Submitted:
24 August 2025
Posted:
26 August 2025
You are already at the latest version
Abstract
The rising global prevalence of inflammatory bowel diseases (IBD), including Crohn’s disease and ulcerative colitis, is paralleled by an increased risk of colitis-associated colorectal cancer (CAC). Persistent intestinal inflammation promotes genetic instability and epigenetic reprogramming within epithelial and immune cells, driving the multistep transition from inflammation to neoplasia. This review integrates findings from preclinical animal models and RNA-seq data to dissect the molecular events underpinning CAC, a subset of colorectal cancer (CRC) driven by chronic inflammation. We highlight how pro-inflammatory cytokines (e.g., TNF-α, IL-6), oxidative stress, and microbial dysbiosis converge on key transcriptional regulators such as NF-κB and STAT3, inducing DNA methylation and histone modifications (e.g., H3K27me3), altering chromatin dynamics, gene expression, and non-coding RNA networks (e.g., miR-21, MALAT1, CRNDE), ultimately reshaping pathways involved in proliferation, apoptosis, and immune evasion. This review updates new potential associations of entities with these diseases, in their networks of interaction, summarizing major aspects of genetic and chromatin-level regulatory mechanisms in IBD and CRC, and emphasizing how these interactions drive the inflammatory-to-neoplastic transition. By underscoring the reversibility of epigenetic changes, we explore their translational potential in early detection, surveillance, and precision epigenetic therapy. Understanding the interplay between genetic mutations and chromatin remodeling provides a roadmap for improving diagnostics and personalized treatments in IBD-associated colorectal carcinogenesis.
Keywords:
inflammatory bowel disease (IBD)
; colorectal cancer (CRC)
; colitis-associated colorectal cancer (CAC)
; autophagy dysfunction
; epigenetic
; DNA methylation
; histone modifications
; noncoding RNAs (ncRNA)
; pro-inflammatory cytokines
; translational biomarkers
1. Introduction
1.1. The Impact of Chronic Inflammation in Carcinogenesis
Inflammation is a physiological process initiated by the immune system following pathogenic and inflammatory cytokine stimulation and is crucial for host protection from invasive pathogens [1,2,3]. It is a beneficial immune defense response to curtail pathogenic infection and tissue damage [4]. It is the immediate response of host tissues and cells to pathogens, harmful stimuli (e.g., chemicals, toxins, other environmental factors, etc.) or physical damage. The innate immune system responds rapidly to inflammation and under normal circumstances inflammation quickly ends after the clearance of infection and injurious agents. There is precise control of the complex networks of inflammatory pathways to limit tissue damage during inflammation, while continued activation of the immune system can lead to inflammatory dysregulation [3]. Though inflammation is a necessary immune defense response against infection and tissue damage, it depends on the balance and crosstalk between pro- and anti- inflammatory [4] immune signals (cytokines and chemokines) to produce a beneficial non-tumorigenic outcome to the host [5]. Prolonged activation of inflammatory signaling results in chronic inflammation [3]. Growing evidence suggests a close link between inflammation and many chronic health conditions, including autoimmune diseases (such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA)), neurodegenerative diseases (ND), viral infections (such as coronavirus 2019 (COVID-19)) and cancer [3].
With respect to cancer, prolonged activation of inflammatory signaling results in chronic inflammation that can induce malignant cellular transformation [4]. Chronic inflammation can be tumor-promoting and is considered a hallmark of cancer. It plays a crucial role in tumor initiation, malignant transformation, invasion and metastasis [6,7,8]. Chronic inflammation predisposes patients to the development of cancer and facilitates practically all stages of carcinogenesis. The cytokines and chemokines produced by immune cells have both pro-tumor and anti-tumor roles and their complex interaction determines the fate of carcinogenesis [8,9]. These pro-inflammatory cytokines produced as an outcome of the inflammatory processes lead to immunosuppression, promotion of angiogenesis, invasion and metastasis [10].
Chronic inflammation provokes alterations and/or dysregulation or dysfunction of molecular events leading to aberrant or altered signaling pathways. More specifically, chronic inflammation can provoke or induce the accumulation of mutations, aberrant changes in cellular, genetic and epigenetic processes leading to aberrant alterations in signaling events favoring neoplastic transformation and tumor initiation. Altered signaling involves the inactivation and activation of tumor suppression and oncogenic pathways, respectively [11]. Proliferation and survival of cancerous cells will be continuously enhanced by the formation of a tumor microenvironment (TME) that is a complex ecosystem of carcinoma-associated immune cells, signaling molecules, fibroblasts, blood cells (angiogenesis), extracellular matrix components, and other factors. Crosstalk between factors of this microenvironment can lead to the survival and growth of the malignant phenotype and the progression of the stages in the process of carcinogenesis, i.e., cancer initiation, promotion and progression at the expense of normal cells and tissues [5].
1.2. Inflammatory Bowel Disease
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), comprises chronic, relapsing inflammatory disorders of the gastrointestinal tract. They are remitting disorders which usually result in repeated abdominal pain, diarrhea, bloody purulent stool and weight loss. These disorders subsequently reduce the quality of life and increase the economic burden of IBD patients. The pathogenesis of IBD remains incompletely understood but current data support the hypothesis that IBD is the result of a complex interplay of genetic predisposition, environmental factors and aberrant immune responses, such as an inappropriate gut mucosal response towards the constituents of the gut microbiota which cross an impaired epithelial barrier [12,13]. The intestinal epithelium is a highly dynamic tissue whose functional integrity is indispensable for proper gut homeostasis. The intestinal epithelium lines the inner walls of the Gastrointestinal (GI) tract and establishes the first line of defense from potential pathogens. A dysfunctional intestinal epithelium barrier can lead to severe dysregulation of gut homeostasis and allows microbial antigens to cross the barrier membrane, triggering inappropriate immune activation. This dysregulation, combined with gut microbiota dysbiosis, contributes to the chronic inflammatory state characteristic of IBD. The latter is a hallmark of the chronically relapsing exaggerated inflammation of IBD that involves drastic alterations in microbiome and epithelial barrier [14]. Recent research highlights the role of genetic loci, epigenetic modifications, and environmental triggers in disease development. Furthermore, chronic inflammation increases the risk of malignancies, notably colorectal cancer, necessitating vigilant monitoring and management. IBD is an extremely complicated chronic disease with unclear pathogenesis and despite its rising incidence rates worldwide and extensive research, precise etiology is still unclear. Both genetic and environmental factors appear to be important in the development of IBD [15]. IBD-related disorders also confer a high risk of development of a number of malignancies, especially CRC. With respect to cancer, it remains a major public health concern globally, with ≈20 million new cases and 9.7 million deaths worldwide in 2022, while colorectal cancer (CRC) ranked third in incidence (≈1.93 million cases) and fourth in mortality (~0.90 million deaths) globally according to GLOBOCAN estimates (for details see Section 4.1), making research efforts for causes and its relationship with IBD and inflammation of major importance.
2. The Role of Infectious Agents in Inflammation
The following section provides a concise overview of the hierarchical sequence of events that occur following the initial infection, focusing on the physiological and immunological mechanisms governing host–pathogen interactions. It outlines how pathogens navigate and manipulate the host environment to establish a successful niche. This dynamic cascade involves the pathogen’s strategies to subvert innate and adaptive immune responses, evade detection, and overcome host defense mechanisms, ultimately tipping the balance in favor of pathogen survival and persistence within the host.
2.1. The Physiology of Infection: A Brief Overview
The term "infection" refers to any situation in which a micro-organism, which is not a member of the local flora, settles and grows in a host, with or without damage to the host. Pathogens, organisms causing disease, initiate infection through mechanisms known as pathogenesis. However, infection is not synonymous with disease, as the presence of a microorganism, even a pathogen, does not always result in harm. Opportunistic pathogens, typically harmless members of normal microflora, can cause disease in hosts with compromised resistance, such as in cancer or AIDS [16]. The physiology of infection involves the body's reaction to microbial invasions and the immune system's role in combating pathogens. Upon exposure to pathogens such as bacteria and viruses, the adaptive immune system mounts an antibody response, with neutralizing antibodies that block pathogen entry or flag them for elimination (binding) by immune cells. At the same time, pathogens have evolved diverse immune evasion strategies, including antigenic variation, decoy epitopes, interference with antibody function, and secretion of immunosuppressive surface proteins - all employed to avoid detection and neutralization. Thus, the host response and pathogen countermeasures engage in a complex evolutionary arms race [17].
2.1.1. Infection Stages
The initial stages of infection involve pathogen entry through routes such as the respiratory and gastrointestinal tracts, or skin and mucous membranes. Mechanisms include inhalation, ingestion, direct contact, or vector-borne transmission. The respiratory system is a common entry point, with pathogens spread through coughing, sneezing, or talking [18]. The digestive system allows entry via contaminated food, water, or direct deposition of agents. Pathogens can also penetrate the body through skin-to-skin contact, mucous membranes, or insect bites. The host body counters these invasions with barriers like skin, mucous membranes, and stomach acidity. However, pathogens evade these defenses through mechanisms like surface proteins and enzymes. Once inside, pathogens attach to host cells, avoiding immune detection [17,18].
2.1.2. The Immune Response
The innate immune system serves as the first line of defense, offering non-specific protection through cells like macrophages, neutrophils, monocytes, natural killer cells, dendritic cells, and molecular components like cytokines and complement proteins [19]. These cells can recognize conserved pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs) present on immune and epithelial cells. Examples of PAMPs include lipopolysaccharides (LPS) in Gram-negative bacteria, flagellin in bacterial flagella, and viral double-stranded RNA. Cytokines such as interferons, interleukins, and tumor necrosis factors also play vital roles in immune responses [19].
Phagocytosis, a critical innate mechanism, involves specialized cells like monocytes, neutrophils, and macrophages engulfing extracellular material, including pathogens [19] Signals from the Toll-like receptors (TLRs) are further activating innate immunity, with specific TLRs recognizing distinct bacterial components, e.g., TLR2 binds peptidoglycan, TLR4 recognizes LPS, and TLR5 detects flagellin. TLRs trigger signaling pathways involving nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), mitogen-activated protein kinases (MAPKs), and interferon regulatory factors (IRFs), leading to the production of pro-inflammatory cytokines and chemokine family members [20].
Adaptive immunity, mediated by T and B lymphocytes, is a highly specific response against pathogens and retains immunological memory for rapid reactivation upon re-exposure. Unlike innate immunity, the adaptive arm engages only upon antigen detection, providing targeted and efficient host defense [21]. Pathogens and their metabolites, including lipopolysaccharide (LPS) and short-chain fatty acids (SCFAs), serve as pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). These molecules bind to pattern recognition receptors (PRRs), initiating downstream signaling cascades that orchestrate protective inflammation. While inflammation is crucial for pathogen clearance, uncontrolled or chronic inflammation - triggered by overactivation of PRRs by microbial molecules such as LPS or SCFAs - can contribute to the development of chronic diseases and potentially fatal immunopathology [22]
2.1.3. Factors Influencing the Outcome of the Infection
The outcome of an infection is influenced by factors such as pathogen virulence, host immune response, age, health status as well as environmental conditions. Infectivity varies among pathogens, with highly virulent agents causing severe disease and less virulent ones often leading to mild or asymptomatic infections [16]. A robust immune response can clear pathogens and prevent disease, whereas a weakened response may result in persistent infections, particularly in immunocompromised individuals, such as those with HIV or undergoing immunosuppressive therapy [22]. Host factors like age and malnutrition also affect susceptibility to severe infections, with poor nutrition impairing immune functions.
Environmental factors, including seasonal variations, influence infectious disease dynamics. For instance, influenza is more prevalent in cold and dry conditions. Stress, genetics, diet and underlying health conditions further modify infection outcomes [23]. Immune dysfunctions, such as allergies, autoimmunity, and immunodeficiencies, occur upon immune system deregulation, highlighting the need to understand infection physiology for effective prevention and treatment strategies [22].
2.2. Physiological Human-Bacteria Interactions
The human microbiome comprised of trillions of microorganisms residing on or within the human body, forming a symbiotic relationship with the host [24]. These microbes, collectively known as the normal microflora, adapt to the host's physiology to create body-specific ecosystems. Introduced at birth, the microbiome includes skin, oral, and gastrointestinal microbiota, with intestinal microbes performing critical metabolic functions, such as producing vitamins B12 and K, which humans cannot synthesize [24].
The microbiome supports host health by producing beneficial compounds and inhibiting harmful microorganisms, while the host provides microenvironments for microbial growth. Dysbiosis, or an imbalance in the microbiome, has been associated with diseases like inflammatory bowel disease, diabetes, asthma, and cancer, highlighting the critical role of a balanced and stable microbiome in maintaining overall health [24].
2.3. Antigens and Subversion of Immune Response
Bacterial pathogens have developed sophisticated mechanisms to evade phagocytosis and survive within host cells. These pathogens, in order to counteract phagocytes, employ strategies such as escaping the phagosome, blocking phagosome-lysosome fusion or surviving within phagolysosomes. For example, Shigella sp, Listeria monocytogenes and some Rickettsia species secrete lysins to dissolve vacuolar membranes, facilitating their escape [25].
Many intracellular pathogens reside in modified vacuoles that deviate from typical microbicidal phagolysosomes. These modifications often rely on type III and IV secretion systems to disrupt host vesicle trafficking. For example, Legionella pneumophila employs the Dot/ICM system, where its effector RalF activates ARF-1 GTPase, facilitating the pathogen’s intracellular survival. Similarly, Salmonella uses Spi-2 secretion to release effectors like SifA, altering vacuole composition. Mycobacterium tuberculosis (discussed in a later section), a highly successful pathogen, prevents phagosome acidification through surface glycolipids and carbohydrates [25]. Beyond evasion strategies, pathogens manipulate host inflammatory responses for survival. While these strategies block inflammatory signaling pathways, such as MAP kinase and NF-κB, others actively induce inflammation to recruit host cells that serve as proliferation sites. Certain pathogens even secrete molecules to attenuate excessive inflammation, displaying their adaptive capabilities [25].
Bacterial pathogens have developed mechanisms to modulate the host immune response, including altering downstream Toll-like receptor (TLR) signaling and cytokine responses critical to innate immunity [25]. Endogenous antimicrobial peptides like defensins and cathelicidins play a vital role in controlling infections by disrupting bacterial membranes and regulating immunity. However, pathogens counteract with these defenses by modifying surface structures to prevent peptide binding, encoding transport systems for peptide removal and secreting proteases to degrade them [25].
Phagocytes combat intracellular pathogens by producing oxygen reactive species, such as nitric oxide (NO), mediated by inducible nitric oxide synthase (iNOS). NO serves both as a microbicidal agent and a signaling molecule. Pathogens evade NO-mediated killing by detoxifying reactive nitrogen intermediates, repairing damage, or inhibiting iNOS activity, demonstrating their evolutionary adaptation to host defenses [25].
2.4. Bacterial Infection and Immune Dysregulation
Bacterial infections pose a serious threat to human health, progressing through host cell adhesion, bacteria growth and multiplication, tissue penetration of the host, and toxin-induced damage. Host defenses counteract these stages, but excessive immune responses can worsen the outcome [26]. Furthermore, bacterial toxins trigger cytokine overproduction, leading to conditions like septic shock and toxic shock syndrome. Gram-negative endotoxins stimulate macrophages to release IL-1 and TNF-α, while staphylococcal exotoxins act as superantigens, inducing excessive cytokine release by T cells [26].
Sepsis progresses through hyperinflammatory and hypo-inflammatory phases. The initial “cytokine storm” leads to clinical symptoms, followed by monocyte dysfunction and lymphocyte apoptosis, impairing infection control. Certain bacteria, such as Mycobacterium tuberculosis, evade immunity by surviving intracellularly, causing chronic activation of CD4+ T cells, macrophage activation, and granuloma formation, often leading to tissue necrosis [26]. Deregulated and uncontrolled inflammation, while crucial for pathogen elimination, can escalate to systemic damage and septic shock, emphasizing the importance of balanced immune responses. In the following sections, we examine specific infectious diseases that serve as key examples of bacterial infections in the gastrointestinal tract, where the human immune system rarely succeeds in completely eliminating the pathogen, which remains active in most cases.
2.5. Gastrointestinal Tuberculosis
Gastrointestinal tuberculosis (GITB) is a form of extrapulmonary tuberculosis that can affect any organ of the gastrointestinal tract [27,28,29,30]. GITB may occur from primary or secondary infection [31]. Primary infection consists of ingestion of food or milk that contains the bovine bacillus [32]. Secondary infection arises from swallowing of contaminated sputum in a patient with active pulmonary tuberculosis, through the spread of the bacteria via the bloodstream and lymphatic system or reactivation of latent tuberculosis infection (LTBI) [9,30]. Once in the gastrointestinal tract, Mycobacterium penetrates the mucosal layer and invades into the intestinal submucosa [32]. The bacillus colonizes the Peyer’s patches and triggers an inflammatory response which leads to the formation of granulomas [32]. These granulomas undergo caseous necrosis, releasing bacteria to the neighboring lymph nodes. . While the granulomas grow in size, the bowel wall thickens and papillary elevations appear in the mucosa [32] . Consequently, mucosa becomes edematous and ulcerative which can either progress to perforation or heal through fibrosis [27,32].
Tuberculosis (TB) can affect any part of the gastrointestinal tract from esophagus to the rectum [27,31]. The most common site of involvement is the ileocecal region due to the abundance of lymphoid tissue at this site [7,30,33]. The diagnosis of GITB is often delayed due to its varying and non-specific clinical manifestations making it hard to distinguish from other intestinal [27,29,34]. GITB in most patients results in chronic intestinal inflammation with the following symptoms: abdominal pain, fever, weight loss, loss of appetite, nausea/vomiting, diarrhea, change in bowel habits and blood in stool [27,28,29,34]. However, some patients may appear asymptomatic [34]. Clinical examination may reveal ascites, splenomegaly or a palpable abdominal mass in the lower quadrant area. If GITB is not treated promptly and properly, complications such as intestinal bleeding, fistula and perforation may occur [34].
2.6. Colonic Tuberculosis
Colonic tuberculosis is rare and can affect any part of the colon including the cecum, anus and rectum [30]. It appears that the cecum is the most common site of involvement [30]. Anal tuberculosis is also uncommon depicting only 1% of the abdominal tuberculosis cases [29]. The reported symptoms of colonic TB often include intestinal obstruction, perforation, fistulae, bleeding, fever, weight loss, diarrhea and the presence of a palpable abdominal mass [27,30]. Colonic perforation is a serious complication that requires surgical intervention [30]. Colonic TB is difficult to diagnose as it can mimic other abdominal diseases, tumors and Crohn’s disease [30,35]. Endoscopy, colonoscopy and CT scan are used to diagnose colonic TB, although the final diagnosis should be based on histological or bacteriological findings [28,36,37]. Differentiating colonic TB from Crohn’s disease is crucial because an immunosuppressive treatment to a TB patient may lead to detrimental effects such as miliary TB [28,36]. It is also noteworthy to mention that colonic TB may mimic or masquerade as precancerous or cancerous states [38]. Furthermore, people with TB have increased risk of both pulmonary and gastrointestinal cancers. For example, there are reports of cancers developing in about 10% of gastric TB cases [39]. Therefore, it is very important for clinicians to keep in mind this association between TB and neoplastic lesions.
2.7. Gastrointestinal Tuberculosis in Animals
TB in mammals is caused by bacteria that belong to the Mycobacterium tuberculosis complex (MTBC) [40]. Gastrointestinal tuberculosis in domestic animals such as cattle and goats can provoke detrimental economic and public health problems to the local communities [40,41]. Companion animals such as dogs have been reported to be infected with Mycobacterium tuberculosis and subsequently developing gastrointestinal tuberculosis [42,43,44]. Dogs are infected by M. tuberculosis by swallowing human sputum or ingestion of food that contains the tubercle bacilli, thereby the main infection site is the abdomen and intestine [43]. There are no reported cases of tuberculosis spreading from dogs to human. Thus, the disease is thought to be anthropozoonosis [42]. Tuberculosis in birds caused by M. avium and M. genavense mainly affect organs like the spleen and intestine and rarely involve the lungs. M. avium is also known to colonize the GI tract of HIV infected patients [45]. Granuloma formation in the gastrointestinal tract following a mycobacterial infection has been observed in a series of animal species such as dogs, rabbits (Oryctolagus cuniculus), birds, cattle and Kenyan sand boas (Eryx colubrinus loveridgei) [45,46,47]. Moreover, intestinal perforation has been reported in a free-ranging Australian Sea Lion (Neophoca cinerea) subsequent to Mycobacterium pinnipedii infection [48]. Infection with M. bovis in cattle may cause intermittent diarrhea and constipation which is considered the causative (bacterial) agent of tuberculosis in the cattle (known as bovine TB) (ICD-10 A16), although it can produce infection in other animals [40].
3. Animal Models of IBD and Colitis-Associated Colorectal cancer (CAC)
A comprehensive understanding of the pathogenesis and progression of inflammatory bowel disease (IBD) and its transition into colitis-associated colorectal cancer (CAC) necessitates the use of various mouse models for IBD, UC etc. These models are tailored to mimic different facets of human disease, including innate and adaptive immune dysfunction, epithelial barrier disruption, and inflammation-induced carcinogenesis. In addition, this is an overview of the experimental animal models that have been developed and utilized to investigate the pathogenesis, progression, and therapeutic responses associated with gastrointestinal cancers (GIC), with a focus on inflammation-associated colorectal cancer. It highlights the strengths and limitations of both chemically induced models (e.g., AOM/DSS), genetically engineered models (e.g., APCMin/+ mice), and xenograft systems. Emphasis is placed on how these models recapitulate key features of human disease - including tumor microenvironment, immune responses, and molecular alterations - making them indispensable tools for preclinical research into cancer biology, drug testing, and biomarker discovery. The section also outlines the translational relevance of these models for studying the interplay between chronic inflammation, epigenetic regulation, and gastrointestinal tumorigenesis. Below, Table 1 highlights a summarized overview of widely employed murine models used in IBD and colitis-associated colorectal cancer (CAC). It contains information about the model, the method, which is the immune system mainly involved, the advantages, limitations, and supported by previous work (PMID)
3.1. Murine Models of Gastrointestinal Cancer (GIC) Through Pathogen Infection
Bacteroides fragilis (B. fragilis), though representing only 0.1% of the normal colonic flora, is present in 80% of children and adults. However, enterotoxigenic B. fragilis (ETBF) strains producing the metalloprotease fragilysin are elevated in stool and colonic mucosal tissues of CRC patients. B. fragilis disrupts cell-cell adhesion by cleaving E-cadherin, a suppressor of invasion [49]. In vitro studies demonstrated that B. fragilis toxin stimulates cell proliferation via the β-catenin pathway, leading to the transcription of oncogenes c-MYC and cyclin D1. Mutations in Adenomatous Polyposis Coli (APC) complex proteins that activate β-catenin signaling are linked to hereditary and sporadic CRC forms in humans. Clinical studies found higher expression of the enterotoxin gene in mucosal samples from CRC patients. ETBF induced CRC in Min mice through STAT3 activation and TH17 cell response, with tumor growth inhibited by blocking IL-17 and IL-23 receptors [49]. Escherichia coli (E. coli), part of the normal colonic flora, shows increased carriage in adenomas and carcinomas of CRC patients. E. coli produces cytotoxic necrotizing factor (Cnf), cytotoxic distending toxin (Cdt), and colibactin, a polypeptide genotoxin associated with CRC. E. coli strains from phylogenetic group B2 produce colibactin via the enzyme complex "PKS". Animal studies showed E. coli with PKS enzymes induced sporadic CRC in mice, with colibactin promoting epithelial cell proliferation through DNA damage and genomic instability [49].
Fusobacterium nucleatum (F. nucleatum) is linked to colorectal adenomas and CRC, with higher levels in CRC tissues and stool samples compared to controls [50]. It is associated with high CRC mortality, low overall survival, and increased metastasis. F. nucleatum stimulates CRC expansion via the Fap2 protein, which interferes with the immune system's antitumor activity. The virulence factor FadA mediates adhesion to E-cadherin, activates β-catenin signaling, and enhances inflammatory and tumorigenic responses. F. nucleatum promotes proliferation and invasion of CRC cell lines through TLR4 signaling, while there is also NF-κB stimulation, and increased of miR-21 marker expression [49].
Enterococcus faecalis (E. faecalis) is a human pathogen found at higher levels in CRC patients' stool samples. It generates reactive oxygen and nitrogen species (RONS), causing DNA breakage, mutations, and chromosomal instability, contributing to its oncogenic activity [49].
Helicobacter pylori (H. pylori) and its role in CRC is less clear but statistically significant associations exist. H. pylori infection is increased in patients with colon cancer and adenomatous polyps. Cytotoxin-associated gene A (cagA)-positive or CagA seropositivity correlates with severe gastrointestinal disease and higher CRC risk [49]. Alterations in other bacterial species, such as Bacteroides/Prevotella, Coriobacteridae, Roseburia, and Fusobacterium, are noted in CRC patients. Studies suggest, that miRNAs may influence gut microbes' gene expression and growth, impacting cancer pathogenesis [49]. Gastrointestinal cancers have high incidence and mortality rates, with bacterial infections playing a significant role in their development [49,51].
3.2. Non-infectious Animal Models for Gastrointestinal Cancer (GIC)
In addition to pathogen-induced models, several non-infectious animal models have been extensively used to study the initiation, progression, and treatment responses of gastrointestinal cancers, particularly colorectal cancer (CRC). These models broadly fall into three categories: chemically induced models, genetically engineered mouse models (GEMMs), and xenograft systems. Each model recapitulates different facets of human disease and serves unique experimental objectives.
3.2.1. Chemically Induced Models: AOM/DSS
The azoxymethane (AOM)/dextran sulfate sodium (DSS) model is one of the most widely used systems for studying inflammation-associated colorectal carcinogenesis. AOM is a potent procarcinogen that induces DNA alkylation, resulting in O6-methylguanine (O6-meG) adducts and subsequent G:C → A:T transitions, often leading to activating mutations in oncogenes such as Kras. When combined with DSS - an irritant that induces colitis - the model mimics the pathophysiological features of colitis-associated CRC [52]. In the context of modeling inflammation-driven colorectal cancer, DSS (dextran sulfate sodium)-induced colitis and the AOM/DSS (azoxymethane combined with DSS) model remain widely adopted experimental systems due to their simplicity, reproducibility, and close histopathological resemblance to human disease. This model faithfully reproduces key steps of tumor development, including crypt abscesses, epithelial injury and acute inflammation in the distal colon, hyperplasia, inflammatory cell infiltration, particularly useful for studying ulcerative colitis-like damage, dysplasia, and adenocarcinoma formation. Furthermore, it allows investigation of molecular events such as cytokine signaling (e.g., IL-6, TNF-α) and epigenetic alterations, such as promoter methylation and histone modification [53].
In this study, we leveraged publicly available RNA-seq datasets derived from these murine models - DSS-induced colitis and AOM/DSS-induced CAC – in comparison to human CRC (Triantaphyllopoulos et al., manuscript in preparation). Thus, by performing a RNA-seq meta-analysis of differentially expressed genes in the AOM/DSS model and cross-species comparison to the human UC, we found strong similarities. The aim was to decode the molecular landscape associated with inflammation-induced colorectal tumorigenesis and identify evolutionarily conserved, upregulated gene signatures that may act as potential biomarkers or therapeutic targets. A selection of commonly upregulated protein-coding genes and ncRNAs was further prioritized for their mechanistic involvement in CRC pathogenesis (see Subsections, 8.5. Inflammation-driven ncRNA Modulation in CRC and 9.5. Comparative Human-Mouse Evidence).
3.2.2. Genetically Engineered Mouse Models (GEMMs)
GEMMs offer the advantage of dissecting the functional roles of specific genes implicated in GIC. The most common knockout (KO) genes used in the murine model of intestinal inflammation are IL-10, IL-23R, CD4+CD25+, NOD2/CARD15, TGF-β1, RAG, ATG16L1, APCMin/+, IL-2, TNF-α, STAT3, NFκB, Muc2, IFN-γ, MyD88 and TLR [54]. Among these, the APCMin/+ mouse is the most commonly used model for studying sporadic and familial adenomatous polyposis (FAP) [55]. These mice carry a heterozygous truncating mutation in the tumor suppressor gene Apc, leading to constitutive activation of the Wnt/β-catenin signaling pathway, and develop multiple intestinal neoplasms spontaneously [56]. Although tumors predominantly arise in the small intestine, combinations with other mutations (e.g., Kras, p53, or Smad4) or with inflammatory agents can shift tumorigenesis toward the colon and more accurately reflect human CRC. Importantly, GEMMs enable time- and tissue-specific gene modifications using Cre-loxP technology, allowing precise modeling of multistage tumor development and microenvironmental interactions.
3.2.3. Xenograft and Patient-Derived Xenograft (PDX) Models
Xenograft models, involving transplantation of human CRC cell lines into immunocompromised mice (e.g., NOD/SCID or nude mice), are widely used for preclinical drug testing and evaluation of tumor growth dynamics. Subcutaneous xenografts offer ease of monitoring tumor size, while orthotopic models - involving implantation into the cecum or colon - better simulate tumor microenvironment and metastatic spread. More recently, patient-derived xenografts (PDX) have gained popularity, as they preserve the genetic, epigenetic, and histopathological characteristics of the original human tumors, offering enhanced predictive value for personalized medicine approaches [57]. However, the lack of a functional immune system in these models limits their utility for immuno-oncology studies.
Animal models have played a pivotal role in dissecting the complex pathophysiology of inflammatory bowel disease (IBD) and its progression towards colitis-associated colorectal cancer (CAC). These animal models allow for controlled experimentation on genetic, environmental, and immunological contributors to disease progression; more importantly, they provide complementary systems for studying the complex interactions between genetic mutations, epigenetic changes, inflammation, and tumor progression. Their continued development and refinement remain essential for translational research aimed at identifying therapeutic targets and validating biomarkers for GICs.
Below, the sidebar infographic Figure 1 presents a concise schematic overview of the more detailed Table 1, for the commonly used murine models of IBD and colitis-associated colorectal cancer (CAC), including method names, and supportive citations.
Below is the more detailed information of commonly used animal models for studying IBD and CAC, as shown in Table 1 about the model, the method, which is the immune system mainly involved, the advantages, limitations, and supported by previous work (PMID).
4. Immune Defense in Gastrointestinal (GI) Chronic Inflammation and Carcinogenesis
There have been theories linking chronic inflammation to cancer dating back to the seventeenth century. German physician Rudolf Virchow postulated in 1863 that tumor formation is the consequence of recurring inflammatory responses after seeing immune cells penetrate cancerous tissue [58]. Based on Virchow's findings, Japanese physician Katsusaburo Yamagiwa showed in 1915 that artificially generated chronic inflammation may promote tumor formation in an animal model [59].
According to Harold F. Dvorak, tumors are “wounds that do not heal,” and the same molecular pathways that promote wound healing and tissue regeneration after damage may also be responsible for the genesis of tumors [60,61]. The cellular and molecular architecture of inflammation in cancer has been better understood because of developments in molecular biology and the creation of genetically engineered mice. In addition to the many tasks carried out by distinct immune cell subtypes, this also involves intricate signaling route networks controlled by an extensive range of cytokines, chemokines, and growth factors [6]. Understanding the molecular pathways relating inflammation to the development and advancement of cancer is necessary to develop viable treatment options. In the next subsection, updated information is provided, through statistics and epidemiology search, the severity of the most prevalent cancer incidents, the rise of their occurrence and death rates as recently recorded and compared to the incidences of GI cancers internationally.
4.1. Inflammation-Induced Cancers Associated with the GI Tract.
Studies suggest that around 20% of cancers are associated with chronic inflammation that is linked to different stages of oncogenesis: cellular transformation, tumor progression, invasion, angiogenesis and metastasis [5]. Approximately 15-20% of all cancer cases develop at the same tissue or organ site that previously had some type of serious infection and/or chronic inflammation. In these cases, inflammation which promotes cancer is induced and exists long before tumor formation [62]. The most prominent examples include inflammatory bowel disease, chronic hepatitis, helicobacter-induced gastritis or schistostoma-induced bladder inflammation which increase the risk of colorectal cancer (CRC), liver cancer, stomach cancer or bladder cancer, respectively [11]. These particular cancers that are associated with inflammatory disorders are cancers of organs of the GI tract.
The GI tract, also known as the gut or the digestive tract, is where food and liquids travel through and are processed i.e., swallowed, digested, absorbed and wastes expelled from the body. The GI tract is made up of the hollow organs (mouth, esophagus, stomach, small intestine, large intestine (bowel, colon), rectum and anus. The solid organs of the GI tract are the liver, pancreas and gallbladder.
Generally speaking for GI tract cancers, i.e., colorectal cancer (cancers of the colon and / or rectum, CRC), liver cancer, pancreatic cancer and stomach cancer are leading causes of cancer-related deaths worldwide [63]. More specifically, worldwide, the three major cancer types in 2022 were lung, breast and colorectal cancers. The new estimates available on IARC’s Global Cancer Observatory (International Agency for Research on Cancer, cancer agency of the World Health Organization - https://gco.iarc.fr/en) show that 10 types of cancers collectively comprised around two-thirds of new cases and cancer deaths globally in 2022, while data covers 185 countries and 36 types of cancer. Lung cancer was the most commonly occurring cancer worldwide with 2.5 million new cases accounting for 12.4% of the total new cases. Female breast cancer ranked second (2.3 million cases, 11.6%), followed by colorectal cancer (1.9 million cases, 9.6%), prostate cancer (1.5 million cases, 7.3%), and stomach cancer (970 000 cases, 4.9%). Lung cancer was the leading cause of cancer death (1.8 million deaths, 18.7% of the total cancer deaths) followed by colorectal cancer (900 000 deaths, 9.3%), liver cancer (760 000 deaths, 7.8%), breast cancer (670 000 deaths, 6.9%) and stomach cancer (660 000 deaths, 6.8%) [64]. Lung cancer’s re-emergence as the most common cancer is likely related to persistent tobacco use in Asia. With respect to GI tract cancers (i.e., colorectal, liver, gall bladder, pancreas and stomach) only, CRC ranked first for new cases of cancers. Moreover, CRC ranked first for leading causes of cancer death, followed by liver and stomach cancer [65,66]. In fact, projections indicate that the CRC burden will rise sharply to 3.2 million new cases and 1.6 million deaths by 2040 [67].
Moreover, though it is known that the risk of CRC increases with age, with most cases affecting people over the age of 50, the incidence of new cases and deaths of CRC in younger age groups under the age of 50 has been rising steadily. In fact, the disease has become a leading cause of cancer deaths for Americans 20 to 49 years old according to the National Cancer Institute (https://www.cancer.gov/types). This may be due to changes brought about by modern lifestyle which add an increased burden to the risk factors already involved (e.g., infections/pathogens). These include diet and industrialized food (e.g. meat processed food etc) rather than home cooking, as well as other environmental risk factors (chemicals, toxins, newly evolved pathogens, atmosphere – atmospheric changes – e.g., increased UV irradiation etc). The environmental factors which may be causal elements in inflammation, combined with genetic predisposition, may partly explain the increasing incidence of early onset CRC [65,66].
All cancers may or may not have genetic and/or epigenetic predispositions and all may or may not be inflammation-induced. However, data support that chronic inflammation can induce carcinogenesis in individuals with susceptibility to infection, which increases the cancer risk, but also in those without. Specifically, CRC can develop with genetic susceptibility only, also taking into account its location, the colon/bowel, which is prone to numerous inflammatory conditions; it is also a prime candidate to be induced by chronic inflammation without genetic risk. As such, CRC may be considered a typical inflammation-dependent cancer and the risk of developing CRC increases in patients with IBD [68].
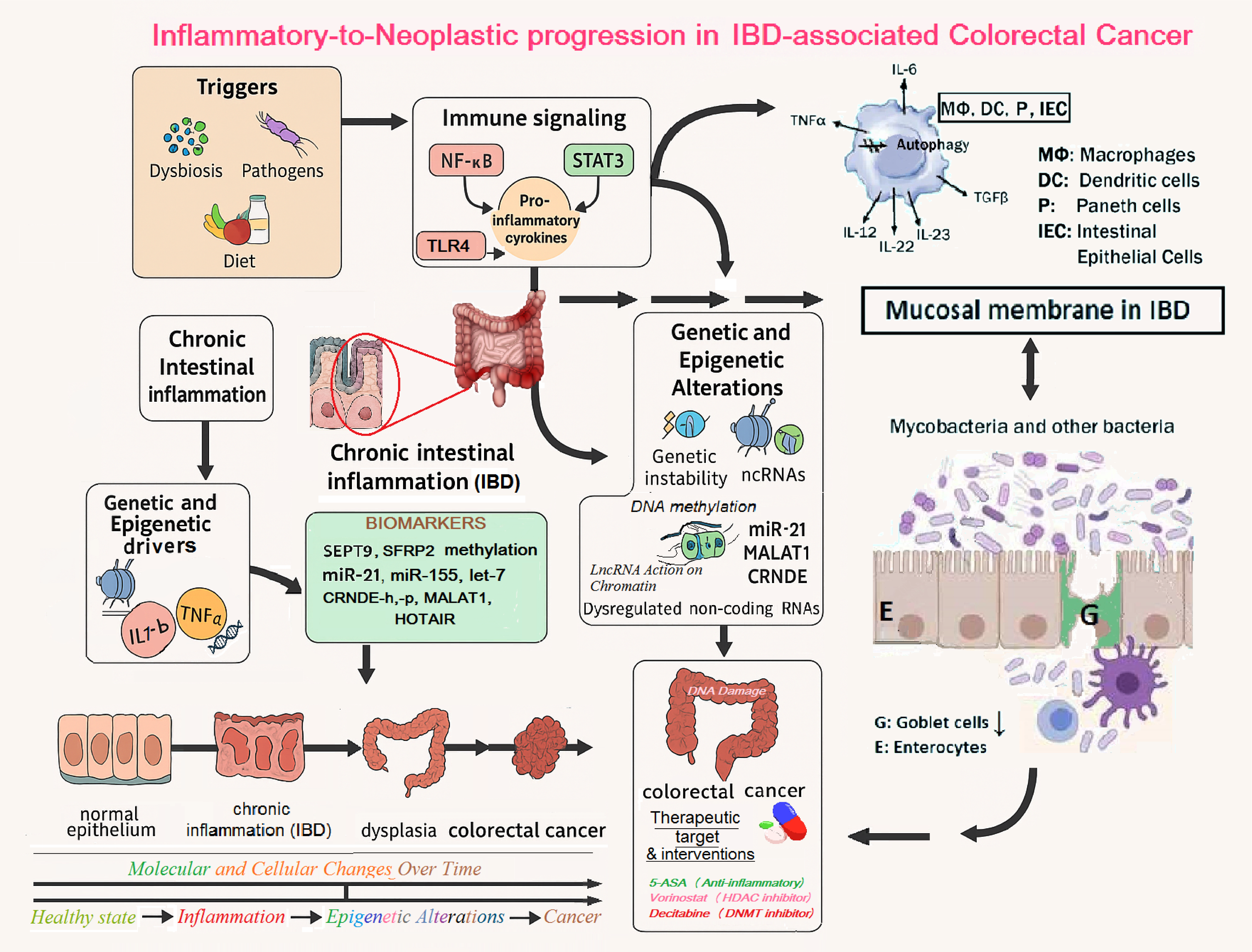
Figure 2 presents a schematic mechanism of inflammation–dysplasia–carcinoma sequence model, anchoring, 1) Microbial triggers (etc dysbiosis etc) 2) immune cell recruitment (macrophages, T cells etc), cytokines (e.g. TNF-α, IL-6 etc), 3) Persistent immune activation and cytokine overproduction compromise the viability of intestinal epithelial cells (IECs) and impair their autophagy machinery. This is accompanied by the loss of goblet cells and disruption of their mucin-secreting function, which weakens the mucosal barrier and facilitates bacterial infiltration and tumor-promoting inflammation. The breakdown of goblet cell–mediated mucosal protection is a hallmark of both active IBD and the transition to colorectal neoplasia [69,70,71,72]. 4) Epigenetic marks leading to silencing of tumor suppressors or activation of oncogenes and 5) Transition to cancer, as the main components leading normal colon to IBD and dysplasia and progressing to colorectal cancer.
In more detail, Figure 2, graphically asigns and emphasize the importance of the following factors in the inflammatory signalling involvement towards colorectal carcinogenesis, as summarized below:
Initiating factors such as pathogenic bacteria (Helicobacter pylori, Mycobacterium tuberculosis) and gut microbiota dysbiosis activate host immune surveillance via pattern recognition receptors, promoting sustained immune activation. This leads to the secretion of pro-inflammatory cytokines including TNF-α, IL-6, and IL-1β, which in turn activate intracellular signaling pathways (e.g., NF-κB, STAT3), maintaining a chronic inflammatory microenvironment within the colonic mucosa.
Persistent inflammation results in accumulated genetic mutations and epigenetic reprogramming. Epigenetic alterations include promoter hypermethylation of tumor suppressor genes (e.g., SEPT9, CDKN2A), histone modifications such as H3K27 trimethylation and histone deacetylation, and dysregulated expression of non-coding RNAs. Upregulation of oncogenic microRNAs (e.g., miR-21, miR-155, miR-214) and aberrant expression of long non-coding RNAs (e.g., HOTAIR, MALAT1, CRNDE) [69], disrupt gene regulation, chromatin accessibility, and epithelial differentiation.
Critically, the sustained inflammatory response impairs autophagy and apoptosis within intestinal epithelial cells (IECs), undermining mucosal homeostasis. Loss of autophagic control contributes to abnormal cell survival, dysregulated turnover, and unrestrained epithelial cell proliferation. Goblet cell dysfunction - characterized by reduced mucin production - further weakens the intestinal barrier, increasing susceptibility to microbial translocation and perpetuating the pro-tumorigenic immune cascade. These epithelial impairments have been increasingly implicated in IBD pathogenesis and the development of colitis-associated colorectal [69,70,71,72].
Figure 2 also depicts the stepwise events and molecules linking chronic intestinal inflammation to colorectal cancer (CRC). Key initiators include microbial pathogens such as Helicobacter pylori and Mycobacterium tuberculosis, as well as gut microbiota dysbiosis. These factors activate inflammatory pathways leading to sustained production of cytokines and transcription factors (IL-6, TNF-α, and NF-κB activation), resulting in a state of chronic inflammation. There are also highlighted key molecular biomarkers (e.g., SEPT9 methylation, miR-21 expression) and emerging therapeutic targets such as 5-ASA (anti-inflammatory agent), vorinostat (histone deacetylase inhibitor), and decitabine (DNA methyltransferase inhibitor), which intervene at various stages of the inflammation – epigenetics - tumorigenesis axis. Understanding this complex interplay provides a dynamic framework for novel diagnostic and potential therapeutic strategies in inflammation-induced colorectal cancer.
More importantly, Figure 2 also emphasizes how chronic inflammation and epigenetic dysregulation intersect to drive CRC progression and identifies critical intervention points for clinical application. Important insights in the complex mechanisms are highlighted below:
Persistent inflammation promotes epigenetic changes, including:
DNA methylation of tumor suppressor genes (e.g., MLH1, CDKN2A/p16),
Histone modifications such as hypoacetylation and trimethylation of histone H3 on lysine 27 (H3K27me3),
Altered expression of non-coding RNAs (miRNAs and lncRNAs), which regulate key inflammatory and tumorigenic genes.
These molecular alterations drive the transition states from a healthy colon epithelium to inflammatory bowel disease (IBD), dysplasia, and ultimately colorectal carcinoma. Clinical markers that can be detected at transitional stages include SEPT9 gene methylation (plasma biomarker) and miR-21 expression (in tissue or circulation), both of which have diagnostic and prognostic potential. Current therapeutic interventions are also illustrated:
Anti-inflammatory agents, such as mesalamine (5-ASA) and infliximab, which reduce inflammatory cytokine activity,
Epigenetic drugs, including decitabine (a DNA methyltransferase inhibitor) and vorinostat (a histone deacetylase inhibitor), which are being explored for their potential to reverse aberrant epigenetic states in cancer and inflammation.
According to the Genetic Testing Registry (GTR) resource (NCBI), in the inherited colon cancer (https://www.ncbi.nlm.nih.gov/gtr/tests/552303/) the following genes have been involved:
Tumor suppressors and mismatch repair genes: APC (5q22.2), MLH1 (3p22.2), MSH2 (2p21-16.3), MSH6 (2p16.3), PMS2 (7p22.1)
Polymerases and modifiers: POLD 1 (19q13.33), POLE
Other associated loci: MUTYH (1p34.1), EPCAM (2p21), GREM1 (15q13.3)
As shown in Figure 2, the aforementioned genes highlight the hereditary predisposition to colorectal neoplasia, interacting with both environmental and epigenetic drivers. Arrows indicate multidirectional crosstalk between genetics, epigenetics, and inflammation, emphasizing the multifactorial nature of IBD-to-CRC transition and the importance of integrated, personalized interventions.
4.2. The Involvement of Bacteria in the Mechanisms of Carcinogenesis
Infections contribute significantly to human tumors, as mentioned in the previous subsection. Most of these infections are attributed to viruses, leading to the oversight of bacterial contributions. While bacterial infections are epidemiologically linked to certain cancers, inflammation resulting from these infections has been traditionally considered the primary cause of tumor formation. However, bacteria can directly manipulate host cells during their infection cycles, impacting cellular integrity and potentially contributing to cancer development [73].
Cancer progression involves genetic alterations disrupting normal cell growth and survival controls. Viral genomes found in tumors and epidemiological studies establish strong links between viruses and cancers, such as human papillomavirus with cervical cancer and hepatitis B/C viruses with liver cancer. These viruses are part of a broader microbiome that interacts with host cells to ensure their survival. Although microbial infections like bacteria, molds, and helminths do not leave genetically identifiable marks in host genes, strong links exist between these infections and cancers [74]. Notable examples include Schistosoma haematobium with bladder cancer, Helicobacter pylori (H. pylori) with gastric cancer, and chronic Salmonella typhi infections with gallbladder carcinoma.
Studies in animals highlight the carcinogenic effects of microbiota, with germ-free or antibiotic-treated models showing reduced tumor development, underscoring the role of the microbiome in cancer [74]. H. pylori is the most documented bacterium with epidemiological data linking it to carcinogenesis, although other bacteria have been also associated with human cancers through interactions within the human microbiome [73].
The bacterial protein CagL is a type IV pilus adhesin of H. pylori that ensures the attachment of H. pylori to gastric epithelial cells. Notably, CagL from H. pylori, binds to gastric epithelial cells and then controls a signaling cascade that increases gastrin secretion, resulting in hypergastrinemia, an important risk factor for the development of gastric adenocarcinoma [75]. Bacteria can interfere with p53 activities and DNA repair mechanisms, promoting DNA damage accumulation and tumor growth [73]. Animal studies show reduced tumor burden when gut microbiota are manipulated with antibiotics, emphasizing the potential role of bacteria in cancer [73]. Clinical studies link Fusobacterium nucleatum with colorectal cancer, Chlamydia trachomatis with cervical cancer, and mycoplasmas with prostate and colorectal cancer, as well as with non-Hodgkin's lymphoma in HIV-seropositive subjects [73].
Mycoplasma infections, in particular, have been shown to inhibit p53 and cooperate with oncogenic Ras, leading to oncogenic transformation in vitro. This strongly suggests that they can be the leading candidate bacteria with oncogenic potential. Persistent mycoplasma infections can lead to decreased expression of tumor suppressors p53 and p21, causing pathological changes and potentially facilitating tumorigenesis. Mycoplasma fermentans, for example, induces chromosomal alterations leading to malignant properties [73].
Recent research on the tumor microbiome highlights the impact of bacteria on tumor progression and therapy. Bacteria employ immune evasion strategies and intracellular infection mechanisms to survive and propagate. For instance, a protein from M. fermentans, DnaK, impairs DNA repair by reducing PARP1 activity, promoting cellular transformation [73]. DnaK also interacts with Ubiquitin Specific Peptidase 10 (USP10), reducing p53-dependent antitumor functions and counteracting anticancer drugs reliant on p53. Phylogenetic analysis suggests a common mechanism of cell transformation among bacteria like Mycoplasmas, Helicobacter. pylori, Fusobacterium nucleatum, and Chlamydia trachomatis.
It was shown that, exogenous DnaK induces inappropriate protein phosphorylation, adding to current knowledge about the role of bacteria in the tumor microenvironment in dysregulating cellular functions to ultimately promote cancer progression. These findings indicate that bacteria with similar DnaK proteins might contribute to tumor progression and therapy resistance by altering DNA repair and anticancer drug actions [73]. Bacterial manipulation of host cells likely results in cancer as an unintended consequence of infection cycles, as cancer typically arises after the bacteria have left the host [74]. While viral proteins involved in carcinogenesis are well-documented, bacterial mechanisms remain less understood. However, similarities between cancer-associated bacteria and oncogenic viruses are becoming clearer, suggesting that also bacteria alter critical cellular proteins and DNA repair processes, leading to cancer [73]. Understanding these mechanisms could enhance knowledge of cancer origins and be of benefit to preventive, diagnostic, and therapeutic strategies [73].
4.3. Chronic Inflammation as a Driver of Colorectal Carcinogenesis
Chronic inflammation is a critical driver of colorectal carcinogenesis, particularly in the setting of inflammatory bowel diseases (IBD) such as ulcerative colitis and Crohn's disease. Patients with long-standing IBD exhibit a significantly increased risk of developing colitis-associated colorectal cancer (CAC), which is characterized by inflammation-induced genetic and epigenetic alterations in colonic epithelial cells [76,77]. Unlike sporadic CRC, where the adenoma - carcinoma sequence predominates, inflammation-driven CRC follows an inflammation - dysplasia - carcinoma pathway, rooted in persistent immune activation and tissue injury [78].
A hallmark of this inflammatory environment is the dysregulation of cytokines and immune signaling cascades. Pro-inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and interleukin-1β (IL-1β) are markedly elevated in the inflamed colonic mucosa and contribute to epithelial cell transformation. These cytokines activate downstream transcription factors, notably nuclear factor-kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3), which orchestrate the transcription of genes involved in cell survival, proliferation, angiogenesis, and immune evasion [6]. NF-κB plays a central role in sustaining chronic inflammation and promoting oncogenesis. Under normal conditions, NF-κB is held inactive in the cytoplasm by IκB proteins. However, in the presence of inflammatory stimuli - including cytokines, bacterial products, and oxidative stress - NF-κB is rapidly activated and translocated to the nucleus. There, it induces the expression of anti-apoptotic genes (e.g., Bcl-xL, XIAP), inflammatory mediators (e.g., COX-2, IL-6), and enzymes that contribute to genomic instability (e.g., iNOS, ROS-generating enzymes) [79,80].
Similarly, IL-6 activation of the JAK/STAT3 pathway further exacerbates carcinogenic signaling. Activated STAT3 enhances the transcription of cyclin D1, c-Myc, and survivin, thereby promoting cellular proliferation and inhibiting apoptosis [81]. Notably, the crosstalk between NF-κB and STAT3 creates a positive feedback loop, reinforcing a tumor-promoting inflammatory niche. This loop not only supports early tumor development but also fosters immune tolerance and resistance to therapy.
In addition to epithelial alterations, the inflammatory microenvironment recruits various immune cells, including macrophages, neutrophils, and Th17 cells, which release additional cytokines and reactive oxygen species (ROS). ROS induce oxidative DNA damage, telomere shortening, and DNA methylation changes, contributing to genomic instability [82]. Over time, this inflammatory pressure leads to architectural and functional distortion of the mucosa, transitioning from chronic inflammation to low-grade dysplasia, then to high-grade dysplasia, and ultimately to the invasive carcinoma stage.
4.4. Effect of bacterial infection on gastrointestinal cancer
The human gastrointestinal tract (GIT) is a highly intricate system housing trillions of microorganisms, including bacteria, archaea, fungi, parasites, and viruses [24]. Among these, bacteria represent the predominant microflora colonizing the GIT. Cancers of the GIT are recognized as a significant global health challenge, with high incidence and mortality rates, as reported in the World Cancer Statistics 2018 [51].
Compelling evidence highlights the role of bacterial infections in the development and progression of various GIT diseases, including cancers. Additionally, emerging research suggests that the GIT microbiota plays a critical role in influencing tumor responses to anticancer therapies, such as conventional chemotherapy and molecularly targeted treatments. As a result, targeting the bacterial microbiota offers promising potential for the prevention and treatment of GIT cancers [49].
4.4.1. Colorectal cancer
Colorectal cancer (CRC) ranks as the third most commonly diagnosed cancer in men and the second in women, with 1.8 million new cases and 881,000 deaths reported in 2018 [51]. Adenocarcinoma is the prevalent histopathological subtype. While the exact causes remain unclear, environmental factors such as smoking, diet, and lifestyle are known risk factors. Age increases CRC incidence, with certain genetic disorders like Adenomatous Polyposis Coli (APC) and family history being significant contributors. Conditions like ulcerative colitis and Crohn's disease also elevate CRC risk, though 80% of cases are sporadic [49].
The human gut hosts over 500 bacterial species, predominantly anaerobes like Bacteroides, Eubacterium, and Fusobacterium, with the colon having the highest concentration. Facultative anaerobes like Enterococci and Lactobacilli form a smaller portion. Dysbiosis, an imbalance in the gut microbiome, is implicated in colon diseases, including CRC. Despite extensive research, the specific mechanisms by which intestinal flora induce CRC remain unclear [49].
Studies have highlighted the role of the gut microbiome in CRC. McCoy and Mason first linked enterococcal endocarditis with cecal carcinoma, suggesting Streptococcus gallolyticus (formerly S. bovis) as a contributing factor. A significant percentage of S. gallolyticus bacteremia patients also have CRC, with prevalence rates in CRC patients ranging from 33% to 100%, compared to 2.5% to 15% in the normal population. Animal studies show that S. gallolyticus increases proliferation markers and polyamines, with colonic adenomas observed in 50% of affected rats. Increased IL-8 production, promoted by S. gallolyticus, enhances free radical generation, aiding the neoplastic process. S. gallolyticus colonizes colonic tissues through collagen-binding proteins and histone-like protein A. Consequently, patients with S. gallolyticus bacteremia are recommended to undergo complete colonoscopy [49]. Collectively, these findings support the notion that chronic inflammation is not merely the milieu to cancer development but an active participant in tumor initiation and progression. Understanding these immune and cytokine networks is therefore pivotal for identifying preventive and therapeutic targets in inflammation-associated CRC.
5. Overview of Chromatin and Epigenetic Modulations
Nuclear DNA is organized into chromatin which consists of nucleic acids (genomic DNA and different types of RNAs), the histone proteins H2A, H2B, H3, H4 and H1 and non-histone chromatin-associated proteins [12,83,84,85]. The basic structural and functional unit of chromatin is the nucleosome. The nucleosome consists of a core histone octamer (two H2A -H2B dimers and one (H3-H4)2 tetramer) around which are wrapped 146 base pairs (approximately 1.65 turns of DNA). Histone H1 is found outside of the nucleosome on the linker DNA region and seals the entrance and exit of the DNA around the nucleosome [86,87]. All biological processes such as replication and transcription take place on the DNA template which must be in an ‘open’ structural form so that proteins of the replication machinery and transcription factors and other proteins involved in transcription can have access. Thus, chromatin and nucleosomal structure must be (and is) dynamic in order for proteins of the transcriptional machinery to have access or be blocked as necessary. Epigenetics is the study of heritable changes in gene expression and function without changes in DNA sequence [88]. Epigenetic mechanisms are responsible for the regulation of transcription, i.e., what genes are expressed, or dynamically have the structural potential to be expressed or what genes are silenced (permanently or temporarily).. These epigenetic mechanisms are the histone post translational modifications (PTMs), changes in the histone variant constitution of the nucleosome, DNA methylation, nucleosomal remodeling and positioning factors (activating complexes such as SWI/SNF). More importantly, interactions with proteins of the nuclear matrix (scaffold proteins) and regulation via long non-coding RNAs (lncRNAs) and microRNAs (miR) and other non-coding RNAs ,complicates the picture of the multifactorial network of interactors that are involved in genomic regulation at the chromatin level [12,85,89,90,91]. These mechanisms of transcriptional regulation establish epigenetic heritable patterns of differential gene expression and silencing profiles from the same genome which are cell-type specific. Cells can change these gene expression signatures in response to stimuli, such as the changing conditions due to changes in the micro and macro environments [12,91,92].
Post-translational modifications of the histone proteins (histones H1, H2A, H2B, H3, H4) take place mostly on their N-terminal tails which protrude from the nucleosome. Notably, some histone modifications also occur on the C-terminal tails which do not protrude from the nucleosome but embedded inside the octamer core in the globular domain of the histone, e.g., H3K79 methylation [93]. These modifications are reversible reactions and include acetylation, methylation, phosphorylation, ubiquitination, poly(A)ribosylation and sumoylation, among other more recently identified histone modifications [12,89,94,95], which have not been thoroughly investigated (i.e., GlcNAcylation, citrullination, crotonylation and isomerization) [88]. They can function alone, or in combination with other histone modifications. The latter has been referred to as the ‘histone code’. Two or more histone modifications, e.g., on the promoter of a gene, can either enhance, reduce/inhibit or alter the function of another histone modification. The ‘histone code’ is a hypothesis which states that DNA transcription is largely regulated by post-translational modifications to the histone proteins [96,97] .
Histone modifications occur at specific amino acid residues. Histone acetylation is one of the most studied and also, most prevalent histone modification [4]. Acetylation occurs only on specific lysine residues of all histones. This modification reduces the positive charge of the histone lysine residues, thus weakening the DNA-histone interactions establishing an ‘open’, permissive towards transcription, chromatin structure and/or a transcriptionally active chromatin landscape. Acetylation, in fact, is a prerequisite for the activation of gene expression. Acetylated chromatin is ‘poised’ chromatin, ready for transcription [98]. The enzymes responsible for the transfer of the acetyl group from acetyl-coenzyme A are the histone acetyltransferases (HATs – comprised of at least six groups of acetyltransferases) and responsible for their removal are the histone deacetylases (HDACs – comprised of four families). The enzymatic activity of HATs and HDACs alter chromatin configuration so as to allow activation or inactivation of a gene, respectively. Histone methylation also occurs at specific amino acids (lysines 4, 36, 79 of histone H3 at active chromatin sites and lysines 9, 27, 20 of histone H3 at inactive chromatin sites and lysines 5 and 20 of histone H4) or arginines (arginines 2, 8, 17, 26 of histone H3 and 3 of histone H4). To increase the complexity, lysines may be mono-, di- or trimethylated, whereas arginine residues may be mono- or dimethylated (symmetric or asymmetric). Unlike acetylation, histone methylation does not alter the charge of the histone protein. A variety of enzymes catalyze the addition or removal of the methyl group: the histone methyltransferases (HMTs) and the histone demethylases (HDMs), respectively. Histone phosphorylation occurs in tyrosine, serine and threonine residues of the N-terminal histone tails. Specifically, in histone H3 residues serine 10, 28, threonine 3, 6, 11, 45, and tyrosine 41, as well as serine 32 of histone H2B [4]. A phosphate group from ATP is transferred to the hydroxyl group of a target amino acid, leading to a build up of negative charge on histones which in turn weakens the histone-DNA interaction and facilitates an ‘open’ transcriptionally permissive chromatin structure. Protein kinases and phosphatases add or remove, respectively, the phosphate group from the histone proteins (as well as from many other cellular proteins) [4]. Histone ubiquitination can be found in all core histone subtypes. Most prominent are histone H2A ubiquitination on lysine 118 or 119 (H2AK118119/ub) and H2B lysine 120 (H2BK120ub), which account for 5-15% of H2A and 1% of H2B, respectively [99]. It is mediated by the sequential interactions of the E1, E2 and E3 ligase enzymes. Histone ubiquitination plays a role in chromatin compaction and transcriptional regulation and can also interact with other histone modifications. Similarly, the reactions leading to the aforementioned histone modifications are catalyzed by other modification-specific enzymes. Various histone modifications, alone or in combination, alter the three dimensional (3D) structure of the nucleosome and affect the transcriptional control of genes by inducing either an ‘inactive’ closed heterochromatin conformation, inaccessible to the transcriptional machinery, or an ‘active’ open euchromatin conformation [100,101,102,103,104,105], or a facultative heterochromatin conformation (forms the poised chromatin with the potential to become euchromatin). Notably, environmental factors can induce changes in histone modifications, thereby altering gene expression signatures.
DNA methylation is the covalent transfer of a methyl group to the carbon atom at position 5 of cytosine. This forms the 5-methylcytosine (5mC), which occurs most frequently at the dinucleotide CG [12,91,106,107]. DNA regions that are ≥ 200 bp long and show a CG:GC ratio ≥0.6 are defined as CpG islands [12,108]. Methylated DNA is a closed structure and transcription factors cannot reach gene promoters. Genes in such methylated DNA are silenced [12,109]. CpG islands are dinucleotide repeats prevalent in mammalian genomes, typically unmethylated and associated with gene promoters located in genetic regulatory elements. DNA methylation starts at one end of the islands and continues to gene promoters and initiation sites, altering the three-dimensional configuration of the DNA and inhibiting its interaction with transcription factors, ultimately silencing gene expression (hypermethylation). In contrast, hypomethylation promotes gene expression [105]. The enyzmes that catalyze the addition of methyl groups to DNA is carried out by a family of enzymes known as DNA methyltransferases (DNMTs) comprising of DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L. DNMT1 catalyzes DNA methylation during DNA replication and cell division, DNMT3A/3B (de novo methylation) are responsible for methylation of DNA during development and differentiation, while DNMT3L is an “aide” in DNA methylation interacting with DNMT3A/3B to stimulate the de novo reactions, as it lacks the conserved catalytic domain, thus, is not directly involved in methylation [110] Methyl groups are transferred from S-adenosyl-L-methionine (SAM) to the cytosine residues of the DNA molecules [111]. The DNA demethylation reaction is catalyzed by the ten-eleven translocation (TET) enzymes, which add a hydroxyl group onto the methyl group of 5mC to form 5hmC (5-hydroxymethyl cytosine) [12,91]. The TET enzymes catalyze the hydroxylation of DNA 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), and can further catalyse oxidation of 5hmC to 5-formylcytosine (5fC) and then to 5-carboxycytosine (5caC). 5fC and 5caC can be removed from the DNA base sequence by base excision repair and replaced by cytosine in the base sequence. TET enzymes have central roles in DNA demethylation required during embryogenesis, gametogenesis, memory, learning, addiction and pain perception [112,113]. Deregulation of DNMTs or demethylases can cause widespread cellular detrimental effects, leading to global and gene-specific hypomethylation, as well as regional hypermethylation, which is linked to cancer [114,115].
Epigenetic regulation can also involve noncoding RNAs (ncRNAs), which are RNAs that are not translated into proteins. The well-known microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) are short molecules with a length of approximately 18-25 nucleotides, while lncRNAs are over 200 bases long, respectively [116]. Although long non-coding RNAs (lncRNAs) may span an open reading frame (ORF) and contain a single exon - a minimal distinguishing feature from other non-coding RNAs - they share similarities with protein-coding genes. However, lncRNAs are generally shorter, composed of fewer but longer exons, and exhibit low evolutionary conservation. This limited conservation complicates the identification of functional domains and hinders comparative studies across species, even when lncRNAs are located within highly conserved genomic regions.
In more detail concerning the classification of ncRNAs, major characteristic classes and subclasses of non-coding RNAs that have been described are: transfer RNAs (tRNAs); ribosomal RNAs (rRNAs); small RNAs, such as microRNAs, siRNAs, piRNAs, snoRNAs, snRNAs, and exRNAs; long ncRNAs (lncRNAs); long intergenic non-coding RNA (lincRNA) lincRNAs; circular RNAs (circRNAs); and examples of ncRNAs, such as the well-known Xist and HOTAIR. CircRNAs are stable, evolutionarily conserved, and single-stranded RNA molecules [69]. Unlike linear RNAs, circRNAs are closed-loop type RNAs with joined 3′ and 5′ ends [117]. Strictly speaking, for circRNAs, four types have been discovered, namely exonic circRNAs (ecircRNAs), circular intronic RNAs (ciRNAs), exon–intron circRNAs (EIciRNAs), and intergenic circRNAs [118,119]. CircRNAs function as miRNA sponges and can regulate RNA expression by consuming miRNA targets [120]. Furthermore, circRNAs can interact with RNA-binding proteins (RBPs) to influence certain physiological processes [121] and can also act as gene transcription regulators [122].
Both, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) act as post-transcriptional regulators and chromatin remodelers, respectively. These molecules can regulate gene expression by interfering with messenger RNA (mRNA) translations by way of degrading the mRNAs or through interactions with protein complexes involved in the regulation of gene expression [12,116,123]. Chronic inflammation modulates the expression of several oncogenic miRNAs, such as miR-21, which is overexpressed in IBD and CRC tissues and associated with inhibition of tumor suppressors like Programmed cell death protein 4 (PDCD4) and Phosphatase and Tensin homolog (PTEN) [124,125]. Micro RNA-21 is currently under investigation as a diagnostic biomarker and therapeutic target.
Similarly, inflammation-sensitive lncRNAs such as HOTAIR and LINC-PINT participate in epigenetic gene silencing through interaction with histone-modifying complexes, contributing to the persistence of an oncogenic transcriptional landscape.
5.1. Integration of Epigenetic Alterations and Inflammatory Pathways
The requirement to expand our view of the contributors in carcinogenesis by integrating the epigenetic alterations and inflammatory pathways, provides the foundation for the growing understanding of inflammation-driven epigenetic alterations, which has been conceptually summarized in Figure 2. The latter graphical annotation portray the interconnected steps leading from microbial triggers and immune activation to tumorigenesis through layered epigenetic reprogramming. It also emphasizes how chronic inflammatory signaling not only reshapes immune and epithelial responses but also establishes persistent chromatin changes that underpin the dysplastic transformation of colonic mucosa. Moreover, the Mechanistic Pathway – From Inflammation to Colorectal Carcinogenesis shows that, epigenetic changes form a crucial axis which are linking early inflammatory triggers - such as Helicobacter pylori, Mycobacterium tuberculosis, or gut dysbiosis – leading to sustained epithelial transformation. This proposed model illustrates how persistent cytokine signaling (e.g., IL-6, TNF-α, NF-κB) drives epigenetic remodeling, leading to progressive histopathological stages from normal mucosa to IBD, to dysplasia and to CRC.
5.2. Epigenetic Alterations in Inflammation-Associated Pathologies of the GI Tract – an overview
Many of these epigenetic mechanisms contribute to the development, progression and maintenance of IBD. They are usually triggered by a variety of environmental factors. Three critical periods have been mentioned during which the environment can favour the onset of the disease (1) during the prenatal period (in response to maternal lifestyle), (2) in the early postnatal period (during gut microbiota colonization) and (3) just before the disease onset [126]. As already mentioned, chronic inflammation can promote the occurrence and progression of colorectal cancer and epigenetic mechanisms, both inherited and acquired by environmental factors, participate in the transformation of inflammation into CRC.
In CRC, histone marks of aberrant acetylation and methylation levels on specific residues have been found, along with a plethora of deregulated enzymes that catalyze these reactions. Mutations, deletions or altered expression patterns transform the function of several histone-modifying proteins. This supports the crucial role of epigenetic effectors in CRC oncogenesis, their involvement in inactivation and activation of tumor suppressor genes and oncogenes, respectively, and their potential as biomarkers [127]. Moreover, in CRC, the commonly observed types of DNA methylation include hypermethylation of anti-oncogene DNA and hypomethylation of oncogene DNA [105]. Non-coding RNAs have also been found to be associated with the transformation of inflammation and the transition towards CRC. MicroRNAs have been found to be involved in the aforementioned, as well as in chemotherapeutic resistance [105], while lncRNAs have also been implicated with the transformation of chronic inflammation into CRC [105].
Moreover in GITB, it has been shown that non-coding RNAs have emerged as crucial regulators of various infectious diseases including tuberculosis [128]. In patients with GITB, miR-375-3p expression levels were noted higher in the plasma but lower in the ileal/ileocecal tissue compared to those who suffered from Crohn’s disease [129]. To date, few studies have explored the role of ncRNAs in the development and progress of gastrointestinal tuberculosis. However, research has revealed that gut microbiota influences immunological responses to tuberculosis by regulating non-coding RNAs [130,131]. For example, a study conducted by Yang and colleagues revealed that Bacteroides fragilis regulates lncRNA CGB which in turn modulates IFN-γ expression, enhancing anti-TB immunity [130].
The rising incidence of IBDs, their difficulties in diagnosis and treatment and their link to CRC, which is a high risk cancer, both as to its occurrence, its increasing incidence in younger age-groups (under 50 years old) and its high rank in cancer deaths, underscore the need for a better understanding of the molecular mechanisms underlying these diseases and their association. At the molecular level, both genetic (gene mutations) and epigenetic alterations have been found or implicated in IBDs and in CRCs (with and without genetic predisposition) [13,63,88,105]. These epigenetic alterations can drive initiation and progression of the inflammatory, or the precancerous and cancer state(s) by altering the gene expression profile(s) of noncancerous and cancer cells of the Tumor Microenvironment (TME) and elsewhere [63]. GI cancer syndromes, can arise from germline (inherited) epigenetic alterations [63]. However, familial epigenetic syndromes are rare and appear to be transmitted to offspring [63]. On the other hand, environmental factors have the potential to modify epigenetic states. These environmental factors, as previously mentioned, include infectious pathogens, diet, smoking, atmosphere, etc, thus, can alter the epigenome. These epigenetic alterations (depending on the changed epigenetic factor and/or profile may also be referred to as ‘aberrant’) can be part of the inflammation and cancer profile, either as causative factors or as resulting factors of the cancer phenotype [63]. The prevailing consensus suggest that epigenetic alterations in cancer occur and are more common than genetic alterations (mutations). Compared with gene mutations which are irreversible, epigenetic alterations, either inherited or acquired, are at large reversible by intervention. Advances in the genomic and epigenomic analyses technologies have led to the identification of epigenetic alterations in IBD and CRC. These epigenetic changes can have significant roles as biomarkers in the clinical setting and as important tools for the early detection, diagnosis, prognosis and management of precancer and cancer states in IBD and CRC [63].
6. Epigenetic Mechanisms Linking Inflammation in IBD to Colorectal Carcinogenesis.
This section explores the role of epigenetic modifications as critical intermediaries in the transition from chronic inflammation to colorectal cancer. It highlights key changes such as aberrant DNA methylation with special emphasis on how inflammatory signaling cascades modulate the epigenome, and how these alterations influence gene expression, immune evasion, and malignant transformation.
6.1. An Overview of Inflammation-driven Carcinogenic Transition in Humans and Murine Models
Inflammation is a well-known risk factor for cancer and epigenetic modifications such as DNA methylation and acetylation play crucial roles in this process [3,4]. In humans, chronic inflammation often leads to global DNA hypomethylation and regional hypermethylation, which can result in chromosomal instability and altered gene expression [3,4]. For instance, in colitis-associated cancer (CAC), oxidative stress and pro-inflammatory cytokines like IL-6 and TNF-α induce DNA methylation changes that contribute to malignant transformation [4]
In mouse models (for details see section 3), research has shown that inflammation-driven changes in DNA methylation and hydroxymethylation patterns can lead to an imbalance in DNA methylation-demethylation dynamics [54,55,112]. This imbalance can shift histone acetylation patterns, further promoting cancer initiation and progression [112]. Two examples, supporting the imbalance are: a) There is compelling evidence indicating that altered methylation and demethylation dynamics contribute to the pathophysiology of acute kidney injury (AKI). Also, in mouse models of ischemia–reperfusion injury (IRI), endotoxin, or maleate-induced AKI, a global reduction in 5hmC has been observed, whereas overall 5mC levels remain largely unchanged [113]. b) Moreover, aberrant DNA methylation is a hallmark of cancer, driving abnormal gene expression through hypermethylation and silencing of tumor suppressors, alongside hypomethylation and activation of prometastatic genes [114],
Histone modification abnormalities arise during the transformation of inflammation into CRC [105]. Recent work has shown that dysregulation of histone modifications is closely associated with pathogenesis of gastrointestinal disorders [88]. Aberrant histone modifications have been identified in colonic mucosa of patients with IBD which may contribute to the chronic inflammation which characterizes these diseases [132,133]. Similarly, alterations in histone modifications have been associated with the development and progression of CRC [134,135].
For example, transcriptomic analyses showed increased expression of inflammatory pathways in UC patient-derived organoids and tissues. More specifically, profiling for active enhancers enriched in the H3K27ac histone modification revealed UC-derived organoid enrichment for pathways indicative of gastrointestinal cancer, including S100 calcium-binding protein P (S100P), and also revealed novel markers for GI cancer, including both LYZ and neuropeptide S receptor 1 (NPSR1). Immunolocalization showed increased levels of LYZ, S100P, and NPSR1 proteins in UC and CAC. The aforementioned results from these models found precancerous molecular pathways that are already activated in UC [136].
The above work focused on the genome-wide enhancer state in cells and tissues derived from CRC patients. However, Chen et al., [137] wanted to further elucidate on how the dynamic states of chromatin contribute to the inflammation-cancer transition in colitis-associated CRC. To this end, they performed epigenomic and transcriptomic studies in a colitis-associated CRC mouse model [138] induced by azoxymethane (AOM) and dextran sodium sulfate (DSS) [138]. Combining the data from the above analyses, they generated a genome-wide landscape of chromatin states during inflammation-cancer transition. They support that their work provides important datasets for CRC studies and reveals new regulatory mechanisms and potential targets for clinical investigations. Their results are not only interesting but more importantly, reveal key modifications and chromatin positions and states in the inflammation-cancer transformation. Functional analysis of different expressed genes (DEGs) compared with control tissues showed that DEGs of the 2- and 4-week samples were enriched in inflammation pathways, and those of the 7- and 10-week samples were enriched in inflammation and cancer-related pathways. Thus, they considered that the 2- and 4-week represented the inflammation stage and the 7- and 10-week represented the tumor stage. This was confirmed by comparing their data with a previous study with mouse models which showed that their 2- and 4-week data were grouped with the inflammatory bowel disease samples and the 7- and 10-week data were grouped with the CRC samples of the previous study.
To investigate the chromatin states in their model, Chen et al., [137] performed ChIP-seq of H3K27ac, H3K4me1, H3K4me3, H3K27me3 and H3K9me3 with tissues collected at the 5 time points. To determine the landscape of chromatin states, they defined the states with different combinations of modifications: quiescent state regions (no detected modifications), heterochromatin state regions (dominant H3K9me3), transcriptionally repressed regions (dominant strong or weak H3K27me3), active enhancer state regions (high H3K4me1 and H3K27ac), poised enhancer state regions (high H3K4me1 and low H3K27ac), bivalent enhancer state regions (H3K4me1, H3K27ac, and H3K27me3), weakly active enhancer state regions (low H3K4me1 and H3K27ac), active promoter state regions (totally four types of regions were identified which all have H3K4me3 and are close to TSS), and poised promoter state regions (high H3K4me3 and H3K27me3). They then compared the chromatin states at different time points and found that all the above chromatin state regions were highly dynamic during the inflammation-cancer transition. The enhancer state regions kept increasing during transition, especially at the late tumor stage.
With respect to histone phosphorylation, Xiao et al., [139] found that reduced levels of phosphorylated histone H3 at Ser 10 (H3S10ph) in mouse and human cancer cell lines. Their work showed that phosphorylation events with T-LAK cell-originated protein kinase (TOPK) facilitated carcinogenesis of colon cancer [139]. Moreover, histone phosphorylation does not act alone, but partners with other histone modifications to control gene regulatory processes. In vitro studies with mouse and human cancer cell lines, showed that the HAT, GCN5 exhibits a preference for H3S10ph, compared to non-phosphorylated histones [4,140] H3S10ph can also stabilize histone H4 acetylation, while dephosphorylation of H3S10ph collaborates with HDAC1, 2 and 3-induced deacetylation of histone H4 under stress [141]. It has also been reported that H3S10ph assists in expanding genomic domains which have H3K4 methylations (open chromatin marker), and concomitantly restricts the propagation of heterochromatin enriched in H3K9me2 and DNA methylation (closed chromatin marks) [142]. Thus, extensive crosstalk takes place between histone phosphorylation and other histone modifications to regulate gene expression in both inflammation and cancer [4].
Histone H2B ubiquitination of lysine 120 (H2BK120ub) has been shown to have a role in inflammation-related colorectal cancer. Specifically, in both human colonic tissue cultures and mouse animal models, reduced levels of H2BK120ub and its E3 ligase, RNF20, were found to activate colonic inflammation and tumorigenesis by way of recruiting NF-κB, a major transcription factor regulating inflammation signaling in both mice and humans [143]. Other studies also demonstrated that dysregulated H2BS120ub causes genomic instability and promotes tumorigenesis and cancer progression in other cancer types [127,144]. Similar to phosphorylation, which has been shown to interact with other histone modifications or histone-modifying enzymes in inflammatory-associated cancers, histone ubiquitination can also crosstalk and influence other histone modifications. For example, H2BK120ub helps in the methylation of H3K79 and H3K4 at promoter regions to induce gene transcription [145,146]. All in all, histone ubiquitination possesses roles in both transcriptional regulation and inflammation-induced tumorigenesis [4].
7. DNA Metylation and Histone Modifications During Tumorigenesis of the GI
This section delves into how chronic inflammation mediates long-term epigenetic reprogramming of intestinal epithelial cells, serving as a bridge between environmental stress and genetic dysregulation. Epigenetic mechanisms such as DNA hypermethylation of tumor suppressor genes (e.g., MLH1, SEPT9), histone modifications (loss of acetylation, H3K27me3), and the dysregulated expression of non-coding RNAs (e.g., miR-21, lncRNA HOTAIR) are discussed in the sections below. The contribution of inflammation-induced reactive oxygen species (ROS) and immune cell infiltrates to these alterations is also discussed. These changes not only disrupt epithelial homeostasis and promote carcinogenesis, but also provide biomarkers and therapeutic targets.
7.1. DNA methylation in IBD, CAC and CRC
In GI cancers, and most other cancer types, global DNA hypomethylation is observed as well as aberrant regional DNA hypermethylation. The latter has been studied extensively in virtually all cancers and is believed to contribute to cancer formation via repression of tumor suppressor gene expression. Hypomethylation is also a hallmark of cancer, however its functional role in cancer is less well understood. It contributes to cancer formation via induction of genomic instability, which may induce the expression of parasitic elements, or the expression of oncogenes or cancer germline genes (germline genes with mutation(s) that are inherited) [63,147,148,149,150]. However, despite the lack of knowledge as to its functional role in cancer, results have shown that global hypomethylation which generally occurs on transposable elements (e.g., LINE1 or L1 or SINE/Alu), occurs in many cancer types, including CRC. Studies have demonstrated that L1 hypomethylation occurs widely in CRC patients and is associated with clinically relevant bio-pathological features [151] and correlates with poor prognosis and early onset (<60 years) [151,152,153,154,155]. In fact, LINE-1 hypomethylation was found to be significantly correlated with shorter overall survival (OS), disease-free survival (DFS) and cancer-specific survival (CSS). Importantly, a shorter OS that was found to be associated with L1 hypomethylation was also identified in early-stage colorectal cancers [151,156]. These correlative findings represent promising tools for prognosis prediction. Moreover, a large study performed on colon and rectum carcinoma cases (1317 cases) highlighted the association between L1 hypomethylation and higher colorectal-specific mortality, which was stronger in proximal colon cancer as compared to distal or rectal cancer [157]. Moreover, patients with low LINE-1 methylation who were treated with adjuvant chemotherapy survived longer than those treated by surgery alone, suggestive of a survival benefit from the use of oral fluoropyrimidines. In contrast, a survival benefit from chemotherapy was not observed for patients with high LINE-1 methylation. This suggests that L1 hypomethylation versus L1 hypermethylation can be used as a predictive marker for the survival benefit of adjuvant chemotherapy with oral fluoropymidines [158]. Furthermore, detection of L1 hypomethylation levels in plasma cell-free DNA (cfDNA) was recently proposed as a novel biomarker for detection of CRC in the early stages (biomarker for CRC, particularly for early-stage detection).
DNA methylation in physiological cells occurs predominantly in repetitive genomic regions, including satellite DNA and parasitic elements, such as long interspersed transposable elements (LINEs) and short interspersed transposable elements, maintaining genomic integrity [63,159,160]. Unlike the rest of the genome, CpG islands, particularly those associated with promoters, are generally unmethylated in normal cells, providing access to transcription factors and chromatin-associated proteins for the expression of most housekeeping genes and other genes that are regulated, although some of them become methylated in a tissue-specific manner during early development and/or in tissues under differentiation (i.e., in which some of the CpGs level of DNA methylation is approximately 6%) [63,161].
DNA hypermethylation promotes tumorigenesis and progression of colitis-associated CRC (CAC). IBD patients show DNA methylation changes both at the cell and at the tissue level [12]. These changes also differ between UC and CD patients [162,163,164,165,166,167,168]. In the following paragraphs of this section, examples of changes in DNA methylation levels and DNA methyltranferases associated with IBD and inflammation-associated CRCs will be summarized. Table 2 lists the DNA methylation status of certain genes which have been associated with certain inflammatory bowel conditions and their use as diagnostic and/or therapeutic biomarkers.
The expression of DNA methyltransferase 1 (DNMT1) which has a crucial role in maintaining DNA methylation patterns in the cell generations, is higher in CAC samples than in those tumor tissue samples of patients with sporadic CRC. Generally speaking, sporadic cancer refers to cancer that arises due to random DNA damage and subsequent genetic mutations in cells, acquired during a person's lifetime, rather than being inherited from a parent. These mutations typically occur in somatic cells (non-reproductive cells), don't have a clear pattern of inheritance within families and are not passed on to future generations. Specifically, sporadic CRCs are cancers that arise from the colorectum without known contribution from germline causes (germline inherited mutation(s)) or significant family history (inherited, familial) or inflammatory bowel disease. The increased levels of the DNA methyltransferase, DNMT1 indicate increased DNA methylation levels in CAC tumor tissues [105,169].
Examples of genes of importance which are tumor suppressor genes and related to cell cycle events that were found to be hypermethylated in tumor tissues of CAC patients are the cell cycle inhibitor gene, p16 [170,171] and the gene involved in the regulation of p53 and p14 [172]. P14 binds to MDM2 and stabilizes the MGM2-p53 complex which holds inactive p53. There is an inverse relationship between p14 expression and p53 function in tumor cell lines. Indeed, down-regulation of the p14 gene by DNA methylation is a relatively early event in ulcerative colitis-associated colorectal carcinogenesis [173]. Furthermore, the DNA methylation levels of the genes TFP12 (tissue factor pathway inhibitor), ITGA4 (integrin alpha 4) and VIM (vimentin) are increased in inflamed colon tissue. These results strongly imply a high risk for development of inflammation – induced CRC and that the methylation levels of these genes can be used as risk markers for inflammation-associated colon cancer [174]. Altered DNA methylation patterns in tumors have been termed DNA methylation valleys (DMV). These regions extend over several kilobases of DNA, are strongly hypomethylated in most normal tissues, and are enriched in genes for transcription factors and development [172]. DMVs have been shown to become hypermethylated in colorectal cancer and may thus contribute to the aberrant epigenetic programming of tumor cells [175]. Specifically, in a colitis-induced mouse colon cancer model, investigators found hypermethylation of DMVs leading to silencing of the DMV-related genes, thus facilitating inflammation-induced cell transformation. Based on the above, the authors [172] proposed that the DNA methylation status of a specific subset of DMVs may be a promising early detection biomarker of inflammation-induced CRC.
Other tumor suppressor genes that have been reported to be hypermethylated in colorectal cancer, have also been proposed to be used as early diagnostic markers of CRC for detection in stool or blood samples of patients. Among the genes that undergo repression in CRC due to CpG hypermethylation, the most extensively studied for their impact in cancer diagnosis or prognosis are MGMT, SEPT9, HLTF, NDRG4, BMP3, CDH13, APC, MLH1, CDKN2A, RASSF1A and RUNX3 [151].
More importantly, and in terms of the mechanisms and signalling pathways involved, inflammation-induced oxidative stress, reactive nitrogen species, and cytokine-driven transcriptional activity promote aberrant DNA methylation patterns in epithelial cells. Hypermethylation of CpG islands in promoter regions leads to transcriptional silencing of tumor suppressor genes such as MLH1, CDKN2A (p16), and SEPT9 [176,177]. Notably, Septin 9 (SEPT9) promoter methylation has been utilized as a clinical biomarker for early CRC detection and has been linked to increased inflammatory signaling via NF-κB activation [178].
In IBD, global DNA hypomethylation is also observed, contributing to genomic instability. Simultaneously, promoter hypermethylation selectively targets genes involved in DNA repair and apoptosis, tipping the balance toward uncontrolled cell survival [179]. Furthermore, DNA repair protein, O6-methylguanine-DNA methyltransferase (MGMT) promoter hypermethylation has been detected early in CRC, while hypermethylation of Helicase-like Transcription Factor (HLTF) was detected in the serum of CRC patients and is associated with an increased risk of disease recurrence and death. Another study confirmed the positive correlation of serum positive HLTF and transmembrane protein containing epidermal growth factor and follistatin domains (HPP1/TPEF) DNA hypermethylation with tumor size, stage, grade and metastatic disease [151,180]. Hypermethylation of N-myc down-regulated family member 4 (NDRG4) was correlated with CRC clinical features [181], while Bone Morphogenetic Protein 3 (BMP3) hypermethylation was correlated with microsatellite instability [182]. Cyclin Dependent Kinase Inhibitor 2A (CDKN2A/p16) hypermethylation has been associated with worse prognosis in CRC, i.e., reduced OS, presence of lymph node metastasis and lymphovascular invasion [151,183,184].
More studies have also shown that methylation of specific genes is associated with inflammatory conditions, dysplasia, and malignant transformation, indicating that it is involved in inflammation-induced cell transformation. Many proinflammatory cytokines secreted as a result of NF-κB and STAT3 (transcription factors) signaling pathway activation are activated and promote the transformation of inflammation into CRC [185]. For example, IL-6 sliences the expression of the suppressor of cytokine signaling 3 (SOCS3) by inducing high expression levels of DNMT1. SOCS3 is an important negative regulator of cytokine-induced STAT3 signaling, and its silencing ultimately contributes to the occurrence of CRC [186]. Il-6 has been demonstrated to increase the levels of methylation of the promoter regions of genes related to tumor inhibition, cell adhesion and apoptosis resistance. These increases in methylation levels could be prevented by treatment with the DNMT1 inhibitor, 5-azadeoxycytidine (Figure 2) [169]. IL-6 produced during intestinal inflammation can modulate the expression of certain genes (CYP2E1, CYP1B1, cytochrome P450 enzymes). This alters the metabolic capability of epithelial cells, which in turn may increase dietary carcinogen activation and DNA injury, leading to the occurrence of CRC [105].
7.2. Histone Modifications in Inflammation-related Cancer Progression
Histone modifications are also major epigenetic determinants of chromation structure and function that can be dysregulated by inflammatory cytokines such as IL-6 and TNF-α. Pro-inflammatory conditions often result in loss of histone acetylation (see 7.2.1 for more details), particularly at tumor suppressor gene loci, through the upregulation of histone deacetylases (HDACs). For instance, reduced H3K9 and H4K16 acetylation have been reported in colonic tissues during chronic inflammation and dysplasia [187].
Moreover, repressive methylation marks like H3K27me3, catalyzed by the Polycomb Repressive Complex 2 (PRC2), are enriched at loci encoding differentiation and apoptotic regulators during early CRC development. These modifications, initially reversible, become fixed under continued inflammatory pressure, locking cells into a dedifferentiated, proliferative state [188].
7.2.1. Histone Acetylation in CRC
Aberrant acetylation marks have been associated with CRC pathogenesis. Among the first histone marks that were found to be deregulated in CRC was the global loss of acetylation of histone H4 at lysine 16 (H4K16ac) in cell lines and primary tumors [127,189]. Also, global hypoacetylation of H4K12 and H3K18 correlated with poorly differentiated colorectal adenocarcinomas [190] whereas, global acetylation was found to be increased in moderately differentiated tumors, increasing progressively from normal tissue to carcinoma (e.g., H4K12ac). H3K9 deacetylation was linked to the inactivation of the tumor suppressor gene E-cadherin in CRC cell lines [191,192]. Interestingly, in a CRC cell line stably transformed with the oncogenic form (mutated) of the Harvey-Ras protein that triggers epithelial to mesenchymal transition (EMT), global H3K9/14 acetylation was observed in the promoters of e-cadherin and cyclin D1 (an EMT and cell cycle gene, respectively) that induced the down-regulation of the corresponding protein expression via the Ras signaling pathway [193]. Moreover, H3 and H4 acetylation is crucial for transcriptional activation of 15-lipoxygenase-1 (15-LOX-1), whose product is silenced in CRC cells [194]. Down-regulation of p21waf1 transcription (a tumor suppressor gene that inhibits cell cycle progression by inhibiting cyclin-dependent kinases of the cell cycle) that is also involved in CRC has been associated with H3 hypoacetylation and histone enzymes [195]. Notably, reduced acetylation was associated with the silencing of N-myc down-regulated family member 1 (NDRG1), a specific metastasis suppressor gene, in a highly metastatic colon cancer cell line SW620 as compared to the colon cancer cell line, SW480 that has a low metastatic ability, in which higher levels of H4 acetylation were found [127,196]. Moreover, enhancers of genes with high expression rates in the cancer state of colon tumorigenesis in mouse model showed elevated H3K27ac levels [137].
7.2.2. Histone methylation in inflammatory signaling and CRC progression
Histone methylation is also important in inflammatory signaling. For example, H3K9 methyl transferases (HMTs) and demethylases (HDMs) are counteracting to balance the status of H3K9. Moreover, an important histone demethylase, Jmjd3 - also known as KDM6B - is responsible for the deletion of histone marks and control of differentiation and cell identity in macrophages. Thus Jmjd3 functions as a link between inflammation and epigenetic reprogramming [5,197]. Macrophages exposed to bacterial products and inflammatory cytokines induce Jmjd3, which in turn binds to polycomb group target genes and regulates their (repressive) H3K27me3 levels and thus transcriptional activity [198]. Continuous IL-4 cytokine treatment activates the Jmjd3 demethylase which removes the H3K27me3 repressive mark from the STAT6 promoter. Jmjd3 is then positively regulated by the now activated STAT6 through promoter binding. This demethylase also triggers the expression of other specific inflammatory genes by removal of their repressive H3K27me marks [5,199].
Thus, histone methylation can affect inflammatory signaling through the above as well as through other inflammatory signaling pathways in many forms of cancer. Cooperative interactions between DNA methylation and histone methylation have been also shown in severe systemic inflammation (SSI). In many forms of cancer, including CRC, deregulation of these inflammatory pathways, through deregulated histone methylation has been ascertained. In colon cancer, production of the Th1-type chemokines, CXC chemokine ligand 9 (CXCL9), and CXCL10, which mediate T cell trafficking, is inhibited by H3K27me3 in their gene promoters [200]. However, CXC chemokine receptor 4 (CXCL4) is upregulated by EZH2-mediated loss of miR-622, thus establishing a favorable environment for the evasion of the immune surveillance system [5,201,202].
In CRC (among other malignancies), a classic histone methylation mark is the loss of tri-methylation at lysine 20 of histone H4 (H4K20me3), along with the global loss of DNA methylation and acetylation at lysine 16 [189]. In addition, mono-, di- and tri-methylation of H3K4 (activating) are targets of SMYD3 (SET and MYND domain-containing protein 3) HMT and LSD1 (lysine-specific demethylase 1) HDM, both of which are highly expressed in CRC [127]. Moreover, genome-wide profiling concerning histone methylation in CRC showed similar profiles for H3K4me3 (activating) and H3K27me3 (repressive) in normal colon and tumour tissue, differing only in CRC cell lines. Tumor genes that were positive for H3K4me3 already in normal tissue became hyperactivated in tumors, whereas genes with H3K27me3 and low expression in normal colon cells became hypersilenced in CRC tumors [127,203]. H3K4me3 and the loss of H3K27me3, along with increased H3 acetylation, were also associated with reactivation of silenced genes in CRC. With respect to CRC metastasis, a decreased level of H3K4me3 (along with decreased acetylation ,see above in 7.2.1) “Histone acetylation in CRC”) in the coding region of the NDRG1 gene in the highly metastatic colon cancer cell line, SW620, was found and associated with the gene’s down-regulated expression [127,196].
A crucial histone mark of heterochromatin linked to transcriptional repression, H3K9me3, was found to be increased in cancer types, possibly promoting gene silencing of tumor suppressor genes [204]. Overexpression of the protein-lysine methyltransferase G9a, and H3K9-specific p53 methyltransferase, have been reported in CRC, as well as in other cancer types, associated with suppressive alterations in gene expression [127].
7.2.3. Histone Phosphorylation in CRC
Histone phosphorylation is aberrantly regulated in CRC, causing an imbalanced state of gene transcription [205]. It affects the transcription of CRC-related genes, promoting the development and progression of the disease. Researchers [205,206] have reported that EZH2 and the anti-silencing factor 1 phosphorylate histone H2B and H4, respectively, activate autophagy-related gene transcription, induce autophagy in colorectal cancer cells, and promote its development and progression in tissues. Moreover, VprBP, a kinase that is excessively expressed in cells of CRC, is directly involved in epigenetic gene silencing via histone H2A phosphorylation-mediated regulation of growth regulating gene transcription, significantly increasing carcinogenesis and cancer cell proliferation [205,207]. Moreover, researchers showed that phosphorylation of H2A.X (an H2A class subtype) is elevated in CRC tissues and was correlated with a more aggressive type of tumor and poor CRC patient survival [208,209]. Another example of aberrantly regulated histone phosphorylation in CRC is the observation of the downregulation of the expression of the dual specificity phosphatase 22 (DUSP22) in CRC specimens and reduced DUSP22 expression in stage IV patients who mainly showed poor survival outcomes [209,210]. Moreover, Chen and co-workers [211] found that PKCε is a kinase that phosphorylates MIIP-S303. EGF stimulation induces PKCε-dependent phosphorylation of migration and invasion inhibitory protein (MIIP) at Ser303; this phosphorylation promotes the interaction between MIIP and RelA in the nucleus, by which MIIP prevents histone deacetylase 6 (HDAC6)-mediated RelA deacetylation, and thus enhances transcriptional activity of RelA, facilitating tumor metastasis. Meanwhile PP1, which functions as a phosphatase, is found to mediate MIIP-S303 dephosphorylation and its expression level inversely correlates with metastatic capability of tumor cells (colon tumor cell lines HCT116 and CaCo2). Moreover, clinical analyses indicate the level of MIIP-S303 phosphorylation correlates with colorectal cancer (CRC) metastasis and prognosis [209,211].
Table 3. shows representative histone modifications whose levels were found to change in different inflammatory bowel conditions (different samples), including CAC and CRC and thus, could be considered potential biomarkers.
8. Exploring the Role of Non-coding RNAs in Epigenetic Regulation of IBD and CRC
MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) act as post-transcriptional regulators and chromatin remodelers, respectively. Chronic inflammation modulates the expression of several oncogenic miRNAs, such as miR-21, which is overexpressed in IBD and CRC tissues and associated with inhibition of tumor suppressors such as PDCD4 and PTEN [124,125]. MicroRNA-21 is now under investigation as a diagnostic biomarker and therapeutic target.
Similarly, inflammation-sensitive lncRNAs such as HOTAIR and LINC-PINT participate in epigenetic gene silencing through interaction with histone-modifying complexes, contributing to the persistence of an oncogenic transcriptional landscape. The next subsections shed light to related research in this subject that have been reported in scientific literature.
Additionally, in the current review recent versions of ncRNA-oriented databases and biotools were employed by the first author, such as LncRNA2Target versions V2.0 and V3.0, Open Targets Genetics v22.10, The Human Reference Protein Interactome Mapping Project (HuRI) NONECODE V6, lncRNAfunc, LncRNADisease v2.0, starBase v2.0, EVLncRNAs2.0, lncRNAfunc, LncRNAWiki 2.0, and the updated LNCipedia_5.2, for the investigation of ncRNA contributions to the disease related to this study [212,213,214,215,216,217,218,219,220].
8.1. MicroRNAs - molecular insights of microRNA dysregulation or aberrant function and its involvement in IBDs and CRC
Dysregulated expression of microRNAs and other non-coding RNAs have been reported in many tumor types, including CRC [151,221,222,223,224] In colorectal cancer, part of this dysregulation is due to epigenetic mechanisms acting on miRNA and ncRNA gene regulatory regions. More specifically, investigators [225] have reported that hypermethylation of miRNA-124-1/3 in colon cancer and other solid tumors, show reduced expression of mature miRNA-124a, along with a concomitant increase of target CDK6 gene and Rb protein phosphorylation (regulatory genes/regulatory protein factors of cell cycle progression) [225]. In CRC tissues, the hypermethylation of microRNA-124 family genes was observed in more than 70% of the cases. Other miRNAs were also found to be aberrantly methylated in the early stages of CRC, such as miR-137 [226], miR-200 family [227], miR-129 and miR-9 [228], which underscores the importance of epigenetic regulation of miRNAs expression in tumor suppression [151].
Evidence has also accumulated showing miRNAs’ critical contribution to the disease onset and progression of IBD, supporting further investigation as to the possible role(s) of miRNA as markers in differential diagnosis. MicroRNA expression patterns have been found to differ significantly between IBD patients and healthy controls, between CD patients and UC patients, as well as between patients in remission and those in the active stages of the illness [13]. Moreover, CD patients always displayed increased levels of miR-340 in peripheral blood. In another study, four specific miRNAs (miR-20b, miR-98, miR-125b-1 and let-7e) were identified in colonic mucosa of UC patients which were differentially upregulated by more than 5-fold in active UC as compared to inactive UC, active CD, inactive CD and healthy controls. These results not only corroborate accumulated evidence that microRNAs contribute to disease onset and progression, but also support the use of specific miRNAs as diagnostic markers of the differential states of colon inflammatory conditions [229] .
In line with disease onset and progression, numerous microRNAs have been implicated or identified as advancing transformation by participating in NF-κB and STAT3 signaling pathways that play an important role in transformation of inflammation into cancer [230]. NF-κB and STAT3 are transcription factors that regulate the expression of a variety of genes that coordinate innate and adaptive immune responses, and responses to cellular stimuli, respectively. MicroRNAs roles in GI tract cancers, namely, colon, gastric and liver cancers have been investigated., and found to play key roles in cell growth and apoptosis [231]. Their activation and the interaction of their signaling pathways play vital roles in control of the communication between cancer cells and inflammatory cells [231]. This interaction can lead to the transformation of inflammation into cancer [105]. Numerous microRNAs promote this transformation by participating in these signaling pathways. TNF-α (Tumor Necrosis Factor-alpha, a multifunctional cytokine) increases the expression of miR-105, which targets RAP2C (is a Ras-related protein subfamily of the Ras GTPase superfamily that regulates cell proliferation, differentiation and apoptosis) and activates NF-κΒ signal transduction by IKK (central core element of the NF-κΒ cascade), which ultimately contributes to CRC progression [232]. Another study showed that TNF-α leads to high expression levels of miR-19a, which can also activate NF-κΒ signaling thus enhancing colitis and the occurrence of CAC. STAT3 is a downstream target of IL-6 and can interact with miR-21, miR-181b-1, PTEN and CYLD. This may be an epigenetic switch (one of many) connecting inflammation to cancer [233]. By activating miR-21, it also promotes TGF-β-dependent epithelial-mesenchymal transition (EMT) in CRC [234]. Additionally, CRC samples and cell lines, increased STAT3 expression levels are accompanied by elevated miR-572. Elevated levels of miR-572 have been found to reduce the expression of the pro-apoptotic protein, Modulator of Apoptosis 1 (MOAP-1), which can contribute to CRC progression [235]. Moreover, natural killer (NK) cells promote tumor cell apoptosis by secreting high levels of cytokines, such as IFN-γ and TNF-α [236]. MicroRNA-24 levels were found to be increased in NK cells from CRC patients. This is thought to play a causative role in the decreased levels of cytokines, including TNF-α and IFN-γ. In this manner, the increased miR-24 levels inhibit the cytotoxic effects of NK cells on CRC cells [237].
8.2. MicroRNAs as Biomarkers in GI diseases
MicroRNAs circulate in human peripheral blood in a stable form and are also present in other body fluids such as urine, saliva, milk, cerebrospinal fluid, and feces [238]. Changes in miRNA expression profiles have been considered for applications in early detection, prognosis, and diagnostic classification of IBD. The most recent studies in the field have measured circulating miRNAs in body fluids and in homogenized tissue biopsies using microarray, RT-qPCR, and next-generation sequencing (NGS) techniques (Table 4) [239,240]. Notably, miRNAs are increasingly being investigated and will be more scrutinized in the future, as non-invasive markers for CRC.
MicroRNA-21 and miR-155 have been identified repeatedly and appear to be the most studied miRNAs associated with IBD [240,241]. MicroRNA-21 is potentially the most interesting miRNA involved in IBD, with associations between miR-21 and disease It has been replicated in several studies and functional significance has been reported in mouse models of IBD [242]. MicroRNA-21 is elevated in both UC and CD patients and is involved in several proinflammatory functions, such as modulating T-cell responses and controlling epithelial tight junction proteins [241]. It should be emphasized that deactivation of miR-21, reduced inflammatory responses and improved survival rate in a mouse model of DSS-induced colitis [243].
In another study, only miR-150 was downregulated out of the 25 miRNAs specifically expressed in the serum of UC patients. Additionally, a significant increase in miR-29a was observed in the blood of UC patients, which regulates innate and adaptive immune responses by targeting interferon (IFN)-γ. Consistent with these findings, two studies have demonstrated an increase in miR-29a expression in the colon of both active and inactive UC patients. Furthermore, it is speculated that serum miR-29a has strong potential as a novel non-invasive biomarker for the early detection of colorectal cancer. In addition, colorectal cancer is known to represent a well-defined complication of long-standing UC. It has been demonstrated that miR-29a is associated with active and inactive UC and is considered a good biomarker for the early detection of colorectal cancer [244,245], along with the increased expression of miR-127-3p in both UC and CD patients, suggesting that miR-127-3p could be a potential biomarker for IBD [246].
Two more studies reported that a) serum microRNA146b2-5p (miR2-146b2-5p) expression was 2.872- and 2.722-fold higher in CD and UC patients than in healthy controls [247], and b) a study, which is dealing with the miRNA family, miR-125, consisting of miR-125a and miR-125b found that only miR-125a is reduced in patients with active disease and negatively correlates with disease severity and inflammatory cytokines in patients with CD [248].
Rashid and co-workers showed that patients with active disease exhibit a distinct miRNA profile and that miR-223 and miR-1246 are generally present at high levels in feces and are upregulated in active patients with IBD. However, the results are not the same in serum samples from the same patients (in serum samples, miR-223 shows a greater increase in patients with CD, as discussed above). This increase was seen in patients with UC as well as CD; it is thus concluded that these miRNAs are generally associated with intestinal inflammation [240].
Moreover, Schaefer and co-workers found that serum samples from patients with IBD showed higher levels of miR-16, miR-21, and miR-223 than controls and were higher in CD patients. In more detail, distinctive changes in miRNA expression were observed in stool samples from patients with IBD for all tested miRNAs with the highest expression of miR-155 and miR-223 in the control groups. In conclusion, miR-21, miR-155, and miR-223 exhibit significant levels and could potentially be considered biomarkers for IBD [249].
Additionally, research has identified a cluster of nine miRNAs altered in the rectal tissue of pediatric patients with IBD. Among the miRNAs selected for validation, four were significantly decreased (miR-192, miR-194, miR-200b, and miR-375), and four were significantly increased (miR-21, miR-142-3p, miR-146a, and let-7i) in pediatric patients compared to healthy controls. Differentiating UC from CD in the pediatric population remains challenging, as the diagnosis may be unclear even after endoscopy. Three serum miRNAs were significantly altered in children with UC, including miR-192 and miR-21, which have previously been shown to be elevated in pediatric CD [250]. The third miRNA was miR-142-3p. However, the study found that colitis-associated miRNA levels could not distinguish UC from CD [251].
Furthermore, regarding the utility of miRNA biomarkers in the treatment of IBD, a recent study was able to identify 8 miRNAs in serum samples that are associated with clinical response to anti-TNF-α and glucocorticoid (GC) therapy [252]. These are miR-146a, miR-146b, miR-320a, miR-126, and let-7c. Although miR-146a and miR-146b are elevated in serum and biopsies of individuals with IBD, they appear to be reduced by anti-TNF and GC treatment. As mentioned above, these miRNAs have also been reported as diagnostic biomarkers of IBD, showing a high correlation with endoscopic disease activity. In parallel, miR-320a, miR-126, and let-7c also show downregulation [253]. However, research in this area is limited, and results are mixed. Therefore, further studies are needed to fully investigate and validate the utility of miRNAs as predictive markers for treatment outcomes in IBD [254].
Importantly, Shi and co-workers showed that alterations in miR-31 levels in TNBS-induced colitis and in IL-10 knockout mice could regulate the IL-12/23 pathway, resulting in improvement or aggravation of colitis. Furthermore, the therapeutic effects of miR-31 inhibitor were eliminated after inducing IL-25 (interleukin-25) overexpression in the colon in mice [255]. In addition, it was proven that miR-223 interacts with the IL-23 pathway by targeting claudin-8 (CLDN8), which is involved in the formation of tight junctions in the gastrointestinal tract. Intraperitoneal injection of antagomiR-223 activated CLDN8 and reduced intestinal permeability in mice with colitis [256].
Interestingly, using a DSS-induced IBD mouse model, a study on miR-133a and its target UCP2 (mitochondrial uncoupling protein 2) revealed that miR-133a levels were reduced following DSS treatment. The DSS-induced IBD was also alleviated by introducing a miR-133a mimic, indicating that miRNA mimics could also serve as therapeutics for IBD [257].
MicroRNA-223 and miR-451 have been identified as strong markers for distinguishing CRC patients from healthy individuals, while miR-223 was also highlighted as biomarker for IBD [240,258]. Additionally, miR-135b has been identified as a marker of cancer origin, further suggesting its potential role in the detection of CRC [259]. Furthermore, miR-421, miR130b-3p, and miR27a-3p were shown to be elevated in CRC patients' fecal samples. An important addition has demonstrated, that a developed algorithm has supported the identification of patients with advanced colorectal neoplasia based on a 5 criteria task force, i.e., fecal levels of two microRNAs (miR-421 and miR-27a-3p), fecal hemoglobin concentration, patient age, and patient sex [260,261].
8.3. LncRNAs in IBD and CRC- A General Overview
LncRNAs are important in cancer biology, generally causing abnormal expression of gene products, which can be involved in the progression of various human tumors [226,262]. They are also involved in the transformation of inflammation into CRC. An example of this is indicated in the study by Hanisch et al. [263] who showed that the interaction between lncRNA PRINS and miR-491-5p regulated the pro-apoptotic factor PMAIP1, a pro-apoptotic member of the Bcl-2 protein family with a specific domain (BH3 domain) that allows it to interact with other Bcl-2 family proteins, ultimately triggering apoptosis. Moreover, the aforementioned interaction enhanced the anti-apoptotic effect of a small peptide known for its role in mucosal protection and wound healing, particularly in the gastrointestinal tract, Trefoil Factor 3 (TFF3), against the pro-apoptotic effects of IFN-γ and TNF-α in CRC cells [263]. Moreover the lncRNA, FEZF1-AS1 expression was found to be higher in CRC tissue than in normal tissue and this was associated with poor prognosis [264]. FEZF1-AS1 can bind to the pyruvate kinase 2 (PKM2) protein which increases its stability, thus increasing its levels and activity in the cytoplasm and nucleus. This upregulation induced by the FEZF1-AS1 lncRNA, further activates STAT3 signal transduction and accelerates the transformation of inflammation into cancer [264]. Research work has found that the lncRNA, AB073614 induced EMT in CRC by regulating the JAK/STAT3 pathway [265]. Moreover, recent studies have shown that lncRNAs not only play a role in the transformation of inflammation into cancer but can also induce the resistance of CRC to chemotherapy by regulating inflammatory signaling pathways [266], showed that the lncRNA, HOTAIR contributes to 5-fluorouracil (5FU) resistance by suppressing miR-218 and activating NF-κB/TS signaling in CRC. HOTAIR has been positively correlated with progression, survival and poor prognosis in different types of cancers, including CRC. Therefore, researchers have endorsed HOTAIR as a novel prognostic indicator and therapeutic target for CRC [105,267,268].
Further on the importance of lncRNA - miRNA interactions, a detailed functional annotation of the new characterized lncRNAs, FIGNL2-DT and GAS5-AS1, is presented in Table S1 (supplementary material), and lncRNA – miRNA interactions are visualized (for 3 important miRNAs) in Figure S1 (supplementary material). Table S1 illustrate the plethora of lncRNA - miRNA interactions involved in CD, with different levels of significance for the disease, according to teir evaluation by the miRNA Recognition Element (MRE) score [269].
In more detail, the first author performed the analysis and mapped lncRNA – miRNA interactions, through the ncFANs-NET module of the ncFAN v2.0 network analysis biotool, which is an updated and full-featured platform for noncoding RNA (ncRNA) functional annotation, that comprises the following three major modules for the lncRNA analysis, i.e. lncFANs-CHIP, ncFANs-NET and ncFANs-eLnc [269], Conducting this research, three (3) ncRNA transcripts were found in our query list by this platform, as shown in Table S1 i.e. FIGNL2-DT, GAS5-AS1 and Pseudogene GOLGA2P8 (data not shown). Notably, the results of the two lncRNAs involved in the CD, such as GAS5-AS1 and FIGNL2-DT displayed high centrality and high Max_miRANDA_score, in the lncRNA-miRNA interactions’ list in the subnetwork (while the estimated edge number in the network was 169 miRNA ID targets), and linked to significant targets, as shown in Table S1, e.g., a) hsa-mi-205-3p - a pro-inflammatory miRNA in IBD (miRANDA_score: 162) and b) hsa-miR-23b-3p (miRANDA_score: 152), significant to inhibiting gastric cancer by regulating miR-23 and/or hsa-miR-200c (miRANDA_score: 155), which is related to epithelial–mesenchymal transition (EMT) in inflammation and carcinogenesis, respectively,. In Table S1, are also presented the interacting miRNAs, hsa-miR-1184, hsa-miR-8082 and hsa-miR-6741-5p, with high Max_miRANDA_score (157, 157 and 163 respectively), as visualized in the lncRNA-miRNA network (Figure S1). Interpretating these reults and their impact, we summarize that GAS5 directly interacts with miR-23a/b, as evidenced by pull-down and Ago2-RIP assays, acting to de-repress tumor suppressors such as GSK3β and PTEN in both oncological and neuronal contexts [270]. Given the involvement of miR-23 family in intestinal epithelial barrier maintenance and cytokine signaling, the potential regulatory role of GAS5-AS1 or GAS5 through these miRNA axes may represent a novel layer of post-transcriptional control during the inflammation-to-cancer transition in IBD. The latter findings are both critical in CD-associated mucosal immunity that may function as tumor suppressors or anti-inflammatory regulators. These lncRNA hubs may also act as molecular sponges, fine-tuning miRNA availability during chronic intestinal inflammation. Importantly, these findings support their potential as regulators of epithelial immune responses but warrant further experimental validation in gastrointestinal disease models to confirm their functional impact and biomarker potential in intestinal pathology.
Furthermore, in order to explore the epigenetic modulation of lncRNA expression in gastrointestinal tumorigenesis, the DNA methylation profiles of cancer-associated lncRNA loci were also searched using the Lnc2Meth database [271]. The analysis highlighted widespread differential methylation of promoters and gene bodies of key lncRNAs in colorectal, colon cancer, gastric cancer, colorectal neoplasia and gastrointestinal stromal tumor (Table S2, supplementary material). Notably, ZNF582-AS1, TP53TG1, MEG3, showed consistent promoter hypermethylation, correlated with transcriptional silencing in tumor tissues of esophageal cancer, suggesting epigenetic inactivation during the malignant progression [272,273,274].
Conversely, hypomethylation of H19 promoter was associated with lncRNA overexpression, indicating potential oncogenic roles especially in esophageal and gastric cancer [275,276,277]. These patterns underscore the relevance of methylation-dependent lncRNA dysregulation in the inflammation-to-cancer axis and offer potential for targeted epigenetic diagnostics.
8.3.1. LncRNA as Biomarkers in GI diseases
The lncRNA profile data of colonic biopsy and blood samples differs significantly between patients with IBD and healthy groups. This suggests that lncRNAs have potential as valuable diagnostic biomarkers for IBD [69,278]. Table 5 shows representative lncRNA biomarkers in IBD, CD, UC and CRC, and their gene expression changes compared to healthy individuals. Thus, as shown in Table 5, reports demonstrate that lncRNA Mirt2 and lncRNA IFNG-AS1 were inversely correlated, with Mirt2 being downregulated and IFNG-AS1 being upregulated in the plasma of UC patients compared to control subjects [69,279]. Furthermore, IL-22, known for its anti-inflammatory properties, is critical in inhibiting intestinal inflammation. Notably, the positive correlation between IL-22 and Mirt2 in UC patients is particularly interesting. More importantly, analysis of the relationship between IL-22/Mirt2 and plasma C-reactive protein (CRP) levels revealed a significant and inverse correlation between plasma IL-22 and Mirt2 levels with CRP levels. CRP, an acute phase protein (APP), is commonly used to assess inflammatory status. Considering these findings, the combined assessment of plasma levels of IL-22, Mirt2, and CRP holds promise to improve the diagnosis of UC [69,280].
One long ncRNA which has attracted attention is the Hox transcript antisense intergenic RNA, which regulates various target genes via sponging and epigenetic mechanisms and controls various oncogenic cellular and signaling mechanisms including metastasis and drug resistance. HOTAIR is reported to reprogram chromatin organization and promote breast cancer metastasis. It is also associated with a genome-wide reprogramming of Polycomb Repressive Complex (PRC2) function not only in breast cancer but also in CRC, where upregulation of this long ncRNA may be a critical element in metastatic progression [267].
It has been reported that the expression level of intestinal mucosa and peripheral blood mononuclear cells (PBMCs) lnc-ITSN1-2 was higher in patients with active UC and patients with UC in remission compared to healthy controls, showing excellent prognostic value in active UC and efficiency in distinguishing patients with active UC from patients with UC in remission [281]. Similar results were obtained with the intestinal mucosal inflammation where upregulated ITSN1-2 levels from CD patients, showing positive effects in predicting the risk of active CD compared to healthy controls (Table 5). Another interesting observation was that lnc-ITSN1-2 was decreased after infliximab treatment in active CD patients [281]. Likewise, lncRNA THRIL is upregulated in UC as well as in CD patients which is implicated in innate immunity by regulating the expression level of TNF-α forming a complex that binds to the promoter region of the TNF-α gene resulting in its induction [69,282]. Owing to its wide range of cellular processes, including cell proliferation, survival and death, THRIL could be used as a potential biomarker for the diagnosis and prognosis of UC and CD [69,283].
Although the dysregulation of lncRNAs in IBD tissue or plasma samples is a potentially valuable diagnostic biomarker, the pathophysiology of IBD is very complex and has not been fully elucidated; currently, there is not a single gold standard for the diagnosis of IBD. Therefore, a combination of several ncRNAs may be necessary to provide an accurate diagnosis. For example, lncRNA CDKN2B-AS1 (Figure 5), which negatively correlated with increased expressions of inflammatory mediators specific to UC (TNF-α, IL-6 and sIL-2R), was an excellent marker in distinguishing UC as well as CD patients from healthy controls. However, CDKN2B-AS1 in UC, and in combination with miR-16-5p and miR-195-5p, could greatly improve the diagnostic efficiency for UC (Table 5) [69,284]. In addition, changes in CDKN2B-AS1 expression are associated with response to infliximab treatment in patients with CD, as CDKN2B-AS1 expression in infliximab treatment responders increased, whereas that of non-responders remained stable. Thereby, lncRNA CDKN2B-AS1 may serve as a biomarker to assess the response of patients to this therapy [69,285].
Also, of equal importance is lncRNA H19, which has been the subject of several studies due to its association with the development of inflammatory diseases, such as osteoarthritis. H19 lncRNA is transcribed from the H19 gene on chromosome 11, is highly expressed in multiple tissues during the embryonic stage but is largely inactivated after birth, and is upregulated in mouse models of colitis and in inflammatory colonic tissues from CD patients. An important aspect of H19's role in disease is its interaction with the vitamin D receptor (VDR), i.e. 1,25 (OH)2D3 (calcitriol), the active form of vitamin D, which is crucial in protecting the intestine from certain damaging agents. VDR also plays a significant role in regulating inflammation and carcinogenesis in various tissues. In the context of UC, overexpression of lncRNA H19 can decrease VDR levels, disrupting the intestinal epithelial barrier's function, which contributes to UC pathogenesis. Therefore, the interaction between lncRNA H19 and VDR signaling may offer potential targets for future therapeutic intervention in UC [69,286].
Additionally, it was found that lncRNA CRNDE in colonic epithelial cell apoptosis in IBD and was highly expressed in tissues from DSS-induced murine colitis models and human colon epithelial cells. In the DSS-induced murine model, CRNDE was found to suppress miRNA-495 and increase the suppressor of cytokine signaling (SOCS1). MicroRNA-495, which is reduced in UC, normally helps prevent apoptosis of IECs via the JAK/STAT3 signaling pathway, while SOCS1, on the other hand, restricts cytokine receptor signaling and promotes IFN-γ-induced apoptosis in these cells. After CRNDE intervention in a murine model, the clinical signs were reduced, showing improvement in weight loss and reduction in bloody stools, suggest that CRNDE could be a promising target for the treatment of IBD [68,69].
Moreover, lncRNA NEAT1 (Table 5), is a remarkable therapeutic biomarker, and NEAT1 expression was found to be involved in the inflammatory response and elevated in serum and tissue samples from mouse models of IBD. This response is mediated through the regulation of the intestinal epithelial barrier and the exosome-mediated polarization of macrophages. Notably, downregulation of NEAT1 suppressed the inflammatory response by affecting the same pathways. These findings suggest that targeting NEAT1 could be a promising strategy for the treatment of IBD [69,287].
As far as the pediatric UC patients are concerned, investigation has shown an association between lncRNA GAS5 (growth arrest-specific 5) (Table 5 and Table S3) and the response to glucocorticoid therapy. It was found that lncRNA GAS5 was upregulated in PBMCs of UC patients who exhibited an adverse response to glucocorticoid therapy. This suggests that GAS5 lncRNA holds promise as a novel pharmacogenomic marker that could be used for personalized glucocorticoid therapy in those patients [69,288].
Additionally to the Table 5, the aforementioned lncRNAs as above (i.e.CDKN2B-AS1,H19,IFNG-AS1,MALAT1,Mirt2,TUG1) have been a subject of vigorous research and an update with more, in detail, information is provided by the Table S3 (supplementary material). For the new characterized lncRNA mentioned above, various analysis platforms have been employed by the first author, such as NONECODE V6, EVLncRNAs2.0 [218,219] Ensembl (https://www.ensembl.org/index.html), ,NCBI (https://www.ncbi.nlm.nih.gov/ gene/) (biotool details in section 8), for more updated information. Thus, details are shown in Table S3 and include, chromosome location, exons, interaction type, interaction target, NCBI accession numbers and description for interaction and function, supported by citation ID (PMID). The aforementioned data (Table S3) are also associated to Figure S2 (supplementary material). Furthermore, the UC network shown in Figure S2, built under the EVlncRNAs 2.0 database by the first author, presents the following lncRNAs CDKN2B-AS1, H19, IFNG-AS1, MALAT1, Mirt2 and TUG1 with their first degree interaction molecules, i.e. significant miRNA interaction targets, coding genes, proteins, protein complexes etc, as first degree node interactors. The interaction column distinguish the types (i.e. U: unknown type; Regulation: shows that lncRNA can regulate the expression of other biomolecules in the same physiological process; Co-expression: to indicate positively or negatively correlated expression of lncRNA with other molecules in the same physiological process; Binding: indicates that lncRNA has direct physical contact with other biomolecules).
As an example of pointing out important interactors in these direct networks, we refer to lncRNA, MALAT1 which promotes UC by upregulating lncRNA CDKN2B-AS1 (ANRIL), since both lncRNAs are significantly and positively correlated in UC patients but not in healthy controls, while the last (CDKN2B-AS1) relieves inflammation of ulcerative colitis via sponging miR-16 and miR-195 [284,289]. Notably, the later reported diagnostic miRNAs in UC (miR-16 and miR-195), - and potentially therapeutic - are shown in Table 4 [cited by their PMIDs:29668922 and 21546856, respectively], are also serum biomarkers for IBD (Table 4). Additionally, miR-200 [227] interaction with MALAT1 was inferred in our network analysis and can be observed in the MALAT1-miR interaction targets network (Figure S2).
The lncRNA known as Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1),(Table 5), misregulation has been linked with a lot of autoimmune diseases as well as it is abundantly expressed in many tissues in various biological processes, including cancer development and metastasis. A study also shows potential interaction between MALAT1 and IL-6, which was found upregulated contributing to inducing apoptosis and inflammation; thus, it shows MALAT1's potential value as a target in diagnosis and treatment for IBD patients [69,289]. MALAT1 was also reported to play a role in CRC, while a study revealed that the inflammation-related MALAT1 and miR-663a constitute a competing endogenous RNA (ceRNA) network in CRC cells through sequence-dependent binding. MALAT1 reduced miR-663a expression via a ceRNA mechanism to prevent the degradation of most of miR-663a’s targets (e.g. P53, PIK3CD, P21, CXCR4, TGFB1, and JUND) in CRC cells and tissues. This shows that MALAT1 and miR-663a may be involved in CRC development and inflammatory formation [69,290].
In addition, the lncRNAs, KIF9-AS1, LINC01272, and DIO3OS (Table 5) were selected for evaluation as potential diagnostic biomarkers for IBD in a study and the findings showed that tissue and plasma samples from IBD patients had considerably higher levels of KIF9-AS1 and LINC01272 mRNA expression than the healthy controls. In contrast, tissue and plasma samples from IBD patients had considerably lower levels of DIO3OS mRNA expression than the healthy controls [69,291]. Furthermore, MIR4435-2HG (Table 5) suppression inhibits macrophage M1 polarization while promoting M2 polarization, thereby alleviating intestinal inflammation in DSS-induced mice with ulcerative colitis through JAK1/STAT1 signaling and can be considered as a potential therapeutic target for UC treatment [69,292].
LncRNAs are also being investigated as non-invasive markers for CRC. A bioinformatic study showed that FHIP1A-DT was downregulated in CRC and patients with low FHIP1A-DT expression had a worse prognosis. Therefore, lncRNA FHIP1A-DT (Table 5) is associated with epigenetic modification and regulates many cancer-related pathways, suggesting a potentially significant direction for future research in the diagnosis and treatment of CRC [293]. Also, several studies were engaged with lncRNA PVT1 (Table 5), which is highly upregulated in CRC patients. This upregulation positively correlates with cell proliferation, invasion, tumor stages, and lymph node metastasis. A study showed PVT1 is highly expressed in CRC patients, and its level is closely related to vascular invasion and lymph node metastasis, while down-regulation of PVT1 induces apoptosis and inhibits proliferation in CRC cells [294].
Collectively, epigenetic dysregulation plays a pivotal role in the stepwise transformation from inflammation to CRC. To better illustrate the stage-specific accumulation of epigenetic disruptions during colitis-to-carcinoma progression, the following Table 6 summarizes key alterations in DNA methylation, histone modifications, and non-coding RNA profiles across each pathological stage. In more detail, Table 6 summarizes stage-specific epigenetic alterations observed during the progression from normal intestinal epithelium to colitis, dysplasia, and colitis-associated colorectal cancer (CAC). Importantly, Table 6 illustrates how specific epigenetic changes emerge and accumulate across the stages of IBD-associated colorectal cancer, offering a roadmap for biomarker discovery and targeted therapies.
Key molecular events are categorized into four major epigenetic regulatory layers: DNA methylation, histone acetylation, histone methylation, and non-coding RNA modulation. For each pathological stage, representative changes are presented based on recent literature, including promoter hypermethylation of tumor suppressor genes (e.g., p16INK4a, MLH1) in dysplasia, decreased global acetylation of histones H3 and H4 during inflammation, aberrant H3K27me3 deposition in advanced lesions, and upregulation of oncogenic microRNAs (e.g., miR-21) and lncRNAs (e.g., MALAT1). Progression is illustrated using directional arrows that reflect temporal and molecular transitions between disease stages.
Albeit, this time-dependent presentation of the pathological stages is not entirely confined to the specific conditions depicted in Table 6, it highlights the sequential accumulation of epigenetic aberrations and their potential use as biomarkers or therapeutic targets in inflammation-driven colon carcinogenesis. In this respect, Table 6 illustrates how specific epigenetic changes emerge and accumulate across the stages of IBD-associated colorectal cancer, offering a blueprint for biomarker discovery and targeted therapies. This multi-layered approach, which takes into account the associations of the diverse entities with the disease, e.g., causal genes, epigenetic modulators and environmental cues in their networks of interactions, which are the most critically important for the pathophysiology of the disease phenotype, are summarized on the whole, in Table 6.
8.4. Other ncRNAs in GI diseases
Small RNAs are abundant in eukaryotic organisms and play a crucial role in regulating gene expression through mRNA degradation or gene silencing. Another class of small RNAs, the Piwi sub-family of Argonaute proteins, specifically binds to piRNAs [295]. The latter gene regulation complex is vital because it is involved in biological processes such as cell renewal and differentiation of stem cells [296], animal development [297], germline cell development [298], and certain types of human cancer [299].
8.4.1. PiRNAs
PIWI-interacting RNAs (piRNAs) are small RNA sequences approximately 26 to 31 nucleotides long [300]. Their functions include regulating mRNA expression of transposable elements through degradation, possibly inhibiting the translation process, and altering chromatin [301]. This surveillance system plays a role in controlling and degrading cancer cells [302]. Additionally, piRNAs are involved in silencing by methylation of transposable elements, deadenylation of Drosophila transcripts [303], de novo DNA methylation [304], and Rat sarcoma guanine nucleotide releasing factor 1 (Rasgrf1) imprinting in rats [305]. PIWI-interacting RNAs originate from single-strand transcripts of non-coding sequences, containing a high concentration of truncated transposable element sequences or transcripts from 3' UTR protein-coding genes. Most of these transcripts come from regions frequently producing piRNA precursors, known as clusters [306]. These clusters are related to the transposable elements' repertoire, which is recognized and silenced. The cluster transcripts are selected and processed randomly within the cell into a large quantity of piRNAs. Despite sequence differences between species and individuals, some patterns are conserved, such as the presence of a uridine at the 5' terminal and an adenosine at the 10th nucleotide position [307]. The specific localization of piRNA clusters in the mammalian genome also appears conserved [308]. There are two models describing piRNA production. In the first model, a piRNA cluster transcript is cleaved, generating a 5' terminal. This transcript can be broken at virtually any position, with a preference for a uridine at the 5' terminal. After binding to the specific Piwi protein, a second cleavage generates the RNA's 3' terminal [309]. The second model, called the ping-pong amplification cycle, starts with forming a large quantity of piRNAs from the sense and antisense strands of the cluster, similar to the first model. When the Piwi-piRNA complex finds its specific target, it cleaves the target transcript 10 nucleotides from the 5' terminal of the piRNA. This process not only inactivates the target mRNA but also creates a new fragment with a 5' terminal that binds to the AGO3 Argonaute protein. This new complex can bind to complementary elements, such as a cluster transcript, resulting in the production of multiple fragments identical to the original piRNAs. This amplification cycle generates many piRNAs from a low source of initial sequence [309]. The Piwi-piRNA complex forms in the cytoplasm and is then imported into the nucleus to repress transposons and guide histones to these loci. The continuous activity of the Piwi-piRNA complex is required to maintain transposon repression, as reduced Piwi activity quickly leads to the reactivation of transposable elements [306].
Small RNAs, like Piwi, are found in eukaryotic organisms with abundance of activities on gene regulation by mRNA degradation or gene silencing. Argonauts proteins guide these mature small RNAs to their targets [310]. The Piwi sub-family of Argonauts are more specific bound to the piRNAs [295]. The role of this gene regulation complex is important by the fact that are related to biologic process of cell renovation and differentiation of stem-cells [296], animal development [297], germline cell development 298] and some types of human cancer [299].
Table 7. below shows representative results of the involvement of piRNAs in IBD-related syndromes as well as relevant citations for further reading. All the databases employed by the first author in these analyses of piRNAs, concerning the associations between piRNAs and disease phenotypes, were a few, e.g., piRDisease v1.0, piRNAQuest V.2, piRPheno [311,312,313], and based on the manually curated associations of experimentally supported piRNAs with CRC. The piRNAs involved in colorectal cancer, are listed with their main characteristics (i.e., SNP information, SNP expression info, function and supportive publication reference (PMID)). For the piRNA expression profile of the top 5 most abundant piRNAs in colorectal-related carcinogenesis, the reader can refer to the Table S4 (supplementary material), as explained below.

Table 7.
piRNAs, involved in colorectal cancer, showing SNP information and function, curated by piRNA databases(#) and reported in PubMed (PMID).
Table 7.
piRNAs, involved in colorectal cancer, showing SNP information and function, curated by piRNA databases(#) and reported in PubMed (PMID).
| Name | SNP Expression info | Function | Database (#) | PubMed (PMID) |
|---|---|---|---|---|
| piR-hsa-679 | rs34383331, base change: A>T | May be involved in the development of CRC | piRBase | 25740697 |
| piR-hsa-7400 | rs2070766f, base change: C>G | -“- | piRBase | 25740697 |
| piR-hsa-21417 | rs2070766f, base change: C>G | -“- | piRBase | 25740697 |
| piR-hsa-29786 | rs2070766f, base change: C>G | -“- | piRBase | 25740697 |
| piR-hsa-21517 | rs11776042, base change: T>C | May be involved in the development of CRC | piRBase | 25740697 |
| piR-hsa-29056 | rs9368782, base change: A>G | -“- | piRBase | 25740697 |
| piR-hsa-2363 | rs12483859, base change: A>G | -“- | piRBase | 25740697 |
| piR-hsa-8401 | rs10433310, base change: C>T | -“- | piRBase | 25740697 |
| piR-hsa-3789 | rs12910401, base change:G>A | -“- | piRBase | 25740697 |
| piR-hsa-1245 | up-regulated | It is a novel oncogene and a potential prognostic biomarker in colorectal cancer | piRBase | 29382334 |
| piR-hsa-1282 | up-regulated | It interacts with HSF1 to promote Ser326 phosphorylation and HSF1 activation, enhancing CRC cell proliferation and suppressing cell apoptosis | piRBase | 28618124 |
| piR-hsa-17444 | up-regulated | Formation of PIWIL2/STAT3/phosphorylated-SRC (p-SRC) complex, which activates STAT3 signaling and promotes proliferation, metastasis and chemoresistance of CRC cells | piRBase | 30555542 |
| piR-hsa-1077(*) | up-regulated | Ontology ID: EFO_1001951 | piRNAdb, piRPheno v2.0 | 16751776 |
Appendix: (*) associated with colorectal carcinoma; (#) piRBase v2.0 and piR Pheno v2.0 were mainly employed for the analysis (see details in the text).
Further research, can be provided by piRNA online databases and biotools for analysis and updated curated information. The latter approach has become increasingly critical due to the importance of relationships between piRNAs and disease phenotypes, [313]. Therefore, the first author employed piRBase V2.0 database, which systematically integrates epigenetic and post-transcriptional regulation data to support piRNA functional analysis, [314,315]. Also, piRPheno v2.0, a manually curated database platform, which provides experimentally supported associations between piRNAs and disease phenotypes with validated novel class of biomarkers and potential drug targets for disease diagnosis, therapy and prognosis. The work performed on the aforementioned databases, including also search and analysis of available and pertinent GEO datasets to colorectal-associated neoplasmatic pathologies at the gene expression level, as shown in Table S4 (Supplementary material). Results of the piRNA expression profile of the top 5 among 200 most abundant piRNAs which were identified in the samples are shown in Table S4, under the column piRNA ID. In more detail, Table S4 presents the piRNA expression profile of the top 5 most abundant piRNAs detected in the samples (GEO, NCBI) by RNA-seq analysis in colorectal-related carcinogenesis. For the specific analysis, piRNAQuest V.2 database was additionally employed, which is a comprehensive updated resource for piRNAs of 28 species with 9277689 unique piRNAs, density based cluster prediction and piRNA expression, corresponding to different tissues and diseases [312]. These new findings suggest that further research is of utmost importance as a goal to elucidate the nature and involvement of piRNAs in diagnosis and/or potential therapy of these diseases by scrutinizing their action as clinical markers.
8.4.2. CircRNAs
Another subclass of small RNAs are the circular RNAs (circRNAs), a novel type of ncRNAs, which display cell or tissue-specific expression and are conserved across species [316]. The expression of circRNAs is highly stable in comparison with their linear counterparts, and is predominantly localized in the cytoplasm, indicating important functions in human diseases and development [316].
CircRNA is a type of RNA molecule which forms a covalently closed continuous loop by back-splicing or lariat [316]. Although circRNA was discovered decades ago, it was initially perceived as RNA splicing errors [316]. With the advent of next-generation sequencing (NGS), circRNAs were found to be abundant, stable and evolutionarily conserved among eukaryotes [316]. So far, tens of thousands of circRNAs have been predicted by bioinformatics methods [316]. These circRNAs are largely classified into three categories, including exonic circRNAs, circular intronic RNAs (ciRNAs), and retained-intron circRNAs or exon-intron circRNAs (EIciRNAs) [316]. They have been implicated in a wide range of physiological and pathological processes, including tumorigenesis, neurodegeneration, cardiovascular diseases, immune dysregulation, and metabolic disorders, and are also known to accumulate during aging. In the GI tract, circRNAs have recently garnered attention for their potential roles in epithelial homeostasis, inflammation-driven signaling, and CRC progression, functioning as miRNA sponges, RBP scaffolds, or even templates for translation. Given their structural stability and conservation, circRNAs are increasingly investigated as diagnostic biomarkers and therapeutic targets in GI diseases. In this section, are integrated pre-analyzed human colon tissue expression datasets by the first author, to highlight the Top 30 most abundantly expressed circRNAs, quantified as CPM (counts per million) in a one-step RNA-seq analysis pipeline (Figure 3), searched under the circAtlas database [317]. These circRNAs represent highly transcribed and potentially functional molecules relevant to mucosal biology, inflammation, and colorectal carcinogenesis. CircAtlas 3.0, integrates more than over 3.1 million circRNAs across 10 species (human, macaque, mouse, rat, pig, chicken, dog, sheep, cat, rabbit) as well as a variety of tissues, thus integrating the most comprehensive circRNAs, their expression and functional profiles in vertebrates.
The profile of thetop 30 most highly expressed circRNAsin normal human colon tissue (Figure 3), shows abundances ranging from approximately 5.5 to 1 CPM. While many of these circRNAs have not yet been characterized in the context of GI pathology, circRNAs such ascircITGA7, (and others not included inFigure 3, such ashsa_circ_0000711,hsa_circ_0000231, andcirc_0062682)- though not necessarily among the most expressed in normal tissue - have well-documented roles in colorectal cancer. For instance,circITGA7is downregulated and correlates with better prognosis (AUC ≈ 0.88), spongingmiR-370-3pto inhibit Ras signaling via NF1;hsa_circ_0000231is significantly upregulated (~4.6-fold) in CRC and promotes proliferation;circ_0062682is overexpressed across multiple CRC cohorts and associates with poor survival, via miR-940/PHGDH regulatory circuit [318,319,320,321].
InTable 8below are presented circRNAs involved in colorectal cancer. These findings underscore a potential functional divergence: highly expressed circRNAs in normal colon may maintain tissue homeostasis, whereas dysregulated circRNAs in CRC often show altered expression rather than absolute abundance. Future work should map overlaps between the expressed circRNAs in this dataset and the dysregulated CRC-associated circRNAs to identify candidates for functional validation and biomarker utility.
Table 8presents examples of circRNAs involved in colorectal cancer, which are listed with their main characteristics (i.e., circRNA names, expression pattern, function, database recorded, method of detection and supported by publication reference ID (PMID) from NCBI)).
Table 8.
circRNAs involved in colorectal cancer (CRC), Predicted interacting RBP, their expression pattern, method of detection, employed database and reported in PubMed (PMID).
Table 8.
circRNAs involved in colorectal cancer (CRC), Predicted interacting RBP, their expression pattern, method of detection, employed database and reported in PubMed (PMID).
| circRNA name | Synonyms |
Predicted interacting RBP (Nοof binding sites) |
Methods | Expression pattern | PubMed ID |
| hsa_circ_0020397 | hsa_circRNA_100722 |
EIF4A3(32); HuR(3); IGF2BP1(2); AGO2(2); SFRS1(1); PTB(1); LIN28A(1); IGF2BP2(1); FUS(1) |
qRT-PCR; dual luciferase reporter assay; in vitro knockdown; in vitro overexpression; western blot; etc. | up-regulated | 28707774 |
| circ-BANP | hsa_circRNA_101902; hsa_circ_0003098 | EIF4A3(12); HuR(1); AGO2(1) | Microarray; RT-PCR; qRT-PCR; in vitro knockdown; ISH; western blot; etc. | up-regulated | 28103507 |
| hsa_circ_0000069 | hsa_circRNA_100213; hsa_circ_001061 | EIF4A3(10); AGO2(9); IGF2BP3(7); PTB(6); IGF2BP2(6); IGF2BP1(4); HuR(2); FMRP(2); SFRS1(1); FXR2(1) |
qRT-PCR; in vitro knockdown; etc. | up-regulated | 28003761 |
| hsa_circ_001569 | N/A | N/A | in vitro knockdown; in vitro overexpression; qRT-PCR; western blot; ; luciferase reporter assay etc. | up-regulated | 27058418 |
| hsa_circ_0001451 | hsa_circ_001988 | EIF4A3(6); HuR(2); LIN28A(1); IGF2BP3(1); IGF2BP2(1); DGCR8(1); AGO2(1) |
RNA-seq; qRT-PCR | down-regulated | 26884878 |
|
circHlPK3 |
circ_0000284, hsa_circ_0000284 | IGF2BP1 (5), HuR (ELAVL1) (8), FUS (4) | RNA-seq; Microarray | up-regulated | 33536039 |
|
circCCDC66 |
circ_0001313 |
HuR (ELAVL1) (6), PTBP1 (7), FUS (5) |
RNA-seq; Microarray; droplet digital PCR | down-regulated | 33536039 |
| circZFR | hsa_circRNA_103809; hsa_circ_0072088 | FMRP(21); EIF4A3(7); AGO2(7); IGF2BP3(6); HuR(6); IGF2BP1(4); AGO1(4); ZC3H7B(1); U2AF65(1); PTB(1); LIN28B(1); IGF2BP2(1) |
Microarray; qRT-PCR | down-regulated | 28349836 |
| circPTK2 | hsa_circRNA_104700; hsa_circ_0005273 | AGO2(5); EIF4A3(2); IGF2BP3(1); IGF2BP2(1); FUS(1) | Microarray; qRT-PCR | down-regulated | 28349836 |
| CDR1as | Cdr1as; ciRS-7; hsa_circRNA_105055; hsa_circ_0001946 |
AGO2(43); FUS(26); IGF2BP1(11); IGF2BP2(10); IGF2BP3(9); AGO1(6); TNRC6(2); TDP43(2) |
qRT-PCR; in vitro overexpression; ; in vivo overexpression; IHC; western blot; etc. | up-regulated | 28174233 |
8.4.3. Functions and Implications of circRNAs
CircRNAs have become of great interest in the field of transcriptional regulation [317]. For instance, they can act as miRNA sponges, or bind to RNA-associated proteins to form RNA-protein complexes that regulate gene transcription [316]. Some ciRNAs and EIciRNAs can interfere with pre-mRNA splicing that is a critical step in the post-transcriptional regulation of gene expression [316]. Recent studies have identified circRNAs as novel regulators in many disorders such as Alzheimer’s disease, diabetes mellitus, gastrointestinal diseases (e.g., IBD, colitis), and cancer progression, particularly colorectal cancer (CRC) [322,323]. Furthermore, due to the lack of 3′ termini, circRNAs are more resistant to degradation by exonuclease RNase R and possess greater stability than linear RNAs. CircRNAs are abundant in blood samples, saliva, and exosomes, making them promising diagnostic biomarkers for complex diseases such as cancer [322,323].
To further explore the landscape of circRNAs in GI inflammation and tumorigenesis, the first author searched and analyzed circRNA profiles derived from extracellular vesicles (EVs), which are known mediators of intercellular communication, by the online biotool exoRBase 2.0 [324]. Thus, Table S5 (supplementary material) presents circRNAs identified from EVs in healthy colon and small intestine tissues, while Table S6 highlights circRNAs enriched in EVs isolated from colorectal cancer (CRC) samples, all detected through RNA-seq analysis searched and retrieved from exoRBase 2.0. These datasets provide insight into the differential expression, genomic origin, and potential regulatory roles of circRNAs in both homeostatic and malignant contexts. Their EV-based localization suggests roles in systemic signaling and possibly in non-invasive biomarker development. The aforementioned tables show and expand the biological relevance of circRNAs beyond intracellular activity, supporting their potential as circulating molecular indicators in IBD and CRC progression.
Notably, it is clear that circHlPK3, is the highest CPM signal, as shown in Figure 3, along with high fold-changes or altered expression in CRC compared to normal tissues (Table 8). The latter provides dual evidence, - i.e. circHlPK3 baseline high abundance and disease-specific overexpression (Table 8) – which underscores its biological relevance in gastrointestinal tissue homeostasis and malignant transformation. According to a key study (PMID: 33536039, Table 8), circHIPK3 promotes CRC progression by sponging miR-7, thereby modulating the expression of multiple oncogenes, including Focal Adhesion Kinase (FAK), epidermal growth factor receptor (EGFR), and the transcription factor (TF), Yin Yang 1 (YY1). These findings suggest that circHIPK3 may act as an endogenous miRNA sponge, contributing to tumor cell proliferation, migration, and invasion.
8.4.4. Circular RNA - Protein Interactions: Functional Significance and Binding Site Density
Furthermore, circRNAs modulate gene expression and cellular pathways through interactions with TFs and RBPs. Strictly speaking, for circRNA binding capacity, the number and affinity of RBP binding sites on circRNAs significantly influence their regulatory potential: multiple binding sites enable circRNAs to act as molecular “scaffolds,” sequestering RBPs away from pre-mRNAs, modulating splicing, translation, or protein stability [325,326,327,328].
These interactions often hinge on the number of binding sites, which can modulate the efficiency and strength of circRNA–protein interactions. For instance, a circRNA with multiple HuR (Human Antigen R) -binding motifs may more effectively sequester HuR, altering mRNA stability and downstream gene expression [327]. This ribonucleoprotein assembly could influence key processes including RNA splicing, translation, and cellular localization, thereby contributing to oncogenic or tumor-suppressive pathways in inflammation-driven CRC. The density of RBP binding sites correlates with competitive binding kinetics - higher counts increase the likelihood of functional RBP sequestration or scaffold formation. Additionally, some RBPs may recruit chromatin-modifying complexes, linking circRNA–protein interactions to epigenetic regulation. For instance, abundant binding sites on oncogenic circRNAs retain RBPs involved in tumor suppression or RNA processing, thus tipping the cellular balance toward proliferation and survival. Moreover, circRNAs may compete with parental linear RNAs for RBP binding, further altering gene expression profiles. Therefore, mapping RBP-binding landscapes on circRNAs is critical to understanding of circRNA's functional complexity in tumor progression and their roles in inflammation-induced cancers like IBD - CAC and CRC, while RBPs may serve as novel biomarkers or targets in CRC therapeutics [329,330,331].
In Table 8, information is also shown concerning the Predicted interacting RBP (Nο of binding sites), as a scored-dependent evaluation of the circRNAs potential binding; thus, correlating with their potential competitive binding affinity to RBPs and sequentially recruiting chromatin-modifying complexes, as linking circRNA–protein interactions expand epigenetic regulation.
8.5. Inflammation-driven ncRNA Modulation in CRC
Chronic inflammation is a well-established contributor to the initiation and progression of colorectal cancer (CRC), particularly in the context of inflammatory bowel disease (IBD). Among the key mediators of inflammation-driven tumorigenesis, various types of non-coding RNAs (ncRNAs) are involved, which some already discussed, mainly include, miRNAs, lncRNAs, piRNAs and circRNAs. These ncRNAs act as post-transcriptional and epigenetic regulators of gene expression, responding to pro-inflammatory signals and reshaping the cellular transcriptome to favor survival, proliferation, and malignant transformation.
In the summerizing Table 9, are highlighted known examples of inflammation-associated ncRNA clinical markers of various types, involved in inflammation-driven colorectal cancer, altered in patients with CRC, their functions, and supporting evidence (PMID). Thus, Table 9 contains a concise selection list of ncRNAs that have revealed potential or are under investigation for validation as biomarkers/clinical markers, emanated from Table 4, Table 5, Table 6 and Table 7, in IBD/CRC conditions.
Table 9.
Overview of Inflammation-Associated key clinical markers of ncRNAs in Colorectal Cancer (CRC).
Table 9.
Overview of Inflammation-Associated key clinical markers of ncRNAs in Colorectal Cancer (CRC).
| ncRNA Type | Name | Function in CRC | References(PMID) |
| lncRNA | HOTAIR | Recruits PRC2 to silence tumor suppressor genes (e.g., CDKN1A); promotes invasion and metastasis. | 21862635; 28701486 |
| lncRNA | MALAT1 | Enhances β-catenin nuclear translocation; regulates alternative splicing and epithelial-mesenchymal transition (EMT). | 12970751; 34144008 |
| circRNA | circHIPK3 |
Acts as a miR-1207 sponge which is downregulated in CRC; enhanced formin like 2 (FMNL2) in CRC; contributes to chemoresistance and proliferation. |
32046858 |
| circRNA | circCCDC66 | Functions as a ceRNA; sequesters tumor-suppressive miRNAs (e.g., miR-33b); enhances c-MYC and YAP1 pathways. | 28249903 |
| miRNA | miR-21 | Overexpressed in CRC; targets PTEN, PDCD4, suppressing apoptosis; regulated by NF-êB and IL-6 inflammatory stimuli. |
17968323; 20797623; 34771727 |
| miRNA | miR-155 | Induced by NF-κB and STAT3; promotes tumor cell survival and immune evasion by targeting SOCS1 and TP53INP1. |
17242365; 17911593;32702393 |
| mRNA target | PTEN | Tumor suppressor inhibited by miR-21; regulates PI3K/AKT signaling. | 32104279 |
| mRNA target | BCL2 | Anti-apoptotic protein regulated by multiple miRNAs (e.g., miR-15, miR-16); supports resistance to cell death. | 16166262; 28984869 |
In the inflamed colonic microenvironment, immune cells release cytokines such as IL-6, IL-1β, and TNF-α. These cytokines activate transcriptional programs through pathways like NF-κB, STAT3, and Wnt/β-catenin, which in turn influence the expression of oncogenic or tumor-suppressive ncRNAs. For example, miR-21, a highly upregulated miRNA in both IBD and CRC, is transcriptionally induced by NF-κB and STAT3 activation. More importantly, it targets tumor suppressors such as PTEN, PDCD4, and Reversion Inducing Cysteine Rich Protein With Kazal Motifs (RECK), thereby enhancing PI3K/AKT signaling and resistance to apoptosis [332,333].
Similarly, miR-155 is induced in macrophages and epithelial cells during chronic inflammation. It modulates immune tolerance and tumor progression by targeting negative regulators of cytokine signaling like Suppressor Of Cytokine Signaling 1 (SOCS1) and Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase 1 (SHIP1), contributing to a feed-forward loop that sustains STAT3 activation and inflammatory gene expression [334,335].
Beyond miRNAs, inflammation-responsive lncRNAs play pivotal roles in chromatin remodeling and transcriptional repression of tumor suppressor genes. The lncRNA HOTAIR, for instance, is upregulated in colonic epithelial cells under inflammatory stress. It recruits PRC2 to silence genes involved in epithelial homeostasis and immune control, such as CDH1 (E-cadherin), facilitating epithelial–mesenchymal transition (EMT) and metastasis [267]. The inflammation-sensitive lncRNA, MALAT1, is elevated in CRC and modulates alternative splicing of genes linked to migration and invasion [336]., Notably, both HOTAIR and MALAT1 participate in chromatin remodeling and transcriptional regulation, further enhancing inflammation-linked oncogenic transformation.
Importantly, circular RNAs have recently emerged as key regulatory molecules in the inflammation–cancer axis. Circular RNA HIPK3 (Table 8 and Table 9), is highly expressed in CRC, and acts as a sponge for 1207-5p, thereby derepressing oncogenes like formin like 2 (FMNL2) [337]. In more detail, circHlPK3 is reported to confer chemoresistance via sponging miR-637 and promoting autophagy in CRC, suggesting its interaction with RBPs like IGF2BP1 and HuR, which may stabilize target mRNAs related to autophagy and survival pathways (Table 8 and Table 9). Furthermore, circCCDC66, another circRNA elevated during chronic inflammation, sequesters tumor-suppressive miRNAs and enhances the expression of MYC Proto-Oncogene, BHLH Transcription Factor (MYC), Zinc finger E-box binding homeobox 1 (ZEB1), and other targets promoting tumor growth [338]. It has oncogenic roles in proliferation, migration, therapy resistance, and promotes autophagy by sponging miR-3140. Binding to HuR and Polypyrimidine Tract Binding Protein 1 (PTBP1) may similarly modulate mRNA stability of proliferative or apoptotic genes (Table 8).
Taken together, inflammation-driven modulation of ncRNAs constitutes a critical epigenetic layer of carcinogenic regulation in IBD-associated CRC. These ncRNAs function both upstream and downstream of canonical signaling pathways, creating a dynamic regulatory network that shapes cell fate in the inflamed epithelium, illustrated in Figure 4.
Figure 4 summarizes key molecular circuits where inflammation-induced epigenetic events interface with non-coding RNAs to regulate tumorigenesis in colorectal cancer (CRC). Inflammatory cytokines such as IL-6 and TNF-α, often elevated in IBD and tumor microenvironments, activate transcription factors including NF-κB and STAT3. These factors orchestrate oncogenic signaling cascades like the Wnt/β-catenin pathway, central to CRC progression. The diagram shows how epigenetic silencing of tumor suppressors, including PTEN, through promoter hypermethylation or histone deacetylation (e.g., H3K27me3 loss), intersects with dysregulated ncRNA networks.
MicroRNAs such as miR-21 and miR-155 - frequently upregulated in CRC - are downstream targets of NF-κB/STAT3 signaling and are known to repress tumor suppressors, including PTEN and pro-apoptotic genes. In more detail, miR-21, commonly upregulated in various cancers, promotes cell survival by suppressing Apoptotic Protease Activating Factor 1 (Apaf-1) and Fas cell surface death receptor (Fas) ligand, key mediators of intrinsic and extrinsic apoptosis [339]. Its anti-apoptotic role is supported by evidence that ectopic miR-21 expression protects cells from gemcitabine-induced apoptosis [340]. Similarly, miR-155, often overexpressed in malignancies, is activated by TGF-β/SMAD4 signaling and facilitates epithelial-to-mesenchymal transition (EMT) by targeting RhoA GTPase, a key regulator of cell polarity and junctions. Silencing miR-155 inhibits TGF-β-induced EMT and cell invasion [341]. In contrast, miR-200 and miR-203 [227] - suppressors of EMT - are downregulated by TGF-β, highlighting the dual regulatory role of miRNAs in metastasis. These miRNAs are further regulated by circular RNAs like circHIPK3 and circCCDC66, which function as competing endogenous RNAs (ceRNAs), sequestering oncogenic miRNAs and thus fine-tuning gene expression, but also inhibiting the expression of TGF-β, inactivating SMAD signaling pathway, and reversing EMT in gastric cancer (GC) cells; this is suggesting that circCCDC66 is an important regulator of EMT in gastric cancer [338]. circCCDC66, which contains target sites for miR-33b, miR-93, miR-510-5p and miR-338-3p, by sponging different miRNAs may perform various functions, while promotes CRC growth and metastasis including induction of drug-resistance [322]. Interestingly, 88% of randomly chosen patients with colon cancer have higher levels of circCCDC66 than randomly chosen normal subjects, indicating that the expression level of circCCDC66 is a good indicator for the detection of colon cancer [322]. circCCDC66 has been also reported to protect PTEN by sponging miRNAs such as miR-93 or miR-33b, and circHIPK3 that may promote tumorigenesis via NF-κB feedback loops. The latter can be explained and supported by the fact, that the knockdown of hsa_circ_0000284 (circHIPK3) markedly suppresses CRC cell proliferation, migration, and invasion, inducing apoptosis in vitro and inhibiting CRC growth and metastasis in vivo [322]. The diagram (Figure 4) supports the central thesis that non-coding RNAs serve as both downstream effectors and modulators of inflammation-driven carcinogenesis. These interactions - combined with stable epigenetic changes - create a self-reinforcing tumor-promoting microenvironment. Identifying and targeting these RNA-mediated axes could offer translational potential for diagnostic biomarkers and therapeutic interventions in CRC.
To expand upon the network logic depicted in Figure 4, the first author explored the complex interactions of ncRNAs, by using the LncACTdb3.0 platform [342], assisted by aforementioned biotools such as, EVlncRNAs 2.0, NDE IQuery and FunCoup 5 [219,343,344], to construct a large-scale, colorectal cancer-centered ceRNA interaction network. For the latter gene interaction network analysis, LncACTdb3.0 was selected as the main biotool, for its validated and experimentally supported ceRNA interactions across human and mouse models.
This interaction network is presented in Figure S3 (Supplementary Material), by integrating three molecular tiers: long non-coding RNAs (lncRNAs), microRNAs (miRNAs), and protein-coding genes, all connected through disease-associated regulatory interactions. Our query was informed by candidate protein coding genes and non-coding RNAs identified from our unpublished RNA-seq meta-analysis results of AOM/DSS-induced CAC mouse model datasets (Triantaphyllopoulos et al.,, manuscript in preparation), incorporating key gastrointestinal inflammatory disease phenotypes, such as Colon Adenocarcinoma, Colorectal Cancer, Colorectal Adenocarcinoma, Ulcerative Colitis, and Colorectal Cancer Liver Metastases. The inclusion of only human and mouse orthologs for the complex network construction was taken into account. The total protein coding gene list and the lncRNA genes that were used for the complex network construction, derived from the first author's unpublished RNA-seq meta-analysis (results of the AOM/DSS), related to CRC-related carcinogenesis.
The strong overrepresented candidate genes which were used for the network construction such as CTNNB1, CD44, EZH2, AXIN2, MMP10, and WIF1 (False Discovery Rate (FDR) -adjusted p-values < 0.001, ***p <0.001), consist of a subset of a larger discovered gene list, which formed the core input for the ceRNA network analysis (Figure S3). These top enriched genes with well-established mechanistic roles in colorectal tumor biology, show concomitant upregulation in both human CRC sources (TCGA, GEP etc) as well as in the studied AOM/DSS mouse model (Triantaphyllopoulos et al.,, manuscript in preparation). The findings include transcription factors, pro-inflammatory mediators, angiogenic drivers, and enzymes critical to tissue remodeling, in the constructed network, accompanied by reference, underscoring their validated translational significance in CRC biology.The final network map highlights densely connected regulatory hubs and crosstalk nodes involving known CRC drivers (e.g., TP53, CTNNB1, CD44, EZH2 etc) and inflammation-associated lncRNAs and miRNAs targeting mRNAs, (e.g. lncRNAs such as, BCYRN1, NEAT1, ZEB1-AS1 and (e.g. miR-34b-5p, miR-221, miR-204-3p, miR-139-5p), respectively (Figure S3). Additional technical details of the node types and biological context are provided in the accompanying Figure S3 (Supplementary material). Notably, the inferred network revealed multiple lncRNAs acting as competing endogenous RNAs (ceRNAs), potentially sequestering disease-relevant miRNAs known to regulate pro-inflammatory and immune-modulatory transcripts.
Finally, the complex gene interactions could serve as a molecular bridge between preclinical and clinical settings, providing a foundation for planning future studies of emerging strategies, targeting the most significant pathways by therapeutic interventions in colitis-associated CRC.
9. Diagnostic and Therapeutic Implications: Biomarkers and Epigenetic Drugs
This section integrates and highlights the translational potential of inflammation-related epigenetic changes in colorectal cancer, outlining actionable diagnostic and therapeutic strategies. As graphically depicted in Figure 2, (subsection 4.1) it reviews clinically validated and experimental key biomarkers such as, methylated SEPT9 promoter (approved for blood-based CRC screening), overexpressed miR-21 (linked to disease severity and prognosis), as well as experimental lncRNA signatures such as CRNDE, HOTAIR and MALAT1. From a therapeutic standpoint, the section discusses anti-inflammatory agents like 5-aminosalicylic acid (5-ASA - Mesalamine), and epigenetic drugs including HDAC inhibitors (e.g., Vorinostat) and DNMT inhibitors (e.g., Decitabine), which are under clinical evaluation for CRC treatment.
One important of our scopes, in the current review, is to present an overview of how understanding the epigenetic landscape in inflammation-associated colorectal cancer can inform precision medicine approaches to improve patient outcomes. Moreover, this integrative approach underscores how inflammation-epigenetic pathways offer novel opportunities for early detection and personalized therapeutic intervention.
9.1. Translational Epigenetics and RNA-Based Therapeutics in IBD/CRC
The increasing understanding of chromatin epigenetics and non-coding RNAs (ncRNAs) in the pathogenesis of inflammatory bowel disease (IBD) and its progression to colorectal cancer (CRC) has opened new avenues for clinical translation. The therapeutic strategies in IBD-associated colorectal cancer (IBD-CRC) now include molecular tools that directly target epigenetic regulators and non-coding RNAs (ncRNAs), particularly miRNAs and lncRNAs. Among the most promising of these are epigenetic-targeting drugs and RNA-based therapeutics, which are reshaping personalized medicine strategies in gastrointestinal oncology. These interventions are being investigated both preclinical and in early-phase clinical trials to halt chronic inflammation or reverse the neoplastic transformation of inflamed intestinal tissues. Within this context, Table 10 below shows top translational therapies targeting epigenetic regulators in colitis-associated colorectal cancer (CAC), including emerging agents such as 5-azacytidine and their molecular targets, also visualized in Figure 2.
9.2. Epigenetic Drugs and Clinical Trials in IBD-Associated CRC
DNA methyltransferase inhibitors (DNMTis) such as azacitidine and decitabine, historically used in hematologic malignancies, have shown potential in reactivating silenced tumor suppressor genes in solid tumors, including CRC. Although not yet standard in IBD-associated CRC, preclinical studies demonstrate that DNMTis can reverse inflammation-induced methylation patterns and modulate immune responses -crucial in the dysplastic transformation of IBD lesions [345]. Similarly, HDACis, including vorinostat and romidepsin, have demonstrated anti-inflammatory and anti-tumor effects by altering histone acetylation states and suppressing pro-inflammatory gene transcription [346].
Of particular interest are BET (bromodomain and extra-terminal) inhibitors, which target chromatin "readers" like BRD4. These proteins regulate oncogenic transcriptional programs, including Myc-driven pathways in CRC. BET inhibitors (e.g., JQ1, Pelabresib (CPI-0610) etc) are undergoing clinical trials and have been shown to suppress cytokine-mediated inflammation and reduce tumor growth in preclinical models of colitis and CAC [347]. BET proteins also modulate T-cell responses, offering additional therapeutic potential in chronic inflammation and immune dysregulation.
9.3. RNA-Based Therapeutics in Preclinical and Clinical Use
Several ncRNAs implicated in IBD-CRC serve as both diagnostic biomarkers and therapeutic targets. MicroRNA-21, upregulated in inflamed and cancerous colonic tissues, targets tumor suppressors like PDCD4 and is currently under investigation for anti-miR-21 oligonucleotide-based therapies [333,348]. For instance, miR-21, frequently upregulated in colonic inflammation and early neoplasia, enhances NF-κB signaling and suppresses tumor suppressor PDCD4 [332]. Antagomirs targeting miR-21 have demonstrated efficacy in reducing colitis severity and tumor burden in murine models [349]. MicroRNA-92a, another oncogenic miRNA elevated in serum and stool samples of CRC patients, is also being explored as a therapeutic target and non-invasive biomarker [350]. These RNA therapeutics are being evaluated using mouse models of DSS- and AOM/DSS-induced colitis/CAC – mouse model analogous to the human disease –, human CRC organoids, and patient-derived xenograft models that preserve tumor heterogeneity [348].
In parallel, miR-155 and miR-223 are under evaluation as modulators of immune cell polarization and epithelial integrity [351,352], while lncRNAs like HOTAIR and CCAT1 have shown oncogenic properties in CRC. Therapeutic silencing of these molecules using antisense oligonucleotides (ASOs) or locked nucleic acid (LNA)-modified inhibitors is currently in early-phase clinical trials or advanced preclinical stages [353].
Furthermore, RNA aptamers and small interfering RNAs (siRNAs) offer precise gene silencing potential and are being explored to target inflammatory mediators, oncogenes, and DNA methyltransferases directly [354]. For example, the siRNA-based drug ALN-PCS, although initially developed for hypercholesterolemia, exemplifies the clinical viability of RNA interference-based platforms [355].
9.4. Diagnostic and Prognostic Utility of Epigenetic and RNA Biomarkers
Several circulating epigenetic and RNA markers have shown promise as non-invasive tools for early detection and surveillance. Stool- (Cologuard in USA and ColoAlert in Europe, approved by FDA) or blood-based assays detecting methylated SEPT9, VIM, or SFRP2 genes [356,357], as well as panels combining miR-21, miR-92a, and miR-135b, have demonstrated high sensitivity for early CRC detection [350]. Stool DNA methylation testing, specifically Cologuard, is an FDA-approved assay for colorectal cancer (CRC) screening. It detects DNA markers and occult hemoglobin in stool samples, helping identify the presence of colorectal cancer or advanced precancerous lesions. The test has been updated with Cologuard Plus, a next-generation version, that improves sensitivity and stability. Moreover, patterns of miRNA expression are increasingly being integrated into risk models for dysplasia surveillance in patients with UC and CD.
9.5. Comparative Human-Mouse Evidence
Preclinical mouse models of colitis and colorectal cancer remain indispensable for dissecting molecular mechanisms that are often challenging to study directly in patients. By mirroring key pathological features of human IBD and CAC, these systems provide a platform to interrogate genetic, epigenetic, and transcriptomic changes under controlled conditions. Importantly, cross-species comparisons allow validation of candidate pathways and biomarkers, strengthening their translational relevance for diagnostics and therapeutics.
For example, in DSS-induced mouse colitis models, HDAC inhibition reduced NF-κB activity and prevented histological inflammation, paralleling human biopsy studies showing HDAC1/2 overexpression in UC patients [345,358]. Similarly, miR-21 overexpression has been validated in both AOM/DSS murine CAC (Figure 1), zebrafish tissues as well as human colorectal carcinoma biopsies, linking inflammation and carcinogenesis via shared transcriptomic patterns [332,333,348].
Referring to the expanded overview of murine models in Section 3 and our RNA-seq meta-analysis, and by contextualizing the DSS and AOM/DSS model (combines genetic mutation induction with inflammation-driven carcinogenesis), one of the most widely adopted chemical models for acute/chronic colitis and CAC, we explored the implicated protein-coding genes and non-coding RNAs (Triantaphyllopoulos et al.,, manuscript in preparation). AOM/DSS model,. The aforementioned models allowed us to identify overlapping protein-coding gene signatures with human colorectal cancer, enhancing its translational relevance for both diagnostic and therapeutic implications in IBD and CRC.
10. Concluding Remarks and Future Outlook
The interplay between genetic predisposition, chronic inflammation, and epigenetic dysregulation is central to the pathogenesis of inflammatory bowel disease (IBD)-associated colorectal cancer (CRC). As detailed in this review, chromatin-modifying enzymes, DNA methylation machinery, and regulatory non-coding RNAs orchestrate dynamic transcriptional changes in both immune and epithelial compartments, driving the progression from inflammation to neoplasia.
At the forefront of translational breakthroughs are several clinically actionable genetic markers such as TP53, KRAS, APC, BRAF, and SMAD4, which not only serve as predictors of malignant transformation but also guide treatment decisions. Simultaneously, epigenetic regulators such as DNMT1, HDACs, EZH2, and BET proteins are gaining attention as both biomarkers and druggable targets, particularly as next-generation epigenetic inhibitors advance through clinical pipelines. Adding another dimension, proteomic studies have unveiled dysregulated pathways involving key inflammation-related proteins (e.g., IL-6, STAT3, TNF-α), tight junction components (e.g., claudins, occludin), and tumor microenvironment factors (e.g., MMP9, VEGF), many of which are now being profiled longitudinally to monitor disease progression and therapeutic response. Moreover, the incorporation of advanced imaging modalities - including CT, MRI, positron emission tomography (PET)/CT, and emerging Electron Paramagnetic Resonance (EPR)-based molecular imaging - into clinical practice allows for early lesion detection and real-time biomarker mapping. Novel imaging biomarkers, particularly those targeting integrin expression or metabolic pathways, are now integrated into multimodal biomarker discovery platforms, offering a powerful complement to genetic and transcriptomic profiling.
Importantly, the convergence of multi-omics technologies, liquid biopsy tools (e.g., cell-free DNA methylation and circulating ncRNAs), and artificial intelligence-based imaging analytics is transforming biomarker discovery from siloed datasets to holistic, integrative diagnostics. These advancements enable not only early CRC detection in IBD patients but also real-time stratification and tailored therapeutic decisions.
Future efforts must prioritize clinical validation of composite biomarker panels, refinement of RNA delivery technologies, and the integration of omics data with radiogenomic signatures. Together, these efforts will bridge the bench-to-bedside gap, allowing for truly personalized, predictive, and preventive strategies in IBD-associated CRC.
Socioeconomic and Health System Impact:
Beyond scientific innovation, the integration of epigenetic and genetic biomarkers into routine clinical care carries profound socioeconomic implications. In the sidebar infographic below is shown a brief review of the Socioeconomic impact of precision medicine in IBD and CRC.
Figure 5.
Sidebar Infographic: Socioeconomic Impact and Precision Medicine in IBD and CRC.

This infographic summarizes key socioeconomic metrics and the potential of precision medicine in IBD and CRC management. Most precision interventions are cost-effective compared with standard care (PMID: 31650223). Evidence also shows that tailored precision strategies, such as targeted antibiotic-based interventions, can reduce hospitalization (PMID: 40001454). In Europe, financial incentives and innovative reimbursement frameworks are emerging as important drivers of sustainable implementation (PMID: 37623911). Broader value frameworks further highlight the importance of capturing patient-centered, long-term, and societal benefits when assessing returns on investment in precision medicine (PMID: 32389217). Together, these findings suggest that precision approaches can minimize unnecessary interventions, improve resource allocation, and strengthen healthcare system resilience.
The integration of epigenetic and genetic biomarkers into clinical practice carries profound socioeconomic implications. As illustrated in Figure 5, precision medicine in IBD and CRC promises to alleviate rising healthcare costs, reduce disparities in access to molecular diagnostics, and optimize therapeutic efficacy. Evidence suggests that precision approaches can be cost-effective compared to standard care (PMID: 31650223). In the European context, financial incentives and innovative reimbursement frameworks are emerging as crucial catalysts for sustainable implementation (PMID: 37623911). Broader value frameworks emphasize the need to capture patient-centered, long-term, and societal benefits when evaluating return on investment (PMID: 32389217). Beyond IBD and CRC, the wider applicability of targeted care has been demonstrated in infectious disease settings, such as urinary tract infections, where biomarker- and precision-based strategies can reduce hospitalization and improve cost efficiency (PMID: 40001454).
Although upfront investments in infrastructure - including interoperable digital health platforms, genomic repositories, and workforce training - are substantial, the long-term benefits are considerable. By decreasing ICU admissions, shortening hospital stays, and minimizing recurrence rates, precision medicine strengthens system resilience. The Organisation for Economic Co-operation and Development (OECD) has emphasized that targeted technologies can be cost-effective and that policy modernization is essential for efficient resource allocation. Taken together, these considerations highlight how predictive and preventive medicine can redefine cost-efficiency, moving healthcare systems toward early intervention strategies that are not only relevant for IBD and CRC but broadly applicable across human disease.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/, Table S1: LncRNA–miRNA interactions inferred by ncFANs-NET network analysis in Crohn’s disease; Table S2: DNA methylation landscape of lncRNA transcripts in gastrointestinal carcinogenesis; Table S3_ New characterized human lncRNAs in UC; Table S4: Expression profile of the top 5 piRNAs in colorectal-related carcinogenesis; Table S5: circRNA characteristics from extracellular vesicles (EVs) in colon and small intestine tissue; Table S6: circRNA characteristics from EVs in colorectal cancer; Figure S1: Functional Annotation of FIGNL2-DT and GAS5-AS1 in CD; Figure S2: LncRNA-miRNA levels of interaction in UC; Figure S3: LncRNA-Gene-miRNA-disease (ceRNA) interaction network created in LncACTdb3.0 highlighting human GI carcinogenesis targets.
Author Contributions
Conceptualization, investigation, Data Analysis, In Silico studies, Figures and Graphics design, reviewing and editing, Tables, project administration, K.A.T.; Writing - original draft, resources, reviewing and editing, supervision,; K.A.T. and T.G.S.; Table 3, Table 4 and Table 5, drafting text in the sections pertinent to the Table 3, Table 4 and Table 5, N.D.R.; drafting chapters in section 2, M-C.E.P.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Further data in this study are available upon request from the authors.
Acknowledgments
Thanks for the patience of all authors during this period of time of the manuscript in preparation.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
IBD: inflammatory bowel disease; CD: Crohn’s disease; UC: ulcerative colitis; APC: adenomatous polyposis coli; CRC: colorectal cancer; CAC: colitis-associated cancer; GI: gastrointestinal; GIT: gastrointestinal tract; GIC: gastrointestinal cancer; ncRNA: non-coding RNA; lncRNA: long non-coding RNA; lincRNA: long intergenic RNA; piRNA: PIWI-interacting RNA; circRNA: circular RNA; RBP: RNA-binding protein; MTBC: mycobacterium tuberculosis complex; GITB: gastrointestinal tuberculosis; TB: tuberculosis; GEMM: genetically engineered mouse models; AOM/DSS: azomethane dextran sulfate sodium; NFκB: nuclear factor-kappa B; IECs: intestinal epithelial cells; PDX: patient-derived xenografts; TF: transcription factor.
References
- Chen, G.Y.; Nunez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10(12), 826-837. [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21(7), 677-687. [CrossRef]
- Zhang, S.; Meng, Y.; Zhou, L., Qiu, L.; Wang, H.; Su, D.; Zhang, B.; Chan, K.-M.; Han, J. Targeting epigenetic regulators for inflammation: Mechanisms and intervention therapy. MedComm 2022, 3(4), e173. [CrossRef]
- Tan, S.Y.X., Zhang, J.; Tee, W.W. Epigenetic Regulation of Inflammatory Signaling and Inflammation-Induced Cancer. Front. Cell Dev. Biol. 2022, 10, 931493. PMID: 35757000; PMCID: PMC9213816 . [CrossRef]
- Das, D.; Karthik, N.; Taneja, R. Crosstalk between inflammatory signaling and methylation in cancer. Front. Cell Dev. Biol. 2021, 9, 756458. [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140(6), 883-899. [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420(6917), 860-867. [CrossRef]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Bhol, C.S.; Praharaj, P.P.; Panigrahi, D.P.; et al. Inflammasomes in cancer: effect of epigenetic and autophagic modulations. Semin. Cancer Biol. 2022, 83, 399-412. [CrossRef]
- Shi, L.; Wang, L.; Hou, J.; Zhu, B.; Min, Z., Zhang, M.; Song, D.; Cheng, Y.; Wang, X. Targeting roles of inflammatory microenvironment in lung cancer and metastasis. Cancer Metastasis Rev. 2015, 34(2), 319-331. [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454(7203), 436-444. [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51(1), 27-41. [CrossRef]
- Vieujean, S.; Caron, B.; Haghnejad, V.; Jouzeau, J.Y.; Netter, P.; Heba, A.C.; Ndiaye, N.C.; Moulin, D.; Barreto, G.; Danese, S.; Peyrin-Biroulet, L. Impact of the exposome on the epigenome in inflammatory bowel disease patients and animal models. Int. J. Mol. Sci. 2022, 23(14). [CrossRef]
- Zeng, Z.; Mukherjee, A.; Zhang, H. From genetics to epigenetics, roles of epigenetics in inflammatory bowel disease. Front. Genet. 2019, 10, 1017. [CrossRef]
- Gerbeth, L.; Glauben, R. Histone Deacetylases in the inflamed intestinal epithelium-Promises of new therapeutic strategies. Front. Med. (Lausanne) 2021, 8, 655956. [CrossRef]
- Jawad, N.; Direkze, N.; Leedham, S.J. Inflammatory bowel disease and colon cancer. Recent Results Cancer Res. 2011, 185, 99-115. [CrossRef]
- Thomson, P.D.; Smith, D.J. Jr. What is infection? Am. J. Surg. 1994, 167(1A), 7S-10S; discussion 10S-11S. PMID: 8109689 . [CrossRef]
- Qerqez, A.N.; Silva, R.P.; Maynard, J.A. Outsmarting pathogens with antibody engineering. Annu. Rev. Chem. Biomol. Eng. 2023 , 14, 217-241. PMID: 36917814; PMCID: PMC10330301. [CrossRef]
- van Seventer, J.M.; Hochberg, N.S. Principles of infectious diseases: transmission, diagnosis, prevention, and control. In International Encyclopedia of Public Health, 2nd ed.; Quah, S.R., Ed.; Academic Press, Elsevier. Cambridge, MA., U.S.A. 2017, 22–39. PMCID: PMC7150340. [CrossRef]
- Burrell, C.J.; Howard, C.R.; .Murphy, F.A. Innate Immunity. In Fenner and White's Medical Virology, 5th ed.; Academic Press, Elsevier, Oxford, U.K. 2017, 57–64. PMCID: PMC7173408 . [CrossRef]
- Schertzer, J.D.; Lam, T.K.T. Peripheral and central regulation of insulin by the intestine and microbiome. Am. J. Physiol. Endocrinol. Metab. 2021, 320(2), E234-E239. [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target Ther. 2021, 6(1), 291. [CrossRef]
- Carretta, M.D.; Quiroga, J.; López, R.; Hidalgo, M.A.; Burgos, R.A. Participation of short-chain fatty acids and their receptors in gut inflammation and colon cancer. Front. Physiol. 2021, 12, 662739. [CrossRef]
- Madison, A.; Kiecolt-Glaser, J.K. Stress, depression, diet, and the gut microbiota: Human-bacteria interactions at the core of psychoneuroimmunology and nutrition. Curr. Opin. Behav. Sci. 2019, 28,105-110. [CrossRef]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8(1), 51. [CrossRef]
- Finlay, B.B.; McFadden, G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006, 124(4), 767-82. [CrossRef]
- Giamarellos-Bourboulis, E.J.; Raftogiannis, M. The immune response to severe bacterial infections: Consequences for therapy. Expert Rev. Anti. Infect. Ther. 2012, 10(3), 369-80. [CrossRef]
- Al-Zanbagi, A.B.; Shariff, M.K. Gastrointestinal tuberculosis: A systematic review of epidemiology, presentation, diagnosis and treatment. Saudi J Gastroenterol. 2021, 27(5), 261-274. [CrossRef]
- Choudhury ,A.; Dhillon, J.; Sekar, A.; Gupta, P.; Singh, H.; Sharma, V. Differentiating gastrointestinal tuberculosis and Crohn's disease- a comprehensive review. BMC Gastroenterol. 2023, 23(1), 246. [CrossRef]
- Eraksoy, H. Gastrointestinal and abdominal tuberculosis. Gastroenterol. Clin. North Am. 2021, 50(2), 341-360. [CrossRef]
- Malikowski T, Mahmood M, Smyrk T, Raffals L, Nehra V. Tuberculosis of the gastrointestinal tract and associated viscera. J. Clin. Tuberc. Other Mycobact. Dis. 2018, 12, 1-8. . Erratum in: J Clin Tuberc Other Mycobact Dis. 2020 Sep 09;21:100177. doi: 10.1016/j.jctube.2020.100177. PMID: 31720391; PMCID: PMC6830173. [CrossRef]
- Gopalaswamy, R.; Dusthackeer, V.N.A.; Kannayan, S.; Subbian, S. Extrapulmonary Tuberculosis—An update on the diagnosis, treatment and drug resistance. J. of Resp. 2021, 1(2), 141–164. [CrossRef]
- Choi, E.; Coyle, W. Gastrointestinal tuberculosis. In Tuberculosis and Nontuberculous Mycobacterial Infections. 7th ed.; Schlossberg, D., Ed.; ASM Press, Wiley. Washington, D.C., U.S.A. 2017, 411–432. [CrossRef]
- Dasgupta, A.; Singh, N.; Bhatia, A. Abdominal tuberculosis: A histopathological study with special reference to intestinal perforation and mesenteric vasculopathy. J. Lab. Physicians 2009, 1(2), 56-61. [CrossRef]
- Maulahela, H.; Simadibrata, M.; Nelwan, E.J.; Rahadiani, N.; Renesteen, E.; Suwarti, S. W.T.; Anggraini, Y.W. Recent advances in the diagnosis of intestinal tuberculosis. BMC Gastroenterol. 2022, 22(1). [CrossRef]
- Chatzicostas, C.; Koutroubakis, I.E.; Tzardi, M.; Roussomoustakaki, M.; Prassopoulos, P.; Kouroumalis, E.A. Colonic tuberculosis mimicking Crohn’s disease: Case report. BMC Gastroenterol. 2002, 2, 10. http://www.biomedcentral.com/1471-230X/2/10.
- Arhan, M.; Köksal, A.Ş.; Özin, Y.; Mesut, Z.; Kılıç, Y.; Tunç, B.; Ülker, A. Colonic tuberculosis or Crohn’s disease ? An important differential diagnosis. Acta Gastroenterol. Belg. 2013, 76(1), 59-61. PMID: 23650785.
- González-Puga, C.; Palomeque-Jiménez, A.; García-Saura, P.L.; Pérez-Cabrera, B.; Jiménez-Ríos, J.A. Colonic tuberculosis mimicking Crohn’s disease: An exceptional cause of massive surgical rectal bleeding. Medecine et Maladies Infectieuses 2015, 45(1–2), 44–46. doi.org/10.1016/j.medmal.2014.11.005.
- Adhikari, G.; Buda, B., Shah, J.K.; Ghimire, B.; Kansaker, P.B.S. Colonic tuberculosis masquerading as ascending colon carcinoma in a patient of FIGO Stage IIB cervical carcinoma following chemo-radiotherapy: A case report. Int. J. Surg. Case Rep. 2022, 93, 106943. [CrossRef]
- Luczynski, P.; Poulin, P.; Romanowski, K. Johston, J.C. Tuberculosis and risk of cancer: A systematic review and meta-analysis. PLO One 2022, 17(12), e0278661. [CrossRef]
- Saidu, A.S.; Okolocha, E.C.; Gamawa, A.A.; Babashani, M.; Bakari, N.A. Occurrence and distribution of bovine tuberculosis (Mycobacterium bovis) in slaughtered cattle in the abattoirs of Bauchi State, Nigeria. Veterinary World, 2015, 8(3), 432–437. [CrossRef]
- Luboya, L.W.; Malangu, M.; Kaleka, M.; Ngulu, N.; Nkokele, B.; Maryabo, K.; Pourrut, X.; Vincent, T.; Gonzalez, J.P. An assessment of caprine tuberculosis prevalence in Lubumbashi slaughterhouse, Democratic Republic of Congo. Tropical Animal Health and Production, 2017, 49(4), 875–878. [CrossRef]
- Engelmann, N.; Ondreka, N.; Michalik, J.; Neiger, R. Intra-abdominal Mycobacterium tuberculosis infection in a dog. J. Vet. Intern. Med. 2014, 28(3), 934-8. https://doi.10.1111/jvim12347.
- Mentula, S.; Karkamo, V.; Skrzypczak, T.; Seppänen, J.; Hyyryläinen, H.L.; Haanperä, M.; Soini, H. Emerging source of infection - Mycobacterium tuberculosis in rescue dogs: a case report. Access Microbiol. 2020, 2(11). [CrossRef]
- Ribeiro, M.G.; Lima, M.C.F.; Franco, M.M.J.; Megid, J.; Soares, L.M.; Machado, L.H.A.; Miyata, M., Pavan, F.R.; Heinemann, M.B.; Souza Filho, A.F.; Lara, G.H.B.; Sanches, O.C.; Sanches, C.D.C.; Listoni, F.J.P.; Paes, A.C. Pre-multidrug-resistant Mycobacterium tuberculosis infection causing fatal enteric disease in a dog from a family with history of human tuberculosis. Transbound. Emerg. Dis. 2017, 64(5), e4-e7. [CrossRef]
- Dhama, K.; Mahendran, M.; Tiwari, R.; Dayal Singh, S.; Kumar, D.; Singh, S.; Sawant, P.M. Tuberculosis in Birds: Insights into the Mycobacterium avium Infections. Vet. Med. Int. 2011, 712369. [CrossRef]
- Bertram, C.A.; Barth, S.A.; Glöckner, B.; Lübke-Becker, A.; Klopfleisch, R. Intestinal Mycobacterium avium Infection in Pet Dwarf Rabbits (Oryctolagus cuniculus). J. Comp. Pathol. 2020, 180, 73-78. [CrossRef]
- Vetere, A.; Bertocchi, M.; Pagano, T.B.; Di Ianni, F.; Nardini G. First case of systemic fatal mycobacteriosis caused by Mycobacterium goodii in a pet Kenyan sand boa (Eryx colubrinus loveridgei). BMC Vet. Res. 2022, 18(1), 291. [CrossRef]
- Lindsay, S.A.; Gray, R. A Novel Presentation of tuberculosis with intestinal perforation in a free-ranging Australian sea lion (Neophoca cinerea). J. Wildl. Dis. 2021, 57(1), 220-224. [CrossRef]
- Elsalem, L.; Jum'ah, A.A.; Alfaqih, M.A.; Aloudat O. The Bacterial Microbiota of Gastrointestinal Cancers: Role in Cancer Pathogenesis and Therapeutic Perspectives. Clin. Exp. Gastroenterol. 2020, 13,151-185. [CrossRef]
- Bundgaard-Nielsen, C.; Baandrup, U.T.; Nielsen L.P.; Sørensen S. The presence of bacteria varies between colorectal adenocarcinomas, precursor lesions and non-malignant tissue. BMC Cancer 2019 19(1), 399. [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68(6), 394-424. . Erratum in: CA Cancer J. Clin. 2020, 70(4), 313. doi: 10.3322/caac.21609. PMID: 30207593. [CrossRef]
- Neufert, C.; Becker, C.; Neurath, M.F. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007, 2(8), 1998-2004. [CrossRef]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Science, 2003, 94(11), 965–973. [CrossRef]
- Mizoguchi, A.; Takeuchi, T.; Himuro, H.; Okada, T.; Mizoguchi, E. Genetically engineered mouse models for studying inflammatory bowel disease. J. Pathol. 2016, 238(2), 205-19. [CrossRef]
- Ren, J.; Sui, H.; Fang, F.; Li, Q.; Li, B. The application of ApcMin/+ mouse model in colorectal tumor researches. J. Cancer Res. Clin. Oncol. 2019, 145(5), 1111-1122. [CrossRef]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247(4940), 322–324. [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; Roman-Roman, S.; Seoane, J.; Trusolino, L.; Villanueva, A. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discovery 2014, 4(9), 998–1013. [CrossRef]
- Balkwill, F.; Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001, 357(9255), 539-45. [CrossRef]
- Fujiki, H. Gist of Dr. Katsusaburo Yamagiwa's papers entitled "Experimental study on the pathogenesis of epithelial tumors" (I to VI reports). Cancer Sci. 2014, 105(2),143-9. [CrossRef]
- Dvorak, H.F. Tumors: wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3(1), 111. [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal-a historical perspective with a focus on the fundamental roles of increased vascular permeability and clotting. Semin. Thromb. Hemost. 2019, 45(6), 576-592. [CrossRef]
- Vanoli, A.; Parente, P.; Fassan, M.; Mastracci, L.; Grillo, F. Gut inflammation and tumorigenesis: every site has a different tale to tell. Intern. Emerg. Med. 2023, 18(8), 2169-2179. [CrossRef]
- Grady, W. M.; Yu, M.; Markowitz, S. D. Epigenetic alterations in the gastrointestinal tract: current and emerging use for biomarkers of cancer. Gastroenterol. 2021, 160(3), 690-709. [CrossRef]
- Filho, A.M.; Laversanne, M.; Ferlay, J.; Colombet, M.; Piñeros, M.; Znaor, A.; Parkin, D.M.; Soerjomataram, I.; Bray, F. The GLOBOCAN 2022 cancer estimates: Data sources, methods, and a snapshot of the cancer burden worldwide. Int. J. Cancer 2025, 156(7),1336-1346. [CrossRef]
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: the trends projection analysis. Chem. Biol. Lett. 2023, 10(1), 451.
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74(3), 229-263. [CrossRef]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasaq, C.J.; Laversanne, M.;Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020and 2040: incidence and mortality estimates from GLOBOCAN. Gut 2023, 72(2), 338-344. [CrossRef]
- Yang, F.; Li, X.F.; Cheng, L.N.; Li, X.L. Long non-coding RNA CRNDE promotes cell apoptosis by suppressing miR-495 in inflammatory bowel disease. Exp. Cell Res. 2019, 382(2), 111484. [CrossRef]
- Triantaphyllopoulos, K.A. Long non-coding RNAs and their "discrete" contribution to IBD and Johne's disease-what stands out in the current picture? A comprehensive review. Int. J. Mol. Sci. 2023, 24(17), 13566. [CrossRef]
- Vetrano, S.; Borroni, E.M.; Sarukhan, A.; Savino, B.; Bonecchi, R.; Correale, C.; Arena, V.; Fantini, M.; Roncalli, M.; Malesci, A.; Mantovani, A.; Locati, M.; Danese, S. The lymphatic system controls intestinal inflammation and inflammation-associated Colon Cancer through the chemokine decoy receptor D6. Gut, 2010 59(2), 197-206. [CrossRef]
- Pelaseyed,T.; Hansson, G.C. Membrane mucins of the intestine at a glance. J. Cell. Sci. 2020, 133(5), jcs240929. [CrossRef]
- Tang, W.; Liu, J.; Ma, Y.; Wei, Y.; Liu, J.; Wang H. Impairment of intestinal barrier function induced by early weaning via autophagy and apoptosis associated with gut microbiome and metabolites. Front. Immunol. 2021, 12, 804870. [CrossRef]
- Zella, D.; Gallo, R.C. Viruses and bacteria associated with cancer: an overview. Viruses 2021, 13(6), 1039. [CrossRef]
- van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19(11), e46632. [CrossRef]
- Vogelmann, R.; Amieva, M.R. The role of bacterial pathogens in cancer. Curr. Opin. Microbiol. 2007, 10(1), 76-81. [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001, 48(4), 526-35. [CrossRef]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10(6),639-45. [CrossRef]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol Gastrointest. Liver Physiol. 2004, 287(1), G7-17. [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5(10),749-59. [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431(7007), 461-6. [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; Matthews, V.; Schmid, R.M.; Kirchner, T., Arkan, M.C.; Ernst, M.; Greten, F.R. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15(2), 91-102. [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009, 30(7), 1073-81. [CrossRef]
- Rubio, K.; Dobersch, S.; Barreto, G. Functional interactions between scaffold proteins, noncoding RNAs, and genome loci induce liquid-liquid phase separation as organizing principle for 3-dimensional nuclear architecture: implications in cancer. FASEB J. 2019, 33(5), 5814-5822. [CrossRef]
- Zhang, Y. Recent progress in the epigenetics and chromatin field. Cell Res. 2011, 21(3), 373-374. [CrossRef]
- Ozturk, N.; Singh, I.; Mehta, A.; Braun, T.; Barreto, G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014, 2, 5. [CrossRef]
- Bentley, G.A.; Lewis-Bentley; A.; Finch, J.T.; Podjarny, A.D.; Roth, M. Crystal structure of the nucleosome core particle at 16 A resolution. J. Mol. Biol. 1984, 176(1), 5-75. [CrossRef]
- Richmond, T.J.; Finch, J.T; Rushton, B.; Rhodes, D.; Klug A. Structure of the nucleosome core particle at 7 A resolution. Nature 1984, 311(5986), 532-7. [CrossRef]
- Liang, B.; Wang, Y.; Xu, J.; Shao, Y.; Xing, D. Unlocking the potential of targeting histone-modifying enzymes for treating IBD and CRC. Clin. Epigenetics 2023, 15(1), 146. [CrossRef]
- Singh, I.; Contreras, A.; Cordero, J.; Rubio, K.; Dobersch, S.; Gunther, S.; Jeratsch S, Mehta A, Krüger M, Graumann J, Seeger W, Dobreva G, Braun T, Barreto, G. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization.. Nat. Genet. 2018, 50(7), 990-1001. [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2018, 10(5), 295-304. [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25(10), 1010-1022. [CrossRef]
- Skinner, M.K. Environmental epigenetics and a unified theory of the molecular aspects of evolution: a neo-lamarckian concept that facilitates neo-darwinian evolution. Genome Biol. Evol. 2015, 7(5), 1296-1302. [CrossRef]
- Kealy, L.; Runting, J.; Thiele, D.; Scheer, S. An emerging maestro of immune regulation: how DOT1L orchestrates the harmonies of the immune system. Front. Immunol. 2024, 15, 1385319. [CrossRef]
- Dobersch, S.; Rubio, K.; Barreto, G. Pioneer Factors and Architectural proteins mediating embryonic expression signatures in cancer. Trends Mol. Med. 2019, 25(4), 287-302. [CrossRef]
- Zhang, M.; Zhao, J.; Lv, Y.; Wang, W.; Feng, C.; Zou, W.; Su, L.; Jiao, J. Histone variants and histone modifications in neurogenesis. Trends Cell Biol. 2020, 30(11), 869-880. [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent modifications. Nature 2000, 403(6765), 41-5. [CrossRef]
- Hake, S.B.; Xiao, A.; Allis, C.D. Linking the epigenetic 'language' of covalent histone modifications to cancer. Br. J. Cancer 2007, 96 Suppl, R31-9. PMID: 17393583.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011 21(3), 381-95. [CrossRef]
- Mattiroli, F.; Penengo, L. Histone Ubiquitination: An integrative signaling platform in genome stability. Trends Genet. 2021, 37(6), 566-581. [CrossRef]
- Andrews, A. J.; Luger, K. Nucleosome structure(s) and stability: variations on a theme Annu. Rev. Biophys. 2011, 40, 99-117. [CrossRef]
- Kouzarides, T. Chromatin modifcations and their function. Cell 2007, 128(4), 693-705. [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code Science 2001, 93(5532),1074-80. [CrossRef]
- Jones, P. A.; Baylin, S. B. The epigenomics of cancer. Cell 2007, 128(4), 683-692. [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Ting, C.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20(1-2), 35–47. [CrossRef]
- Yang, Z.H.; Dang, Y.-Q.; Ji, G. Role of epigenetics in transformation of inflammation into colorectal cancer. World J. Gastroenterol. 2019, 25(23), 2863-2877. [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8(4), 286-98. [CrossRef]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517(7534), 321-326. [CrossRef]
- Saif, I.; Kasmi, Y.; Allali, K.; Ennaji, M.M. Prediction of DNA methylation in the promoter of gene suppressor tumor. Gene 2018, 651,166-173. [CrossRef]
- Yang, I.V.; Schwartz, D.A. Epigenetic control of gene expression in the lung. Am. J. Respir. Crit. Care Med. 2011, 183(10), 1295-1301. [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28(8), 812-28. [CrossRef]
- Jin, B.; Robertson, K. D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3-29. [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology, 2013, 38, 23-38. [CrossRef]
- Chakraborty, A.; Viswanathan, P. Methylation-Demethylation Dynamics: Implications of Changes in Acute Kidney Injury. Anal. Cell. Pathol. (Amst) 2018, 2018, 8764384. [CrossRef]
- Szyf, M. DNA methylation and cancer therapy. Drug Resist Updat. 2003, 6(6), 341-53. [CrossRef]
- Greenberg, M.V.C.; Bourc'his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 2019 20(10), 590-607. [CrossRef]
- Nie, L.; Wu, H.J.; Hsu, J.M.; Chang, S.S.; Labaff, A.M.; Li, C.W.; Wang, Y.; Hsu, J.L. Long non-coding RNAs: versatile master regulators of gene expression and crucial players in cancer. Am. J. Transl. Res. 2012, 4(2),127-50. PMID: 22611467; PMCID: PMC3353529.
- Panda, A.C. Circular RNAs Act as miRNA Sponges. Adv. Exp. Med. Biol. 2018, 1087, 67-79. [CrossRef]
- Li, J.; Xu, Q.; Huang, Z.J.; Mao, N.; Lin, Z.T.; Cheng, L.; Sun, B.; Wang, G. CircRNAs: A new target for the diagnosis and treatment of digestive system neoplasms. Cell Death Dis. 2021, 12, 205. [CrossRef]
- Wang,F.; Nazarali, A.J.; Ji, S. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am. J. Cancer Res. 2016, 6, 1167–1176. PMID: 27429839; PMCID: PMC4937728.
- Zhou,Z.; Wang, X.; Hu, Q.; Yang, Z. CircZfp609 contributes to cerebral infarction via sponging miR-145a-5p to regulate BACH1. Metab. Brain Dis. 2023, 38, 1971–1981. doi: 10.1007/s11011-023-01208-4. PMID: 37097437.
- Mowel, W.K.; Kotzin, J.J.; McCright, S.J.; Neal, V.D.; Henao-Mejia, J. Control of immune cell homeostasis and function by lncRNAs. Trends Immunol. 2018, 39(1), 55-69. [CrossRef]
- Lei, K.; Bai, H.; Wei, Z; Xie, C.; Wang, J.; Li, J.; Chen, Q. The mechanism and function of circular RNAs in human diseases. Exp. Cell Res. 2018, 368, 147-158. [CrossRef]
- Aalto, A.P.; Pasquinelli, A. E. Small non-coding RNAs mount a silent revolution in gene expression. Curr. Opin. Cell Biol. 2012, 24(3), 333-340. [CrossRef]
- Li, J.; Zhong, Y.; Cai, S.; Zhou, P.; Yao, L. MicroRNA expression profiling in the colorectal normal-adenoma-carcinoma transition. Oncol. Lett. 2019, 18(2), 2013-2018. [CrossRef]
- Peng, Q.; Zhang, X.; Min, M.; Zou, L.; Shen, P.; Zhu, Y. The clinical role of microRNA-21 as a promising biomarker in the diagnosis and prognosis of colorectal cancer: a systematic review and meta-analysis. Oncotarget 2017, 8(27), 44893-44909. [CrossRef]
- Kellermayer, R.; Zilbauer, M. The gut microbiome and the triple environmental hit concept of inflammatory bowel disease pathogenesis. J. Pediatr. Gastroenterol. Nutr. 2020, 71(5), 589-595. [CrossRef]
- Gargalionis, A. N.; Piperi, C.; Adamopoulos, C.; Papavassiliou, A. G. Histone modifications as a pathogenic mechanism of colorectal tumorigenesis. Int. J. Biochem. Cell Biol. 2012, 44(8), 1276-1289. [CrossRef]
- Tamgue, O.; Mezajou, C. F.; Ngongang, N. N.; Kameni, C.; Ngum, J. A.; Simo, U. S. F.; Tatang, F. J.; Akami, M.; Ngono, A. N. Non-Coding RNAs in the Etiology and Control of Major and Neglected Human Tropical Diseases. Front. Immunol. 2021, 12, 703936. [CrossRef]
- Roy, S.; Ghosh, S.; Banerjee, M.; Laha, S.; Bhattacharjee, D.; Sarkar, R.; Ray, S.; Banerjee, A.; Ghosh, R.; Halder, A.; Ghosh, A.; Chatterjee, R.; Datta, S.; Dhali, G. K.; Banerjee, S. A combination of circulating microRNA-375-3p and chemokines CCL11, CXCL12, and G-CSF differentiate Crohn’s disease and intestinal tuberculosis. Sci. Rep. 2021, 11(1), 23303. [CrossRef]
- Yang, F.; Yang, Y.; Chen, L.; Zhang, Z.; Liu, L.; Zhang, C.; Mai, Q.; Chen, Y.; Chen, Z.; Lin, T.; Chen, L.; Guo, H.; Zhou, L.; Shen, H.; Chen, X.; Liu, L.; Zhang, G.; Liao, H.; Zeng, L.; Zeng, G. The gut microbiota mediates protective immunity against tuberculosis via modulation of lncRNA. Gut Microbe 2022, 14(1), 2029997. [CrossRef]
- Zhuo, Q.; Zhang, X.; Zhang, K.; Chen, C.; Huang, Z.; Xu, Y. The gut and lung microbiota in pulmonary tuberculosis: susceptibility, function, and new insights into treatment. Expert Review of Anti-Infective Therapy 2023, 21(12), 1355-1364. [CrossRef]
- Xu, J.; Xu, H.M.; Yang, M.F.; Liang, Y.J.; Peng, Q.Z.; Zhang, Y.; Tian, C.M.; Wang, L.S.; Yao, J.; Nie, Y.Q.; Li. D.F. New insights into the epigenetic regulation of inflammatory bowel disease. Front. Pharmacol. 2022, 13, 813659. [CrossRef]
- Lin, Y.; Qiu, T.; Wei, G.; Que, Y.; Wang, W.; Kong, Y.; Xie, T.; Chen, X. Role of histone post-translational modifications in inflammatory diseases. Front. Immunol. 2022, 13, 852272. [CrossRef]
- Goossens-Beumer, I.J.; Bernard, A.; van Hoesel, A.Q.; Zeestraten, E.C.; Putter, H.; Böhringer, S.; Liefers, G.J.; Morreau, H.; van de Velde, C.J.; Kuppen, P.J. Age-dependent clinical prognostic value of histone modifications in colorectal cancer. Transl. Res. 2015, 165(5), 578-88. [CrossRef]
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li. H. Histone modifications and their role in colorectal cancer. Pathol. Oncol. Res. 2020, 26(4), 2023-2033. [CrossRef]
- Sarvestani, S. K.; Signs, S. A.; Lefebvre, V.; Mack, S.; Ni, Y., Morton, A.; Chan, E.R.; Li, X.; Fox, P.; Ting, A.; Kalady, M.F.; Cruise, M.; Ashburn, J.; Stiene, J.; Lai, W.; Liska, D.; Xiang, S.; Huang, E. H. Cancer-predicting transcriptomic and epigenetic signatures revealed for ulcerative colitis in patient-derived epithelial organoids. Oncotarget 2018 9(47), 28717-28730. [CrossRef]
- Chen, L.; Luo, Z.; Zhao, C.; Li, Q.; Geng, Y.; Xiao, Y.; Chen, M.K.; Li, L.; Chen, Z.X.; Wu, M. Dynamic chromatin states coupling with key transcription factors in colitis-associated colorectal cancer. Adv. Sci. (Weinh) 2022, 9(23), e2200536. [CrossRef]
- Parang, B.; Barrett C.W.; Williams, C.S. AOM/DSS Model of colitis-associated cancer. Methods Mol. Biol. 2016, 1422, 297-307. [CrossRef]
- Xiao, J.; Duan, Q.; Wang, Z.; Yan, W.; Sun, H.; Xue, P.; Fan, X.; Zeng, X.; Chen, J.; Shao, C.; Zhu, F. Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon. Oncotarget 2016, 7(17), 24483-94. [CrossRef]
- Cheung, P., T. K.; Cheung, W.L.; Sassone-Corsi, P.; Denu, J.M.; Allis, C.D. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 2000, 5(6):905-15. [CrossRef]
- Hu, X.; L. X.; Liu, R.; Ai, N.; Cao, Z.; Li, Y.; Liu, J.; Yu, B.; Liu, K.; Wang, H.; Zhou, C.; Wang, Y.; Han, A.; Ding, F.; Chen, R. Histone cross-talk connects protein phosphatase 1α (PP1α) and histone deacetylase (HDAC) pathways to regulate the functional transition of bromodomain-containing 4 (BRD4) for inducible gene expression. J. Biol. Chem. 2014, 289(33), 23154-23167. [CrossRef]
- Komar, D.; Juszcynski, P. Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin. Epigenetics 2020, 12(1), 147. [CrossRef]
- Tarcic ,O.; Pateras. I.S.; Cooks, T.; Shema, E.; Kanterman, J.; Ashkenazi, H.; Boocholez, H.; Hubert, A.; Rotkopf, R.; Baniyash, M.; Pikarsky, E.; Gorgoulis, V.G.; Oren. M. RNF20 Links Histone H2B ubiquitylation with inflammation and inflammation-associated cancer. Cell Rep. 2016, 14(6), 1462-1476. [CrossRef]
- Jeusset, L.M.; McManus, K.J. Characterizing and exploiting the many roles of aberrant H2B monoubiquitination in cancer pathogenesis. Seminars Cancer Biol. 2021, 22(4), 1044–1579. [CrossRef]
- Worden, E.J.; Wolberger, C. Activation and regulation of H2B-ubiquitin-dependent histone methyltransferases. Curr. Opin. Struct. Biol. 2020, 59, 98-106. [CrossRef]
- Worden, E.J.; Zhang, X.; Wolberger C. Structural basis for COMPASS recognition of an H2B-ubiquitinated nucleosome. Elife, 2020, 9, e53199. [CrossRef]
- Van Tongelen, A.; Loriot, A.; De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Letters 2017, 396, 130-137. [CrossRef]
- Dokun, O. Y.; Florl, A. R.; Seifert, H. H.; Wolff, I.; Schulz, W. A. Relationship of SNCG, S100A4, S100A9 and LCN2 gene expression and DNA methylation in bladder cancer. Int. J. Cancer 2008, 123(12), 2798-2807. [CrossRef]
- Dokun, O. Y.; Florl, A. R.; Seifert, H. H.; Wolff, I.; Schulz, W. A. Relationship of SNCG, S100A4, S100A9 and LCN2 gene expression and DNA methylation in bladder cancer. Int. J. Cancer 2008, 123(12), 2798-2807. [CrossRef]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. BBA, 2007, 1775, 138–162. [CrossRef]
- Porcellini, E.; Laprovitera, N.; Riefolo, M.; Ravaioli, M.; Garajova, I.; Ferracin, M. Epigenetic and epitranscriptomic changes in colorectal cancer: Diagnostic, prognostic, and treatment implications. Cancer Lett. 2018, 419, 84-95. [CrossRef]
- Antelo, M.; Balaguer, F.; Shia, J.; Shen, Y.; Hur, K.; Moreira, L.; Cuatrecasas, M.; Bujanda, L.; Giraldez, M.D.; Takahashi, M.; Cabanne, A.; Barugel, M.E.; Arnold, M.; Roca, E.L.; Andreu, M.; Castellvi-Bel, S.; Llor, X.; Jover, R.; Castells, A.; Boland C.R.; Goel, A. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer PLoS One 2012, 7(9), e45357. [CrossRef]
- Ahn, J.B.; Chung, W.B. Maeda, O.; Shin, S.J.; Kim, H.S.; Chung, H.C.; Kim, N.K.; Issa, J.P. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011, 117(9), 1847-1854. [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100(23), 1734-8. [CrossRef]
- Baba, Y.; Huttenhower, C.; Nosho, K.; Tanaka, N.; Shima, K.; Hazra, A.; Schernhammer, E.S.; Hunter, D.J.; Giovannucci, E.L.; Fuchs, C.S.; Ogino. S. Epigenomic diversity of colorectal cancer indicated by LINE-1 methylation in a database of 869 tumors. Mol. Cancer 2010, 9, 125. [CrossRef]
- Swets, M.; Zaalberg, A.; Boot, A.; van Wezel, T.; Frouws, M. A.; Bastiaannet, E.; Gelderblom, H.; van de Velde, C.J.; Kuppen, P.J. Tumor LINE-1 Methylation level in association with survival of patients with stage II colon cancer. Int. J. Mol. Sci. 2016, 18(1). [CrossRef]
- Mima, K.; Nowak, J.A.; Qian, Z.R.; Cao, Y.; Song, M.; Masugi, Y.; Shi, Y.; da Silva, A.; Gu, M.; Li, W.; Hamada, T.; Zhang, X.; Wu, K.; Meyerhardt, J.A.; Baba, H.; Giovannucci, E.L.; Chan, A.T.; Fuchs, C.S.; Ogino, S.; Nishihara, R. Tumor LINE-1 methylation level and colorectal cancer location in relation to patient survival. Oncotarget 2016, 7(34), 55098-55109. [CrossRef]
- Kawakami, K.; Matsunoki, A.; Kaneko, M.; Saito, K.; Watanabe, G.; Minamoto, T. Long interspersed nuclear element-1 hypomethylation is a potential biomarker for the prediction of response to oral fluoropyrimidines in microsatellite stable and CpG island methylator phenotype-negative colorectal cancer. Cancer Sci. 2011, 102(1), 166-174. [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6(8), 597-610. [CrossRef]
- Ambrosi, C.; Manzo, M.; Baubec, T. Dynamics and context-dependent roles of dna methylation. J. Mol. Biol. 2017, 429(10), 1459-1475. [CrossRef]
- Kanwal, R.; Gupta, S. Epigenetics and cancer. J. Appl. Physiol. 2010, 109, 598–605. [CrossRef]
- Nimmo, E.R.; Prentergast, J.G.; Aldhous, M.C.; Kennedy, N.A., Henderson, P.; Drummond, H.E.; Ramsahoye, B.H.; Wilson, D.C.; Semple, C.A.; Satsangi, J. Genome-wide methylation profiling in Crohn's disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm. Bowel Dis. 2012, 18(5), 889-99. [CrossRef]
- Hasler, R.; Feng, Z.; Backdahl, L.; Spehlmann, M.E.; Franke, A.; Teschendorff, A.; Rakyan, V.K.; Down, T.A.; Wilson, G.A.; Feber, A.; Beck, S.; Schreiber, S.; Rosenstiel, P. A functional methylome map of ulcerative colitis. Genome Res. 2012, 22(11), 2130-2137. [CrossRef]
- Cooke, J.; Zhang, H.; Greger, L.; Silva, A.L.; Massey, D.; Dawson, C.; Metz, A.; Ibrahim, A.; Parkes, M. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18(11), 2128-2137. doi.org/10.1002/ibd.22942.
- Saito, S.; Kato, J.; Hiraoka, S.; Horii, J.; Suzuki, H.; Higashi, R.; Kaji, E.; Kondo, Y.; Yamamoto, K. DNA methylation of colon mucosa in ulcerative colitis patients: correlation with inflammatory status. Inflamm. Bowel Dis. 2011, 17(9), 1955-1965. [CrossRef]
- Harris, R.A.; Nagy-Szakal, D.; Mir, S.A.; Frank, E.; Szigeti, R.; Kaplan, J.L.; Bronsky, J.; Opekun, A.; Ferry, G.D., Winter, H., Kellermayer, R. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics 2014, 9(8), 1131-7. [CrossRef]
- Koizumi, K.; Alonso, S.; Miyaki, Y.; Okada, S.; Ogura, H.; Shiiya, N.; Konishi, F.; Taya, T.; Perucho, M.; Suzuki, K. Array-based identification of common DNA methylation alterations in ulcerative colitis. Int. J. Oncol. 2012, 40(4), 983-94. [CrossRef]
- Lin, Z.; Hegarty, J.P.; Yu, W.; Cappel, J.A.; Chen, X.; Faber, P.W.; Wang, Y.; Poritz, L.S.; Fan, J.B.; Koltun, W.A. Identification of disease-associated DNA methylation in B cells from Crohn's disease and ulcerative colitis patients. Dig. Dis. Sci. 2012, 57(12), 3145-3153. [CrossRef]
- Foran, E.; Garrity-Park, M.M.; Mureau, C.; Newell, J.; Smyrk, T.C.; Limburg, P.J.; Egan, L.J. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol. Cancer Res. 2010, 8(4), 471-481. [CrossRef]
- Wang, F.Y.; Arisawa, T.; Tahara, T.; Takahama, K.; Watanabe, M.; Hirata, I.; Nakano, H. Aberrant DNA methylation in ulcerative colitis without neoplasia. Hepatogastroenterol. 2008, 55(81), 62-5.
- Hsieh, C.J.; Klump, B.; Holzmann, K.; Borchard, F.; Gregor, M.; Porschen, R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998, 58(17), 942-5.
- Abu-Remaileh, M.; Bender, S.; Raddatz, G.; Ansari, I.; Cohen, D.; Gutekunst, J.; Musch, T.; Linhart, H.; Breiling, A.; Pikarsky, E.; Bergman, Y.; Lyko, F. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res. 2015, 75(10), 2120-2130. [CrossRef]
- Sato, F.; Harpaz, N.; Shibata, D.; Xu, Y.; Yin, J.; Mori, Y.; Zou, T.T.; Wang, S.; Desai, K.; Leytin, A.; Selaru, F.M.; Abraham, J.M.; Meltzer, S.J. Hypermethylation of the p14(ARF) gene in ulcerative colitis-associated colorectal carcinogenesis. Cancer Res. 2002, 62(4), 1148-51.
- Gerecke, C.; Scholtka, B.; Lowenstein, Y.; Fait, I.; Gottschalk, U.; Rogoll, D.; Melcher, R.; Kleuser, B. Hypermethylation of ITGA4, TFPI2 and VIMENTIN promoters is increased in inflamed colon tissue: putative risk markers for colitis-associated cancer. J. Cancer Res. Clin. Oncol. 2015, 141(12), 2097-2107. [CrossRef]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; Yang, H.; Wang, T.; Lee, A.Y.; Swanson, S.A.; Zhang, J.; Zhu, Y.; Kim, A.; Nery, J.R.; Urich, M.A.; Kuan, S.; Yen, C.A.; Klugman, S.; Yu, P.; Suknuntha, K.; Propson, N.E.; Chen, H.; Edsall, L.E.; Wagner, U., Li, Y.; Ye, Z.; Kulkarni, A.; Xuan, Z.; Chung, W.Y.; Chi, N.C.; Antosiewicz-Bourget, J.E.; Slukvin, I.; Stewart, R.; Zhang, M.Q.; Wang, W.; Thomson, J.A.; Ecker, J.R.; Ren, B. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153(5), 1134-48. [CrossRef]
- Issa, J.P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4(12), 988–993. [CrossRef]
- Dong D, Zhang J, Zhang R, Li F, Li Y, Jia Y. Multiprobe Assay for Clinical SEPT9 Methylation Based on the Carbon Dot-Modified Liquid-Exfoliated Graphene Field Effect Transistor with a Potential to Present a Methylation Panorama. ACS Omega 2020, 5(26), 16228-16237. [CrossRef]
- Song, L; Li, Y. SEPT9: A Specific Circulating Biomarker for Colorectal Cancer. Adv. Clin. Chem. 2015, 72, 171-204. [CrossRef]
- Kondo, Y.; Issa, J.P. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004, 23(1-2), 29-39. [CrossRef]
- Philipp, A.B.; Stieber, P.; Nagel, D.; Neumann, J.; Spelsberg, F.; Jung, A.; Lamerz, R.; Herbst, A.; Kolligs, F.T. Prognostic role of methylated free circulating DNA in colorectal cancer. Int. J. Cancer, 2012, 131(10), 2308-2319. [CrossRef]
- Melotte, V.; Lentjes, M. H.; van den Bosch, S.M.; Hellebrekers, D.M.; de Hoon, J.P.; Wouters, K.A.; Daenen, K.L.; Partouns-Hendriks, I.E.; Stessels, F.; Louwagie, J.; Smits, K.M.; Weijenberg, M.P.; Sanduleanu, S.; Khalid-de Bakker, C.A.; Oort, F.A.; Meijer, G.A.; Jonkers, D.M.; Herman, J.G.; de Bruïne, A.P; van Engeland, M. N-Myc downstream-regulated gene 4 (NDRG4): a candidate tumor suppressor gene and potential biomarker for colorectal cancer. J. Natl. Cancer Inst. 2009, 101(13), 916-927. [CrossRef]
- Loh, K.; Chia, J.A.; Greco, S.; Cozzi, S.J.; Buttenshaw, R.L.; Bond, C.E.; Simms, L.A.; Pike, T.; Young, J.P.; Jass, J.R.; Spring, K.J.; Leggett, B.A; Whitehall, V. L. Bone morphogenic protein 3 inactivation is an early and frequent event in colorectal cancer development. Genes Chromosomes Cancer 2008, 47(6), 449-460. [CrossRef]
- Xing, X.; Cai, W.; Shi, H.; Wang, Y.; Li, M.; Jiao, J.; Chen, M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: a meta-analysis. Br. J. Cancer 2013, 108(12), 2542-2548. [CrossRef]
- Jiang, W.; Wang, P.G.; Zhan, Y.; Zhang, D. Prognostic value of p16 promoter hypermethylation in colorectal cancer: a meta-analysis. Cancer Invest. 2014, 32(2), 43-52. [CrossRef]
- West, N.R.; McCuaig. S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015, 15(10), 615-29. [CrossRef]
- Li, Y.; Deuring, J.; Peppelenbosch, M.P.; Kuipers, E.J.; de Haar, C.; van der Woude, C. J. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis 2012, 33(10), 1889-1896. [CrossRef]
- Clawson, G.A. Histone deacetylase inhibitors as cancer therapeutics. Ann. Transl. Med. 2016, 4(15), 287. PMID: 27568481; PMCID: PMC4980376. [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13(7), 497-510. [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; Iyer, N.G.; Pérez-Rosado, A.; Calvo, E.; Lopez, J.A.; Cano, A.; Calasanz, M.J.; Colomer, D.; Piris, M.A.; Ahn, N.; Imhof, A.; Caldas, C.; Jenuwein, T.; Esteller, M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37(4), 391-400. [CrossRef]
- Ashktorab, H.; Belgrave, K.; Hosseinkhah, F.; Brim, H.; Nouraie, M.; Takkikto, M.; Hewitt, S.; Lee, E.L.; Dashwood, R.H.; Smoot D. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig. Dis. Sci. 2009, 54(10), 2109-17. [CrossRef]
- Tamagawa, H.; Oshima, T.; Shiozawa, M.; Morinaga, S.; Nakamura, Y.; Yoshihara, M.; Sakuma, Y.; Kameda, Y.; Akaike, M.; Masuda, M.; Imada, T.; Miyagi, Y. The global histone modification pattern correlates with overall survival in metachronous liver metastasis of colorectal cancer. Oncol. Rep. 2012 , 27(3), 637-42. [CrossRef]
- Liu,Y.; Hong, Y.; Zhao, Y.; Ismail, T.M.; Wong, Y.; Eu, K.W. Histone H3 (lys-9) deacetylation is associated with transcriptional silencing of E-cadherin in colorectal cancer cell lines. Cancer Invest. 2008 , 26(6), 575-82. [CrossRef]
- Peláez, I.M.; Kalogeropoulou, M.; Ferraro, A.; Voulgari, A.; Pankotai, T.; Boros, I.; Pintzas, A. Oncogenic RAS alters the global and gene-specific histone modification pattern during epithelial-mesenchymal transition in colorectal carcinoma cells. Int. J. Biochem. Cell Biol. 2010, 42(6), 911-20. [CrossRef]
- Zuo, X.; Morris, J.S.; Shureiqi, I. Chromatin modification requirements for 15-lipoxygenase-1 transcriptional reactivation in colon cancer cells. J. Biol. Chem. 2008, 283(46), 31341-7. [CrossRef]
- Chen, Y.X.; Fang, J.Y.; Lu, R.; Qiu, D.K. Expression of p21(WAF1) is related to acetylation of histone H3 in total chromatin in human colorectal cancer. World J. Gastroenterol. 2007, 13(15), 2209-13. [CrossRef]
- Li, Q.; Chen, H. Transcriptional silencing of N-Myc downstream-regulated gene 1 (NDRG1) in metastatic colon cancer cell line SW620. Clin. Exp. Metastasis. 2011, 28(2), 127-35. [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F. 4th; Cavassani, K.A.; Li, X.; Lukacs, N.W.; Hogaboam, C.M.; Dou, Y.; Kunkel, S.L. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114(15), 3244-54. [CrossRef]
- De Santa, F.; Totaro, M.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli. G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130(6), 1083-94. [CrossRef]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90(1), 9-17. [CrossRef]
- Nagarsheth, N.; Peng, D.; Kryczek, I.; Wu, K.; Li, W.; Zhao, E.; Zhao, L.; Wei, S.; Frankel, T.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; Marquez, V.; Tao, K.; Huang, E.; Wang, G.; Zou W. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. . Cancer Res. 2016, 76(2), 275-82. [CrossRef]
- Ghanem, I.; Riveiro, M.; Paradis, V.; Faivre, S.; de Parga, P.M.; Raymond, E. Insights on the CXCL12-CXCR4 axis in hepatocellular carcinoma carcinogenesis. Am. J. Transl. Res. 2014, 6(4), 340-52.
- Liu, H.; Liu, Y.; Liu, W.; Zhang, W.; Xu, J. EZH2-mediated loss of miR-622 determines CXCR4 activation in hepatocellular carcinoma. Nat. Commun. 2015, 6, 8494. [CrossRef]
- Enroth, S.; Alvaro, R.I.; Andersson, R.; Wallerman, O.; Wanders, A.; Påhlman, L.; Komorowski, J.; Wadelius C. Cancer associated epigenetic transitions identified by genome-wide histone methylation binding profiles in human colorectal cancer samples and paired normal mucosa. BMC Cancer 2011, 11, 450. [CrossRef]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; Gonzales, F.A.; Lin, J.C.; Liang, G.; Jones, P.A. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2'-deoxycytidine. Cancer Res. 2002, 62(22), 6456-61. PMID: 12438235.
- An, X.; Lan, X.; Feng, Z.; Li, X.; Su, Q. Histone modification: Biomarkers and potential therapies in colorectal cancer. Ann. Hum. Genet. 2023, 87(6), 274-284. [CrossRef]
- Qiu, F.; Wang, Y.; Chu, X.; Wang, J. ASF1A regulates H4Y72 phosphorylation and promotes autophagy in colon cancer cells via a kinase activity. Artif .Cells Nanomed. Biotechnol. 2019, 47(1), 2754-2763. [CrossRef]
- Ghate, N.B.; Kim, S.; Spiller, E.; Kim, S.; Shin, Y.; Rhie, S.K.; Smbatyan, G., Lenz, H.J.; Mumenthaler, S.M.; An, W. VprBP directs epigenetic gene silencing through histone H2A phosphorylation in colon cancer. Mol. Oncol. 2021, 15(10), 2801-2817. [CrossRef]
- Lee, Y.C.; Yin, T.C.; Chen, Y.T.; Chai, C.Y.; Wang, J.Y.; Liu, M.C.; Lin, Y.C.; Kan, J.Y. High expression of phospho-H2AX predicts a poor prognosis in colorectal cancer. Anticancer Res. 2015, 35(4), 2447-53.
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li, H. Histone modifications and their role in colorectal cancer. Pathol. Oncol. Res. 2020, 26(4), 2023-2033. [CrossRef]
- Yu, D.; Li, Z.; Gan, M.; Zhang, H.; Yin, X.; Tang, S.; Wan, L.; Tian, Y.; Zhang, S.; Zhu, Y.; Lai, M.; Zhang, D. Decreased expression of dual specificity phosphatase 22 in colorectal cancer and its potential prognostic relevance for stage IV CRC patients. Tumour Biol. 2015, 36(11), 8531-5. [CrossRef]
- Chen, T.; Li, J.; Xu, M.; Zhao, Q.; Hou, Y.; Yao, L.; Zhong, Y.; Chou, P.C.; Zhang, W.; Zhou, P.; Jiang, Y. PKCε phosphorylates MIIP and promotes colorectal cancer metastasis through inhibition of RelA deacetylation. Nat. Commun. 2017, 8(1), 1-11. [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034– D1037. [CrossRef]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G; Jiang, H.; Jiang, Q. LncRNA2Target v2.0: a comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019, 47(D1), D140-D144. [CrossRef]
- Ghoussaini, M.; Mountjoy, E.; Carmona, M.; Peat, G.; Schmidt, E.M.; Hercules, A.; Fumis, L.; Miranda, A.; Carvalho-Silva, D.; Buniello, A.; et al. Open Targets Genetics: Systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 2021, 49(D1), D1311–D1320. [CrossRef]
- Luck, K; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580(7803), 402–408. [CrossRef]
- Volders, P.J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47(D1), D135–D139. [CrossRef]
- Yang, M.; Lu, H.; Liu, J.; Wu, S.; Kim, P.; Zhou, X. lncRNAfunc: A knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 2022, 50(D1), D1295–D1306. [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: an updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49(D1), D165–D171. [CrossRef]
- Zhou, B.; Ji, B.; Liu, K.; Hu, G.; Wang, F.; Chen, Q.; Yu, R.; Huang, P.; Ren, J.; Guo, C.; et al. EVLncRNAs 2.0: An updated database of manually curated functional long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 2021, 49(D1), D86–D91. [CrossRef]
- Lin, L.; Zhou, G.; Chen, P.; Wang, Y.; Han, J.; Chen, M.; He, Y.; Zhang, S. Which long noncoding RNAs and circular RNAs contribute to inflammatory bowel disease? [Cell Death Dis. 2020, 11(6), 456. [CrossRef]
- Cummins, J.M.; He, Y.; Leary, R.J.; Pagliarini, R.; Diaz, L.A.; Jr.; Sjoblom, T.; Barad, O.; Bentwich, Z.; Szafranska, A.E.; Labourier, E.; Raymond, C.K.; Roberts, B.S.; Juhl. H.; Kinzler, K.W.; Vogelstein, B,;, Velculescu, V.E. The colorectal microRNAome. Proc. Natl. Acad. Sci. U. S. A. 2006, 103(10), 3687-3692. [CrossRef]
- Lanza, G.; Ferracin, M.; Gafa, R.; Veronese, A.; Spizzo, R.; Pichiorri, F.; Liu, C.G.; Calin, G,A,; Croce, C.M,; Negrini, M. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Mol. Cancer 2007, 6, 54. [CrossRef]
- Garajova, I.; Ferracin, M.; Porcellini, E.; Palloni, A.; Abbati, F.; Biasco, G.; Brandi, G. Non-coding RNAs as predictive biomarkers to current treatment in metastatic colorectal cancer. Int. J. Mol. Sci. 2017, 18(7), 1547. [CrossRef]
- Ragusa, M.; Barbagallo, C.; Statello, L.; Condorelli, A.G.; Battaglia, R.; Tamburello, L.; Barbagallo, D.; Di Pietro, C.; Purrello, M. Non-coding landscapes of colorectal cancer. World J. Gastroenterol. 2015, 21(41), 11709-11739. [CrossRef]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; Spiteri, I.; Das, P.P.; Caldas, C.; Miska, E.; Esteller, M. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67(4), 1424-1429. [CrossRef]
- Balaguer, F.; Link, A.; Lozano, J.J.; Cuatrecasas, M.; Nagasaka, T.; Boland, C.R.; Goel, A. Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res. 2010, 70(16), 6609-6618. [CrossRef]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2012, 31(16), 2062-2074. [CrossRef]
- Bandres, E.; Agirre, X.; Bitarte, N.; Ramirez, N.; Zarate, R.; Roman-Gomez, J.; Prosper, F.; Garcia-Foncillas, J. Epigenetic regulation of microRNA expression in colorectal cancer. Int. J. Cancer 2009, 125(11), 2737-2743. [CrossRef]
- Coskun, M.; Bjerrum, J.T.; Seidelin, J.B.; Troelsen, J.T.; Olsen, J.; Nielsen, O.H. miR-20b, miR-98, miR-125b-1*, and let-7e* as new potential diagnostic biomarkers in ulcerative colitis. World J. Gastroenterol. 2013, 19(27), 4289-4299. [CrossRef]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2005, 8(1), 33-40. [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4(3), 176-85. [CrossRef]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.; Samowitz, W.S.; Wolff, R.K.; Stevens, J.R.; Herrick, J.S. The NF-kappaB signalling pathway in colorectal cancer: associations between dysregulated gene and miRNA expression. J. Cancer Res. Clin. Oncol. 2018, 144(2), 269-283. [CrossRef]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39(4), 493-506. [CrossRef]
- Wang, H.; Nie, L.; Wu, L.; Liu, Q.; Guo, X. NR2F2 inhibits Smad7 expression and promotes TGF-beta-dependent epithelial-mesenchymal transition of CRC via transactivation of miR-21. Biochem. Biophys. Res. Commun. 2017, 485(1), 181-188. [CrossRef]
- Wang, N.; He, X.; Zhou, R.; Jia, G.; Qiao, Q. STAT3 induces colorectal carcinoma progression through a novel miR-572-MOAP-1 pathway. Onco. Targets Ther. 2018, 11, 3475-3484. [CrossRef]
- Wang, R.; Jaw, J.J.; Stutzman, N.C.; Zou, Z.; Sun, P.D. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J. Leukoc. Biol. 2012, 91(2), 299-309. [CrossRef]
- Zhang, L.L.; Zhang, L.F.; Shi, Y.B. MiR-24 inhibited the killing effect of natural killer cells to colorectal cancer cells by downregulating Paxillin. Biomed. Pharmacother. 2018, 101, 257-263. [CrossRef]
- Alamdari-Palangi, V.; Vahedi, F.; Shabaninejad, Z.; Dokeneheifard, S.; Movehedpour, A.; Taheri-Anganeh, M.; Savardashtaki, A. MicroRNA in inflammatory bowel disease at a glance. Eur. J. Gastroenterol. Hepatol. 2021, 32(2), 140-148. doi.org/10.1097/MEG.0000000000001815.
- Correia, C.N.; Nalpas, N.C.; McLoughlin, K.E.; Browne, J.A.; Gordon, S.V.; MacHugh, D.; Shaughnessy, R.G. Circulating microRNAs as Potential Biomarkers of Infectious Disease. Front. Immunol. 2017, 8, 118. [CrossRef]
- Rashid, H.; Hossain, B.; Siddiqua, T.; Kabir, M.; Noor, Z.; Ahmed, M.; Haque, R. Fecal microRNAs as potential biomarkers for screening and diagnosis of intestinal diseases. Front. Mol. Biosci. 2020, 7, 181. [CrossRef]
- Thorlacius-Ussing, G.; Schnack Nielsen, B.; Andersen, V.; Holmstrom, K.; Pedersen, A.E. Expression and localization of mir-21 and mir-126 in mucosal tissue from patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2017, 23(5), 739-752. doi.org/10.1097/MIB.0000000000001086.
- Feng, Y.H; Tsao, C.J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5(4), 395-402. [CrossRef]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu ,W.; Gao, R.; Qin, H. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury. PLoS One 2013, 8(6), e66814. [CrossRef]
- Wang, L.G.; Gu, J. Serum microRNA-29a is a promising novel marker for early detection of colorectal liver metastasis. Cancer Epidemiol. 2012, 36(1), e61-67. [CrossRef]
- Zhu, Y.; Xu, A.; Li, J.; Fu, J.; Wang, G.; Yang, Y.; Sun, J. Fecal miR-29a and miR-224 as the noninvasive biomarkers for colorectal cancer. Cancer Biomark. 2016, 16(2), 259-264. [CrossRef]
- Iborra, M.; Bernuzzi, F.; Correale, C.; Vetrano, S.; Fiorino, G.; Beltran, B.; Marabita, F.; Locati, M.; Spinelli, A.; Nos, P.; Invernizzi, P.; Danese, S. Identification of serum and tissue micro-RNA expression profiles in different stages of inflammatory bowel disease. Clin. Exp. Immunol. 2013, 173(2), 250-258. [CrossRef]
- Chen, P.; Li, Y.; Li, L.; Yu, Q.; Chao, K.; Zhou, G.; Qiu, Y.; Feng, R.; Huang, S.; He, Y.; Chen, B.; Chen, M.; Zeng, Z.; Zhang, S. Circulating microRNA146b-5p is superior to C-reactive protein as a novel biomarker for monitoring inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 49(6), 733-743. [CrossRef]
- Sun, C.M.; Wu, J.; Zhang, H.; Shi, G.; Chen, Z.T. Circulating miR-125a but not miR-125b is decreased in active disease status and negatively correlates with disease severity as well as inflammatory cytokines in patients with Crohn's disease. World J. Gastroenterol. 2017, 23(44), 7888-7898. [CrossRef]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus. C.; Klein, J. R. MicroRNA signatures differentiate Crohn's disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [CrossRef]
- Zahm, A.M.; Thayu, M.; Hand, N.J.; Horner, A.; Leonard, M.B.; Friedman, J.R. Circulating microRNA is a biomarker of pediatric Crohn disease. J. Pediatr. Gastroenterol. Nutr. 2011, 53(1), 26-33. [CrossRef]
- Zahm, A.M.; Hand, N.J.; Tsoucas, D.M.; Le Guen, C.L.; Baldassano, R.N.; Friedman, J.R. Rectal microRNAs are perturbed in pediatric inflammatory bowel disease of the colon. J. Crohns Colitis 2014, 8(9), 1108-1117. [CrossRef]
- Heier, C.R.; Fiorillo, A.A.; Chaisson, E.; Gordish-Dressman, H.; Hathout, Y.; Damsker, J. M.; Conklin, L.S. Identification of pathway-specific serum biomarkers of response to glucocorticoid and infliximab treatment in children with inflammatory bowel disease. Clin. Transl. Gastroenterol. 2016, 7(9), e192. [CrossRef]
- Batra, S.K.; Heier, C.R.; Diaz-Calderon, L.; Tully, C.B.; Fiorillo, A.A.; van den Anker, J.; Conklin, L.S. Serum miRNAs are pharmacodynamic biomarkers associated with therapeutic response in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 2020, 26(10), 1597-1606. [CrossRef]
- Kumar, M.; Murugesan, S.; Ibrahim, N.; Elawad, M.; Al Khodor, S. Predictive biomarkers for anti-TNF alpha therapy in IBD patients. J. Transl. Med. 2024, 22(1), 284. [CrossRef]
- Shi, T.; Xie, Y.; Fu, Y.; Zhou, Q.; Ma, Z.; Ma, J.; Chen, J. The signaling axis of microRNA-31/interleukin-25 regulates Th1/Th17-mediated inflammation response in colitis. Mucosal. Immunol. 2017, 10(4), 983-995. [CrossRef]
- Wang, H.; Chao, K.; Ng, S.C.; Bai, A.H.; Yu, Q.; Yu, J.; Zhang, S. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016, 17, 58. [CrossRef]
- Jin, X.; Chen, D.; Zheng, R.H.; Zhang, H.; Chen, Y.P.; Xiang, Z. MiRNA-133a-UCP2 pathway regulates inflammatory bowel disease progress by influencing inflammation, oxidative stress and energy metabolism. World J. Gastroenterol. 2017, 23(1), 76-86. [CrossRef]
- Schonauen, K.; Le, N.; von Arnim, U.; Schulz, C.; Malfertheiner, P.; Link, A. Circulating and fecal microRNAs as biomarkers for inflammatory bowel diseases. Inflamm. Bowel Dis. 2018, 24(7), 1547-1557. [CrossRef]
- Phua, L.C.; Chue, X.P.; Koh, P.K.; Cheah, P.Y.; Chan, E.C.; Ho, H.K. Global fecal microRNA profiling in the identification of biomarkers for colorectal cancer screening among Asians. Oncol. Rep. 2014, 32(1), 97-104. [CrossRef]
- Duran-Sanchon, S.; Moreno, L.; Auge, J.M.; Serra-Burriel, M.; Cuatrecasas, M.; Moreira, L.; Martín, A.; Serradesanferm, A.; Pozo, À.; Costa, R.; Lacy, A.; Pellisé, M.; Lozano, J.J.; Gironella M.; Castells, A. Identification and validation of microrna profiles in fecal samples for detection of colorectal cancer. Gastroenterol. 2020, 158(4), 947-957 e944. [CrossRef]
- Duran-Sanchon, S.; Moreno, L.; Gomez-Matas, J.; Auge, J.M.; Serra-Burriel, M.; Cuatrecasas, M.; Moreira, L.; Serradesanferm, A.; Pozo, À.; Grau, J.; Pellisé, M.; Gironella, M.; Castells, A. Fecal microRNA-based algorithm increases effectiveness of fecal immunochemical test-based screening for colorectal cancer. Clin. Gastroenterol. Hepatol. 2021, 19(2), 323-330 e321. [CrossRef]
- Ma, Y.; Yang, Y.; Wang, F.; Moyer, M.P.; Wei, Q.; Zhang, P.; Yang, Z.; Liu, W.; Zhang, H.; Chen, N.; Wang, H.; Wang, H.; Qin, H. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/β-catenin signalling pathway via suppression of activator protein 2α. Gut 2016, 65(9), 1494-504. [CrossRef]
- Buda, A.; Jepson, M.A.; Pignatelli, M. Regulatory function of trefoil peptides (TFF) on intestinal cell junctional complexes. Cell Commun. Adhes. 2012, 19(5-6), 63-68. [CrossRef]
- Bian, Z.; Zhang, J.; Li, M.; Feng, Y.; Wang, X.; Yao, S.; Jin, G.; Du, J.; Han, W.; Yin, Y.; Huang, S.; Fei, B.; Zou, J.; Huang, Z. LncRNA-FEZF1-AS1 promotes tumor proliferation and metastasis in colorectal cancer by regulating PKM2 signaling. Clin. Cancer Res. 2018, 24(19), 4808-4819. [CrossRef]
- Xue, J.; Liao, L.; Yin, F.; Kuang, H.; Zhou, X.; Wang, Y. LncRNA AB073614 induces epithelial- mesenchymal transition of colorectal cancer cells via regulating the JAK/STAT3 pathway. Cancer Biomark. 2018, 21(4), 849-858. [CrossRef]
- Li, C.Y.; Liang, G.Y.; Yao, W.Z.; Sui, J.; Shen, X.; Zhang, Y.Q.; Peng, H.; Hong, W.W.; Ye, Y.C.; Zhang, Z.Y.; Zhang, W.H.; Yin, L.H.; Pu, Y.P. Integrated analysis of long non-coding RNA competing interactions reveals the potential role in progression of human gastric cancer. Int. J. Oncol. 2016, 48(5), 1965-76. [CrossRef]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; Miyano, S.; Mori, M. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71(20), :6320-6. . Erratum in: Cancer Res. 2012 Feb 15;72(4), 1039. PMID: 21862635. [CrossRef]
- Svoboda, M.; Slyskova, J.; Schneiderova, M.; Makovicky, P.; Bielik, L.; Levy, M.; Lipska, L.; Hemmelova, B.; Kala, Z.; Protivankova, M.; Vycital, O.; Liska, V.; Schwarzova, L.; Vodickova, L.; Vodicka, P. HOTAIR long non-coding RNA is a negative prognostic factor not only in primary tumors, but also in the blood of colorectal cancer patients. Carcinogenesis 2014, 35(7), 1510-5.PMID: 24583926. [CrossRef]
- Zhang, Y.; Bu, D.; Huo, P.; Wang, Z.; Rong, H.; Li, Y.; Liu, J.; Ye, M.; Wu, Y.; Jiang, Z.; Liao, Q.; Zhao, Y. ncFANs v2.0: an integrative platform for functional annotation of non-coding RNAs. Nucleic Acids Res. 2021, 49(W1), W459-W468. [CrossRef]
- Zeng, L.; Zhao, K.; Liu, J.; Liu, M.; Cai, Z.; Sun, T.; Li Z, Liu R. Long noncoding RNA GAS5 acts as a competitive endogenous RNA to regulate GSK-3β and PTEN expression by sponging miR-23b-3p in Alzheimer's disease. Neural. Regen. Res. 2024, 21(1), 392-405. [CrossRef]
- Zhi, H.; Li, X.; Wang, P.; Gao, Y.; Gao, B.; Zhou, D.; Zhang, Y.; Guo, M.; Yue, M.; Shen, W.; Ning, S.; Jin, L.; Li, X. Lnc2Meth: a manually curated database of regulatory relationships between long non-coding RNAs and DNA methylation associated with human disease. Nucleic Acids Res. 2018, 46(D1), D133-D138. [CrossRef]
- Li, Z.; Li, Z.; Zhang, Y.; Zhou, L.; Xu, Q.; Li, L.; Zeng, L.; Xue, J.; Niu, H.; Zhong, J.; Yu, Q.; Li, D.; Gui, M.; Huang, Y.; Tu, S.; Zhang, Z.; Song, C.Q.; Wu, J.; Shen, E.Z. Mammalian PIWI-piRNA-target complexes reveal features for broad and efficient target silencing. Nat. Struct. Mol. Biol. 2024, 31(8), 1222-1231. [CrossRef]
- Kumegawa, K.; Maruyama, R.; Yamamoto, E.; Ashida, M.; Kitajima, H.; Tsuyada, A.; Niinuma, T.; Kai, M.; Yamano, H.O.; Sugai, T.; Tokino, T., Shinomura, Y.; Imai, K.; Suzuki, H. A genomic screen for long noncoding RNA genes epigenetically silenced by aberrant DNA methylation in colorectal cancer. Sci. Rep. 2016, 6, 26699. [CrossRef]
- Diaz-Lagares, A.; Crujeiras, A.B.; Lopez-Serra, P.; Soler, M.; Setien, F.; Goyal, A.; Sandoval, J.; Hashimoto, Y.; Martinez-Cardús, A.; Gomez, A.; Heyn, H.; Moutinho, C.; Espada, J.; Vidal, A.; Paúles, M.; Galán, M.; Sala, N.; Akiyama, Y.; Martínez-Iniesta, M.; Farré, L.; Villanueva, A.; Gross, M.; Diederichs, S.; Guil, S.; Esteller M. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc. Natl. Acad. Sci. U. S. A. 2016, 113(47), E7535-E7544. [CrossRef]
- Hibi, K.; Nakamura, H.; Hirai, A.; Fujikake, Y.; Kasai, Y.; Akiyama, S.; Ito, K.; Takagi, H. Loss of H19 imprinting in esophageal cancer. Cancer Res. 1996, 56(3), 480-2. PMID: 8564957.
- Tian, F.; Tang, Z.; Song, G.; Pan, Y.; He, B.; Bao, Q.; Wang S. Loss of imprinting of IGF2 correlates with hypomethylation of the H19 differentially methylated region in the tumor tissue of colorectal cancer patients. Mol. Med. Rep. 2012, 5(6), 1536-40. [CrossRef]
- Hidaka, H.; Higashimoto, K.; Aoki, S.; Mishima, H.; Hayashida, C.; Maeda, T.; Koga, Y.; Yatsuki, H.; Joh, K.; Noshiro, H.; Iwakiri, R.; Kawaguchi, A.; Yoshiura, K.I.; Fujimoto, K.; Soejima H. Comprehensive methylation analysis of imprinting-associated differentially methylated regions in colorectal cancer. Clin. Epigenetics 2018, 10(1), 150. [CrossRef]
- Yarani, R.; Mirza, A.H.; Kaur, S.; Pociot, F. The emerging role of lncRNAs in inflammatory bowel disease. Exp. Mol. Med. 2018, 50(12), 1-14. [CrossRef]
- Ding, G.; Ming, Y.; Zhang, Y. LncRNA Mirt2 is downregulated in ulcerative colitis and regulates IL-22 expression and apoptosis in colonic epithelial cells. Gastroenterol. Res. Pract. 2019, 2019, 8154692. [CrossRef]
- Li, F.; Liu, H.; Fu, J.; Fan, L.; Lu, S.; Zhang, H.; Liu, Z. Knockdown of long non-coding RNA NEAT1 relieves inflammation of ulcerative colitis by regulating the miR-603/FGF9 pathway. Exp. Ther. Med. 2022, 23(2), 131. [CrossRef]
- Nie, J.; Zhao, Q. Lnc-ITSN1-2, derived from RNA sequencing, correlates with increased disease risk, activity and promotes CD4(+) T cell activation, proliferation and Th1/Th17 cell differentiation by serving as a ceRNA for IL-23R via sponging mir-125a in inflammatory bowel disease. Front. Immunol. 2020, 11, 852. [CrossRef]
- Hur, K.; Kim, S.H.; Kim, J.M. Potential implications of long noncoding RNAs in autoimmune diseases. Immune Netw. 2019, 19(1), e4. [CrossRef]
- Elamir, A.; Shaker, O.; Kamal, M.; Khalefa, A.; Abdelwahed, M.; Abd El Reheem, F.; Ahmed, T.; Hassan, E.; Ayoub, S. Expression profile of serum LncRNA THRIL and MiR-125b in inflammatory bowel disease. PLoS One 2022, 17(10), e0275267. [CrossRef]
- Tian, Y.; Cui, L.; Lin, C.; Wang, Y.; Liu, Z.; Miao, X. LncRNA CDKN2B-AS1 relieved inflammation of ulcerative colitis via sponging miR-16 and miR-195. Int. Immunopharmacol. 2020, 88, 106970. [CrossRef]
- Ge, Q.; Dong, Y.; Lin, G.; Cao, Y. Long noncoding RNA antisense noncoding RNA in the INK4 locus correlates with risk, severity, inflammation and infliximab efficacy in Crohn's disease. Am. J. Med. Sci. 2019, 357(2), 134-142. [CrossRef]
- Chen, S.W.; Wang, P.Y.; Liu, Y.C.; Sun, L.; Zhu, J.; Zuo, S.; Ma, J.; Li, T.Y.; Zhang, J.L.; Chen, G.W.; Wang, X.; Zhu, Q.R.; Zheng, Y.W.; Chen, Z.Y.; Yao, Z.H.; Pan, Y.S. Effect of long noncoding RNA H19 overexpression on intestinal barrier function and its potential role in the pathogenesis of ulcerative colitis. Inflamm. Bowel Dis. 2016, 22(11), 2582-2592. [CrossRef]
- Liu, R.; Tang, A.; Wang, X.; Chen, X.; Zhao, L.; Xiao, Z.; Shen, S. Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int. J. Mol. Med. 2018, 42(5), 2903-2913. [CrossRef]
- Lucafo, M.; Di Silvestre, A.; Romano, M.; Avian, A.; Antonelli, R.; Martelossi, S.; Naviglio, S.; Tommasini, A.; Stocco, G.; Ventura, A.; Decorti, G.; De Iudicibus, S. Role of the long non-coding RNA growth arrest-specific 5 in glucocorticoid response in children with inflammatory bowel disease. Basic Clin. Pharmacol. Toxicol. 2018, 122(1), 87-93. [CrossRef]
- Nemati Bajestan, M.; Piroozkhah, M.; Chaleshi, V.; Ghiasi, N.E.; Jamshidi, N.; Mirfakhraie, R.; Balaii, H.; Shahrokh, S.; Asadzadeh Aghdaei, H.; Salehi, Z.; Nazemalhosseini Mojarad, E. Expression analysis of long noncoding RNA-MALAT1 and interleukin-6 in inflammatory bowel disease patients. Iran. J. Allergy Asthma Immunol. 2023, 22(5), 482-494. [CrossRef]
- Tian, W.; Du, Y.; Ma, Y.; Gu, L.; Zhou, J.; Deng, D. MALAT1-miR663a negative feedback loop in colon cancer cell functions through direct miRNA-lncRNA binding. Cell Death Dis. 2018, 9(9), 857. [CrossRef]
- Wang, S.; Hou, Y.; Chen, W.; Wang, J.; Xie, W.; Zhang, X.; Zeng, L. KIF9-AS1, LINC01272 and DIO3OS lncRNAs as novel biomarkers for inflammatory bowel disease. Mol. Med. Rep. 2018, 17(2), 2195-2202. [CrossRef]
- Cao, L.; Tan, Q.; Zhu, R.; Ye, L.; Shi, G.; Yuan, Z. LncRNA MIR4435-2HG suppression regulates macrophage M1/M2 polarization and reduces intestinal inflammation in mice with ulcerative colitis. Cytokine 2023, 170, 156338. [CrossRef]
- Yang, Y.; Xiong, Z.; Li, W.; Lin, Y.; Huang, W.; Zhang, S. FHIP1A-DT is a potential novel diagnostic, prognostic, and therapeutic biomarker of colorectal cancer: A pan-cancer analysis. Biochem. Biophys. Res. Commun. 2023, 679, 191-204. [CrossRef]
- Guo, K.; Yao, J.; Yu, Q.; Li, Z.; Huang, H.; Cheng, J.; Zhu., Y. The expression pattern of long non coding PVT1 in tumor tissues and in extracellular vesicles of colorectal cancer correlates with cancer progression. Tumour Biol. 2017, 39(4), 1010428317699122. PMID: 28381186. [CrossRef]
- Mani, S.R.; Juliano, C.E. Untangling the Web: The diverse functions of the PIWI/piRNA pathway. Mol. Reprod. Dev. 2013, 80(8), 632-664. [CrossRef]
- Gangaraju, V.K.; Lin, H. MicroRNAs: Key regulators of stem cells. Nat. Rev. Mol. Cell Biol. 2009, 10, 116–125. [CrossRef]
- Stefani, G.; Slack, F.J. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol. 2008, 9, 219–230. [CrossRef]
- Saxe, J.P.; Lin, H. Small noncoding RNAs in the germline. Cold Spring Harb. Perspect. Biol. 2011, 3, a002717. [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [CrossRef]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N.; Morris, P.; Brownstein, M.J.; Kuramochi-Miyagawa, S.; Nakano, T.; Chien, M.; Russo, J.J.; Ju, J.; Sheridan, R.; Sander, C.; Zavolan, M.; Tuschl, T. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006, 442, 203–207. [CrossRef]
- Watanabe, T.; Lin, H. Posttranscriptional regulation of gene expression by Piwi proteins and piRNAs. Mol. Cell 2014, 56, 18– 27. [CrossRef]
- Moyano, M.; Stefani, G. piRNA involvement in genome stability and human cancer. J. Hematol. Oncol. 2015, 8, 38. [CrossRef]
- Rouget, C.; Papin, C.; Boureux, A.; Meunier, A.; Franco, B.; Robine, N.; Lai, E.; Pelisson, A.; Simonelig, M. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature 2010, 467, 1128–1132. [CrossRef]
- Aravin, A.; Sachidanandam, R.; Bourc’his, D.; Schaefer, C.; Pezic, D.; Toth, K.; Bestor, T.; Hannon, G. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 2008, 31(6), 785-799. [CrossRef]
- Watanabe, T.; Tomizawa, S.; Mitsuya, K.; Totoki, Y.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Iida, N.; Hoki, Y.; Murphy, P.; Toyoda, A.; Gotoh, K.; Hiura, H.; Arima, T.; Fujiyama, A.; Sado, T.; Shibata, T.; Nakano, T.; Lin, H.; Ichiyanagi, K.; Soloway, P.; Sasaki, H. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 2011, 332(6031), 848-852. [CrossRef]
- Yamanaka, S.; Siomi, M.C; Siomi, H. piRNA clusters and open chromatin structure. Mobile DNA 2014, 5, 22. [CrossRef]
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: the vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 2011, 12, 246–258. [CrossRef]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.T.; He, N.; Chuva de Sousa Lopes, S.M.; van der Westerlaken, L.A.; Zischler, H.; Butter, F.; Roelen, B.A.; Ketting, R.F. Piwi Proteins and piRNAs in Mammalian Oocytes and Early Embryos. Cell Rep. 2015, 10(12), 2069-82. [CrossRef]
- Aravin, A.; Hannon, G.; Brennecke, J.; The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318(5851), 761-764. [CrossRef]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [CrossRef]
- Muhammad, A.; Waheed, R.; Khan, N.A.; Jiang, H.; Song, X. piRDisease v1.0: a manually curated database for piRNA associated diseases. Database (Oxford) 2019, 2019, baz052. [CrossRef]
- Ghosh, B.; Sarkar, A.; Mondal, S.; Bhattacharya, N.; Khatua, S.; Ghosh, Z. piRNAQuest V.2: an updated resource for searching through the piRNAome of multiple species. RNA Biol. 2022, 19(1), 12-25. [CrossRef]
- Zhang, W.; Wu, S.; Zhang, H.; Guan, W.; Zeng, B., Wei, Y.; Chi-Fung Chan, G.; Li, W. piRPheno: a manually curated database to prioritize and analyze human disease related piRNAs. bioRxiv (The preprint server for biology). 2020. [CrossRef]
- Zhang, P.; Si, X.; Skogerbø, G.; Wang, J.; Cui, D.; Li, Y.; Sun, X.; Liu, L.; Sun, B.; Chen, R.; He, S.; Huang, D.W. piRBase: a web resource assisting piRNA functional study. Database (Oxford) 2014, 2014, bau110. [CrossRef]
- Wang, J.; Zhang, P.; Lu, Y.; Li, Y. Zheng, Y.; Kan, Y.; Chen, R.; He, S. piRBase: a comprehensive database of piRNA sequences. Nucleic Acids Res. 2019, 47(D1), D175-D180. [CrossRef]
- Greene, J.; Baird, A.M.; Brady, L.; Lim, M.; Gray, S.G.; McDermott, R.; Finn, S.P. Circular RNAs: biogenesis, function and role in human diseases. Front. Mol. Biosci. 2017, 4, 38. [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21(1), 101. [CrossRef]
- Hao, S.; Cong, L.; Qu, R.; Liu, R.; Zhang, G.; Li, Y. Emerging roles of circular RNAs in colorectal cancer. Onco. Targets Ther. 2019, 12, 4765-4777. [CrossRef]
- Li, J.; Ni, S.; Zhou, C.; Ye, M. The expression profile and clinical application potential of hsa_circ_0000711 in colorectal cancer. Cancer Manag. Res. 2018, 10, 2777-2784. [CrossRef]
- Zhang, W.; Wang, B.; Lin, Y.; Yang, Y.; Zhang, Z.; Wang, Q.; Zhang, H.; Jiang, K.; Ye, Y.; Wang, S.; Shen, Z. Hsa_circ_0000231 promotes colorectal cancer cell growth through upregulation of CCND2 by IGF2BP3/miR-375 dual pathway. Cancer Cell Int. 2022, 22(1), 27. [CrossRef]
- Sun, S.; Li, C.; Cui, K.; Liu, B.; Zhou, M.; Cao, Y.; Zhang, J.; Bian, Z.; Fei, B.; Huang, Z. Hsa_circ_0062682 promotes serine metabolism and tumor growth in colorectal cancer by regulating the miR-940/PHGDH axis. Front. Cell Dev. Biol. 2021, 9, 770006. [CrossRef]
- Long, F.; Lin, Z.; Li, L.; Ma, M.; Lu, Z.; Jing, L.; Li, X.; Lin, C. Comprehensive landscape and future perspectives of circular RNAs in colorectal cancer. Mol. Cancer 2021, 20(1), 26. [CrossRef]
- Sun, Z.; Chen, C.; Su, Y.; Wang, W.; Yang, S.; Zhou, Q.; Wang, G.; Li, Z.; Song, J.; Zhang, Z.; Yuan, W.; Liu, J. Regulatory mechanisms and clinical perspectives of circRNA in digestive system neoplasms. J. Cancer 2019, 10(13), 2885-2891. [CrossRef]
- Lai, H.; Li, Y.; Zhang, H.; Hu, J.; Liao, J.; Su, Y.; Li, Q.; Chen, B.; Li, C.; Wang, Z.; Li, Y.; Wang, J.; Meng, Z.; Huang, Z.; Huang, S. ExoRBase 2.0: an atlas of mRNA, lncRNA and circRNA in extracellular vesicles from human biofluids. Nucleic Acids Res. 2022, 50(D1), D118-D128. [CrossRef]
- Wen, J.; Liao, J.; Liang, J.; Chen, X.P.; Zhang, B.; Chu, L. Circular RNA HIPK3: a key circular rna in a variety of human cancers. Front. Oncol. 2020, 10, 773. [CrossRef]
- Viralippurath Ashraf, J.; Sasidharan Nair, V.; Saleh, R.; Elkord, E. Role of circular RNAs in colorectal tumor microenvironment. Biomed. Pharmacother. 2021, 137, 111351. [CrossRef]
- Liang, X.; Long, L.; Guan, F.; Xu, Z.; Huang, H. Research status and potential applications of circRNAs affecting colorectal cancer by regulating ferroptosis. Life Sci. 2024, 352, 122870. [CrossRef]
- Camandona, A.; Gagliardi, A.; Licheri, N.; Tarallo, S.; Francescato, G.; Budinska, E.; Carnogurska, M.; Zwinsová, B.; Martinoglio, B.; Franchitti, L.; Gallo, G.; Cutrupi, S.; De Bortoli, M.; Pardini, B.; Naccarati, A.; Ferrero, G. Multiple regulatory events contribute to a widespread circular RNA downregulation in precancer and early stage of colorectal cancer development. Biomark. Res. 2025, 13(1), 30. [CrossRef]
- Mao, J., Lu, Y. Roles of circRNAs in the progression of colorectal cancer: novel strategies for detection and therapy. Cancer Gene Ther. 2024, 31(6), 831-841. [CrossRef]
- Li, L., Wei, C., Xie, Y., Su, Y.; Liu, C.; Qiu, G., Liu, W.; Liang, Y.; Zhao, X.; Huang, D.; Wu, D. Expanded insights into the mechanisms of RNA-binding protein regulation of circRNA generation and function in cancer biology and therapy. Genes Dis. 2024, 12(4), 101383. [CrossRef]
- Hussen, B.M.; Abdullah, S.R.; Mohammed, A.A.; Rasul, M.F.; Hussein, A.M.; Eslami, S.; Glassy, M.C.; Taheri, M. Advanced strategies of targeting circular RNAs as therapeutic approaches in colorectal cancer drug resistance. Pathol. Res. Pract. 2024, 260, 155402. [CrossRef]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27(15), 2128-36. [CrossRef]
- Lai, C.Y.; Yeh, K.Y.; Liu, B.F.; Chang, T.M.; Chang, C.H.; Liao, Y.F.; Liu, Y.W.; Her, G.M. MicroRNA-21 plays multiple oncometabolic roles in colitis-associated carcinoma and colorectal cancer via the PI3K/AKT, STAT3, and PDCD4/TNF-α signaling pathways in zebrafish. Cancers (Basel) 2021, 13(21), 5565. [CrossRef]
- O'Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 2007, 104(5), 1604-9. [CrossRef]
- Jabłońska, E.; Białopiotrowicz, E.; Szydłowski, M.; Prochorec-Sobieszek, M.; Juszczyński, P.; Szumera-Ciećkiewicz, A. DEPTOR is a microRNA-155 target regulating migration and cytokine production in diffuse large B-cell lymphoma cells. Exp. Hematol. 2020, 88, 56-67.e2. [CrossRef]
- Uthman, Y.A., Ibrahim, K.G., Abubakar, B., Bello, M.B.; Malami, I.; Imam, M.U.; Qusty, N.; Cruz-Martins, N.; Batiha, G.E.; Abubakar, M.B. MALAT1: a promising therapeutic target for the treatment of metastatic colorectal cancer. Biochem. Pharmacol. 2021, 190, 114657. [CrossRef]
- Yan, Y.; Su, M.; Qin, B. CircHIPK3 promotes colorectal cancer cells proliferation and metastasis via modulating of miR-1207-5p/FMNL2 signal. Biochem. Biophys. Res. Commun. 2020, 524(4), 839-846. [CrossRef]
- Hsiao, K.Y.; Lin, Y.C.; Gupta, S.K.; Chang, N.; Yen, L.; Sun, H.S.; Tsai, S.J. Noncoding effects of circular RNA CCDC66 promote colon cancer growth and metastasis. Cancer Res. 2017, 77(9), 2339-2350. [CrossRef]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by microRNA-21. Cancer Cell 2010, 18(3), 282-93. [CrossRef]
- Wang, P.; Zhuang, L.; Zhang, J.; Fan, J.; Luo, J.; Chen, H.; Wang, K.; Liu, L.; Chen, Z.; Meng, Z. The serum miR-21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR-21 expression confers chemoresistance by targeting FasL. Mol. Oncol. 2013, 7(3), 334-45. [CrossRef]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28(22), 6773-84. [CrossRef]
- Wang, P.; Guo, Q.; Qi, Y.; Hao, Y.; Gao, Y.; Zhi, H.; Zhang, Y.; Sun, Y.; Zhang, Y.; Xin, M.; Zhang, Y.; Ning, S.; Li, X. LncACTdb 3.0: an updated database of experimentally supported ceRNA interactions and personalized networks contributing to precision medicine. Nucleic Acids Res. 2022, 50(D1), D183-D189. [CrossRef]
- Pillich, R.T.; Chen, J.; Churas, C.; Fong, D.; Gyori, B.M.; Ideker, T.; Karis, K.; Liu, S.N.; Ono, K.; Pico, A.; Pratt, D. NDEx IQuery: a multi-method network gene set analysis leveraging the network data exchange. Bioinformatics 2023, 39(3), btad118. [CrossRef]
- Persson, E.; Castresana-Aguirre, M.; Buzzao, D.; Guala, D.; Sonnhammer, E.L.L. FunCoup 5: functional association networks in all domains of life, supporting directed links and tissue-specificity. J. Mol. Biol. 2021, 433(11), 166835. [CrossRef]
- Ilango, S.; Paital, B.; Jayachandran, P.; Padma, P.R.; Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. (Landmark Ed) 2020, 25(6), 1058-1109. [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6(10), a026831. [CrossRef]
- Gajjela, B.K.; Zhou, M.M. Bromodomain inhibitors and therapeutic applications. Curr. Opin. Chem. Biol. 2023, 75, 102323. PMID: 37207401; PMCID: PMC10524616. [CrossRef]
- Martínez-Gutierrez, A.; Carbajal-Lopez, B.; Bui, T.M.; Mendoza-Rodriguez, M.; Campos-Parra, A.D.; Calderillo-Ruiz, G.; Cantú-De Leon, D.; Madrigal-Santillán, E.O.; Sumagin, R.; Pérez-Plasencia, C.; Pérez-Yépez, E.A. A microRNA panel that regulates proinflammatory cytokines as diagnostic and prognosis biomarkers in colon cancer. Biochem. Biophys. Rep. 2022, 30,101252. PMID: 35313644; PMCID: PMC8933814. [CrossRef]
- Xie, X.; Liu, P.; Wu, H.; Li, H.; Tang, Y.; Chen, X.; Xu, C.; Liu, X.; Dai, G. MiR-21 antagonist alleviates colitis and angiogenesis via the PTEN/PI3K/AKT pathway in colitis mice induced by TNBS. Ann. Transl. Med. 2022, 10(7), 413. PMID: 35530951; PMCID: PMC9073802 . [CrossRef]
- Carter, J.V.; Galbraith, N.J.; Yang, D.; Burton, J.F.; Walker, S.P.; Galandiuk, S. Blood-based microRNAs as biomarkers for the diagnosis of colorectal cancer: a systematic review and meta-analysis. Br. J. Cancer 2017, 116(6), 762-774. PMID: 28152545; PMCID: PMC5355921 . [CrossRef]
- Kalkusova, K.; Taborska, P.; Stakheev, D.; Smrz, D. The role of miR-155 in antitumor immunity. Cancers (Basel) 2022, 14(21), 5414. PMID: 36358832; PMCID: PMC9659277. [CrossRef]
- Zhang, J.; Wang, C.; Guo, Z.; Da, B.; Zhu, W.; Li, Q. miR-223 improves intestinal inflammation through inhibiting the IL-6/STAT3 signaling pathway in dextran sodium sulfate-induced experimental colitis. Immun. Inflamm. Dis. 2021, 9(1), 319-327. PMID: 33332758; PMCID: PMC7860526 . [CrossRef]
- Zhao, W.; Song, M.; Zhang, J.; Kuerban, M.; Wang, H. Combined identification of long non-coding RNA CCAT1 and HOTAIR in serum as an effective screening for colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8(11), 14131-40. PMID: 26823726; PMCID: PMC4713512.
- Liu, J.; Guo, B. RNA-based therapeutics for colorectal cancer: Updates and future directions. Pharmacol. Res. 2020, 152, 104550. PMID: 31866285; PMCID: PMC7027327 . [CrossRef]
- Fitzgerald, K.; Frank-Kamenetsky, M.; Shulga-Morskaya, S.; Liebow, A.; Bettencourt, B.R.; Sutherland, J.E.; Hutabarat, R.M.; Clausen, V.A.; Karsten, V.; Cehelsky, J.; Nochur, S.V.; Kotelianski, V.; Horton, J.; Mant, T.; Chiesa, J.; Ritter, J.; Munisamy, M.; Vaishnaw, A.K.; Gollob, J.A.; Simon, A. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 2014, 383(9911) 60-68. PMID: 24094767; PMCID: PMC4387547 . [CrossRef]
- Hitchins, M.P.; Vogelaar, I.P.; Brennan, K.; Haraldsdottir, S.; Zhou, N.; Martin, B.; Alvarez, R; Yuan, X.; Kim, S.; Guindi, M.; Hendifar, A.E.; Kalady, M.F.; DeVecchio, J.; Church, J.M.; de la Chapelle, A.; Hampel, H.; Pearlman, R.; Christensen, M.; Snyder, C.; Lanspa, S.J.; Haile, R.W.; Lynch, H.T. Methylated SEPTIN9 plasma test for colorectal cancer detection may be applicable to Lynch syndrome. BMJ Open Gastroenterol. 2019, 6(1), e000299. . PMID: 31275589; PMCID: PMC6577308. [CrossRef]
- Sui, C.; Ma, J.; Chen, Q.; Yang, Y. The variation trends of SFRP2 methylation of tissue, feces, and blood detection in colorectal cancer development. Eur. J. Cancer Prev. 2016, 25(4), 288-98. . PMID: 26258809. [CrossRef]
- Gonneaud, A.; Gagné, J.M.; Turgeon, N.; Asselin, C. The histone deacetylase Hdac1 regulates inflammatory signalling in intestinal epithelial cells. J. Inflamm. (Lond.) 2014, 11(1), 43. . PMID: 25606026; PMCID: PMC4299484. [CrossRef]
Figure 1.
Sidebar Infographic: Commonly Used Animal Models for Studying Inflammatory Bowel Disease and CAC. A schematic overview of widely employed murine models used in IBD and colitis-associated colorectal cancer (CAC) research. These include chemically induced models (DSS, TNBS, Oxazolone, AOM/DSS), genetically engineered mice (IL-10⁻/⁻, TRUC, APCMin/+), and adoptive T cell transfer models. Each model has distinct immune engagement and phenotypic features, supporting diverse aspects of inflammation and tumorigenesis. P number, corresponds to the published reference (PMID).
Figure 1.
Sidebar Infographic: Commonly Used Animal Models for Studying Inflammatory Bowel Disease and CAC. A schematic overview of widely employed murine models used in IBD and colitis-associated colorectal cancer (CAC) research. These include chemically induced models (DSS, TNBS, Oxazolone, AOM/DSS), genetically engineered mice (IL-10⁻/⁻, TRUC, APCMin/+), and adoptive T cell transfer models. Each model has distinct immune engagement and phenotypic features, supporting diverse aspects of inflammation and tumorigenesis. P number, corresponds to the published reference (PMID).
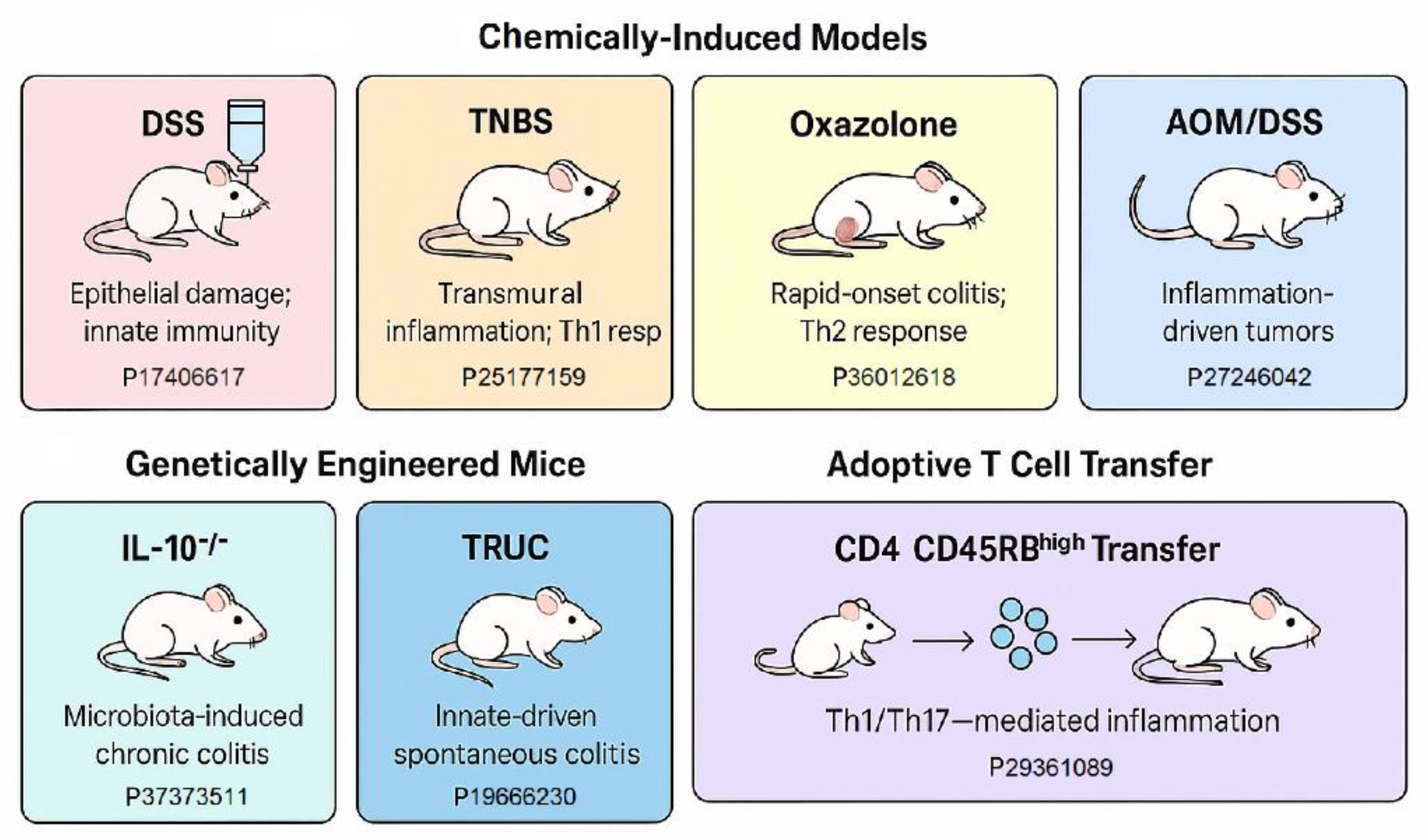
Figure 2.
Mechanistic Pathway – From Inflammation to Colorectal Carcinogenesis.. This schematic illustrates the multi-step cascade of molecular and cellular mechanisms involved in the transition from chronic intestinal inflammation to inflammation-driven colorectal cancer (CRC) development, particularly in the context of inflammatory bowel disease (IBD) with a focus on genetic and epigenetic regulation. Appendix: Hereditary Susceptibility genes to colorectal neoplasia (in white background); Familial disease (in pink background); CRC* predisposition of tumor suppressor genes promoted by epigenetic changes (in orange background) (see details in the text).
Figure 2.
Mechanistic Pathway – From Inflammation to Colorectal Carcinogenesis.. This schematic illustrates the multi-step cascade of molecular and cellular mechanisms involved in the transition from chronic intestinal inflammation to inflammation-driven colorectal cancer (CRC) development, particularly in the context of inflammatory bowel disease (IBD) with a focus on genetic and epigenetic regulation. Appendix: Hereditary Susceptibility genes to colorectal neoplasia (in white background); Familial disease (in pink background); CRC* predisposition of tumor suppressor genes promoted by epigenetic changes (in orange background) (see details in the text).
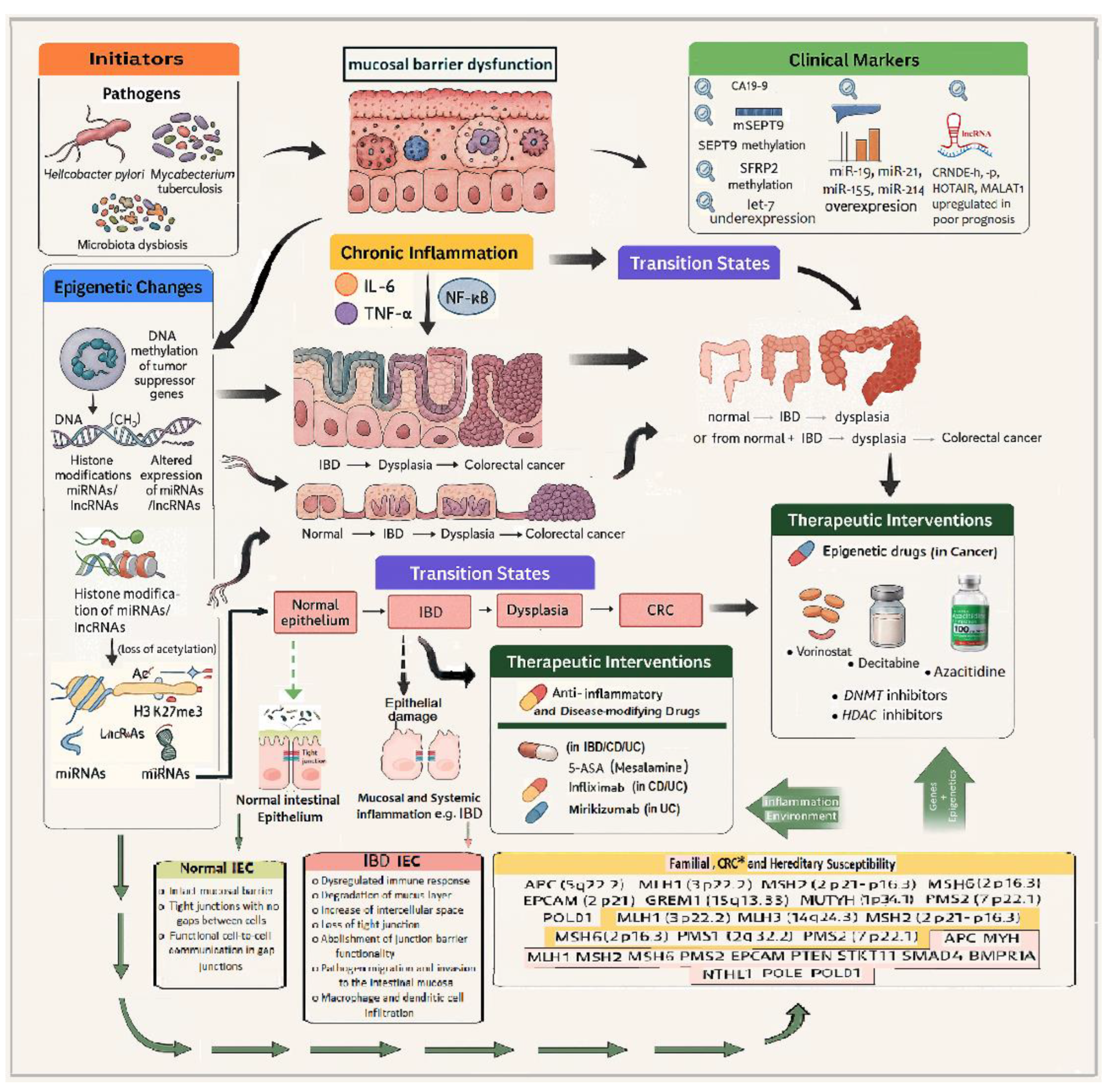
Figure 3.
Top 30 highly expressed circRNAs from preanalyzed datasets of human colon tissue (one-step analysis) in CPM (counts per million). Data Accessed June 16th, 2025, from circAtlas 3.0 database [317] (see details in the text).
Figure 3.
Top 30 highly expressed circRNAs from preanalyzed datasets of human colon tissue (one-step analysis) in CPM (counts per million). Data Accessed June 16th, 2025, from circAtlas 3.0 database [317] (see details in the text).
Figure 4.
ncRNA Networks in Inflammation-Associated Colorectal Cancer. The above diagram illustrates the interactions between key ncRNAs and inflammation-related signaling cascades within the colorectal tumor microenvironment. The upper region depicts inflammatory cytokines (e.g., IL-6, TNF-α) activating intracellular pathways such as NF-κB, STAT3, in CRC epithelial cells, which in turn regulate the expression of oncogenic miRNAs and interact with Wnt/β-catenin in colonic epithelial cells. These signals modulate the transcriptional expression of lncRNAs (HOTAIR, MALAT1), miRNAs (miR-21, miR-155) and circRNAs (circHIPK3, circCCDC66), in CRC pathogenesis. Circular RNAs act as sponges modulating miRNA activity, such as circCCDC66 protecting PTEN from miRNA-mediated repression, while circHIPK3 affecting NF-κB signaling. Downstream key targets include tumor suppressor genes (e.g., PTEN, PDCD4) and oncogenes (e.g., BCL2, Wnt/β-catenin), leading to increased cell survival, proliferation, and evasion of apoptosis. The diagram portrays chromatin-level epigenetic changes, including DNA methylation and histone modification (e.g., H3K27me3), which are orchestrated by ncRNA-interacting complexes such as PRC2. This integrated view underscores the central role of ncRNAs in mediating inflammation-associated carcinogenesis. The references in brackets refer to PMIDs, which are supporting the interactions. Appendix: PMIDs: [1], [35305641]; [2], [35961438]; [3] [29263891]; [4], [36733201]; [5], [24165275]; [6], [32380907].
Figure 4.
ncRNA Networks in Inflammation-Associated Colorectal Cancer. The above diagram illustrates the interactions between key ncRNAs and inflammation-related signaling cascades within the colorectal tumor microenvironment. The upper region depicts inflammatory cytokines (e.g., IL-6, TNF-α) activating intracellular pathways such as NF-κB, STAT3, in CRC epithelial cells, which in turn regulate the expression of oncogenic miRNAs and interact with Wnt/β-catenin in colonic epithelial cells. These signals modulate the transcriptional expression of lncRNAs (HOTAIR, MALAT1), miRNAs (miR-21, miR-155) and circRNAs (circHIPK3, circCCDC66), in CRC pathogenesis. Circular RNAs act as sponges modulating miRNA activity, such as circCCDC66 protecting PTEN from miRNA-mediated repression, while circHIPK3 affecting NF-κB signaling. Downstream key targets include tumor suppressor genes (e.g., PTEN, PDCD4) and oncogenes (e.g., BCL2, Wnt/β-catenin), leading to increased cell survival, proliferation, and evasion of apoptosis. The diagram portrays chromatin-level epigenetic changes, including DNA methylation and histone modification (e.g., H3K27me3), which are orchestrated by ncRNA-interacting complexes such as PRC2. This integrated view underscores the central role of ncRNAs in mediating inflammation-associated carcinogenesis. The references in brackets refer to PMIDs, which are supporting the interactions. Appendix: PMIDs: [1], [35305641]; [2], [35961438]; [3] [29263891]; [4], [36733201]; [5], [24165275]; [6], [32380907].
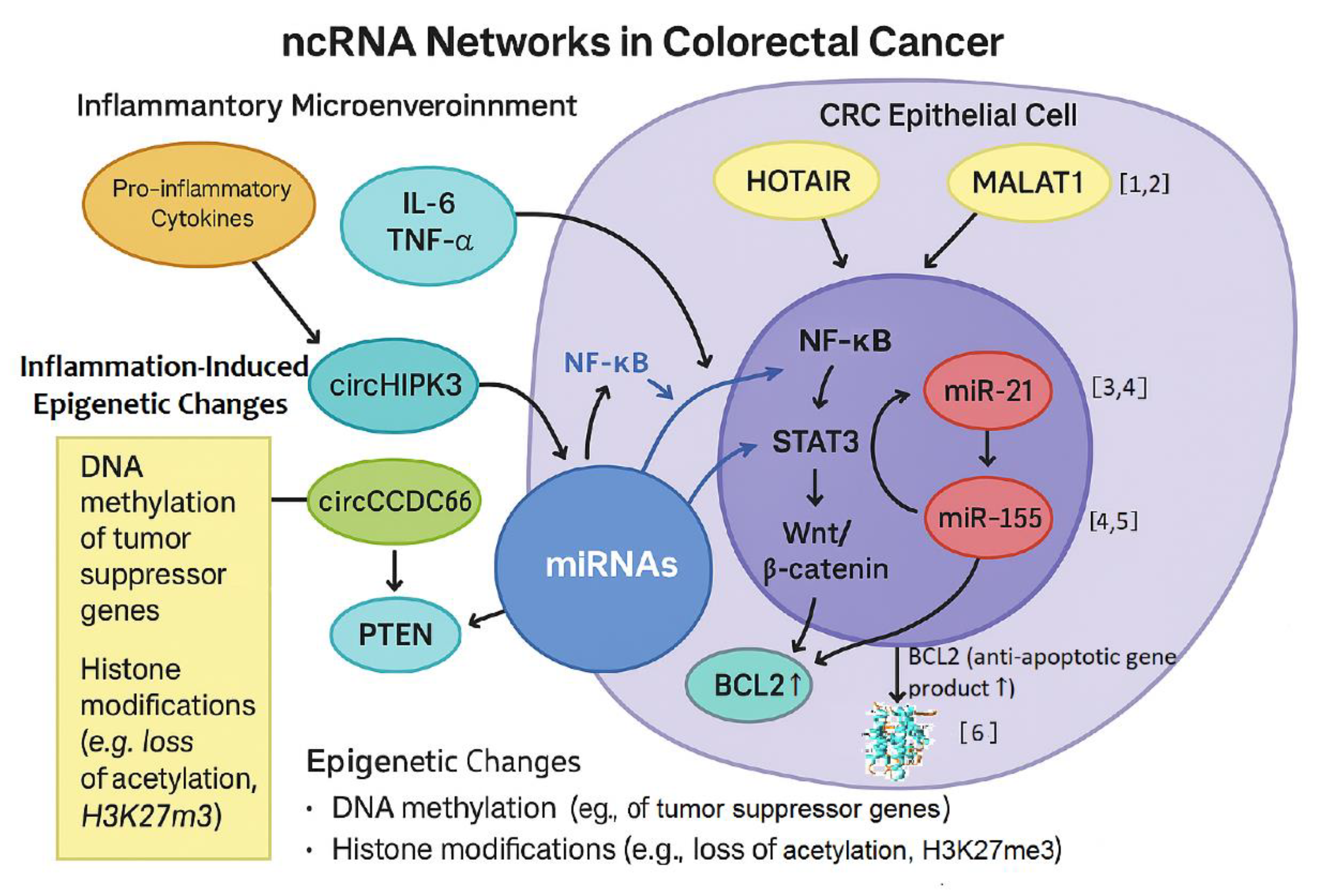
Table 1.
Murine Models of Inflammatory Bowel Disease (IBD) and Colitis-Associated Colorectal Cancer (CAC).
Table 1.
Murine Models of Inflammatory Bowel Disease (IBD) and Colitis-Associated Colorectal Cancer (CAC).
| Model | Method | Immune System Involvement | Advantages | Limitations | Reference (PMID) |
|---|---|---|---|---|---|
| DSS | Oral DSS in drinking water | Innate | Rapid, simple, epithelial injury | No adaptive immune involvement | 17406617 36012618 34440615 |
| AOM/DSS | AOM injection + DSS cycles | Innate + DNA damage | Models CAC pathogenesis | Less suitable for sporadic CRC | 27246042 |
| TNBS | Rectal instillation of TNBS | Adaptive (Th1) | Crohn's-like inflammation | Variability, toxic risk | 25177159 34440615 |
| Oxazolone | Chemical | Adaptive (Th2) | Mimics UC; rapid onset | Short-lived; strain-dependent | 36012618 |
| IL-10⁻/⁻ | Genetic deletion of IL-10 | Adaptive | Spontaneous colitis | Microbiota dependence | 37373511 |
| TRUC (T-bet⁻/⁻ × RAG⁻/⁻) | Double knockout | Innate | Innate immunity-driven model progressing to colonic dysplasia and rectal adenocarcinoma | Complex breeding, microbiota sensitive | 19666230 |
| APCMin/+ | Genetic (multiple intestinal neoplasia) | Sporadic Intestinal Tumors |
FAP model; Wnt pathway activation | Small intestine focus | 30887153 |
| CD4+CD45RBhigh T Cell Transfer to RAG⁻/⁻ Mice |
Adaptive cell transfer | Adaptive | T-cell driven colitis | Requires expertise, chronic model | 29361089 34440615 25989337 |
Table 2.
DNA Methylation as diagnostic and therapeutic biomarkers according to the subtype of IBD.
| Disease Type | Sample Type | Methylated markers | Methylation status | Reference (PMID) |
| IBD | Rectal biopsies | THRAP2, FANCC and GBGT1 | ↑ | 22419656 |
| CD | Blood | WDR8 and ITGB2 | ↑ | 27886173 |
| CD | Rectal biopsies | DOK2 and TNFSF4 | ↓ | 22419656 |
| CD | Blood | VMP1 | ↓ | 27886173 |
| UC | Rectal biopsies | CARD9 and CDH1 | ↑ | 22419656 |
| UC | Blood | WDR8 | ↑ | 27886173 |
| UC | Rectal biopsies | ICAM3, DOK2 and TNFSF4 | ↓ | 22419656 |
| UC | Blood | VMP1 | ↓ | 27886173 |
| UC | Colon biopsies | EBI3 | ↓ | 22419656 |
| CRC | Rectal biopsies | TGFB2, SLIT2, HS3ST2, TMEFF2, | ↑ | 27886173 |
| CRC | Colon biopsies | FOXE1, SYNE1 | ↑ | 22419656 |
| CRC | Colonic mucosa | APC, CDH13, MGMT, RUNX3 and MLH1 | ↑ | 27886173 |
| CRC | Colon biopsies | ITGA4 | ↑ | 34069352 |
Appendix: ↑, DNA methylation increase; ↓, DNA methylation decrease.
Table 3.
Histone modifications as diagnostic and therapeutic biomarkers according to the subtype of IBD.
Table 3.
Histone modifications as diagnostic and therapeutic biomarkers according to the subtype of IBD.
| Disease Type | Sample Type |
Histone modifying enzymes/histone markers |
Modification type/ status | Reference (PMID) | |
| IBD | Intestinal tissue | KAT2B | Acetylation ↓ | 26802082 | |
| UC/CAC | Colon Tissue | H3K27ac | Acetylation ↑ | 29983891 | |
| CD | Colonic biopsy | H2Bub1 | Ubiquitination ↓ | 34088983 | |
| IBD | Colon Tissue | HDAC | Acetylation ↑ | 38903915 | |
| IBD | Colon Tissue | HDAC8 | Acetylation ↑ | 36558966 | |
| CRC | Primary cancer tissue | H3K9me2 | Methylation ↑ | 22076537 |
Appendix: ↑, increase of Post-translational modification; ↓, decrease of Post-translational modification.
Table 4.
MicroRNAs as diagnostic and therapeutic biomarkers according to the subtype of IBD, and CRC.
Table 4.
MicroRNAs as diagnostic and therapeutic biomarkers according to the subtype of IBD, and CRC.
| Disease Type | Sample Type | miRNAs | Gene Expression change | Reference (PMID) | |
| UC | Serum | miR-29a, miR-196b, miR-127-3p | ↑ | 23607522 | |
| CD | Serum | miR-140-3p, miR-127-3p | ↑ | 23607522 | |
| UC | Serum | miR-150 | ↓ | 23607522 | |
| CD | Blood | miRNA-125a | ↓ | 29209130 | |
| UC | Blood | miRNA-19a | ↑ | 25886994 | |
| UC | Blood | miRNA-146a | ↓ | 25886994 | |
| CD | Blood | miR-31 | ↓ | 25886994 | |
| CD | Blood | miR-101 | ↑ | 25886994 | |
| IBD | Serum | miR-146b-5p | ↑ | 30734320 | |
| IBD | Feces | miR-223, miR-1246 | ↑ | 32850969 | |
| IBD | Serum | miR-16, miR-21, and miR-223 | ↑ | 29668922 | |
| IBD | Feces | miR-223, miR-155 | ↑ | 29668922 | |
| IBD | Serum / Feces | miR-21, miR-142-3p, miR-146a and let-7i | ↑ | 24613022 | |
| IBD | IECs | miR-192, miR-194, miR-200b, miR-375 | ↓ | 24613022 | |
| IBD | Serum | miR-192, miR-195, miR-20a, miR-30a, miR-484 and let-7b | ↑ | 21546856 | |
| IBD | Serum | miR-146a, miR-146b, miR-320a, miR-126 and let-7c | ↓ | 32793975 | |
| IBD | Colonic tissue | miR-133a | ↓ | 28104982 | |
| CRC | Feces | miR-135b, miR-223 and miR-451 | ↑ | 24841830 | |
| CRC | Feces | miR-29a, miR-223 and miR-224 | ↓ | 26756616 | |
| CRC | Feces | miR-421, miR130b-3p miR27a-3P | ↑ | 31622624 |
Appendix: ↑, increase of expression; ↓, decrease of expression.
Table 5.
LncRNAs(#) as diagnostic and therapeutic biomarkers according to the subtype of IBD and CRC.
Table 5.
LncRNAs(#) as diagnostic and therapeutic biomarkers according to the subtype of IBD and CRC.
| Disease Type | Sample Type | lncRNAs | Gene Expression change | Reference (PMID) | |
| UC | Plasma | Mirt2 | ↓ | 31687015 | |
| CRC | Blood | HOTAIR | ↑ | 21862635, 24583926 | |
| CRC | COAD tissue | FHIP1A-DT | ↓ | 37703762 | |
| UC | Plasma | IFNG-AS1 | ↑ | 34970354 | |
| UC | Plasma | ITSN1-2 | ↑ | 32547537 | |
| IBD | Serum | THRIL | ↑ | 36206229 | |
| UC | Blood | CDKN2B-AS1 | ↓ | 33182065 | |
| CD | Blood | CDKN2B-AS1 | ↑ | 30665494 | |
| UC | Colonic tissue | H19 | ↑ | 27661667 | |
| IBD | Colonic tissue | CRNDE | ↑ | 31251902 | |
| IBD | Serum | NEAT1 | ↑ | 30132508 | |
| UC | Blood | GAS5 | ↑ | 28722800 | |
| IBD/ CRC | Intestinal tissue | MALAT1 | ↑ | 38085149 | |
| IBD | Plasma/ Tissue | KIF9-AS1 and LINC01272 | ↑ | 29207070 | |
| IBD | Plasma/ Tissue | DIO3OS | ↓ | 29207070 | |
| UC | Human intestinal epithelial Caco2 cells and murine macrophage RAW264.7 cells | MIR4435-2HG | ↑ | 37597495 | |
| CRC | Colon/Rectal biopsies | RVT1 | ↑ | 28381186 |
Appendix: (#) Adapted from [69], ↑, Up-regulated; ↓ Down-regulated.
Table 6.
Epigenetic Alterations Across the Stages of Colon Disease Progression.
| Condition | DNA Methylation | Histone Acetylation | Histone Methylation | Non-coding RNAs (miRNAs/lncRNAs) |
References (PMID) |
| Normal Colon | Homeostatic balance of methylation | Balanced H3/H4 acetylation regulating gene expression | Physiological levels of H3K4me3 and H3K27me3 | Normal expression of regulatory miRNAs and lncRNAs | 17447863, 15800552 |
| Inflammatory Bowel Disease (IBD) | ↑ Promoter hypermethylation of anti-inflammatory genes | ↑ H3K27ac | ↑ H3K4me3, ↑ H3K9me3 | ↑ Inflammatory miRNAs (e.g., miR-155), altered lncRNAs | 27886173 |
| Colitis (IBD) | Global hypomethylation; hypermethylation of SOCS3 | ↓ H3/H4 acetylation; HDAC overexpression | ↑ H3K9me3 (heterochromatinization); ↓ H3K27me3 | ↑ miR-155, miR-21; ↓ let-7; ↑ lncRNA HOTAIR | 26573286, 27385797, 20404267 |
| Dysplasia | ↑ CIMP phenotype, ↑ Promoter hypermethyl-ation of MLH1, CDKN2A, APC | Loss of histone acetylation at tumor suppressor loci | ↑ H3K27me3, H3K9me3 by EZH2; ↓ H3K4me3 at differentiation genes | ↑ MALAT1, CRNDE, ↓ MEG3; ↑ oncogenic lncRNAs, miRNAs, ↓ tumor-suppressive circRNAs | 24833482, 29559771 |
| Colorectal Cancer | Global hypomethylation; ↑ Promoter CpG island hypermethylation | Aberrant HDAC recruitment; hypoacetylation at TSGs; ↓ H4K16ac | ↑ H3K9me2/3; ↑ EZH2-mediated H3K27me3; ↓ H3K4me3 | ↑ Oncogenic miRNAs (e.g. miR-21, miR-135b); ↑ lncRNAs NEAT1, PVT1, CCAT1; ↓ lncRNAs like GAS5 | 31483834, 33081851, 31230041 |
Appendix: ↑, increase; ↓, decrease.Abbreviations: TSG, tumor suppressor gene; HDAC, histone deacetylase; EZH2, Enhancer of Zeste Homolog 2; SOCS3, suppressor of cytokine signaling 3.PubMed IDs (PMIDs) are provided for traceability independent of citation numbering.
Table 10.
Top Therapeutics in Development Through Epigenetic Research for Cancer.
| Drug / RNA | Type | Target | Disease Phase | Function/Application Notes | Reference (PMID) |
| OTX015 | BET inhibitor | MYC, NF-κB pathways | Phase I/II clinical trials | Solid tumors, anti-inflammatory profile | 37207401 |
| 5-Azacytidine | DNMT inhibitor | DNA methylation reversal | Approved (hematologic); off-label, investigational in CRC | Reverses methylation silencing | 24583822; 26317465, 33359448 |
| Anti-miR-21 Oligonucleotides | Antisense oligo | miR-21 suppression | Preclinical studies | Targets PDCD4, inhibits tumor invasion | 17968323 |
| miR-92a sponge | miRNA decoy | miR-92a suppression | Preclinical studies | Biomarker and therapeutic target | 28957811; 33620640 |
| Romidepsin | HDAC inhibitor | HDAC1/2 (Histone deacetylation) | Approved (T-cell lymphoma); clinical trials for CRC | Suppresses inflammation and tumor growth | 27599530 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



