
Submitted:
26 August 2025
Posted:
27 August 2025
You are already at the latest version
Abstract
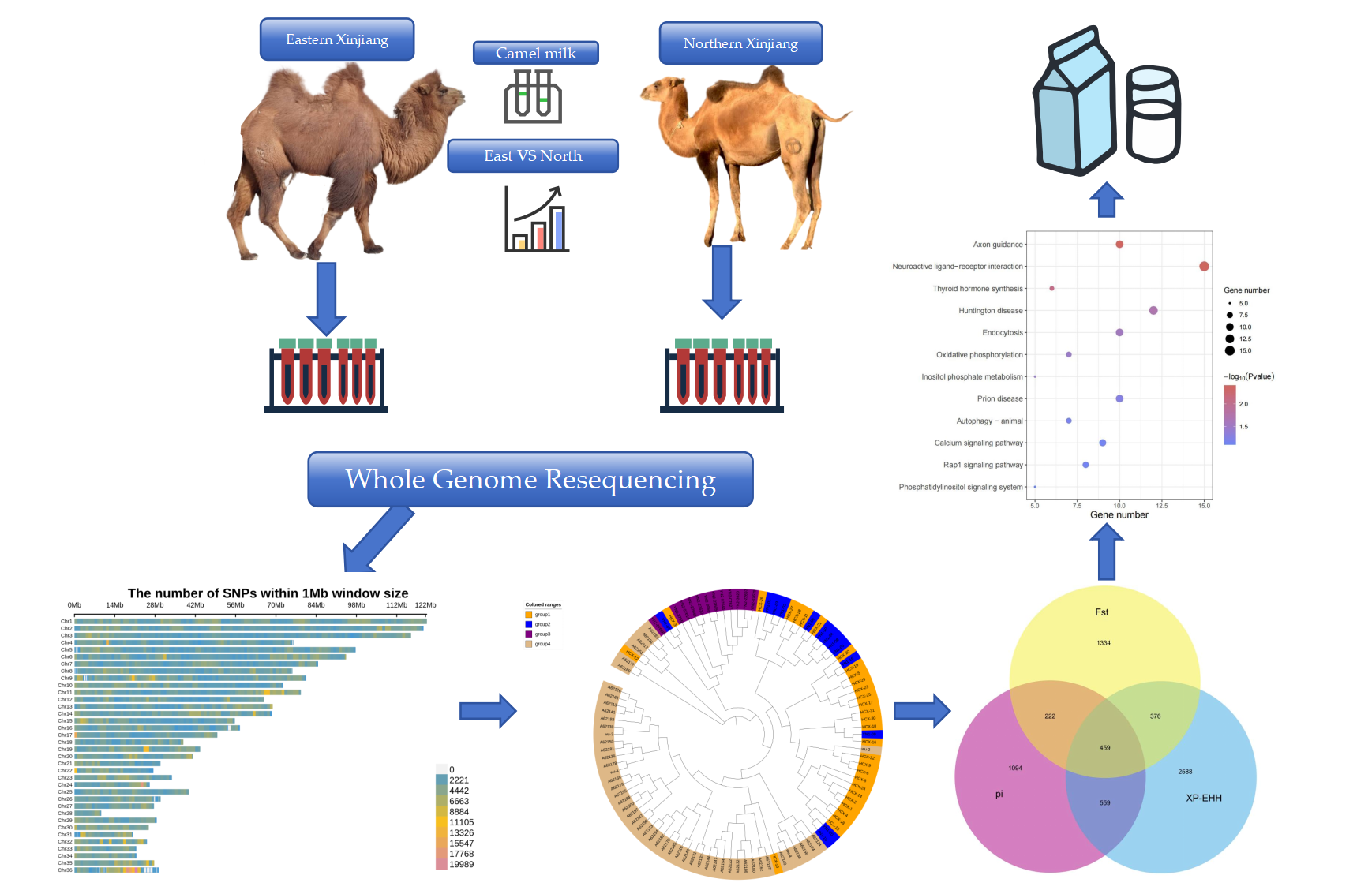
Camel milk is rich in nutrients and bioactive factors, which are crucial for human nutrition, health, and disease treatment. However, there is limited research on the genes related to lactation traits in Bactrian camels and their regulatory mechanisms. This lack of research prevents the provision of systematic molecular breeding guidance to improve milk production traits and enhance milk quality in Bactrian camels. Previous research has shown a significant difference in milk production traits between the eastern and northern regions of Xinjiang. In this study, 106 Bactrian camels were collected—55 from three camel farms in the northern region and 51 from one farm in the eastern region—for whole-genome resequencing. Through variant detection, population structure, and selection signal analysis, a total of 6,451,453 SNPs were identified. Principal component analysis and phylogenetic tree analysis revealed that the Bactrian camel populations from the eastern and northern regions can be clearly distinguished. GO analysis and KEGG enrichment analysis revealed that, compared to Bactrian camels from the northern region, candidate genes from the eastern region were mainly enriched in two pathways: plasma membrane and thyroid hormone synthesis. Whole-genome selection signal analysis identified eight candidate genes potentially related to lactation traits: DUOX1, DUOX2, CPQ, CGA, PLCG1, FYN, GNRHR, and TRHR. These genes can serve as potential marker sites for molecular breeding in the future.
Keywords:
Bactrian Camels
; Whole Genome Resequencing
; Milk Production Traits
1. Introduction
The ancestors of camels originated in Canada, North America [1], and later migrated to Central Asia due to changes in the natural environment, where they gradually evolved humps, giving rise to dromedary and Bactrian camels [2].
In the 19th century, camels were primarily used for transportation. Since then, their economic value has increasingly been reflected in milk production, meat production, wool production, tourism, and cosmetics [3]. Camel milk is rich in nutrients such as fat, protein, minerals, unsaturated fatty acids, and amino acids, and also contains various bioactive compounds such as lactoferrin, lysozyme, immunoglobulin, lactoperoxidase, and insulin [4]. These compounds mediate immune regulation [5], antibacterial and anti-inflammatory effects [6], anticancer properties [7,8], anti-Parkinson’s effects [9], antioxidant activity [7], neuroprotective effects [10], and therapeutic effects on diabetes. Moreover, studies have shown that camel milk does not contain β-lactoglobulin and has a high content of α-lactalbumin, which is similar to human milk. Therefore, camel milk can be considered an alternative to cow’s milk for infants and young children prone to allergies [11].
According to the Food and Agriculture Organization of the United Nations (FAO) statistics for 2023, there are approximately 1.3 million Bactrian camels worldwide, with the largest populations in China (579,700), Mongolia (473,900), Kazakhstan (264,900), and Uzbekistan (18,000) [12]. In 2024, China’s “National Catalogue of Livestock and Poultry Genetic Resources” included five local breeds of Bactrian camels: Alxa Bactrian camel and Sunite Bactrian camel from Inner Mongolia, Tarim Bactrian camel and Dzungarian Bactrian camel from Xinjiang, and Qinghai camel. According to data from the “National Statistical Yearbook of China” (ISBN 978-7-5037-8743-1), there were 579,700 camels in China in 2023, distributed across Xinjiang, Inner Mongolia, Gansu, Qinghai, and other regions. Among them, Xinjiang and Inner Mongolia had the largest populations, with 316,000 and 206,000 camels, respectively. Currently, the development of the Bactrian camel industry faces several prominent issues, including insufficient milk supply, significant variations in the nutrient and functional substance content of milk from different breeding regions, and unclear quality characteristics of the milk. Additionally, compared to other dairy animals, there has been less research on the molecular breeding of Bactrian camels. Studies on genes related to lactation traits and their regulatory mechanisms are not yet sufficiently in-depth, and a large number of genes remain functionally unverified. Therefore, selecting camel groups with high milk yield and good milk quality for breeding will help produce high-quality camel milk and promote the healthy and sustainable development of the camel milk industry.
Screening lactation-related genes and candidate gene regions in Bactrian camels is of great significance for improving their lactation traits [13]. Currently, in camel molecular breeding, studies have shown that casein genes [9] and PICALM genes (phosphatidylinositol-binding clathrin assembly protein) [14] are associated with lactation traits in dromedary camels. Gubin et al. [15] found that two SNP sites in the intergenic regions of OSBPL8, MRPL37, SSBP3, and LOC102516351 genes in Gobi Red Camels from Inner Mongolia, China, are linked to the content of conventional nutrients (fat, protein, and lactose) in camel milk. In addition, Yao et al. [16] discovered that the secretion of GnRH (gonadotropin-releasing hormone), mTOR, PI3K-Akt, and MAPK signaling pathways is related to the regulation of lactation traits in Bactrian camels. Relatively few genes have been reported to regulate lactation traits in Bactrian camels. Milk production traits are typically regulated by multiple genes. Therefore, this study employed whole-genome resequencing to analyze the population structure and identify selection signals in two Bactrian camel populations: those from Northern Xinjiang and Eastern Xinjiang. The goal was to screen for lactation-related genes and regulatory signaling pathways, providing important molecular markers for the genetic selection and breeding of Bactrian camels in the Xinjiang region.
2. Materials and Methods
2.1. Selection of Experimental Population and Genomic DNA Extraction
The experimental population consisted of 106 Bactrian camels, including 51 from the eastern part of Xinjiang and 55 from the northern part of Xinjiang. The northern population was sourced from three different farms, while the eastern population came from a single farm. Both populations were raised under stall-feeding conditions. Information on the experimental population is provided in Table 1. Blood samples were collected from the carotid artery between March and October 2024. All blood samples were stored in anticoagulant tubes and immediately frozen at -80°C. Previous analyses of the nutritional components in camel milk from the eastern and northern regions revealed differences in nutrient content. Genomic DNA was extracted from the blood samples using a magnetic bead method. The concentration of DNA samples was measured using a Qubit fluorometer. The integrity and purity of DNA samples were assessed by 1% agarose gel electrophoresis. Only qualified samples were used for library preparation.
2.2. Whole-Genome Resequencing
The purified genomic DNA from Bactrian camel blood was sent to Huazhi Biotechnology Co., Ltd. for sequencing. Sequencing was performed on the MGI DNBSEQ-T7 platform by BGI Genomics. The library construction process included DNA fragmentation, end repair, adapter ligation, amplification, fragment selection, and circularization to generate high-quality DNA fragments ranging from 300 to 500 bp. The size of the library fragments was verified using a Qsep400 Bioanalyzer. After completing the library construction, sequencing was conducted on the platform.
2.3. Data Processing and Alignment
To ensure the removal of sequencing adapter sequences, low-quality bases, and undetermined bases, the raw sequencing data were filtered, checked for contamination, and assessed for quality. The quality control process included the following steps: (1) Filtering of low-quality reads, where read pairs were removed if more than 50% of the bases had a quality score of Q ≤ 20; (2) Removal of adapter sequences from the reads; (3) Filtering out reads with excessive N bases, specifically those containing more than 5 Ns; (4) Elimination of reads shorter than 100 bases.
For the reference genome, the study selected the genome sequence (GCA_048773025.1) from the NCBI database (https://www.ncbi.nlm.nih.gov/), with a reference genome size of 2.5 Gb. The clean reads obtained after quality control were aligned to the reference genome. Sentieon was used to detect variant sites in each sample, followed by joint calling to analyze the gVCF (genomic Variant Call Format) files of all samples collectively, yielding variant results for each individual in the population.
2.4. Variant Detection
The SNPs obtained from the joint analysis were subjected to initial hard filtering based on the following criteria: QD < 2.0, FS > 60.0, MQ < 40.0, SOR > 3.0, MQRankSum < -12.5, and ReadPosRankSum < -8.0. Subsequently, Vcftools software was used for further filtering of the SNPs with the following criteria: missing rate > 0.1 and MAF < 0.05.
2.5. Population Structure Analysis
Principal component analysis (PCA) was performed using PLINK based on the SNPs detected through whole-genome sequencing. Genetic distances were calculated, and a phylogenetic tree was constructed using the Neighbor-Joining (NJ) method.
2.6. Detection of Selection Signals
The composite likelihood ratio (CLR) was employed to detect selection signals within populations. The detection principle was based on the Site Frequency Spectrum (SFS). In addition to detecting selection signals within individual populations, signals were also explored across different populations to identify molecular regions associated with adaptation to diverse ecological environments and the formation of high-yield phenotypes. Three methods were used for this purpose: fixation index (Fst), nucleotide diversity (π), and cross-population extended haplotype homozygosity (XP-EHH). Furthermore, KEGG pathway analysis and Gene Ontology (GO) enrichment analysis were conducted.
3. Results
3.1. Data Quality Control
3.1.1. Raw Data Preprocessing
To ensure data quality, the raw data were filtered and assessed for quality before analysis. After filtering low-quality data using FASTP, the total amount of clean data generated from sequencing for all samples was 2770.3 Gb, with an average of 26.13 Gb per sample. The average Q30 value across all samples was 96.64%, and the average GC content was 40.77%. These results indicate that the sequencing data are authentic and reliable. The data quality for some samples are shown in Table 2.
3.1.2. Reference Genome Alignment
The alignment rate of sequencing data (Clean reads) to the reference genome (MappedReadsRate), sequencing depth (MapDepth), and coverage (Cov1X) are key indicators reflecting the quality of samples, library construction, sequencing, and the reference sequence. The results show that the average MappedReadsRate reached 99.01%, indicating a high alignment success rate, with a coverage depth of 96.14%, which meets the requirements for alignment analysis. The alignment results of some samples’ sequencing data to the reference genome are shown in Table 3.
3.1.3. Genetic Variant Analysis
A total of 6,451,453 SNPs were detected across the entire genome. The distribution of these SNPs is shown in Figure 1. The majority of mutations are located in intergenic regions (3,003,030, 46.55%), followed by intronic regions (2,422,399, 37.55%). A smaller proportion of SNPs are found in the 1 kb upstream region of genes (111,624, 1.73%) and the 1 kb downstream region of genes (124,037, 1.92%), as well as in exonic regions (63,231, 0.98%). A small number of SNPs are located at splice sites (the 2 bp near the exon/intron boundary within introns) (277, 0.004%), and in regions that are both the 1 kb upstream region of one gene and the 1 kb downstream region of another gene (9,838, 0.15%). The remaining SNPs are distributed in other regions (717,017, 11.11%).
Functional annotations were performed on the SNPs located in exonic regions. The results revealed that the majority of SNPs were synonymous mutations (32,739, 54.16%) and non-synonymous mutations (27,289, 45.14%). A smaller proportion of SNPs were mutations that introduced stop codons (366, 0.61%) or removed stop codons (60, 0.10%).
3.2. Population Genetic Structure Analysis
3.2.1. Principal Component Analysis
To investigate the genetic relationships and clustering patterns among the two major Bactrian camel populations in Eastern and Northern Xinjiang, as well as among different camel populations from various farms within the same region, we performed Principal Component Analysis (PCA) using PLINK. The first three principal components, which explained 10.12%, 7.11%, and 6.71% of the total variance, were selected for analysis. These three components effectively distinguished the Bactrian camel population from Group 3 in Northern Xinjiang and the Bactrian camel population from Group 4 in Eastern Xinjiang from the other populations (Groups 1 and 2). However, the three populations in Northern Xinjiang showed significant admixture and could not be clearly separated by clustering, indicating a close genetic relationship. The PCA results showed that the overall genetic structure could differentiate Bactrian camels from Northern and Eastern Xinjiang, although there was some admixture between the two regions. The PCA results are shown in Figure 2a.
3.2.2. Phylogenetic Tree Construction
Genetic distances were calculated using PLINK, and a phylogenetic tree was constructed using the Neighbor-Joining (NJ) method (Figure 2b). In conjunction with the PCA results, the phylogenetic tree shows that the Bactrian camels from Eastern Xinjiang and Northern Xinjiang are distinctly separated into different clusters, indicating a relatively distant genetic relationship but with some gene flow between them. In contrast, the Bactrian camels from the three camel farms in Northern Xinjiang are closely related.
3.3. Detection of Genomic Selection Signals
3.3.1. Single-Population Selection Signals
We treated the 55 camels from Northern Xinjiang and the 51 camels from Eastern Xinjiang as two distinct populations. The SWEED software was used to detect selection signals within each of these populations. This method relies on the composite likelihood ratio (CLR) test to identify selection signals within populations, based on the site frequency spectrum (SFS). The genome was divided into 10-kb windows. For single-population selection signal detection in the camels from Northern and Eastern Xinjiang, we selected the top 5% of regions. This process identified 5,017 genes in the Northern Xinjiang population and 4,405 genes in the Eastern Xinjiang population, accounting for approximately 12.93% and 11.35% of the genome, respectively. The Manhattan plots of selection signals within the Bactrian camel populations from Eastern and Northern Xinjiang are shown in Figure 3.
For the 4,405 candidate genes from the 51 Bactrian camels in Eastern Xinjiang and the 5,017 candidate genes from the 55 Bactrian camels in Northern Xinjiang, we performed GO and KEGG enrichment analyses using the DAVID software. Selected GO and KEGG terms are presented, with the visualization results shown in Figure 4. The GO analysis results indicate that the 51 Bactrian camels from Eastern Xinjiang are enriched in 20 significant GO terms (Figure 4a), covering three major categories: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). Among them, 447 genes are enriched in protein binding. After PDR correction of the P-values, the terms signal transduction, axon, and protein binding show highly significant enrichment. To understand the metabolic pathways in which the candidate genes of the Eastern Xinjiang Bactrian camels are involved, we performed KEGG pathway enrichment analysis on the 4,405 candidate genes. A total of 20 significantly enriched pathways were identified in the Eastern Xinjiang Bactrian camel population (Figure 4b). Among them, 67 genes were enriched in the Calcium signaling pathway. The three pathways of Glutamatergic synapse, Long-term depression, and Calcium signaling pathway reached highly significant P-values. The GO analysis results show that the 55 Bactrian camels from Northern Xinjiang are enriched in 20 significant GO terms (Figure 4c), covering three major categories: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). Among them, 634 genes are enriched in the plasma membrane. After PDR correction of the P-values, the terms axon guidance, cell adhesion, and intracellular signal transduction show highly significant enrichment. To explore the metabolic pathways in which the candidate genes of the 55 Bactrian camels from Northern Xinjiang are involved, we conducted KEGG pathway enrichment analysis on the 5,017 candidate genes. A total of 20 significantly enriched pathways were identified in the Northern Xinjiang Bactrian camel population (Figure 4d). Among them, 73 genes were enriched in the Calcium signaling pathway. The P-values for four pathways—Calcium signaling pathway, signaling pathway, Pancreatic secretion, and Axon guidance—reached highly significant levels.
3.3.2. Inter-Population Selection Signals
To identify the molecular regions underlying adaptation to different geographical environments and the formation of superior milk quality traits in Bactrian camels from different populations in Northern and Eastern Xinjiang, China, we employed three methods—fixation index (Fst), genomic heterozygosity (π), and cross-population extended haplotype homozygosity (XP-EHH)—to detect inter-population selection signals. Candidate genes were identified and subjected to pathway enrichment analysis.
3.3.2.1. Fst-Based Method
Using the fixation index (Fst) method, we divided the genome into 50-kb windows with a step size of 25 kb and selected the top 5% of differentiated regions as candidate regions. A total of 2,391 candidate genes were identified between the Bactrian camels from Eastern Xinjiang and those from Northern Xinjiang (Figure S1a). We performed GO and KEGG enrichment analyses on these differentiated genes. We selected the top 20 GO terms for display (Figure S2a). The GO enrichment results showed significant enrichment in three major categories: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). Three highly significant GO terms were identified: adherens junction, phosphatidylinositol phospholipase C activity, and cysteine-type endopeptidase inhibitor activity, mainly concentrated in CC and MF. A large number of candidate genes were enriched in protein binding. The KEGG enrichment analysis results (Figure S2b) showed that the majority candidate genes were enriched in Pathways in cancer. Three significant pathways (P < 0.05) were identified: Axon guidance, Pathways in cancer, and Ras signaling pathway.
3.3.2.2. π-Based Method
Using the genomic heterozygosity (π) method, we divided the genome into 50-kb windows with a step size of 25 kb and selected the top 5% of differentiated regions as candidate regions. Between the Bactrian camel populations from Eastern Xinjiang and Northern Xinjiang, a total of 2,334 differentiated candidate genes were identified (Figure S1b). We performed GO and KEGG enrichment analyses on these differentiated genes. The GO enrichment results (Figure S2c) showed that 135 candidate genes were enriched in the ATP binding term. The candidate genes were highly significantly enriched (P < 0.01) in three terms: one-carbon metabolic process, plasma membrane tubulation, and regulation of axonogenesis. The KEGG enrichment analysis results (Figure S2d) indicated that the majority of candidate genes were enriched in the Axon guidance pathway, which was the only highly significant pathway (P < 0.01).
3.3.2.3. XP-EHH-Based Method
Using the cross-population extended haplotype homozygosity (XP-EHH) method, the top 5% of differentiated regions were selected as candidate regions. In the 51 camel samples, a total of 3,982 genes were identified in the selected regions (Figure S1c). GO and KEGG enrichment analyses were performed on these differentiated genes.The top 20 GO terms were selected for display. The analysis results showed that the plasma membrane term had the highest number of enriched genes, reaching 522. A total of 17 GO terms were highly significantly enriched (P < 0.01), as shown in Figure S2e. The KEGG enrichment analysis results (Figure S2f) indicated that the Calcium signaling pathway had the highest number of candidate genes. Axon guidance was the only highly significant pathway (P < 0.01).
3.3.2.4. Joint Detection by Fst, π, and XP-EHH
Given Xinjiang’s vast land area and diverse geographical environments, the long-term differences in natural environments and selective breeding practices between Eastern and Northern Xinjiang have led to significant changes and differences in the physical characteristics and economic traits of Bactrian camels in these regions, which enhances the production efficiency of Bactrian camels. To identify candidate regions in the Bactrian camel population of Eastern Xinjiang compared to those of Northern Xinjiang, we used three different population selection signal methods—Fst, π, and XP-EHH (Figure 5a)—to detect inter-population selection signals. These three methods collectively identified 459 overlapping genes. By intersecting these genes with those related to Bactrian camel lactation, we annotated eight genes associated with lactation traits in Bactrian camels (see Table 4), namely DUOX1, DUOX2, CPQ, CGA, PLCG1, FYN, GNRHR, and TRHR.
To understand the signaling pathways in which the candidate genes are involved in camels, we performed GO and KEGG analyses on the 459 candidate genes. The results of the GO enrichment analysis (Figure 5b) showed that 11 pathways were included in BP (Biological Process), 4 pathways were included in CC (Cellular Component), and 5 pathways were included in MF (Molecular Function). Eight pathways were highly significant, with the thyroid hormone generation term showing a particularly significant difference. Additionally, a large number of genes were enriched in the plasma membrane term. The results of the KEGG enrichment analysis (Figure 5c) showed that the Neuroactive Ligand-Receptor Interaction pathway had the highest number of enriched genes (15). The pathways related to Axon guidance, Neuroactive Ligand-Receptor Interaction, and Thyroid Hormone Synthesis showed high significance levels. Among these, the Thyroid Hormone Synthesis pathway is associated with lactation traits.
4. Discussion
4.1. Differences in Nutritional Composition of Camel Milk from East and North Xinjiang Regions
Research suggests that the nutrient and functional substance content in camel milk is influenced by various factors, such as breed [17], lactation period [18], feeding methods, sampling season [19], and genetics. Additionally, the tools and methods used for milk testing and analysis [20] may also affect the measured nutrient content in the milk. In this study, samples were collected from camel farms in Hami City, East Xinjiang, as well as Yining County and Huocheng County in North Xinjiang. The camels in both regions were of the Xinjiang Junggar Bactrian breed. However, the two regions are approximately 1,500 kilometers apart, with significant differences in geography, climate, and feeding management practices. At the start of the study, the conventional nutritional components of camel milk from both regions were analyzed. The results revealed that the average values for conventional nutritional components in North Xinjiang camel milk were 4.44% milk fat, 4.04% milk protein, 5.83% lactose, and 15.00% total solids. In contrast, the average values for East Xinjiang camel milk were 5.55% milk fat, 4.11% milk protein, 5.76% lactose, and 16.86% total solids. Based on these results, it can be preliminarily concluded that camel milk from East Xinjiang has slightly higher nutritional content than that from North Xinjiang.
4.2. Molecular Marker Progress
This study identified eight genes: DUOX1, DUOX2, CPQ, CGA, PLCG1, FYN, GNRHR, and TRHR. Existing research suggests that DUOX1 and DUOX2 are primarily involved in the thyroid hormone synthesis signaling pathway. There is cross-regulation between thyroid hormones and prolactin (PRL), and hypothyroidism can reduce sensitivity to PRL. In dairy cows, thyroid hormone levels (T3/T4) are positively correlated with the content of conventional nutrients in milk, within the normal range [21]. This suggests that the DUOX1 and DUOX2 genes may play a role in the lactation process of Bactrian camels.
Moreover, the expression level of the DUOX1 gene is significant in normal human keratinocytes [22]. In Iranian Markhoz goats, both the DUOX1 and DUOX2 genes are located within the quantitative trait locus (QTL) region associated with shearing weight at one year of age [23]. Through transcriptome sequencing and protein interaction network analysis of Bactrian camel populations with and without mastitis, the PLCG1 gene was identified as a major immune core gene in Bactrian camel mastitis [24]. Additionally, the PLC gene family has been shown to play a role in targeted therapies for breast cancer [25]. In this study, the PLCG1 gene was enriched in multiple pathways, with the axon guidance signaling pathway reaching a highly significant level (P < 0.01). In the study of lactation mechanisms in Holstein dairy cows, it was found that the axon guidance signaling pathway influences lactation performance [26], further indirectly validating that the PLCG1 gene could serve as a candidate gene for lactation traits in Bactrian camels. Hussain Bahbahani et al. [27] conducted whole-genome sequencing of dromedary camels and found that the FYN gene plays a role in fat metabolism and energy expenditure, consistent with the results of this study. The GNRHR gene, which encodes the gonadotropin-releasing hormone receptor, plays a significant role in mammalian reproduction [28] and regulates hormone levels, such as estrogen and progesterone. These hormones significantly impact the development of mammary glands and lactation regulation in mammals. Therefore, the GNRHR gene is likely associated with lactation traits in Bactrian camels, suggesting that the findings of this study are highly reliable. The roles of the CGA and TRHR genes in regulating camel phenotypic traits and their mechanisms of action have not yet been reported.
4.3. Pathway Selection and Analysis in This Study
In this study, the enriched pathways of the differentially selected candidate genes between the East Xinjiang Bactrian camel and the North Xinjiang Bactrian camel primarily focus on axon guidance, neuroactive ligand-receptor interactions, and thyroid hormone synthesis. In mammals, signaling pathways associated with lactation traits include RAP1, Wnt, MTOR, PI3K-AKT, and MAPK pathways, which are mainly involved in mammogenesis, mammary gland development, and lactation [29,30,31]. However, compared to dairy cows and other mammals, Bactrian camels exhibit significantly different mammary gland structures and lactation patterns. There are also notable differences in the signaling pathways involved. The findings of this study provide support for this perspective. Additionally, based on previously measured differences in the nutrient content of milk from East and North Xinjiang Bactrian camels, the camels were categorized into two groups. The relatively small sample size of the Bactrian camels selected is a limiting factor. Nonetheless, this study offers valuable biological insights into the unique lactation mechanisms of Bactrian camels and lays the groundwork for further research on the nutritional characteristics of Bactrian camel milk.
5. Conclusions
This study utilized whole-genome resequencing technology to analyze the lactation-related genetic differences between Bactrian camel populations in East Xinjiang (from a single camel farm) and North Xinjiang (from three camel farms across two counties and cities), identifying a total of 6,451,453 SNPs. Principal component analysis revealed a clear distinction between the two populations. GO and KEGG enrichment analyses indicated that, compared to North Xinjiang, the candidate genes in East Xinjiang Bactrian camels were primarily enriched in the plasma membrane and thyroid hormone synthesis pathways. Whole-genome selection signals identified eight candidate genes potentially associated with lactation traits: DUOX1, DUOX2, CPQ, CGA, PLCG1, FYN, GNRHR, and TRHR. These genes may serve as key markers for future molecular breeding.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Li Yongqing; methodology, Li Yongqing; software, Li Yongqing; validation, Liu Shihao, Lin Changchun; formal analysis, Li Yongqing; investigation, Cai Shudong; resources, Cai Shudong; data curation, Hanikezi Tulafu; writing—original draft preparation, Li Yongqing; writing—review and editing, Liu Shihao, Lin Changchun; visualization, Li Yongqing; supervision, Wu WeiWei; project administration, Wu WeiWei; funding acquisition, Wu WeiWei; project administration, Huang Juncheng. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Xinjiang Uygur Autonomous Region Academy of Animal Science [grant number 2022TSYCCX0045] and the Basic Research Business Fund for Public Welfare Scientific Research Institutes in Xinjiang Uygur Autonomous Region [grant number ky202473] and The APC was funded by 2022TSYCCX0045.
Institutional Review Board Statement
The animal study protocol was approved by Science and Technology Ethics Committee of Xinjiang Academy of Animal Science (LLSC0026, March 10(th), 2024).
Informed Consent Statement
Animal owners were informed and consented to the study.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| SNPs | Single Nucleotide Polymorphism |
| PCA | Principal component analysis |
| NJ | Neighbor-Joining |
| CLR | Composite Likelihood Ratio |
| SFS | Site Frequency Spectrum |
| Fst | Fixation index |
| XP-EHH | Cross-population extended haplotype homozygosity |
| BP | Biological Process |
| CC | Cellular Component |
| MF | Molecular Function |
References
- Lapidge, S.J.; Eason, C.T.; Humphrys, S.T. A review of chemical, biological and fertility control options for the camel in Australia. Rangeland Journal 2010, 32, 95–115. [Google Scholar] [CrossRef]
- D, K.N. History of camel in Indian context. Asian Agri History 1997, 1, 15–19. [Google Scholar]
- Zarrin, M.; Riveros, J.L.; Ahmadpour, A.; de Almeida, A.M.; Konuspayeva, G.; Vargas-Bello-Pérez, E.; Faye, B.; Hernández-Castellano, L.E. Camelids: new players in the international animal production context. Trop Anim Health Prod 2020, 52, 903–913. [Google Scholar] [CrossRef]
- Alhassani, W.E. Camel milk: Nutritional composition, therapeutic properties, and benefits for human health. Open Veterinary Journal 2024, 14, 3164–3180. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.Y., C.L., Liu J. K., et al. Research progress on the nutritional value of camel milk and its application and mechanism in disease prevention. Food Science 2022, 43, 392–401. [Google Scholar]
- Rasheed, Z. Medicinal values of bioactive constituents of camel milk: A concise report. Int J Health Sci (Qassim) 2017, 11, 1–2. [Google Scholar]
- Khatoon, H.a.N., R. Bioactive components in camel milk: their nutritional value and therapeutic application; Academic Press: MA, 2017. [Google Scholar]
- Muthukumaran, M.S.; Mudgil, P.; Baba, W.N.; Ayoub, M.A.; Maqsood, S. A comprehensive review on health benefits, nutritional composition and processed products of camel milk. Food Reviews International 2022, 39, 1–37. [Google Scholar] [CrossRef]
- Seifu, E. Recent advances on camel milk: Nutritional and health benefits and processing implications—A review. AIMS Agriculture and Food 2022, 7, 777–804. [Google Scholar] [CrossRef]
- Behrouz, S.; Saadat, S.; Memarzia, A.; Sarir, H.; Folkerts, G.; Boskabady, M.H. The Antioxidant, Anti-Inflammatory and Immunomodulatory Effects of Camel Milk. Front Immunol 2022, 13, 855342. [Google Scholar] [CrossRef]
- El-Hatmi, H.; Girardet, J.-M.; Gaillard, J.-L.; Yahyaoui, M.H.; Attia, H. Characterisation of whey proteins of camel (Camelus dromedarius) milk and colostrum. Small Ruminant Res 2007, 70, 267–271. [Google Scholar] [CrossRef]
- FAOSTAT. Food and Agricultural Organisation, United Nations statistics. http://faostat.fao.org/. 2022.
- Ayalew, W.; Wu, X.; Tarekegn, G.M.; Sisay Tessema, T.; Naboulsi, R.; Van Damme, R.; Bongcam-Rudloff, E.; Edea, Z.; Chu, M.; Enquahone, S.; et al. Whole Genome Scan Uncovers Candidate Genes Related to Milk Production Traits in Barka Cattle. International Journal of Molecular Sciences 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Bahbahani, H.; Musa, H.H.; Wragg, D.; Shuiep, E.S.; Almathen, F.; Hanotte, O. Genome Diversity and Signatures of Selection for Production and Performance Traits in Dromedary Camels. Front Genet 2019, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Gubin, H.L., M.; Wusutiannan; Qimude; Lihushan; Liyi; Song, H.; Liang, M.; Hasi; Jirenduribu. Genome wide association studies for milk nutrition traits in Gobi red Bactrian camel. Journal of Camel Practice and Research 2023, 30, 273–282. [Google Scholar] [CrossRef]
- Yao, H.; Pan, Z.; Ma, W.; Zhao, Z.; Su, Z.; Yang, J. Whole-Genome Resequencing Analysis of the Camelus bactrianus (Bactrian Camel) Genome Identifies Mutations and Genes Affecting Milk Production Traits. International Journal of Molecular Sciences 2024, 25. [Google Scholar] [CrossRef]
- Alhaj, O.A.; Lajnaf, R.; Jrad, Z.; Alshuniaber, M.A.; Jahrami, H.A.; Serag El-Din, M.F. Comparison of Ethanol Stability and Chemical Composition of Camel Milk from Five Samples. Animals 2022, 12, 615. [Google Scholar] [CrossRef]
- Oselu, S.; Ebere, R.; Arimi, J.M.; Amante, E. Camels, Camel Milk, and Camel Milk Product Situation in Kenya in Relation to the World. International Journal of Food Science 2022, 2022, 1–15. [Google Scholar] [CrossRef]
- Nagy, P.; Juhasz, J.; Reiczigel, J.; Csaszar, G.; Kocsis, R.; Varga, L. Circannual changes in major chemical composition of bulk dromedary camel milk as determined by FT-MIR spectroscopy, and factors of variation. Food Chemistry 2019, 278, 248–253. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Y.; Gao, J.; Liu, L.; Cao, L.; Hu, B.; Abula, Z.; Xieermaola, Y.; Wang, H.; Chu, C.; et al. Rapid detection and spectroscopic feature analysis of mineral content in camel milk using fourier-transform mid-infrared spectroscopy and traditional machine learning algorithms. Food Control 2025, 169. [Google Scholar] [CrossRef]
- Miao, S.J.; Han, M.T.; Li, Y.J.; Luo, Y.Z.; Jiang, Z.D. Study on serum T4 and T3 concentration changes in mid-late lactation period of dairy cows with different milk production levels. Heilongjiang Animal Science and Veterinary Medicine.
- Choi, H.; Park, J.Y.; Kim, H.J.; Noh, M.; Ueyama, T.; Bae, Y.; Lee, T.R.; Shin, D.W. Hydrogen peroxide generated by DUOX1 regulates the expression levels of specific differentiation markers in normal human keratinocytes. J Dermatol Sci 2014, 74, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Ghadikolaei, A.; Mehrabani-Yeganeh, H.; Miarei-Aashtiani, S.R.; Staiger, E.A.; Rashidi, A.; Huson, H.J. Genome-Wide Association Studies Identify Candidate Genes for Coat Color and Mohair Traits in the Iranian Markhoz Goat. Frontiers in Genetics 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Yao, H.; Zhang, L.; Zhang, Y.; Wang, Y.; Wang, W.; Liu, Y.; Zhao, X.; Tong, P.; Su, Z. Transcriptomics-Based Study of Immune Genes Associated with Subclinical Mastitis in Bactrian Camels. Veterinary Sciences 2025, 12. [Google Scholar] [CrossRef]
- Ommolbanin Asadpour, P.D., Fatemeh Rahbarizadeh, Ph.D. Phospholipase-Cγ1 Signaling Protein Down-Regulation by Oligoclonal-VHHs based Immuno-Liposome: A Potent Metastasis Deterrent in HER2 Positive Breast Cancer Cells. Cell Journal 2020, 22, 30–39. [Google Scholar] [CrossRef]
- Fang Chenhui, Z.Z., Zhu Mengting, Zheng Wei, Xie Mengting, Qi Xingdong, Zhang Yong. Resequencing screening and validation of differential genes in Holstein cows with different lactation performances. hinese Journal of Animal Science 2024, 60, 176–182. [Google Scholar]
- Bahbahani, H.; Mohammad, Z.; Al-Ateeqi, A.; Almathen, F. A comprehensive map of copy number variations in dromedary camels based on whole genome sequence data. Scientific Reports 2024, 14. [Google Scholar] [CrossRef]
- Desaulniers, A.T.; Cederberg, R.A.; Lents, C.A.; White, B.R. Expression and Role of Gonadotropin-Releasing Hormone 2 and Its Receptor in Mammals. Frontiers in Endocrinology 2017, 8. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.-E.; Lee, J.-S.; Park, J.-S.; Moon, J.-O.; Lee, H.-G. Phenylalanine and valine differentially stimulate milk protein synthetic and energy-mediated pathway in immortalized bovine mammary epithelial cells. Journal of Animal Science and Technology 2020, 62, 263–275. [Google Scholar] [CrossRef]
- Du, A.; Zhao, F.; Liu, Y.; Xu, L.; Chen, K.; Sun, D.; Han, B. Genetic polymorphisms of PKLR gene and their associations with milk production traits in Chinese Holstein cows. Front Genet 2022, 13, 1002706. [Google Scholar] [CrossRef]
- Mumtaz, P.T.; Bhat, B.; Ibeagha-Awemu, E.M.; Taban, Q.; Wang, M.; Dar, M.A.; Bhat, S.A.; Shabir, N.; Shah, R.A.; Ganie, N.A.; et al. Mammary epithelial cell transcriptome reveals potential roles of lncRNAs in regulating milk synthesis pathways in Jersey and Kashmiri cattle. BMC Genomics 2022, 23, 176. [Google Scholar] [CrossRef]
Figure 1.
SNP Detection and Annotation Results. (a) Distribution of SNPs Across Different Genomic Region; (b) Functional Annotation of Exonic SNPs; (c) SNP Density on Each Chromosome.
Figure 1.
SNP Detection and Annotation Results. (a) Distribution of SNPs Across Different Genomic Region; (b) Functional Annotation of Exonic SNPs; (c) SNP Density on Each Chromosome.
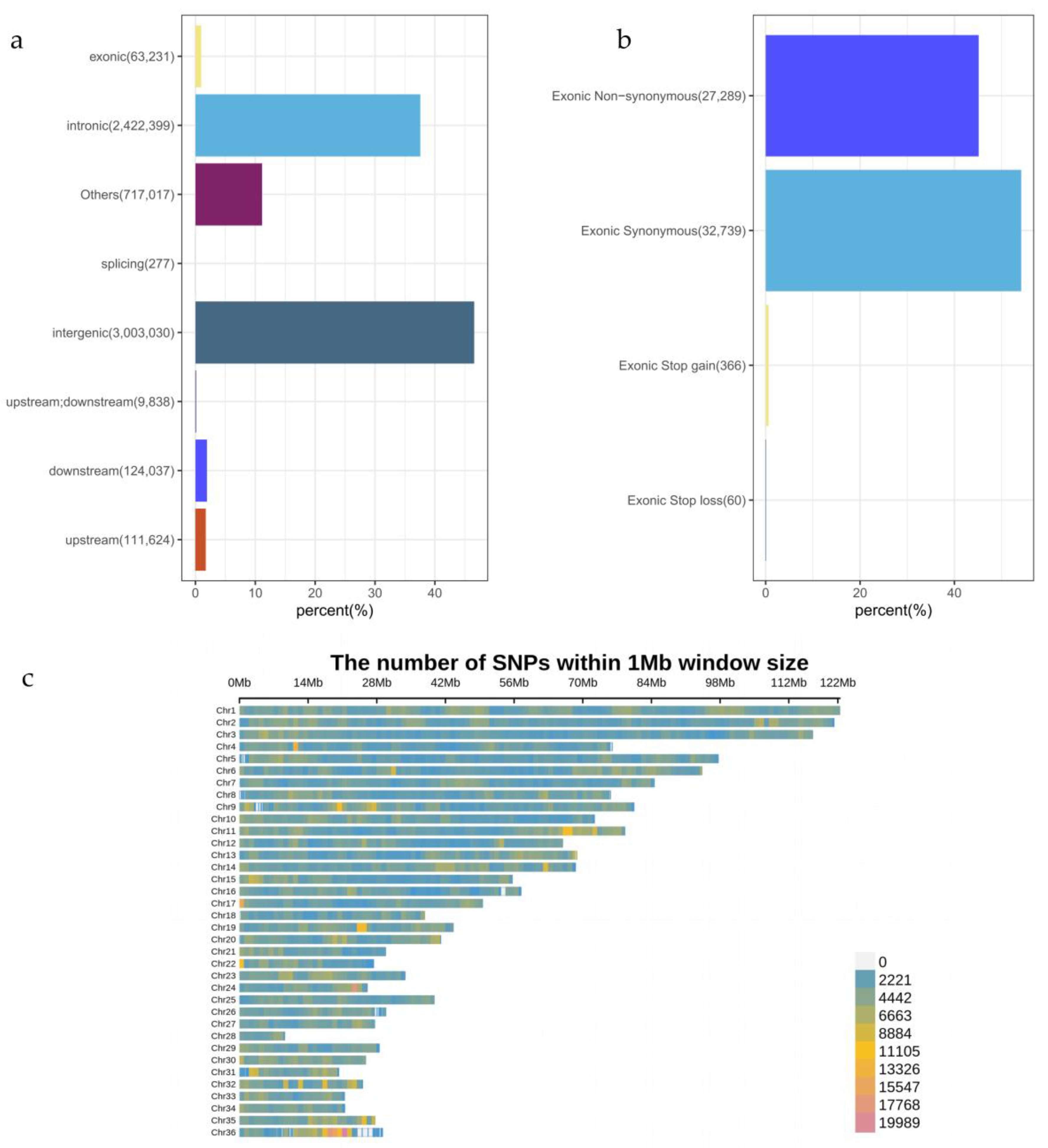
Figure 2.
Population Structure Analysis. (a) PCA Plot; (b) Phylogenetic Tree.
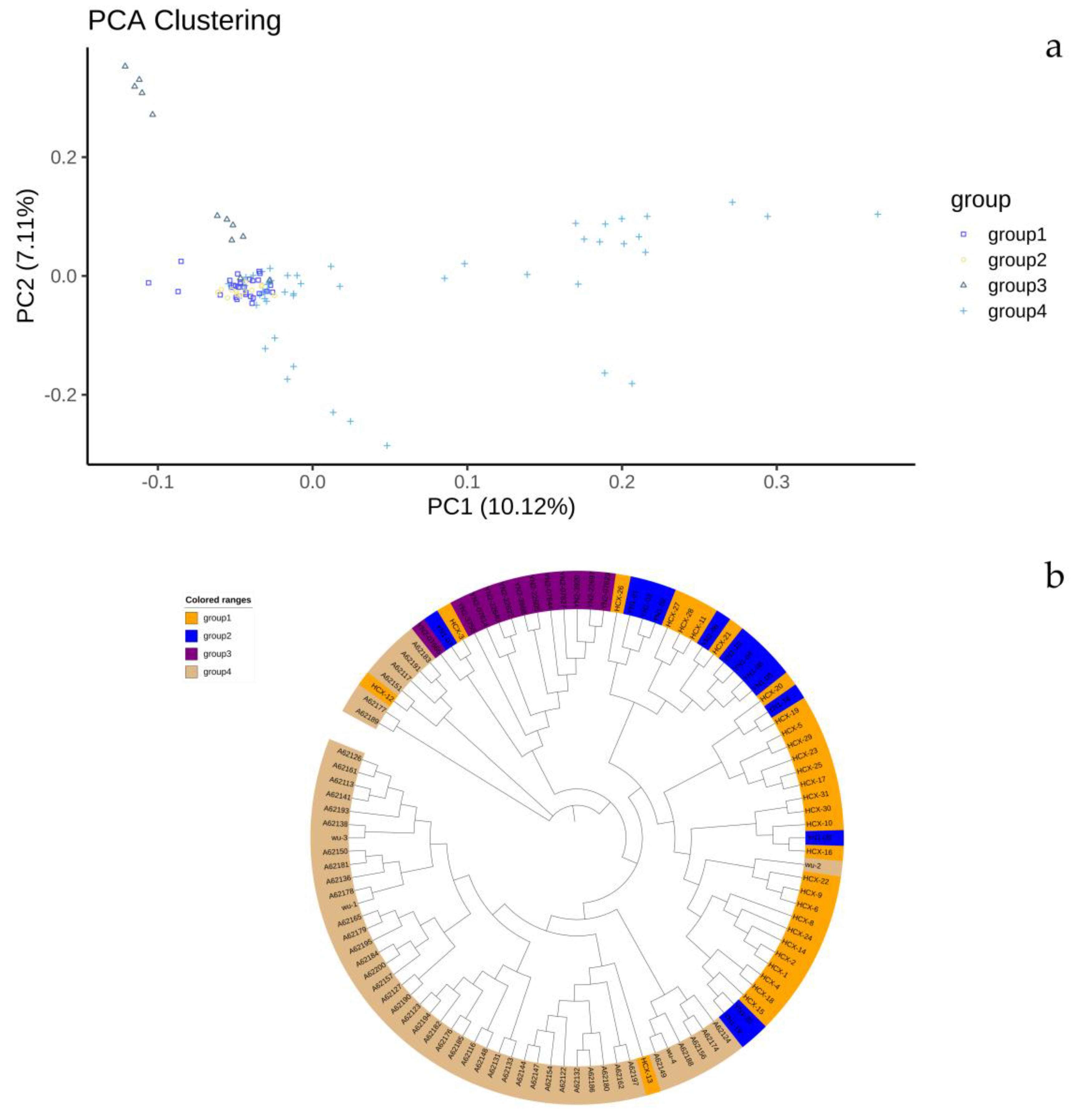
Figure 3.
Manhattan Plot of iHS Values for Bactrian Camels from Eastern Xinjiang vs. Northern Xinjiang. (a) Eastern Xinjiang Bactrian camel population; (b) Northern Xinjiang Bactrian camel population.
Figure 3.
Manhattan Plot of iHS Values for Bactrian Camels from Eastern Xinjiang vs. Northern Xinjiang. (a) Eastern Xinjiang Bactrian camel population; (b) Northern Xinjiang Bactrian camel population.
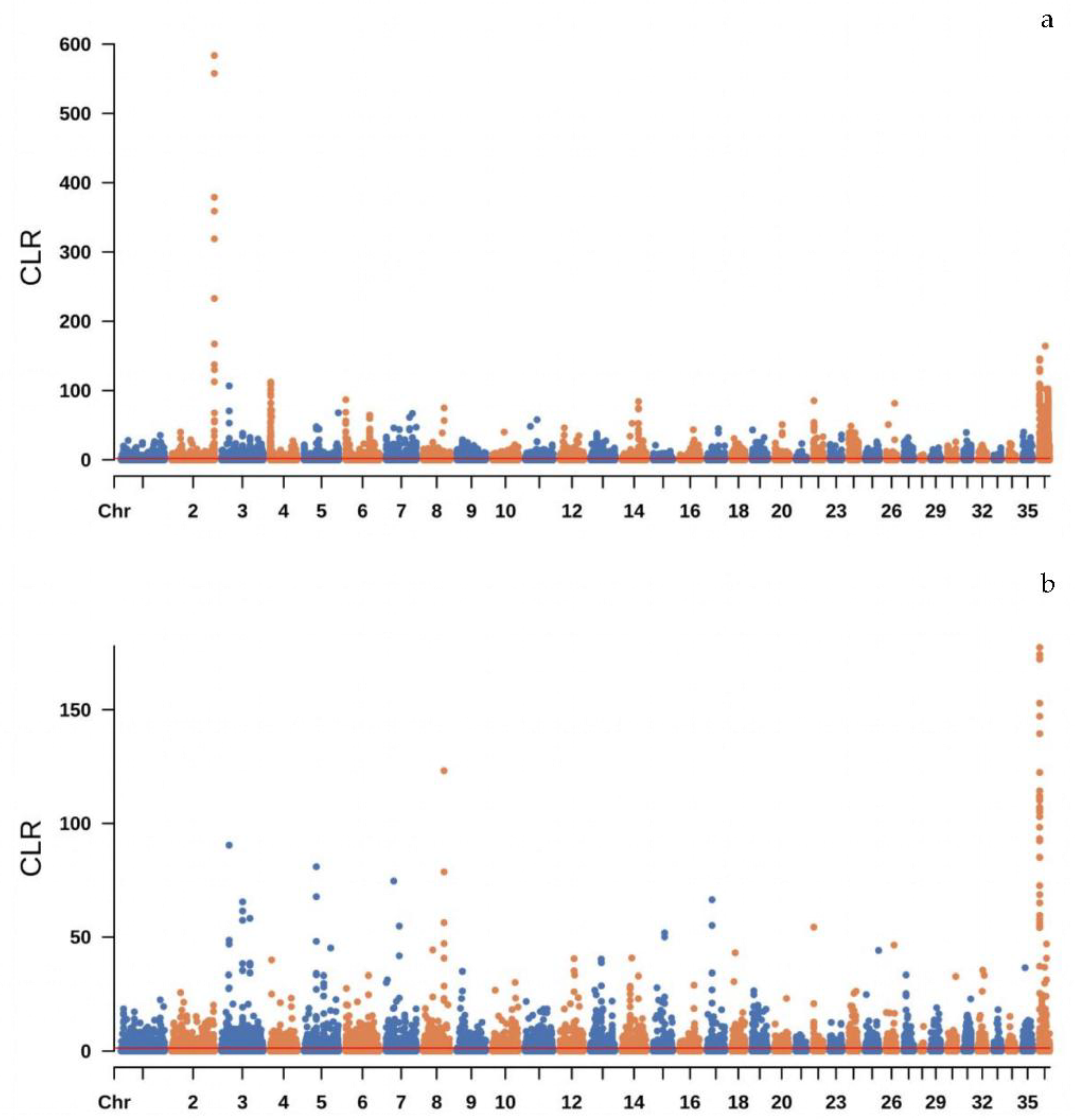
Figure 4.
GO and KEGG Enrichment Analysis Results of SWEED Candidate Genes. (a) GO Enrichment Analysis Results of SWEED Candidate Genes in the 51 Bactrian Camels from Eastern Xinjiang; (b) KEGG Enrichment Analysis Results of SWEED Candidate Genes in the 51 Bactrian Camels from Eastern Xinjiang; (c) GO Enrichment Analysis Results for SWEED Candidate Genes in 55 Bactrian Camels from Northern Xinjiang; (d) KEGG Enrichment Analysis Results for SWEED Candidate Genes in 55 Bactrian Camels from Northern Xinjiang.
Figure 4.
GO and KEGG Enrichment Analysis Results of SWEED Candidate Genes. (a) GO Enrichment Analysis Results of SWEED Candidate Genes in the 51 Bactrian Camels from Eastern Xinjiang; (b) KEGG Enrichment Analysis Results of SWEED Candidate Genes in the 51 Bactrian Camels from Eastern Xinjiang; (c) GO Enrichment Analysis Results for SWEED Candidate Genes in 55 Bactrian Camels from Northern Xinjiang; (d) KEGG Enrichment Analysis Results for SWEED Candidate Genes in 55 Bactrian Camels from Northern Xinjiang.
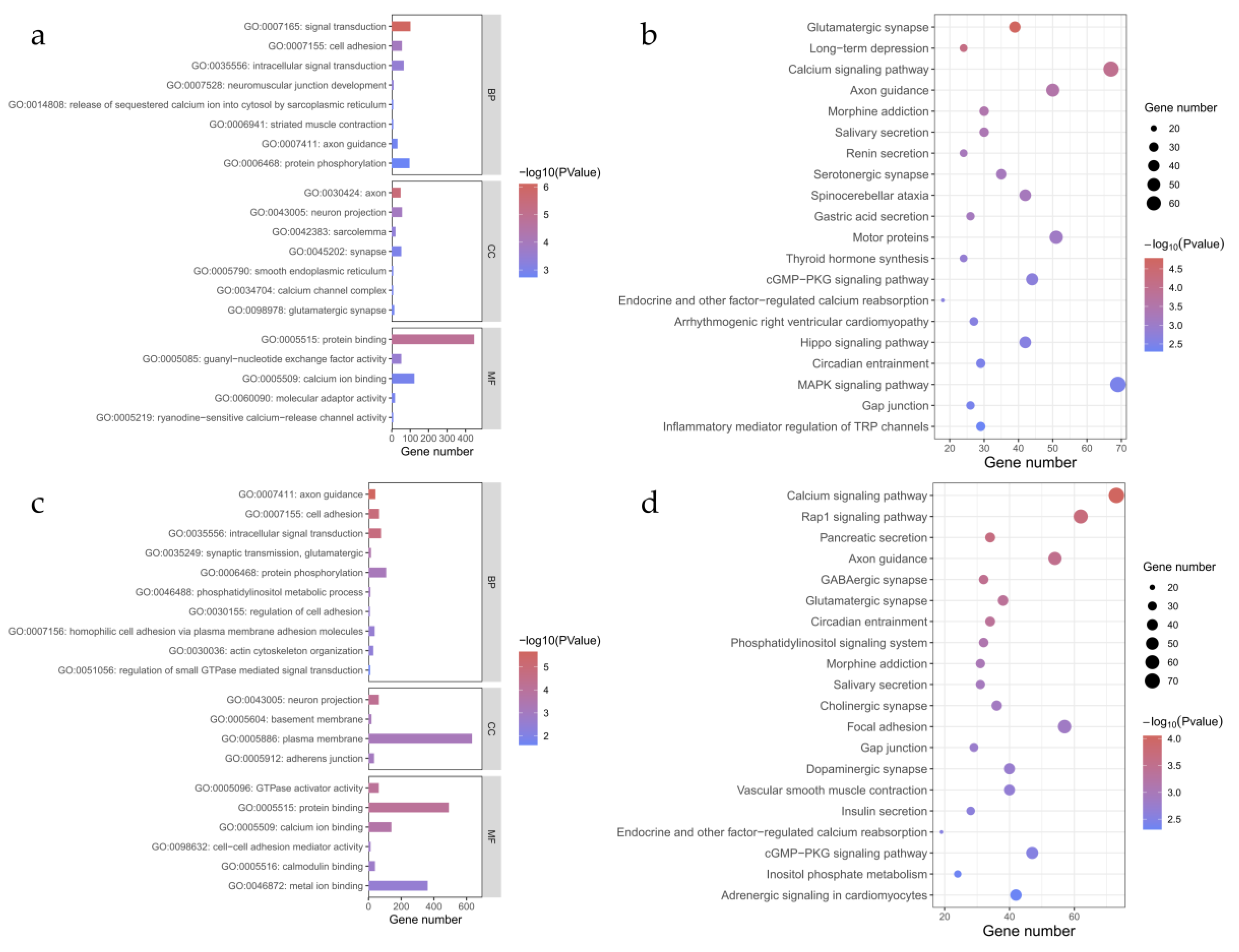
Figure 5.
Joint Detection Results Based on Fst, π, and XP-EHH. (a) Overlapping Genes from Three Population Selection Signal Methods; (b) GO Enrichment Analysis of Common Candidate Genes; (c) KEGG enrichment analysis results of common candidate genes.
Figure 5.
Joint Detection Results Based on Fst, π, and XP-EHH. (a) Overlapping Genes from Three Population Selection Signal Methods; (b) GO Enrichment Analysis of Common Candidate Genes; (c) KEGG enrichment analysis results of common candidate genes.
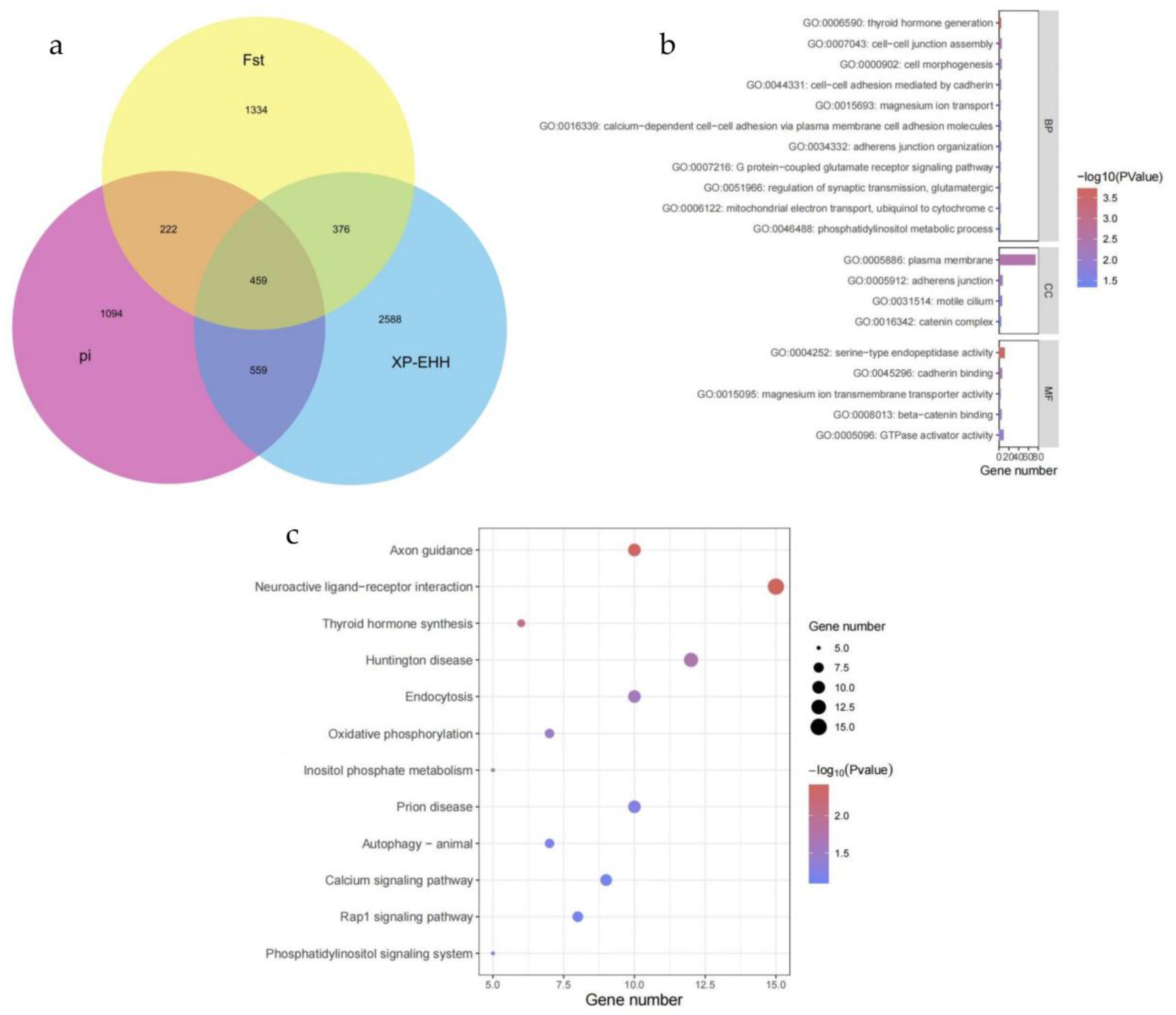

Table 1.
Sample Source Information Table.
| Sampling region | Research farms | Breed | N | Age (years) | Days in milk | Sampling month | Sample type |
|---|---|---|---|---|---|---|---|
| North Xinjiang region | Group1 | Junggar Bactrian camel | 13 | 5–10 | 28-59 | 4 | Individual camels |
| Group2 | 12 | 5–10 | 31-62 | 4 | |||
| Group3 | 30 | 4–10 | 88-121 | 6 | |||
| East Xinjiang region | Group4 | 51 | 5–10 | 183–215 | 10 |
Table 2.
Summary of Data Quality Output.
| Samples | Rawreads | Rawbases | Cleanreads | Cleanbases | Cleanrate/% | CleanQ20/% | CleanQ30/% | Depth | GC_rate/% |
|---|---|---|---|---|---|---|---|---|---|
| HCX-1 | 167,104,712 | 25,065,706,800 | 161,262,758 | 23,818,023,015 | 95.02 | 98.90 | 96.61 | 23.82 | 40.65 |
| HCX-2 | 165,684,538 | 24,852,680,700 | 160,309,470 | 23,691,436,199 | 95.33 | 98.98 | 96.80 | 23.69 | 40.62 |
| HCX-3 | 187,549,766 | 28,132,464,900 | 181,014,810 | 26,696,183,861 | 94.89 | 99.04 | 97.00 | 26.7 | 40.63 |
| HCX-4 | 190,501,754 | 28,575,263,100 | 183,527,246 | 27,056,383,370 | 94.68 | 99.06 | 97.04 | 27.06 | 40.56 |
| HCX-5 | 180,226,658 | 27,033,998,700 | 175,431,442 | 26,000,863,876 | 96.18 | 98.94 | 96.65 | 26 | 40.52 |
| HCX-6 | 167,333,676 | 25,100,051,400 | 162,668,074 | 24,102,338,444 | 96.03 | 98.90 | 96.49 | 24.1 | 40.61 |
| HCX-8 | 174,520,428 | 26,178,064,200 | 169,001,152 | 24,996,978,982 | 95.49 | 98.98 | 96.81 | 25 | 40.45 |
| HCX-9 | 179,506,744 | 26,926,011,600 | 173,424,156 | 25,592,661,111 | 95.05 | 98.88 | 96.53 | 25.59 | 40.38 |
| HCX-10 | 208,411,996 | 31,261,799,400 | 203,436,646 | 30,224,428,668 | 96.68 | 98.75 | 96.17 | 30.22 | 40.34 |
| HCX-11 | 196,912,568 | 29,536,885,200 | 189,281,688 | 27,810,085,619 | 94.15 | 99.03 | 96.92 | 27.81 | 40.68 |
| Mean | 180,193,129 | 27,028,969,358 | 175,866,655 | 26,134,933,609 | 96.70 | 98.95 | 96.64 | 26.13 | 40.77 |
Note: Samples: Sample names; Raw reads: Total number of raw reads; Raw bases: Total number of raw bases; Clean reads: Number of reads remaining after filtering; Clean bases: Number of bases remaining after filtering; Clean rate: Ratio of clean reads to raw reads; Clean Q20: Percentage of bases with a quality score ≥ 20; Clean Q30: Percentage of bases with a quality score ≥ 30; Depth: Sequencing depth; GC: GC content. Table 1 shows sequencing quality metrics for the first 10 samples only, where the “Mean” row represents the average sequencing quality across all 106 samples.
Table 3.
Statistics of Sequence Alignment.
| Sample | TotalReads | TotalBases | MappedReads | MappedBases | MappedReadsRate/% | Cov1X/% |
|---|---|---|---|---|---|---|
| HCX-10 | 203436646 | 30224428668 | 201447488 | 29926302598 | 99.02 | 96.23 |
| HCX-11 | 189281688 | 27810085619 | 188004845 | 27618957254 | 99.33 | 96.08 |
| HCX-12 | 176324418 | 25889820476 | 174889251 | 25674893336 | 99.19 | 96.23 |
| HCX-13 | 175388702 | 25782573959 | 174091269 | 25588267541 | 99.26 | 96.09 |
| HCX-14 | 189018312 | 27748357075 | 187378160 | 27502789784 | 99.13 | 96.37 |
| HCX-15 | 187586578 | 27589227760 | 186099158 | 27366460408 | 99.21 | 96.23 |
| HCX-16 | 170180476 | 25070724998 | 168218103 | 24776840869 | 98.85 | 96.03 |
| HCX-17 | 177420988 | 26083825150 | 175919484 | 25858979012 | 99.15 | 96.12 |
| HCX-18 | 193465218 | 28604335830 | 191574249 | 28321015891 | 99.02 | 96.43 |
| HCX-19 | 166366004 | 24536648981 | 164847919 | 24309236926 | 99.09 | 96.18 |
| Mean | 175866654 | 26134933609 | 174129340 | 25874605895 | 99.01 | 96.14 |
Note: Total Reads: Number of clean reads; Total Bases: Number of clean bases; Mapped Reads: Reads aligned to the reference genome; Mapped Bases: Bases aligned to the reference genome; Mapped Reads Ratio: Alignment rate of reads, calculated as Mapped Reads / Total Clean Reads; Map Depth: Sequencing depth after alignment. Table 1 presents the alignment results for the first 10 samples only. The “Mean” row represents the average alignment results for the sequencing data of all 106 samples.
Table 4.
Candidate Genes List.
| Serial Number | Chromosome Number | Gene Start Position | Gene End Position | Gene Name |
|---|---|---|---|---|
| 1 | 6 | 19589586 | 19625820 | DUOX1 |
| 2 | 6 | 19638040 | 19659885 | DUOX2 |
| 3 | 25 | 32700070 | 33089923 | CPQ |
| 4 | 8 | 23344490 | 23356944 | CGA |
| 5 | 19 | 42320197 | 42358748 | PLCG1 |
| 6 | 8 | 31721195 | 31886562 | FYN |
| 7 | 2 | 54411098 | 54424743 | GNRHR |
| 8 | 25 | 23865074 | 24343870 | TRHR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



