Submitted:
12 September 2025
Posted:
12 September 2025
You are already at the latest version
Abstract
Epithelial-mesenchymal transition (EMT) and inflammasome signaling are interconnected processes which underpin tumour progression, metastasis, and therapeutic resistance. Inflammasomes such as NLRP3 encourage pro-inflammatory states (IL-1β, IL-18, NF-κB) and the activation of signalling pathways like TGF-β that promote mesenchymal traits crucial for EMT. EMT transcriptional programs can then in turn modulate the inflammasome via NF-κB/TGF-β signaling, creating self-perpetuating mechanisms of cellular plasticity and dysregulated therapeutic response. We have reviewed the mechanistic evidence for EMT–inflammasome crosstalk in cancer and discussed the potential therapeutic implications. The function of the ENT-inflammasome axis is clearly context-dependent, with the cancer type, stage, and the complexity of the tumour microenvironment heavily contributing. The between EMT and the inflammasome is an overlooked mechanism of tumour evolution and targeting inflammasomes like NLRP3, or their downstream signalling pathways offer a promising therapeutic avenue, with the therapeutic objective of reducing metastasis and overcoming drug resistance.
Keywords:
inflammasome
; epithelial‐to‐mesenchymal transition
; metastasis
; fibrosis
; therapeutic targeting
1. Introduction: Two Molecular Processes Controlling Tumour Cell Plasticity
Cancer progression is dependent on more than simply the accumulation of genomic lesions. Indeed, it requires cellular plasticity and phenotypic changes that allows malignant cells to invade, evade, and adapt to conditions at their primary and distant sites. The epithelial–mesenchymal transition (EMT) and inflammasome signalling have been identified as two key components of tumour cell versatility, yet are often studied as separate pathological processes. EMT is a dynamic cellular programme by which epithelial cells lose their polarity and cell-cell adhesion, yielding a more mesenchymal profile that enable motility and tissue infiltration [1]. This phenotypic plasticity is dependent on a complex network of transcriptional regulators, epigenetic modifiers, and extracellular cues within the tumour microenvironment [2,3,4]. In physiological states, EMT is involved in processes such as embryogenic gastrulation, wound healing, and tissue remodelling [5]. Conversely, EMT is exploited by cancer cells during the metastatic cascade, as it confers invasive and migratory potential upon epithelial cells [6]. EMT is also implicated in the pathogenesis of various non-malignant diseases, such as chronic fibrotic disorders [7], inflammatory rheumatic disorders [8], age-related macular degeneration [9], and reproductive disorders such as adenomyosis and endometriosis [10,11]. Emerging evidence suggests that components of the innate immune system, including the inflammasome complex, play a significant regulatory role in EMT [12]. Inflammasomes are cytosolic multimeric complexes that respond to pathogenic or stress signals through the activation of caspases and the maturation of pro-inflammatory cytokines [13]. The inflammasome plays a crucial physiological role in innate immunity by activating inflammatory responses to pathogens [14], yet its dysregulation contributes to the development of various diseases such as cancer, gout, atherosclerosis, and neurodegeneration [15]. This review aims to critically examine the emerging interplay between inflammasome signalling and EMT in malignant and non-malignant diseases and consider its potential as a therapeutic target.
1.1. Epithelial-Mesenchymal Transition: A Complex Molecular and Transcriptional Programme
The changes to cellular identity seen in EMT ensue as a result of molecular changes and the activity of various transcriptional regulators. Molecular changes identified include the repression of epithelial markers such as E-cadherin, occludin, and specific cytokeratins, with concomitant upregulation of mesenchymal genes including N-cadherin, vimentin, fibronectin, and matrix metalloproteinases (MMPs) [16] [17]. These changes are facilitated by EMT transcription factors (EMT-TFs) such as SNAI1 (SNAIL), SNAI2 (SLUG) [4,18,19]. Notch signalling is also involved in EMT, mainly due to its role in upregulating transcriptional repressors like SNAIL and SLUG [20,21].
These transcriptional events are accompanied by histological changes. Cells undergoing EMT demonstrate a transition from a cobblestone-like epithelial morphology to an elongated, spindle-shaped mesenchymal phenotype with front-rear polarity, extensive actin cytoskeletal reorganisation, and loss of basement membrane association [16,22]. These morphological changes are due to hemidesmosome loss and switching from integrin β4 to integrins α5β1 and αvβ6 [23]. It is important to acknowledge that EMT is not a binary switch, but instead a spectrum with transitional states that yield hybrid epithelial/mesenchymal phenotypes [24]. EMT has also been linked to the acquisition of cancer stem cell (CSC)-like properties [25]. CSCs are a group of cells characterised by enhanced tumourigenicity, plasticity, and therapeutic resistance, and they are considered the apex of the cellular hierarchy of the tumour [26].
EMT is traditionally divided into three distinct subtypes which perform specific biological functions. Type 1 EMT creates cell movement that is not associated with fibrosis or an invasive phenotype, and has roles in embryo formation, implantation, and organogenesis [27,28]. Type 2 EMT is associated with repair events such as wound healing, tissue regeneration and organ fibrosis. This EMT subgroup is associated with inflammation and can lead to tissue fibrosis and organ destruction [27,28]. Type 3 EMT occurs in malignant cells that have undergone genetic and epigenetic changes to promote growth of tumours at their primary site [27,28] with further genetic changes and environmental factors promoting invasion and metastasis [28]. Figure 1 illustrates the dynamic and reversible process of epithelial–mesenchymal transition (EMT) and mesenchymal–epithelial transition (MET), highlighting the shift between epithelial, partial/hybrid, and mesenchymal cell states, characterised by distinct morphological features, marker expression, and regulatory pathways.
1.2. Inflammasomes in Cancer
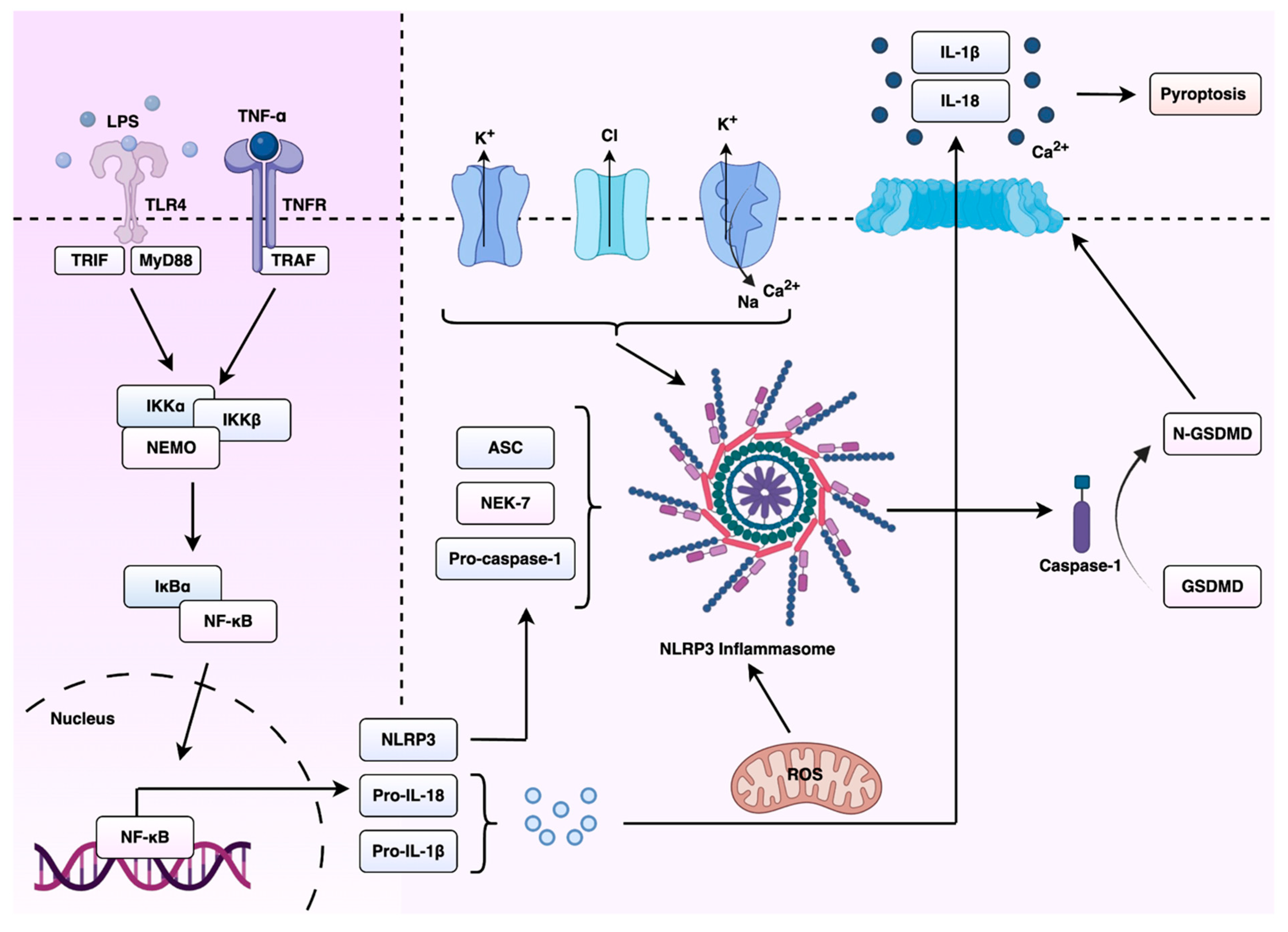
Inflammasomes are multimeric protein complexes that play a crucial role in innate immunity. These molecules assemble in the cytosol after sensing pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [29]. Three critical components make up canonical inflammasomes, an activated pattern recognition receptor (PRR), apoptosis-associated speck-like protein (ASC) and caspase-1 [15]. PRRs can be classified as cytosolic or transmembrane receptors, serving as sensor proteins [30]. Transmembrane PRRs can recognise PAMPs and DAMPs, triggering a signalling cascade which recruits cytosolic PRRs, although the cytosolic counterpart can also be activated by intracellular pathogens alone. Multiple PRRs can form inflammasome complexes, including NLR family pyrin domain-containing 1 (NLRP1), NLRP3, NLR family CARD domain-containing protein 4 (NLRC4) and absent in melanoma 2 (AIM2) [31]. Activation of the NLRP3 inflammasome begins with a priming step, where a cytokine receptor or (TLR) signalling induces the activation and nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), which in turn initiates the transcription and translation of NLRP3 and pro-IL1β. Other intracytosolic signals, including lysosomal or mitochondrial disruption, cause NLRP3 to undergo a conformational change via post-translational modifications which allows it to associate with the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) through pyrtin domain (PYD) or caspase recruitment domain (CARD) interactions [32,33]. ASC can then recruit caspase-1 through CARD-CARD interactions [34]. Active caspase-1 is a cystine-dependent protease which can cleave precursor cytokines pro-IL-1β and pro-IL-18 to their active states, generating biologically active cytokines [35]. Active caspase-1 can also contribute to an inflammatory form of cell death formally titled pyroptosis [36].
The NLRP3 inflammasome is activated in response to a wide range of stimuli, including nucleic acids, monosodium urate crystals, potassium and chloride flux, and B-glucans [37,38,39,40]. It represents the best studied inflammasome-nucleating sensor and is associated with a variety of inflammatory and autoimmune disorders, including gout, type 2 diabetes [41,42], Alzheimer’s disease [43] and atherosclerosis [44]. Chronic activation of the NLRP3 inflammasome can result in increased production of pro-inflammatory cytokines (IL-1 β and IL-18), causing tissue damage and exacerbation of disease symptoms [45]. The understanding of the molecular mechanisms that control NLRP3 activation has become a major research focus, including the therapeutic potential of this complex. A summary of the priming and activation mechanisms of the NLRP3 inflammasome, and its downstream effects on cytokine maturation and pyroptosis, are shown in Figure 2. The targeting of NLRP3 or its downstream signalling pathways offers promise for the development of novel anti-inflammatory drugs, mitigating the progression of inflammasome-associated diseases and improving patient outcomes.
2. Molecular Mechanisms Linking Inflammasome Signalling to Epithelial–Mesenchymal Transition in Cancer
Previous work has linked inflammasome activation and signalling to the process of EMT across several disease states. Early research demonstrated that Il-1β enhances TGF-β activity and in turn promotes EMT in human bronchial epithelial cells [46]. Further work illustrated that inflammasome-independent NLRP3 signalling can augment TGF-B signalling in kidney epithelium. This demonstrated that EMT can be triggered independently of inflammasome oligomerisation, with only the presence of the NLRP3 protein [47].IL-1β was shown to promote changes associated with EMT, such as E-cadherin downregulation and Snail expression in both gastric and oral cancer cells [48,49]. Previous research has also suggested that Angiotensin II drives hepatocyte EMT by inducing NOX4-dependent H₂O₂ production, resulting in activation of the NLRP3 inflammasome [50] and IL-1β release. IL-1β engages Smad signalling pathways, orchestrating the epithelial-to-mesenchymal shift. Critically, this work showed that NLRP3 and caspase-1 are required for angiotensin II-induced EMT. The peptide Ang-(1–7) was later discovered to act as a counterregulatory protein that attenuated this process through NLRP3 suppression. Several lines of evidence link inflammasome/EMT crosstalk to the pathogenesis of various malignancies and benign diseases, such as fibrotic disorders, diabetic nephropathy, and endometriosis. Thus, the mechanistic crosstalk between inflammasomes and EMT is best viewed as a dynamic feedback circuit: inflammasomes activate EMT through cytokines and signalling networks, while EMT factors remodel inflammasome activity to sustain inflammatory plasticity. This symbiosis produces self-reinforcing loops that underpin lethal properties of cancer including invasion, metastasis, and therapy resistance.
2.1. Context-Specific Mechanisms Across Tumour Types: Lung Cancer
Inflammasome pathways modulate EMT in different tissues, but the consequences are highly context-dependent. In lung cancer, chronic carcinogen exposure fosters a persistently inflamed tumour microenvironment which is partially driven by inflammasome action. Specifically, inflammasomes release IL-1β and IL-18, activating NF-κB and HIF-1a signalling in the tumour microenvironment. The literature recognises HIF-1a as a major regulator of transcription factors which are associated with a mesenchymal prototype, including TWIST, SLUG, SNAIL, ZEB1 and SIP1 [51]. There is also a known correlation between IL-1β and the induction of EMT in lung cancer [52]. Tumour-derived small extracellular vesicles (TEVs) from cells treated with IL-1β have been shown to activate a5β1 signalling, resulting in the upregulation of vimentin and SMAD3, cell migration and the EMT [53]. Additionally, exposure to IL-1β decreases PTEN expression, subsequently activating PI3K/AKT signalling and inducing EMT in lung cancer [54].
Wang et al. in 2016 [55] demonstrated that activation of the NLRP3 inflammasome in A549 lung adenocarcinoma cells, achieved via lipopolysaccharide (LPS) priming followed by adenosine triphosphate (ATP) stimulation, led to a significant upregulation of SNAIL and a downregulation of E-cadherin. This effect was dependent on NLRP3 expression, and indeed, neutralization of NLRP3 via shRNA reversed these EMT-associated transcriptional and molecular changes. Pharmacological inhibition of downstream inflammasome signalling through caspase-1 inhibition, IL-1Ra and 18BP produced similar cellular consequences [55]. Additionally, inhibition of the AIM2 inflammasome (a DNA-sensing inflammasome) via luteolin, a natural flavonoid, reduced mesenchymal markers like vimentin and MMP-9 while increasing E-cadherin. These findings indicate that both NLRP3 and AIM2 inflammasome signalling actively induce the EMT program in lung cancer [56].
2.2. Colorectal Cancer: Dualistic and Context-Dependent Roles
Recent investigations into the interplay between EMT and inflammasome signalling in colorectal cancer (CRC) have shown contrasting results, which highlight the nuanced role of the inflammasome in tumour progression. A study by Marandi et al. [57] examined tissue samples from 43 CRC patients and demonstrated that the activation of the NLRP3 inflammasome correlated with EMT induction and tumour grade. In addition, the expression levels of NLRP3, caspase-1, IL-1β, and NF-κB were markedly elevated in CRC, particularly in high-grade tumours. EMT-associated proteins such as N-cadherin, vimentin, and MMP-9 were upregulated, whereas E-cadherin was negatively correlated with NLRP3 protein levels [57]. In contrast, a mechanistic study by Qian et al. using murine in situ models and co-culture systems provided opposing findings. Here, the inhibition of NLRP3 inflammasome activity by transmembrane protein 176B (TMEM176B) facilitated the EMT while knockdown of TMEM176B in CT26 murine CRC cells increased the expression of NLRP3 inflammasome proteins and epithelial markers, while reducing the expression of mesenchymal markers [58]. The partial reversal of these effects was observed upon the concurrent knockdown of NLRP3 [58]. This work suggests that in certain contexts, inflammasome activation may instead exert an anti-metastatic effect by reinforcing epithelial integrity. Complementing these findings, Yang et al. [59] demonstrated that the inflammasome sensor AIM2 plays a tumour-suppressive role in human CRC cells by inhibiting EMT via both Akt-dependent and inflammasome-mediated mechanisms. Overexpression of AIM2 led to increased E-cadherin and reduced vimentin and SNAIL expression, correlating with decreased migratory and invasive capacity. Conversely, AIM2 silencing promoted mesenchymal marker expression and Akt phosphorylation. The EMT marker shifts were reversed by inhibition of Akt or caspase-1, highlighting a dual axis through which AIM2 inhibits EMT [59]. Overall, while clinical data associates inflammasome activation with EMT progression, experimental findings indicate that relieving inflammasome inhibition can suppress EMT. This dichotomy highlights the need for a more refined understanding of the cellular context and tissue-specific inflammatory dynamics driving EMT in cancer.
2.3. Breast Cancer: NLRP3 as a Promoter of EMT and Therapy Resistance
NLRP3 is aberrantly overexpressed in invasive breast ductal carcinoma, particularly in the claudin-low subtype of triple-negative breast cancer (TNBC) [60]. This subtype is known for mesenchymal traits, immune evasion, and poor clinical outcomes [61,62,63]. High NLRP3 expression correlated with worse patient survival, highlighting potential prognostic relevance in breast cancer [60]. In this context, NLRP3 was found to drive EMT and metastatic progression through autocrine IL-1β signalling via a caspase-1-dependent manner. Knockdown of NLRP3 in claudin-low MDA-MB-231 cells suppressed migration, invasion, and EMT. Conversely, overexpression of NLRP3 in MCF-7 cells induced EMT-like changes and invasive behaviour. Rescue experiments supported a causal relationship, as treatment with IL-1β restored EMT in NLRP3-silenced cells. Similarly, IL-1β antibody treatment reversed EMT in NLRP3-overexpressing cells [60]. In vivo, NLRP3 knockdown reduced metastasis in both zebrafish and nude mouse models, whereas its overexpression promoted the formation of metastatic foci in the lungs of mice [60].
Zheng et al. [64] demonstrated that NLRP3 also contributes to chemoresistance in TNBC by modulating epithelial plasticity and inflammatory signalling. Using gemcitabine-resistant TNBC cell lines, they found that NLRP3 expression was significantly elevated compared to parental lines. Agonism of NLRP3 further reduced sensitivity to gemcitabine. Conversely, inhibition of NLRP3 using the selective antagonist CY-09 restored drug sensitivity and reduced cell viability. NLRP3 upregulation was found to induce a mesenchymal phenotype and to enhance the secretion of IL-1β and activation of Wnt/β-catenin signalling [64]. This was supported by increased phosphorylation of GSK-3β at Ser9, indicative of decreased GSK-3β activity, as well as accumulation of β-catenin. Inhibition of Wnt/β-catenin signalling downregulated NLRP3 expression, suppressed IL-1β production, reversed EMT marker expression, and impaired cell survival. This indicates the presence of a regulatory pathway that enables drug resistance through NLRP3-IL-1β-Wnt/β-catenin signalling [64]. These findings reveal that inflammasome activity confers EMT properties and chemoresistance in TNBC, yet also highlight NLRP3 as a potential target to enhance therapeutic efficacy.
2.4. Pancreatic Cancer: Regulation of inflammasome–EMT Axis by lncRNA
Local inflammation seen in pancreatic cancer (PC) subtypes, such as pancreatic ductal adenocarcinoma (PDAC) is implicated in the process of metastasis [65,66]. Recent findings highlight long non-coding RNA (lncRNA) XLOC_000647 and its impact on inflammasome signalling and EMT in the pathogenesis of PC. XLOC_000647 was found to be significantly downregulated in PC tissues and cell lines. Its expression also inversely correlated with tumour stage, lymph node metastasis, and overall survival [67]. Overexpression of XLOC_000647 suppressed proliferation, invasion, and EMT-related changes in vitro, while attenuating tumour growth in vivo. Mechanistically, luciferase reporter assays in human embryonic kidney cells (293T) demonstrated that XLOC_000647 repressed NLRP3 by reducing promoter activity. Knockdown of NLRP3 inhibited proliferation, invasion, and mesenchymal marker expression such as vimentin, while restoring epithelial traits such as E-cadherin expression. Conversely, ectopic expression of NLRP3 reversed the inhibitory effects of XLOC_000647 on EMT and invasion. These findings affirm NLRP3’s functional role as a downstream effector and novel lncRNA-based epigenetic circuit modulating inflammasome activity in PC [67]. Moreover, lncRNA-inflammasome interactions were identified as a potential target for the modulation of PC dissemination.
2.5. The Tumour Microenvironment as a Driver of the Inflammasome/EMT Axis
The tumour microenvironment is deeply involved in the crosstalk between EMT and inflammasome signalling in lung cancer. For instance, tumour-secreted TGF-β shifts neutrophil differentiation from a tumour-suppressive N1 phenotype into a tumour-promoting N2 phenotype [68]. Wang et al. previously demonstrated that neutrophil extracellular traps (NETs) produced in the microenvironment promote NSCLC cell invasion and migration by inducing EMT. NETs are thought to induce the NLRP3 signalling pathway and the metastatic process by downregulating the expression of a specific tumour-suppressing long-coding RNA, MIR503HG [69]. Additional studies have supported these findings, as pre-treatment with NET inhibitors (anti-NE antibody and DNase) effectively reduces the migration and invasion of lung cancer cells [70].
3. Therapeutic Targeting of the Inflammasome/EMT Axis
Therapeutic targeting of the inflammasome–EMT axis in cancer reveals a convergent theme. Previous work has shown that the inhibition of NLRP3–IL-1β and downstream MAPK/NF-κB/TGF-β networks can attenuate EMT-driven invasion and metastasis. However, several factors in tumour biology limit the translational applicability of these approaches, namely: tumour type, stage, and a more complex cellular niche compared to that observed in non-malignant fibrotic disorders. Recent evidence has explored the potential of inhibiting inflammasome signalling to attenuate EMT-driven metastatic progression in non-small-cell lung cancer (NSCLC). Arjsri et al. [71] investigated the anti-inflammatory and anti-metastatic properties of morin, a polyphenolic flavonoid, in LPS and ATP-stimulated A549 and H1299 NSCLC cell lines. Morin significantly suppressed the expression and secretion of pro-inflammatory cytokines IL-1β, IL-18, and IL-6 in a dose-dependent manner. In addition, several key inflammasome components including NLRP3, ASC, and cleaved caspase-1 were downregulated in morin-treated conditions. These molecular changes were associated with a significant reduction in EMT, evidenced by reduced expression of mesenchymal markers and proteolytic proteins (MMP-2, MMP-9, u-PA, u-PAR, and MT1-MMP). Functional assays demonstrated that morin significantly inhibited cancer cell migration and invasion, likely mediated by the parallel shut-down of the MAPK pathway [71]. As such, NLRP3/MAPK axis is likely a critical player in inflammation-driven EMT in NSCLC. This also demonstrates that morin may be a promising therapeutic candidate capable of disrupting this axis to limit metastatic potential, although this needs to be validated using in-vivo studies.
3.1. Insights from Non-Malignant Conditions: Pulmonary Fibrosis
Recent studies have demonstrated the potential of targeting the inflammasome-EMT axis in various benign disorders, primarily those involving pathological visceral fibrosis. Pulmonary fibrosis (PF) is a progressive interstitial lung disease characterised by excessive extracellular matrix (ECM) deposition and subsequent architectural distortion, reduced lung compliance, and inefficient gas exchange [72]. PF encompasses a heterogeneous group of disorders, with idiopathic pulmonary fibrosis (IPF) being the most prevalent subtype [73]. Patients with IPF are believed to have an increased risk of lung cancer [74], and concomitant disease is associated with poorer clinical outcomes [74]. Aetiologically, PF can also occur secondary to known insults, including autoimmune diseases such as rheumatoid arthritis and systemic sclerosis, environmental and occupational exposures including silica and asbestos, and medications like amiodarone and bleomycin [75,76,77,78]. Key signalling pathways implicated in the pathogenesis of PF include TGF-β, Wnt/β-catenin, and the PI3K/Akt/mTOR axis. These networks collectively promote fibrogenesis through the modulation of cellular senescence, EMT, and apoptosis resistance [79,80,81].
Experimental and clinical evidence supports the role of EMT in PF. TGF-β1 treatment in vitro robustly induces EMT in alveolar epithelial cells [81]. Interestingly, Tanjore et al. revealed that approximately one-third of fibroblast-specific protein FSP1-positive fibroblasts in bleomycin-induced fibrotic lungs are of epithelial origin [82]. Inflammasome-associated cytokines have demonstrated their role in the pathogenesis of bleomycin-induced PF. For instance, the inhibition of IL-18 using an IL-18 binding protein prevented the induction of EMT, shown by a reduction in alpha-smooth muscle actin (α-SMA) and an increase in E-cadherin [83].
One study [84] on silica-induced EMT in human bronchial epithelial cells demonstrated that the inhibition of the NLRP3 inflammasome pathway through MCC950 (selective inhibitor), Z-YVAD-FMK (caspase-1 inhibitor), and shRNA knockdown could suppress EMT. This was mechanistically linked to blockade of the TAK1-MAPK-SNAIL and NF-κB signalling pathways in the case of MCC950. The use of pirfenidone, an antifibrotic agent approved for IPF, also mitigated silica-induced EMT by inhibiting NLRP3 activation [84].
Berberine is a bioactive alkaloid that has been shown to modulate various biological pathways including PI3K/AKT, mTOR, and Wnt/β-catenin [85,86,87,88]. Demethyleneberberine (DMB) is the primary metabolite of berberine and exhibits higher bioavailability and mitochondrial antioxidant properties [89,90]. In bleomycin-induced PF models, DMB suppressed the activation of the NLRP3 inflammasome, attenuated IL-1β and IL-18 secretion, and reversed mesenchymal marker expression. Importantly, overexpression of NLRP3 partially diminished the therapeutic effects of DMB, suggesting a causal link between inflammasome suppression and EMT inhibition. DMB also inhibited the expression of caspase-11 and gasdermin D, leading the authors to conclude that DMB acts on both the non-canonical and canonical pathways of NLRP3 inflammasome activation [91].
Another agent which has shown efficacy in bleomycin-induced PF is Iguratimod. Iguratimod is a disease modifying agent approved for the treatment of rheumatoid arthritis in China and Japan [92]. Its exact mechanism of action is not fully understood, but it has been shown to reduce the production or activity of various pro-inflammatory cytokines such as IL-1β and IL-6 [93]. In one study, Iguratimod treatment reduced hydroxyproline content and fibrosis scores in a bleomycin-induced PF mouse model [94]. Iguratimod was shown to inhibit NLRP3, reverse EMT, and suppress the production of ROS [94]. While this study did not specifically establish causation between NLRP3 inhibition and EMT reversal, this link seems highly plausible given the evidence provided by other studies. For instance, a similar effect is seen with scutellarin, a flavonoid derivative extracted from the Erigeron breviscapus plant. In both in vivo and in vitro models of bleomycin-induced PF, scutellarin treatment dose-dependently suppressed NF-κB activation and canonical NLRP3 inflammasome components, reducing downstream IL-1β and IL-18 production [95]. This was accompanied with EMT marker reversal and restoration of E-cadherin expression, alongside reduced collagen I and α-SMA expression and deposition [95].
From a cell therapy perspective, exosomes may provide a promising avenue in bleomycin-induced PF. Exosomes are nano-sized extracellular vesicles released by a variety of cells into the extracellular space, carrying diverse cargo consisting of proteins, lipids, and nucleic acids [96,97]. Thus, exosomes are central players in cell-cell communication and are capable of influencing various biological processes [98]. In a recent study [99], mesenchymal stem cell-derived exosomes (MSC-exos) have shown anti-fibrotic activity. In both in vivo and in vitro models, MSC-exos suppressed the activation of NOD1/NF-κB/NLRP3 signalling. In addition, they reduced expression of IL-1β and IL-18, and reversed the expression of mesenchymal markers. Overexpression of NLRP3 significantly attenuated the therapeutic effects of MSC-exos on EMT and ECM deposition [99]. These findings highlight the potential of MSC-exos as a cell-free therapeutic approach targeting NLRP3-driven inflammasome/EMT signalling in PF.
3.2. Peritoneal Fibrosis
Peritoneal fibrosis is a major complication and leading cause of discontinuation of peritoneal dialysis in end-stage renal disease [100,101]. The peritoneal mesothelium is a monolayer of specialised epithelial-like cells that line the peritoneal cavity [102]. In peritoneal dialysis, repeated exposure to bioincompatible solutions and inflammatory insults leads to the denudation of this monolayer [103]. EMT has been shown to play a role in this fibrotic condition [104,105]. A study by Ko et al. [106] provided evidence that EMT in peritoneal mesothelial cells is critically mediated by the NLRP3 inflammasome. Stimulation with TGF-β1 triggered EMT in peritoneal cells, as demonstrated by the loss of epithelial markers and acquisition of mesenchymal traits. TGF-β1 stimulation led to increased expression and activation of NLRP3, ASC, and cleaved caspase-1, resulting in elevated IL-1β and IL-18 secretion. Blocking NLRP3, ASC, or neutralising IL-1β/IL-18 significantly reduced EMT-related alterations, indicating that the activation of the inflammasome and its related cytokines drive this phenotypic transition. Mechanistically, TGF-β1 induced both membrane-bound NOX1 activation and mitochondrial NOX4 upregulation, promoting oxidative stress that was believed to act as a proximal signal for inflammasome activation. Crucially, treatment with paricalcitol, a vitamin D receptor agonist, suppressed NOX expression, NLRP3 expression, and EMT [106]. To our knowledge this work is the first to establish an inflammasome-EMT axis in peritoneal cells, and also suggest a therapeutic avenue for the targeting of this pathway.
3.3. Adenomyosis and Endometriosis
Endometriosis is a chronic inflammatory disorder characterised by the growth of endometrial-like tissue outside the uterine cavity [107]. Endometriosis may be associated with an increased risk of mainly ovarian cancer [108], but also various other malignancies such as endometrial and breast cancer [74]. Adenomyosis is a related condition defined by the presence of endometrial glands and stroma within the myometrium, resulting in uterine enlargement, heavy menstrual bleeding, and dysmenorrhea [109]. EMT has emerged as a key driver in the development and progression of adenomyosis. Mounting evidence indicates that innate inflammatory signalling, including the inflammasome complex, is upregulated in adenomyosis and may increase disease progression [110]. EMT is also known to be a key driver of adenomyosis in endometrial cells [111]. 17β-oestradiol, or E2 exposure has been demonstrated to cause endometrial glandular cells to adopt a fibroblast-like morphology, downregulate epithelial markers, upregulate mesenchymal markers and gain migratory and invasive capabilities [110,112]. Increased expression of oestrogen receptor beta (ER-β) has been demonstrated to activate the inflammasome, promoting cell invasion and proliferation [113].
Liu et al. previously demonstrated that blockage of the NLRP3 inflammasome by MCC950, a diary sulfonylurea-containing compound, attenuated the invasive capacity of endometrial cells. Additionally, in a murine model of adenomyosis, MCC950 reduced the severity of adenomyosis [114]. Other studies have confirmed these effects in similar models, demonstrating that the downregulation of GRIM19, a mitochondrial protein in macrophages, triggers NLRP3 inflammasome activation, which results in macrophage pyroptosis and elevated IL-1β secretion, promoting proliferation and migration of endometrial cells. Neutralising IL-1B reversed these effects, confirming the inflammasome’s role in adenomyosis and endometrial cell migration [115].
Additionally, the NLRP3 inflammasome has been implicated in the process of invasion and migration in endometriosis. In patients with endometriosis, peritoneal fluid washings demonstrate higher levels of caspase 1, IL-1β and gasdermin D (GSDMD) compared with healthy individuals. IL-1β concentrations are also associated with increased disease severity. The treatment of endometrial stromal cells with IL-1 also increased the migratory ability of ESCs, increasing concentrations of matrix metallopeptidase (MMP1 and 3)[116]. TRIM24 is a negative regulator of NLRP3/caspase-1/IL-1B mediated migration. In cells stimulated by the inhibition of TRIM24 with TRIM24-small-interfering RNA, the promoted pyroptosis and cell migration was reversed by CY-09, a specific inhibitor of NLRP3. This further implicates this inflammasome in the process of EMT in endometriosis [117].
3.4. Diabetic Nephropathy
Targeting the inflammasome/EMT interface has shown promise in the treatment of diabetic nephropathy (DN) in pre-clinical studies. Using a DN rat model induced by unilateral nephrectomy and intraperitoneal streptozotocin injection, Han et al demonstrated an upregulation of the NLRP3 inflammasome, its downstream effectors, and the expression of mesenchymal markers [118]. The study further revealed that treatment with Huangkui capsule, a traditional anti-inflammatory phytomedicine extracted from the Abelmoschus Manihot plant, significantly attenuated renal tubular EMT by suppressing NLRP3 inflammasome activation. This inhibition was likely mediated by the upstream blockade of the TLR4/NF-κB signalling pathway [118]. However, although the study suggests that TLR4/NF-κB signalling acts upstream of NLRP3, causal relationships were inferred rather than directly demonstrated. Therefore, we suggest that the use of pathway-specific inhibitors or gene knockdown models in the future would strengthen mechanistic conclusions in DN. In a separate study on DN rat models, Huangkui capsule was shown to activate the peroxisome proliferator-activated receptor (PPAR)-α/γ and attenuated endoplasmic reticulum stress in rats [119]. Further studies should aim to elucidate the pathways modulated by the Huangkui capsule in DN more clearly, particularly with regards to modulation of the EMT/inflammasome axis.
Another possibility for the targeting of EMT/inflammasome signalling in DN is through berberine. One study laid out in vivo and in vitro evidence that berberine ameliorates diabetic nephropathy by inhibiting renal tubular EMT through suppression of the NLRP3 inflammasome [120]. In a streptozotocin-induced diabetic rat model, berberine treatment significantly recued pathological findings on kidney samples as shown by decreased markers of interstitial fibrosis, and the reversal EMT-associated changes. In vitro experiments in high glucose-stimulated HK-2 cells confirmed that BBR downregulated both protein and mRNA levels of NLRP3, ASC, caspase-1, and IL-1β in a dose-dependent manner [120]. These findings link NLRP3 inflammasome activation and EMT progression in DN. They also support berberine’s therapeutic potential as an inhibitor of inflammasome-mediated EMT and fibrosis.
In summary, across cancer and fibrotic diseases, inflammasome inhibition can curb EMT by disrupting NLRP3-dependent IL1β signalling and its engagement of MAPK, NFκB, and TGFβ/Smad pathways in order to achieve particular treatment goals. In cancer, the therapeutic benefit is to halt EMT driven invasion, metastasis, and associated therapy resistance, with a focus on tumour type, microenvironmental modulators (e.g., NETs, TEVs), and sensor specificity (NLRP3 versus AIM2/AIM2axis) while also preserving normal host immune defences. In non-malignant diseases, the priority is to prevent or reverse EMT associated fibrosis and organ remodelling, often with broader safety margins and potential for combination with antifibrotic agents or cell-based therapies.
4. Beyond EMT: The Inflammasome as a Driver of Endothelial-Mesenchymal Transition
The endothelial-to-mesenchymal transition (EndoMT) has emerged as another NLRP3-driven source of mesenchymal cells in fibrosis. One study [121] investigated this phenomenon using a bleomycin-induced lung injury model in C57BL/6 mice and complementary in vitro experiments in human pulmonary microvascular endothelial cells (HPMECs). Bleomycin markedly upregulated NLRP3, cleaved caspase-1, and IL-1β. These changes were joined by loss of endothelial markers VE-cadherin and CD31 and the acquisition of mesenchymal properties. Moreover, pharmacological inhibition of caspase-1 with Ac-YVAD-cmk (YVAD) decreased NLRP3 and mesenchymal marker expression, restored the expression of endothelial markers, and improved pathological indices of lung injury and fibrosis on histological specimens. Mechanistically, the study identified focal adhesion kinase (FAK) phosphorylation as a key downstream effector in this process. Bleomycin increased the p-FAK/FAK ratio in vivo and in vitro, whereas YVAD treatment reversed these effects [121]. These findings suggest a pathogenic sequence in which NLRP3 inflammasome activation promotes caspase-1-dependent FAK activation, driving EndoMT and contributing to fibrotic remodelling. Similar endothelial involvement was observed in a study on mechanical stretch-induced fibrosis, where ventilator overdistension triggered NLRP3 activation, EndoMT marker shifts, and collagen accumulation. These effects were largely prevented in NLRP3-deficient mice or following NLRP3 knockdown in stretched endothelial cells [122]. These results suggest a NLRP3-EndoMT pathway in ventilator-associated fibrosis.
5. Inflammasome-EMT Signalling as a Danger-Plasticity Axis
It is widely acknowledged that chronic low-grade inflammation is a major enabling characteristic of cancer [123,124]. Tumour-promoting inflammation supports the malignant phenotype by enhancing genomic instability, cell proliferation, angiogenesis, invasion, and immune evasion through the sustained release of secreted factors and ROS [125,126]. We therefore propose that the discussed interplay between EMT and inflammasome activation can be conceptualised as a ‘danger-plasticity’ axis of evolutionarily conserved cellular programmes. That is, in cases of severe injury, cells respond by activating inflammasomes, which in turn initiate the pyroptosis pathway. This response can be protective in acute injury, and may function to clear damaged or potentially malignant cells. In contrast, under sublethal cellular stress and a survivable level of inflammation, activation of the EMT programme may influence cells towards a mesenchymal phenotype capable of tissue remodelling and repair. Within this adaptive shift, loss of E-cadherin occurs, which would typically push epithelial cells into a form of programmed cell death known as anoikis [127,128]. However, in EMT, loss of E-cadherin is accompanied by the upregulation of various anti-apoptotic mediators like BCL-xL and survivin [129,130]. These transcriptional programmes enhance the capacity of these mesenchymal-like cells to persist and function despite sustained inflammatory or metabolic stress. This evolutionary trade-off may serve an important physiological role in wound healing and tissue remodelling, but may similarly be advantageous to cancer cells, allowing them to acquire invasive and metastatic capabilities. Harold F. Dvorak famously likened tumours to ‘wounds that do not heal’ [131], a description that resonates strongly with the persistent, dysregulated interplay between inflammation and phenotypic adaptation seen in cancer. As such, the inflammasome could operate as a central decision-making node on this axis, by integrating the intensity and persistence of stress cues in order to direct cells towards either elimination or adaptive reprogramming.
6. Conclusions
The inflammasome-EMT axis is a critical regulator of cancer progression. NLRP3 activity is central to this relationship in multiple tumour types, due to its role in coupling inflammasome activation to EMT via IL-1β–dependent signalling that engages NF-κB, TGF-β/Smad, and Wnt/β-catenin pathways. The AIM2 inflammasome, on the other hand, can either promote or attenuate the EMT process depending on the tumour type. This phenomenon is well-characterized in colorectal cancer where inflammasome activity sometimes promotes the EMT, while on other occasions can restrict EMT. These tissue-specific changes are probably explained by differences in the tumour niche, as well as molecular regulators such as RNAs, epigenetic changes and post-transcriptional modifications which control inflammasome components. The tumour microenvironment also contributes to this bidirectional inflammasome-EMT relationship, with inflammatory mediators and neutrophil extracellular traps amplifying inflammasome signalling in tumour and stromal cells, and in turn, promoting dissemination. From a clinical point of view, the effects of EMT driven by inflammasome activity have been linked to therapeutic resistance. For instance, in triple negative breast cancer, NLRP3–IL1β–Wnt signalling promotes both EMT and chemoresistance. The translational of these findings into a safe and effective therapeutic depends on a tumour-specific approach which suppresses EMT, while upkeeping the role of inflammasome in host defense and tissue repair.
References
- Dongre:, A. : Weinberg, R. A., New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nature Reviews Molecular Cell Biology 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. , Epithelial-Mesenchymal Transition in tumor microenvironment. Cell & Bioscience 2011, 1, 29. [Google Scholar] [CrossRef]
- Sun, L.; Fang, J. , Epigenetic regulation of epithelial–mesenchymal transition. Cellular and Molecular Life Sciences 2016, 73, 4493–4515. [Google Scholar] [CrossRef] [PubMed]
- Debnath, P. ; Huirem, Rohit S.; Dutta, P.; Palchaudhuri, S., Epithelial–mesenchymal transition and its transcription factors. Bioscience Reports 2021, 42, BSR20211754. [Google Scholar] [CrossRef]
- Acloque, H.; Thiery, J. P.; Nieto, M. A. , The physiology and pathology of the EMT. EMBO reports 2008, 9, 322-326–326. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. , Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Translational Oncology 2020, 13, 100773. [Google Scholar] [CrossRef]
- Liu, L.; Sun, Q.; Davis, F.; Mao, J.; Zhao, H.; Ma, D. , Epithelial–mesenchymal transition in organ fibrosis development: current understanding and treatment strategies. Burns & Trauma 2022, 10, tkac011. [Google Scholar] [CrossRef]
- Sarrand, J.; Soyfoo, M. S. , Involvement of Epithelial-Mesenchymal Transition (EMT) in Autoimmune Diseases. International Journal of Molecular Sciences 2023, 24, 14481. [Google Scholar]
- Shu, D. Y.; Butcher, E.; Saint-Geniez, M. , EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. International Journal of Molecular Sciences 2020, 21, 4271. [Google Scholar]
- Hu, Y.; Yuan, M.; Cheng, L.; Wang, G. , Extracellular vesicles contribute to EMT in adenomyosis by inducing macrophage polarization†. Biology of Reproduction 2023, 108, 584–596. [Google Scholar] [CrossRef]
- Yang, Y.-M.; Yang, W.-X. , Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Alyaseer, A. A. A.; de Lima, M. H. S.; Braga, T. T. , The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process During the Fibrosis. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- de Zoete, M. R.; Palm, N. W.; Zhu, S.; Flavell, R. A. , Inflammasomes. Cold Spring Harb Perspect Biol 2014, 6, a016287. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. , Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discovery 2020, 6, 36. [Google Scholar] [CrossRef]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. , The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduction and Targeted Therapy 2024, 9, 10. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. , Molecular mechanisms of epithelial–mesenchymal transition. Nature Reviews Molecular Cell Biology 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, W.; Wei, X. , The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. Journal of Hematology & Oncology 2022, 15, 129. [Google Scholar] [CrossRef]
- Herranz, N.; Pasini, D.; Díaz, V. M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K. , et al., Polycomb Complex 2 Is Required for E-cadherin Repression by the Snail1 Transcription Factor. Molecular and Cellular Biology 2008, 28, 4772–4781. [Google Scholar] [CrossRef]
- Xue, W.; Yang, L.; Chen, C.; Ashrafizadeh, M.; Tian, Y.; Sun, R. , Wnt/β-catenin-driven EMT regulation in human cancers. Cellular and Molecular Life Sciences 2024, 81, 79. [Google Scholar] [CrossRef]
- Timmerman, L. A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J. M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J. C. , et al., Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004, 18, 99–115. [Google Scholar] [CrossRef]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P. A.; McFadden, D.; Karsan, A. , Slug is a direct Notch target required for initiation of cardiac cushion cellularization. Journal of Cell Biology 2008, 182, 315–325. [Google Scholar] [CrossRef]
- Sinha, A.; Mehta, P.; Fan, C.; Zhang, J.; Marvin, D. L.; van Dinther, M.; Ritsma, L.; Boukany, P. E.; Ten Dijke, P. , Visualizing Dynamic Changes During TGF-β-Induced Epithelial to Mesenchymal Transition. Methods Mol Biol 2022, 2488, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Banyard, J.; Bielenberg, D. R. , The role of EMT and MET in cancer dissemination. Connective Tissue Research 2015, 56, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, E. J.; Bronner, M. E. , A Spectrum of Cell States During the Epithelial-to-Mesenchymal Transition. Methods Mol Biol 2021, 2179, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Mani, S. A.; Guo, W.; Liao, M.-J.; Eaton, E. N.; Ayyanan, A.; Zhou, A. Y.; Brooks, M.; Reinhard, F.; Zhang, C. C.; Shipitsin, M. , et al., The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Doherty, M. R.; Smigiel, J. M.; Junk, D. J.; Jackson, M. W. , Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8, 8. [Google Scholar] [CrossRef]
- Marconi, G. D.; Fonticoli, L.; Rajan, T. S.; Pierdomenico, S. D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. , Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R. A. , The basics of epithelial-mesenchymal transition. J Clin Invest 2009, 119, 1420–8. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V. M. , Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–22. [Google Scholar] [CrossRef]
- Chen, R.; Zou, J.; Chen, J.; Zhong, X.; Kang, R.; Tang, D. , Pattern recognition receptors: function, regulation and therapeutic potential. Signal Transduct Target Ther 2025, 10, 216. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. , Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 2020, 6, 36. [Google Scholar] [CrossRef]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q. X.; Halfmann, R.; Chen, Z. J. , Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef]
- Akbal, A.; Dernst, A.; Lovotti, M.; Mangan, M. S. J.; McManus, R. M.; Latz, E. , How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell Mol Immunol 2022, 19, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Kersse, K.; Lamkanfi, M.; Bertrand, M. J. M.; Vanden Berghe, T.; Vandenabeele, P. , Interaction patches of procaspase-1 caspase recruitment domains (CARDs) are differently involved in procaspase-1 activation and receptor-interacting protein 2 (RIP2)-dependent nuclear factor κB signaling. J Biol Chem 2011, 286, 35874–35882. [Google Scholar] [CrossRef] [PubMed]
- Makoni, N. J.; Nichols, M. R. , The intricate biophysical puzzle of caspase-1 activation. Arch Biochem Biophys 2021, 699, 108753. [Google Scholar] [CrossRef] [PubMed]
- Miao, E. A.; Rajan, J. V.; Aderem, A. , Caspase-1-induced pyroptotic cell death. Immunol Rev 2011, 243, 206–14. [Google Scholar] [CrossRef]
- Swanson, K. V.; Deng, M.; Ting, J. P. , The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. , Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Kumar, H.; Kumagai, Y.; Tsuchida, T.; Koenig, P. A.; Satoh, T.; Guo, Z.; Jang, M. H.; Saitoh, T.; Akira, S.; Kawai, T. , Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J Immunol 2009, 183, 8061–7. [Google Scholar] [CrossRef]
- Pan, Y. G.; Huang, M. T.; Sekar, P.; Huang, D. Y.; Lin, W. W.; Hsieh, S. L. , Decoy Receptor 3 Inhibits Monosodium Urate-Induced NLRP3 Inflammasome Activation via Reduction of Reactive Oxygen Species Production and Lysosomal Rupture. Front Immunol 2021, 12, 638676. [Google Scholar] [CrossRef]
- Nițulescu, I. M.; Ciulei, G.; Cozma, A.; Procopciuc, L. M.; Orășan, O. H. , From Innate Immunity to Metabolic Disorder: A Review of the NLRP3 Inflammasome in Diabetes Mellitus. J Clin Med 2023, 12. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Mohammadi, M. T.; Rezaee, R.; Sahebkar, A. , Fenofibrate improves renal function by amelioration of NOX-4, IL-18, and p53 expression in an experimental model of diabetic nephropathy. J Cell Biochem 2018, 119, 7458–7469. [Google Scholar] [CrossRef]
- Heneka, M. T.; Kummer, M. P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T. C. , et al., NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–8. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Takahashi, M. , Role of NLRP3 Inflammasomes in Atherosclerosis. J Atheroscler Thromb 2017, 24, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T. S.; Stutz, A. , Activation and regulation of the inflammasomes. Nat Rev Immunol 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Doerner, A. M.; Zuraw, B. L. , TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res 2009, 10, 100. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N. A.; Trpkov, K.; Bonni, S.; Duff, H. J. , et al., Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol 2013, 190, 1239–49. [Google Scholar] [CrossRef]
- Lee, C.-H.; Chang, J. S.-M.; Syu, S.-H.; Wong, T.-S.; Chan, J. Y.-W.; Tang, Y.-C.; Yang, Z.-P.; Yang, W.-C.; Chen, C.-T.; Lu, S.-C. , et al., IL-1β Promotes Malignant Transformation and Tumor Aggressiveness in Oral Cancer. Journal of Cellular Physiology 2015, 230, 875–884. [Google Scholar] [CrossRef]
- Jee, Y. S.; Jang, T. J.; Jung, K. H. , Prostaglandin E(2) and interleukin-1β reduce E-cadherin expression by enhancing snail expression in gastric cancer cells. J Korean Med Sci 2012, 27, 987–92. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Huang, S.; Ma, X.-X.; Zhang, W.-Y.; Wang, D.; Jin, S.-Y.; Zhang, Y.-P.; Li, Y.; Li, X. , Angiotensin(1–7) attenuated Angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Radical Biology and Medicine 2016, 97, 531–543. [Google Scholar] [CrossRef]
- Tam, S. Y.; Wu, V. W. C.; Law, H. K. W. , Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front Oncol 2020, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ong, S. L.; Tran, L. M.; Jing, Z.; Liu, B.; Park, S. J.; Huang, Z. L.; Walser, T. C.; Heinrich, E. L.; Lee, G. , et al., Chronic IL-1β-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer. Scientific Reports 2020, 10, 377. [Google Scholar] [CrossRef]
- Heydari Sheikhhossein, H.; Amato, L.; De Rosa, V.; De Rosa, C.; Ariano, A.; Critelli, S.; Omodei, D.; Nele, V.; Tuccillo, C.; Franco, P. , et al., Effect of IL-1β on NSCLC-Derived Small Extracellular Vesicles as Actors in Mediating Cancer Progression and Evading Immune System. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lan, X.; Wang, T.; Lu, H.; Cao, M.; Yan, S.; Cui, Y.; Jia, D.; Cai, L.; Xing, Y. , Targeting the IL-1β/EHD1/TUBB3 axis overcomes resistance to EGFR-TKI in NSCLC. Oncogene 2020, 39, 1739–1755. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. , Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol Rep 2016, 35, 2053–64. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, M.; Ying, Q.; Xie, X.; Yue, S.; Tong, B.; Wei, Q.; Bai, Z.; Ma, L. , Decrease of AIM2 mediated by luteolin contributes to non-small cell lung cancer treatment. Cell Death & Disease 2019, 10, 218. [Google Scholar] [CrossRef]
- Marandi, Y.; Hashemzade, S.; Tayebinia, H.; Karimi, J.; Zamani, A.; Khodadadi, I. , NLRP3-inflammasome activation is associated with epithelial-mesenchymal transition and progression of colorectal cancer. Iran J Basic Med Sci 2021, 24, 483–492. [Google Scholar] [CrossRef]
- Qian, W.; Xu, C. Y.; Hong, W.; Li, Z. M.; Xu, D. G. , Transmembrane protein 176B promotes epithelial-mesenchymal transition in colorectal cancer through inflammasome inhibition. World J Gastrointest Oncol 2025, 17, 97673. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, M.; Jin, C.; Ding, Y.; Yang, M.; Wang, R.; Zhou, Y.; Zhou, Y.; Li, T.; Wang, K. , et al., Absent in melanoma 2 suppresses epithelial-mesenchymal transition via Akt and inflammasome pathways in human colorectal cancer cells. J Cell Biochem 2019, 120, 17744–17756. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Xu, Y.; Peng, T.; Meng, X.; Zou, F. , NLRP3 induces the autocrine secretion of IL-1β to promote epithelial–mesenchymal transition and metastasis in breast cancer. Biochemical and Biophysical Research Communications 2021, 560, 72–79. [Google Scholar] [CrossRef]
- Grasset, E. M.; Dunworth, M.; Sharma, G.; Loth, M.; Tandurella, J.; Cimino-Mathews, A.; Gentz, M.; Bracht, S.; Haynes, M.; Fertig, E. J. , et al., Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Science Translational Medicine 14, eabn7571. [CrossRef]
- Dias, K.; Dvorkin-Gheva, A.; Hallett, R. M.; Wu, Y.; Hassell, J.; Pond, G. R.; Levine, M.; Whelan, T.; Bane, A. L. , Claudin-Low Breast Cancer; Clinical & Pathological Characteristics. PLoS One 2017, 12, e0168669. [Google Scholar] [CrossRef]
- Dogra, A. K.; Prakash, A.; Gupta, S.; Gupta, M. , Prognostic Significance and Molecular Classification of Triple Negative Breast Cancer: A Systematic Review. Eur J Breast Health 2025, 21, 101–114. [Google Scholar] [CrossRef]
- Zheng, Q.; Yao, D.; Cai, Y.; Zhou, T. , NLRP3 augmented resistance to gemcitabine in triple-negative breast cancer cells via EMT/IL-1β/Wnt/β-catenin signaling pathway. Biosci Rep 2020, 40. [Google Scholar] [CrossRef]
- Shadhu, K.; Xi, C. , Inflammation and pancreatic cancer: An updated review. Saudi J Gastroenterol 2019, 25, 3–13. [Google Scholar] [CrossRef]
- Padoan, A.; Plebani, M.; Basso, D. , Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, Y.; Ding, X.; He, Y.; Lu, Z.; Wu, P.; Tian, L.; Yuan, H.; Liu, D.; Shi, G. , et al., Long non-coding RNA XLOC_000647 suppresses progression of pancreatic cancer and decreases epithelial-mesenchymal transition-induced cell invasion by down-regulating NLRP3. Molecular Cancer 2018, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z. G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G. S.; Albelda, S. M. , Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell 2009, 16, 183–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, F.; Chen, L.; Fang, C.; Li, S.; Yuan, S.; Qian, X.; Yin, Y.; Yu, B.; Fu, B. , et al., Neutrophil Extracellular Traps (NETs) Promote Non-Small Cell Lung Cancer Metastasis by Suppressing lncRNA MIR503HG to Activate the NF-κB/NLRP3 Inflammasome Pathway. Front Immunol 2022, 13, 867516. [Google Scholar] [CrossRef]
- Zhang, L.; Yi, H.; Chen, J.; Li, H.; Luo, Y.; Cheng, T.; Yang, H.; Jiang, Z.; Pan, C. , Neutrophil Extracellular Traps Facilitate A549 Cell Invasion and Migration in a Macrophage-Maintained Inflammatory Microenvironment. BioMed Research International 2022, 2022, 8316525. [Google Scholar] [CrossRef]
- Arjsri, P.; Srisawad, K.; Umsumarng, S.; Thippraphan, P.; Anuchapreeda, S.; Dejkriengkraikul, P. , Anti-Inflammatory and Anti-Migratory Effects of Morin on Non-Small-Cell Lung Cancer Metastasis via Inhibition of NLRP3/MAPK Signaling Pathway. Biomolecules 2025, 15, 103. [Google Scholar] [CrossRef]
- Zisman, D. A.; Keane, M. P.; Belperio, J. A.; Strieter, R. M.; Lynch, J. P. Pulmonary Fibrosis. In Fibrosis Research: Methods and Protocols; Varga, J., Brenner, D. A., Phan, S. H., Eds.; Humana Press: Totowa, NJ, 2005; pp. 3–44. [Google Scholar]
- Barratt, S. L.; Creamer, A.; Hayton, C.; Chaudhuri, N. , Idiopathic Pulmonary Fibrosis (IPF): An Overview. J Clin Med 2018, 7. [Google Scholar] [CrossRef]
- Ye, J.; Peng, H.; Huang, X.; Qi, X. , The association between endometriosis and risk of endometrial cancer and breast cancer: a meta-analysis. BMC Womens Health 2022, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Diesler, R.; Cottin, V. , Pulmonary fibrosis associated with rheumatoid arthritis: from pathophysiology to treatment strategies. Expert Review of Respiratory Medicine 2022, 16, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Budin, C. E.; Cocuz, I. G.; Sabău, A. H.; Niculescu, R.; Ianosi, I. R.; Ioan, V.; Cotoi, O. S. , Pulmonary Fibrosis Related to Amiodarone—Is It a Standard Pathophysiological Pattern? A Case-Based Literature Review. Diagnostics 2022, 12, 3217. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M. S.; Wynn, T. A. , Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunology 2009, 2, 103–121. [Google Scholar] [CrossRef]
- Herzog, E. L.; Mathur, A.; Tager, A. M.; Feghali-Bostwick, C.; Schneider, F.; Varga, J. , Review: Interstitial Lung Disease Associated With Systemic Sclerosis and Idiopathic Pulmonary Fibrosis: How Similar and Distinct? Arthritis & Rheumatology 2014, 66, 1967–1978. [Google Scholar] [CrossRef]
- Yue, X.; Shan, B.; Lasky, J. A. , TGF-β: Titan of Lung Fibrogenesis. Curr Enzym Inhib 2010, 6. [Google Scholar] [CrossRef]
- Wang, J.; Hu, K.; Cai, X.; Yang, B.; He, Q.; Wang, J.; Weng, Q. , Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharmaceutica Sinica B 2022, 12, 18–32. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J. T.; Mason, R. M.; Kamimura, T.; Zhang, Z. , TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respiratory Research 2005, 6, 56. [Google Scholar] [CrossRef]
- Tanjore, H.; Xu, X. C.; Polosukhin, V. V.; Degryse, A. L.; Li, B.; Han, W.; Sherrill, T. P.; Plieth, D.; Neilson, E. G.; Blackwell, T. S. , et al., Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 2009, 180, 657–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. M.; Zhang, Y.; Fei, C.; Zhang, J.; Wang, L.; Yi, Z. W.; Gao, G. , Neutralization of IL-18 by IL-18 binding protein ameliorates bleomycin-induced pulmonary fibrosis via inhibition of epithelial-mesenchymal transition. Biochem Biophys Res Commun 2019, 508, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, X.; Wang, Y.; Wang, J.; Zhou, F.; Wang, H.; Xie, W.; Kong, H. , NLRP3 inflammasome inhibition attenuates silica-induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells. Experimental Cell Research 2018, 362, 489–497. [Google Scholar] [CrossRef]
- Almatroodi, S. A.; Alsahli, M. A.; Rahmani, A. H. , Berberine: An Important Emphasis on Its Anticancer Effects through Modulation of Various Cell Signaling Pathways. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Wu, K.; Yang, Q.; Mu, Y.; Zhou, L.; Liu, Y.; Zhou, Q.; He, B. , Berberine inhibits the proliferation of colon cancer cells by inactivating Wnt/β-catenin signaling. Int J Oncol 2012, 41, 292–8. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Z.; Ni, J.; Ye, L.; Huang, X.; Jing, D.; Lu, Y.; Yue, L. , Berberine triggers apoptosis through the PI3K/Akt pathways and Nrf2 by inducing ROS in papillary thyroid cancer. Arch Biochem Biophys 2025, 771, 110481. [Google Scholar] [CrossRef]
- Park, G. S.; Park, B.; Lee, M. Y. , Berberine Induces Autophagic Cell Death by Inactivating the Akt/mTOR Signaling Pathway. Planta Med 2022, 88, 1116–1122. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q.; Chen, Y.; Lu, C.; Tong, Y. , Pharmacokinetics, Tissue Distribution and Excretion of Demethyleneberberine, a Metabolite of Berberine, in Rats and Mice. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Zhang, P.; Qiang, X.; Zhang, M.; Ma, D.; Zhao, Z.; Zhou, C.; Liu, X.; Li, R.; Chen, H.; Zhang, Y. , Demethyleneberberine, a natural mitochondria-targeted antioxidant, inhibits mitochondrial dysfunction, oxidative stress, and steatosis in alcoholic liver disease mouse model. J Pharmacol Exp Ther 2015, 352, 139–47. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Zhou, L.; Lin, Y.; Tang, Z.; Chen, C.; Wang, B.; Lin, Y. , Demethyleneberberine ameliorates pulmonary fibrosis by inhibiting the NLRP3 inflammasome and the epithelial-mesenchymal transition. Int Immunopharmacol 2025, 161, 115003. [Google Scholar] [CrossRef]
- Xie, S.; Li, S.; Tian, J.; Li, F. , Iguratimod as a New Drug for Rheumatoid Arthritis: Current Landscape. Front Pharmacol 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Aikawa, Y.; Kawasaki, H.; Asaoka, K.; Inaba, T.; Yoshida, C. , Pharmacological studies on 3-formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-one (T-614), a novel antiinflammatory agent. 4th communication: inhibitory effect on the production of interleukin-1 and interleukin-6. J Pharmacobiodyn, 1992; 15, 649-55. [Google Scholar] [CrossRef]
- Liu, W.; Han, X.; Li, Q.; Sun, L.; Wang, J. , Iguratimod ameliorates bleomycin-induced pulmonary fibrosis by inhibiting the EMT process and NLRP3 inflammasome activation. Biomedicine & Pharmacotherapy 2022, 153, 113460. [Google Scholar] [CrossRef]
- Peng, L.; Wen, L.; Shi, Q. F.; Gao, F.; Huang, B.; Meng, J.; Hu, C. P.; Wang, C. M. , Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis 2020, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Xavier, J.; Kumar, N.; Ahmad, M. Z.; Ranjan, O. P. , Exosomes as natural nanocarrier-based drug delivery system: recent insights and future perspectives. 3 Biotech 2023, 13, 101. [Google Scholar] [CrossRef]
- Di Bella, M. A. , Overview and Update on Extracellular Vesicles: Considerations on Exosomes and Their Application in Modern Medicine. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Lee, Y. J.; Shin, K. J.; Chae, Y. C. , Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp Mol Med 2024, 56, 877–889. [Google Scholar] [CrossRef]
- Chen, W.; Peng, J.; Tang, X.; Ouyang, S. , MSC-derived exosome ameliorates pulmonary fibrosis by modulating NOD 1/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Heliyon 2025, 11, e41436. [Google Scholar] [CrossRef]
- Taheri, S.; Thiagaraj, S. S.; Shukla, T. S.; Gutlapalli, S. D.; Farhat, H.; Muthiah, K.; Pallipamu, N.; Hamid, P. , A Review on Major Pathways Leading to Peritoneal Fibrosis in Patients Receiving Continuous Peritoneal Dialysis. Cureus 2022, 14, e31799. [Google Scholar] [CrossRef]
- Suryantoro, S. D.; Thaha, M.; Sutanto, H.; Firdausa, S. , Current Insights into Cellular Determinants of Peritoneal Fibrosis in Peritoneal Dialysis: A Narrative Review. J Clin Med 2023, 12. [Google Scholar] [CrossRef]
- Yung, S.; Li, F. K.; Chan, T. M. , Peritoneal mesothelial cell culture and biology. Perit Dial Int 2006, 26, 162–73. [Google Scholar] [CrossRef]
- Yung, S.; Chan, T. M. , Pathophysiology of the peritoneal membrane during peritoneal dialysis: the role of hyaluronan. J Biomed Biotechnol 2011, 2011, 180594. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, R.; Moreno-Vicente, R.; Battistelli, C.; Cicchini, C.; Noce, V.; Amicone, L.; Marchetti, A.; Del Pozo, M. A.; Tripodi, M. , Molecular Mechanisms Underlying Peritoneal EMT and Fibrosis. Stem Cells Int 2016, 2016, 3543678. [Google Scholar] [CrossRef] [PubMed]
- del Peso, G.; Jiménez-Heffernan, J. A.; Bajo, M. A.; Aroeira, L. S.; Aguilera, A.; Fernández-Perpén, A.; Cirugeda, A.; Castro, M. J.; de Gracia, R.; Sánchez-Villanueva, R. , et al., Epithelial-to-mesenchymal transition of mesothelial cells is an early event during peritoneal dialysis and is associated with high peritoneal transport. Kidney International 2008, 73, S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Kang, H. J.; Kim, D. A.; Ryu, E. S.; Yu, M.; Lee, H.; Lee, H. K.; Ryu, H. M.; Park, S. H.; Kim, Y. L. , et al., Paricalcitol attenuates TGF-β1-induced phenotype transition of human peritoneal mesothelial cells (HPMCs) via modulation of oxidative stress and NLRP3 inflammasome. Faseb j 2019, 33, 3035–3050. [Google Scholar] [CrossRef]
- Bulun, S. E.; Yilmaz, B. D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. , Endometriosis. Endocr Rev 2019, 40, 1048–1079. [Google Scholar] [CrossRef]
- Barnard, M. E.; Farland, L. V.; Yan, B.; Wang, J.; Trabert, B.; Doherty, J. A.; Meeks, H. D.; Madsen, M.; Guinto, E.; Collin, L. J. , et al., Endometriosis Typology and Ovarian Cancer Risk. Jama 2024, 332, 482–489. [Google Scholar] [CrossRef]
- Schrager, S.; Yogendran, L.; Marquez, C. M.; Sadowski, E. A. , Adenomyosis: Diagnosis and Management. Am Fam Physician 2022, 105, 33–38. [Google Scholar]
- Chen, Y. J.; Li, H. Y.; Huang, C. H.; Twu, N. F.; Yen, M. S.; Wang, P. H.; Chou, T. Y.; Liu, Y. N.; Chao, K. C.; Yang, M. H. , Oestrogen-induced epithelial-mesenchymal transition of endometrial epithelial cells contributes to the development of adenomyosis. J Pathol 2010, 222, 261–70. [Google Scholar] [CrossRef]
- Liu, X.; Shen, M.; Qi, Q.; Zhang, H.; Guo, S. W. , Corroborating evidence for platelet-induced epithelial-mesenchymal transition and fibroblast-to-myofibroblast transdifferentiation in the development of adenomyosis. Hum Reprod 2016, 31, 734–49. [Google Scholar] [CrossRef]
- Khan, K. N.; Kitajima, M.; Hiraki, K.; Fujishita, A.; Nakashima, M.; Masuzaki, H. , Involvement of hepatocyte growth factor-induced epithelial-mesenchymal transition in human adenomyosis. Biol Reprod 2015, 92, 35. [Google Scholar] [CrossRef]
- Dong, W.; Peng, Q.; Liu, Z.; Xie, Z.; Guo, X.; Li, Y.; Chen, C. , Estrogen plays an important role by influencing the NLRP3 inflammasome. Biomed Pharmacother 2023, 167, 115554. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, Z.; Zhang, L.; Tian, W.; Lin, A.; Li, M. , Blockage of the NLRP3 inflammasome by MCC950 inhibits migration and invasion in adenomyosis. Reprod Biomed Online 2024, 49, 104319. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Y.; Yang, Y.; Huang, W.; Chao, L. , GRIM19 downregulation-induced pyroptosis of macrophages through NLRP3 pathway in adenomyosis. Reprod Biomed Online 2022, 44, 211–219. [Google Scholar] [CrossRef]
- Zhou, F.; Zhao, F.; Huang, Q.; Lin, X.; Zhang, S.; Dai, Y. , NLRP3 activated macrophages promote endometrial stromal cells migration in endometriosis. J Reprod Immunol 2022, 152, 103649. [Google Scholar] [CrossRef] [PubMed]
- Hang, Y.; Tan, L.; Chen, Q.; Liu, Q.; Jin, Y. , E3 ubiquitin ligase TRIM24 deficiency promotes NLRP3/caspase-1/IL-1β-mediated pyroptosis in endometriosis. Cell Biology International 2021, 45, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Ma, Q.; Liu, Y.; Wu, W.; Tu, Y.; Huang, L.; Long, Y.; Wang, W.; Yee, H.; Wan, Z. , et al., Huangkui capsule alleviates renal tubular epithelial-mesenchymal transition in diabetic nephropathy via inhibiting NLRP3 inflammasome activation and TLR4/NF-κB signaling. Phytomedicine 2019, 57, 203–214. [Google Scholar] [CrossRef]
- Ge, J.; Miao, J. J.; Sun, X. Y.; Yu, J. Y. , Huangkui capsule, an extract from Abelmoschus manihot (L.) medic, improves diabetic nephropathy via activating peroxisome proliferator-activated receptor (PPAR)-α/γ and attenuating endoplasmic reticulum stress in rats. J Ethnopharmacol 2016, 189, 238–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhu, L.; Wang, S.; Guo, X.; Sun, B.; Wang, Q.; Chen, L. , Berberine protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition involving the inactivation of the NLRP3 inflammasome. Ren Fail 2022, 44, 923–932. [Google Scholar] [CrossRef]
- Chen, W. C.; Yu, W. K.; Su, V. Y.; Hsu, H. S.; Yang, K. Y. , NLRP3 Inflammasome Activates Endothelial-to-Mesenchymal Transition via Focal Adhesion Kinase Pathway in Bleomycin-Induced Pulmonary Fibrosis. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, Y.; Liu, Y. J.; Mao, Y. F.; Dong, W. W.; Ding, Z. N.; Meng, G. X.; Jiang, L.; Zhu, X. Y. , NLRP3 Inflammasome Activation Contributes to Mechanical Stretch-Induced Endothelial-Mesenchymal Transition and Pulmonary Fibrosis. Crit Care Med 2018, 46, e49–e58. [Google Scholar] [CrossRef]
- Hanahan, D. ; Weinberg, Robert A., Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Greten, F. R.; Grivennikov, S. I. , Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Andoh, A. , The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells 2025, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. , Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduction and Targeted Therapy 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, S.; Lugo-Martínez, V. H.; Faussat, A. M.; Renaud, F.; Cardot, P.; Chambaz, J.; Pinçon-Raymond, M.; Thenet, S. , Early loss of E-cadherin from cell-cell contacts is involved in the onset of Anoikis in enterocytes. J Biol Chem 2004, 279, 43061–9. [Google Scholar] [CrossRef]
- Kim, Y. N.; Koo, K. H.; Sung, J. Y.; Yun, U. J.; Kim, H. , Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol 2012, 2012, 306879. [Google Scholar] [CrossRef]
- Lee, J.; Choi, J. H.; Joo, C. K. , TGF-β1 regulates cell fate during epithelial–mesenchymal transition by upregulating survivin. Cell Death & Disease, 2013; 4, e714–e714. [Google Scholar] [CrossRef]
- Keitel, U.; Scheel, A.; Thomale, J.; Halpape, R.; Kaulfuß, S.; Scheel, C.; Dobbelstein, M. , Bcl-xL mediates therapeutic resistance of a mesenchymal breast cancer cell subpopulation. Oncotarget 2014, 5, 11778–91. [Google Scholar] [CrossRef]
- Dvorak, H. F. , Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986, 315, 1650–9. [Google Scholar] [CrossRef]
Figure 1.
Overview of the phenotypic, molecular, and transcriptional changes in epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET). This diagram depicts the bidirectional and dynamic nature of EMT, in which epithelial cells with apio-basolateral polarity and strong intercellular junctions transition through partial/hybrid phenotypes to acquire mesenchymal traits such as front-rear polarity, basement membrane detachment, and increased migratory capacity. Conversely, mesenchymal-epithelial transition (MET) allows mesenchymal cells to regain epithelial morphology and polarity. Abbreviations: CDH1 (E-cadherin); EMT, epithelial-to-mesenchymal transition; FSP-1, fibroblast-specific protein 1; MET, mesenchymal-to-epithelial transition; MMPs, matrix metalloproteinases; TGF-β, transforming growth factor beta; ZO-1, zonula occludens-1. Figure created using BioRender.com and Microsoft PowerPoint.
Figure 1.
Overview of the phenotypic, molecular, and transcriptional changes in epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET). This diagram depicts the bidirectional and dynamic nature of EMT, in which epithelial cells with apio-basolateral polarity and strong intercellular junctions transition through partial/hybrid phenotypes to acquire mesenchymal traits such as front-rear polarity, basement membrane detachment, and increased migratory capacity. Conversely, mesenchymal-epithelial transition (MET) allows mesenchymal cells to regain epithelial morphology and polarity. Abbreviations: CDH1 (E-cadherin); EMT, epithelial-to-mesenchymal transition; FSP-1, fibroblast-specific protein 1; MET, mesenchymal-to-epithelial transition; MMPs, matrix metalloproteinases; TGF-β, transforming growth factor beta; ZO-1, zonula occludens-1. Figure created using BioRender.com and Microsoft PowerPoint.
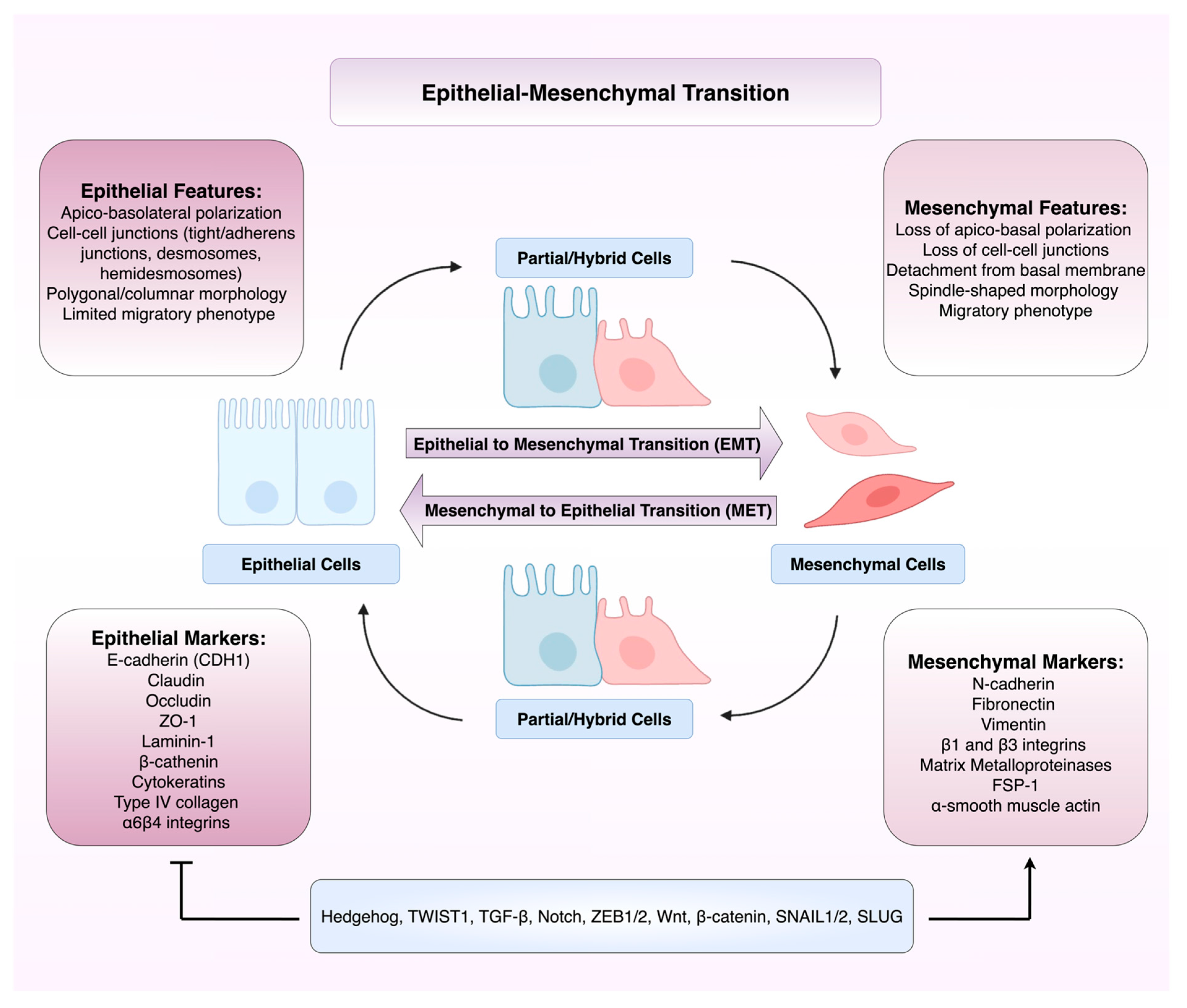
Figure 2.
Overview of NLRP3 inflammasome activation and downstream signalling in cancer. This schematic illustrates the canonical activation pathway of the NLRP3 inflammasome. Upon sensing cellular stressors such as ROS, DNA damage, oncogenic mutations, the NLRP3 inflammasome assembles and activates caspase-1. This leads to the cleavage and maturation of pro-inflammatory cytokines IL-1β and IL-18, which shape the inflammatory milieu of carcinomas. Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; GSDMD, gasdermin D; IKKα, inhibitor of nuclear factor kappa-B kinase subunit alpha; IKKβ, inhibitor of nuclear factor kappa-B kinase subunit beta; IκBα, inhibitor of kappa B alpha; IL-1β, interleukin-1 beta; IL-18, interleukin-18; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response 88; NEMO, NF-κB essential modulator; NEK7, never in mitosis gene A-related kinase 7; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; N-GSDMD, N-terminal fragment of gasdermin D; NLRP3, NLR family pyrin domain containing 3; pro-IL-1β, pro-interleukin-1 beta; pro-IL-18, pro-interleukin-18; pro-caspase-1, procaspase-1; ROS, reactive oxygen species; TLR4, Toll-like receptor 4; TNF-α, tumour necrosis factor alpha; TNFR, tumour necrosis factor receptor; TRAF, TNF receptor-associated factor; TRIF, Toll/interleukin-1 receptor domain-containing adaptor-inducing interferon-β. Figure created using BioRender.com and Microsoft PowerPoint.
Figure 2.
Overview of NLRP3 inflammasome activation and downstream signalling in cancer. This schematic illustrates the canonical activation pathway of the NLRP3 inflammasome. Upon sensing cellular stressors such as ROS, DNA damage, oncogenic mutations, the NLRP3 inflammasome assembles and activates caspase-1. This leads to the cleavage and maturation of pro-inflammatory cytokines IL-1β and IL-18, which shape the inflammatory milieu of carcinomas. Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; GSDMD, gasdermin D; IKKα, inhibitor of nuclear factor kappa-B kinase subunit alpha; IKKβ, inhibitor of nuclear factor kappa-B kinase subunit beta; IκBα, inhibitor of kappa B alpha; IL-1β, interleukin-1 beta; IL-18, interleukin-18; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response 88; NEMO, NF-κB essential modulator; NEK7, never in mitosis gene A-related kinase 7; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; N-GSDMD, N-terminal fragment of gasdermin D; NLRP3, NLR family pyrin domain containing 3; pro-IL-1β, pro-interleukin-1 beta; pro-IL-18, pro-interleukin-18; pro-caspase-1, procaspase-1; ROS, reactive oxygen species; TLR4, Toll-like receptor 4; TNF-α, tumour necrosis factor alpha; TNFR, tumour necrosis factor receptor; TRAF, TNF receptor-associated factor; TRIF, Toll/interleukin-1 receptor domain-containing adaptor-inducing interferon-β. Figure created using BioRender.com and Microsoft PowerPoint.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



