Submitted:
18 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
Advancements in genomic technologies have transformed prenatal genetic testing, offering more accurate, comprehensive, and noninvasive approaches to reproductive care. This review provides an in-depth overview of current methodologies and emerging innovations, including expanded carrier screening (ECS), cell-free DNA (cfDNA) testing, chromosomal microarray analysis (CMA), and sequencing-based diagnostics. We highlight how next-generation sequencing (NGS) technologies have revolutionized carrier screening and fetal genome analysis, enabling detection of a broad spectrum of genetic conditions. The clinical implementation of cfDNA has expanded from common aneuploidies to include copy number variants (CNVs), and single-gene disorders. Diagnostic testing has similarly evolved, with genome sequencing outperforming traditional CMA and exome sequencing through its ability to detect both sequence and structural variants in a single assay. Emerging tools such as optical genome mapping, RNA sequencing, and long-read sequencing further enhance diagnostic yield and variant interpretation. This review summarizes major technological advancements, assesses their clinical utility and limitations, and outlines future directions in prenatal genomics.
Keywords:
Prenatal genetic testing
; sequencing
; cfDNA
; genomics
1. Introduction
Over the past decade, rapid advancements in prenatal genetics have transformed the landscape of reproductive care, largely driven by high-throughput next-generation sequencing (NGS). This technology has revolutionized genomic screening and diagnostics by enabling parallel sequencing of multiple genomic regions [1]. Several key assays now form the foundation of modern prenatal screening and diagnosis. Expanded carrier screening (ECS) enables pan-ethnic detection of a broad range of autosomal recessive and X-linked conditions in prospective parents [2]. Cell-free DNA (cfDNA) screening noninvasively assesses fetal risk for common aneuploidies, and with advancing technology, has expanded to include sex chromosome aneuploidy (SCA)[3], rare autosomal trisomies (RATs)[4], both common and rare copy number variants (CNVs)[5], and single-gene disorders [6]. Among diagnostic tests performed prenatally, chromosomal microarray analysis (CMA) which detects aneuploidy, and CNVs, including submicroscopic CNVs not visible through conventional karyotyping, is the recommended diagnostic test for pregnancies complicated by fetal anomalies [7]. More recently, low-pass genome sequencing using NGS has been shown to detect CNVs with broader coverage compared to traditional CMA in the prenatal setting [8]. Additionally, NGS-based tests can target a selected panel of genes, the exome, or the entire genome at nucleotide level resolution, enabling the detection of a wide array of genetic conditions during pregnancy [9]. Genetic variants are classified as either pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign [10]. Based on American College of Medical Genetics/Association for Molecular Pathology (ACMG/AMP) guidelines, laboratories are recommended to report only pathogenic and likely pathogenic variants; however, variants of uncertain significance (VUS) may be reported in certain clinical contexts [10]. In parallel with these technologies, emerging methods such as optical genome mapping, RNA sequencing, and long-read sequencing are beginning to expand diagnostic possibilities and, along with advancements in bioinformatics, can enhance variant interpretation. These emerging tools, while not yet routine, mark the next frontier in prenatal genomics. This review outlines the evolution of key assays, recent technological advances, and their clinical utility and limitations in prenatal screening and diagnosis.
2. Expanded Carrier Screening
2.1. Overview of Carrier Screening
Carrier screening identifies usually asymptomatic, individuals and their partners who carry pathogenic variants for autosomal recessive or X-linked conditions. While ideally performed preconceptionally, it is often offered in early pregnancy as part of routine obstetric care to assess the risk of having an affected offspring [11,12]. Historically, carrier screening was limited to conditions with high prevalence in specific populations, such as Tay-Sachs disease in Ashkenazi Jewish communities and β-thalassemia in Mediterranean populations [12]. The discovery of the CFTR gene prompted widespread cystic fibrosis screening now recommended pan-ethnically. Spinal Muscular Atrophy (SMA) screening followed a similar path [13]. Traditional criteria for screening included severe phenotype, high carrier frequency, reliable testing, clear genotype–phenotype correlation, and access to prenatal diagnosis and reproductive choices [2,11].
2.2. Technological Evolution, Challenges and Limitations
How we approach genetic screening has been reshaped by the rapidly advancing technology used for DNA sequencing, allowing for high throughput with rapid turnaround times and lower costs of genome sequencing [14]. Traditional carrier screening relied on simpler methods, including polymerase-chain-reaction-based techniques, multiplex ligation-dependent probe amplification, microarrays, Sanger sequencing, and genotyping. NGS now enables simultaneous screening of large number of genes in carrier screening panels covering tens to hundreds of genes. In practice, panels often incorporate additional methods, such as copy number analysis for genes where copy number changes are a significant cause of disease, with assay selection tailored to each gene or variant based on feasibility, reliability, and cost [15].
Although NGS enables carrier screening for many recessive or X-linked diseases simultaneously, developing a multigene panel that meets carrier screening criteria, along with known positive and negative predictive values for each can be challenging [16]. Each test should be thoroughly validated to define its analytical sensitivity, specificity, and accuracy, and establish confidence in the detection, analysis and reporting of genetic variants [17]. Additionally, certain clinically significant genes present technical challenges for NGS analysis due to presence of pseudogenes (for example GBA for Gaucher disease), repeat expansions (FMR1 for Fragile X Syndrome), or DNA structural variations [18].
2.3. Guidelines and Ethical Considerations
The American College of Obstetricians and Gynecologists (ACOG) recommends universal screening for cystic fibrosis and SMA, and hemoglobinopathies via complete blood count, with additional screening based on ethnicity or family history (e.g., Tay-Sachs disease and fragile X syndrome). ECS for conditions with a population carrier frequency of ≥1/100 is considered acceptable, though providers are advised to standardize their approach for each patient [11]. In contrast, ACMG recommends pan-ethnic carrier screening to promote equity and proposed a tier-based classification for carrier screening [2]. Tier 1 aligns with ACOG’s recommendations; Tier 2 includes conditions with a carrier frequency ≥1/100; Tier 3 includes conditions with a frequency ≥1/200 (including some X-linked conditions); and Tier 4 covers rare conditions without frequency thresholds [11,19,20]. ACMG recommends Tier 3 screening for all individuals and Tier 4 for those with higher-risk factors such as family history or consanguinity [2]. Despite these guidelines, ECS panel content varies across laboratories [21]. Increasing multiethnicity and population admixture, inaccuracies in self-reported ethnicity, and growing emphasis on patient autonomy all support using larger pan-ethnic ECS panels [12,22,23,24]. However, larger panels may yield uncertain results, increase parental anxiety, and lead to additional testing and costs [25]. Furthermore, accurate residual risk counseling remains challenging due to limited data on disease-specific carrier frequencies across diverse populations. These factors highlight the importance of clear, well-documented counseling and informed consent before carrier screening [26].
2.4. Carrier Screening by Genome Sequencing
Data on genome sequencing for carrier screening are still limited, but improved detection rates for certain conditions compared to traditional panels have been reported [27]. In one study 202 individuals underwent preconception carrier screening using genome sequencing, but result reporting was limited to a curated list of 728 single gene disorders. Although the test relied on genome sequencing, reporting was constrained to the predefined panel, limiting its scope and making it not a true application of comprehensive genome sequencing. Nonetheless, this approach identified 304 clinically significant variants demonstrating improved sensitivity over traditional panels [28]. A separate study used duo exome sequencing of parents after fetal or embryonic loss to identify shared carrier status for pathogenic variants, offering “molecular autopsy by proxy” when fetal DNA was unavailable. This approach enabled diagnosis of autosomal recessive conditions underlying the pregnancy loss and mimics the concept of genome-wide carrier screening. While both studies highlight potential applications of broad sequencing, the clinical utility of genome sequencing for routine carrier screening remains to be fully explored [29].
3. Cell-Free DNA
3.1. Overview of cfDNA Screening for Aneuploidies and Copy Number Variants
Prenatal screening by cfDNA analysis for the detection of aneuploidies became clinically available in 2011 and has been rapidly introduced into prenatal care to detect common aneuploidies (chromosomes 13, 18, and 21) [30,31,32]. Circulating fetal cfDNA in maternal plasma was initially described in 1997 [33], and was shown to be trophoblast derived . The fetal fraction, describes the proportion of all cfDNA circulating in maternal blood that is of placental (trophoblast) origin, comprises about 3-13% of total cfDNA, and increases throughout gestation [34,35]. Multiple studies have consistently shown the superior performance of prenatal cfDNA screening for common aneuploidies compared to traditional maternal serum screening, and it has been endorsed by professional societies for that use [3,5,32].
cfDNA screening for aneuploidy uses two NGS-based approaches. In the first, massively parallel sequencing, randomly sequences cfDNA from maternal plasma, map reads to the reference genome, and uses counting statistics to detect chromosomal imbalances [30]. The second, targeted sequencing, focuses on single nucleotide polymorphism (SNP) rich regions using capture probes or multiplex PCR, with statistical analysis of read counts and allele distributions to identify chromosomal abnormalities [36,37]. With advancements in cfDNA screening, the test has also been validated for use in twin pregnancies, with detection rates for trisomy 21 approaching those seen in singletons, while test performance for trisomies 18 and 13 is slightly lower. cfDNA screening is also available for higher order multiples [5,38]. Additionally, analysis of SNPs and fetal-specific alleles can determine zygosity in twin pregnancies [39]. Circulating cfDNA from a vanishing twin can persist for weeks and confound screening results due to the persistence of cfDNA from a demised fetus [40].
Non-invasive fetal CNV screening from cfDNA in maternal blood typically follows one of two approaches: targeted analysis of known microdeletion/duplication syndromes or broader genome-wide screening. In the United States, genome-wide CNV screening offered by one commercial provider is designed to detect CNVs larger than 7 megabases (Mb), but includes selected known clinically relevant microdeletions smaller than 7 Mb [5], but many other pathogenic CNVs also fall below this size threshold [7]. Test performance of cfDNA screening depends on several factors, including the prevalence of chromosomal abnormality, presence of maternal CNVs, placental mosaicism, fetal fraction, sequencing depth, and CNV size [4,5,41]. Most individual CNVs are rare, resulting in lower sensitivity and specificity of cfDNA screening compared to common trisomies and SCAs [3]. Among pathogenic CNVs, 22q11.2 deletion syndrome (22q11.2DS), is the most common one identified prenatally [42], resulting in better performance metrics for this condition. A recent study of 18,289 pregnancies assessed SNP-based cfDNA screening for 22q11.2DS and detected 10 of 12 confirmed cases using an updated algorithm. With a 1-in-100 risk cutoff, the false positive rate was 0.05%, and the positive predictive value (PPV) was 52.6% [43]. Currently, there is insufficient evidence to support routine screening for CNVs other than 22q11.2DS [5].
Genome-wide CNV screening with cfDNA analysis can detect RATs which are trisomies involving chromosomes other than 13, 18, 21, X, or Y. Liveborn infants with non-mosaic RATs are extremely rare, and when detected prenatally, RATs are most often mosaic[5] . A recent meta-analysis of 31 studies reviewed results from 1,703 high-risk cfDNA screening results for RATs and reported a pooled PPV of 11.46% [44]. Recent studies have shown that RATs, when present as confined placental mosaicism (CPM), are associated with adverse pregnancy outcomes, including fetal growth restriction, low birth weight, and preeclampsia, emphasizing the need for optimal antenatal surveillance and counseling in pregnancies when RATs are detected by cfDNA testing [45,46]. Despite these technical capabilities, evidence regarding the clinical validity of genome-wide CNV or aneuploidy screening by cfDNA remains limited [45], and its use in pregnant individuals has been subject of scientific debate [47]. It is currently not recommended by professional societies in the United States [5,32]. cfDNA screening has expanded beyond common autosomal trisomies to routinely include SCAs. Given the unique clinical complexities of SCA, including variable phenotypic expression, mosaicism, and lower PPV, several reports have underscored the importance of thorough pre- and post-test counseling and need for confirmatory testing [5,48].
3.2. Maternal cfDNA Testing and Screening for Fetal Single Gene Disorders
Single-gene disorders (SGD) affect approximately 1% of births [49]. cfDNA testing has been developed for the non-invasive prenatal diagnosis of SGD, most commonly through targeted assays for known familial or suspected variants, encompassing both autosomal dominant and recessive conditions. Additionally, cfDNA-based screening panels have been primarily designed to detect dominant, often de novo, pathogenic variants associated with severe, early-onset phenotypes [50]. More recently, genome-wide non-invasive fetal sequencing approaches have been explored for the comprehensive detection of monogenic disorders [51]. It is important to distinguish between: (1) non-invasive prenatal diagnosis (NIPD) by cfDNA analysis for SGD diagnosis, targeted for known variants; (2) cfDNA analysis for SGD screening, primarily for dominant conditions [50,51].
In one study that evaluated 422 cases using the cfDNA-SGD assay that targets 30 genes associated with dominant SGD, there were 20 true-positive and 127 true-negative among 147 cases with available confirmatory results. Notably, no false-positive or false-negative results were identified in this cohort [52]. In a separate study of 2,208 women with high-risk pregnancies, cfDNA-based SGD testing with the same panel of 30 genes, 125 (5.7%) tested positive. The highest detection rates were observed for pregnancies referred for fetal long-bone abnormalities (33.7%), craniofacial malformations (28.6%), and a family history of a disorder included in the panel (15.2%). Among those with confirmatory diagnostic testing, no false-positive or false-negative results were reported [6]. In a study of 2,745 women with low-risk pregnancies screened with this cfDNA-based SGD assay, 14 (0.51%) had high-risk results. While no false positives were observed, there were discrepancies in variant classification between cfDNA results and diagnostic testing for two of them. The study highlighted both the potential for early detection and the counseling challenges posed by variable expressivity, uncertain genotype–phenotype correlations, and interpretive differences [53]. While these findings are promising, broader adoption will require population-based validation, equitable access, and critical assessment of ethical, economic and policy-related challenges. A comprehensive framework, encompassing rigorous test design, standardized counseling, and informed consent is essential to support responsible implementation and improve clinical outcomes [50].
3.3. Developments in cfDNA Analysis
cfDNA tests for non-invasive fetal Rhesus (Rh) genotyping have been introduced into obstetric care in different countries. In the U.S., one multicenter cohort study evaluated the accuracy of cfDNA-based fetal red blood cell antigen genotyping in 156 alloimmunized pregnancies. Testing between 10–37 weeks’ gestation showed complete concordance with postnatal neonatal genotyping across 465 antigen calls, yielding 100% sensitivity, specificity, and accuracy. These findings support the use of cfDNA testing from 10 weeks’ gestation to guide management and reduce unnecessary interventions [54]. The evidence base for cfDNA-based Rh testing is extensive and has a long-standing track record in European clinical practice prior to its introduction in the United States. Broader adoption also enables targeted Rh immunoglobulin administration by identifying RhD-negative pregnancies carrying RhD-positive fetuses [55].
In a recent report, one U.S. commercial laboratory developed and evaluated the effectiveness of a maternal carrier screening test for cystic fibrosis, hemoglobinopathies, and spinal muscular atrophy followed by cfDNA-based single-gene testing when the mother is identified as a carrier for one of these conditions [56]. This strategy can provide a fetal risk assessment for these recessive conditions without the requirement for paternal carrier screening. This facilitates prenatal counseling and management, while addressing logistical challenges of obtaining paternal carrier status [56]. They reported a sensitivity of 96%, specificity of 95.2%, positive predictive value of 50.0% and negative predictive value of 99.8% in a study of 42,067 individuals [57]. However, the number of genes included in this carrier screening with cfDNA analysis approach is well below the ACMG recommended tier 3 panel.
Fragmentomics refers to the analysis of cfDNA fragmentation patterns. Fetal (placental) cfDNA fragments are typically shorter than maternal cfDNA, and this size difference has been leveraged to estimate fetal fraction and detect fetal aneuploidies [58,59]. In cases of fetal trisomy, the affected chromosome contributes to a shift in the fragment length distribution, depending on whether there is a chromosomal gain or loss. Integrating size-based and count-based analytical approaches improves the precision of cfDNA screening by aiding in the interpretation of CNVs [60]. Current NGS-based cfDNA screening typically requires a fetal fraction greater than 4%. Fetal fraction amplification, which enriches placental cfDNA in maternal plasma based on fragment size, can help overcome maternal DNA background interference [61]. This technique may be especially useful in cases where fetal fraction is reduced, such as with high maternal body mass index, or at earlier gestational ages. Studies have demonstrated that fetal fraction amplification improves the analytical performance of cfDNA, reduces test failures due to low fetal fraction, enables higher-resolution detection of fetal chromosomal abnormalities and CNVs, and enables cfDNA screening at an earlier gestational age [62,63].
A newer promising development is genome-wide non-invasive fetal sequencing (NIFS) on maternal plasma cfDNA [50,51]. Two proof-of-concept studies demonstrated the feasibility of exome-based noninvasive prenatal testing using cfDNA for detecting single nucleotide variants (SNVs), indels, and select CNVs. Sensitivity was highest for de novo and paternally inherited variants, with reduced accuracy at low fetal fractions and for maternally inherited alleles. Trio-based sequencing achieved 100% concordance with invasive testing in high-risk pregnancies. These findings support the potential future expansion of NIFS to detect monogenic disorders, but also underscore the need for confirmatory testing of any detected variants, larger validation studies, and technical and cost improvements [64,65].
There are instances when cfDNA screening can yield unexpected results. Retrospective studies from large commercial and national laboratories have associated unusual sequencing patterns, such as multiple aneuploidies or autosomal monosomies, with underlying maternal malignancy [66,67]. When a tumor is present, it may shed cfDNA into maternal plasma, contributing to the total cfDNA pool and distorting expected chromosomal ratios, thereby complicating interpretation. Such discordant results often lead to a nonreportable outcome, as fetal aneuploidy status cannot be confidently determined. A recent study of 107 pregnant individuals with unusual or nonreportable cfDNA results found that 52 (48.6%) were subsequently diagnosed with occult cancer. Among 49 participants whose sequencing showed copy number alterations across three or more chromosomes, 47 (95.9%) had a confirmed malignancy, which was best detected by whole body MRI [68]. As cfDNA screening expands in scope, understanding its limitations and incidental findings becomes increasingly important.
4. Chromosomal Microarray and Copy Number Variant Detection
CMA is the recommended test for patients undergoing prenatal diagnostic testing when one or more major fetal structural abnormalities are identified by ultrasonography or pregnancies with an intrauterine fetal demise or stillbirth [69]. CMA detects clinically significant deletions and duplications, can reveal regions of homozygosity (ROH), which represent copy-neutral loss of heterozygosity (CN-LOH), and may suggest uniparental disomy or parental consanguinity.
Among samples with a non-diagnostic karyotype, CMA identifies clinically significant deletions or duplications in 6.0% of fetuses with structural anomalies with higher yields when there are multiple anomalies, and in 1.7% of fetuses where testing was done for advanced maternal age or positive screening results [7,70]. The interpretation of VUS remains a challenge, potentially complicating genetic counseling and decision-making during pregnancy. CMA has additional limitations: it cannot detect balanced chromosomal rearrangements, such as balanced translocations or inversions, has limited sensitivity for low-level mosaicism (<10-20%), and cannot reliably detect triploidy unless SNP probes are included [69]. Furthermore, CMA cannot reveal the underlying mechanism of a chromosomal imbalance, such as trisomies. These still require conventional karyotyping to determine if the imbalance resulted from meiotic non-disjunction or an unbalanced Robertsonian translocation, which is critical for assessing recurrence risk in future pregnancies [7,69]. CMA also does not detect SNVs, small indels, and small CNVs below platform resolution [69]. The general consensus for the reportable size threshold in prenatal CMA is 200 to 400 Kb, with some suggesting even more conservative cutoffs (e.g., 500 Kb for deletions and 1 Mb for duplications) to reduce the detection of VUS that could result in associated parental anxiety [71]. A 2023 study using high-resolution CMA platforms found that reducing the size threshold to 20 Kb can uncover pathogenic CNVs below the standard reporting cutoff, while keeping VUS low at 4%, supporting that higher-resolution platforms can improve diagnostic yield without significantly increasing uncertainty [72]. Given the rapidly increasing number of gene–disease associations and continuous updates in genetic databases, adopting higher-resolution CMA testing in prenatal diagnostics is essential for accurately identifying gene-level CNVs.
Although CMA is widely used for prenatal CNV detection, it is limited by a preset probe design, and a workflow that does not readily incorporate new gene-disease associations. As more laboratories adopt NGS, low-pass genome sequencing (LP-GS) has emerged as a promising alternative, offering flexible CNV detection with reported coverage in prenatal studies ranging from 0.25x to 5x [8,73,74,75]. Although the fundamental principles and clinical utility of LP-GS have been established, broader adoption may require further validation and endorsement by professional guidelines. In the long run, LP-GS holds potential as a more cost-effective and efficient diagnostic tool, particularly when integrated into a comprehensive sequencing strategy.
5. Diagnostic Sequencing
Since the advent of NGS, prenatal genetic diagnostics have undergone significant transformation. NGS-based tests can target a selected panel of genes, the exome, or the entire genome at a nucleotide level, enabling the detection of a wide array of genetic conditions during pregnancy.
5.1. Targeted Gene Panels
Targeted gene panels focus on sequencing specific sets of genes associated with particular phenotypes or conditions, such as a skeletal dysplasia [76] and RASopathies [77]. However, their diagnostic utility can be limited by gene content. In a cohort of 127 NIHF cases, exome sequencing (ES) identified pathogenic variants in 29% of fetuses, while targeted panels would have captured only 11% to 62% of these variants, depending on the panel composition [78]. In addition, selecting an appropriate panel testing strategy heavily depends on ultrasound findings and the suspected diagnosis. This becomes particularly challenging when prenatal phenotypes are non-specific, such as ventriculomegaly, fetal growth restriction or oligohydramnios, which can result from a wide range of genetic and non-genetic causes. Therefore, non-targeted approaches such as ES and genome sequencing (GS) may be more suitable in such cases.
5.2. Exome Sequencing
ES is an increasingly utilized tool in prenatal diagnostics, particularly for fetuses with ultrasound anomalies and non-diagnostic karyotype and CMA results. ES targets the coding exons and exon-intron boundaries, typically covering up to ±20 base pairs into the intronic regions to capture potential splice-site variants. These cover approximately 1% to 2% of the genome but contain most known disease-causing variants [79]. ES is typically performed using a capture-based enrichment method followed by high-throughput sequencing, achieving an average sequencing depth of approximately 100x to ensure reliable variant detection.
Several studies have evaluated the diagnostic yield of ES in prenatal settings, demonstrating wide variability based on fetal phenotype and selection criteria [9,80,81,82]. Higher yields have been reported in fetuses with multisystem anomalies or in specific organ system involvement, such as skeletal dysplasias [9,81], compared to those with isolated findings [80,82]. Two of the largest prospective studies by Lord et al. [83] and Petrovski et al. [84] evaluated the clinical utility of trio-based prenatal ES in fetuses with structural anomalies detected by ultrasound and non-diagnostic karyotype and CMA, reporting diagnostic yields of 8.5% and 10.3%, respectively. Both studies underscore that yield varies by the type of anomaly. A systematic review and meta-analysis further support these findings, with an overall diagnostic yield of 31%, increasing to 42% in cases with a suspected monogenic etiology, compared to 15% in unselected cases [9]. Consistent with these findings, the International Society for Prenatal Diagnosis (ISPD) recommends genome-wide (exome or genome) sequencing when karyotype and CMA are non-diagnostic, especially in fetuses with multiple anomalies or have a suspected monogenic condition [85].
5.3. Genome Sequencing
GS extends beyond the exome to include most non-coding regions, offering a more comprehensive view of the genome. This broader scope allows for the detection of variants that may be missed by ES, including deep intronic variants, noncoding regulatory variants, and copy number changes. GS is typically performed at an average depth of 30-40x and does not rely on capture-based enrichment, reducing bias and improving uniformity across the genome. GS offers a comprehensive diagnostic approach by enabling the simultaneous detection of SNVs and CNVs at a high resolution, down to the exonic level of coding genes. Unlike traditional sequential testing workflows, in which CMA is followed by ES if negative, GS can consolidate the capabilities of both into a single, streamlined assay, but this has yet to be fully validated on prenatal clinical samples. Beyond SNV and CNV detection, GS can also detect regions of homozygosity (ROH) and uniparental disomy (UPD) when parental samples are available, which is crucial for diagnosing recessive conditions and imprinting disorders. Furthermore, GS has the added advantage of detecting tandem repeat expansions and mitochondrial DNA abnormalities, both of which are typically missed by ES and CMA [86].
Prenatal GS for fetal anomalies has a comparable diagnostic yield to the combined performance of CMA and ES [87]. A recent study by Westenius et al. reported that clinical prenatal trio GS detected a causative variant in 26% (13/50) of fetuses with congenital malformations after non-diagnostic testing for CNVs and aneuploidy [88]. It is important to note that the analyses in these GS studies primarily focused on detecting SNVs and CNVs. As such, the reported diagnostic yield may underestimate the full potential of GS, since repeat expansions, mitochondrial DNA abnormalities, and structural variants were not systematically evaluated in these prenatal GS studies.
Although GS has clear technical advantages, its widespread adoption is currently limited by less mature bioinformatics pipelines, particularly for repeat detection due to the limitations of short-read sequencing. Additionally, the higher costs of sequencing, data storage and interpretation remain significant challenges for widespread adoption in laboratories. As sequencing costs continue to decline, GS is expected to become more widely used in prenatal diagnostics and may eventually replace existing testing strategies, serving as a single comprehensive platform for detecting both chromosomal and single-gene disorders.
5.4. Considerations for Data Interpretation in Prenatal Sequencing
The interpretation of variants detected on prenatal ES/GS can be challenging because the prenatal-onset phenotypes of many disorders are poorly characterized, and the phenotype information available from fetal imaging is often limited [89]. To address this gap, the Human Phenotype Ontology consortium has actively collaborated with an international network of fetal medicine experts to expand and refine ontology terms specific to prenatal development. This global initiative significantly enhances the capability to systematically capture, analyze, and interpret prenatal phenotypic data [90]. Additionally, broader data sharing through initiatives like the Fetal Sequencing Consortium is critical to improve our understanding of both causative and incidental findings in fetal ES and GS [90,91,92].
To improve diagnostic yield, trio-based sequencing is often preferred to singleton because it enables identification of de novo variants, determination of inheritance patterns, and improved interpretation of variant penetrance [9]. Trio analysis also significantly reduces the number of VUS identified by ES, decreasing from 15% in proband-only analysis to 6% with trio analysis. Most guidelines, including ACMG, the ISPD, and the Canadian College of Medical Geneticists (CCMG), recommend reporting only VUS that are strongly associated with the fetal phenotype, typically after consensus is reached through multidisciplinary team review [74,93]. However, criteria and practice for reporting VUS can vary widely between different laboratories. ES/GS might generate incidental or secondary findings. Secondary findings are pathogenic or likely pathogenic variants in a defined list of medically actionable genes (e.g., those linked to hereditary cancer or cardiovascular disease), where early intervention can reduce morbidity and mortality [94]. Incidental findings also involve pathogenic variants unrelated to the testing indication but includes genes that fall outside the secondary findings list. They can involve genes associated with childhood conditions without a (distinct) prenatal phenotype as well as late-onset conditions such as neurological or neuromuscular disorders. As outlined in the ISPD position statement [85], these findings can raise important ethical considerations in the prenatal context. These highlight the importance of comprehensive genetic counseling both before and after testing to support informed decision-making and appropriate result disclosure.
Reanalysis of fetal ES and GS may be considered in cases with only VUS reported or with nondiagnostic or negative results, especially when new phenotypic features are observed after birth. However, it is often constrained by challenges related to cost, logistics, and the timing of reanalysis.
6. Emerging and Future Technologies
6.1. Optical Genome Mapping
Optical Genome Mapping (OGM) is a new cytogenetics tool that labels ultra-high molecular weight (UHMW) DNA molecules and analyzes their pattern to detect copy number and structural variants across the genome [95]. OGM achieves average read lengths exceeding 200 Kb, allowing for detection of CNVs, balanced translocations, inversions, and complex chromosomal rearrangements as small as 500 bp . This technology has been clinically validated for detecting prenatal chromosomal aberrations, with a high sensitivity (99.5%) and specificity (100%), closely approximating that of conventional methods including karyotyping, CMA, and FISH [96]. OGM can also screen for Fragile X Syndrome (FXS) expansions using the EnFocus Fragile X analysis pipeline, which can detect full CGG repeat expansions, holding promise for future expanded screening. It has been proposed that by consolidating multiple cytogenomic analyses into a single assay, OGM could streamline prenatal diagnostic workflows [96], but this has not yet been validated in large studies. The requirement for UHMW DNA precludes using OGM on DNA extracted by commonly used protocols in clinical laboratories.
6.2. RNA Sequencing
RNA sequencing (RNA-seq) is also gaining traction as a functional prenatal diagnostic tool that can complement DNA sequencing to resolve VUS by detecting aberrant splicing, expression outliers, and monoallelic expression. It holds future potential for transcriptome-driven diagnosis, especially when DNA sequencing alone yields inconclusive results [97]. Such a reflex RNA-seq approach can clarify the functional impact of splicing variants, with approximately one-third of suspected cases demonstrating aberrant splicing [98,99], but translating it to the prenatal setting is challenging because of the added time required to obtain interpretable results and current limited knowledge about gene expression and splicing patterns in amniocytes or chorionic villi. Further development and validation before routine integration into prenatal diagnostics will be needed.
6.3. Long-Read Sequencing
Another emerging technology is long-read sequencing, which depending on the sequencing platform used, can achieve read lengths ranging from 10–15 Kb to as high as 50–100 Kb, substantially longer than the standard short reads of a few hundred nucleotides [100]. This enables detection of complex structural variants, tandem repeat expansions, and methylation patterns at kilobase resolution. Long-read sequencing can also distinguish highly homologous genes from their pseudogenes and detect inversions, including those involving genes frequently included in carrier screening, such as CYP21A2, SMN1/SMN2, and F8 that present challenges for conventional sequencing methods [101,102,103,104]. In addition, long read technology enables the detection and characterization of long cfDNA fragments (>600 bp) and has potential to enhance resolution for fragmentomics [105]. Specifically, the analysis of long cfDNA fragments shows promise for early detection of preeclampsia through size-based classifiers and offers potential for assessing fetal inheritance patterns using methylation markers [106]. However, long cfDNA fragments make up less than 10% of total cfDNA in maternal plasma, while short fragments account for over 90%, which presents analytical challenges but efforts to enrich these underrepresented fragments are ongoing [107].
6.4. Cell-Based Noninvasive Prenatal Testing
Despite developments in cfDNA analysis, cell-based noninvasive prenatal testing (cbNIPT) using intact fetal cells, primarily extravillous trophoblasts (EVTs), or fetal nucleated red blood cells (fNRBCs), isolated from maternal blood for genetic analysis is actively investigated. The principal advantage of cbNIPT is its ability to analyze pure fetal DNA, free from maternal DNA admixture, which is not possible with cfDNA analysis [108,109,110]. Extravillous trophoblasts (EVTs) can also be retrieved from the cervix using Pap smear or cytobrush techniques and has been investigated for its diagnostic potential [111,112]. Immunoaffinity and microchip-based technologies, including antibody- and aptamer-mediated enrichment, are the leading methods for isolating trophoblasts and fNRBCs, with microfluidic platforms showing promise for clinical scalability [113,114]. Key technical challenges across all these fetal cell types include optimizing cell recovery, minimizing maternal cell contamination, and addressing issues such as allelic dropout during single-cell genomic analysis, which can impact diagnostic accuracy. Overall, cbNIPT holds clinical potential for early, noninvasive, and comprehensive prenatal genetic diagnosis, but further technological refinement and validation are required before widespread clinical implementation [108,115].
7. Conclusions
Rapid developments in prenatal genomic technologies are reshaping the landscape of prenatal and reproductive testing and screening by enhancing the ability to identify a wide array of genetic conditions during pregnancy. Methods such as prenatal cfDNA-based testing and screening, CMA, and exome sequencing are now well-established. Genome sequencing is increases and holds the promise of a streamlined and more comprehensive prenatal diagnostic alternative. Innovations like optical genome mapping, RNA-seq, and long-read sequencing promise to address remaining gaps in variant interpretation and detection of complex genomic abnormalities. As these tools advance toward broader clinical use, challenges in areas such as variant interpretation, equitable access, counseling frameworks, and ethical considerations will need to be addressed. Future integration of emerging technologies will require careful validation, guideline development, and robust bioinformatics support to translate their potential into routine prenatal care.
Figure 1.
Timeline and Current Assays Used for Prenatal Genetic Testing. Schematic illustrating the timing and range of genetic testing options from preconception through pregnancy. Expanded carrier screening (yellow bar) is typically offered preconception or any time in pregnancy. Cell-free DNA (cfDNA) can be offered from 10 weeks’ gestation onwards, while diagnostic procedures such as chorionic villus sampling (CVS) and amniocentesis (yellow boxes) are available later. Examples of downstream laboratory methods include targeted gene panels, exome sequencing, and genome sequencing (left), as well as chromosomal microarray (CMA) analysis (right). CMA plot from de-identified patient tested through Baylor Genetics.
Figure 1.
Timeline and Current Assays Used for Prenatal Genetic Testing. Schematic illustrating the timing and range of genetic testing options from preconception through pregnancy. Expanded carrier screening (yellow bar) is typically offered preconception or any time in pregnancy. Cell-free DNA (cfDNA) can be offered from 10 weeks’ gestation onwards, while diagnostic procedures such as chorionic villus sampling (CVS) and amniocentesis (yellow boxes) are available later. Examples of downstream laboratory methods include targeted gene panels, exome sequencing, and genome sequencing (left), as well as chromosomal microarray (CMA) analysis (right). CMA plot from de-identified patient tested through Baylor Genetics.
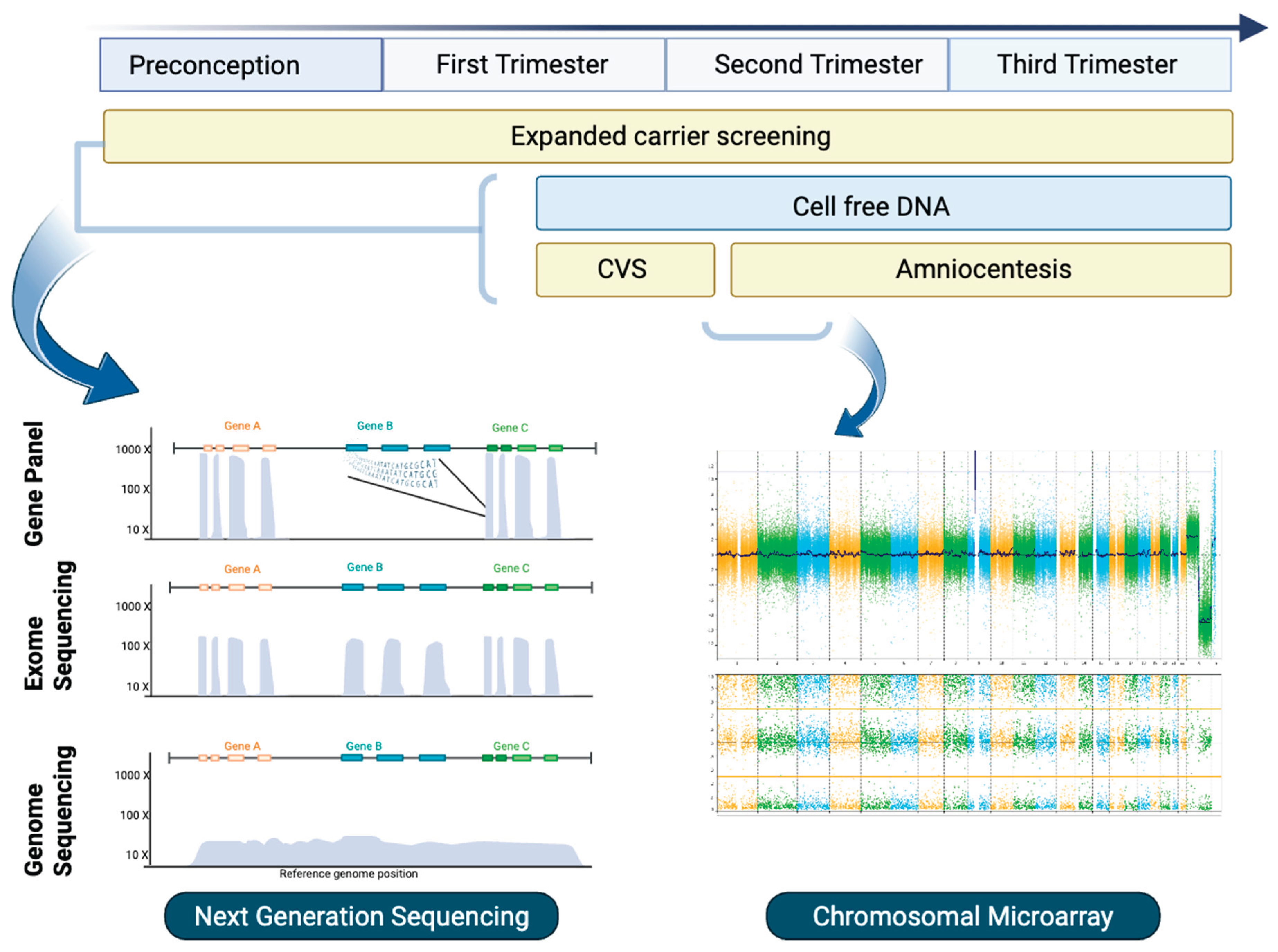
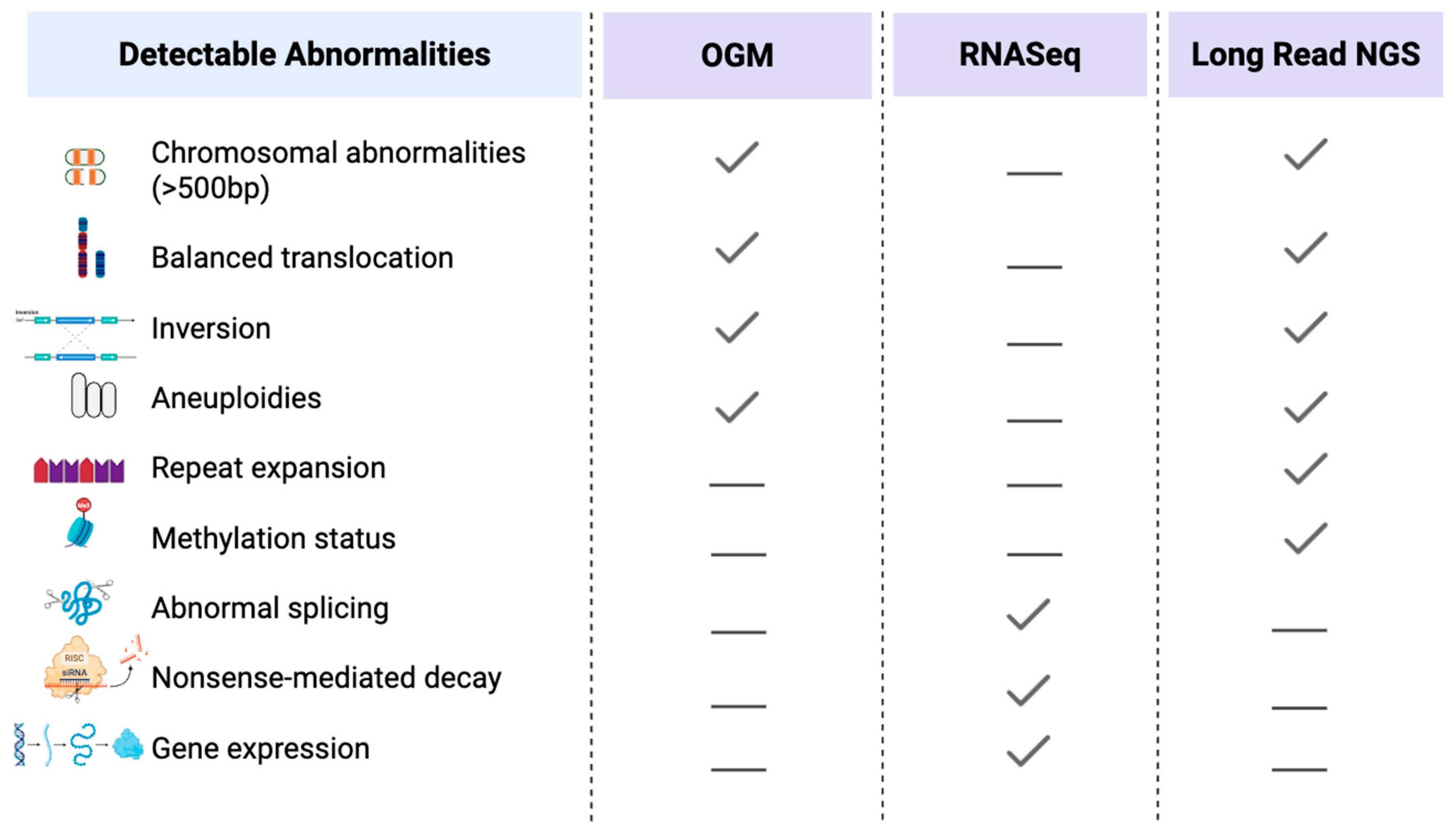
Figure 2.
Emerging Technologies and Corresponding Genomic Abnormalities Detectable in the Prenatal Setting. Comparison of three emerging genomic technologies: optical genome mapping (OGM), RNA sequencing (RNASeq), and long-read next-generation sequencing (NGS) and their ability to detect various types of abnormalities.
Figure 2.
Emerging Technologies and Corresponding Genomic Abnormalities Detectable in the Prenatal Setting. Comparison of three emerging genomic technologies: optical genome mapping (OGM), RNA sequencing (RNASeq), and long-read next-generation sequencing (NGS) and their ability to detect various types of abnormalities.
Acknowledgments
This work was supported in part through the Duncan Neurological Research Institute and the Research Vision at Texas Children’s Hospital.
References
- Mardis, E.R. , Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 2013, 6, 287–303. [Google Scholar] [CrossRef]
- Gregg, A.R. , et al., Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021, 23, 1793–1806. [Google Scholar] [CrossRef]
- Rose, N.C. , et al., Systematic evidence-based review: The application of noninvasive prenatal screening using cell-free DNA in general-risk pregnancies. Genet Med 2022, 24, 1379–1391. [Google Scholar] [CrossRef]
- van der Meij, K.R.M. , et al., TRIDENT-2: National Implementation of Genome-wide Non-invasive Prenatal Testing as a First-Tier Screening Test in the Netherlands. Am J Hum Genet 2019, 105, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Dungan, J.S. , et al., Noninvasive prenatal screening (NIPS) for fetal chromosome abnormalities in a general-risk population: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2023, 25, 100336. [Google Scholar] [CrossRef]
- Mohan, P. , et al., Clinical experience with non-invasive prenatal screening for single-gene disorders. Ultrasound Obstet Gynecol 2022, 59, 33–39. [Google Scholar] [CrossRef]
- Wapner, R.J. , et al., Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012, 367, 2175–2184. [Google Scholar] [CrossRef]
- Chau, M.H.K. , et al., Low-pass genome sequencing: a validated method in clinical cytogenetics. Hum Genet 2020, 139, 1403–1415. [Google Scholar] [CrossRef]
- Mellis, R. , et al., Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat Diagn 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Richards, S. , et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–424. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists, Carrier screening for genetic conditions. Committee opinion no. 691. Obstet Gynecol 2017, 129, e41–e55. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.G. , et al., Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol 2015, 125, 653–662. [Google Scholar] [PubMed]
- Gregg, A.R. , Expanded Carrier Screening. Obstet Gynecol Clin North Am 2018, 45, 103–112. [Google Scholar] [CrossRef] [PubMed]
- National Human Genome Research Institute. The Cost of Sequencing a Human Genome. 2023 [cited 2025 ]; Available from: https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost. 23 May.
- Guo M.H., G. A., Carrier Screening for Genetic Conditions, in Perinatal Genetics, M.E. Norton, Kuller, J.A., Dugoff, L., Editor. 2019, ELSEVIER.
- Rehder, C. , et al., Next-generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021, 23, 1399–1415. [Google Scholar] [CrossRef]
- Fridman, H. , et al., Preconception carrier screening yield: effect of variants of unknown significance in partners of carriers with clinically significant variants. Genet Med 2020, 22, 646–653. [Google Scholar] [CrossRef]
- Beauchamp, K.A. , et al., Systematic design and comparison of expanded carrier screening panels. Genet Med 2018, 20, 55–63. [Google Scholar] [CrossRef]
- Ben-Shachar, R. , et al., A data-driven evaluation of the size and content of expanded carrier screening panels. Genet Med 2019, 21, 1931–1939. [Google Scholar] [CrossRef]
- Guo, M.H. and A.R. Gregg, Estimating yields of prenatal carrier screening and implications for design of expanded carrier screening panels. Genet Med 2019, 21, 1940–1947. [Google Scholar] [CrossRef]
- Chokoshvili, D., D. F. Vears, and P. Borry, Growing complexity of (expanded) carrier screening: Direct-to-consumer, physician-mediated, and clinic-based offers. Best Pract Res Clin Obstet Gynaecol 2017, 44, 57–67. [Google Scholar] [CrossRef]
- Haque, I.S. , et al., Modeled Fetal Risk of Genetic Diseases Identified by Expanded Carrier Screening. Jama 2016, 316, 734–742. [Google Scholar] [CrossRef]
- Lazarin, G.A. , et al., An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med 2013, 15, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Westemeyer, M. , et al., Clinical experience with carrier screening in a general population: support for a comprehensive pan-ethnic approach. Genet Med 2020, 22, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Rink, B.D. , Informed consent for expanded carrier screening: Past, present, and future. Prenatal Diagnosis 2023, 43, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, R.L., R. N. Slotnick, and N.J. Risch, Challenges in providing residual risks in carrier testing. Prenatal Diagnosis 2021, 41, 1049–1056. [Google Scholar] [CrossRef]
- Yang, Y. , et al., Applications of genome sequencing as a single platform for clinical constitutional genetic testing. Genet Med Open 2024, 2, 101840. [Google Scholar] [CrossRef]
- Punj, S. , et al., Preconception Carrier Screening by Genome Sequencing: Results from the Clinical Laboratory. Am J Hum Genet 2018, 102, 1078–1089. [Google Scholar] [CrossRef]
- Shamseldin, H.E. , et al., Molecular autopsy in maternal-fetal medicine. Genet Med 2018, 20, 420–427. [Google Scholar] [CrossRef]
- Fan, H.C. , et al., Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A 2008, 105, 16266–16271. [Google Scholar] [CrossRef]
- Palomaki, G.E. , et al., DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011, 13, 913–920. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists, Screening for Fetal Chromosomal Abnormalities: ACOG Practice Bulletin, Number 226. Obstet Gynecol 2020, 136, e48–e69. [CrossRef]
- Lo, Y.M. , et al., Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, E. , et al., Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat Diagn 2013, 33, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W. and R.W.K. Chiu, Sequencing of Circulating Cell-free DNA during Pregnancy. N Engl J Med 2018, 379, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.J. , et al., Noninvasive prenatal diagnosis of fetal trisomy 21 by allelic ratio analysis using targeted massively parallel sequencing of maternal plasma DNA. PLoS One 2012, 7, e38154. [Google Scholar] [CrossRef]
- Nicolaides, K.H. , et al., Validation of targeted sequencing of single-nucleotide polymorphisms for non-invasive prenatal detection of aneuploidy of chromosomes 13, 18, 21, X, and Y. Prenat Diagn 2013, 33, 575–579. [Google Scholar] [CrossRef]
- Dugoff, L. , et al., Cell-free DNA screening for trisomy 21 in twin pregnancy: a large multicenter cohort study. Am J Obstet Gynecol 2023, 229, e1–e435. [Google Scholar] [CrossRef]
- Wang, Y. , et al., Noninvasive Evaluation of Fetal Zygosity in Twin Pregnancies Involving a Binary Analysis of Single-Nucleotide Polymorphisms. J Mol Diagn 2023, 25, 682–691. [Google Scholar] [CrossRef]
- van Riel, M. , et al., Performance and Diagnostic Value of Genome-Wide Noninvasive Prenatal Testing in Multiple Gestations. Obstet Gynecol 2021, 137, 1102–1108. [Google Scholar] [CrossRef]
- Ehrich, M. , et al., Genome-wide cfDNA screening: clinical laboratory experience with the first 10,000 cases. Genet Med 2017, 19, 1332–1337. [Google Scholar] [CrossRef]
- Grati, F.R. , et al., Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn 2015, 35, 801–809. [Google Scholar] [CrossRef]
- Dar, P. , et al., Cell-free DNA screening for prenatal detection of 22q11.2 deletion syndrome. Am J Obstet Gynecol 2022, 227, e1–e79. [Google Scholar] [CrossRef] [PubMed]
- Acreman, M.L. , et al., The predictive value of prenatal cell-free DNA testing for rare autosomal trisomies: a systematic review and meta-analysis. Am J Obstet Gynecol 2023, 228, 292–305.e6. [Google Scholar] [CrossRef] [PubMed]
- van Prooyen Schuurman, L. , et al., Clinical impact of additional findings detected by genome-wide non-invasive prenatal testing: Follow-up results of the TRIDENT-2 study. Am J Hum Genet 2022, 109, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Eggenhuizen, G.M. , et al., Confined placental mosaicism and the association with pregnancy outcome and fetal growth: a review of the literature. Hum Reprod Update 2021, 27, 885–903. [Google Scholar] [CrossRef] [PubMed]
- Chitty, L.S., L. Hudgins, and M.E. Norton, Current controversies in prenatal diagnosis 2: Cell-free DNA prenatal screening should be used to identify all chromosome abnormalities. Prenat Diagn 2018, 38, 160–165. [Google Scholar] [CrossRef]
- Grati, F.R. , et al., Implications of fetoplacental mosaicism on cell-free DNA testing for sex chromosome aneuploidies. Prenat Diagn 2017, 37, 1017–1027. [Google Scholar] [CrossRef]
- Chitty, L.S. , Advances in the prenatal diagnosis of monogenic disorders. Prenat Diagn 2018, 38, 3–5. [Google Scholar] [CrossRef]
- Liao, J. , et al., Advances in Prenatal Cell-Free DNA Screening for Dominant Monogenic Conditions: A Review of Current Progress and Future Directions in Clinical Implementation. Prenat Diagn 2025, 45, 445–452. [Google Scholar] [CrossRef]
- Hayward, J. and L.S. Chitty, Beyond screening for chromosomal abnormalities: Advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing. Semin Fetal Neonatal Med 2018, 23, 94–101. [Google Scholar] [CrossRef]
- Zhang, J. , et al., Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med 2019, 25, 439–447. [Google Scholar] [CrossRef]
- Adams, S. , et al., Routine Prenatal cfDNA Screening for Autosomal Dominant Single-Gene Conditions. Clinical Chemistry 2025, 71, 129–140. [Google Scholar] [CrossRef]
- Rego, S. , et al., Cell-Free DNA Analysis for the Determination of Fetal Red Blood Cell Antigen Genotype in Individuals With Alloimmunized Pregnancies. Obstet Gynecol 2024, 144, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.R., B. Goodhue, and N.L. Vora, Rho(D) immune globulin shortage and fetal Rh(D) screening with cell-free DNA. Curr Opin Obstet Gynecol 2025, 37, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Hoskovec, J. , et al., Maternal carrier screening with single-gene NIPS provides accurate fetal risk assessments for recessive conditions. Genet Med 2023, 25, 100334. [Google Scholar] [CrossRef] [PubMed]
- Wynn, J. , et al., Performance of single-gene noninvasive prenatal testing for autosomal recessive conditions in a general population setting. Prenat Diagn 2023, 43, 1344–1354. [Google Scholar] [CrossRef]
- Lo, Y.M. , et al., Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010, 2. [Google Scholar] [CrossRef]
- Yu, S.C. , et al., Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci U S A 2014, 111, 8583–8588. [Google Scholar] [CrossRef]
- Yu, S.C.Y. , et al., Combined Count- and Size-Based Analysis of Maternal Plasma DNA for Noninvasive Prenatal Detection of Fetal Subchromosomal Aberrations Facilitates Elucidation of the Fetal and/or Maternal Origin of the Aberrations. Clinical Chemistry 2017, 63, 495–502. [Google Scholar] [CrossRef]
- Yang, Q. , et al., Size-selective separation and overall-amplification of cell-free fetal DNA fragments using PCR-based enrichment. Sci Rep 2017, 7, 40936. [Google Scholar] [CrossRef]
- Acevedo, A. , et al., Fetal fraction amplification within prenatal cfDNA screening enables detection of genome-wide copy-number variants at enhanced resolution. Genet Med 2025, 27, 101269. [Google Scholar] [CrossRef]
- Welker, N.C. , et al., High-throughput fetal fraction amplification increases analytical performance of noninvasive prenatal screening. Genet Med 2021, 23, 443–450. [Google Scholar] [CrossRef]
- Brand, H. , et al., High-Resolution and Noninvasive Fetal Exome Screening. N Engl J Med 2023, 389, 2014–2016. [Google Scholar] [CrossRef]
- Miceikaite, I. , et al., Comprehensive prenatal diagnostics: Exome versus genome sequencing. Prenat Diagn 2023, 43, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W. , et al., Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. Jama 2015, 314, 162–169. [Google Scholar] [CrossRef]
- Ji, X. , et al., Identifying occult maternal malignancies from 1.93 million pregnant women undergoing noninvasive prenatal screening tests. Genet Med 2019, 21, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Turriff Amy, E. , et al., Prenatal cfDNA Sequencing and Incidental Detection of Maternal Cancer. New England Journal of Medicine 2024, 391, 2123–2132. [Google Scholar] [CrossRef]
- Shao, L. , et al., Chromosomal microarray analysis, including constitutional and neoplastic disease applications, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021, 23, 1818–1829. [Google Scholar] [CrossRef]
- Elron, E. , et al., The Diagnostic Yield of Chromosomal Microarray Analysis in Third-Trimester Fetal Abnormalities. Am J Perinatol 2024, 41, 2232–2242. [Google Scholar]
- Armour, C.M. , et al., Practice guideline: joint CCMG-SOGC recommendations for the use of chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss in Canada. J Med Genet 2018, 55, 215–221. [Google Scholar] [CrossRef]
- Mitrakos, A. , et al., Prenatal Chromosomal Microarray Analysis: Does Increased Resolution Equal Increased Yield? Genes (Basel).
- Dong, Z. , et al., Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med 2016, 18, 940–948. [Google Scholar] [CrossRef]
- Monaghan, K.G. , et al., The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2020, 22, 675–680. [Google Scholar] [CrossRef]
- Wang, H. , et al., Low-pass genome sequencing versus chromosomal microarray analysis: implementation in prenatal diagnosis. Genet Med 2020, 22, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X. , et al., Prenatal diagnosis of skeletal dysplasias using a targeted skeletal gene panel. Prenat Diagn 2018, 38, 692–699. [Google Scholar] [CrossRef]
- Scott, A. , et al., When to test fetuses for RASopathies? Proposition from a systematic analysis of 352 multicenter cases and a postnatal cohort. Genet Med 2021, 23, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Norton, M.E. , et al., Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol 2022, 226, 128–e1. [Google Scholar] [CrossRef]
- Yaldiz, B. , et al., Twist exome capture allows for lower average sequence coverage in clinical exome sequencing. Hum Genomics 2023, 17, 39. [Google Scholar] [CrossRef]
- Mellis, R. , et al., Fetal exome sequencing for isolated increased nuchal translucency: should we be doing it? BJOG 2022, 129, 52–61. [Google Scholar] [CrossRef]
- Pauta, M., R. J. Martinez-Portilla, and A. Borrell, Diagnostic yield of exome sequencing in fetuses with multisystem malformations: systematic review and meta-analysis. Ultrasound Obstet Gynecol 2022, 59, 715–722. [Google Scholar] [CrossRef]
- Mone, F. , et al., Should we offer prenatal exome sequencing for intrauterine growth restriction or short long bones? A systematic review and meta-analysis. Am J Obstet Gynecol 2023, 228, 409–417. [Google Scholar] [CrossRef]
- Lord, J. , et al., Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 2019, 393, 747–757. [Google Scholar] [CrossRef]
- Petrovski, S. , et al., Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 2019, 393, 758–767. [Google Scholar] [CrossRef]
- Van den Veyver, I.B. , et al., International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat Diagn 2022, 42, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.H. , et al., Genome Sequencing for Diagnosing Rare Diseases. N Engl J Med 2024, 390, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J. , et al., Whole Genome Sequencing in the Evaluation of Fetal Structural Anomalies: A Parallel Test with Chromosomal Microarray Plus Whole Exome Sequencing. Genes (Basel).
- Westenius, E. , et al., Whole-genome sequencing in prenatally detected congenital malformations: prospective cohort study in clinical setting. Ultrasound Obstet Gynecol 2024, 63, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S. , et al., Exome sequencing for perinatal phenotypes: The significance of deep phenotyping. Prenat Diagn 2020, 40, 260–273. [Google Scholar] [CrossRef]
- Dhombres, F. , et al., Prenatal phenotyping: A community effort to enhance the Human Phenotype Ontology. Am J Med Genet C Semin Med Genet 2022, 190, 231–242. [Google Scholar] [CrossRef]
- Van den Veyver, I.B., Y. Yaron, and Z.C. Deans, International Society for Prenatal Diagnosis 2022 debate 3-Fetal genome sequencing should be offered to all pregnant patients. Prenat Diagn 2023, 43, 428–434. [Google Scholar] [CrossRef]
- Giordano, J.L. and R.J. Wapner, The fetal sequencing consortium: The value of multidisciplinary dialog and collaboration. Prenat Diagn 2022, 42, 807–810. [Google Scholar] [CrossRef]
- Jeanne, M. and W.K. Chung, Prenatal genomic sequencing: Navigating uncertainty. Semin Perinatol 2025, 49, 152058. [Google Scholar] [CrossRef]
- Lee, K. , et al., ACMG SF v3.3 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2025, 27, 101454. [Google Scholar] [CrossRef]
- Dremsek, P. , et al., Optical Genome Mapping in Routine Human Genetic Diagnostics-Its Advantages and Limitations. Genes (Basel).
- Levy, B. , et al., Multisite Evaluation and Validation of Optical Genome Mapping for Prenatal Genetic Testing. J Mol Diagn 2024, 26, 906–916. [Google Scholar] [CrossRef]
- Liu, P. and L. Vossaert, Emerging technologies for prenatal diagnosis: The application of whole genome and RNA sequencing. Prenat Diagn 2022, 42, 686–696. [Google Scholar] [CrossRef]
- Truty, R. , et al., Spectrum of splicing variants in disease genes and the ability of RNA analysis to reduce uncertainty in clinical interpretation. Am J Hum Genet 2021, 108, 696–708. [Google Scholar] [CrossRef]
- Wai, H.A. , et al., Correction: Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet Med 2020, 22, 1129. [Google Scholar] [CrossRef]
- Mahmoud, M. , et al., Utility of long-read sequencing for All of Us. Nat Commun 2024, 15, 837. [Google Scholar] [CrossRef] [PubMed]
- Long, J. , et al., Evaluating the clinical efficacy of a long-read sequencing-based approach for carrier screening of spinal muscular atrophy. Hum Genomics 2024, 18, 110. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. , et al., Comprehensive SMN1 and SMN2 profiling for spinal muscular atrophy analysis using long-read PacBio HiFi sequencing. Am J Hum Genet 2023, 110, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. , et al., Comprehensive Analysis of Hemophilia A (CAHEA): Towards Full Characterization of the F8 Gene Variants by Long-Read Sequencing. Thromb Haemost 2023, 123, 1151–1164. [Google Scholar]
- Wang, R. , et al., Long-Read Sequencing Solves Complex Structure of CYP21A2 in a Large 21-Hydroxylase Deficiency Cohort. J Clin Endocrinol Metab 2025, 110, 406–416. [Google Scholar] [CrossRef]
- Yu, S.C.Y., L. Y.L. Choy, and Y.M.D. Lo, ‘Longing’ for the Next Generation of Liquid Biopsy: The Diagnostic Potential of Long Cell-Free DNA in Oncology and Prenatal Testing. Mol Diagn Ther 2023, 27, 563–571. [Google Scholar] [CrossRef]
- Yu, S.C.Y. , et al., Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proc Natl Acad Sci U S A.
- Vong, J.S.L. , et al., Enrichment of fetal and maternal long cell-free DNA fragments from maternal plasma following DNA repair. Prenat Diagn 2019, 39, 88–99. [Google Scholar] [CrossRef]
- Maktabi, M.A., L. Vossaert, and I.B. Van den Veyver, Cell-based Noninvasive Prenatal Testing (cbNIPT)-A Review on the Current Developments and Future Prospects. Clin Obstet Gynecol 2023, 66, 636–648. [Google Scholar] [CrossRef]
- Jeppesen, L.D. , et al., Clinical interpretation of cell-based non-invasive prenatal testing for monogenic disorders including repeat expansion disorders: potentials and pitfalls. Front Genet 2023, 14, 1188472. [Google Scholar] [CrossRef] [PubMed]
- Vossaert, L. , et al., Validation Studies for Single Circulating Trophoblast Genetic Testing as a Form of Noninvasive Prenatal Diagnosis. Am J Hum Genet 2019, 105, 1262–1273. [Google Scholar] [CrossRef] [PubMed]
- Imudia, A.N. , et al., Retrieval of trophoblast cells from the cervical canal for prediction of abnormal pregnancy: a pilot study. Hum Reprod 2009, 24, 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Kadam, L. , et al., Endocervical trophoblast for interrogating the fetal genome and assessing pregnancy health at five weeks. Eur J Med Genet 2019, 62, 103690. [Google Scholar] [CrossRef]
- Jou, H.J., P. H. Lo, and P.Y. Ling, Recent Advances of Microfluidic Platform for Cell Based Non-Invasive Prenatal Diagnosis. Int J Mol Sci.
- Li, X. , et al., Aptamer-Mediated Enrichment of Rare Circulating Fetal Nucleated Red Blood Cells for Noninvasive Prenatal Diagnosis. Anal Chem 2023, 95, 5419–5427. [Google Scholar] [CrossRef]
- Feng, C. , et al., The path winds along isolation and analyses of fetal nucleated red blood cells in maternal peripheral blood: Past, present, and future toward non-invasive prenatal diagnosis. Life Sci 2025, 369, 123530. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



