Submitted:
31 August 2025
Posted:
01 September 2025
You are already at the latest version
Abstract
Small Ruminant Lentiviruses are one of the common viral agents among livestock animals mainly sheeps and goats and are responsible for fatal diseases such as Maedi-Visna and Caprine Arthritis Encephalitis, hence resulting in compromised livestock outcomes. Developing a safe and effective vaccine against these viruses, that can provide protection in broad range of host species has been a great challenge, hence we employed different bioinformatic tools to design a novel multi-epitope vaccine construct based on the envelope gene of the virus. After predicting the secondary and tertiary structure of the vaccine construct, the structural accuracy was further enhanced by refinement and the stability/binding affinity of the vaccine construct was validated by using cutting-edge computational tools such as molecular docking and molecular dynamic simulation. Furthermore, the immune system activation potential was validated via immune simulation studies followed by the codon optimization and in-silico cloning. The vaccine construct showed good levels of immunogenicity and antigenicity without any allergic/toxic effects. The study revealed that the multi-epitope vaccine shows significant potency to combat SRLVs among goats and sheep simultaneously.
Keywords:
Reverse vaccinology
; Small Ruminant Lentiviruses
; molecular docking
; molecular dynamics simulation
1. Introduction
Maedi-Visna (MV) is a common viral infection mainly prevalent among sheeps and it progresses slowly throughout the life cycle leading to the development of severe pneumonia along with neurological aberrations such as tremors, weakness and paralysis etc. (Mosa and Zenad 2020). The infected sheeps also experience chronic indurative mastitis (Blacklaws and diseases 2012). Another common disease associated with the MV is Caprine Arthritis Encephalitis (CAE), however in most cases the host organism is goat. CAE infected goats exhibit a range of symptoms such as young goats have incomplete muscles development leading to ‘encephalitis’ where the infected goat experiences paresis, dizziness, seizures, difficulty in coordination ultimately leading to paralysis (Moroz, Czopowicz et al. 2022). While the older goats usually have ‘arthritis’, exhibition of enlarged and painful limbs. Both MV and CAE affected sheeps and goats experience sudden weight loss, lower milk production along with poor fur coat development (ENACHE, BARAITAREANU et al. 2017). Two common routes of MV and CAE transmission are: respiratory/oral (inhalation of the infective agents) and colostrum milk (Rahman, Ahmed et al. 2022).
The viral agents responsible for the incidence of MV and CAE are Maedi-Visna Virus (MVV) and Caprine Arthritis Encephalitis Virus (CAEV) respectively (Blacklaws, Berriatua et al. 2004) and it has been recently revealed that both these species are a viral continuum and can infect goat and sheeps without any specie barriers and are collectively called Small Ruminant Lentiviruses (SRLVs) (Minardi da Cruz, Singh et al. 2013). The genomic size of these SRLVs is around 8.4-9.2 kb (Santry, de Jong et al. 2013, Arcangeli, Torricelli et al. 2022). The viral genome encodes both structural and non-structural components. Majorly three structural genes: gag, pol and env make up the viral capsid, however the regulatory functions are performed by tat, vif and ref genes (Querat, Audoly et al. 1990, Pépin, Vitu et al. 1998, Gomez-Lucia, Barquero et al. 2018, Olech, Valas et al. 2018). Based on the genetic sequence information (gag and pol genes) from different viral SRLV isolates, these SRLVs have been divided into four genotypes namely A-C, E. Genotype-A includes the MVV whereas the CEAV isolates are included in the Genotype B, C and E (Molaee, Bazzucchi et al. 2020, Davaasuren, Molaee et al. 2024).
Timely diagnosis of SRLVs is a crucial parameter while designing any preventive measurement or treatment as the viral agent stays dormant in the host organism for a long period without the appearance of any notable clinical symptoms and hence remains undetected (Michiels, Van Mael et al. 2018). Various preventive/eradication measures have been employed to limit the spread of viral diseases among ungulates however, vaccination stands out as the most reliable treatment. Likewise, SRLV based vaccine development has also been targeted in the past (See Table 1), however currently, there is no commercially available vaccine against SRLVs that could provide protection against MVV and CAEV continuum simultaneously. Hence the aim of the study is to utilize the advancements in the field of bioinformatics, reverse vaccinology and protein structure predictions to develop a vaccine construct that could simultaneously provide protection against MV and CAE induced by SRLVs in diverse goats/sheep breeds, while using the coding sequence of the envelope gene of the virus: mainly involved in viral attachment and epitope presentation, so that the resulting vaccine construct is capable to boost sufficient immunity and can provide protection against diverse serotypes of the viral SRLV agents.
2. Material and Methods
Protein Sequence Retrieval
The protein sequences (amino acid sequences) of the envelope gene of the prevalent SRLV serotypes namely A1(NCBI Accession No. M60609), A1(NCBI Accession No. OL436272), A2(NCBI Accession No. AY101611), B1(NCBI Accession No.436260) and B2(NCBI Accession No. OL436262) were retrieved from the protein server of the National Center of Biotechnology Information (NCBI) (for details see Table 2) in the FASTA format and were subjected to multiple sequence alignment aided by Clustal Omega (Sievers and Higgins 2014). The conserved regions were further analyzed using JalView: a tool commonly used to analyze the multiple sequence alignment results and the assigned conservation scores were used to generate a consensus sequence that served as a representative of the SRLVs envelope gene sequence (Martin, Procter et al. 2020). The consensus sequence was also verified with the BioEdit software to ensure the accuracy of the conservation analysis (Hall, Biosciences et al. 2011). A flow chart describing the methodology of the study can be seen in the Supplementary Figure 1.
BCL Epitope Prediction
Since antibodies play a crucial role in stimulating the specific immunity, identifying the BCL epitopes is an essential part of designing a reserve vaccinology-based vaccine. BCL epitopes are generally classified into two types: conformational and linear. Various publicly accessible tools including the ABCPRED; predicts all the possible BCL epitopes on the basis of respective antigenicity scores (Saha, Raghava et al. 2006), BCEPRED; predicts the flexibility of the predicted BCL epitopes (Saha and Raghava 2004) and BEPIPRED; predicts the possible linear epitopes, were utilized for linear BCL epitopes prediction (Jespersen, Peters et al. 2017). Furthermore, the antigenicity of the selected epitopes was again validated by using VaxiJen 2.0 by keeping default parameters and a threshold value of >0.4 (Doytchinova and Flower 2007). All the peptide sequences that had lower antigenicity scores were discarded. Determination of the allergenicity and toxicity of the short-listed BCL epitopes was done by using AllerTOP: used to determine the allergen BCL epitopes (Dimitrov, Bangov et al. 2014) and ToxinPred: used for the determination of toxicity for the predicted BCL epitopes respectively (Sharma, Naorem et al. 2022). Both of the above tools were used with default parameters in order to find the potential linear BCL epitopes which have the ability to generate a potential and a robust immune response.
CTL and HTL Based Epitopes Prediction
Prediction of CD8+T Cell Epitopes
A comprehensive computational tool-based approach was employed to predict the potential CD8+ T cell epitopes, hence a combination of ProPred-I (Singh and Raghava 2001), EpiJen (Doytchinova, Guan et al. 2006), IEDB MHC-I Binding Prediction tools based on the NetCpan EL 4.1 updated versions was utilized. IEDB MHC-I Binding Prediction is based on the machine learning approaches and has been updated to NNAlign_MA to encompass the multiallelic dataset (Trolle, Metushi et al. 2015). The available alleles for BoLA on IEDB database (see Table 4) were utilized to match them with the query SRLV envelope sequence. Based on percentile score and other parameters, the predicted epitopes were further validated by AllerTOP, VaxiJen 2.0 and ToxinPred respectively to select those CTL epitopes that were antigenic, immunogenic, non-toxic and non-allergenic.
Prediction of CD4+T Cell Epitopes
The HTL epitopes were predicted by utilizing the publicly available NetMHCIIpan 2.1 tool which has been efficiently trained by making use of the artificial neural networks (ANN) and includes the Bovine leukocytes alleles of the MHC-II (Yang, Wei et al. 2024). The predicted epitopes were selected on the basis of their binding affinities (IC50: less than 50nm), percentile rank (<0.5) and prediction scores (>0.9) respectively. Furthermore, the NET-CTL 1.2 based tools like VaxiJen 2.0 and EpiJen were used to check the antigenicity of the predicted epitopes. Whereas the commonly used tools like ToxinPred and AllerTOP were used to determine the toxicity and allergenicity of the predicted epitopes.
Prediction of Discontinuous Epitopes
Discontinuous epitopes are of significant importance as they help us to predict the antigen residues that might go to interact strongly with the antibody of the host organism. Hence, ElliPro webserver was used to make the discontinuous epitopes predictions using default parameters (Ponomarenko, Bui et al. 2008).
Design of the SRLV Based Vaccine
All the selected BCL (ten), CTL (four) and HTL (four) epitopes were fused together to design the SRLV based vaccine candidate. All these epitopes were highly conserved, highly antigenic, non-toxic and non-allergenic. At the N-terminal end of the construct, a TLR2 agonist adjuvant namely Pam3CSK4 was used to enhance the immune response and it was connected with the first CTL epitope by using the ‘‘EAAAK’’ linker that provided flexibility between the adjuvant and the multiepitopic regions. Further, CTL epitopes were linked together via the ‘‘AAY’’ linkers whereas they were joined further to the HTL epitopes using the ‘‘EAAAK’’ linker molecule. Special linker sequences such as the ‘‘GPGPG’’ were used for linking all the HTL epitopes, they were further joined with the BCL epitopes that were linked together by the ‘‘GPGPG’ linkers. Furthermore, the ‘‘EAAAK’’ linker was used to connect the rest of vaccine construct with the Histidine 6x-Tag at the N-terminal of the vaccine construct to assist the purification/recovery of the vaccine construct after expression.
Physiochemical Properties
The physiochemical properties of the generated vaccine candidate were determined by using the Expasy ProtParam online server and as a result various properties of the vaccine construct such as molecular weight, molecular formula, isoelectric point (PI), instability index (II), estimated half-life, number of atoms and Grand average of hydropathicity (GRAVY) were calculated (Gasteiger, Hoogland et al. 2005). Furthermore, the solubility of the vaccine candidate was also evaluated using SolPro to ensure its viability inside the living cell (Magnan, Randall et al. 2009).
Secondary Structure Prediction
The secondary structure of the vaccine construct was predicted by using the SOPMA online server with default parameters and the percentage of different structural parameters such as alpha helix (H), beta sheets (E), random coils (C) and beta-turns was estimated (Geourjon and Deleage 1995). Additionally, the protein structure of the vaccine was also analyzed on a neural network tool PSIPRED to reveal different type of chain elements and types of amino acid (McGuffin, Bryson et al. 2000).
Tertiary Structure Prediction
The tertiary structure of the vaccine construct was predicted by using the widely utilized protein prediction server called I-TASSER (Zhang 2008). It mainly relies on the multiple threading approach to predict the 3-D structures of the proteins by using the iterative assembly tools. The linear sequence of the vaccine construct was uploaded on the I-TASSER server to get the tertiary structure of the protein. All the predicted models were analyzed by using the ProCheck server (Laskowski, MacArthur et al. 1993). Based on the Ramachandran plots and other outcomes, the best model was chosen and further refinement of the chosen model was performed using the Galaxy Refine server (Heo, Park et al. 2013). The refined models were again analyzed using the ProCheck server, and the best model was chosen for further studies on the basis of residues in the favored region, clash scores, Mol Probity and Root Mean Square Deviation (RMSD) of atoms. In addition, the overall quality of the chosen model was also verified with a protein structure analysis ProSa webserver (Wiederstein and Sippl 2007).
Molecular Docking Analysis of the SRLV Based Vaccine Construct with TLR-2 and TLR-4 Receptors
The interactions between the potential SRLV based vaccine construct and the most common transmembrane pattern recognition receptors such as TLR-2 and TLR-4 were predicted at the protein-protein level by using the ClusPro webserver (Kozakov, Hall et al. 2017). ClusPro works around three algorithms involving combination of rigid-body docking, energy-based scoring and clustering. It employs the use of FFT based technique; a modified PIPER algorithm, exploring billions of protein interacting/docking orientations based on the energy calculations of Van der Waals, electrostatic and desolvation forces. Post-docking, the server then ranks them based on multiple scoring schemes be it balanced, electrostatic favored or hydrophobic favored followed by grouping the similar ones based on their iRMSD (interface Root Mean Square Deviation). The largest cluster score indicates the most probable docked site. For this purpose, the structures of the human TLR-2 complex (PDB ID:6NIG) and TLR-4 complex (PDB ID: 4G8A) were retrieved from the Protein Data Bank and their extracellular domains were utilized to check their possible interactions with the vaccine construct.
The output of ClusPro webserver was analyzed and the best models showing the interaction between extracellular domains of the ligands and receptor molecule were studied further based on their highest cluster scores and lowest energy values. Large cluster scores indicate the appearance of many similar confirmations, hence increasing the confidence to choose it as the best docked pose and lowest energy values suggests a strong and a favorable interaction between the two proteins. Hence, together a higher cluster score and low energy confirmation works best together and the best docking complexes for each ligand molecule were selected.
Furthermore, energy and stability linked properties of the docked complexes were analyzed by using PRODIGY (Xue, Rodrigues et al. 2016). PRODIGY gives a valuable output regarding the docked models binding affinity (delta G) and interfacial properties like interface area, interfacial contacts and interacting residues along with the dissociation constant (Kd). Furthermore, PDBePISA was used to evaluate if our docked model is biologically meaningful. It also identified the interfaces between chains and evaluates interaction energies (Krissinel and Henrick 2007). The chosen docked complex structures were also visualized manually by using Chimera (Pettersen, Goddard et al. 2004) and PyMol (Seeliger and de Groot 2010). Furthermore, the properties of the complex such as the number of chains, no. of bonds, interacting residues etc. were also identified by confirmed by PDBSum (Laskowski, Jabłońska et al. 2018).
Molecular Dynamic Simulations
After completing the docking studies, the vaccine construct containing the TLRs was subjected to additional examination for stability within the binding pocket. The Schrodinger Maestro software package was utilized to perform a 100 ns molecular dynamics simulation of the complexes (Kosloff and Kosloff 1983). The vaccine construct containing the TLRs complex was prepared using the Protein Preparation tool available in the SCHRODINGER suite. The OPLS4 force field and TIP3P water model were utilized under constant temperature and pressure conditions, with a periodic boundary condition implemented via the System Builder tool. Equilibrium simulations were conducted at a temperature of 300 K and a pressure of 1 bar, utilizing 100 ns for both NVT and NPT ensembles. The molecular dynamics simulations were performed under NPT conditions for a period of 100 ns to analyse the complex’s RMSD, RMSF, Rg, and SASA.
Immune Simulation Studies
The natural immune stimulating potential of the generated vaccine construct was tested using the C-ImmSim server: a machine learning based tool that visualizes the host humoral immune system against the administered vaccine construct (Rapin, Lund et al. 2011). LPS-free injection with a random seed of 12.643 was chosen whereas the simulation volume was kept at 50. Furthermore, the number of simulation steps were kept at 1000 with intervals of 1, 84 and 168.
Codon Optimization and In-Silico Cloning
Efficient expression of the vaccine construct in a suitable prokaryotic organism such as E. coli is essential to ensure the large-scale production of the protein product. To ensure this, a combination of tools was utilized. First, the codon optimization was carried out using the JCat Server (Grote, Hiller et al. 2005) which ensured the best expression of the construct along with the prediction of the GC content percentage and CAI scores. To facilitate the proper insertion of the linear construct into the cloning vector, restriction sites were added for XhoI and BamHI restriction enzymes at the N and C terminals of the vaccine sequence respectively. The construct was later cloned into the pET28a (+) cloning vector by using the SnapGene tool (https://www.snapgene.com/).
3. Results
Sequence Retrieval
The amino acid sequences coding for the envelope protein of the selected SRLV isolates responsible for the incidence of MVV and CAEV were successfully retrieved from NCBI and multiple sequence alignment was done to visualize the conserved residues by JalView and BioEdit software. As a result, a conserved consensus sequence based on the envelope gene sequence of the most prevalent SRLV isolates was generated.
Linear BCL Epitopes Prediction
Linear BCL epitopes were successfully predicted through the use of highly interactive and authentic servers such as ABCPRED, BCEPRED, BEPIPRED respectively. Furthermore, VaxiJen 2.0 was utilized for the prediction of immunogenicity of epitopes whereas their allergenicity and toxicity was validated via AllerTop and ToxinPred respectively. As a result, ten BCL epitopes (15-mer) were selected. Furthermore, the antigenicity scores of the selected epitopes ranged 0.51 to 1.26 whereas the toxicity predicting SVM scores ranged -0.57 to -1.40. The outcomes of the linear BCL epitopes prediction are listed in the Table 3.
CTL Epitopes
CTL epitopes play a vital role in any vaccine construct preparation as they elicit cell mediated-immunity. At first, a total of twenty-one epitopes were predicted by the ProPred-I server out of which only four (9-mer) epitopes were filtered out to be further used in the vaccine construct. The selected epitopes had a minimal percentile rank ranging from 0.02 to 0.07, scores ranging from 0.76 to 0.94 and exhibited strong binding affinity (IC50 <50 nM) towards all 4 different BOLA alleles (BoLA-T2c, BoLA-HD6, BoLA-1:02301, BoLA-2:00801) and proved to be non-allergen, non-toxic and immunogenic. The results of the CTL epitope predictions are listed in the Table 4.

Table 4.
List of selected CTL Epitopes for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
Table 4.
List of selected CTL Epitopes for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
| Allele | Epitope Sequence | Score | Percentile Rank | AllerTOP | VaxiJen | ToxinPred |
|---|---|---|---|---|---|---|
| BoLA-T2c | SMMHQQMLL | 0.933478 | 0.04 | Non-Allergen | Non-Immunogen (100%) | Non-Toxin |
| BoLA-HD6 | RQQEQEKLL | 0.945614 | 0.03 | Non-Allergen | Immunogen (100%) | Non-Toxin |
| BoLA-1:02301 | RQQEQEKLL | 0.768478 | 0.07 | Non-Allergen | Immunogen (100%) | Non-Toxin |
| BoLA-2:00801 | YIAGGKQFW | 0.859677 | 0.02 | Non-Allergen | Immunogen (66%) | Non-Toxin |
| This first column shows the respective allele used to predict the CTL epitope sequence for the vaccine construct whereas the epitope sequence length, score and percentile length are listed in the second, third and fourth column respecttively. Furthermore, the results of allergenicity analysis, antigenicity, toxicity are depicted in column 4-7 respectively. | ||||||
HTL Epitopes
Being a crucial player in cellular and humoral based immunity in the host, a combination of tools was utilized for the successful prediction of the HTL epitopes. As a result, four HTL epitopes having highest binding affinity (IC50: <50nm), percentile rank (<0.5) and prediction scores (>0.9) were selected. Note that the Net Chop results showed the C-term of 3.0 and a threshold level of 0.5. Furthermore, it was also made sure that the selected epitopes were non-toxic and non-allergenic. The predicted HTL epitopes are listed in the Table 5.
Prediction of Discontinuous Epitopes
Epitopes Selection Criteria
All the selected candidate epitopes possessed three desirable attributes: non-toxic, non-allergen and highly antigenic/immunogenic as confirmed by the results of the above bioinformatics tools such as ToxinPred, AllerTop and VexiJen. The tools utilized in the current study are mentioned in the Table 6.
Design of the SRLV Based Vaccine
A multi-epitope vaccine construction involved the assembly of ten BCL epitopes linked together by GPGPG linkers. In the same manner four CTL and four HTL epitopes were linked by using the AAY and GPGPG linkers respectively. The adjuvant was kept at the N-terminal of the construct and it was joined with the CTL epitopes (MHC-I) molecules by using the EAAAK linkers, they were further connected with the HTL (MHC-II) epitopes (via the EAAAK linkers). Further, the HTL epitopes were linked with BCL epitopes by using the EAAAK linker. The C-terminal end of the vaccine construct had 6x His-Tag molecules to enable identification and purification of expressed portion of vaccine. The histidine tag was joined to the BCL epitopes by using the EAAA linker. As a result, a final construct with an amino acid length of 361 was generated. The one-code amino acid sequence and overall geometry of the final vaccine construct is shown in the Figure 1.
Physiochemical Properties
The physiochemical properties of the vaccine construct via ProtParam revealed that there are 5426 atoms in the vaccine construct with molecular formula C1723H2679N505O500S19. The extinction coefficient for the construct measured in water is 108440 M-1 cm-1. The estimated half life for the vaccine construct is 30 hours when measured in mammalian reticulocytes (in-vitro). The instability index (II) of 35.15 indicated that the protein construct is stable, further validated by the aliphatic index (53.02) and GRAVY (-0.840). SolPro analysis also showed that the generated vaccine candidate is soluble with a score of 0.978 upon overexpression. However, further details of the physiochemical properties of the vaccine construct are listed in the Table 7.
Secondary Structure Prediction
In order to have a complete understanding about the functional and structural properties of our vaccine construct, its secondary structure was predicted using SOPMA that uses homology bases and statistical methods. It revealed that there is an abundance of structure elements such as alpha-helix (26.04%) providing structural stability to the vaccine construct, along with turns, extended strands (18.28%) and loops hence allowing enough room for evolutionary changes and functional binding stuff. The structural composition not only gave insights about the potential antigenic regions rather its stability too. Supplementary Table 2 shows the results of the SOPMA analysis of the vaccine construct that predicted the distribution of the secondary structure. PSIPRED also validated the above fact that there exists different kind of secondary structure elements such as helix, chains and coils, along with hydrophobic, hydrophilic amino acid residues as illustrated in the Figure 1 (for more details see Supplementary Figure 3).
Tertiary Structure Prediction
The tertiary structure of the vaccine construct was predicted by the help of I-TASSER and the best model was chosen on the basis of different considerations, further it showed more than 90% of the residues lie in the favored region when analyzed through the Ramachandran plot. Hence, to further improve or enhance the efficiency/accuracy of the predicted model, the model was refined through the Galaxy Refine server and the model with the finest results (95% residues in the allowed region) was chosen for further studies and was viewed with the help of visualization tools such as Chimera and PyMol. The results of Galaxy Refine modelling are shown in the Supplementary Table 3 and the 3-D structure of the vaccine construct with Ramachandran plot for both unrefined and refined model are illustrated in the Figure 1. Furthermore, the selected model was also validated with the help of ProSa webserver that predicted the overall model quality Z score of -2.0 and the geometry of the 3-D structure was also validated via the ProCheck webserver (See Supplementary Figure 4).
Molecular Docking Analysis of the SRLV Based Vaccine Construct with TLR-2 and TLR-4 Receptors
Understanding the interactions between the proposed vaccine construct and the immune stimulators in the body holds great significance hence molecular docking analysis between the TLR-2 and TLR-4 receptor molecules was done using the ClusPro webserver. ClusPro generated a range of models that were individually visualized to observe the interacting residues between the ligands and the SRLV based vaccine construct. The best model was chosen on the basis of cluster size, energy values, interacting residues, location and type of bonding (Supplementary Table 4 and Supplementary Table 5). The analysis of the docking complex through PDBSum interaction analysis revealed some useful insights highlighting the major interacting residues and bond-types in the docked complex. The docked complex based on vaccine construct and the TLR-2 ligand (6NIG) revealed that there exists a good level of binding as evident by the presence of 16 hydrogen bonds, 3 salt bridges and 205 non-bonding contacts. Whereas, the TLR-4 (4G8A) based docked complex revealed that there are 10 salt bridges, 33 hydrogen bonds along with 340 non-bonded atoms, hence revealing a much stronger interaction between the suggested vaccine construct and the TLR-4 as compared to the TLR-2 molecule as shown in the Figure 2. The same was supported via the PRODIGY outcome which revealed that TLR-4 based ligand molecule has more stronger binding ability with vaccine construct as suggested by binding affinity of -22.5 kcal/mol compared to the -17.4 kcal/mol for the TLR-2 as it had more interfacial residues (30) as compared to 22 and more hydrogen bonds 31 versus 16. The disassociation constant also shows that 4G8A binds 10,000 times more tightly as compared to the 6NIG molecule. The results of the PRODIGY analysis are listed in the Table 8(a) and 8(b) respectively.
Table 8.
(a): PRODIGY Analysis for the Docked Vaccine Construct with 6NIG Ligand.
| Protein-Protein Complex | ΔG (kcal mol-1) | Kd (M) at °C |
ICs charged-charged | ICs charge-polar | ICs charged-apolar | ICs polar-polar | ICs polar-apolar | ICs apolar-apolar | NIS charged | NIS apolar |
|---|---|---|---|---|---|---|---|---|---|---|
| 6NIG_ Construct |
-17.4 | 1.7e-13 | 22 | 21 | 50 | 16 | 33 | 39 | 27.14 | 33.27 |
| The above table shows the outcome of the PRODIGY analysis for the docked vaccine construct for the TLR-2 based ligand 6NIG. Various affinity binding parameters are listed in the respective columns. | ||||||||||
Table 8.
(b): PRODIGY Analysis for the Docked Vaccine Construct with 4G8A Ligand.
| Protein-Protein Complex | ΔG (kcal mol-1) | Kd (M) at °C |
ICs charged-charged | ICs charged-polar | ICs charged-apolar | ICs polar-polar | ICs polar-apolar | ICs apolar-apolar | NIS charged | NIS apolar | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4G8A-Construct | -22.5 | 3e-17 | 30 | 38 | 96 | 13 | 31 | 20 | 23.69 | 37.85 | |
| The above table shows the outcome of the PRODIGY analysis for the docked vaccine construct for the TLR-4 based ligand 4G8A. Various affinity binding parameters are listed in the respective columns. | |||||||||||
MD Simulations
A molecular dynamics simulation was performed for 100 ns to analyze the dynamic motion and stability of the developed vaccine complex. The assessment of the docked complexes stability was conducted through the analysis of RMSD, RMSF, and Rg throughout the simulation duration. The RMSD graph for the TLR2-vaccine complex shows slight variations ranging from 2.76 nm to 5.91 nm, ultimately reaching stability after 40 ns. The TLR4-vaccine complex exhibited commendable stability ranging from 2.68 nm to 14.39 nm, reflecting only a slight variation after 25 ns. RMSF analysis was utilized to assess the flexibility and stability of the complexes. The TLR2-vaccine complex exhibited slight variations, with peak and lowest RMSF values recorded at 13.72 nm and 1.06 nm, respectively. In a similar manner, the TLR4-vaccine complex demonstrated maximum and minimum RMSF values of 3.96 nm and 0.72 nm, respectively. The analysis of Rg also assessed the structural equilibrium and protein compactness over the course of the simulation period. The simulation run revealed an average Rg value of 4.69 nm² for the vaccine-TLR2 complex, whereas the vaccine-TLR4 complex exhibited an average Rg value of 3.21 nm². Additionally, the SASA analysis indicated that vaccine-TLR2 and vaccine-TLR4 yielded average values of 1.13 nm2 and 0.37 nm2, respectively. The results of the MD simulation are listed in the Figure 3.
Immune Simulations
Both primary and secondary immune responses were elevated upon the administration of the vaccine candidate in the host body, as evident by a robust immune response as shown in the Figure 4. During the primary or humoral immune response, the IgM was the first antibody to be produced and peaked around 20-30 days, clearing out the antigen in around 50 days however the levels of IgM declined as soon as the response shifted towards secondary immune response components IgG (IgG1 and IgG2) indicating B cell and affinity maturation. IgG1 rose after IgM decline but persisted longer, whereas the IgG2 rose after IgG1 indicting its possible activation by the Th-1 factors indicating pathogen clearance. The persistent high levels of IgG antibodies reveal robust immune memory as shown in the Figure. Furthermore, the immune simulation studies also revealed the robust activation of the B cell as soon the pathogen gets detected in the body, whereas the presenting B cells rise later showing presentation of antigens to T-cells. During the transition phase, duplicating cells declined hence shifting to anergic cells activation to ensure that there occurs no over antibody production/excess immune activation. However, the B cell level persists showing efficient memory cells. Furthermore, it was also evident that there was efficient presentation of B cell epitopes to the T cell as the levels/concentration both the active state cytotoxic and helper T cells elevated gradually until 100 days post-injection (with good memory cells concentration) and the same was noted for both natural killer cells and the dendritic cells. The results of the immune simulations are illustrated in the Figure 4.
SRLV Vaccine Molecular Cloning and Codon-Optimization
The vaccine construct was codon optimized using the JCat Codon Optimization server and the optimized codon had a length of 361 amino acids. The CAI value was measured at 1.0 whereas the GC percentage of the optimized construct was 54.85. Following that, after the addition of the XhoI and BamHI restriction enzymes sites at both N and C terminal of the construct respectively, it was successfully cloned into the pET28(a) + cloning vector using the SnapGene tool. The length of the generated vaccine construct-Pet 28a (+) clone was 6430 base pairs. The finally generated clone is shown in the Figure 5.
4. Discussion
MV and CAE caused by SRLVs are among the most prevalent viral infections of livestock animals including goats and sheeps. SRLVs possess longer incubation periods leading to identification ineffectiveness. Furthermore, they have a RNA genome that undergoes rapid mutation events hence limiting the efficacy of the available treatments. Vaccination stands out as the most effective treatment against viral diseases as it has the potential to stimulate the host cell's immune response and in turn it helps the body to defend itself against the invading foreign pathogens, however the traditional vaccine designing process is complex, tedious and expensive. Reverse Vaccinology is an advanced vaccine development process that utilizes cutting edge bioinformatics and computational biology tools to screen datasets from different proteomics, genomics, transcriptomics platforms to screen out those antigens that have the ability to stimulate hosts immune response at the cellular level hence making vaccine generation less complex, fast, more specific and an economical process. However, RV validates vaccine development computationally by validating that the generated vaccine is immunogenic, non-allergenic and non-toxic. However, the validation of these parameters through experiments in wet-lab is essential for confirming the efficacy of the vaccine construct before going into clinical trials and hence the relevant experiments could be conducted in the future studies.
In this study, we have used the RV strategy to formulate a potential multi-epitope vaccine construct that could provide efficient protection against SRLVs in both goats and sheeps simultaneously. Hence, the envelope gene involved in the viral host attachment was screened among five most prevalent SRLV isolates to find the conserved regions of envelope gene among different genotypes to ensure broader species protection with enhanced specificity. On the basis of the selected sequences, we first predicted the linear B cell epitopes on the basis of their higher antigenicity scores followed by the prediction of discontinuous B cell epitopes. Furthermore, CTL and HTL epitopes were predicted using specific alleles. It was made sure that all the selected epitopes were antigenic, non-toxic and non-allergenic as these epitopes had the ability to produce robust immune response in the host organisms. The final vaccine construct was generated by joining ten BCL, four CTL, four HTL epitopes linked together via different linker molecules. An adjuvant was added on the C-terminal to elicit sufficient immune response, whereas the 6x-Histidine Tag was added at the N-terminal to have ease during protein purification steps later on. The generated vaccine construct has a length of 361 amino acids and the vaccine showed good solubility while remaining antigenic, non-toxic and non-allergenic. The secondary structure prediction revealed the presence of secondary structure elements such as alpha helix (26.04%), extended strands (18.28%) and random coils (55.68%). Furthermore, the tertiary structure was generated via the I-TASSER and the results were further refined with the GalaxyRefine tool. Best refined model was chosen based on the outcomes of PROCHECK and ProSA server. The molecular docking analysis revealed that the generated vaccine construct had a strong interaction with the targeted TLR-4 receptor molecule (binding energy: -22.5 kcal/mol) as compared to the binding with the TLR-2 receptor (binding energy: -17.4 kcal/mol). This was also validated by the interaction analysis which revealed that there are 31 hydrogen bonds and 30 interacting residues between the TLR-4 receptor and vaccine construct whereas 16 hydrogen bonds and 22 interacting residues were present between TLR-2 receptor and the vaccine construct. The stability of the vaccine complex in the binding pockets and its motion was analyzed via the molecular dynamics simulation studies which revealed that both the TLR-2 and TLR-4 bounded vaccine complexes were stable however there exist more stability and better interactions for the TLR-4 vaccine construct complex as compared to the TLR-2 vaccine construct complex. Additionally, the immune simulating potential of the generated vaccine construct was also validated via the immune simulation studies which showed that the vaccine has the potential to activate the host cell immune system and there was enough concentration of B cell lymphocytes, which in turn activated the other immune cells such as cytotoxic and helper T cells along with natural killer cells. Meanwhile, good concentration levels for memory cells were noted ensuring long lasting immune effect. All this ensured that the vaccine has a potential to provide immunity against the SRLV strains among goats and sheep. Also, to facilitate the protein production in bacterial system, the optimization of codon was done through in-silico cloning via the SNAPGene tool and the optimized codon (361 amino acid) was successfully cloned in the commercially used vector. The length of the generated vaccine construct-Pet 28a (+) clone was 6430 base pairs. However, despite showing potentials of an effective vaccine, the above vaccine candidate must need to go under in-vitro validation experiments toe ensure its safety and efficacy for the livestock.
5. Limitations
The current study utilized the highly conserved epitopes however different bioinformatic tools mainly focus on those epitopes that are easily expressed hence many other factors linked with underlying immunity may get ignored. Furthermore, the linkers used in the study may need some further validation as they may lead to the instability of the vaccine construct and pose issues with the safety construct.
6. Conclusions
SRLVs the main responsible agent behind MV and CAE incidence among goats and sheeps, leading to loss in milk and meat production globally. A novel vaccine candidate was developed to combat diverse SRLV strains by using bioinformatic and immunoinformatic approaches. The vaccine was generated by combining ten BCL epitopes, four CTL and four HTL epitopes joined to each other by respective linkers. Furthermore, an adjuvant Pam3CSK4 and histidine tag was added to generate a more robust immune response and purified product respectively. The vaccine showed good antigenicity while being non-cytotoxic and non-allergenic. Furthermore, the vaccine showcased stable and high-affinity interaction when docked/simulated with TLR-2 and TLR-4 receptors. From above findings, the generated vaccine serves as a potential candidate against the SRLV caused MV and CEC. However, these findings need further support via in-vitro and animal experiments to further elucidate the safety and the efficacy of the vaccine candidate.
Author Contributions
Muhammad Zeeshan Shabbir (MZS): Research idea, study design, data collection, interpretation, write-up, coordination and data management. Rumesa Ghazanfar (RG): data screening, epitopes selection and interpretation, figure designing, in-silico technical work related to molecular docking. Zainab Nagari (ZN): epitope selection, writeup, figures designing and in-silico technical work related to protein structure determination. Muhammad Atiq Ur Rehman (MAR): study design, write-up, data management and immune simulation studies. Chandra Sourav (CS): Molecular Dynamic Simulation, Writeup.
Funding
This research did not receive any funding or research grants from any commercial, public or non-profit organization.
Data Availability Statement
The data that support the findings of this study are available on request.
Acknowledgments
The authors have nothing to report.
Ethics Statement/Consent Information
This study was entirely in-silico based while using the publicly available data and did not involve any animal or human participation. Therefore, ethical approval and consent to participate were not required.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arcangeli, C., M. Torricelli, C. Sebastiani, D. Lucarelli, M. Ciullo, F. Passamonti, M. Giammarioli and M. J. V. Biagetti (2022). "Genetic characterization of small ruminant lentiviruses (SRLVs) circulating in naturally infected sheep in Central Italy." 14(4): 686.
- Blacklaws, B., E. Berriatua, S. Torsteinsdottir, N. Watt, D. De Andres, D. Klein and G. J. V. M. Harkiss (2004). "Transmission of small ruminant lentiviruses." 101(3): 199-208.
- Blacklaws, B. A. J. C. i., microbiology and i. diseases (2012). "Small ruminant lentiviruses: Immunopathogenesis of visna-maedi and caprine arthritis and encephalitis virus." 35(3): 259-269.
- Cheevers, W., D. Knowles, T. McGuire, T. Baszler, G. J. V. i. Hullinger and immunopathology (1994). "Caprine arthritis-encephalitis lentivirus (CAEV) challenge of goats immunized with recombinant vaccinia virus expressing CAEV surface and transmembrane envelope glycoproteins." 42(3-4): 237-251.
- Davaasuren, N., V. Molaee, T.-O. Erdene-Ochir, G. Nyamdavaa, S. Ganzorig, M. Mazzei, Y. Sakoda, G. Lühken and S. J. V. R. C. Tumenjargal (2024). "Phylogenetic analysis of small ruminant lentiviruses in Mongolian sheep supports an ancient east-west split for the genotype A." 48(3): 1955-1962.
- De Andres, X., R. Reina, J. Ciriza, H. Crespo, I. Glaria, H. Ramírez, M. J. Grilló, M. M. Pérez, V. Andresdottir and S. J. V. Rosati (2009). "Use of B7 costimulatory molecules as adjuvants in a prime-boost vaccination against Visna/Maedi ovine lentivirus." 27(34): 4591-4600.
- Dimitrov, I., I. Bangov, D. R. Flower and I. J. J. o. m. m. Doytchinova (2014). "AllerTOP v. 2—a server for in silico prediction of allergens." 20: 1-6.
- Doytchinova, I. A. and D. R. J. B. b. Flower (2007). "VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines." 8: 1-7.
- Doytchinova, I. A., P. Guan and D. R. J. B. b. Flower (2006). "EpiJen: a server for multistep T cell epitope prediction." 7: 1-11.
- ENACHE, D. A., S. BARAITAREANU, M. DAN, M. R. GURAU, F. OTELEA, A. DOBRE and D. J. S. W. S. C. DANES, Veterinary Medicine (2017). "Preliminary results of MVV and CAEV seroprevalence in Romanian sheep and goats." 63(1).
- Gasteiger, E., C. Hoogland, A. Gattiker, S. e. Duvaud, M. R. Wilkins, R. D. Appel and A. J. T. p. p. h. Bairoch (2005). "Protein identification and analysis tools on the ExPASy server." 571-607.
- Geourjon, C. and G. J. B. Deleage (1995). "SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments." 11(6): 681-684.
- Gomez-Lucia, E., N. Barquero, A. J. V. M. R. Domenech and Reports (2018). "Maedi-Visna virus: current perspectives." 11-21.
- González, B., R. Reina, I. García, S. Andrés, I. Glaria, M. Alzueta, M. I. Mora, B. M. Jugo, I. Arrieta-Aguirre and J. M. P. J. V. de la Lastra (2005). "Mucosal immunization of sheep with a Maedi-Visna virus (MVV) env DNA vaccine protects against early MVV productive infection." 23(34): 4342-4352.
- Grote, A., K. Hiller, M. Scheer, R. Münch, B. Nörtemann, D. C. Hempel and D. J. N. a. r. Jahn (2005). "JCat: a novel tool to adapt codon usage of a target gene to its potential expression host." 33(suppl_2): W526-W531.
- Hall, T., I. Biosciences and C. J. G. b. b. Carlsbad (2011). "BioEdit: an important software for molecular biology." 2(1): 60-61.
- Heo, L., H. Park and C. J. N. a. r. Seok (2013). "GalaxyRefine: Protein structure refinement driven by side-chain repacking." 41(W1): W384-W388.
- Jespersen, M. C., B. Peters, M. Nielsen and P. J. N. a. r. Marcatili (2017). "BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes." 45(W1): W24-W29.
- Koçkaya, E. S., H. Can, Y. Yaman and C. J. B. Ün (2023). "In silico discovery of epitopes of gag and env proteins for the development of a multi-epitope vaccine candidate against maedi visna virus using reverse vaccinology approach." 84: 101715.
- Kosloff, D. and R. J. J. o. C. P. Kosloff (1983). "A Fourier method solution for the time dependent Schrödinger equation as a tool in molecular dynamics." 52(1): 35-53.
- Kozakov, D., D. R. Hall, B. Xia, K. A. Porter, D. Padhorny, C. Yueh, D. Beglov and S. J. N. p. Vajda (2017). "The ClusPro web server for protein–protein docking." 12(2): 255-278.
- Krissinel, E. and K. J. J. o. m. b. Henrick (2007). "Inference of macromolecular assemblies from crystalline state." 372(3): 774-797.
- Laskowski, R. A., J. Jabłońska, L. Pravda, R. S. Vařeková and J. M. J. P. s. Thornton (2018). "PDBsum: Structural summaries of PDB entries." 27(1): 129-134.
- Laskowski, R. A., M. W. MacArthur, D. S. Moss and J. M. J. A. C. Thornton (1993). "PROCHECK: a program to check the stereochemical quality of protein structures." 26(2): 283-291.
- Magnan, C. N., A. Randall and P. J. B. Baldi (2009). "SOLpro: accurate sequence-based prediction of protein solubility." 25(17): 2200-2207.
- Martin, D., J. Procter, B. Soares, A. Waterhouse, S. Shehata, N. Giang, M. Carstairs, C. Ofoegbu, K. Mourão, S. J. M. Duce and I. Tutorial (2020). "Jalview 2.11.".
- McGuffin, L. J., K. Bryson and D. T. J. B. Jones (2000). "The PSIPRED protein structure prediction server." 16(4): 404-405.
- Michiels, R., E. Van Mael, C. Quinet, N. R. Adjadj, A. B. Cay and N. J. V. De Regge (2018). "Comparative analysis of different serological and molecular tests for the detection of small ruminant lentiviruses (SRLVs) in Belgian sheep and goats." 10(12): 696.
- Minardi da Cruz, J. C., D. K. Singh, A. Lamara and Y. J. V. Chebloune (2013). "Small ruminant lentiviruses (SRLVs) break the species barrier to acquire new host range." 5(7): 1867-1884.
- Molaee, V., M. Bazzucchi, G. M. De Mia, V. Otarod, D. Abdollahi, S. Rosati and G. J. S. r. Lühken (2020). "Phylogenetic analysis of small ruminant lentiviruses in Germany and Iran suggests their expansion with domestic sheep." 10(1): 2243.
- Moroz, A., M. Czopowicz, M. Sobczak-Filipiak, I. Dolka, M. Rzewuska, M. Kizerwetter-Świda, D. Chrobak-Chmiel, M. Mickiewicz, L. Witkowski and O. J. P. Szaluś-Jordanow (2022). "The prevalence of histopathological features of pneumonia in goats with symptomatic caprine arthritis-encephalitis." 11(6): 629.
- Mosa, A. H. and M. M. Zenad (2020). "Serological and histopathological detection of Maedi-Visna virus in middle Iraq regions.".
- Olech, M., S. Valas and J. J. P. O. Kuźmak (2018). "Epidemiological survey in single-species flocks from Poland reveals expanded genetic and antigenic diversity of small ruminant lentiviruses." 13(3): e0193892.
- Pépin, M., C. Vitu, P. Russo, J.-F. Mornex and E. J. V. r. Peterhans (1998). "Maedi-visna virus infection in sheep: a review." 29(3-4): 341-367.
- Pettersen, E. F., T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. J. J. o. c. c. Ferrin (2004). "UCSF Chimera—a visualization system for exploratory research and analysis." 25(13): 1605-1612.
- Pétursson, G., S. Matthíasdóttir, V. Svansson, V. Andrésdóttir, G. Georgsson, A. H. Martin, G. Agnarsdóttir, E. Gísladóttir, S. Árnadóttir and S. J. V. Högnadóttir (2005). "Mucosal vaccination with an attenuated maedi–visna virus clone." 23(24): 3223-3228.
- Ponomarenko, J., H.-H. Bui, W. Li, N. Fusseder, P. E. Bourne, A. Sette and B. J. B. b. Peters (2008). "ElliPro: a new structure-based tool for the prediction of antibody epitopes." 9: 1-8.
- Querat, G., G. Audoly, P. Sonigo and R. J. V. Vigne (1990). "Nucleotide sequence analysis of SA-OMVV, a visna-related ovine lentivirus: phylogenetic history of lentiviruses." 175(2): 434-447.
- Rahman, M. H., E. Ahmed, M. N. Haque, M. Z. Hassan and M. Z. J. B. J. o. L. R. Ali (2022). "Major respiratory diseases of goat and their epidemiology, prevention and control." 1-20.
- Rapin, N., O. Lund and F. J. B. Castiglione (2011). "Immune system simulation online." 27(14): 2013-2014.
- Saha, S. and G. P. S. Raghava (2004). BcePred: prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. International conference on artificial immune systems, Springer.
- Saha, S., G. P. S. J. P. S. Raghava, Function, and Bioinformatics (2006). "Prediction of continuous B-cell epitopes in an antigen using recurrent neural network." 65(1): 40-48.
- Santry, L. A., J. de Jong, A. C. Gold, S. R. Walsh, P. I. Menzies and S. K. J. V. R. Wootton (2013). "Genetic characterization of small ruminant lentiviruses circulating in naturally infected sheep and goats in Ontario, Canada." 175(1): 30-44.
- Seeliger, D. and B. L. J. J. o. c.-a. m. d. de Groot (2010). "Ligand docking and binding site analysis with PyMOL and Autodock/Vina." 24(5): 417-422.
- Sharma, N., L. D. Naorem, S. Jain and G. P. J. B. i. b. Raghava (2022). "ToxinPred2: an improved method for predicting toxicity of proteins." 23(5): bbac174.
- Sievers, F. and D. G. J. C. p. i. b. Higgins (2014). "Clustal omega." 48(1): 3.13. 11-13.13. 16.
- Singh, H. and G. J. B. Raghava (2001). "ProPred: prediction of HLA-DR binding sites." 17(12): 1236-1237.
- Torsteinsdóttir, S., H. M. Carlsdóttir, V. Svansson, S. Matthíasdóttir, A. H. Martin and G. J. V. Pétursson (2007). "Vaccination of sheep with Maedi-visna virus gag gene and protein, beneficial or harmful?" 25(37-38): 6713-6720.
- Trolle, T., I. G. Metushi, J. A. Greenbaum, Y. Kim, J. Sidney, O. Lund, A. Sette, B. Peters and M. J. B. Nielsen (2015). "Automated benchmarking of peptide-MHC class I binding predictions." 31(13): 2174-2181.
- Wiederstein, M. and M. J. J. N. a. r. Sippl (2007). "ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins." 35(suppl_2): W407-W410.
- Xue, L. C., J. P. Rodrigues, P. L. Kastritis, A. M. Bonvin and A. J. B. Vangone (2016). "PRODIGY: a web server for predicting the binding affinity of protein–protein complexes." 32(23): 3676-3678.
- Yang, Y., Z. Wei, G. Cia, X. Song, F. Pucci, M. Rooman, F. Xue and Q. J. F. i. i. Hou (2024). "MHCII-peptide presentation: an assessment of the state-of-the-art prediction methods." 15: 1293706.
- Zhang, Y. J. B. b. (2008). "I-TASSER server for protein 3D structure prediction." 9: 1-8.
Figure 2.
Molecular Docking Analysis for the SRLV Vaccine Construct. A. Illustrates the results of the molecular interaction/docking analysis between the SRLV vaccine construct and the TLR-2 receptor molecule (PDB ID:6NIG). The results were analyzed with PDBSum webserver. Here, the Chain A presents the vaccine construct whereas the Chain B stands for the receptor molecule while the different colored boxes show interacting amino acid residues in the respective chains. Whereas, the different colored lines show different intermolecular forces such as hydrogen bonds and salt-bridges etc. B. Depicts the results of the molecular interaction/docking analysis between the SRLV vaccine construct and the TLR-4 receptor molecule (PDB ID:4G8A). The results were analyzed with PDBSum webserver. Here, the Chain A is for the receptor molecule whereas the Chain B represents the vaccine construct sequence. Different colored boxes show the interacting amino acid residues in the respective chains, whereas the different colored lines show different intermolecular forces such as hydrogen bonds and salt-bridges etc. The results collectively reveal the presence of a stronger molecular interaction between the generated SRLV vaccine construct and the TLR-4 receptor molecule as compared to the TLR-2 receptor.
Figure 2.
Molecular Docking Analysis for the SRLV Vaccine Construct. A. Illustrates the results of the molecular interaction/docking analysis between the SRLV vaccine construct and the TLR-2 receptor molecule (PDB ID:6NIG). The results were analyzed with PDBSum webserver. Here, the Chain A presents the vaccine construct whereas the Chain B stands for the receptor molecule while the different colored boxes show interacting amino acid residues in the respective chains. Whereas, the different colored lines show different intermolecular forces such as hydrogen bonds and salt-bridges etc. B. Depicts the results of the molecular interaction/docking analysis between the SRLV vaccine construct and the TLR-4 receptor molecule (PDB ID:4G8A). The results were analyzed with PDBSum webserver. Here, the Chain A is for the receptor molecule whereas the Chain B represents the vaccine construct sequence. Different colored boxes show the interacting amino acid residues in the respective chains, whereas the different colored lines show different intermolecular forces such as hydrogen bonds and salt-bridges etc. The results collectively reveal the presence of a stronger molecular interaction between the generated SRLV vaccine construct and the TLR-4 receptor molecule as compared to the TLR-2 receptor.
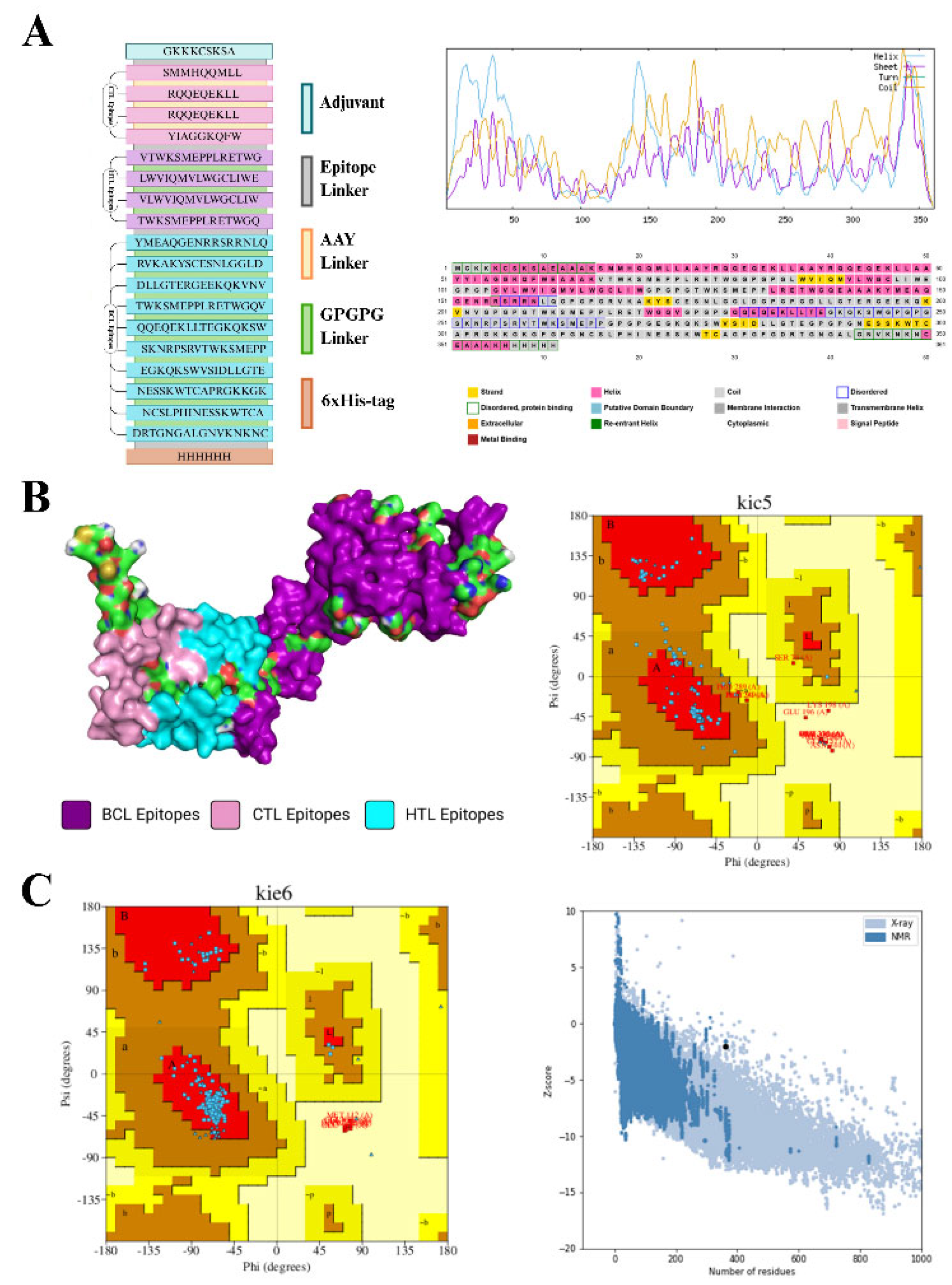
Figure 1.
Schematic Diagram Illustrating the SRLV Based Vaccine Designing Strategy. A. Illustrates the geometry of the generated vaccine construct by joining adjuvant, BCL, CTL and HTL together with the help of linker molecules. All the components of the construct are represented in different colors. Furthermore, the results of SOPMA secondary structure analysis show the presence of different elements such as helix, turns and coils in the generated vaccine construct. Whereas the other elements of the structure are highlighted with different colored boxes. B. The tertiary structure of the protein construct is shown in different colors (highlighting different epitopes) when generated using the I-TASSER protein prediction server. The visualization of the prediction structure was aided by Chimera and PyMol. Furthermore, the quality of the predicted model was analyzed through the generation of the Ramachandran plot for the predicted model via the ProCheck webserver. C. Shows the Ramachandran plot generated via ProCheck server for the selected protein model (refined with Galaxy Refine tool) showing >90% of the residues in the allowed regions. The quality of the predicted model was also analyzed via the ProSa webserver via seeking help of the predicted Z-scores for each amino acid residue in the vaccine construct.
Figure 1.
Schematic Diagram Illustrating the SRLV Based Vaccine Designing Strategy. A. Illustrates the geometry of the generated vaccine construct by joining adjuvant, BCL, CTL and HTL together with the help of linker molecules. All the components of the construct are represented in different colors. Furthermore, the results of SOPMA secondary structure analysis show the presence of different elements such as helix, turns and coils in the generated vaccine construct. Whereas the other elements of the structure are highlighted with different colored boxes. B. The tertiary structure of the protein construct is shown in different colors (highlighting different epitopes) when generated using the I-TASSER protein prediction server. The visualization of the prediction structure was aided by Chimera and PyMol. Furthermore, the quality of the predicted model was analyzed through the generation of the Ramachandran plot for the predicted model via the ProCheck webserver. C. Shows the Ramachandran plot generated via ProCheck server for the selected protein model (refined with Galaxy Refine tool) showing >90% of the residues in the allowed regions. The quality of the predicted model was also analyzed via the ProSa webserver via seeking help of the predicted Z-scores for each amino acid residue in the vaccine construct.
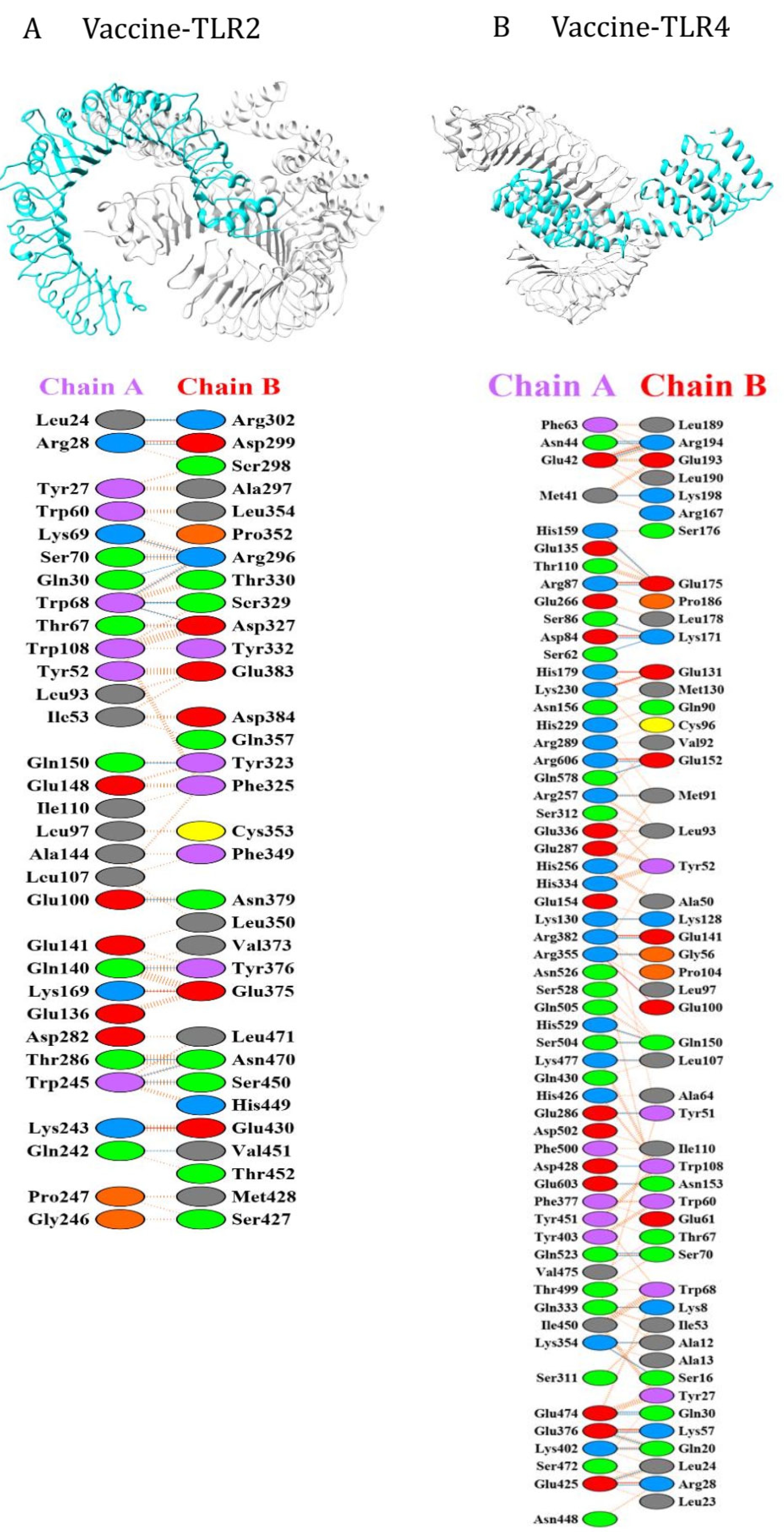
Figure 3.
Molecular Dynamic Simulations for the SRLV vaccine construct. The figure shows the outcomes of the molecular dynamic simulations when carried out using the generated vaccine construct and two receptor molecules i.e. TLR-2 (6NIG) and TLR-4 (4G8A) using the Schrodinger Maestro software package. Different parameters such as CA, Radius of Gyration, SASA and RMSD were calculated and compared for both ligands with the construct molecule.
Figure 3.
Molecular Dynamic Simulations for the SRLV vaccine construct. The figure shows the outcomes of the molecular dynamic simulations when carried out using the generated vaccine construct and two receptor molecules i.e. TLR-2 (6NIG) and TLR-4 (4G8A) using the Schrodinger Maestro software package. Different parameters such as CA, Radius of Gyration, SASA and RMSD were calculated and compared for both ligands with the construct molecule.
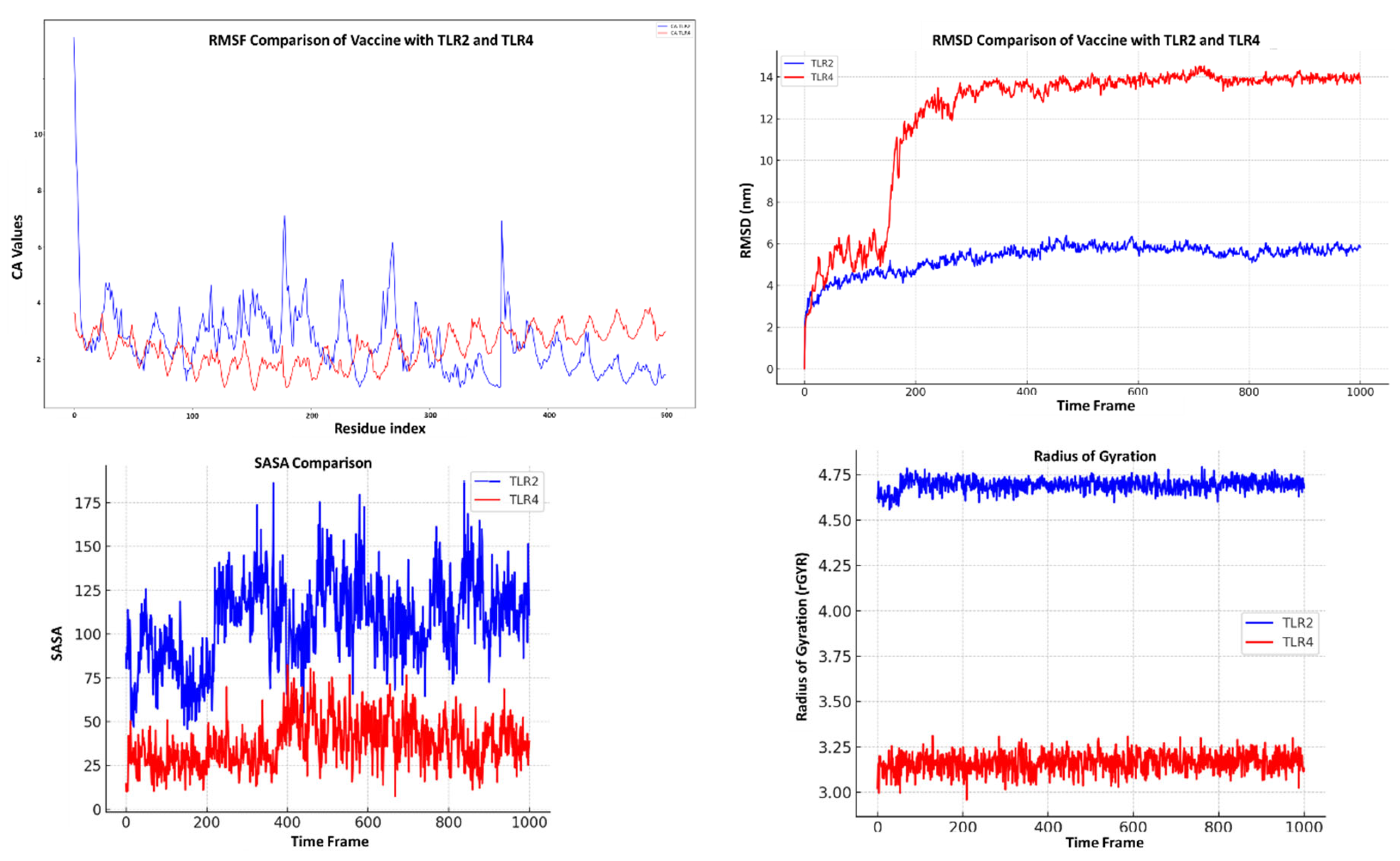
Figure 4.
Immune Simulations for the SRLV vaccine construct. The figure shows the outcomes for the immune simulations when carried out using the generated vaccine construct via the online C-Imm Sim server. Different parameters such as B cell population, helper and cytotoxic T cell populations were determined along with concentration of memory cells.
Figure 4.
Immune Simulations for the SRLV vaccine construct. The figure shows the outcomes for the immune simulations when carried out using the generated vaccine construct via the online C-Imm Sim server. Different parameters such as B cell population, helper and cytotoxic T cell populations were determined along with concentration of memory cells.
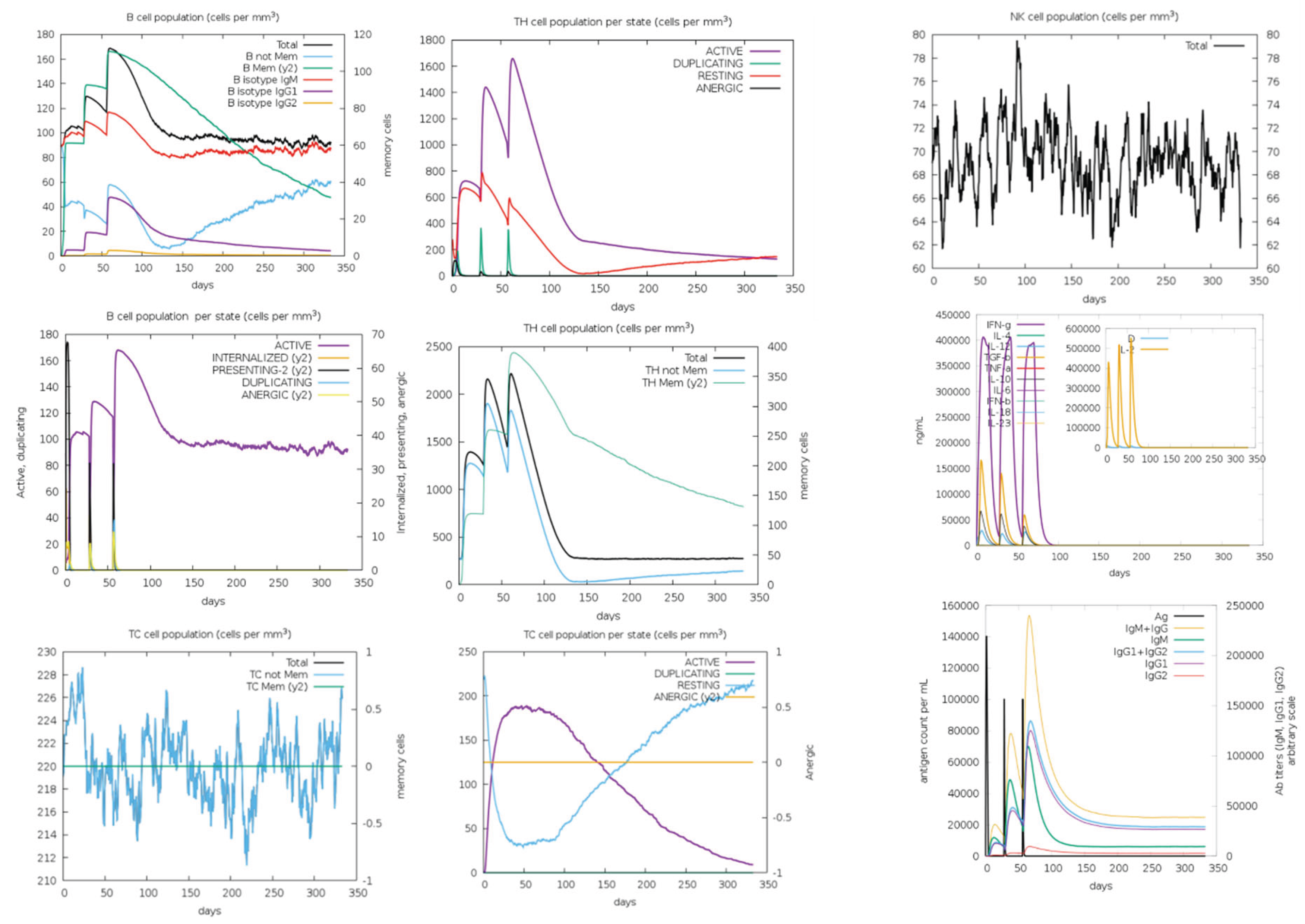
Figure 5.
In-Silico Cloning and Codon Optimization for the SRLV vaccine construct. The figure shows the results of in-silico cloning, first the codon was optimized using the JCat Server, later the vaccine construct was cloned into the commercially available E. coli (Pet28a+) vector molecule by using the SNAPGene tool (clone is highlighted in red color). The generated clone had a length of 6430 base pairs.
Figure 5.
In-Silico Cloning and Codon Optimization for the SRLV vaccine construct. The figure shows the results of in-silico cloning, first the codon was optimized using the JCat Server, later the vaccine construct was cloned into the commercially available E. coli (Pet28a+) vector molecule by using the SNAPGene tool (clone is highlighted in red color). The generated clone had a length of 6430 base pairs.
Table 1.
Major Vaccine Candidates Against MV and CAE.
| TYPE OF VACCINE | TARGETED VIRAL AGENT | TARGETED SPECIE | STUDY PARAMETERS | OUTCOMES | REFERENCES |
|---|---|---|---|---|---|
| Attenuated molecular clone of Maedi Visna virus (MVV) (2005) |
Maedi-Visna Virus (MVV) |
Sheep |
Four sheep were infected with attenuated molecular clone of maedi visna virus (MVV) (LV1-1KS1); while comparing them with control group (n=4) | Low viral titer was detected in the ELISA test indicating partial protection. | (Pétursson, Matthíasdóttir et al. 2005) |
| DNA based vaccine based on the envelope gene (2005) |
Maedi-Visna Virus (MVV) | Sheep | pcDNA plasmid (pcDNA-env) encoding the env glycoproteins of MVV, boosted with combined pcDNA-env and pCR3.1-IFN-γ plasmid inoculations were introduced in both test and control groups | Early protective effect was observed in the vaccinated sheeps that restricted the virus replication however it disappeared after two years. | (González, Reina et al. 2005) |
| DNA vaccine containing the Maedi-Visna virus (MVV) gag gene (2007) |
Maedi-Visna Virus (MVV) | Sheep |
Sheep were immunized eight times over a period of 2.5 years with the Maedi-Visna (MVV) gag gene, VR1012-gag-CTE and pcDNA3.1-gag-CTE. | No positive outcome was observed. In fact, the vaccine may have made them more susceptible to the virus. | (Torsteinsdóttir, Carlsdóttir et al. 2007) |
| Use of B7 costimulatory molecules as adjuvants in a prime-boost vaccination against Visna/Maedi ovine lentivirus (2009) |
Visna/Maedi ovine lentivirus |
Sheep (Llewyn sheep breed |
Sheep were primed with particle-mediated epidermal bombardment (PMED) using plasmids containing the VMV gag and env genes. | Enhanced early cellular immune response. | (De Andres, Reina et al. 2009) |
| In silico discovery of epitopes of gag and env proteins for the development of a multi-epitope vaccine candidate against Maedi Visna Virus (2023) | Maedi-Visna | Sheep | Retrieved the DNA sequence for gag and env gene of MVV KV1514 isolate from NCBI, and a multi-epitope vaccine was designed based on the 19 epitopes. | Gag protein was detected to be more conserved and a strong affinity was shown between the final multi-epitope vaccine and TLR-2/4. | (Koçkaya, Can et al. 2023) |
| Recombinant Vaccinia Virus rWR63 Expressing CAEV Envelope Glycoproteins | CEAV | Goat | Goats (n=6) were vaccinated thrice with rWR63, followed by different booster immunizations. Control group received rWRSC11 | All vaccinated goats developed antibodies to CAEV glycoproteins, but no neutralizing antibodies were detected. | (Cheevers, Knowles et al. 1994) |
| The table depicts the various types of vaccine candidates that have been reported over the years against MVV and CAEV infections. The type of vaccine candidate is shown in the first column, whereas the targeted viral agent and specie are mentioned in the second and third column. Furthermore, the parameters chosen for the specific study and the outcomes are depicted in the fourth and fifth column respectively. Moreover, the reference articles of the study are mentioned in the sixth column. | |||||
Table 2.
Reference Isolates for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
Table 2.
Reference Isolates for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
| Serotype | NCBI Accession Number | Region | Gene Type | Accession Links |
|---|---|---|---|---|
| A1 | M60609 | Iceland | Envelope | https://www.ncbi.nlm.nih.gov/nuccore/M60609 |
| A1 | OL436272 | Poland | Envelope | https://www.ncbi.nlm.nih.gov/nuccore/OL436272 |
| A2 | AY101611 | USA | Envelope | https://www.ncbi.nlm.nih.gov/nuccore/AY101611 |
| B1 | OL436260 | Poland | Envelope | https://www.ncbi.nlm.nih.gov/nuccore/OL436260 |
| B2 | OL436262 | Poland | Envelope | https://www.ncbi.nlm.nih.gov/nuccore/OL436262 |
| This first column shows the serotype of the selected isolates whereas its accession number is given in the second column. The third column contains the information about the region and followed by its targeted gene type in fourth column. All the information is retrieved through NCBI for which the link is provided in the last column. | ||||
Table 3.
List of selected BCL Epitopes for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
Table 3.
List of selected BCL Epitopes for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
| Sequence | Sequence Length | Score | AllerTop | VexiJen | ToxinPred | SVM Score | Molecular Weight |
|---|---|---|---|---|---|---|---|
| YMEAQGENRRSRRNLQ | 16 | 0.80 | Non-Allergen | Antigen (0.6151) |
Non-Toxin | -1.04 | 1880.28 |
| RVKAKYSCESNLGGLD | 16 | 0.77 | Non-Allergen | Antigen (1.1465) |
Non-Toxin | -0.59 | 1625.09 |
| DLLGTERGEEKQKVNV | 16 | 0.89 | Non-Allergen | Antigen (0.7215) |
Non-Toxin | -1.40 | 1716.11 |
| TWKSMEPPLRETWGQV | 16 | 0.80 | Non-Allergen | Antigen (0.5101) |
Non-Toxin | -0.80 | 1846.31 |
| QQEQEKLLTEGKQKSW |
16 | 0.79 | Non-Allergen | Antigen (0.5243) |
Non-Toxin | -1.29 | 1744.20 |
| SKNRPSRVTWKSMEPP |
16 | 0.70 | Non-Allergen | Antigen (0.8433) |
Non-Toxin | -0.99 | 1803.27 |
| EGKQKSWVSIDLLGTE |
16 | 0.60 | Non-Allergen | Antigen (1.0230) |
Non-Toxin | -1.01 | 1661.12 |
| NESSKWTCAPRGKKGK | 16 | 0.89 | Non-Allergen | Antigen (0.9891) |
Non-Toxin | -0.57 | 1649.06 |
| NCSLPHINESSKWTCA | 16 | 0.70 | Non-Allergen | Antigen (0.5145) |
Non-Toxin | -0.57 | 1719.13 |
| DRTGNGALGNVKNKNC | 16 | 0.82 | Non-Allergen | Antigen (1.2684) |
Non-Toxin | 0.73 | 1557.91 |
| The table shows the selected BCL epitopes for vaccine development. The first column shows the sequence of the selected BCL epitope sequence whereas the sequence length and scores are listed in the second and third column respectively. Furthermore, the results of allergenicity analysis, antigenicity, toxicity are depicted in column 4-6 respectively. SMV score and molecular weight information is mentioned in the seventh and eighth column. | |||||||
Table 5.
List of selected HTL Epitopes for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
Table 5.
List of selected HTL Epitopes for the Generation of In-silico Based Vaccine Candidate Against MV and CAE.
| Sequence | Length | Allele | Affinity nM | Percentile Rank | Binding Level | AllerTOP | VaxiJen | Toxin Pred (SVM, Molecular Weight) |
|---|---|---|---|---|---|---|---|---|
| VTWKSMEPPLRETWG |
16 | BoLA-DRB3*0301 |
0.644 |
30.63 | Strong Binder | Non-Allergen | Immunogen (100%) |
Non-Toxin (-0.89, 1817.31) |
| LWVIQMVLWGCLIWE |
16 | DRB1*0102 |
4.41 |
7.00 | Strong Binder | Non-Allergen | Immunogen (66%) |
Non-Toxin (-0.14, 1889.64) |
|
VLWVIQMVLWGCLIW |
16 | DRB1*0104 |
0.761 |
10.00 | Strong Binder | Non-Allergen | Immunogen (66%) |
Non-Toxin (-0.30, 1859.62) |
| TWKSMEPPLRETWGQ |
16 | DRB1*0120 |
0.648 |
29.46 | Strong Binder | Non-Allergen | Immunogen (66%) |
Non-Toxin (-0.80, 1846.31) |
| The predicted HTL epitope sequence for the vaccine construct is shown in the first column whereas the epitope sequence length, respective allele, binding affinity and percentile rank are depicted in the column and percentile length are listed in the second, third and fourth column respecttively. Furthermore, the results of allergenicity analysis, antigenicity, toxicity are depicted in column 4-7 respectively. | ||||||||
Table 6.
List of tools used in the study.
| Tool Used | Link |
|---|---|
| Clustal Omega | https://www.ebi.ac.uk/jdispatcher/msa/clustalo |
| JalView | https://www.jalview.org/ |
| BioEdit | https://bioedit.software.informer.com/7.2/ |
| ABCPRED | https://webs.iiitd.edu.in/raghava/abcpred/ |
| BCEPRED | https://webs.iiitd.edu.in/raghava/bcepred/bcepred_submission.html |
| BEPIPRED | https://services.healthtech.dtu.dk/services/BepiPred-2.0/ |
| VAXIJEN 2.0 | https://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html |
| AllerTOP | https://www.ddg-0pharmfac.net/allertop_test/ |
| ToxinPred | https://webs.iiitd.edu.in/raghava/toxinpred/ |
| ProPred-I | http://crdd.osdd.net/raghava/propred1/ |
| EpiJen | https://www.ddg-pharmfac.net/epijen/ |
| ElliPro | http://tools.iedb.org/ellipro/ |
| IEDB MHC-I Binding Prediction tools | http://tools.iedb.org/mhci/ |
| NetMHCIIpan 2.1 server | https://services.healthtech.dtu.dk/services/NetMHCIIpan-2.1/ |
| Expasy ProtParam | https://web.expasy.org/cgi-bin/protparam/protparam |
| SolPro | https://scratch.proteomics.ics.uci.edu/ |
| SOPMA | https://npsa.lyon.inserm.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html |
| PSIPRED | https://bioinf.cs.ucl.ac.uk/psipred/ |
| I-TASSER | https://zhanggroup.org/I-TASSER/ |
| ProCheck server | https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ |
| Galaxy Refine server | https://bio.tools/galaxyrefine |
| ProSa webserver | https://prosa.services.came.sbg.ac.at/prosa.php |
| ClusPro | https://cluspro.bu.edu/ |
| Protein Data Bank | https://www.rcsb.org/ |
| PRODIGY | https://rascar.science.uu.nl/prodigy/ |
| PDBePISA | https://www.ebi.ac.uk/pdbe/pisa/ |
| PDBSum | https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/ |
| Schrodinger Maestro software package | https://www.schrodinger.com/platform/products/maestro/ |
| C-ImmSim server | https://wwwold.iac.rm.cnr.it/ |
| The table describes the tools utilized in the current study. The first column depicts the name of the tool whereas the respective link is mentioned in the second column. | |
Table 7.
Physiochemical Properties of the SRLV based Vaccine Construct.
| Physiochemical Property | Result |
|---|---|
| No. of amino acids | 361 |
| Theoretical pI | 9.38 |
| Molecular Weight | 39077.45 |
| Total number of negatively charged residues (Asp + Glu) | 33 |
| Total number of positively charged residues (Arg + Lys) | 47 |
| Total no. of atoms | 5426 |
| Formula | C1723H2679N505O500S19 |
| Extinction coefficients | Ext. coefficient 108440 M-1 cm-1 Abs 0.1% (=1 g/l) 2.775, assuming all pairs of Cys residues form cystines |
| Estimated half-life | 30 hours (mammalian reticulocytes, in vitro) >20 hours (yeast, in vivo) >10 hours (Escherichia coli, in vivo) |
| Instability Index | 35.15 (Stable) |
| Aliphatic index |
53.02 |
| Grand average of hydropathicity (GRAVY) | -0.840 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



