovulation, high 3βHSD2 levels in the corpus luteum convert pregnenolone to P4 (47).
The two-cell/two gonadotropin interplay between theca and granulosa cells ensures precise control of androgen production by LH and its conversion to estrogens under FSH influence, fine-tuning hormone output for folliculogenesis and endometrial cycling (46,48). Ultimately, ovarian hormones enter the systemic circulation and exert specific molecular actions on endometrial cells. E2-ER signalling upregulates paracrine growth factors such as and cyclin proteins that regulate cell division (49) and insulin-like growth Factor-1 (IGF-1) (50) that drive cell proliferation during the follicular phase (51,52). In addition, E2 signalling increases vascular endothelial growth factor (VEGF) and angiogenesis (53,54) and alters glandular epithelium and stromal cell function (55). Following ovulation, P4-PR signalling induces secretory changes in the endometrium followed by decidualization in stromal cells around cycle day 23 (e.g., FOXO1, IGFBP-1, prolactin) (56). P4-PR signalling also suppresses transforming growth factor β (TGFβ)-mediated increases in matrix metalloproteinases (MMP) (57) and proinflammatory cytokines (54). In the absence of pregnancy, P4 withdrawal activates nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), which elevates inflammatory cytokines (IL-1β, TNF-α) and MMP’s, induces spiral arteriolar vasoconstriction and focal hypoxia in the functional layer, triggering endometrial tissue breakdown, repair and regeneration. Disruption to sequential sex steroid production in the ovaries from adverse systemic factors such as IR and CSI, makes a significant contribution to endometrial dysfunction (see section 4.3.1).
3.3.2. Intracrine metabolism in the endometrium—local hormonal control
Beyond the influence of circulating hormones, the endometrium orchestrates its own steroid environment by expressing local intracrine enzymes (47,58). These tissue-specific steroidogenic and metabolic enzymes convert inactive precursors—like dehydroepiandrosterone (DHEA) and DHEA-sulphate (DHEAS)—into bioactive sex steroids within selected endometrial cell populations, enabling the endometrium precise, site-specific control over hormone-driven processes (59).
Advances in liquid chromatography-tandem mass spectrometry have allowed more accurate profiling of intracrine sex steroids (60). Endometrial tissue levels of E2 can be 5-8 times higher than in serum during the proliferative phase, and half the concentration in the secretory phase (61). Levels of P4 are similar in tissue and serum, suggesting that local levels are determined by passive diffusion (62). Testosterone levels are lower in endometrial tissue than serum and do not show cyclical variation (62). This may reflect the dual role of androgens as ligands for AR and substrates for E2 biosynthesis. Nevertheless, AR signalling has been found to regulate genes involved in cytoskeletal organization, cell motility, and cell cycle progression (63). Testosterone also inhibits the production of MMP-1 in cultured human endometrial stromal cells in a similar manner to P4 (64). MMP-1 is involved in the regulation of menstruation (65) and implantation (66), therefore, elevated androgens in PCOS, may have pathophysiological significance (see section 4.3.2).
3.4. The endometrium as a component of the mucosal immune system
The mucosal immune system is an extensive network of cells and tissues that forms the interface between the internal and external environments of the human body (67). It is estimated to have a surface area of 400 square meters, which is more than 200 times that of the skin (2 square meters) (67). The distribution of the mucosal immune system includes the conjunctiva, respiratory tract, gastrointestinal system, and urogenital tract, with the endometrium being a key component. The mucosal immune system performs a delicate balance between tolerance of some antigens—such as commensal microbes and dietary proteins—and defense against invasive pathogens. This dual functionality preserves barrier health, prevents unnecessary inflammation, and sustains mucosal integrity throughout the body (68).
The mucosal immune system and the human MB form an interconnected network that spans the entire body (67). Each mucosal interface has its own unique MB—which refers to the entire habitat of microorganisms including the genome and surrounding environmental conditions—and microbiota (collection of microorganisms) (69,70). Microorganisms therefore colonize diverse niches throughout the human body and establish an intricate symbiotic relationship at both a local and systemic level (69,71). Furthermore, the MB is now recognized as an integral component of human biology that has direct effects on mucosal function at a local level, and indirect effects on systemic physiology. Therefore, the mucosal immune system and MB act as an adaptive interface between the host and the environment, constantly recalibrating to maintain health (72). Disruptions in this symbiosis, through diet, environmental influences (e.g., EDCs, microparticulate air pollution, or microplastics), infection, or antibiotics, can trigger mucosal immune dysregulation, and contribute to a range of local and systemic conditions, such as infertility, pregnancy complications, and PCOS (13,73–76). In addition, disturbances in the MB at one mucosal site can contribute to IR, CSI, and hormonal imbalance that have pathological influences in apparently unrelated tissues and organs, such as the ovary and endometrium (see section 4.9).
The endometrial mucosal immune system and its resident MB form a dynamic, interdependent ecosystem crucial for normal menstrual function and reproductive success (77). The endometrium is lined by a single layer of columnar epithelial cells that is joined by tight junctions and covered by mucous which forms a selective barrier that prevents pathogen entry while permitting nutrient and hormone exchange (78). The endometrial mucosal associated immune tissue consists of numerous effector cells from the innate and adaptive immune systems. Together, endometrial epithelial cells and mucosal leukocytes produce antimicrobial proteins that vary throughout the menstrual cycle and pregnancy (B-defensins, secretory protease inhibitors, immunoglobulins) (79). In addition to its protective function, the endometrial immune system has a unique role in allowing fertilization, implantation, placental function and pregnancy, and menstruation (78).
E2 and P4 tightly regulate endometrial immunity and immune-stroma crosstalk (80). E2 enhances epithelial barrier integrity, modulates cytokine production, and promotes inflammation (e.g., leukemia inhibitory factor) (LIF). P4, acting via P4 and glucocorticoid receptor, dampens proinflammatory signals during the secretory phase of the menstrual cycle, in part via inhibition of NFκB. Immune cells (T cells, natural killer cells, and macrophages) engage in bidirectional crosstalk with stromal and epithelial cells, ensuring immunological tolerance to semen and embryo, while maintaining defense against pathogens (78). Dysregulated sex steroid signals and adverse systemic responses—such as IR, CSI, and E2/P4/androgen imbalance in PCOS can impair mucosal immune defenses altering cytokine secretion and immune cell recruitment in the endometrium, thereby contributing to reproductive and menstrual disturbances (see section 4.5) (81).
3.5. Endometrial inflammation and leukocyte function
Menstruation represents a cascade of inflammatory events orchestrated by hormone withdrawal, immune cell recruitment, and the release of inflammatory mediators. P4 withdrawal releases the inhibition on NFκB in decidualized stromal cells, promoting transcription of proinflammatory genes (82). This initiates inflammasome formation—which involves the assembly of the NLRP3-ASC-caspase-1 complex—leading to release of chemokines IL-1B and IL-18 that promote leukocyte influx into the endometrium. Recruited immune cells secrete MMPs that break down the extracellular matrix and enable tissue breakdown at the onset of menstrual bleeding (83).
The inflammatory process appears to be self-limited in physiological menstruation. For example, IL-1 (pro-inflammatory) increases expression of 11βHSD1 which converts cortisone to cortisol (anti-inflammatory). Glucocorticoids downregulate inflammation by increasing transcription of anti-inflammatory genes, decreasing pro-inflammatory transcription factors, and limiting production of cytokines (84). In addition, P4 withdrawal promotes phospholipase A2 activity releasing arachidonic acid—from endometrial cell membranes—that is converted by cyclooxygenase-2 into prostaglandins (PG), which drive myometrial contractions and spiral arteriolar vasoconstriction (85). Low-grade CSI can have direct and indirect effects on the endometrium and contributes to endometrial dysfunction and AUB in women with PCOS (see section 4.4).
3.6. Role of vasoconstriction, hypoxia, and hemostasis in menstruation
Menstrual bleeding commences after a tightly coordinated series of events that constrict spiral arterioles to limit bleeding, followed by repair signals generated by short-lived low oxygen levels, and finally from activation of the coagulation cascade to seal ruptured vessels (86). Early in menses, local vasoconstrictors—such as PGF₂α and endothelin-1—cause transient vasoconstriction of spiral arterioles in the functional layer (86). The resulting local hypoxia prevents the degradation of hypoxia-inducible factor-1α (HIF-1α), which up-regulates genes for new vessel growth and tissue regeneration such as VEGF and chemokines such as CXCR4 (87). As vessels rupture, decidual stromal cells express high levels of tissue factor, triggering the extrinsic clotting pathway (88). Platelets adhere to exposed collagen forming a fibrin matrix, while thrombin stops bleeding and facilitates extracellular matrix remodeling that is essential for repair (89). Disruption at any point can manifest as dysmenorrhea, HMB or delayed endometrial repair (see section 4.6).
3.7. Endometrial stem cells
Over the reproductive lifetime, the uterine lining renews itself hundreds of times through a monthly cycle of shedding and restoration. This repeated turnover depends on stem and progenitor cells residing within the basal stratum, which survive the menstrual breakdown and regenerate the functional compartment (90). In the first two days after menstruation, these precursors rapidly expand to reform the epithelial surface (91). During the proliferative phase, E2 drives their proliferation and orchestrates scar-free rebuilding of the endometrium. These stem cells also secrete growth factors and signaling molecules—such as VEGF and cysteine-rich angiogenesis inducer 61 (CYR61)—that promote new vessel growth, fine-tune immune cell trafficking, and support stromal expansion along with matrix remodeling (92). Disruption of this communication network impairs decidualization and predisposes to reproductive disorders and AUB (see sections 4.7 and 4.8).
Dysfunction of endometrial stem cells can underlie disorders such as Asherman’s syndrome (impaired repair) and HMB (excessive breakdown), and stem cell therapy may have a future role in the treatment of PCOS (93). Stem cell therapy has been investigated in a number of reproductive disorders (93). In PCOS, the majority of studies have examined the impact of stem cell treatment on ovarian function (94). Mesenchymal stem cell therapy has been found to improve endocrine (95) and metabolic function (via the PI3K/AKT and FOXO signaling pathway) (96), enhance oocyte quality (97), and reduce the synthesis of proinflammatory cytokines (IL-1β, IL-10, TNF-α, and IFN-γ) (98), in cell and animal models. Human endometrial stem cells can also be used to develop organoid systems that mimic native endometrial tissue and permit detailed molecular investigation of endometrial physiology and dysfunction (99) (see section 4.10).
4. Pathophysiology of Dysfunctional Endometrium
4.1. Classification of abnormal uterine bleeding (AUB) and heavy menstrual bleeding (HMB)
A normal menstrual cycle is regular and has a frequency of 24 to 38 days, duration <8 days, and bleeding volume that does not interfere with quality of life (100,101). Abnormal uterine bleeding refers to uterine bleeding that occurs in non-gestational reproductive-age women, that is abnormal in regularity, frequency, length, or volume (100). Approximately one third of women are affected by AUB, which can have a significant impact on quality of life (102). The etiology of AUB is categorized into structural and non-structural causes using the International Federation of Gynecology and Obstetrics (FIGO) PALM-COEIN system (100).
HMB occurs in up to 25% of women and is the most common form of AUB (103). HMB is defined objectively by having measured menstrual blood loss of >80mL (104,105). However, in a clinical setting, HMB is usually defined subjectively using visual methods, and is reported in up to 50% of women (101,104,106). This aligns with the more person-centered definition of HMB as—“excessive menstrual blood loss that interferes with a woman’s physical, social, emotional, and/or material quality of life”—outlined by FIGO (100), and adopted by the National Institute for Health and Care Excellence (NICE) guidelines (107) and by the Australian Commission on Safety and Quality in Health Care Clinical Standards (108).
HMB can be associated with oligomenorrhea, iron-deficiency (present in 60% of women at presentation), iron deficiency anemia—which occurs in one in four people with HMB—lost productivity from work (estimated at 30 days per year) (109), decreased physical activity and sexual function, reduced overall quality of life, and is a substantial healthcare burden (110,111). Although most women with PCOS have AUB (70-80%), there is a scarcity of data on the prevalence of HMB in this patient group. A cross-sectional study from China examining the relationship between pubertal timing and menstrual characteristics, did not show an association with PCOS and HMB, although the majority of women with PCOS had regular menstrual cycles and normal BMI (112). The management of HMB by General Practitioners is increasing (113), and a large longitudinal cohort survey found that women with PCOS were more likely to experience HMB (114). In addition, HMB was found to increase with age (17.6% at age 22 years to 32.1% at 48 years), and body mass index (BMI). The increased prevalence of HMB in women with elevated BMI, and in PCOS, suggests a role for modifiable risk factors in the pathogenesis and management of HMB.
Elevated BMI is also associated with an increased risk of structural causes of HMB, including uterine polyps (115) and fibroids (116,117). A recent systematic review of 33 articles reported that high BMI was the most significant risk factor for fibroids (117). Healthy nutrition (high fruit and vegetable intake), high sun exposure, and increased vitamin D intake, were protective for uterine fibroid development. In addition, a large phenome-wide association study revealed that women with fibroids had a statistically significant increase in multiple coexisting chronic diseases, suggesting shared pathophysiological processes (118). Given that overweight and obesity are often associated with similar underlying metabolic dysfunction to that found in PCOS, such as IR, hyperinsulinemia, CSI, and hormone imbalance, there may be shared molecular pathways that contribute to HMB in both structural and non-structural causes of AUB (see section 4.8).
Importantly, nearly all metabolic and endocrine features related to PCOS also influence endometrial function, independent of PCOS (119). As a result, there is a significant overlap in the observed molecular changes in the endometrium of women with PCOS and unexplained AUB and HMB (e.g., IL, TNFα, MMP, PG, VEGF) (see sections 4.2-4.9). These include molecular changes in the endometrium in the PALM-COEIN non-structural categories of AUB-Ovulatory dysfunction type-4 (PCOS) and AUB-Endometrial (E). AUB-E is a diagnosis of exclusion after structural lesions (e.g. polyps, fibroids, adenomyosis, cancer), coagulopathy (e.g. von Willebrand disease), and not otherwise classified causes (e.g. arteriovenous malformation and isthmocele) have been ruled out (100). Therefore, there may be common modifiable risk factors that contribute to many seemingly unrelated causes of AUB and HMB.
The uterine endometrium is an end organ and can be impacted by direct and indirect factors. Preexisting factors like IR, elevated insulin levels, and CSI, can impair ovarian aromatase activity and skew the E2-androgen ratio (120). Such imbalance may have an indirect impact on the endometrium by disrupting follicle development, inhibiting ovulation and corpus luteum formation, and ultimately blocking the P4 surge required for endometrial decidualization during the secretory phase of the menstrual cycle. In addition, IR, CSI, and hormone imbalances can have direct effects on signalling networks in endometrial cells and is discussed in the following sections (121). There are range of clinical problems related to abnormal endometrial function in PCOS (Table 1).
4.2. Association between IR, hyperinsulinemia, and menstrual dysfunction
Previous studies have demonstrated a relationship between irregular menstrual cycles and the severity of IR in women with (143–146) and without PCOS (147–149). As a result, the degree of menstrual dysfunction has been proposed as a surrogate marker of IR in PCOS (143,149). Other factors—such as elevated body weight and HA—may also exacerbate both the degree of IR and menstrual disturbance (144).
Menstrual disturbance has also been reported to be a risk factor for type 2 diabetes mellitis (T2DM), which is characterised by IR plus elevated blood glucose levels. Although not all studies report an elevated risk (150), many studies—including several large-scale longitudinal cohorts—have demonstrated an association (147,151). The Nurses’ Health Study II—a large prospective cohort study with 1,639,485 person-years of follow-up—reported a greater risk of T2DM in women with long or irregular menstrual cycles, with greater risk in those with unhealthy lifestyles (152). The study showed an additive effect of excess weight, inactivity, and poor-quality diet, and menstrual cycle dysfunction, with risk of T2DM. The authors emphasized the need to consider menstrual cycle disturbance as an independent predictor of metabolic risk, and aligns with the recommendations from professional colleges and other reports (37,38,153).
4.3. Impact of IR and hyperinsulinemia on endometrial dysfunction
It is well established that metabolic risk factors—such as IR, obesity, and metabolic syndrome—are associated with structural causes of AUB such as polyps (124,154,155) and leiomyoma (156–159). Similarly, non-structural causes of AUB—such as AUB-O type 4 (PCOS) and AUB-E—are also associated with metabolic risk factors such as IR, CSI, and obesity, via both indirect and direct mechanisms (see section 4.2). It is estimated that almost half of all individuals with HMB referred for secondary care do not have an identifiable cause (160). Preventable and reversible metabolic risk factors may therefore contribute to the majority of cases of AUB, including structural and non-structural (e.g., PCOS and unexplained HMB).
4.3.1. Indirect effect of IR and hyperinsulinemia on endometrial dysfunction via altered ovarian hormone production
Hyperinsulinemia and IR significantly disrupt ovarian function via a number of local and systemic mechanisms. Insulin increases GnRH pulse frequency and LH release, and enhances theca cell responsiveness to LH via the PI3K, MAPK, and inositolglycan signalling systems, enhancing ovarian androgen production (161–164). IR, hyperinsulinemia, and related defects in postreceptor signalling pathways—such as serine phosphorylation of insulin receptor substrate-1 (IRS-1), AKT phosphorylation, and GLUT4 translocation—have been shown to disturb glucose metabolism and enhance androgen biosynthesis in ovarian theca and granulosa cells (165,166). Hyperinsulinemia directly inhibits FSH-mediated phosphorylation of AKT in cultured luteinized granulosa cells from women with anovulatory PCOS, and causes reduced glucose uptake (165).
Insulin-mediated impairment of FSH also downregulates aromatase expression, contributing to androgen excess and decreased ovarian E2 production (167). As a result, women with PCOS often have serum E2 levels that are in the low to mid part of the normal range, but the window of action can be prolonged due to P4 resistance, or anovulation and lack of P4 antagonism (see section 4.5) (168). Furthermore, women with PCOS and IR have elevated advanced glycation end-products in follicular fluid that disrupt granulosa cell steroidogenesis, contribute to mitochondrial oxidative stress, and impair ovulation and oocyte quality (169,170). Together, IR and hyperinsulinemia alter ovarian steroid production and indirectly contribute to endometrial dysfunction.
4.3.2. Direct effect of IR and hyperinsulinemia on endometrial dysfunction
IR and hyperinsulinemia promote endometrial inflammation and proliferation (171,172). At a cellular level, hyperinsulinemia activates or disrupts multiple signalling pathways that regulate metabolism, cell survival, apoptosis, proliferation, and endometrial homeostasis (173). Differentiating the effects of elevated insulin from those of androgens in the endometrium is complex, as they often co-exist and have a bidirectional relationship in PCOS (9,174). For example, in human endometrium, HA alters gene expression related to insulin signalling, glucose metabolism, and GLUT4 transport (175). Furthermore, in PCOS, elevated inflammatory cytokines such as TNF, suppress insulin receptor substrate-1 (IRS1) activa tion and impair glucose uptake in the endometrial stromal cells—an effect that is further amplified by rising levels of insulin and testosterone (26). Nevertheless, many studies provide evidence of an independent effect of IR and hyperinsulinemia on endometrial cells in PCOS.
Insulin and IGF-1 receptors share common signaling pathways but also regulate distinct cellular processes (176). In endometrial tissue, PI3K/AKT serves as a key conduit for signals from both insulin and IGF-1 (124). When insulin engages its receptor, PI3K is activated, which in turn phosphorylates AKT. Activated AKT then enhances survival and proliferation of endometrial cells while blocking apoptotic pathways (177). Aberrant regulation of the AKT-NR4A1 cascade by hyperinsulinemia has also been linked to impaired decidualization in PCOS (178). IGF-1 feeds into the same insulin signaling network, boosting cell-growth and anti-apoptotic effects (179). IGF-1 also has an important role in decidualization, and hyperinsulinemia—along with unopposed E2 and/or HA—may augment the mitogenic activity of IGF-1 in PCOS (16).
Increased insulin exposure has also been shown to increase the expression of heterogenous paired box6 (PAX6)—a master transcription regulator of proliferation in endometrial epithelial cells—in IR-induced cell cultures (171). Increased PAX6 repressed CDKNIB gene expression of p27 protein—an important negative regulator of endometrial cell cycle progression—in an insulin-dependent manner. In addition, PAX6 mRNA expression was higher in PCOS endometrial tissue than controls (171). These findings provide a mechanistic explanation for the link between hyperinsulinemia and endometrial epithelial cell proliferation in PCOS.
In summary, observational, cross-sectional, and retrospective studies support a robust association between menstrual disturbance and IR (Table 2)—which is completely reversible (180,181). These data are supported by interventional and mechanistic studies showing improved menstrual function following treatment of IR (see section 4.11) (14,157). Diet, physical activity, and other lifestyle interventions are the recommended first-line treatment for metabolic dysfunction and IR in PCOS, and additional medical treatment with metformin or inositol supplementation is supported by current evidence (7). Emerging therapeutic options provide innovations for prevention and treatment of IR (181). These include spexin (neuropeptide), sitagliptin (incretin inhibitor), thrombomodulin (coagulation regulator), exenatide, semiglutide, liraglutide (GLP-1 receptor agonists), Tirzepatide (GLP1-/GIP), gliflozins (sodium-glucose cotransporter (SGLT2)-inhibitors), clenbuterol (β2-agonist), baicalin (GABA receptor modulator), minerals (Magnesium sulphate, Calcium), vitamins (K2, D3), nitric oxide donors, and nature-derived compounds (marine oil, flaxseed oil, isoflavones, triterpenes, anthocyanins). The investigation of these therapeutics has revealed further insight into detailed molecular pathways involved in IR and can be found elsewhere in excellent reviews (14,181).
4.4. Chronic systemic inflammation (CSI) and endometrial dysfunction
Low-grade CSI is a key component in the pathophysiology of PCOS (9,184). Women with PCOS have higher levels of inflammatory cytokines including interleukins (IL-1,2,6,8,17,18) and TNF-α, and increased C-reactive protein and leukocytes (184–186). CSI can have an indirect effect on endometrial function by altering the inflammatory milieu in the ovary—and ovarian hormone production—or via a direct effect on endometrial cells and tissues (186–188). There is a bidirectional relationship between inflammatory cell imbalances and production of inflammatory cytokines in ovarian and endometrial tissue, and the systemic circulation, making it difficult to determine the direction of causality (189). Nevertheless, the number and composition of immune cell subsets, cytokines, and chemokines in the PCOS endometrium has consistently been found to differ from non-PCOS controls (187,189). As a result, activation of proinflammatory factors in the endometrium disrupts immune homeostasis, exaggerates normal endometrial inflammation, delays hemostasis and repair, and contributes to HMB (172).
Inflammation plays a significant role in normal menstruation and PGs perform a number of important regulatory functions in this process (see sections 3.5 and 3.6) (82,83,85). Cyclooxygenase (COX) enzymes—COX-1 and COX-2—convert arachidonic acid to prostaglandins. COX-PG signalling pathways have been found to be upregulated in the endometrium of women with HMB (190). Gene expression analysis of biopsies from secretory endometrium of women with measured HMB showed a significant elevation of COX-1 and COX-2 mRNA compared to women with normal blood loss (190). PGs mediate their actions via membrane-bound G-protein coupled receptors and increase cAMP after ligand-receptor binding (191). In-vitro tissue culture of endometrium from women with HMB showed a significant elevation of cAMP after PGE2 stimulation (190). PG inhibitors have been found to decrease menstrual blood loss by 30-50% (192,193) via inhibition of PG synthesis and decreased PGE2 binding to its receptors (194). These data suggest that increased endometrial inflammatory changes are important in the pathophysiology of HMB as described in FIGO AUB-E.
Many factors can contribute to the etiology of CSI in PCOS including diet (74), environmental (EDC, microplastics, microparticulate air pollution) (195,196), microbial, gastrointestinal dysbiosis-induced metabolic endotoxemia (74,197), and other lifestyle factors (198). These factors can directly modulate CSI, IR, and systemic metabolite levels, and are likely to contribute to endometrial dysfunction and HMB (199). Given that many of these factors are preventable and reversible—and provide increased autonomy and individual control over health—research priority should be given to appropriate intervention trials.
4.5. Molecular changes in the dysfunctional endometrium in ovulatory and anovulatory PCOS
4.5.1. Endometrial changes in ovulatory PCOS
Clinical trials comparing reproductive outcomes in women with PCOS that ovulate with healthy matched controls are not available. Nevertheless, they have a higher rate of implantation failure and reduced pregnancy rates following ovulation induction and in-vitro fertilization, suggesting oocyte competence and endometrial dysfunction may play a role (200,201). Experimental data indicate that ovulatory women with PCOS have alterations in molecular pathways in the ovary and endometrium that are independent of ovulation (119). These changes lead to reduced oocyte quality and altered endometrial competence.
Despite ovulation, women with PCOS may experience excess E2 exposure due to a short luteal phase or subtle P4 resistance in endometrial tissues. The resulting imbalance of E2 and P4 can disrupt cell regulatory mechanisms and lead to continued E2 stimulation and P4 resistance (168). Progesterone resistance refers to the reduced ability of the endometrial lining to respond to P4 and occurs in both ovulatory and anovulatory PCOS, although it may be more pronounced when ovulation is absent (202,203). Mechanisms underlying P4 resistance include altered P4 receptor expression and/or ratios, receptor gene polymorphisms and epigenetic changes, disrupted co-regulators and microRNA (miRNA), IR, and chronic inflammation (204). E2 activates Wnt/β-catenin signalling and induces epithelial cell proliferation in the proliferative phase of the menstrual cycle, then elevated P4 levels suppress proliferation during the secretory phase (204,205). Normally, progesterone acts via progesterone receptors (PR-A and PR-B) to suppress proliferation, drive decidualization, and reduce inflammation (168,206). When P4 resistance occurs, these processes become dysregulated, tipping the balance toward E2-driven growth and a proinflammatory state.
The endometrial transcriptome of ovulatory obese PCOS women was found to be significantly different to that of normal weight subjects in ovulatory menstrual cycles (207). PCOS women had 610 differentially expressed genes, with biological processes related to inflammation (TNFR1), insulin signalling (PI3K/AKT), fatty acid metabolism, and lipotoxicity being most prominent. The most important differences in PCOS endometrium were therefore related to inflammation and metabolism (207).
4.5.2. Endometrial changes in anovulatory PCOS
In chronic anovulatory PCOS, the endometrium is continuously exposed to E2 without the antiproliferative and differentiating effects of P4. This imbalance leads to altered expression of P4 receptor isoform ratios (PR-A/PR-B) and dysregulated ER-α levels, impairing endometrial decidualization and perpetuating unopposed proliferative signals. In addition, women with anovulatory PCOS have differences in gene expression in mid-secretory endometrium—compared to controls with normal endometrium—that demonstrate P4 resistance, further exacerbating E2/P4 imbalance (208). As a result, the combination of sustained low E2 and P4, elevated androgens and anti-Mullerian hormone, low-grade CSI, hyperinsulinemia, and IR, collectively impair decidualization and contribute to AUB, implantation failure, reduced fertility, increased risk of pregnancy complications, and postmenopausal hyperplasia (see Table 1) (13,173).
Normal endometrial decidualization relies on progesterone-driven upregulation of homeobox transcription factors such as HOXA10 and HOXA11 (206,209). In PCOS, hyperandrogenism directly represses HOXA10, while low P4 and PR resistance further blunt stromal differentiation (210). Concurrently, endometrial levels of LIF and downstream STAT3 phosphorylation are reduced, compromising the decidual response and trophoblast invasion if pregnancy ensues (211). In addition, women with anovulatory PCOS exhibit marked downregulation of integrin αvβ3 complex—which is essential for blastocyst attachment and invasion—during the implantation window (202). Many of these signalling pathways and transcription factors have not been investigated for their role in HMB where no cause has been identified.
In PCOS, the endometrium is the target of endocrine, immune, biochemical, and metabolic factors present in affected women (14). Both CSI and IR appear to exacerbate the impact of hormonal imbalance on the endometrium in PCOS. Endometrial IR in PCOS is characterized by impaired glucose transporter expression and dysregulated PI3K-AKT signaling (212). Simultaneously, the endometrial microenvironment shows elevated pro-inflammatory cytokines (e.g., IL-6, TNF-α), reduced uterine perfusion, and oxidative stress, creating an energy-deficient, hostile environment that undermines receptivity (187). Many of these aberrations, especially within epithelial and stromal cell subclusters, partially normalize after interventions such as metformin or lifestyle modification (17,212). Therefore, in common with many other conditions—such as PCOS, T2DM, metabolic syndrome—some cases of HMB may result from an interaction between lifestyle and environmental factors superimposed on genetic susceptibility variants (see section 4.7) (1).
4.6. Reduced vasoconstriction, angiogenesis, and matrix remodeling in HMB
Women with HMB have lower levels of PG-F2α (PGF2α) receptor (190), PG imbalance (excess PGE2/PGI2 relative to thromboxane) (190,213), and higher levels of vasodilatory PGE2 (214). They also have reduced endothelin-1 (215), altered spiral arteriole differentiation (216) and inadequate arteriolar vasoconstriction (217) in the endometrium, compared to women with normal menstrual bleeding. In addition, women with HMB have excessive local fibrinolysis (increased tissue plasminogen activator, decreased plasminogen activator inhibitor-1) (36,218), imbalanced MMP and tissue inhibitors of MMP (219), and altered angiogenic and inflammatory mediators (e.g., VEGF, cytokines) (172,220). The additive effect of these changes make a significant contribution to the pathophysiology of HMB (86).
Glucocorticoids promote vasoconstriction and inhibit angiogenesis in vitro and in vivo and contribute to normal menstruation (221). Cortisol is inactivated by 11βHSD1 and 11βHSD2, and women with HMB have elevated endometrial 11βHSD2 expression (222). Inactivation of cortisol by 11βHSD2 may promote unrestrained vessel proliferation and contribute to HMB. Novel therapies for HMB may include inhibition of 11βHSD2 or glucocorticoid replacement. A placebo-controlled randomized controlled trial (RCT) showed that 1.8mg of dexamethasone given twice daily for 5 days in the mid-luteal phase of the menstrual cycle, reduced measured blood loss by 25mL more than placebo (95% credible interval 1 to 49mL) (109).
To our knowledge, there are no published studies that directly compare endometrial PGF₂α receptor expression, fibrinolysis, MMP, or VEGF mRNA or protein levels, in women with PCOS versus controls. Similarly, there are no published histologic studies that specifically examine spiral artery differentiation or arteriolar vasoconstriction in the endometrium of women with PCOS. The development of endometrial organoid models should provide an experimental model to investigate whether similar changes contribute to endometrial dysfunction, AUB, and HMB, in women with and without PCOS (see section 4.10).
4.7. Genetic insights into endometrial changes in PCOS
Endometrial cells from women with PCOS differentially express genes related to endometrial function (e.g., steroid hormone receptors for E2, P4, and androgens) (223), endometrial receptivity (e.g., HOXA10, IGFBP1) (224), and inflammation (IL-8, TNFα, NFκB, CCL2, CCL5, CCL7) (224,225). Single-cell nuclear RNA sequencing of proliferative phase endometrium reveals a PCOS-specific endometrial signature that is characterized by an increased epithelial-to-stromal cell ratio, diminished lymphoid cell populations, and widespread dysregulation of genes involved in cell adhesion (e.g., NLGN1), cytokine signaling (e.g., integrin genes), and metabolic pathways (e.g., NRCAM, CNTN1, CD44, ITGA6) (17). Cell-type differentially expressed genes (DEG) correlate with metabolic and endocrine features of PCOS—such as HOMA-IR and androstenedione—suggesting that IR and HA contribute to endometrial changes in PCOS (17). Eriksson et al, compared DEG profiles before and after treatment with lifestyle and metformin in overweight and obese IR women with and without PCOS (17). Notably, after 16 weeks treatment with lifestyle and metformin, there was extensive recovery of disease-specific DEGs in endometrial cells from women with PCOS.
In PCOS, elevated circulating androgens have an adverse impact on decidualization, the process in which endometrial stromal cells undergo functional transformation in preparation for implantation (22,226). WT1 is a key transcription factor that performs a gatekeeper role as a regulator of a large network of genes involved in decidualization in human endometrial stromal cells (22,227). Women with PCOS have HA, increased AR in stromal cells, and reduced WT1. Genome-wide chromatin immunoprecipitation experiments reveal that AR binds to regulatory elements on DNA normally occupied by WT1, displacing WT1 from its target promotor sites (227). This competitive binding disrupts WT1’s control of genes governing cell differentiation, immune response, and angiogenesis pathways critical for decidualization. This study provides mechanistic insight into how HA dysregulates decidualization, potentially leading to symptoms such as infertility and AUB, commonly associated with PCOS.
Taken together, genetic, transcriptomic, and histological analysis of endometrial biopsies reveal PCOS-specific endometrial changes that correlate with serum endocrine and metabolic profiles. These data provide evidence that PCOS-related IR and HA have an adverse impact on endometrial function. Management with lifestyle and metformin partially restored pathways that reflect treatment-related reversal of endometrial dysfunction. Importantly, these insights provide opportunities for further research into pathway-specific treatments involving extracellular matrix organization, collagen metabolism, integrin signalling, immune cell function, and lifestyle interventions.
4.8. Genetic insights into endometrial changes in AUB and HMB
Gene array analysis of endometrial biopsies taken during menstruation from women with and without HMB and normal menstrual cycles—and in women with fibroids— identified genes and functional gene clusters suggesting different mechanisms cause excess bleeding in the different clinical groups (123). These data suggest that HMB may reflect alterations in the immune and inflammatory response in women with regular menstrual cycles (123). A recent large meta-analysis of GWAS from 5 biobanks revealed 36 gene variants that were significantly associated with HMB. HMB shared a portion of its genetic risk with other female reproductive disorders including endometriosis, fibroids, and ovarian cysts, suggesting shared biological pathways (228). Factor 5 Leiden variant, genes involved in hormone production, and signals near genes involved in the Wnt/β-catenin signaling pathway, were found to be protective. The discordant results between genetic analysis of endometrial biopsy samples and GWAS needs further clarification.
Recent studies have also identified a role for exosomes (extracellular vesicle involved in communication) derived from human leiomyoma on the function of endometrium-related cell lines (229). Cells exposed to exosomes from leiomyoma increased proliferation by 60% compared to untreated control cells, and upregulated angiogenesis markers (C-MYC and VEGFA). miRNA profiling revealed significant changes in miRNA regulation in treated cells compared to controls. The investigators concluded that leiomyoma-derived exosomes contain factors that enhance endometrial proliferation and angiogenesis that may contribute to HMB (229).
Whole exome sequencing of adolescents diagnosed with low von Willebrand factor (VWF) levels has shed some light on possible gene variants that may be involved in unexplained HMB (230). Adolescents with low VWF and HMB were also found to have an excess of pathogenic genes variants (e.g., FERMT2) involved in the regulation of angiogenesis and hemostasis. Variants of these genes require further investigation as they may be involved in unexplained HMB, and their identification may help with risk stratification.
In summary, HMB is increasingly recognized as a genetically complex, multifactorial trait, reflecting the interplay of genetic predispositions, hormonal dysregulation, hemostatic imbalance, and extracellular matrix dynamics. The suggestion of shared biological pathways raises the possibility that common pathophysiological features—such IR, hyperinsulinemia, CSI, and hormonal imbalances—may play important roles in the pathogenesis of the many different types of AUB. Future studies using endometrial organoids could be a valuable tool for investigating these pathways and interactions (see section 4.10) (99,231).
4.9. Role of the microbiome (MB) in PCOS, AUB and HMB
4.9.1. Role of the microbiome in PCOS
As previously discussed, the endometrium in PCOS is characterized by molecular changes resulting from systemic factors such as CSI, IR, and HA, that disrupt cyclical remodeling and alter the mucosal immune system towards a more proinflammatory state. The gut MB is recognized to play a significant role in the pathogenesis of PCOS (74). The central role of gastrointestinal “dysbiosis”—or imbalance of the MB—in PCOS has been supported and expanded by many publications investigating mechanisms involved in the described pathogenic pathways (69,232,233). This theory proposes that diet, environmental, and lifestyle factors are superimposed on inherited genetic susceptibility genes that together result in the symptoms, biochemical and endocrine changes in PCOS (73,74). The theory aligns with evolutionary theories of PCOS (1), recommendations of the international guidelines for lifestyle interventions as the first line of treatment (7), and is supported by an extensive body of research into the role of the MB in many other chronic systemic diseases (234).
Nutritional and environmental factors modify the course and prognosis of PCOS by altering the composition and function of beneficial gut microbial species, inducing oxidative stress and CSI, promoting metabolic changes and IR, and disrupting hormonal balance (74,197,235). Diet quality and composition have a significant impact on gut microbial physiology that in turn regulates molecular pathways and alters systemic physiology in diverse tissues, including the endometrium (13,17). The dysbiosis theory of the pathogenesis of PCOS proposed that a high saturated fat/high sugar, low fibre diet causes increased gut mucosal permeability, allowing lipopolysaccharide (LPS) to traverse the gut barrier. LPS binds with LPS-binding protein which activates toll-like receptors on innate immune cells, upregulating NFκB-mediated inflammatory cytokine production. This cascade of events promotes CSI and IR, and impacts normal physiology in systemic tissues, including the ovary and endometrium (74). Similar diet-induced changes are also recognized to play a part in many chronic diseases, in addition to PCOS (72).
While there is ongoing debate regarding the specific dietary recommendations for the management of PCOS, there is general agreement about the features of a high-quality healthy diet (7,236). Diet index, diet composition, diet pattern and metabolomic studies have identified a whole-foods diet containing carbohydrates (CHO), fats, proteins, vitamins, minerals, and contingent nutrients (e.g., polyphenols)—derived from plant-based sources and free-range or pasture raised animals—that has the potential to reduce symptoms and metabolic and endocrine changes in women with PCOS (237,238). This type of diet is high in fibre and low in ultraprocessed food and environmental chemicals and has a beneficial impact on the gut MB and systemic physiology.
Recent developments in environmental epigenetics provide details of the interaction between environmental factors and genetics that highlight the dynamic interaction between environmental exposures and molecular and systemic pathways (239,240). Detailed discussion of the molecular impact of dietary and lifestyle modification in PCOS can be found in other reviews (241,242).
Since the original publication by Tremellen and Pearce (74), further research has identified a potential role for other mucosal microbiomes, beyond the gut MB, in the pathogenesis of PCOS (196,243–245). It is now clear that CSI, IR, and hormonal imbalance can be initiated at any mucosal surface via a variety of environmental risk factors (246). A range of environmental chemicals impact the MB (247) and are absorbed, ingested or inhaled into the human body at diverse mucosal sites, initiating CSI, metabolic and endocrine dysfunction (Figure 3) (248).
For example, nano- and microplastics enter the body via mucosal sites and can disrupt the MB (249), alter HPO function, induce oxidative stress and inflammation in reproductive tissues, and impair ovulation and fertility (250). In addition, ingested or inhaled nanoplastics, microplastics, and microparticulate air pollution have been identified in the human endometrium and placenta, and may contribute directly and indirectly to reproductive dysfunction and/or AUB (251–253). Studies in mice showed that long-term exposure to microplastics increased endometrial inflammatory cytokines (e.g., TNFα, IL-1β, and IL-6), and reduced fertility (253). Human endometrial organoids exposed to microplastics showed a significantly distorted shape and increased apoptosis (253). Nano- and microplastic particles have also been found to accumulate in human endometrial stromal cells, where they induced morphological changes and cell death (252). This is clearly concerning given the significant role of endometrial stromal cells in many aspects of normal reproductive function.
Alterations in the mucosal microbiota can result from exposure to a range of air pollutants (254). These include particulate matter (PM 2.5, PM 5, PM 10), polycyclic aromatic hydrocarbons, heavy metals, pesticides, gases, and many others. There has been more extensive research on ecosystem changes in the respiratory and gastrointestinal MBs, and the gut MB has been found to be particularly sensitive to diet, environmental chemicals and medications (255). Population-based cohort studies have provided preliminary data that microparticulate air pollution may increase the risk of PCOS (196). Observational studies have suggested the mechanisms may include pulmonary-induced CSI (245,256), elevated fasting insulin (257), and elevated androgens (258). A cross-sectional study of 34,832 women reported an association between irregular menstrual cycles and total suspended particulate matter in young women with PCOS diagnosed by self-reported oligomenorrhoea and androgen excess (259).
There is a significant body of evidence linking EDC to dysbiosis of the MB, gut barrier dysfunction, CSI, metabolic disturbance and IR, endocrine imbalance, obesity, and many systemic diseases, including infertility, pregnancy complications, PCOS and endometrial dysfunction. EDC enter the body via air, water, and food (e.g., pesticides, herbicides, ultraprocessed food additives). Detailed analysis is beyond the scope of the present review and excellent reviews are available (260–263).
4.9.2. Mechanistic links between the gut microbiota and endometrial dysfunction
The microbiome functions as an endocrine messenger that can directly regulate the HPO axis and alter ovarian and endometrial function (264). Emerging evidence from animal studies now recognizes a reciprocal gut-gonadal communication network that contributes to reproductive health (265). Key mediators include short-chain fatty acids (e.g., acetate and butyrate), neurotransmitters (e.g., serotonin, gamma-aminobutyric acid), inflammatory cytokines (e.g., IL-6, TNFα), the estrabolome (collection of gut microbial genes involved in the metabolism of E2), and alterations in the HPO axis (266). Disruptions in intestinal microbial balance (dysbiosis) can alter ovarian function, affect oocyte development, disrupt hormone production, and reduce fertility through diverse molecular mechanisms (266). In PCOS, for example, an imbalanced gut ecosystem augments inflammatory signaling (NF-κB) and creates a feed-forward loop of inflammation (74,267). Lipopolysaccharide from Gram-negative bacteria can bind TLR4 on epithelial and stromal cells, activating NF-κB and escalating proinflammatory cytokine release (268,269). This heightened inflammatory milieu can disrupt decidualization by dampening PR signaling in stromal fibroblasts (270). In addition, randomized controlled trials have demonstrated that supplementation with probiotics (197), resistant starch (271), and synbiotics (probiotic plus prebiotic) (272), can reduce IR, drive down testosterone and increase menstrual cycle regularity.
In summary, current evidence provides a mechanistic link between diet and environmental effects on both ovarian and endometrial dysfunction and provides the background for further investigation of the molecular mechanisms involved. It is anticipated that future research will reveal detailed molecular mechanisms regarding the role of the mucosal MB-immune network in endometrial dysfunction in PCOS. In addition, understanding the role of the distant and endometrial mucosal microbiomes may lead to novel prevention and management strategies for reproductive dysfunction and AUB.
4.9.3. Role of the endometrial microbiome in endometrial dysfunction
Although the vaginal MB has been extensively characterised, there is ongoing debate regarding the typical composition of the core endometrial MB (77,273). For instance, some studies report abundant phylum Firmicutes, with Lactobacillus as the predominant genus, followed by Bacteroidetes, Proteobacteria, and Actinobacteria. Other studies have found higher proportions of Bacteroidetes or Proteobacteria and lower levels of Lactobacillus. Nevertheless, most studies confirm a protective effect of lactobacillus spp and there is ongoing research directed at characterizing the dysbiotic endometrium. Recent studies have reported differences between fertile and infertile endometrial microbiota and a possible role for the endometrial MB in other gynecological pathology (e.g., polyps, endometriosis, fibroids, cancer) (274,275). Determining the characteristics of the endometrial MB is a growing field that should improve scientific understanding of specific bacterial and host pathways in reproductive physiology and disease, as has occurred at other mucosal immune sites.
4.9.4. Role of the microbiome in AUB and HMB
There has been a paucity of research into the role of the endometrial MB in AUB and HMB. Pelzer et al. compared 16 rRNA sequencing results of endometrial samples taken from women with menorrhagia or dysmenorrhea (276). The authors reported diverse microbial populations dominated by Lactobacillus spp. in both cohorts, with no significant differences in diversity. Acinetobacter spp. were more abundant in women with menorrhagia and Lactobacillus spp. were significantly lower in nulliparous women with menorrhagia. The significance of these findings in relation to endometrial function and AUB is not known. It is anticipated that future studies will uncover a bidirectional relationship between the endometrial MB and endometrial epithelial and stromal cells as occurs at all other mucosal sites.
4.10. PCOS endometrium-derived organoids and endometrial dysfunction
Historically, studies of the PCOS endometrium have used two-dimensional cell cultures and animal models for providing insights into human physiology (277). The emergence of three-dimensional organoid systems now permits the assembly of endometrial structures that self-organize and retain features of the original epithelium (20). Human endometrial organoids can be developed from primary endometrial cells or from pluripotent stem cells induced to form endometrial stromal fibroblasts (278,279). Endometrial organoids form gland-like units with apico-basal polarity and undergo steroid-driven differentiation in response to E2 and P4 (99). More recent organoid co-culture models—or “assembloids”—incorporate epithelial organoids with other autologous cells (e.g., stromal, immune, and/or endothelial cells) allowing exploration of bidirectional cross-talk between different cell-types, and mechanisms of immune tolerance (280). Patient-derived endometrial epithelial organoids (EEOs) from women with PCOS will provide a human disease-relevant system for investigating mechanisms of endometrial dysfunction. Importantly, such cultures can originate from non-invasive menstrual fluid (281), routine biopsies, or endometrial tissue from PCOS-affected women (282).
Luyckx et al. recently reported the successful establishment of an EEO model in women with PCOS. Endometrial biopsies from overweight/obese-PCOS and lean-PCOS women were compared to BMI-matched controls in both groups (282). Morphological assessment was performed using immunostaining and the size of the EEO determined after 6 days of hormone exposure. Bulk RNA-sequencing was performed to determine differences in gene expression. PCOS organoids from obese and lean samples revealed increased inflammatory gene expression (OSMR, ICAM 1), and decreased size (diameter), compared to BMI matched controls in both groups. EEO from obese-PCOS women displayed an altered response to E2 and P4 with reduced expression of receptivity-related genes (LIF, PAEP). Addition of dihydrotestosterone did not alter the EEO transcriptome, in accordance with the minimal expression of ARs present in epithelial cells (282).
In summary, this EEO study supports previous in vivo findings that PCOS endometrium has an aberent inflammatory gene expression and an altered response to sex hormones (208,283). Notably, there was no difference in inflammatory gene profile or EEO size between obese- and lean-PCOS EEO. The authors hypothesized that the reduced size of both PCOS EEOs may be due to the increased inflammatory nature, and/or altered mitochondrial function, as there were no differences in the levels of apoptosis markers (CC3), apoptosis gene expression, or cellular proliferation. Given the low expression of ARs in endometrial epithelial cells, coupled with their high level in stromal cells, the authors recommended that future organoid models use a co-culture system containing both cell types to investigate the effects of a hyperandrogenic environment.
4.11. Impact of lifestyle changes on endometrial dysfunction where no cause is identified
Modifiable risk factors—including diet, exercise, and other lifestyle factors—have been demonstrated to have a significant impact on the symptoms, biochemical, and endocrine changes in PCOS (7,284,285). As a result, the international evidence-based guidelines recommend lifestyle modification as the first line of management for PCOS (7). Similarly, most chronic health conditions have modifiable risk factors that can be addressed for prevention and treatment of symptoms and underlying pathophysiology (286,287). Therefore, it is important to consider the role of lifestyle modification in the management of AUB in PCOS and otherwise unexplained HMB (AUB-E).
Most of the research on AUB in PCOS has been focused on restoring cycle regularity to improve cycle control and fertility. Abundant evidence demonstrates that irregular menstruation can be improved—and regular menstruation restored—following diet and exercise interventions, as outlined in the international guidelines and many other publications (7,288). As discussed in previous sections, there has been very little attention directed at the prevalence, molecular causes, and treatment of HMB in PCOS. It is likely that the symptom of HMB is also improved following lifestyle interventions that restore cycle regularity, but this requires further investigation in appropriately designed studies.
For example, a RCT that compared a low-glycemic index (GI) diet to a conventional healthy diet, showed a significant improvement in menstrual cyclicity with a low-GI diet, in women with PCOS (95% versus 63%; P = 0.03), but did not report data on HMB (289). A 2013 systematic review of diet composition studies on anthropomorphic, metabolic, psychological, and reproductive outcomes in PCOS, reported that there were no differences for most outcomes between different diets (290). The investigators concluded that a diet that reduced weight was beneficial—regardless of composition—with greater improvement in menstrual cycle regularity with a low-GI diet, and greater reductions in IR for a low-GI or low-CHO diet. Prevalence or changes in HMB was not assessed. To date, no controlled dietary intervention has specifically quantified changes in menstrual blood volume loss following insulin-sensitizing low-GI or low-CHO diets.
Dietary recommendations for the treatment of IR and T2DM have changed significantly over time with a recent resurgence of the recommendation for a CHO restricted diet (CRD) (291). Low CHO diets have been conclusively shown to achieve T2DM remission in ten meta-analyses (292). Low CHO diets have been defined as <26% of total energy from CHO (or 130 grams/day) and low CHO ketogenic diets as <10% (20-50 grams/day) (293). The American Diabetes Association, American Heart Association, Diabetes United Kingdom, European Association for the Study of Diabetes, and Diabetes Australia, all recommend a CHO restricted diet (CRD) as a treatment option for prediabetes and T2DM (294–296). A CRD is also recommended by the Society of Metabolic Health Practitioners for treatment of obesity, hypertension, metabolic syndrome, metabolic-associated steatotic liver disease, cardiovascular disease, and T2DM (297). A CRD is a central component of the dietary recommendations for PCOS, although no specific diet is advised. Diet, environmental exposures, and other lifestyle recommendations for the treatment of AUB and HMB are completely lacking in current guidelines and should be addressed as a matter of priority.
5. Conclusions
This review has identified the molecular pathways by which IR, CSI, and hormonal imbalance drive endometrial dysfunction and/or AUB in women with PCOS. Systemic metabolic and inflammatory changes disturb key signaling cascades in the ovary and endometrium, disrupting normal menstrual cycling and bleeding patterns, impairing receptivity and implantation, altering placentation and normal pregnancy development, and predisposing to hyperplasia and endometrial cancer. Women presenting with unexplained irregular and/or HMB share overlapping risk factors and endometrial molecular alterations, pointing to common underlying pathophysiology. Given the high prevalence of PCOS and AUB among reproductive-age women, these disorders represent substantial personal and public health burdens. Importantly, the pathophysiological drivers highlighted in this review—IR, CSI, and hormonal imbalance—are amenable to intervention in a large subset of patients. Prospective trials that target and reverse these metabolic and endocrine derangements should be prioritized, as they have the potential to restore normal endometrial function and improve reproductive health outcomes.
Author Contributions
Conceptualization, J.P., C.O.B., T.U., K.T.; methodology, J.P., C.O.B., T.U., K.T.; writing—original draft preparation, J.P.; writing—review and editing, J.P., C.O.B., T.U., K.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
During the preparation of this manuscript the authors used Microsoft Copilot (GPT-4; Microsoft 2025) for the purposes of optimizing the literature search and language editing. The authors have independently checked each reference, reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AKT | Serene/threonine specific kinase (also known as protein kinase B) |
| AR | Androgen receptor |
| ASC | Apoptosis-associated speck-like protein containing a caspase recruitment |
| domain | |
| AUB | Abnormal uterine bleeding |
| BMI | Body mass index |
| cAMP | Cyclic adenosine 3’5’-monophosphate |
| CDKN1B | Cyclin-dependent kinase inhibitor 1B |
| CHO | Carbohydrate |
| COEIN | Coagulopathy, Ovulatory Dysfunction, Endometrial, Iatrogenic, |
| Not Otherwise Classified | |
| COX-2 | Cyclooxygenase-2 |
| CRP | C-reactive protein |
| CSI | Chronic systemic inflammation |
| CXCR4 | Chemokine receptor type 4 |
| CYP19A1 | Aromatase |
| CYR61 | Cysteine-rich angiogenesis inducer 61 |
| DEG | Differentially expressed genes |
| DHEA | Dehydroepiandrosterone |
| DHEAS | Dehydroepiandrosterone sulphate |
| EEO | Endometrial epithelial organoid |
| ER | Estrogen receptor |
| E2 | 17β-estradiol |
| FIGO | International Federation of Gynecology and Obstetrics |
| FOXO | Forkhead box protein O |
| FSH | Follicle stimulating hormone |
| GABA | Gamma-aminobutyric acid |
| GWAS | Genome-wide association studies |
| GLUT4 | glucose transporter type 4 |
| GnRH | Gonadotropin releasing hormone |
| HA | Hyperandrogenism |
| HIF | Hypoxia inducible factor |
| HMB | Heavy menstrual bleeding |
| HOXA10 | Homeobox 10 |
| HPO | Hypothalamic-pituitary-ovarian |
| HSD | Hydroxysteroid dehydrogenase |
| ICAM 1 | Intercellular adhesion molecule 1 |
| IFN | Interferon |
| IGF-1 | Insulin-like growth factor-1 |
| IGFBP | Insulin-like growth factor binding protein |
| IL | Interleukin |
| IR | Insulin resistance |
| IRS1 | Insulin receptor substrate 1 |
| KNDy | Kisspeptin, neurokinin B, dynorphin-y |
| LIF | Leukemia inhibitory factor |
| LH | Luteinizing hormone |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MB | Microbiome |
| MMP | Matrix metalloproteinase |
| mRNA | Messenger ribose nucleic acid |
| miRNA | microRNA |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NICE | National Institute for Health and Care Excellence |
| NLRP3 | Nod-Like receptor family pyrin domain containing 3 |
| OSMR | Oncostatin M receptor |
| PALM | Polyp, Adenomyosis, Leiomyoma, Malignancy or Hyperplasia |
| PAX6 | Heterogenous paired box 6 |
| PCOS | Polycystic ovary syndrome |
| PG | Prostaglandin |
| PI3K | Phosphoinositide 3-kinase |
| PKA | Protein Kinase A |
| PAEP | Progesterone-associated endometrial protein |
| PR | Progesterone receptor |
| P4 | Progesterone |
| RNA | Ribose nucleic acid |
| STAT3 | Signal transducer and activator of transcription 3 |
| T cells | T lymphocytes |
| TGF | Transforming growth factor |
| TNF-α | Tumour necrosis factor-α |
| T2DM | Type 2 diabetes mellitis |
| VEGF | Vascular endothelial growth factor |
| VWF | von Willebrand factor |
| WT1 | Wilms tumour-1 |
| Wnt | Wingless-related integration site |
References
- Parker J, O’Brien C, Hawrelak J, Gersh FL. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int J Environ Res Public Health 2022, 19, 1336. [CrossRef] [PubMed]
- Dumesic DA, Abbott DH, Chazenbalk GD, Scholar G. An Evolutionary Model for the Ancient Origins of Polycystic Ovary Syndrome. J Clin Med. 2023;12(6120):1–16.
- Dumesic DA, Padmanabhan V, Abbott DH. Polycystic ovary syndrome : an evolutionary metabolic adaptation. Reproduction. 2025;169(e250021).
- Shaw LMA, Elton S. Polycystic ovary syndrome: A transgenerational evolutionary adaptation. BJOG An Int J Obstet Gynaecol. 2008;115(2):144–8.
- Azziz R, Dumesic DA, Goodarzi MO. Polycystic ovary syndrome: An ancient disorder? Fertil Steril [Internet]. 2011;95(5):1544–8. [CrossRef]
- Charifson MA, Trumble BC. Evolutionary origins of polycystic ovary syndrome: An environmental mismatch disorder. Evol Med Public Heal. 2019;(1):50–63.
- Teede HJ, Misso ML, Costello MF, Dokras A, Laven J, Moran L, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Eur J Endocrinol [Internet]. 2023;189:G43–64. [CrossRef]
- Wechsung K, Neumann U, Balint N, Wiegand S, Klinikum CV, Entwicklung P Der, et al. Polycystic Ovary Syndrome – Support and Prevention in Adolescence. Geburtsh Frauenheilk [Internet]. 2025;August:1–7. Available from. Available online: https://www.thieme-connect.de/products/ejournals/abstract/10.1055/a-2622-6321.
- Parker, J. Pathophysiological Effects of Contemporary Lifestyle on Evolutionary-Conserved Survival Mechanisms in Polycystic Ovary Syndrome. Life. 2023;13(4):1056.
- Balen A, Rajkowha M. Polycystic ovary syndrome - A systemic disorder? Best Pract Res Clin Obstet Gynaecol. 2003;17(2):263–74.
- Teede HJ, Moran LJ, Morman R, Gibson M, Dokras A, Berry L, et al. Polycystic ovary syndrome perspectives from patients and health professionals on clinical features, current name, and renaming: a longitudinal international online survey. eClinicalMedicine [Internet]. 2025;84:103287. [CrossRef]
- Zhang J, Zhang J, Zheng Z, Zhang R. Evolution of polycystic ovary syndrome and related infertility in women of child-bearing age: A global burden of disease study 2021 analysis. Eur J Obstet Gynecol Reprod Biol [Internet]. 2025;314(August):114659. [CrossRef]
- Parker J, O’Brien C, Yeoh C, Gersh FL, Brennecke S. Reducing the Risk of Pre-Eclampsia in Women with Polycystic Ovary Syndrome Using a Combination of Pregnancy Screening, Lifestyle, and Medical Management Strategies. J Clin Med. 2024;13(1774):1–33.
- Palomba S, Piltonen TT, Giudice LC. Endometrial function in women with polycystic ovary syndrome: A comprehensive review. Hum Reprod Update. 2021;27(3):584–618.
- Mousa, A; Tay, CT; Teede H. Technical Report for the 2023 International Evidence-based Guideline for the Assessment and Management of Polycystic Ovary Syndrome. Monash University; 2023.
- Giudice, LC. Endometrium in PCOS: Implantation and predisposition to endocrine CA. Best Pract Res Clin Endocrinol Metab. 2006;20(2):235–44.
- Eriksson G, Li C, Sparovec TG, Dekanski A, Torstensson S, Risal S, et al. Single-cell profiling of the human endometrium in polycystic ovary syndrome. Nat Med. 2025;(Epub ahead of print).
- Wong FC, Kim CE, Garcia-Alonso L, Vento-Tormo R. The human endometrium: atlases, models, and prospects. Curr Opin Genet Dev. 2025;92(102341):1–14.
- Yang J, Yang L, Zhou Y, Cao F, Fang H, Ma H, et al. Molecular subtype of recurrent implantation failure reveals distinct endometrial etiology of female infertility. J Transl Med. 2025;23(1):1–18.
- Lou L, Kong S, Sun Y, Zhang Z, Wang H. Human Endometrial Organoids: Recent Research Progress and Potential Applications. Front Cell Dev Biol. 2022;10(February):1–7.
- Fornes R, Ormazabal P, Rosas C, Gabler F, Vantman D, Romero C, et al. Changes in the expression of insulin signaling pathway molecules in endometria from polycystic ovary syndrome women with or without hyperinsulinemia. Mol Med. 2010;16(3–4):129–36.
- Gonzalez D, Thackeray H, Lewis PD, Mantani A, Brook N, Ahuja K, et al. Loss of WT1 expression in the endometrium of infertile PCOS patients: A hyperandrogenic effect? J Clin Endocrinol Metab. 2012;97(3):957–66.
- Mehdinejadiani S, Amidi F, Mehdizadeh M, Barati M. Effects of letrozole and clomiphene citrate on Wnt signaling pathway in endometrium of polycystic ovarian syndrome and healthy women. Biol Reprod. 2019;100(3):641–8.
- Xue, Z, Li, J, Feng, J et al. Research Progress on the Mechanism Between Polycystic Ovary Syndrome and Abnormal Endometrium. Front physio. 2021;12(788772).
- Sanchez-Garrido MA, Tena-Sempere M. Metabolic dysfunction in polycystic ovary syndrome: Pathogenic role of androgen excess and potential therapeutic strategies. Mol Metab [Internet]. 2020;35(February):100937. [CrossRef]
- Oróstica L, Poblete C, Romero C, Vega M. Pro-Inflammatory Markers Negatively Regulate IRS1 in Endometrial Cells and Endometrium from Women with Obesity and PCOS. Reprod Sci. 2020;27(1):290–300.
- Retis-Resendiz AM, González-García IN, León-Juárez M, Camacho-Arroyo I, Cerbón M, Vázquez-Martínez ER. The role of epigenetic mechanisms in the regulation of gene expression in the cyclical endometrium. Clin Epigenetics [Internet]. 2021;13(1):1–23. [CrossRef]
- Khatun M, Meltsov A, Lavogina D, Loid M, Kask K, Arffman RK, et al. Decidualized endometrial stromal cells present with altered androgen response in PCOS. Sci Rep [Internet]. 2021;11(1):1–11. [CrossRef]
- Ding N, Wang R, Wang P, Wang F. Metabolism-related proteins as biomarkers for predicting prognosis in polycystic ovary syndrome. Proteome Sci. 2024;22(1):1–13.
- Reavey JJ, Walker C, Murray AA, Brito-Mutunayagam S, Sweeney S, Nicol M, et al. Obesity is associated with heavy menstruation that may be due to delayed endometrial repair. J Endocrinol. 2021;249(2):71–82.
- Parker J, Hofstee P, Brennecke S. Prevention of Pregnancy Complications Using a Multimodal Lifestyle, Screening, and Medical Model. J Clin Med. 2024;13(15):4344.
- Hoffman, MK. The great obstetrical syndromes and the placenta. BJOG An Int J Obstet Gynaecol. 2023;130(S3):8–15.
- Hopkins BD, Goncalves MD, Cantley LC. Insulin–PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol. 2020;16(5):276–83.
- Burton GJ, Jauniaux E. The human placenta: new perspectives on its formation and function during early pregnancy. Proc R Soc B Biol Sci. 2023;290:20230191.
- Dias Da Silva I, Wuidar V, Zielonka M, Pequeux C. Unraveling the Dynamics of Estrogen and Progesterone Signaling in the Endometrium: An Overview. Cells. 2024;13(15):1–30.
- Vannuccini S, Ph D, Jain V, Sc M, Critchley H, Sc D. From menarche to menopause, heavy menstrual bleeding is the underrated compass in reproductive health. Fertil Steril [Internet]. 2022;118(4):625–36. [CrossRef]
- American Academy of Pediatrics Committee on Adolescence AC of O and GC on AH. Menstruation in Girls and Adolescents : Using the Menstrual Cycle as a Vital Sign. Pediatrics [Internet]. 2006;118(5):2245–50. Available from. Available online: www.pediatrics.org/cgi/doi/10.1542/%0Apeds.2006-2481.
- American College of Obstetricians and Gynecologists. Menstruation in Girls and Adolescents: Using the Menstrual Cycle as a Vital Sign. Committee Opinion No. 651. Obstet Gynecol. 2015;126(6):e143–6.
- Houghton, LC. Menstruation as the Next Vital Sign. JAMA Netw open. 2024;7(5):e2412778.
- Vollmar AKR, Ph D, Mahalingaiah S, Jukic AM, Ph D. The menstrual cycle as a vital sign: a comprehensive review. Fertil Steril Rev [Internet]. 2025;6(1):100081. [CrossRef]
- Wang YX, Arvizu M, Rich-Edwards JW, Stuart JJ, Manson JAE, Missmer SA, et al. Menstrual cycle regularity and length across the reproductive lifespan and risk of premature mortality: prospective cohort study. BMJ. 2020;371:m3464.
- Moore AM, Coolen LM, Porter DT, Goodman RL, Lehman MN. KNDy cells revisited. Endocrinology. 2018;159(9):3219–34.
- Stevenson H, Bartram S, Charalambides MM, Murthy S, Petitt T, Pradeep A, et al. Kisspeptin-neuron control of LH pulsatility and ovulation. Front Endocrinol (Lausanne). 2022;13(November):1–9.
- Nagae M, Uenoyama Y, Okamoto S, Tsuchida H, Ikegami K, Goto T, et al. Direct evidence that KNDy neurons maintain gonadotropin pulses and folliculogenesis as the GnRH pulse generator. Proc Natl Acad Sci U S A. 2021;118(5):1–11.
- Marques P, Skorupskaite K, George JT, Anderson RA. Physiology of GNRH and Gonadotropin Secretion. Endotext [Internet]. 2024;13 October:1–35. Available from: http://www.ncbi.nlm.nih. 2590.
- Hillier SG, Whitelaw PF, Smyth CD. Follicular oestrogen synthesis: the “two-cell, two-gonadotrophin” model revisited. Mol Cell Endocrinol. 1994;100(1–2):51–4.
- Konings G, Brentjens L, Delvoux B, Linnanen T, Cornel K, Koskimies P, et al. Intracrine regulation of estrogen and other sex steroid levels in endometrium and non-gynecological tissues; Pathology, physiology, and drug discovery. Front Pharmacol. 2018;Sep 19(9):940.
- Liu T, Huang Y, Lin H. Estrogen disorders: Interpreting the abnormal regulation of aromatase in granulosa cells (Review). Int J Mol Med. 2021;47(5):1–12.
- Das, SK. Cell cycle regulatory control for uterine stromal cell decidualization in implantation. Reproduction. 2009;137(6):889–99.
- Hewitt SC, Winuthayanon W, Lierz SL, Hamilton KJ, Donoghue LJ, Ramsey JT, et al. Role of ERα in mediating female uterine transcriptional responses to IGF1. Endocrinology. 2017;158(8):2427–35.
- Zhu H, Hou CC, Luo LF, Hu YJ, Yang WX. Endometrial stromal cells and decidualized stromal cells: Origins, transformation and functions. Gene [Internet]. 2014;551(1):1–14. [CrossRef]
- Vasquez YM, Wang X, Wetendorf M, Franco HL, Mo Q, Wang T, et al. FOXO1 regulates uterine epithelial integrity and progesterone receptor expression critical for embryo implantation. PLoS Genet. 2018;14(11):1–27.
- Mueller MD, Vigne JL, Minchenko A, Lebovic DI, Leitman DC, Taylor RN. Regulation of vascular endothelial growth factor (VEGF) gene transcription by estrogen receptors α and β. Proc Natl Acad Sci U S A. 2000;97(20):10972–7.
- King AE, Critchley HOD. Oestrogen and progesterone regulation of inflammatory processes in the human endometrium. J Steroid Biochem Mol Biol [Internet]. 2010;120(2–3):116–26. [CrossRef]
- Yilmaz BD, Sison CAM, Yildiz S, Miyazaki K, Coon V J, Yin P, et al. Genome-wide estrogen receptor-α binding and action in human endometrial stromal cells. F S Sci [Internet]. 2020;1(1):59–66. [CrossRef]
- Nakamura M, Takakura M, Fujii R, Maida Y, Bono Y, Mizumoto Y, et al. The PRB-dependent FOXO1/IGFBP-1 axis is essential for progestin to inhibit endometrial epithelial growth. Cancer Lett [Internet]. 2013;336(1):68–75. [CrossRef]
- Itoh H, Kishore AH, Lindqvist A, Rogers DE, Word RA. Transforming growth factor β1 (TGFβ1) and progesterone regulate matrix metalloproteinases (MMP) in human endometrial stromal cells. J Clin Endocrinol Metab. 2012;97(6):888–97.
- Labrie, F. Intracrinology. Mol Cell Endocrinol. 1991;78(3):13–8.
- Gibson DA, Simitsidellis I, Collins F, Saunders PTK. Endometrial intracrinology: Oestrogens, androgens and endometrial disorders. Int J Mol Sci. 2018;19(10):3276.
- Alanazi, S. Recent Advances in Liquid Chromatography–Mass Spectrometry (LC–MS) Applications in Biological and Applied Sciences. Anal Sci Adv. 2025;6(1):1–12.
- Huhtinen K, Desai R, Stah̊le M, Salminen A, Handelsman DJ, Perheentupa A, et al. Endometrial and endometriotic concentrations of estrone and estradiol are determined by local metabolism rather than circulating levels. J Clin Endocrinol Metab. 2012;97(11):4228–35.
- Huhtinen K, Saloniemi-Heinonen T, Keski-Rahkonen P, Desai R, Laajala D, Stahle M, et al. Intra-tissue steroid profiling indicates differential progesterone and testosterone metabolism in the endometrium and endometriosis lesions. J Clin Endocrinol Metab. 2014;99(11):E2188–97.
- Cloke B, Huhtinen K, Fusi L, Kajihara T, Yliheikkilä M, Ho KK, et al. The androgen and progesterone receptors regulate distinct gene networks and cellular functions in decidualizing endometrium. Endocrinology. 2008;149(9):4462–74.
- Ishikawa T, Harada T, Kubota T, Aso T. Testosterone inhibits matrix metalloproteinase-1 production in human endometrial stromal cells in vitro. Reproduction. 2007;133(6):1233–9.
- Grzechocińska B, Dabrowski F, Cyganek A, Panek G, Wielgoś M. The role of metalloproteinases in endometrial remodelling during menstrual cycle. Ginekol Pol. 2017;88(6):337–42.
- Staun-Ram E, Goldman S, Gabarin D, Shalev E. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod Biol Endocrinol. 2004;2:1–13.
- Zhou X, Wu Y, Zhu Z, Lu C, Zhang C, Zeng L, et al. Mucosal immune response in biology, disease prevention and treatment. Signal Transduct Target Ther [Internet]. 2025;10(1):1–32. [CrossRef]
- Silva-sanchez A, Randall TD. Anatomical Uniqueness of the Mucosal for the Induction and Regulation of Mucosal Immunity and Tolerance [Internet]. Mucosal Vaccines. Elsevier Inc.; 2020. 19–54 p. [CrossRef]
- Kim S, Ndwandwe C, Devotta H, Kareem L, Yao L, O’Mahony L. Role of the microbiome in regulation of the immune system. Allergol Int [Internet]. 2025;74(2):187–96. [CrossRef]
- Lunjani N, Ahearn-Ford S, Dube FS, Hlela C, O’Mahony L. Mechanisms of microbe-immune system dialogue within the skin. Genes Immun [Internet]. 2021;22(5–6):276–88. [CrossRef]
- Park, JC. Chang, L. Kwon, HK. Im S. Beyond the gut: decoding the gut–immune–brain axis in health and disease. Cell Mol Immunol [Internet]. 2025;14 August(On-line ahead of print). [CrossRef]
- Choden T, Cohen NA. The gut microbiome and the immune system. Explor Med. 2022;3(3):219–33.
- Parker J, O’Brien C, Hawrelak J. A narrative review of the role of gastrointestinal dysbiosis in the pathogenesis of polycystic ovary syndrome. Obstet Gynecol Sci. 2021;Epub ahead():1–15.
- Tremellen K, Pearce K. Dysbiosis of Gut Microbiota (DOGMA) - A novel theory for the development of Polycystic Ovarian Syndrome. Med Hypotheses [Internet]. 2012;79(1):104–12. [CrossRef]
- Colonetti T, Limas Carmo Teixeira D, Grande AJ, Rodrigues Uggioni ML, Generoso J, Harding S, et al. The role of intestinal microbiota on pre-eclampsia: Systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2023;291(April):49–58.
- Ishimwe, JA. Maternal microbiome in preeclampsia pathophysiology and implications on offspring health. Physiol Rep. 2021;9(10):1–19.
- Medina-Bastidas D, Camacho-Arroyo I G-GE. Current findings in endometrial microbiome: impact on uterine diseases. Reproduction. 2022;163(5):R81–96.
- Monin L, Whettlock EM, Male V. Immune responses in the human female reproductive tract. Immunology. 2020;160(2):106–15.
- Bister J, Crona Guterstam Y, Strunz B, Dumitrescu B, Haij Bhattarai K, Özenci V, et al. Human endometrial MAIT cells are transiently tissue resident and respond to Neisseria gonorrhoeae. Mucosal Immunol. 2021;14(2):357–65.
- Wira CR, Rodriguez-Garcia M, Patel M V. The role of sex hormones in immune protection of the female reproductive tract. Nat Rev Immunol. 2015;15(4):217–30.
- Chatterjee SK, Saha S, Munoz MNM. Activation of mucosal immunity and novel prophylactic and therapeutic strategy in combating COVID-19. Explor Immunol. 2021;1(5):374–97.
- Evans J, Salamonsen LA. Inflammation, leukocytes and menstruation. Rev Endocr Metab Disord. 2012;13(4):277–88.
- Azlan A, Salamonsen LA, Hutchison J, Evans J. Endometrial inflammasome activation accompanies menstruation and may have implications for systemic inflammatory events of the menstrual cycle. Hum Reprod. 2020;35(6):1363–76.
- Maybin JA, Critchley HOD. Menstrual physiology: Implications for endometrial pathology and beyond. Hum Reprod Update. 2015;21(6):748–61.
- Grzybowska ME, Barcikowska Z. Inflammatory Markers in Dysmenorrhea and Therapeutic Options. Int J Environ Res Public Health. 2020;17(1991):1–14.
- Critchley HOD, Maybin JA, Armstrong GM, Williams ARW. Physiology of the endometrium and regulation of menstruation. Physiol Rev. 2020;100(3):1149–79.
- Maybin JA, Murray AA, Saunders PTK, Hirani N, Carmeliet P, Critchley HOD. Hypoxia and hypoxia inducible factor-1α are required for normal endometrial repair during menstruation. Nat Commun [Internet]. 2018;9(1). [CrossRef]
- Davies J, Kadir RA. Endometrial haemostasis and menstruation. Rev Endocr Metab Disord. 2012;13(4):289–99.
- Gelety TJ, Chaudhuri G. Haemostatic mechanism in the endometrium: role of cyclo-oxygenase products and coagulation factors. Br J Pharmacol. 1995;114(5):975–80.
- Cousins FL, Filby CE, Gargett CE. Endometrial Stem/Progenitor Cells–Their Role in Endometrial Repair and Regeneration. Front Reprod Heal. 2021;3(January):1–14.
- Gargett CE, Nguyen HPT, Ye L. Endometrial regeneration and endometrial stem/progenitor cells. Rev Endocr Metab Disord. 2012;13(4):235–51.
- Kong Y, Shao Y, Ren C, Yang G. Endometrial stem/progenitor cells and their roles in immunity, clinical application, and endometriosis. Stem Cell Res Ther [Internet]. 2021;12(1):1–16. [CrossRef]
- Nair R, Agarwal P, Gadre MA, Vasanthan KS, Seetharam RN. Stem cell treatments for female reproductive disorders : a comprehensive review. J Ovarian Res. 2025;18(161):1–27.
- Sarvestani M, Rajabzadeh A, Mazoochi T, Samimi M, Navari M, Moradi F. Use of placental-derived mesenchymal stem cells to restore ovarian function and metabolic profile in a rat model of the polycystic ovarian syndrome. BMC Endocr Disord. 2024;24(1):1–11.
- Chugh RM, Park HS, Esfandyari S, Elsharoud A, Ulin M, Al-Hendy A. Mesenchymal stem cell-conditioned media regulate steroidogenesis and inhibit androgen secretion in a PCOS cell model via BMP-2. Int J Mol Sci. 2021;22(17):1–15.
- Choi JH, Seok J, Lim SM, Kim TH, Kim GJ. Microenvironmental changes induced by placenta-derived mesenchymal stem cells restore ovarian function in ovariectomized rats via activation of the PI3K-FOXO3 pathway. Stem Cell Res Ther. 2020;11(1):1–13.
- Jafarzadeh H, Nazarian H, Ghaffari Novin M, Shams Mofarahe Z, Eini F, Piryaei A. Improvement of oocyte in vitro maturation from mice with polycystic ovary syndrome by human mesenchymal stromal cell–conditioned media. J Cell Biochem. 2018;119(12):10365–75.
- Xie Q, Xiong X, Xiao N, He K, Chen M, Peng J, et al. Mesenchymal Stem Cells Alleviate DHEA-Induced Polycystic Ovary Syndrome (PCOS) by Inhibiting Inflammation in Mice. Stem Cells Int. 2019;Sep 12:201(9782373).
- Ciprietti M, Bueds C, Vankelecom H, Vriens J. Organoids as powerful models of endometrium epithelium in transcriptomic, cellular and functional mimicry. Cell Mol Life Sci. 2025;82(1):1–19.
- Munro MG, Critchley HOD, Fraser IS, Haththotuwa R, Kriplani A, Bahamondes L, et al. The two FIGO systems for normal and abnormal uterine bleeding symptoms and classification of causes of abnormal uterine bleeding in the reproductive years: 2018 revisions. Int J Gynecol Obstet. 2018;143(3):393–408.
- Rodriguez MB, Dias S, Jordan V, Lethaby A, Lensen SF, Wise MR, et al. Interventions for heavy menstrual bleeding; overview of Cochrane reviews and network meta-analysis. Cochrane Database Syst Rev. 2022;5(5):CD013180.
- Pouraliroudbaneh S, Marino J, Riggs E, Saber A, Jayasinghe Y, Peate M. Heavy menstrual bleeding and dysmenorrhea in adolescents: A systematic review of self-management strategies, quality of life, and unmet needs. Int J Gynecol Obstet. 2024;167(1):16–41.
- Munro MG, Critchley HOD, Fraser IS. The FIGO systems for nomenclature and classification of causes of abnormal uterine bleeding in the reproductive years: Who needs them? Am J Obstet Gynecol [Internet]. 2012;207(4):259–65. [CrossRef]
- Comishen KJ, Bhatt M, Yeung K, Irfan J, Zia A, Sidonio RF, et al. Etiology and diagnosis of heavy menstrual bleeding among adolescent and adult patients: a systematic review and meta-analysis of the literature. J Thromb Haemost [Internet]. 2025;23(3):863–76. [CrossRef]
- Cole SK, Billewicz WZ, Thomson AM. Sources of Variation in Menstrual Blood Loss. BJOG An Int J Obstet Gynaecol. 1971;78(10):933–9.
- Sinharoy SS, Chery L, Patrick M, Conrad A, Ramaswamy A, Stephen A, et al. Prevalence of heavy menstrual bleeding and associations with physical health and wellbeing in low-income and middle-income countries: a multinational cross-sectional study. Lancet Glob Heal [Internet]. 2023;11(11):e1775–84. [CrossRef]
- National Institute for Health and Care Excellence (NICE). Heavy Menstrual Bleeding: Assessment and Management [Internet]. [NICE Guideline NG88]: Updated . Available from: http://www.nice.org.uk/guidance/ng88. Accessed 10 August 2025.
- Australian Commission on Safety and Quality in health Care. Heavy Menstrual Bleeding Clinical Care Standard: June 2024. [Internet]. Available from: https://www.safetyandquality.gov.au/standards/clinical-care-standards/heavy-menstrual-bleeding-clinical-care-standard. Accessed.
- Warner P, Harriet L, Whitaker R, Anthony R, John C, Douglas A, et al. Low dose dexamethasone as treatment for women with heavy menstrual bleeding: A response-adaptive randomised placebo-controlled dose-finding parallel group trial ( DexFEM ). EBioMedicine [Internet]. 2021;69:103434. [CrossRef]
- Mardon, AK. White, S. Howe, D. O’Shea, M. Eathorne, A. Gannott, M. Schott, A. Armour M. Problematic Periods Costing Young Women—The Impact of Menstrual Symptoms on Work and Study. ANZJOG. 2024;Dec 19(on line ahead of print.).
- Ponzo S, Wickham A, Bamford R, Radovic T, Zhaunova L, Peven K, et al. Menstrual cycle-associated symptoms and workplace productivity in US employees: A cross-sectional survey of users of the Flo mobile phone app. Digit Heal. 2022;8:1–12.
- Bigambo FM, Wang D, Zhang Y, Mzava SM, Dai R, Wang X. Current situation of menstruation and gynecological diseases prevalence among Chinese women: a cross-sectional study. BMC Womens Health [Internet]. 2022;22(1):1–12. [CrossRef]
- Ashworth, G. Management of heavy menstrual bleeding in Australian general practice An analysis of BEACH data. Aust J Gen Pract. 2021;50(8):573–9.
- Wilson L, Copp T, Hickey M, Jenkinson B, Jordan SJ, Thompson R, et al. Women who experience heavy menstrual bleeding: prevalence and characteristics from young adulthood to midlife, Australia, 2000–2021: a longitudinal cohort survey study. Med J Aust. 2025;222(4):191–7.
- Serhat E, Cogendez E, Selcuk S, Asoglu MR, Arioglu PF, Eren S. Is there a relationship between endometrial polyps and obesity, diabetes mellitus, hypertension? Arch Gynecol Obstet. 2014;290(5):937–41.
- Harmon QE, Patchel S, Denslow S, Wegienka G, Baird DD. Body Mass Index and Uterine Fibroid Development: A Prospective Study. J Clin Endocrinol Metab [Internet]. 2024;109(11):e2016–23. [CrossRef]
- Keizer AL, Semmler A, Kok HS, van Kesteren PJM, Huirne JAF, Hehenkamp WJK. Modifiable prognostic factors in uterine fibroid development: a systematic review of literature. J Obstet Gynaecol (Lahore) [Internet]. 2024;44(1):2288225. [CrossRef]
- Jasper EA, Mautz BS, Hellwege JN, Piekos JA, Jones SH, Zhang Y, et al. A phenome-wide association study of uterine fibroids reveals a marked burden of comorbidities. Commun Med. 2025;5(1):1–15.
- Palomba, S. Is fertility reduced in ovulatory women with polycystic ovary syndrome? An opinion paper. Hum Reprod. 2021;36(9):2421–8.
- Patel, S. Disruption of aromatase homeostasis as the cause of a multiplicity of ailments: A comprehensive review. J Steroid Biochem Mol Biol [Internet]. 2017;168:19–25. [CrossRef]
- Szukiewicz D, Trojanowski S, Kociszewska A, Szewczyk G. Modulation of the Inflammatory Response in Polycystic Ovary Syndrome (PCOS)—Searching for Epigenetic Factors. Int J Mol Sci. 2022;23(14663):1–27.
- Hapangama DK, Bulmer JN. Pathophysiology of heavy menstrual bleeding. Women’s Heal. 2016;12(1):3–13.
- Girling JE, Lockhart MG, Olshansky M, Paiva P, Woodrow N, Marino JL, et al. Differential Gene Expression in Menstrual Endometrium from Women with Self-Reported Heavy Menstrual Bleeding. Reprod Sci. 2017;24(1):28–46.
- Li X, Wang F, Chen M, Ling L, Zhao F, Peng D. The association between endometrial polyps and insulin resistance from the expression of PI3K and AKT proteins perspective. BMC Womens Health [Internet]. 2024;24(366):1–7. [CrossRef]
- Oliva MM, Gambioli R, Forte G, Porcaro G, Aragona C, Unfer V. Unopposed estrogens: current and future perspectives. Eur Rev Med Pharmacol Sci. 2022;26(8):2975–89.
- Nijkang NP, Anderson L, Markham R, Manconi F. Endometrial polyps: Pathogenesis, sequelae and treatment. SAGE Open Med. 2019;7:1–12.
- Zhong⋅ X, Li⋅ Y, Liang W, Hu Q, Zeng A, Ding M, et al. Clinical and metabolic characteristics of endometrial lesions in polycystic ovary syndrome at reproductive age. BMC Womens Health. 2023;23(1):1–9.
- Cakmak H, Taylor HS. Implantation failure: Molecular mechanisms and clinical treatment. Hum Reprod Update. 2011;17(2):242–53.
- Yusuf M, Amri MF, Ugusman A, Hamid AA, Wahab NA, Mokhtar MH. Hyperandrogenism and Its Possible Effects on Endometrial Receptivity : A Review. int J Mol Sci. 2023;24:12026.
- Salamonsen LA, Dimitriadis E. Infertility and the Endometrium. Clin Exp Obstet Gynecol. 2022;49(9):1–10.
- Skliutė G, Baušytė R, Ramašauskaitė D, Navakauskienė R. Characterization of Epigenetic and Molecular Factors in Endometrium of Females with Infertility. Biomedicines. 2022;10(6):1–12.
- Khodaei MM, Noori Z, Zare F, Meshkin A. Ferroptosis and recurrent miscarriage: a critical review of pathophysiology and emerging therapeutic targets. Front Cell Dev Biol. 2025;13:1559300.
- Gusarova TA, Nizyaeva N V., Mikhalev SA, Tikhonova NB, Orgadeeva DA, Mikhaleva LM, et al. Morphological and molecular features of decidual endometrial cells in miscarriage. Morphology. 2023;161(1):37–49.
- Muter J, Kong CS, Nebot MT, Tryfonos M, Vrljicak P, Brighton PJ, et al. Stalling of the endometrial decidual reaction determines the recurrence risk of miscarriage. Sci Adv. 2025;11(26):1–14.
- Bacon SJ, Zhu Y, Ghosh P. Early spiral arteriole remodeling in the uterine–placental interface: A rat model. J Anat. 2024;244(6):1054–66.
- Elawad T, Scott G, Bone JN, Elwell H, Lopez CE, Filippi V, et al. Risk factors for pre-eclampsia in clinical practice guidelines: Comparison with the evidence. BJOG An Int J Obstet Gynaecol. 2022;131(1):46–62.
- Kibret KT, Chojenta C, Gresham E, Tegegne TK, Loxton D. Maternal dietary patterns and risk of adverse pregnancy (hypertensive disorders of pregnancy and gestational diabetes mellitus) and birth (preterm birth and low birth weight) outcomes: A systematic review and meta-analysis. Public Health Nutr. 2019;22(3):506–20.
- Paula WO, Patriota ESO, Gonçalves VSS, Pizato N. Maternal Consumption of Ultra-Processed Foods-Rich Diet and Perinatal Outcomes: A Systematic Review and Meta-Analysis. Nutrients. 2022;14:3242.
- Sanderson PA, Critchley HOD, Williams ARW, Arends MJ, Saunders PTK. New concepts for an old problem: The diagnosis of endometrial hyperplasia. Hum Reprod Update. 2017;23(2):232–54.
- Damor, CB. Damor P. Molecular Profiling of Endometrial Hyperplasia and its Progression to Endometrial Carcinoma: A Histopathological and Genomic Correlation Study. Int J Life Sci Biotechnol Pharma Res [Internet]. 2025;14(6):150–3. Available from: https://www.ijlbpr.com/uploadfiles/28vol14issue6pp150-153.20250607043048.
- Bostan IS, Mihaila M, Roman V, Radu N, Neagu MT, Bostan M, et al. Landscape of Endometrial Cancer: Molecular Mechanisms, Biomarkers, and Target Therapy. Cancers (Basel). 2024;16(11):1–24.
- Balhara N, Yadav R, Chauhan MB. Role of signaling pathways in endometrial cancer. Mol Biol Rep. 2025;52(1):1–18.
- Brower M, Brennan K, Pall M, Azziz R. The severity of menstrual dysfunction as a predictor of insulin resistance in PCOS. J Clin Endocrinol Metab. 2013;98(12):E1967–71.
- Li X, Yang D, Pan P, Azziz R, Yang D. The Degree of Menstrual Disturbance Is Associated With the Severity of Insulin Resistance in PCOS. Front Endo. 2022;13:873726.
- Ezeh, U. Ezeh, C. Pisarska, MD. Azziz R. Menstrual dysfunction in polycystic ovary syndrome: association with dynamic state insulin resistance rather than hyperandrogenism. Fertil Steril [Internet]. 2021;115(6):1557–68. [CrossRef]
- Salcido, AC, Shehata, H. Berry, A. Riba C. Insulin resistance and other risk factors of cardiovascular disease amongst women with abnormal uterine bleeding. J Insul Resist [Internet]. 2022;5(1):1–7. [CrossRef]
- Solomon CG, Hu FB, Dunaif A, Rich-edwards J, Hunter DJ, Colditz GA, et al. Long or Highly Irregular Menstrual Cycles as a Marker for Risk of Type 2 Diabetes Mellitus. Jama. 2001;286(19):2421–6.
- Roumain, J. Charles, M. A. Courten, M. P. D. Hanson, R. L. Brodie, T. D. Pettitt, D. J. Knowler WC. The Relationship of Menstrual Irregularity to Type 2 Diabetes in Pima Indian Women. Diabetes Care. 1998;21(3):1–4.
- Strowitzki T, Capp E, Eye H Von. The degree of cycle irregularity correlates with the grade of endocrine and metabolic disorders in PCOS patients. Eur J Obstet Gynecol [Internet]. 2010;149(2):178–81. [CrossRef]
- Cooper GS, Ephross SA, Sandler DP. Menstrual patterns and risk of adult-onset diabetes mellitus ଝ. J Clin Epidemiol. 2000;53:1170–3.
- Wang Y, Shan Z, Arvizu M, Pan A, Manson JE, Missmer SA. Associations of Menstrual Cycle Characteristics Across the Reproductive Life Span and Lifestyle Factors With Risk of Type 2 Diabetes. JAMA Netw Open. 2020;3(12):e2027928.
- Wang YX, Shan Z, Arvizu M, Pan A, Manson JAE, Missmer SA, et al. Associations of Menstrual Cycle Characteristics Across the Reproductive Life Span and Lifestyle Factors With Risk of Type 2 Diabetes. JAMA Netw open. 2020;3(12):e2027928.
- Foryś E, Baran A, Dziurdzia A, Jarosz-wójcik E, Matusik P, Gawlik A, et al. Are menstrual disorders in adolescent girls related to metabolic disorders? Paediatr Endocrinol Diabetes, Metab [Internet]. 2023;29(2):75–82. [CrossRef]
- Onalan R, Onalan G, Tonguc E, Ozdener T. Body mass index is an independent risk factor for the development of endometrial polyps in patients undergoing in vitro fertilization. Fertil Steril. 2009;91(4):1056–60.
- Kacalska-Janssen O, Rajtar-Ciosek A, Zmaczyński A, Wyroba J, Milewicz T, Krzyczkowska-Sendrakowska M KJ. Markers of insulin resistance in perimenopausal women with endometrial pathology. Ginekol Pol. 2013;84(11):922–9.
- He Y, Zeng Q, Li X, Liu B, Wang P. The Association between Subclinical Atherosclerosis and Uterine Fibroids. PLoS One. 2013;8(2):1–10.
- Salcedo AC, Yun J, Carter C, Hart E. Therapeutic Carbohydrate Restriction as a Metabolic Modality for the Prevention and Treatment of Abnormal Uterine Bleeding. Nutrients [Internet]. 2023;15:3760. [CrossRef]
- Uimari O, Auvinen J, Jokelainen J, Puukka K, Ruokonen A, Järvelin M, et al. Uterine fibroids and cardiovascular risk. Hum Reprod. 2016;31(12):2689–703.
- Laughlin-tommaso SK, Fuchs EL, Wellons MF, Lewis CE, Calderon-margalit R, Stewart EA, et al. Uterine Fibroids and the Risk of Cardiovascular Disease in the Coronary Artery Risk Development in Young Adult Women ’s Study. J Women’s. 2019;28(1):46–52.
- Final Annual Report RCOG. NHMBA. Final Annual Report: National Heavy Menstrual Bleeding Audit [Internet]. 2014. Available from: https://www.hqip.org.uk/wp-content/uploads/2018/02/HwNYNM.
- Soldani R, Cagnacci A, Yen SSC. Insulin insulin-like growth factor I (IGF-I) and IGF-II enhance basal and gonadotrophin-releasing hormone-stimulated luteinizing hormone release from rat anterior pituitary cells in vitro. Eur J Endocrinol. 1994;131(6):641–5.
- Sliwowska JH, Fergani C, Gawałek M, Skowronska B, Fichna P, Lehman MN. Insulin: Its role in the central control of reproduction. Physiol Behav. 2014;133:197–206.
- Nestler JE, Jakubowicz DJ, De Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83(6):2001–5.
- Baillargeon JP, Nestler JE. Commentary: Polycystic ovary syndrome: A syndrome of ovarian hypersensitivity to insulin? J Clin Endocrinol Metab. 2006;91(1):22–4.
- S Rice, A Khalid, S Khan, M Lacey RH. The effect of hyperinsulinemia on FSH-mediated signalling pathways in granulosa cells - implications for follicle growth in women with PCOS. Hum Reprod [Internet]. 2023;38(Supplement-1):P-643. [CrossRef]
- Nelson-degrave VL, Wickenheisser JK, Hendricks KL, Asano T, Fujishiro M, Legro RS, et al. Alterations in Mitogen-Activated Protein Kinase Kinase and Extracellular Regulated Kinase Signaling in Theca Cells Contribute to Excessive Androgen Production in Polycystic Ovary Syndrome. Mol Endocrinol. 2015;19(2):379–90.
- Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr Rev. 2012;33(6):981–1030.
- Maclean JA, Hayashi K. Progesterone Actions and Resistance in Gynecological Disorders. Cells. 2022;11:647.
- Garg D, Merhi Z. Relationship between Advanced Glycation End Products and Steroidogenesis in PCOS. Reprod Biol Endocrinol [Internet]. 2016;14(1):1–13. [CrossRef]
- Zuo T, Zhu M, Xu W. Roles of oxidative stress in polycystic ovary syndrome and cancers. Oxid Med Cell Longev. 2016;2016:8589318.
- Zheng X, Pan X, Zhang J, Cao X. Hyperinsulinemia-induced PAX6 expression promotes endometrial epithelial cell proliferation via negatively modulating p27 signaling. Biomed Pharmacother [Internet]. 2018;97(97):802–8. [CrossRef]
- Maybin JA, Critchley HOD, Jabbour HN. Inflammatory pathways in endometrial disorders. Mol Cell Endocrinol [Internet]. 2011;335(1):42–51. [CrossRef]
- Matsuyama S, Whiteside S, Li SY. Implantation and Decidualization in PCOS: Unraveling the Complexities of Pregnancy. Int J Mol Sci. 2024;25:1203.
- Bremer AA, Miller WL. The serine phosphorylation hypothesis of polycystic ovary syndrome: a unifying mechanism for hyperandrogenemia and insulin resistance. Fertil Steril. 2008;89(5):1039–48.
- Lee M, Yoon J, Kim H, Kim YS, Lyu SW, Lee BS. Hyperandrogenic Milieu Dysregulates the Expression of Insulin Signaling Factors and Glucose Transporters in the Endometrium of Patients with Polycystic Ovary Syndrome. Reprod Sci. 2020;27(8):1637–47.
- Nagao H, Cai W, Wewer NJ, Steger M, Batista TM, Pan H. Distinct signaling by insulin and IGF-1 receptors and their extra- and intracellular domains. PNAS. 2021;118(17):e2019474118.
- Wang Y, Hua S, Tian W, Zhang L, Zhao J, Zhang H, et al. Mitogenic and anti-apoptotic effects of insulin in endometrial cancer are phosphatidylinositol 3-kinase/Akt dependent. Gynecol Oncol [Internet]. 2012;125(3):734–41. [CrossRef]
- Jiang N, Zhao W, Shen H, Du D, Li X. Hyperinsulinemia impairs decidualization via AKT-NR4A1 signaling: new insight into polycystic ovary syndrome ( PCOS ) -related infertility. J Ovarian Res [Internet]. 2024;17(1):31. [CrossRef]
- Kooijman, R. Regulation of apoptosis by insulin-like growth factor ( IGF ) -I. Cytokine Growth Factor Rev. 2006;17:305–23.
- Foley, PJ. Effect of low carbohydrate diets on insulin resistance and the metabolic syndrome. Curr Opin Endocrinol Diabetes Obes. 2021;28(5):463–8.
- Wolosowicz M, Prokopiuk S, Kaminski TW. Recent Advances in the Treatment of Insulin Resistance Targeting Molecular and Metabolic Pathways: Fighting a Losing Battle? Med. 2022;58:472.
- Robinson S, Kiddy D, Gelding S V., Willis D, Niththyananthan R, Bush A, et al. The relationship of insulin insensitivity to menstrual pattern in women with hyperandrogenism and polycystic ovaries. Clin Endocrinol (Oxf). 1993;39(3):351–5.
- Niu J, Lu M, Liu B. Association between insulin resistance and abnormal menstrual cycle in Chinese patients with polycystic ovary syndrome. J Ovarian Res [Internet]. 2023;16(1):1–11. [CrossRef]
- Dey R, Bhattacharya K, Basak AK, Paul N, Bandyopadhyay R, Chaudhuri GR, et al. Inflammatory perspectives of polycystic ovary syndrome: role of specific mediators and markers. Middle East Fertil Soc J [Internet]. 2023;28(33):1–12. [CrossRef]
- Vasyukova E, Zaikova E, Kalinina O, Gorelova I, Pyanova I, Bogatyreva E, et al. Inflammatory and Anti-Inflammatory Parameters in PCOS Patients Depending on Body Mass Index: A Case-Control Study. Biomedicines. 2023;11(10):1–13.
- Zhai Y, Pang Y. Systemic and ovarian inflammation in women with polycystic ovary syndrome. J Reprod Immunol [Internet]. 2022;151(April):103628. [CrossRef]
- Ye Z, Zhao J, Li R. Effects of immune cells and cytokines on the endometrial immune microenvironment in polycystic ovary syndrome. Gynecol Obstet Clin Med [Internet]. 2022;2(4):181–5. [CrossRef]
- Wang J, Yin T, Liu S. Dysregulation of immune response in PCOS organ system. Front Immunol. 2023;14(May):1–13.
- Liu S, Hong L, Mo M, Xiao S, Chen C, Li Y, et al. Evaluation of endometrial immune status of polycystic ovary syndrome. J Reprod Immunol [Internet]. 2021;144(20):103282. [CrossRef]
- Smith OPM, Jabbour HN, Critchley HOD. Cyclooxygenase enzyme expression and E series prostaglandin receptor signalling are enhanced in heavy menstruation. Hum Reprod. 2007;22(5):1450–6.
- Milne SA, Perchick GB, Boddy SC, Jabbour HN. Expression, Localization, and Signaling of PGE2 and EP2/EP4 Receptors in Human Nonpregnant Endometrium across the Menstrual Cycle. J Clin Endocrinol Metab. 2015;86(October):4453–9.
- Eijkeren MA Van, Christiaens GCML. Effects of mefenamic acid on menstrual hemostasis in essential menorrhagia. Am J Obstet Gynecol [Internet]. 1992;166(5):1419–28. [CrossRef]
- Khajehei M, Abdali K, Tabatabaee H. The effect of mefenamic acid and naproxen on heavy menstrual bleeding : A placebo-controlled study. South African J Obstet Gynecolgy. 2013;19(2):31–4.
- Rees MCP, Bernal SL, Ca R, Turnbull AC, Hospital JR, Ox O. Effect of Fenamates on Prostaglandin E Receptor Binding. Lancet. 1988;September:541–2.
- Shetty SS, Deepthi D, Harshitha S, Sonkusare S, Naik PB, N SK, et al. Environmental pollutants and their effects on human health. Heliyon [Internet]. 2023;9(9):e19496. [CrossRef]
- Stegehuis N, Kotsirilos V, Parker J. The Impact of Microparticulate Air Pollution in Polycystic Ovary Syndrome: A Narrative Review. Clin Exp Obstet Gynecol. 2024;51(10):233.
- Parker, J. O’Brien C, Hawrelak J. A narrative review of the role of gastrointestinal dysbiosis in the pathogenesis of polycystic ovary syndrome. Obstet Gynecol Sci. 2022;65(1):14–28.
- Ameho S, Klutstein M. The effect of chronic inflammation on female fertility. Reproduction [Internet]. 2025;169(e240197). [CrossRef]
- Drizi A, Djokovic D, Laganà AS, Herendael B Van. Impaired inflammatory state of the endometrium: A multifaceted approach to endometrial inflammation. Current insights and future directions. Menopause Rev. 2020;19(2):90–100.
- Palomba S, Daolio J, La Sala GB. Oocyte Competence in Women with Polycystic Ovary Syndrome. Trends Endocrinol Metab [Internet]. 2017;28(3):186–98. [CrossRef]
- Piltonen, TT. Polycystic ovary syndrome: Endometrial markers. Best Pract Res Clin Obstet Gynaecol [Internet]. 2016;37:66–79. [CrossRef]
- Quezada S, Avellaira C, Johnson MC, Gabler F, Fuentes A, Vega M. Evaluation of steroid receptors, coregulators, and molecules associated with uterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil Steril [Internet]. 2006;85(4):1017–26. [CrossRef]
- Li X, Feng Y, Lin JF, Billig H, Shao R. Endometrial progesterone resistance and PCOS. J Biomed Sci. 2014;21(1):1–7.
- Marquardt RM, Kim TH, Shin JH, Jeong JW. Progesterone and estrogen signaling in the endometrium: What goes wrong in endometriosis? Int J Mol Sci. 2019;20(15):1–28.
- van der Horst PH, Wang Y, van der Zee M, Burger CW, Blok LJ. Interaction between sex hormones and WNT/β-catenin signal transduction in endometrial physiology and disease. Mol Cell Endocrinol [Internet]. 2012;358(2):176–84. [CrossRef]
- Gellersen B, Brosens JJ. Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr Rev. 2014;35(6):851–905.
- Salamun V, Rizzo M, Lovrecic L, Hocevar K, Papler Burnik T, Janez A, Jensterle M, Vrtacnik Bokal E, Peterlin B MA. The Endometrial Transcriptome of Metabolic and Inflammatory Pathways During the Window of Implantation Is Deranged in Infertile Obese Polycystic Ovarian Syndrome Women. Metab Syndr Relat Disord. 2022;20(7):384–94.
- Savaris RF, Groll JM, Young SL, DeMayo FJ, Jeong JW, Hamilton AE, et al. Progesterone resistance in PCOS endometrium: A microarray analysis in clomiphene citrate-treated and artificial menstrual cycles. J Clin Endocrinol Metab. 2011;96(6):1737–46.
- Das, SK. Regional development of uterine decidualization: Molecular signaling by Hoxa-10. Mol Reprod Dev. 2010;77(5):387–96.
- Cermik D, Selam B, Taylor HS. Regulation of HOXA-10 expression by testosterone in vitro and in the endometrium of patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(1):238–43.
- Ali R, Ahmed T, Gul H, Rehman R. An interplay of Progesterone, Leukemia Inhibitor Factor and Interleukin-6 in the window of implantation ; Impact on fertility. Cytokine [Internet]. 2023;170(May):156332. [CrossRef]
- Ujvari D, Hulchiy M, Calaby A, Nybacka A, Byström B, Hirschberg AL. Lifestyle intervention up-regulates gene and protein levels of molecules involved in insulin signaling in the endometrium of overweight/obese women with polycystic ovary syndrome. Hum Reprod. 2014;29(7):1526–35.
- Hagenfeldt, K. The role of prostaglandins and allied substances in uterine haemostasis. Contraception. 1987;36(1):23–35.
- Smith SK, Abel MH, Kelly RW, Baird DT. Prostaglandin Synthesis in the Endometrium of Women with Ovulatory Dysfunctional Uterine Bleeding. Brithish J Obstet Gynaecol. 1981;88(April):434–432.
- Marsh MM, Malakooti N, Taylor NH. Endothelin and neutral endopeptidase in the endometrium of women with menorrhagia. Hum Reprod [Internet]. 1997;12(9):2036–40. [CrossRef]
- Abberton KM, Healy DL, Rogers PAW. Smooth muscle alpha actin and myosin heavy chain expression in the vascular smooth muscle cells surrounding human endometrial arterioles. Hum Reprod [Internet]. 1999;14(12):3095–100. [CrossRef]
- Abberton KM, Taylor NH, Healy DL, Rogers PAW. Vascular smooth muscle cell proliferation in arterioles of the human endometrium. Hum Reprod. 1999;14(4):1072–9.
- Gleeson, N. Devitt, M. Sheppard, BL. Bonnar N. Endometrial fibrinolytic enzymes in women with normal menstruation and dysfunctional uterine bleeding. Br J Obstet Gynaecol. 1993;100(8):768–71.
- Sylus AM, Nandeesha H, Chitra T. Matrix metalloproteinase-9 increases and interleukin-10 reduces with increase in body mass index in polycystic ovary syndrome: A cross-sectional study. Int J Reprod Biomed. 2020;18(8):605–10.
- Middelkoop MA, Don EE, Hehenkamp WJK, Polman NJ, Griffioen AW, Huirne JAF. Angiogenesis in abnormal uterine bleeding: a narrative review. Hum Reprod Update. 2023;29(4):457–85.
- Critchley HOD, Maybin JA, Ch MBB. Molecular and Cellular Causes of Abnormal Uterine Bleeding of Endometrial Origin. Semin Reprod Med. 2011;29(5):400–9.
- Rae M, Mohamad A, Price D, Hadoke PWF, Walker BR, Mason JI, et al. Cortisol Inactivation by 11β-Hydroxysteroid dehydrogenase-2 May Enhance Endometrial Angiogenesis via Reduced Thrombospondin-1 in Heavy Menstruation. J Clin Endocrinol Metab. 2009;94(4):1443–50.
- Villavicencio A, Bacallao K, Avellaira C, Gabler F, Fuentes A, Vega M. Androgen and estrogen receptors and co-regulators levels in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. Gynecol Oncolo. 2006;103:307–14.
- Guo F, Huang Y, Fernando T, Shi Y. Altered Molecular Pathways and Biomarkers of Endometrial Receptivity in Infertile Women with Polycystic Ovary Syndrome. Reprod Sci [Internet]. 2022;29:3335–45. [CrossRef]
- Koc O, Ozdemirici S, Acet M, Soyturk U. Nuclear factor-κB expression in the endometrium of normal and overweight women with polycystic ovary syndrome. J Obstet Gynaecol (Lahore) [Internet]. 2017;37(7):924–30. [CrossRef]
- Younas K, Quintela M, Thomas S, Garcia-Parra J, Blake L, Whiteland H, et al. Delayed endometrial decidualisation in polycystic ovary syndrome; the role of AR-MAGEA11. J Mol Med. 2019;97(9):1315–27.
- James DW, Quintela M, Lucini L, Alkafri NK, Healey GD, Younas K, et al. Homeobox regulator Wilms Tumour 1 is displaced by androgen receptor at cis-regulatory elements in the endometrium of PCOS patients. Front Endocrinol (Lausanne). 2024;15(April):1–13.
- Thibord F, Cunha J, Džigurski J, Tuftin B, Huffman JE, Pujol-Gualdo N, Cho K, Wilson PWF, Johnsen JM, Raffield LM, Chen MH, Laisk T JA. Genome-wide meta-analysis of heavy menstrual bleeding reveals 36 risk loci. Blood. 2025;146(6):745–58.
- Navarro A, Bariani MV, Park HS, Zota AR, Al-Hendy A. Report of Exosomes Isolated from a Human Uterine Leiomyoma Cell Line and Their Impact on Endometrial Vascular Endothelial Cells. Pharmaceuticals. 2022;15(577):1–15.
- Sadler B, Minard CG, Haller G, Gurnett CA, O’Brien SH, Wheeler A, et al. Whole-exome analysis of adolescents with low VWF and heavy menstrual bleeding identifies novel genetic associations. Blood Adv. 2022;6(2):420–8.
- Wang X, Yang D, Peng H. Female Reproductive Tract Organoids: Applications from Physiology to Pathology. Biomolecules. 2025;15:925.
- Zhao X, Jiang Y, Xi H, Chen L, Feng X. Exploration of the Relationship between Gut Microbiota and Polycystic Ovary Syndrome (PCOS): A Review. Geburtshilfe Frauenheilkd. 2020;80(2):161–71.
- Rizk MG, Thackray VG. Intersection of Polycystic Ovary Syndrome and the Gut Microbiome. J Endocr Soc. 2021;5(2):1–16.
- Hou K, Wu ZX, Chen XY, Wang JQ, Zhang D, Xiao C, et al. Microbiota in health and diseases. Signal Transduct Target Ther. 2022;7(1):1–28.
- Yurtdaş G, Akdevelioğlu Y. A New Approach to Polycystic Ovary Syndrome: The Gut Microbiota. J Am Coll Nutr [Internet]. 2020;39(4):371–82. [CrossRef]
- PARKER JIM, HAWRELAK J, GERSH FL. Nutritional Role of Polyphenols As a Component of a Wholefood Diet in the Management of Polycystic Ovary Syndrome. Australas Coll Nutr Environ Med J [Internet]. 2021;40(2):6–12. Available online: https://search.ebscohost.com/login.aspx?direct=true&db=rzh&AN=154032844&lang=ja&site=ehost-live.
- Parker, J. Hawrelak, J. Gersh F. Nutritional Role of Polyphenols As a Component of a Wholefood Diet in the Management of Polycystic Ovary Syndrome. Australas Coll Nutr Environ Med J [Internet]. 2021;40(2):6–12. Available online: https://search.ebscohost.com/login.aspx?direct=true&db=rzh&AN=154032844&lang=ja&site=ehost-live.
- Tremellen KPK. Nutrition, Fertility, and Human Reproductive Function. In: Nutrition, Fertility, and Human Reproductive Function. Adelaide: CRC Press; 2015. p. 27–50.
- Wu Y, Lin Z, Li C, Lin X, Shan S, Guo B. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduct Target Ther. 2023;8:98.
- Faraji J, Metz GAS. Environmental Epigenetics: New Horizons in Redefining Biological and Health Outcomes. PREPRINT. 2025.
- Scarfo G, Daniele S, Fusi J, Gesi M, Martini C, Franzoni F, et al. Metabolic and Molecular Mechanisms of Diet and Physical Exercise in the Management of Polycystic Ovarian Syndrome. Biomedicines. 2022;10:1305.
- Szczuko M, Kikut J, Szczuko U, Szydłowska I, Nawrocka-rutkowska J, Zi M. Nutrition Strategy and Life Style in Polycystic Ovary Syndrome — Narrative Review. Nutrients. 2021;13:2452.
- Gu Y, Zhou G, Zhou F, Li Y, Wu Q, He H, et al. Gut and Vaginal Microbiomes in PCOS: Implications for Women’s Health. Front Endocrinol (Lausanne). 2022;13(February):1–9.
- Sola-Leyva A, Pérez-Prieto I, Molina NM, Vargas E, Ruiz-Durán S, Leonés-Baños I, et al. Microbial composition across body sites in polycystic ovary syndrome: a systematic review and meta-analysis. Reprod Biomed Online. 2023;47(1):129–50.
- Lin SY, Yang YC, Chang CYY, Lin CC, Hsu WH, Ju SW, et al. Risk of polycystic ovary syndrome in women exposed to fine air pollutants and acidic gases: A nationwide cohort analysis. Int J Environ Res Public Health. 2019;16(23):4816.
- Leceta J, Del Campo R, Jordan S, Klose CSN. Editorial: Immunoregulation at mucosal surfaces. Front Immunol. 2022;13(August):1–5.
- Hampl R, Starka L. Endocrine Disruptors and Gut Microbiome Interactions. Physiol Res. 2020;69(suppl. 2):S211–23.
- Bachmann MC, Bellalta S, Basoalto R, Gómez-Valenzuela F, Jalil Y, Lépez M, et al. The Challenge by Multiple Environmental and Biological Factors Induce Inflammation in Aging: Their Role in the Promotion of Chronic Disease. Front Immunol. 2020;11(October):1–19.
- Thin ZS, Chew J, Yu T, Ong Y, Affendi R, Ali R, et al. Impact of microplastics on the human gut microbiome : a systematic review of microbial composition, diversity, and metabolic disruptions. BMC Gastroenterol. 2025;25(583):1–18.
- Balali H, Morabbi A, Karimian M. Concerning influences of micro/nano plastics on female reproductive health: focusing on cellular and molecular pathways from animal models to human studies. Reprod Biol Endocrinol [Internet]. 2024;22(1):1–35. [CrossRef]
- Sun J, Sui M, Wang T, Teng X, Sun J, Chen M. Detection and quantification of various microplastics in human endometrium based on laser direct infrared spectroscopy. Sci Total Environ [Internet]. 2024;906(23):167760. [CrossRef]
- Kim N, Lee JH, Lee I, Park JH, Jung GS, Lee MJ, et al. Investigation of potential toxic effects of nano- and microplastics on human endometrial stromal cells. Reprod Toxicol [Internet]. 2025;132(24):108848. [CrossRef]
- Qin X, Cao M, Peng T, Shan H, Lian W, Yu Y, et al. Features, Potential Invasion Pathways, and Reproductive Health Risks of Microplastics Detected in Human Uterus. Environ Sci Technol. 2024;58(24):10482–93.
- Gupta N, Yadav VK, Gacem A, Al-Dossari M, Yadav KK, Abd El-Gawaad NS, et al. Deleterious Effect of Air Pollution on Human Microbial Community and Bacterial Flora: A Short Review. Int J Environ Res Public Health. 2022;19(23):1–16.
- Jin Y, Wu S, Zeng Z, Fu Z. Effects of environmental pollutants on gut microbiota. Environ Pollut [Internet]. 2017;222:1–9. [CrossRef]
- Chuang KJ, Chan CC, Su TC, Lee C Te, Tang CS. The effect of urban air pollution on inflammation, oxidative stress, coagulation, and autonomic dysfunction in young adults. Am J Respir Crit Care Med. 2007;176(4):370–6.
- Zhang S, Mwiberi S, Pickford R, Breitner S, Huth C, Koenig W, et al. Longitudinal associations between ambient air pollution and insulin sensitivity: results from the KORA cohort study. Lancet Planet Heal [Internet]. 2021;5(1):e39–49. [CrossRef]
- Li J, Wu Q, Wu XK, Zhou ZM, Fu P, Chen XH, et al. Effect of exposure to second-hand smoke from husbands on biochemical hyperandrogenism, metabolic syndrome and conception rates in women with polycystic ovary syndrome undergoing ovulation induction. Hum Reprod. 2018;33(4):617–25.
- Mahalingaiah S, Missmer SE, Cheng JJ, Chavarro J, Laden F, Hart JE. Perimenarchal air pollution exposure and menstrual disorders. Hum Reprod. 2018;33(3):512–9.
- Liang Y, Lu Q, Chen M, Zhao X, Chu C, Zhang C, et al. Impact of endocrine disrupting chemicals ( EDCs ) on epigenetic regulation in the uterus : a narrative review. Reprod Biol Endocrinol. 2025;23(80):1–23.
- Tricotteaux-Zarqaoui S, Lahimer M, Abou Diwan M, Corona A, Candela P, Cabry R, et al. Endocrine disruptor chemicals exposure and female fertility declining: from pathophysiology to epigenetic risks. Front Public Heal. 2024;12(December):1–12.
- Parent, AS. Damdimopoulou, P. Johansson, HKL. Bouftas, N. Draskau, MK. Franssen, D. Fudvoye, J. van Duursen, M. Svingen T. Endocrine-disrupting chemicals and female reproductive health: a growing concern. Nat Rev Endocrinol [Internet]. 2025;22 May(Online ahead of print). [CrossRef]
- Paramasivam A, Murugan R, Jeraud M, Dakkumadugula A, Periyasamy R, Arjunan S. Additives in Processed Foods as a Potential Source of Endocrine-Disrupting Chemicals: A Review. J Xenobiotics. 2024;14(4):1697–710.
- Qi X, Yun C, Pang Y, Qiao J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes [Internet]. 2021;13(1):1–22. [CrossRef]
- Ashonibare VJ, Akorede BA, Ashonibare PJ, Akhigbe TM, Akhigbe RE. Gut microbiota-gonadal axis: the impact of gut microbiota on reproductive functions. Front Immunol. 2024;15(February):1–14.
- Moustakli E, Stavros S, Katopodis P, Potiris A, Drakakis P, Dafopoulos S, et al. Gut Microbiome Dysbiosis and Its Impact on Reproductive Health: Mechanisms and Clinical Applications. Metabolites. 2025;15(6):1–15.
- Kong FS, Huang P, Chen JH, Ma Y. The Novel Insight of Gut Microbiota from Mouse Model to Clinical Patients and the Role of NF-κB Pathway in Polycystic Ovary Syndrome. Reprod Sci [Internet]. 2024;31(11):3323–33. [CrossRef]
- Sheldon IM, Roberts MH. Toll-like receptor 4 mediates the response of epithelial and stromal cells to lipopolysaccharide in the endometrium. PLoS One. 2010;5(9):1–10.
- Krikun G, Trezza J, Shaw J, Rahman M, Guller S, Abrahams VM LC. LPS appears to activate human endometrial endothelial cells through TLR-4-dependent and TLR-4-independent mechanisms. Am J Reprod Immunol. 2012;68(3):233–7.
- Yu J, Berga SL, Zou W, Taylor RN. Interleukin-1β inhibits estrogen receptor-α, progesterone receptors A and B and biomarkers of human endometrial stromal cell differentiation: Implications for endometriosis. Mol Hum Reprod. 2019;25(10):625–37.
- Gholizadeh Shamasbi S, Dehgan P, Mohammad-Alizadeh Charandabi S, Aliasgarzadeh A, Mirghafourvand M. The effect of resistant dextrin as a prebiotic on metabolic parameters and androgen level in women with polycystic ovarian syndrome: a randomized, triple-blind, controlled, clinical trial. Eur J Nutr [Internet]. 2019;58(2):629–40. [CrossRef]
- Martinez Guevara D, Vidal Cañas S, Palacios I, Gómez A, Estrada M, Gallego J, et al. Effectiveness of Probiotics, Prebiotics, and Synbiotics in Managing Insulin Resistance and Hormonal Imbalance in Women with Polycystic Ovary Syndrome (PCOS): A Systematic Review of Randomized Clinical Trials. Nutr. 2024;16(22):1–28.
- Kaluanga Bwanga P, Tremblay-Lemoine PL, Timmermans M, Ravet S, Munaut C, Nisolle M, et al. The Endometrial Microbiota: Challenges and Prospects. Med. 2023;59(9):1–10.
- Balla B, Illés A, Tobiás B, Pikó H, Beke A, Sipos M, et al. The Role of the Vaginal and Endometrial Microbiomes in Infertility and Their Impact on Pregnancy Outcomes in Light of Recent Literature. Int J Mol Sci. 2024;25(23):1–23.
- Abdalla W, Nabeel W, Atiyeh I. The role of the microbiome in endometrial carcinoma: Pathogenesis, biomarkers, and therapeutic prospects. J Obs Gynaecol Res. 2025;51:e70070.
- Pelzer ES, Willner D, Buttini M, Huygens F. A role for the endometrial microbiome in dysfunctional menstrual bleeding. Antonie van Leeuwenhoek, Int J Gen Mol Microbiol [Internet]. 2018;111(6):933–43. [CrossRef]
- Stener-Victorin E, Padmanabhan V, Walters KA, Campbell RE, Benrick A, Giacobini P, et al. Animal Models to Understand the Etiology and Pathophysiology of Polycystic Ovary Syndrome. Endocr Rev. 2020;41(4):538–76.
- Hibaoui Y, Feki A. Organoid Models of Human Endometrial Development and Disease. Front Cell Dev Biol. 2020;8(February):1–6.
- Miyazaki K, Dyson MT, Coon V JS, Furukawa Y, Yilmaz BD, Maruyama T, et al. Generation of Progesterone-Responsive Endometrial Stromal Fibroblasts from Human Induced Pluripotent Stem Cells: Role of the WNT/CTNNB1 Pathway. Stem Cell Reports [Internet]. 2018;11(5):1136–55. [CrossRef]
- Gnecco JS, Brown A, Buttrey K, Ives C, Goods BA, Baugh L, et al. Organoid co-culture model of the human endometrium in a fully synthetic extracellular matrix enables the study of epithelial-stromal crosstalk. Med [Internet]. 2023;4(8):554-579.e9. [CrossRef]
- Filby CE, Wyatt KA, Mortlock S, Cousins FL, McKinnon B, Tyson KE, et al. Comparison of organoids from menstrual fluid and hormone-treated endometrium: Novel tools for gynecological research. J Pers Med. 2021;11(12):1314.
- Luyckx L, Wei M, Saarela U, Myllykangas M, Kinnunen J, Arffman R, et al. PCOS endometrium-derived epithelial organoids as a novel model to study endometrial dysfunction. Hum Reprod. 2025;40(8):1535–49.
- Piltonen TT, Chen J, Erikson DW, Spitzer TLB, Barragan F, Rabban JT, et al. Mesenchymal stem/progenitors and other endometrial cell types from women with polycystic ovary syndrome (PCOS) display inflammatory and oncogenic potential. J Clin Endocrinol Metab. 2013;98(9):3765–75.
- Shishehgar F, Ramezani Tehrani F, Mirmiran P, Hajian S, Baghestani AR, Moslehi N. Comparison of Dietary Intake between Polycystic Ovary Syndrome Women and Controls. Glob J Health Sci. 2016;8(9):302.
- Lim SS, Hutchison SK, Van Ryswyk E, Norman RJ, Teede HJ, Moran LJ. Lifestyle changes in women with polycystic ovary syndrome. Cochrane Database Syst Rev. 2019;2019(3).
- Benton, ML. The influence of evolutionary history on human health and disease. Nat Rev Genet [Internet]. 2021. [Google Scholar] [CrossRef]
- Gakidou E, Afshin A, Abajobir AA, Abate KH, Abbafati C, Abbas KM, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1345–422.
- Sabag A, Patten RK, Moreno-Asso A, Colombo GE, Dafauce Bouzo X, Moran LJ, et al. Exercise in the management of polycystic ovary syndrome: A position statement from Exercise and Sports Science Australia. J Sci Med Sport [Internet]. 2024;27(10):668–77. [CrossRef]
- Marsh KA, Steinbeck KS, Atkinson FS, Petocz P, Brand-miller JC. Effect of a low glycemic index compared with a conventional healthy diet on polycystic ovary syndrome. Am J Clin Nutr [Internet]. 2010;92(1):83–92. [CrossRef]
- Moran LJ, Ko H, Misso M, Marsh K, Noakes M, Talbot M, et al. Dietary Composition in the Treatment of Polycystic Ovary Syndrome: A Systematic Review to Inform Evidence-Based Guidelines. J Acad Nutr Diet. 2013;113(4):520–45.
- Medeiros FL, Fernandes AC, Padovan M, Kraemer MV, Bernardo GL, Uggioni PL, et al. The evolution of carbohydrate-restricted diets for type 2 diabetes mellitus: a scoping review. Acad Nutr Diet. 2025;2(2):1–13.
- Nicholas AP, Soto-Mota A, Lambert H, Collins AL. Restricting carbohydrates and calories in the treatment of type 2 diabetes: A systematic review of the effectiveness of “low-carbohydrate” interventions with differing energy levels. J Nutr Sci. 2021;10(e76):1–15.
- Feinman RD, Pogozelski WK, Astrup A, Bernstein RK, Fine EJ, Westman EC, et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition [Internet]. 2015;31(1):1–13. [CrossRef]
- Evert AB, Dennison M, Gardner CD, Timothy Garvey W, Karen Lau KH, MacLeod J, et al. Nutrition therapy for adults with diabetes or prediabetes: A consensus report. Diabetes Care. 2019;42(5):731–54.
- Joseph JJ, Deedwania P, Acharya T, Aguilar D, Bhatt DL, Chyun DA, et al. Comprehensive Management of Cardiovascular Risk Factors for Adults with Type 2 Diabetes: A Scientific Statement from the American Heart Association. Circulation. 2022;145(9):722–59.
- Stranks, Stephen N. Lawlor-Smith L. Managing Type 2 Diabetes with Therapeutic Carbohydrate Reduction [Internet]. 2023. p. 1–9. Available from: https://www.diabetessociety.com.au/wp-content/uploads/2023/11/Managing-Type-2-Diabetes-with-Therapeutic-Carbohydrate-reduction-TCR-November-2023_Final.
- Hite A, Cavan D, Cucuzella M, Cywes R, Ede G. Clinical Guidelines for Therapeutic Carbohydrate Restriction [Internet]. Society of Metabolic Health Practitioners. 2025. p. 1–23. Available from: https://thesmhp.
Figure 1.
Direct and indirect impact of metabolic and hormonal factors on the endometrium in reproductive age women with PCOS. Abbreviations: PCOS = polycystic ovary syndrome; IL= interleukin; TNF = tumour necrosis factor; PI3K = phosphoinositide 3-kinase; AKT = serene/threonine specific kinase; MAP = mitogen activated protein kinase; Wnt/ -catenin = wingless-related integration site; CYP19A1 = aromatase gene; HSD = hydroxysteroid dehydrogenase; COX = cyclooxygenase; PGE2 = prostaglandin E2; HOX = homeobox; WT = Wilms tumour; IRS = insulin receptor substrate; hsCRP = high sensitivity C-reactive protein.
Figure 1.
Direct and indirect impact of metabolic and hormonal factors on the endometrium in reproductive age women with PCOS. Abbreviations: PCOS = polycystic ovary syndrome; IL= interleukin; TNF = tumour necrosis factor; PI3K = phosphoinositide 3-kinase; AKT = serene/threonine specific kinase; MAP = mitogen activated protein kinase; Wnt/ -catenin = wingless-related integration site; CYP19A1 = aromatase gene; HSD = hydroxysteroid dehydrogenase; COX = cyclooxygenase; PGE2 = prostaglandin E2; HOX = homeobox; WT = Wilms tumour; IRS = insulin receptor substrate; hsCRP = high sensitivity C-reactive protein.
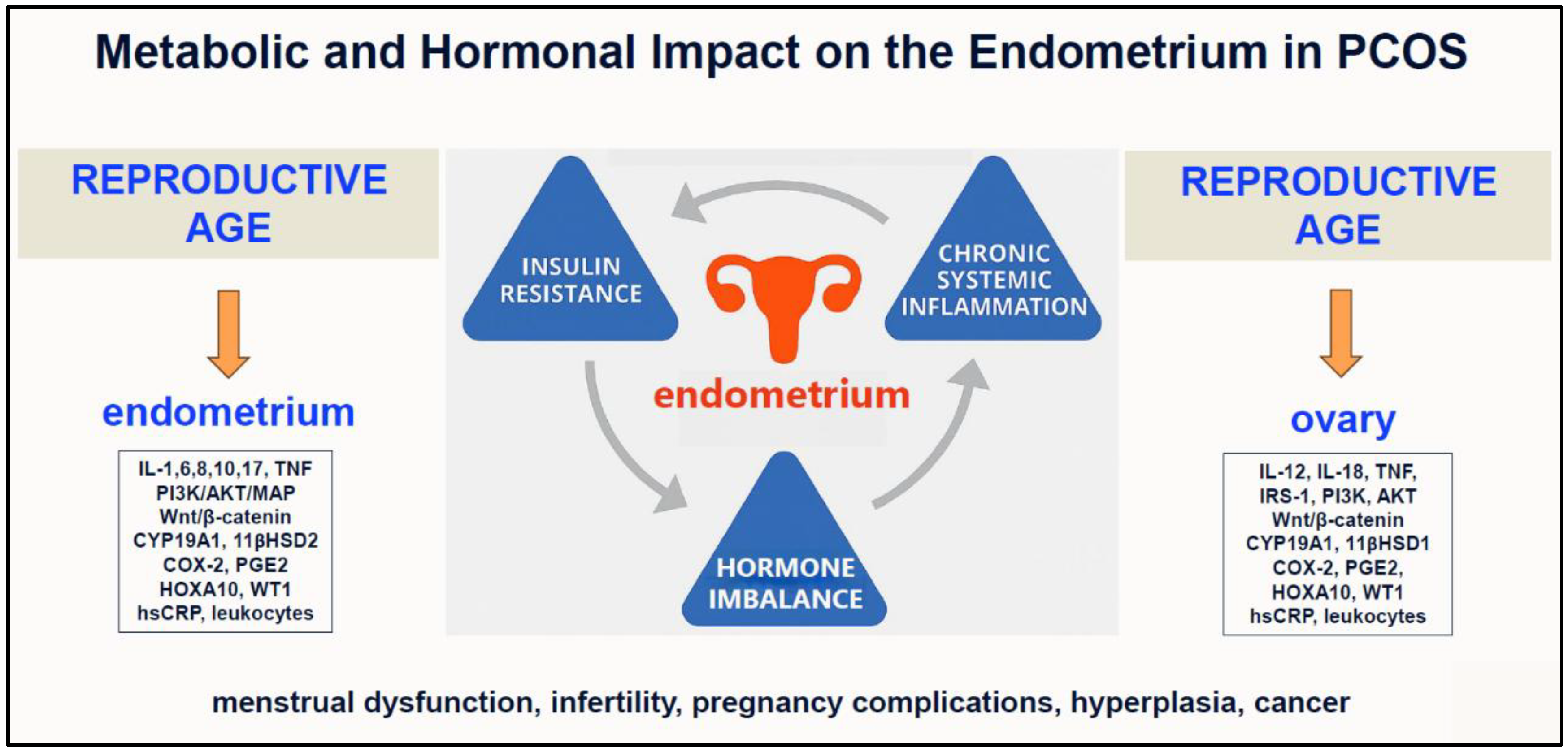
Figure 2.
Spheres of influence that determine menstrual function in polycystic ovary syndrome. Abbreviations: HPO = hypothalamic pituitary-ovarian; ANS = autonomic nervous system; EDC = endocrine disrupting chemicals; MAP = microparticulate air pollution.
Figure 2.
Spheres of influence that determine menstrual function in polycystic ovary syndrome. Abbreviations: HPO = hypothalamic pituitary-ovarian; ANS = autonomic nervous system; EDC = endocrine disrupting chemicals; MAP = microparticulate air pollution.
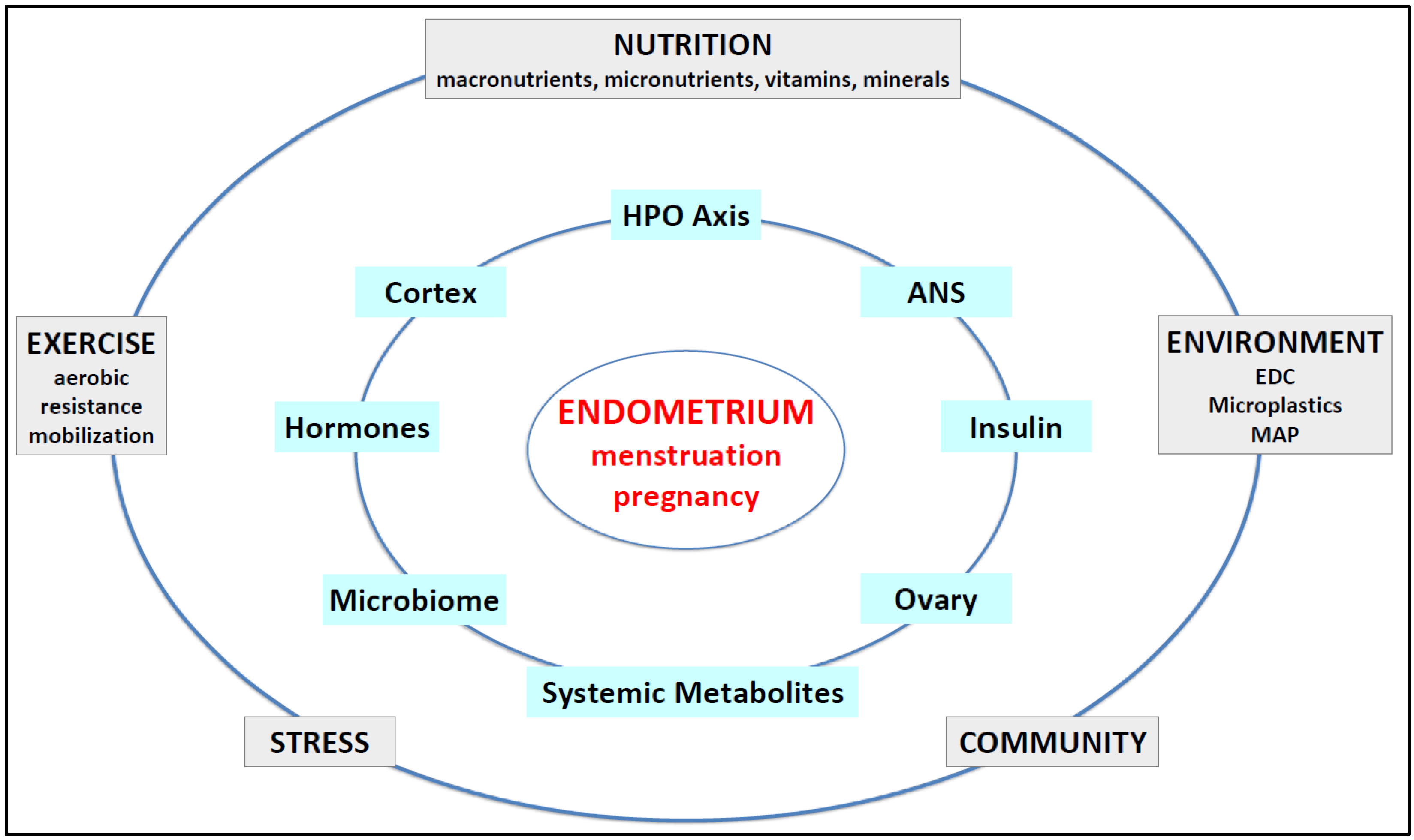
Figure 3.
Pathogenesis of polycystic ovary syndrome. Abbreviations: HPO = hypothalamic-pituitary-ovarian; ANS = autonomic nervous system. Adapted with permission from Ref. (9). 2023 Life.
Figure 3.
Pathogenesis of polycystic ovary syndrome. Abbreviations: HPO = hypothalamic-pituitary-ovarian; ANS = autonomic nervous system. Adapted with permission from Ref. (9). 2023 Life.
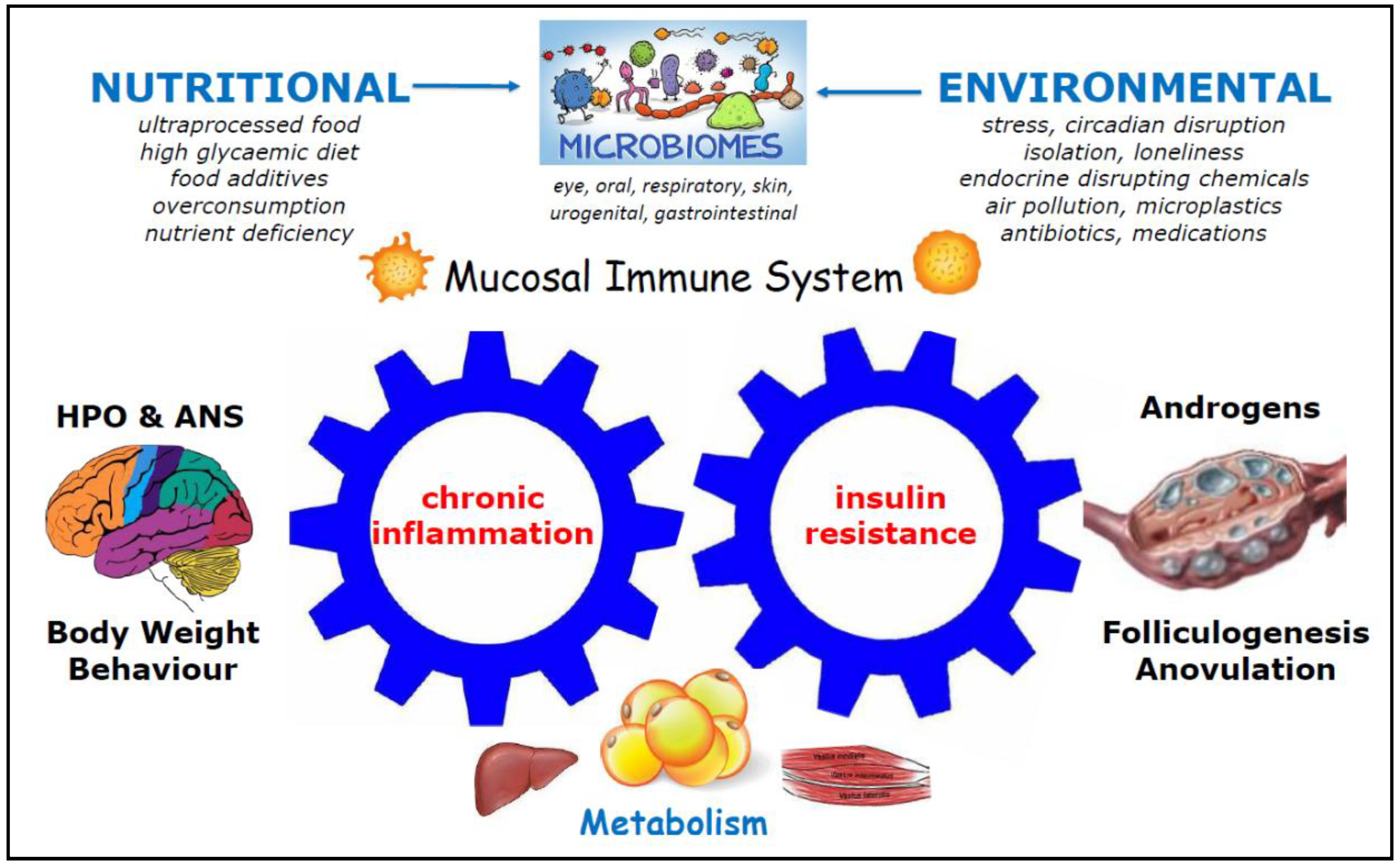

Table 1.
Clinical Relevance and Pathological Mechanisms of Endometrial Dysfunction in PCOS.
| Endometrial Problem | Pathological Findings and Mechanisms | References |
|---|---|---|
| Heavy menstrual bleeding |
Irregular breakdown of a thickened, hyperplastic endometrium due to unopposed E2 and/or P4 deficiency, vascular fragility, and low-grade inflammation. Alterations in inflammatory mediators, hemostasis, fibrinolysis, tissue and vascular remodeling. |
(122,123) |
| Polyps | Chronic unopposed E2 from anovulatory cycles together with insulin-mediated growth factors (VEGF, TGFβ-1)—and possibly HA—drive excess inflammation and focal endometrial proliferation, cystic glandular changes, stromal fibrosis, and increased vascularity. |
(124–127) |
| Implantation failure |
Defective decidualization, altered epithelium, inadequate spiral artery remodeling, immune cell imbalance, and defective extracellular matrix remodeling. Altered endometrial receptivity markers (reduced LIF, HOXA10, αvβ3 integrin and pinopode formation) driven by HA, IR, inflammation, and obesity. Lifestyle strategies (weight loss, diet, physical activity, circadian alignment) targeting obesity and IR play an important role. |
(19,128,129) |
|
Infertility |
Combined effects of chronic anovulation and impaired endometrial receptivity reduces conception rates. Key drivers include P4 resistance, low LIF/HOXA10/αvβ3-integrin, defective pinopodes, IR, inflammation, and HA. Upregulated genes involving decidualization (HAND2, MUC1, CSF2), angiogenesis (PDGFA), and inflammation (RELA, CXCL10). Altered epigenetic expression of miRNAs. Lifestyle factors, particularly elevated BMI, are significantly associated with infertility. |
(130,131) |
| Miscarriage | Impaired decidualization and trophoblast invasion resulting from P4 resistance, imbalanced cytokines, chronic inflammation, and metabolic dysfunction. Endometrial cells have heightened oxidative stress and dysregulated iron metabolism leading to increased ferroptosis. Depleted antioxidant defenses (impaired glutathione peroxidase 4) compromise endometrial cell viability and placental development. |
(132–134) |
| Pregnancy complications |
Elevated risk of GDM, PE, FGR, PTB, and stillbirth. Placental abnormalities include defective spiral artery remodeling, spiral artery thrombosis, atherosis of basal arterioles, and failure of deep placentation, with co-existing maternal endothelial dysfunction. Underlying maternal CSI, IR, and HA, alter placental physiology and development. Lifestyle factors modify risk of pregnancy complications. |
(13,31,135–138) |
| Hyperplasia | Prolonged estrogen exposure (unopposed by P4), obesity, decreased SHBG, dyslipidemia, elevated FAI, and IR promote abnormal growth of endometrial glands in relation to stroma +/- cytological atypia (EIN). Loss of PTEN expression, PI3K3CA mutations, MMR deficiencies. Modifiable risks include obesity, diet and exercise. |
(122,139,140) |
| Endometrial Cancer |
Progression from untreated atypical hyperplasia under chronic E2 stimulation, compounded by hyperinsulinemia, inflammation, and genetic mutations. Dysregulated signaling pathways (Notch, Wnt/β-catenin, PI3K/AKT/mTOR, MAPK, JAK/STAT, HER2). |
(122,141,142) |
Abbreviations: PCOS = polycystic ovary syndrome; E2 = 17β estradiol; P4 = progesterone; VEGF = vascular endothelial growth factor; TGFβ-1; HA = hyperandrogenism; LIF = leukemia inhibitory factor; HOXA10 = homeobox A10; IR = insulin resistance; mRNA = messenger ribose nucleic acid; BMI = body mass index; GDM = gestational diabetes mellitis; PE = pre-eclampsia; FGR = fetal growth restriction; PTB = preterm birth; FAI = free androgen index; CSI = chronic systemic inflammation; SHBG = sex hormone binding globulin; EIN = endometrial intraepithelial neoplasia; PTEN = phosphatase and tensin homologue; MMR = mismatch repair; PI3K = phosphoinositide 3-kinase; AKT = Serene/threonine Specific Kinase; mTOR = mammalian target of rapamycin; MAPK = mitogen-activated protein kinase; JAK = Janus kinase; STAT = signal transducers and activators of transcription; HER2 = human epidermal growth factor receptor 2.
Table 2.
Studies investigating the relationship between IR and menstrual disturbance in PCOS.
| Study (Year) |
Study Design Population (Country) |
Key Findings | Insulin Resistance Metrics |
Menstrual Cycle Length |
Citation |
|---|---|---|---|---|---|
| Robinson et al. (1993) |
Cross-sectional 72 PCOS 31 Controls (UK) |
↓Insulin sensitivity in PCOS with oligomenorrhea cw controls (p<0.01), but normal in PCOS with eumenorrhea |
IV Insulin Tolerance Test |
>35 days | (182) |
| Strowitzki et al. (2010) |
Cross-sectional 118 HA PCOS (Germany) |
↑HOMA-IR with amenorrhea (4.6) cw eumenorrhea (2.8) (p=0.019) |
HOMA-IR | >35 days | (149) |
| Brower et al. (2013) |
Cross-sectional 494 PCOS 138 Controls (USA) |
Higher mean HOMA-IR (2.2) in PCOS cw controls (1.41), after adjusting for age, BMI, and race |
HOMA-IR fasting insulin |
>35 days | (143) |
| Ezeh et al. (2021) |
Cross-sectional 57 HA PCOS 57 Controls (USA) |
↑Plasma glucose disappearance rate constant (kITT) in amenorrhea (1.98 +/- 0.28) cw eumenorrhea (3.33 +/- 0.51), after adjusting for age, BMI, and ethnicity |
Short Insulin Tolerance Test |
>35 days | (145) |
| Li et al. (2022) |
Cross-sectional 527 PCOS 565 Controls (China) |
↑HOMA-IR, ↑HOMA-β, and ↓QUICKI in women with cycles 45-90 days cw cycle >90 days and controls. No significant difference between cycles <45 and 45-90 days |
HOMA-IR HOMA-β QUICKI |
45-90 days | (144) |
| Niu et al. (2023) |
Retrospective 140 PCOS (China) |
Dose-response relationship between ↑HOMA-IR and cycle length: eumenorrhea (1.61: CI 1.3-1.85), oligomenorrhea (2.02: CI 1.61-2.445) amenorrhea (2.35: CI 1.96-2.75) |
HOMA-IR QUICKI ISI |
>35 days | (183) |
Abbreviations: PCOS = polycystic ovary syndrome; IV = intravenous; HA = hyperandrogenism; HOMA = homeostatic model of assessment; IR = insulin resistance; QUICKI = quantitative insulin sensitivity check index; ISI = insulin sensitivity index; BMI = body mass index; kITT = plasma glucose disappearance rate constant; cw – compared with; CI = confidence interval; UK = United Kingdom; USA = United States of America.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



