Submitted:
22 August 2025
Posted:
25 August 2025
You are already at the latest version
Abstract
Several genetic diseases affecting the human nervous system are incurable and insufficiently understood. Among them, nine rare diseases form the polyglutamine (polyQ) family: Huntington's disease (HD), spinocerebellar ataxia types 1, 2, 3, 6, 7, and 17, dentatorubral pallidoluysian atrophy, and spinal and bulbar muscular atrophy. In most patients, these diseases progress over decades to cause severe movement incoordination and neurodegeneration. Although their inherited genes with CAG repeat elongations and the encoded polyQ-containing proteins have been extensively studied, the cell-type-specific pathologies and their long pre-symptomatic latency have received less attention. However, recent advances in detecting the single-cell transcriptome alongside the length of tandem repeats in HD post-mortem brains have enabled the identification of the repeat size that triggers extensive transcriptional dysregulation and cell death in specific neurons. The first challenge is to understand better the complexity of movement coordination circuits, including the basal ganglia and cerebellum, and which of their neurons are most vulnerable to the CAG expansion in each disease. The second challenge is to detect the dynamic increase in CAG repeat length at single-cell resolution in vulnerable neurons. This will offer hope for identifying primary and secondary pathological events and developing targeted therapies for all tandem-repeat expansion diseases.
Keywords:
neurodegenerative genetic diseases
; movement disorders
; tandem repeat expansion
; polyQ diseases
; movement coordination circuitry
; single-cell sequencing
; genetic instability
; neuronal mosaicism
1. Nine Human-Specific Diseases Need Better-Defined Primary Pathogenicity
The human genome contains nine evolutionarily conserved genes (HTT, ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, TBP, ATN1, and AR) with cytosine-adenine-guanine (CAG) or cytosine-adenine-adenine (CAA) triplet repeats that encode polyglutamine (polyQ) tracks in the respective proteins. The process of CAG-repeat expansion in these nine genes, both within and across generations, can lead to pathological expansions of DNA, RNA and polyQ traits in the encoded proteins, resulting in the human-specific rare diseases known as the polyQ disease family: Huntington’s Disease (HD), Spinocerebellar Ataxia (SCA) types 1, 2, 3, 6, 7, and 17, Dentato-Rubral Pallidoluysian Atrophy (DRPLA), and Spinal and Bulbar Muscular Atrophy (SBMA) (Figure 1; and reviewed in [1,2,3,4,5,6,7]).
Some of these genes and the proteins they encode were discovered and named after the disease caused by the expansion of their tandem repeats: the Huntingtin gene (HTT) and protein (HTT), mutated in HD; the ataxin genes (ATXN1, ATXN2, ATXN3 and ATXN7) and proteins (ATXN1, ATXN2, ATXN3 and ATXN7), mutated in SCA1, 2, 3, and 7 respectively; and the Atrophin 1 gene (ATN1) and protein (ATN1), mutated in DRPLA. For the other two polyQ SCAs (SCA6 and SCA17) and SBMA, the names of the mutated genes derive from their encoded protein‘s function or structure: the CACNA1A gene mutated in SCA6 is bicistronic, encoding two proteins, the first being the main subunit of the calcium channel Cav2.1, called the protein α1A, and the second a small nuclear protein, named based on its structure, α1ACT (representing the C-terminal fragment of α1A); the TBP gene mutated in SCA17 encodes the TATA-box binding protein (TBP), while the AR gene mutated in SBMA encodes the androgen receptor (AR) protein.
The polyQ diseases typically manifest in an autosomal dominant manner, except for SBMA, which is X-linked. In most patients, the tandem repeat is elongated in only one allele. However, rare homozygous patients, described in HD, SCA1, SCA3, SCA6, SCA17, and DRPLA, exhibit earlier onset, faster disease progression, and more severe symptoms than heterozygotes [9,10,11,12].
The nine polyQ diseases share not only the nature of the genetic mutation but also certain clinical and neurodegenerative features. However, the severity of the symptoms and progression of brain pathologies show considerable variability between patients, even for a similar gene mutation. While most polyQ disease symptoms onset in adults, rare cases of juvenile or even infantile forms have been documented, which exhibit longer inherited repeats and faster disease progression [1,3,4,5,6,7].
1.1. Dysfunctions and Degeneration of the Motor Coordination Network Stations
All polyQ patients develop symptoms related to uncontrollable movements (such as in chorea, ataxia and saccades), along with specific symptoms that vary depending on the nature of the mutation and disease stage. These symptoms are associated with dysfunctions and degeneration in different central nervous system (CNS) regions, including at least one station of the motor coordination network. This comprises multiple layers and follows a hierarchical control principle, which is highly conserved among mammals. In this network, the cerebral cortex sends outputs to the effector motor neurons in the brainstem and spinal cord (which project their axons to effector muscles) after integrating input from various regulatory units, mainly via the thalamus. The regulatory units are the basal ganglia and cerebellum, which also receive direct or indirect cortical output. The cerebellum receives cortical inputs via the brainstem integration nuclei (such as the red nuclei, pons nuclei and inferior olives) and projects outputs to the thalamus and red nuclei (which further project outputs to the thalamus) (Figure 2A; and reviewed in [13,14]).
The accumulated knowledge on the connectivity and heterogeneity of brain areas responsible for motor coordination provides new directions for understanding the dynamics of polyQ pathology. PolyQ diseases present both common and specific symptomatic and anatomopathological features, some of which are reflected in their names, such as ataxia with cerebellar and spinal cord atrophy in SCAs, atrophy of the cerebellar dentate nuclei and midbrain red nuclei, the basal ganglia nuclei (GP and STN, also known as corpus Luysii) in DRPLA, and atrophy of the spinal cord, medulla oblongata (also called bulbus), and muscles in SBMA.
The overall organisation and function of the human basal ganglia, cerebellum, pons, inferior olives, and red nuclei, along with their central interconnections, is reviewed in several publications [13,14,15,16,17,18,19,20,21]. The basal ganglia consist of a group of interconnected subcortical nuclei (striata, globi pallidi, substantia nigrae, and subthalamic nuclei) that relay cortical motor commands to ensure proper control of action selection and execution. Each striatum comprises the caudate nucleus and putamen, separated by the internal capsule, the nucleus accumbens and the olfactory tubercle. Each globus pallidus (GP) contains a “pars externa” (GPe) and a “pars interna” (GPi), and each substantia nigra (SN) includes a pars compacta (SNc) and a pars reticulata (SNr). The striatum and subthalamic nucleus (STN) receive the excitatory inputs from the cerebral cortex, while the GPi transmits the inhibitory output of the basal ganglia to the thalamus. Between these structures lies an internal network of inhibitory neurons (GPe, SNr, SNc) and excitatory neurons of the STN, as well as a dopaminergic regulatory network between the striatum and SNc (nigrostriatal pathway) (Figure 2B).
About half of the projection neurons in the striatum, which are inhibitory, send signals to the GPi inhibitory projection neurons. Because these two inhibitory synapses are linked in series, there is a decrease in the inhibitory effect of the GPi on the thalamic neurons, a process known as disinhibition of the thalamus. Consequently, following the principle of positive feedback, this so-called direct pathway excites the motor cortex and boosts motor activity. The other half of the projection neurons in the striatum send their axons to the inhibitory projection neurons of the GPe, which then send inhibitory projections to the STN. The excitatory projection neurons of the STN extend their axons to the GPi neurons, which in turn send inhibitory signals to the thalamus. Thus, this pathway, known as the indirect pathway, produces inhibition of the GPe neurons, leading to disinhibition of the STN. Consequently, the neurons of the STN can excite the GPi/SNr, thereby suppressing thalamic activation of the cerebral cortex and spontaneous movements. Alongside the indirect pathway, there is a smaller route (so-called the hyper-direct pathway) that bypasses the striatum and inhibits spontaneous movements. It consists of neurons projecting directly from the cortex and thalamus to the STN (Figure 2B and reviewed in [17]).
Alongside the basal ganglia, the motor pattern produced by the cerebral cortex is modulated by the cerebellum, especially for fine-tuning (reviewed in [13,14,15,16,17,18,19,20,21]). The cerebellum consists of two lateral hemispheres connected by the vermis, all covered by the cerebellar cortex. The cerebellar deep nuclei (CDN) are located within the white matter of each hemisphere; the largest of these are the dentate nuclei, followed by the interposed nuclei, with the smallest being the fastigial nuclei. The cerebellar cortex receives excitatory signals from the cerebral cortex via the pons, red nuclei (RN) and inferior olives (IO) (Figure 2C). These signals are processed by the inhibitory Purkinje neurons, which relay the results of this processing to excitatory neurons in the CDN. Each excitatory neuron in the CDN receives inputs from multiple Purkinje neurons. These neurons form the primary cerebellar output and project back to the motor cortex, mainly via the RN, and further to the thalamus [18] (Figure 2C). RN are essential hubs in the cerebro-cerebellar pathway. They are large paired subcortical structures with a roughly spherical shape, encased by the superior cerebellar peduncles and having two histologically distinct subregions: a small caudal magnocellular region (mRN), composed of large, sparse neurons, and a larger rostral parvocellular part (pRN), mainly characterised by small and medium-sized neurons. Most projections from bilateral motor, premotor, and supplementary motor cortices target the pRN, with fewer from the ipsilateral motor cortex reaching the mRN. The mRN receives afferents from the interposed nucleus, while the pRN receives fibres from the dentate nucleus; the pRN primarily projects to the IO. IO modulates Purkinje neurons, causing the inhibition of the CDN and further of the RN, thalami and cortex; meanwhile, pRN receives excitatory input from the cerebral cortex, sends excitatory fibres to the IO, creating a feedforward loop (Figure 2C; and reviewed in [22]).
The early stages of any of the polyQ diseases affect one or more segments of the motor coordination network, especially the basal ganglia and cerebellum, while later stages further impact these areas, as well as other CNS regions such as the neocortex, thalami, retinae, red nuclei, pons, inferior olives, and motor nuclei in the brainstem or spinal cord, with both common and different distribution between the polyQ diseases (Table 1) [3,5,7,23].
HD generally begins with involuntary movements that gradually worsen, accompanied by rigidity, psychiatric symptoms, and cognitive loss over 10 to 20 years until death. In the early stages of the disease, adult HD patients experience minor chorea and oculomotor impairments, such as saccadic eye movements. During the middle stages, chorea is prominent and is associated with increasing difficulty in voluntary motor tasks, dysarthria, postural and gait instability, and ataxia. In the late stages, chorea is often replaced by rigidity, dystonia, and bradykinesia. Brain imaging during disease progression detects significant atrophy in the striatum and neocortex, followed by the GP and thalamus, with moderate atrophy of the brainstem nuclei and cerebellum in late clinical stages. In all HD disease cases, postmortem investigations revealed bilateral, symmetrical and massive neuronal loss in the striatum, sometimes accompanied by astrogliosis. The cerebral cortex exhibits significant neurodegeneration, but the cerebellum may also show atrophy in the cortex and CDN. Interestingly, while the disease progression occurs much earlier, massive cerebellar atrophy was detected in infantile or juvenile forms. Degeneration of the thalamus includes regions associated with the striatum and the cerebellum (i.e., the motor ventrolateral nucleus). The pontine nuclei, as well as the IO in the medulla oblongata and vestibular nuclei in both segments, may also be affected (Table 1; and reviewed in [24,25,26,27,28]).
The polyQ SCAs share clinical features of cerebellar ataxia, dysarthria, and ocular disturbances, but also have specific aspects. Some forms of SCAs share many symptoms with HD, mostly SCA3 and SCA17, and to a lesser extent, SCA1 and SCA2. In contrast, SCA6 and SCA7 are characterised by predominant cerebellar ataxia. Brain imaging reveals moderate to severe atrophy of the cerebellum and brainstem nuclei, as well as volume loss in the cerebral cortex. Anatomopathologically, while the cerebellum is the most affected, other regions involved in motor coordination may also exhibit atrophic features (Table 1; and reviewed in [7]). The cerebral cortex is generally spared in most SCAs during the early stages of the disease, with the primary motor cortex and other areas being impacted in the late stages of SCA1, SCA6, and SCA17 [29].
SCA1 typically begins with ataxia, speech difficulties, spasticity, and abnormal eye movements. In later stages, after approximately 10 to 30 years from onset, patients develop muscular atrophy, cognitive impairments, and bulbar dysfunction, which may lead to respiratory failure. The infantile and juvenile forms progress rapidly, exhibiting severe disease features, with massive degeneration occurring in the cerebellar cortex and CDN, IO, and motor nuclei in the brainstem and spinal cord [7].
SCA2 begins with progressive involuntary movements of the limbs and slow eye movements. The juvenile forms may present with pigmentary retinopathy, seizures, dysphagia and cognitive regression. Brain imaging may reveal a markedly small cerebellum and vermis, along with atrophy of the pontocerebellar regions, brainstem, and cerebral cortex. Postmortem examination shows profound cell loss in the cerebellar cortex [7,30,31,32].
SCA3 begins and progresses with ataxia, pyramidal signs, and dystonia. The brain pathology is the most severe among polyQ SCAs; in addition to cerebellar atrophy, a widespread atrophy is detected in GP, STN, as well as the cerebral cortex, thalamus, and cranial and spinal motor nuclei. Juvenile SCA3 patients also exhibit degeneration in the striatum, SN, pontine nuclei, and cranial nerve nuclei, with massive loss of fibres in the superior and middle cerebellar peduncles and spinocerebellar tracts [7,33].
SCA6 has milder clinical signs at onset, whereas brain imaging shows central cerebellar and brainstem atrophy during disease progression [7,34]. Postmortem studies reveal massive degeneration in the cerebellar cortex, but also widespread neurodegeneration similar to that seen in SCA1, 2, 3, and 7. However, extracerebellar neuronal loss tends to be less severe than in other SCAs, and the basal ganglia are mainly spared [7]. The juvenile form of SCA6 shows severe neurodegeneration in the cerebellar cortex, dentate nucleus, and IO. Purkinje neurons are the most affected cell type in both adult and juvenile SCA6 [35].
SCA7 manifests as progressive cerebellar ataxia and vision loss. Neurodegeneration mainly affects the cerebellum and brainstem, especially the inferior olive, as well as the retina, leading to a gradual decline in visual acuity that often results in blindness. Juvenile and infantile SCA7 show significant atrophy of the cerebrum and cerebellum, absent or reduced deep tendon reflexes, seizures, dysphagia, myoclonus, head lag, absence of cough reflex, and severe hypotonia. The most affected cell types in SCA7 are retinal, cerebellar, and medullary neurons. The condition also presents less common symptoms for polyQ diseases, such as hepatomegaly, haemangiomas, atrial septal defect, and congestive heart failure [7,36,37,38,39].
SCA17 shares typical features of SCAs, including ataxic gait, dysarthria, spasticity, tremor, oculomotor dysfunctions, as well as symptoms with HD, such as dysphagia, chorea, dystonia, rigidity, pyramidal signs, psychiatric symptoms, and cognitive decline. Neuroimaging showed prominent cerebellar atrophy and milder cerebral atrophy. Neurohistopathological analyses reveal neuronal loss in the striatum, ventral thalamic nuclei, cerebellar cortical layers, the dentate nucleus, SN, and IO (reviewed in [40]).
DRPLA also exhibits many of the symptoms associated with both HD and SCAs. Most patient manifests with choreic movements, ataxia, myoclonus, epilepsy, and dementia. Brain imaging demonstrates severe atrophy of the cerebrum and cerebellum, with mild atrophy of the brainstem and spinal cord. The postmortem brains appear atrophic, exhibiting severe degeneration of the GP, STN, and dentate nuclei, as well as moderate degeneration of the RN, with mild neuronal loss and gliosis in the cerebral cortex [41]. Patients with juvenile-onset DRPLA often experience progressive myoclonic epilepsy as one of the initial symptoms, with onset occurring in the first years of life. Disease onset can be as early as 6 months of age, when hyperkinetic and involuntary movements, difficulty controlling head movements, and seizures develop. Juvenile-onset cases tend to show more pronounced pallidoluysian degeneration compared to dentatorubral degeneration, which is opposite to the pattern observed in late-adult onset [41,42].
SBMA, also known as Kennedy disease, is a form of medullary and spinal degeneration associated with muscular atrophy. Brain imaging reveals changes in the white matter of the corticospinal tracts and the cerebellum. Neurodegeneration primarily involves the loss of motor neurons in the spinal cord and brainstem. Anatomopathologically, there is massive degeneration in the anterior horn of the spinal cord and of brainstem motor nuclei, except for the third, fourth, and sixth cranial nerves [43,44,45].
1.2. Expression and Dysfunction of the polyQ-Related Genes and Proteins
Numerous investigations have sought to elucidate the mechanisms of polyQ diseases that lead to both the common and specific dysfunctions and degeneration aspects in polyQ diseases. Several studies focused on examining the expression and function of the affected genes and proteins. These studies primarily rely on animal and cellular models, supplemented by human tissue samples. They explored the roles of normal and mutated polyQ proteins, as well as their distribution in tissues and cells. The primary structures, functions, and expression profiles of normal polyQ-containing proteins have been extensively interrogated (reviewed in [8,46,47]). The levels of normal polyQ proteins generally mirror the respective RNA expression levels, which vary across tissues and cells (Gene Search | HTCA; Human Protein Atlas proteinatlas.org; reviewed in [8] and Table 2)
None of the polyQ-related genes is expressed exclusively in a single tissue type; however, several show high expression levels in the adult brain (CACNA1A and ATN1). Their expression across adult brain regions reveals some common patterns and differences. In the neocortex, ATN1 exhibits the highest expression, followed by HTT and CACNA1A. Expression levels in the basal ganglia, thalamus, and brainstem are generally similar to those in the neocortex, except for ATN1. The highest expression in the cerebellum is observed for CACNA1A and ATN1, with moderate to high levels also for HTT and ATXN2. Regarding neural cell types, polyQ-related genes show differential expression in neurons and glial cells across most brain regions. For example, HTT expression level is higher in neurons than in glial cells in the striatum. In the neocortex, ATN1 is highly expressed in both neurons and astrocytes, while oligodendrocytes show low expression; ATXN3 is highly expressed in both astrocytes and microglia but at very low levels in neurons; HTT is moderately expressed in neurons, astrocytes, and microglia, yet at very low levels in oligodendrocytes. CACNA1A is highly expressed in neurons but at very low levels in all glial cells (Table 2).
When CAG repeats expand beyond a certain threshold for each gene, they cause structural, functional, and clearance alterations in the encoded proteins [3,5,7,8,11]. Several studies have explored the dysfunctions of the proteins with elongated polyQ, revealing that all of them gain a toxic function, while some act by sequestering RNA-binding proteins, losing their normal function, or a combination of these. Many cells exhibit nuclear inclusions during the disease progression in all polyQ diseases (Table 3 and [49,50,51]).
Several reports have identified common and specific functions for proteins associated with polyQ diseases, mainly involved in essential cellular functions such as transcriptional, post-transcriptional, splicing, and translational regulation, as well as intracellular transport. The small proteins ATXN1, α1ACT, ATXN7, and ATN1, along with several proteolytically derived HTT fragments, move to and from the nucleus via nuclear localisation signals or nuclear export signals; the even smaller ATXN3 and TBP proteins pass freely through nuclear pores and are often found in the nucleoplasm (Figure 1; and reviewed in [8]). The primary functions of the polyQ-containing proteins α1A, TBP, AR, ATXN2, and HTT are more clearly defined [46,48], whereas ATXN1, ATXN3, ATXN7, ATN1, and α1ACT have complex and not yet fully understood functions.
Interestingly, among the more than 80 billion neurons in the human CNS [52,53], all containing CAG repeat expansions in polyQ disease patients, many expressing high levels of the normal or mutated proteins, and several having nuclear inclusions, only a small proportion are extensively dysfunctional and degenerate in any of the affected regions. The mechanisms of selective degeneration have been thoroughly studied in various animal and cellular models over the past 30 years [54,55]. Most of the observed expression levels of elongated polyQ proteins are close to those of their normal counterparts in both animal models [54,55] and patient cells [1,3,4,5,6]. Consequently, the distribution of polyQ proteins or the related RNAs in human tissues and neural cell types does not explain the high vulnerability of neurons in the basal ganglia and cerebellum to various polyQ diseases, nor the differential neurodegeneration across tissues (Table 1 and 2; and reviewed in [1,3,4,5,6]). Although most polyQ-related genes are highly expressed in the brain, their expression levels are not the highest in the tissues most vulnerable to polyQ diseases. The expression levels of most elongated polyQ proteins tend to remain relatively stable throughout the progression of the disease. Many neurons in motor coordination networks that show high levels of mutated proteins do not display obvious dysfunction, while others are affected early, and their condition progressively and massively deteriorates. The same applies to intranuclear and cytoplasmic neuronal inclusions found in different brain regions, as well as in several cellular models. Their nature and role in pathology remain a subject of debate [49,50,51,54,55].
Although various organelle dysregulations have been identified, the role of polyQ aggregations in the disease mechanism remains a matter of controversy. Both the long latency before symptoms, often attributed to biological processes with slowly cumulative toxicity or a decades-long lag phase in forming protein aggregates, and the cell-specific toxicity, which is unrelated to the amount of the toxic protein or RNA, are not fully supported by the polyQ protein-related theories [49]. In addition, several strategies targeting the polyQ aggregates have not shown benefits in recent clinical trials [56,57].
1.3. The Expansion of the Repeat Elongation is the New Challenge for the Primary Neuronal Pathogenesis
The observations regarding disease progression and the initial length of the inherited mutation (the number of CAG repeats in the respective gene), as well as the intergenerational expansion of the mutation in germ cells, support the influence of expansion length on the onset and severity of the disease. This expansion predicts earlier and more severe disease onset in successive generations, guiding research towards a deeper understanding of the dynamic CAG elongation in various somatic cell populations [8,60]. Several studies have underlined that the length of a CAG repeat that is not interrupted by CAA (both coding for Q) is directly correlated to the onset and severity of HD disease [58,59].
The biological significance of differential tandem repeat elongation that leads to somatic mosaicism (meaning that the sequence of the inherited genes is selectively modified during lifetime in different cells) has been debated for 30 years, with a prevailing view that genetic instability alters the inherent toxicity of HD-causing alleles [58]. Although several studies have reported relatively modest CAG expansions during the patient’s disease progression in accessible cell populations such as blood cells, the extent of elongation is significantly greater in non-dividing cells like specific long-projection neurons in several animal models. Notably, significant increases in elongation were detected in striatal neurons of HD post-mortem patient brains, indicating hyperelongation as the primary mechanism underlying HD-related neurodegeneration [61]. These important findings, along with the related research methodology (briefly overviewed in the next section), pave the way for a deeper understanding of neuronal-specific genetic instability in polyQ and other tandem-repeat diseases, offering new tools and hope for elucidating their pathological mechanisms and finding treatments.
2. Approaches for Linking Neuronal-Type-Specific Vulnerability and Genetic Instability
Histopathological examinations of post-mortem brains from all nine polyQ disease patients revealed that neurons that are part of the corticobasal ganglia and corticocerebellar circuits are more severely affected (Figure 2 and Table 2). Some neuronal populations in these circuits are more vulnerable than others to CAG repeat elongations. Technological advances in single-cell mRNA sequencing (scRNA-seq) have enabled detailed characterisation of cellular diversity and phenotypic heterogeneity within tissues, aiding the identification of distinct cell subtypes in the mouse and human cortex, cerebellum, basal ganglia, and other CNS regions. Several human cell atlases and databases are now available for the scientific community, supporting the deep characterisation of cells vulnerable to diseases [62,63]. The cellular complexity of the neuronal populations affected in polyQ diseases, their cell-specific vulnerability to CAG elongations, as well as the new findings regarding the mechanisms of cell-specific dysfunction and degeneration, are briefly overviewed here.
2.1. The Neuronal Complexity, Excitability and Vulnerability to CAG Repeat Elongation in the Basal Ganglia.
At the centre of the basal ganglia circuits are various types of striatal projection neurons (SPNs), which are inhibitory (GABAergic). The SPNs are the most affected in HD, but they are also affected in SCA3 and SCA17, especially in the juvenile forms. Due to their morphological features, SPNs are also known as medium spiny neurons (MSNs). They comprise the majority (approximately 90%) of the striatal neuronal population, with the remainder consisting of GABAergic and cholinergic interneurons, which modulate the activity of the projection neurons [64]. SPNs have two main subtypes with different functions: one subtype expresses D1 dopamine receptors and substance P, which enhance cell excitability through dopamine released from the SNc neurons and participate in the direct pathway (dSPNs); another subtype expresses the Drd2 (D2) dopamine receptor and enkephalin, with dopamine decreasing their excitability, acting via the indirect pathway (iSPNs). iSPNs are more excitable than dSPNs [15]. Dopamine plays a fundamental role in balancing the regulation of the direct and indirect pathways, which is crucial for motor control (Figure 2B; and reviewed in [15,16]). Single-nuclei transcriptomics further classified the canonical neuronal types of the human striatum, including iSPNs and dSPNs, as well as several less abundant cell types, such as a few D1/D2-hybrid projection neurons, subclasses of striatal interneurons, and glial cells [55].
The neuronal population in the GP mainly comprises GABAergic projection neurons, which are generally larger than SPNs and are approximately 100 times fewer in number. This indicates a convergence in the striatal-pallidal projection, with a single pallidal cell receiving input from about 100 SPNs. The axon of one pallidal neuron can extend over long distances, innervating various neuronal types in the ventral thalamic and subthalamic nuclei. The morphology of GPi neurons resembles that of GPe neurons. Neurochemically, GPi and GPe can be distinguished by the neuropeptide expression, which reflects the fact that striatal afferents to the GPi originate from a different population of striatal neurons, containing either substance P (for GPi) or enkephalin (for GPe). Three types of pallidal projection neurons have been identified based on their neurochemistry and morphology. Types 1 and 2 are large and the most numerous, constituting 80–90% of pallidal neurons. Type 1 neurons contain the calcium-binding protein parvalbumin, representing around 10% of the large pallidal neuron population. Type 2 cells are characterised by their double immunoreactivity for parvalbumin and calretinin and account for 90% of large pallidal neurons. Type 3 neurons are medium-sized, intensely immunoreactive for calretinin, and make up roughly 10–20% of the pallidal neurons.
SNr shares several features with the GPi and mainly contains GABAergic neurons. The SNr includes several neuronal subpopulations classified by their molecular signatures, phenotypes, or projection targets. The motor control is supported by parvalbumin-expressing SNr neurons. The SNc contains dopaminergic neurons that send excitatory input to the striatum via the D1 receptors, thereby enabling movement initiation through the direct pathway, as well as via the D2 receptors, enabling involuntary movement blocking through the indirect pathway. The STN contain projection glutamatergic neurons, along with a minor population of GABAergic interneurons.
Interestingly, the firing rate is increased in SPN in several HD models, both in pre-symptomatic and early symptomatic stages; however, the excitation-inhibition balance shifts towards greater inhibition as the disease progresses [65]. Among the striatal neurons, iSPNs, which are more excitable than dSPNs, are the most vulnerable in HD and are the first to degenerate. This leads to the disinhibition of targeted thalamic neurons, resulting in hyperkinetic symptoms such as saccadic eye movements and chorea. Later, when dSPNs are also significantly affected, dyskinesia is replaced by akinesia and muscle stiffness. These functional changes have been studied in various HD animal models and are considered crucial to the early imbalance between the direct and indirect pathways [21]. An interesting aspect of the function of SPN is their modulation by the dopaminergic projection neurons from SNc. These neurons fire spontaneously in a highly rhythmic manner, providing continual release of their transmitters. Thalamic neurons have also particularly striking intrinsic electrical properties, which are silenced by steady depolarisation. Interestingly, other key cell types in the basal ganglia also show intrinsic pacemaking activity, including both the excitatory glutamatergic neurons of the STN and GABAergic inhibitory neurons in the GPi and SNr, which maintain tonic activity by inhibiting the target cells in the thalamic nuclei [66,67]. Altered firing patterns of STN neurons were detected in HD animal models [68,69].
2.2. The Neuronal Complexity, Excitability and Vulnerability to CAG Repeat Elongation in the Cerebellar Circuits
Several neurons involved in rapid computation and rapid motor control are spontaneously active, often at relatively high frequencies. Purkinje neurons are a striking example. The cerebellum is critical for controlling precise, rapid motor movements, in addition to more broadly regulating other rapid brain functions, and has a unique architecture. The cortical cerebellar neurons are arranged in a highly regular pattern, forming three distinct layers: the inner granular layer, the middle Purkinje cell layer, and the outer molecular layer. The granular layer consists of many tightly packed small granule neurons (also known as granule cells) and fewer unipolar brush neurons (also known as unipolar brush cells), which are excitatory. In contrast, the Purkinje cell layer contains the Purkinje neurons (also known as Purkinje cells), which are inhibitory and the sole output of the cerebellar cortex. The molecular layer primarily consists of cell projections from Purkinje, granule, and unipolar brush neurons, along with a few small inhibitory interneurons, including basket and stellate neurons. The axons of granule neurons form the parallel fibres, which cross the expansive dendritic trees of Purkinje neurons at right angles; in humans, there are approximately 3000 granule neurons per Purkinje neuron. Inhibitory interneurons in this layer also influence circuit topography by forming synapses with the dendritic trees of Purkinje neurons and regulating their activity.
The granule and unipolar brush neurons receive excitatory signals from the cerebral cortex via the mossy fibres, mainly with a station in the pons nuclei. Information from approximately 25 million mossy fibres is distributed to around 50 billion granule neurons and about 25 million unipolar brush neurons. Each of their dendrites forms a synapse with a single mossy fibre, while their axons branch locally within the granular layer, where an intrinsic system overlays the canonical extrinsic mossy fibre system. Activation of a parallel fibre produces an excitatory response in the Purkinje cell called a “simple spike”.
Apart from receiving input from granule and unipolar brush neurons, each Purkinje neuron also gets extrinsic excitatory signals from IO neurons located in the superior medulla, just inferior to the pons. The IO nuclei have the shape of a crenated "C" and include the principal olive, as well as the medial and dorsal accessory olives. IO neurons are large, with spherical dendritic trees; the dendrites of several IO form the so-called “ glomeruli”, which facilitate their electric coupling via gap junctions. Their axons form the “climbing fibres” that reach the contralateral cerebellar cortex and synapse directly on Purkinje dendrites, creating the most powerful synaptic contact in the brain with a 1:1 ratio. Activation of a climbing fibre produces an all-or-none excitatory response in the Purkinje neurons called a “complex spike”. There are about ten times more Purkinje neurons than IO neurons, meaning each IO neuron generates an average of ten climbing fibres.
A detailed characterisation of cerebellar cells, RN and IO was recently carried out using high-throughput single-cell sequencing [19,20]. These high-throughput results indicate that there is still much to be learned about the composition and functions of human cerebellar networks. Despite their generally regular shape, cerebellar neurons within each subclass form a heterogeneous group, with different subsets identified by various molecular markers, such as co-neurotransmitters, neuromodulators, and regional indicators [19,63].
Purkinje neurons have the remarkable property of firing spontaneously at extremely high frequencies in a highly regular manner, even when isolated from all excitatory synaptic input—other spontaneously active cerebellar neurons, including unipolar brush cells and deep nuclei neurons. Several studies have revealed abnormalities in the excitability of several neuronal populations of the cerebellum in polyQ diseases. Purkinje neurons at early time points of the SCA1 and other SCAs showed the highest enrichment in genes related to synaptic signalling. Synapses between the parallel fibre and Purkinje neurons require type 1 glutamatergic signalling, which has been previously reported to be dysfunctional in multiple cerebellar disorders [70]. The most affected cerebellar cells in both adult and juvenile SCA1, SCA2, SCA6, SCA7, and SCA17 are Purkinje neurons, which are the only inhibitory projection neurons in the cortico-cerebellar pathways [35]. By contrast, in SCA3, DRPLA, and SBMA, the most affected are the projection excitatory neurons of the CDN, as well as those in the RN, STN, thalamus and neocortex. Again, the level of expression of the affected gene and protein cannot explain the increased vulnerability of the respective neuronal type (Table 1 and Table 2).
2.3. DNA Repair and Somatic Instability in Long-Projection Neurons Make the Motor Coordination Network
The long projection neurons located in all the stations of the motor coordination network, which form the cortico–basal ganglia and cortico–cerebellar circuits, should function well throughout life to facilitate quick and efficient motor coordination. From an energetic standpoint, the spontaneous firing of the many neurons in the motor coordination network needs a high level of energy. At the same time, sustained neuronal excitability comes with a cost of high DNA damage throughout life. Nonetheless, since the long projection neurons are destined to live as long as the organism itself, there is a high demand for the lifelong maintenance of their integrity. Multiple protective mechanisms enable them to function effectively throughout the organism’s lifetime, including neurotrophic factors and prominent DNA repair complexes.
Recent findings support the theory that genetic instability caused by defective DNA repair is the primary driver of neurodegeneration in many neurodegenerative diseases, including polyQ diseases. New data from high-throughput analysis of HD postmortem brains reinforce the role of cell-specific genetic instability, helping to identify crucial stages in disease progression [61,71].
In post-mitotic cells such as neurons, DNA-repair elongation is believed to result from occasional strand misalignment (mispaired repeats) after transcription or transient helix destabilisation. Briefly, during transcription in neurons, single-stranded DNA can form small extrahelical extrusions, especially when highly expanded repeats are present. The formation of these structures depends on various factors, including sequence motifs, their “purity”, expansion length, and possibly methylation status. Long CAG repeats, in particular, can produce complex, higher-order non-canonical structures, such as R-loops (slip-outs). Once these structures form, they are recognised by DNA mismatch repair (MMR) proteins, which attempt to repair the DNA damage; repair pathways that involve nicking, excision, and resynthesis of one of the two strands. Resynthesis may result in a length-change mutation—an expansion or contraction, depending on which strand has been nicked and excised. Repeat expansion typically occurs in small increments. However, instead of repairing the damage, these proteins can contribute to the somatic expansion of pathogenic repeats, as demonstrated in the SPN carrying the CAG expansions in the HTT gene [59,61,71,72].
The concept of genetic instability and somatic mosaicism in HD and similar diseases with tandem repeats is well recognised. More than 30 years ago, Telenius et al. measured the level of somatic mosaicism of the expanded HD allele in adult patients with late-onset HD. Larger expansions were linked to increased mosaicism. Furthermore, the degree of mosaicism is high in the basal ganglia and cerebral cortex, intermediate levels in the blood and liver, and at low levels in the cerebellum. Additionally, greater instability was seen in sperm compared to blood. This pioneering study provides evidence that somatic mosaicism in HD varies by organ and correlates with neuropathology [73,74,75].
The first study reporting somatic hyperexpansions in the HD post-mortem brain identified molecules with long CAG-repeat tracts, up to 1000 repeats, mainly in the neocortex. The solid-phase PCR (SP-PCR) used in this study relies on amplifying dilutions of DNA from bulk tissue containing heterogeneous cell types. SP-PCR has attracted considerable interest across various research fields since it enables direct DNA amplification on the surface of a solid substrate [76]. However, a more precise correlation of mutation length variability with specific cell types depended on the development of sequencing methodologies at the single-cell level. One method involved implementing fluorescence-activated nuclear sorting to isolate different cell populations from five post-mortem HD brains based on marker gene expression. Subsequently, the isolated cells were sequenced, including the determination of CAG size in HTT transcripts, providing matched transcriptional and instability profiles at the subpopulation level. Similar studies have examined somatic instability in striatal and cortical neurons in post-mortem brains of individuals with HD. Nevertheless, their methods were limited by short-read sequencing, which could detect a maximum of 110 CAG repeats. They conclude that although elongation increase may be significant, it is not sufficient to determine cell death [77,78].
What makes iSPN and dSPN so vulnerable yet remain relatively normal for many years in a patient with a mutation in HTT before experiencing a dramatic decline and cell death? The development of new techniques for measuring long CAG expansions at the single-neuron level has significantly refined previous data and revealed the disease’s dynamics. Handsaker and colleagues [61] advanced the technological frontier by developing a sophisticated single-cell RNA-sequencing method and analysis based on long reads, enabling the simultaneous acquisition of transcriptional profiles and HTT CAG sizes from the same cell. This allowed for the grouping of cells by both transcriptional profile and CAG length in the HTT model. Somatic expansion was allele-specific, strongly affecting the mutated HTT allele but not the other inherited allele or the alleles of unaffected individuals. While 95%–98% of each HD donor’s SPNs had expanded beyond the inherited (germline) length with around 20–31 CAGs, only a minority exhibited much longer expansions (100–500+ CAGs). A tiny proportion of SPNs exhibited extreme expansions, with more than 800 CAG repeats. These were massively transcriptionally dysregulated, losing both their marker identity and markers of cell death pathways. Once again, iSPNs were found to be lost earlier than dSPNs. Striatal interneurons and glial cells displayed modest CAG-repeat instability.
Surprisingly, no significant effects on the transcriptome of SPNs were observed due to CAG-repeat expansion from 36 to 150 CAGs; however, SPNs with longer expansions (>150 CAGs) showed significantly altered gene expression involving hundreds of genes. Remarkably, the gene expression changes in SPNs with expansions beyond 150 CAGs were highly consistent across patients. Once again, the expression levels of HTT did not change with CAG repeat expansion. The altered gene expression exhibited two types of relationships to CAG-repeat length: one set of genes showed continuous changes as the CAG repeat length extended beyond 150 CAGs. In contrast, another set displayed more discrete and dramatic changes in both iSPN and dSPN with a CAG repeat length greater than 250 CAGs. The genes whose expression decreased in SPNs with a repeat length over 150 CAGs were among the most highly expressed in SPNs, suggesting that a core biological change involves eroding the SPN identity features that distinguish them from other neurons. Many of these genes also perform essential physiological functions, such as those encoding potassium channel subunits [61].
A specific set of over 100 genes, normally suppressed in SPNs, becomes active in those with 350 or more CAG repeats. This set includes genes typically expressed in other neural cell types but not in SPNs, as well as two genes (CDKN2A and CDKN2B) that encode proteins (p16(INK4a) and p15(INK4b)) promoting senescence and apoptosis. Interestingly, inactivation of the polycomb repressor complex 2 (PRC2) in adult mice results in similar gene expression changes, leading to SPN loss, a decline in motor function, and death within months. The findings suggest that transcriptional changes in SPNs with very long repeats may contribute to their death. These findings are consistent with analyses of a specific HD mouse model (Q175), which begins life with a CAG-repeat tract exceeding 170 CAGs in all cells, as well as with other studies analysing the transcriptome in patients’ striatum at the single-cell level. SPNs showed reduced expression of genes that differentiate them from other neurons [59,79,80,81].
The breakthrough results of Handsaker and colleagues [61] support a multi-phase model called ‘ELongATE’, in which somatic expansion rates in neurons increase significantly once they exceed 80 CAG repeats, with a threshold of approximately 150 CAG repeats needed to trigger cell-autonomous transcriptional dysregulation in SPNs, leading to even faster expansion and cell death. The proposed ‘ELongATE’ model provides a plausible explanation for some unresolved questions. Firstly, the progressive loss of SPNs during HD neurodegeneration may result from the time needed for extreme expansion events to accumulate in these neurons over a lifetime. Secondly, the extended period of degeneration observed in HD patients—typically 10–20 years from diagnosis to death—seems to align with the rapid yet asynchronous neuronal degeneration suggested by the ‘ELongATE’ model. According to this model, although all cells in the patient’s brain produce mHTT, only those with more than 150 CAG repeats are truly toxic. Consequently, at any given moment, only a few SPNs might generate highly expanded and toxic forms of DNA, RNA, and HTT protein, contributing to the slow, progressive degeneration of the neurons.
The asynchronicity in HD mutation elongation in SPNs occurs because length-change mutations were initially rare events (occurring less than once a year per cell across 36–55 CAGs). Still, once they occur, they increase the likelihood of subsequent elongations. Future work will be required to determine whether the repeat-expansion-driven dynamics also follow the dysfunction and neurodegeneration in other brain regions and neuronal types, or these areas are secondarily affected due to the dysfunctions of the neuronal networks in which they are part (Figure 2). Extensive expansions were previously identified in the cerebral cortex of post-mortem HD brains, as well as in animal models of HD. These studies demonstrate that the vast expansion of the mHTT CAG tract occurs in many deep-layer pyramidal cell types, despite the selective loss of L5a corticostriatal projection neurons. However, the association with the most affected long projection excitatory neurons was not performed due to technical limitations at the time [78].
Interestingly, the somatic mHTT CAG expansion was also detected in Purkinje neurons [10,82]. However, the confirmation of the increased expansions is expected in further studies for other cell populations with high electrical and metabolic activities, with well-developed projections and spontaneous firing. It included neurons in the other basal ganglia nuclei, such as GPi, GPe, STN, and SNr, as well as in the different hubs of motor coordination, such as the thalamus, RN, IO, and DCN. All these regions are affected in late stages of HD (Table 1), but the pathological mechanisms remain unclear.
Somatic instability and mosaicism have been reported across all polyQ diseases, including in animal models and patient tissue samples (reviewed in 81). Studies on somatic mosaicism in SCA1 soon followed those in HD. Kacher et al. reported that the somatic instability of CAG repeats correlates with clinical progression in individuals with SCA1. Alleles of intermediate size without CAT interruption in the ATXN1 gene are also mutable and exhibit somatic instability. The somatic instability of both alleles in an SCA1 patient may therefore have contributed to modifying the age at disease onset, progression, or phenotypic severity [83]. Adding further descriptions to confirm or challenge these results will be necessary.
The somatic instability of expanded SCA1 was examined in patients’ blood, sperm, and neuronal tissues. CAG repeats in ATXN1 are more unstable in sperm than in blood, supporting the HD findings. Moreover, the neuronal mosaicism patterns in SCA1 resembled those in HD. The most significant instability was observed in the cerebrum, while the least was seen in the cerebellum [84,85]. Although this finding contradicts the idea that tissue mosaicism does not relate to neuronal vulnerability, we must consider the technical limitations in detecting mosaicism in Purkinje neurons, which are a minor population in the cerebellum. The correlation between neuronal vulnerability and genetic instability has also been examined in SCA1 “knock-in” mice carrying 154 CAG repeats (154Q) on one allele and two CAG repeats (2Q) on the other allele. Expansions exceeding 200 repeats were observed in the striatum and spinal cord, but no expansions larger than 174 repeats were found in the cerebellum. These tissues show different levels of mosaicism, indicating no clear link between mosaicism and selective neuronal vulnerability in SCA1. However, technical limitations should be considered before taking any further conclusions. Moreover, repeat instability in SCA1 was age-dependent. At 7 weeks, the knock-in allele remained stable; however, small expansions and contractions appeared by 30 weeks. Most larger expansions occurred after 30 weeks, following the onset of neuronal dysfunction, indicating that repeat instability relates to neuronal vulnerability [85].
Subsequent research revealed similar patterns of neuronal mosaicism in SCA3 and DRPLA, despite several differences in neuropathology and neuronal vulnerabilities. The preferential expansion of the mATXN3 CAG tract found in SPN nuclei isolated from SCA3 donors suggests that these neurons generally tend to expand long CAG tracts. Notably, repeat expansion was observed in specific cerebellar cells in both early- and late-onset DRPLA cerebellum. These findings indicate that expansions were more common in Purkinje neurons than in granule neurons, regardless of age at onset. White matter glial cells showed more expansions than Purkinje neurons in late-onset tissue but had similar levels in early-onset tissue. The study was conducted on one early-onset case and one late-onset case; thus, further studies are required to draw definitive conclusions [86].
Further single-cell studies are required to address the CAG elongation dynamics in Purkinje neurons in several polyQ diseases affecting predominantly this cell type, such as SCA2, SCA6, SCA7 and SCA17. The projection neurons in the CDN, IO, pons, and RN should also be addressed in the polyQ SCAs, as well as in HD, DPRLA and SBMA. The same is valid for the long projection neurons in the basal ganglia nuclei, other than SPN.
3. Conclusion and Outlook
Understanding the complexity of networks that regulate motor coordination in the normal human brain is essential for recognising their dysfunctions, but it also presents an important challenge. Significant advances have been made in recent years to uncover the cellular complexity of the mammalian brain and its specific features, particularly in the human brain. While the circuits and mechanisms for motor coordination are mainly conserved in vertebrates, several human-specific aspects are only partly understood. However, over the past decade, findings from neuroanatomical studies in non-human primates and human neuroimaging studies have provided new insights into conceptualising the Human Connectome Project (HCP) repository [87]. The basal ganglia and the cerebellum are not seen as independent subcortical systems but, instead, form a densely interconnected network. Thus, changes at one node can propagate throughout the entire network, influencing operations at other nodes [13,14]. However, the precise role of cortico-rubro-olivary projections remains unclear. Several studies also indicate robust cooperation between the RN and basal ganglia circuitry (STN, GP, SN, dorsal thalamus) in initiating and terminating motor tasks. The integrated network perspective provides a new way of conceptualising the functional organisation of basal ganglia and cerebellar circuits in relation to the cerebral cortex, as well as to understand better the primary and secondary pathology in movement disorders, including polyQ diseases. The connections between neural regions affected by polyQ diseases and their cellular heterogeneity are crucial topics, and understanding these links helps distinguish cell-autonomous from non-cell-autonomous mechanisms in these diseases, as well as the disease progression. However, while neuronal networks may explain why different areas and cells are affected secondarily, they cannot clarify why the CAG expansions in specific genes primarily impact certain neurons.
The mechanisms behind region- and cell-specific vulnerability in most polyQ diseases have largely remained unexplored for a long time, as have the degenerative processes in affected cells. Although many aspects still need further understanding, the hyperexpansion of repeats in neurons is now recognised as a key primary pathogenic mechanism in HD, with significant implications for other neurodegenerative disorders involving tandem repeats. Accurate measurement of CAG size in individual brain cells at the single-cell level, along with their corresponding transcriptional profiles, has become a central focus in the field of polyQ diseases, as it is increasingly important for understanding the vulnerability of specific neuronal types. Using advanced single-cell technologies and long-read sequencing to monitor somatic instability in vulnerable neuronal populations is becoming essential for confirming the extent of pathological CAG elongation in the most susceptible neurons in other polyQ diseases. While the SPN, Purkinje neurons, and cortical projection neurons have already been examined for dynamic elongations, other vital cells in the motor coordination network remain unaddressed. These include at least the long projection neurons in the GPi, GPe, SNc, SNr, STN, IO, pRN, and CDN (Figure 2).
Still, elucidating the mechanisms by which the very long expansions led to neuronal loss is a work in progress, and it is expected that new findings will come from novel human neuronal models and humanised animal models. The same is true for therapy addressing neuronal-specific hyperelongations. Ultimately, the final challenge is to halt the pathological process before neuronal degeneration occurs. While several studies have shown an efficient reduction of HD symptoms and neurodegeneration after editing the mutation or interfering with DNA repair complexes in animal models [88,89,90], the translation of these techniques to patients requires caution, and it is still in its very early stages. However, by focusing on the primary cause of the disease and targeting the relevant cells, new therapeutic strategies can be developed to slow or even halt the progression of the elongations. With advances in genome editing technologies, the ability to stop the CAG expansion could soon lead to revolutionary treatments.
Funding
The author declares no external funding for this work.
Acknowledgments
The author acknowledges Professor Lars Klimaschewski for the impactful comments on the manuscript.
Conflicts of Interest
The author declares no conflicts of interest.
References
- La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010, 11, 247–258. [Google Scholar] [CrossRef]
- La Spada AR, Taylor JP. Polyglutamines placed into context. Neuron. 2003, 38, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Lieberman AP, Shakkottai VG, Albin RL. Polyglutamine Repeats in Neurodegenerative Diseases. Annu Rev Pathol. 2019, 14, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Stoyas CA, La Spada AR. The CAG-polyglutamine repeat diseases: a clinical, molecular, genetic, and pathophysiologic nosology. Handb Clin Neurol. 2018, 147, 143–170. [Google Scholar]
- Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019, 5, 24. [Google Scholar] [CrossRef]
- Rüb U, Schöls L, Paulson H, Auburger G, Kermer P, Jen JC, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef]
- Johnson SL, Tsou WL, Prifti MV, Harris AL, Todi SV. A survey of protein interactions and posttranslational modifications that influence the polyglutamine diseases. Front Mol Neurosci. 2022, 15, 974167. [Google Scholar] [CrossRef]
- McLoughlin HS, Moore LR, Paulson HL. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol Dis. 2020, 134, 104635. [Google Scholar]
- Rüb U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D, et al. Degeneration of the cerebellum in Huntington’s disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol. 2013, 23, 165–177. [Google Scholar] [CrossRef]
- Buijsen RAM, Toonen LJA, Gardiner SL, van Roon-Mom WMC. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics. 2019, 16, 263–286. [Google Scholar] [CrossRef]
- Cubo E, Martinez-Horta SI, Santalo FS, Descalls AM, Calvo S, Gil-Polo C, et al. Clinical manifestations of homozygote allele carriers in Huntington’s disease. Neurology. 2019, 92, e2101–e2108. [Google Scholar]
- Bostan AC, Strick PL. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci. 2018, 19, 338–350. [Google Scholar] [CrossRef]
- Groenewegen, HJ. The basal ganglia and motor control. Neural Plast. 2003, 10, 107–120. [Google Scholar] [CrossRef]
- Surmeier DJ, Graves SM, Shen W. Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr Opin Neurobiol. 2014, 29, 109–117. [Google Scholar] [CrossRef]
- Vogt Weisenhorn DM, Giesert F, Wurst W. Diversity matters - heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s Disease. J Neurochem. 2016, 139 (Suppl. S1), 8–26. [Google Scholar] [CrossRef]
- Prasad AA, Wallén-Mackenzie Å. Architecture of the subthalamic nucleus. Commun Biol. 2024, 7, 78. [Google Scholar]
- De Zeeuw CI, Simpson JI, Hoogenraad CC, Galjart N, Koekkoek SK, Ruigrok TJ. Microcircuitry and function of the inferior olive. Trends Neurosci. 1998, 21, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Chen X, Du Y, Broussard GJ, Kislin M, Yuede CM, Zhang S, et al. Transcriptomic mapping uncovers Purkinje neuron plasticity driving learning. Nature. 2022, 605, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Kozareva V, Martin C, Osorno T, Rudolph S, Guo C, Vanderburg C, et al. A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature. 2021, 598, 214–219. [Google Scholar] [CrossRef]
- Reiner A, Deng YP. Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci Ther. 2018, 24, 250–280. [Google Scholar] [CrossRef]
- Stacho M, Häusler AN, Brandstetter A, Iannilli F, Mohlberg H, Schiffer C, et al. Phylogenetic reduction of the magnocellular red nucleus in primates and inter-subject variability in humans. Front Neuroanat. 2024, 18, 1331305. [Google Scholar] [CrossRef]
- Landwehrmeyer GB, McNeil SM, Dure LS, Ge P, Aizawa H, Huang Q, et al. Huntington’s disease gene: regional and cellular expression in the brain of normal and affected individuals. Ann Neurol. 1995, 37, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, Barbagallo G, et al. Biological and clinical manifestations of juvenile Huntington’s disease: a retrospective analysis. Lancet Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef]
- Latimer CS, Flanagan ME, Cimino PJ, Jayadev S, Davis M, Hoffer ZS, et al. Neuropathological Comparison of Adult Onset and Juvenile Huntington’s Disease with Cerebellar Atrophy: A Report of a Father and Son. J Huntingtons Dis. 2017, 6, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Hedjoudje A, Nicolas G, Goldenberg A, Vanhulle C, Dumant-Forrest C, Deverrière G, et al. Morphological features in juvenile Huntington disease associated with cerebellar atrophy - magnetic resonance imaging morphometric analysis. Pediatr Radiol. 2018, 48, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Sakazume S, Yoshinari S, Oguma E, Utsuno E, Ishii T, Narumi Y, et al. A patient with early onset Huntington disease and severe cerebellar atrophy. A patient with early onset Huntington disease and severe cerebellar atrophy. Am J Med Genet A. 2009, 149A, 598–601. [Google Scholar]
- Nance MA, Myers RH. Juvenile onset Huntington’s disease--clinical and research perspectives. Ment Retard Dev Disabil Res Rev. 2001, 7, 153–157. [Google Scholar] [CrossRef]
- Nolan M, Scott C, Hof PR, Ansorge O. Betz cells of the primary motor cortex. J Comp Neurol. 2024, 532, e25567. [Google Scholar] [CrossRef]
- Babovic-Vuksanovic D, Snow K, Patterson MC, Michels VV. Spinocerebellar ataxia type 2 (SCA 2) in an infant with extreme CAG repeat expansion. Am J Med Genet. 1998, 79, 383–387. [Google Scholar] [CrossRef]
- Paciorkowski AR, Shafrir Y, Hrivnak J, Patterson MC, Tennison MB, Clark HB, et al. Massive expansion of SCA2 with autonomic dysfunction, retinitis pigmentosa, and infantile spasms. Neurology. 2011, 77, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Ramocki MB, Chapieski L, McDonald RO, Fernandez F, Malphrus AD. Spinocerebellar ataxia type 2 presenting with cognitive regression in childhood. J Child Neurol. 2008, 23, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Donis KC, Saute JA, Krum-Santos AC, Furtado GV, Mattos EP, Saraiva-Pereira ML, et al. Spinocerebellar ataxia type 3/Machado-Joseph disease starting before adolescence. Neurogenetics. 2016, 17, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa K, Tanaka H, Saito M, Ohkoshi N, Fujita T, Yoshizawa K, et al. Japanese families with autosomal dominant pure cerebellar ataxia map to chromosome 19p13.1-p13.2 and are strongly associated with mild CAG expansions in the spinocerebellar ataxia type 6 gene in chromosome 19p13.1. Am J Hum Genet. 1997, 61, 336–346. [Google Scholar] [CrossRef]
- Wang X, Wang H, Xia Y, Jiang H, Shen L, Wang S, et al. A neuropathological study at autopsy of early onset spinocerebellar ataxia 6. J Clin Neurosci. 2010, 17, 751–755. [Google Scholar] [CrossRef]
- Ansorge O, Giunti P, Michalik A, Van Broeckhoven C, Harding B, Wood N, et al. Ataxin-7 aggregation and ubiquitination in infantile SCA7 with 180 CAG repeats. Ann Neurol. 2004, 56, 448–452. [Google Scholar] [CrossRef]
- Donis KC, Mattos EP, Silva AA, Furtado GV, Saraiva-Pereira ML, Jardim LB, et al. Infantile spinocerebellar ataxia type 7: Case report and a review of the literature. J Neurol Sci. 2015, 354, 118–121. [Google Scholar] [CrossRef]
- Horton LC, Frosch MP, Vangel MG, Weigel-DiFranco C, Berson EL, Schmahmann JD. Spinocerebellar ataxia type 7: clinical course, phenotype-genotype correlations, and neuropathology. Cerebellum. 2013, 12, 176–193. [Google Scholar] [CrossRef]
- Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, Holmberg M. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet. 1998, 7, 171–176. [Google Scholar] [CrossRef]
- Toyoshima Y, Takahashi H. Spinocerebellar Ataxia Type 17 (SCA17). Adv Exp Med Biol. 2018, 1049, 219–231. [Google Scholar]
- Nowak B, Kozlowska E, Pawlik W, Fiszer A. Atrophin-1 Function and Dysfunction in Dentatorubral-Pallidoluysian Atrophy. Mov Disord. 2023, 38, 526–536. [Google Scholar] [CrossRef]
- Takeda S, Takahashi H. Neuropathology of dentatorubropallidoluysian atrophy. Neuropathology; 1996. p. 48-55.
- Breza M, Koutsis G. Kennedy’s disease (spinal and bulbar muscular atrophy): a clinically oriented review of a rare disease. J Neurol. 2019, 266, 565–573. [Google Scholar] [CrossRef]
- Echaniz-Laguna A, Rousso E, Anheim M, Cossée M, Tranchant C. A family with early-onset and rapidly progressive X-linked spinal and bulbar muscular atrophy. Neurology. 2005, 64, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Grunseich C, Kats IR, Bott LC, Rinaldi C, Kokkinis A, Fox D, et al. Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscul Disord. 2014, 24, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron; 2016. p. P910-26.
- Gerbich TM, Gladfelter AS. Moving beyond disease to function: Physiological roles for polyglutamine-rich sequences in cell decisions. Curr Opin Cell Biol. 2021, 69, 120–126. [Google Scholar] [CrossRef]
- Lee J, Kim M, Itoh TQ, Lim C. Ataxin-2: A versatile posttranscriptional regulator and its implication in neural function. Wiley Interdiscip Rev RNA. 2018, 9, e1488. [Google Scholar] [CrossRef] [PubMed]
- Bäuerlein FJB, Saha I, Mishra A, Kalemanov M, Martínez-Sánchez A, Klein R, et al. In Situ Architecture and Cellular Interactions of PolyQ Inclusions. Cell. 2017, 171, 179–187.e10. [Google Scholar] [CrossRef]
- Hasegawa A, Ikeuchi T, Koike R, Matsubara N, Tsuchiya M, Nozaki H, et al. Long-term disability and prognosis in dentatorubral-pallidoluysian atrophy: a correlation with CAG repeat length. Mov Disord. 2010, 25, 1694–1700. [Google Scholar] [CrossRef]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997, 19, 333–344. [Google Scholar] [CrossRef]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Lent R, Azevedo FA, Andrade-Moraes CH, Pinto AV. How many neurons do you have? Some dogmas of quantitative neuroscience under revision. Eur J Neurosci. 2012, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Figiel M, Szlachcic WJ, Switonski PM, Gabka A, Krzyzosiak WJ. Mouse models of polyglutamine diseases: review and data table. Part I. Mol Neurobiol. 2012, 46, 393–429. [Google Scholar] [CrossRef] [PubMed]
- Switonski PM, Szlachcic WJ, Gabka A, Krzyzosiak WJ, Figiel M. Mouse models of polyglutamine diseases in therapeutic approaches: review and data table. Part II. Mol Neurobiol. 2012, 46, 430–466. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron. 2019, 101, 801–819. [Google Scholar] [CrossRef]
- Wadman, M. S: drug for Huntington disease fails in major trial, 2021.
- gusella@helix. mgh.harvard.edu GMoHsDG-HCEa, Consortium GMoHsDG-H. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell. 2019, 178, 887–900.e14. [Google Scholar] [CrossRef]
- Gusella JF, Lee JM, MacDonald ME. Huntington’s disease: nearly four decades of human molecular genetics. Hum Mol Genet. 2021, 30, R254–R63. [Google Scholar] [CrossRef]
- Tezenas du Montcel S, Durr A, Bauer P, Figueroa KP, Ichikawa Y, Brussino A, et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain. 2014, 137 Pt 9, 2444–55. [Google Scholar]
- Handsaker RE, Kashin S, Reed NM, Tan S, Lee WS, McDonald TM, et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease. Cell. 2025, 188, 623–639.e19. [Google Scholar] [CrossRef]
- Siletti K, Hodge R, Mossi Albiach A, Lee KW, Ding SL, Hu L, et al. Transcriptomic diversity of cell types across the adult human brain. Science. 2023, 382, eadd7046. [Google Scholar] [CrossRef]
- Apsley EJ, Becker EBE. Purkinje Cell Patterning-Insights from Single-Cell Sequencing. Cells.
- Gritton HJ, Howe WM, Romano MF, DiFeliceantonio AG, Kramer MA, Saligrama V, et al. Unique contributions of parvalbumin and cholinergic interneurons in organizing striatal networks during movement. Nat Neurosci. 2019, 22, 586–597. [Google Scholar] [CrossRef]
- Blumenstock S, Dudanova I. Cortical and Striatal Circuits in Huntington’s Disease. Front Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef]
- Plotkin JL, Goldberg JA. Thinking Outside the Box (and Arrow): Current Themes in Striatal Dysfunction in Movement Disorders. Neuroscientist. 2019, 25, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Callahan JW, Abercrombie ED. Relationship between subthalamic nucleus neuronal activity and electrocorticogram is altered in the R6/2 mouse model of Huntington’s disease. J Physiol. 2015, 593, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Callahan JW, Abercrombie ED. Age-dependent alterations in the cortical entrainment of subthalamic nucleus neurons in the YAC128 mouse model of Huntington’s disease. Neurobiol Dis. 2015, 78, 88–99. [Google Scholar] [CrossRef]
- Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004, 13, 2535–2543. [Google Scholar] [CrossRef]
- Flower MD, Tabrizi SJ. The breaking point where repeat expansion triggers neuronal collapse in Huntington’s disease. Cell Genom. 2025, 5, 100816. [Google Scholar] [CrossRef]
- Iyer RR, Pluciennik A. DNA Mismatch Repair and its Role in Huntington’s Disease. J Huntingtons Dis. 2021, 10, 75–94. [Google Scholar] [CrossRef]
- Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. 1994, 6, 409–414. [Google Scholar] [CrossRef]
- Malik I, Kelley CP, Wang ET, Todd PK. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Bunting EL, Hamilton J, Tabrizi SJ. Polyglutamine diseases. Curr Opin Neurobiol. 2022, 72, 39–47. [Google Scholar] [CrossRef]
- Kennedy L, Evans E, Chen CM, Craven L, Detloff PJ, Ennis M, et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. 2003, 12, 3359–3367. [Google Scholar] [CrossRef]
- Mätlik K, Baffuto M, Kus L, Deshmukh AL, Davis DA, Paul MR, et al. Cell Type Specific CAG Repeat Expansions and Toxicity of Mutant Huntingtin in Human Striatum and Cerebellum. Cell Type Specific CAG Repeat Expansions and Toxicity of Mutant Huntingtin in Human Striatum and Cerebellum. bioRxiv. 2023.
- Pressl C, Mätlik K, Kus L, Darnell P, Luo JD, Paul MR, et al. Selective vulnerability of layer 5a corticostriatal neurons in Huntington’s disease. Neuron. 2024, 112, 924–941.e10. [Google Scholar] [CrossRef] [PubMed]
- Gao R, Matsuura T, Coolbaugh M, Zühlke C, Nakamura K, Rasmussen A, et al. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet. 2008, 16, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Consortium GMoHsDG-H. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell. 2015, 162, 516–526. [Google Scholar] [CrossRef]
- Malaiya S, Cortes-Gutierrez M, Herb BR, Coffey SR, Legg SRW, Cantle JP, et al. Single-Nucleus RNA-Seq Reveals Dysregulation of Striatal Cell Identity Due to Huntington’s Disease Mutations. J Neurosci. 2021, 41, 5534–5552. [Google Scholar] [CrossRef]
- Singh-Bains MK, Mehrabi NF, Sehji T, Austria MDR, Tan AYS, Tippett LJ, et al. Cerebellar degeneration correlates with motor symptoms in Huntington disease. Ann Neurol. 2019, 85, 396–405. [Google Scholar] [CrossRef]
- Kacher R, Lejeune FX, David I, Boluda S, Coarelli G, Leclere-Turbant S, et al. CAG repeat mosaicism is gene-specific in spinocerebellar ataxias. Am J Hum Genet. 2024, 111, 913–926. [Google Scholar] [CrossRef]
- Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR, et al. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1995, 10, 344–350. [Google Scholar] [CrossRef]
- Zühlke C, Dalski A, Hellenbroich Y, Bubel S, Schwinger E, Bürk K. Spinocerebellar ataxia type 1 (SCA1): phenotype-genotype correlation studies in intermediate alleles. Eur J Hum Genet. 2002, 10, 204–209. [Google Scholar] [CrossRef]
- Hashida H, Goto J, Suzuki T, Jeong S, Masuda N, Ooie T, et al. Single cell analysis of CAG repeat in brains of dentatorubral-pallidoluysian atrophy (DRPLA). J Neurol Sci. 2001, 190, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Elam JS, Glasser MF, Harms MP, Sotiropoulos SN, Andersson JLR, Burgess GC, et al. The Human Connectome Project: A retrospective. Neuroimage. 2021, 244, 118543. [Google Scholar] [CrossRef] [PubMed]
- Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. 1999, 23, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Phadte AS, Bhatia M, Ebert H, Abdullah H, Elrazaq EA, Komolov KE, et al. FAN1 removes triplet repeat extrusions via a PCNA- and RFC-dependent mechanism. Proc Natl Acad Sci U S A. 2023, 120, e2302103120. [Google Scholar] [CrossRef] [PubMed]
- Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, St Claire J, et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. 2013, 9, e1003930. [Google Scholar]
Figure 1.
Ten human proteins containing polyglutamine (polyQ) traits are associated with nine rare genetic diseases. These proteins have varying numbers of glutamine (Q) repeats at different positions (marked in orange). When the inherited elongation of the CAG tandem repeats in the corresponding gene, and the related polyQ trait, exceeds a threshold value (marked in red for each protein), it leads over several decades to the respective polyQ disease: Huntington’s Disease (HD), Spinocerebellar Ataxia (SCA) types 1, 2, 3, 6, 7, and 17, Dentato-Rubral Pallidoluysian Atrophy (DRPLA), and Spinal and Bulbar Muscular Atrophy (SBMA). Most of these proteins translocate into the nucleus, some containing sequences that act as nuclear localisation signals or nuclear export signals (marked in light and dark green, respectively). The only polyQ protein located within the cell membrane is the longest isoform of the α1A protein; however, its gene is bicistronic and produces also the nuclear protein α1ACT, which shares the same sequence as its intracellular terminal domain elongated in SCA6 (modified from [5,8]).
Figure 1.
Ten human proteins containing polyglutamine (polyQ) traits are associated with nine rare genetic diseases. These proteins have varying numbers of glutamine (Q) repeats at different positions (marked in orange). When the inherited elongation of the CAG tandem repeats in the corresponding gene, and the related polyQ trait, exceeds a threshold value (marked in red for each protein), it leads over several decades to the respective polyQ disease: Huntington’s Disease (HD), Spinocerebellar Ataxia (SCA) types 1, 2, 3, 6, 7, and 17, Dentato-Rubral Pallidoluysian Atrophy (DRPLA), and Spinal and Bulbar Muscular Atrophy (SBMA). Most of these proteins translocate into the nucleus, some containing sequences that act as nuclear localisation signals or nuclear export signals (marked in light and dark green, respectively). The only polyQ protein located within the cell membrane is the longest isoform of the α1A protein; however, its gene is bicistronic and produces also the nuclear protein α1ACT, which shares the same sequence as its intracellular terminal domain elongated in SCA6 (modified from [5,8]).
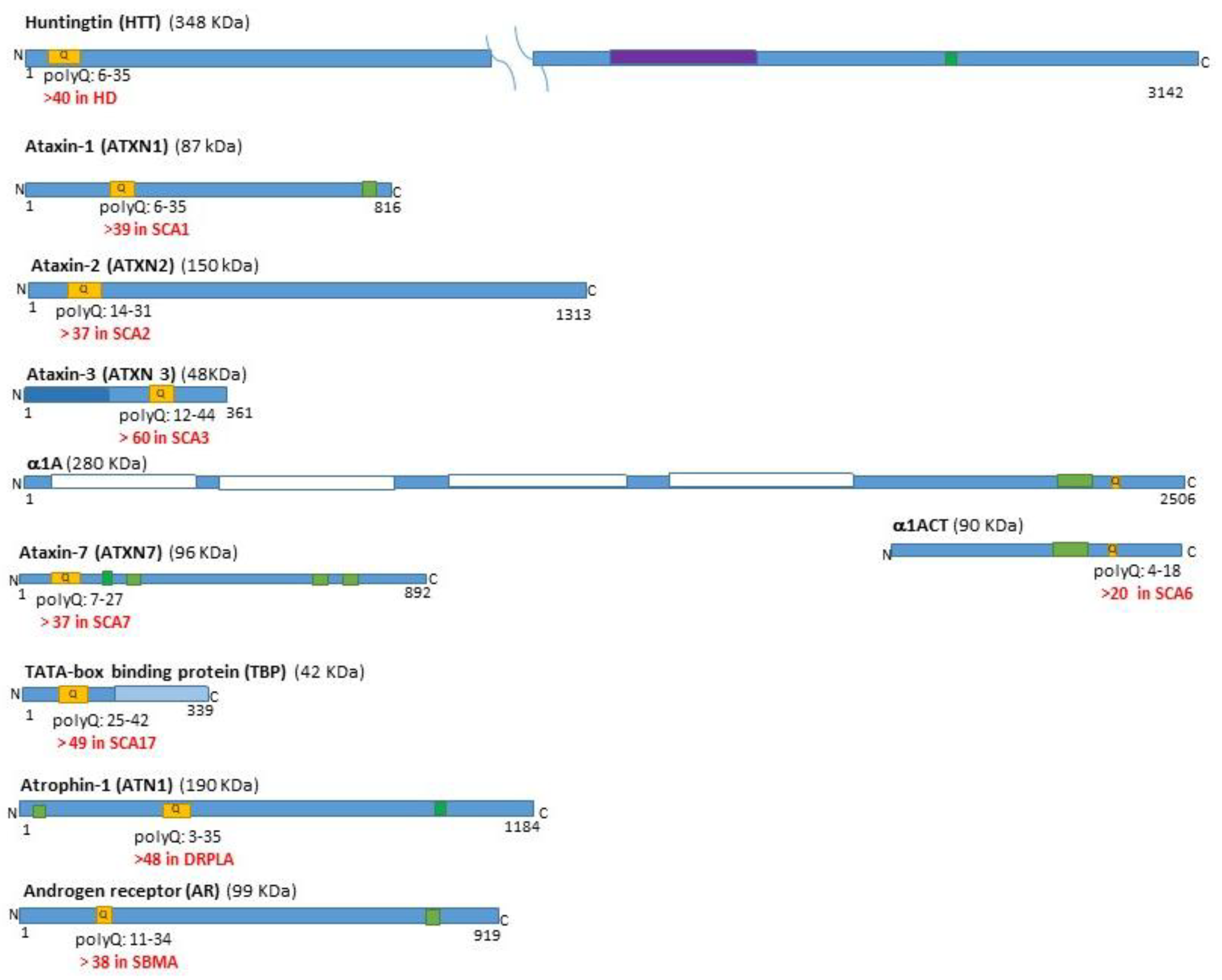
Figure 2.
The main stations and pathways in the human motor coordination network. A. The cortical-basal ganglia and cortical-cerebellar pathways, along with their connecting stations. B. The human cortico-basal ganglia pathways. The striatum (STR) contains neurons that project to the globus pallidus externus (GPe), globus pallidus internus (GPi), substantia nigra pars reticulata (SNr), and pars compacta (SNc). SNr and GPi act as the output regions of the basal ganglia, sending projections to the thalamus. The striatal neurons that project to SNr/GPi express dopamine D1 receptors (D1), while those that project to GPe express dopamine D2 receptors (D2). The dopamine input from the SNc to the striatum is indicated in blue. C. The human cortico-cerebellar pathways. The connections between the cerebral cortex and the cerebellum have several intermediate stations, including the red nuclei (RN), inferior olives (IO), pontine nuclei and thalami. The primary excitatory input from the cerebral cortex arrives at the larger rostral parvocellular part of the red nucleus (pRN) and further to the IO and the cerebellar cortex. The output of the cerebellar cortex is inhibitory and primarily targets the cerebellar deep nuclei (CDN), including the dentate nucleus (DN) and the interposed nucleus (IN). From them, excitatory projections extend to the thalamus and return to the cerebral cortex, as well as to the RN. Excitatory pathways are shown in green, while inhibitory pathways are depicted in red.
Figure 2.
The main stations and pathways in the human motor coordination network. A. The cortical-basal ganglia and cortical-cerebellar pathways, along with their connecting stations. B. The human cortico-basal ganglia pathways. The striatum (STR) contains neurons that project to the globus pallidus externus (GPe), globus pallidus internus (GPi), substantia nigra pars reticulata (SNr), and pars compacta (SNc). SNr and GPi act as the output regions of the basal ganglia, sending projections to the thalamus. The striatal neurons that project to SNr/GPi express dopamine D1 receptors (D1), while those that project to GPe express dopamine D2 receptors (D2). The dopamine input from the SNc to the striatum is indicated in blue. C. The human cortico-cerebellar pathways. The connections between the cerebral cortex and the cerebellum have several intermediate stations, including the red nuclei (RN), inferior olives (IO), pontine nuclei and thalami. The primary excitatory input from the cerebral cortex arrives at the larger rostral parvocellular part of the red nucleus (pRN) and further to the IO and the cerebellar cortex. The output of the cerebellar cortex is inhibitory and primarily targets the cerebellar deep nuclei (CDN), including the dentate nucleus (DN) and the interposed nucleus (IN). From them, excitatory projections extend to the thalamus and return to the cerebral cortex, as well as to the RN. Excitatory pathways are shown in green, while inhibitory pathways are depicted in red.
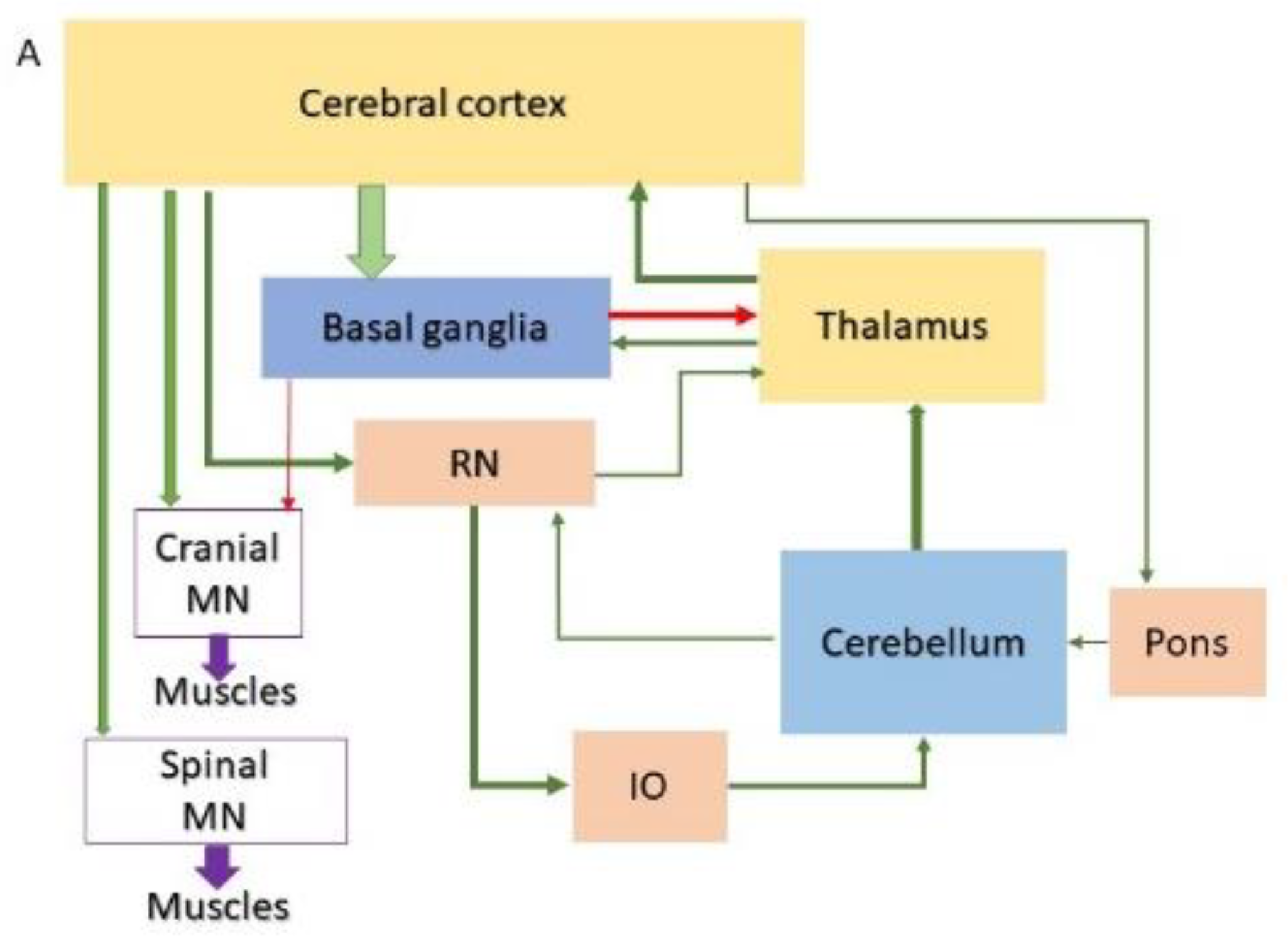
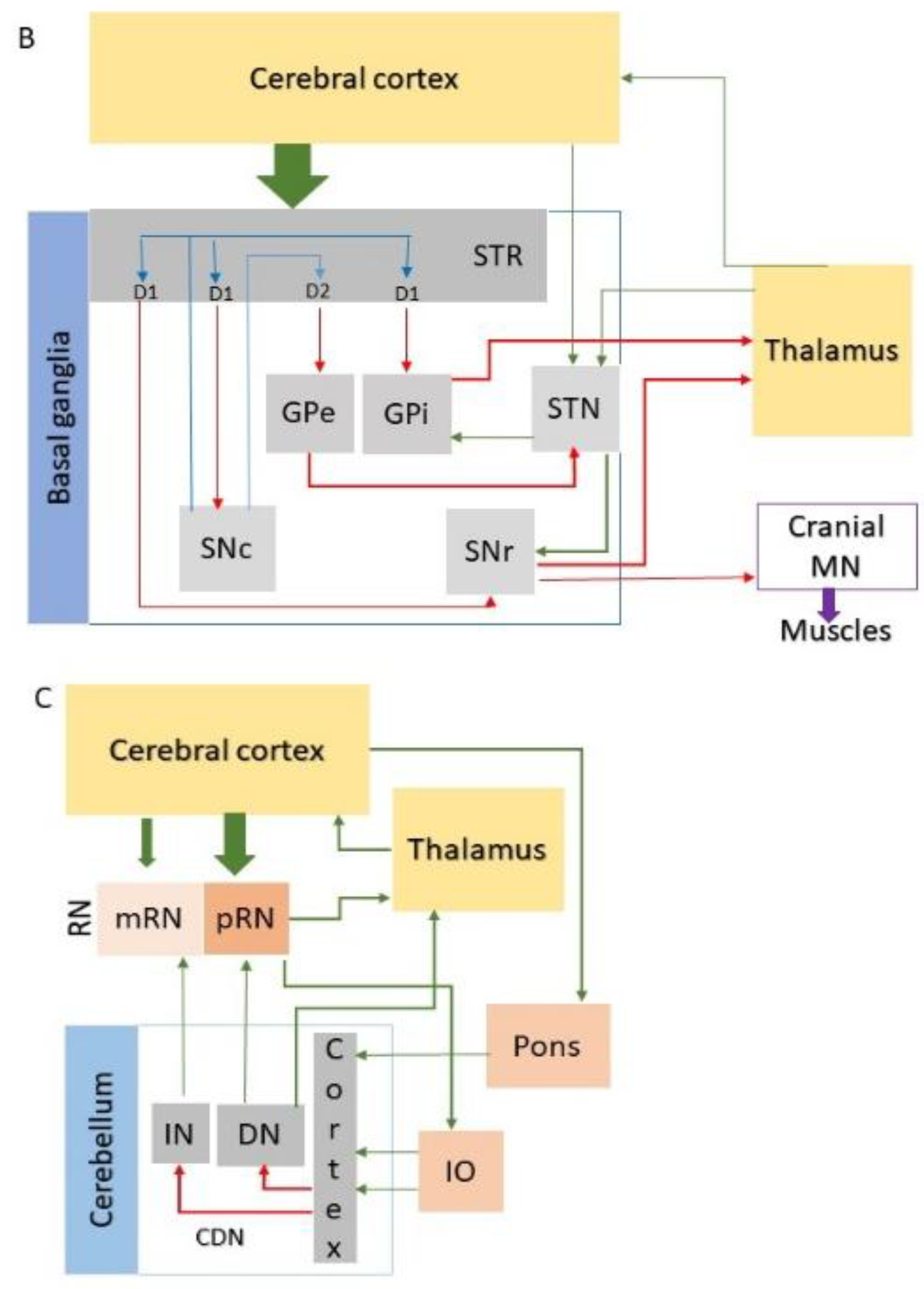

Table 1.
Distribution of degenerated regions in the central nervous system of polyQ disease patients.
Table 1.
Distribution of degenerated regions in the central nervous system of polyQ disease patients.
| Disease | Forebrain | Midbrain | Hindbrain | SC | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTX | TH |
R | Basal ganglia | RN | CN | Pons | Cerebellum | MO | MN | WM | |||||||
| STR | PL | STN | SN | PN | CN | CBC | CDN | IO | CN | ||||||||
| HD | |||||||||||||||||
| SCA1 | |||||||||||||||||
| SCA2 | |||||||||||||||||
| SCA3 | |||||||||||||||||
| SCA6 | |||||||||||||||||
| SCA7 | |||||||||||||||||
| SCA17 | |||||||||||||||||
| DRPLA | |||||||||||||||||
| SBMA | |||||||||||||||||
Affected subdivisions in polyQ diseases include the cerebral cortex (CC), thalamus (TH), retina (R), basal ganglia (divided into striatum (STR), pallidum (PL), subthalamic nucleus (STN), and substantia nigra (SN)), red nucleus (RN), cranial nerve nuclei (CN) in the midbrain, pons, and medulla oblongata (MO), pontine nuclei (PN), the inferior olive (IO), cerebellum (with cerebellar cortex (CBC) and deep nuclei (CDN)), and spinal cord (SC) with motor nuclei (MN) and white matter (WM). Colour intensities indicate the extent of degeneration in affected areas, ranging from severe to moderate and mild. Regions that are relatively spared are not coloured. Huntington’s Disease (HD), Spinocerebellar Ataxia (SCA) types 1, 2, 3, 6, 7, and 17, Dentato-Rubral Pallidoluysian Atrophy (DRPLA), and Spinal and Bulbar Muscular Atrophy (SBMA) [3,5,7].
Table 2.
The expression of polyQ disease-related genes in several human tissues and neural cells.
| Genes | Brain | CNS regions | Neural cell types | H | K | B | Ms | S | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NC | BG | R | TH | MB | CB | P | MO | SC | N | A | O | M | ||||||||
| HTT | ||||||||||||||||||||
| ATXN1 | ||||||||||||||||||||
| ATXN2 | ||||||||||||||||||||
| ATXN3 | ||||||||||||||||||||
| CACNA1A | ||||||||||||||||||||
| ATXN7 | ||||||||||||||||||||
| TBP | ||||||||||||||||||||
| ATN1 | ||||||||||||||||||||
| AR | ||||||||||||||||||||
The relative expression levels of the genes in different tissues and central nervous system (CNS) regions (NC-neocortex, BG-basal ganglia, TH-thalamus, R-retina, MB-midbrain, CB-cerebellum, P-pons, MO-medulla oblongata, SC-spinal cord) are shown as colour intensities, based on median values from Gene Search | HTCA and the Human Protein Atlas (proteinatlas.org); the values for the neural cell types (neurons (N), astrocytes (A), oligodendrocytes (O) and microglia (M)) correspond to the NC; H-heart, K-kidney, B-blood, Ms-striatal muscles, S-skin.
Table 3.
Proposed pathomechanisms due to the nuclear proteins with elongated polyQ traits.
| PolyQ disease | PolyQ protein | GOF | RBP sequestration | LOF | Nuclear inclusions |
|---|---|---|---|---|---|
| HD | HTT | + | + | nd | + |
| SCA1 | ATXN1 | + | nd | + | + |
| SCA2 | ATXN2 | + | + | + | + |
| SCA3 | ATXN3 | + | + | nd | + |
| SCA6 | 1ACT | + | nd | nd | + |
| SCA7 | ATXN7 | + | nd | nd | + |
| SCA17 | TBP | + | nd | nd | + |
| DRPLA | ATN1 | + | + | nd | + |
| SBMA | AR | + | nd | + | + |
An elongated polyQ may disturb the protein function by a gain-of-function (GOF), RNA-binding protein (RBP) sequestration or by a loss-of-function (LOF). Huntington’s Disease (HD), Spinocerebellar Ataxia (SCA) types 1, 2, 3, 6, 7, and 17, Dentato-Rubral Pallidoluysian Atrophy (DRPLA), and Spinal and Bulbar Muscular Atrophy (SBMA); nd-not determined (modified from [49,50,51].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



