Submitted:
25 August 2025
Posted:
26 August 2025
You are already at the latest version
Abstract
Although the ectopic expression of genes that regulate differentiation has been observed in various cancers, its significance has not been fully investigated. In this article, based on the biochemical premise that “new biological responses do not fundamentally arise from mutations,” I redefine the role of ectopic expression in carcinogenesis and propose the “carcinogenic chimera cell (CCC) hypothesis.” The CCC hypothesis posits that ectopic expression of genes contributes to carcinogenesis via four mechanisms: (1) execution of their original functions, (2) expression of latent functions, (3) dysregulation of differentiation, and (4) inhibition of differentiation. This hypothesis provides a comprehensive explanation for the characteristics of cancer, including atypia, glycosylation defects, invasiveness, tissue-type diversity, and plasticity. Additionally, it complements and clarifies the molecular mechanisms of the traditional aberrant differentiation hypotheses and offers a perspective that integrates and encompasses existing theories such as the mutation theory, multistage carcinogenesis theory, epigenetic theory, cancer stem cell theory, and clonal evolution theory. Furthermore, this hypothesis introduces the concept of “non-mutational drivers,” a new type of cancer gene, expanding the definition of malignancy. This study provides a theoretical foundation for the development of molecular markers and new molecule-targeted drugs for diagnostic and therapeutic applications.
Keywords:
ectopic expression
; epigenetics
; carcinogenesis
; cancer plasticity
; transdifferentiation
; cancer stem cell
; Twist
; atypia
1. Introduction
The mechanism underlying cancer development remains unclear, and no definitive theory has been established to date. Previously proposed theories, such as the mutation theory, epigenetic theory, aberrant differentiation hypothesis, tumor microenvironment theory, and cancer stem cell theory, explain certain aspects of cancer but are insufficient to comprehensively account for the highly diverse nature of cancer, cellular atypia, invasive and metastatic potential, recurrence, and plasticity.
Recent reports have increasingly suggested that transcription factors and genes that normally regulate differentiation are involved in the onset and progression of cancer via ectopic expression outside their normal expression domains. Among these, there are cases where even “normal-type” genes, which do not harbor mutations, can induce tumorigenic phenotypes by expressing in a context inconsistent with their cellular lineage. Although these are not defined as traditional driver mutations, they should clearly be considered factors involved in the development of cancer.
In this article, we focus on cancer-related genes exhibiting ectopic expression and propose a new hypothesis, the “carcinogenic chimera cell (CCC) hypothesis.” This hypothesis suggests that genes regulating differentiation, when expressed in a cellular environment different from their original environment, disrupt cellular functions such as cell structure, adhesion, glycan modification, secretory capacity, intercellular communication, and motility, leading to the acquisition of atypia and malignant properties by executing their original function. This theory goes beyond the conventional concept of cancer genes, which assumes that mutations result in “gain of function,” and instead posits “function-retaining ectopic expression” as the primary cause of carcinogenesis.
This theory provides a unified interpretation of various phenomena that were previously difficult to explain, such as the diverse tissue and differentiation patterns of cancer as well as the existence of undifferentiated cancers and fetal-type tumors. Additionally, ectopically expressed cancer-related genes, such as Kit [1], Mer [2], Hox11 [3], mGluR5 [4], and Wnt1 [5], have been reported, analysis of which contributes to the validation of these hypotheses and has high potential for clinical and research applications, including the development of molecule-targeted drugs targeting genes showing ectopic expression, exploration of diagnostic markers, and identification of the physiological functions of genes with unknown functions. In this article, based on these hypotheses, we discuss the relationship between ectopically expressed genes and carcinogenesis using specific examples and propose experimental methods to verify this theory.
2. Hypothesis
I propose the CCC hypothesis, which focuses on the ectopic expression of differentiation-controlling genes during cancer development. Ectopically expressed differentiation-controlling genes are expressed in the parent cells of cancer cells while retaining their original physiological functions, giving rise to tumor characteristics such as atypia, diversity, invasion, and plasticity . The CCC hypothesis posits that two molecular mechanisms are essential for cancer development. The first is the “constant activation of cell proliferation-related genes,” which has been proposed in the traditional mutation theory and is caused by gain-of-function mutations. The other is “ectopic expression of differentiation-controlling genes,” where genes that should normally be expressed in specific differentiation lineages are expressed instead in different cells, leading to differentiation dysfunction. A combination of these two events is believed to trigger cancer onset and progression.
2.1. Supporting Evidence: Examples of How Ectopic Expression of Genes Controlling Differentiation Contribute to Carcinogenesis.
To illustrate the core concept of the CCC hypothesis, Table 1 summarizes the representative genes that are ectopically expressed in cancers. These genes, which are normally restricted to specific lineages, are expressed in unrelated cell types and contribute to carcinogenesis by retaining or misapplying their original functions.
This table lists ten genes that are normally expressed in specific developmental or tissue contexts but are ectopically expressed in cancers. The normal expression of each gene associated cancer types and their functional implications are shown, demonstrating how ectopic expression may disrupt differentiation programs and contribute to carcinogenesis according to the CCC hypothesis.
There are numerous examples in which genes regulating differentiation are ectopically expressed and are possibly involved in carcinogenesis. This article presents 10 examples, with additional 50 examples in the supplement. The details of erbB, ALK, CDX2, NKX2-1, VAV1, REST, Shh, OCT4, PAX8, and Twist are presented below.
2.1.1. erbB
erbB is an important cancer-related gene that inspired the author to conceive the CCC hypothesis. This gene was first identified as a virus-derived cancer gene (v-erbB) encoded by the avian erythroblastosis virus (avian erythroblastosis virus) [6]. It is one of the first cancer genes the function of which was elucidated, following the example of “sis (the viral version of PDGF)” [7].
v-erbB possesses the structure of a receptor tyrosine kinase belonging to the epidermal growth factor receptor family, but lacks a ligand-binding site, resulting in “constant activation independent of ligands”. This structural change leads to the abnormal activation of cell proliferation signals and is involved in carcinogenesis.
Normal erbB expression is primarily limited to epithelial cells and is not expressed in erythroid cells. Therefore, erbB expression in erythroid cells represents ectopic expression in a cell lineage where it is not normally expressed. This can be considered an early example of carcinogenesis due to ectopic expression, which is the central concept of this hypothesis.
In erythrocytes, when cellular information (cytoskeleton, cell adhesion molecules, glycosyltransferases, and expression of epithelial cell-specific genes) is regulated by erbB, the cytoskeleton is disrupted, leading to atypia, disruption of cell adhesion molecules, and glycosylation defects, resulting in invasion and metastasis. This leads to the formation of defective cancer cells.
2.1.2. Anaplastic Lymphoma Kinase (ALK)
ALK, a receptor tyrosine kinase belonging to the insulin receptor superfamily, is primarily transiently expressed in specific neurons of the brain and nervous system during development, where it regulates neuronal proliferation and differentiation [8].
In adults, ALK expression is typically suppressed; however, in certain cancers, such as anaplastic large cell lymphoma [9] and non-small cell lung cancer [10] (particularly adenocarcinoma in younger individuals), ALK is reactivated by chromosomal rearrangements (e.g., ALK/EML4 or NPM1/ALK), point mutations, or gene amplification, leading to the expression of fusion or abnormally active ALK proteins.
In these tumors, ALK is ectopically expressed in non-neural cells where it is not normally expressed, and its active protein disrupts the control of cell differentiation, contributing to tumor initiation and progression. Therefore, ALK provides a classic example of oncogenesis due to ectopic expression, according to the CCC hypothesis. In contrast, ALK abnormalities in neuroblastomas, which originate from neural tissue, are classified as activation abnormalities within the original differentiation lineage, rather than ectopic expression.
2.1.3. Caudal Type Homeobox 2 (CDX2)
CDX2 is an intestinal-specific homeobox transcription factor that is expressed in the postembryonic intestinal region and is highly expressed in the small and large intestinal epithelia in adults, maintaining intestinal differentiation and homeostasis [11]. Since it is barely expressed in the normal gastric mucosa or hematopoietic cells, CDX2 expression in the lesions described below is considered to be ectopic, supporting the CCC hypothesis.
2.1.3.1. Gastrointestinal Epithelial Metaplasia and Gastric Cancer
In gastric mucosa-specific CDX2 transgenic mice created by Mutoh et al., ectopic expression of CDX2 alone induced intestinal epithelial metaplasia, converting the gastric epithelium into intestinal-type cells [12,13]. In a large-scale analysis of The Cancer Genome Atlas data, CDX2 mRNA induction accounted for 54.5% of gastric cancers, with intestinal-type/mixed-type histology being predominant [14]. CDX2-positive gastric cancers are often associated with low-stage well-differentiated tumors [15].
2.1.3.2. Acute Myeloid Leukemia (AML)
CDX2 is overexpressed in over 90% of patients, and ectopic expression of Cdx2 induces lethal myelodysplastic syndrome to leukemia in mouse hematopoietic stem/progenitor cells [16]. Recombination of ETV6 (t(12;13)(p13;q12)) has been shown to cause ectopic expression of CDX2 as the primary conversion event, leading to lethal AML [17].
2.1.3.3. Relationship with the CCC Hypothesis
CDX2 is a transcription factor that drives the intestinal cell program, and its ectopic expression in the gastric mucosa or hematopoietic system converts surrounding tissues into intestinal-type or undifferentiated intestinal-like cells while maintaining their original differentiation functions, consistent with the mechanism proposed by the CCC hypothesis, which leads to tissue heterogeneity and tumorigenesis. In particular, intestinal metaplasia in the stomach demonstrates that CDX2 can induce differentiation of the gastric epithelium into intestinal-type cells, positioning it as a model for differentiation dysregulation patterns.
2.1.4. NKX2-1
NKX2-1 (thyroid transcription factor-1; TTF-1) is a transcription factor that regulates the differentiation of the thyroid and lungs. In the thyroid, it binds to the promoters of thyroid-specific gene clusters such as thyroglobulin, thyroid peroxidase, and sodium-iodine cotransporter and activates their expression. In the lungs, it is expressed in alveolar type II cells and induces the expression of surfactant proteins (SFTPB, SFTPC) and SCGB3A2, thereby contributing to lung morphogenesis and functional maintenance [18].
In gastric adenocarcinoma of the fundic gland type, NKX2-1 is reported to be strongly expressed ectopically in gastric tissue where it is normally not expressed; furthermore, in tumor cells of gastric adenocarcinoma of fundic gland type, NKX2-1 is induced alongside the lung-specific surfactant proteins, SFTPB and SFTPC [19]. This is a classic example of the CCC hypothesis, which posits that genes regulating differentiation are ectopically expressed, inducing pleomorphism while maintaining their original function.
Furthermore, ectopic expression of NKX2-1 has been observed in T cell acute lymphoblastic leukemia (T-ALL). Although NKX2-1 is not normally expressed in hematopoietic cells, in some cases of T-ALL, chromosomal translocations or enhancer hijacking leads to ectopic expression of NKX2-1, contributing to tumor formation [20]. This example clearly supports the CCC hypothesis, which proposes that ectopically expressed differentiation-related genes contribute to tumorigenesis.
Ectopic expression of NKX2-1 not only explains morphological heterogeneity and abnormal differentiation patterns in tumors but has also been reported to influence tumor invasion and metastasis in some cases. Thus, the effects of the ectopic expression of differentiation regulatory factors on tumor biology are multifaceted [19].
2.1.5. VAV1
VAV1 is a member of the VAV gene family, which functions as a guanine nucleotide exchange factor (GEF) for Rho family GTPases and is involved in cellular functions such as actin cytoskeletal remodeling and transcription regulation. VAV1 is primarily expressed in hematopoietic cells and plays an essential role in the differentiation and activation of T cells and B cells [21].
In contrast, ectopic expression of VAV1 has been reported in pancreatic ductal adenocarcinoma, which is caused by hypomethylation of the promoter region. Ectopically expressed VAV1 contributes to proliferation, transformation, and survival of tumor cells and has been shown to be involved in the progression of pancreatic cancer [22]. Such ectopic expression corroborates the CCC hypothesis as it is expressed in non-hematopoietic tissues where it induces carcinogenesis while maintaining its original function in the hematopoietic system.
2.1.6. RE1-Silencing Transcription Factor (REST)
REST is a transcription repressor that suppresses the expression of neuron-specific genes in non-neuronal cells; it has been shown in many studies to function as a tumor suppressor gene by binding to the RE1 sequence (or NRS: neuron-restrictive silencer element) present in the promoter region of neuron-specific genes and suppressing their transcription. This function prevents non-neuronal cells from differentiating into neurons and allows them to maintain their original cell lineage—such as epithelial cells or muscle cells.
Interestingly, suppression or loss of REST expression has been reported in epithelial tumors, particularly small cell lung cancer, which is in agreement with the phenomenon of tumorigenesis due to ectopic activation of neural gene information [23]. This is a classic example of the CCC hypothesis, which proposes that tumorigenesis progresses through an influx of differentiation programs from other cell lineages.
Although the position of REST, a tumor suppressor gene, is different from that of “oncogenes with ectopic expression” discussed in the CCC hypothesis, it is an important molecule with respect to the CCC hypothesis as loss of REST function leads to the ectopic expression of neural differentiation genes that were originally suppressed. If genes with mechanisms of action similar to that of REST are identified in future, they may be considered as candidates for the CCC hypothesis.
2.1.7. Sonic Hedgehog (Shh)
Shh encodes a morphogen that plays a central role in pattern formation during embryonic development. Shh is involved in the induction of the ventral neural tube, formation of the anterior-posterior axis of the limbs, and ventralization of somites, and it performs specific and critical functions during embryogenesis [24]. Therefore, the reactivation (ectopic expression) of Shh in adult somatic cells is representative of functional expression that deviates from its physiological context and thereby fits in with the CCC hypothesis. Ectopic expression or pathway activation of Shh has been reported in various cancer types, including basal cell carcinoma, pancreatic cancer, lung cancer, hepatocellular carcinoma, glioma, gastric cancer, prostate cancer, and breast cancer [25].
2.1.8. OCT4
OCT4 (POU5F1) is a transcription factor necessary for maintaining pluripotency and self-renewal in embryonic stem cells that belongs to the POU homeodomain transcription factor family [26]. Normal expression of OCT4 is primarily limited to pre-implantation embryos and embryonic stem cells and is not detected in postnatal somatic cells. Therefore, expression of OCT4 in somatic cell-derived tumors is considered ectopic, which is in agreement with the CCC hypothesis.
Ectopic expression of OCT4 is widely recognized in testicular germ cell tumors (particularly germ cell carcinomas and seminomas) [27] and has also been reported in somatic tumors such as pancreatic [28], gastric, and colorectal cancers [29]. In these cancers, OCT4 expression has been suggested to contribute to differentiation inhibition, tumorigenicity, and treatment resistance, implying that the ectopic expression of pluripotency maintenance factors may be involved in the acquisition of malignant properties in cancer.
2.1.9. PAX8
PAX8 encodes a transcription factor that regulates early thyroid development and functional differentiation, playing a role in inducing and maintaining specific gene expression in the thyroid. It is primarily expressed in the thyroid, kidneys, ureters, and fallopian tubes of the urogenital system, and is not expressed in the surface epithelium of the ovaries [30].
However, PAX8 expression is frequently observed in epithelial tumors of the ovaries, particularly in serous adenocarcinomas, which represents ectopic expression in cell types that normally do not express PAX8 [31]. This is an example of genes that control differentiation outside their normal differentiation lineage, providing support for the CCC hypothesis.
2.1.10. Twist
The transcription factor, Twist, is involved in embryonic development and cancer and plays a crucial role in cellular morphogenesis and motility acquisition via its ability to induce epithelial-mesenchymal transition (EMT) [32]. While Twist is essential for neural crest cell migration and organogenesis during embryonic development, its ectopic expression has been observed in somatic cells and various tumors, including breast, ovarian, bladder, and gastric cancers [33]. Twist promotes EMT in cancer cells, causing them to lose their epithelial characteristics and acquire invasive and metastatic properties, thereby enhancing their malignant properties [34]. This represents an ectopic reproduction of its original function during embryonic development and is a classic example supporting the CCC hypothesis.
Figure 1 shows how several representative differentiation-controlling genes, including CDX2, PAX8, OCT4, NKX2-1, and VAV1, are ectopically expressed in unrelated tissues and contribute to cancer development via dysregulation of differentiation. These real-world examples exemplify the mechanistic basis of CCC.
In general, while mutations may cause genes to become inactive, constantly ac tivated, or undergo changes in substrate specificity, the emergence of new biological responses within the body is unlikely. The CCC hypothesis assumes that cancer genes created by mutations retain their original functions.
3. Genes with Carcinogenic Potential Exist Even Without Mutations
CDX2 is a homeobox gene normally expressed in intestinal epithelial cells. However, it has been reported to be ectopically expressed in the hematopoietic system because of t(12;13)(p13;q12). CDX2 is expressed in hematopoietic stem cells without genetic mutations and has been shown to induce lethal AML in a mouse model [17]. This is a representative example of “tumor formation due to ectopic expression of normal differentiation-controlling genes,” as assumed by the CCC hypothesis.
VAV1 is an important example of a gene without any mutations that supports the CCC hypothesis. VAV1, a guanine nucleotide exchange factor, is specifically expressed in the hematopoietic system, but is expressed ectopically in pancreatic epithelial tumors (pancreatic ductal adenocarcinoma). VAV1 expressed in these pancreatic cancer cells is wild type, with no structural mutations or abnormalities in the gene sequence.
However, epigenetic abnormalities such as promoter demethylation induce ectopic expression, activating the Rac1–Pak1–NF-κB–Cyclin D1 signaling pathway and contributing to tumor cell proliferation and tumor formation [22]. Thus, the fact that normal differentiation-related genes can promote tumor formation when expressed in cell lines other than their original ones supports the core of the CCC hypothesis, i.e, the “ectopic expression of differentiation-related genes contributes to carcinogenesis while maintaining their original function. The discovery of iPS cells has proven that somatic cells have the potential to differentiate into other cell types without harboring mutations (totipotency of differentiation) [35].
3.1. Four Criteria of the CCC Hypothesis
In embryology, ectopic expression experiments are employed to elucidate two aspects of gene function: the execution of their original functions and the expression of latent functions.In the CCC hypothesis, the first of the four mechanisms includes execution of the original function, while the second includes execution of the potential function. As oncogenes cause cancer via ectopic expression, they perform their original function in ectopically expressing cells and contribute to carcinogenesis, targeting the cytoskeleton, cell adhesion, glycosyltransferase activity, and cell-specific proteins. When these activities conflict with those of cancer-initiating cells, the cytoskeleton is disrupted and becomes atypical, glycosylation is impaired, the activity of cell adhesion molecules is perturbed, leading to invasion, and defective cancer cells are formed.
There are actual examples in which ectopically expressed genes transmit the original information. For example, CDX2 expression in the stomach leads to MUC2 expression and intestinal metaplasia [36]. Other examples include NKX2-1 expression in gastric fundic gland adenocarcinoma (which leads to surfactant expression in the lungs) [19], hepatoid adenocarcinoma of the stomach, and squamous cell carcinoma of the stomach.
Transdifferentiation is a phenomenon via which mature cells with specific functions are transformed into different types of mature cells without reverting to an undifferentiated state [37]. This is often artificially induced by the introduction of specific transcription factors. These manipulations activate the internal switches of cells, enabling them to express the characteristic functions of different cell types. Here, we present three examples, while ten examples are provided in Supplementary Material.
Based on this hypothesis, we propose that ectopic expression of genes regulating differentiation confers new properties to cells, leading to atypia and structural disorganization, which may contribute to carcinogenesis. Relatedly, studies have shown that ectopic expression of PDX1 in the liver induces the conversion of hepatocytes into pancreatic cells [38]. Thus, the fact that ectopic expression of differentiation-controlling genes alters the differentiation fate of cells is considered to be an example supporting the biological validity of this hypothesis.
Liu et al. reported that ectopic expression of MyoD induces the conversion of fibroblasts into myoblasts [39]. This suggests that cellular lineage-determining factors can fundamentally rewrite cellular fate. The ectopic expression of development-related factors in tissues where they are not normally expressed may disrupt normal differentiation and lead to tumor formation, providing another example that supports our CCC hypothesis.
Studies have shown that differentiation-inducing factors, such as Neurog2, when ectopically expressed in astrocytes, forcibly rewrites the cellular differentiation fate [40]. This finding supports the possibility that ectopic expression of development-specific cancer-related genes in somatic cells can lead to the malfunction of differentiation programs and contribute to carcinogenesis. Ectopic hormone-producing tumors, immunoglobulin-expressing cancer cells [41], and various tumor markers (AFP, CEA) are the consequences of ectopically expressed molecules.
Experimentally, c-Maf is a differentiation factor normally expressed in the Th2 lineage; when expressed ectopically, it induces IL-4 production in Th1 and non-lymphoid cells [42]. Carcinogenesis experiments using transgenic mice support this function of c-Maf.
A-myb is typically not expressed in hematopoietic tissues such as the bone marrow. In transgenic mice with ectopic systemic expression of A-myb, hyperplasia of B cells has been observed in the spleen and lymph nodes [43]. Thus, the function of wild type transcription factors in cells outside their normal differentiation lineage can induce tumor-like proliferation.
Rhombotin-2/LMO2 (RBTN-2) is not normally expressed in T cells; however, in transgenic mice with ectopic systemic expression of RBTN-2, only cells of T cell lineage undergo tumorigenesis [44]. CD4⁻ CD8⁻ immature T cells proliferated selectively and underwent clonal expansion, which were eventually followed by tumorigenesis and metastasis. This phenomenon provides evidence supporting the core principle of the CCC hypothesis and acts as an important model demonstrating the potential of ectopic expression of wild type genes in inducing tumor formation.
T cell tumors were selectively induced when LMO-2 (RBTN-2) was ectopically expressed in transgenic mice. This indicates that high LMO-2 expression disrupts T cell differentiation, inhibits the proliferation of CD4⁻ CD8⁻ cells and their differentiation into DP cells, and subsequently ensures progression to leukemia and lymphoma [45]. This finding supports the CCC hypothesis that the ectopic expression of factors that regulate differentiation determines tumor diversity and malignancy.
Studies have reported functions of genes unrelated to their original function; hence, we have added “dysfunction of differentiation” as the third criterion. The transcription factor, PBX1, normally forms heterodimers with tissue-specific partners such as Hox proteins to precisely regulate development and differentiation. However, the E2a-PBX1 fusion protein forms structurally distinct transcription factor complexes and activates gene sets that differ from those normally regulated by PBX1. In experiments in which E2a-PBX1 was introduced into NIH3T3 fibroblasts, the induced genes were found to be different from the original PBX1 targets, exhibiting tissue-or developmental stage-specific expression patterns. These included immune system genes such as those coding Igκ, epithelial differentiation-related factors such as villin, ion channel-related genes, and PDE1A (3',5'-cyclic nucleotide phosphodiesterase), which are not typically expressed in fibroblasts [46].
This suggests that the ectopic expression of differentiation-related genes forms an alternative transcription factor network, leading to dysregulation of differentiation. Fusion proteins, such as E2a-PBX1, not only induce ectopic expression but also erroneous target gene selection, disrupt cellular differentiation programs, and potentially contribute to carcinogenesis. This is an important example supporting the core concept of the CCC hypothesis, which posits that “differentiation program crosstalk (malfunction of differentiation switches)” plays a key role in carcinogenesis.
Based on studies reporting inhibition of differentiation, we have added differentiation inhibition as the fourth criterion. PAX5 is a B cell–specific transcription factor that is not normally expressed in T cells. In a mouse model mimicking human IgH-PAX5 translocation, ectopic PAX5 expression in T cell progenitors caused partial developmental arrest at the DN1 stage. PAX5 represses T-lineage genes and induces B-lineage genes such as CD19, disrupting normal T cell differentiation. This aberrant transcriptional program leads to the development of immature T cell lymphoblastic lymphomas. Mice with higher Pax5 expression at the Ikaros locus developed more aggressive tumors with shorter latencies. These results showed that the ectopic expression of a lineage-specific factor can block differentiation and drive oncogenesis, supporting the CCC hypothesis [47].
3.1.2. Tal1 Is Ectopically Expressed in T Cell Acute Lymphoblastic Leukemia
Ectopically expressedTAL1 retains its original transcriptional regulatory function in cells of muscle differentiation lineage, inducing inhibition of differentiation by suppressing the transcriptional activity of myogenin and E2A. This suggests that TAL1 functions outside its original expression site, disrupts differentiation control, and supports the core of the CCC hypothesis, which posits that “ectopic expression leads to disruption of differentiation control and hence tumor formation” [48]. The four mechanisms via which heterotopic expression of genes control differentiation have been established: 1. expression of original function 2. expression of latent functions; 3. dysfunction of differentiation, and 4. inhibition of differentiation.
4. Implication of the Hypothesis
4.1. Explanation of Atypia
The CCC hypothesis explains the pathological features of cancer cells, known as atypia. Assuming that a gene regulating differentiation is a cell proliferation factor receptor (e.g., erbB), according to the CCC hypothesis, ectopically expressed cell proliferation factor receptors transmit information regarding the cytoskeleton and nucleus to the cancer-initiating cells. The cytoskeletal information transmitted by cell proliferation factor receptors merges with the cytoskeletal information of the cancer precursor cells, leading to changes in cell morphology. Additionally, the nuclear information transmitted by cell proliferation factor receptors merges with the nuclear information of the cancer precursor cells, resulting in nuclear atypia. Regarding structural atypia, in hepatoid adenocarcinoma of the stomach, information from hepatocytes flows into gastric cells, causing them to adopt a hepatocyte-like structure. This explains atypia based on the CCC hypothesis.
4.2. Explanation of Cancer Malignancy
The CCC hypothesis also explains the pathological malignancy of cancers. The higher the degree of cellular atypia, the lower the degree of differentiation, and the faster the cell proliferation rate, the higher the malignancy. In gastric cancer, highly differentiated tumors exhibit higher CDX2 expression and better prognosis. CDX2 expression decreases with reduction in differentiation, worsening prognosis [49]. CDX2 is expressed in the intestinal tract (small and large intestines), the cellular lineage of which is similar to that of the stomach. Low-differentiated cancers express embryonic genes such as NANOG, OCT4, and SOX2, which are associated with poor prognosis [50]. The cellular lineage of NANOG, OCT4, and SOX2 is distant those of gastric cells. In AML, ectopically expressed CDX2 is detected in cells of a lineage distant from its original lineage and may function as an oncogene [51]. The higher the differentiation grade of a tumor, the more is the flow of information from genes with close cellular lineage, resulting in lower atypia and malignancy. Conversely, tumors with lower differentiation grades exhibit greater atypia and higher malignancy due to the flow of information from cells with a distant cellular lineage. According to the CCC hypothesis, heterogeneity is due to the ectopic expression of genes that regulate differentiation. Malignancy is also influenced by cell proliferation rate, which is associated with the constitutive activation of cell proliferation-related genes. The CCC hypothesis assumes that two types of genes are essential for carcinogenesis: genes that confer cell proliferation-related advantages through the constitutive activation of cell proliferation-related genes and genes that regulate differentiation through ectopic expression.
Figure 2 provides a schematic representation of the CCC hypothesis, summarizing how ectopic expression of differentiation-controlling genes leads to carcinogenesis via four distinct mechanisms.
5. Non-Mutated Drivers of Carcinogenesis
In classical cancer genetics, driver genes are defined as those that confer a proliferative advantage to cells via mutations. In contrast, the CCC hypothesis proposes an alternative mechanism: non-mutated driver genes, which retain their wild-type gene sequences but are ectopically expressed in cancer cells. These genes are involved in normal cell differentiation; however, when inappropriately expressed in the wrong cellular context, they provide abnormal structural and differentiational signals to cancer cells, acting as functional drivers. For example, CDX2, essential for intestinal epithelial differentiation, is often ectopically expressed in gastric cancer and is associated with intestinal-type morphology. VAV1 is also expressed ectopically in pancreatic cancer without mutations. Additionally, genes such as NKX2-1 [19], OCT4 [26], PAX8 [30], and TAL1 [52] are expressed ectopically in cancers without mutations and are involved in differentiation traits and phenotypes. These gene clusters also support the concept of “non-mutational drivers” in the CCC hypothesis. The existence of such non-mutational drivers supports the idea that the identity and structure of cancer cells are significantly influenced not only by the mutational state of genes but also by the spatiotemporal control of their expression, i.e., “when and where” they are expressed. Therefore, the CCC hypothesis extends the concept of carcinogenic drivers from mutation-dependent to include the role of spatiotemporal abnormal expression of differentiation-related genes. Non-mutated drivers are activated by epigenetic mechanisms and chromosomal translocations.
6. Pathological Diversity and Progression of Cancer
Even cancers originating in the same organ show diversity in morphology, histological type, and differentiation. For example, in gastric cancer, well-differentiated adenocarcinomas and moderately differentiated adenocarcinomas are common, and these exhibit high expression of CDX2, which is associated with intestinal epithelial differentiation. CDX2 expression declines as differentiation decreases. Instead, genes associated with undifferentiated cell lineages expressed during fetal development, such as NANOG, OCT4, and SOX2, are expressed ectopically.
Furthermore, in hepatoid adenocarcinoma of the stomach, genes associated with the hepatocyte lineage are believed to be expressed ectopically, whereas in squamous cell carcinoma of the stomach, genes associated with squamous cell differentiation are expressed ectopically. According to the CCC hypothesis, this histological diversity is explained by differences in the types of differentiation-regulating genes that are expressed ectopically.
Unlike iPS cells, cancer cells derived from somatic cells are subject to certain constraints in their direction of differentiation. Therefore, the histological types observed in gastric cancer are limited. The following assumptions were made regarding the direction of cancer progression (progression). First, in highly differentiated adenocarcinomas, ectopic expression of genes with relatively close cell lineages, such as CDX2, is more likely to occur. With further genetic and environmental changes, ectopic expression of genes from more distant (undifferentiated) embryonic lineages is gradually added, leading to increased malignancy. In fact, poorly differentiated adenocarcinomas account for about 10% of all gastric cancers; however, in these cases, CDX2 expression is low, while the expression of NANOG, SOX2, and OCT4 is observed. This observation supports the cancer progression model based on the phylogenetic distance of ectopic expression discussed in the CCC hypothesis.
7. Explanation of the CCC Hypothesis Regarding Glycosylation Defect and Invasion/Metastasis
Glycosylation defect in cancer refers to a state in which the structure or expression pattern of glycans present on the surface or within cancer cells differs from that of normal cells. According to the CCC hypothesis, this glycosylation defect arises when the glycan transferase system normally regulated by ectopically expressed differentiation control genes (e.g., erbB) interfere with the glycan transferase system inherent in cancer-initiating cells, leading to abnormal glycan modification. This glycosylation defect disrupts cell-cell adhesion and interactions with the extracellular matrix, reducing cell adhesion and increasing the tendency for invasion. In addition, the CCC hypothesis may explain some molecular mechanisms underlying invasion and metastasis. In cancer cells, factors such as Snail, Slug, Zeb, and Twist, which are involved in EMT during embryonic development, are expressed ectopically, causing cells to acquire mesenchymal-like properties, including motility, invasive ability, and metastatic potential. Details regarding EMT are available in the section discussing Twist.
8. Relationship Between Various Carcinogenesis Theories and the CCC Hypothesis
8.1. Relationship Between the Mutation Theory and the CCC Hypothesis
The mutation theory of carcinogenesis posits that cancer arises from the accumulation of genetic changes (DNA mutations) in cells primarily via two mechanisms: oncogene activation mutations and loss-of-function mutations in tumor suppressor gene. The CCC hypothesis states that among these, the phenomenon of constant activation of cancer genes related to cell proliferation includes activation caused by mutations. However, regarding genes that control differentiation, rather than assuming that mutations lead to the acquisition of new functions, the hypothesis suggests that the expression of these genes in a different differentiation environment due to ectopic expression contributes to carcinogenesis. Therefore, the CCC hypothesis differs from the mutation theory in this regard. In contrast, regarding loss-of-function mutations in tumor suppressor genes, the CCC hypothesis is compatible with examples such as the loss of function of REST, an inhibitor of neuro-specific genes, leading to ectopic expression of the neural differentiation program in non-neural cells. In this way, the CCC hypothesis does not deny the mutation theory of carcinogenesis but rather complements and expands it by adding the perspective of ectopic expression.
8.2. Relationship Between the Multistage Carcinogenesis Hypothesis and CCC Hypothesis
According to the multistage carcinogenesis hypothesis, multiple gene mutations accumulate over time in a stepwise manner before normal cells transform into cancer cells. In particular, it is believed that sequential acquisition of cancer gene mutations and loss of tumor suppressor gene function gradually transform cells, leading to tumor formation. In a model of colorectal cancer, mutations in the APC gene are followed by sequential accumulation of mutations in genes such as KRAS and TP53, leading to progression from adenoma to early-stage cancer and then to advanced cancer. In gastric cancer, intestinal metaplasia caused by ectopic expression of CDX2 marks the preliminary stage [53].
The CCC hypothesis posits that both “constant activation of cell proliferation-related genes” and “ectopic expression of genes that regulate differentiation” are essential for cancer development. In other words, a single gene mutation alone is insufficient for carcinogenesis; rather, malignant transformation progresses via the two-pronged action of dysfunction in the differentiation program and sustained activation of cell proliferation signals. From this perspective, APC/KRAS mutations in colorectal cancer models correspond to constant activation of cell proliferation signals, while ectopic expression of CDX2 in gastric cancer corresponds to malfunction of the differentiation program. Which of these occur first may vary depending on the organ or case. For example, in gastric cancer, there are cases where adenomatous proliferative lesions precede malignant transformation, in which case the sequence is proliferative advantage → differentiation abnormality.
In this way, the CCC hypothesis introduces ectopic expression, which represents a deviation from the differentiation program, as an independent and essential axis within the multistage carcinogenesis framework, thereby complementing and expanding existing multistage models.
8.3. Relationship Between the Epigenetic Hypothesis and CCC Hypothesis
Traditionally, cancer has been understood as a genetic disease caused by changes in the DNA base sequence, i.e., gene mutations. However, recent studies have shown that epigenetic changes, such as DNA methylation and histone modifications, play an important role in the onset and progression of cancer, even without changes in the DNA base sequence. This is the so-called epigenetic theory of carcinogenesis [54].
The CCC hypothesis posits that the essence of cancer diversity and heterogeneity is the ectopic expression of genes that regulate differentiation. This further suggests that epigenetic regulatory abnormalities, including DNA demethylation, are involved as triggers for this ectopic expression. In other words, the CCC hypothesis posits malfunction of the differentiation program due to epigenetic abnormalities as one cause, and it partially incorporates and complements the epigenetic theory.
8.4. Relationship Between the Aberrant Differentiation Hypothesis and CCC Hypothesis
This hypothesis inherits the spirit of the conventional aberrant differentiation hypothesis while refining its concept through the specific molecular mechanism of ectopic expression. This explains the abstract disruption of differentiation previously described in the aberrant differentiation hypothesis from the perspective of the ectopic expression of differentiation-controlling genes, enabling a consistent understanding of the morphological heterogeneity, tissue type diversity, and plasticity of cancer cells.
8.5. Relationship Between the Cancer Stem Cell Hypothesis and CCC Hypothesis
The cancer stem cell hypothesis posits that a small number of cells with self-renewal and multipotent differentiation capabilities, similar to those found in normal tissue stem cells, exist within cancerous tissue and play a central role in the initiation, maintenance, recurrence, and metastasis of cancer [55]. Embryonic transcription factors such as OCT4, NANOG, and SOX2 are known molecular markers of cancer stem cells [56].
According to the CCC hypothesis, these embryonic genes are normally expressed only during embryonic development, and their expression in somatic cells is considered ectopic. Therefore, cancer cells expressing OCT4, NANOG, or SOX2 in an ectopic manner are interpreted as being in a “less differentiated state” under the CCC hypothesis and can be considered candidates for cancer stem cells. Additionally, the CCC hypothesis explains the direction of cancer progression based on “proximity along the cell lineage”. For example, in gastric cancer, well-differentiated adenocarcinomas express the intestinal-type marker, CDX2, whereas poorly differentiated adenocarcinomas express OCT4, NANOG, and SOX2, which are associated with a more undifferentiated cell lineage. If cells ectopically expressing embryonic genes already exist in the early stages of carcinogenesis, they may correspond to cancer stem cells. Even in the advanced stages of carcinogenesis, the expression of OCT4, NANOG, and SOX2 may indicate the presence of cancer stem cell-like cells.
Therefore, the CCC hypothesis does not negate the cancer stem cell theory but rather provides a new perspective on its molecular mechanisms, complementing and expanding our understanding regarding the definition and origin of cancer stem cells.
8.6. Relationship Between the Clonal Evolution Hypothesis and CCC Hypothesis
The CCC hypothesis does not negate the conventional clonal evolution hypothesis [57] but rather adds a new driver, the ectopic expression of differentiation control genes, as part of its evolutionary process.
9. Applications of the CCC Hypothesis
The CCC hypothesis offers a new perspective regarding the understanding of differentiation control and tumor diversity in cancer research and several applications are anticipated. First, it can be used to elucidate unknown differentiation mechanisms. For example, analyzing molecular clusters that are ectopically expressed in hepatoid adenocarcinoma, which occurs in the stomach, may provide clues regarding the mechanisms controlling hepatocyte differentiation, and investigating molecules expressed in an ectopic manner in gastric squamous cell carcinoma may advance our understanding regarding the differentiation mechanisms of the squamous epithelium. These approaches utilize the abnormal state of cancer cells to elucidate the transcriptional control networks and epigenetics underlying normal development and differentiation. Second, analysis of ectopic hormone-producing tumors may advance our understanding regarding the regulatory mechanisms of hormone secretion and endocrine cell differentiation. Clinically, ectopic ACTH-producing tumors are already known; however, reinterpreting these from the perspective of the CCC hypothesis may lead to a more systematic understanding of differentiation abnormalities. Third, the discovery of new molecular markers can be highlighted. Molecular markers are extremely important for cancer diagnosis, prognosis prediction, treatment effect monitoring, and early detection of recurrence. In particular, differentiation-related genes expressed ectopically and their downstream transcription products and secreted products are promising candidates as cancer cell-specific markers. This is expected to lead to a more precise classification of cancer subtypes and promote the development of personalized medicine.
Finally, new molecule-targeted drugs are being developed. Research is already underway on inhibitors targeting NANOG [58], OCT4 [59], SOX2 [60], and other cancer stem cell markers with stem cell properties. Within the framework of the CCC hypothesis, in which these molecules are expressed ectopically, their role as cancer-specific targets caused by differentiation malfunction is becoming clear. The inhibition of these molecules may affect the self-replicative and plastic properties of cancer cells, thereby opening new avenues for therapeutic strategies.
Thus, the CCC hypothesis is not limited to understanding the mechanisms of cancer development but is a theoretical framework applicable to multiple fields, including diagnostics, pharmacology, and regenerative medicine, and its development in the future is highly anticipated.
10. Differences Between Benign and Malignant Tumors and the Inevitability of Carcinogenesis in Multicellular Organisms
The CCC hypothesis explains the fundamental difference between benign and malignant tumors in terms of the regulatory state of genes involved in cell proliferation and differentiation. Benign tumors are characterized by low cellular atypia, slow proliferation, and the ability to invade surrounding tissues without infiltration or metastasis. This possibly occurs because genes that promote cell proliferation are constantly activated but restricted to their normal expression sites, thereby maintaining the order of differentiation; in other words, the cellular differentiation program remains intact and the tissue's identity is preserved. In addition, in malignant tumors, genes that regulate differentiation are expressed ectopically, disrupting the direction of cellular differentiation and structural characteristics, leading to the emergence of malignant features, such as atypia, invasiveness, and metastasis, according to the CCC hypothesis.
Malfunctional differentiation disrupts the morphological and functional unity of cells, resulting in the diversity and plasticity of cancer cells. Furthermore, the CCC hypothesis explains why multicellular organisms develop cancer. Multicellular organisms possess precise differentiation programs and diverse gene clusters that control differentiation such that individual cells can perform different functions. This is because the ectopic expression and malfunction of such a control system can lead to the pathological state of cancer. In other words, the occurrence of cancer can be positioned as a phenomenon that exists with a certain inevitability as the reverse side of the “mechanism of differentiation” unique to multicellular organisms.
11. Metastasis and Organ Specificity
The CCC hypothesis may also provide insights into organ specificity of metastasis. In particular, differentiation-related genes that are ectopically expressed in cancer cells may influence the selection of metastatic sites on the basis of their affinity for the organ environment in which the gene is normally functional. For example, lung cancer with ectopic expression of ALK, which is normally expressed in the nervous system, is more likely to metastasize to the brain [61], while breast cancer expressing Runx2, which is involved in bone differentiation, is more likely to metastasize to bone [62] and hepatoid adenocarcinoma of the stomac expressing hepatocyte-related genes will possibly metastasize to the liver [63]. These findings do not prove causality and are based solely on observational facts or trends; however, they suggest that the pattern of ectopic expression may be involved in the organ specificity of metastasis and that the CCC hypothesis does not fully explain the molecular mechanisms of metastasis but may provide a theoretical framework offering insights into the selectivity of metastasis sites.
12. Methods for Verifying the CCC Hypothesis
The following experimental approaches can be considered to verify the CCC hypothesis. First, when a cancer gene (e.g., ALK, PAX8, and CDX2) was ectopically expressed, we verified whether the wild type protein retained the same function as in its originally expressing cells from the perspective of molecular function (e.g., transcription activity, signal transduction, and cell morphological changes). Next, we compared and analyzed the downstream gene clusters induced by ectopic expression with those induced in cells where they are normally expressed using RNA-seq or similar methods and used transgenic mouse models with ectopic expression of differentiation-related genes to observe their carcinogenicity (tumor formation, histology, and atypia). These approaches may provide evidence supporting the validity of the CCC hypothesis, which proposes that the ectopic expression of genes with preserved original functions can lead to carcinogenesis.
13. Relationship Between Cancer Plasticity and the CCC Hypothesis
Cancer plasticity refers to the ability of cancer cells to flexibly change their morphology, function, and differentiation. These include EMT, mesenchymal-epithelial transition (MET), and acquisition of stem cell-like properties and drug resistance. [64]
According to the CCC hypothesis, ectopic expression of genes that regulate differentiation is assumed to be one of the main mechanisms underlying cancer plasticity. Typically, differentiation-controlling genes are strictly regulated during development and organ-specific differentiation stages; however, in cancer cells, the ectopic expression of these genes leads to dysregulation of differentiation. For example, embryonic stem cell-related genes such as OCT4, NANOG, and SOX2, as well as genes regulating EMT such as TWIST, SNAIL, and ZEB, are well known to be ectopically expressed in somatic cancers. This allows cancer cells to acquire the ability to freely alter their differentiation state and gain plasticity. This phenomenon is believed to be involved in determining the properties of cancer stem cells, treatment resistance, metastasis site selection, and formation of pathological diversity. The CCC hypothesis posits that the plasticity of cancer is not merely due to mutations or epigenetic changes but rather the disruption of cellular identity caused by the ectopic expression of genes that regulate differentiation.
14. Repositioning Tumor Suppressor Genes in the CCC Hypothesis
Traditionally, tumor suppressor genes have been identified as passive regulators of cell proliferation. The CCC hypothesis redefines their roles, especially those involved in lineage repression, as active guardians of cellular identity. For example, the loss of REST leads to the ectopic expression of neuronal genes in non-neural cancers, contributing to cellular transformation and malignancy. Rather than being mere loss-of-function events, such disruptions promote inappropriate differentiation programs, which are key drivers in the CCC model. Thus, tumor suppressor genes play a dual role in preventing overgrowth and lineage misexpression, making them central players in carcinogenesis via ectopic gene expression.
15. Conclusion
In this study, we propose a new hypothesis, the CCC hypothesis, regarding the role of ectopic expression of differentiation-controlling genes in cancer development. The CCC hypothesis provides a unified framework explaining complex pathological phenomena, such as cancer diversity, atypia, invasion, metastasis, and plasticity, via the disruption of cellular differentiation programs caused by ectopic expression. In particular, this hypothesis explains the phenomenon that more than 200 diverse cancer-related genes, while retaining their original functions, exhibit ectopic expression and lead to the common outcome of cancer initiation and progression. This provides a new perspective that the essence of cancer lies not in simple gene mutations or acquisition of new functions but in the disruption of cellular programs due to errors in differentiation. Future studies are required to establish a causal relationship between the ectopic expression of differentiation-controlling genes and carcinogenesis. The CCC hypothesis has the potential to act as a new theoretical foundation for advancing our understanding of cancer and for redefining diagnostic and therapeutic strategies. The occurrence of cancer can be positioned as a phenomenon that exists with a certain inevitability as the reverse side of the mechanism underlying differentiation unique to multicellular organisms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1: List of cancer-related genes with ectopic expression patterns supporting the CCC hypothesis.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors acknowledge the use of ChatGPT (OpenAI, GPT-4) for improving the clarity and structure of the language of the manuscript. The final content was reviewed and edited by the authors to ensure accuracy and originality. The author gratefully acknowledges Editage (Cactus Communications) for English language editing.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CCC | Carcinogenic Chimera Cell |
| EMT | Epithelial–Mesenchymal Transition |
| CSC | Cancer Stem Cell |
| HPA | Human Protein Atlas |
| EGFR | Epidermal Growth Factor Receptor |
| AML | Acute Myeloid Leukemia |
| T-ALL | T-cell Acute Lymphoblastic Leukemia |
| iPS | Induced Pluripotent Stem Cell |
| MET | Mesenchymal–Epithelial Transition |
| DN | Double Negative (T cell developmental stage) |
| DP | Double Positive (T cell developmental stage) |
References
- Hida, T.; Ueda, R.; Sekido, Y.; Hibi, K.; Matsuda, R.; Ariyoshi, Y.; Sugiura, T.; Takahashi, T.; Takahashi, T. Ectopic expression of c-kit in small-cell lung cancer. Int. J. Cancer Suppl. 1994, 8, 108–109. [Google Scholar] [CrossRef]
- Graham, D.K.; Salzberg, D.B.; Kurtzberg, J.; Sather, S.; Matsushima, G.K.; Keating, A.K.; Liang, X.; Lovell, M.A.; Williams, S.A.; Dawson, T.L.; Schell, L.J.; Anwar, A.A.; Snodgrass, H.R.; Earp, H.S. Ectopic expression of the proto-oncogene Mer in pediatric T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2006, 12, 2662–269. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hatano, M.; Iitsuka, Y.; Mahyar, N.S.; Yamamoto, M.; Tokuhisa, T. Two forms of Hox11 a T cell leukemia oncogene, are expressed in fetal spleen but not in primary lymphocytes. Mol. Immunol. 1995, 32, 1177–1182. [Google Scholar] [CrossRef]
- Choi, K.Y.; Chang, K.; Pickel, J.M.; Badger, J.D. 2nd; Roche, K.W. 2nd; Roche, K.W. Expression of metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc. Natl. Acad. Sci. USA. 2011, 108, 15219–15224. [Google Scholar] [CrossRef] [PubMed]
- Keilholz, U.; Menssen, H.D.; Gaiger, A.; Menke, A.; Oji, Y.; Oka, Y. Scheibenbogen, C.; Stauss, H.; Thiel, E.; Sugiyama, H. Wilms tumor gene 1 (WT1) in human neoplasia. Leukemia. 2005, 19, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Toyoshima, K. The ErbB-related proto-oncogene genes encoding growth factor receptors. Gan To Kagaku Ryoho. 1987, 14, 2075–2082. [Google Scholar] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- The Human Protein Atlas. ALK [Internet]. Available from: https://www.proteinatlas.org/ENSG00000171094-ALK.
- Tsuyama, N.; Sakamoto, K.; Sakata, S.; et al. . Anaplastic large cell lymphoma: pathology,genetics, and clinical aspects. J. Clin. Exp. Hematop. 2017, 57, 120―142. [Google Scholar] [CrossRef]
- Du, X.; Shao, Y.; Qin, H-F.; Tai, Y-H.; Gao, H-J. ALK rearrangements in non-small-cell lung cancer (NSCLC). Transl. Lung Cancer Res. 2018, 9, 423–430. [Google Scholar] [CrossRef]
- The Human Protein Atlas. CDX2 [Internet]. Available from: https://www.proteinatlas.org/ENSG00000165556-CDX2.
- Mutoh, H.; Hakamata, Y.; Sato, K.; Eda, A.; Yanaka, I.; Honda, S.; Osawa, H.; Kaneko, Y.; Sugano, K. Conversion of gastric mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice. Biochem. Biophys. Res. Commun. 2002, 294, 470–479. [Google Scholar] [CrossRef]
- Mutoh, H.; Sakurai, S.; Satoh, K.; Tamada, K.; Kita, H.; Osawa, H.; Tomiyama, T.; Sato, Y.; Yamamoto, H.; Isoda, N.; Yoshida, T.; Ido, K.; Sugano, K. Development of gastric carcinoma from intestinal metaplasia in Cdx2-transgenic mice. Cancer Res. 2004, 64, 7740–7747. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Gastric adenocarcinomas with CDX2 induction show a higher frequency of TP53 and KMT2B mutations and MYC amplification but similar survival compared to cancers without CDX2 induction. J. Clin. Med. 2024, 13, 7635. [Google Scholar] [CrossRef] [PubMed]
- Khayyat, A.; Esmaeil Pour, M.A.; Poursina, O.; Zohouri, S.A.; Jian, P.V.; Patel, N.; Amin, A. Evaluation of the biomarkers CDX1 and CDX2 in gastric cancer prognosis: A meta-analysis. Int. J. Mol. Cell Med. 2024, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Scholl, C.; Bansal, D.; Döhner, K.; Eiwen, K.; Huntly, B.J.P.; Lee, B.H.; Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Döhner, H.; Gilliland, D.G.; Fröhling, S. The homeobox gene, CDX2, is aberrantly expressed in most AML cases of acute myeloid leukemia and promotes leukemogenesis. J. Clin. Invest. 2007, 117, 1037–1048. [Google Scholar] [CrossRef]
- Rawat, V.P.S.; Cusan, M.; Deshpande, A.; Hiddemann, W.; Quintanilla-Martinez, L.; Humphries, R.K.; Bohlander, S.K.; Feuring-Buske, M.; Buske, C. Ectopic expression of the homeobox gene Cdx2 is the transforming event in a mouse model of t(12;13)(p13;q12) acute myeloid leukemia. Proc. Natl. Acad. Sci. USA. 2004, 101, 817–822. [Google Scholar] [CrossRef]
- The Human Protein Atlas. NKX2-1 [Internet]. Available from: https://www.proteinatlas.org/ENSG00000136352-NKX2-1.
- Fukagawa, K.; Takahashi, Y.; Yamamichi, N.; Kageyama-Yahara, N.; Sakaguchi, Y.; Obata, M.; Cho, R.; Sakuma, N.; Nagao, S.; Miura, Y.; Tamura, N.; Ohki, D.; Mizutani, H.; Yakabi, S.; Minatsuki, C.; Niimi, K.; Tsuji, Y.; Yamamichi, M.; Shigi, N.; Tomida, S.; Abe, H.; Ushiku, T.; Koike, K.; Fujishiro, M. Transcriptome analysis has revealed the essential role of NK2 homeobox 1/thyroid transcription factor 1 (NKX2-1/TTF-1) in fundic gland-type gastric adenocarcinoma. Gastric Cancer. 2023, 26, 44–54. [Google Scholar] [CrossRef]
- Homminga, I.; Pieters, R.; Langerak, A.W.; de Rooi, J.J.; Stubbs, A.; Verstegen, M.; Vuerhard, M.; Buijs-Gladdiness, J.; Kooi, C.; Klous, P.; van Vlierberghe, V.; Ferrando, A.A.; Cayuela, J.M.; Verhaaf, B.; Berna Beverloo, H.; Horstmann, M.; de Haas, V.; Weikmeijer, A.-S.; Pike-Overzet, K.; Staal, F.J.T.; de Laat, W.; Soulier, J.; Sigaux, F.; Meijerink, J.P.P. Integrated transcript and genome analyses have revealed that NKX2-1 and MEF2C are potential oncogenes in T-cell acute lymphoblastic leukemia. Cancer Cell. 2011, 19, 484–497. [Google Scholar] [CrossRef]
- The Human Protein Atlas. VAV1 [Internet]. Available from: https://www.proteinatlas.org/ENSG00000141968-VAV1.
- Fernandez-Zapico, M.E.; Gonzalez-Paz, N.C.; Weiss, E.; Savoy, D.N.; Molina, J.R.; Fonseca, R.; Smyrk, T.C.; Chari, S.T.; Urrutia, R.; Billadeau, B.B. The ectopic expression of VAV1 plays an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005, 7, 39–49. [Google Scholar] [CrossRef]
- Coulson, J.M. Transcriptional regulation: Cancer, neurons, and REST. Review Curr. Biol. 2005, 15, R665–R668. [Google Scholar] [CrossRef]
- The Human Protein Atlas. SHH [Internet]. Available from: https://www.proteinatlas.org/ENSG00000164690-SHH.
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Lee, S.; Kwon, H.Y. Hedgehog signaling in cancer: A prospective therapeutic target for eradicating cancer stem cells. Cells. 2018, 7, 208. [Google Scholar] [CrossRef]
- The Human Protein Atlas. POU5F1 (OCT4 [Internet]. Available at: https://www.proteinatlas.org/ENSG00000204531-POU5F1.
- von Eyben, F.E.; Kristiansen, K.; Kapp, D.S.; Hu, R.; Preda, O.; Nogales, F.F. Epigenetic regulation of driver genes involved in testicular tumorigenesis. Int. J. Mol. Sci. 2023, 24, 4148. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Bujanda, L.; Billadeau, D.D.; Zhang, J.-S. Embryonic stem cell factors and pancreatic cancer. World J. Gastroenterol. 2014, 20, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Pádua, D.; Figueira, P.; Ribeiro, I.; Almeida, R.; Mesquita, P. Relevance of transcription factors in gastric and colorectal cancer stem cell identification and eradication. Front. Cell Dev. Biol. 2020, 8, 442. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. PAX8 [Internet]. Available from: https://www.proteinatlas.org/ENSG00000125618-PAX8.
- Di Palma, T.; Zannini, M. PAX8 as a potential target for ovarian cancer: what we know so far. Onco Targets Ther. 2022, 15, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Shin, D.M. Multiple biological functions of Twist1 in various cancers. Oncotarget. 2017, 8, 20380–20393. [Google Scholar] [CrossRef]
- The Human Protein Atlas. PAX8 [Internet]. Available from: https://www.proteinatlas.org/ENSG00000125618-PAX8. [CrossRef]
- Takahashi,K.;Yamanaka,S.;Induction of pluripotent stem cells from mouse embryonic and adult fibroblast culyures by defined factors. Cell. 2006, 126, 663-676. [CrossRef]
- Silberg, D.G.; Sullivan, J.; Kang, E.; Swain, G.P.; Moffett, J.; Sund, N.J.; Sackett, S.D.; Kaestner, K.H. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology. 2002, 122, 689–696. [Google Scholar] [CrossRef]
- Graf, T. Historical origins of transdifferentiation and reprogramming. Cell Stem Cell. 2011, 9504–516. [Google Scholar] [CrossRef]
- Horb, M.E.; Shen, C.N.; Tosh, D.; Slack, J.M. Experimental conversion of liver to pancreas. Curr. Biol. 2003, 13, 105–115. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, H.; Li, Y.; Zheng, S.G. Experimental studies on the differentiation of fibroblasts into myoblasts induced by MyoD genes in vitro. Int. J. Biomed. Sci. 2008, 4, 14–19. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Y.; Chen, F.; Yuan, J.; Li, S.; Han, S.; Lu, D.; Geng, J.; Rao, Z.; Sun, L.; Xu, J.; Shi, Y.; Wang, X.; Liu, Y. Neurog2 directly converts astrocytes into functional neurons in midbrain and spinal cord. Cell Death Dis. 2021, 12, 225. [Google Scholar] [CrossRef]
- Cui, M.; Huang, J.; Zhang, S.; Liu, Q.; Liao, Q.; Qiu, X. Immunoglobulin expression in cancer cells and its critical roles in tumorigenesis. Front. Immunol. 2021, 12, 613530. [Google Scholar] [CrossRef]
- Ho, I.C.; Hodge, M.R.; Rooney, J.W.; Glimcher, L.H. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996, 85, 973–983. [Google Scholar] [CrossRef] [PubMed]
- DeRocco, S.E.; Iozzo, R.; Ma, X.P.; Schwarting, R.; Peterson, D.; Calabretta, B. Ectopic expression of A-myb in transgenic mice causes follicular hyperplasia and enhanced B lymphocyte proliferation. Proc. Natl. Acad. Sci. USA. 1997, 94, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Neale, G.A.; Rehg, J.E.; Goorha, R.M. Ectopic expression of rhombotin-2 causes selective expansion of CD4-CD8- lymphocytes in the thymus and T-cell tumors in transgenic mice. Blood. 1995, 86, 3060–3071. [Google Scholar] [CrossRef]
- Neale, G.A.; Rehg, J.E.; Goorha, R.M. Disruption of T-cell differentiation precedes T-cell tumor formation in LMO-2 (rhombotin-2) transgenic mice. Leukemia. 1997, 11 Suppl 3, 289–290. [Google Scholar]
- Fu, X.; Kamps, M.P. E2a-Pbx1 induces aberrant expression of tissue-specific and developmentally regulated genes when expressed in NIH 3T3 fibroblasts. Mol. Cell Biol. 1997, 17, 1503–1512. [Google Scholar] [CrossRef]
- Souabni, A.; Jochum, W.; Busslinger, M. Oncogenic role of Pax5 in the T-lymphoid lineage upon ectopic expression from the immunoglobulin heavy-chain locus. Blood. 2007, 109, 281–289. [Google Scholar] [CrossRef]
- Hofmann, T.J.; Cole, M.D. The TAL1/Scl basic helix-loop-helix protein blocks myogenic differentiation and E-box dependent transactivation. Oncogene. 1996, 13, 617–624. [Google Scholar]
- Wang, X.T.; Wei, W.Y.; Kong, F.B.; Lian, C.; Luo, W.; Xiao, Q.; Xie, Y-B. Prognostic significance of Cdx2 immunohistochemical expression in gastric cancer: a meta-analysis of published literatures. J. Exp. Clin. Cancer Res. 2012, 31, 98. [Google Scholar] [CrossRef]
- Basati, G.; Mohammadpour, H.; Razavi, A.E. Association of high expression levels of SOX2, NANOG, and OCT4 in gastric cancer tumor tissues with progression and poor prognosis. J. Gastrointest. Cancer. 2020, 51, 41–47. [Google Scholar] [CrossRef]
- Scholl, C.; Bansal, D.; Döhner, K.; Eiwen, K.; Huntly, B.J.P.; Lee, B.H.; Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Dohner, H.; Gilliland, D.G.; Frohling, S. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J. Clin. Invest. 2007, 117, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.K.; Zhang, C.; Sanda, T. Oncogenic transcriptional program driven by TAL1 in T-cell acute lymphoblastic leukemia. Int. J. Hematol. 2019, 109, 5–17. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell. 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev.Cancer. 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N.Engl.J.Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R; Dirks, P.; Eaves, C.J. Cancer stem cells:an evolving concept. Nat. Rev. Cancer. 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Swanton, C.; Bernard, E.; Abbosh, C.; André, F.; Auwerx, J.; Balmain, A.; Bar-Sagi, D.; Bernards, R.; Bullman, S.; DeGregori, J.; Elliott, C.; Erez, A.; Evan, G.; Febbraio, M.A.; Hidalgo, A.; Jamal-Hanjani, M.; Joyce, JA.; Kaiser, M.; Lamia, K.; Locasale, J.W.; Loi, S.; Malanchi, I.; Merad, M.; Musgrave, K.; Patel, K.J.; Quezada, S.; Wargo, J.A.; Weeraratna, A.; White, E.; Winkler, F.; Wood, J.N.; Vousden, K.H.; Hanahan, D. Embracing cancer complexity:Hallmarks of systemic disease. Cell. 2024, 187, 1589–1616. [Google Scholar] [CrossRef]
- Wang, M.L.; Chiou, S.H.; Wu, C.W. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013, 6, 1207–1220. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. The role and specific mechanism of OCT4 in cancer stem cells: a review. Int. J. Stem Cells. 2020, 13, 312–325. [Google Scholar] [CrossRef]
- Swain, N.; Thakur, M.; Pathak, J.; Swain, B. SOX2, OCT4 and NANOG: The core embryonic stem cell pluripotency regulators in oral carcinogenesis. J. Oral Maxillofac. Pathol. 2020, 24, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.A.; Wang, N. Targeting lung cancer brain metastases: a narrative review of emerging insights for anaplastic lymphoma kinase (ALK)-positive disease. Transl. Lung Cancer Res. 2023, 12, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Lu, J.T.; Tan, C.C.; Wang, Q.S.; Feng, Y.M. RUNX2 promotes breast cancer bone metastasis by increasing integrin α5-mediated colonization. Cancer Lett. 2016, 380, 78–86. [Google Scholar] [CrossRef]
- Nagai, E.; Ueyama, T.; Yao, T.; Tsuneyoshi, M. Hepatoid adenocarcinoma of the stomach. A clinicopathologic and immunohistochemical analysis. Cancer. 1993, 72, 1827–1835. [Google Scholar] [CrossRef] [PubMed]
- Laisne,M,;lupien,M,;vallot.C,;Epigenomic heterogeneity as a source of tumour evolution.Nat.Rev.cancer. 2025, 25, 7-26. [CrossRef]
Figure 1.
Representative examples of ectopic expression of differentiation-controlling genes (e.g., CDX2, PAX8, OCT4, NKX2-1, VAV1) in cancer. Each gene is normally expressed in lineage-specific tissues, but when aberrantly expressed in unrelated tissues, it may induce dysdifferentiation or block normal differentiation, contributing to carcinogenesis, as postulated in the CCC hypothesis.
Figure 1.
Representative examples of ectopic expression of differentiation-controlling genes (e.g., CDX2, PAX8, OCT4, NKX2-1, VAV1) in cancer. Each gene is normally expressed in lineage-specific tissues, but when aberrantly expressed in unrelated tissues, it may induce dysdifferentiation or block normal differentiation, contributing to carcinogenesis, as postulated in the CCC hypothesis.
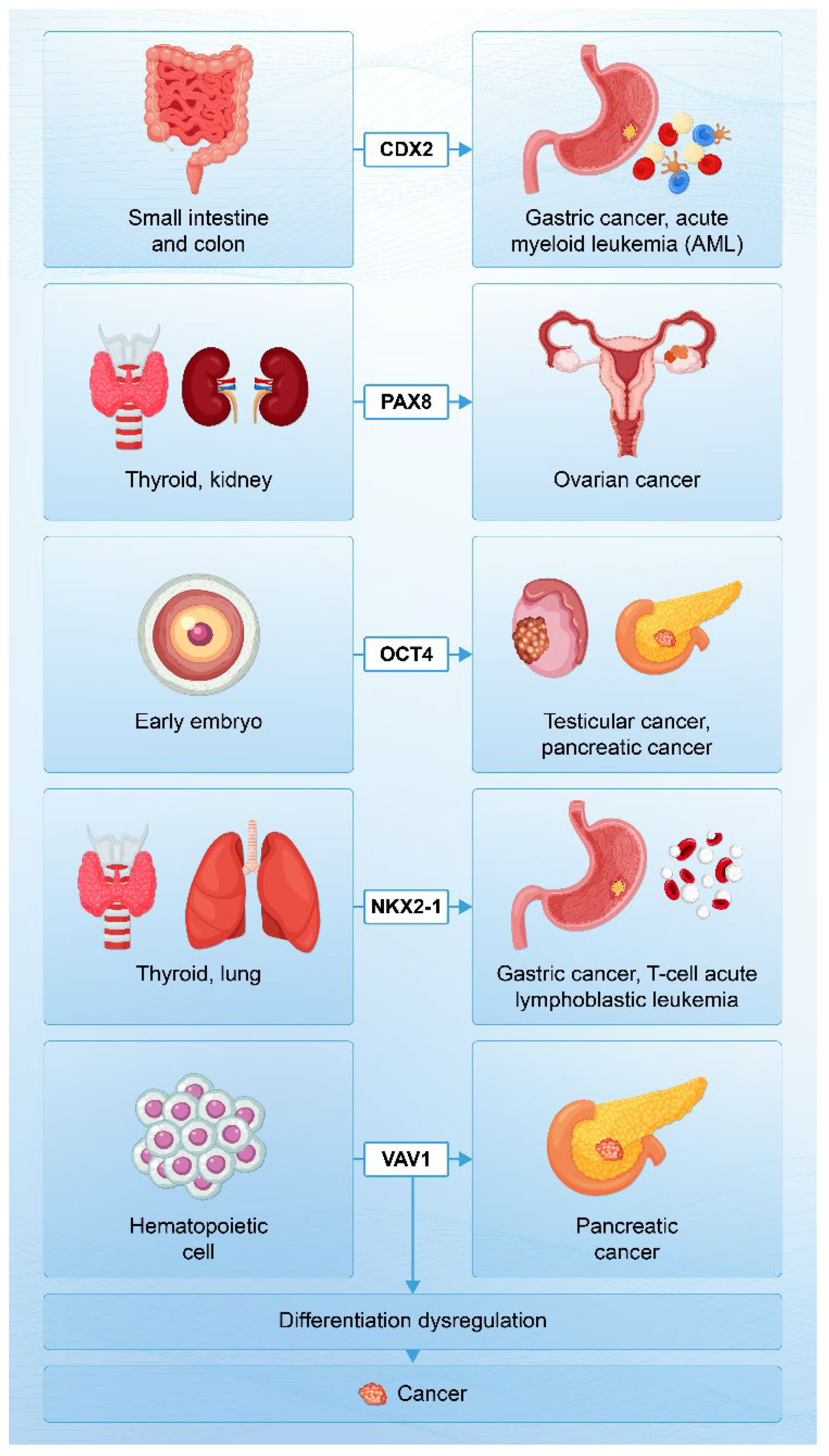
Figure 2.
Two pathways to cancer: Ectopic expression of differentiation-controlling genes and constitutive activation of proliferation-related genes. The diagram illustrates the interaction between differentiation-controlling genes (e.g., transcription factors, embryonic genes) and proliferation-related genes. Ectopic expression of differentiation-controlling genes in non-native cellular contexts leads to the expression of original or latent functions, dysregulation of differentiation, or inhibition of differentiation. In parallel, constitutive activation of proliferation-related genes enhances growth potential. These two processes result in atypia, invasion (including epithelial-mesenchymal transition), glycosylation defects, and tissue-type diversity on one hand and permanent proliferation on the other. Together, they synergistically contribute to malignant transformation, as postulated by the CCC hypothesis.
Figure 2.
Two pathways to cancer: Ectopic expression of differentiation-controlling genes and constitutive activation of proliferation-related genes. The diagram illustrates the interaction between differentiation-controlling genes (e.g., transcription factors, embryonic genes) and proliferation-related genes. Ectopic expression of differentiation-controlling genes in non-native cellular contexts leads to the expression of original or latent functions, dysregulation of differentiation, or inhibition of differentiation. In parallel, constitutive activation of proliferation-related genes enhances growth potential. These two processes result in atypia, invasion (including epithelial-mesenchymal transition), glycosylation defects, and tissue-type diversity on one hand and permanent proliferation on the other. Together, they synergistically contribute to malignant transformation, as postulated by the CCC hypothesis.
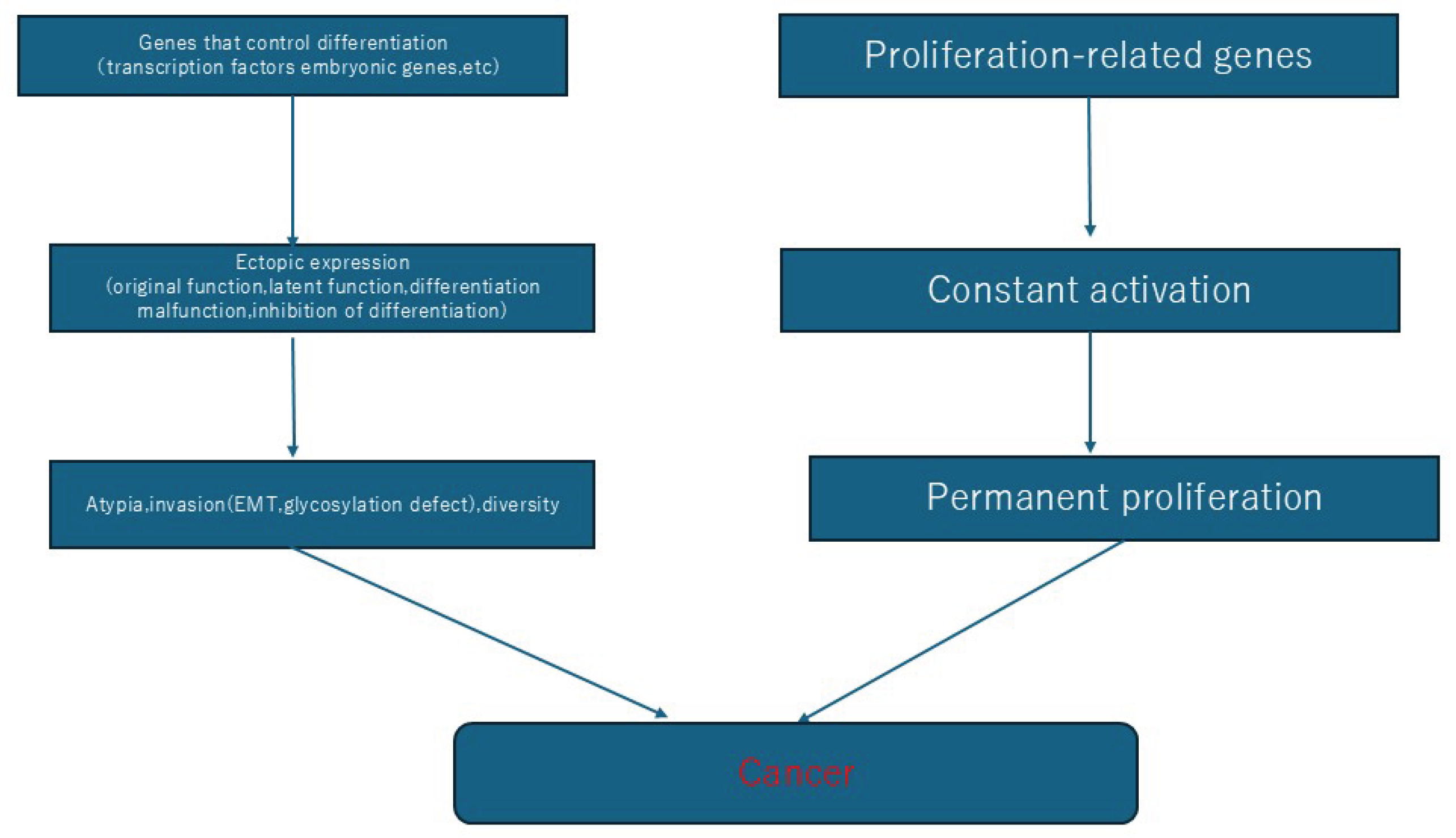

Table 1.
Representative Genes Exhibiting Ectopic Expression in Cancers and Supporting the Carcinogenic Chimera Cell (CCC) Hypothesis.
Table 1.
Representative Genes Exhibiting Ectopic Expression in Cancers and Supporting the Carcinogenic Chimera Cell (CCC) Hypothesis.
| Gene name | Normal expression site | Related cancers | Notes |
| erbB | Epithelial |
Erythroblastosis |
Not expressed in erythroblasts |
| ALK | Specific neurons in the brain and nervous system during development |
Anaplastic large cell lymphoma, small cell lung carcinoma | Small cell lung carcinoma expressing ALK are prone to metastasizing to the brain. |
| CDX2 | Small intestine, colon |
Gastric cancer, acute myeloid leukemia |
Not expressed in bone marrow and gastric mucosa |
| NKX2-1 | Thyroid, lung |
Gastric adenocarcinoma of fundic gland, T-cell acute lymphoblastic leukemia | Gastric adenocarcinoma of fundic gland also induces lung-specific surfactant proteins. |
| VAV1 | Expressed in hematopoietic cells |
Pancreatic cancer |
Expressed as wild type due to low methylation |
| REST | A transcription repressor that suppresses the expression of neuro-specific genes in non-neural cells |
Small cell lung carcinoma | Tumor suppressor gene |
| Shh | Expressed during embryogenesis |
Basal cell carcinoma, pancreatic cancer, lung cancer |
Not expressed in somatic cells |
| OCT4 | Embryonic |
Testicular germ cell tumors, pancreatic cancer | Not expressed in somatic cells |
| PAX8 | Thyroid, kidney, ureter, etc. | Ovarian cancer | Not expressed in the surface epithelium of the ovary |
| Twist | Embryonic | Breast cancer, ovarian cancer, prostate cancer, pancreatic cancer, etc. | Induces epithelial-mesenchymal transition |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



