Submitted:
26 August 2025
Posted:
26 August 2025
You are already at the latest version
Abstract
The nucleation of coherent L12 precipitates in aluminum alloys is investigated by first-principles supercell modeling, with density functional theory (DFT) calculations in the generalized gradient approximation, employing a plane wave basis set and pseudopotentials. Distance-resolved solute–solute interaction maps were obtained for Zr in Al, revealing a strong energetic preference for the 2nd coordination shell, consistent with the atomic arrangement in the metastable L12-Al3Zr phase. Stepwise cluster assembly shows a linear stabilization energy gain, while configurations corresponding to the equilibrium D023 structure are unfavorable at early nucleation stages. Similar 2nd-shell attraction was confirmed for Sc, Ti, and Hf, in contrast to V, Cr, and Cu, which lack strong ordering tendencies. These results provide atomistic insight into precipitate formation pathways and establish predictive criteria for identifying alloying elements with a high propensity to form thermally stable, coherent L12 phases in aluminum alloys.
Keywords:
aluminum alloys
; L12 precipitates
; density functional theory (DFT)
; solute–solute interactions
; nucleation pathways
; thermal stability
; cluster formation
1. Introduction
Aluminum is an abundant element in the earth’s crust [1,2,3]. As a structural material, aluminum is used in extensive industrial applications [4,5,6]. However, pure aluminum (Al) exhibits a yield strength of approximately 32 MPa at room temperature [7]. In contrast, the high-strength aluminum alloys can achieve yield strengths exceeding 460 MPa [8].
[4,9,10]. Aluminum alloys are typically composed of alloying elements such as magnesium (Mg) [11], silicon (Si) [12], zirconium (Zr) [13,14], copper (Cu) [15], zinc (Zn) [16], manganese (Mn) [17], titanium (Ti) [18], chromium (Cr) [19], vanadium (V) [20], and iron (Fe) [21], scandium (Sc) [22] and hafnium (Hf) [23], etc. Through various processing technologies, these alloying elements significantly improve the mechanical properties of aluminum materials.
In industrial applications, the primary methods for enhancing the strength of aluminum alloys include solid solution strengthening [24], work hardening [25], precipitation strengthening [26], dispersion strengthening [27], grain-boundary strengthening [28] and so on. These mechanisms often act synergistically, each impeding dislocation motion through distinct pathways, thereby improving the alloy’s overall strength. Precipitation strengthening is a particularly effective approach for increasing the yield strength of aluminum alloys [29]. This process involves the formation of finely distributed, coherent precipitates within the aluminum crystal lattice [26]. These precipitates act as obstacles to dislocation motion, substantially improving the mechanical properties of the alloy. For instance, the formation of Al3Zr precipitates within an aluminum matrix contributes significantly to dispersion strengthening via the Orowan mechanism, effectively hindering dislocation motion [30].
The alloys containing rare-earth elements or transition metals commonly form two distinct types of precipitate structures: the L12 (cubic) and D023 (tetragonal) phases. Elements such as Zr, Sc, Ti, Hf, and certain f-electron metals are known to promote the formation of these precipitates as shown in Table 1.
In an aluminum matrix, zirconium (Zr) exhibits preferential enrichment within dendrite cells or grain interiors [32,37]. The partition coefficient of Zr in Al, approximately 2.5, results in significantly elevated Zr concentrations in the dendrite core compared to the grain boundary regions [37]. This heterogeneous distribution influences the nucleation driving force, leading to distinct nucleation mechanisms in different microstructural regions [37]. In the dendrite core, high Zr supersaturation promotes homogeneous nucleation, yielding a high density of fine, coherent L12-Al3Zr precipitates with sizes below 10 nm [32]. Conversely, in the interdendritic or grain boundary regions, lower Zr supersaturation favors heterogeneous nucleation at defects such as dislocations or grain boundaries, producing larger precipitates (approximately 25 nm) with a reduced number density [32].
The addition of 0.2 wt.% Zr to an aluminum matrix significantly enhances the mechanical properties of aluminum alloys. In 6063 aluminum alloy with 0.2 wt.% Zr addition, the Vickers hardness and tensile strength increase by 12.96% and 7.86%, respectively, after 6 hours of aging treatment [38]. Similarly, for AlSi5Cu2Mg alloy containing 0.2 wt.% Zr, both yield strength and tensile strength demonstrate notable improvement after the heat treatment [39]. The formation of fine, coherent L12-Al3Zr precipitates results in a ∼60% increase in microhardness and nearly doubles the yield strength at room temperature [40]. These fine coherent Al3Zr dispersoids also impede grain boundary migration, enhancing resistance to recrystallization through the Zener pinning effect. Notably, the maximum pinning force exerted by coherent dispersoids is twice that of incoherent dispersoids of equivalent size [41,42]. The effectiveness of resistance to this recrystallization and microstructure coarsening depends on the size, volume fraction, and coherency of the precipitates within the matrix. Overall, the formation of alloy precipitates significantly enhances the mechanical performance of aluminum alloys [41].
Beyond mechanical properties, precipitates in aluminum alloys can improve other material characteristics, such as thermal stability and high-temperature performance [43]. Al3Zr precipitates, for example, exhibit exceptional thermal stability due to the low diffusivity of Zr in the aluminum matrix [44]. At 300∘C, Al-Zr alloys demonstrate superior creep resistance compared to conventional aluminum alloys, attributable to the presence of thermally stable precipitates [43]. In comparison of L12 precipitates, the stable equilibrium phase in the Al-Zr system is the D023-structured Al3Zr. When aged at temperatures above approximately 450–500∘C, the metastable L12 Al3Zr particles gradually transform into the disk-shaped D023 morphology, often nucleating heterogeneously along dislocations [32].
In addition to the aforementioned alloying elements, other elements in aluminum alloys exhibit distinct precipitation behaviors. In Al-Cu alloys, the precipitation sequence is well reported that it initiates with the formation of Guinier-Preston (GP) zones, followed by the development of metastable intermediate phases such as (GP II zones) and [45], ultimately leading to the stable equilibrium phase -Al2Cu. Among these, the plate-shaped (Al2Cu) precipitates are recognized as the primary strengthening phases in these alloys. The Cu-Al alloy exhibits a body-centered tetragonal crystal structure (space group I4/mmm) and maintains a specific orientation relationship with the face-centered cubic (FCC) aluminum matrix [45]. Moreover, magnesium and silicon in Al-Mg-Si alloys combine to form Mg2Si precipitates, which crystallize in a face-centered cubic lattice, specifically adopting the fluorite structure (space group Fmm). Similarly, chromium and vanadium form discrete precipitate phases within aluminum matrices, typically in combination with additional alloying elements. The incorporation of chromium into aluminum-manganese alloys has been observed to slow precipitation kinetics while promoting the stabilization of Al12Mn-type precipitates [46]. In Al-Zr-V alloy systems, solute vanadium and zirconium co-precipitate to form L12-ordered Al3(Zr,V) particles. These coherent precipitates exhibit excellent lattice matching with the aluminum matrix and provide significant strengthening effects [47].
On the other hand, certain alloying elements primarily contribute to the strength of wrought aluminum alloys through solid solution strengthening, rather than the formation of coherent or semi-coherent precipitates. Although some of these elements may form intermetallic phases under specific conditions, their dominant strengthening mechanism arises from their dissolution within the aluminum matrix. Alloying elements that achieve strengthening through this mechanism include Mg, Si, Zn. Mg exhibits substantial solid solubility in the aluminum matrix at 0.3–0.7 wt.% at room temperature [24]. Due to the larger atomic radii of Mg, the dissolved Mg atoms create lattice distortion fields that effectively impede dislocation motion, thereby significantly enhancing both yield strength and ultimate tensile strength of the alloy system [24]. Both Si and Zn also exhibit solid solution strengthening effects in -Al matrices [48]. Tokuteru et al. [48] employed first-principles calculations to determine the misfit strains induced by these solute atoms (Si and Zn) and subsequently quantified their contributions to yield strength through the Friedel model. The computational results revealed misfit strains of approximately for Si and for Zn in -Al, demonstrating good agreement with experimental measurements from Al–Zn alloy systems.
This study employs density functional theory (DFT) to calculate and analyze the influence of interatomic distances between various alloying elements in the aluminum matrix on the overall energy change of the aluminum alloy system. The study further seeks to correlate and predict these distance-resolved interactions with the formation of distinct precipitate phases in aluminum alloys.
2. Computational Details
Density-functional theory calculations were performed using the ABINIT code [49,50,51], incorporating generalized gradient approximations (GGA) for the exchange-correlation functional as parameterized by Perdew, Burke, and Ernzerhof (PBE) [52]. The simulations employed a supercell of the face-centered cubic (fcc) aluminum unit cell, containing 108 atoms (Figure 1a), with periodic boundary conditions applied in all three spatial dimensions. The supercell dimensions were Å Å Å. A real-space k-point grid parameter (kptrlen) of Å was utilized, and Brillouin zone sampling was performed using a Monkhorst-Pack grid with a shift of .
Core electrons were treated implicitly using norm-conserving pseudopotentials following the Hamann scheme [53], while valence electrons were expanded in a plane-wave basis with a kinetic energy cutoff of 1088 eV. The electronic configurations for the explicitly treated valence states were as follows: for aluminum matrix atoms, and for alloying species: Sc–[Ar], Ti–[Ar], V–[Ar], Cr–[Ar], Cu–[Ar], Zr–[Kr], and Hf–[Xe].
The self-consistent field (SCF) calculations were considered converged when the energy difference between successive iterations fell below eV. Structural relaxations proceeded until the maximum residual force on any atom was less than eV/atom. All calculations were performed without spin polarization.
3. Results
Understanding impurity interactions in aluminum alloys is critically important, as these interactions fundamentally govern the nucleation of precipitates, cluster formation pathways, and, ultimately, the mechanical performance of engineering alloys. Solute clusters act as precursors to strengthening precipitates, and the strength and range of solute–solute interactions determine the stability, morphology, and coarsening behavior of these clusters, thereby directly influencing the age-hardening response and thermal stability of the material.
Numerous computational and experimental studies have addressed this topic; however, the conventional first-principles modeling approach typically involves two configurations: a single impurity atom in a supercell and a pair of impurity atoms at a fixed separation. The energy difference between these configurations is then interpreted as the interaction energy. While this approach yields valuable information for a given impurity configuration, it provides only limited insight into the concentration and distance dependence of impurity interactions and the way these interactions evolve spatially during early-stage clustering.
In the present work, we follow an alternative approach we employed earlier for vacancy clustering in SiC [54], and used supercell models containing a pair of impurity atoms and with various sizes to represent different impurity concentrations. We position the impurities at multiple relative positions corresponding to the 1st–4th coordination shells as indicated in Figure 1a. This distance-resolved methodology enables the construction of an interaction energy profile as a function of interatomic separation, offering a more comprehensive atomistic picture of the binding energetics. By systematically varying the distance between impurity atoms, we identify:
- Optimal separation distances that are energetically most favorable for a given solute pair.
- Energy barriers associated with cluster rearrangement and growth.
- The transition from attractive to repulsive interaction regimes with distance.
Such detailed interaction maps capture the energetic evolution from isolated solutes to clustered configurations, which act as precursors for ordered precipitate phases. The obtained results provide essential input parameters for higher-scale modeling frameworks, such as cluster dynamics and kinetic Monte Carlo simulations, thereby improving the predictive capability for precipitation kinetics and microstructure evolution in complex Al-based alloy systems.
In detail, Figure 1 presents the atomic configurations investigated in this study for the step-by-step (atom-by-atom) assembly of the initial Zr clusters in an Al matrix. Panel (a) shows the 108-atom supercell with the position #1 occupied by the first (isolated) Zr atom. Positions #5, #7, and #9 correspond to sites in the 2nd coordination shell relative to the first Zr atom, which were identified in Figure 2 as energetically favorable and match the relative Zr–Zr arrangement in the metastable L12 ZrAl3 phase (mp-569775) shown in Figure 1b. This cubic phase, fully coherent with the FCC Al matrix, is known to form as fine precipitates in Al–Zr alloys during aging at moderate temperatures.
Figure 2 presents the calculated interaction energies between two Zr atoms in an Al matrix for separations corresponding to the 1st–4th coordination shells, using both 108-atom (open symbols) and 48-atom (filled symbols) supercells. The zero-energy reference corresponds to the configuration with the atoms located in relative 4th coordination shell positions, while the blue arrow marks the thermal excitation energy window of meV at room temperature for scale comparison. The proposed distance-resolved approach clearly reveals that the 2nd coordination shell represents an energetically favorable position for the second Zr atom regardless of the supercell size. Notably, this relative positioning corresponds to the atomic arrangement of Zr atoms in the cubic L12-Al3Zr phase (see Figure 1a,b), a metastable trialuminide structure well known to form in Al–Zr alloys. L12-type Al3M compounds are highly coherent with the FCC Al matrix, ensuring low interfacial energy and strong resistance to coarsening, thereby maximizing the dispersion-strengthening effect and promoting exceptional recrystallization resistance.
The fact that our first-principles calculations reproduce this favorable configuration from fundamental energetic considerations demonstrates the capability of the method to predict the propensity of alloying elements to form precipitates of a given crystal structure. Furthermore, the effect becomes even more pronounced at higher local Zr concentrations, which in our simulations can be modeled using a smaller 48-atom supercell () with the solute pair aligned along the longest supercell dimension to minimize periodic boundary condition effects. In this configuration the interaction strength considerably amplified with the 2nd coordination sphere being even the absolute minimum of all investigated structures. This suggests that increasing the local solute content enhances the thermodynamic driving force for the nucleation of L12-structured Al3Zr precipitates.
Figure 3 quantify the energetic evolution during the atom-by-atom nucleation of the first L12-like cluster by sequentially placing additional Zr atoms into positions #5, #7, and #9 (Figure 1a). A nearly linear decrease in total energy is observed relative to the sum of isolated solute atoms (Figure 3a), along with a corresponding linear gain in binding energy for each newly added atom (Figure 3b). This indicates a strong and cumulative thermodynamic driving force for the formation of such clusters, fully consistent with experimental observations of L12 ZrAl3 precipitate nucleation.
To examine the possibility of direct nucleation of the stable tetragonal DO23 ZrAl3 phase (mp-395, space group I4/mmm), which is the equilibrium phase in the Al–Zr system but forms in Al only after prolonged exposure at elevated temperatures (>500 ∘C), we replaced the fourth Zr atom position (#9) with position #21 (Figure 1c), corresponding to the DO23 atomic arrangement shown in Figure 1d. In full agreement with experimental data, this configuration was found to be energetically unfavorable—both for the individual atom (Figure 3b) and for the cluster as a whole (Figure 3a)—indicating that DO23-type ordering is not favored at the earliest nucleation stages.
We then investigated multiple possible configurations for attaching the fifth Zr atom to the growing cluster. Figure 1f illustrates positions #33 (fully equivalent to #37) and #77, which correspond to a more cubic cluster morphology. As seen in Figure 3a, this pathway does not provide the maximum energetic stabilization, while Figure 3b reveals that positions #33 and #37 are in fact energetically unfavorable for a Zr atom compared to the isolated solute reference.
We also examined the placement of a Zr atom at position #84, located further along the (111) plane passing through positions #5, #7, and #9, and representing a continuation of a close-packed atomic layer. According to our calculations, this pathway also fails to deliver the most energetically favorable growth mode for the nucleating precipitate.
The energetically most favorable pathway for continued growth was found to involve the formation of a “planar” cluster morphology, initiated by placing the fifth Zr atom at position #36 (Figure 1g). Subsequent addition at position #39—lying in the (111) plane that passes through positions #1 and #36, parallel to the plane containing atoms at #5, #7, and #9—leads to a nearly linear decrease in the total cluster energy relative to the isolated-atom reference (Figure 3a). As shown in Figure 3b, in such a planar arrangement, positions #9 and #39 yield the highest binding energies for incoming Zr atoms, with the largest gains observed when the newly added atom forms the maximum number of triangular connections with the existing cluster, reaching approximately half of the maximum possible for Zr atoms in the fully ordered L12 ZrAl3 phase (horizontal reference line in Figure 3b). This morphology is reminiscent of the metastable, atom-layer thin GP I platelets observed by transmission electron microscopy in the Guinier-Preston zones of Cu-rich fcc Al [55] and modeled by DFT-enhanced Monte-Carlo simulations [56].
Given the finding that the nucleation of initial precipitate clusters in a planar morphology is energetically more favorable than in a cubic one, it is of interest to assess whether this geometric preference has implications for the mechanical properties of the alloy. To obtain a first-order estimate, we evaluated the effect of such clusters on the bulk modulus (B) of the Al matrix. Specifically, we performed ab initio calculations for a 108-atom supercell containing a five-atom Zr cluster arranged in either a more cubic (Figure 1f) or a more planar configuration (Figure 1g). The bulk modulus was determined by computing the pressure as a function of the supercell volume, followed by a linear fit of the resulting data. The calculated values were GPa for the cubic configuration and GPa for the planar configuration, both notably higher than the corresponding value for pure fcc Al obtained using the same approach ( GPa). Within the accuracy of this simple evaluation, no significant difference in the bulk modulus was observed between the two cluster morphologies, although both clearly contribute to an overall stiffening of the Al matrix.
The preceding analysis for Zr (Figure 1, Figure 2 and Figure 3) demonstrated that the most energetically favorable configurations for the initial stages of cluster nucleation occur when successive solute atoms occupy sites in the 2nd coordination shell, consistent with the atomic arrangement in the metastable L12-Al3Zr phase. This observation raises the question of whether similar interaction patterns exist for other transition metals known to form coherent L12-type trialuminide precipitates in aluminum alloys.
Following this step-by-step investigation of Zr cluster formation, Figure 4a extends the analysis to Sc, Ti, and Hf, for which the Al3M L12 structure has been reported [23,31,32,33,34,35,36]. The calculated interaction energy profiles for these solutes in a 108-atom supercell reveal pronounced minima for configurations where the second solute atom resides in the 2nd coordination shell relative to the first one—mirroring the Zr case. This spatial preference directly corresponds to the arrangement of solute atoms in the cubic L12 crystal structure, as illustrated earlier for Zr in Figure 1a,b.
The L12-Al3M precipitates formed by these elements share key features responsible for their strengthening efficiency: a high degree of coherency with the fcc Al matrix, low interfacial energy, and excellent thermal stability. Scandium forms the thermodynamically stable L12-Al3Sc phase, producing a fine, dense distribution of precipitates that significantly enhance strength and recrystallization resistance [57]. Hafnium and titanium can also form metastable L12 phases, often in combination with Zr or Sc, thereby contributing to the thermal stability and coarsening resistance of the precipitate population [43].
Importantly, the interaction energies in the 2nd coordination shell for all four L12-forming elements (Zr, Sc, Ti, and Hf) are substantially larger than the thermal excitation energy kT (indicated by the blue arrow in Figure 4a), indicating a strong thermodynamic driving force for such specific atomic arrangements even at moderate temperatures. The consistency of this energetic preference across multiple L12-forming elements suggests that first-principles mapping of distance-dependent solute–solute interactions can serve as a predictive tool for identifying alloying additions with a high propensity to form coherent L12 precipitates in aluminum alloys.
Having established the characteristic interaction patterns for L12-forming elements (Figure 4a), it is instructive to contrast these with other transition metals that can also form precipitates in aluminum alloys, but with crystal structures different from the coherent L12 trialuminides. At first glance, one might expect the interaction profiles of all transition-metal solutes in Al to follow a universal trend, potentially linked to the symmetry characteristics of their valence d-orbitals. However, Figure 4b presents the calculated interaction energies for V, Cr, and Cu—three elements which represent the early, central and late transition metals.
While these elements are capable of forming precipitates with Al under specific conditions, the resulting phases differ structurally from the cubic L12 trialuminides and are typically incoherent or only partially coherent with the aluminum matrix. For example, Cu in Al–Cu alloys forms plate-shaped (Al2Cu) precipitates with a body-centered tetragonal structure [45], V forms cubic Al10V or related phases [58], and Cr can form several complex intermetallics such as cubic Al13Cr4Si4 or hexagonal Al4Cr [59].
The interaction energy profiles for these elements do not exhibit as clear or deep minima at the 2nd coordination shell as seen for L12-forming solutes. This suggests that while solute–solute attraction still plays a role in the nucleation of their respective precipitate phases, the spatial arrangement of atoms in the early clusters is less directly tied to the final precipitate crystal structure. Consequently, our distance-resolved interaction analysis reveals not only the thermodynamic driving forces for clustering, but also qualitative differences in the nucleation pathways between coherent L12 phases and other, less coherent precipitate structures.
4. Conclusions
In summary, our first-principles analysis of distance-dependent solute–solute interaction energies reveals clear distinctions between several characteristic groups of alloying elements in aluminum:
- L12-forming elements — such as Zr, Sc, Ti, and Hf — which exhibit strong, well-defined attraction in the 2nd coordination shell, directly matching the atomic arrangement in coherent L12 trialuminide precipitates. This group also includes several rare-earth elements (e.g., Er, Lu, Tm, Yb) known to form similar coherent L12-type phases under appropriate conditions.
- Elements forming other structured precipitates — for instance, V, Cr, and Cu — which are among the most widely used in practice to produce semi-coherent or incoherent strengthening phases or dispersoids. While these are representative examples, other transition metals, though less frequently employed, can also form non-L12 intermetallics with aluminum.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
The authors gratefully acknowledge funding by the German Research Foundation (DFG) within the Research Training Group GRK 2868 D3 – project number 493401063.
References
- McLeod, C.L.; Shaulis, B.J. Rare Earth Elements in Planetary Crusts: Insights from Chemically Evolved Igneous Suites on Earth and the Moon. Minerals 2018, 8, 455. [Google Scholar] [CrossRef]
- Andersen, S.J.; Marioara, C.D.; Friis, J.; Wenner, S.; Holmestad, R. Precipitates in aluminium alloys. Adv. Phys. X 2018, 3, 1479984. [Google Scholar] [CrossRef]
- Mookherjee, M. Aluminum. In Encyclopedia of Geochemistry; Springer, 2016; pp. 1–3.
- Stojanovic, B.; Bukvic, M.; Epler, I. Application of Aluminum and Aluminum Alloys in Engineering. SCIDAR-Digit. Arch. Univ. Kragujev. 2018. [CrossRef]
- Dyvik, S.; Manum, B.; Mork, J.; Luczkowski, M. Structural aluminum in architecture—The history and future of aluminum as a structural material. Structures and architecture, CRC Press 2019, 1. [Google Scholar]
- De Matteis, G.; Brando, G.; Mazzolani, F.M. Pure aluminium: An innovative material for structural applications in seismic engineering. Constr. Build. Mater. 2012, 26, 677–686. [Google Scholar] [CrossRef]
- Guo, S.; Yu, H.; Wang, Z.; Yu, W.; Cheng, W.; Huang, L.; Liu, C.; Yin, F.; Zhao, W.; Qin, C. Microstructural evolution and mechanical properties of pure aluminum upon multi-pass caliber rolling. Materials 2022, 15, 1206. [Google Scholar] [CrossRef]
- Dong, X.; Chen, F.; Chen, S.; Liu, Y.; Huang, Z.; Chen, H.; Feng, S.; Zhao, L.; Wu, Z.; Zhang, X. Microstructure and microhardness of hot extruded 7075 aluminum alloy micro-gear. J. Mater. Process. Technol. 2015, 219, 199–208. [Google Scholar] [CrossRef]
- Rana, R.; Purohit, R.; Das, S. Reviews on the influences of alloying elements on the microstructure and mechanical properties of aluminum alloys and aluminum alloy composites. Int. J. Sci. Res. Publ. 2012, 2, 1–7. [Google Scholar]
- Bishop, D.; Cahoon, J.; Chaturvedi, M.; Kipouros, G.; Caley, W. On enhancing the mechanical properties of aluminum P/M alloys. Mater. Sci. Eng. A 2000, 290, 16–24. [Google Scholar] [CrossRef]
- Murray, J.L. The Al- Mg (aluminum- magnesium) system. J. Phase Equilibria 1982, 3, 60–74. [Google Scholar] [CrossRef]
- Murray, J.; McAlister, A. The Al-Si (aluminum-silicon) system. Bull. Alloy Phase Diagrams 1984, 5, 74–84. [Google Scholar] [CrossRef]
- Plyushchay, I.; Wei, Z.; Bulut, N.; Gemming, S. First-Principles Modeling of Solid Solution Softening and Hardening Effects in Al–Mg–Zr–Si Aluminum Alloys. Adv. Eng. Mater. 2025, p. 2500321.
- Murray, J.; Peruzzi, A.; Abriata, J. The Al-Zr (aluminum-zirconium) system. J. Phase Equilibria 1992, 13, 277–291. [Google Scholar] [CrossRef]
- Meng, Q.; Frankel, G. Effect of Cu content on corrosion behavior of 7xxx series aluminum alloys. J. Electrochem. Soc. 2004, 151, B271. [Google Scholar] [CrossRef]
- Chuvil’deev, V.; Nokhrin, A.; Kopylov, V.; Gryaznov, M.Y.; Shotin, S.; Likhnitskii, C.; Kozlova, N.; Shadrina, Y.S.; Berendeev, N.; Melekhin, N.; et al. Investigation of mechanical properties and corrosion resistance of fine-grained aluminum alloys Al-Zn with reduced zinc content. J. Alloys Compd. 2022, 891, 162110. [Google Scholar] [CrossRef]
- Nam, S.W.; Lee, D.H. The effect of Mn on the mechanical behavior of Al alloys. Met. Mater. 2000, 6, 13–16. [Google Scholar] [CrossRef]
- Truax, D.; McMahon Jr, C. Plastic behavior of titanium-aluminum alloys. Mater. Sci. Eng. 1974, 13, 125–139. [Google Scholar] [CrossRef]
- Murray, J. The Al-Cr (aluminum-chromium) system. J. Phase Equilibria Diffus. 1998, 19, 368. [Google Scholar] [CrossRef]
- Murray, J. Al-V (aluminum-vanadium). Bull. Alloy Phase Diagrams 1989, 10, 351–357. [Google Scholar] [CrossRef]
- Taylor, A.; Jones, R.M. Constitution and magnetic properties of iron-rich iron-aluminum alloys. J. Phys. Chem. Solids 1958, 6, 16–37. [Google Scholar] [CrossRef]
- Eskin, D.G. Sc applications in aluminum alloys: Overview of Russian research in the 20th century. In Proceedings of the TMS Annual Meeting & Exhibition, Springer; 2018; pp. 1565–1572. [Google Scholar]
- Jia, Z.H.; Huang, H.L.; Wang, X.L.; Xing, Y.; Liu, Q. Hafnium in aluminum alloys: a review. Acta Metall. Sin. (Engl. Lett.) 2016, 29, 105–119. [Google Scholar] [CrossRef]
- Ryen, ∅. ; Holmedal, B.; Nijs, O.; Nes, E.; Sjölander, E.; Ekström, H.E. Strengthening mechanisms in solid solution aluminum alloys. Metall. Mater. Trans. A 2006, 37, 1999–2006. [Google Scholar] [CrossRef]
- Pedersen, K.O.; Westermann, I.; Furu, T.; Børvik, T.; Hopperstad, O.S. Influence of microstructure on work-hardening and ductile fracture of aluminium alloys. Mater. Des. 2015, 70, 31–44. [Google Scholar] [CrossRef]
- Fine, M.E. Precipitation hardening of aluminum alloys. Metall. Trans. A 1975, 6, 625–630. [Google Scholar] [CrossRef]
- Bloch, E. Dispersion-strengthened aluminium alloys. Metall. Rev. 1961, 6, 193–240. [Google Scholar] [CrossRef]
- Nes, E.; Holmedal, B.; Evangelista, E.; Marthinsen, K. Modelling grain boundary strengthening in ultra-fine grained aluminum alloys. Mater. Sci. Eng. A 2005, 410, 178–182. [Google Scholar] [CrossRef]
- Ma, K.; Wen, H.; Hu, T.; Topping, T.D.; Isheim, D.; Seidman, D.N.; Lavernia, E.J.; Schoenung, J.M. Mechanical behavior and strengthening mechanisms in ultrafine grain precipitation-strengthened aluminum alloy. Acta Mater. 2014, 62, 141–155. [Google Scholar] [CrossRef]
- Gupta, R.; Daniel, B. Strengthening mechanisms in Al3Zr-reinforced aluminum composite prepared by ultrasonic assisted casting. J. Mater. Eng. Perform. 2021, 30, 2504–2513. [Google Scholar] [CrossRef]
- Harada, Y.; Dunand, D. Microstructure of Al3Sc with ternary transition-metal additions. Mater. Sci. Eng. A 2002, 329, 686–695. [Google Scholar] [CrossRef]
- Knipling, K.E.; Dunand, D.C.; Seidman, D.N. Precipitation evolution in Al–Zr and Al–Zr–Ti alloys during aging at 450–600 C. Acta Mater. 2008, 56, 1182–1195. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, F.; Liu, H.; Ren, X.; Xu, H.; Wang, M.; Fu, Y. Exploration of D022 type Al3TM (TM = Sc, Ti, V, Zr, Nb, Hf, Ta): elastic anisotropy, electronic structures, work function and experimental design. Materials 2021, 14, 2206. [Google Scholar] [CrossRef] [PubMed]
- Knipling, K.E.; Karnesky, R.A.; Lee, C.P.; Dunand, D.C.; Seidman, D.N. Precipitation evolution in Al–0.1 Sc, Al–0.1 Zr and Al–0.1 Sc–0.1 Zr (at.%) alloys during isochronal aging. Acta Mater. 2010, 58, 5184–5195. [Google Scholar] [CrossRef]
- Knipling, K.E.; Dunand, D.C.; Seidman, D.N. Nucleation and precipitation strengthening in dilute Al-Ti and Al-Zr alloys. Metall. Mater. Trans. A 2007, 38, 2552–2563. [Google Scholar] [CrossRef]
- Liu, S.; Liu, F.; Yan, Z.; Nie, B.; Fan, T.; Chen, D.; Song, Y. Nucleation of L12-Al3M (M = Sc, Er, Y, Zr) nanophases in aluminum alloys: a first-principles thermodynamics study. Crystals 2023, 13, 1228. [Google Scholar] [CrossRef]
- Elasheri, A.; Elgallad, E.M.; Parson, N.; Chen, X.G. Nucleation and transformation of Zr-bearing dispersoids in Al–Mg–Si 6xxx alloys. J. Mater. Res. 2023, 38, 696–707. [Google Scholar] [CrossRef]
- Jassim, A.H.M. Influence of Zirconium Additions and Ageing Time on Some Mechanical Properties of (6063) Aluminum Alloy: The Influence of Zirconium Additions and the Aging Time on Some Mechanical Properties. J. Univ. Kerbala 2016, 14, 3. [Google Scholar]
- Bolibruchová, D.; Sỳkorová, M.; Brüna, M.; Matejka, M.; Širanec, L. Effect of Zr addition on selected properties and microstructure of aluminum alloy AlSi5Cu2Mg. Int. J. Met. 2023, 17, 2598–2611. [Google Scholar] [CrossRef]
- Šmalc, J.; Zaky, A.; Markoli, B.; Šturm, R. Microstructural Stability and High-Temperature Mechanical Behavior of Al–Ni–Zr Alloy Strengthened by L12-Al3Zr Precipitates. Materials 2025, 18, 3068. [Google Scholar] [CrossRef]
- Elasheri, A.; Elgallad, E.; Parson, N.; Chen, X.G. Evolution of Zr-bearing dispersoids during homogenization and their effects on hot deformation and recrystallization resistance in Al-0.8% Mg-1.0% Si alloy. J. Mater. Eng. Perform. 2021, 30, 7851–7862. [Google Scholar] [CrossRef]
- Liu, F.; Chen, S.; Dong, Q.; Qin, J.; Li, Z.; Zhang, B.; Nagaumi, H. Tailoring microstructure and mechanical properties of Al-Mg-Si-Cu alloy with varying Mn and/or Cr additions. Mater. Sci. Eng. A 2024, 892, 146053. [Google Scholar] [CrossRef]
- Knipling, K.E.; Dunand, D.C. Creep resistance of cast and aged Al–0.1 Zr and Al–0.1 Zr–0.1 Ti (at.%) alloys at 300–400 C. Scr. Mater. 2008, 59, 387–390. [Google Scholar] [CrossRef]
- Fuller, C.B.; Murray, J.L.; Seidman, D.N. Temporal evolution of the nanostructure of Al (Sc, Zr) alloys: Part I–Chemical compositions of Al3(Sc1-xZrx) precipitates. Acta Mater. 2005, 53, 5401–5413. [Google Scholar] [CrossRef]
- Miyoshi, H.; Kimizuka, H.; Ishii, A.; Ogata, S. Competing nucleation of single-and double-layer Guinier–Preston zones in Al–Cu alloys. Sci. Rep. 2021, 11, 4503. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.; Frisk, K.; Nyborg, L. Role of Cr in Mn-rich precipitates for Al–Mn–Cr–Zr-based alloys tailored for additive manufacturing. Calphad 2024, 84, 102667. [Google Scholar] [CrossRef]
- Fan, Y.; Makhlouf, M.M. Precipitation strengthening of aluminum by transition metal aluminides. Master of Science Thesis, Worcester Polytechnic Institute, Worcester, MA 2015, 1609. [Google Scholar]
- Uesugi, T.; Higashi, K. Modeling Solid Solution Strengthening Using First-Principles Results of Misfit Strain with Friedel Model in Al-Based Alloys. In Proceedings of the 12th International Conference on Aluminium Alloys, Yokohama, Japan, September 5-9 2010; pp. 1421–1425. [Google Scholar]
- Gonze, X.; Beuken, J.M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.M.; Sindic, L.; Verstraete, M.; Zerah, G.; Jollet, F.; et al. First-principles computation of material properties : the ABINIT software project. Comp. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Goedecker, S. Fast radix 2, 3, 4 and 5 kernels for Fast Fourier Transformations on computers with overlapping multiply-add instructions. SIAM J. Sci. Comput. 1997, 18, 1605. [Google Scholar] [CrossRef]
- Gonze, X.; Amadon, B.; Anglade, P.M.; Beuken, J.M.; et al. The ABINIT code, 2024.
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Hamann, D. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 2013, 88, 085117. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Wendler, E.; Hübner, R.; Anwand, W.; Wang, G.; Chen, X.; Tong, W.; Yang, Z.; Munnik, F.; et al. Defect-induced magnetism in SiC: Interplay between ferromagnetism and paramagnetism. Phys. Rev. B 2015, 92, 174409. [Google Scholar] [CrossRef]
- Berlin, J.; Stegmüller, T.; Haider, F. Observation of in-plane oriented Guinier-Preston zones in Al-Cu. Micron 2023, 173, 103504. [Google Scholar] [CrossRef]
- Suzuki, T.; Yabe, T.; Enoki, M.; Ohtani, H. Thermodynamic investigation of Guinier–Preston zone formation in the Al–Cu binary system. Scr. Mater. 2025, 265, 116746. [Google Scholar] [CrossRef]
- Ahmad, Z.; Aleem, A.A.B.; Abbas, M. Effect of scandium doping on the corrosion resistance and mechanical behavior of Al-3Mg alloy in neutral chloride solutions. Mater. Sci. Appl. 2011, 2, 244–250. [Google Scholar] [CrossRef]
- Wang, F.; Chiu, Y.L.; Eskin, D.; Du, W.; Shearing, P.R. A grain refinement mechanism of cast commercial purity aluminium by vanadium. Mater. Charact. 2021, 181, 111468. [Google Scholar] [CrossRef]
- Wang, S.; Dong, L.; Han, X.; Fan, Y.; Chen, B. Orientations and interfaces between α’-Al13Cr4Si4 and the matrix in Al-Si-Cr-Mg alloy. Mater. Charact. 2020, 160, 110096. [Google Scholar] [CrossRef]
Figure 1.
Atomic configurations used in the study. (a) 108-atom Al supercell showing positions #3, #5, and #9 corresponding to the 2nd coordination shell around the first solute atom (#1). (b) Atomic structure of ZrAl3 precipitate (mp-569775) with labeled atomic sites equivalent to those in (a). (c) Fragment of the supercell indicating position #21, corresponding to the stable ZrAl3 phase (mp-395) shown in (d). (f) and (g) Fragments of the supercell with labeled atomic sites corresponding to a more cubic and a more planar cluster shape, respectively; in (g), the (111) plane containing position #39 is indicated.
Figure 1.
Atomic configurations used in the study. (a) 108-atom Al supercell showing positions #3, #5, and #9 corresponding to the 2nd coordination shell around the first solute atom (#1). (b) Atomic structure of ZrAl3 precipitate (mp-569775) with labeled atomic sites equivalent to those in (a). (c) Fragment of the supercell indicating position #21, corresponding to the stable ZrAl3 phase (mp-395) shown in (d). (f) and (g) Fragments of the supercell with labeled atomic sites corresponding to a more cubic and a more planar cluster shape, respectively; in (g), the (111) plane containing position #39 is indicated.
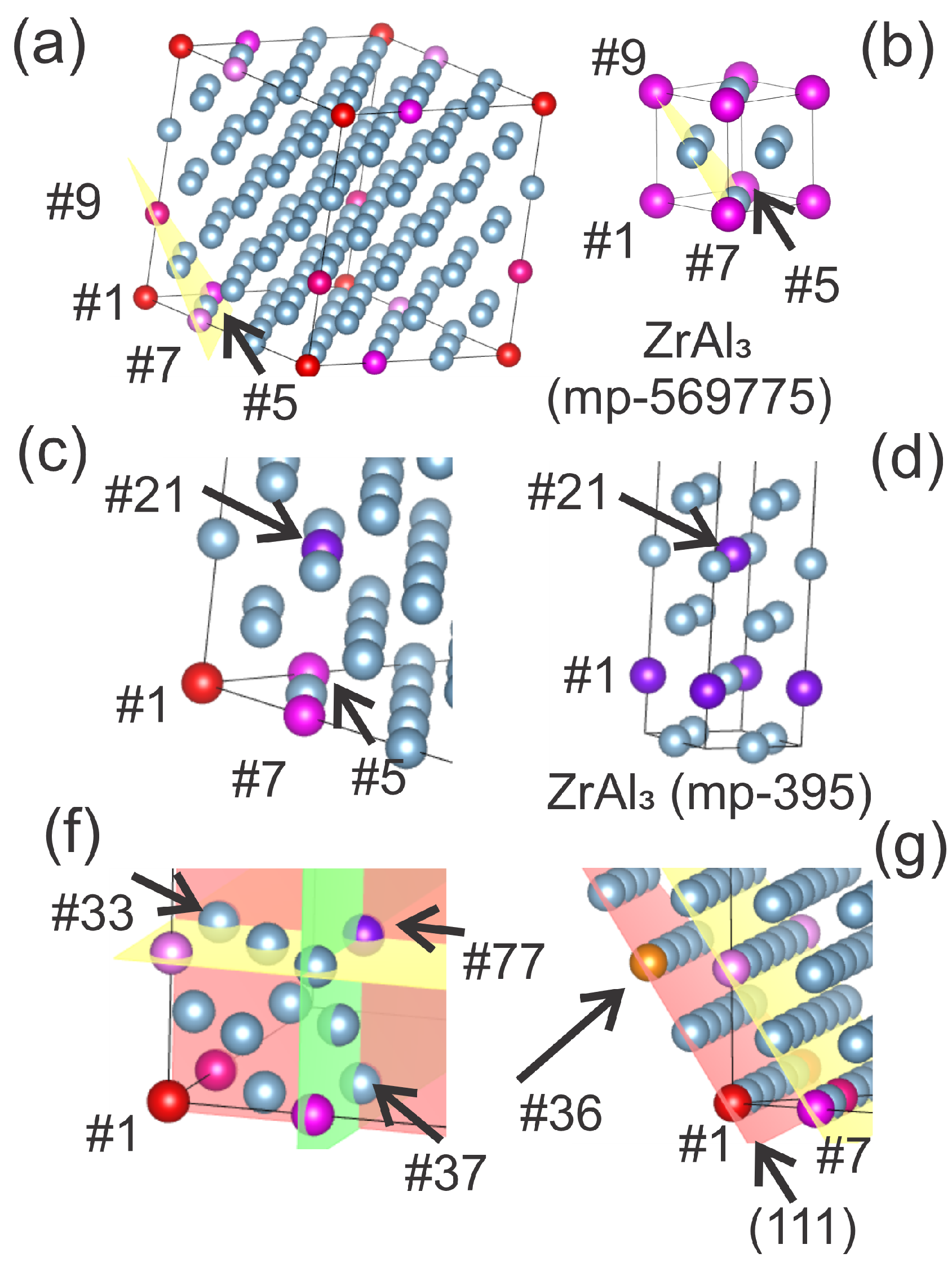
Figure 2.
Interaction energy between two Zr atoms in an Al matrix as a function of their separation (1st–4th coordination shell). Zero energy is referenced to the 4th shell configuration. Results are shown for 108-atom and 48-atom supercells; the blue arrow indicates the thermal energy scale () for reference.
Figure 2.
Interaction energy between two Zr atoms in an Al matrix as a function of their separation (1st–4th coordination shell). Zero energy is referenced to the 4th shell configuration. Results are shown for 108-atom and 48-atom supercells; the blue arrow indicates the thermal energy scale () for reference.
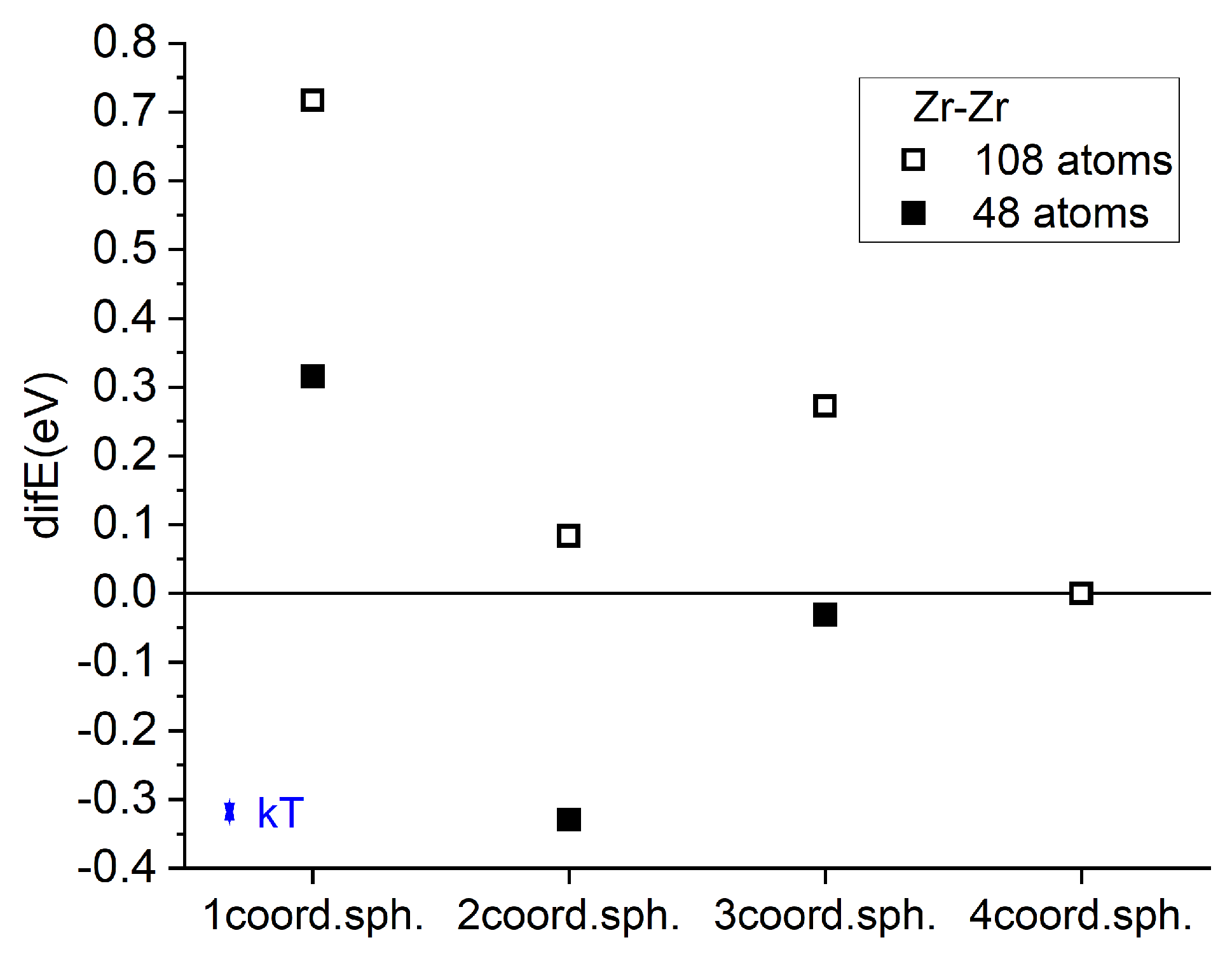
Figure 3.
(a) Total energy gain relative to isolated Zr atoms as a function of the number of Zr atoms in the cluster. The zero level corresponds to the reference state of an isolated Zr atom in the Al matrix. Numbers next to the data points indicate the lattice positions where the next Zr atom is added (see Figure 1). (b) Energy gain per Zr atom relative to isolated Zr atoms as a function of the number of Zr atoms in the cluster. The zero level corresponds to the reference state of an isolated Zr atom in the Al matrix.
Figure 3.
(a) Total energy gain relative to isolated Zr atoms as a function of the number of Zr atoms in the cluster. The zero level corresponds to the reference state of an isolated Zr atom in the Al matrix. Numbers next to the data points indicate the lattice positions where the next Zr atom is added (see Figure 1). (b) Energy gain per Zr atom relative to isolated Zr atoms as a function of the number of Zr atoms in the cluster. The zero level corresponds to the reference state of an isolated Zr atom in the Al matrix.
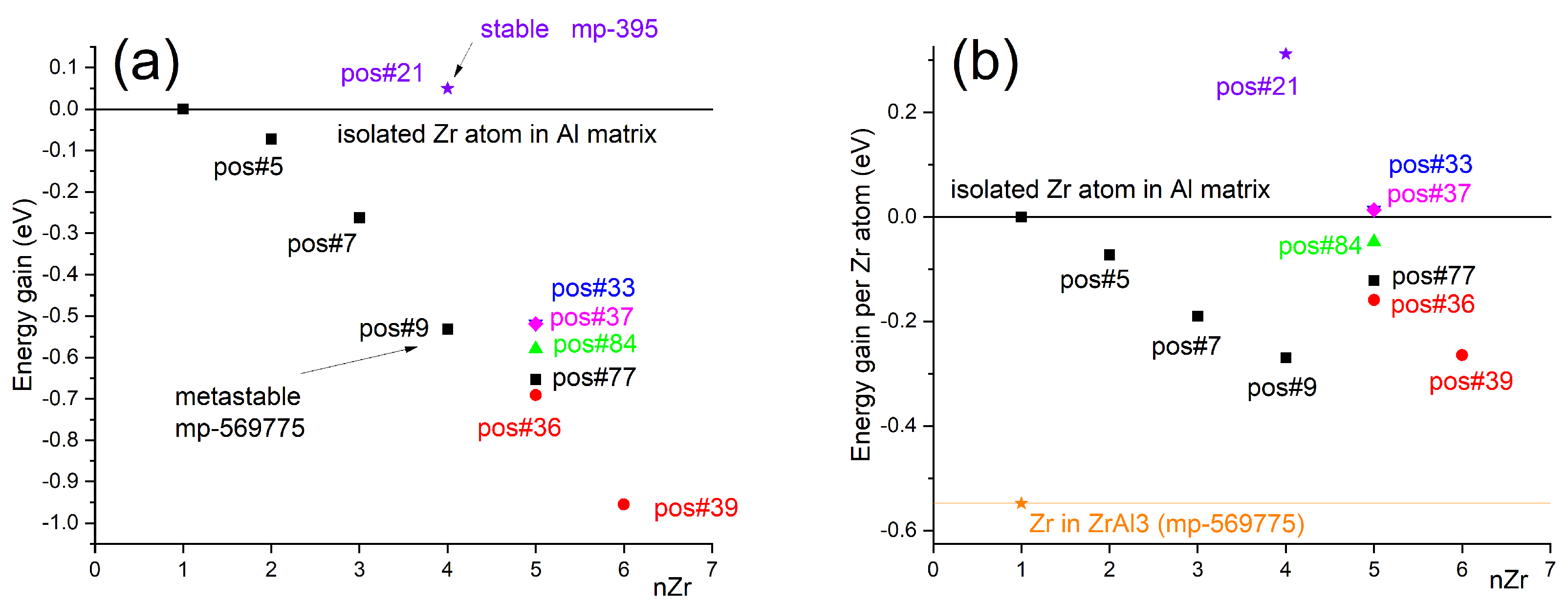
Figure 4.
Interaction energy between two solute atoms in an Al matrix as a function of their separation (1st–4th coordination shell) for a 108-atom supercell: (a) Zr, Sc, Ti, and Hf; (b) V, Cr, and Cu. Zero energy is referenced to the 4th shell configuration; the blue arrow indicates the thermal energy scale () for comparison.
Figure 4.
Interaction energy between two solute atoms in an Al matrix as a function of their separation (1st–4th coordination shell) for a 108-atom supercell: (a) Zr, Sc, Ti, and Hf; (b) V, Cr, and Cu. Zero energy is referenced to the 4th shell configuration; the blue arrow indicates the thermal energy scale () for comparison.


| Precipitate | Phase | Condition | Stability | Lattice constant |
|---|---|---|---|---|
| Al3Zr | L12 | <425 ∘C | metastable | - Å |
| D023 | >450 ∘C | stable | Å, Å, | |
| Al3Sc | L12 | 200-650 ∘C | stable | Å |
| Al3Ti | L12 | rapid cooling | metastable | Å, Å, |
| D022 | 500-650 ∘C | stable | Å, Å, | |
| Al3Hf | L12 | 250-350 ∘C | metastable | - Å |
| D022 | 450-550 ∘C | stable | – | |
| D023 | >550 ∘C | stable | Å, Å |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



