Submitted:
02 September 2025
Posted:
02 September 2025
You are already at the latest version
Abstract
We present the first preparation of novel 6-O-(fluoroalkyl)-6-O-desmethyl-diprenorphine analogues and 6-O-(tosyloxyalkyl)-6-O-desmethyl-3-O-trityl-diprenorphine type precur-sors for the radiosynthesis of 6-O-([18F]fluoroalkyl)-6-O-desmethyl-diprenorphine radio-tracers for molecular imaging by positron emission tomography (PET). The synthesis se-quence to the new opioid receptor ligands consists of eleven steps starting from the poppy alkaloid thebaine. The precursor molecules were prepared in a three-step synthesis from the «Luthra-precursor» (TDDPN). We report the complete 1H- and 13C-NMR assignment of the new 6-O-(substituted)-6-O-desmethyl-diprenorphine derivatives as well as the results of docking studies in silico for diverse novel opioid receptor ligands, including a new se-ries of 6-O-(fluoroalkyl)- and 6-O-(hydroxyalkyl)-6-O-desmethyl-diprenophine derivatives.
Keywords:
opioid receptors
; 6
; 14-ethenomorphinans
; diprenorphine
; 6-O-fluoroalkyl-6-O-desmethyl-diprenorphine
; in silico docking
; positron emission tomography
1. Introduction
The word opium [1] derives from the ancient Greek term (οπιον, opion) originally describing the juice of any plant, but today refering specifically to the air-dried latex juice obtained after incising of the unripe seed capsule of the opium poppy (Papaver somniferum). [2] The opium poppy, which presumably originated in Asia Minor [3], is one of the few plants that was already domesticated in the neolithic period [2,4], and remains under cultivation today for its tasty seeds and oil as food, and for the medicinal properties of its extruded latex. Sumerian cuneiform tablets discovered in Mesopotamia (mainly modern Iraq), describe a method for the preparation of poppy juice and present its medical indications. Archaeological, botanical, and literary [5,6] sources show that cultivation of the opium poppy was widespread in the ancient Near East, Mesopotamia, Egypt, the Mediterranean basin, and Central Europe [7,8]. Morphine [9], was first isolated from opium[10] in 1804 by the Paderborn apothecary Friedrich Wilhelm Adam Sertürner, and remains to this day an important pharmaceutical for the treatment of chronic and severe pain. Since the time of Sertürner, more than eighty alkaloids have been isolated from opium poppy [11].
Opioid receptors (ORs, μ, κ, δ, NOP), are members of the G-protein-coupled receptor family (GPCR) of plasma membrane proteins [12,13], which have widespread expression in the central nervous system and in peripheral tissues and neurons, including the enteric nervous system. The ORs control diverse physiological and pathophysiological processes, with notable involvement in pain modulation, and are implicated in diverse neuropsychiatric disorders such as, Alzheimer’s disease (AD), epilepsy, and substance abuse. The activation of ORs by endogenous opioid peptides (EOPs), and by opiate alkaloids and several other classes of agonist drugs results in intracellular signal transduction via inhibition of the adenyl cyclase enzyme [13]. The resultant decrease in the second messenger cAMP level leads to inhibition of certain calcium ion channels and activation of inwardly-rectifying K⊕ channels and various other signal transduction pathways [14]. The resulting neuronal hyperpolarization dampens the release of various neurotransmitters (i.e., γ-aminobutyric acid (GABA), acetylcholine, dopamine, and norepinephrine), which net physiological effects such as euphoria, analgesia, sedation, and respiratory inhibition.
Molecular imaging of ORs by positron emission tomography (PET) [15,16,17,18,19] began four decades ago with the advent of the μ-OR selective agonist ligand [11C]carfentanil (1, [11C]Caf, Figure 1) [20] and the μ/κ-OR selective [18F]cyclofoxy ([18F]FcyF, 2) [21]. Other PET tracers include the δ-selective antagonist naltrindole (NTI) derivatives developed by Lever et al., namely N1’-[11C]methyl-naltrindole (3, [11C]MeNTI) [22], and N1’-(2-[18F]fluoroethyl)naltrindole (4, [18F]FE-NTI, BU97001) [23]. Ravert et al. [24] reported the radiosynthesis of the κ-OR selective agonist arylacetamidopiperazine derivative (R)-(-)-[11C]GR103545 (5) in 1999. Subsequently Pike et al. [25] synthesized the labelled NOP (ORL1) ligand [11C]NOP-1A (6).
Several non-selective orvinol ligands [15] have served for PET imaging of ORs in living brain, beginning with the [11C]-labeling of N17-cyclopropylmethyl-dihydronororvinone in position-20 with [11C]methyllithium, as reported by Burns et al. in 1984 [26]. Subsequent years saw the development of tracers based on the 6,14-ethenomorphinan skeleton (Figure 2). For brain imaging by single photon emission computer tomography (SPECT), Tafani et al [27] synthesized the gamma-emitting position-2 [125I]-labeled diprenorphine. Several position-N17 labelled diprenorphine derivatives (Figure 2) for PET imaging were synthesized from N17-nor-diprenorphine: [N17-cyclopropylmethyl-11C]-nordiprenorphine [28], N17-(2-[18F]fluoroethyl)-nordiprenorphine [29], N17-(3-[18F]fluoropropyl)-nordiprenorphine [30], and N17-((S)-3-[18F]fluoro-2-methylpropyl)-nordiprenorphine [29].
Since the publication of the fundamental study by Lever et al. [31] there were numerous efforts to develop metabolically more stable orvinol-type tracers labelled in position-6: 6-O-([125I]iodoallyl)-6-O-desmethyl-diprenorphine ([125I]-O-IA-DPN) [32]; [11C]-labelled orvinols: [11C]diprenorphine ([11C]DPN, 7) [31,33], [11C]buprenorphine ([11C]BPN, 8) [33], [11C]phenethyl-orvinol ([11C]PEO, 9) [34]), and also [18F]fluoroethyl orvinol derivatives: (6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN, 10) [35,36,37,38,39,40], 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-buprenorphine ([18F]FE-BPN, 11) [37], 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-phenethylorvinol ([18F]FE-PEO, 12)) [37].
6,14-Ethenomorphinans are semisynthetic, thebaine and/or oripavine derivatives, that possess a ring-C 6,14-etheno bridge originating from a stereoselective [4+2] cycloaddition reaction of different asymmetric substituted olefinic dienophiles (e.g., acrolein, methyl vinyl ketone) to morphinan-6,8-dienes (e.g., thebaine, oripavine) [19,41,42,43,44,45]. The first members of this family of Diels-Alder adducts were prepared by Sandermann [46] and Schöpf et al. [47] in the 1930s. Starting in the mid-1950s, Bentley and associates made such substantial increments in the synthesis of 6,14-ethenomorphinans that they are often referred to as Bentley compounds [41]. 6,14-Ethenomorphinans[48] and their fluorinated analogues [49] are important targets of current investigations for the discovery of novel potent and more selective OR ligands, with potential for application as PET ligands. Very recently, we provided a comprehensive survey of the organic chemistry of 6,14-ethenomorphinans dating back nine decades [19]. Our longstanding program on the synthesis of labelled 6,14-ethenomorphinans for the molecular brain imaging resulted in numerous tracers: [11C]PEO[34], [18F]FE-DPN [35,36,37,38,39,40], [18F]FE-BPN [37], and [18F]FE-PEO [37,50].
In this paper, we describe the organic synthesis of a novel type of OR ligand with fluoroalkyl and hydroxyalkyl substituents in position-6 of the 6,14-ethenomorphinan scaffold. We prepared 6-O-(fluoroalkyl)-6-O-desmethyl-diprenorphine (28a–d, (CH2)nF, n = 2–5) and 6-O-(hydroxyalkyl)-6-O-desmethyl-diprenorphine derivatives (29a–d, (CH2)nOH, n = 2–5) in multistep syntheses starting from the well-known «Luthra precursor» (TDDPN, 23) [33]. Furthermore, we synthesized the corresponding 3-O-trityl-6-O-(tosyloxyalkyl)-6-O-desmethyl-diprenorphine precursors (26a–d) for the direct nucleophilic radiosynthesis of 6-O-([18F]fluoroalkyl)-6-O-desmethyl-diprenorphine ([18F]28a–d, (CH2)nF, n = 2–5) derivatives. We also performed in silico docking analyses of 6-O-(fluoroalkyl)- and 6-O-(hydroxyalkyl)-diprenorphine derivatives (n = 2–5), together with further 6,14-ethenomorphinans and another relevant OR ligands.
2. Results and Discussion
2.1. Chemistry
2.1.1. Diprenophine
Diprenorphine (17, DPN Revivon®, RX-5050M), a prominent representative of the orvinol family of OR ligands, is a semisynthetic thebaine derivative belonging to the class of ring-C bridged morphinan derivatives (6,14-ethenomorphinans, or Bentley compounds) [19,41]. According to Chemical Abstracts, diprenorphine (17) has the systematic name: (5R,6R,7R,9R,13S,14S)-(5α,7α)-17-cyclopropylmethyl-4,5-epoxy-18,19-dihydro-3-hydroxy-6-methoxy-α,α-dimethyl-6,14-ethenomorphinan-7-methanol (CAS RN [14357-78-9]). DPN (17) is a nonselective OR antagonist that is approximately 100 times more potent than N17-allyl-normorphine (nalorphine), with nanomolar range affinities: Ki (μOR) = 0.07 nM, Ki (δOR) = 0.23 nM, Ki (κOR) = 0.02 nM [51]. [3H]DPN ([3H]17) is used for binding studies in vitro due to its high affinity. DPN (17) serves in veterinary medicine for remobilizing large animals (elephant, lion, tiger, rhinoceros), which had been previously sedated with the ultra-potent OR-agonists carfentanil (Caf) or etorphine (Immobilon®).
2.1.2. Synthesis of Diprenorphine
An eight-step procedure for the synthesis of diprenorphine (17) was already described by Bentley et al. in the late 1960s (route: 13 → 14 → 15 → 16a → 16b → 16c → 16d → 16e → 17, Figure 3) [52,53,54]. As noted above, the characteristic sub-structural unit of 17, the 6,14-ethano-bridge in ring-C, originates from a Diels-Alder cycloaddition of asymmetric substituted ethylene dienophiles into the conjugated morphinan-6,8-dienes. In the first step of the synthesis, thebaine (13) was reacted in a steroselective [2+4] cycloaddition reaction with methyl vinyl ketone to result in a mixture of the 6,14-etheno-bridged 7α-acetyl- (14, 7α-thevinone, 98 %) and 7β-acetyl derivative (7β-thevinone, 2 %) [52]. Thereafter the Δ6,14-double bond was hydrogenated by heterogenous catalytic reduction (H2, 10 % Pd-C, EtOH, 4 bar, 50 °C) to dihydrothevinone (15). The Grignard addition of methylmagnesium iodide into 15 gave 20-methyl-dihydrothevinol (16a). Next, N-demethylation by application of the von Braun method via the cyanamide (16b) resulted in 20-methyl-dihydronorthevinol (16c). The N17-cyclopropylmethyl substituent was introduced by acylation of the secondary amine (16c) with cyclopropylcarbonyl chloride and subsequent reduction of the N17-acyl derivative with LiAlH4 in THF or by direct alkylation with cyclopropylmethyl bromide. In the last step, diprenorphine-3-O-methylether (16e) was 3-O-demethylated with potassium hydroxide under strong/harsh conditions (diethylene glycol, 210–220 °C) to yield diprenorphine (17, DPN).
In the early 1990s, the Makleit research group developed several alternative routes for the synthesis of diprenorphine, buprenorphine and their N17-substituted analogues [55,56]. In the first approach (13 → 18a–b, → 19a–b → 20a–b → 21 → 16e → 17, Figure 3) N17-formyl-northebaine (18a) and N17-benzyl-northebaine (18b) [57,58] was reacted with methyl vinyl ketone to yield the corresponding cycloadducts, N17-formyl-northevinone (19a) and N17-benzyl-northevinone (19b), respectively. The hydrogenation of 19a in the presence of 10 % Pd-C in ethanol (60 °C, 5 bar) gave N17-formyl-dihydronorthevinone (20a), which was converted to dihydronorthevinone (20b) by hydrolysis with HCl/EtOH (reflux, 4 h). Subsequently, 20b was alkylated with cyclopropylmethyl bromide to N17-cyclopropylmethyl-dihydronorthevinone (21). The Grignard reaction of 21 with methylmagnesium iodide in a toluene-diethylether mixture resulted in 20-methyl-N17-cyclopropylmethyl-dihydronorthevinol (16e) in 94 % yield. Thereafter compound 16e was 3-O-demethylated to diprenorphine (17). In the second approach (route: 13 → 14 → 15 → 20b → 21 → 16e → 17, Figure 3), dihydrothevinone (15) was N-demethylated with diethylazodicarboxylate (DEAD) to yield dihydronorthevinone (20b). Analogously to the previous route, dihydronorthevinone (20b) was converted to diprenorphine (17) via N17-alkylation, Grignard addition, and 3-O-demethylation.
2.1.3. 6-O-Desmethyl-6,14-ethenomorphinans
6-O-Desmethyl-6,14-ethenomorphinans was unknown until the mid-1980s. The fortuitous/serendipitous discovery of Kopcho and Schaeffer [59] that the reaction of 7α-aminomethyl-6,14-ethenomorphinans with 15 equiv. LiAlH4 in THF containing CCl4 (4 equiv.) co-solvent can selectively result in the 6-O-demethylated derivatives was the first mention of this type of reaction: a selective 6-O-demethylation. Subsequently Lever et al. [31] successfully extended this method for the synthesis of orvinol derivatives. 6-O-Desmethyl-buprenorphine (DBPN) and 6-O-desmethyl-diprenorphine (DDPN) were prepared and applied for the synthesis of PET precursor molecules: 3-O-tert-butyldimethylsilyl-6-O-desmethyl-diprenorphine (TBDMS-DDPN, «Lever precursor» [31]) and 3-O-trityl-6-O-desmethy-diprenorphine (TDDPN, «Luthra precursor»[33]). Despite 6-O-desmethyl-orvinols being successfully applied for the synthesis of PET precursors, their first biochemical investigations were only reported by the opioid research group of Benyhe [60] at HUN-REN BRC Szeged more than three decades after the first preparation of DDPN (22, Figure 4) by Lever et al. [31]. The 6-O-desmethyl derivatives (DBPN, DDPN, DDHE, and DPEO) of buprenorphine (BPN), diprenorphine (DPN), dihydroetorphine (DHE) and phenethyl-orvinol (PEO) showed higher binding affinity to μORs compared to the respective parent 6-O-methyl compounds [60].
2.1.4. Synthesis of 6-O-Substituted-6-O-desmethyl-diprenorphine derivatives
There are only a few reports available regarding the synthesis of 6-O-substituted-orvinols with other than methyl substituents. In 1992, Musachio and Lever [32] developed 6-O-(3-tri-n-butylstannyl-prop-2-enyl)-diprenorphine for the preparation of 6-O-([125I]iodoallyl)-6-O-desmethyl-diprenorphine. In 2000, Wester et al. [35] described the radiosynthesis of 6-O-(2-[18F]fluoroethyl-6-O-desmethyl-diprenorphine ([18F]FE-DPN, 10) starting from the «Luthra precursor» (TDDPN). Investigations seeking an improved radiosynthesis of [18F]FE-DPN (10) via indirect radiofluorination from TDDPN were reported later by Schoultz et al. [36,37].
Subsequently, Czakó et al. [61] synthesized 6-O-substituted-20-methyl-orvinols (R = Et, nPr, cyclopropylmethyl) starting from 6-O-substituted-northebaine derivatives. [4+2] Cycloaddition of 6-O-substituted-6,8-morphinandienes with methyl vinyl ketone gave the corresponding 6-O-substituted-thevinone analogues. Next, the 7α-ketons were reacted with methylmagnesium iodide to 6-O-alkyloxy-20-methylthevinols which were 3-O-demethylated with potassium hydroxide in diethylene glycol to the target 6-O-substituted-20-methyl-orvinols [61]. 6-Phenyl-20-methylorvinol was also prepared from 6-bromo-6-demethoxythebaine, which is available from the poppy alkaloid thebaine in five subsequent reaction steps. 6-Bromo-6-demethoxythebaine was converted to 6-phenyl-6-demethoxythebaine in a Suzuki-Miyaura cross-coupling reaction (PhB(OH)2, Pd(PPh3)4, Ba(OH)2 . 8 H2O) in good yield. Next, the target orvinol was prepared in three consecutive steps analogously to the route described above: Diels-Alder reaction of the morphinandiene with methyl vinyl ketone, Grignard addition of MeMgI to the 7α-acetyl compound, and finally 3-O-demethylation [61]. In 2016, Lever et al. [62] synthesized 6-O-propylamido- and 6-O-hexylamido-[111In]-DOTA-diprenorphine conjugates for SPECT imaging of ORs in a six-step procedure starting from diprenorphine (17).
Very recently, we developed a new precursor 6-O-(2-tosyloxyethyl)-6-O-desmethyl-diprenorphine (TE-TDDPN (26a), «Henriksen precursor») for the preparation of [18F]FE-DPN (10) via direct nucleophile radiofluorination [38]. In 2023, the research group of Mikecz [40] in Debrecen performed optimalization studies of the [18F]FE-DPN radiosynthesis from TE-TDDPN. In the same year, the research group of Michaelides [39] at NIH performed PET experiments in mice by application of [18F]FE-DPN (10) prepared from the new precursor TE-TDDPN thorough direct one-pot, two step nucleophilic radiosynthesis. Their PET investigation μ-opioid receptor knock-out (μOR KO) compared with wildtype mice confirmed that [18F]FE-DPN (10) preferentially binds to μORs in vivo, despite its incomplete selectivity in vitro. [39]
Continuing our research [38,39,40] in the field of radiolabelled orvinol derivatives, especially in the light of results of the Michaelides group concerning the unexpected selectivity of [18F]FE-DPN (10) for μORs in vivo [39], we set about to synthesize 6-O-fluoroalkyl-6-O-desmethyl-diprenorphine derivatives with longer side chains (i.e., 3-fluoropropyl, 4-fluorobutyl, and 5-fluoropentyl). This investigation also served to make 6-O-substituted-6-O-desmethyl-orvinol OR ligands for studying the influence of the 6-O-side chain on the OR affinity and subtype selectivity.
As the starting point to our investigation, we used the «Luthra precursor» (TDDPN, 23) [33], which was prepared from diprenorphine (17) in two steps. 6-O-(Fluoroalkyl)-6-O-desmethyl-diprenorphine derivatives (28a–d, Figure 5) were then prepared from TDDPN (23) in two steps. First, TDDPN (23) was alkylated with the corresponding fluoroalkyl toluene sulfonate (33a–c, Figure 6) to yield 6-O-fluoroalkyl-6-O-desmethyl-3-O-trityl-diprenorphine derivatives (27a–d), with yields in the range of 43–66 %. Fluoroalkyl toluene sulfonate (33a–c, Figure 6) reagents were synthesized according to previously developed protocols: 33a [63], 33b [64], 33c [65]. Subsequently the trityl protecting group was removed by acidic hydrolysis with acetic acid -water (27:7 (v/v)) at 100 °C for 5 min to give the target fluoroalkylated derivatives (28a–d) in 71–78 % yields.
Tosyloxyalkyl precursor molecules (26a–d) for direct nucleophilic one pot, two step nucleophilic radiofluorination for 6-O-([18F]fluoroalkyl)-6-O-desmethyl-diprenorphines ([18F]28a–d) were synthesized in a three-step procedure. The direct introduction of the tosyloxyalkyl side chain into position-6 by reacting TDDPN (23) with bistosyloxy alkanes (TosO(CH2)nOTos, n = 2–5) proved unfeasible in our hands [50], yielding undesired heterocyclic products. Consequently, we reverted to our previous strategy [38,50] for introduction of the desired tosyloxyalkyl side chain at the 6-O-position in three consecutive reaction steps (route: 24a–d → 25a–d → 26a–d). First, the «Luthra» precursor (TDDPN, 23) was alkylated with 2.3 equiv. of the corresponding tert-butyldiphenysilyl- (TBDPS) protected bromoalkanol (34a–c, Br(CH2)nOTBDPS, n = 3–5, Figure 6) in DMF in the presence of 10 equiv. sodium hydride to yield 6-O-(tert-butyldiphenylsilyloxy-alkyl)-6-O-desmethyl-3-O-trityl-diprenorphine derivatives (24a–d), with yields in a range of 40–71 %. TBDPS-protected bromoalkanols (34a–c, Figure 6) were synthesized from the corresponding bromoalkanols (30d–f) with tert-butyldiphenylsilyl chloride (TBDPSCl) in dichloromethane in the presence of DMAP and DIPEA. Subsequently the TBDPS protecting group of the compounds 24a–d was removed by treatment with 1.28 equiv. of 1M tetrabutylammonium fluoride in THF (RT, argon, 4 h) to give 6-O-(hydroxyalkyl)-6-O-desmethyl-3-O-trityl-diprenorphines (25a–d), with 84–93 % yields.
For introduction of the tosyloxy leaving group, the hydroxyalkyl-TDDPN compounds (25a–d) were reacted with 3.4 equiv. toluenesulfonic anhydride in dichloromethane in the presence of 4 equiv. pyridine (RT, 4 h) to give the desired precursors (26a–d), with 60–87 % yileds. Complete 1H-, and 13C-NMR assignments of the 6-O-(tosyloxyalkyl)-6-O-desmethyl-diprenorphine analogues (26b–d) are presented in Table 1. 6-O-(Hydroxyalkyl)-6-O-desmethyl-diprenorphine derivatives (29a–d) were also prepared from the corresponding 6-O-(hydroxyalkyl)-6-O-desmethyl-3-O-trityl-diprenorphines (25a–d) by acidic hydrolysis of the 3-O-trityl protecting group with acetic acid – water 4:1 (v/v) (100 °C, 10 min). The 6-O-(hydroxyalkyl)- compounds were obtained with yields in range of 72–77 %.
The syntheses of fluoroalkyl-tosylates (33a–c) and bromoalkoxy-tert-butyl-diphenylsilanes (34a–c) were performed according to the reaction sequences depicted in Figure 6. We verified the identity of the target compounds by NMR spectroscopy (24b–d, 25b–d, 26b–d, 27b–d, 28b–d, 29b–d) and by HRMS (26b–d, 28b–d, 29b–d). The 1H and 13C-NMR assignments were facilitated by previously reported NMR studies of Bentley compounds [66,67,68,69]. Complete 1H-, and 13C-NMR assignments of the 6-O-fluoroalkyl-6-O-desmethyl-diprenorphine analogues (28b–d) are presented in Table 2. Complete 1H-, and 13C-NMR chemical shifts and assignments for 6-O-fluoroalyl-6-O-desmethyl-3-O-trityl-diprenorphine analogues (27b–d) and for 6-O-hydroxyalkyl-6-O-desmethyl-diprenorphine derivatives (29b–d) are presented in Supplementary Material (Table S1 and Table S2).
High Resolution Mass Spectra measurements were carried out for compounds 26b–d, 28b–d, and 29b–d similarly to the previous method of Biri et al. in the field of buprenorphine analogues. [70]
2.2. In Silico Studies
To investigate the binding dynamics and conformational stability of series 28 (28a–d) and series 29 (29a–d) ligands, multiple molecular dynamics (MD) simulations were conducted for each compound in complex with both the active and inactive states of the receptor. The stability of the ligand-receptor complexes was assessed by monitoring two key metrics throughout the simulation trajectories: the root mean square deviation (RMSD) of the ligands’ heavy atoms and the center of mass (COM) distance between the ligand and the receptor’s binding site. The ligand RMSD was calculated with respect to its initial docked pose after a rotational and translational fit to the receptor’s backbone, providing a quantitative measure of the ligand’s conformational stability within the binding pocket. A low, equilibrated RMSD value is indicative of a stable binding mode, whereas high or continuously increasing values suggest significant conformational changes or instability [71]. The COM distance was measured between the geometric centers of the ligand and a defined set of binding site residues. This metric evaluates the ligand’s positional stability; a stable COM distance signifies that the ligand remains localized, while large fluctuations can indicate movement within the pocket or dissociation events [72].
2.2.1. Ligand Binding to the Active Receptor State
In the simulations involving the active receptor conformation, all ligands from both series demonstrated stable binding behaviour. As shown in Figure 7, the ligand RMSD values for most compounds equilibrated within the first few frames and subsequently fluctuated around a stable mean, generally below 3.5 Å. This suggests that the ligands maintained a consistent binding pose throughout the simulation. The COM distances were similarly stable, clustering within a range of approximately 9.0 to 12.0 Å, which confirms the persistent localization of the ligands within the active site.
2.2.2. Ligand Binding to the Inactive Receptor State
The simulations with the inactive receptor state revealed a significant difference in binding stability, particularly for ligand 28a (FE-DPN, Figure 8). While most ligands exhibited stable RMSD and COM distance profiles comparable to those in the active state, ligand 28a (FE-DPN) displayed marked instability. Its RMSD value increased sharply, reaching over 6 Å, and remained highly volatile. This conformational instability was in harmony of its COM distance, which showed large-scale fluctuations and a shift to greater distances (>13.5 Å), indicating that 28a (FE-DPN) was unable to maintain a stable pose and position within the inactive binding pocket. In contrast, the ligands from series 29 remained stable in the inactive state, with RMSD values consistently below 4.0 Å.
2.2.3. Ligand Binding to the Partial Agonist Receptor State
Simulations with the partial agonist receptor state (PDB ID: 7U2K) revealed a complex and distinct dynamic profile, providing further insight into the ligands’ state-selectivity (Figure 9). The most critical finding from this new set of simulations involves ligand 28a (FE-DPN). In stark contrast to its stability in the fully active state, 28a (FE-DPN) now displays significant conformational instability in the partial agonist conformation. Its RMSD value is highly volatile, fluctuating between 2.5 Å and 4.0 Å throughout the trajectory. Interestingly, this conformational instability is paired with a Center of Mass (COM) distance that is consistently lower than its series analogs, suggesting that 28a (FE-DPN) occupies a deeper region of the binding pocket but in a strained and unstable manner. This behavior mirrors that of ligand 29, which also exhibits a high RMSD (~4.0 Å) coupled with a similarly low COM distance. The remaining ligands in both series demonstrated relatively stable binding profiles, with COM distances clustering in an intermediate range between those observed for the active and inactive states.
2.2.4. Molecular Docking Reveals a Clear Energetic Preference for the Active State
To complement the dynamic analysis from MD simulations, we first assessed the static binding predictions from molecular docking. The calculated binding affinities (in kcal/mol) for each ligand across the three receptor states—agonist (active), antagonist (inactive), and partial agonist—are presented in Figure 10. The results reveal a clear and consistent energetic hierarchy. Altogether, all compounds show a distinct preference for the active receptor state, which yielded the most favorable binding affinities for every ligand tested. In the active state, scores were consistently around -9.5 kcal/mol, with series 28 ligands showing slightly more favorable energies than those in series 29. Within series 29, a trend was observed where ligands with longer chains achieved somewhat better scores. A similar pattern was observed for the inactive state, which showed intermediate affinities of approximately -8.5 kcal/mol. Here too, series 28 was slightly better than series 29, and longer chains again appeared to confer a modest energetic advantage. The partial agonist state consistently produced the least favorable scores, ranging from -7.5 to -8.0 kcal/mol, yet the same structural trends persisted: series 28 ligands docked more favorably than series 29, and longer-chained compounds were preferred. This overarching trend indicates that, even in a static model, all ligands are energetically predisposed to bind most favorably to the fully active receptor conformation. This provides an initial, static validation for the state-selective behavior observed in the more detailed molecular dynamics simulations.
2.2.5. Conclusion and Implications
The combination of molecular docking and molecular dynamics simulations provides a comprehensive and compelling rationale for the state-selective behavior of the tested ligands. The static docking scores establish an initial energetic basis for selectivity, revealing that all compounds are intrinsically favored to bind to the fully active receptor conformation. The MD simulations then provide the crucial dynamic context, illustrating how this initial energetic preference translates into conformational stability over time.
The collective computational results strongly suggest that ligand 28a (FE-DPN) possesses a highly specific binding preference for the fully active conformation of the receptor. Its stability in the active state contrasts sharply with its profound instability in both the inactive and the partial agonist states. [73]. The compounds of series 29 display more complex, state-dependent behaviour. While appearing conformationally tolerant in the active and inactive states, our results show that binding of the parent compound 29 is uniquely destabilized by the partial agonist conformation. This suggests that subtle changes in OR structure can induce distinct stability profiles even within a closely related chemical series, which may translate to differences in functional activity.
The observed COM distances provide a clear structural rationale for the different functional states, revealing a gradient in the binding site topology across the receptor conformations. In general, the ligands bound most closely in the active state (~9.0-12.0 Å), at an intermediate distance in the partial agonist state (~10.5-12.5 Å), and most distally in the inactive state (~13.0 Å). Intriguingly, within this trend, the unstably-binding ligands (28a and 29) adopted a closer COM distance in the partial agonist state, suggesting a common mechanistic feature. This may indicate that the partial agonist binding pocket forces these ligands into a strained, high-energy conformation that is nevertheless “deep” in the pocket, a phenomenon that could be central to the mechanism of partial agonism. These findings provide a nuanced structural and dynamic rationale for the observed activities of the compounds and strengthen the case for 28a (FE-DPN) as a potent candidate for further investigation as a μOR selective ligand.
3. Materials and Methods
3.1. General Methods
Reagents and solvents were purchased from major commercial suppliers, and were used without further purification. Melting points were determined on a Büchi-535 melting point apparatus, without correction. Column chromatography (CC) was performed on Kieselgel 60 Merck 1.09385 (0.040-0.063 mm). Analytical TLC was accomplished on Macherey-Nagel Alugram® Sil G/UV254 40x80 mm aluminum sheets (0.25 mm silica gel with fluorescent indicator) with the following eluent systems (each (v/v)): A: dichloromethane-methanol 9:1, B: hexane-ethyl acetate 7:3, C: hexane-ethyl acetate 1:1, D: ethyl acetate-methanol 8:2, E: dichloromethane-methanol 95:5. The spots were visualized with a 254 nm UV lamp or with 5% phosphomolybdic acid in ethanol.
Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AV 500 (Avance 500 MHz) spectrometer at 298 K, using a BBO probehead (HP workstation XW 5000, software: Bruker TOPSPIN 1.3). For 1H- and 19F-NMR experiments: 10 mg of the appropriate orvinol were dissolved in 500 μL of deuterated chloroform (CDCl3). We measured the 13C-NMR spectra after dissolving a 20 mg sample of the corresponding oripavine in 500 μL CDCl3. Chemical shifts were reported as δ values in parts per million (ppm), and coupling constants (J) reported in Hertz. 1H- and 13C-NMR chemical shifts were referenced to the residual peak of CDCl3 at δ 7.26 for proton and 77.16 ppm for carbon.
During our high resolution mass spectroscopic (HRMS, 26b–c, 28b–d, 29b–d) inves-tigation, the LC–MS system consisted of a Q-Exactive® Plus mass spectrometer (Thermo Scientific, Bremen, Germany) was hyphenated to a Vanquish Neo HPLC system (Thermo Scientific, Germering, Germany). Chromatographic separation was performed on the Ac-claim PepMap® RSLC C18 column (2 μm × 15 cm × 1 mm). The mobile phases were 0.1% formic acid in water (A) and acetonitrile (B). The gradient elution was conducted by line-arly increasing B from 1% to 95% over 14 minutes, followed by a 4-minute wash and and re-equilibration. The total run time was 18 minutes with a flow rate of 50 µL/min. Column and autosampler temperatures were 25 and 7 °C, respectively. All samples were analyzed using the positive electrospray ionization (ESI) mode. Each sample was injected in dupli-cates with same source settings. The source settings were optimized as follows: sheath gas flow rate, 30 arbitrary units; auxiliary gas flow rate, 15 arbitrary units; spray voltage, 3.5 kV; S-lens, 50 arbitrary units; capillary temperature, 325 °C; and auxiliary heater temper-ature, 75 °C. Full MS scan data were obtained from m/z 200 to 1500 at a resolution of 70000 with the AGC target of 3×106, and data-dependent MS/MS spectra were acquired at a res-olution of 17500 by using stepped normalized collision energy (NCE), either 20, 25, and 30%, or 45, 50 and 55, both with Top-5 MS/MS experiments. Microscans were set to 1 for both MS and MS/MS. The AGC target was set to 1×105, the intensity threshold to 8×103, with the maximum injection time of 50 ms. The precursor ions were filtered by the quad-rupole, which operates at an isolation window of m/z 4. All the obtained mass spectra were evaluated by the Compound Discoverer 3.3 SP2 software from Thermo Fisher Scientific.
3.2. Chemistry
We described the preparation of 24a, 25a, 26a, 27a, 28a and 29a derivatives (n = 2, -CH2CH2-) in our previous studies [38,40,69].
3.2.1. General Procedure for the Synthesis of 6-O-(tert-Butyldiphenylsilyloxyalkyl)-6-O-desmethyl-3-O-trityl-diprenorphine (24b–d) Derivatives
To a stirred suspension of sodium hydride (60 % dispersion in mineral oil, 490 mg, 12.2 mmol, 10 equiv.) in anhydrous N,N-dimethylformamide (6 mL) was added a solution of TDDPN (23, CAS RN: 157891-92-4, 800 mg, 1.22 mmol) in anhydrous N,N-dimethylformamide (7 mL) at 0 °C under an argon atmosphere. The reaction mixture was stirred for 30 min at the same temperature. A solution of the corresponding tert-butyldiphenylsilyl (TBDPS) protected bromoalkanol (34a–c, 2.3 mmol) in dry N,N-dimethylformamide (2.5 mL) was then added drop-wise. After stirring at RT for 20 h, the mixture was poured into water (50 mL). The suspension was extracted with dichloromethane (4 x 40 mL) and the combined organic extracts washed with brine (40 mL), and dried over Na2SO4. The solvent was removed under reduced pressure and the resulting crude product mixture was purified by column chromatography on silica gel.
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(3-tert-butyldiphenylsilyloxypropyl)-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(3-TBDPSOP)-6-O-desmethyl-3-O-trityl-diprenorphine, 24b, TBDPS-OP-TDDPN).
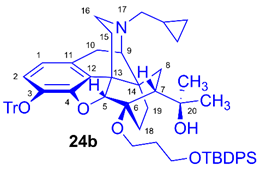
Compound 24b was prepared from TDDPN (23, 800 mg, 1.22 mmol) using the general procedure 3.2.1. CC: Kieselgel: 100 g, eluent: hexane-ethyl acetate 7:3 (v/v) to ethyl acetate, fractions: 50 mL. Yield: 470 mg (40 %), mp. 98-102 °C, RfA = 0.80, RfB = 0.25, RfC = 0.71. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.42 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.89 (td, 2J19anti,19syn = 13.3 Hz, 3J19anti,18syn = 6.1 Hz, 1H, 19-Hanti), 0.97 (dd, 2J8α,8β = 13.3 Hz, 3J8α,7β = 9.7 Hz, 1H, 8α-H), 1.03 (s, 9H, (CH3)3CSi(Ph)2), 1.15 (s, 3H, 20CH3), 1.31 (s, 3H, 20CH3), 1.38 (dd, 2J15eq,15ax = 12.9 Hz, 3J15eq,16ax = 2.7 Hz, 1H, 15-Heq), 1.43 (td, 2J18syn,18anti = 13.0 Hz, 3J18syn,19anti = 5.6 Hz, 1H, 18-Hsyn), 1.55 (m, 1H, 18-Hanti), 1.73-1.78 (m, 2H, CH2CH2CH2OTBDPS), 1.79 (app t, 3J7β,8α = 9.6 Hz, 1H, 7β-H), 1.87 (td, 2J15ax,15eq = 13.7 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15-Hax), 2.04 (dd, 2J10α,10β = 18.3 Hz, 3J10α,9α = 6.4 Hz, 1H, 10α-H), 2.13 (td, 2J16ax,16eq = 13.1 Hz, 3J16ax,15eq = 3.7 Hz, 1H, 16-Hax), 2.18 (dd, JAB = 12.9 Hz, JAX = 6.7 Hz, 1H, NCH2 (a)), 2.32 (dd, JBA =12.9 Hz, JBX = 5.9 Hz, NCH2 (b)), 2.54 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.75 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 12.0 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H ), 2.85 (d, 2J10β,10α = 18.3 Hz, 1H, 10β-H), 2.89 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H), 3.59-3.67 (m, 2H, 6-O-CH2CH2CH2OTBDPS), 3.70-3.80 (m, 2H, 6-O-CH2CH2CH2OTBDPS), 4.04 (d, 4J5β,18anti = 1.3 Hz, 1H, 5β-H), 5.14 (br s, 1H, 20-OH), 6.19 (d, J1,2 = 8.4 Hz, 1H, 1-H), 6.48 (d, J2,1 = 8.4 Hz, 1H, 2-H), 7.16-7.18 (m, 9H, Tr(m,p)), 7.33-7.66 (m, 16H, Tr(o) and (CH3)3CSi(Ph)2). 13C-NMR (CDCl3): δ = 3.3 (cProp(a)), 4.1 (cProp(b)), 9.4 (cPropCH), 17.5 (C-19), 19.2 ((CH3)3CSi(Ph)2), 22.5 (C-10), 24.8 (20CH3), 26.9 ((CH3)3CSi(Ph)2), 29.7 and 29.8 (C-18 and 20CH3), 32.1 (C-8), 36.9 (OCH2CH2CH2O), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.1 (C-7), 58.0 (C-9), 59.8 (NCH2), 60.7 and 61.2 (6-O-CH2CH2CH2OTBDPS and 6-O-CH2CH2CH2OTBDPS), 74.1 (C-20), 80.3 (C-6), 91.5 (TrCO), 96.4 (C-5), 117.8 (C-2), 123.1 (C-1), 127.2 (pCTr), 127.3 (mCTr), 127.6 (TBDPS-Ph-3,5), 129.4 (oCTr), 129.5 and 129.6 (TBDPS-Ph-C4), 130.7 (C-11), 132.0 (C-12), 133.9 and 134.0 (TBDPS-Ph-C1), 135.5 and 135.6 (TBDPS-Ph-C2,6), 137.2 (C-3), 144.2 (TrC-1), 151.4 (C-4). C63H71NO5Si (950.33).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(4-tert-butyldiphenylsilyloxybutyl))-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(4-tert-butyldiphenylsilyloxybutyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 24c, TBDPS-OB-TDDPN)
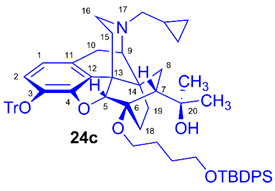
Compound 24c was synthesized from TDDPN (23, 400 mg, 0.61 mmol) using the general procedure 3.2.1. CC: Kieselgel: 60 g, eluent: hexane-ethyl acetate 7:3 (v/v), fractions: 25 mL. Yield: 419 mg (71 %), mp. 77-88 °C, RfA = 0.81, RfB = 0.33, RfC = 0.64. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.43 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.89 (td, 2J19anti,19syn = 13.8 Hz, 3J19anti,18syn = 5.7 Hz, 1H, 19-Hanti), 0.97 (dd, 2J8α,8β = 13.5 Hz, 3J8α7β = 9.6 Hz, 1H, 8α-H), 1.05 (s, 9H, (CH3)3CSi(Ph)2), 1.15 (s, 3H, 20CH3), 1.34 (s, 3H, 20CH3), 1.37 (m, 1H, 18-Hsyn), 1.42 (dd, 2J15eq,15ax = 12.5 Hz, 3J15eq,16ax = 2.5 Hz, 1H, 15-Heq), 1.73-1.78 (m, 5H, 6-O-CH2CH2CH2CH2OTBDPS and 18-Hanti), 1.82 (app t, 3J7β,8α = 9.9 Hz, 1H, 7β-H), 1.89 (td, 2J15ax,15eq = 13.0 Hz, 3J15ax,16eq = 5.5 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.0 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 13.0 Hz, 3J16ax,15eq = 3.6 Hz, 1H, 16-Hax), 2.18 (dd, JAB = 12.9 Hz, JAX = 6.7 Hz, 1H, NCH2 (a)), 2.33 (dd, JBA =12.9 Hz, JBX = 6.0 Hz, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 5.1 Hz, 1H, 16-Heq), 2.76 (ddd, 2J8β,8α = 13.3 Hz, 3J8β,7β = 12.1 Hz, 4J8β,19syn = 3.9 Hz, 1H, 8β-H ), 2.85 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.0 Hz, 1H, 9α-H), 3.44-3.82 (m, 4H, 6-O-CH2CH2CH2CH2OTBDPS), 4.09 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.26 (br s, 1H, 20-OH), 6.19 (d, J1,2 = 8.0 Hz, 1H, 1-H), 6.48 (d, J2,1 = 8.0 Hz, 1H, 2-H), 7.16-7.22 (m, 9H, Tr(m,p)), 7.35-7.67 (m, 16H, Tr(o) and (CH3)3CSi(Ph)2). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.6 (C-19), 19.2 ((CH3)3CSi(Ph)2), 22.5 (C-10), 24.8 (20-CH3), 26.9 ((CH3)CSi(Ph)2), 27.2 and 29.3 (6-O-CH2CH2CH2CH2OTBDPS), 29.7 and 29.8 (C-18 and 20CH3), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 63.6 and 64.9 (6-O-(CH2)3CH2OTBDPS and 6-O-CH2(CH2)3OTBDPS), 74.1 (C-20), 80.2 (C-6), 91.4 (TrCO), 96.6 (C-5), 117.8 (C-2), 122.9 (C-1), 127.2 (pCTr), 127.3 (mCTr), 127.6 (TBDPS-Ph-3,5), 129.4 (oCTr), 129.5 (TBDPS-Ph-C4), 130.7 (C-11), 132.0 (C-12), 134.0 and 134.1 (TBDPS-Ph-C1), 135.5 (TBDPS-Ph-C2,6), 137.2 (C-3), 144.2 (Tr-C1), 151.4 (C-4). C64H73NO5Si (964.35).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(5-tert-butyldiphenylsilyloxypentyl))-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(5-tert-butyldiphenylsilyloxypentyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 24d, TBDPS-OPe-TDDPN)
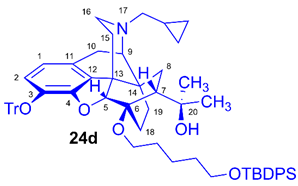
Compound 24d was synthesized from TDDPN (23, 625 mg, 0.95 mmol) using the general procedure 3.2.1. CC: Kieselgel: 70 g, eluent: hexane-ethyl acetate 7:3 (v/v), fractions: 50 mL. Yield: 573 mg (61 %), mp. 70-81 °C, RfA = 0.90, RfB = 0.24, RfC = 0.70. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.42 (m, 1H, 19-Hsyn), 0.47 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.89 (td, 2J19anti,19syn = 13.3 Hz, 3J19anti,18syn = 5.5 Hz, 1H, 19-Hanti), 0.97 (dd, 2J8α,8β = 13.8 Hz, 3J8α7β = 8.6 Hz, 1H, 8α-H), 1.05 (s, 9H, (CH3)3CSi(Ph)2), 1.16 (s, 3H, 20CH3), 1.24-1.32 (m, 2H, (CH2)2CH2(CH2)2), 1.34 (s, 3H, 20CH3), 1.37-1.58 (m, 7H, 18-Hsyn, 15-Heq, 18-Hanti, 6-O-CH2CH2CH2CH2CH2OTBDPS), 1.81 (app t, 3J7β,8α = 9.7 Hz, 1H, 7β-H), 1.89 (td, 2J15ax,15eq = 13.5 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.7 Hz, 3J10α,9α = 6.3 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 12.8 Hz, 3J16ax,15eq = 3.6 Hz, 1H, 16-Hax), 2.18 (dd, JAB = 12.8 Hz, JAX = 6.7 Hz, 1H, NCH2 (a)), 2.33 (dd, JBA =12.8 Hz, JBX = 5.9 Hz, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 11.9 Hz, 3J16eq,15ax = 4.9 Hz, 1H, 16-Heq), 2.76 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 12.3 Hz, 4J8β,19syn = 3.8 Hz, 1H, 8β-H ), 2.85 (d, 2J10β,10α = 18.7 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.3 Hz, 1H, 9α-H), 3.39-3.79 (m, 4H, 6-O-CH2(CH2)3CH2OTBDPS), 4.10 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.29 (br s, 1H, 20-OH), 6.19 (d, J1,2 = 8.4 Hz, 1H, 1-H), 6.47 (d, J2,1 = 8.4 Hz, 1H, 2-H), 7.15-7.21 (m, 9H, Tr(m,p)), 7.36-7.68 (m, 16H, Tr(o) and (CH3)3CSi(Ph)2). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.6 (C-19), 19.2 ((CH3)3CSi(Ph)2), 22.3 ((CH2)2CH2(CH2)2), 22.5 (C-10), 24.8 (20-CH3), 26.9 ((CH3)CSi(Ph)2), 29.7 and 29.8 (C-18 and 20CH3), 30.5 and 32.4 (6-O-CH2CH2CH2CH2CH2OTBDPS), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 63.9 and 64.8 (6-O-(CH2)4CH2OTBDPS and 6-O-CH2(CH2)4OTBDPS), 74.2 (C-20), 80.1 (C-6), 91.4 (TrCO), 96.6 (C-5), 117.8 (C-2), 122.9 (C-1), 127.2 (pCTr), 127.3 (mCTr), 127.6 (TBDPS-Ph-3,5), 129.4 (oCTr), 129.5 (TBDPS-Ph-C4), 130.7 (C-11), 132.1 (C-12), 134.1 (TBDPS-Ph-C1), 135.5 (TBDPS-Ph-C2,6), 137.2 (C-3), 144.2 (Tr-C1), 151.3 (C-4). C65H75NO5Si (978.38).
3.2.2. General Procedure for the Synthesis of 6-O-Hydroxyalkyl-6-O-desmethyl-3-O-trityl-diprenorphine (25b–d) Derivatives
The corresponding TBDPS-protected TDDPN derivatives (24b–d, 0.42 mmol) were dissolved in tetrahydrofuran (15 mL) under argon atmosphere. A 1M solution of tetrabutylammonium fluoride in tetrahydrofuran (0.54 mL, 0.54 mmol, 1.28 equiv.) was added, and the mixture was stirred for 4 h. The solvent was removed under reduced pressure. Water (50 mL) was added to the residue and the mixture was extracted with dichloromethane (3 x 40 mL). The combined organic extracts were dried over anhydrous Na2SO4 and the solution was concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel.
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(3-hydroxypropyl)-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(3-hydroxypropyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 25b, HP-TDDPN)
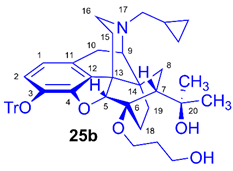
Compound 25b was synthesized from TBDPS-OP-TDDPN (24b, 400 mg, 0.42 mmol) using the general procedure 3.2.2. CC: Kieselgel: 40 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 254 mg (84 %), mp. 110-121 °C, RfC = 0.09, RfD = 0.77, RfE = 0.23. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.42 (tt, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.90 (td, 2J19anti,19syn = 13.4 Hz, 3J19anti,18syn = 5.7 Hz, 1H, 19-Hanti), 0.98 (dd, 2J8α,8β = 13.4 Hz, 3J8α,7β = 9.3 Hz, 1H, 8α-H), 1.16 (s, 3H, 20CH3), 1.35 (s, 3H, 20CH3), 1.38-1.47 (m, 2H, 18-Hsyn, 15-Heq), 1.61 (tt, 1H, 18-Hanti), 1.72-1.75 (m, 2H, CH2CH2CH2OH), 1.83 (app t, 3J7β,8α = 10.1 Hz, 1H, 7β-H), 1.90 (td, 2J15ax,15eq = 13.5 Hz, 3J15ax,16eq = 5.5 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.4 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 13.4 Hz, 3J16ax,15eq = 3.9 Hz, 1H, 16-Hax), 2.18 (dd, JAB = 12.8 Hz, JAX = 6.7 Hz, 1H, NCH2 (a)), 2.32 (dd, JBA =12.8 Hz, JBX = 5.7 Hz, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 12.1 Hz, 3J16eq,15ax = 4.6 Hz, 1H, 16-Heq), 2.77 (ddd, 2J8β,8α = 13.2 Hz, 3J8β,7β = 12.0 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H ), 2.85 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H), 3.57-3.92 (m, 4H, 6-O-CH2CH2CH2OH, 6-O-CH2CH2CH2OH), 4.14 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.12 (br s, 1H, 20-OH), 6.18 (d, J1,2 = 8.4 Hz, 1H, 1-H), 6.47 (d, J2,1 = 8.4 Hz, 1H, 2-H), 7.20-7.26 (m, 9H, Tr(m,p)), 7.42-7.45 (m, 4H, Tr(o)). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp (b)), 9.3 (cPropCH), 17.7 (C-19), 22.5 (C-10), 24.9 (20CH3), 29.8 (20CH3, C-18), 32.2 (C-8), 33.4 (6-O-CH2CH2CH2O), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 60.1 and 62.0 (6-O-CH2CH2CH2OH and 6-O-CH2CH2CH2OH), 74.2 (C-20), 80.4 (C-6), 91.3 (TrCO), 96.7 (C-5), 117.9 (C-2), 122.8 (C-1), 127.2 (pCTr), 127.3 (mCTr), 129.3 (oCTr), 130.6 (C-11), 132.0 (C-12), 137.3 (C-3), 144.2 (TrC-1), 151.1 (C-4). C47H53NO5 (711.93).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(4-hydroxybutyl))-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(4-hydroxybutyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 25c, HB-TDDPN)
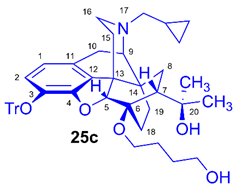
Compound 25c was synthesized from TBDPS-OB-TDDPN (24c, 486 mg, 0.50 mmol) using general procedure 3.2.2. CC: Kieselgel: 50 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 340 mg (93 %), mp. 105-116 °C, RfC = 0.09, RfD = 0.85, RfE = 0.25. 1H-NMR (CDCl3): δ = 0.05 (m, 2H, cPropCH2syn), 0.43 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.90 (td, 2J19anti,19syn = 13.8 Hz, 3J19anti,18syn = 5.4 Hz, 1H, 19-Hanti), 0.98 (dd, 2J8α,8β = 13.8 Hz, 3J8α,7β = 9.9 Hz, 1H, 8α-H), 1.16 (s, 3H, 20CH3), 1.35 (s, 3H, 20CH3), 1.37-1.46 (m, 2H, 15-Heq, 18-Hsyn), 1.51-1.60 (m, 5H, 6-O-CH2CH2CH2CH2OH and 18-Hanti), 1.83 (app t, 3J7β,8α = 10.0 Hz, 1H, 7β-H), 1.90 (td, 2J15ax,15eq = 13.7 Hz, 3J15ax,16eq = 5.5 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.7 Hz, 3J10α,9α = 6.4 Hz, 1H, 10α-H), 2.12-2.20 (m, 2H, 16-Hax, NCH2 (a)), 2.32 (dd, JBA =12.5 Hz, JBX = 5.7 Hz, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 4.7 Hz, 1H, 16-Heq), 2.77 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 12.8 Hz, 4J8β,19syn = 2.7 Hz, 1H, 8β-H), 2.85 (d, 2J10β,10α = 18.7 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H), 3.46-3.84 (m, 4H, 6-O-CH2CH2CH2CH2OH), 4.12 (d, 4J5β,18anti = 1.8 Hz, 1H, 5β-H), 5.28 (br s, 1H, 20-OH), 6.18 (d, J1,2 = 8.4 Hz, 1H, 1-H), 6.47 (d, J2,1 = 8.4 Hz, 1H, 2-H), 7.22-7.26 (m, 9H, Tr(m,p)), 7.43-7.44 (m, 6H, Tr(o)). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.7 (C-19), 22.5 (C-10), 24.8 (20-CH3), 26.9 and 29.3 (6-O-CH2CH2CH2CH2OH), 29.7 (C-18 and 20CH3), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 62.4 and 64.5 (6-O-(CH2)3CH2OH and 6-O-CH2(CH2)3OH), 74.3 (C-20), 80.3 (C-6), 91.4 (TrCO), 96.6 (C-5), 117.8 (C-2), 122.8 (C-1), 127.2 (pCTr), 127.3 (mCTr), 129.3 (oCTr), 130.6 (C-11), 132.0 (C-12), 137.2 (C-3), 144.2 (Tr-C1), 151.2 (C-4). C48H55NO5 (725.95).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(5-hydroxypentyl))-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(5-hydroxypentyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 25d, HPe-TDDPN)
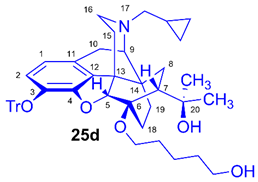
Compound 25d was synthesized from TBDPSOPe-TDDPN (24d, 562 mg, 0.57 mmol) using the general procedure 3.2.2. CC: Kieselgel: 70 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 385 mg (90 %), mp. 101-109 °C, RfA = 0.15, RfD = 0.49, RfE = 0.35. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.43 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.89 (td, 2J19anti,19syn = 13.2 Hz, 3J19anti,18syn = 5.9 Hz, 1H, 19-Hanti), 0.98 (dd, 2J8α,8β = 13.4 Hz, 3J8α7β = 9.6 Hz, 1H, 8α-H), 1.16 (s, 3H, 20CH3), 1.32-1.38 (m, 2H, (CH2)2CH2(CH2)2), 1.35 (s, 3H, 20CH3), 1.39-1.61 (m, 7H, 18-Hsyn, 15-Heq, 18-Hanti, 6-O-CH2CH2CH2CH2CH2OTBDPS), 1.82 (app t, 3J7β,8α = 10.0 Hz, 1H, 7β-H), 1.89 (td, 2J15ax,15eq = 13.7 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.3 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 12.7 Hz, 3J16ax,15eq = 3.7 Hz, 1H, 16-Hax), 2.18 (dd, JAB = 12.7 Hz, JAX = 6.7 Hz, 1H, NCH2 (a)), 2.32 (dd, JBA =12.7 Hz, JBX = 6.0 Hz, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 11.9 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.76 (ddd, 2J8β,8α = 13.4 Hz, 3J8β,7β = 12.2 Hz, 4J8β,19syn = 3.8 Hz, 1H, 8β-H ), 2.85 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.3 Hz, 1H, 9α-H), 3.42-3.83 (m, 4H, 6-O-CH2(CH2)3CH2OTBDPS), 4.11 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.36 (br s, 1H, 20-OH), 6.18 (d, J1,2 = 8.0 Hz, 1H, 1-H), 6.47 (d, J2,1 = 8.0 Hz, 1H, 2-H), 7.20-7.26 (m, 9H, Tr(m,p)), 7.42-7.44 (m, 6H, Tr(o)). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.6 (C-19), 22.2 ((CH2)2CH2(CH2)2), 22.5 (C-10), 24.8 (20-CH3), 29.7 and 29.8 (C-18 and 20CH3), 30.3 and 32.4 (6-O-CH2CH2CH2CH2CH2OH), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 62.6 and 64.6 (6-O-(CH2)4CH2OH and 6-O-CH2(CH2)4OH), 74.3 (C-20), 80.2 (C-6), 91.3 (TrCO), 96.7 (C-5), 117.8 (C-2), 122.9 (C-1), 127.2 (pCTr), 127.3 (mCTr), 129.4 (oCTr), 130.7 (C-11), 132.0 (C-12), 137.2 (C-3), 144.2 (Tr-C1), 151.3 (C-4). C49H57NO5 (739.98).
3.2.3. General Procedure for the Synthesis of 6-O-(Tosyloxyalkyl)-6-O-desmethyl-diprenorphine (26b–d) Derivatives
The corresponding 6-O-(hydroxyalkyl)-6-O-desmethyl-3-O-trityl-diprenorphine (25b–d, 0.67 mmol) derivative was dissolved in dry dichloromethane (10 mL) under argon. The solution was cooled to 0 °C and pyridine (220 μL, 215 mg, 2.72 mmol, 4 equiv.) was added. After stirring for 15 min, toluenesulfonic anhydride (750 mg, 2.29 mmol, 3.4 equiv.) was added in small portions. After stirring at RT for 4 h, the reaction mixture was poured into water (50 mL). The resulting suspension was extracted with dichloromethane (3 x 60 mL). The combined organic layer was dried with anhydrous Na2SO4 and the solvent removed under reduced pressure. The resulting crude product mixture was separated by column chromatography on silica gel.
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(3-(4-toluene sulfonyloxy)propyl)-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(3-tosyloxypropyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 26b, TP-TDDPN)
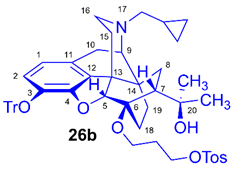
Compound 26b was prepared from HP-TDDPN (25b, 480 mg, 0.67 mmol) using the general procedure 3.2.3. CC: Kieselgel: 70 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 349 mg (60 %), mp. 87-102 °C, RfC = 0.28, RfD = 0.85, RfE = 0.61. 1H-NMR (CDCl3): δ = 0.05 (m, 2H, cPropCH2syn), 0.43 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.75 (m, 1H, cPropCH), 0.88 (td, 2J19anti,19syn = 12.4 Hz, 3J19anti,18syn = 5.7 Hz, 1H, 19-Hanti), 0.95 (dd, 2J8α,8β = 13.3 Hz, 3J8α7β = 9.7 Hz, 1H, 8α-H), 1.13 (s, 3H, 20CH3), 1.26 (s, 3H, 20CH3), 1.30-1.41 (m, 2H, 18-Hsyn, 15-Heq), 1.49 (m, 1H, 18-Hanti), 1.75 (app t, 3J7β,8α = 10.1 Hz, 1H, 7β-H), 1.79-1.84 (m, 2H, CH2CH2CH2OH), 1.86 (td, 2J15ax,15eq = 13.2 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.7 Hz, 3J10α,9α = 6.1 Hz, 1H, 10α-H), 2.14 (td, 2J16ax,16eq = 13.0 Hz, 3J16ax,15eq = 3.6 Hz, 1H, 16-Hax), 2.18 (dd, JAB = 12.9 Hz, JAX = 6.7 Hz, 1H, NCH2 (a)), 2.32 (dd, JBA =12.9 Hz, JBX = 5.6 Hz, NCH2 (b)), 2.41 (s, 3H, Tos-CH3), 2.54 (dd, 2J16eq,16ax = 11.9 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.74 (ddd, 2J8β,8α = 13.4 Hz, 3J8β,7β = 12.4 Hz, 4J8β,19syn = 3.9 Hz, 1H, 8β-H ), 2.85 (d, 2J10β,10α = 18.7 Hz, 1H, 10β-H), 2.89 (d, 3J9α,10α = 6.1 Hz, 1H, 9α-H), 3.49-4.12 (m, 4H, 6-O-CH2CH2CH2OH, 6-O-CH2CH2CH2OH), 3.99 (d, 4J5β,18anti = 2.3 Hz, 1H, 5β-H), 4.83 (br s, 1H, 20-OH), 6.20 (d, J1,2 = 8.4 Hz, 1H, 1-H), 6.49 (d, J2,1 = 8.4 Hz, 1H, 2-H), 7.21-7.24 (m, 9H, Tr(m,p)), 7.31 (d, J = 8.1 Hz, 2H, Tos-3,5), 7.39-7.42 (m, 4H, Tr(o)), 7.78 (d, J = 8.1 Hz, 2H, Tos-2,6). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp (b)), 9.3 (cPropCH), 17.4 (C-18), 21.6 (Tos-CH3), 22.5 (C-10), 24.8 (20CH3), 29.7 (20CH3), 30.3 (C-19), 32.1 (C-8), 33.4 (6-O-CH2CH2CH2O), 35.4 (C-15), 35.7 (C-14), 43.5 (C-16), 46.8 (C-13), 48.1 (C-7), 57.9 (C-9), 59.8 (NCH2), 60.4 and 67.4 (6-O-CH2CH2CH2OH and 6-O-CH2CH2CH2OH), 74.2 (C-20), 80.8 (C-6), 91.5 (TrCO), 96.2 (C-5), 118.0 (C-2), 123.2 (C-1), 127.3 (pCTr), 127.4 (mCTr), 127.9 (Tos-2,6), 129.4 (oCTr), 129.8 (Tos-3,5), 130.7 (C-11), 131.9 (C-12), 133.1 (Tos-C1), 137.2 (C-3), 144.2 (TrC-1), 144.6 (Tos-C4), 151.2 (C-4). Calculated for C54H59NO7S (865.4012), found: 866.4104 ([M+H]+).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(4-(4-toluenesulfonyloxy)butyl))-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(4-tosyloxybutyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 26c, TB-TDDPN)
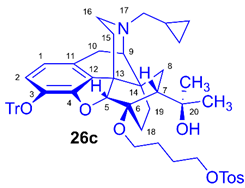
Compound 26c was synthesized from HB-TDDPN (25c, 320 mg, 0.44 mmol) using the general procedure 3.2.3. CC: Kieselgel: 60 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 337 mg (87 %), mp. 80-98 °C, RfC = 0.38, RfD = 0.85, Rf E = 0.63. 1H-NMR (CDCl3): δ = 0.05 (m, 2H, cPropCH2syn), 0.41 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.75 (m, 1H, cPropCH), 0.88 (td, 2J19anti,19syn = 13.6 Hz, 3J19anti,18syn = 5.7 Hz, 1H, 19-Hanti), 0.96 (dd, 2J8α,8β = 13.3 Hz, 3J8α7β = 9.7 Hz, 1H, 8α-H), 1.14 (s, 3H, 20CH3), 1.31 (s, 3H, 20CH3), 1.32-1.37 (m, 1H, 18-Hsyn), 1.42 (dd, 2J15eq,15ax = 13.1 Hz, 3J15eq,16ax = 2.7 Hz, 1H, 15-Heq), 1.40-1.63 (m, 5H, 6-O-CH2CH2CH2CH2OTos and 18-Hanti), 1.78 (app t, 3J7β,8α = 9.4 Hz, 1H, 7β-H), 1.87 (td, 2J15ax,15eq = 13.9 Hz, 3J15ax,16eq = 5.6 Hz, 1H, 15-Hax), 2.04 (dd, 2J10α,10β = 18.8 Hz, 3J10α,9α = 6.0 Hz, 1H, 10α-H), 2.14 (td, 2J16ax,16eq = 11.5 Hz, 3J16ax,15eq = 3.8 Hz, 1H, 16-Hax), 2.17 (dd, JAB =12.8 Hz, JAX = 6.7 Hz, NCH2 (a)), 2.32 (dd, JBA =12.8 Hz, JBX = 5.7 Hz, NCH2 (b)), 2.42 (s, 3H, Tos-CH3), 2.54 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 5.4 Hz, 1H, 16-Heq), 2.75 (ddd, 2J8β,8α = 13.8 Hz, 3J8β,7β = 12.4 Hz, 4J8β,19syn = 3.9 Hz, 1H, 8β-H), 2.84 (d, 2J10β,10α = 18.8 Hz, 1H, 10β-H), 2.89 (d, 3J9α,10α = 6.0 Hz, 1H, 9α-H), 3.36-3.99 (m, 4H, 6-O-CH2CH2CH2CH2OTos), 4.05 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.07 (br s, 1H, 20-OH), 6.18 (d, J1,2 = 8.0 Hz, 1H, 1-H), 6.46 (d, J2,1 = 8.0 Hz, 1H, 2-H), 7.19-7.24 (m, 9H, Tr(m,p)), 7.33 (d, J = 8.1 Hz, 2H, Tos-3,5), 7.38-7.42 (m, 6H, Tr(o)), 7.79 (d, J = 8.1 Hz, 2H, Tos-2,6). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.6 (C-18), 21.6 (Tos-CH3), 22.5 (C-10), 24.9 (20-CH3), 25.7 and 26.7 (6-O-CH2CH2CH2CH2OTos), 29.7 (C-19 and 20CH3), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 57.9 (C-9), 59.8 (NCH2), 64.0 and 70.2 (6-O-CH2CH2CH2CH2OTos and 6-O-CH2CH2CH2CH2OTos), 74.2 (C-20), 80.5 (C-6), 91.4 (TrCO), 96.5 (C-5), 117.9 (C-2), 122.9 (C-1), 127.3 (pCTr), 127.4 (mCTr), 127.9 (Tos-2,6), 129.3 (oCTr), 129.8 (Tos-3,5), 130.6 (C-11), 131.9 (C-12), 133.1 (Tos-C1), 137.2 (C-3), 144.2 (TrC-1), 144.6 (Tos-C4), 151.2 (C-4). Calculated for C55H61NO7S (879.4169), found 880.4255 ([M+H]+).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(5-(4-toluenesulfonyloxy)pentyl))-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(5-tosyloxypentyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 26d, TPe-TDDPN)
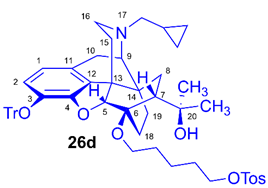
Compound 26d was synthesized from HPe-TDDPN (25d, 380 mg, 0.51 mmol) using the general procedure 3.2.3. CC: Kieselgel: 60 g, eluent: dichloromethane-methanol 98:2 (v/v), fractions: 25 mL. Yield: 311 mg (67 %), mp. 77-88 °C, RfC = 0.50, RfD = 0.87, RfE = 0.70. 1H-NMR (CDCl3): δ = 0.05 (m, 2H, cPropCH2syn), 0.42 (tt, J = 12.7 Hz, J = 5.7 Hz, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.75 (m, 1H, cPropCH), 0.88 (td, 2J19anti,19syn = 13.3 Hz, 3J19anti,18syn = 5.9 Hz, 1H, 19-Hanti), 0.97 (dd, 2J8α,8β = 13.1 Hz, 3J8α7β = 9.8 Hz, 1H, 8α-H), 1.14 (s, 3H, 20CH3), 1.32 (s, 3H, 20CH3), 1.26-1.62 (m, 9H, 18-Hsyn, 15-Heq, 6-O-CH2CH2CH2CH2CH2OTos and 18-Hanti), 1.80 (app t, 3J7β,8α = 9.7 Hz, 1H, 7β-H), 1.89 (td, 2J15ax,15eq = 13.7 Hz, 3J15ax,16eq = 5.7 Hz, 1H, 15-Hax), 2.04 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.4 Hz, 1H, 10α-H), 2.14 (td, 2J16ax,16eq = 13.2 Hz, 3J16ax,15eq = 3.8 Hz, 1H, 16-Hax), 2.18 (dd, JAB =12.6 Hz, JAX = 6.7 Hz, NCH2 (a)), 2.32 (dd, JBA =12.6 Hz, JBX = 5.7 Hz, NCH2 (b)), 2.43 (s, 3H, Tos-CH3), 2.55 (dd, 2J16eq,16ax = 12.1 Hz, 3J16eq,15ax = 4.7 Hz, 1H, 16-Heq), 2.76 (ddd, 2J8β,8α = 13.1 Hz, 3J8β,7β = 11.9 Hz, 4J8β,19syn = 3.9 Hz, 1H, 8β-H), 2.85 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 2.89 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H), 3.36-3.98 (m, 4H, 6-O-CH2(CH2)3CH2OTos), 4.08 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.18 (br s, 1H, 20-OH), 6.18 (d, J1,2 = 8.4 Hz, 1H, 1-H), 6.46 (d, J2,1 = 8.4 Hz, 1H, 2-H), 7.18-7.22 (m, 9H, Tr(m,p)), 7.33 (d, J = 8.4 Hz, 2H, Tos-3,5), 7.39-7.44 (m, 6H, Tr(o)), 7.79 (d, J = 8.4 Hz, 2H, Tos-2,6). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.6 (C-19), 21.6 (Tos-CH3), 22.0 (6-O-O(CH2)2CH2(CH2)2OTos), 22.5 (C-10), 24.8 (20-CH3), 29.7 and 29.8 (C-18 and 20CH3), 28.6 and 30.0 (6-O-CH2CH2CH2CH2CH2OTos), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 57.9 (C-9), 59.8 (NCH2), 64.3 and 70.3 (6-O-(CH2)4CH2OTos and 6-O-CH2(CH2)4OTos), 74.2 (C-20), 80.3 (C-6), 91.4 (TrCO), 96.6 (C-5), 117.8 (C-2), 122.8 (C-1), 127.2 (pCTr), 127.3 (mCTr), 127.9 (Tos-2,6), 129.3 (oCTr), 129.8 (Tos-3,5), 130.6 (C-11), 132.0 (C-12), 133.2 (Tos-C1), 137.2 (C-3), 144.2 (TrC-1), 144.6 (Tos-C4), 151.2 (C-4). Calculated for C56H63NO7S (893.4325), found: 894.4405 ([M+H]+).
3.2.4. General Procedure for the Preparation of 6-O-Fluoroalkyl-6-O-desmethyl-3-O-trityl-diprenorphine (27b–d) Derivatives
A solution of TDDPN (23, CAS RN: 157891-92-4, 650 mg, 1 mmol) in dry N,N-dimethylformamide (3 ml) was added dropwise to a suspension of sodium hydride (60 % dispersion in mineral oil, 50 mg, 1.2 mmol, 1.2 equiv.) in anhydrous N,N-dimethylformamide (3 mL) under argon atmosphere. The mixture was stirred at RT for 15 min. Then a solution of the appropriate fluoroalkyl toluenesulfonate (33a–c, 1.6 mmol) in anhydrous N,N-dimethylformamide (3 mL) was added via syringe, and the mixture was stirred at RT for 20 h. The product mixture was poured into water (30 mL) and the suspension was extracted with dichloromethane (4 x 70 mL). The combined organic extracts were washed with brine (40 mL) and dried over anhydrous Na2SO4. The resulting crude prodcut was purified by column chromatography on silica gel.
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(3-fluoropropyl)-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(3-fluoropropyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 27b, FP-TDDPN)
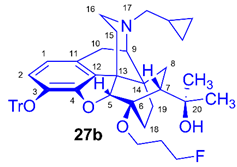
Compound 27b was synthesized from TDDPN (23, 650 mg, 1 mmol) using the general procedure 3.2.4. CC: Kieselgel: 60 g, eluent: hexane-ethyl acetate 7:3 to 6:4 (v/v), fractions: 25 mL. Yield: 310 mg (43 %), mp. 80-105 °C, RfA = 0.42, RfB = 0.18, RfD = 0.83. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.42 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.90 (td, 2J19anti,19syn = 12.6 Hz, 3J19anti,18syn = 5.8 Hz, 1H, 19-Hanti), 0.98 (dd, 2J8α,8β = 13.3 Hz, 3J8α,7β = 9.7 Hz, 1H, 8α-H), 1.16 (s, 3H, 20-CH3), 1.35 (s, 3H, 20-CH3), 1.38 (td, 2J18syn,18anti = 13.9 Hz, 3J18syn,19anti = 5.3 Hz, 1H, 18-Hsyn); 1.43 (dd, 2J15eq,15ax = 13.0 Hz, 3J15eq,16ax = 3.7 Hz, 1H, 15-Heq), 1.55-1.60 (m, 1H, 15-Hax), 1.84 (m, 1H, 7β-H), 1.85-1.92 (m, 3H, 18-Hanti, CH2CH2CH2F(b,b’)), 2.05 (dd, 2J10α,10β = 18.3 Hz, 3J10α,9α = 6.3 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 11.7 Hz, 3J16ax,15eq = 3.7 Hz, 1H, 16-Hax), 2.19 (dd, JBA = 12.6 Hz, JBX = 6.8 Hz, 1H, NCH2 (a)), 2.32 (dd, JBA = 12.7 Hz, JBX = 5.7 Hz, 1H, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 11.7 Hz, 3J16eq,15ax = 4.9 Hz, 1H, 16-Heq), 2.77 (ddd, 2J8β,8α = 13.4 Hz, 3J8β,7β = 11.7 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H), 2.86 (d, 2J10β,10α = 18.3 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.3 Hz, 1H, 9α-H), 3.60 (dt, J = 9.5 Hz, 6.5 Hz, 1H, CH2CH2CH2F (a)), 3.89 (dt, J = 9.6 Hz, 5.9 Hz, 1H, CH2CH2CH2F (a’)), 4.10 (d, 4J5β,18anti = 1.9 Hz, 1H, 5β-H), 4.42 (m, 1H, CH2CH2CH2F(c)), 4.51 (m, 1H, CH2CH2CH2F(c’), 5.08 (br s, 1H, 20-OH), 6.20 (d, 3J1,2 = 8.1 Hz, 1H, 1-H), 6.49 (d, 3J2,1 = 8.1 Hz, 1H, 2-H), 7.20-7.26 (m, 9H, Tr(m,p)), 7.40-7.45 (m, 6H, oTr). 13C-NMR (CDCl3): δ = 3.2 (cProp(a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.5 (C-18), 22.5 (C-10), 24.8 (20-CH3), 29.7 (20-CH3), 29.8 (C-19), 31.7 (d, 2JC,F = 20.2 Hz, CH2CH2CH2F), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.8 (C-13), 48.1 (C-7), 57.9 (C-9), 59.8 (NCH2), 61.0 (d, 3JC,F = 5.5 Hz, CH2CH2CH2F), 74.2 (C-20), 80.6 (C-6), 81.2 (d, 1JC,H = 165.9 Hz, CH2CH2CH2F), 91.4 (TrCO), 96.5 (C-5), 117.9 (C-2), 123.0 (C-1), 127.2 (pCTr), 127.3 (mCTr), 129.4 (oCTr), 130.7 (C-11), 131.9 (C-12), 137.2 (C-3), 144.2 (Tr-C1), 151.3 (C-4). 19F-NMR δ = - 220.7 dq (J = 26.1 Hz, 48.1 Hz). C47H52FNO4 (713.92).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(4-fluorobutyl)-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(4-fluorobutyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 27c, FB-TDDPN)
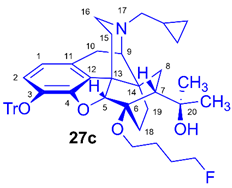
Compound 27c was synthesized from TDDPN (23, 650 mg, 1 mmol) using general procedure 3.2.4. CC: Kieselgel: 60 g, eluent: hexane-ethyl acetate 6:4 (v/v), fractions: 25 mL. Yield: 440 mg (60 %), mp. 93-102 °C, RfA = 0.75, RfB = 0.22, RfD = 0.88. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.43 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.90 (td, 2J19anti,19syn = 12.6 Hz, 3J19anti,18syn = 5.7 Hz, 1H, 19-Hanti), 0.98 (dd, 2J8α,8β = 13.6 Hz, 3J8α,7β = 9.7 Hz, 1H, 8α-H), 1.16 (s, 3H, 20-CH3), 1.35 (s, 3H, 20-CH3), 1.40 (td, 2J18syn,18anti = 13.2 Hz, 3J18syn,19anti = 5.4 Hz, 1H, 18-Hsyn); 1.44 (dd, 2J15eq,15ax = 13.2 Hz, 3J15eq,16ax = 2.6 Hz, 1H, 15-Heq), 1.57-1.72 (m, 5H, 18-Hanti, CH2CH2CH2CH2F(b,b’), CH2CH2CH2CH2F(c,c’),), 1.83 (m, 1H, 7β-H), 1.90 (td, 2J15ax,15eq = 12.8 Hz, 3J15ax,16eq = 5.5 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.5 Hz, 3J10α,9α = 6.3 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 10.4 Hz, 3J16ax,15eq = 3.7 Hz, 1H, 16-Hax), 2.18 (dd, JBA = 12.9 Hz, JBX = 6.7 Hz, 1H, NCH2 (a)), 2.33 (dd, JBA = 12.9 Hz, JBX = 5.8 Hz, 1H, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 4.7 Hz, 1H, 16-Heq), 2.77 (ddd, 2J8β,8α = 13.6 Hz, 3J8β,7β = 9.9 Hz, 4J8β,19syn = 3.8 Hz, 1H, 8β-H), 2.86 (d, 2J10β,10α = 18.5 Hz, 1H, 10β-H), 2.90 (d, 3J9α,10α = 6.3 Hz, 1H, 9α-H), 3.48 (dt, J = 9.5 Hz, 6.5 Hz, 1H, CH2CH2CH2CH2F (a)), 3.83 (dt, J = 9.6 Hz, 5.9 Hz, 1H, CH2CH2CH2CH2F (a’)), 4.11 (d, 4J5β,18anti = 2.0 Hz, 1H, 5β-H), 4.35 (m, 1H, CH2CH2CH2CH2F(d)), 4.48 (m, 1H, CH2CH2CH2CH2F(d’), 5.21 (br s, 1H, 20-OH), 6.19 (d, 3J1,2 = 8.0 Hz, 1H, 1-H), 6.47 (d, 3J2,1 = 8.0 Hz, 1H, 2-H), 7.21-7.24 (m, 9H, Tr(m,p)), 7.41-7.44 (m, 6H, oTr). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.7 (C-18), 22.5 (C-10), 24.9 (20-CH3), 26.4 (d, 3JC,F = 4.6 Hz, CH2CH2CH2CH2F), 27.2 (d, 2JC,F = 20.2 Hz, CH2CH2CH2CH2F), 29.7 and 29.8 (20-CH3 and C-19), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 64.2 (d, 4JC,F = 1.9 Hz, CH2CH2CH2CH2F), 74.2 (C-20), 80.4 (C-6), 83.7 (d, 1JC,H = 165.1 Hz, CH2CH2CH2CH2F), 91.4 (TrCO), 96.6 (C-5), 117.9 (C-2), 122.9 (C-1), 127.2 (pCTr), 127.3 (mCTr), 129.3 (oCTr), 130.6 (C-11), 132.0 (C-12), 137.2 (C-3), 144.2 (Tr-C1), 151.2 (C-4). 19F-NMR δ = - 218.5 ddd (J = 25.2 Hz, 47.4 Hz, 72.8 Hz). C48H54FNO4 (727.95).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(5-fluoropentyl)-18,19-dihydro-α,α-dimethyl-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (6-O-(5-fluoropentyl)-6-O-desmethyl-3-O-trityl-diprenorphine, 27d, FPe-TDDPN)
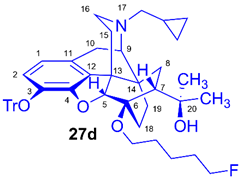
Compound 27d was synthesized from TDDPN (23, 650 mg, 1 mmol) using the general procedure 3.2.4. CC: Kieselgel: 50 g, eluent: hexane-ethyl acetate 6:4 (v/v), fractions: 25 mL. Yield: 487 mg (66 %), mp. 82-92 °C, RfA = 0.87, RfB = 0.20, RfD = 0.90. 1H-NMR (CDCl3): δ = 0.06 (m, 2H, cPropCH2syn), 0.41 (m, 1H, 19-Hsyn), 0.46 (m, 2H, cPropCH2anti), 0.76 (m, 1H, cPropCH), 0.90 (td, 2J19anti,19syn = 12.5 Hz, 3J19anti,18syn = 5.8 Hz, 1H, 19-Hanti), 0.98 (dd, 2J8α,8β = 13.1 Hz, 3J8α,7β = 9.6 Hz, 1H, 8α-H), 1.16 (s, 3H, 20-CH3), 1.35 (s, 3H, 20-CH3), 1.37-1.70 (m, 9H, 18-Hsyn, 15-Heq, 18-Hanti, (CH2)3), 1.83 (app t, 3J7β,8α = 10.0 Hz, 1H, 7β-H), 1.90 (td, 2J15ax,15eq = 12.7 Hz, 3J15ax,16eq = 5.5 Hz, 1H, 15-Hax), 2.05 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.4 Hz, 1H, 10α-H), 2.15 (td, 2J16ax,16eq = 13.6 Hz, 3J16ax,15eq = 3.5 Hz, 1H, 16-Hax), 2.18 (dd, JBA = 12.7 Hz, JBX = 6.7 Hz, 1H, NCH2 (a)), 2.32 (dd, JBA = 12.7 Hz, JBX = 5.7 Hz, 1H, NCH2 (b)), 2.55 (dd, 2J16eq,16ax = 11.9 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.77 (ddd, 2J8β,8α = 13.1 Hz, 3J8β,7β = 12.1 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H), 2.85 (d, 2J10β,10α = 18.4 Hz, 1H, 10α-H), 2.90 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H), 3.45 (dt, J = 9.1 Hz, 6.3 Hz, 1H, 6-O-CH2(CH2)4F (a)), 3.81 (dt, J = 9.1 Hz, 6.3 Hz, 1H, 6-O-CH2(CH2)4F (a’)), 4.11 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 4.35 (t, J = 6.1 Hz, 1H, CH2F (a)), 4.45 (t, J = 6.1 Hz, 2H, CH2F (a’)), 5.27 (br s, 1H, 20-OH), 6.18 (d, 3J1,2 = 8.0 Hz, 1H, 1-H), 6.47 (d, 3J2,1 = 8.0 Hz, 1H, 2-H), 7.20-7.25 (m, 9H, Tr(m,p)), 7.41-7.45 (m, 6H, oTr). 13C-NMR (CDCl3): δ = 3.2 (cProp (a)), 4.1 (cProp(b)), 9.3 (cPropCH), 17.7 (C-18), 21.8 (d, 3JC,F = 5.5 Hz, (CH2)2CH2(CH2)2F), 22.5 (C-10), 24.8 (20-CH3), 29.7 (20-CH3), 29.8 (C-19), 30.1 (d, 2JC,F = 20.2 Hz, (CH2)3CH2CH2F), 30.3 (6-O-CH2CH2(CH2)3F), 32.1 (C-8), 35.4 (C-15), 35.7 (C-14), 43.6 (C-16), 46.7 (C-13), 48.0 (C-7), 58.0 (C-9), 59.8 (NCH2), 64.5 (6-O-CH2(CH2)4F), 74.2 (C-20), 80.3 (C-6), 83.9 (d, 1JC,H = 164.8 Hz, (CH2)4CH2F), 91.4 (TrCO), 96.7 (C-5), 117.8 (C-2), 122.9 (C-1), 127.2 (pCTr), 127.3 (mCTr), 129.4 (oCTr), 130.7 (C-11), 132.0 (C-12), 137.2 (C-3), 144.2 (Tr-C1), 151.3 (C-4). 19F-NMR δ = - 218.0 m. C49H56FNO4 (741.97).
3.2.5. General Procedure for the Preparation of 6-O-Fluoroalkyl-6-O-desmethyl-diprenorphine (28b–d) Derivatives
6-O-Fluoroalkyl-6-O-desmethyl-3-O-trityl-diprenorphine derivative (27b–d, 0.35 mmol) was dissolved in a mixture of acetic acid (27 mL) and water (7 mL) under argon atmosphere. The solution was stirred at 100 °C for 5 min. The reaction mixture was cooled to RT and then poured into ice-water (40 mL). The solution was basified with a 25 mass % aqueous ammonia solution (pH 9). The mixture was extracted with dichloromethane (4 x 50 mL). The combined organic phase was dried over Na2SO4 and the solvent was removed under reduced pressure. The resulting crude product was purified by column chromatography on silica gel.
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(3-fluoropropyl)-18,19-dihydro-α,α-dimethyl-6,14-ethenomorphinan-7-methanol (6-O-(3-fluoropropyl)-6-O-desmethyl-diprenorphine, 28b, FP-DPN)
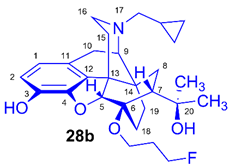
Compound 28b was synthesized from FP-TDDPN (27b, 250 mg, 0.35 mmol) using the general procedure 3.2.5. CC: Kieselgel: 50 g, eluent: hexane-ethyl acetate 7:3 (v/v), fractions: 25 mL. Yield: 129 mg (78 %), mp. 98-115 °C, RfA = 0.58, RfB = 0.21, RfC = 0.28. 1H-NMR (CDCl3): δ = 0.10 (m, 2H, cPropCH2syn), 0.49 (m, 2H, cPropCH2anti), 0.74 (m, 1H, 19-Hsyn), 0.80 (m, 1H, cPropCH), 1.02-1.10 (m, 2H, 19-Hanti, 8α-H), 1.20 (s, 3H, 20-CH3), 1.39 (s, 3H, 20-CH3), 1.67 (dd, 2J15eq,15ax = 13.1 Hz, 3J15eq,16ax = 2.7 Hz, 1H, 15-Heq), 1.76-1.79 (m, 2H, 18-Hsyn, 18-Hanti), 1.89-2.05 (m, 4H, 7β-H, 10α-H, CH2CH2CH2F(b,b’)), 2.18-2.30 (m, 3H, NCH2 (a), 15-Hax, 16-Hax), 2.37 (dd, JBA = 12.6 Hz, JBX = 5.7 Hz, 1H, NCH2 (b)), 2.63 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 5.4 Hz, 1H, 16-Heq), 2.87 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 12.0 Hz, 4J8β,19syn = 4.0 Hz, 1H, 8β-H), 2.98 (d, 2J10β,10α = 18.7 Hz, 1H, 10β-H), 3.01 (d, 3J9α,10α = 6.7 Hz, 1H, 9α-H), 3.78 (dt, J = 9.3 Hz, 6.3 Hz, 1H, CH2CH2CH2F (a)), 4.07 (dt, J = 9.3 Hz, 5.9 Hz, 1H, CH2CH2CH2F (a’)), 4.42 (d, 4J5β,18anti = 1.2 Hz, 1H, 5β-H), 4.51 (m, 1H, CH2CH2CH2F(c)), 4.60 (m, 1H, CH2CH2CH2F(c’), 4.94 (br s, 1H, 3-OH), 5.13 (br s, 1H, 20-OH), 6.50 (d, 3J1,2 = 8.0 Hz, 1H, 1-H), 6.69 (d, 3J2,1 = 8.0 Hz, 1H, 2-H). 13C-NMR (CDCl3): δ = 3.3 (cProp (a)), 4.2 (cProp(b)), 9.4 (cPropCH), 17.8 (C-18), 22.6 (C-10), 24.8 (20-CH3), 29.7 (20-CH3), 29.9 (C-19), 31.6 (d, 2JC,F = 20.2 Hz, CH2CH2CH2F), 32.3 (C-8), 35.5 (C-15), 35.9 (C-14), 43.7 (C-16), 47.3 (C-13), 48.2 (C-7), 58.1 (C-9), 59.8 (NCH2), 60.8 (d, 3JC,F = 4.6 Hz, CH2CH2CH2F), 74.4 (C-20), 80.8 (C-6), 81.1 (d, 1JC,H = 165.3 Hz, CH2CH2CH2F), 97.6 (C-5), 116.3 (C-2), 119.5 (C-1), 128.3 (C-11), 132.3 (C-12), 137.2 (C-3), 145.4 (C-4). 19F-NMR δ = - 221.1 tt (J = 26.4 Hz, 47.2 Hz). Calculated for C28H38FNO4 (471.2785), found: 472.2866 ([M+H]+).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(4-fluorobutyl)-18,19-dihydro-α,α-dimethyl-3-hydroxy-6,14-ethenomorphinan-7-methanol (6-O-(4-fluorobutyl)-6-O-desmethyl-diprenorphine, 28c, FB-DPN)
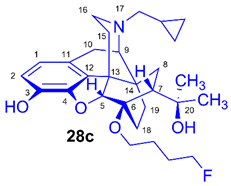
Compound 28c was synthesized from FB-TDDPN (27c, 400 mg, 0.55 mmol) using the general procedure 3.2.5. Column chromatography: Kieselgel: 50 g, eluent: hexane-ethyl acetate 6:4 (v/v), fractions: 25 mL. Yield: 190 mg (71 %), mp. 81-96 °C, RfA = 0.60, RfB = 0.13, RfC = 0.38. 1H-NMR (CDCl3): δ = 0.09 (m, 2H, cPropCH2syn), 0.49 (m, 2H, cPropCH2anti), 0.73 (m, 1H, 19-Hsyn), 0.80 (m, 1H, cPropCH), 1.01-1.09 (m, 2H, 19-Hanti, 8α-H), 1.20 (s, 3H, 20-CH3), 1.39 (s, 3H, 20-CH3), 1.66 (dd, 2J15eq,15ax = 13.3 Hz, 3J15eq,16ax = 2.5 Hz, 1H, 15-Heq), 1.77-1.82 (m, 6H, 18-Hsyn, 18-Hanti, CH2CH2CH2CH2F(b,b’), CH2CH2CH2CH2F(c,c’),), 1.94 (app t, 1H, 7β-H), 2.03 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.5 Hz, 1H, 10α-H), 2.17-2.30 (m, 3H, 15-Hax, 16-Hax, NCH2 (a)), 2.37 (dd, JBA = 12.7 Hz, JBX = 6.0 Hz, 1H, NCH2 (b)), 2.63 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.86 (ddd, 2J8β,8α = 13.7 Hz, 3J8β,7β = 12.2 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H), 2.97 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 3.01 (d, 3J9α,10α = 6.5 Hz, 1H, 9α-H), 3.67 (dt, J = 9.3 Hz, 6.1 Hz, 1H, CH2CH2CH2CH2F (a)), 3.99 (dt, J = 9.3 Hz, 6.1 Hz, 1H, CH2CH2CH2CH2F (a’)), 4.40 (d, 4J5β,18anti = 1.3 Hz, 1H, 5β-H), 4.42 (m, 1H, CH2CH2CH2CH2F(d)), 4.51 (m, 1H, CH2CH2CH2CH2F(d’), 5.13 (br s, 1H, 3-OH), 5.30 (br s, 1H, 20-OH), 6.50 (d, 3J1,2 = 8.0 Hz, 1H, 1-H), 6.68 (d, 3J2,1 = 8.0 Hz, 1H, 2-H). 13C-NMR (CDCl3): δ = 3.3 (cProp (a)), 4.1 (cProp(b)), 9.4 (cPropCH), 17.9 (C-18), 22.6 (C-10), 24.8 (20-CH3), 26.6 (d, 3JC,F = 4.6 Hz, CH2CH2CH2CH2F), 27.3 (d, 2JC,F = 20.1 Hz, CH2CH2CH2CH2F), 29.7 and 29.9 (20-CH3 and C-19), 32.2 (C-8), 35.3 (C-15), 35.9 (C-14), 43.7 (C-16), 47.3 (C-13), 48.1 (C-7), 58.1 (C-9), 59.8 (NCH2), 64.2 (d, 4JC,F = 1.0 Hz, CH2CH2CH2CH2F), 74.5 (C-20), 80.6 (C-6), 83.8 (d, 1JC,F = 165.0 Hz, CH2CH2CH2CH2F), 97.6 (C-5), 116.3 (C-2), 119.4 (C-1), 128.2 (C-11), 132.3 (C-12), 137.2 (C-3), 145.4 (C-4). 19F-NMR δ = - 218.4 ddd (J = 25.2 Hz, 47.9 Hz, 93.6 Hz). Calculated for C29H40FNO4 (485.2941), found: 486.3023 ([M+H]+).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(5-fluoropentyl)-18,19-dihydro-α,α-dimethyl-3-hydroxy-6,14-ethenomorphinan-7-methanol (6-O-(5-fluoropentyl)-6-O-desmethyl-diprenorphine, 28d, FPe-DPN)
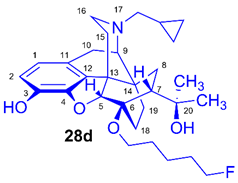
Compound 28d was prepared from FPe-TDDPN (27d, 430 mg, 0.58 mmol) using the general procedure 3.2.5. CC: Kieselgel: 50 g, eluent: hexane-ethyl acetate 6:4 (v/v), fractions: 25 mL. Yield: 220 mg (76 %), mp. 81-90 °C, RfA = 0.67, RfB = 0.09, RfC = 0.43. 1H-NMR (CDCl3): δ = 0.09 (m, 2H, cPropCH2syn), 0.49 (m, 2H, cPropCH2anti), 0.73 (m, 1H, 19-Hsyn), 0.80 (m, 1H, cPropCH), 1.01-1.09 (m, 2H, 19-Hanti, 8α-H), 1.20 (s, 3H, 20-CH3), 1.39 (s, 3H, 20-CH3), 1.46-1.79 (m, 9H, 18-Hsyn, 15-Heq, 18-Hanti, (CH2)3), 1.93 (app t, 3J7β,8α = 10.0 Hz, 1H, 7β-H), 2.03 (td, 2J15ax,15eq = 13.8 Hz, 3J15ax,16eq = 5.7 Hz, 1H, 15-Hax), 2.20 (dd, 2J10α,10β = 18.7 Hz, 3J10α,9α = 6.7 Hz, 1H, 10α-H), 2.22-2.30 (m, 2H, 16-Hax, NCH2 (a)), 2.37 (dd, JBA = 12.6 Hz, JBX = 6.0 Hz, 1H, NCH2 (b)), 2.63 (dd, 2J16eq,16ax = 12.1 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.86 (ddd, 2J8β,8α = 13.8 Hz, 3J8β,7β = 12.1 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H), 2.97 (d, 2J10β,10α = 18.7 Hz, 1H, 10β-H), 3.00 (d, 3J9α,10α = 6.7 Hz, 1H, 9α-H), 3.63 (dt, J = 8.9 Hz, 6.4 Hz, 1H, 6-O-CH2(CH2)4F (a)), 3.94 (dt, J = 8.9 Hz, 6.4 Hz, 1H, 6-O-CH2(CH2)4F (a’)), 4.38 (t, J = 6.0 Hz, 1H, CH2F (a)), 4.40 (d, 4J5β,18anti = 1.1 Hz, 1H, 5β-H), 4.48 (t, J = 6.0 Hz, 2H, CH2F (a’)), 5.10 (br s, 1H, 3-OH), 5.34 (br s, 1H, 20-OH), 6.49 (d, 3J1,2 = 8.0 Hz, 1H, 1-H), 6.68 (d, 3J2,1 = 8.0 Hz, 1H, 2-H). 13C-NMR (CDCl3): δ = 3.3 (cProp (a)), 4.1 (cProp(b)), 9.4 (cPropCH), 17.7 (C-18), 22.0 (d, 3JC,F = 5.5 Hz, (CH2)2CH2(CH2)2F), 22.6 (C-10), 24.8 (20-CH3), 29.7 (20-CH3), 29.9 (C-19), 30.1 (d, 2JC,F = 20.3 Hz, (CH2)3CH2CH2F), 30.2 (6-O-CH2CH2(CH2)3F), 32.2 (C-8), 35.5 (C-15), 35.9 (C-14), 43.7 (C-16), 47.2 (C-13), 48.0 (C-7), 58.1 (C-9), 59.8 (NCH2), 64.5 (6-O-CH2(CH2)4F), 74.5 (C-20), 80.5 (C-6), 83.9 (d, 1JC,H = 164.2 Hz, (CH2)4CH2F), 97.7 (C-5), 116.3 (C-2), 119.4 (C-1), 128.3 (C-11), 132.3 (C-12), 137.2 (C-3), 145.5 (C-4). 19F-NMR δ = - 218.0 m. Calculated for C30H42FNO4 (499.3098), found: 500.3180 ([M+H]+).
3.2.6. General Procedure for the Preparation of 6-O-(Hydroxyalkyl)-6-O-desmethyl-diprenorphine (29b–d) Derivatives
6-O-Hydroxyalkyl-6-O-desmethyl-3-O-trityl-diprenorphine (25b–d) derivatives were dissolved in a 4:1 (v/v) acetic acid-water mixture (1 mg substrate (25b–d) in 140 μL solvent). The reaction mixture was stirred at 100 °C for 10 min. The absence of the starting material was confirmed by analytical TLC. The solution was cooled to RT, poured into ice-water, and basified with a 25 mass % aqueous ammonia solution (pH = 9). The product was extracted with dichloromethane. The combined organic layer was washed with brine, dried with anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel.
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(3-hydroxypropyl)-18,19-dihydro-α,α-dimethyl-3-hydroxy-6,14-ethenomorphinan-7-methanol (6-O-(3-hydroxypropyl)-6-O-desmethyl-diprenorphine, 29b, HP-DPN)
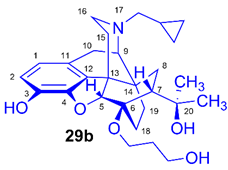
Compound 29b was prepared from FP-TDDPN (25b, 235 mg, 0.33 mmol) using the general procedure 3.2.6. CC: Kieselgel: 30 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 120 mg (77 %), mp. 208-210 °C (dec.), RfA = 0.33, RfD = 0.68, RfE = 0.14. 1H-NMR (CDCl3): δ = 0.09 (m, 2H, cPropCH2syn), 0.49 (m, 2H, cPropCH2anti), 0.73 (m, 1H, 19-Hsyn), 0.79 (m, 1H, cPropCH), 1.01 (m, 1H, 19-Hanti), 1.06 (dd, 2J8α,8β = 13.4 Hz, 3J8α7β = 9.3 Hz, 1H, 8α-H), 1.20 (s, 3H, 20CH3), 1.39 (s, 3H, 20CH3), 1.63 (dd, 2J15eq,15ax = 13.1 Hz, 3J15eq,16ax = 2.3 Hz, 1H, 15-Heq), 1.75-1.85 (m, 4H, 18-Hsyn, 18-Hanti, CH2CH2CH2OH), 1.93 (app t, 3J7β,8α = 10.1 Hz, 1H, 7β-H), 2.01 (td, 2J15ax,15eq = 13.6 Hz, 3J15ax,16eq = 5.7 Hz, 1H, 15-Hax), 2.17-2.29 (m, 3H, 10α-H, 16-Hax, NCH2 (a)), 2.36 (dd, JBA =12.9 Hz, JBX = 5.7 Hz, NCH2 (b)), 2.61 (dd, 2J16eq,16ax = 12.1 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.85 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 12.3 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H ), 2.96 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 3.00 (d, 3J9α,10β = 6.4 Hz, 1H, 9α-H), 3.13 (br s, 1H, 6-OCH2CH2CH2OH), 3.69-4.17 (m, 4H, 6-O-CH2CH2CH2OH, 6-O-CH2CH2CH2OH), 4.39 (d, 4J5β,18anti = 1.4 Hz, 1H, 5β-H), 5.34 (br s, 1H, 20-OH), 6.49 (d, J1,2 = 8.0 Hz, 1H, 1-H), 6.69 (d, J2,1 = 8.0 Hz, 1H, 2-H), 6.76 (br s, 1H, 3-OH). 13C-NMR (CDCl3): δ = 3.3 (cProp (a)), 4.1 (cProp (b)), 9.4 (cPropCH), 18.0 (C-18), 22.6 (C-10), 24.9 (20CH3), 29.7 (20CH3), 29.9 (C-19), 32.3 (C-8), 33.1 (6-O-CH2CH2CH2O), 35.4 (C-15), 35.9 (C-14), 43.7 (C-16), 47.0 (C-13), 48.0 (C-7), 58.2 (C-9), 59.2 (NCH2), 59.8 and 61.4 (6-O-CH2CH2CH2OH and 6-O-CH2CH2CH2OH), 74.7 (C-20), 80.6 (C-6), 97.4 (C-5), 116.8 (C-2), 119.5 (C-1), 127.7 (C-11), 132.2 (C-12), 137.6 (C-3), 145.5 (C-4). Calculated for C28H39NO5 (469.2828), found: 470.2907 ([M+H]+).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(4-hydroxybutyl)-18,19-dihydro-α,α-dimethyl-3-hydroxy-6,14-ethenomorphinan-7-methanol (6-O-(4-hydroxybutyl)-6-O-desmethyl-diprenorphine, 29c, HB-DPN)
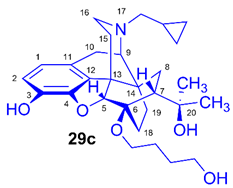
Compound 29c was prepared from FB-TDDPN 25c, 285 mg, 0.39 mmol) using the general procedure 3.2.6. CC: Kieselgel: 40 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 145 mg (77 %), mp. 209-210 °C (dec.), RfA = 0.45, RfD = 0.75, RfE = 0.15. 1H-NMR (CDCl3): δ = 0.09 (m, 2H, cPropCH2syn), 0.49 (m, 2H, cPropCH2anti), 0.72-0.81 (m, 2H, 19-Hsyn, cPropCH), 1.02 (m, 1H, 19-Hanti), 1.07 (dd, 2J8α,8β = 12.4 Hz, 3J8α,7β = 9.3 Hz, 1H, 8α-H), 1.20 (s, 3H, 20CH3), 1.39 (s, 3H, 20CH3), 1.58-1.66 (m, 2H, 15-Heq, 18-Hsyn), 1.69-1.79 (m, 5H, 6-O-CH2CH2CH2CH2OH and 18-Hanti), 1.93 (app t, 3J7β,8α = 10.0 Hz, 1H, 7β-H), 2.02 (td, 2J15ax,15eq = 13.6 Hz, 3J15ax,16eq = 5.7 Hz, 1H, 15-Hax), 2.17-2.30 (m, 3H, 10α-H, 16-Hax, NCH2 (a)), 2.37 (dd, JBA =12.6 Hz, JBX = 5.7 Hz, NCH2 (b)), 2.40 (br s, 1H, 6-O-(CH2)4OH), 2.62 (dd, 2J16eq,16ax = 11.9 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.85 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 11.9 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H), 2.97 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 3.00 (d, 3J9α,10α = 6.7 Hz, 1H, 9α-H), 3.60-4.06 (m, 4H, 6-O-CH2CH2CH2CH2OH), 4.39 (d, 4J5β,18anti = 1.0 Hz, 1H, 5β-H), 5.31 (br s, 1H, 20-OH), 6.40 (br s, 1H, 3-OH), 6.50 (d, J1,2 = 8.1 Hz, 1H, 1-H), 6.69 (d, J2,1 = 8.1 Hz, 1H, 2-H). 13C-NMR (CDCl3): δ = 3.3 (cProp (a)), 4.2 (cProp(b)), 9.4 (cPropCH), 18.1 (C-18), 22.6 (C-10), 24.9 (20-CH3), 26.9 and 28.7 (6-O-CH2CH2CH2CH2OH), 29.8 and 29.9 (C-19 and 20CH3), 32.3 (C-8), 35.5 (C-15), 35.9 (C-14), 43.7 (C-16), 47.1 (C-13), 48.0 (C-7), 58.2 (C-9), 59.8 (NCH2), 62.1 and 63.9 (6-O-CH2CH2CH2CH2OH and 6-O-CH2CH2CH2CH2OH), 74.5 (C-20), 80.6 (C-6), 97.3 (C-5), 116.6 (C-2), 119.6 (C-1), 127.9 (C-11), 132.3 (C-12), 137.5 (C-3), 145.2 (C-4). Calculated for C29H41NO5 (483.2985), found: 484.3065 ([M+H]+).
(5R,6R,7R,9R,13S,14S)-(5α,7α)-17-Cyclopropylmethyl-4,5-epoxy-6-O-(5-hydroxypentyl))-18,19-dihydro-α,α-dimethyl-3-hydroxy-6,14-ethenomorphinan-7-methanol (6-O-(5-hydroxypentyl)-6-O-desmethyl-diprenorphine, 29d, HPe-DPN)
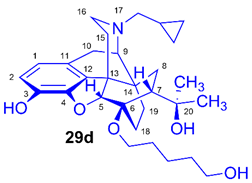
Compound 29d was prepared from FB-TDDPN 25d, 216 mg, 0.29 mmol) using the general procedure 3.2.6. Column chromatography: Kieselgel: 30 g, eluent: dichloromethane-methanol 95:5 (v/v), fractions: 25 mL. Yield: 105 mg (72 %), mp. 86-116 °C, RfA = 0.51, RfD = 0.71, RfE = 0.20. 1H-NMR (CDCl3): δ = 0.09 (m, 2H, cPropCH2syn), 0.49 (m, 2H, cPropCH2anti), 0.75 (tt, 1H, 19-Hsyn), 0.78 (m, 1H, cPropCH), 1.02 (m, 1H, 19-Hanti), 1.07 (dd, 2J8α,8β = 13.7 Hz, 3J8α7β = 9.7 Hz, 1H, 8α-H), 1.20 (s, 3H, 20CH3), 1.39 (s, 3H, 20CH3), 1.42-1.59 (m, 3H, (CH2)2CH2(CH2)2, 18-Hsyn), 1.62-1.87 (m, 6H, 15-Heq, 18-Hanti, 6-O-CH2CH2CH2CH2CH2OH), 1.94 (app t, 3J7β,8α = 10.0 Hz, 1H, 7β-H), 2.03 (td, 2J15ax,15eq = 13.6 Hz, 3J15ax,16eq = 5.3 Hz, 1H, 15-Hax), 2.16 (br s, 1H, 6-O-(CH2)5OH), 2.20 (dd, 2J10α,10β = 18.4 Hz, 3J10α,9α = 6.3 Hz, 1H, 10α-H), 2.23-2.30 (m, 2H, 16-Hax and NCH2 (a)), 2.37 (dd, JBA =12.7 Hz, JBX = 5.8 Hz, NCH2 (b)), 2.62 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 5.0 Hz, 1H, 16-Heq), 2.85 (ddd, 2J8β,8α = 13.5 Hz, 3J8β,7β = 12.2 Hz, 4J8β,19syn = 3.7 Hz, 1H, 8β-H ), 2.97 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H), 3.00 (d, 3J9α,10α = 6.3 Hz, 1H, 9α-H), 3.60-3.97 (m, 4H, 6-O-CH2CH2CH2CH2CH2OH), 4.38 (d, 4J5β,18anti = 1.7 Hz, 1H, 5β-H), 5.38 (br s, 1H, 20-OH), 6.16 (br s, 1H, 3-OH), 6.50 (d, J1,2 = 8.0 Hz, 1H, 1-H), 6.68 (d, J2,1 = 8.0 Hz, 1H, 2-H). 13C-NMR (CDCl3): δ = 3.3 (cProp (a)), 4.1 (cProp(b)), 9.4 (cPropCH), 17.9 (C-18), 21.8 ((CH2)2CH2(CH2)2), 22.6 (C-10), 24.9 (20-CH3), 29.7 and 29.9 (C-19 and 20CH3), 30.0 and 32.3 (6-O-CH2CH2CH2CH2CH2OH), 32.3 (C-8), 35.5 (C-15), 35.9 (C-14), 43.7 (C-16), 47.0 (C-13), 48.0 (C-7), 58.2 (C-9), 59.8 (NCH2), 62.4 and 64.2 (6-O-(CH2)4CH2OH and 6-O-CH2(CH2)4OH), 74.6 (C-20), 80.5 (C-6), 97.5 (C-5), 116.7 (C-2), 119.5 (C-1), 127.9 (C-11), 132.3 (C-12), 137.5 (C-3), 145.4 (C-4). Calculated for C30H43NO5 (497.3141), found: 498.3221 ([M+H]+).
3.2.7. Sythesis of Fluoroalkyl-Tosylates (33a–c, Figure 6)
3-Fluoro-1-(toluene-p-sulfonyloxy)propane (33a, FPOTos, CAS RN: 312-68-5)
Compound 33a was prepared from 3-fluoro-propan-1-ol (30a, 5.0 g, 64 mmol) with tosyl chloride (95 mmol) in dichloromethane in the presence of pyridine and DMAP applying the procedure of Goswami et al. [63]. CC: Kieselgel: 300 g, eluent: hexane-ethyl acetate 10:1 (v/v), fractions: 250 mL. Yield: 11.46 g (77 %) colourless oil. Rf (hexane-ethyl acetate 10:1) = 0.20. 1H-NMR (CDCl3): δ = 2.04 (dpent, J1 = 25.7 Hz, J2 = 6.0 Hz, 2H, FCH2CH2CH2O), 2.45 (s, 3H, Tos-CH3), 4.16 (t, J = 6.0 Hz, 2H, FCH2CH2CH2), 4.48 (dt, J1 = 46.9 Hz, J2 = 5.6 Hz, 2H, FCH2CH2CH2O), 7.35 (d, J = 8.1 Hz, 2H, Tos-3,5), 7.79 (d, J = 8.1 Hz, 2H, Tos-2,6). 13C-NMR (CDCl3): δ = 21.5 (Tos-CH3), 29.9 (d, 2JC,F = 20.4 Hz, OCH2CH2CH2F), 66.1 (d, 3JC,F = 4.8 Hz, OCH2CH2CH2F), 79.5 (d, 1JC,F = 165.8 Hz, OCH2CH2CH2Br), 127.8 (Tos-2,6), 129.8 (Tos-3,5), 132.7 (Tos-1), 144.9 (Tos-4). 19F-NMR (CDCl3): δ = - 223.4 m. C10H13FO3S (232.27).
4-Fluoro-1-(toluene-p-sulfonyloxy)butane (33b, FBOTos, CAS RN: 433-10-3)
Compound 33b was synthesized from 1,4-butandiol (30b, 3.46 g, 38.4 mmol) via 1,4-butandiol-ditosylate (31) using the method of van Wieringen et al. [64]. CC: Kieselgel: 200 g, eluent: hexane-ethyl acetate 7:3 (v/v), fractions: 100 mL. Yield: 620 mg (31 → 33b: 41 %). 1H-NMR (CDCl3): δ = 1.67-1.82 (m, 4H, FCH2CH2CH2CH2O), 2.44 (s, 3H, Tos-CH3), 4.07 (t, J = 6.0 Hz, 2H, F(CH2)3CH2O), 4.40 (dt, J1 = 47.3 Hz, J2 = 5.7 Hz, 2H, FCH2(CH2)3O), 7.34 (d, J = 8.4 Hz, 2H, Tos-3,5), 7.78 (d, J = 8.4 Hz, 2H, Tos-2,6). 13C-NMR (CDCl3): δ = 21.6 (Tos-CH3), 25.0 (d, 3JC,F = 4.6 Hz, OCH2CH2CH2CH2F), 26.4 (d, 2JC,F = 20.1 Hz, O(CH2)2CH2CH2F), 69.8 (OCH2CH2CH2CH2F), 83.0 (d, 1JC,F = 165.1 Hz, O(CH2)3CH2F), 127.8 (Tos-2,6), 129.8 (Tos-3,5), 133.0 (Tos-1), 144.8 (Tos-4). 19F-NMR (CDCl3): δ = - 219.5 m. C11H15FO3S (246.30).
5-Fluoro-1-(toluene-p-sulfonyloxy)pentane (33c, FPeOTos, CAS RN: 565-44-6)
Compound 33c was prepared from 5-bromo-pentan-1-ol (30c, 4.0 g, 38.4 mmol) via 5-tosyloxy-pentanol (32) according to the method of Galante et al. [65]. CC: Kieselgel: 350 g, eluent: eluent: hexane-ethyl acetate 8:2 (v/v), fractions: 250 mL. Yield: 2.09 g (32 → 33c: 64 %) colourless oil. 1H-NMR (CDCl3): δ = 1.40-1.47 (m, 2H, F(CH2)2CH2(CH2)2O), 1.58-1.66 (m, 2H, FCH2CH2(CH2)3O), 1.67-1.72 (m, 2H, F(CH2)3CH2CH2O), 2.44 (s, 3H, Tos-CH3), 4.03 (t, J = 6.4 Hz, 2H, F(CH2)4CH2O), 4.39 (dt, J1 = 47.2 Hz, J2 = 6.0 Hz, 2H, FCH2(CH2)4O), 7.34 (d, J = 8.4 Hz, 2H, Tos-3,5), 7.78 (d, J = 8.4 Hz, 2H, Tos-2,6). 13C-NMR (CDCl3): δ = 21.3 (d, 3JC,F = 4.6 Hz, O(CH2)2CH2(CH2)2F), 21.6 (Tos-CH3), 29.6 (d, 2JC,F = 19.2 Hz, O(CH2)3CH2CH2F), 70.2 (OCH2(CH2)4F), 83.6 (d, 1JC,F = 165.3 Hz, O(CH2)4CH2F), 127.8 (Tos-2,6), 129.8 (Tos-3,5), 133.0 (Tos-1), 144.7 (Tos-4). 19F-NMR (CDCl3): δ = - 218.7 m. C12H17FO3S (260.33).
3.2.8. General Procedure for Preparation of tert-Butyldiphenylsilyl (TBDPS) Protected bromoalkanols (34a–c, Figure 6)
The corresponding bromoalkanol (30d–f, 25 mmol) was dissolved in dry dichloromethane (5 mL) under argon atmosphere. N-ethyldiisopropylamine (3.59 g, 4.6 mL, 27.7 mmol) was added and the mixture stirred at RT for 15 min. tert-Butyldiphenylsilyl chloride (TBDPSCl, 7.71 g, 7.3 mL, 28 mmol) and DMAP (20 mg) were added, and the reaction mixture stirred for 48 h at RT. The solvent was removed under reduced pressure and the crude product purified by CC on silica gel. CC: Kieselgel: 200 g, eluent: hexane-ethyl acetate 50:1 (v/v), fractions: 250 mL.
(3-Bromopropoxy)-tert-butyldiphenylsilane (34a, CAS RN: 177338-13-5)
Compound 34a was prepared from 3-bromo-propanol (30d, 3.52 g, 2.3 mL, 25 mmol) using general procedure 3.2.8. Yield: 7.4 g (77 %) colourless oil. 1H-NMR (CDCl3): δ = 1.05 (s, 9H, (CH3)3CSiPh2), 2.03 (m, 2H, BrCH2CH2CH2O), 3.59 (t, J = 6.7 Hz, 2H, BrCH2CH2CH2), 3.72 (t, J = 6.4 Hz, 2H, BrCH2CH2CH2O), 7.37–7.45 (m, 6H, 2 x Ph-4 and 2 x Ph-3,5), 7.65-7.68 (m, 4H, 2 x Ph-2,6). 13C-NMR (CDCl3): δ = 19.2 ((CH3)3C), 26.8 ((CH3)3C), 30.5 (OCH2CH2CH2Br), 35.4 (OCH2CH2CH2Br), 61.3 (OCH2CH2CH2Br), 127.7 (Ph-3,5), 129.6 and 133.6 (Ph-4 and Ph-1), 135.5 (Ph-2,6). C19H25BrOSi (377.39).
(4-Bromobutoxy)-tert-butyldiphenylsilane (34b, CAS RN: 125010-58-4)
Compound 34b was prepared from 4-bromo-butan-1-ol (30e, 3.82 g, 25 mmol) using the general procedure 3.2.8. Yield: 5.6 g (57 %) colourless oil. 1H-NMR (CDCl3): δ = 1.05 (s, 9H, (CH3)3CSiPh2), 1.67-1.72 (m, 4H, BrCH2CH2CH2CH2O), 3.42 (t, J = 6.7 Hz, 2H, BrCH2(CH2)3O), 3.69 (t, J = 6.0 Hz, 2H, Br(CH2)3CH2O), 7.37–7.44 (m, 6H, 2 x Ph-4 and 2 x Ph-3,5), 7.65-7.67 (m, 4H, 2 x Ph-2,6). 13C-NMR (CDCl3): δ = 19.2 ((CH3)3C), 26.8 ((CH3)3C), 29.4 and 31.0 (OCH2CH2CH2CH2Br), 33.8 (O(CH2)3CH2Br), 62.9 (OCH2(CH2)3Br), 127.6 (Ph-3,5), 129.6 and 133.8 (Ph-4 and Ph-1), 135.5 (Ph-2,6). C20H27BrOSi (391.42).
(4-Bromopentoxy)-tert-butyldiphenylsilane (34c, CAS RN: 125010-60-8)
Compound 34c was prepared from 5-bromo-pentan-1-ol (30f, 4.17 g, 25 mmol) using the general procedure 3.2.8. Yield: 2.9 g (28 %) colourless oil. 1H-NMR (CDCl3): δ = 1.05 (s, 9H, (CH3)3CSiPh2), 1.48-1.60 (m, 4H, OCH2CH2CH2(CH2)Br), 1.74-1.85 (m, 2H, O(CH2)3CH2CH2Br), 3.38 (t, J = 6.7 Hz, 2H, BrCH2(CH2)3O), 3.68 (t, J = 6.4 Hz, 2H, Br(CH2)3CH2O), 7.36–7.44 (m, 6H, 2 x Ph-4 and 2 x Ph-3,5), 7.66-7.67 (m, 4H, 2 x Ph-2,6). 13C-NMR (CDCl3): δ = 19.2 ((CH3)3C), 24.5 (O(CH2)2CH2(CH2)2Br), 26.8 ((CH3)3C), 31.6 and 32.5 (O(CH2CH2CH2CH2CH2)2Br), 33.8 (O(CH2)4CH2Br), 63.5 (OCH2(CH2)4Br), 127.6 (Ph-3,5), 129.5 and 133.9 (Ph-4 and Ph-1), 135.5 (Ph-2,6). C21H29BrOSi (405.45).
3.3. Computational Part
3.3.1. Ligand and Receptor Preparation for Docking
The initial 3D structures for the eight ligands under examination were provided as MOL2 files. These structures were then processed using Open Babel (v3.1.1) [74] to generate a diverse set of conformers for each ligand. A conformational search was performed using a genetic algorithm (--conformer) to generate a maximum of fifty conformers (--nconf 50). Each of these conformers was subsequently energy-minimized using the MMFF94s force field [75], and the resulting ensemble of low-energy conformers for each ligand was exported into a single SDF file.
Initial crystal structures of the µOR in its inactive (PDB ID: 4DKL), partial agonist (PDB ID: 7U2K) and active (PDB ID: 5C1M) states were downloaded from the Protein Data Bank. For docking purposes, the receptors were prepared using AutoDockTools (ADT). This involved removing all non-protein atoms, adding polar hydrogens, and assigning Gasteiger charges. The final prepared receptor structures and the ligand conformers were then converted to PDBQT format to meet the criteria for AutoDock Vina [76].
3.3.2. Molecular Docking Protocol
An initial docking simulation was performed for the pool of conformers for each of the eight ligands into the 4DKL, 5C1M, and 7U2K receptor structures using AutoDock Vina [76]. Following this initial screening, the conformers yielding the most favourable binding affinity energies were selected. These conformers then underwent a more exhaustive docking simulation consisting of five repeats. From these repeats, the single pose with the best binding energy for each ligand-receptor pair was selected as the starting structure for the subsequent molecular dynamics simulations.
3.3.3. System Preparation for Molecular Dynamics
Receptor Preparation: The original PDB structures of the inactive (4DKL), active (5C1M), and partial agonist (7U2K) receptors were used as the starting point for MD-specific preparation. All non-protein atoms were removed, and the PDBFixer library [77] was employed to repair the three protein structures by adding all missing heavy atoms and hydrogens, assuming a physiological pH of 7.4 to assign appropriate protonation states. The final, complete receptor models were then processed using the pdb4amber utility in AmberTools22 [78] to ensure Amber-compatible atom and residue naming conventions and to apply standard N-terminal and C-terminal charged patches.
Ligand Parameterization: For each of the selected ligand poses, atom types were assigned from the second-generation General Amber Force Field (GAFF2) and partial atomic charges were calculated using the AM1-BCC method, as implemented in the antechamber module of AmberTools22 [79,80,81]. Any missing force field parameters for the ligands were generated using the parmchk2 utility, creating the corresponding force field modifications (.frcmod) files.
3.3.4. Molecular Dynamics Simulation Protocol
All simulations were performed using the Amber22 simulation package with the sander engine [78].
System Assembly and Membrane Embedding: The tleap module was used to assemble each simulation system. The proteins were described by the ff19SB force field [78] and the ligands by the GAFF2 force field [79], and the lipid bilayer by the Lipid 21 force field. Prior to membrane insertion, each receptor-ligand complex was oriented using a custom Python script that aligned the principal axis of its Cα at-oms with the Z-axis. The pre-oriented complex was then embedded into a POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine) bilayer, consisting of 200 lipids per leaflet, using the packmol-memgen tool [78]. The resulting protein-membrane system was subsequently solvated with TIP3P water molecules [82] in an orthogonal box with fixed lateral dimensions of 80 Å × 80 Å. The net charge of each system was neutralized by adding a minimal number of chloride (Cly) counter-ions.
Minimization, Heating, and Equilibration: A two-stage energy minimization was first performed to relax each system. Following minimization, systems were gradually heated from 0 K to 300 K over 200 picoseconds (ps) using the Langevin thermostat [83] under constant volume (NVT) conditions, with a gentle restraint on the complex. Subsequently, each system was equilibrated for 500 ps under constant temperature (300 K) and pressure (1 bar) conditions (NPT ensemble), using a restraint on the protein backbone heavy atoms to allow the solvent density to stabilize.
Production MD: Production simulations were run in the NPT ensemble at 300 K and 1 bar. The SHAKE algorithm was used to constrain bonds involving hydrogen, allowing for a 2 femtosecond (fs) timestep [84]. To sample the binding site, the backbone heavy atoms (@C,CA,N) of the receptor located further than 8.0 Å from any ligand atom were gently restrained. Coordinates and restart files were saved every 10,000 steps (20 ps) to generate a continuous trajectory for analysis.
Summary
We synthesized new 6-O-substituted-6-O-desmethyl-analogues (24–27b–d) of diprenorphine (17) from 3-O-trityl-6-O-desmethyl-diprenorphine (TDDPN, 23, «Luthra precursor»). To study the impact of the 6-O-side chain we prepared numerous derivatives with varying the chain length (n = 3,4,5). 6-O-Fluoroalyl-6-O-desmethyl-diprenorphine (28b–d), 6-O-(hydroxyalkyl)-6-O-desmethyl-diprenorphine (29b–d), and 6-O-tosyloxyalkyl)-6-O-desmethyl-diprenorphine (26a–d) derivatives with longer 6-O-side chains (n = 3,4,5). Further biochemical/pharmacological investigations of the synthesized novel derivatives could shed light on the influence of 6-O-side chain on the OR affinity and subtype selectivity and could deliver new knowledges for structure-activity relationships. The compounds with 6-O-(tosyloxyaklyl)- substituent (26b–d) can be suitable precursors for the synthesis of 6-O-([18F]fluoroalkyl)-6-O-desmethyl-diprenorphine type ([18F]28b–d) radiotracers through direct aliphatic nucleophilic substitution (SN2) with [18F]fluoride under mild basic conditions.
Computational investigations: Altogether, according to docking energy, all compounds show a preference for the active receptor state. Actives state: series 28 is slightly better than series 29; and in series 29: longer chain is shomewat better. Inactive state: series 28 is slightly better than series 29 and longer chains are somewhat better. Partially active state: series 28 is slightly better than series 29; and longer chains are somewhat better. Root mean square deviation (RMSD): series 29: lowest values were observed in the active receptor state. The rank order is active > inactive > partially active; series 28: the trend is as for series 29. The most satble one in the active were 28a and 28b. Center of mass (COM): displacement of COM is generally is least in the active and partially active receptor states. Smallest displacement of COM observed: active state 28; inactive state 28, 28b, 28c; partially active state: 28a. Our final conclusion is that, 28a is the lead compound, but it is not exclusively the only candidate for further pharmacological investigations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Initial idea and conceptualization: J.M., P.C., F.W., B.W.S., F.Ö.; Supervision: P.C., J.M., F.Ö., S.B.; Investigation: organic chemistry: B.B., J.M.; HRMS measurements: A.M.; Resources: J.M., B.B., T.F., F.Ö., A.M.; Writing – original draft preparation: J.M., P.C., D.G., F.Ö; Writing – review and editing: J.M., P.C., F.Ö, S.B.; Molecular modelling: D.G., F.Ö., S.B., T.F.; Software: F.Ö.; Conduction of the initial radiosynthesis: B.W.S., F.W. This manuscript was writen with contributions of all the authors. All authors have read and agreed to the published version of the manuscript.
Funding
The in silico modelling part of this work was supported by the National Biotechnology Laboratory Grant of Hungary (NRDIO Grant No. 2022-2.1.1-NL-2022-00008, F.Ö.).
Conflicts of Interest
Beate Bauer, Alexander Milentyev, and János Marton were employed by the company ABX Advanced Biochemical Compounds Biomedizinische Forschungsreagenzien GmbH, Radeberg, Germany. The in silico modelling part of this work, performed by D.G., T.F., S.B., and F.Ö., was supported by the National Biotechnology Laboratory Grant of Hungary (NRDIO Grant No. 2022-2.1.1-NL-2022-00008, F.Ö.). The remaining authors (P.C., F.W., and B.W.S.) declare that their research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Presley, C. C.; Lindsley, C. W. , Dark classics in chemical neuroscience: Opium, a historical perspective. ACS Chem. Neurosci. 2018, 9, 2503–2518. [Google Scholar] [CrossRef]
- Bernáth, J. , Poppy, The Genus Papaver. Harwood Academic Publishers: Amsterdam, The Netherlands, 1998.
- Brownstein, M. J. , A brief history of opiates, opioid peptides, and opioid receptors. Proc. Natl. Acad. Sci. 1993, 90, 5391–5393. [Google Scholar] [CrossRef]
- Schiff, P. L. , Opium and its alkaloids. Am. J. Pharm. Educat. 2002, 66, 188–196. [Google Scholar]
- Carod-Artal, F. J. , Psychoactive plants in ancient Greece. Neurosci. Hist. 2013, 1, 28–38. [Google Scholar]
- Molina-Venegas, R.; Verano, R. , The quest for Homer’s moly: exploring the potential of an early ethnobotanical complex. J. Ethnobiol. Ethnomed. 2024, 20, 1–11. [Google Scholar]
- Chovanec, Z.; Rafferty, S.; Swiny, S. , Opium for the Masses: An experimental archaeological approach in determining the antiquity of the opium poppy. Ethnoarchaeology 2012, 4, 5–36. [Google Scholar] [CrossRef]
- Sinclair, A., High times in ancient Egypt. In Alternative Egyptology: Critical essays on the relation between academic and alternative interpretations of ancient Egypt, van den Bercken, B. J. L., Ed. Sidestone Press: Leiden, 2024; pp 76–94.
- Devereaux, A. L.; Mercer, S. L.; Cunningham, C. W. , Dark classics in chemical neuroscience: morphine. ACS Chem. Neurosci. 2018, 9, 2395–2407. [Google Scholar] [CrossRef]
- Schmitz, R. , Friedrich Wilhelm Sertürner and the discovery of morphine. Pharmacy in History 1985, 27, 61–74. [Google Scholar]
- Knutsen, H. K.; Alexander, J; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Nebbia, C. S.; Oswald, I. P.; Petersen, A.; Rose, M.; Roudot, A.-C.; Schwerdtle, T.; Vollmer, G.; Wallace, H.; Benford, D.; Calò, G.; Dahan, A.; Dusemund, B.; Mulder, P.; Németh-Zámboriné, É.; Arcella, D.; Baert, K.; Cascio, C.; Levorato, S.; Schutte, M.; Vleminckx, C. Update of the Scientific Opinion on opium alkaloids in poppy seeds. EFSA Journal 2018, 16, 5, 5243, 119 pp. [CrossRef]
- Borsodi, A.; Bruchas, M.; Caló, G.; Chavkin, C.; Christie, M. J.; Civelli, O.; Connor, M.; Cox, B. M.; Devi, L. A.; Evans, C.; Höllt, V.; Henderson, G.; Husbands, S.; Kelly, E.; Kieffer, B.; Kitchen, I.; Kreek, M. J.; Liu-Chen, L.-Y.; Massot, D.; Meunier, J.-C.; Portoghese, P. S.; Schulz, S.; Shippenberg, T. S.; Simon, E. J.; Toll, L.; Traynor, J. R.; Ueda, H.; Wong, Y. H.; Zaveri, N.; Zimmer, A., Opioid receptors (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. In IUPHAR/BPS GtoPdb CITE 2019, 4. [CrossRef]
- Benyhe, S.; Zádor, F.; Ötvös, F. , Biochemistry of opioid (morphine) receptors: binding, structure and molecular modelling. Acta Biol. Szeged 2015, 59 Suppl 1, 17–37. [Google Scholar]
- Lamberts, J. T.; Traynor, J. R. , Opioid receptor interacting proteins and the control of opioid signaling. Curr. Pharm. Design 2013, 19, 7333–7347. [Google Scholar] [CrossRef] [PubMed]
- Lever, J. R. , PET and SPECT imaging of the opioid system: Receptors, radioligands and avenues for drug discovery and development. Curr. Pharm. Design 2007, 13, 33–49. [Google Scholar] [CrossRef]
- Henriksen, G.; Willoch, F. , Imaging of opioid receptors in the central nervous system. Brain 2008, 131, 1171–1196. [Google Scholar] [CrossRef]
- Dannals, R. F. , Positron emission tomography radioligands for the opioid system J. Label. Compd. Radiopharm. 2013, 56, 187–195. [Google Scholar] [CrossRef]
- Cumming, P.; Marton, J.; Lilius, T. O.; Olberg, D. E.; Rominger, A. , A Survey of Molecular Imaging of Opioid Receptors. Molecules 2019, 24, 4190. [Google Scholar] [CrossRef]
- Marton, J.; Fekete, A.; Cumming, P.; Hosztafi, S.; Mikecz, P.; Henriksen, G. , Diels–Alder adducts of morphinan-6, 8-dienes and their transformations. Molecules 2022, 27, 2863. [Google Scholar] [CrossRef]
- Dannals, R. F.; Ravert, H. T.; Frost, J. J.; Wilson, A. A.; Burns, H. D.; Wagner, H. N. , Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Radiat. Isot. 1985, 36, 303–306. [Google Scholar] [CrossRef]
- Pert, C. B.; Danks, J. A.; Channing, M. A.; Eckelman, W. C.; Larson, S. M.; Bennett, J. M.; Burke, T. R.; Rice, K. C. , S-[18F]Acetylcyclofoxy: a useful probe for the visualization of opiate receptors in living animals. FEBS Letters 1984, 177, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Lever, J. R.; Kinter, C. M.; Ravert, H. T.; Musachio, J. L.; Mathews, W. B.; Dannals, R. F. , Synthesis of N1′-([11C]methyl)naltrindole ([11C]MeNTI): A radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1995, 36, 137–145. [Google Scholar] [CrossRef]
- Mathews, W. B.; Kinter, C. M.; Palma, J.; Daniels, R. V.; Ravert, H. T.; Dannals, R. F.; Lever, J. R. , Synthesis of N1′-([18F]fluoroethyl)naltrindole ([18F]FEtNTI): a radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1999, 42, 43–54. [Google Scholar] [CrossRef]
- Ravert, H. T.; Mathews, W. B.; Musachio, J. L.; Scheffel, U.; Finley, P.; Dannals, R. F. , [11C]-methyl 4-[(3, 4-dichlorophenyl) acetyl]-3-[(1-pyrrolidinyl) methyl]-1- piperazinecarboxylate ([11C] GR89696): synthesis and in vivo binding to kappa opiate receptors. Nucl. Med. Biol. 1999, 26, 737–741. [Google Scholar] [CrossRef]
- Pike, V. W.; Rash, K. S.; Chen, Z.; Pedregal, C.; Statnick, M. A.; Kimura, Y.; Hong, J.; Zoghbi, S. S.; Fujita, M.; Toledo, M. A.; Diaz, N.; Gackenheimer, S. L.; Tauscher, J. T.; Barth, V. N.; Innis, R. B. Synthesis and evaluation of radioligands for imaging brain nociceptin/orphanin FQ peptide (NOP) receptors with positron emission tomography. J. Med. Chem. 2011, 54, 2687–2700. [Google Scholar] [CrossRef]
- Burns, H. D.; Lever, J. R.; Dannals, R. F.; Frost, J. J.; Wilson, A. A.; Ravert, H. T.; Subramanian, B.; Zeyman, S. E.; Langstrom, B.; Wagner Jr., H. N., Synthesis of ligands for imaging opiate receptors by positron emission tomography: carbon-11 labelled diprenorphine. J. Labell. Compds. Radiopharm. 1984, 22, 1167–1169. [Google Scholar]
- Tafani, J.-A. M.; Francés, B.; Coulais, Y.; Méjan-Galzi, A.; Goeldner, M.; Hirth, C.; Guiraud, R.; Zajac, J.-M. , In vivo binding of [125I]NH2-carfentanil to mu opioid receptors in mouse brain. Nucl. Med. Biol. 1994, 21, 231–238. [Google Scholar] [CrossRef]
- Luthra, S. K.; Pike, V. W.; Brady, F. , The preparation of carbon-11 labelled diprenorphine: a new radioligand for the study of the opiate receptor system in vivo. J. Chem. Soc., Chem. Commun. 1985; 1423–1425. [Google Scholar]
- Chesis, P. L.; Hwang, D.-R.; Welch, M. J. , N-(3-[18F]Fluoropropyl)-N-nordiprenorphine: Synthesis and characterization of a new agent for imaging opioid receptors with positron emission tomography. J. Med. Chem. 1990, 33, 1482–1490. [Google Scholar] [CrossRef]
- Bai, L.-Q.; Teng, R.-R.; Shiue, C.-Y.; Wolf, A. P.; Dewey, S. L.; Holland, M. J.; Simon, E. J. , No-carrier-added (NCA) N-(3-[18F]fluoropropyl-N-norbuprenorphine and N-(3-[18F]fluoropropyl-N-nordiprenorphine - Synthesis distribution in mice and rats, and tomographic studies in baboon. Int. J. Radiat. Appl. Instr. Part B, Nucl. Med.Chem. 1990, 17, 217–227. [Google Scholar] [CrossRef]
- Lever, J. R.; Dannals, R. F.; Wilson, A. A.; Ravert, H. T.; Wagner Jr., H. N. , Synthesis of carbon-11 labeled diprenorphine: A radioligand for positron emission tomographic studies of opiate receptors Tetrahedron Lett. 1987, 28, 4015–4018. [Google Scholar]
- Musachio, J. L.; Lever, J. R. , Vinylstannylated alkylating agents as prosthetic groups for radioiodination of small molecules: Design, synthesis and application to iodoallyl analogues of spiperone and diprenorphine. Bioconjugate Chem. 1992, 3, 167–175. [Google Scholar] [CrossRef]
- Luthra, S. K.; Brady, F.; Turton, D. R.; Brown, D. J.; Dowsett, K.; Waters, S. L.; Jones, A. K. P.; Matthews, R. W.; Crowder, J. C. , Automated radiosyntheses of [6-O-methyl-11C]diprenorphine and [6-O-methyl-11C]buprenorphine from 3-O-trityl protected precursors. Appl. Radiat. Isot. 1994, 45, 857–873. [Google Scholar] [CrossRef]
- Marton, J.; Schoultz, B. W.; Hjørnevik, T.; Drzezga, A.; Yousefi, B. H.; Wester, H.-J.; Willoch, F.; Henriksen, G. , Synthesis and evaluation of a full-agonist orvinol for PET-Imaging of opioid receptors: [11C]PEO. J. Med. Chem. 2009, 52, 5586–5589. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.-J.; Willoch, F.; Tölle, T. R.; Munz, F.; Herz, M.; Øye, I.; Schadrack, J.; Schwaiger, M.; Bartenstein, P. , 6-O-(2-[18F]Fluoroethyl-6-O-desmethyl diprenorphine ([18F]DPN): Synthesis, biologic evaluation, and comparison with [11C]DPN in humans J. Nucl. Med. 2000, 41, 1279–1286. [Google Scholar]
- Schoultz, B. W.; Reed, B. J.; Marton, J.; Willoch, F.; Henriksen, G. , A fully automated radiosynthesis of [18F]fluoroethyl-diprenorphine on a single module by use of SPE cartridges for preparation of high quality 2-[18F]fluoroethyl tosylate. Molecules 2013, 18, 7271–7278. [Google Scholar] [CrossRef]
- Schoultz, B. W.; Hjørnevik, T.; Reed, B. J.; Marton, J.; Coello, C. S.; Willoch, F.; Henriksen, G. , Synthesis and evaluation of three structurally related 18F-labeled orvinols of different intrinsic activities: 6-O-[18F]Fluoroethyl-diprenorphine ([18F]FDPN), 6-O-[18F]fluoroethyl-buprenorphine ([18F]FBPN), and 6-O-[18F]fluoroethyl-phenethyl-orvinol ([18F]FPEO). J. Med. Chem. 2014, 57, 5464–5469. [Google Scholar]
- Marton, J.; Cumming, P.; Bauer, B.; Henriksen, G. , A new precursor for the radiosynthesis of 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) by nucleophilic radiofluorination. Lett. Org. Chem. 2021, 18, 344–352. [Google Scholar] [CrossRef]
- Levinstein, M. R.; Ventriglia, E. N.; Gomez, J. L.; Budinich, R. C.; Marton, J.; Henriksen, G.; Holt, D. P.; Dannals, R. F.; Pomper, M. G.; Zarate, C. A.; Jr, B., J.; Michaelides, M. , 6-O-(2-[18F]Fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) preferentially binds to mu opioid receptors in vivo. Mol. Imag. Biol. 2023, 25, 2, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Németh, E.; Gyuricza, B.; Forgács, V.; Cumming, P.; Henriksen, G.; Marton, J.; Bauer, B.; Mikecz, P.; Fekete, A. , Optimization of a nucleophilic two-step radiosynthesis of 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) for PET imaging of brain opioid receptors. Int. J. Mol. Sci. 2023, 24, 13152. [Google Scholar] [CrossRef]
- Bentley, K. W., The morphine alkaloids. In The Alkaloids: Chemistry and Physiology, Manske, R. H. F., Ed. Academic Press Inc.: New York, London 1971, Vol. 13.
- Linders, J. T. M.; Maat, L. , Face selectivity of the Diels-Alder reaction of thebaine-like morphinandienes, A computational approach (Chemistry of opium alkaloids, Part XXXI). Bull. Soc. Chim. Belg. 1989, 98, 265–276. [Google Scholar] [CrossRef]
- Maat, L. Novel thebainelike morphinan-dienes and their Diels-Alder adducts. In National Institute on Drug Abuse, Research Monograph Series, Drugs of Abuse: Chemistry, Pharmacology, Immunology, and AIDS, Pham, P. T. K.; Rice, K. C., Eds. 1990; pp 35–49.
- Lewis, J. W.; Husbands, S. M. , The Orvinols and Related Opioids - High Affinity Ligands with Diverse Efficacy Profiles Curr. Pharm. Design 2004, 10, 717–732. [Google Scholar]
- Husbands, S. H. Buprenorphine and related orvinols. In Research and Development of Opioid-Related Ligands, 2013; pp 127–144.
- Sandermann, W. , Diën-Anlagerungsverbindunden des Thebains. Ber. Dtsch. Chem. Ges. 1938, 71, 648–650. [Google Scholar] [CrossRef]
- Schöpf, C.; von Gottberg, K.; Petri, W. , Über Thebain-maleinsäureanhydrid, Thebainchinon, Thebainhydrochinon und dessen Säreumlagerungsprodukt, das Flavothebaon. Justus Liebigs Ann. Chem. 1938, 536, 216–257. [Google Scholar] [CrossRef]
- Li, Z.; Ye, R.; He, Q.; Lu, J.; Sun, Y.; Sun, X.; Tang, S.; Hu, S.; Chai, J.; Kong, L.; Liu, X.; Chen, J.; Fang, Y.; Lan, Y.; Xie, Q.; Liu, J.; Shao, L.; Fu, W.; Wang, Y.; Li, W. , Discovery of an ortho-substituted N-cyclopropylmethyl-7α-phenyl-6,14-endoethano-tetrahydronorthebaine derivative as a selective and potent kappa opioid receptor agonist with subsided sedative effect. J. Med. Chem. 2024. [Google Scholar] [CrossRef]
- Sandulenko, I. V.; Ambartsumyan, A. A.; Moiseev, S. K. , Fluorinated and [18F]fluorinated morphinan based opioid ligands. Org. Biomol. Chem. 2020, 18, 5533–5557. [Google Scholar] [CrossRef] [PubMed]
- Marton, J.; Henriksen, G. , Design and synthesis of an 18F-labeled version of phenylethyl orvinol ([18F]FE-PEO) for PET-imaging of opioid receptors. Molecules 2012, 17, 11554–11569. [Google Scholar] [CrossRef] [PubMed]
- Raynor, K.; Kong, H.; Chen, Y.; Yasuda, K.; Yu, L.; Bell, G. I.; Reisine, T. , Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994, 45, 330–334. [Google Scholar] [CrossRef]
- Bentley, K. W.; Hardy, D. G. , Novel analgesics and molecular rearrangements in the morphine-thebaine group I. Ketones derived from 6,14-endo-ethenotetrahydrtothebaine J. Am. Chem. Soc. 1967, 89, 3267–3273. [Google Scholar]
- Bentley, K. W.; Hardy, D. G.; Meek, B. , Novel analgesics and molecular rearrangements in the morphine-thebaine group. II. Alcohols derived from 6,14-endo-etheno- and 6,14-endo-ethanotetrahydrothebaine. J. Am. Chem. Soc. 1967, 89, 3273–3280. [Google Scholar] [CrossRef]
- Bentley, K. W.; Hardy, D. G. , Novel analgesics and molecular rearrangements in the morphine-thebaine group. III. Alcohols of the 6,14-endo-ethenotetrahydrooripavine series and derived analogs of N-allylnormorphine and -norcodeine. J Am Chem Soc. 1967, 89, 3281–3292. [Google Scholar] [CrossRef]
- Marton, J.; Szabó, Z.; Hosztafi, S. , Herstellung von 6,14 Ethenomorphinan- Derivaten. Liebigs Ann. Chem. 1993, 8, 915–919. [Google Scholar] [CrossRef]
- Marton, J.; Hosztafi, S.; Berényi, S.; Simon, C.; Makleit, S. , Herstellung von 6,14-Ethenomorphinan- Derivaten. Monatsh. Chem. 1994, 125, 1229–1239. [Google Scholar] [CrossRef]
- Hosztafi, S.; Timár, T.; Makleit, S. , Morfin alkaloidok N-demetilezése III. Magy. Kém. Foly. 1985, 91, 126–130. [Google Scholar]
- Fleischhacker, W.; Richter, B. , 14,17-Ethano-norcodeinones. Monatsh. Chem. 1992, 123, 837–848. [Google Scholar] [CrossRef]
- Kopcho, J. J.; Schaeffer, J. C. , Selective O-demethylation of 7.alpha.-(aminomethyl)6,14-endo-ethenotetrahydrothebaine. J. Org. Chem. 1986, 51, 1620–1622. [Google Scholar] [CrossRef]
- Szűcs, E.; Marton, J.; Szabó, Z.; Hosztafi, S.; Kékesi, G.; Tuboly, G.; Bánki, L.; Horváth, G.; Szabó, P. T.; Tömböly, C.; Varga, Z. K.; Benyhe, S.; Ötvös, F. , Synthesis, biochemical, pharmacological characterization and in silico profile modelling of highly potent opioid orvinol and thevinol derivatives. Eur. J. Med. Chem. 2020, 191, 112145. [Google Scholar] [CrossRef] [PubMed]
- Czakó, B.; Marton, J.; Berényi, S.; Gach, K.; Fichna, J.; Storr, M.; Tóth, G.; Sipos, A.; Janecka, A. , Synthesis and opioid activity of novel 6-substituted-6-demethoxy-ethenomorphinans. Bioorg. Med. Chem. 2010, 18, 3535–3542. [Google Scholar] [CrossRef]
- Srivastava, S.; Fergason-Cantrell, E. A.; Nahas, R. I.; Lever, J. R. , Synthesis and opioid receptor binding of indium (III) and [111In]-labeled macrocyclic conjugates of diprenorphine: novel ligands designed for imaging studies of peripheral opioid receptors. Tetrahedron Lett. 2016, 72, 6127–6135. [Google Scholar] [CrossRef]
- Goswami, R.; Ponde, D. E.; Kung, M.-P.; Hou, C.; Kilbourn, M. R.; Kung, H. F. , Fluoroalkyl derivatives of dihydrotetrabenazine as positron emission tomography imaging agents targeting vesicular monoamine transporters. Nucl. Med. Biol. 2006, 33, 685–694. [Google Scholar] [CrossRef]
- van Wieringen, J.-P.; Shalgunov, V.; Janssen, H. M.; Fransen, M.; Janssen, A. G. M.; Michel, M. C.; Booij, J.; Elsinga, P. H. , Synthesis and characterization of a novel series of agonist compounds as potential radiopharmaceuticals for imaging dopamine D2/3 receptors in their high-affinity state. J. Med. Chem. 2014, 57, 391–410. [Google Scholar] [CrossRef]
- Galante, E.; Okamura, T.; Sander, K.; Kikuchi, T.; Okada, M.; Zhang, M.-R.; Robson, M.; Badar, A.; Lythgoe, M.; Koepp, M.; Arstad, E. , Development of purine-derived 18F-labeled pro-drug tracers for imaging of MRP1 activity with PET. J. Med. Chem. 2014, 57, 1023–1032. [Google Scholar] [CrossRef]
- Fulmor, W.; Lancaster, J. E.; Morton, G. O.; Brown, J. J.; Howell, C. F.; Nora, C. T.; Hardy, J. A. , Nuclear magnetic resonance studies in the 6, 14-endo-ethenotetrahydrothebaine Series. J. Am. Chem. Soc. 1967, 89, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Uff, B. C.; Mallard, A. S.; Davis, J. A.; Henson, R. , NMR spectra and stereochemistry of some 7-substituted 6,14-bridged thebaine derivatives. Magn. Reson. Chem. 1985, 23, 454–459. [Google Scholar] [CrossRef]
- Mazza, S. M.; Erickson, R. H.; Blake, P. R.; Lever, J. R. , Two-dimensional homonuclear and heteronuclear correlation NMR studies of diprenorphine: A prototypic 6α, 14α-endoethanotetrahydrothebaine. Magn. Reson. Chem. 1990, 28, 675–681. [Google Scholar] [CrossRef]
- Marton, J.; Sipos, A.; Henriksen, G.; Cumming, P.; Berényi, S.; Schmitt, B. M.; Szabó, Z. , NMR Analysis of a series of 6,14-ethenomorphinan derivatives as PET precursors and reference substances. ChemistrySelect 2021, 6, 5994–6005. [Google Scholar] [CrossRef]
- Biri, B.; Kalmár, J.; Nagy, L.; Sipos, A.; Zsuga, M.; Kéki, S. Energy-dependent collision-induced dissociation study of buprenorphine and its synthestic precursors. Rapid. Commun. Mass Spectrom. 2011, 25, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. PNAS 2002, 99, 2799–2804. [Google Scholar] [CrossRef]
- Dror, R. O.; Dirks, R. M.; Grossman, J. P.; Xu, H.; Shaw, D. E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Kenakin, T. P. Principles of drug-receptor interaction. in Goodman & Gilman’s The Pharmacological Basis of Therapeutics. (eds. Brunton, L.L., Hilal-Dandan, R. & Knollmann, B.C.), McGraw-Hill Education, 2018.
- O’Boyle, N. M.; Banck, M.; James, C. A.; Morley, C.; Vandermeersch, T.; Hutchison, G. R. Open Babel: An open chemical toolbox. J. Cheminformatics, 2011, 3, 1–33. [Google Scholar] [CrossRef]
- Halgren, T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eastman, P; Swails, J.; Chodera, J. D.; McGibbon, R. T.; Zhao, Y.; Beauchamp, K. A., et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 2017, 13, 7: e1005659.
- Case, D. A.; Duke, R. E.; Walker, R. C.; et al. AMBER 2022 Reference Manual. University of California, San Francisco, United States – California, 2022.
- Wang, J.; Wolf, R. M.; Caldwell, J. W.; Kollman, P. A.; Case, D. A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Bush, B. L.; Jack, D. B.; Bayly, C. I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K. A. A.; Raguette, L.; et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Loncharich, R. J.; Brooks, B. R.; Pastor, R. W. An impulse implementation of Langevin dynamics in a prime power method. Biopolymers 1992, 32, 523–535. [Google Scholar]
- Ryckaert, J. P.; Ciccotti, G.; Berendsen, H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
Figure 1.
Structures of selected opioid receptor radiotracers.
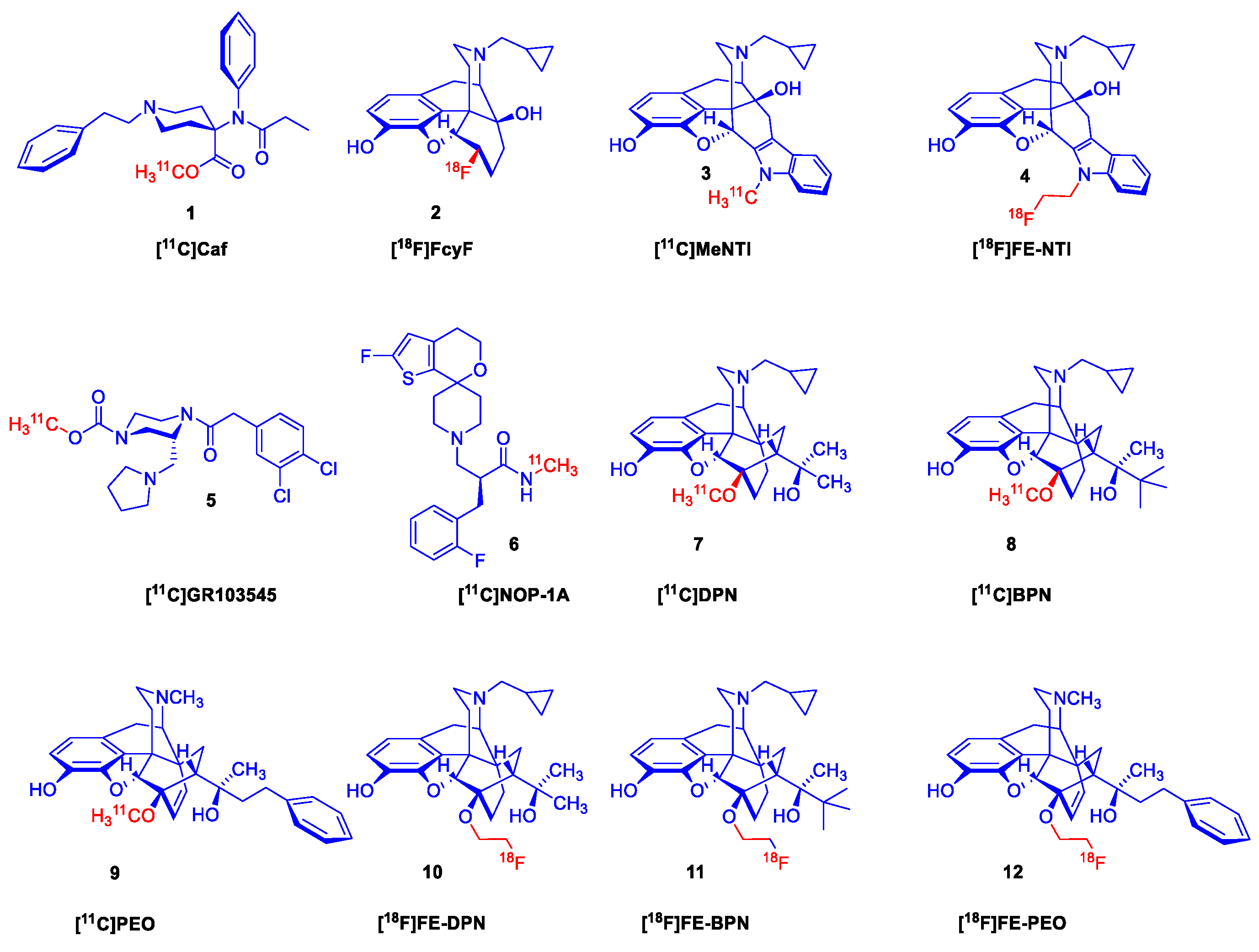
Figure 2.
Strategies for the synthesis of radiolabeled diprenorphine derivatives.
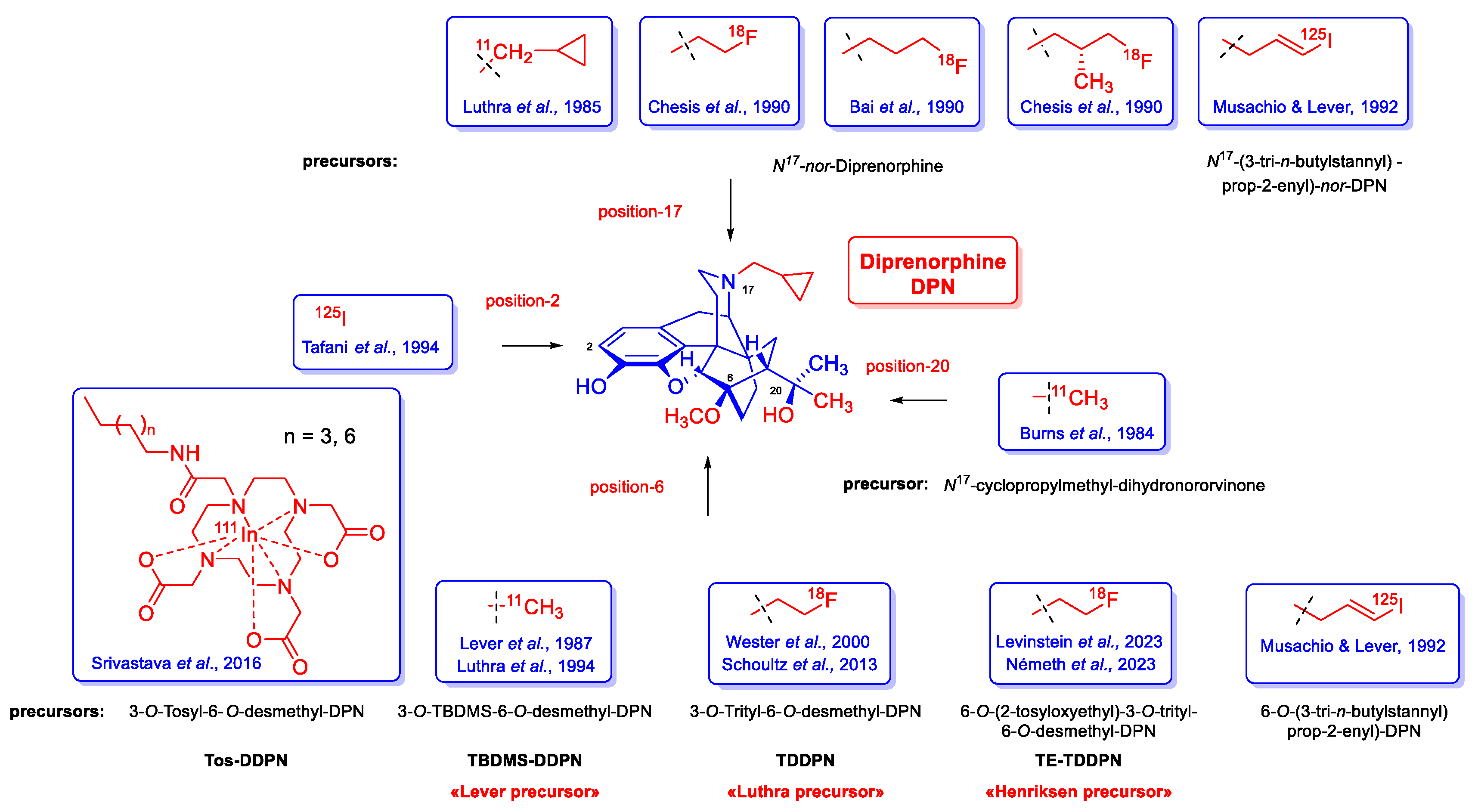
Figure 3.
Synthesis of diprenorphine (17). Reagents and conditions: (i): methyl vinyl ketone, reflux, 1 h, 93%; (ii): H2, 10% Pd-C, EtOH, 4 bar, 50 °C, 10h, 75%; (iii): methylmagnesium iodide, diethylether - benzene, reflux, 2 h; (iv): cyanogen bromide, CH2Cl2, room temperature, 16 h; (v): KOH, diethylene glycol, 165-170 °C, 75 min.; (vi): cyclopropylcarbonyl chloride, CH2Cl2, Et3N, 2 days; (vii): LiAlH4, THF, reflux, 4 h; (viii): KOH, diethylene glycol, 210-220 °C, nitrogen atmosphere, 2 h; (ix): (1) N-demethylation: A. 1.6 equiv. DEAD, benzene, reflux, 16 h, B. pyridine hydrochloride, EtOH, room temperature, 24 h; (2) N-alkylation: cyclopropylmethyl bromide, NaHCO3, DMF, 90 °C, 20 h; (x): methyl vinyl ketone, reflux, 1 h, 85 % (19a), 87 % (19b); (xi): A: H2, Pd-C, MeOH, 60 °C, 6 bar, 1 h, 91 % (20a), 7 h, 71 % (20b), B: 20a → 20b, saturated HCl in EtOH, reflux, 4 h, 81 %; (xii): cyclopropylmethyl bromide, NaHCO3, DMF, 90 °C, 20 h, 63 %; (xiii): methylmagnesium iodide, diethylether – toluene, reflux, 2 h, 94 %.
Figure 3.
Synthesis of diprenorphine (17). Reagents and conditions: (i): methyl vinyl ketone, reflux, 1 h, 93%; (ii): H2, 10% Pd-C, EtOH, 4 bar, 50 °C, 10h, 75%; (iii): methylmagnesium iodide, diethylether - benzene, reflux, 2 h; (iv): cyanogen bromide, CH2Cl2, room temperature, 16 h; (v): KOH, diethylene glycol, 165-170 °C, 75 min.; (vi): cyclopropylcarbonyl chloride, CH2Cl2, Et3N, 2 days; (vii): LiAlH4, THF, reflux, 4 h; (viii): KOH, diethylene glycol, 210-220 °C, nitrogen atmosphere, 2 h; (ix): (1) N-demethylation: A. 1.6 equiv. DEAD, benzene, reflux, 16 h, B. pyridine hydrochloride, EtOH, room temperature, 24 h; (2) N-alkylation: cyclopropylmethyl bromide, NaHCO3, DMF, 90 °C, 20 h; (x): methyl vinyl ketone, reflux, 1 h, 85 % (19a), 87 % (19b); (xi): A: H2, Pd-C, MeOH, 60 °C, 6 bar, 1 h, 91 % (20a), 7 h, 71 % (20b), B: 20a → 20b, saturated HCl in EtOH, reflux, 4 h, 81 %; (xii): cyclopropylmethyl bromide, NaHCO3, DMF, 90 °C, 20 h, 63 %; (xiii): methylmagnesium iodide, diethylether – toluene, reflux, 2 h, 94 %.
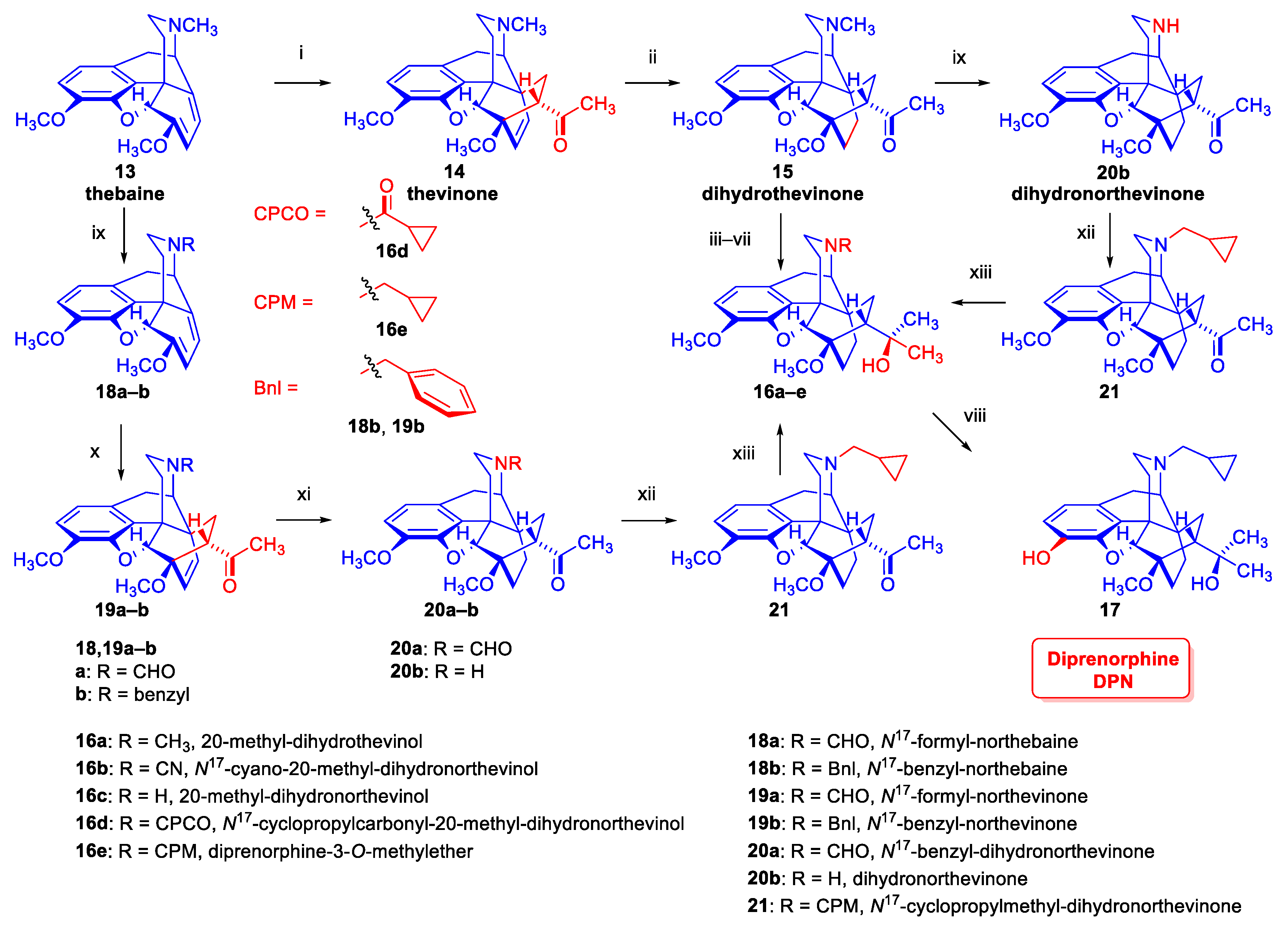
Figure 4.
Synthesis of the «Luthra precursor»: 3-O-trityl-6-O-desmethyl-diprenorphine. Reagents and conditions: (i): 15 equiv. LiAlH4, 4 equiv. CCl4, THF, reflux, 48 h; (ii): trityl chloride, Et3N, CH2Cl2, room temperature, 48 h.
Figure 4.
Synthesis of the «Luthra precursor»: 3-O-trityl-6-O-desmethyl-diprenorphine. Reagents and conditions: (i): 15 equiv. LiAlH4, 4 equiv. CCl4, THF, reflux, 48 h; (ii): trityl chloride, Et3N, CH2Cl2, room temperature, 48 h.
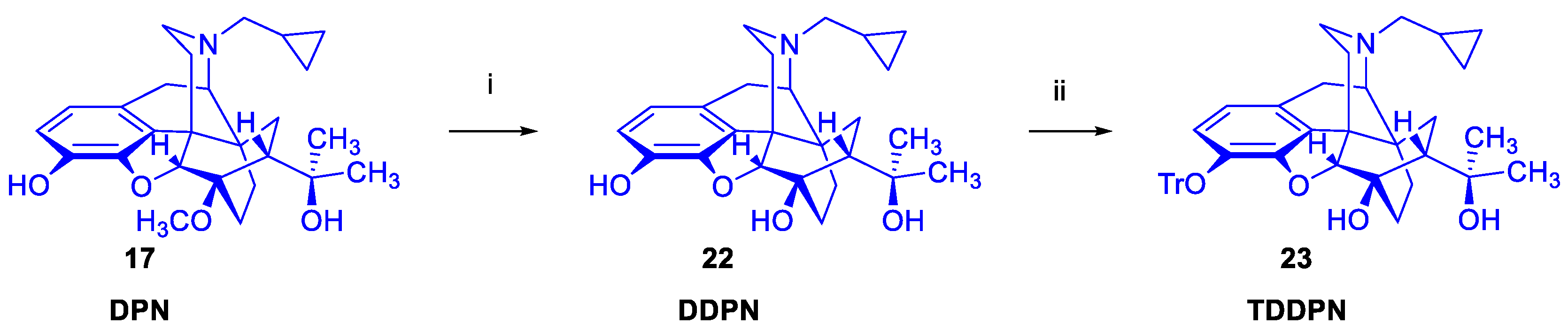
Figure 5.
Synthesis of 3-O-trityl-6-O-(tosyloxyalkyl)-6-O-desmethyl-diprenorphine derivatives for use as precursors for radiosyntheses of 18F-fluroalkylated-DPN radiotracers. Reagents and conditions: (i): TBDPSO(CH2)nBr, NaH, DMF, RT; (ii): 1M Bu4NF in THF, RT; (iii): Tos2O, pyridine, CH2Cl2; (iv): [K+⊂K222]18Fy, CH3CN, 90 °C, 10 min; (v): 1M HCl in EtOH, 40 °C, 5–10 min; (vi): TosO(CH2)nF, NaH, DMF, RT; (vii): AcOH-H2O, 100 °C, 5–10 min.
Figure 5.
Synthesis of 3-O-trityl-6-O-(tosyloxyalkyl)-6-O-desmethyl-diprenorphine derivatives for use as precursors for radiosyntheses of 18F-fluroalkylated-DPN radiotracers. Reagents and conditions: (i): TBDPSO(CH2)nBr, NaH, DMF, RT; (ii): 1M Bu4NF in THF, RT; (iii): Tos2O, pyridine, CH2Cl2; (iv): [K+⊂K222]18Fy, CH3CN, 90 °C, 10 min; (v): 1M HCl in EtOH, 40 °C, 5–10 min; (vi): TosO(CH2)nF, NaH, DMF, RT; (vii): AcOH-H2O, 100 °C, 5–10 min.
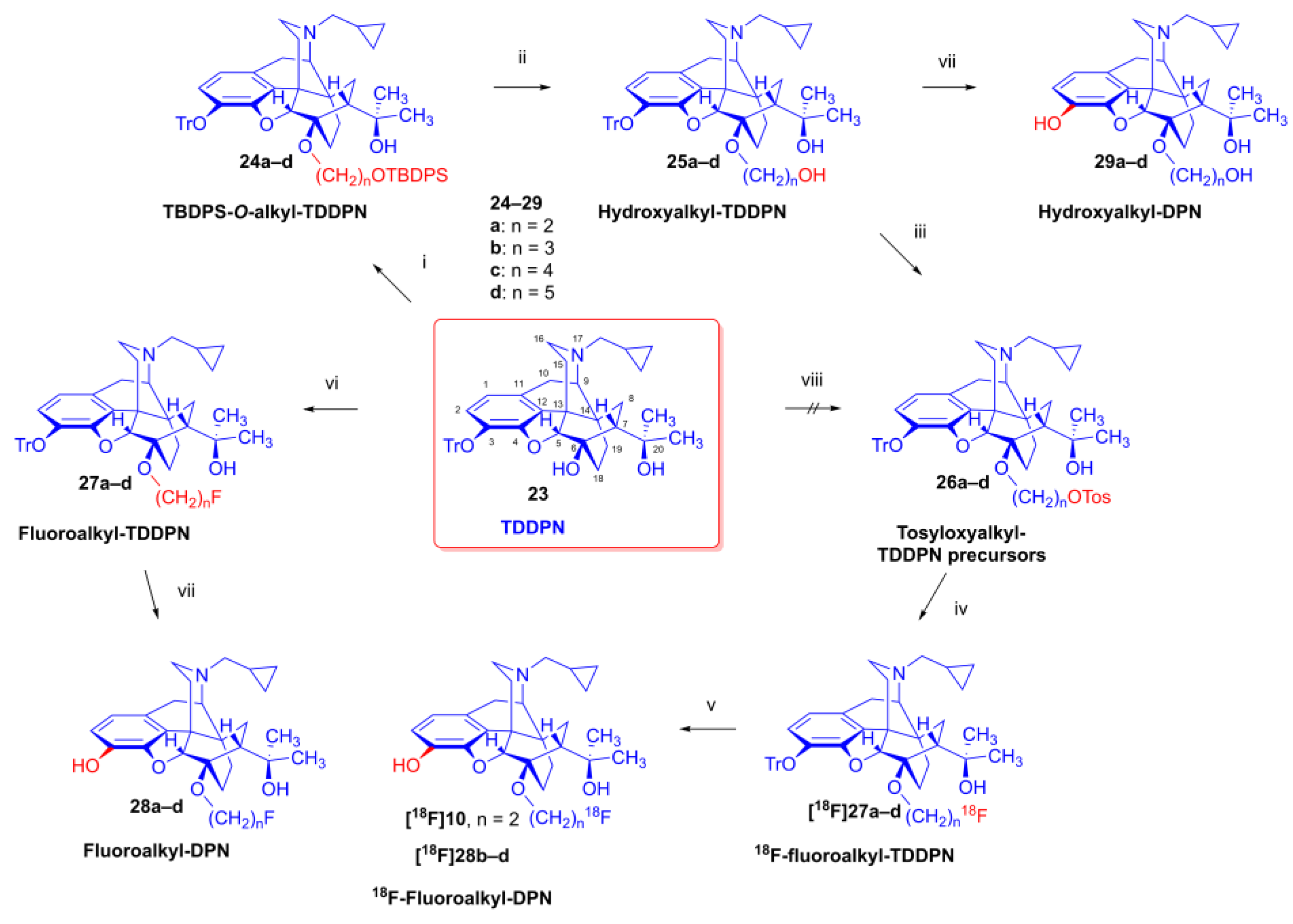
Figure 6.
Synthesis of fluoroalkyl-tosylates and bromoalkoxy-tert-butyl-diphenylsilanes. Reagents and conditions: (i): TosCl, DMAP, pyridine, CH2Cl2, 16 h, RT; (ii): TosCl, Et3N, CH2Cl2, 16 h, RT; (iii): TosCl, pyridine, CH2Cl2, 16 h, RT; (iv): 1M Bu4NF, acetonitrile, 24 h, reflux; (v): DAST, CH2Cl2, 4 h, RT; (vi): TBDMSCl, DIPEA, DMAP, CH2Cl2, 16 h, RT.
Figure 6.
Synthesis of fluoroalkyl-tosylates and bromoalkoxy-tert-butyl-diphenylsilanes. Reagents and conditions: (i): TosCl, DMAP, pyridine, CH2Cl2, 16 h, RT; (ii): TosCl, Et3N, CH2Cl2, 16 h, RT; (iii): TosCl, pyridine, CH2Cl2, 16 h, RT; (iv): 1M Bu4NF, acetonitrile, 24 h, reflux; (v): DAST, CH2Cl2, 4 h, RT; (vi): TBDMSCl, DIPEA, DMAP, CH2Cl2, 16 h, RT.
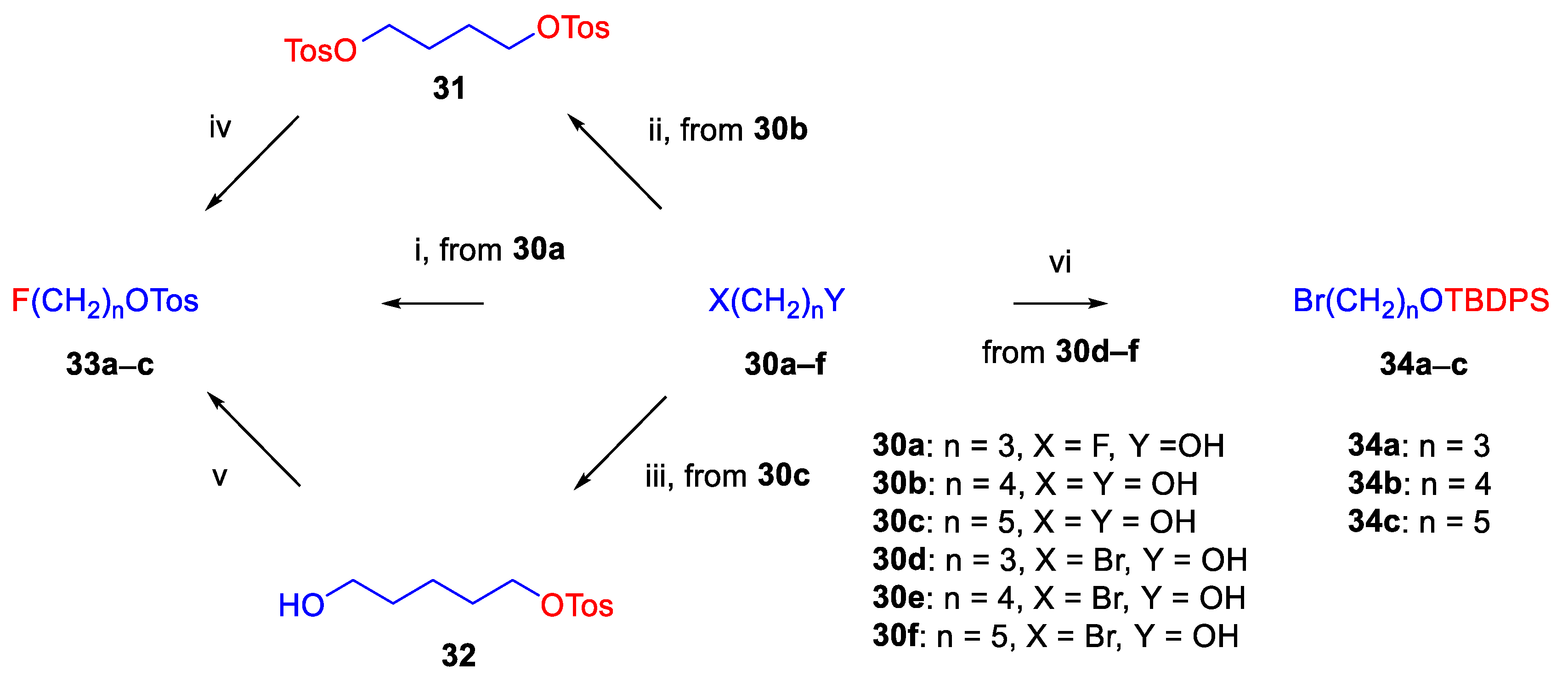
Figure 7.
Ligand RMSD (top) and COM Distance (bottom) for series 28 (left) and series 29 (right) compounds in complex with the active receptor state.
Figure 7.
Ligand RMSD (top) and COM Distance (bottom) for series 28 (left) and series 29 (right) compounds in complex with the active receptor state.
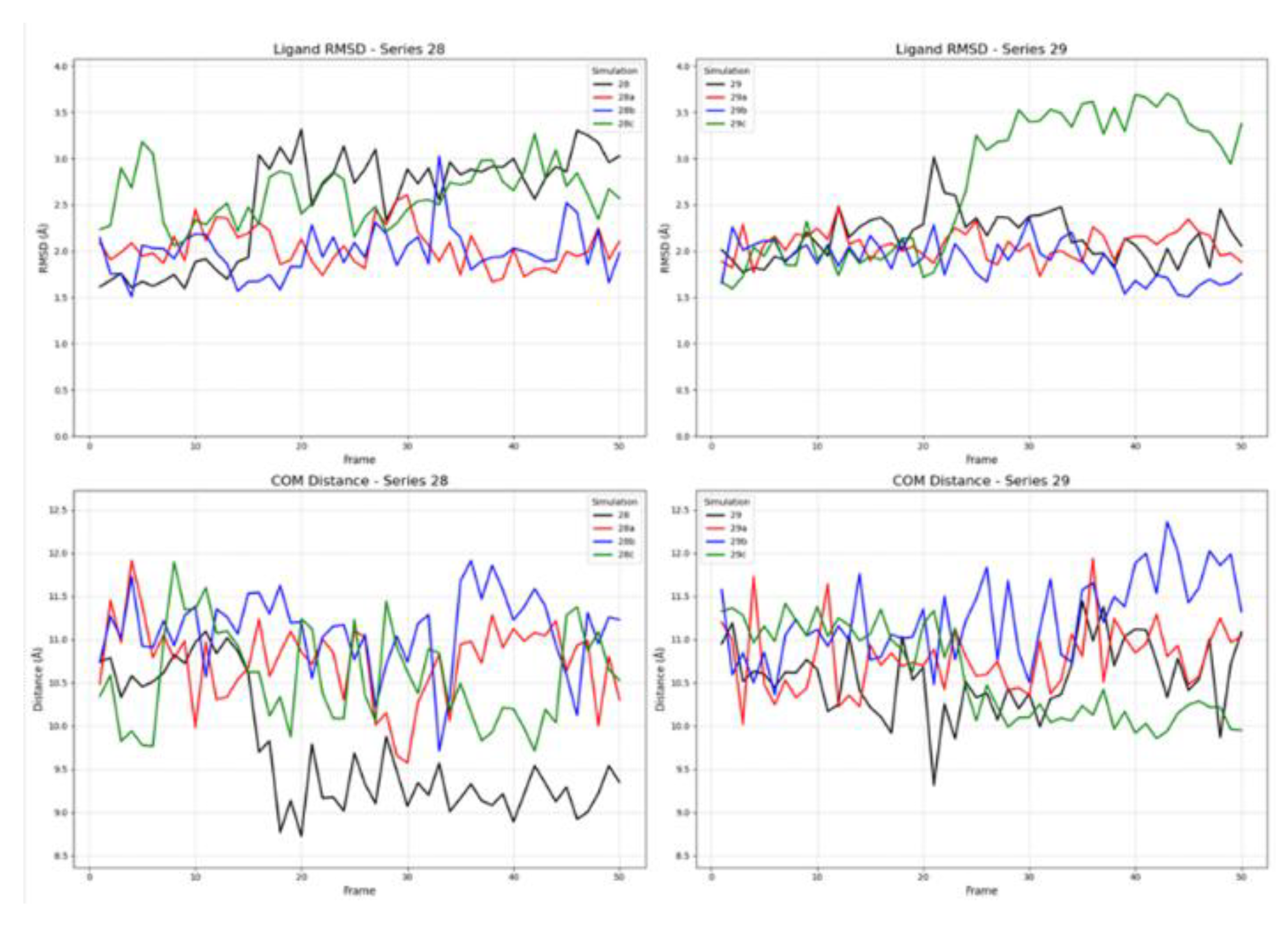
Figure 8.
Ligand RMSD (top) and COM Distance (bottom) for series 28 (left) and series 29 (right) compounds in complex with the inactive receptor state. The instability of ligand 28a (red trace) is evident.
Figure 8.
Ligand RMSD (top) and COM Distance (bottom) for series 28 (left) and series 29 (right) compounds in complex with the inactive receptor state. The instability of ligand 28a (red trace) is evident.
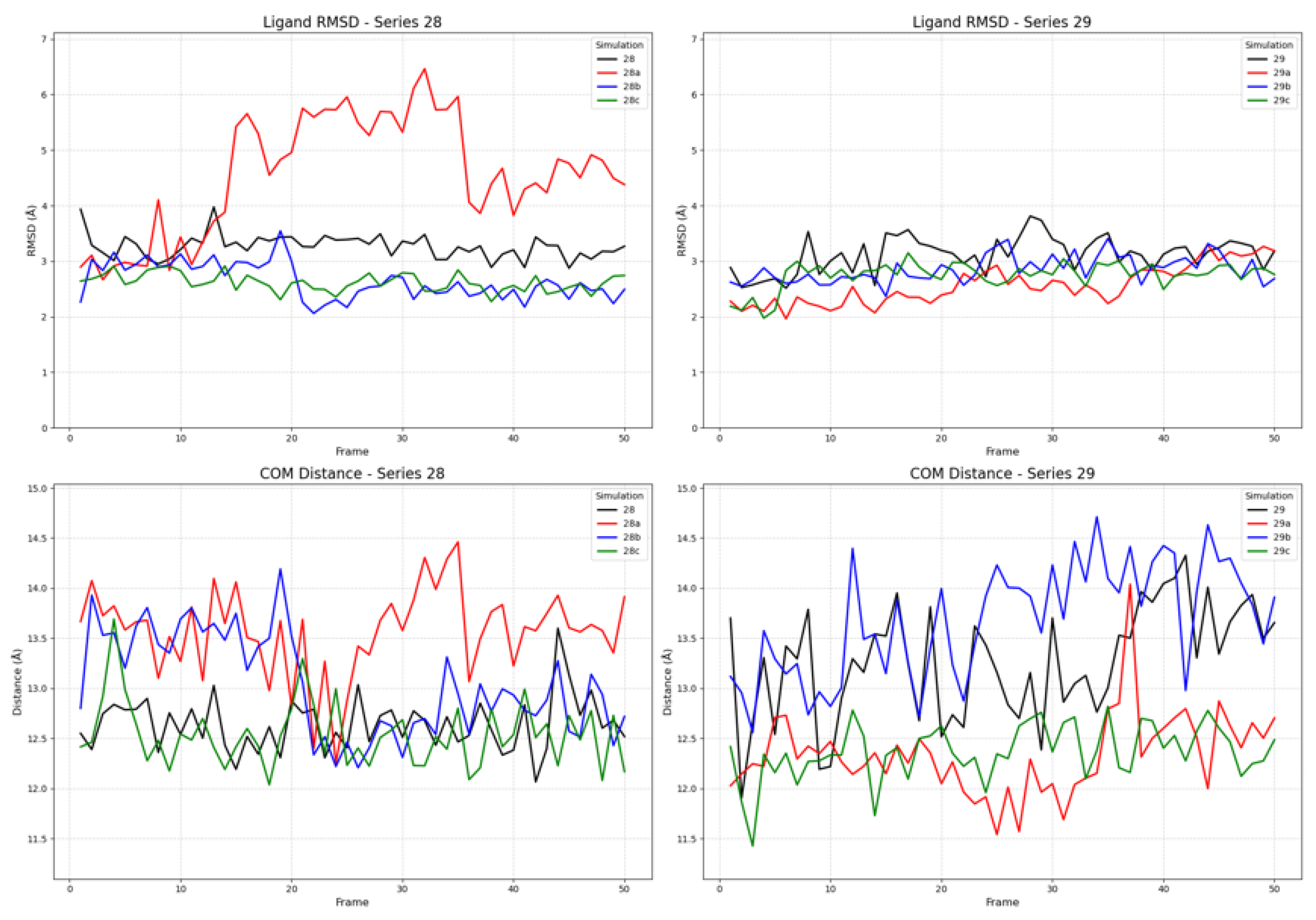
Figure 9.
Ligand RMSD (top) and COM Distance (bottom) for series 28 (left) and series 29 (right) compounds in complex with the partial agonist receptor state. The conformational instability of ligand 28a (red trace, top left) and ligand 29 (black trace, top right) is evident.
Figure 9.
Ligand RMSD (top) and COM Distance (bottom) for series 28 (left) and series 29 (right) compounds in complex with the partial agonist receptor state. The conformational instability of ligand 28a (red trace, top left) and ligand 29 (black trace, top right) is evident.
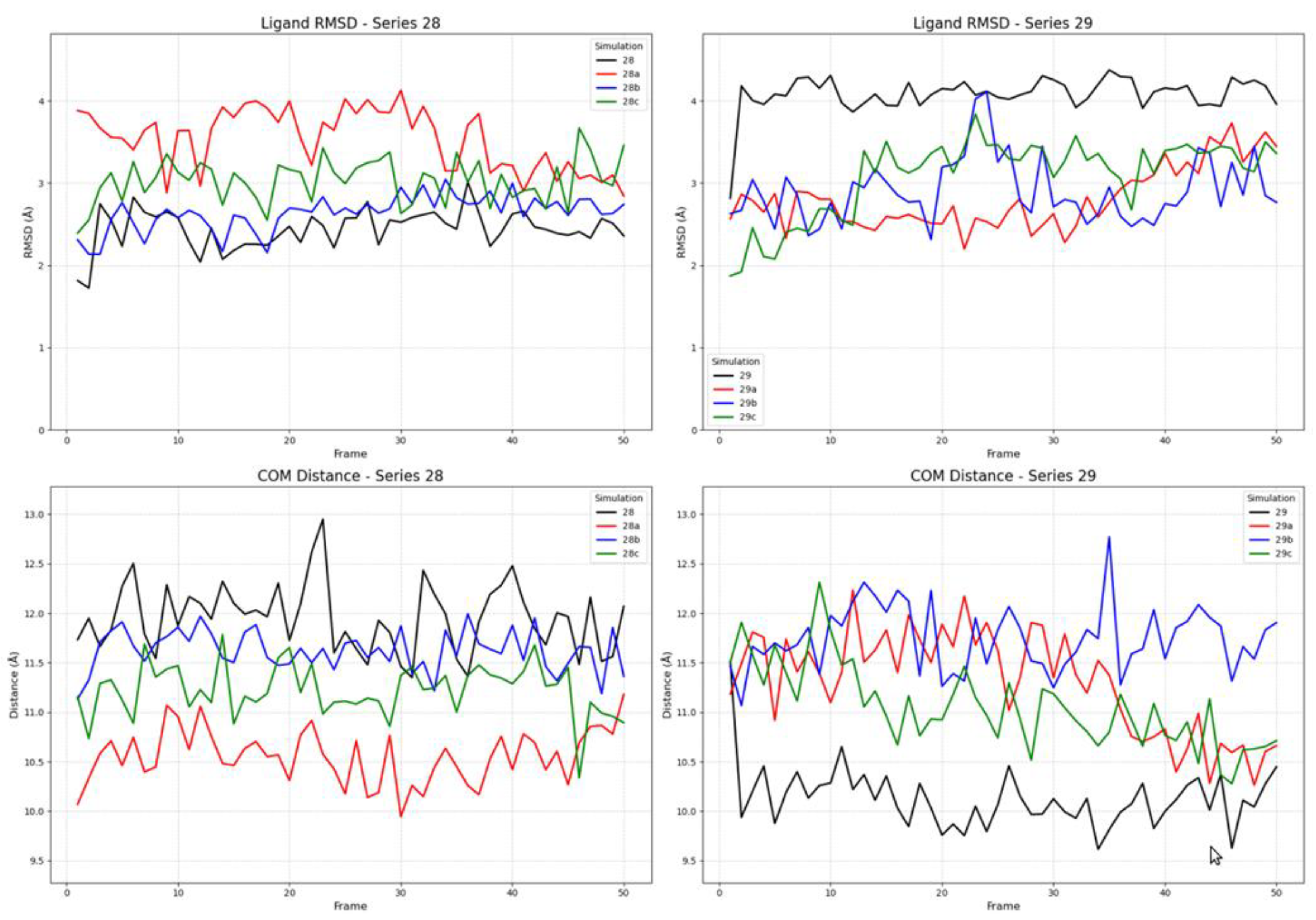
Figure 10.
Docking affinity scores (kcal/mol) for all ligands across the three receptor states. The agonist (active) state consistently shows the most favorable (most negative) binding energy for all compounds.
Figure 10.
Docking affinity scores (kcal/mol) for all ligands across the three receptor states. The agonist (active) state consistently shows the most favorable (most negative) binding energy for all compounds.
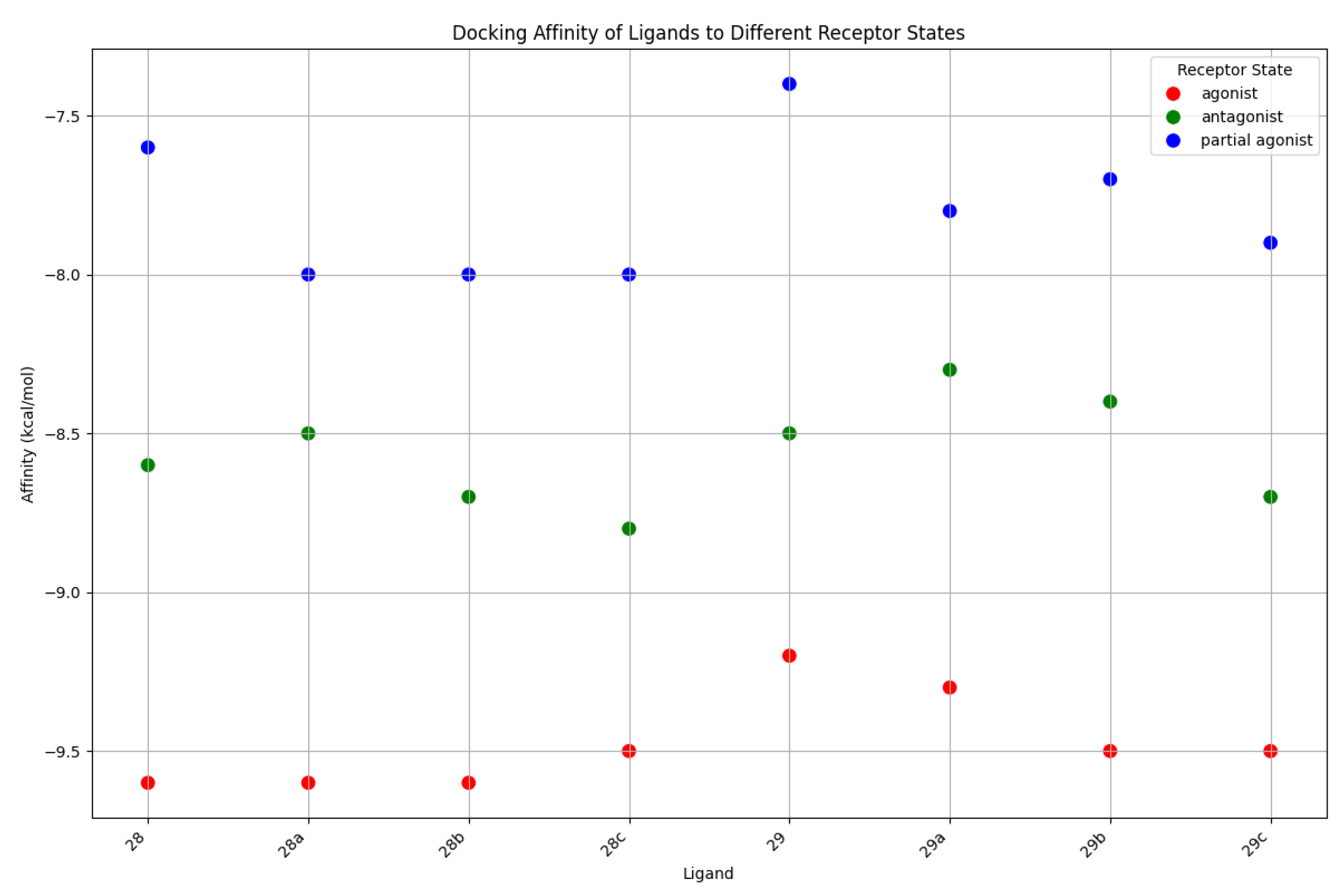

Table 1.
1H and 13C NMR chemical shifts and coupling constants of TP-TDDPN (26b), TB-TDDPN (26c) and TPe-TDDPN (26d)a in CDCl3.
Table 1.
1H and 13C NMR chemical shifts and coupling constants of TP-TDDPN (26b), TB-TDDPN (26c) and TPe-TDDPN (26d)a in CDCl3.
| Position | TP-TDDPN (26b) | TB-TDDPN (26c) | TPe-TDDPN (26d) | |||
| δ13C | δ1H(m, J in Hz) | δ13C | δ1H(m, J in Hz | δ13C | δ1H(m, J in Hz) | |
| 1 | 123.2 | 6.20 (d, 8.4) | 122.9 | 6.18 (d, 8.0) | 122.8 | 6.18 (d, 8.4) |
| 2 | 118.0 | 6.49 (d, 8.4) | 117.9 | 6.46 (d, 8.0) | 117.8 | 6.46 (d, 8.4) |
| 3 | 137.2 | - | 137.2 | - | 137.2 | - |
| 4 | 151.2 | - | 151.2 | - | 151.2 | - |
| 5β | 96.2 | 3.99 (d, 2.3) | 96.5 | 4.05 (d, 1.7) | 96.6 | 4.08 (d, 1.7) |
| 6 | 80.8 | - | 80.5 | - | 80.3 | - |
| 7β | 48.1 | 1.75 (app t, 10.1) | 48.0 | 1.78 (app t, 9.4) | 48.0 | 1.80 (app t, 9.7) |
| 8α | 32.1 | 0.95 (dd, 13.3, 9.7) | 32.1 | 0.96 (dd, 13.3, 9.7) | 32.1 | 0.97 (dd, 13.1, 9.8) |
| 8β | 2.74 (ddd, 13.4, 12.4, 3.9) | 2.75 (ddd, 13.8, 12.4, 3.9) | 2.76 (ddd, 13.1, 11.9, 3.9) | |||
| 9α | 57.9 | 2.89 (d, 6.1) | 57.9 | 2.89 (d, 6.0) | 57.9 | 2.89 (d, 6.4) |
| 10α | 22.5 | 2.05 (dd, 18.7, 6.1) | 22.5 | 2.04 (dd, 18.8, 6.0) | 22.5 | 2.04 (dd, 18.4, 6.4) |
| 10β | 2.85 (d, 18.7) | 2.84 (d, 18.8) | 2.85 (d, 18.4) | |||
| 11 | 130.7 | - | 130.6 | - | 130.6 | - |
| 12 | 131.9 | - | 131.9 | - | 132.0 | - |
| 13 | 46.8 | - | 46.7 | - | 46.7 | - |
| 14 | 35.7 | - | 35.7 | - | 35.7 | - |
| 15ax | 35.4 | 1.86 (td, 13.2, 5.4) | 35.4 | 1.87 (td, 13.9, 5.6) | 35.4 | 1.89 (td, 13.7, 5.7) |
| 15eq | 1.30–1.41(mb) | 1.42 (dd, 13.1, 2.7) | 1.26–1.62 (md) | |||
| 16ax | 43.5 | 2.14 (td, 13.0, 3.6) | 43.6 | 2.14 (td, 11.5, 3.8) | 43.6 | 2.14 (td, 13.2, 3.8) |
| 16eq | 2.54 (dd, 11.9, 5.0) | 2.54 (dd, 12.0, 5.4) | 2.55 (dd, 12.1, 4.7) | |||
| 18syn | 17.4 | 1.30–1.41 (mb) | 17.6 | 1.32–1.37 (m) | 17.6 | 1.26–1.62 (md) |
| 18anti | 1.49 (m) | 1.40–1.63 (mc) | 1.26–1.62 (md) | |||
| 19syn | 30.3 | 0.43 (m) | 29.7 | 0.41 (m) | 29.8 | 0.42 (tt, 12.7, 5.7) |
| 19anti | 0.88 (td, 12.4, 5.7) | 0.88 (td, 13.6, 5.7) | 0.88 (td, 13.3, 5.9) | |||
| 20 | 74.2 | - | 74.2 | - | 74.2 | - |
| Others | ||||||
| 20-CH3 | 24.8 | 1.13 (s) | 24.9 | 1.14 (s) | 24.8 | 1.14 (s) |
| 20-CH3 | 29.7 | 1.26 (s) | 29.7 | 1.31 (s) | 29.7 | 1.32 (s) |
| 6-O-(CH2)nOTos | ||||||
| Tos-CH3 | 21.6 | 2.41 (s) | 21.6 | 2.42 (s) | 21.6 | 2.43 (s) |
| Tos-3,5 | - | 7.31 (d, 8.1) | - | 7.33 (d, 8.1) | - | 7.33 (d, 8.4) |
| Tos-2,6 | - | 7.78 (d, 8.1) | - | 7.79 (d, 8.1) | - | 7.79 (d, 8.4) |
| Tos-C1 | 133.1 | - | 133.1 | - | 133.2 | - |
| Tos-C2,6 | 129.9 | - | 127.9 | - | 127.9 | - |
| Tos-C3,5 | 129.8 | - | 129.8 | - | 129.8 | - |
| Tos-C4 | 144.6 | - | 144.6 | - | 144.6 | - |
| cPropCH2syn | 3.2 | 0.05 (m) | 3.2 | 0.05 (m) | 3.2 | 0.05 (m) |
| cPropCH2anti | 4.1 | 0.43 (m) | 4.1 | 0.46 (m) | 4.1 | 0.46 (m) |
| cPropCH | 9.3 | 0.75 (m) | 9.3 | 0.75 (m) | 9.3 | 0.75 (m) |
| NCH2 (a) | 59.8 | 2.18 (dd, 12.9, 6.7) | 59.8 | 2.17 (dd, 12.8, 6.7) | 59.8 | 2.18 (dd, 12.6, 6.7) |
| NCH2 (b) | 2.32 (dd, 12.9, 5.6) | 2.32 (dd, 12.8, 5.7) | 2.32 (dd, 12.6, 5.7) | |||
| Trityl (Tr) | ||||||
| Tr (m,p) | - | 7.21–7.24 (m) | - | 7.19–7.24 (m) | - | 7.18–7.22 (m) |
| Tr (o) | - | 7.39–7.42 (m) | - | 7.38–7.42 (m) | - | 7.39–7.44 (m) |
| Ph3CO | 91.5 | - | 91.4 | - | 91.4 | - |
| pCTr | 127.3 | - | 127.3 | - | 127.2 | - |
| mCTr | 127.4 | - | 127.4 | - | 127.3 | - |
| oCTr | 129.4 | - | 129.3 | - | 129.3 | - |
| TrC1 | 144.2 | - | 144.2 | - | 144.2 | - |
| 20-OH | - | 4.83 (br s) | - | 5.07 (br s) | - | 5.18 (br s) |
| 6-O-(CH2)3OTos | 33.4 | 1.79–1.84 (m) | - | - | - | - |
| 6-O-(CH2)3OTos | 60.4, 67.4 | 3.49–4.12 (m) | - | - | - | - |
| 6-O-(CH2)4OTos | - | - | 25.7, 26.7 | 1.40–1.63 (mc) | - | - |
| 6-O-(CH2)4OTos | - | - | 64.0, 70.2 | 3.36–3.99 (m) | - | - |
| 6-O-(CH2)5OTos | - | - | - | - | 64.3, 70.3 | 1.26–1.62 (m) |
| 6-O-(CH2)5OTos | - | - | - | - | 28.6, 30.0 | 3.36–3.98 (m) |
a: observation frequency: 500.130 MHz (for 1H-NMR), 125.758 MHz (for 13C-NMR), in CDCl3, δ in ppm; b: overlapping signals of H-15eq and H-18syn; c: overlapping multiplets of H-18anti and 6-O-(CH2)4 OTos; d: overlapping multiplets of H-15eq and H-18syn.
Table 2.
1H and 13C NMR chemical shifts and coupling constants of FP-DPN (28b), FB-DPN (28c) and FPe-DPN (28d)a in CDCl3.
Table 2.
1H and 13C NMR chemical shifts and coupling constants of FP-DPN (28b), FB-DPN (28c) and FPe-DPN (28d)a in CDCl3.
| Position | TP-DPN (28b) | TB-DPN (28c) | TPe-DPN (28d) | |||
| δ 13C | δ 1H (m, J in Hz) | δ 13C | δ 1H (m, J in Hz) | δ 13C | δ 1H (m, J in Hz) | |
| 1 | 119.5 | 6.50 (d, 8.0) | 119.4 | 6.50 (d, 8.0) | 119.4 | 6.49 (d, 8.0) |
| 2 | 116.3 | 6.69 (d, 8.0) | 116.3 | 6.68 (d, 8.0) | 116.3 | 6.68 (d, 8.0) |
| 3 | 137.2 | - | 137.2 | - | 137.2 | - |
| 4 | 145.4 | - | 145.4 | - | 145.5 | - |
| 5β | 97.6 | 4.42 (d, 1.2) | 97.6 | 4.40 (d, 1.3) | 97.7 | 4.40 (d, 1.1) |
| 6 | 80.8 | - | 80.6 | - | 80.5 | - |
| 7β | 48.2 | 1.89–2.05 (me) | 48.1 | 1.94 (m) | 48.0 | 1.93 (app t, 10.0) |
| 8α | 32.3 | 1.02–1.10 (mf) | 32.2 | 1.01–1.09 (mm) | 32.2 | 1.01–1.09 (ms) |
| 8β | 2.87 (ddd, 13.5, 12.0, 4.0) | 2.86 (ddd, 13.7, 12.2, 3.7) | 2.86 (ddd, 13.8, 12.1, 3.7) | |||
| 9α | 58.1 | 3.01 (d, 6.7) | 58.1 | 3.01 (d, 6.5) | 58.1 | 3.00 (d, 6.7) |
| 10α | 22.6 | 1.89–2.05 (me) | 22.6 | 2.03 (dd, 18.4, 6.5) | 22.6 | 2.20 (dd, 18.7, 6.7) |
| 10β | 2.98 (d, 18.7) | 2.97 (d, 18.4) | 2.97 (d, 18.7) | |||
| 11 | 128.3 | - | 128.2 | - | 128.3 | - |
| 12 | 132.3 | - | 132.3 | - | 132.3 | - |
| 13 | 47.3 | - | 47.3 | - | 47.2 | - |
| 14 | 35.9 | - | 35.9 | - | 35.9 | - |
| 15ax | 35.5 | 2.18–2.30 (mg) | 35.3 | 2.17–2.30 (mn) | 35.5 | 2.03 (td, 13.8, 5.7) |
| 15eq | 1.67 (dd, 13.1, 2.7) | 1.66 (dd, 13.3, 2.5) | 1.46–1.79 (mt) | |||
| 16ax | 43.7 | 2.18–2.30 (mg) | 43.7 | 2.17–2.30 (mn) | 43.7 | 2.22–2.30 (m)u |
| 16eq | 2.63 (dd, 12.0, 5.4) | 2.63 (dd, 12.0, 5.0) | 2.63 (dd, 12.1, 5.0) | |||
| 18syn | 17.8 | 1.76–1.79 (mh) | 17.9 | 1.77–1.82 (mo) | 17.7 | 1.46–1.79 (mt) |
| 18anti | 1.76–1.79 (mh) | 1.77–1.82 (mo) | 1.46–1.79 (mt) | |||
| 19syn | 29.9 | 0.74 (m) | 29.7 | 0.73 (m) | 29.9 | 0.73 (m) |
| 19anti | 1.02–1.10 (mf) | 1.01–1.09 (mm) | 1.01–1.09 (ms) | |||
| 20 | 74.4 | - | 74.5 | - | 74.5 | - |
| Others | ||||||
| 20-CH3 | 24.8 | 1.20 (s) | 24.8 | 1.20 (s) | 24.8 | 1.20 (s) |
| 20-CH3 | 29.7 | 1.39 (s) | 29.9 | 1.39 (s) | 29.7 | 1.39 (s) |
| cPropCH2syn | 3.3 | 0.10 (m) | 3.3 | 0.09 (m) | 3.3 | 0.09 (m) |
| cPropCH2anti | 4.2 | 0.49 (m) | 4.1 | 0.49 (m) | 4.1 | 0.49 (m) |
| cPropCH | 9.4 | 0.80 (m) | 9.4 | 0.80 (m) | 9.4 | 0.80 (m) |
| NCH2 (a) | 59.8 | 2.18–2.30 (mg) | 59.8 | 2.17–2.30 (mn) | 59.8 | 2.22–2.30 (m)u |
| NCH2 (b) | 2.37 (dd, 12.6, 5.7) | 2.37 (dd, 12.7, 6.0) | 2.37 (dd, 12.6, 6.0) | |||
| 20-OH | - | 5.13 (br s) | - | 5.30 (br s) | - | 5.34 (br s) |
| 3-OH | 4.94 (br s) | 5.13 (br s) | 5.10 (br s) | |||
| 6-O-CH2CH2CH2F (b,b’) | 31.6 db | 1.89–2.05 (me) | - | - | - | - |
| 6-O-CH2CH2CH2F (a) | 60.8 dc | 3.78 (dt, 9.3, 6.3, 1H) | - | - | - | - |
| 6-O-CH2CH2CH2F (a’) | 4.07 (dt, 9.3, 5.9, 1H) | - | - | - | - | |
| 6-O-CH2CH2CH2F (c,c’) | 81.1 dd | 4.51–4.60 (m, 2H) | - | - | - | - |
| 6-O-CH2CH2CH2CH2F (a,a’) | - | - | 64.2 di | 3.67 (dt, 9.3, 6.1) | - | - |
| - | - | 3.99 (dt, 9.3, 6.1) | - | - | ||
| 6-O-CH2CH2CH2CH2F (b,b’) | - | - | 26.6 dj | 1.77–1.82 (mo) | - | - |
| 6-O-CH2CH2CH2CH2F (c,c’) | - | - | 27.3 dk | 1.77–1.82 (mo) | - | - |
| 6-O-CH2CH2CH2CH2F (d,d’) | - | - | 83.8 d | 4.42–4.51 (m) | - | - |
| 6-O-CH2CH2CH2CH2CH2F (a) | - | - | - | - | 64.5 | 3.63 (dt, 8.9, 6.4) |
| 6-O-CH2CH2CH2CH2CH2F (a’) | - | - | - | - | 3.94 (dt, 8.9, 6.4) | |
| 6-O-CH2CH2CH2CH2CH2F (b,b’) | - | - | - | - | 22.0 dp | 1.46–1.79 (mt) |
| 6-O-CH2CH2CH2CH2CH2F (c,c’) | - | - | - | - | 30.1 dq | 1.46–1.79 (mt) |
| 6-O-CH2CH2CH2CH2CH2F (d,d’) | - | - | - | - | 30.2 | 1.46–1.79 (mt) |
| 6-O-CH2CH2CH2CH2CH2F (e) | - | - | - | - | 83.9 dr | 4.38 (t, 6.0) |
| 6-O-CH2CH2CH2CH2CH2F (e’) | - | - | - | - | 4.48 (t, 6.0) | |
a: observation frequency: 500.130 MHz (for 1H-NMR), 125.758 MHz (for 13C-NMR), in CDCl3, δ in ppm; b: 2JC,F = 20.2 Hz; c: 3JC,F = 4.6 Hz; d: 1JC,F = 97.6 Hz; e: overlapping signals of H-7β, H-10α and 6-O-CH2CH2CH2F (b,b’); f: overlapping signals of H-8α and H-19anti; g: overlapping multiplets of H-15ax, H-16ax and NCH2 (a); h: overlapping signals of H-18syn and H-18anti; i: 4JC,F = 1.0Hz; j: 3JC,F = 4.6 Hz; k: 2JC,F = 20.1 Hz; l: 1JC,F = 165.0 Hz; m: overlapping signals of H-8α and H-19anti; n: overlapping multiplets of H-15ax, H-16ax and NCH2 (a); o: overlapping signals of H-18anti, H-18syn, 6-O-CH2CH2CH2CH2F (b,b’) and 6-O-CH2CH2CH2CH2F (c,c’); p: 3JC,F = 5.5 Hz; q: 2JC,F = 20.3 Hz; r: 1JC,F = 164.2 Hz; s: overlapping signals of H-8α and H-19anti; t: overlapping multiplets of H-18syn, H-15eq, H-18anti, and 6-O-CH2CH2CH2CH2CH2F; u: overlapping multiplets of H-16ax and NCH2 (a).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



