Submitted:
27 August 2025
Posted:
29 August 2025
You are already at the latest version
Abstract
Auranofin (AF) is an oral gold(I) compound with a well-known pharmacological profile, currently used in the treatment of some severe forms of rheumatoid arthritis. Over the last twenty years, AF has also been repurposed as antitumor, antiviral, and antibacterial drug. In this context, this review provides an updated overview of all clinical trials investigating AF for the treatment of various pathologies, either as monotherapy or in combination with other agents. We started summarizing the rationale behind repurposing AF in oncology, including its ability to inhibit thioredoxin reductase (TrxR) and disrupt redox homeostasis, leading to selective cytotoxicity in cancer cells. Clinical data from trials across a range of tumors are reviewed, highlighting safety profiles, dosing regimens, pharmacokinetics, and observed therapeutic outcomes. Then, we discussed the synergistic effects observed when AF is combined with chemotherapeutics, targeted therapies, or immune modulators. Then, an overview concerning the trials involving AF in non-oncological setting is also provided. Despite promising preclinical results, clinical translation remains in early stages, with most trials still in phase I or II. Nevertheless, emerging evidence supports continued exploration of AF-based therapies to address unmet medical needs.
Keywords:
auranofin
; clinical trials
; drug repurposing
; cancer
1. Introduction
Auranofin (AF, CAS 34031-32-8, molecular weight of 678.5 g/mol) is a linear gold(I)-based coordination complex that includes triethylphosphine and tetraacetylated thioglucose as ligands (Figure 1). Developed for the treatment of rheumatoid arthritis as an orally administered drug, AF received approval from the U.S. Food and Drug Administration (FDA) in 1985 as a first-line therapeutic agent [1]. However, its clinical application in rheumatology has diminished over time, mostly due to the advent of more effective synthetic and biological disease-modifying antirheumatic drugs (DMARDs), such as methotrexate, sulfasalazine, and adalimumab [2]. Recently, AF has gathered renewed attention due to its therapeutic potential in areas beyond rheumatology. Over the past decade, it has been widely repurposed and studied in multiple disease contexts, demonstrating potential efficacy in antibacterial, antiviral, antiparasitic, and particularly anticancer therapies [3,4,5,6].
AF is a prodrug, which activates in biological media releasing its bioactive gold(I) species, mainly consisting of the [AuPEt3]+ cation. Importantly, the thioglucose moiety, though essential for solubility and transport, as well as for an improved pharmacokinetics (PK) when orally administered, does not directly contribute to the therapeutic activity. In fact, it is the released [AuPEt3]+ cation that targets and inhibits thioredoxin reductase (TrxR), a selenoprotein deputed to the reduction of thioredoxin (Trx) [7]. There are two major isoforms of TrxR: TrxR1, located in the cytoplasm, and TrxR2, found in mitochondria. TrxRs are critical enzymes involved in maintaining cellular redox balance and supporting a range of physiological processes, such as DNA replication, regulation of gene expression, cell proliferation, and protection against oxidative stress-induced apoptosis [8]. The gold(I) ion released from AF strongly binds to the redox-active selenocysteine residue into the catalytic site of TrxRs, leading to irreversible enzyme inhibition. Mass spectrometry analyses indicated that each TrxR enzyme can coordinate up to four [AuPEt3]⁺ units, while biochemical studies confirm a profound disruption of the active site upon binding [9,10]. AF has also been shown to interact with hydroselenyl radicals (HSe)⁻, key intermediates in the biosynthesis of selenoproteins. These radicals are essential for the incorporation of selenium into selenocysteine, which is required for the proper function of a variety of selenium-dependent enzymes. AF can form a stable complex with HSe⁻, effectively sequestering these reactive selenium species. By trapping hydroselenyl radicals, it may reduce the intracellular selenium pool, thereby hindering the synthesis of selenoproteins. Since many organisms, including human cells and various pathogens, rely on selenoproteins for vital redox-regulating and metabolic functions, this interference can lead to impaired cellular function and reduced viability [11].
The TrxR/Trx system has gained significant attention in cancer biology due to its frequent upregulation in tumor tissues compared to healthy ones. This enhanced activity enables cancer cells to sustain a highly reduced intracellular environment, which facilitates survival under oxidative conditions, promotes tumor progression, and contributes to chemoresistance [12,13]. Notably, overexpression of the TrxR/Trx system has been associated with increased tumor aggressiveness and has emerged as a negative prognostic marker in several malignancies. These observations underscore the importance of redox-regulating enzymes not only in supporting malignant phenotypes, but also in influencing clinical outcomes, highlighting the therapeutic potential of targeting redox homeostasis in oncology [13,14].
Based on these evidences, several clinical trials investigating AF have been initiated to evaluate its safety, efficacy, and therapeutic value in various cancer types (e.g. epithelial ovarian, primary peritoneal, fallopian tube cancer [NCT01747798, NCT03456700], chronic lymphocytic leukemia (CLL) [NCT01419691], non-small cell lung cancer (NSCLC) [NCT01737502], etc.), either as monotherapy or in combination with other agents (Table 1). Beyond oncology, AF is currently being investigated in different clinical trials targeting non-oncological diseases, including parasitic infections and chronic viral diseases such as human immunodeficiency virus (HIV) (Table1).
In this context, this review explores clinical evidence from both early-phase and ongoing trials involving various solid tumors and hematologic malignancies, examining key aspects including the safety and tolerability of AF, optimal dosing strategies, PK, and preliminary therapeutic efficacy observed in diverse cancer types. Furthermore, clinical trials investigating AF in non-oncological diseases are discussed.
2. Auranofin-based Monotherapy for Cancer Management
NCT01747798. Auranofin in Treating Patients with Recurrent Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer https://clinicaltrials.gov/study/NCT01747798
Rationale. Recurrent epithelial ovarian, primary peritoneal, and fallopian tube cancers remain a major clinical challenge due to the limited efficacy of standard treatments in the recurrent setting [15]. Ovarian cancer cells often exhibit a pro-oxidative status, meaning they have a higher level of Reactive Oxygen Species (ROS) than normal cells. This oxidative stress can contribute to the development and progression of the disease, including drug resistance and tumor metastasis [16]. AF is known to act as a pro-oxidant agent by inhibiting TrxR, with the ability to disrupt this antioxidant defense mechanism, leading to an accumulation of ROS and potentially inducing cancer cells death [17].
Objectives. The main objective of this phase 0 clinical trial was to evaluate the feasibility, tolerability, safety and preliminary efficacy of AF in patients with recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer, particularly focusing on those who were asymptomatic but exhibited elevated cancer antigen 125 (CA 125) levels, a biomarker associated with ovarian cancer recurrence [18]. More specifically, the trial aimed to assess the biochemical response rate, as measured by changes in CA 125 levels. By focusing on patients who showed rising CA 125 but no overt clinical symptoms, the study sought to determine if early intervention with AF could delay clinical progression.
Treatment. Patients received AF orally twice daily on days 1-28. No detailed information regarding the dosage of AF were provided. Courses repeated every 28 days in the absence of disease progression or unacceptable toxicity. After completion of study treatment, patients were followed up every 6 months for two years.
Eligibility. The study enrolled 10 female patients aged 18 years or older who completed their initial treatment for epithelial ovarian, primary peritoneal, or fallopian tube cancer, which may have included surgery and/or chemotherapy, and who did not receive any further treatment for disease progression. A key inclusion criterion involved an elevated CA 125 tumor marker, which must meet one of two definitions: either an increase of at least 100 units/mL following normalization during first-line chemotherapy, or a doubling of CA 125 levels beyond the upper limit of normal (ULN) after initial normalization, confirmed within four weeks. Patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status, a standardized scale used to evaluate a cancer patient's level of functioning, of 0–2 (patients were fully active to moderately limited but still self-sufficient and ambulatory) and must have sufficient bone marrow, liver, and kidney function according to standard laboratory values. Women of childbearing potential were required to have a negative pregnancy test prior to enrollment and must agree to use effective contraception for the duration of the study. In addition, all participants must be willing to provide informed written consent, take part in telephone interviews related to changes in CA 125 levels, and provide tissue samples for research purposes.
Results and Conclusions. The study successfully demonstrated the feasibility of enrolling and treating 10 asymptomatic patients with elevated CA 125 levels using AF. While specific efficacy outcomes were not detailed, the trial provided valuable insights into patient perceptions of CA 125 elevation and the potential role of AF in stabilizing or reducing CA 125 levels. These findings supported further investigation of AF as a maintenance therapy option for patients at high risk of recurrent disease.
NCT01419691. Phase I and II Study of AF in Chronic Lymphocytic Leukemia (CLL) https://clinicaltrials.gov/study/NCT01419691
Rationale. CLL is the most common adult leukemia in Western countries, characterized by the accumulation of functionally incompetent B lymphocytes, which reside in the blood, bone marrow, lymph nodes, and spleen [19]. Although current therapies have improved patient outcomes, CLL remains incurable, and many patients eventually relapse or develop resistance to existing treatments. In this scenario, there is an ongoing need for new therapeutic strategies, particularly those that target the specific vulnerabilities of CLL cells [19].
AF showed preclinical efficacy against CLL by inducing apoptosis in CLL cells. This effect is achieved by triggering oxidative and endoplasmic reticulum stress, even in CLL cells with high-risk cytogenetic features like 11q or 17p deletions [20,21]. Given AF’s established safety profile in non-oncologic settings [22], repurposing it for use in CLL could represent a potentially cost-effective and innovative approach for treating this challenging disease.
Objectives. This clinical trial is structured in two phases—Phase I and Phase II—each with distinct but complementary goals aimed at evaluating the safety, tolerability, and preliminary efficacy of repurposing AF in patients with relapsed or refractory CLL, small lymphocytic lymphoma (SLL), and prolymphocytic leukemia (PLL). In the Phase I, the primary objective was to determine the maximum tolerated dose (MTD) of AF when given orally to patients with CLL, by including careful assessment of the drug’s safety profile, potential toxicities, and dose-limiting side effects. In fact, establishing the appropriate dosage is essential to ensure patient safety and to define the optimal therapeutic window for subsequent evaluation. Following this, Phase II focused on assessing the clinical activity of AF at the identified dose, by measuring the overall response rate (ORR), which included partial and complete responses based on established criteria for CLL. Additional objectives included evaluating progression-free survival (PFS) and overall survival (OS), as well as monitoring changes in biological markers that may reflect the drug’s mechanism of action—particularly its effects on oxidative stress, apoptosis, and redox regulation in leukemic cells.
Treatment. Patients received AF orally, 6 mg in the morning and 6 mg in the evening.
Eligibility. This study enrolled 15 patients who must have a histologically confirmed diagnosis of B-cell CLL, SLL, PLL arising from CLL/SLL, or Richter’s transformation. According to World Health Organization (WHO) classification, Richter's transformation is a serious complication of CLL, where the leukemia transforms into a more aggressive lymphoma, typically diffuse large B-cell lymphoma or, less commonly, Hodgkin lymphoma [23]. In addition, patients must meet the criteria for requiring therapy as defined by the 2008 revised guidelines of the International Workshop on Chronic Lymphocytic Leukemia (IWCLL). Only patients with relapsed or refractory disease who have received at least one prior line of treatment for CLL were eligible. Participants must be 18 years of age or older, have an ECOG performance status of 0 or 1, and a life expectancy of at least 2 months. Furthermore, they must have adequate organ and bone marrow function, including a total bilirubin level ≤ 1.5 times the ULN, alanine aminotransferase (ALT) ≤ twice the ULN, and serum creatinine ≤ 1.5 times the ULN.
Results and Conclusions. While specific results regarding response rates and adverse events (AEs) were not detailed in the available sources, the trial's completion suggested that AF was administered safely at the specified dosage. Further studies would be necessary to conclusively determine its efficacy in this patient population.
NCT02063698. Auranofin in Decreasing Pain in Patients with Paclitaxel-Induced Pain Syndrome https://clinicaltrials.gov/study/NCT02063698
Rationale. Paclitaxel is a widely used chemotherapy drug, particularly in the treatment of breast, ovarian, and lung cancers [24]. Classified as a taxane, it works by interfering with the normal function of microtubules, which are essential for cell division, thereby preventing cancer cells from dividing and ultimately causing them to die [25]. One of its most common and distressing side effects is the development of paclitaxel-induced pain syndrome (PIPS), a form of neuropathic pain that typically arises within a few days after treatment and can severely impact a patient’s quality of life. This pain, often described as aching or burning, typically affects the hands, feet, legs, or back and may persist for several days to weeks after each cycle, limiting both patient comfort and treatment adherence [26]. Currently, there are no standard or effective treatments to prevent or alleviate PIPS, highlighting the urgent need to explore new therapeutic approaches to better manage this condition.
In this context, AF has been shown in preclinical studies to possess anti-inflammatory and neuroprotective properties, thanks to its ability to modulate oxidative stress and inhibit key pro-inflammatory pathways such as TrxR and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a protein complex that acts as a transcription factor regulating gene expression in response to various stimuli. It plays a crucial role in cellular processes like inflammation, immune responses, cell survival, and proliferation. NF-κB is involved in both normal physiological functions and pathological conditions, including atherosclerosis and cancer [27]. Given the suspected role of inflammation and oxidative damage in the development of PIPS, AF could represent a promising candidate for repurposing in this context.
Objectives. The primary objective of this clinical trial was to determine whether AF could effectively reduce the severity of PIPS in patients receiving chemotherapy. This study sought to evaluate whether one dose of AF could alleviate the intensity of pain symptoms, particularly in the days following chemotherapy, when pain tends to peak. To assess this, patients were asked to daily complete the Modified Brief Pain Inventory (mBPI) for 7 days [28]. The mBPI is a modification of the Brief Pain Inventory (BPI), a questionnaire used to evaluate the severity of a patient's pain and the impact of this pain on the patient's daily functioning, often used in clinical trials and research settings [28].
In addition to its primary goal, the study also included secondary objectives, such as evaluating the tolerability and safety profile of AF in this population, monitoring for any side effects or adverse reactions. Furthermore, the trial might explore whether AF improves functional outcomes, such as physical activity levels, sleep quality, and the ability to carry out daily tasks during chemotherapy.
Treatment. Patients were randomly assigned to receive orally either AF or a placebo in a double-blind fashion. No specific details about the dose of AF have been published in the registry sources. Participants in the treatment arm received AF orally, starting within 24 hours after paclitaxel administration. The drug was taken once daily for 7 days, which is the period when PIPS typically peaks. The control group followed the same schedule but received a placebo tablet instead of the active drug. The short treatment window was designed to specifically target the acute pain flare associated with each paclitaxel cycle, without interfering with the ongoing chemotherapy regimen. Severity and duration of pain between patients who received AF and those who received a placebo were compared to identify a statistically and clinically meaningful reduction in pain.
Eligibility. This trial enrolled 30 patients undergoing chemotherapy with paclitaxel for cancer. To be eligible for this study, participants must meet specific laboratory and clinical criteria. These included having an absolute neutrophil count (ANC) of at least 1500/mm³, a platelet count of at least 100,000/mm³, and hemoglobin levels ≥ of 9 mg/dL. Kidney and liver function must also fall within acceptable limits, defined as a creatinine level ≤ twice the ULN, ALT or Aspartate Aminotransferase (AST) levels ≤ 1.5 times the ULN, total or direct bilirubin ≤ 1.5 times the ULN, and alkaline phosphatase also ≤ 1.5 times the ULN. Women of childbearing potential must have a negative urine or serum pregnancy test performed within 7 days prior to registration, and all participants must be able to complete study questionnaires independently or with assistance. Importantly, participants must have previously experienced pain associated with paclitaxel administration, either during current or past treatment. Formal documentation of this prior pain episode was not required. Additionally, patients must be scheduled to receive paclitaxel at a dose of at least 70 mg/m² within 14 days of randomization.
Results and Conclusions. In the phase II trial NCT02063698, eight distinct outcome measures were posted to evaluate the effect of a single dose of oral AF on PIPS. Although 30 patients were enrolled and randomly assigned to receive either a single oral dose of AF or a placebo on the day after paclitaxel administration, the study did not demonstrate a statistically significant reduction in overall pain scores. Although these differences were not large enough to meet conventional significance thresholds, the observed trend suggests a potential delayed benefit of AF over time.
3. Combinatory Regimens for Cancer Management Involving Auranofin
NCT01737502. Sirolimus and Auranofin in Treating Patients with Advanced or Recurrent Non-Small Cell Lung Cancer or Small Cell Lung Cancer https://clinicaltrials.gov/study/NCT01737502
Rationale. The interplay between AF and sirolimus is particularly relevant in cancer, as AF may enhance the effects of sirolimus. Sirolimus suppresses cell growth and proliferation by inhibiting the mechanistic target of rapamycin (mTOR) pathway, commonly aberrant in lung cancer and contributing to tumor progression and therapy resistance [29].The oxidative stress triggered by AF may sensitize tumor cells to mTOR inhibition, enhancing the antiproliferative effects of sirolimus [30]. This synergistic effect is particularly relevant in chemoresistant tumors, where alternative survival pathways and redox adaptations contribute to treatment failure, and represents a promising strategy to overcome resistance and improve outcomes in patients with advanced or recurrent NSCLC and small cell lung cancer (SCLC).
Objectives. This Phase I/II study was designed to explore the therapeutic potential of combining AF and sirolimus in patients with advanced or recurrent lung cancer.
The primary objective of the Phase I of the trial was to determine the MTD of the combination therapy, in order to establish a safe and effective dosage regimen that can be carried out into further evaluation. In the Phase II, the focus shifted to assess the PFS at 4 months of treatment, providing an early measure of the treatment’s clinical efficacy in this heavily pretreated population. The study also outlined several secondary objectives. These included evaluating OS of participants compared to controls, assessing the safety profile and AEs of the regimen, and determining the ORR, as well as duration of response, in patients with measurable disease, based on Response Evaluation Criteria in Solid Tumors (RECIST), a standardized system that assess treatment efficacy through changes in tumor size on imaging studies. In addition, a key correlative research objective was to investigate the association between molecular biomarkers and clinical outcomes such as PFS, OS, treatment response, and toxicity. This component aims to identify potential predictive markers that could guide future patient selection and personalized therapeutic approaches.
Treatment. The treatment protocol involved oral administration of AF every day from day 1 to day 28 of the treatment cycle. Sirolimus was also given orally from day 1 to day 28 but only starting from day 8 of the first course. No detailed information regarding the dosage of AF and sirolimus were provided. Courses repeated every 28 days in the absence of disease progression or unacceptable toxicity. Following completion of therapy, patients entered a long-term follow-up phase with assessments scheduled every 3 to 6 months for up to five years. This extended follow-up period allowed for comprehensive monitoring of survival outcomes and late-onset adverse effects.
Eligibility. The study enrolled 29 patients. Eligible participants for this trial were required to have histologically or cytologically confirmed lung cancer, specifically squamous cell carcinoma, Ras-mutated adenocarcinoma, or SCLC. All patients must have previously received at least one line of platinum-based chemotherapy and must lack further standard treatment options. Patients were allowed to have had prior radiation therapy, provided they had sufficiently recovered from its toxic effects (excluding alopecia) before entering the study. Key hematologic criteria included an ANC ≥ 1500/μL, platelets ≥ 100,000/μL, and hemoglobin ≥ 9 g/dL, all confirmed within 14 days prior to registration. Liver function requirements included total bilirubin ≤ 1.5 times the ULN (or direct bilirubin within normal limits), and AST and ALT ≤ 3 times the ULN, with a relaxed threshold ≤ 5 times the ULN permitted in cases of liver involvement by tumor. Patients had to have an ECOG performance status of 0–2 and a life expectancy of at least 12 weeks. Women of childbearing potential were required to have a negative serum pregnancy test within 7 days prior to enrollment. Additional criteria included providing informed consent, a commitment to return enrolling in Mayo Clinic institution for follow-up, and a willingness to provide tumor tissue for related biomarker research.
Results and Conclusions. Although the clinical trial NCT01737502 has been completed, as of the latest update on March 25, 2024, no results have been posted.NCT03456700. Auranofin and Sirolimus in Treating Participants with Ovarian Cancer https://clinicaltrials.gov/study/NCT03456700
Rationale. As previously mentioned, AF induces oxidative stress and apoptosis in cancer cells by disrupting redox homeostasis, while sirolimus inhibits mTOR signaling. Preclinical data suggest that the simultaneous disruption of redox balance and growth signaling may exert synergistic antitumor effects. Importantly, this trial also enrolled patients based on the overexpression of protein kinase C-iota (PKC-ι), an oncogenic kinase implicated in tumor progression and poor clinical outcomes across multiple cancer types. PKC-ι has been shown to regulate both redox-sensitive and mTOR-related pathways, providing a biologically relevant target for dual inhibition using AF and sirolimus [31,32].
Objectives. The primary objective was to estimate the ORR across the entire study population. Secondary objectives included assessing the ORR specifically in the subgroup of patients whose tumors overexpress PKC-ι, as well as estimating PFS, OS, and documenting AEs associated with the treatment. In addition, the study included correlative analyses to explore whether PKC-ι related biomarkers in tumor tissue are associated with treatment outcomes, including ORR, PFS, and OS.
Treatment. The treatment regimen consisted of oral administration of AF and sirolimus once daily in 28-day cycles, which were continued until disease progression or unacceptable toxicity. After treatment completion, participants were monitored every 6 months for up to 3 years to assess long-term outcomes. No detailed information regarding the dosage of AF and sirolimus were provided.
Eligibility. This study enrolled 22 patients with incurable serous ovarian, fallopian tube, or primary peritoneal cancer, who had measurable disease and an ECOG performance status of 0 or 1, indicating they were fully active or only mildly symptomatic. Patients were required to have adequate hematologic, hepatic, and renal function, including an ANC ≥ 1500/μL, platelets ≥ 100,000/μL, hemoglobin ≥ 9 g/dL, total bilirubin and liver enzymes within acceptable limits, and creatinine ≤ 1.5 times the ULN. Additionally, metabolic parameters such as fasting glucose, total cholesterol, and triglyceride levels had to be ≤ 1.5 times the ULN. A life expectancy of at least 12 weeks was also required. Importantly, participants needed to be willing to provide archival tumor tissue for biomarker analysis.
Results and Conclusions. Of the 22 patients enrolled, 21 completed the study, with one participant discontinuing early. The median OS was 4.4 months (95% Confidence Interval 2.6–12.5 months), and the PFS was 2.1 months, suggesting limited efficacy of the combination therapy in this patient population. Regarding safety, 61.9% of participants experienced at least one Grade 3 or higher AE. Common serious AEs included small intestinal obstruction (14.3%), vomiting (9.5%), and sepsis (9.5%). Non-serious AEs were also prevalent, with fatigue (90.5%), diarrhea (81%), and anemia (76.2%) being the most frequently reported. In summary, while the combination of AF and sirolimus was generally tolerable, the modest clinical benefits observed did not support its use as a standard treatment for recurrent serous ovarian cancer.
NCT02770378. A Proof-of-concept Clinical Trial Assessing the Safety of the Coordinated Undermining of Survival Paths by 9 Repurposed Drugs Combined with Metronomic Temozolomide (CUSP9v3 Treatment Protocol) for Recurrent Glioblastoma https://clinicaltrials.gov/study/NCT02770378
Rationale. Recurrent glioblastoma multiforme poses a major clinical challenge due to its aggressive nature, poor prognosis, and limited response to conventional therapies [33,34]. This Clinical Trial investigated the CUSP9v3 protocol (Coordinated Undermining of Survival Paths, version 3), an innovative treatment approach that combines low-dose temozolomide with nine repurposed non-cancer drugs, including AF. Temozolomide, an alkylating agent, remains the backbone of glioblastoma treatment due to its ability to induce DNA damage [35]. However, its efficacy is often limited by intrinsic and acquired resistance mechanisms, including DNA repair and metabolic adaptation. The combination of AF and temozolomide in the CUSP9v3 protocol is based on a strategy to overcome glioblastoma’s resistance to conventional therapies by simultaneously targeting complementary vulnerabilities within tumor cells. The inclusion of AF among the nine repurposed drugs used in combination with temozolomide was based on the observation that glioblastoma cells possess strong antioxidant mechanisms to survive in hypoxic and metabolically stressful environments, while also displaying a notable sensitivity to disruptions in redox balance [35]. Thus, AF has been included in this trial due to its ability to disrupt redox homeostasis, and increase oxidative stress in tumor cells, leading to apoptosis and reduced proliferation.
Objectives. The primary objective of this phase I/II clinical trial was to determine whether this multi-drug approach can be administered safely over time, with particular focus on identifying dose-limiting toxicities and establishing an optimal regimen that can be tolerated by most patients. Safety was monitored by tracking the incidence and severity of treatment-emergent AEs, which were evaluated using the Common Terminology Criteria for Adverse Events, version 4.03 (CTCAE v4.03), a standardized system developed by the U.S. National Cancer Institute to classify and grade the intensity of AEs. In addition to safety, the trial includes several important secondary objectives aimed at exploring the preliminary efficacy of the combination therapy, including measuring OS and PFS, as well as evaluating objective tumor responses.
Treatment. In this investigational protocol, patients diagnosed with glioblastoma received temozolomide in combination with a multi-drug regimen composed of nine repurposed agents, each with a distinct pharmacologic profile and potential anticancer activity. The treatment began with an induction phase lasting 35 days, characterized by a gradual and sequential introduction and up-titration of each individual drug. This phase was carefully structured to monitor tolerability, and adjustments were permitted based on individual patient responses and toxicity profiles.
The induction cycle initiated with temozolomide, administered at a low dose of 20 mg/m² body surface area twice daily, and continued at this dosage throughout the entire treatment course. Aprepitant, a Neurokinin-1 (NK1) receptor antagonist traditionally used as an antiemetic [36], was introduced on day 1 at 80 mg once daily and maintained continuously. Subsequently, over the course of the induction cycle, additional agents were added in a staggered way. Minocycline, a tetracycline antibiotic with anti-inflammatory and neuroprotective properties [37], was initiated at 50 mg twice daily and escalated to 100 mg twice daily. Disulfiram, an aldehyde dehydrogenase inhibitor with potential anti-tumor activity [38], followed a similar titration from 250 mg once daily to twice daily dosing. Celecoxib, a selective COX-2 inhibitor known for its anti-inflammatory and anti-angiogenic effects [39], was introduced and escalated to 400 mg twice daily. Sertraline, a selective serotonin reuptake inhibitor with reported pro-apoptotic effects in glioma cells [40], was increased from 50 mg to 100 mg twice daily. Captopril, an Angiotensin-Converting Enzyme (ACE) inhibitor that may impact tumor vasculature and immune modulation [41], was titrated to 50 mg twice daily. Itraconazole, an antifungal agent with anti-hedgehog pathway and anti-angiogenic properties [42], was increased to 200 mg twice daily. Ritonavir, a protease inhibitor with potential to inhibit P-glycoprotein and affect tumor drug resistance [43], was escalated to 400 mg twice daily. Lastly, AF, was introduced and increased to 3 mg twice daily. Following the induction period, patients proceeded to up to 12 treatment cycles, each lasting 28 days, during which the full combination was maintained at the target doses established during induction. The protocol allowed for individualized modification during the early treatment phases to optimize balance between treatment intensity and patient safety.
Eligibility. The eligibility criteria were designed to enroll 10 patients with histologically confirmed WHO grade IV glioblastoma, including those with a prior diagnosis of lower-grade glioma who subsequently experienced histological transformation. Eligible participants had to demonstrate radiologically confirmed progression of disease, as defined by Response Assessment in Neuro-Oncology (RANO) criteria, standardized guidelines used to evaluate how brain tumors respond to treatment, following prior standard treatment with radiotherapy and temozolomide. To maintain consistency in disease staging, only patients who had experienced no more than three episodes of tumor progression were allowed to enroll. To mitigate the potential impact of recent therapies on trial outcomes, patients were required to have completed chemotherapy or surgical resection at least 4 weeks prior to enrollment and radiotherapy at least 12 weeks beforehand. Additional criteria included being over 18 years of age, having a Karnofsky Performance Status (KPS) of 70% or higher, a clinical scale used to measure a cancer patient's functional ability and overall well-being, and being on a stable corticosteroid dose for at least one week prior to participation. Key hematologic and biochemical values also needed to fall within specific ranges to ensure safety and treatment tolerability, including hemoglobin ≥ 10 g/dL, adequate neutrophil and platelet counts, and acceptable renal and hepatic function. Furthermore, patients had to be appropriately vaccinated (e.g., Pneumovax and varicella) and agree to contraceptive measures if of reproductive potential. All participants provided written informed consent and demonstrated an ability to comply with the study protocol.
Results and Conclusions. Of the 10 patients enrolled in the study, 9 were evaluable for the primary endpoint, which was safety. All these 9 patients successfully met the predefined safety criteria. Among the agents used in the combination regimen, ritonavir, temozolomide, captopril, and itraconazole commonly required dose adjustments or temporary discontinuation, primarily due to toxicity. The most frequently reported AEs included nausea, headache, fatigue, diarrhea, and ataxia, consistent with known side effects of the included agents. Importantly, the PFS rate at 12 months was 50%, which is notable in the context of recurrent glioblastoma. Based on these findings, the CUSP9v3 regimen appeared to be tolerable and feasible under close clinical supervision. These encouraging results have led to the planning of a randomized phase II trial to formally evaluate the regimen’s efficacy in a larger patient population [44].
4. Auranofin in Non-Oncological Diseases
While much of the current research has focused on its anticancer activity, AF is also being actively investigated in non-oncological settings. Three clinical trials, namely NCT02089048, NCT02736968, and NCT02961829, have explored its use in treating protozoal infections such as giardiasis and amebiasis, as well as chronic viral conditions like Human Immunodeficiency Virus (HIV), where its immunomodulatory and latency-reversing effects may offer therapeutic benefits [6,45].
NCT02089048. Auranofin pharmacokinetic (PK) Following Oral Dose Administration https://clinicaltrials.gov/study/NCT02089048
Rationale. Although AF is still in use for rheumatoid arthritis, most of the available pharmacokinetic data dated back to the 1980s and were based on long-term administration in chronically ill patients [6]. Moreover, older analytical methods lacked the sensitivity to accurately characterize gold distribution and elimination after short-course dosing [6].
Considering AF’s established mechanism of action and therapeutic potential, it became important to evaluate gold levels in healthy individuals following short-term administration, which better represents its intended use for parasitic infections more accurately than long-term dosing typically applied in autoimmune disorders. Building on strong preclinical findings and increasing interest in AF as a repurposed antiparasitic agent, including its orphan drug designation for the treatment of amebiasis, clinical investigation has become essential to guide its development for infectious indications [46].
Objectives. The primary objective of this Phase I clinical trial was to assess the PK of gold following a short-term dosing regimen in healthy adult subjects. This included determining key pharmacokinetic parameters, such as peak plasma concentration (Cₘₐₓ), elimination half-life (t₁/₂), and fecal excretion, during the treatment period and through subsequent follow-up. The secondary objective was to evaluate the safety and tolerability of short-course oral AF in healthy volunteers. This involved monitoring AEs, clinical laboratory values, and vital signs throughout and after the dosing period. The trial also aimed to quantify fecal and plasma gold concentrations to estimate drug distribution and elimination. These data are essential for establishing dosing strategies in future trials targeting gastrointestinal or systemic parasitic infections.
Treatment. Participants were enrolled at a single study site for this Phase I trial. Each subject received 6 mg of AF orally once daily for 7 consecutive days, which corresponds to the standard dosing approved for rheumatoid arthritis. The total duration of individual subject participation extended up to 23 weeks, which included a 7-day dosing period followed by long-term safety follow-up visits to monitor for delayed adverse effects and track the elimination of gold from the body. This extended monitoring was particularly important given gold’s known pharmacological persistence and tissue accumulation profile.
Eligibility. To ensure safety and uniformity in pharmacokinetic assessment, the trial enrolled 15 healthy adult volunteers who met clearly defined inclusion criteria. Participants were required to be male or female of non-childbearing potential, aged between 18 and 45 years, and in overall good health as determined by a comprehensive medical history, physical examination, and standard laboratory evaluations. Women were considered of non-childbearing potential if they were postmenopausal for at least two years with elevated FSH levels or had undergone surgical sterilization procedure. This restriction helped eliminate risks related to potential teratogenicity. Participants needed to have a body mass index (BMI) between 18 and 30 kg/m², and a body weight between 50 and 122 kg. In addition, male participants were required to agree to use appropriate contraception for the duration of the study to minimize reproductive risks associated with the investigational drug. All participants had to demonstrate a clear ability and willingness to comply with study procedures, including scheduled clinic visits, drug administration, laboratory testing, and safety assessments over the full duration of the study. These strict eligibility criteria ensured reliable pharmacokinetic data collection while maintaining subject safety in this first-in-population trial for antiparasitic use.
Results and Conclusions. This Phase I clinical trial demonstrated that AF is well tolerated in healthy adult volunteers and provides a favorable PK profile for repurposing as an antiparasitic agent when administered during a limited treatment period. No serious AEs were reported, and all treatment-emergent adverse effects were mild and self-limiting. PK analysis revealed a plasma Cₘₐₓ of 0.312 µg/mL on day 7 and a t₁/₂ of approximately 35 days, indicating slow systemic clearance. Importantly, high fecal concentrations of gold were achieved (up to 13 μM, or ≥ 25 times the IC₅₀ for E. histolytica), suggesting that oral AF delivers therapeutically relevant levels to the intestinal lumen, the primary site of infection in amebiasis and giardiasis. These results support the feasibility and safety of short-course AF treatment in humans and provide essential pharmacological data to guide dose selection for future clinical trials targeting intestinal protozoan infections [6].
NCT02736968. Auranofin for Giardia Protozoa and Entamoeba histolytica http://clinicaltrials.gov/study/NCT02736968
Rationale. The continued reliance on metronidazole as first-line therapy for anaerobic protozoan infections, including Giardia intestinalis and Entamoeba histolytica, has been challenged by emerging resistance, suboptimal efficacy against cystic forms, and the need for prolonged treatment regimens [47]. These limitations, combined with documented cases of clinical failure and laboratory-induced resistance [48], highlight an urgent need for alternative antiparasitic agents.
In this context, AF emerged as a promising candidate. It demonstrated potent in vitro activity against both G. intestinalis and E. histolytica, including metronidazole-resistant strains, with IC₅₀ values significantly lower than those of metronidazole. Additionally, in vivo efficacy has been established in rodent models of amoebic colitis and liver abscess [49] as well as in Giardia infection models [50] As already mentioned, AF targets TrxR, which is essential for protozoan survival and its repurposing as a broad-spectrum antiparasitic agent is further supported by efficacy data in other parasitic diseases [51,52].
Objectives. This Phase IIa clinical trial was designed to assess the efficacy and safety of AF in adults affected by two major protozoan infections: giardiasis and amebiasis. The primary objectives of the study were two. First, for patients with G. intestinalis, the trial aimed to determine the proportion of individuals who achieved clinical resolution of diarrhea (defined as fewer than three loose stools within 24 hours) by day 5 of treatment. Similarly, for those with E. histolytica, the primary outcome was clinical resolution by day 7. These assessments were limited to individuals with confirmed infection via rapid enzyme immunoassay (EIA) and antigen detection assays at enrollment. The secondary objectives were more extensive and included both parasitological and clinical endpoints. For both infections, the study aimed to evaluate: (i) parasitological clearance, defined as the absence of trophozoites on microscopic examination or antigen detection at various time points (days 3, 5, 7, 14, and 28); (ii) reduction in trophozoite and cyst load in stool as measured by quantitative PCR (qPCR); (iii) sustained cure and relapse or reinfection, using molecular genotyping to distinguish between persistent or newly acquired strains; (iv) the time to resolution of diarrhea as an additional measure of clinical response. These secondary outcomes allowed for a deeper understanding of the dynamics of infection clearance and treatment durability over a 28-day follow-up period. By combining traditional microscopy, molecular tools (qPCR, genotyping), and antigen assays, the trial was well-equipped to provide robust data on the effectiveness of a short course AF regimen.
Ultimately, this trial aimed to position AF as a novel, short-duration, oral therapeutic for protozoan infections, potentially overcoming the limitations of current standard-of-care treatments and offering a repurposed solution with a good safety profile.
Treatment. This Phase IIa trial used a randomized, single-blind, placebo-controlled design in adult patients diagnosed with either giardiasis or amebiasis. Participants were randomly assigned to receive 6 mg of AF orally once daily or a visually identical placebo. The treatment course was 5 days for those with giardiasis and 7 days for those with amebiasis. The total study participation for each subject spanned approximately 30 days, including a pre-enrollment screening period of up to 4 days and subsequent in-person follow-up visits.
Eligibility. A total of 136 participants (68 with giardiasis and 68 with amebiasis) were enrolled and randomized to receive either AF or a placebo. Participants eligible for inclusion in this trial were adults aged 18 to 65 years, including males and non-pregnant females. All subjects were required to have a confirmed diagnosis of either G. intestinalis or E. histolytica infection, verified through stool antigen detection via rapid EIA and confirmatory laboratory testing at enrollment. In cases of dual infection, enrollment was directed to the E. histolytica arm unless that cohort was already full.
Participants were required to have recent diarrheal symptoms (≥ 3 loose stools in the past 24 hours) while remaining clinically stable. Stable chronic conditions were permitted if unchanged in medication type, dosage, or frequency over the past 3 months and clinically well-managed in the last 6 months. Routine medications, including topical, inhaled, and contraceptive therapies, were allowed if not deemed to pose additional safety risk. Subjects had to present with normal vital signs and laboratory parameters within protocol-defined ranges, including assessments of renal and liver function, hematology, and urinalysis. Women of reproductive potential needed a negative pregnancy test within 72 hours prior to study medication initiation and were required to use contraception throughout the study and for 4 months after enrollment.
Results and Conclusions. Although full trial data have not yet been published, early reports indicate that AF demonstrated a good safety profile, with no serious AEs reported and good overall tolerability among participants. Clinically, AF showed a clear benefit over placebo in achieving resolution of diarrhea by the designated endpoints, day 5 for giardiasis and day 7 for amebiasis, defined as fewer than three loose stools in 24 hours. Additionally, parasitological outcomes, including stool antigen clearance and microscopic evaluation, supported the efficacy of the treatment. These findings suggested that AF may be an effective and safe alternative therapy for protozoan gastrointestinal infections, especially in contexts where resistance to traditional drugs, like metronidazole, is a growing concern. The trial results provide a strong basis for future Phase III studies aimed at confirming the drug’s clinical utility and broadening its role as a repurposed antiparasitic agent.
NCT02961829. Multi Interventional Study Exploring HIV-1 Residual Replication: a Step Towards HIV-1 Eradication and Sterilizing Cure https://clinicaltrials.gov/study/NCT02961829
Rationale. Despite the remarkable success of antiretroviral therapy (ART) in suppressing viral replication and restoring immune function, a definitive cure for HIV remains elusive. This is primarily due to the persistence of latent viral reservoirs, mainly located in long-lived CD4⁺ memory T cells and certain tissue compartments. For this reason, a multifaceted approach is increasingly considered essential to achieve HIV cure.
Therefore, a combination strategy that simultaneously targets different mechanisms of viral persistence appears to be the most promising approach.
Objectives. This pilot proof-of-concept study aimed to explore the use of a combination of drugs to eliminate residual plasma viremia and reduce HIV reservoirs in patients on long-term ART. These include: (i) enhancing antiretroviral therapy with Maraviroc and/or Dolutegravir, aimed at blocking viral entry and replication [53]; (ii) stimulating the immune system with a personalized dendritic cell vaccine derived from autologous HIV; (iii) reactivating latent virus using a latency-reversing approach focused on Class III histone deacetylase (HDAC) inhibition, like Sirtuin-1 [54]; (iv) selectively reducing long-lived HIV reservoir cells, mainly located in central and transitional memory CD4⁺ T cells, by using AF [45,55].
Treatment. Adults with chronic HIV infection and stable viral suppression under ART were selected and assigned to one of six groups, each receiving a different combination of drugs over 48 weeks. The six groups were: (i) the control group which continued with the standard ART regimen therapy without any additional interventions, serving as a baseline for comparison; (ii) the ART Intensification group, which received dolutegravir and maraviroc; (iii) the ART Intensification + Nicotinamide group which received the same ART intensification (dolutegravir + maraviroc), along with nicotinamide, Sirtuin HDAC inhibitor; (iv) the ART Intensification + AF group for which the intensified ART was combined with AF; (v) the ART Intensification + Therapeutic Dendritic Cell Vaccine group for which the intensified ART was supplemented with an autologous dendritic cell-based vaccine; (vi) the Multimodal Intervention Group which received the most comprehensive treatment, combining all components: intensified ART, nicotinamide, AF, and the dendritic cell vaccine.
Eligibility. The study enrolled 30 patients. Participants eligible for inclusion in the study were adults aged 18 years or older with a documented diagnosis of HIV-1 infection. To qualify, participants needed to be on a stable Highly Active AntiRetroviral Therapy (HAART) regimen for at least two years, with no changes in their ART during the 24 weeks immediately preceding the screening.
In addition, all candidates had to demonstrate sustained virologic suppression, defined as an HIV-1 viral load below 50 copies/mL, without any two consecutive measurements exceeding this threshold in the past two years.
Results and Conclusions. While a full “cure” was not achieved yet, the early results obtained suggest that combining ART intensification, immune stimulation, and latency reversal could be a promising pathway toward reducing the HIV reservoir. However, more comprehensive analysis is needed.
5. Conclusions and Future Perspectives
It is nowadays widely recognized that improvement of currently available clinical protocols for treating cancer or different diseases can extensively benefit from multiple approaches. Among them the repurposing of approved drugs for indications different from the original ones represents a reliable and efficient strategy coupling the likely rapid translation in the clinic with economically sustainable profile. In this frame inorganic approved medicines are suitable because of their chemical versatility and ability to target multiple biological substrates. In this view, the well-known high reactivity of metal centers can be conveniently exploited for the tailored treatment of various pathologies [56,57].
In such context, this review aims to provide an updated overview of the clinical trials investigating the repurposing of AF in oncological and non-oncological settings [58].
In detail, a search conducted on ClinicalTrials.gov using the entry "auranofin" as “other terms” identified fifteen studies. Out of these, six studies were excluded for the following reasons: two had been withdrawn prior to patient enrollment (the reasons are unknown), one was listed with an unknown status, one had not yet begun recruiting at the time of our search, and two were unrelated to the primary scope of this review (AF is used as reference drug in rheumatoid arthritis condition). The remaining nine trials were herein analyzed and discussed.
Among these, six trials repurposed AF in various cancer types in mono- (Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer, Chronic Lymphocytic Leukemia, paclitaxel-induced pain syndrome) and in combination-(Lung, ovarian cancer and glioblastoma) therapy.
The other three trials target non-oncological diseases, namely amoebiasis, giardiasis and HIV.
Three trials posted clinical results, NCT02063698, NCT 03456700 and NCT02736968.
The NCT02063698 focused on the potential of AF to counteract paclitaxel side-effects, aiming to offer preliminary evidence for its use as a supportive care option in oncology. By comparing the outcomes between the AF and placebo groups, the study aimed to determine whether short-term use of AF could meaningfully prevent or reduce chemotherapy-induced pain. Although no conclusive evidence on the effectiveness of AF, the delay in the insurgence of pain observed in the treatment group provided proof that may warrant further investigation, perhaps with adjusted dosing regimens or extended treatment duration.
The NCT03456700 is a Phase II study conducted by the Mayo Clinic evaluating the efficacy of AF in combination with sirolimus in patients with recurrent serous ovarian cancer. It was evidenced that, while the combination of AF and sirolimus was generally well tolerated, the modest clinical benefits observed do not support its adoption as a standard treatment for this patient population.
Trial NCT02736968 is a phase IIa study assessing the efficacy of AF in patients with either giardiasis or amebiasis, sponsored by the National Institute of Allergy and Infectious Diseases (NIAID). The obtained results indicate the potential of AF as a safe and effective treatment option for gastrointestinal protozoal infections, particularly in settings where resistance to standard therapies such as metronidazole is becoming more prevalent. The trial outcomes offer solid support for advancing to Phase III studies to further assess its clinical effectiveness and expand its application as a repurposed antiparasitic drug.
Overall, data from these AF trials support continued investigation, as the safety and PK findings are strong enough to justify advancement to patient trials, though further rigorous, controlled clinical studies are still needed to determine AF’s tolerability profile, optimal dosing, and long-term efficacy for each specific indication. Nevertheless, these trials illustrate the therapeutic versatility of AF and reflect a growing interest in drug repurposing to address unmet medical needs.
Author Contributions
Conceptualization, E.B., S.T., T.M.; supervision, E.B., S.T., T.M.; methodology, D.G., L.C., E.B; investigation, D.G., L.C., E.B; writing, D.G., L.C., E.B; M.V, R.B., F.D.S., S.T., T.M., E.B. All authors have read and agreed to the published version of the manuscript.
Funding
D.G.: L.C., M.V. and E.B. thank the financial support from Ministero Italiano dell’Università e della Ricerca (MUR) under the program PRIN 2022 PNRR -Progetti di Rilevante Interesse Nazionale P202252C7C MILESTONE: Mitochondrial delIvery of goLd-bearing complExeS with Tspo ligands for Ovarian caNcEr treatment. T.M. and E.B. thank the financial support from Ministero Italiano dell’Università e della Ricerca (MUR) under the program PRIN 2022-Progetti di Rilevante Interesse Nazionale, project code: 2022ALJRPL “Biocompatible nanostructures for the chemotherapy treatment of prostate cancer.”.
References
- Yamashita, M. Auranofin: Past to Present, and Repurposing. Int. Immunopharmacol. 2021, 101, 108272. [Google Scholar] [CrossRef]
- Betts, K.A.; Griffith, J.; Ganguli, A.; Li, N.; Douglas, K.; Wu, E.Q. Economic Burden and Treatment Patterns of Cycling between Conventional Synthetic Disease-Modifying Antirheumatic Drugs among Biologic-Treated Patients with Rheumatoid Arthritis. Clin. Ther. 2016, 38, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Messori, L. A Role for Metal-Based Drugs in Fighting COVID-19 Infection? The Case of Auranofin. ACS Med. Chem. Lett. 2020, 11, 1067–1068. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, Y.; Xu, Z.; Ma, X.; Chen, X.; Liu, W. Repurposing of the Gold Drug Auranofin and a Review of Its Derivatives as Antibacterial Therapeutics. Drug Discov. Today 2022, 27, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Gamberi, T.; Chiappetta, G.; Fiaschi, T.; Modesti, A.; Sorbi, F.; Magherini, F. Upgrade of an Old Drug: Auranofin in Innovative Cancer Therapies to Overcome Drug Resistance and to Increase Drug Effectiveness. Med. Res. Rev. 2022, 42, 1111–1146. [Google Scholar] [CrossRef]
- Capparelli, E. V.; Bricker-Ford, R.; Rogers, M.J.; McKerrow, J.H.; Reed, S.L. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.; Linder, S. Repurposing of Auranofin: Thioredoxin Reductase Remains a Primary Target of the Drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Bjørklund, G.; Zou, L.; Wang, J.; Chasapis, C.T.; Peana, M. Thioredoxin Reductase as a Pharmacological Target. Pharmacol. Res. 2021, 174, 105854. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P.; Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Thioredoxin Reductase: A Target for Gold Compounds Acting as Potential Anticancer Drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar] [CrossRef]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of Thioredoxin Reductase by Auranofin Induces Apoptosis in Cisplatin-Resistant Human Ovarian Cancer Cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Jackson-Rosario, S.; Cowart, D.; Myers, A.; Tarrien, R.; Levine, R.L.; Scott, R.A.; Self, W.T. Auranofin Disrupts Selenium Metabolism in Clostridium Difficile by Forming a Stable Au-Se Adduct. J. Biol. Inorg. Chem. 2009, 14, 507–519. [Google Scholar] [CrossRef]
- Arnér, E.S.J.; Holmgren, A. The Thioredoxin System in Cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The Role of Thioredoxin System in Cancer: Strategy for Cancer Therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Gencheva, R.; Arner, E.S.J. Thioredoxin Reductase Inhibition for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2021, 62, 177–196. [Google Scholar] [CrossRef]
- Berek, J.S.; Renz, M.; Kehoe, S.; Kumar, L.; Friedlander, M. Cancer of the Ovary, Fallopian Tube, and Peritoneum: 2021 Update. Int. J. Gynecol. Obstet. 2021, 155, 61–85. [Google Scholar] [CrossRef]
- Ding, D.N.; Xie, L.Z.; Shen, Y.; Li, J.; Guo, Y.; Fu, Y.; Liu, F.Y.; Han, F.J. Insights into the Role of Oxidative Stress in Ovarian Cancer. Oxid. Med. Cell. Longev. 2021, 2021, 8388258. [Google Scholar] [CrossRef]
- Abdalbari, F.H.; Telleria, C.M. The Gold Complex Auranofin: New Perspectives for Cancer Therapy. Discov. Oncol. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Momenimovahed, Z.; Mazidimoradi, A.; Allahqoli, L.; Salehiniya, H. The Role of CA-125 in the Management of Ovarian Cancer: A Systematic Review. Cancer Rep. 2025, 8, e70142. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Ferreri, A.M.; Galigaris-Cappio, F. Chronic Lymphocytic Leukemia. Crit. Rev. Oncol. Hematol. 2007, 64, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S. Das; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Saba, N.; Shen, M.; Ghias, M.; Farooqui, M.; Austin, C.; Schorno, K.; Weir, S.; Bhalla, K.; Wiestner, A. The Gold Compound Auranofin Induces Oxidative Stress and Apoptosis in Primary CLL Cells Independent of Classic Prognostic Markers and the Protective Effect of the Tissue Microenvironment. Blood 2012, 120, 865–865. [Google Scholar] [CrossRef]
- Blodgett, R.C.; Pietrusko, R.G. Long-Term Efficacy and Safety of Auranofin: A Review of Clinical Experience. Scand. J. Rheumatol. 1987, 16, 67–78. [Google Scholar] [CrossRef]
- Hleuhel, M.H.; Ben-Dali, Y.; Da Cunha-Bang, C.; Brieghel, C.; Clasen-Linde, E.; Niemann, C.U.; Andersen, M.A. Risk Factors Associated with Richter’s Transformation in Patients with Chronic Lymphocytic Leukaemia: Protocol for a Retrospective Population-Based Cohort Study. BMJ Open 2019, 9, e023566. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Quispe, C.; Patra, J.K.; Singh, Y.D.; Panda, M.K.; Das, G.; Adetunji, C.O.; Michael, O.S.; Sytar, O.; Polito, L.; et al. Paclitaxel: Application in Modern Oncology and Nanomedicine-Based Cancer Therapy. Oxid. Med. Cell. Longev. 2021, 2021, 3687700. [Google Scholar] [CrossRef]
- Alqahtani, F.Y.; Aleanizy, F.S.; El Tahir, E.; Alkahtani, H.M.; AlQuadeib, B.T. Paclitaxel. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press Inc., 2019; Vol. 44, pp. 205–238 ISBN 9780128171653.
- Yan, X.; Maixner, D.W.; Yadav, R.; Gao, M.; Li, P.; Bartlett, M.G.; Weng, H.R. Paclitaxel Induces Acute Pain via Directly Activating Toll like Receptor 4. Mol. Pain 2015, 11. [Google Scholar] [CrossRef]
- Da̧bek, J.; Kułach, A.; Ga̧sior, Z. Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-ΚB): A New Potential Therapeutic Target in Atherosclerosis? Pharmacol. Reports 2010, 62, 778–783. [Google Scholar] [CrossRef]
- Chen, W.H.; Chan, K.S.; Gan, T.J.; Chen, C.; Lakshminarayanan, M.; Revicki, D.A. Validation of the Modified Brief Pain Inventory-Exploratory Form in Surgery Patients. Health Outcomes Res. Med. 2010, 1, e17–e28. [Google Scholar] [CrossRef]
- Rehan, M. An Anti-Cancer Drug Candidate OSI-027 and Its Analog as Inhibitors of MTOR: Computational Insights Into the Inhibitory Mechanisms. J. Cell. Biochem. 2017, 118, 4558–4567. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chen, J.; Yu, Y.; Wu, F.; Shen, X.; Qiu, C.; Zhang, T.; Hong, L.; Zheng, P.; Shao, R.; et al. Compensatory Combination of MTOR and TrxR Inhibitors to Cause Oxidative Stress and Regression of Tumors. Theranostics 2021, 11, 4335–4350. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hill, K.S.; Fields, A.P. PKCi Maintains a Tumor-Initiating Cell Phenotype That Is Required for Ovarian Tumorigenesis. Mol. Cancer Res. 2013, 11, 1624–1635. [Google Scholar] [CrossRef]
- Tyagi, K.; Roy, A.; Mandal, S. Protein Kinase C Iota Promotes Glycolysis via PI3K/AKT/MTOR Signalling in High Grade Serous Ovarian Cancer. Mol. Biol. Rep. 2024, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro. Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Jezierzański, M.; Nafalska, N.; Stopyra, M.; Furgoł, T.; Miciak, M.; Kabut, J.; Gisterek-Grocholska, I. Temozolomide (TMZ) in the Treatment of Glioblastoma Multiforme—A Literature Review and Clinical Outcomes. Curr. Oncol. 2024, 31, 3994–4002. [Google Scholar] [CrossRef]
- Pendergrass, K.; Hargreaves, R.; Petty, K.J.; Carides, A.D.; Evans, J.K.; Horgan, K.J. Aprepitant: An Oral NK1 Antagonist for the Prevention of Nausea and Vomiting Induced by Highly Emetogenic Chemotherapy. Drugs of Today 2004, 40, 853–863. [Google Scholar] [CrossRef]
- Elewa, H.F.; Hilali, R.; Hess, D.C.; Machado, L.S.; Fagan, S.C. Minocycline for Short-Term Neuroprotection. Pharmacotherapy 2006, 26, 515–521. [Google Scholar] [CrossRef]
- Caminear, M.W.; Harrington, B.S.; Kamdar, R.D.; Kruhlak, M.J.; Annunziata, C.M. Disulfiram Transcends ALDH Inhibitory Activity When Targeting Ovarian Cancer Tumor-Initiating Cells. Front. Oncol. 2022, 12, 762820. [Google Scholar] [CrossRef]
- Rosas, C.; Sinning, M.; Ferreira, A.; Fuenzalida, M.; Lemus, D. Celecoxib Decreases Growth and Angiogenesis and Promotes Apoptosis in a Tumor Cell Line Resistant to Chemotherapy. Biol. Res. 2014, 47, 1–9. [Google Scholar] [CrossRef]
- Fayyaz, S.; Atia-Tul-Wahab; Irshad, R. ; Siddiqui, R.A.; Choudhary, M.I. Antidepressant Sertraline Hydrochloride Inhibits the Growth of HER2+ AU565 Breast Cancer Cell Line through Induction of Apoptosis and Cell Cycle Arrest. Anticancer. Agents Med. Chem. 2024, 24, 1038–1046. [Google Scholar] [CrossRef]
- Wysocki, P.J.; Kwiatkowska, E.P.; Kazimierczak, U.; Suchorska, W.; Kowalczyk, D.W.; Mackiewicz, A. Captopril, an Angiotensin-Converting Enzyme Inhibitor, Promotes Growth of Immunogenic Tumors in Mice. Clin. Cancer Res. 2006, 12, 4095–4102. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Tsai, T.F. Itraconazole in the Treatment of Nonfungal Cutaneous Diseases: A Review. Dermatol. Ther. (Heidelb). 2019, 9, 271–280. [Google Scholar] [CrossRef]
- Laurence, J.; Elhadad, S.; Gostynska, S.; Yu, Z.; Terry, H.; Varshney, R.; Fung, K.M.; Choi, M.E.; Ahamed, J. HIV Protease Inhibitor Ritonavir Induces Renal Fibrosis and Dysfunction: Role of Platelet-Derived TGF-SS1 and Intervention via Antioxidant Pathways. AIDS 2020, 34, 989–1000. [Google Scholar] [CrossRef]
- Halatsch, M.E.; Kast, R.E.; Karpel-Massler, G.; Mayer, B.; Zolk, O.; Schmitz, B.; Scheuerle, A.; Maier, L.; Bullinger, L.; Mayer-Steinacker, R.; et al. A Phase Ib/IIa Trial of 9 Repurposed Drugs Combined with Temozolomide for the Treatment of Recurrent Glioblastoma: CUSP9v3. Neuro-Oncology Adv. 2021, 3. [Google Scholar] [CrossRef]
- Diaz, R.S.; Shytaj, I.L.; Giron, L.B.; Obermaier, B.; della Libera, E.; Galinskas, J.; Dias, D.; Hunter, J.; Janini, M.; Gosuen, G.; et al. Potential Impact of the Antirheumatic Agent Auranofin on Proviral HIV-1 DNA in Individuals under Intensified Antiretroviral Therapy: Results from a Randomised Clinical Trial. Int. J. Antimicrob. Agents 2019, 54, 592–600. [Google Scholar] [CrossRef]
- Coscione, F.; Zineddu, S.; Vitali, V.; Fondi, M.; Messori, L.; Perrin, E. The Many Lives of Auranofin: How an Old Anti-Rheumatic Agent May Become a Promising Antimicrobial Drug. Antibiotics 2024, 13, 652. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, C.; Hellberg, A.; Tannich, E.; Bruchhaus, I. Metronidazole Resistance in the Protozoan Parasite Entamoeba Histolytica Is Associated with Increased Expression of Iron-Containing Superoxide Dismutase and Peroxiredoxin and Decreased Expression of Ferredoxin 1 and Flavin Reductase. J. Biol. Chem. 1999, 274, 26051–26056. [Google Scholar] [CrossRef] [PubMed]
- Upcroft, P.; Upcroft, J.A. Drug Targets and Mechanisms of Resistance in the Anaerobic Protozoa. Clin. Microbiol. Rev. 2001, 14, 150–164. [Google Scholar] [CrossRef]
- Debnath, A.; Parsonage, D.; Andrade, R.M.; He, C.; Cobo, E.R.; Hirata, K.; Chen, S.; García-Rivera, G.; Orozco, E.; Martínez, M.B.; et al. A High-Throughput Drug Screen for Entamoeba Histolytica Identifies a New Lead and Target. Nat. Med. 2012, 18, 956–960. [Google Scholar] [CrossRef]
- Tejman-Yarden, N.; Miyamoto, Y.; Leitsch, D.; Santini, J.; Debnath, A.; Gut, J.; McKerrow, J.H.; Reed, S.L.; Eckmann, L. A Reprofiled Drug, Auranofin, Is Effective against Metronidazole-Resistant Giardia Lamblia. Antimicrob. Agents Chemother. 2013, 57, 2029–2035. [Google Scholar] [CrossRef]
- Ocholaid, E.A.; Karanja, D.M.S.; Elliott, S.J. The Impact of Neglected Tropical Diseases (Ntds) on Health and Wellbeing in Sub-Saharan Africa (Ssa): A Case Study of Kenya. PLoS Negl. Trop. Dis. 2021, 15, 1–19. [Google Scholar] [CrossRef]
- Ilari, A.; Baiocco, P.; Messori, L.; Fiorillo, A.; Boffi, A.; Gramiccia, M.; Di Muccio, T.; Colotti, G. A Gold-Containing Drug against Parasitic Polyamine Metabolism: The X-Ray Structure of Trypanothione Reductase from Leishmania Infantum in Complex with Auranofin Reveals a Dual Mechanism of Enzyme Inhibition. Amino Acids 2012, 42, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Auclair, M.; Guénantin, A.C.; Fellahi, S.; Garcia, M.; Capeau, J. HIV Antiretroviral Drugs, Dolutegravir, Maraviroc and Ritonavir-Boosted Atazanavir Use Different Pathways to Affect Inflammation, Senescence and Insulin Sensitivity in Human Coronary Endothelial Cells. PLoS One 2020, 15, e0226924. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Chavez, L.; Hakre, S.; Calvanese, V.; Verdin, E. Reactivation of Latent HIV by Histone Deacetylase Inhibitors. Trends Microbiol. 2013, 21, 277–285. [Google Scholar] [CrossRef]
- de Almeida Baptista, M.V.; da Silva, L.T.; Samer, S.; Oshiro, T.M.; Shytaj, I.L.; Giron, L.B.; Pena, N.M.; Cruz, N.; Gosuen, G.C.; Ferreira, P.R.A.; et al. Immunogenicity of Personalized Dendritic-Cell Therapy in HIV-1 Infected Individuals under Suppressive Antiretroviral Treatment: Interim Analysis from a Phase II Clinical Trial. AIDS Res. Ther. 2022, 19, 1–15. [Google Scholar] [CrossRef]
- Cirri, D.; Chiaverini, L.; Pratesi, A.; Marzo, T. Is the Next Cisplatin Already in Our Laboratory? https://doi.org/10.1080/02603594.2022.2152016 2022, 1–14. [CrossRef]
- Chiaverini, L.; Leo, R. Di; Famlonga, L.; Pacini, M.; Baglini, E.; Barresi, E.; Peana, M.F.; Tolbatov, I.; Marrone, A.; Mendola, D. La; et al. The Metal(Loid)s’ Dilemma. What’s the next Step for a New Era of Inorganic Molecules in Medicine? Metallomics 2025, 17, 13. [Google Scholar] [CrossRef]
- Cirri, D.; Bartoli, F.; Pratesi, A.; Baglini, E.; Barresi, E.; Marzo, T. Strategies for the Improvement of Metal-Based Chemotherapeutic Treatments. Biomedicines 2021, 9, 504. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical Structure of Auranofin.
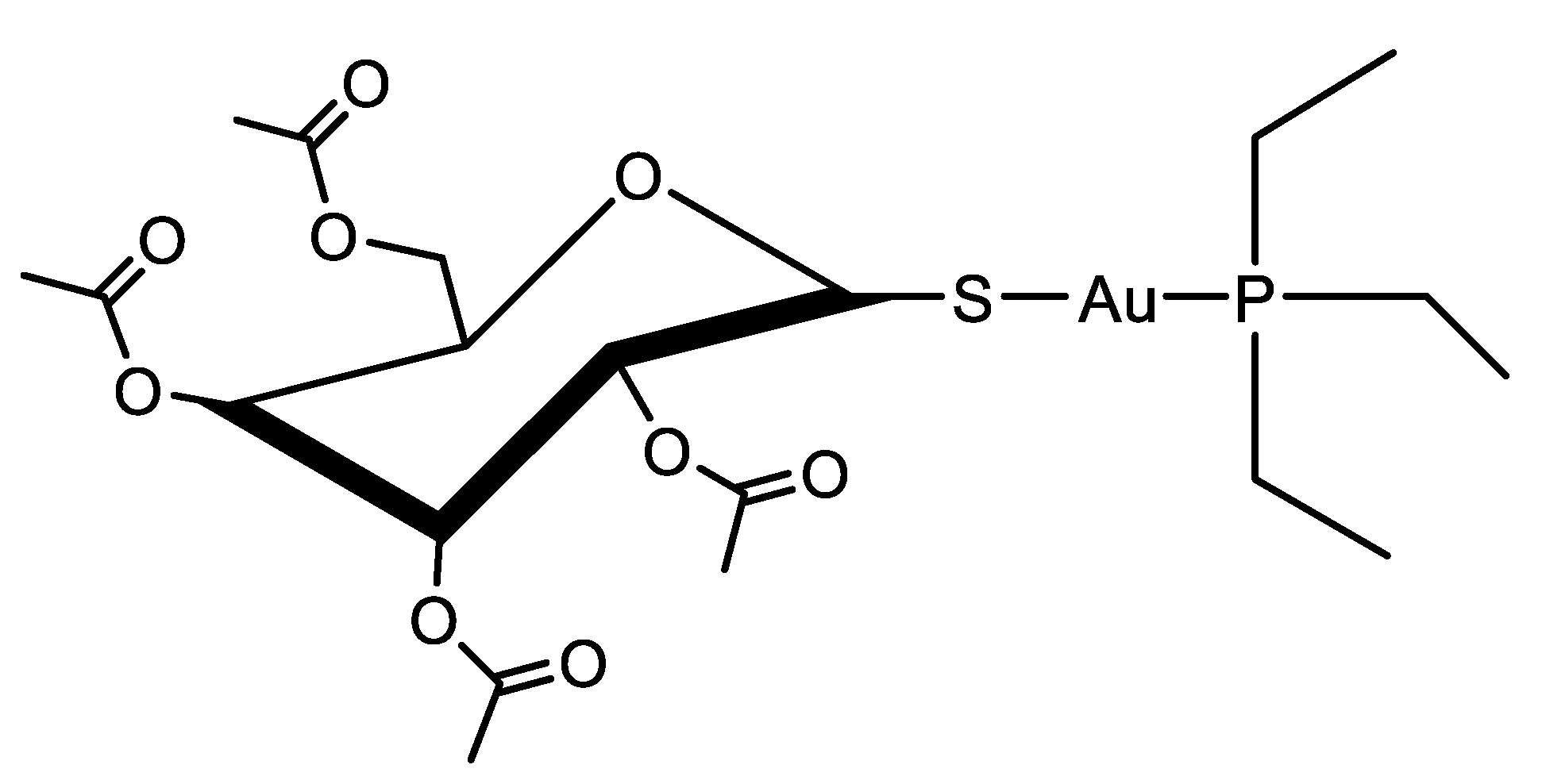

Table 1.
Clinical Trials involving Auranofin (from https://clinicaltrials.gov/, accessed on july 2025).
Table 1.
Clinical Trials involving Auranofin (from https://clinicaltrials.gov/, accessed on july 2025).
| ClinicalTrials.gov Identifier | Indication | Sponsor | Statusa | Phaseb | Resultsc |
| NCT01747798 | Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | Mayo Clinic | C(2019) | Early 1 (0) | N/A |
| NCT01419691 | Chronic Lymphocytic Leukemia (CLL) | University of Kansas Medical Center | C(2016) | 2 | N/A |
| NCT02063698 | Paclitaxel-Induced Pain Syndrome | Mayo Clinic | C(2019) | 2 | Y |
| NCT01737502 | Lung cancer | Mayo Clinic | C(2024) | 1/2 | N/A |
| NCT03456700 | Ovarian Cancer | Mayo Clinic | T(2025) | 2 | Y |
| NCT02770378 | Glioblastoma | University of Ulm | C(2021) | 1/2 | N/A |
| NCT02089048 | Amoebiasis | National Institute of Allergy and Infectious Diseases (NIAID) | C(2017) | 1 | N/A |
| NCT02736968 | amoebiasis or giardiasis | National Institute of Allergy and Infectious Diseases (NIAID) | C(2023) | 2 | Y |
| NCT02961829 | HIV | Federal University of São Paulo | C(2020) | N/A | N/A |
aC = completed (The study has ended normally, and participants are no longer being examined or treated); T = terminated (The study has stopped early and will not start again. Participants are no longer being examined or treated). The year of the last update is reported in bracket in the “Status” column.bN/A = Not ApplicablecY = Results published; N/A = Results not Available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



