Submitted:
13 August 2025
Posted:
13 August 2025
You are already at the latest version
Abstract
Aging is a complex biological process that involves a gradual decline in the human body's structure and function and increases the risks of aging-related diseases such as Alzheimer's disease (AD) and type 2 diabetes. Type 2 diabetes accelerates all clinical manifestations of aging. Metabolic disorders in type 2 diabetes are unfavorably associ-ated with all hallmarks of aging such as inflammation and mitochondrial dysfunction. AMP-activated protein kinase (AMPK) and the mammalian target of rapamycin com-plex 1 (mTORC1) are key players in cellular metabolism. AMPK activation and mTORC1 inhibition improve all hallmarks of aging. Furthermore, AMPK activation and mTORC1 inhibition are favorably associated with diabetic complications such as diabetic neuropathy, retinopathy, nephropathy and cardiovascular diseases and AD. Nutrition such as resveratrol and astaxanthin have AMPK activating and mTORC1 in-hibitory effects and improve metabolic abnormalities in type 2 diabetes. Metformin, sodium-glucose co-transporter-2 inhibitors, glucagon-like peptide 1 receptor agonists (GLP-1RAs), and dual glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 RAs, have been reported to have AMPK activating and mTORC1 inhibitory effects. The therapeutic interventions that activate AMPK and inhibit mTORC1 may be optimal treatments for type 2 diabetes from the perspective of anti-aging medicine.
Keywords:
Aging
; AMP-activated protein kinase
; mammalian target of rapamycin complex 1
; metformin
; type 2 diabetes
1. Introduction
Aging is a complex biological process that involves a gradual decline in the human body's structure and function and increases the risks of various aging-related diseases, including neurodegenerative diseases such as Alzheimer's disease (AD), cardiovascular diseases (CVD), metabolic diseases such as type 2 diabetes, musculoskeletal diseases, and immune system diseases [1]. Recent studies have identified several key hallmarks, or characteristics, of aging at the molecular, cellular, and systemic levels [2,3]. These include oxidative stress [4], inflammation [5], advanced glycation end products (AGEs) [6], dysbiosis [7,8], DNA damage [9,10], telomere shortening [11], epigenetic alterations [12,13], loss of protein balance [14], deregulated nutrient-sensing [15], altered intercellular communication [16,17], mitochondrial dysfunction [18,19], loss of nicotinamide adenine dinucleotide (NAD+) levels [20,21], cellular senescence [22], and stem cell exhaustion [23,24], impaired macro-autophagy [25,26], and hormonal changes [27,28].
Current research on aging focuses on elucidating how various endogenous and exogenous stresses participate in the regulation of aging [1]. Furthermore, thorough research on the pathogenesis of aging to identify clinical treatment methods for aging-related diseases could decrease the incidence and development of aging-related diseases and, in turn, promote healthy aging and longevity [1]. Here, we will report the influence of type 2 diabetes on the clinical manifestations of aging and hallmarks of aging.
A protein complex called AMP-activated protein kinase (AMPK) regulates energy balance within cells and controls cellular metabolism. AMPK signaling declines with aging, enhancing the aging process [29]. AMPK activity is reduced in type 2 diabetes [30]. Therefore, numerous studies have highlighted the remarkable potential of AMPK and its activators in treating type 2 diabetes. The mammalian target of rapamycin complex 1 (mTORC1) is a key component of cellular metabolism that integrates nutrient sensing with cellular processes that fuel cell growth and proliferation, and inhibition of mTORC1 with therapeutic rapamycin promotes health and longevity in diverse model organisms [31]. Hyperactive mTORC1 has been proposed to offer a novel perspective for type 2 diabetes in terms of pathogenesis, clinical focus, and treatment strategy [32].
We will consider effects of AMPK activation and mTORC1 inhibition on hallmarks of aging and the development and progression of type 2 diabetes and its complications. Furthermore, we searched the influence of nutritional interventions and drugs that improve aging hallmarks by AMPK activation and mTORC1 inhibition on type 2 diabetes and its complications. We would like to propose optimal treatments for type 2 diabetes from the perspective of anti-aging medicine.
2. The Influence of Type 2 Diabetes on the Clinical Manifestations of Aging
The clinical manifestations of aging are shown in Figure 1.
2.1. Hair Aging
Dysregulation in the balance of the melanocyte spectrum within the hair follicle is the underlying cause of physiological and pathological grey hair [33]. During the progressive deterioration of cellular functions with aging, melanin synthesis is impeded after tyrosinase activity is reduced [34]. Type 2 diabetes is usually accompanied by premature grey hair [35].
2.2. Skin Aging
Diabetes causes increased production of AGEs, which may lead to irreversible damage to collagen fibers and early and more accentuated signs of skin aging [36]. In fact, higher trans-epidermal water loss was observed in patients with diabetes, who also showed lower skin elasticity and wrinkles with greater volume, area, and depth [36]. In addition, patients with diabetes presented polycyclic papillae and deformed and amorphous collagen fibers [36].
2.3. Voice Aging
Voice aging changes include a weak, breathy, or hoarse voice, and such changes are called presbyphonia. A scoping review including 188 published articles reported that laryngeal aging results in calcifications of the hyaline cartilages, decreased vocal folds lubrication, decreased neuromotor transmission, decreased hyaluronic acid, increased lamina propria stiffness, and changes in the thyroarytenoid muscles [37]. Concomitant aging of the respiratory system, hormonal changes, concomitant presbycusis, and medical comorbidities interact with the presbylarynx to induce presbyphonia [37].
2.4. Ear Aging
Type 2 diabetes and sensorineural hearing loss are common health problems manifested with aging [39]. Individuals with diabetes have twice the incidence of hearing loss compared to those without diabetes, and those with prediabetes have a 30% higher rate of hearing loss [39]. Whether hearing loss is associated with diabetes independent of glycemic control remains to be determined.
2.5. Brain Aging
In the meta-analysis using the UK Biobank, compared to age, sex, education, and hypertension-matched healthy controls, patients with type 2 diabetes were associated with marked cognitive deficits, particularly in executive functioning and processing speed [40]. Type 2 diabetes was significantly associated with gray matter atrophy, primarily within the ventral striatum, cerebellum, and putamen [40]. The structural and functional changes associated with type 2 diabetes show marked overlap with the effects correlating with age but appear earlier.
2.6. Bone Aging
The meta-analysis revealed no significant difference in overall bone mineral density (BMD) between the nondiabetic and type 2 diabetic groups (mean difference [MD], -0.07; 95% confidence interval [CI], -0.17 to 0.03; P = 0.15) [41]. However, BMD at the lumbar vertebrae was significantly higher in nondiabetic individuals compared with those with type 2 diabetes (MD, -0.14; 95% CI, -0.22 to -0.06; P = 0.0009), as was the case with femoral neck BMD (MD, -0.11; 95% CI, -0.18 to -0.04; P = 0.002) [41]. Reduced BMD at the lumbar vertebrae and femoral neck can be a marker for bone aging in patients with type 2 diabetes.
2.7. Vascular Aging
Increased arterial stiffness has been shown to be predictive of CVD and all-cause mortality [42,43,44]. Arterial stiffening is known to increase with age, and arterial stiffening is especially accelerated in obese and diabetic females who tend to lose the normal protection afforded by female sex hormones against vascular disease and show an increase in CVD events relative to lean and non-diabetic premenopausal women [45,46,47]. The current standard for clinically assessing vascular stiffness is measurement of carotid-femoral pulse wave velocity (PWV) [48,49]. The meta-analysis showed that PWV was a useful noninvasive early marker for vascular dysfunction detection in patients with type 2 diabetes [50].
2.8. Muscular Aging
Insulin resistance has been closely associated with increased muscle protein degradation [51]. Accumulation of AGEs induces skeletal muscle atrophy and dysfunction, while oxidative stress and mitochondrial dysfunction caused by type 2 diabetes can lead to myocyte apoptosis. Therefore, patients with type 2 diabetes have a higher risk of developing sarcopenia, a progressive and generalized skeletal muscle disease linked to a higher risk of negative consequences such as falls, fractures, physical disability, and mortality [52]. Calf circumference was the most effective screening tool for sarcopenia in patients with type 2 diabetes [53].
2.9. Effect of Type 2 Diabetes on the Clinical Manifestations of Aging
Type 2 diabetes accelerates all clinical manifestations of aging.
3. The Hallmarks of Aging and Their Association with Type 2 Diabetes
3.1. Oxidative Stress
Oxidative stress, an imbalance between the production of reactive oxygen species (ROS) and the body's ability to neutralize them, is strongly linked to aging and age-related diseases [54]. Cumulative damage to mitochondria and mitochondrial DNA caused by ROS is one of the causes of aging [55]. Oxidative damage affects replication and transcription of mitochondrial DNA and results in a decline in mitochondrial function, which in turn leads to enhanced ROS production and further damage to mitochondrial DNA [55].
ROS are highly reactive and can damage cellular components, including DNA, proteins, and lipids, and such accumulated damage can impair cellular function and contribute to the aging process [56]. ROS can also cause mutations and breaks in DNA strands, leading to genomic instability. DNA damage can accumulate with age and is associated with an increased risk of age-related diseases [57]. Oxidative stress can trigger inflammation and accelerate the shortening of telomeres, which are associated with cellular aging and senescence [58].
Oxidative stress has been implicated in the progression of prediabetes to type 2 diabetes [59]. Oxidative stress contributes to insulin resistance, beta-cell dysfunction, and diabetic complications. Glutathione, a major antioxidant, is crucial for neutralizing ROS, but glutathione levels are notably low in type 2 diabetes, exacerbating oxidative stress and inflammation [60]. Elevated interleukin-6 (IL-6) levels further intensify inflammation and oxidative stress, disrupting insulin signaling and worsening complications such as nephropathy, retinopathy, and neuropathy. Type 2 diabetes is a chronic metabolic disease in which patients are prone to inflammation and oxidative stress [61,62], and hyperglycemia, hyperglycemia-induced oxidative stress, and inflammation are closely associated with the development and progression of type 2 diabetes and its complications.
3.2. Inflammation
Aging is associated with persistent activation of the immune system, witnessed by a high circulating level of inflammatory markers and activation of immune cells in the circulation and in tissue, a condition called "inflammaging [63]." Like aging, inflammaging is associated with increased risk of many age-related pathologies and disabilities, as well as frailty and death.
Low-grade inflammation represents a key driver of type 2 diabetes and CVD [64]. Inflammatory markers such as high-sensitive C-reactive protein (hs-CRP) and IL-6 predict the development of type 2 diabetes and its complications. Overnutrition, unhealthy diets, physical inactivity, obesity, and aging are all recognized triggers of inflammation, promoting insulin resistance and sustaining the pathogenesis of type 2 diabetes. The infiltration of proinflammatory cells in adipose tissue is associated with an increased production of key chemokines such as C-C motif chemokine ligand 2, proinflammatory cytokines including tumor necrosis factor α and IL-6 as well as reduced expression of the key insulin-sensitizing adipokine, adiponectin [65]. Adipose tissue inflammation is a key factor in the development of insulin resistance and type 2 diabetes in obesity.
In addition to adipose tissue, liver, muscle, and pancreas are the sites of inflammation in the presence of obesity [66]. An infiltration of macrophages and other immune cells is observed in these tissues associated with a cell population shift from an anti-inflammatory to a pro-inflammatory profile [66]. These cells are crucial to produce pro-inflammatory cytokines, which act in an autocrine and paracrine manner to interfere with insulin signaling in peripheral tissues or induce beta-cell dysfunction and subsequent insulin deficiency.
3.3. AGEs
AGEs arise from the nonenzymatic glycosylation of proteins and lipids [67]. Studies have demonstrated a clear correlation between AGE accumulation in tissues and blood glucose levels [68]. Accumulation of AGEs on nucleotides, lipids, and peptides/proteins is an inevitable component of the aging process in all eukaryotic organisms, including humans [69]. A substantial body of evidence shows that AGEs and their functionally compromised adducts are linked to changes seen during aging and the development of many age-related morbidities.
Even after correcting hyperglycemia, AGEs levels in diabetic tissues often fail to return to normal, leading to the concept of “metabolic memory” [70]. AGEs are not merely byproducts of hyperglycemia but are also implicated in the development of diabetes [71]. AGEs bind to the receptor for AGEs (RAGE), thereby activating proinflammatory signaling pathways [72]. AGEs bind RAGE, triggering multiple intracellular signaling pathways, including the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that leads to increased production of ROS, thus contributing to generating a pro-oxidant environment [73]. These processes are also thought to contribute to the development of diabetic microvascular complications [74].
3.4. Dysbiosis
The gut microbiome plays a crucial role in modulating health and disease across the lifespan. As individuals age, the gut microbiome undergoes significant compositional and functional changes, often leading to dysbiosis—an imbalance associated with increased inflammation, weakened immunity, and metabolic disorders [75]. Inflammaging is closely linked to gut microbial alterations and contributes to neurodegenerative diseases, CV conditions, and metabolic disorders such as obesity and diabetes [75]. Additionally, the gut-brain axis is emerging as a critical pathway in neurocognitive aging.
A systematic review has reported a major gut microbiota alteration in obesity, characterized by a specific decrease in butyrate-producing bacteria and the production of metabolites and components that lead to metabolic impairments and affect the progression of various diseases associated with obesity through distinct signaling pathways, including insulin resistance, type 2 diabetes, and CVD [76]. Inflammation plays a major role as a link between gut microbiota dysbiosis and obesity-associated metabolic complications. Emerging evidence indicates that gut microbiota dysbiosis may contribute to the development of type 2 diabetes [77]. Individuals with type 2 diabetes exhibit notable changes in gut microbiota composition, including shifts in the balance between Firmicutes and Bacteroidetes, a reduction in butyrate-producing bacteria, and an increase in opportunistic pathogens [77]. Gut dysbiosis induces inflammation and consequently provokes both peripheral and cerebral insulin resistance and can amplify processes promoting AD. Additionally, the risk of increased progression of AD was revealed due to pre-diabetes, type 2 diabetes, and gut dysbiosis [78].
3.5. DNA Damage
Genomic instability is an important driver of aging. The accumulation of DNA damage contributes to aging by inducing cell death, senescence, and tissue dysfunction [9]. Inflammation is a hallmark of aging and the driver of multiple age-related diseases. DNA damage induces inflammation through activation of the cyclic GMP-AMP synthase-stimulator of interferon genes axis and nuclear factor-kappa B (NF-κB) activation [9]. Loss of efficient DNA damage repair can lead to accelerated aging phenotypes [10]. High levels of mutations in DNA repair mutants often lead to excessive cell death and stem cell exhaustion, which may promote premature aging [10]. Increasing DNA damage and mutation accumulation are the hallmarks of aging.
Cell senescence is defined as a state of irreversible cell cycle arrest combined with DNA damage and the induction of a senescence-associated secretory phenotype (SASP). This includes increased secretion of many inflammatory agents, proteases, and microRNAs, which have recently emerged as important regulators of cellular senescence and aging [79]. Recent evidence demonstrates that SASP may also play important roles in diabetes. White adipose tissue (WAT) cells are highly susceptible to becoming senescent both with aging but also with obesity and type 2 diabetes, independently of chronological age. WAT senescence is associated with hypertrophy of adipocytes, insulin resistance, and dyslipidemia [79]. Insulin resistance, high blood glucose levels, and increased body fat are linked to a chronically elevated inflammatory state [80]. This amplifies oxidative stress, might lead to oxidative DNA damage, impairs the cellular proliferation process, and results in mutations; all of which increase the possibility for the development of dysfunctional cells, tissue, and organs. The meta-analysis showed that significantly increased frequencies of chromosomal aberrations were seen in type 2 diabetic subjects [80].
3.6. Telomere Shortening
Telomere is a part of a chromosome that prevents the tangling of the chromosome during DNA replication. When a cell divides, the telomere gets slightly shorter each time, and this process continues until the telomere becomes too short, at which point the cell cannot divide anymore, and the cell dies [81]. The main reason for telomere shortening is oxidative stress and incomplete DNA replication [82]. When the shortening of telomeres gets elevated and reaches a critical length, apoptosis is triggered and contributes to cellular senescence or the aging process, potentially impacting overall cellular function [83].
In patients with diabetes or impaired glucose tolerance, excessive oxidative stress induces damage to telomeres and shortens their length [84]. Telomere length is a good surrogate marker for mortality and diabetic complications in diabetic patients. Telomere length in pancreatic beta-cells is also shortened in diabetic patients, potentially leading to an impaired capacity for proliferation and insulin secretion, and accelerated cell death [84]. In an animal model, it has been shown that telomere attrition in adipose tissue induces insulin resistance. Type 2 diabetes is characterized by increased oxidative stress, increased oxidative DNA damage, and senescent retinal and renal phenotypes [85]. Increased oxidative DNA damage and telomere attrition in type 2 diabetes have been suggested to lead to senescent retinal and renal phenotypes (diabetic retinopathy and nephropathy) and senescent vascular endothelial, monocyte-macrophage, and vascular smooth muscle cells (endothelial dysfunction and accelerated atherogenesis) [85].
3.7. Epigenetic Alterations
In epigenetic alterations, DNA methylation, involvement of non-coding RNAs, and modifications to histones play an important role in senescence [86]. DNA methylation is a type of heritable modification that can inhibit gene expression through recruiting gene repression proteins and suppressing the normal transcription factor binding, without altering the original DNA sequences. Aberrant DNA methylation influences cellular functions and may lead to diseases such as CVD, metabolic diseases, and cancer. Cellular senescence, aging, and age-related pathogenesis are the major hallmarks of long non-coding RNAs in aging [87]. A histone is a protein that provides structural support for a chromosome. Each chromosome contains a long molecule of DNA, which must fit into the cell nucleus. To do that, the DNA wraps around complexes of histone proteins, giving the chromosome a more compact shape. In general, histone levels are decreased, and aberrant nucleosome occupancies occur during aging. The accumulation of histone variants is another common feature in the aging process [88]. Methylation and acetylation of histone play an important role in regulating the chromatin structure [89], and such structure changes influence the gene expression patterns associated with senescence [90]. Improper dietary habits and lack of physical exercise are important behavioral factors that increase the risk of obesity, which can affect gene expression through epigenetic modifications [91].
Epigenetic alterations can occur in the early stage of obesity and lead to obesity-related complications [91]. Patients with type 2 diabetes exhibit epigenetic alterations linked to mitochondrial dysfunction in pancreatic islets [92]. The epigenetic mechanisms in type 2 diabetes mainly consist of DNA methylation, histone modifications, and regulation by noncoding RNAs [93]. Emerging evidence suggests that oxidative stress can induce aberrant DNA methylation patterns in beta-cells, leading to altered gene expression profiles associated with insulin secretion and cell survival [94]. High glucose induces DNA methyltransferase 1-dependent epigenetic reprogramming of the endothelial exosome proteome in type 2 diabetes [95]. In middle-aged and older Australians, epigenetic age was positively associated with prevalent and incident type 2 diabetes [96].
3.8. Loss of Protein Balance
Aging is associated with a decline in muscle mass, strength, and function, a process known as sarcopenia [97]. This decline is largely attributed to an imbalance between muscle protein synthesis and breakdown, leading to a net loss of muscle protein [97]. This imbalance is influenced by factors like reduced anabolic response to stimuli like food intake and exercise, as well as changes in protein digestion and absorption. The muscle protein synthetic response to the main anabolic stimuli, food intake and physical activity, appears to be blunted in older individuals [98,99,100]. This anabolic resistance is now considered to be a key factor contributing to progressive loss of skeletal muscle mass throughout our lifespan.
Metabolic stress, such as diabetes, can lead to pronounced physiological and cellular adaptations in protein regulation [101], and such pathological conditions can result in muscle atrophy, a net loss of muscle mass, which is highly associated with morbidity [102]. Muscle strength is reduced in individuals with insulin resistance and type 2 diabetes [103,104]. It is plausible that factors derived from adipose tissue can contribute to muscle proteostasis. Isthmin-1 (Ism1), as an important adipokine discovered in recent years, plays a key role in physiological processes such as cell metabolism, energy balance, and insulin sensitivity [105,106]. Ism1 has dual effects of increasing fat and glucose uptake while inhibiting hepatic fat synthesis. Ism1 mitigates diabetes by increasing adipocyte and skeletal muscle glucose uptake by activating the phosphatidylinositol 3'-kinase-Akt pathway [107]. Many studies have shown that the abnormal expression or function of Ism1 is closely related to the occurrence and development of diabetes and its complications. Physiologically, Ism1 ablation in mice results in altered proteostasis, including lower muscle protein levels under fed and fasted conditions, reduced amino acid incorporation into proteins, and reduced phosphorylation of the key protein synthesis effectors Akt and downstream mTORC1 targets [107].
3.9. Deregulated Nutrient Sensing
Deregulated nutrient sensing is a key hallmark of aging, where cellular and organismal processes become less effective at responding to nutrient availability. This dysregulation, particularly in pathways like mTORC1 signaling, can lead to metabolic dysfunction, increased disease susceptibility, and ultimately, contribute to the aging process [108]. The nutrient-sensing network (NSN) is an interlocking network of signaling pathways centered on insulin and insulin-like growth factor-1 (IGF-1), mTORC1, AMPK, and sirtuins (SIRT), and its perturbations promote aging [109]. Lifestyle factors such as diet can modulate NSN [110,111].
3.10. Altered Intercellular Communication
Altered intercellular communication during aging includes the canonical SASP, direct cell-cell communication through gap junctions or tubule-like structures, and long-distance communication, involving extracellular vesicles and paracrine communication mediated by Connexin-containing hemichannels [112].
Extracellular vesicles (EVs) are submicron-sized lipid envelopes that are produced and released from a parent cell and can be taken up by a recipient cell [113]. EVs are capable of mediating cellular signaling by carrying nucleic acids, proteins, lipids, and cellular metabolites between cells and organs. EVs are produced by cells within metabolic tissues, such as adipose tissue, pancreas, muscle, and liver. EV-dependent communication between adipocytes, the vasculature, and immune cells may play an important role in developing type 2 diabetes [113]. Type 2 diabetes involves the loss or dysfunction of pancreatic beta-cells. Impaired Ca2+ uptake into mitochondria, or collapse of a normally interconnected mitochondrial network, is associated with defective insulin secretion [114]. Altered mitochondrial metabolism may also impair beta-cell-beta-cell communication.
3.11. Mitochondrial Dysfunction
Mitochondrial dysfunction is a hallmark of aging, significantly impacting cellular energy, oxidative balance, and calcium levels [115]. It's closely linked to age-associated diseases and is characterized by impaired oxidative phosphorylation, increased oxidative damage, and reduced mitochondrial quality. Mitochondrial dysfunction can also trigger cellular senescence [115].
Type 2 diabetes is characterized by hyperglycemia, hyperlipidemia, and insulin resistance, and such pathological shifts in both circulating fuel levels and energy substrate utilization by central and peripheral tissues contribute to mitochondrial dysfunction across organ systems [116]. The mitochondrion lies at the intersection of critical cellular pathways such as energy substrate metabolism, ROS generation, and apoptosis. Mitochondrial dysfunction leads to impaired oxidative phosphorylation, increased ROS production, mitochondrial DNA damage, and altered mitochondrial dynamics, exacerbating metabolic dysregulation and contributing to diabetic complications [117]. Therefore, mitochondrial dysfunction induces downstream deficits in vital functions, including hepatocyte metabolism, cardiac output, skeletal muscle contraction, beta-cell insulin production, and neuronal health, in type 2 diabetes [116]. Furthermore, the accumulation of mitochondrial DNA mutations and mitochondrial DNA copy number depletion, as well as epigenetic modification of the mitochondrial genome, are associated with the development of type 2 diabetes.
3.12. A Decline in Nicotinamide Adenine Dinucleotide (NAD+)
NAD+ is a crucial coenzyme involved in numerous cellular processes, including energy production and DNA repair. Most of the cellular NAD+ is generated through Nampt activation, a key rate-limiting enzyme that is involved in the salvage pathway. NAD+ is essential for mitochondrial function, and NAD+ acts as a cofactor for enzymes involved in the electron transport chain, a key part of cellular ATP synthesis [118]. Further, NAD+ is known to benefit and restore the body's physiological mechanisms, including DNA replication, chromatin and epigenetic modifications, and gene expression. A decline in NAD+ is strongly linked to the aging process and the development of age-related diseases [119]. As we age, NAD+ levels naturally decrease, potentially impairing these vital cellular functions and contributing to age-related decline and disease [120].
3.13. Cellular Senescence
Cellular senescence, a state of irreversible cell cycle arrest, is a key hallmark of aging. It's a cellular response to stress and damage that can contribute to age-related diseases and the overall aging process [123]. Cell senescence accumulation with age can also be detrimental, driving chronic inflammation and tissue dysfunction [124].
Senescence of islet beta-cells has a remarkable impact on the pathogenesis of type 2 diabetes. Islet beta-cells isolated from older mice and those with type 2 diabetes have increased the activity of senescence-associated beta-galactosidase [125]. Furthermore, type 2 diabetes model mice have shown a higher expression level of IGF-1 receptor (IGF-1R) in their beta-cells compared with control mice, indicating a close relationship between pancreatic beta-cell senescence and type 2 diabetes [126]. Studies on aging have revealed that senescent islet beta-cells exhibit reduced insulin secretion ability and less sensitivity to high sugar stimulation [127]. A high-calorie diet can promote fat cell senescence and lead to insulin resistance [128], and insulin resistance can further induce the senescence of pancreatic beta-cells, thereby causing type 2 diabetes [129].
3.14. Stem Cell Exhaustion
Stem cell exhaustion is a significant factor in aging, characterized by a decline in the ability of stem cells to regenerate and maintain tissue homeostasis [130]. Stem cell exhaustion contributes to aging-related decline in tissue function and the development of various diseases.
Individuals with type 2 diabetes may have a reduced ability to revascularize ischemic tissues due to abnormal production of circulating pro-vascular progenitor cells [131]. This 'regenerative cell exhaustion' process is intensified by increasing oxidative stress and inflammation, and during type 2 diabetes progression. Chronic exhaustion may be mediated by changes in the bone marrow microenvironment that dysregulate the wingless-related integration site network, a central pathway maintaining the progenitor cell pool.
The discovery that diabetes reduces circulating stem/progenitor cells and impairs their function has opened an entirely new field of study where diabetology encounters hematology and regenerative medicine [132]. From a clinical perspective, pauperization of circulating stem cells predicts adverse outcomes and death. Diabetes impairs the mobilization of bone marrow stem/progenitor cells, with negative consequences for physiologic hematopoiesis, immune regulation, and tissue regeneration.
3.15. Impaired Macro-Autophagy
Autophagy is a fundamental cellular process that eliminates molecules and subcellular elements, including nucleic acids, proteins, lipids, and organelles, via lysosome-mediated degradation to promote homeostasis, differentiation, development, and survival [133]. There is an age-dependent decline in autophagy, and autophagy is a crucial determinant of cellular health and organismal longevity, and impairment or imbalance in autophagy promotes pathological aging and disease [133]. The decline in macro-autophagy with age has been implicated in various aging-related diseases, including neurodegenerative diseases such as Parkinson's disease and AD, cancer, and CVD [134].
Autophagy-deficient mice showed hypoinsulinemia and hyperglycemia, suggesting that autophagy is necessary to maintain the structure, mass, and function of pancreatic beta-cells [135]. The major nutrient-sensing signal pathways and diabetes-induced altered intracellular metabolism and cellular events, including accumulation of AGEs, increased oxidative stress, endoplasmic reticulum stress, hypoxia, and activation of the renin-angiotensin system, which modulate autophagic activity and contribute to the development of diabetic nephropathy [136]. Enhancing macro-autophagy is important to maintain the fidelity of the secretory apparatus in conditions of high insulin demands, and failures in these adaptive mechanisms are likely to contribute to beta-cell failure [137]. Islet macroautophagic impairment was found in human type 1 diabetes [138]. Altered lysosome content may be associated with lysosome dysfunction before clinical hyperglycemia. Endothelial dysfunction is a key determinant in the pathogenesis of diabetes. Autophagy, a lysosomal recycling process, appears to play an important role in endothelial cells, ensuring endothelial homeostasis and functions. Autophagic flux impairment has been observed in patients with type 1 or type 2 diabetes [139]. Diabetes triggers endothelial autophagy impairment, which contributes to diabetes-mediated endothelial dysfunction.
3.16. The Association Between Hallmarks of Aging and Type 2 Diabetes
The association between hallmarks of aging and type 2 diabetes is shown in Figure 2. Metabolic disorders such as hyperglycemia and insulin resistance observed in type 2 diabetes induce harmful factors such as oxidative stress, inflammation, AGEs, and dysbiosis. Such harmful factors induce impaired maintenance of healthy cells by inducing DNA damage, epigenetic alteration, mitochondrial dysfunction, and worsening type 2 diabetes, and developing diabetic complications. Impaired maintenance of healthy cells is induced by an impaired clearance of senescent cells due to stem cell exhaustion and impaired macro-autophagy. Impaired clearance of senescent cells is unfavorably associated with metabolic disorders and diabetic complications. Thus, type 2 diabetes and aging form a vicious cycle, exacerbating each other's pathology.
4. The Significance of AMPK Activation and mTORC1 Inhibition for Anti-Aging Medicine
AMPK activation is strongly linked to anti-aging mechanisms, as it acts as a cellular energy sensor and regulator that maintains cellular homeostasis, metabolism, stress resistance, and promotes processes like autophagy, all of which are crucial for healthy aging and extended lifespan [140]. Aging results from the progressive dysregulation of several molecular pathways, and mTORC1 and AMPK signaling have been suggested to play a role in the complex changes in key biological networks involved in cellular senescence [141]. AMPK activation and mTORC1 inhibition are closely linked mechanisms involved in the aging process and have shown promise in extending lifespan and mitigating aging-related diseases. AMPK, a cellular energy sensor, becomes activated under low ATP levels, leading to the inhibition of mTORC1, a key pathway involved in cell growth and metabolism that is often dysregulated with age [141].
Effects of AMPK activation and mTOR inhibition on aging-related molecules and aging hallmarks were shown in Figure 3.
Nuclear factor E2-related factor 2 (Nrf2) is activated under oxidative stress conditions, which promotes antioxidant gene expression [142]. Peroxisome proliferator-activated receptor- γ Coactivator 1α (PGC1α) and Nrf2 have been shown to promote each other and regulate the expression of antioxidant genes. Nrf2 is the downstream target of AMPK. Activation of the AMPK/Nrf2 signaling pathway is key to protection from aging-related diseases.
Chronic inflammation associated with increased NF-κB signaling is a typical hallmark of several age-related metabolic disorders [143]. Activators of AMPK have anti-inflammatory properties. Several studies have demonstrated that the activation of AMPK can inhibit NF-κB signaling, a master regulator of inflammation, both in vitro and in vivo [144].
AMPK activation counteracts AGEs by restoring signaling pathways that reduce inflammation and cellular damage [145]. AGEs, formed under hyperglycemia and oxidative stress, induce cellular dysfunction and inflammatory responses by downregulating AMPK activity, whereas AMPK activation can restore these pathways [145]. mTORC1 inhibition has been reported to be a new potential therapeutic target to diminish the accumulation of AGEs, which was possibly due to a protective role of autophagy in the clearance of cytotoxic AGEs [146].
AMPK inactivation is an etiological factor in intestinal dysfunction, and AMPK activation favorably influences intestinal health [147].
AMPK acts as a central regulator of epigenetic processes by phosphorylating histones, DNA methyltransferases, and histone modifiers [148]. Epigenetic modifications play a crucial and complex role in the cellular response to DNA damage, influencing both the detection and repair of DNA lesions and potentially leading to long-term alterations in gene expression [149].
Telomerase reverse transcriptase (TERT) has been known for its telomere-lengthening effect [150]. AMPK-dependent PGC1α upregulation is required for the metformin-induced upregulation of TERT and telomere activity and length [151].
mTORC1 signaling pathway regulates cellular processes relevant to aging, such as cellular and organismal energetics, proteostasis, cell stemness, and cellular senescence. Therefore, mTORC1 inhibition clearly extends life span [152].
Experiments in mammals have demonstrated that AMPK controls autophagy through mTORC1 and Unc-51-like autophagy activating kinase 1 (ULK1) signaling, which augments the quality of cellular housekeeping. The ULK1 complex plays a central role in the initiation stage of autophagy [153]. mTORC1 inhibits the activity of the ULK1 complex by phosphorylating ULK1 and Atg13 [154].
AMPK is a highly conserved sensor of low intracellular ATP levels that is rapidly activated after nearly all mitochondrial stresses [155]. AMPK regulates autophagy and mitophagy through activation of the kinase ULK1 [156]. AMPK phosphorylates mitochondrial fission factor and promotes mitochondrial fission upon energetic stress. By simultaneously regulating mitochondrial fission, mitophagy, and transcriptional control of mitochondrial biogenesis, AMPK acts as a signal integration platform to maintain mitochondrial health.
NAD+ is a vital coenzyme crucial for maintaining mitochondrial function, directly participating in key energy-producing pathways like the tricarboxylic acid cycle and oxidative phosphorylation, and playing a role in cellular signaling and overall homeostasis [157]. Declining NAD+ levels are associated with aging and various chronic diseases, often accompanied by mitochondrial dysfunction, making NAD+ boosting strategies a focus of research for restoring mitochondrial health and combating age-related decline [157]. AMPK controls the expression of genes involved in energy metabolism by acting in coordination with another metabolic sensor, the NAD+-dependent type III deacetylase SIRT1 [158]. AMPK enhances SIRT1 activity by increasing cellular NAD+ levels.
The anti-aging effects of SIRT1 have been widely studied [159,160]. SIRT1 regulates transcriptional activity and protein expression levels by altering the acetylation status of its substrates, thereby regulating inflammation and stress resistance and suppressing various aging-related effects, including genomic instability, stem cell exhaustion, mitochondrial dysfunction, and telomere shortening. Activating SIRT1 inhibits inflammation by deacetylating NF-κB [161].
The summary of effects of AMPK activation and mTORC1 inhibition on hallmarks of aging is shown in Figure 4.
AMPK activation and mTORC1 inhibition improve every hallmark of aging.
5. Effects of AMPK Activation and mTORC1 Inhibition on Diabetic Complications
Diabetic complications include microangiopathy, such as neuropathy, retinopathy, and nephropathy, and macroangiopathy, such as CVD. The link between AD and type 2 diabetes has been increasingly revealed by research; the danger of developing both AD and type 2 diabetes rises exponentially with age, with type 2 being especially prone to AD [162]. What about the effects of AMPK activation and mTOR inhibition on these diabetic complications and AD?
5.1. Diabetic Neuropathy
Increased ROS, amplified apoptosis, loss of insulinotropic support, neuroinflammation, and decreased autophagy induce the pathogenesis of diabetic neuropathy [163]. Impaired AMPK signaling in dorsal root ganglia neurons is associated with mitochondrial dysfunction and peripheral neuropathy in diabetic models [164]. Activation of AMPK was shown to reverse established losses in thermal sensitivity and epidermal innervation density in streptozotocin-treated rodents [164].
Tang Bi formula (TBF) has been proven effective for diabetic polyneuropathy, while the underlying mechanism remains unclarified. Very recently, TBF ameliorated diabetic polyneuropathy by rectifying mitochondrial dynamic imbalance and modulating the activation of the AMPK-PGC-1α-mitochondrial fusion protein pathway [165].
The altered mTORC1 activity can impair nerve function by affecting essential components like brain-derived neurotrophic factor (BDNF) in Schwann cells, increasing ROS and AGEs, and suppressing beneficial processes like autophagy [166]. Targeting the mTOR pathway with inhibitors or natural compounds shows promise for novel regenerative therapeutic strategies against diabetic complications like neuropathy [167].
5.2. Diabetic Retinopathy (DR)
AMPK is a master metabolic regulator whose activation is being explored as a therapeutic strategy for diabetic retinopathy because it helps maintain cellular energy homeostasis and protects against DR-related complications by modulating stress, inflammation, and cell death. C1q/tumor necrosis factor-related protein-3 (CTRP3) is a newly discovered adipokine and a vital biomarker, predicting DR severity. CTRP3 prevents diabetes-induced retinal vascular permeability by activating the AMPK signaling pathway [168]
Recently, mTORC1 inhibition has been reported as a novel gene therapeutic strategy for DR [169].
5.3. Diabetic Nephropathy
Renal tubulointerstitial fibrosis was a crucial pathological feature of diabetic nephropathy, and renal tubular injury may be associated with abnormal mitophagy. AMPK agonist metformin ameliorated renal oxidative stress and tubulointerstitial fibrosis in high-fat diet and streptozotocin-induced type 2 diabetic mice via activating the AMPK signal pathway [170].
mTORC1 assumes a pivotal role in cellular division, survival, apoptosis delay, and angiogenesis. It is implicated in diverse signaling pathways and has been observed to partake in the progression of diabetic nephropathy by inhibiting autophagy, promoting inflammation, and increasing oxidative stress [171]. Several studies have reported the inhibitory effect of rapamycin on the development and progression of diabetic kidney disease (DKD) in diabetic animal models [172]. mTORC1 has been considered as a new therapeutic approach for DKD, it is desirable to develop safe mTORC1 modulators in the future.
5.4. CVD
Emerging evidence has suggested that AMPK activators from naturally occurring sources provide notable CV benefits [173]. AMPK activation prevents cardiometabolic disease by its capacity to lower blood pressure, plasma glucose and serum lipids, ROS production, and improve NO bioavailability. AMPK activators thus serve as novel potential drugs in gatekeeping CV health and preventing CVD.
mTORC1 pathway plays a dual role in CVD, promoting processes like atherosclerosis, myocardial infarction, and heart failure through effects on cell growth, metabolism, and inflammation [174]. Inhibiting mTORC1 can offer cardioprotective effects, reducing infarct size and slowing disease progression, making mTORC1 a potential therapeutic target for CVD treatment [175].
5.5. AD
AMPK reduces tau phosphorylation and improves brain function in AD-like model [176]. These findings proved that AMPK might be a new target for AD in the future.
Aberrant mTORC1 signaling is associated with AD pathogenesis, contributing to hallmark pathologies like amyloid beta plaques and tau neurofibrillary tangles through increased production and decreased clearance [177,178]. mTORC1 activation also promotes neuroinflammation, oxidative stress, and cognitive decline, while its inhibition, particularly with drugs like rapamycin, has shown promise in preclinical models for clearing protein aggregates and improving cognitive function [177,178].
6. Effects of Nutritional Interventions and Drugs That Improve Aging Hallmarks by AMPK Activation and mTORC1 Inhibition on Type 2 Diabetes
Effects of nutritional interventions and drugs that improve aging hallmarks by AMPK activation and mTORC1 inhibition on type 2 diabetes are shown in Table 1.
Calorie restriction, vitamin E and C, resveratrol, astaxanthin, and n3-polyunsaturated fatty acids (PUFA) have been reported to have both AMPK-activating and mTORC1-inhibitory effects and to be favorably associated with the incidence, remission, and metabolic parameters of type 2 diabetes.
Drugs used to treat type 2 diabetes, metformin, sodium-glucose co-transporter-2 (SGLT2) inhibitors, glucagon-like peptide 1 receptor agonists (GLP-1RAs), and dual glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 RAs, have been reported to have both AMPK-activating and mTORC1-inhibitory effects.
Imeglimin has been reported to enhance AMPK activity [204], however, the effect of imeglimin on mTORC1 has not yet been reported. However, imeglimin and metformin exert similar pharmacological effects on mitochondrial respiration, AMPK activity, and gene expression [204]. Imeglimin may inhibit mTORC1 in the same way as metformin.
7. Conclusions
Type 2 diabetes accelerates clinical manifestations of aging, such as grey hair and skin aging. Metabolic disorders such as hyperglycemia and insulin resistance observed in type 2 diabetes induce harmful factors such as oxidative stress, inflammation, AGEs, and dysbiosis. Such harmful factors induce impaired maintenance of healthy cells by inducing DNA damage, epigenetic alteration, mitochondrial dysfunction, and worsening type 2 diabetes, and developing diabetic complications. Impaired maintenance of healthy cells is induced by an impaired clearance of senescent cells, due to stem cell exhaustion and impaired macro-autophagy. Impaired clearance of senescent cells is unfavorably associated with metabolic disorders and diabetic complications. Thus, type 2 diabetes and aging form a vicious cycle, exacerbating each other's pathology.
AMPK activation and mTOR inhibition improve every hallmark of aging and are favorably associated with diabetic complications such as diabetic neuropathy, DR, DKD, and AD. Calorie restriction, vitamin E and C, resveratrol, astaxanthin, and n3-PUFA have been reported to have both AMPK-activating and mTORC1-inhibitory effects and to be favorably associated with the incidence, remission, and metabolic parameters of type 2 diabetes. Metformin, SGLT2 inhibitors, GLP-1RAs, and dual GIP/GLP-1 RAs, have been reported to have both AMPK-activating and mTORC1-inhibitory effects. The therapeutic interventions that activate AMPK and inhibit mTORC1 may be optimal treatments for type 2 diabetes.
Author Contributions
H.Y., M.H., H.A., and H.K. conceived the review; H. Y. wrote the paper; H.K. edited the paper and provided critical guidance. All authors read and approved the final version of this paper.
Funding
This review research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest in relation to the present review paper.
Abbreviations
| AD | Alzheimer’s disease |
| AGEs | advanced glycation end products |
| AMPK | AMP-activated protein kinase |
| BDNF | brain-derived neurotrophic factor |
| BMD | bone mineral density |
| CTRP3 | C1q/tumor necrosis factor-related protein-3 |
| CI | confidence interval |
| CVD | cardiovascular diseases |
| DR | diabetic retinopathy |
| DKD | Diabetic Kidney disease |
| EVs | extracellular vesicles |
| GIP | glucose-dependent insulinotropic polypeptide |
| GLP-1RAs | glucagon-like peptide 1 receptor agonists |
| HFD | high-fat diet |
| hs-CRP | high-sensitive C-reactive protein |
| IL-6 | interleukin-6 |
| IGF-1 | insulin-like growth factor 1 |
| MD | mean difference |
| NSN | nutrient-sensing network |
| mTORC1 | mammalian target of rapamycin complex 1 |
| NAD+ | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor-kappa B |
| Nrf2 | nuclear factor E2-related factor 2 |
| PGC1α | peroxisome proliferator-activated receptor- γ Coactivator 1α |
| PWV | pulse wave velocity |
| PUFA | polyunsaturated fatty acids |
| RAGE | Receptors for advanced glycation end products |
| ROS | reactive oxygen species |
| SASP | senescence-associated secretory phenotype |
| SGLT2 | sodium glucose co-transporter-2 |
| SIRT | sirtuin |
| TBF | Tang Bi formula |
| TERT | telomerase reverse transcriptase |
| ULK1 | Unc-51-like autophagy activating kinase 1 |
| WAT | White adipose tissue |
References
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal. Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, X.; Luo, J.; Bao, T.; Wang, S.; Wu, X. Molecular mechanisms of aging and anti-aging strategies. Cell. Commun. Signal. 2024, 22, 285. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell. 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Umetani, M.; Nishio, M.; Shigetomi, H.; Imanaka, S.; Hashimoto, H. Molecular Mechanisms of Cellular Senescence in Age-Related Endometrial Dysfunction. Cells. 2025, 14, 858. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell. 2021, 184, 1990–2019. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Tu, C.; Chen, X.; He, R. Advanced Glycation End Products in Disease Development and Potential Interventions. Antioxidants (Basel). 2025, 14, 492. [Google Scholar] [CrossRef]
- DeJong, E.N.; Surette, M.G.; Bowdish, D.M.E. The gut microbiota and unhealthy aging: disentangling cause from Consequence. Cell. Host. Microbe. 2020, 28, 180–189. [Google Scholar] [CrossRef]
- Alsegiani, A.S.; Shah, Z.A. The influence of gut microbiota alteration on age-related neuroinflammation and cognitive decline. Neural. Regeneration. Res. 2022, 17, 2407–2412. [Google Scholar]
- Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA damage and repair in age-related inflammation. Nat. Rev. Immunol. 2023, 23, 75–89. [Google Scholar] [CrossRef]
- Stead, E.R.; Bjedov, I. Balancing DNA repair to prevent ageing and cancer. Exp. Cell. Res. 2021, 405, 112679. [Google Scholar] [CrossRef]
- Huang, B.; Hu, X. Causality of Aging Hallmarks. Aging. Dis. Online ahead of print. 2025. [Google Scholar] [CrossRef]
- Pang, Y.; Zhang, H.; Li, H.; Wang, X.; Wei, P.; Yi, L.; Lin, S. Epigenetic Insights Into Aging: Emerging Roles of Natural Products in Therapeutic Interventions. Phytother Res. Online ahead of print. 2025. [Google Scholar] [CrossRef] [PubMed]
- Kaltsas, A. Multi-Omics Perspectives on Testicular Aging: Unraveling Germline Dysregulation, Niche Dysfunction, and Epigenetic Remodeling. Cells. 2025, 14, 899. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, A.M.; Paulussen, K.J.M.; Overkamp, M.; Goessens, J.P.B.; Kramer, I.F.; Wodzig, W.K.W.H.; Verdijk, L.B.; van Loon, L.J.C. Dose-Dependent Increases in Whole-Body Net Protein Balance and Dietary Protein-Derived Amino Acid Incorporation into Myofibrillar Protein During Recovery from Resistance Exercise in Older Men. J. Nutr. 2019, 149, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Alic, N.; Foley, A.; Cabecinha, M.; Hoddinott, M.P.; Partridge, L. The Ras-Erk-ETS-Signaling Pathway Is a Drug Target for Longevity. Cell. 2015, 162, 72–83. [Google Scholar] [CrossRef]
- Yang, B.A.; Westerhof, T.M.; Sabin, K.; Merajver, S.D.; Aguilar, C.A. Engineered Tools to Study Intercellular Communication. Adv. Sci (Weinh). 2020, 8, 2002825. [Google Scholar] [CrossRef]
- Fafián-Labora, J.A.; O'Loghlen, A. Classical and Nonclassical Intercellular Communication in Senescence and Ageing. Trends. Cell. Biol. 2020, 30, 628–639. [Google Scholar] [CrossRef]
- Zhu, D.; Li, X.; Tian, Y. Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends. Biochem. Sci. 2022, 47, 645–659. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell. 2020, 183, 76–93. [Google Scholar] [CrossRef]
- Igarashi, M.; Miura, M.; Williams, E.; Jaksch, F.; Kadowaki, T.; Yamauchi, T.; Guarente, L. NAD+ supplementation rejuvenates aged gut adult stem cells. Aging. Cell. 2019, 18, e12935. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal. Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Li, Q.; Qian, Z.; Huang, Y.; Yang, X.; Yang, J.; Xiao, N.; Liang, G.; Zhang, H.; Fu, Y.; Lin, Y.; et al. Mechanisms of endothelial senescence and vascular aging. Biogerontology. 2025, 26, 128. [Google Scholar] [CrossRef]
- Kalamakis, G.; Brüne, D.; Ravichandran, S.; Bolz, J.; Fan, W.; Ziebell, F.; Stiehl, T.; Catalá-Martinez, F.; Kupke, J.; Zhao, S.; et al. Quiescence Modulates Stem Cell Maintenance and Regenerative Capacity in the Aging Brain. Cell. 2019, 176, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Lawton, A.; Tripodi, N.; Feehan, J. Running on empty: Exploring stem cell exhaustion in geriatric musculoskeletal disease. Maturitas. 2024, 188, 108066. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Tasset, I.; Arias, E.; Pampliega, O.; Wong, E.; Martinez-Vicente, M.; Cuervo, A.M. Autophagy and the hallmarks of aging. Ageing. Res. Rev. 2021, 72, 101468. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, L.D.; Narita, M. Autophagy at the intersection of aging, senescence, and cancer. Mol. Oncol. 2022, 16, 3259–3275. [Google Scholar] [CrossRef]
- Rio, P.; Caldarelli, M.; Miccoli, E.; Guazzarotti, G.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Sex Differences in Immune Responses to Infectious Diseases: The Role of Genetics, Hormones, and Aging. Diseases. 2025, 13, 179. [Google Scholar] [CrossRef]
- Matina, S.S.; Cohen, E.; Mokwena, K.; Mendenhall, E. Menopause and aging in sub-Saharan Africa: a narrative review. Climacteric. 2025, 28, 230–241. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing. Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef]
- Aydin, S.; Tekinalp, S.G.; Tuzcu, B.; Cam, F.; Sevik, M.O.; Tatar, E.; Deepak Kalaskarf, D.; Emin Cam, M.E. The role of AMP-activated protein kinase activators on energy balance and cellular metabolism in type 2 diabetes mellitus. Obesity. Medicine. 2025, 53, 100577. [Google Scholar] [CrossRef]
- Mannick, J.B.; Lamming, D.W. Targeting the biology of aging with mTOR inhibitors. Nat. Aging. 2023, 3, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Bar-Tana, J. Type 2 diabetes - unmet need, unresolved pathogenesis, mTORC1-centric paradigm. Rev. Endocr. Metab. Disord. 2020, 21, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Miao, Y.; Hu, Z. Research progress on the regulation of grey hair production by disruption of melanocyte stem cell homeostasis. Chin. J. Plast. Surg. 2017, 33, 313–316. [Google Scholar]
- Yang, K.; Han, X. Lipidomics: Techniques, applications, and outcomes related to biomedical sciences. Trends. Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, S.; He, C. Lipidomics Combined with Network Pharmacology to Explore Differences in the Mechanisms of Grey Hair Development Between Type 2 Diabetes Mellitus and Normal Populations (Female). Int. J. Mol. Sci. 2025, 26, 2034. [Google Scholar] [CrossRef]
- Moraes, V.R.; Melo, M.O.; Maia Campos, P.M.B.G. Evaluation of Morphological and Structural Skin Alterations on Diabetic Subjects by Biophysical and Imaging Techniques. Life (Basel). 2023, 13, 579. [Google Scholar] [CrossRef]
- Haddad, R.; Bogdanski, E.; Mattei, A.; Michel, J.; Giovanni, A. Presbyphonia: A Scoping Review for a Comprehensive Assessment of Aging Voice. J. Voice. 2024, S0892-1997, 432–436. [Google Scholar] [CrossRef]
- Santos, M.; Azevedo, S.; Sousa, F.; Machado, A.S.; Santos, P.C.; Freitas, S.V.; Almeida, E.; Sousa, C.; da Silva, Á.M. Presbylarynx: Is It a Sign of the Health Status of the Elderly? J. Voice. 2023, 37, e1–e304. [Google Scholar] [CrossRef]
- Samocha-Bonet, D.; Wu, B.; Ryugo, D.K. Diabetes mellitus and hearing loss: A review. Ageing. Res. Rev. 2021, 71, 101423. [Google Scholar] [CrossRef]
- Antal, B.; McMahon, L.P.; Sultan, S.F.; Lithen, A.; Wexler, D.J.; Dickerson, B.; Ratai, E.M.; Mujica-Parodi, L.R. Type 2 diabetes mellitus accelerates brain aging and cognitive decline: Complementary findings from UK Biobank and meta-analyses. Elife. 2022, 11, e73138. [Google Scholar] [CrossRef]
- Li, M.; Sun, H.; Chen, H.; Ma, W.; Li, Y. Type 2 diabetes and bone mineral density: A meta-analysis and systematic review. Medicine (Baltimore). 2024, 103, e40468. [Google Scholar] [CrossRef]
- Laurent, S.; Boutouyrie, P.; Asmar, R.; Gautier, I.; Laloux, B.; Guize, L.; Ducimetiere, P.; Benetos, A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001, 37, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, C.; Aznaouridis, K.; Stefanadis, C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 2010, 55, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.F.; Hwang, S.J.; Vasan, R.S.; Larson, M.G.; Pencina, M.J.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010, 121, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.L.; Lee, J.M.; Seo, J.B.; Chung, W.Y.; Kim, S.H.; Zo, J.H.; Kim, M.A. The effects of metabolic syndrome and its components on arterial stiffness in relation to gender. J. Cardiol. 2015, 65, 243–249. [Google Scholar] [CrossRef]
- Masding, M.G.; Stears, A.J.; Burdge, G.C.; Wootton, S.A.; Sandeman, D.D. Premenopausal advantages in postprandial lipid metabolism are lost in women with type 2 diabetes. Diabetes. Care. 2003, 26, 3243–3249. [Google Scholar] [CrossRef]
- Madonna, R.; Balistreri, C.R.; De Rosa, S.; Muscoli, S.; Selvaggio, S.; Selvaggio, G.; Ferdinandy, P.; De Caterina, R. Impact of sex differences and diabetes on coronary atherosclerosis and ischemic heart disease. J. Clin. Med. 2019, 8, 98. [Google Scholar] [CrossRef]
- Laurent, S.; Cockcroft, J.; Van Bortel, L.; Boutouyrie, P.; Giannattasio, C.; Hayoz, D.; Pannier, B.; Vlachopoulos, C.; Wilkinson, I.; Struijker-Boudier, H.; et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur. Heart. J. 2006, 27, 2588–2605. [Google Scholar] [CrossRef]
- Townsend, R.R.; Wilkinson, I.B.; Schiffrin, E.L.; Avolio, A.P.; Chirinos, J.A.; Cockcroft, J.R.; Heffernan, K.S.; Lakatta, E.G.; McEniery, C.M.; Mitchell, G.F.; et al. Recommendations for improving and standardizing vascular research on arterial stiffness: a scientific statement from the American Heart Association. Hypertension. 2015, 66, 698–722. [Google Scholar] [CrossRef]
- Yapei, Y.; Xiaoyan, R.; Sha, Z.; Li, P.; Xiao, M.; Shuangfeng, C.; Lexin, W.; Lianqun, C. Clinical Significance of Arterial Stiffness and Thickness Biomarkers in Type 2 Diabetes Mellitus: An Up-To-Date Meta-Analysis. Med. Sci. Monit. 2015, 21, 2467–2475. [Google Scholar]
- Szablewski, L. Changes in Cells Associated with Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 2397. [Google Scholar] [CrossRef]
- Takahashi, F.; Hashimoto, Y.; Kaji, A.; Sakai, R.; Okamura, T.; Kitagawa, N.; Okada, H.; Nakanishi, N.; Majima, S.; Senmaru, T.; et al. Sarcopenia Is Associated With a Risk of Mortality in People With Type 2 Diabetes Mellitus. Front. Endocrinol (Lausanne). 2021, 12, 783363. [Google Scholar] [CrossRef] [PubMed]
- Laohajaroensombat, O.; Limpaarayakul, T.; Sathavarodom, N.; Boonyavarakul, A.; Samakkarnthai, P. A comparative analysis of sarcopenia screening methods in Thai people with type 2 diabetes mellitus in an outpatient setting. BMC. Geriatr. 2025, 25, 346. [Google Scholar] [CrossRef] [PubMed]
- Iakovou, E.; Kourti, M.A. Comprehensive Overview of the Complex Role of Oxidative Stress in Aging, The Contributing Environmental Stressors and Emerging Antioxidant Therapeutic Interventions. Front. Aging. Neurosci. 2022, 14, 827900. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009, 30, 2–10. [Google Scholar] [CrossRef]
- Erusalimsky, JD. Oxidative stress, telomeres and cellular senescence: What non-drug interventions might break the link? Free. Radic. Biol. Med. 2020, 150, 87–95. [Google Scholar] [CrossRef]
- Yesupatham, A.; Saraswathy, R. Role of oxidative stress in prediabetes development. Biochem. Biophys. Rep. 2025, 43, 102069. [Google Scholar] [CrossRef]
- Dawi, J.; Misakyan, Y.; Affa, S.; Kades, S.; Narasimhan, A.; Hajjar, F.; Besser, M.; Tumanyan, K.; Venketaraman, V. Oxidative Stress, Glutathione Insufficiency, and Inflammatory Pathways in Type 2 Diabetes Mellitus: Implications for Therapeutic Interventions. Biomedicines. 2024, 13, 18. [Google Scholar] [CrossRef]
- Klisic, A.; Karakasis, P.; Patoulias, D.; Khalaji, A.; Ninić, A. Are oxidative stress biomarkers reliable part of multimarker panel in female patients with type 2 diabetes mellitus. Metab. Syndrome. Related. Disord. 2024, 22, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, L.; Liu, J.; Li, D. Litchi pericarp extract treats type 2 diabetes mellitus by regulating oxidative stress, inflammatory response, and energy metabolism. Antioxidants (Basel Switzerland). 2024, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Schurman, S.H.; Bektas, A.; Kaileh, M.; Roy, R.; Wilson, D.M. 3rd.; Sen, R.; Ferrucci, L. Aging and Inflammation. Cold. Spring. Harb. Perspect. Med. 2024, 14, a041197. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, V.; La Grotta, R.; Carreras, F.; Giuliani, A.; Sabbatinelli, J.; Olivieri, F.; Berra, C.C.; Ceriello, A.; Prattichizzo, F. Inflammatory Trajectory of Type 2 Diabetes: Novel Opportunities for Early and Late Treatment. Cells. 2024, 13, 1662. [Google Scholar] [CrossRef]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes. Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Shamsi, A.; Shahwan, M.; Husain, F.M.; Khan, M.S. Characterization of methylglyoxal induced advanced glycation end products and aggregates of human transferrin: Biophysical and microscopic insight. Int. J. Biol. Macromol. 2019, 138, 718–724. [Google Scholar] [CrossRef]
- Li, Q.; Wen, Y.; Wang, L.; Chen, B.; Chen, J.; Wang, H.; Chen, L. Hyperglycemia-induced accumulation of advanced glycosylation end products in fibroblast-like synoviocytes promotes knee osteoarthritis. Exp. Mol. Med. 2021, 53, 1735–1747. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Rajaobelina, K.; Cougnard-Gregoire, A.; Delcourt, C.; Gin, H.; Barberger-Gateau, P.; Rigalleau, V. Autofluorescence of skin advanced glycation end products: marker of metabolic memory in elderly population. J. Gerontol. A. Biol. Sci. Med. Sci. 2015, 70, 841–846. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules. 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules. 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jeong, M.S.; Jang, S.B. Molecular characteristics of RAGE and advances in small-molecule inhibitors. Int. J. Mol. Sci. 2021, 22, 6904. [Google Scholar] [CrossRef] [PubMed]
- Mengstie, M.A.; Chekol Abebe, E.; Behaile Teklemariam, A.; Tilahun Mulu, A.; Agidew, M.M.; Teshome Azezew, M.; Zewde, E.A.; Agegnehu Teshome, A. Endogenous advanced glycation end products in the pathogenesis of chronic diabetic complications. Front. Mol. Biosci. 2022, 9, 1002710. [Google Scholar] [CrossRef]
- Kanimozhi, N.V.; Sukumar, M. Aging through the lens of the gut microbiome: Challenges and therapeutic opportunities. Archives of Gerontology and Geriatrics Plus 2 2025, 100142. [Google Scholar]
- Hamjane, N.; Mechita, M.B.; Nourouti, N.G.; Barakat, A. Gut microbiota dysbiosis-associated obesity and its involvement in cardiovascular diseases and type 2 diabetes. A systematic review. Microvasc. Res. 2024, 151, 104601. [Google Scholar] [CrossRef]
- Yu, Y.; Ding, Y.; Wang, S.; Jiang, L. Gut Microbiota Dysbiosis and Its Impact on Type 2 Diabetes: From Pathogenesis to Therapeutic Strategies. Metabolites. 2025, 15, 397. [Google Scholar] [CrossRef]
- Lazar, E.; Sherzai, A.; Adeghate, J.; Sherzai, D. Gut dysbiosis, insulin resistance and Alzheimer's disease: review of a novel approach to neurodegeneration. Front. Biosci (Schol Ed). 2021, 13, 17–29. [Google Scholar] [CrossRef]
- Smith, U.; Li, Q.; Rydén, M.; Spalding, K.L. Cellular senescence and its role in white adipose tissue. Int. J. Obes (Lond). 2021, 45, 934–943. [Google Scholar] [CrossRef]
- Franzke, B.; Schwingshackl, L.; Wagner, K.H. Chromosomal damage measured by the cytokinesis block micronucleus cytome assay in diabetes and obesity - A systematic review and meta-analysis. Mutat. Res. Rev. Mutat. Res. 2020, 786, 108343. [Google Scholar] [CrossRef]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: history, health, and hallmarks of aging. Cell. 2021, 184, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and cell senescence-size matters not. EBioMedicine. 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care. 2011, 14, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Takubo, K.; Aida, J.; Araki, A.; Ito, H. Telomere attrition and diabetes mellitus. Geriatr. Gerontol. Int. 2016, 16 Suppl 1, 66–74. [Google Scholar] [CrossRef]
- Sampson, M.J.; Hughes, D.A. Chromosomal telomere attrition as a mechanism for the increased risk of epithelial cancers and senescent phenotypes in type 2 diabetes. Diabetologia. 2006, 49, 1726–1731. [Google Scholar] [CrossRef]
- Bure, I.V.; Nemtsova, M.V.; Kuznetsova, E.B. Histone modifications and non-coding RNAs: mutual epigenetic regulation and role in pathogenesis. Int. J. Mol. Sci. 2022, 23, 5801. [Google Scholar] [CrossRef]
- Sherazi, S.A.M.; Abbasi, A.; Jamil, A.; Uzair, M.; Ikram, A.; Qamar, S.; Olamide, A.A.; Arshad, M.; Fried, P.J.; Ljubisavljevic, M.; et al. Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases. Neural. Regen. Res. 2023, 18, 959–968. [Google Scholar]
- Yi, S.J.; Kim, K. New Insights into the Role of Histone Changes in Aging. Int. J. Mol. Sci. 2020, 21, 8241. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 2012, 61, 289–317. [Google Scholar]
- Gharipour, M.; Mani, A.; Amini Baghbahadorani, M.; de Souza Cardoso, C.K.; Jahanfar, S.; Sarrafzadegan, N.; de Oliveira, C.; Silveira, E.A. How Are Epigenetic Modifications Related to Cardiovascular Disease in Older Adults? Int. J. Mol. Sci. 2021, 22, 9949. [Google Scholar] [CrossRef]
- Long, Y.; Mao, C.; Liu, S.; Tao, Y.; Xiao, D. Epigenetic modifications in obesity-associated diseases. MedComm (2020). 2024, 5, e496. [Google Scholar] [CrossRef] [PubMed]
- Rönn, T.; Ofori, J.K.; Perfilyev, A.; Hamilton, A.; Pircs, K.; Eichelmann, F.; Garcia-Calzon, S.; Karagiannopoulos, A.; Stenlund, H.; Wendt, A.; et al. Genes with epigenetic alterations in human pancreatic islets impact mitochondrial function, insulin secretion, and type 2 diabetes. Nat. Commun. 2023, 14, 8040. [Google Scholar] [CrossRef] [PubMed]
- Odimegwu, C.L.; Uwaezuoke, S.N.; Chikani, U.N.; Mbanefo, N.R.; Adiele, K.D.; Nwolisa, C.E.; Eneh, C.I.; Ndiokwelu, C.O.; Okpala, S.C.; Ogbuka, F.N.; et al. Targeting the Epigenetic Marks in Type 2 Diabetes Mellitus: Will Epigenetic Therapy Be a Valuable Adjunct to Pharmacotherapy? Diabetes. Metab. Syndr. Obes. 2024, 17, 3557–3576. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Chakraborty, R.; Faizy, A.F.; Moin, S. Exploring the key role of DNA methylation as an epigenetic modulator in oxidative stress related islet cell injury in patients with type 2 diabetes mellitus: a review. J. Diabetes. Metab. Disord. 2024, 23, 1699–1718. [Google Scholar] [CrossRef]
- Vasishta, S.; Ammankallu, S.; Poojary, G.; Gomes, S.M.; Ganesh, K.; Umakanth, S.; Adiga, P.; Upadhya, D.; Prasad, T.S.K.; Joshi, M.B. High glucose induces DNA methyltransferase 1 dependent epigenetic reprogramming of the endothelial exosome proteome in type 2 diabetes. Int. J. Biochem. Cell. Biol. 2024, 176, 106664. [Google Scholar] [CrossRef]
- Li, D.L.; Hodge, A.M.; Southey, M.C.; Giles, G.G.; Dugué, P.A. Association of Epigenetic Markers of Aging With Prevalent and Incident Type 2 Diabetes. J. Gerontol. A. Biol. Sci. Med. Sci. 2025, 80, glaf085. [Google Scholar] [CrossRef]
- Fry, C.S.; Rasmussen, B.B. Skeletal muscle protein balance and metabolism in the elderly. Curr. Aging. Sci. 2011, 4, 260–268. [Google Scholar] [CrossRef]
- Wall, B.T.; Gorissen, S.H.; Pennings, B.; Koopman, R.; Groen, B.B.L.; Verdijk, L.B.; van Loon, L.J.C. Aging is accompanied by a blunted muscle protein synthetic response to protein ingestion. PLoS. ONE. 2015, 10, e0140903. [Google Scholar] [CrossRef]
- Katsanos, C.S.; Kobayashi, H.; Sheffield-Moore, M.; Aarsland, A.; Wolfe, R.R. Aging is associated with diminished accretion of muscle proteins after the ingestion of a small bolus of essential amino acids. Am. J. Clin. Nutr. 2005, 82, 1065–1073. [Google Scholar] [CrossRef]
- Cuthbertson, D.; Smith, K.; Babraj, J.; Leese, G.; Waddell, T.; Atherton, P.; Wackerhage, H.; Taylor, P.M.; Rennie, M.J. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB. J. 2005, 19, 422–424. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Andersen, H.; Nielsen, S.; Mogensen, C.E.; Jakobsen, J. Muscle strength in type 2 diabetes. Diabetes. 2004, 53, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Goodpaster, B.H.; Strotmeyer, E.S.; Kuller, L.H.; Broudeau, R.; Kammerer, C.; de Rekeneire, N.; Harris, T.B.; Schwartz, A.V.; Tylavsky, F.A.; Cho, Y.; Newman, A.B. Health, Aging, and Body Composition Study Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes. Care. 2007, 30, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.Y.; Wei, H.J.; Tang, Y.Y. Isthmin: A multifunctional secretion protein. Cytokine. 2024, 173, 156423. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhao, M.; Voilquin, L.; Jung, Y.; Aikio, M.A.; Sahai, T.; Dou, F.Y.; Roche, A.M.; Carcamo-Orive, I.; Knowles, J.W.; et al. Isthmin-1 is an adipokine that promotes glucose uptake and improves glucose tolerance and hepatic steatosis. Cell. Metab. 2021, 33, 1836–1852. [Google Scholar] [CrossRef]
- Zhao, M.; Banhos Danneskiold-Samsøe, N.; Ulicna, L.; Nguyen, Q.; Voilquin, L.; Lee, D.E.; White, J.P.; Jiang, Z.; Cuthbert, N.; et al. Phosphoproteomic mapping reveals distinct signaling actions and activation of muscle protein synthesis by Isthmin-1. Elife. 2022, 11, e80014. [Google Scholar] [CrossRef]
- Yang, K.; Hou, R.; Zhao, J.; Wang, X.; Wei, J.; Pan, X.; Zhu, X. Lifestyle effects on aging and CVD: A spotlight on the nutrient-sensing network. Ageing. Res. Rev. 2023, 92, 102121. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell. 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell. Biol. 2022, 23, 56–73. [Google Scholar] [CrossRef]
- Yang, G.; Cao, X.; Li, X.; Zhang, J.; Ma, C.; Zhang, N.; Lu, Q.; Crimmins, E.M.; Gill, T.M.; Chen, X.; Liu, Z. Association of Unhealthy Lifestyle and Childhood Adversity With Acceleration of Aging Among UK Biobank Participants. JAMA. Netw. Open. 2022, 5, e2230690. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Kelly, G.; Korolchuk, V.I.; Girao, H. Intercellular communication and aging. Aging, Fundam. Biol. Soc. Impact 2023, 257–274. [Google Scholar]
- Akbar, N.; Azzimato, V.; Choudhury, R.P.; Aouadi, M. Extracellular vesicles in metabolic disease. Diabetologia. 2019, 62, 2179–2187. [Google Scholar] [CrossRef]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia. 2020, 63, 1990–1998. [Google Scholar] [CrossRef]
- Guo, Y.; Guan, T.; Shafiq, K.; Yu, Q.; Jiao, X.; Na, D.; Li, M.; Zhang, G.; Kong, J. Mitochondrial dysfunction in aging. Ageing Res Rev. 2023, 88, 101955. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Iheagwam, F.N.; Joseph, A.J.; Adedoyin, E.D.; Iheagwam, O.T.; Ejoh, S.A. Mitochondrial Dysfunction in Diabetes: Shedding Light on a Widespread Oversight. Pathophysiology. 2025, 32, 9. [Google Scholar] [CrossRef]
- Prolla, T.A.; Denu, J.M. NAD+ deficiency in age-related mitochondrial dysfunction. Cell. Metab. 2014, 19, 178–180. [Google Scholar] [CrossRef] [PubMed]
- McReynolds, M.R.; Chellappa, K.; Baur, J.A. Age-related NAD+ decline. Exp. Gerontol. 2020, 134, 110888. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell. Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Santhana, S.; Xuan, W.; Bisht, K.; Tipparaju, S. Nampt: a new therapeutic target for modulating NAD(+) levels in metabolic, cardiovascular, and neurodegenerative diseases. Can. J. Physiol. Pharmacol. 2025, 103, 208–224. [Google Scholar] [CrossRef]
- Ruskovska, T.; Bernlohr, D.A. The Role of NAD(+) in Metabolic Regulation of Adipose Tissue: Implications for Obesity-Induced Insulin Resistance. Biomedicines. 2023, 11, 2560. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d'Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell. Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: a key therapeutic target in aging and diseases. J. Clin. Invest. 2022, 132, e158450. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell. Metab. 2019, 30, 129–142. [Google Scholar] [CrossRef]
- Yan, X.; Gao, Z.; Zhou, Y.; Gao, F.; Li, Q. Expressions of IGF-1R and Ki-67 in breast cancer patients with diabetes mellitus and an analysis of biological characteristics. Pak. J. Med. Sci. 2022, 38, 281–286. [Google Scholar] [CrossRef]
- Tudurí, E.; Soriano, S.; Almagro, L.; García-Heredia, A.; Rafacho, A.; Alonso-Magdalena, P.; Nadal, Á.; Quesada, I. (2022). The effects of aging on male mouse pancreatic β-cell function involve multiple events in the regulation of secretion: influence of insulin sensitivity. Journals. Gerontology. Ser. A, Biol. Sci. Med. Sci. 2022, 77, 405–415. [Google Scholar] [CrossRef]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. p53 functions in adipose tissue metabolism and homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef]
- Murakami, T.; Inagaki, N.; Kondoh, H. Cellular senescence in diabetes mellitus: distinct senotherapeutic strategies for adipose tissue and pancreatic β cells. Front. Endocrinol. 2022, 13, 869414. [Google Scholar] [CrossRef]
- Mi, L.; Hu, J.; Li, N.; Gao, J.; Huo, R.; Peng, X.; Zhang, N.; Liu, Y.; Zhao, H.; Liu, R.; et al. The Mechanism of Stem Cell Aging. Stem. Cell. Rev. Rep. 2022, 18, 1281–1293. [Google Scholar] [CrossRef]
- Terenzi, D.C.; Trac, J.Z.; Teoh, H.; Gerstein, H.C.; Bhatt, D.L.; Al-Omran, M.; Verma, S.; Hess, D.A. Vascular Regenerative Cell Exhaustion in Diabetes: Translational Opportunities to Mitigate Cardiometabolic Risk. Trends. Mol. Med. 2019, 25, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Ciciliot, S.; Albiero, M. Concise Review: Perspectives and Clinical Implications of Bone Marrow and Circulating Stem Cell Defects in Diabetes. Stem. Cells. 2017, 35, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging. 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in Age-Associated Neurodegeneration. Cells. 2018, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Lee, M.S. Macroautophagy in homeostasis of pancreatic beta-cell. Autophagy. 2009, 5, 241–243. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef]
- Vivot, K.; Pasquier, A.; Goginashvili, A.; Ricci, R. Breaking Bad and Breaking Good: beta-Cell Autophagy Pathways in Diabetes. J. Mol. Biol. 2020, 432, 1494–1513. [Google Scholar] [CrossRef]
- Muralidharan, C.; Conteh, A.M.; Marasco, M.R.; Crowder, J.J.; Kuipers, J.; de Boer, P.; Linnemann, A.K. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia. 2021, 64, 865–877. [Google Scholar] [CrossRef]
- Salemkour, Y.; Lenoir, O. Endothelial Autophagy Dysregulation in Diabetes. Cells. 2023, 12, 947. [Google Scholar] [CrossRef]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell. Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef]
- Sorrenti, V.; Benedetti, F.; Buriani, A.; Fortinguerra, S.; Caudullo, G.; Davinelli, S.; Zella, D.; Scapagnini, G. Immunomodulatory and Antiaging Mechanisms of Resveratrol, Rapamycin, and Metformin: Focus on mTOR and AMPK Signaling Networks. Pharmaceuticals (Basel). 2022, 15, 912. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Guo, Z.; Song, C. AMPK, a key molecule regulating aging-related myocardial ischemia-reperfusion injury. Mol. Biol. Rep. 2024, 51, 257. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell. Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing. Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell. Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Fernandez, E.; Aragones, G.; Renneburg, C.; Francisco, S.G.; Kageyama, S.; Komatsu, M.; Taylor, A. Targeting mTOR pathway to ameliorate glycation-derived proteotoxicity in retinal and lens cells. Invest. Ophthalmol. Vis. Sci. 2020, 61, 3099. [Google Scholar]
- Sun, X.; Zhu, M.J. AMP-activated protein kinase: a therapeutic target in intestinal diseases. Open. Biol. 2017, 7, 170104. [Google Scholar] [CrossRef]
- Gongol, B.; Sari, I.; Bryant, T.; Rosete, G.; Marin, T. AMPK: An Epigenetic Landscape Modulator. Int. J. Mol. Sci. 2018, 19, 3238. [Google Scholar] [CrossRef]
- Ding, N.; Maiuri, A.R.; O'Hagan, H.M. The emerging role of epigenetic modifiers in repair of DNA damage associated with chronic inflammatory diseases. Mutat. Res. Rev. Mutat. Res. 2019, 780, 69–81. [Google Scholar] [CrossRef]
- Smith, E.M.; Pendlebury, D.F.; Nandakumar, J. Structural biology of telomeres and telomerase. Cell. Mol. Life. Sci. 2020, 77, 61–79. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, S.G.; Park, S.Y.; Kim, J.R.; Choi, H.C. Telomere stabilization by metformin mitigates the progression of atherosclerosis via the AMPK-dependent p-PGC-1α pathway. Exp. Mol. Med. 2024, 56, 1967–1979. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Res. 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel). 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sauve, A.A. NAD+ content and its role in mitochondria. Methods. Mol. Biol. 2015, 1241, 39–48. [Google Scholar]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Begum, M.K.; Konja, D.; Singh, S.; Chlopicki, S.; Wang, Y. Endothelial SIRT1 as a target for the prevention of arterial aging: promises and challenges. J. Cardiovasc. Pharm. 2021, 78, S63–S77. [Google Scholar] [CrossRef]
- Qi, W.; Hu, C.; Zhao, D.; Li, X. SIRT1-SIRT7 in diabetic kidney disease: biological functions and molecular mechanisms. Front. Endocrinol. (Lausanne), 2022, 13, 801303. [Google Scholar] [CrossRef]
- Sun, C.; Bai, S.; Liang, Y.; Liu, D.; Liao, J.; Chen, Y.; Zhao, X.; Wu, B.; Huang, D.; Chen, M.; Wu, D. The role of Sirtuin 1 and its activators in age-related lung disease. Biomed. Pharmacother. 2023, 162, 114573. [Google Scholar] [CrossRef]
- He, S.; Wang, Y.; Liu, J.; Li, P.; Luo, X.; Zhang, B. Activating SIRT1 deacetylates NF-κB p65 to alleviate liver inflammation and fibrosis via inhibiting NLRP3 pathway in macrophages. Int. J. Med. Sci. 2023, 20, 505–519. [Google Scholar] [CrossRef]
- Peng, Y.; Yao, S.Y.; Chen, Q.; Jin, H.; Du, M.Q.; Xue, Y.H.; Liu, S. True or false? Alzheimer's disease is type 3 diabetes: Evidences from bench to bedside. Ageing. Res. Rev. 2024, 99, 102383. [Google Scholar] [CrossRef]
- Yerra, V.G.; Areti, A.; Kumar, A. Adenosine Monophosphate-Activated Protein Kinase Abates Hyperglycaemia-Induced Neuronal Injury in Experimental Models of Diabetic Neuropathy: Effects on Mitochondrial Biogenesis, Autophagy and Neuroinflammation. Mol. Neurobiol. 2017, 54, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Roy Chowdhury, S.K.; Smith, D.R.; Saleh, A.; Schapansky, J.; Marquez, A.; Gomes, S.; Akude, E.; Morrow, D.; Calcutt, N.A.; Fernyhough, P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain. 2012, 135, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yu, H.; Zhou, L.; Su, H.; Li, X.; Qi, W.; Lian, F. Tang Bi formula alleviates diabetic sciatic neuropathy via AMPK/PGC-1α/MFN2 pathway activation. Sci. Rep. 2025, 15, 25069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Lv, X.; Du, W.; Cheng, M.J.; Liu, Y.P.; Zhu, L.; Hao, J. The Akt/mTOR cascade mediates high glucose-induced reductions in BDNF via DNMT1 in Schwann cells in diabetic peripheral neuropathy. Exp. Cell. Res. 2019, 383, 111502. [Google Scholar] [CrossRef]
- Noori, T.; Sureda, A.; Shirooie, S. Role of natural mTOR inhibitors in treatment of diabetes mellitus. Fundam. Clin. Pharmacol. 2023, 37, 461–479. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, C.; Meng, Z.; Gan, L.; Guo, R.; Liu, J.; Bond Lau, W.; Xie, D.; Zhao, J.; Lopez, B.L.; et al. C1q/TNF-Related Protein 3 Prevents Diabetic Retinopathy via AMPK-Dependent Stabilization of Blood-Retinal Barrier Tight Junctions. Cells. 2022, 11, 779. [Google Scholar] [CrossRef]
- Lee, S.H.S.; Lee, J.Y.; Choi, J.S.; Kim, H.J.; Kim, J.; Cha, S.; Lee, K.J.; Woo, H.N.; Park, K.; Lee, H. mTOR inhibition as a novel gene therapeutic strategy for diabetic retinopathy. PLoS. One. 2022, 17, e0269951. [Google Scholar] [CrossRef]
- Han, Y.C.; Tang, S.Q.; Liu, Y.T.; Li, A.M.; Zhan, M.; Yang, M.; Song, N.; Zhang, W.; Wu, X.Q.; Peng, C.H.; et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell. Death. Dis. 2021, 12, 925. [Google Scholar] [CrossRef]
- Shi, J.; Liu, X.; Jiao, Y.; Tian, J.; An, J.; Zou, G.; Zhuo, L. mTOR pathway: A key player in diabetic nephropathy progression and therapeutic targets. Genes. Dis. 2024, 12, 101260. [Google Scholar] [CrossRef]
- Yasuda-Yamahara, M.; Kume, S.; Maegawa, H. Roles of mTOR in Diabetic Kidney Disease. Antioxidants (Basel). 2021, 10, 321. [Google Scholar] [CrossRef]
- Heidary Moghaddam, R.; Samimi, Z.; Asgary, S.; Mohammadi, P.; Hozeifi, S.; Hoseinzadeh-Chahkandak, F.; Xu, S.; Farzaei, M.H. Natural AMPK Activators in Cardiovascular Disease Prevention. Front. Pharmacol. 2022, 12, 738420. [Google Scholar] [CrossRef]
- Kaldirim, M.; Lang, A.; Pfeiler, S.; Fiegenbaum, P.; Kelm, M.; Bönner, F.; Gerdes, N. Modulation of mTOR Signaling in Cardiovascular Disease to Target Acute and Chronic Inflammation. Front. Cardiovasc. Med. 2022, 9, 907348. [Google Scholar] [CrossRef]
- Guo, J.; Wu, Y.; Wan, Z.; Zhang, Z. Post-translational modifications orchestrate mTOR-driven cell death in cardiovascular disease. Front. Cardiovasc. Med. 2025, 12, 1620669. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, N.; Shi, F.X.; Xu, W.Q.; Cao, Y.; Lei, Y.; Wang, J.Z.; Tian, Q.; Zhou, X.W. Upregulation of AMPK Ameliorates Alzheimer's Disease-Like Tau Pathology and Memory Impairment. Mol. Neurobiol. 2020, 57, 3349–3361. [Google Scholar] [CrossRef] [PubMed]
- Mueed, Z.; Tandon, P.; Maurya, S.K.; Deval, R.; Kamal, M.A.; Poddar, N.K. Tau and mTOR: The Hotspots for Multifarious Diseases in Alzheimer's Development. Front. Neurosci. 2019, 12, 1017. [Google Scholar] [CrossRef] [PubMed]
- Rapaka, D.; Bitra, V.R.; Challa, S.R.; Adiukwu, P.C. mTOR signaling as a molecular target for the alleviation of Alzheimer's disease pathogenesis. Neurochem. Int. 2022, 155, 105311. [Google Scholar] [CrossRef]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell. Metab. 2017, 26, 884–896. [Google Scholar] [CrossRef]
- Tulsian, R.; Velingkaar, N.; Kondratov, R. Caloric restriction effects on liver mTOR signaling are time-of-day dependent. Aging (Albany NY). 2018, 10, 1640–1648. [Google Scholar] [CrossRef]
- Jayedi, A.; Zeraattalab-Motlagh, S.; Shahinfar, H.; Gregg, E.W.; Shab-Bidar, S. Effect of calorie restriction in comparison to usual diet or usual care on remission of type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2023, 117, 870–882. [Google Scholar] [CrossRef]
- Pan, J.; Li, Z.; Zhu, M.; Guo, L.; Chen, W.; Yu, L. Vitamin E exerts a mitigating effect on LPS-induced acute lung injury by regulating macrophage polarization through the AMPK/NRF2/NF-κB pathway. Int. Immunopharmacol. 2025, 159, 114893. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hou, L.; Song, H.; Xu, P.; Sun, Y.; Wu, K. Akt/AMPK/mTOR pathway was involved in the autophagy induced by vitamin E succinate in human gastric cancer SGC-7901 cells. Mol. Cell. Biochem. 2017, 424, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Asbaghi, O.; Nazarian, B.; Yousefi, M.; Anjom-Shoae, J.; Rasekhi, H.; Sadeghi, O. Effect of vitamin E intake on glycemic control and insulin resistance in diabetic patients: an updated systematic review and meta-analysis of randomized controlled trials. Nutr. J. 2023, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Min, S.; Hong, R.; Zou, M.; Zhou, D. High-dose Vitamin C inhibits PD-L1 expression by activating AMPK in colorectal cancer. Immunobiology. 2025, 230, 152893. [Google Scholar] [CrossRef]
- Qin, S.; Wang, G.; Chen, L.; Geng, H.; Zheng, Y.; Xia, C.; Wu, S.; Yao, J.; Deng, L. Pharmacological vitamin C inhibits mTOR signaling and tumor growth by degrading Rictor and inducing HMOX1 expression. PLoS. Genet. 2023, 19, e1010629. [Google Scholar] [CrossRef]
- Nosratabadi, S.; Ashtary-Larky, D.; Hosseini, F.; Namkhah, Z.; Mohammadi, S.; Salamat, S.; Nadery, M.; Yarmand, S.; Zamani, M.; Wong, A.; et al. The effects of vitamin C supplementation on glycemic control in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes. Metab. Syndr. 2023, 17, 102824. [Google Scholar] [CrossRef]
- Lan, F.; Weikel, K.A.; Cacicedo, J.M.; Ido, Y. Resveratrol-Induced AMP-Activated Protein Kinase Activation Is Cell-Type Dependent: Lessons from Basic Research for Clinical Application. Nutrients. 2017, 9, 751. [Google Scholar] [CrossRef]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef]
- Delpino, F.M.; Figueiredo, L.M. Resveratrol supplementation and type 2 diabetes: a systematic review and meta-analysis. Crit. Rev. Food. Sci. Nutr. 2022, 62, 4465–4480. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, Y.; Yu, W. Activation of the AMPK-mTOR Pathway by Astaxanthin Against Cold Ischemia-Reperfusion in Rat Liver. Tohoku. J. Exp. Med. 2025, 265, 151–159. [Google Scholar] [CrossRef]
- Urakaze, M.; Kobashi, C.; Satou, Y.; Shigeta, K.; Toshima, M.; Takagi, M.; Takahashi, J.; Nishida, H. The Beneficial Effects of Astaxanthin on Glucose Metabolism and Modified Low-Density Lipoprotein in Healthy Volunteers and Subjects with Prediabetes. Nutrients. 2021, 13, 4381. [Google Scholar] [CrossRef] [PubMed]
- Oppedisano, F.; Macrì, R.; Gliozzi, M.; Musolino, V.; Carresi, C.; Maiuolo, J.; Bosco, F.; Nucera, S.; Caterina Zito, M.; Guarnieri, L.; et al. The Anti-Inflammatory and Antioxidant Properties of n-3 PUFAs: Their Role in Cardiovascular Protection. Biomedicines. 2020, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Chen, J.; Yang, J.; Pei, Q.; Li, J.; Liu, J.; Xu, L.; Li, N.; Chen, Y.; Chen, X.; Luo, H.; Sun, T. Inhibition of mammalian target of rapamycin complex 1 signaling by n-3 polyunsaturated fatty acids promotes locomotor recovery after spinal cord injury. Mol. Med. Rep. 2018, 17, 5894–5902. [Google Scholar] [CrossRef]
- Qian, F.; Ardisson Korat, A.V.; Imamura, F.; Marklund, M.; Tintle, N.; Virtanen, J.K.; Zhou, X.; Bassett, J.K.; Lai, H.; Hirakawa, Y.; et al. n-3 Fatty Acid Biomarkers and Incident Type 2 Diabetes: An Individual Participant-Level Pooling Project of 20 Prospective Cohort Studies. Diabetes. Care. 2021, 44, 1133–1142. [Google Scholar] [CrossRef]
- Goel, S.; Singh, R.; Singh, V.; Singh, H.; Kumari, P.; Chopra, H.; Sharma, R.; Nepovimova, E.; Valis, M.; Kuca, K.; et al. Metformin: Activation of 5' AMP-activated protein kinase and its emerging potential beyond anti-hyperglycemic action. Front. Genet. 2022, 13, 1022739. [Google Scholar] [CrossRef]
- Amin, S.; Lux, A.; O'Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef]
- Safaie, N.; Masoumi, S.; Alizadeh, S.; Mirzajanzadeh, P.; Nejabati, H.R.; Hajiabbasi, M.; Alivirdiloo, V.; Basmenji, N.C.; Derakhshi Radvar, A.; Majidi, Z.; et al. SGLT2 inhibitors and AMPK: The road to cellular housekeeping? Cell. Biochem. Funct. 2024, 42, e3922. [Google Scholar] [CrossRef]
- Schaub, J.A.; AlAkwaa, F.M.; McCown, P.J.; Naik, A.S.; Nair, V.; Eddy, S.; Menon, R.; Otto, E.A.; Demeke, D.; Hartman, J.; et al. SGLT2 inhibitors mitigate kidney tubular metabolic and mTORC1 perturbations in youth-onset type 2 diabetes. J. Clin. Invest. 2023, 133, e164486. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Feng, Y.; Liu, M.; Lu, Z.; Hu, B.; Chen, L.; Zhang, Y.; Liu, J.; Cai, F.; et al. Activation of AMPK by GLP-1R agonists mitigates Alzheimer-related phenotypes in transgenic mice. Nat. Aging. 2025, 5, 1097–1113. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Pandey, S.; Mangmool, S. Exendin-4 exhibits cardioprotective effects against high glucose-induced mitochondrial abnormalities: Potential role of GLP-1 receptor and mTOR signaling. Biochem. Pharmacol. 2024, 229, 116552. [Google Scholar] [CrossRef]
- Xu, J.; Chen, P.; Wu, D.; Zhou, Q.; Chen, S.; Ding, X.; Xiong, H. The novel GLP-1/GIP dual agonist DA3-CH improves rat type 2 diabetes through activating AMPK/ACC signaling pathway. Aging (Albany NY). 2023, 15, 11152–11161. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, N.; Shi, W.; Xing, Y.; Liu, S.; Meng, X.; Li, L.; Zhang, H.; Meng, Y.; Xie, S.; Deng, W. Glucose-dependent insulinotropic polypeptide/glucagon-like peptide 1 receptor agonist tirzepatide promotes branched chain amino acid catabolism to prevent myocardial infarction in non-diabetic mice. Cardiovasc. Res. 2025, 121, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, K.; Sugawara, K.; Ishihara, T.; Ishihara, N.; Ogawa, W. Effects of imeglimin on mitochondrial function, AMPK activity, and gene expression in hepatocytes. Sci. Rep. 2023, 13, 746. [Google Scholar] [CrossRef]
Figure 1.
The clinical manifestations of aging. PWV, pulse wave velocity.
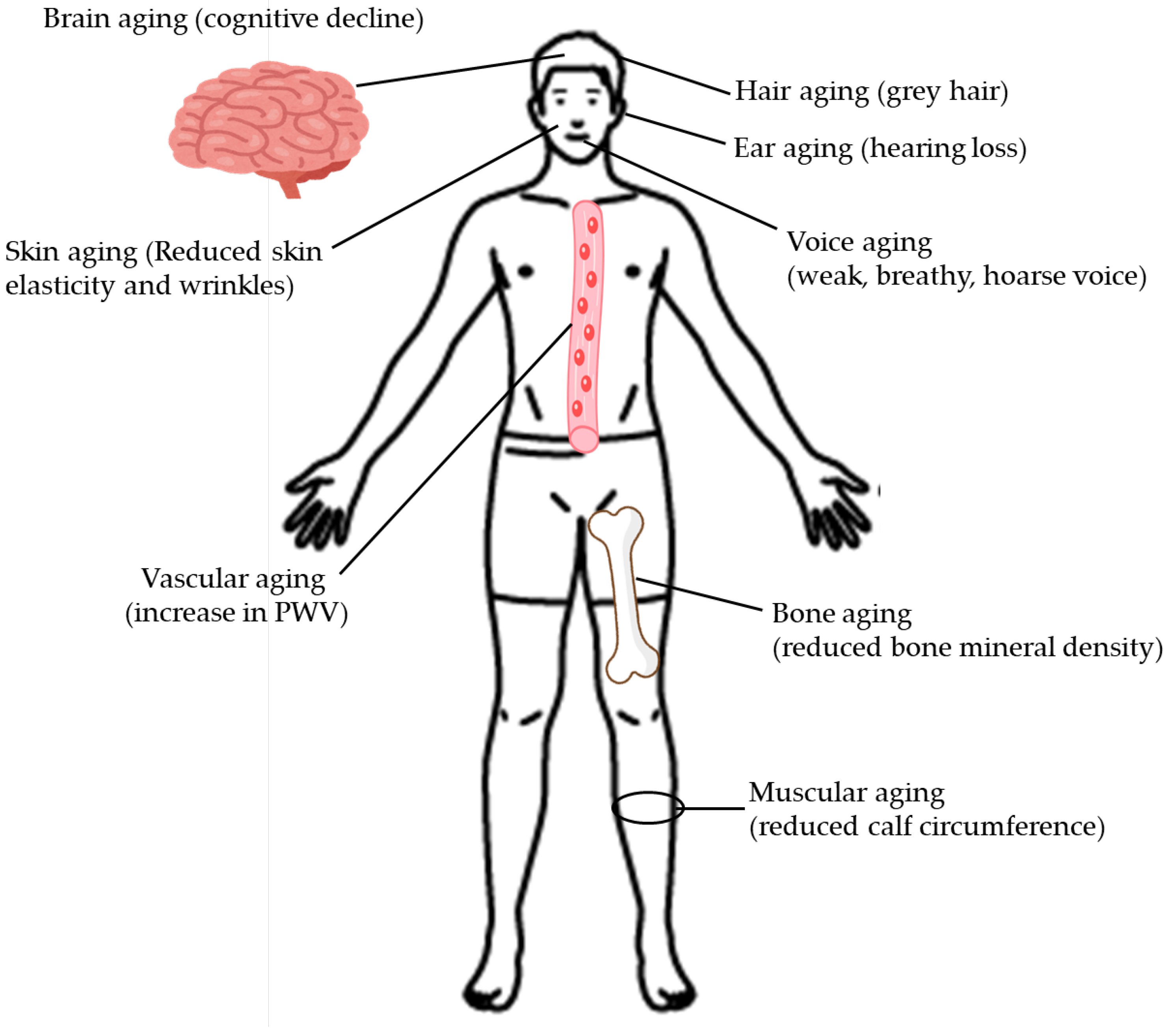
Figure 2.
The association between hallmarks of aging and type 2 diabetes. Pink arrows indicate effects. NAD+, nicotinamide adenine dinucleotide.
Figure 2.
The association between hallmarks of aging and type 2 diabetes. Pink arrows indicate effects. NAD+, nicotinamide adenine dinucleotide.
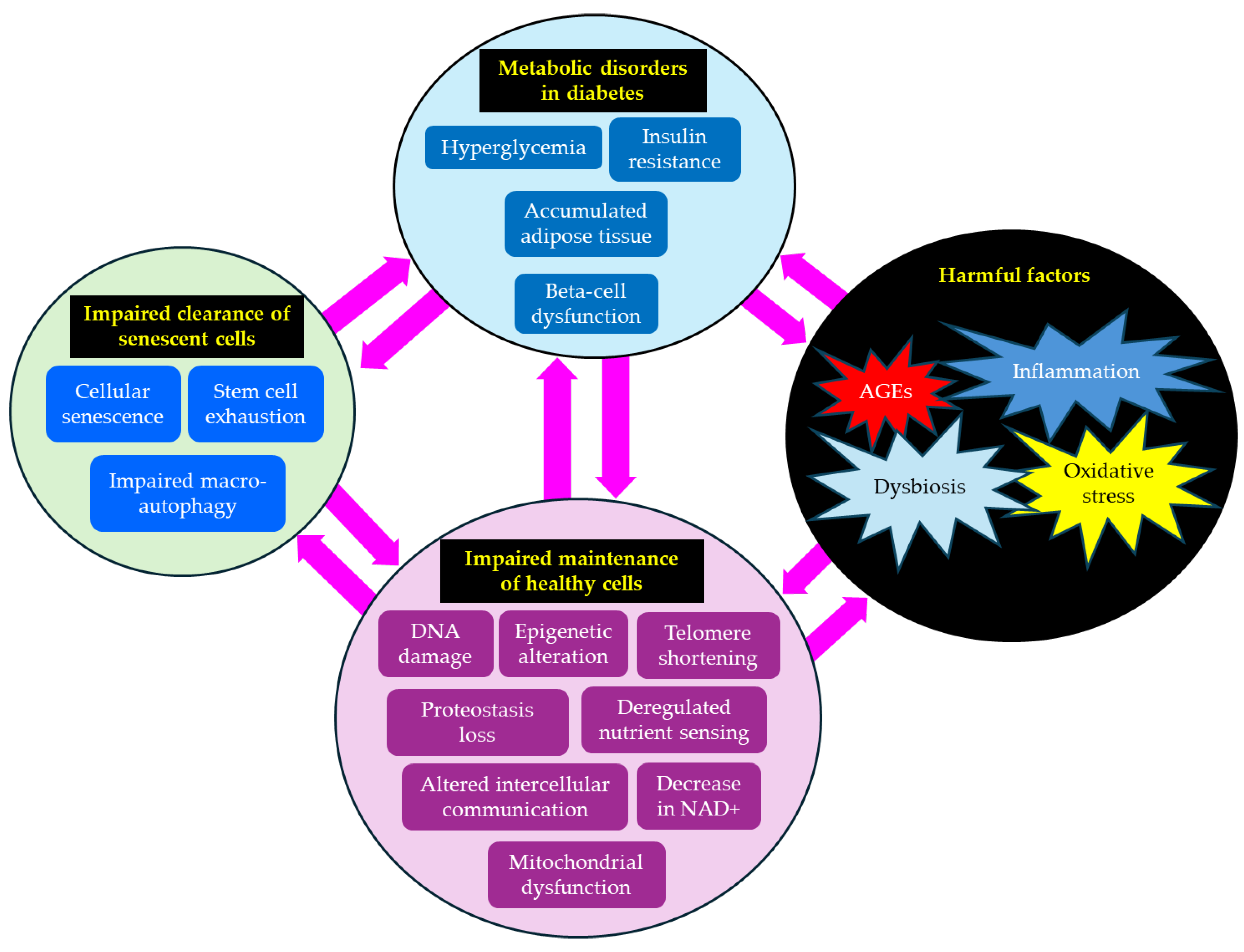
Figure 3.
Effects of AMPK activation and mTOR inhibition on aging-related molecules and aging hallmarks. Thick gray arrows indicate effects, and red upward arrows indicate the increase, and blue downward arrows indicate the decrease. AGEs, advanced glycation end products; AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; NAD+, nicotinamide adenine dinucleotide; Nrf2, nuclear factor E2-related factor 2; NF-κB, nuclear factor-kappa B; PGC1α, peroxisome proliferator-activated receptor- γ Coactivator 1α; ROS, reactive oxygen species; SIRT1, sirtuin 1; ULK1, Unc-51-like autophagy activating kinase 1.
Figure 3.
Effects of AMPK activation and mTOR inhibition on aging-related molecules and aging hallmarks. Thick gray arrows indicate effects, and red upward arrows indicate the increase, and blue downward arrows indicate the decrease. AGEs, advanced glycation end products; AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; NAD+, nicotinamide adenine dinucleotide; Nrf2, nuclear factor E2-related factor 2; NF-κB, nuclear factor-kappa B; PGC1α, peroxisome proliferator-activated receptor- γ Coactivator 1α; ROS, reactive oxygen species; SIRT1, sirtuin 1; ULK1, Unc-51-like autophagy activating kinase 1.
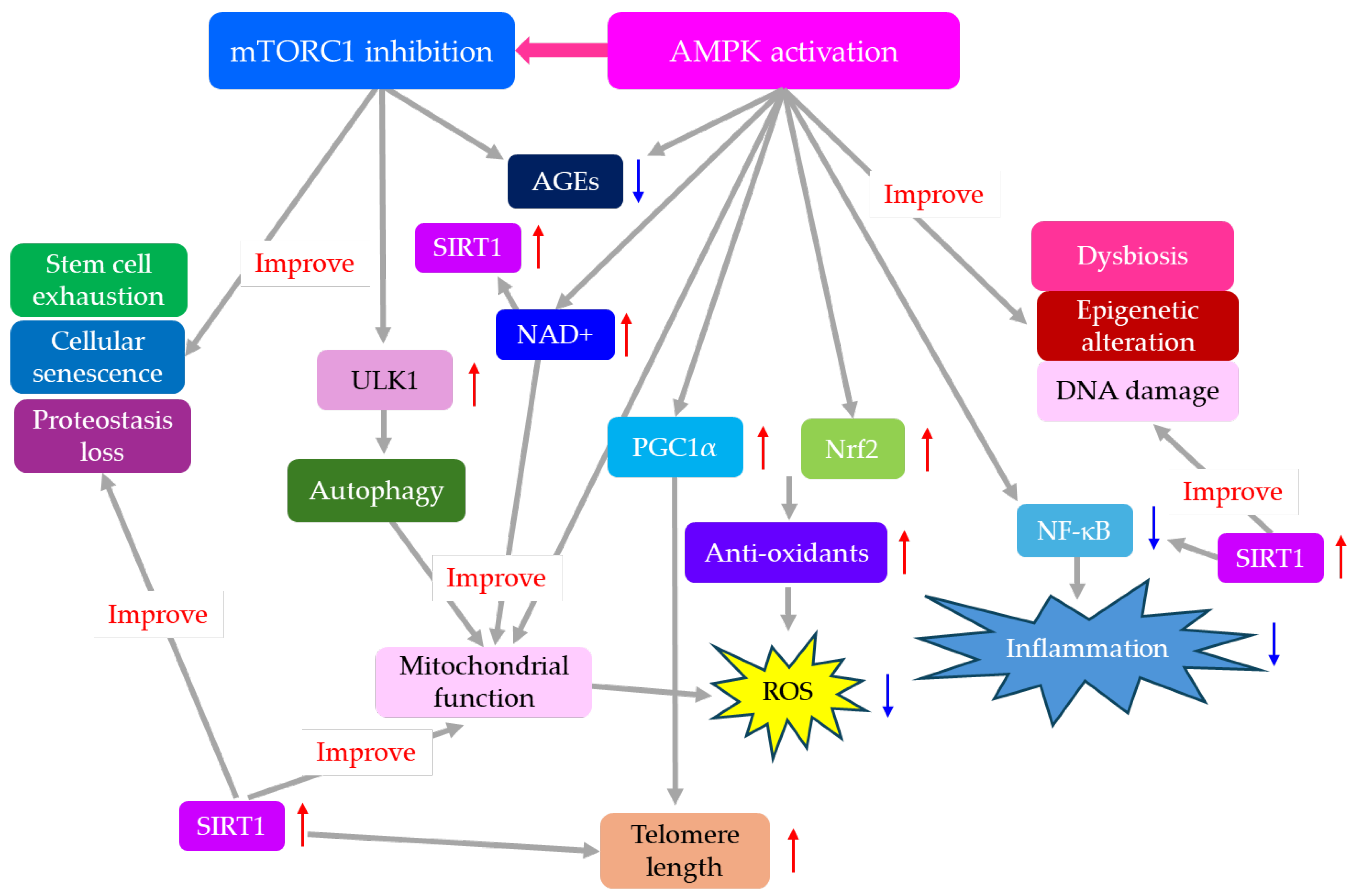
Figure 4.
Effects of AMPK activation and mTOR inhibition on hallmarks of aging. Arrows indicate effects. AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; NAD+, nicotinamide adenine dinucleotide.
Figure 4.
Effects of AMPK activation and mTOR inhibition on hallmarks of aging. Arrows indicate effects. AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; NAD+, nicotinamide adenine dinucleotide.
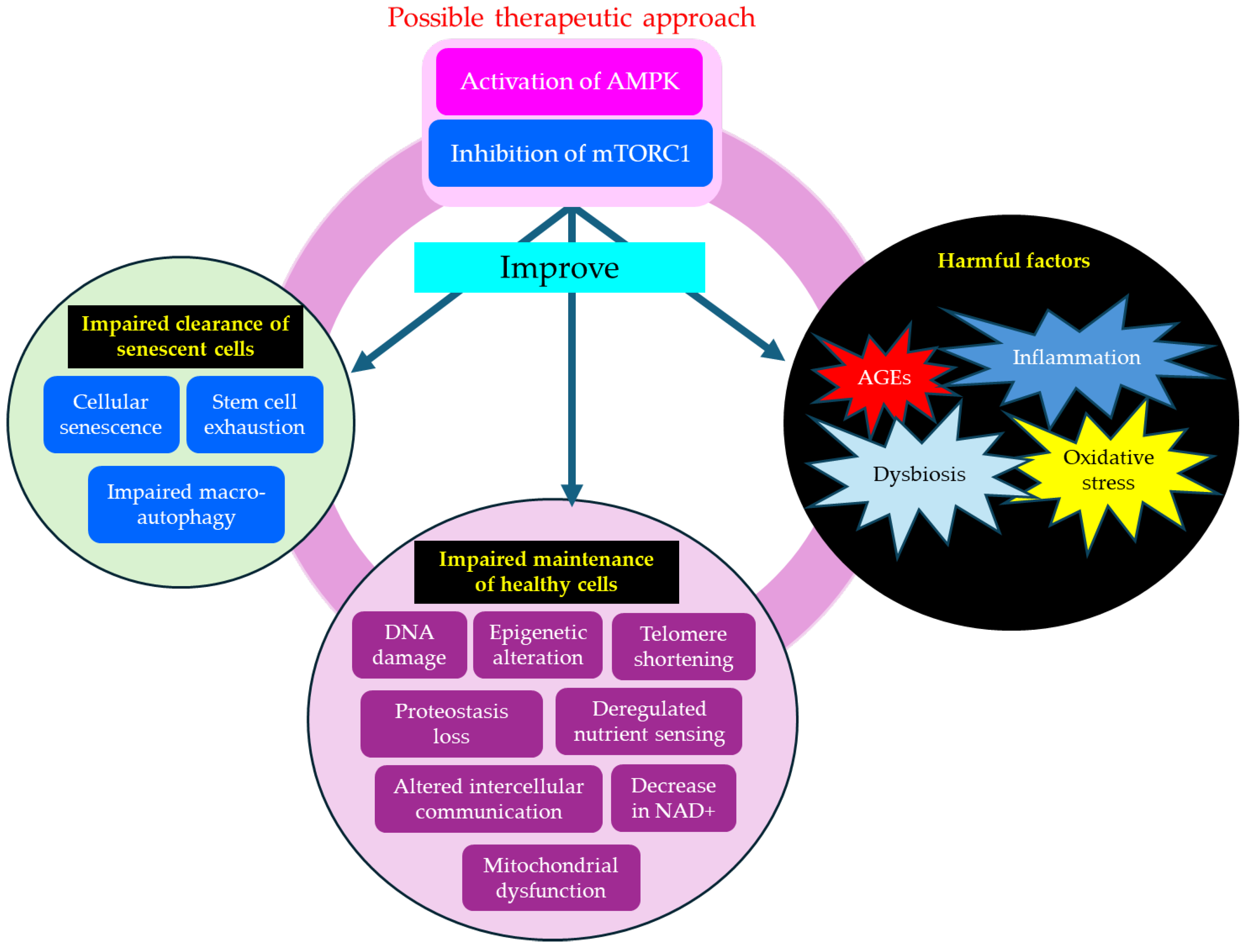

Table 1.
Effects of nutritional interventions and drugs that improve aging hallmarks by AMPK activation and mTORC1 inhibition on type 2 diabetes.
Table 1.
Effects of nutritional interventions and drugs that improve aging hallmarks by AMPK activation and mTORC1 inhibition on type 2 diabetes.
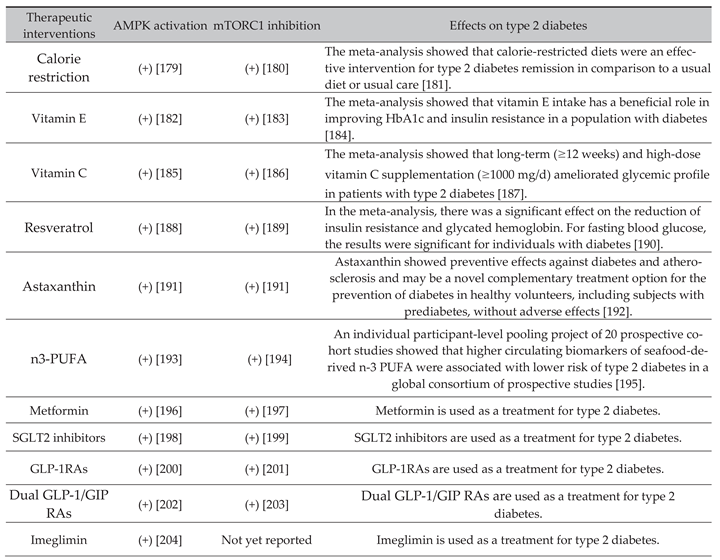 |
AMPK, AMP-activated protein kinase; GIP, glucose-dependent insulinotropic polypeptide; GLP-1RAs, glucagon-like peptide 1 receptor agonists; mTORC1, mammalian target of rapamycin complex 1; PUFA, polyunsaturated fatty acids; SGLT2is, sodium-glucose co-transporter-2 inhibitors; TG, triglyceride.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



