Submitted:
05 August 2025
Posted:
05 August 2025
You are already at the latest version
Abstract
Amorphous solid dispersions (ASDs) represent a promising formulation strategy for improving the solubility and bioavailability of poorly water-soluble drugs, a major challenge in pharmaceutical development. This review provides a comprehensive analysis of the physicochemical principles underlying ASD stability, with a focus on drug–polymer miscibility, molecular mobility, and thermodynamic properties. The main manufacturing techniques—including hot-melt extrusion, spray drying, and KinetiSol® dispersing—are discussed for their impact on formulation homogeneity and scalability. Recent advances in excipient selection, molecular modeling, and in silico predictive approaches have transformed ASD design, reducing dependence on traditional trial-and-error methods. Furthermore, machine learning and artificial intelligence (AI)-based computational platforms are reshaping formulation strategies by enabling accurate predictions of drug–polymer interactions and physical stability. Advanced characterization methods such as solid-state NMR, IR, and dielectric spectroscopy provide valuable insights into phase separation and recrystallization. Despite these technological innovations, ensuring long-term stability and maintaining supersaturation remain significant challenges for ASDs. Integrated formulation design frameworks, including PBPK modeling and accelerated stability testing, offer potential solutions to address these issues. Future research should emphasize interdisciplinary collaboration, leveraging computational advancements together with experimental validation to refine formulation strategies and accelerate clinical translation. The scientists can unlock the full therapeutic potential with emerging technologies and a data–driven approach.
Keywords:
amorphous solid dispersion
; stability
; thermodynamics
; machine learning
; simulation
1. Introduction
Amorphous solid dispersion (ASD) has emerged as an effective formulation strategy for poorly soluble drugs classified under biopharmaceutical classification system (BCS) Class II and Class IV [1]. The bioavailability of poorly soluble drugs primarily depends on their solubility and dissolution in biological fluids [2]. The drug crystals are converted into an amorphous form, which enhances its solubility, dissolution rate, and bioavailability [3]. In ASD systems, the crystalline drug is incorporated into a suitable polymer carrier, resulting in transformation into an amorphous state, which eliminates the need to break the crystal lattice [4]. Consequently, the amorphous form of many poorly soluble drugs attains substantially higher apparent solubility and a markedly faster dissolution rate [5]. However, producing the amorphous form requires significant energy input, rendering it susceptible to phase separation and recrystallization during manufacturing and storage [6].
The molecular mobility of the amorphous state increases upon exposure to ambient temperature and humidity, promoting recrystallization [7]. The crystalline drugs possess a well-defined, ordered structure with strong intermolecular bonds, conferring superior stability and predictable physicochemical behavior such as solubility, drug release, and melting point depression [8]. The intermolecular arrangement within crystals, extending in all directions, results in single or polycrystalline forms at the micron scale [9]. Molecules can adopt different conformations within the lattice, a phenomenon known as polymorphism [10]. Therefore, different polymorphic forms of the same drug exhibit distinct physicochemical properties. The crystalline drug forms are characterized by specific melting points at which the material transitions to the liquid state [11].
The amorphous form lacks long-range order and defined shape. Its molecular arrangement is unpredictable, leading to high intermolecular energy and metastability [12]. The amorphous forms possess higher free energy compared to their crystalline counterparts due to increased Gibbs free energy, which translates to greater apparent solubility and is advantageous for poorly soluble drugs. However, the amorphous form is thermodynamically unstable and tends to recrystallize over time or under stress conditions [13]. The absence of long-range order in the amorphous state results in short-range molecular interactions that promote clustering and nucleus formation [14].
This review investigates the physical stability of ASDs from a physicochemical perspective, considering thermodynamic, kinetic, and environmental aspects. The thermodynamic factors influencing ASD stability include drug solubility in the polymer, phase separation, drug–polymer compatibility, glass transition temperature, and drug–polymer interactions [15]. The kinetic factor associated with stability can estimate the molecular mobility, nucleus formation, and nucleus growth [16]. The environmental factors, such as temperature and humidity, can affect the physical stability of the amorphous form through both thermodynamic and kinetic mechanisms. Environmental conditions may induce polymorphic transformations, altering drug behavior [17]. Additional factors, including suboptimal formulation component selection, thermal and manufacturing stresses, and increased molecular mobility, can promote crystal precipitation, coarsening, and aging, ultimately diminishing dissolution rate and bioavailability [18]. The stable ASD products require careful selection of formulation components, manufacturing procedures, process parameters, and packaging [19].
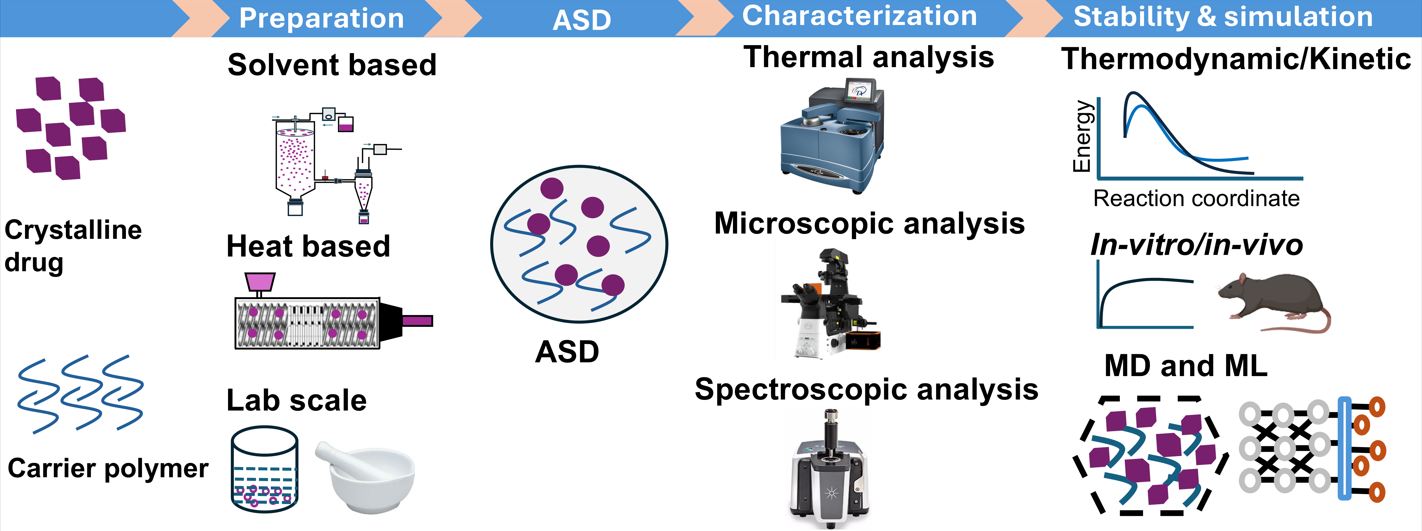
ASDs are commonly classified into three generations: first generation (amorphous drug only), second generation (polymeric carrier), and third generation (amorphous carrier with surfactant) [20]. They are typically prepared by solvent evaporation or heat-congealing. Among these, spray drying, hot-melt extrusion, and KinetiSol® are widely used for industrial-scale production [21]. Currently, 48 drug products containing ASDs, representing 36 unique amorphous drugs, have been approved by the US FDA and are commercially available. These dosage forms include tablets, capsules, and granules. Table 1 summarizes commercially available drug products manufactured using ASD technology [22].
Solid-state characterization techniques are employed to investigate the thermodynamic and kinetic properties of the amorphous form. The crystallinity in ASDs is typically assessed by differential scanning calorimetry (DSC) and powder X-ray diffraction (PXRD) [23]. Advanced imaging technologies such as nano-tomography and terahertz spectroscopy [24,25] enable investigation of intermolecular interactions between polymer and drug at the submicron scale. These innovative characterization methods facilitate prediction of dissolution properties, elucidation of recrystallization patterns, and evaluation of stability outcomes [7,26,27,28]. In addition, classic theories, molecular modeling, and machine learning approaches for assessing physical stability factors are introduced and discussed.
2. Effects of Material and Process Attributes
ASDs can be prepared using the drug alone via different preparation methods; however, such dispersions tend to recrystallize rapidly. Therefore, excipients or stabilizers are incorporated prior to processing to induce the”parachute effect.” The hydrophilic polymers are commonly used as excipients. The choice of polymer plays a crucial role in ASD stability [29]. The polymeric carriers are classified into distinct groups according to their chemical structure and physicochemical properties for use in dosage forms. Table 2 summarizes polymers commonly employed in ASD development. Among these, cellulose polymers derived from natural plant cellulose are widely used to enhance drug solubility, stabilize amorphous forms, and control drug release [30,31,32,33]. Vinyl polymers, synthesized from vinyl pyrrolidone or vinyl caprolactam monomers, exhibit diverse physicochemical properties depending on side chain chemistry [34]. Polymethacrylate polymers, derived from acrylic acid esters (acrylates) and methacrylic acid esters (methacrylate), are used as adhesives, enteric film formers, sustained-release agents, moisture protectants, and for pH-dependent solubility [35,36]. Polyvinyl acetate phthalate (PVAP) is a nonionic produced by polymerization and dissolves at basic pH [37]. Polyacrylic acid (PAA) is a synthetic polymer made from acrylic acid monomers and is notable for its water solubility and high-water absorption capacity [38]. Polyethylene glycol/polyethylene oxide is used as a crosslinker with carriers in ASD formulations [39,40].
Figure 1 provides an overview of principal and advanced manufacturing techniques. The stability of the amorphous form is influenced by process parameters such as speed, temperature, flow rate, drying rate, and residual solvent, all of which affect molecular mobility and crystallization risk [42]. In the case of solvents, even when removed after post-processing, can significantly impact the final product’s properties and stability by modulating drug–polymer interactions, evaporation rates, and the potential for phase separation and recrystallization. Solvents differ in miscibility and interaction strength, thereby affecting ASD stability and dissolution. Additionally, evaporation kinetics vary among solvents. Table 3 presents solvent selection criteria, which depend on boiling point, solubility, and toxicity, in accordance with ICH guidelines [43].
Quality Design and Process Parameter Tools
The quality design and process parameters encompass quality by design (QbD) and process analytical technology (PAT) approaches to ensure product quality throughout manufacturing. QbD incorporates various process parameters, including critical quality attributes (CQAs), critical material attributes (CMAs), critical process parameters (CPPs), and design space (DS), all of which play significant roles in ASD quality. CQAs identify key attributes that affect ASD performance, such as solubility, stability, and dissolution rate. CMAs inform product design and understanding. CPPs define manufacturing parameters that influence CQAs, such as temperature, mixing speed, and solvent evaporation rate, while DS establishes the CPP range that consistently yields high-quality ASDs [44,45]. PAT emphasizes real-time monitoring (on-line, in-line, or at-line) and control of manufacturing processes. Figure 2 illustrates in-line and on-line monitoring with near-infrared (NIR) and Raman spectroscopy, used to assess crystalline state, particle size, and uniformity during production. Such monitoring enhances process understanding and control, ensuring consistent quality and reducing waste [46,47,48,49].
The integration of QbD and process ana PAT offers a robust framework for ASD manufacturing. QbD establishes the design space and CQAs) while PAT maintains process control within this space through real-time monitoring. This constructive collaboration enhances product quality, lowers production costs, and accelerates the time to market for new formulations. Such approaches mark a departure from conventional trial-and-error methods toward more systematic, data-driven strategies. For example, the optimization of extrusion processes for ASD of piroxicam with Kollidon® VA64 using a QbD methodology involved in-line UV–Visible spectroscopy to monitor CQAs in real time during manufacturing, demonstrating that PAT provides a reliable tool for tracking and managing ASD quality during hot-melt extrusion (HME) [50].
The physicochemical properties of polymers significantly influence CQAs, which include nature of polymer type, molecular weight, polydispersity, glass transition temperature (Tg), particle size, mechanical properties, and chemical stability [51]. It is important to understand how the property of polymer influences the process parameters and affects the final product’s likelihood of crystallization in the final product [52]. The monitoring of physical stability during storage, handling, and dissolution testing is critical in early formulation development [53]. In order to guarantee the therapeutic efficacy of the final product remains a primary regulatory objective in ASD development. This objective is addressed through the application of diverse analytical techniques such as differential scanning calorimetry (DSC), polarized light microscopy (PLM), Raman spectroscopy, infrared (IR) spectroscopy, dielectric spectroscopy, and powder X-ray diffraction (PXRD) at each stage of development [54]..
3. Physical Stability of Amorphous Solid Dispersion
Many amorphous drug forms exhibit instability during preparation and storage, which presents a major challenge for formulation scientists [55,56]. Amorphous forms lack a defined structure and exist in a metastable state characterized by increased entropy, enthalpy, free energy, and volume. Consequently, these forms have an inherent thermodynamic tendency to crystallize. However, when functional groups capable of hydrogen bonding are present, stabilizing drug–polymer interactions can occur, increasing the energetic barrier to crystallization [57]. At room temperature, the molecule in a solid dispersion vibrates at their relative position and does not display diffusive mobility. Over long-term storage, the amorphous state gradually evolves toward thermodynamic equilibrium at the storage temperature, altering its physical structure. The drug loading affects solid-state saturation solubility under storage conditions, and over time, this may result in phase separation and recrystallization [58]. The amorphous–amorphous phase separation (AAPS) is characterized by the formation of distinct drug-rich and polymer-rich amorphous domains within the ASD matrix, which initially consisted of a molecularly mixed system. The drug-rich phase is generally more susceptible to crystallization due to limited polymer inhibition, whereas the polymer-rich phase exhibits greater stability because of the uniform distribution of polymer within the ASD matrix [59]. The following sections focus on key factors influencing ASD stability, emphasizing thermodynamic and kinetic properties as well as environmental influences.
3.1. Thermodynamic Factors on Physical Stability
3.1.1. Solubility of Drug in Polymer
The solubility of a drug in a polymer matrix is a critical determinant of ASD stability and performance. It governs the extent to which the drug remains molecularly dispersed, thereby preventing recrystallization and improving bioavailability [57]. When drug concentration surpasses its saturated solubility, the solid dispersion exhibits elevated chemical potential within the metastable system, increasing the likelihood of recrystallization. Thermodynamically, equilibrium between crystalline and dissolved (or dispersed) drug corresponds to the formation of a saturated solution [60]. In this context, the polymer acts as the solvent, and the crystalline drug as the solute; the saturated solubility of the drug in the polymer can be calculated using the solid–liquid equilibrium equation [61].
where, is the mole fraction solubility of the drug in the polymer, is the drug’s melting enthalpy, is the gas constant, is the drug’s melting point, T is the two-phase equilibrium temperature, and is the drug’s activity coefficient. These thermodynamic parameters can be determined from DSC data. Thus, drug solubility in the polymer at various temperatures can be calculated using activity coefficients of ᵧdrug. The activity coefficient is estimated using the extended Hansen model as shown below:
where, is the molar volume of the drug, denotes the solubility parameter for the drug, and denotes the solubility parameter for the combined system (weighted average), with subscripts d, ρ, and h indicating dispersion, polar, and hydrogen bonding forces, respectively, and indicating the mixed volume. The calculations proceed as follows:
where, is the volume fraction of component K.
where, and are the mole fraction and molar volume of component K, respectively.
where, , and are the molecular weight and density of component K, respectively.
The drug solubility in the polymer can also be estimated by plotting melting enthalpy versus drug concentration and extrapolating the enthalpy to zero. In practice, mixtures of drug and polymer at varying ratios are analyzed by DSC. The solubility of cinnarizine in Soluplus® at different drug concentrations was determined using phase diagrams to identify the thermodynamically stable region of the binary system [62]. As drug loading increases, melting enthalpy rises, but solubility in the polymer remains unchanged, indicating a supersaturated and thermodynamically unstable dispersion. The reduced drug loading enables molecular dispersion, forming a solid solution that supports high dissolution rates and improved drug bioavailability [63].
3.1.2. Phase Separation
Phase separation is a critical process in which an initially homogeneous drug–polymer mixture separates into two distinct phases—typically drug-rich and polymer-rich regions—significantly influencing performance, stability, dissolution, and bioavailability. This phenomenon commonly arises when drug loading exceeds the miscibility threshold within the polymer matrix, particularly during storage [64]. The amorphous–amorphous phase separation (AAPS) can be triggered by factors such as temperature, humidity, drug loading, and intrinsic drug–polymer miscibility. The AAPS in ASD can be characterized using various analytical techniques, including DSC, IR spectroscopy, nuclear magnetic resonance (NMR), confocal fluorescence microscopy (CFM), scanning electron microscopy (SEM), PXRD, and dielectric spectroscopy [65,66].
3.1.3. Compatibility of Drug and Polymers
The drug–polymer compatibility is essential for maintaining ASD stability and efficacy. The metastable form supports the maximum drug loading within the polymer and possesses high intermolecular energy; exceeding this limit induces phase separation and ultimately leads to drug crystallization. The Flory–Huggins lattice theory provides a means to estimate drug–polymer compatibility [67].
where, is the gas constant, the absolute temperature of the drug, the volume fraction, is the number of lattice sites occupied by the polymer chain, and is the drug–polymer interaction parameter. The parameter is calculated as follows:
where, and are the molecular weight and density of the polymer or drug, respectively. The interaction parameter can be estimated from solubility parameters [68]:
where, is the drug’s molar volume, and and are the solubility parameters of the drug and polymer, respectively. The spinodal equation is derived by setting the second derivative of the Flory–Huggins free energy ( in equation (7) considered to zero and got the spinodal equation:
The region below the spinodal line denotes where phase separation spontaneously occurs, resulting in drug crystallization. The drug–polymer compatibility can thus be evaluated by the extent of the area bounded by the spinodal region [69]. The accuracy of the spinodal line depends on the precision of the interaction parameter, as this parameter dictates intermolecular interactions and mixing between drug and polymer [70,71].
3.1.4. Glass Transition Temperature
Glass transition temperature (Tg) is a key thermophysical property of amorphous materials. It defines the temperature at which a solid transitions to a rubbery, fluid-like state. The amorphous state’s characteristic Tg makes it a crucial parameter for assessing ASD stability [72]. When homogeneous and heterogeneous intermolecular forces are equivalent, the drug and polymer form an ideal mixture, and the amorphous system displays a single Tg. Under these conditions, the Tg of the ASD follows the Fox equation [73]:
where, Tg is the predicted glass transition temperature, and are the mass fractions of the components, and and are the actual glass transition temperatures of the components. When the measured Tg is similar to the predicted value, the intermolecular forces among like and unlike molecules are equivalent, indicating a homogeneous distribution of the drug within the polymer. If the observed Tg exceeds the predicted value, then the intermolecular forces between like molecules are weaker than those between unlike molecules. Conversely, if the actual Tg is lower than predicted, the intermolecular forces among like molecules are stronger than those among unlike molecules. A large negative deviation between observed and predicted Tg suggests that self-interactions of the drug molecules are stronger than drug–polymer interactions, favoring aggregation into large amorphous clusters and phase separation.
Figure 3 illustrates the conversion from crystalline to amorphous forms and subsequent reconversion. The Tg of the amorphous drug is generally lower than that of the polymer, so the Tg of an ASD system typically falls between those of the drug and the polymer. The increased Tg raises the kinetic barrier to crystallization [74]. This principle underlies the”Tg 50 °C rule” molecular mobility in an amorphous solid decrease markedly and becomes negligible when the temperature is approximately 50 °C below its Tg. This empirical rule is widely applied in material science, particularly for polymers and pharmaceuticals [75]. Therefore, polymers with high Tg are selected for ASD development, as they enhance both drug and polymer solubility. This approach improves bioavailability and maintains drug stability by preventing phase separation and crystallization. In addition, polymeric carriers function as stabilizers for the amorphous form during prolonged storage [76].
3.1.5. Drug–Polymer Interaction
Drug molecules interact with polymers through hydrogen bonding, van der Waals forces, ionic interactions, electrostatic interactions, or hydrophobic interactions, all of which play key roles in ASD physical stability. The hydrogen bonding notably affects the stability of nifedipine with various polymers, with the strength of hydrogen bonding, structural relaxation time, and physical stability following the order: polyvinylpyrrolidone (PVP) > hydroxypropyl methylcellulose succinate (HPMCAS) > polyacrylic acid (PAA) [77]. The strongest drug–polymer interactions were observed in PVP-based ASD, effectively inhibiting amorphous drug crystallization [78]. The introduction of hydrophobic segments into water-soluble polymers can shield drug–polymer interactions from atmospheric moisture, thereby reducing ASD hygroscopicity and drug supersaturation [79]. Pea protein isolate, a novel plant-derived polymer, can replace animal proteins in pharmaceuticals, improving drug precipitation inhibition and stability during dissolution [80,81]. The acidic polymer PAA, when combined with clofazimine to form drug–polymer salts, creates strong ionic interactions, and enhances crystallization resistance [82]. Additionally, halogen bonding influences dissolution profiles by lowering the binding energy between drug and polymer, increasing drug solubility [83]. The drug–polymer interactions reduce molecular mobility, enhancing physical stability, and can be assessed by IR spectroscopy, Raman spectroscopy, and solid-state NMR [84,85].
3.2. Kinetic Factors on Physical Stability
3.2.1. Molecular Mobility
Molecular mobility is a critical factor governing ASD crystallization. In the amorphous state, drug molecules exhibit a strong tendency to migrate and rearrange, promoting crystallization, especially as mobility increases. Amorphous molecules undergo continuous thermal vibrations, leading to the glass transition, or”global mobility.” These molecular motions are categorized as α-relaxation, primarily involving rotation and translation of functional groups in both drug and polymer. When the ambient temperature exceeds Tg, molecular vibrations intensify, and α-relaxation becomes the dominant driver of amorphous material recrystallization (T > Tg). For example, broadband dielectric spectroscopy has been used to evaluate the molecular mobility of itraconazole in ASD containing PVP or HPMCAS [86]. The dielectric spectra revealed that α-relaxation times for itraconazole increased with HPMCAS, while PVP had minimal effect. An isothermal crystallization studies confirmed that HPMCAS inhibited crystallization more effectively than PVP, suggesting a strong relationship between α-relaxation and crystallization kinetics [87].
The local molecular motion in amorphous systems, termed β-relaxation (secondary relaxation or local mobility), involves bond rotation or whole-molecule motion. When the ambient temperature is below Tg, the molecular system is in a rigid or”frozen” state (T < Tg), favoring nucleation and recrystallization. The olanzapine–malic acid film system shows, secondary relaxation in the intermediate frequency range of the dielectric spectrum may result from intermolecular interactions [88]. The models such as Adam–Gibbs–Vogel and Vogel–Tammann–Fulcher equations are commonly applied to describe the structural relaxation of drug–polymer combinations for predicting long-term physical stability [89]. In the supercooled state, molecular mobility, represented as the reciprocal of the relaxation time τ, exhibits pronounced temperature dependence, as described by the VTF equation [90]:
β-relaxation follows Arrhenius temperature dependence, as shown below:
where, τ is the average relaxation time,”τ” _”0” is the relaxation time constant under ideal conditions,”D” is the intensity constant,”T” _”f” is the virtual temperature, and”T” _”0” is the temperature at which molecular mobility becomes zero.”R” is the gas constant, and Eαβ is the activation energy for β-relaxation, typically much lower than for α-relaxation. Notably, β-relaxation involves longer structural relaxation times and requires less energy for molecular mobility.
Thus, when the temperature is below Tg, crystallization of solid dispersions is suppressed, increasing physical stability. For example, crosslinking ketoconazole–PAA amorphous dispersions with PVA progressively reduced molecular mobility and improved physical stability as crosslinker content increased [91].
3.2.2. Nucleus Formation
Nucleus formation generally initiates with molecular mobility within the amorphous matrix, allowing molecules to aggregate and form stable clusters. These clusters function as nuclei, triggering the transition from the amorphous to crystalline state. According to classical nucleation theory (CNT), the formation of a crystal nucleus via phase interaction is described as follows [92]:
where, is the change in free energy during nucleus formation, is the change in chemical potential due to volume change, and is the change in interfacial free energy. The rate of crystal nucleus formation can be calculated as [93]:
where, is the number of nuclei formed per unit time and volume, is the kinetic pre-factor, is the free energy change for nucleus formation within the critical radius, is the Boltzmann constant, and is the absolute temperature. The pre-factor is calculated as [94]:
where, is the vapor pressure, is the surface tension, is the molecular weight, and is the molecular volume. The factor can be calculated as [95]:
where, is the mobility of molecules or atoms at the crystal interface, is the interfacial energy per unit area, and s is the supersaturation. Substituting equations (16) and (17) into (15) yields [96]:
This equation demonstrates that increasing surface tension (σ) slows nucleation, while increasing supersaturation (“s” ) accelerates it. Thus, selecting polymers with good compatibility can reduce nucleation rates and recrystallization. Taylor et al. reported that strong hydrogen bond donors in PAA increased the nucleation rates of amorphous acetaminophen, whereas HPMCAS, which contains weaker hydrogen bond donors and acceptors, most effectively inhibited nucleation. Their study found no direct correlation between nucleation rates and easily identifiable system properties, such as drug–polymer interactions or Tg [97]. The polyethylene terephthalate has been employed as a heteronucleants in the melt crystallization of acetaminophen in PEG to form crystalline solid dispersions. The acetaminophen exhibited two polymorphic forms, form I and form II. The coating of acetaminophen with heteronucleants inhibited polymorphic transitions by 10 % compared to the uncoated substrate [98]. A faster cooling rate during manufacturing increases amorphous yield and stability in highly drug-loaded ASD, ultimately facilitating nucleus formation [99].
3.2.3. Growth of Nucleus
Crystal growth commences immediately after nucleus formation, according to crystal growth and diffusion theory. This process comprises two main steps: drug molecules diffuse from the dispersion medium and accumulate on the surface of the crystal nucleus, after which they are incorporated into the crystal lattice, releasing the heat of crystallization [3]. The crystal growth rate is typically represented by the rate of increase in crystal radius, calculated as follows [73]:
where, D denotes the diffusion coefficient of drug molecules, k+ is the surface coagulation factor,”N” _“A” is the Avogadro constant, and”(C –” 〖” C” 〗_”eq” “)” the difference between the drug concentration in the carrier and the concentration of drug molecules not integrated on the crystal surface. The equation (19) indicates that if”r ≫ D/”“k” _”+” , the crystal growth rate is primarily diffusion-controlled, whereas if”r ≪ D/”“k” _”+” , the growth is primarily governed by the surface aggregation factor. As the crystal expands, the viscosity, molecular migration rate, and “(C –”“C” _”eq” “)” of the system change; thus, equation (19) has inherent limitations [100]. Figure 4 illustrates that an increased crystal growth rate negatively impacts dissolution performance by reducing solubility and promoting crystal seeding, which leads to de-supersaturation [101]. The ASD of itraconazole with PVP–K12 and HPMCAS demonstrated that systems containing soluble polymers exhibit a higher tendency to crystallize than those with insoluble polymers, resulting in a sharp decline in drug concentration in the dissolution medium [102].
3.3. Environmental Factors on Physical Stability
The stability of ASD is influenced by environmental factors that indirectly affect both thermodynamic and kinetic stability. Key environmental factors include temperature, humidity, storage conditions, and light exposure. Molecular mobility is intricately linked to temperature; as temperature approaches Tg, molecular mobility increases, which can significantly impact ASD stability. Enhanced molecular mobility raises the likelihood of phase separation and recrystallization [103,104]. Humidity in the environment is absorbed by the ASD, disrupting hydrogen bonding between the drug and polymer and thereby reducing Tg. For instance, absorption of 1.0 % moisture by an amorphous system can lower its Tg by 10 °C. When a hydrophobic polymer (Eudragit® EPO) and a hydrophilic polymer (PVP–VA64) are co-extruded, phase separation occurs due to their immiscibility. After co-extrusion with the drug, a solid dispersion is formed with a microstructure resembling an emulsion. In this system, the hydrophobic phase contains a small amount of drug as the continuous phase, while the hydrophilic phase contains a larger quantity of drug as the dispersed phase. The hydrophilic (dispersed) phase is coated by the hydrophobic (continuous) phase, which inhibits moisture penetration [105].
A 20 % drug-loaded cinnarizine–Soluplus® ASD was prepared via HME. The samples stored at 40 °C and 75 % humidity, and at 60 °C and 94 % humidity, exhibited similar effects on physical stability. Thus, both temperature and humidity had comparable impacts on the stability of the dispersion system [100]. In another study, individual felodipine, carbamazepine, celecoxib, and fenofibrate were formulated with Eudragit® EPO using the HME method. It was found that high humidity (75 % RH) induced greater crystallization than elevated temperature (40 °C). This effect was observed at both low (10 %, w/w) and high (70 %, w/w) drug loadings, underscoring the importance of humidity control in ASD storage and formulation [106].
4. Molecular Simulation and Statistical Methods
When a drug is distributed within a carrier, various intermolecular forces play significant roles in preventing drug molecule aggregation. These forces include hydrogen bonding, acid–base or ionic interactions, dipole–dipole forces, and van der Waals interactions. Such interactions reduce self-association, enhance drug–carrier binding strength, and contribute to long-term physical stability [107,108,109]. This challenge can be addressed using various molecular techniques. The quantum mechanics (QM), molecular mechanics (MM), and molecular dynamics (MD) approaches are applied to investigate intermolecular interactions, molecular mobility, solubility, and stability. Table 4 summarizes the application of MD, QM, and docking studies in ASD systems to simulate molecular mobility and intermolecular interactions between drug and polymer.
4.1. Quantum Mechanics (QM)
QM methods, such as density functional theory (DFT), offer valuable insights into non-bonding interactions between drugs and carriers. These forces influence stability and binding affinity via molecular complexes and interaction energies. The modeling of non-bonding interactions has emphasized DFT as a rapid and effective computational approach for predicting interaction energies and identifying hydrogen bonding pairs. The periodic DFT computations are increasingly used as a modeling tool to analyze solid-state pharmaceutical compounds. The combined DFT/MD approaches are employed to investigate drug–polymer molecular behavior by utilizing electronic structure information [110,111]. Large, structurally realistic systems are challenging to model accurately; however, molecular modeling can predict a wider range of properties and chemical behaviors. The strong drug–polymer interactions influence miscibility and stability [112]. The excipient and formulation screening can evaluate ASD stability and solubility. For example, cationic drug candidates such as propranolol hydrochloride and diphenhydramine hydrochloride interact with the ionic carriers Eudragit® L100 and Eudragit® L100–55 [113]. The carriers are represented in their monomeric forms with various configurations and polymers, considering both the presence and absence of an explicit chloride anion in the complex. The DFT computations revealed that the hydroxyl and amine groups of the drugs form hydrogen bonds with the ester and hydroxyl groups of the monomer units. The presence of chloride ions disrupts hydroxyl group interactions but does not affect amine group hydrogen bonding. These results indicate optimal interactions occur between the amine group of propranolol or diphenhydramine and the carbonyl group of Eudragit® L100 or Eudragit® L100–55 polymers. The proposed hydrogen bonding motif was confirmed by NMR and X-ray photoelectron spectroscopy. The DFT computations indicated an average increase in binding energy of 5 to 8 kcal/mol for each additional hydrogen bond, with the most stabilizing interactions occurring between tertiary amine and carboxyl groups (20 to 28 kcal/mol) [114]. The interactions between amine group of drug molecules (cetirizine HCl and verapamil HCl) and carboxyl group of polymers (Eudragit® L100 and Eudragit® L100–55) showed the strongest binding after extrusion, indicating high binding energy and the formation of a more stable amorphous form [115]. The partial charge analysis of clofazimine and HPMC phthalate revealed strong donor and acceptor sites, with a simplified model system using acetic acid as a structural substitute for hypromellose phthalate carboxylic groups. The calculations and spectroscopic studies suggested that ion pair complex formation is a key factor for drug–carrier miscibility [116]. PVP inhibited crystallization of resveratrol–PVP and griseofulvin–PVP for storage stability. DFT calculations revealed greater drug–carrier interaction strength in the resveratrol–PVP complex than in the griseofulvin–PVP system. The stronger interaction energies led to higher stability, as evidenced by the sustained high dissolution rate of the PVP–resveratrol system after storage, compared to the lower dissolution rate of the griseofulvin–PVP ASD [117]. The QM insights were used to screen polymer compatibility and process related factors in advanced drug delivery system [118].
4.2. Molecular Mechanics (MM) and Molecular Dynamics (MD)
MD simulations require parameterized system components for molecular mechanics (MM) force fields, which compute total energy by summing empirical potential energy functions [119]. Commonly applied force fields include condensed phase optimized molecular potentials for atomistic simulation studies (COMPASS) [120], polymer consistent force field (PCFF) [121], chemistry at Harvard macromolecular mechanics (CHARMM) [122], assisted model building with energy refinement (AMBER) [123], and optimized potential for liquid simulations (OPLS) [124]. The force fields represent energy as a function of atomic positions, with potential energy divided into two categories: (a) bonded interactions, describing bond lengths, angles, and torsions, and (b) non-bonded interactions, accounting for Coulombic and van der Waals forces. Chemical phenomena are essential for predicting drug–polymer miscibility and molecular mobility, such as flexible chain motion or hydrogen bond formation. These interactions vary during the simulation and can be averaged over its duration or across multiple runs from different initial conditions [125,126].
Classical molecular dynamics (MD) adheres to Newtonian mechanics, monitoring atomic positions, velocities, accelerations, forces, and energies at each simulation step. At any given time, total energy depends on atomic positions, which are updated iteratively at each step based on the forces acting on each atom. Recent advances in computation have made MD simulations of amorphous drug–drug [127,128], drug–micelle [129], polymer–polymer [130,131,132,133], polymer–membrane [134], and polymer–plasticizer [135] systems routine in the scientific literature.
The MD simulations of ASD systems model drug interactions with polymer carriers. The MM-based docking and MD simulations predict a range of phenomena and properties. The docking simulations investigate the drug–carrier interactions, while MD simulations assess drug–carrier miscibility based on solubility and Flory–Huggins interaction parameters. These simulations have shown that ASDs form through hydration and dissolution mechanisms. The drug–excipient solubility parameters can be computed via MD simulation [136]. The miscibility of artemisinin in PEG and PVP was determined by MD simulation at 298 K, revealing a solubility parameter difference (∆δ) of 0.08 for artemisinin–PVP, 0.57 for artemisinin–PEG, and 0.15 for artemisinin–PVP–PEG (50:50 w/w), indicating drug miscibility with the polymer blend [137]. The MD simulation has been used to investigate the drug loading efficiency of gemcitabine at different chitosan concentrations [138]. The MD simulations of copolymer–curcumin systems monitored conformational adjustments and distance changes between components. Initially, the drug and polymer were separated by a distance beyond the van der Waals interaction range; after 100 picoseconds (ps) of MD simulation, continuous interactions formed binding sites on the polymer surface [139]. The hydrogen bonding occurred between the hydroxyl group of the polymer and the chlorine group of lafutidine. The MD simulations showed that lafutidine with Soluplus® and Lutrol® exhibited the lowest energy and strongest bonding interactions, confirming stable ASD formulation [140]. The hydroxyl group of the polymer and the chlorine group of posaconazole established hydrogen bonds. According to MD simulation, the most stable dispersion had the greatest number of bonding contacts and the lowest energy [141]. MD simulations have clarified interactions between ritonavir and Lutrol®, revealing that the oxyethylene moiety in Lutrol® interacts with the hydrophobic group of ritonavir, thereby increasing ritonavir solubility in the molten phase and forming a stable solution [142].
4.3. Docking Studies of Drug in Polymer Carrier
Docking is commonly employed to generate favorable preliminary binding conformations that are subsequently refined through molecular dynamics (MD) simulations. This approach provides an algorithmic method for rapid sampling and scoring of drugs–polymer carrier complexes. The intermolecular interaction of lumefantrine with Soluplus®, Lutrol® F127, Lutrol® F68, and PEG 4000, revealed strong hydrogen bonding between polymer hydroxyl and carbonyl groups and the drug’s chlorine and amine groups [143]. The docking system was also used to determine the binding energy of the tautomeric di–keto and keto–enol forms of curcumin with monomer and dimer units of Eudragit® EPO [144]. The di–keto form exhibited higher binding energy than the keto–enol form, involving van der Waals forces, Coulombic interactions, and hydrogen bonding [144]. MD simulations of clonazepam, ibuprofen, fenofibrate, and alprazolam with polymers such as PVP–VA64, HPMC, and Eudragit® EPO were conducted to assess miscibility and intermolecular interactions. The results showed that ibuprofen/PVP–VA64, ibuprofen/Eudragit® EPO, ibuprofen/HPMC, clonazepam/PVP–VA64, clonazepam/HPMC, fenofibrate/PVP–VA64, fenofibrate/Eudragit® EPO, alprazolam/PVP–VA64, alprazolam/ Eudragit® EPO, and alprazolam/HPMC combinations were miscible. However, hydrogen bond analysis indicated that only ibuprofen/PVP–VA64 and ibuprofen/Eudragit® EPO formed strong hydrogen bonds that stabilized solid dispersions, while the other drug/polymer pairs exhibited weak or no hydrogen bonding [145].
A systematic docking simulation using full–length polymers evaluated the anchoring ability of cyclosporin A across various polymer chain lengths: short (~7 nm), medium (13–14 nm), and long (~20 nm) for L/D polylactide, chitosan, polyglycolic acid, PEG, and cellulose. For each polymer type and chain length, one million complexes were generated between stationary cyclosporin A and a flexible polymer. The interaction energy analysis identified chitosan and cellulose as the most miscible with cyclosporin A [146]. The miscibility of ibuprofen and carbamazepine with Soluplus®/PEG was assessed using the Hoftyzer–Van Krevelen and Hildebrand solubility parameters. The molecular docking images showed uniform distribution of drugs and polymers in ternary systems. The carbamazepine–Soluplus®/PEG system had a more negative binding affinity (−6.2 to −6.7 kcal/mol) than the ibuprofen–Soluplus®/PEG system (−5.3 to −5.5 kcal/mol), indicating stronger interactions between carbamazepine and Soluplus®/PEG. The calculated solubility parameters and DSC experiments confirmed the miscibility of each ternary system. Additionally, FT–IR spectroscopy revealed strong hydrogen bonding among the carbamazepine primary amine, carbonyl, and amide groups, as identified by docking and MD simulations [147].
The dynamic simulations of naproxen, diclofenac sodium, dimethyl fumarate, and omeprazole with polymers (HPMCAS, HPMCP, and Eudragit® L100) were performed to elucidate the molecular interactions between delayed–release drugs and enteric polymeric excipients. The optimal API–polymer pairs identified were naproxen–Eudragit® L100, diclofenac sodium–HPMCP, dimethyl fumarate–HPMCAS, and omeprazole–HPMCAS. All APIs formed hydrogen bonds with the polymeric excipients. However, as API loading increased, API–polymer interactions decreased, resulting in higher API mobility and accelerated release. The increased temperature further enhanced API mobility, leading to faster release [148]. The solubility and Flory–Huggins (FH) interaction parameters for the amorphous indomethacin–PVP system were evaluated by varying drug candidates, polymers, and water content in each MD simulation [149]. While differences in the solubility parameters of indomethacin and PVP predicted borderline miscibility (δIMC–PVP = 6.5 MPa1/2), FH interaction parameters predicted complete miscibility (IMC–PVP = –0.61).
Similarly, FH parameters were calculated for felodipine ASD with HMPC and water. The hydrogen bonding between felodipine and HMPC promoted miscibility, although these bonds were disrupted by added water [150]. The formation of hydrogen bonds between Lutrol® F68 and two carbamazepine molecules, investigated by MD simulation, indicated a high tendency for carbamazepine aggregation and phase separation [151]. MD simulations showed that binding affinity and solvation free energy can inhibit the crystallization of telaprevir with carboxylate–containing polymers, suggesting that MD can serve as a predictive tool for screening suitable polymers [152].
Another study examined indomethacin mixed with PEG and polylactic acid (PLA) polymers, focusing on ASD formation via simulated annealing, API–polymer miscibility using MD-predicted FH interaction parameters, and polymer carrier encapsulation efficiency [153]. The in silico ASD screening predicted drug–polymer compatibility for solubility enhancement [154]. The MD simulation showed that drug release increased with higher PEG concentrations in modified PLA carriers [155]. MD simulations also identified suitable carriers for olmesartan medoxomil, enhancing dissolution with PVP–VA64 and Soluplus®. The interaction between drug and polymer interactions were investigated using atomistic MD simulations [156].
Solubility and FH interaction parameters were determined for tacrine with chitosan and polybutylcyanoacrylate (PBCA) polymers. Tacrine showed greater miscibility with PBCA by both methods, and MD simulations indicated that longer polymer chains yielded higher interaction energies. Computational and experimental studies of simvastatin and PVP predicted miscibility, dynamic Hansen solubility, and FH parameters, as confirmed by DSC experiments [157]. For rivaroxaban with Soluplus®, specific molecular interactions and shrinkage led to a drug–rich amorphous phase, resulting in recrystallization under high humidity [158]. The molecular dynamics indicated weak hydrogen bonding between erlotinib HCl and PVP or PEG individually, but a combination of PVP and PEG enabled hydrogen bond formation and enhanced molecular interactions [159]..
5. Machine Learning for Better Performance
Machine learning (ML) plays a transformative role in the preparation and stabilization of ASDs, improving drug solubility and bioavailability while reducing experimental workload. ML, a branch of artificial intelligence (AI), has driven rapid advances in silico drug development over the past decade. ML uncovers complex, nonlinear relationships between input parameters and target features. It leverages large experimental datasets and data–driven supervised algorithms for drug formulation optimization. ML algorithms such as transfer learning, one–shot, zero–shot, and Bayesian–based optimization have gained popularity for enhancing model performance with sparse data [160,161]. Deep learning (DL), a subset of ML, is typically represented by artificial neural networks (ANNs) that emulate neural connectivity in the brain. In ANNs, nodes are interconnected directly or indirectly through multiple layers. The information enters via the input layer, is processed by hidden layers, and reaches the output layer. ANNs are particularly effective at identifying complex, nonlinear relationships between input and output variables.
ANNs are increasingly utilized in drug development and process optimization. They are prominent in recent ML models for predicting and optimizing ASD composition, stability, and dissolution rates. In addition to ANNs, other ML methods include genetic algorithms (GA), multiple linear regression (MLR), logistic regression (LR), decision trees (DT), random forest (RF), k–nearest neighbor (kNN), Naïve Bayes (NB), and light gradient boosting (LGBM) [162,163]. Statistical learning models predict properties and phenomena occurring between drug molecules and carriers, especially when experimental data for model training is limited. Despite such constraints, ML models designed to predict amorphous API properties provide valuable insights for rational ASD and formulation development. Multiple linear regression (MLR) models have been used to predict the long–term physical stability of the amorphous form for 25 poorly soluble drug candidates by employing physiochemical properties derived from two-dimensional structures, as well as measured thermodynamic and kinetic solid properties such as fusion, relaxation time, and configurational free energy. Features relevant for predicting the amorphous behavior of APIs may also play a significant role in predicting drug–carrier ASD systems [164]. Table 5 summarizes recent ML predictions for physicochemical properties, stability, and formulation strategies of ASDs.
To predict Hansen solubility parameters from a dataset of 130 compounds, the simplified molecular input line entry system (SMILE) was used to generate connectivity features, indices, and physiochemical properties. This diverse collection of features served as input for multivariate adaptive training, which was extended to solvent–polymer miscibility prediction using binary classification and solvent-dependent drug-like solid dissolution models [165]. The logistic regression (LR) models assessed the potential for ASD development using data from 12 drugs with PVP–VA copolymer, which were experimentally characterized for miscibility and stability. Topological and molecular indices were derived from atom connectivity and three–dimensional drug structures, identifying atomic mass–weighted third–order R autocorrelation indices (R3m) and topological distances between oxygen and chlorine atoms [166]. ANN and GA methods were applied to optimize the formulation and drug release properties of effervescent controlled–release floating tablets. The ASD of nimodipine API with PEG polymer carriers was characterized, and ANN and GA models were trained to predict the dissolution rate based on polymer matrix swelling and erosion [167]. The dissolution rate of tibolone improved with all prepared ASDs and was dependent on drug concentration. ANN consisted of an input layer with four units, an intermediate layer with six units, and an output layer with one unit. The predictive ability of the optimal ANN structure was 0.048, indicating a strong correlation between examined factors and measured response [168].
Figure 5 illustrates that machine learning (ML) is increasingly valuable in addressing the long-term physical stability challenges of amorphous solid dispersions (ASD). Han et al. applied molecular modeling and eight distinct ML algorithms to predict ASD stability for three and six months. The random forest (RF) algorithm produced the most accurate model, identifying drug loading ratio, humidity, temperature, and molecular weight as critical stability determinants. This data-driven strategy improves formulation development efficiency and reduces dependence on empirical trial-and-error approaches [169]. The generalized regression (GR) neural network was employed to optimize the solid dispersion of carbamazepine with magnesium aluminosilicate (Neusilin® UFL2) and Kollidon® VA64. The ML approach yielded root mean square error (RMS) values of 0.00029 and 0.1185 for the training and test data sets, respectively, indicating excellent predictive performance of the neural network [170]. ML models have also predicted chemically stable ASD and identified critical attributes during manufacturing [171]. Additionally, a random forest regressor was used to predict the Tg of polymers and drugs, providing insights into the impact of drug loading on Tg and highlighting the importance of drug loading as a key feature in model training [172]..
6. Future Perspectives
Physiologically based pharmacokinetic (PBPK) modeling is a robust tool for predicting drug absorption, distribution, metabolism, and excretion in humans. It integrates physiological and biochemical parameters to simulate drug behavior across populations, accounting for variations in sex, age, disease state, and metabolism [173,174]. PBPK modeling enables simulation of drug disposition in complex scenarios, potentially reducing reliance on human and animal trials. This approach has gained prominence in regulatory science, supporting drug development through evaluation of drug–drug interactions, first-in-human dosing, formulation design, and pharmacokinetics in special populations [173]. PBPK can incorporate the perturbed-chain statistical associating fluid theory (PC-SAFT) method to predict drug absorption and distribution. PC-SAFT is a thermodynamic model that estimates compound solubility and partitioning, which are essential for understanding drug interactions with biological systems. PBPK modeling has been applied to estimate drug solubility, partition coefficients, membrane permeability, drug distribution, and model interactions [175,176]. Studies have predicted pharmacokinetic profiles in rats and extrapolated findings to humans. In rats, pharmacokinetic analysis demonstrated higher C max and AUC, indicating enhanced absorption and brain penetration. PBPK model simulations corresponded with observed data, suggesting improved therapeutic efficacy in humans [177]. PC-SAFT predicts thermodynamic properties such as density, thermal expansion coefficient, glass transition temperature, isothermal compressibility, free energy change, and heat capacity. It is also used for phase equilibria calculations and chemical reaction predictions in ASD systems [178,179].
The emergence of novel artificial intelligence (AI)–based computational platforms, such as PharmSD, is transforming pharmaceutical development by advancing drug formulation and delivery processes through sophisticated computational methodologies. These platforms leverage ML and AI to analyze large datasets, optimize formulations, and predict drug interactions, thereby accelerating drug development and improving patient outcomes [180]. AI algorithms analyze formulation databases to identify optimal excipient combinations and processing parameters, substantially reducing experimental iterations. AI-driven process optimization ensures consistent product quality by implementing quality by design (QbD) principles. These systems utilize historical data to generate innovative formulation strategies and enhance drug efficacy [181]. It identifies the target by analyzing biological data to pinpoint disease-related targets, streamlining drug discovery, and increasing approval rates. Cost reduction is achieved through lead compound optimization, and AI minimizes the need for extensive animal testing [182]. The integration of AI with big data and the Internet of Things (IoT) delivers comprehensive solutions across the drug lifecycle, from discovery to registration [183]. These computational platforms employ various ML techniques, including RF, deep learning, support vector machines (SVM), and gradient boosting algorithms, to predict drug dissolution profiles across different polymers, assess the physical stability of solid dispersions, and provide virtual screening tools to streamline formulation design [184].
Accelerated stability modeling techniques for predicting ASD stability primarily include the accelerated stability assessment program (ASAP) and advanced kinetic modeling (AKM). These approaches enable rapid and accurate prediction of drug stability, especially for ASD formulations prone to crystallization and chemical degradation. ASAP is a predictive stability modeling approach that uses accelerated temperature and humidity conditions to induce rapid degradation, followed by kinetic modeling to estimate shelf life under standard conditions [185]. The ASAPprime software operationalizes ASAP by guiding experimental design with ASAPdesign and modeling degradation kinetics to predict shelf life within one week instead of months or years [186]. It exposes products to multiple combinations of temperature (typically 50–70 °C) and humidity (10–80 % RH), measuring the time-to-fail (isoconversion) at each condition [185]. ASAPprime then fits kinetic parameters such as activation energy (Ea), pre-exponential factor (ln A), and humidity sensitivity constant (B) using appropriate kinetic models, including diffusion kinetics, to accommodate the complex degradation behavior typical of ASD [185]. AKM extends beyond traditional Arrhenius-based kinetics to capture degradation pathways and physical instabilities in ASD, such as recrystallization and phase separation. It primarily assesses drug–polymer miscibility, drug–polymer interactions, manufacturing methods, and storage conditions, including temperature and humidity [187]. These models often incorporate multiparametric kinetic and probabilistic simulations to describe physical and chemical stability phenomena, enabling prediction of crystallization onset and other stability parameters within short experimental periods. AKM is particularly relevant for ASDs due to their complex microenvironment and the influence of excipients and moisture ingress during long-term storage [188]. This approach is less time- and resource-intensive than conventional testing, while enabling accelerated clinical supply and improved quality assurance [189].
7. Conclusions
ASD represent an effective strategy to enhance the solubility and bioavailability of poorly soluble drugs, a persistent challenge in pharmaceutical formulation. This review thoroughly discusses the physical stability considerations highlighting both opportunities and challenges in their development. The main advantage of ASD lies in maintaining the drug in a supersaturated state, thereby increasing the dissolution rate and therapeutic efficacy. However, this benefit is frequently compromised by physical instability, leading to phase separation and recrystallization. Thermodynamic, kinetic, and environmental factors are critical for ensuring the long-term stability. The combination of QbD and PAT frameworks facilitate effective control over material and process attributes, enhancing product consistency. Recent advances in ML and molecular modeling further support prediction of drug–polymer interactions and formulation improvement, minimizing reliance on empirical approaches. Future directions include the integration of AI-driven predictive tools, PBPK modeling, and accelerated stability assessments to refine ASD formulation strategies. The utilization of computational techniques with experimental validation, researchers can address remaining challenges in stability and clinical translation. ASD continues to offer a robust and versatile approach for overcoming poor solubility and maintaining physical stability over time using predictive tools and ML.
Author Contributions
Hari Prasad Bhatta: Conceptualization, Methodology, Data curation, Formal analysis, Writing – original draft. Hyo-Kyung Han: Data curation, Formal analysis. Ravi Maharjan: Conceptualization, Data curation, Formal analysis, Writing – original draft. Seong Hoon Jeong: Conceptualization, Methodology, Supervision, Funding acquisition, Writing – review and editing.
Acknowledgments
This research was supported by the Yonsei University Research Fund of 2025 (2025-22-0161).
References
- Zhang, J.; Guo, M.; Luo, M.; Cai, T. Advances in the development of amorphous solid dispersions: The role of polymeric carriers. Asian J. Pharm. Sci. 2023, 18, 100834. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Crystallization tendency of pharmaceutical glasses: Relevance to compound properties, impact of formulation process, and implications for design of amorphous solid dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Shweta, P.; Upendra, K. Formulation and evaluation of ternary solid dispersion of curcumin. Int. J. Pharm. Pharm. Sci. 2012, 4, 360–365. [Google Scholar]
- Vaka, S.R.K.; Bommana, M.M.; Desai, D.; Djordjevic, J.; Phuapradit, W.; Shah, N. Excipients for amorphous solid dispersions. In Amorphous Solid Dispersions: Theory and Practice; Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., Eds.; Springer New York: New York, NY, 2014; pp. 123–161. [Google Scholar]
- Zhang, M.; Li, H.; Lang, B.; O’Donnell, K.; Zhang, H.; Wang, Z.; Dong, Y.; Wu, C.; Williams III, R.O. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. Eur. J. Pharm. Biopharm. 2012, 82, 534–544. [Google Scholar] [CrossRef]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Del. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Kumari, N.; Ghosh, A. Cocrystallization: Cutting edge tool for physicochemical modulation of active pharmaceutical ingredients. Curr. Pharm. Des. 2020, 26, 4858–4882. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M.; Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D. Why amorphous drugs?; Springer: 2018.
- Zhou, Y.; Wang, J.; Xiao, Y.; Wang, T.; Huang, X. The effects of polymorphism on physicochemical properties and pharmacodynamics of solid drugs. Curr. Pharm. Des. 2018, 24, 2375–2382. [Google Scholar] [CrossRef]
- Jones, W.; Eddleston, M.D. Crystal imperfections in molecular crystals: Physical and chemical consequences. Disord. Pharm. Mater. 2016, 83–102. [Google Scholar]
- Grohganz, H.; Löbmann, K.; Priemel, P.; Jensen, K.T.; Graeser, K.; Strachan, C.; Rades, T. Amorphous drugs and dosage forms. J. Drug Deliv. Sci. Technol. 2013, 23, 403–408. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Rades, T. The role of configurational entropy in amorphous systems. Pharmaceutics 2010, 2, 224–244. [Google Scholar] [CrossRef]
- Sleutel, M.; Van Driessche, A.E. Role of clusters in nonclassical nucleation and growth of protein crystals. PNAS 2014, 111, E546–E553. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical stability of amorphous solid dispersions: A physicochemical perspective with thermodynamic, kinetic and environmental aspects. Pharm. Res. 2018, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, G.G.; Law, D.; Grant, D.J.; Schmitt, E.A. Thermodynamics, molecular mobility and crystallization kinetics of amorphous griseofulvin. Mol. Pharm. 2008, 5, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Sawicki, W.; Kozyra, A.; Tajber, L. Physical stability of solid dispersions with respect to thermodynamic solubility of tadalafil in PVP-VA. Eur. J. Pharm. Biopharm. 2015, 96, 237–246. [Google Scholar] [CrossRef]
- Xie, T.; Taylor, L.S. Effect of temperature and moisture on the physical stability of binary and ternary amorphous solid dispersions of celecoxib. J. Pharm. Sci. 2017, 106, 100–110. [Google Scholar] [CrossRef]
- Huang, L.; Lin, H.; Wang, H.; Ouyang, L.; Zhu, M. Amorphous alloys for hydrogen storage. J. Alloys Compd. 2023, 941, 168945. [Google Scholar] [CrossRef]
- Kaushik, R.; Budhwar, V.; Kaushik, D. An overview on recent patents and technologies on solid dispersion. Recent Pat. Drug Deliv. Formul. 2020, 14, 63–74. [Google Scholar] [CrossRef]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Dmelivery 2020, 27, 1625–1643. [Google Scholar] [CrossRef]
- Moseson, D.E.; Tran, T.B.; Karunakaran, B.; Ambardekar, R.; Hiew, T.N. Trends in amorphous solid dispersion drug products approved by the US Food and Drug Administration between 2012 and 2023. Int. J. Pharm. 2024, 7, 100259. [Google Scholar]
- Shah, H.S.; Chaturvedi, K.; Kuang, S.; Wang, J. Accelerating pre-formulation investigations in early drug product life cycles using predictive methodologies and computational algorithms. Ther. Deliv. 2021, 12, 789–797. [Google Scholar] [CrossRef] [PubMed]
- S’ari, M.; Blade, H.; Cosgrove, S.; Drummond-Brydson, R.; Hondow, N.; Hughes, L.P.; Brown, A. Characterization of amorphous solid dispersions and identification of low levels of crystallinity by transmission electron microscopy. Mol. Pharm. 2021, 18, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Santitewagun, S.; Thakkar, R.; Zeitler, J.A.; Maniruzzaman, M. Detecting crystallinity using terahertz spectroscopy in 3D printed amorphous solid dispersions. Mol. Pharm. 2022, 19, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.V.; Zemlyanov, D.Y.; Cavallaro, A.; Mangal, S.; Taylor, L.S.; Zhou, Q.T. Qualitative and quantitative characterization of composition heterogeneity on the surface of spray dried amorphous solid dispersion particles by an advanced surface analysis platform with high surface sensitivity and superior spatial resolution. Mol. Pharm. 2018, 15, 2045–2053. [Google Scholar] [CrossRef]
- Hancock, B.C.; Chauhan, H.V. Special topic cluster of articles on”Advancement in the formulation, characterization and performance of amorphous solid dispersions (ASDs)”. J. Pharm. Sci. 2021, 110, 1431. [Google Scholar] [CrossRef]
- WY Lee, T.; A Boersen, N.; Hui, H.; Chow, S.; Wan, K.; HL Chow, A. Delivery of poorly soluble compounds by amorphous solid dispersions. Curr. Pharm. Des. 2014, 20, 303–324. [Google Scholar] [CrossRef]
- Mukesh, S.; Mukherjee, G.; Singh, R.; Steenbuck, N.; Demidova, C.; Joshi, P.; Sangamwar, A.T.; Wade, R.C. Comparative analysis of drug-salt-polymer interactions by experiment and molecular simulation improves biopharmaceutical performance. Commun. Chem. 2023, 6, 201. [Google Scholar] [CrossRef]
- Pinket, W.; Aphibanthammakit, C.; Kasemwong, K.; Puttipipatkhachorn, S. Hydroxypropyl methylcellulose phthalate films reinforced with nanocrystalline cassava starch and intended its applications for colonic drug delivery. J. Drug Deliv. Sci. Technol. 2024, 98, 105908. [Google Scholar] [CrossRef]
- Butreddy, A.; Sarabu, S.; Almutairi, M.; Ajjarapu, S.; Kolimi, P.; Bandari, S.; Repka, M.A. Hot-melt extruded hydroxypropyl methylcellulose acetate succinate based amorphous solid dispersions: Impact of polymeric combinations on supersaturation kinetics and dissolution performance. Int. J. Pharm. 2022, 615, 121471. [Google Scholar] [CrossRef]
- Kallakunta, V.R.; Sarabu, S.; Bandari, S.; Batra, A.; Bi, V.; Durig, T.; Repka, M.A. Stable amorphous solid dispersions of fenofibrate using hot melt extrusion technology: Effect of formulation and process parameters for a low glass transition temperature drug. J. Drug Deliv. Sci. Technol. 2020, 58, 101395. [Google Scholar] [CrossRef]
- Nair, A.R.; Lakshman, Y.D.; Anand, V.S.K.; Sree, K.N.; Bhat, K.; Dengale, S.J. Overview of extensively employed polymeric carriers in solid dispersion technology. AAPS PharmSciTech 2020, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, M.; Rao, G.K. Pharmaceutical assessment of polyvinylpyrrolidone (PVP): As excipient from conventional to controlled delivery systems with a spotlight on COVID-19 inhibition. J. Drug Deliv. Sci. Technol. 2020, 60, 102046. [Google Scholar] [CrossRef] [PubMed]
- Nikam, A.; Sahoo, P.R.; Musale, S.; Pagar, R.R.; Paiva-Santos, A.C.; Giram, P.S. A systematic overview of Eudragit® based copolymer for smart healthcare. Pharmaceutics 2023, 15, 587. [Google Scholar] [CrossRef] [PubMed]
- Parikh, T.; Gupta, S.S.; Meena, A.; Serajuddin, A.T. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion-III: Polymethacrylates and polymethacrylic acid based polymers. J. Excip. Food Chem. 2016, 5. [Google Scholar]
- Browne, E.; Worku, Z.A.; Healy, A.M. Physicochemical properties of poly-vinyl polymers and their influence on ketoprofen amorphous solid dispersion performance: A polymer selection case study. Pharmaceutics 2020, 12, 433. [Google Scholar] [CrossRef]
- Terao, K. Poly(acrylic acid) (PAA). In Encyclopedia of polymeric nanomaterials; Kobayashi, S., Müllen, K., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2021; pp. 1–6. [Google Scholar]
- Long, R.; Long, S.; Zou, L.; Huang, Z.; Huang, Y.; Hu, C.; Li, D.; Li, X. Rheology, crystallization, and enhanced mechanical properties of uniaxially oriented ethylene-octene copolymer/polyolefin elastomer blends. Polymer 2022, 243, 124655. [Google Scholar] [CrossRef]
- Braun, S. Encapsulation of cells (cellular delivery) using sol-gel systems. In Comprehensive Biomaterials; Ducheyne, P., Ed.; Elsevier: Oxford, 2011; pp. 529–543. [Google Scholar]
- Sandhu, H.; Shah, N.; Chokshi, H.; Malick, A.W. Overview of amorphous solid dispersion technologies. Amorphous solid dispersions: theory and practice 2014, 91-122.
- Huang, S.; Williams, R.O. Effects of the preparation process on the properties of amorphous solid dispersions. AAPS PharmSciTech 2018, 19, 1971–1984. [Google Scholar] [CrossRef]
- FDA. Impurities: Guideline for residual solvents Q3 (R8) 2021.
- Demmon, S.; Bhargava, S.; Ciolek, D.; Halley, J.; Jaya, N.; Joubert, M.K.; Koepf, E.; Smith, P.; Trexler-Schmidt, M.; Tsai, P. A cross-industry forum on benchmarking critical quality attribute identification and linkage to process characterization studies. Biologicals 2020, 67, 9–20. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, J.H.; Kim, M.-S.; Jeong, S.H.; Choi, D.H. Process analytical technology tools for monitoring pharmaceutical unit operations: a control strategy for continuous process verification. Pharmaceutics 2021, 13, 919. [Google Scholar] [CrossRef]
- Panzitta, M.; Calamassi, N.; Sabatini, C.; Grassi, M.; Spagnoli, C.; Vizzini, V.; Ricchiuto, E.; Venturini, A.; Brogi, A.; Font, J.B. Spectrophotometry and pharmaceutical PAT/RTRT: Practical challenges and regulatory landscape from development to product lifecycle. Int. J. Pharm. 2021, 601, 120551. [Google Scholar] [CrossRef]
- Araújo, A.S.; Andrade, D.F.; Babos, D.V.; Pricylla, J.; Castro, J.A.G.; Sperança, M.A.; Gamela, R.R.; Machado, R.C.; Costa, V.C.; Guedes, W.N. Key information related to quality by design (QbD) applications in analytical methods development. Braz. J. Anal. Chem 2021, 8, 14–28. [Google Scholar]
- Puchert, T.; Holzhauer, C.-V.; Menezes, J.; Lochmann, D.; Reich, G. A new PAT/QbD approach for the determination of blend homogeneity: Combination of on-line NIRS analysis with PC scores distance analysis (PC-SDA). Eur. J. Pharm. Biopharm. 2011, 78, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.; Bezerra, M.; Markl, D.; Berghaus, A.; Borman, P.; Schlindwein, W. Development and validation of an in-line API quantification method using a QbD principles based on UV-vis spectroscopy to monitor and optimise continuous hot melt extrusion process. Pharmaceutics 2020, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.; Petrovska Jovanovska, V.; Berginc, K.; Jaklič, M.; Fabiani, F.; Harlacher, C.; Huzjak, T.; Sanchez-Felix, M.V. Amorphous solid dispersions (ASDs): The influence of material properties, manufacturing processes and analytical technologies in drug product development. Pharmaceutics 2021, 13, 1682. [Google Scholar] [CrossRef]
- Fabijański, M.; Gołofit, T. Influence of processing parameters on mechanical properties and degree of crystallization of polylactide. Materials 2024, 17, 3584. [Google Scholar] [CrossRef]
- Maclean, N.; Khadra, I.; Mann, J.; Williams, H.; Abbott, A.; Mead, H.; Markl, D. Investigating the role of excipients on the physical stability of directly compressed tablets. Int. J. Pharm. 2022, 4, 100106. [Google Scholar] [CrossRef]
- Sihorkar, V.; Dürig, T. Chapter 5 - The role of polymers and excipients in developing amorphous solid dispersions: An industrial perspective. In Drug Delivery Aspects, Shegokar, R., Ed.; Elsevier: 2020; pp. 79–113.
- Pendam, D.; Tomake, P.; Debaje, S.; Guleria, K.; Saha, A.; Thakran, P.; Sangamwar, A.T. Advances in formulation strategies and stability considerations of amorphous solid dispersions. J. Drug Deliv. Sci. Technol. 2025, 106922. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q.T. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B. 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Bookwala, M.; Wildfong, P.L. The implications of drug-polymer interactions on the physical stability of amorphous solid dispersions. Pharm. Res. 2023, 40, 2963–2981. [Google Scholar] [CrossRef]
- Qian, K.; Stella, L.; Jones, D.S.; Andrews, G.P.; Du, H.; Tian, Y. Drug-rich phases induced by amorphous solid dispersion: Arbitrary or intentional goal in oral drug delivery? Pharmaceutics 2021, 13, 889. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Microstructure formation for improved dissolution performance of lopinavir amorphous solid dispersions. Mol. Pharm. 2019, 16, 1751–1765. [Google Scholar] [CrossRef]
- Hiew, T.N.; Zemlyanov, D.Y.; Taylor, L.S. Balancing solid-state stability and dissolution performance of lumefantrine amorphous solid dispersions: The role of polymer choice and drug-polymer interactions. Mol. Pharm. 2021, 19, 392–413. [Google Scholar] [CrossRef]
- Akbari, F.; Didehban, K.; Farhang, M. Solubility of solid intermediate of pharmaceutical compounds in pure organic solvents using semi-empirical models. Eur. J. Pharm. Sci. 2020, 143, 105209. [Google Scholar] [CrossRef]
- Tian, B.; Wang, X.; Zhang, Y.; Zhang, K.; Zhang, Y.; Tang, X. Theoretical prediction of a phase diagram for solid dispersions. Pharm. Res. 2015, 32, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Jacobs, E.; Jones, D.S.; McCoy, C.P.; Wu, H.; Andrews, G.P. The design and development of high drug loading amorphous solid dispersion for hot-melt extrusion platform. Int. J. Pharm. 2020, 586, 119545. [Google Scholar] [CrossRef] [PubMed]
- Sarpal, K.; Delaney, S.; Zhang, G.G.; Munson, E.J. Phase behavior of amorphous solid dispersions of felodipine: Homogeneity and drug-polymer interactions. Mol. Pharm. 2019, 16, 4836–4851. [Google Scholar] [CrossRef]
- Mansuri, A.; Münzner, P.; Heermant, A.; Hänsch, S.; Feuerbach, T.; Fischer, B.r.; Winck, J.; Vermeer, A.W.; Hoheisel, W.; Böhmer, R. Characterizing phase separation of amorphous solid dispersions containing imidacloprid. Mol. Pharm. 2023, 20, 2080–2093. [Google Scholar] [CrossRef]
- Krummnow, A.; Danzer, A.; Voges, K.; Kyeremateng, S.O.; Degenhardt, M.; Sadowski, G. Kinetics of water-induced amorphous phase separation in amorphous solid dispersions via Raman mapping. Pharmaceutics 2023, 15, 1395. [Google Scholar] [CrossRef]
- Pourhakkak, P.; Taghizadeh, A.; Taghizadeh, M.; Ghaedi, M.; Haghdoust, S. Fundamentals of adsorption technology. In Interface Science and Technology, Ghaedi, M., Ed.; Elsevier: 2021; Volume 33, pp. 1–70.
- Rubinstein, M.; Panyukov, S. Elasticity of polymer networks. Macromolecules 2002, 35, 6670–6686. [Google Scholar] [CrossRef]
- Higgins, J.S.; Cabral, J.T. A thorny problem? Spinodal decomposition in polymer blends. 2020, 53, 4137–4140. [Google Scholar]
- Lin, D.; Huang, Y. A thermal analysis method to predict the complete phase diagram of drug-polymer solid dispersions. Int. J. Pharm. 2010, 399, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Stella, L.; Liu, F.; Jones, D.S.; Andrews, G.P.; Tian, Y. Kinetic and thermodynamic interplay of polymer-mediated liquid-liquid phase separation for poorly water-soluble drugs. Mol. Pharm. 2024, 21, 2878–2893. [Google Scholar] [CrossRef] [PubMed]
- Mathers, A.; Pechar, M.; Hassouna, F.; Fulem, M. API solubility in semi-crystalline polymer: Kinetic and thermodynamic phase behavior of PVA-based solid dispersions. Int. J. Pharm. 2022, 623, 121855. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Newman, A.; Zografi, G. What are the important factors that influence API crystallization in miscible amorphous API–excipient mixtures during long-term storage in the glassy state? Mol. Pharm. 2021, 19, 378–391. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Mahlin, D.; Bergström, C.A. Physical stability of drugs after storage above and below the glass transition temperature: Relationship to glass-forming ability. Int. J. Pharm. 2015, 495, 312–317. [Google Scholar] [CrossRef]
- Yu, D.; Li, J.; Wang, H.; Pan, H.; Li, T.; Bu, T.; Zhou, W.; Zhang, X. Role of polymers in the physical and chemical stability of amorphous solid dispersion: A case study of carbamazepine. Eur. J. Pharm. Sci. 2022, 169, 106086. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. The role of drug-polymer hydrogen bonding interactions on the molecular mobility and physical stability of nifedipine solid dispersions. Mol. Pharm. 2015, 12, 162–170. [Google Scholar] [CrossRef]
- Rusdin, A.; Mohd Gazzali, A.; Ain Thomas, N.; Megantara, S.; Aulifa, D.L.; Budiman, A.; Muchtaridi, M. Advancing drug delivery paradigms: Polyvinyl pyrolidone (PVP)-based amorphous solid dispersion for enhanced physicochemical properties and therapeutic efficacy. Polymers 2024, 16, 286. [Google Scholar] [CrossRef]
- Frank, D.S.; Matzger, A.J. Effect of polymer hydrophobicity on the stability of amorphous solid dispersions and supersaturated solutions of a hydrophobic pharmaceutical. Mol. Pharm. 2019, 16, 682–688. [Google Scholar] [CrossRef]
- Du, X.; Zhao, Z.; Li, Y.X. Production of soluble pea protein/sodium caseinate co-dispersions using ultrasonication and their acid coagulation properties. Food Hydrocoll. 2023, 139, 108562. [Google Scholar] [CrossRef]
- Zhi, Z.; Yan, L.; Li, H.; Dewettinck, K.; Van der Meeren, P.; Liu, R.; Van Bockstaele, F. A combined approach for modifying pea protein isolate to greatly improve its solubility and emulsifying stability. Food Chem. 2022, 380, 131832. [Google Scholar] [CrossRef]
- Gui, Y.; McCann, E.C.; Yao, X.; Li, Y.; Jones, K.J.; Yu, L. Amorphous drug-polymer salt with high stability under tropical conditions and fast dissolution: the case of clofazimine and poly (acrylic acid). Mol. Pharm. 2021, 18, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Que, C.; Qi, Q.; Zemlyanov, D.Y.; Mo, H.; Deac, A.; Zeller, M.; Indulkar, A.S.; Gao, Y.; Zhang, G.G.; Taylor, L.S. Evidence for halogen bonding in amorphous solid dispersions. Cryst. Growth Des. 2020, 20, 3224–3235. [Google Scholar] [CrossRef]
- Lu, X.; Li, M.; Huang, C.; Lowinger, M.B.; Xu, W.; Yu, L.; Byrn, S.R.; Templeton, A.C.; Su, Y. Atomic-level drug substance and polymer interaction in posaconazole amorphous solid dispersion from solid-state NMR. Mol. Pharm. 2020, 17, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Huang, C.; Lowinger, M.B.; Yang, F.; Xu, W.; Brown, C.D.; Hesk, D.; Koynov, A.; Schenck, L.; Su, Y. Molecular interactions in posaconazole amorphous solid dispersions from two-dimensional solid-state NMR spectroscopy. Mol. Pharm. 2019, 16, 2579–2589. [Google Scholar] [CrossRef]
- Orszulak, L.; Lamrani, T.; Bernat, R.; Tarnacka, M.; Żakowiecki, D.; Jurkiewicz, K.; Zioła, P.; Mrozek-Wilczkiewicz, A.; Zięba, A.; Kamiński, K. The influence of PVP polymer topology on the liquid crystalline order of itraconazole in binary systems. Mol. Pharm. 2024, 21, 3027–3039. [Google Scholar] [CrossRef]
- Mehta, M.; Ragoonanan, V.; McKenna, G.B.; Suryanarayanan, R. Correlation between molecular mobility and physical stability in pharmaceutical glasses. Mol. Pharm. 2016, 13, 1267–1277. [Google Scholar] [CrossRef]
- Quan, P.; Wan, X.; Tian, Q.; Liu, C.; Fang, L. Dicarboxylic acid as a linker to improve the content of amorphous drug in drug-in-polymer film: Effects of molecular mobility, electrical conductivity and intermolecular interactions. J. Controlled Release 2020, 317, 142–153. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Mizoguchi, R.; Kawakami, K.; Miyazaki, T. Influence of the crystallization tendencies of pharmaceutical glasses on the applicability of the Adam-Gibbs-Vogel and Vogel-Tammann-Fulcher equations in the prediction of their long-term physical stability. Int. J. Pharm. 2022, 626, 122158. [Google Scholar] [CrossRef]
- Diederichsen, K.M.; Buss, H.G.; McCloskey, B.D. The compensation effect in the Vogel–Tammann–Fulcher (VTF) equation for polymer-based electrolytes. Macromolecules 2017, 50, 3831–3840. [Google Scholar] [CrossRef]
- Sahoo, A.; Kumar, N.K.; Suryanarayanan, R. Crosslinking: An avenue to develop stable amorphous solid dispersion with high drug loading and tailored physical stability. J. Controlled Release 2019, 311, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Kashchiev, D. Nucleation: Basic theory with applications. 1st edition. 2000. [Google Scholar]
- Zhang, J.; Jiang, Q.; Xu, Z.; Yang, Q.; Hao, G.; Liu, M.; Zeng, Z. Recent progress on crystal nucleation of amorphous solid dispersion. Cryst. Growth Des. 2024, 24, 8655–8666. [Google Scholar] [CrossRef]
- Schmelzer, J.W. On the determination of the kinetic pre-factor in classical nucleation theory. J. Non-Cryst. Solids 2010, 356, 2901–2907. [Google Scholar] [CrossRef]
- Tournier, R.F. Crystal growth nucleation and Fermi energy equalization of intrinsic spherical nuclei in glass-forming melts. Sci. Technol. Adv. Mater. 2009, 10, 014607. [Google Scholar] [CrossRef]
- Karthika, S.; Radhakrishnan, T.; Kalaichelvi, P. A review of classical and nonclassical nucleation theories. Cryst. Growth Des. 2016, 16, 6663–6681. [Google Scholar] [CrossRef]
- Trasi, N.S.; Taylor, L.S. Effect of polymers on nucleation and crystal growth of amorphous acetaminophen. CrystEngComm 2012, 14, 5188–5197. [Google Scholar] [CrossRef]
- López Burgos, G.; Hernández Espinell, J.R.; Graciani-Massa, T.; Yao, X.; Borchardt-Setter, K.A.; Yu, L.; López-Mejías, V.; Stelzer, T. Role of heteronucleants in melt crystallization of crystalline solid dispersions. Cryst. Growth Des. 2022, 23, 49–58. [Google Scholar] [CrossRef]
- Kolisnyk, T.; Mohylyuk, V.; Fil, N.; Bickerstaff, E.; Li, S.; Jones, D.S.; Andrews, G.P. High drug-loaded amorphous solid dispersions of a poor glass forming drug: The impact of polymer type and cooling rate on amorphous drug behaviour. Int. J. Pharm. 2025, 670, 125095. [Google Scholar] [CrossRef]
- Tian, Y.; Jones, D.S.; Andrews, G.P. An investigation into the role of polymeric carriers on crystal growth within amorphous solid dispersion systems. Mol. Pharm. 2015, 12, 1180–1192. [Google Scholar] [CrossRef]
- Moseson, D.E.; Corum, I.D.; Lust, A.; Altman, K.J.; Hiew, T.N.; Eren, A.; Nagy, Z.K.; Taylor, L.S. Amorphous solid dispersions containing residual crystallinity: competition between dissolution and matrix crystallization. AAPS J. 2021, 23, 69. [Google Scholar] [CrossRef]
- Ojo, A.T.; Ma, C.; Lee, P.I. Elucidating the effect of crystallization on drug release from amorphous solid dispersions in soluble and insoluble carriers. Int. J. Pharm. 2020, 591, 120005. [Google Scholar] [CrossRef]
- Li, W.; Buckton, G. Using DVS-NIR to assess the water sorption behaviour and stability of a griseofulvin/PVP K30 solid dispersion. Int. J. Pharm. 2015, 495, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Almotairy, A.; Almutairi, M.; Althobaiti, A.; Alyahya, M.; Sarabu, S.; Alzahrani, A.; Zhang, F.; Bandari, S.; Repka, M.A. Effect of pH modifiers on the solubility, dissolution rate, and stability of telmisartan solid dispersions produced by hot-melt extrusion technology. J. Drug Deliv. Sci. Technol. 2021, 65, 102674. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nollenberger, K.; Albers, J.; Craig, D.; Qi, S. Microstructure of an immiscible polymer blend and its stabilization effect on amorphous solid dispersions. Mol. Pharm. 2013, 10, 2767–2780. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nollenberger, K.; Albers, J.; Craig, D.; Qi, S. Molecular indicators of surface and bulk instability of hot melt extruded amorphous solid dispersions. Pharm. Res. 2015, 32, 1210–1228. [Google Scholar] [CrossRef]
- Wegiel, L.A.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Crystallization of amorphous solid dispersions of resveratrol during preparation and storage-impact of different polymers. J. Pharm. Sci. 2013, 102, 171–184. [Google Scholar] [CrossRef]
- Yang, F.; Su, Y.; Small, J.; Huang, C.; Martin, G.E.; Farrington, A.M.; DiNunzio, J.; Brown, C.D. Probing the molecular-level interactions in an active pharmaceutical ingredient (API)-polymer dispersion and the resulting impact on drug product formulation. Pharm. Res. 2020, 37, 1–16. [Google Scholar] [CrossRef]
- Telang, C.; Mujumdar, S.; Mathew, M. Improved physical stability of amorphous state through acid base interactions. J. Pharm. Sci. 2009, 98, 2149–2159. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT calculations-review of applications in the pharmaceutical sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef]
- Van Der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Meng, F.; Trivino, A.; Prasad, D.; Chauhan, H. Investigation and correlation of drug polymer miscibility and molecular interactions by various approaches for the preparation of amorphous solid dispersions. Eur. J. Pharm. Sci. 2015, 71, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Morgan, D.J.; Mendham, A.P.; Pang, J.; Snowden, M.J.; Douroumis, D. Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions. Int. J. Pharm. 2013, 443, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Pang, J.; Morgan, D.J.; Douroumis, D. Molecular modeling as a predictive tool for the development of solid dispersions. Mol. Pharm. 2015, 12, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Snowden, M.J.; Bradely, M.S.; Douroumis, D. Studies of intermolecular interactions in solid dispersions using advanced surface chemical analysis. RSC Adv. 2015, 5, 74212–74219. [Google Scholar] [CrossRef]
- Nie, H.; Mo, H.; Zhang, M.; Song, Y.; Fang, K.; Taylor, L.S.; Li, T.; Byrn, S.R. Investigating the interaction pattern and structural elements of a drug-polymer complex at the molecular level. Mol. Pharm. 2015, 12, 2459–2468. [Google Scholar] [CrossRef]
- Wang, B.; Wang, D.; Zhao, S.; Huang, X.; Zhang, J.; Lv, Y.; Liu, X.; Lv, G.; Ma, X. Evaluate the ability of PVP to inhibit crystallization of amorphous solid dispersions by density functional theory and experimental verify. Eur. J. Pharm. Sci. 2017, 96, 45–52. [Google Scholar] [CrossRef]
- Antolović, I.; Vrabec, J.; Klajmon, M. COSMOPharm: Drug–Polymer Compatibility of Pharmaceutical Amorphous Solid Dispersions from COSMO-SAC. Mol. Pharm. 2024, 21, 4395–4415. [Google Scholar] [CrossRef]
- Ma, S.-M.; Zhao, L.; Wang, Y.-L.; Zhu, Y.-L.; Lu, Z.-Y. The coarse-grained models of poly (ethylene oxide) and poly (propylene oxide) homopolymers and poloxamers in big multipole water (BMW) and MARTINI frameworks. PCCP 2020, 22, 15976–15985. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, N.; Almutairy, B.; Kallakunta, V.R.; Sarabu, S.; Thipsay, P.; Bandari, S.; Repka, M.A. Manufacturing strategies to develop amorphous solid dispersions: An overview. J. Drug Deliv. Sci. Technol. 2020, 55, 101459. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Huang, D.; Zhang, T.; Luo, T. Determining influential descriptors for polymer chain conformation based on empirical force-fields and molecular dynamics simulations. Chem. Phys. Lett. 2018, 704, 49–54. [Google Scholar] [CrossRef]
- Mao, Q.; Feng, M.; Jiang, X.Z.; Ren, Y.; Luo, K.H.; van Duin, A.C. Classical and reactive molecular dynamics: Principles and applications in combustion and energy systems. Prog. Energy Combust. Sci. 2023, 97, 101084. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153. [Google Scholar] [CrossRef]
- Shelke, R.; Velagacherla, V.; Nayak, U.Y. Recent advances in dual-drug co-amorphous systems. Drug Discov. Today 2024, 29, 103863. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Suryanarayanan, R. Local mobility in amorphous pharmaceuticals-characterization and implications on stability. J. Pharm. Sci. 2009, 98, 2935–2953. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. Influence of molecular mobility on the physical stability of amorphous pharmaceuticals in the supercooled and glassy states. Mol. Pharm. 2014, 11, 3048–3055. [Google Scholar] [CrossRef]
- Bookwala, M.; DeBoyace, K.; Buckner, I.S.; Wildfong, P.L. Predicting density of amorphous solid materials using molecular dynamics simulation. AAPS PharmSciTech 2020, 21, 1–11. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Molecular dynamics simulation of amorphous indomethacin. Mol. Pharm. 2013, 10, 102–114. [Google Scholar] [CrossRef]
- Kasimova, A.O.; Pavan, G.M.; Danani, A.; Mondon, K.; Cristiani, A.; Scapozza, L.; Gurny, R.; Möller, M. Validation of a novel molecular dynamics simulation approach for lipophilic drug incorporation into polymer micelles. J. Phys. Chem. B. 2012, 116, 4338–4345. [Google Scholar] [CrossRef]
- Chen, S.; Li, J.; Wei, L.; Jin, Y.; Khosla, T.; Xiao, J.; Cheng, B.; Duan, H. A molecular modeling study for miscibility of polyimide/polythene mixing systems with/without compatibilizer. J. Polym. Eng. 2018, 38, 891–898. [Google Scholar] [CrossRef]
- Andrews, J.; Handler, R.A.; Blaisten-Barojas, E. Structure, energetics and thermodynamics of PLGA condensed phases from Molecular Dynamics. Polymer 2020, 206, 122903. [Google Scholar] [CrossRef]
- Muljajew, I.; Erlebach, A.; Weber, C.; Buchheim, J.R.; Sierka, M.; Schubert, U.S. A polyesteramide library from dicarboxylic acids and 2, 2′-bis (2-oxazoline): Synthesis, characterization, nanoparticle formulation and molecular dynamics simulations. Polym. Chem. 2020, 11, 112–124. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Molecular dynamics simulation of amorphous hydroxypropyl-methylcellulose acetate succinate (HPMCAS): Polymer model development, water distribution, and plasticization. Mol. Pharm. 2014, 11, 2400–2411. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Goliaei, A.; Tsereteli, L.; Berkowitz, M.L. Properties of poloxamer molecules and poloxamer micelles dissolved in water and next to lipid bilayers: results from computer simulations. J. Phys. Chem. B. 2016, 120, 5823–5830. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Nunes, C.; Jonnalagadda, S. A molecular dynamics approach for predicting the glass transition temperature and plasticization effect in amorphous pharmaceuticals. Mol. Pharm. 2013, 10, 4136–4145. [Google Scholar] [CrossRef]
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B. 2011, 115, 2014–2023. [Google Scholar] [CrossRef]
- Shahzad, Y.; Sohail, S.; Arshad, M.S.; Hussain, T.; Shah, S.N.H. Development of solid dispersions of artemisinin for transdermal delivery. Int. J. Pharm. 2013, 457, 197–205. [Google Scholar] [CrossRef]
- Razmimanesh, F.; Amjad-Iranagh, S.; Modarress, H. Molecular dynamics simulation study of chitosan and gemcitabine as a drug delivery system. J. Mol. Model. 2015, 21, 1–14. [Google Scholar] [CrossRef]
- Gong, C.; Deng, S.; Wu, Q.; Xiang, M.; Wei, X.; Li, L.; Gao, X.; Wang, B.; Sun, L.; Chen, Y. Improving antiangiogenesis and anti-tumor activity of curcumin by biodegradable polymeric micelles. Biomaterials 2013, 34, 1413–1432. [Google Scholar] [CrossRef]
- Fule, R.; Amin, P. Development and evaluation of lafutidine solid dispersion via hot melt extrusion: Investigating drug-polymer miscibility with advanced characterisation. Asian J. Pharm. Sci. 2014, 9, 92–106. [Google Scholar] [CrossRef]
- Fule, R.; Amin, P. Hot melt extruded amorphous solid dispersion of posaconazole with improved bioavailability: investigating drug-polymer miscibility with advanced characterisation. Biomed. Res. Int. 2014, 2014, 146781. [Google Scholar] [CrossRef] [PubMed]
- Aulifa, D.L.; Al Shofwan, A.A.; Megantara, S.; Fakih, T.M.; Budiman, A. Elucidation of molecular interactions between drug-polymer in amorphous solid dispersion by a computational approach using molecular dynamics simulations. Adv. Appl. Bioinform. Chem. 2024, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fule, R.; Meer, T.; Sav, A.; Amin, P. Solubility and dissolution rate enhancement of lumefantrine using hot melt extrusion technology with physicochemical characterisation. J. Pharm. Investig. 2013, 43, 305–321. [Google Scholar] [CrossRef]
- Gangurde, A.B.; Kundaikar, H.S.; Javeer, S.D.; Jaiswar, D.R.; Degani, M.S.; Amin, P.D. Enhanced solubility and dissolution of curcumin by a hydrophilic polymer solid dispersion and its insilico molecular modeling studies. J. Drug Deliv. Sci. Technol. 2015, 29, 226–237. [Google Scholar] [CrossRef]
- Yani, Y.; Kanaujia, P.; Chow, P.S.; Tan, R.B. Effect of API-polymer miscibility and interaction on the stabilization of amorphous solid dispersion: A molecular simulation study. Ind. Eng. Chem. Res. 2017, 56, 12698–12707. [Google Scholar] [CrossRef]
- Macháčková, M.; Tokarský, J.; Čapková, P. A simple molecular modeling method for the characterization of polymeric drug carriers. Eur. J. Pharm. Sci. 2013, 48, 316–322. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Karagianni, A.; Katopodis, K.; Vardaka, E.; Kachrimanis, K. Molecular modelling and simulation of fusion-based amorphous drug dispersions in polymer/plasticizer blends. Eur. J. Pharm. Sci. 2019, 130, 260–268. [Google Scholar] [CrossRef]
- Gupta, K.M.; Chin, X.; Kanaujia, P. Molecular interactions between APIs and enteric polymeric excipients in solid dispersion: insights from molecular simulations and experiments. Pharmaceutics 2023, 15, 1164. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Molecular dynamics simulation of amorphous indomethacin-poly (vinylpyrrolidone) glasses: Solubility and hydrogen bonding interactions. J. Pharm. Sci. 2013, 102, 876–891. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Molecular dynamics simulation of amorphous hydroxypropylmethylcellulose and its mixtures with felodipine and water. J. Pharm. Sci. 2017, 106, 803–816. [Google Scholar] [CrossRef]
- Medarević, D.P.; Kachrimanis, K.; Mitrić, M.; Djuriš, J.; Djurić, Z.; Ibrić, S. Dissolution rate enhancement and physicochemical characterization of carbamazepine-poloxamer solid dispersions. Pharm. Dev. Technol. 2016, 21, 268–276. [Google Scholar] [CrossRef]
- Mosquera-Giraldo, L.I.; Borca, C.H.; Meng, X.; Edgar, K.J.; Slipchenko, L.V.; Taylor, L.S. Mechanistic design of chemically diverse polymers with applications in oral drug delivery. Biomacromolecules 2016, 17, 3659–3671. [Google Scholar] [CrossRef]
- Erlebach, A.; Ott, T.; Otzen, C.; Schubert, S.; Czaplewska, J.; Schubert, U.S.; Sierka, M. Thermodynamic compatibility of actives encapsulated into PEG-PLA nanoparticles: In silico predictions and experimental verification. J. Comput. Chem. 2016, 37, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Turpin, E.R.; Taresco, V.; Al-Hachami, W.A.; Booth, J.; Treacher, K.; Tomasi, S.; Alexander, C.; Burley, J.; Laughton, C.A.; Garnett, M.C. In silico screening for solid dispersions: The trouble with solubility parameters and χFH. Mol. Pharm. 2018, 15, 4654–4667. [Google Scholar] [CrossRef] [PubMed]
- Iesavand, H.; Rahmati, M.; Afzali, D.; Modiri, S. Investigation on absorption and release of mercaptopurine anticancer drug from modified polylactic acid as polymer carrier by molecular dynamic simulation. Mater. Sci. Eng. C Biomimetic Supramol. Syst. 2019, 105, 110010. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.; Gokarna, V.; Deshpande, V.; Vavia, P. Bioavailability enhancement of olmesartan medoxomil using hot-melt extrusion: In-silico, in-vitro, and in-vivo evaluation. AAPS PharmSciTech 2020, 21, 254. [Google Scholar] [CrossRef]
- Eslami, M.; Nikkhah, S.J.; Hashemianzadeh, S.M.; Sajadi, S.A.S. The compatibility of tacrine molecule with poly (n-butylcyanoacrylate) and chitosan as efficient carriers for drug delivery: A molecular dynamics study. Eur. J. Pharm. Sci. 2016, 82, 79–85. [Google Scholar] [CrossRef]
- Kapourani, A.; Eleftheriadou, K.; Kontogiannopoulos, K.N.; Barmpalexis, P. Evaluation of rivaroxaban amorphous solid dispersions physical stability via molecular mobility studies and molecular simulations. Eur. J. Pharm. Sci. 2021, 157, 105642. [Google Scholar] [CrossRef]
- Hussan, K.S.; Govindaraj, G.; Correia, N.T.; Shinyashiki, N.; Thayyil, M.S.; Babu, T.D. Molecular dynamics and interactions in amorphous solid dispersion of Erlotinib HCl for improved cancer therapy. J. Mol. Struct. 2025, 1336, 142014. [Google Scholar] [CrossRef]
- Mendyk, A.; Szlȩk, J.; Jachowicz, R. A heuristic decision support system for microemulsions formulation development. In Formulation tools for pharmaceutical development; Elsevier: 2013; pp. 39–71.
- Zhang, Z.-h.; Pan, W.-s. Expert system for the development and formulation of push-pull osmotic pump tablets containing poorly water-soluble drugs. In Formulation tools for pharmaceutical development; Elsevier: 2013; pp. 73–108.
- Das, M.K.; Chakraborty, T. ANN in pharmaceutical product and process development. In Artificial neural network for drug design, delivery and disposition; Elsevier: 2016; pp. 277–293.
- Han, R.; Yang, Y.; Li, X.; Ouyang, D. Predicting oral disintegrating tablet formulations by neural network techniques. Asian J. Pharm. Sci. 2018, 13, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Nurzyńska, K.; Booth, J.; Roberts, C.J.; McCabe, J.; Dryden, I.; Fischer, P.M. Long-term amorphous drug stability predictions using easily calculated, predicted, and measured parameters. Mol. Pharm. 2015, 12, 3389–3398. [Google Scholar] [CrossRef] [PubMed]
- Przybyłek, M.; Jeliński, T.; Cysewski, P. Application of multivariate adaptive regression splines (MARSplines) for predicting hansen solubility parameters based on 1D and 2D molecular descriptors computed from SMILES string. J. Chem. 2019, 2019, 9858371. [Google Scholar] [CrossRef]
- Moore, M.D.; Wildfong, P.L. Informatics calibration of a molecular descriptors database to predict solid dispersion potential of small molecule organic solids. Int. J. Pharm. 2011, 418, 217–226. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Kachrimanis, K.; Georgarakis, E. Solid dispersions in the development of a nimodipine floating tablet formulation and optimization by artificial neural networks and genetic programming. Eur. J. Pharm. Biopharm. 2011, 77, 122–131. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Koutsidis, I.; Karavas, E.; Louka, D.; Papadimitriou, S.A.; Bikiaris, D.N. Development of PVP/PEG mixtures as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique and optimization of dissolution using artificial neural networks. Eur. J. Pharm. Biopharm. 2013, 85, 1219–1231. [Google Scholar] [CrossRef]
- Han, R.; Xiong, H.; Ye, Z.; Yang, Y.; Huang, T.; Jing, Q.; Lu, J.; Pan, H.; Ren, F.; Ouyang, D. Predicting physical stability of solid dispersions by machine learning techniques. J. Controlled Release 2019, 311, 16–25. [Google Scholar] [CrossRef]
- Vojinovic, T.; Potpara, Z.; Vukmirovic, M.; Turkovic, N.; Ibric, S. Artificial neural networks and their application in the optimization of carbamazepine solid dispersions. Acta Pol. Pharm. 2022, 79. [Google Scholar] [CrossRef]
- Jiang, J.; Lu, A.; Ma, X.; Ouyang, D.; Williams III, R.O. The applications of machine learning to predict the forming of chemically stable amorphous solid dispersions prepared by hot-melt extrusion. Int. J. Pharm. 2023, 5, 100164. [Google Scholar] [CrossRef]
- Di Mare, E.J.; Punia, A.; Lamm, M.S.; Rhodes, T.A.; Gormley, A.J. Data-driven design of novel polymer excipients for pharmaceutical amorphous solid dispersions. Bioconj. Chem. 2024, 35, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Hartmanshenn, C.; Scherholz, M.; Androulakis, I.P. Physiologically-based pharmacokinetic models: approaches for enabling personalized medicine. J. Pharmacokinet. Pharmacodyn. 2016, 43, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Khalil, F.; Läer, S. Physiologically based pharmacokinetic modeling: Methodology, applications, and limitations with a focus on its role in pediatric drug development. J. Biomed. Biotechnol. 2011, 2011, 907461. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Thomas, S.; Tsang, Y.C.; Almeida, S.; Ashraf, M.; Fotaki, N.; Heimbach, T.; Patel, N.; Shah, H.; Jiang, X. Advances in physiologically based pharmacokinetic (PBPK) modeling and its regulatory utility to support oral drug product development and harmonization. Pharm. Res. 2025, 1–15. [Google Scholar] [CrossRef]
- Miller, N.A.; Reddy, M.B.; Heikkinen, A.T.; Lukacova, V.; Parrott, N. Physiologically based pharmacokinetic modelling for first-in-human predictions: an updated model building strategy illustrated with challenging industry case studies. Clin. Pharmacokinet. 2019, 58, 727–746. [Google Scholar] [CrossRef]
- Bharti, K.; Deepika, D.; Kumar, M.; Jha, A.; Manjit; Akhilesh; Tiwari, V.; Kumar, V.; Mishra, B. Bharti, K.; Deepika, D.; Kumar, M.; Jha, A.; Manjit; Akhilesh; Tiwari, V.; Kumar, V.; Mishra, B. Development and evaluation of amorphous solid dispersion of riluzole with pbpk model to simulate the pharmacokinetic profile. AAPS PharmSciTech 2023, 24, 219. [Google Scholar] [CrossRef]
- Pakravesh, A.; Mohammadi, A.H.; Richon, D. Performance evaluation of PρT-SAFT, PρT-PC-SAFT, PC-SAFT, and CPA equations of state for predicting density, thermal expansion coefficient, isothermal compressibility, isobaric heat capacity, speed of sound, and saturated vapor pressure of three pure ethylene glycols and their mixtures. Int. J. Thermophys. 2025, 46, 30. [Google Scholar]
- Ascani, M.; Sadowski, G.; Held, C. Simultaneous predictions of chemical and phase equilibria in systems with an esterification reaction using PC-SAFT. Molecules 2023, 28, 1768. [Google Scholar] [CrossRef]
- Dong, J.; Gao, H.; Ouyang, D. PharmSD: A novel AI-based computational platform for solid dispersion formulation design. Int. J. Pharm. 2021, 604, 120705. [Google Scholar] [CrossRef]
- Saha, G. Artificial Intelligence (AI) in formulation development. In Computer Aided Drug Development; 2024; pp. 459–481.
- Agrawal, G.; Tushir, S.; Arora, D.; Sangwan, K. Artificial intelligence in pharmaceutical drug delivery. In Proceedings of the 2024 International conference on computational Intelligence and computing applications (ICCICA); 2024; pp. 406–410. [Google Scholar]
- Xiao, L.; Zhang, Y. AI-driven smart pharmacology. Int. Pharm. 2023, 1, 179–182. [Google Scholar] [CrossRef]
- Au-Yeung, L.; Tseng, C.-Y.; Tam, Y.K.; Tsai, P.A. Machine learning based quantitative structure-dissolution profile relationship. J. Chem. Inf. Model. 2025, 65, 6273–6286. [Google Scholar] [CrossRef]
- Modhave, D.; Vrielynck, S.; Roeleveld, K. Assessing drug product shelf life using the accelerated stability asessment pogram: A case study of a GLPG4399 capsule formulation. Pharmaceutics 2024, 16, 1400. [Google Scholar] [CrossRef]
- Flavier, K.; McLellan, J.; Botoy, T.; Waterman, K.C. Accelerated shelf life modeling of appearance change in drug products using ASAP prime®. Pharm. Dev. Technol. 2022, 27, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Lennard, A.; Zimmermann, B.; Clenet, D.; Molony, M.; Tami, C.; Aviles, C.O.; Moran, A.; Pue-Gilchrist, P.; Flores, E.L. Stability modeling methodologies to enable earlier patient access. J. Pharm. Sci. 2024. [Google Scholar] [CrossRef] [PubMed]
- Waterman, K.C. The application of the accelerated stability assessment program (ASAP) to quality by design (QbD) for drug product stability. AAPS PharmSciTech 2011, 12, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Rack, C. An introduction to the accelerated stability assessment program. Am. Pharm. Rev. 2017, 20, 86–90. [Google Scholar]
- Sarabu, S.; Kallakunta, V.R.; Bandari, S.; Batra, A.; Bi, V.; Durig, T.; Zhang, F.; Repka, M.A. Hypromellose acetate succinate based amorphous solid dispersions via hot melt extrusion: Effect of drug physicochemical properties. Carbohydr. Polym. 2020, 233, 115828. [Google Scholar] [CrossRef]
- Roxin, P.; Karlsson, A.; Singh, S.K. Characterization of cellulose acetate phthalate (CAP). Drug Dev. Ind. Pharm. 1998, 24, 1025–1041. [Google Scholar] [CrossRef]
- Gupta, S.S.; Meena, A.; Parikh, T.; Serajuddin, A.T. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion-I: Polyvinylpyrrolidone and related polymers. J. Excip. Food Chem. 2016, 5. [Google Scholar]
- Gong, C.; Xie, Y.; Wu, Q.; Wang, Y.; Deng, S.; Xiong, D.; Liu, L.; Xiang, M.; Qian, Z.; Wei, Y. Improving anti-tumor activity with polymeric micelles entrapping paclitaxel in pulmonary carcinoma. Nanoscale 2012, 4, 6004–6017. [Google Scholar] [CrossRef]
- Kapourani, A.; Chatzitheodoridou, M.; Kontogiannopoulos, K.N.; Barmpalexis, P. Experimental, thermodynamic, and molecular modeling evaluation of amorphous simvastatin-poly (vinylpyrrolidone) solid dispersions. Mol. Pharm. 2020, 17, 2703–2720. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram illustrating manufacturing approaches for preparation of amorphous solid dispersions (ASDs).
Figure 1.
Schematic diagram illustrating manufacturing approaches for preparation of amorphous solid dispersions (ASDs).
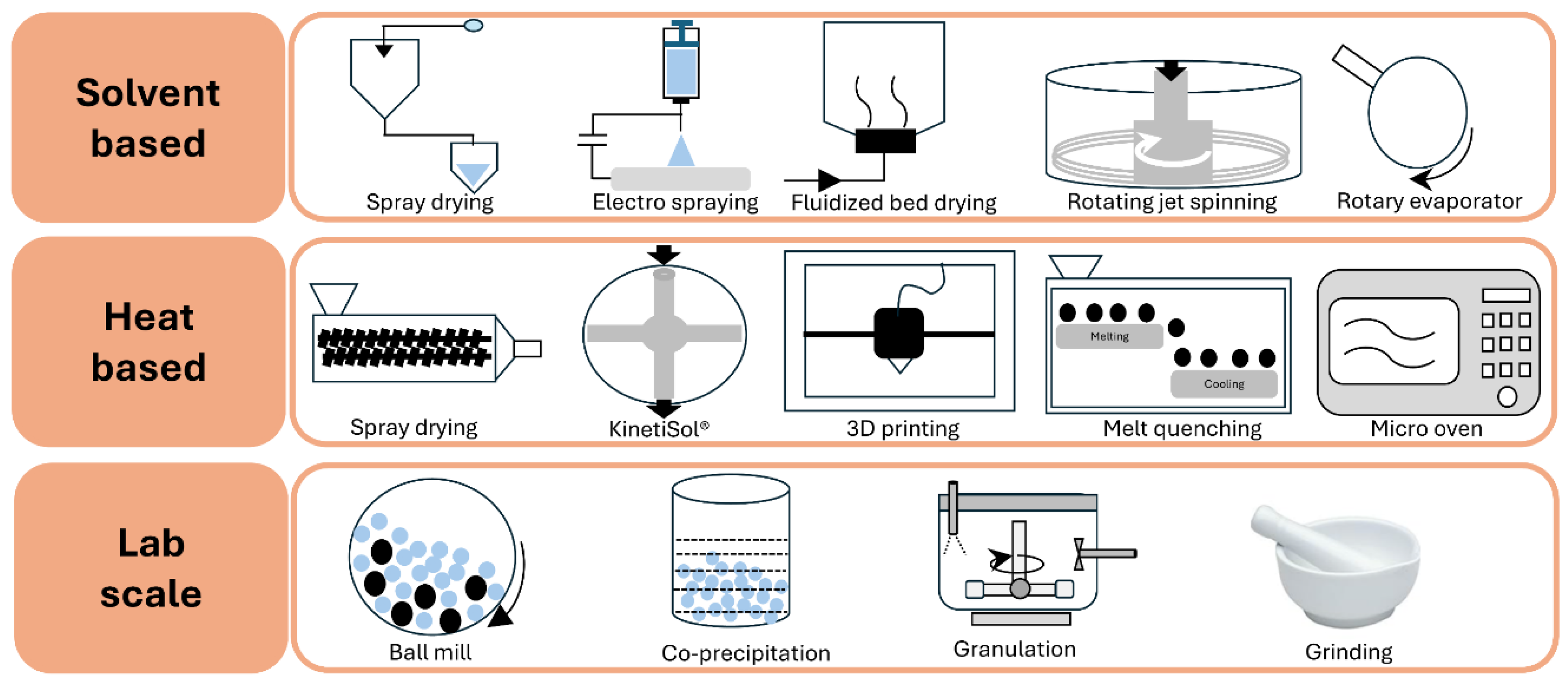
Figure 2.
Schematic diagram depicting the application of combined QbD and PAT tool approaches in the manufacturing of pharmaceutical dosage forms of ASD.
Figure 2.
Schematic diagram depicting the application of combined QbD and PAT tool approaches in the manufacturing of pharmaceutical dosage forms of ASD.
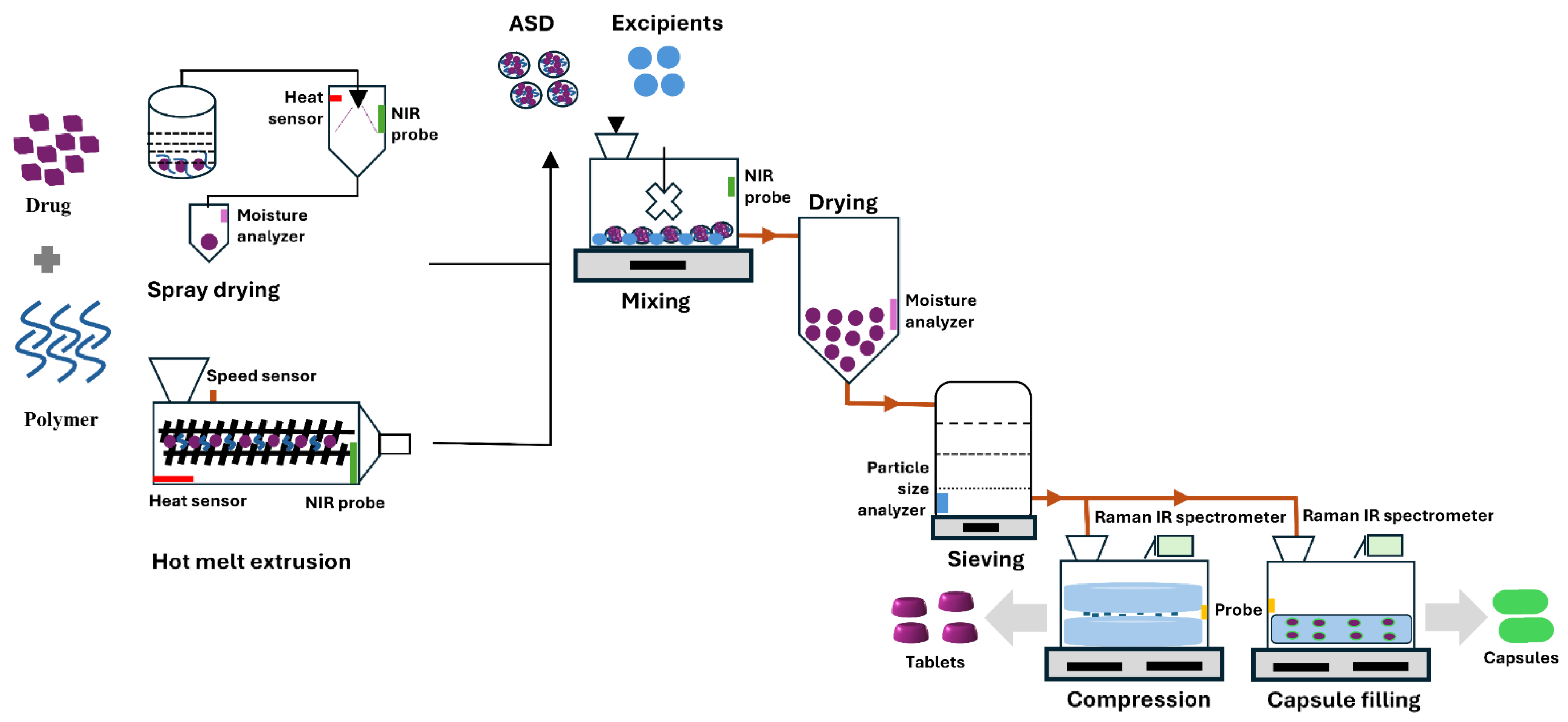
Figure 3.
Schematic diagram showing the effect of temperature on the physical form of materials. Tg and Tm denote glass transition and melting temperatures, respectively. Diagram not to scale.
Figure 3.
Schematic diagram showing the effect of temperature on the physical form of materials. Tg and Tm denote glass transition and melting temperatures, respectively. Diagram not to scale.
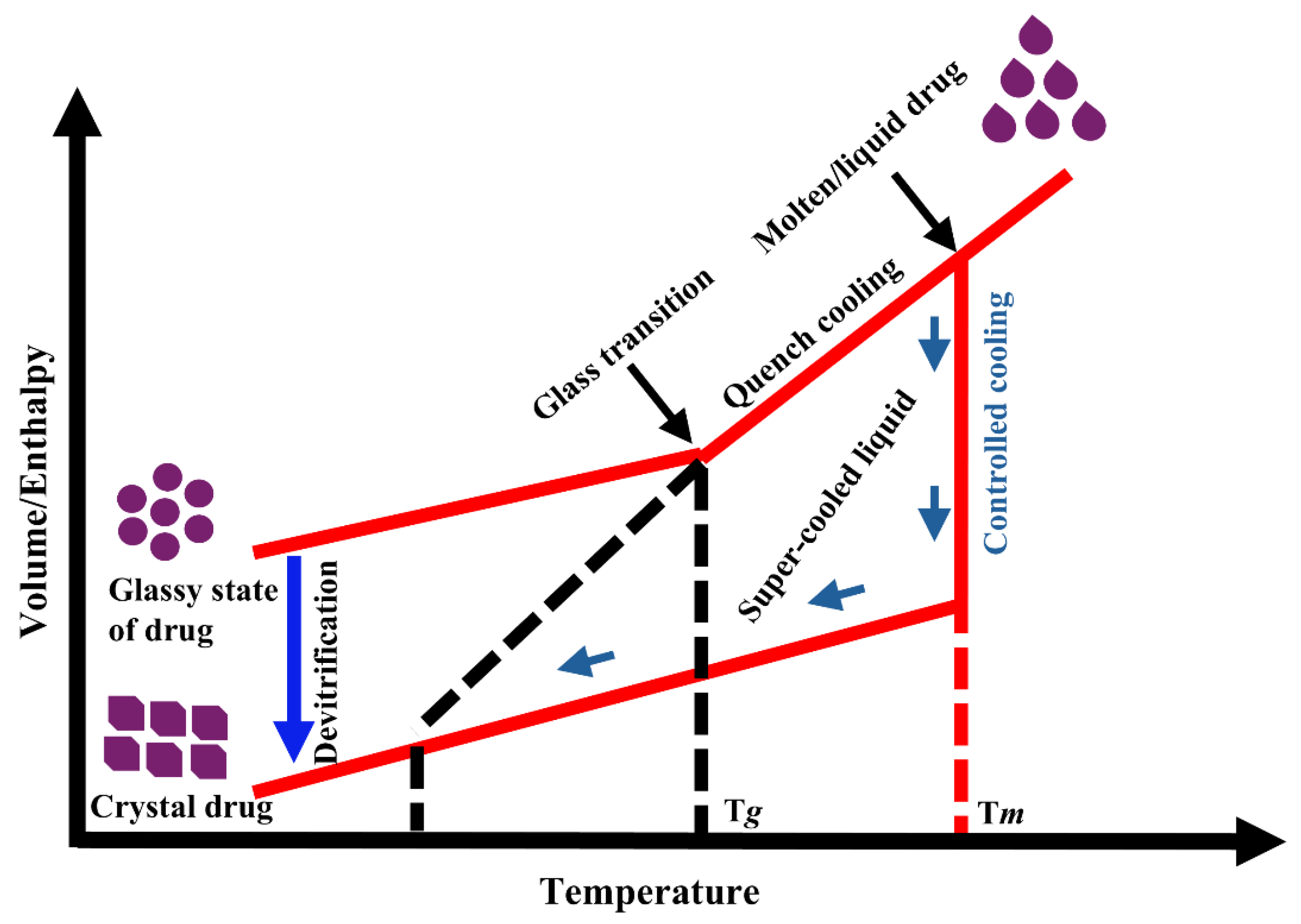
Figure 4.
Schematic representation of molecular mobility and drug recrystallization over time. Diagram not to scale.
Figure 4.
Schematic representation of molecular mobility and drug recrystallization over time. Diagram not to scale.

Figure 5.
Schematic representation of (a) formation of amorphous solid dispersion of a poorly soluble drug in a carrier polymer, (b) assessment of stability parameters of the amorphous solid by various approaches, and (c) prediction of physical behavior using molecular modeling and machine learning.
Figure 5.
Schematic representation of (a) formation of amorphous solid dispersion of a poorly soluble drug in a carrier polymer, (b) assessment of stability parameters of the amorphous solid by various approaches, and (c) prediction of physical behavior using molecular modeling and machine learning.
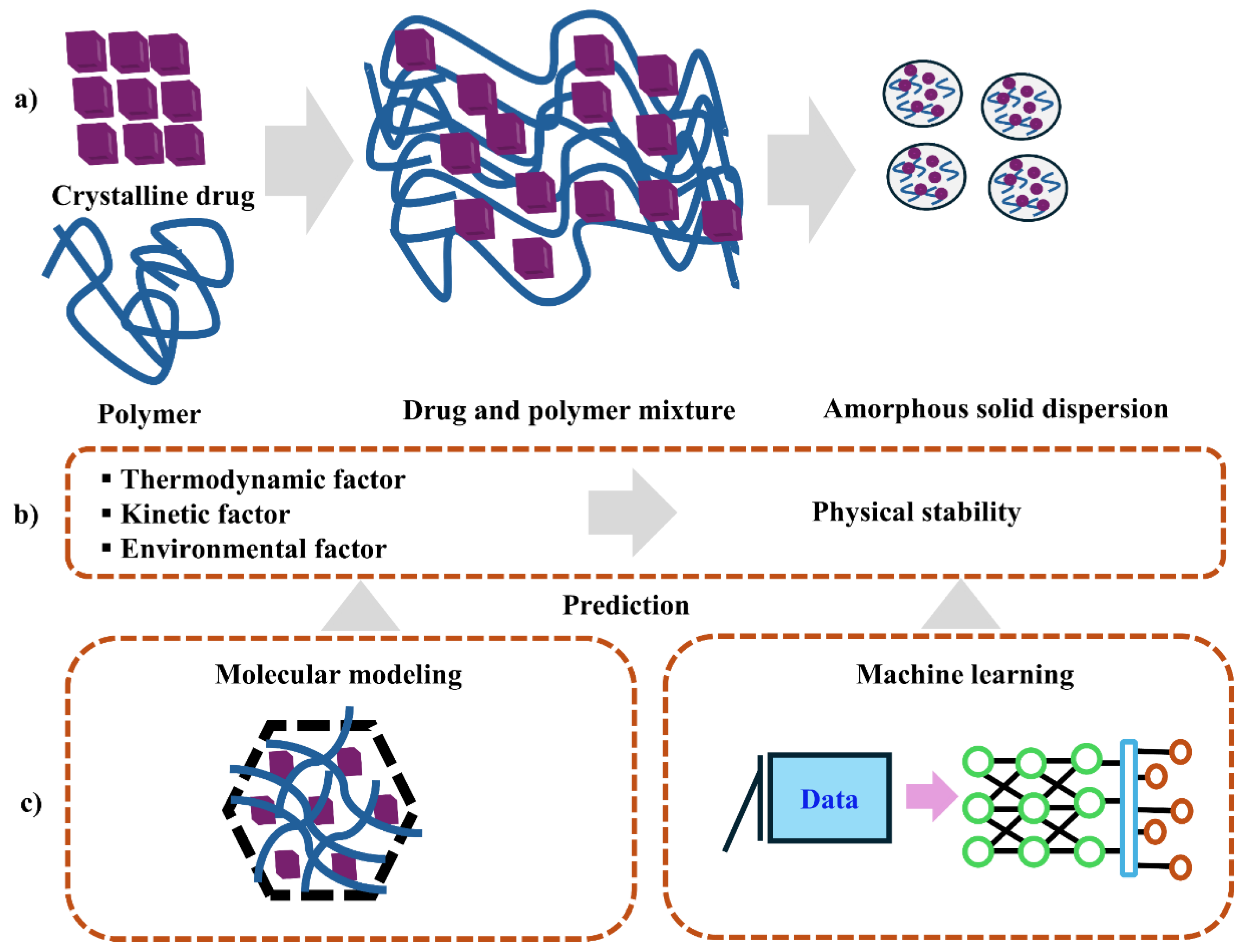

Table 1.
Trends in amorphous solid dispersion drug products approved by the U.S. Food and Drug Administration (FDA) through 2023.
Table 1.
Trends in amorphous solid dispersion drug products approved by the U.S. Food and Drug Administration (FDA) through 2023.
| Trade name | Drug(s) | Polymer(s) | Manufacturing method | Dosage form | Company | Year of approval |
|---|---|---|---|---|---|---|
| Cesamet® | Nabilone | PVP | Solvent evaporation | Tablet | Valeant | 1985 |
| Isoptin® | Verapamil | HPC/HPMC | Hot melt extrusion | Tablet | Abbott | 1987 |
| Rezulin® | Troglitazone | HPMC | Hot melt extrusion | Tablet | Pfizer | 1997 |
| Sporanox® | Itraconazole | HPMC | Fluidized bed layering | Capsule | Janssen | 1992 |
| Prograf® | Tacrolimus | HPMC | Solvent evaporation | Capsule | Astella | 1994 |
| NuvaRing® | Etonogestrel and ethyl estradiol | EVA | Hot melt extrusion | Ring | Merck | 2001 |
| Crestor® | Rosuvastatin | HPMC | Spray drying | Tablet | AstraZeneca | 2002 |
| Cymbalta® | Duloxetine | HPMCAS | Not disclosed | Capsule | Eli Lilly | 2004 |
| Kaletra® | Ritonavir/lopinavir | PVP–VA64 | Hot melt extrusion | Tablet | Abbott | 2007 |
| Intelence® | Etravirine | HPMC | Spray drying | Tablet | Janssen | 2008 |
| Samsca® | Tolvaptan | HPC | Spray drying | Tablet | Otsuka | 2009 |
| Zortress® | Everolimus | HPMC | Spray drying | Tablet | Novartis | 2010 |
| Norvir® | Ritonavir | PVP–VA64 | Hot melt extrusion | Tablet | Abbott | 2010 |
| Onmel® | Itraconazole | HPMC | Hot melt extrusion | Tablet | Merz | 2010 |
| Zelboraf® | Vemurafenib | HPMCAS | Solvent/antisolvent precipitation | Tablet | Roche | 2011 |
| Incivek® | Telaprevir | HPMCAS | Spray drying | Tablet | Vertex | 2011 |
| Kalydeco® | Ivacaftor | HPMCAS | Spray drying | Tablet | Vertex | 2012 |
| Noxafil® | Posaconazole | HPMCAS | Hot melt extrusion | Tablet | Merck | 2013 |
| Astagraf XL® | Tacrolimus | HPMC; EC | Wet granulation | Capsule | Astella | 2013 |
| Belsomra® | Suvorexant | PVP–VA64 | Hot melt extrusion | Tablet | Merck | 2014 |
| Harvoni® | Ledipasvir/sofosbuvir | PVP–VA64 | Spray drying | Tablet | Gilead | 2014 |
| Viekira XR™ | Dasabuvir/ombitasvir/paritaprevir/ritonavir | PVP–VA64; HPMC | Hot melt extrusion | Tablet | AbbVie | 2014 |
| Epclusa® | Sofosbuvir/velpatasvir | PVP–VA64 | Spray drying | Tablet | Gilead | 2016 |
| Orkambi® | Lumacaftor/ivacaftor | HPMCAS | Spray drying | Tablet and granule | Vertex | 2016 |
| Venclexta® | Venetoclax | PVP–VA64 | Hot melt extrusion | Tablet | AbbVie | 2016 |
| Zepatier™ | Elbasvir/grazoprevir | PVP–VA64 | Spray drying | Tablet | Merck | 2016 |
| Mavyret™ | Glecaprevir/pibrentasvir | PVP–VA64 | Hot melt extrusion | Tablet | AbbVie | 2017 |
| Vosevi™ | Sofosbuvir/velpatasvir/voxilaprevir | PVP–VA64 | Spray drying | Tablet | Gilead | 2017 |
| Idhifa® | Enasidenib | HPMCAS | Hot melt extrusion | Tablet | Bristol | 2017 |
| Lynparza® | Olaparib | PVP–VA | Hot melt extrusion | Tablet and capsule | AstraZeneca | 2017 |
| Jynarque® | Tolvaptan | HPC | Spray drying | Tablet | Otsuka | 2018 |
| Tibsovo® | Ivosidenib | HPMCAS | Spray drying | Tablet | Servier | 2018 |
| Pifeltro® | Doravirine | HPMCAS | Spray drying | Tablet | Merck | 2018 |
| Delstrigo® | Doravirine/lamivudine/tenofovir disoproxil fumarate | HPMCAS | Spray drying | Tablet | Merck | 2018 |
| Tolsura® | Itraconazole | HPMCP | Spray drying | Capsule | Mayne | 2018 |
| Erleada® | Apalutamide | HPMCAS | Spray drying | Tablet | Janssen | 2018 |
| Orilissa® | Elagolix | HPMCAS | Wet granulation | Tablet | AbbVie | 2018 |
| Symdeko® | Tezacaftor/ivacaftor and ivacaftor | HPMCAS | Spray drying | Tablet | Vertex | 2018 |
| Braftovi® | Encorafenib | PVP–VA64 | Hot melt extrusion | Capsule | Array | 2018 |
| Trikafta® | Elexacaftor/ivacaftor/tezacaftor | HPMCAS | Spray drying | Tablet | Vertex | 2019 |
| Ubrelvy® | Ubrogepant | PVP–VA64 | Hot melt extrusion | Tablet | AbbVie | 2019 |
| Oriahnn® | Elagolix/estradiol/norethindrone acetate | PVP–VA | Hot melt extrusion | Tablet | AbbVie | 2020 |
| Tukysa® | Tucatinib | PVP–VA | Hot melt extrusion | Tablet | Seagen | 2020 |
| Xtandi® | Enzalutamide | HPMCAS | Hot melt extrusion | Tablet | Astella | 2020 |
| Qinlock® | Ripretinib | PVP–VA | Spray drying | Tablet | Deciphera | 2020 |
| Qulipta® | Atogepant | PVP–VA64 | Hot melt extrusion | Tablet | AbbVie | 2021 |
| Welireg® | Belzutifan | HPMCAS | Hot melt extrusion | Tablet | Merck | 2021 |
| Sotyktu® | Deucravacitinib | HPMCAS | Spray drying | Tablet | Bristol | 2022 |
| Sunlenca® | Lenacapavir | PVP–VA | Spray drying | Tablet | Gilead | 2022 |
| Jaypirca® | Pirtobrutinib | HPMCAS | Spray drying | Tablet | Loxo Oncology | 2023 |
| Phyrago® | Dasatinib | Methacrylic acid–ethyl acrylate copolymer | Electro spraying | Tablet | Nanocopoeia | 2023 |
| Paxlovid® | Nirmatrelvir/ritonavir | PVP–VA | Hot melt extrusion | Tablet | Pfizer | 2023 |
| Alvaiz® | Eltrombopag | PVP–VA | Hot melt extrusion | Tablet | Teva | 2023 |
Table 2.
Commonly used polymers and their physicochemical properties: category, molecular weight, Tg, degradation temperature, hygroscopicity, solubility, and key features.
Table 2.
Commonly used polymers and their physicochemical properties: category, molecular weight, Tg, degradation temperature, hygroscopicity, solubility, and key features.
| Category | Polymer type | Polymer subtype | Mol. wt. (g/mol) |
Tg/Tm (°C) |
Degradation temp. (°C) | Moisture retention | Solubility | Key features | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Cellulose derivative | HPMCAS | HPMCAS LG | 144,700 | 119 | 204 | Low | pH 5.5–6.0 | Anionic | [31,32,190] |
| HPMCAS MG | 103,200 | 120 | 190 | Low | pH 6.0–6.5 | Anionic | [31,32,190] | ||
| HPMCAS HG | 75,100 | 122 | 200 | Low | Above pH 6.8 | Anionic | [31,32,190] | ||
| HPMCP | HPMCP 50 | 37,900 | 137 | 160–190 | Low | Below pH 5 | Amphiphilic | [30] | |
| HPMCP 55 | 45,600 | 133 | 150 | Low | below pH 5.5 | Amphiphilic | [30] | ||
| HPMC | HPMC E | 40,000–150,000 | 141 | NA | High | Water | Nonionic | [33] | |
| HPMC F | 40,000–150,000 | 160 | 240 | High | Water | Nonionic | [33] | ||
| HPMC K | 40,000–150,000 | 172 | 260 | High | Water | Nonionic | [33] | ||
| CAP | 2534.12 | 175 | 200 | Low | Below pH 6 | Nonionic | [191] | ||
| Polyvinyl derivatives | PVP | PVP K12 | 2000–3000 | 72 | 196 | High | Water | Amphiphilic | [34,192] |
| PVP K17 | 7000–11,000 | 140 | 217 | High | Water | Amphiphilic | [34,192] | ||
| PVP K25 | 28,000–34,000 | 153 | 166 | High | Water | Amphiphilic | [34,192] | ||
| PVP K30 | 44,000–54,000 | 160 | 171 | High | Water | Amphiphilic | [34,192] | ||
| PVP K90 | 1,000,000–1,500,000 | 177 | 194 | High | Water | Amphiphilic | [34,192] | ||
| PVP/VA | 45,000–70,000 | 115 | 270 | High | Water | Amphiphilic | [34,192] | ||
| Soluplus® | 90,000–140,000 | 72 | 278 | Moderate | Water | Amphiphilic | [34,192] | ||
| Polymethacrylate derivatives | Eudragit® EPO | 47,000 | 48 | 250 | Low | Below pH 5 | Cationic | [35,36] | |
| Eudragit® L100 | 125,000 | 150 | 176 | Low | Above pH 6 | Anionic | [35,36] | ||
| Eudragit® S100 | 125,000 | >150 | 173 | Low | Above pH 7 | Anionic | [35,36] | ||
| Eudragit® L100–55 | 250,000 | 110 | 176 | Low | Above pH 5.5 | Anionic | [35,36] | ||
| Miscellaneous | PVAP | 47,000–61,000 | 46/116 | 150 | Low | Below pH 6 | Nonionic | [37] | |
| PAA | 1800–450,000 | 126 | 200 | Low | Water | Nonionic | [38] | ||
| PEG/POE | 1000–7,000,000 | 55–66 | >200 | Low | Water | Nonionic | [39] | ||
| Lutrol® | 7600–17,400 | 52–57 | >200 | Low | Water | Nonionic | [40] | ||
Table 3.
Solvents used in preparation of ASD and their boiling point, solubility, density, viscosity, dielectric constant, and toxicity level according to ICH guidelines [43].
Table 3.
Solvents used in preparation of ASD and their boiling point, solubility, density, viscosity, dielectric constant, and toxicity level according to ICH guidelines [43].
| Solvent | Boiling point | Solubility in water (g/mL) | Density at 25 °C (g/mL) | Viscosity (at 25 °C, cP) | Dielectric constant | ICH class (limit, ppm) |
|---|---|---|---|---|---|---|
| Acetone | 56.2 | Miscible | 1.049 | 0.295 | 20.7 | Class 3 |
| Butanone | 79.6 | 29 | 0.805 | 0.4 | 18.51 | Class 3 |
| Butyl acetate | 126.1 | 0.68 | 0.882 | 0.685 | 5.07 | Class 3 |
| Chloroform | 61.7 | 0.795 | 1.498 | 0.536 | 4.81 | Class 2 (60) |
| Dichloromethane | 39.6 | 1.32 | 1.326 | 0.413 | 9.08 | Class 2 (600) |
| Dimethyl acetamide | 165 | Miscible | 0.937 | 0.92 | 37.78 | Class 2 (1090) |
| Dimethyl formamide | 153 | Miscible | 0.944 | 0.97 | 36.7 | Class 2 (880) |
| Dimethyl sulfoxide | 189 | 25.3 | 1.092 | 1.987 | 47 | Class 3 |
| Ethanol | 78.5 | Miscible | 0.789 | 1.04 | 24.6 | Class 3 |
| Ethyl acetate | 77 | 8.7 | 0.895 | 0.428 | 6 | Class 3 |
| Glycerin | 290 | Miscible | 1.261 | 954 | 42.5 | - |
| Isopropanol | 82.6 | Miscible | 0.786 | 1.96 | 18.2 | Class 3 |
| Methanol | 64.6 | Miscible | 0.791 | 0.543 | 32.6 | Class 2 (3000) |
| Tetrahydrofuran | 66 | Miscible | 0.889 | 0.48 | 7.52 | Class 2 (720) |
| Water | 100 | - | 0.998 | 1 | 78.5 | - |
| -, not applicable | ||||||
Table 4.
Application of QM, MM, and docking studies in amorphous solid dispersion systems to simulate drug–polymer interactions.
Table 4.
Application of QM, MM, and docking studies in amorphous solid dispersion systems to simulate drug–polymer interactions.
| Drug candidates | Polymer carrier | Simulation | Software | Force field | Summary | Reference |
|---|---|---|---|---|---|---|
| Indomethacin | Eudragit® PEO, glucose, sucrose | Molecular dynamics | Material Studio 4.0 | COMPASS | Eudragit® PEO was miscible, glucose was immiscible, and sucrose had borderline miscibility with indomethacin, as shown by thermal analysis. | [136] |
| Paclitaxel | PEG, PCL, MPEG–PCL | Molecular dynamics | HyperChem | CHARMM 27 | Paclitaxel binds to the PCL segments of MPEG–PCL copolymer, forming a core–shell micelle structure with PEG surrounding the core. | [193] |
| Curcumin | MPEG–PCL | Molecular dynamics | HyperChem | CHARMM 27 | An increased number of hydrophobic binding sites for curcumin indicate enhanced stability and stronger binding between the copolymer and drug. | [139] |
| Artemisinin | PEG, PVP | Molecular dynamics | Material Studio 6.0 | COMPASS | Polymers were miscible with artemisinin, forming stable solid dispersions and suggesting molecular dispersion of the drug within the polymer matrix. | [137] |
| Lumefantrine | Soluplus®, Kollidon® VA64, Plasdone™ S630 | Molecular dynamics | Maestro Schrodinger | GAUSSIAN | Strong interactions occurred between the hydroxyl and carbonyl groups of the polymers and the chlorine and amine groups of lumefantrine, respectively. | [143] |
| Cyclosporin A | L/D–polylactide, chitosan, polyglycolic acid, PEG, cellulose | Molecular docking | Materials Studio | PCFF | Polycellulose and polychitosan exhibited high miscibility, attributed to their larger open surface area for drug interaction. | [146] |
| Indomethacin | PVP | Molecular dynamics | AMBER | AMBER | The solubility of indomethacin increased when dispersed with PVP, compared to pure indomethacin. | [149] |
| Lafutidine | Soluplus®, PEG 400, Lutrol® F127, Lutrol® F68 | Molecular dynamics | Maestro Schrodinger | GAUSSIAN | Strong interactions occurred between the hydroxyl and carbonyl groups of the polymers and the chlorine and amine groups of lafutidine, respectively. | [140] |
| Posaconazole | Soluplus®, PEG 400, Lutrol® F127, Lutrol® F68, TPGS | Molecular dynamics | Maestro Schrodinger | GAUSSIAN | Strong hydrogen bonding between the drug and polymer resulted in the lowest energy and highest binding interaction. | [141] |
| Propranolol HCl, diphenhydramine HCl, paracetamol, ibuprofen, diclofenac sodium, hydrocortisone | Eudragit® L100, Eudragit® EPO, Eudragit® L100–55, Kollidon® VA64 | Quantum mechanical | Gaussian 09 | GAUSS VIEW | The strength of interactions depended on both the donor and acceptor types, as well as the number of hydrogen bonds formed between drug and polymer, as observed by DSC. | [114] |
| Cetirizine HCl, verapamil HCl | Eudragit® L100, Eudragit® L100–55 | Molecular dynamics | Maestro Schrodinger | GAUSSIAN 09 | The strongest interactions were between the amine groups of cetirizine HCl and verapamil HCl and the carboxylate groups of the polymers, indicating higher binding energy and increased stability. | [115] |
| Gemcitabine | Chitosan | Molecular dynamics | Material Studio 4.3 | COMPASS | Maximum drug loading was suggested to result from the strongest interaction between chitosan and gemcitabine. | [138] |
| Carbamazepine | Lutrol® F68 | Molecular dynamics | XenoView | PCFF | Carbamazepine molecules showed a strong tendency to aggregate, which is a critical step in nucleation and crystal formation. | [151] |
| Telaprevir | Cellulose derivatives | Quantum mechanical | HyperChem 8.0.3 | CHARMM | Effective polymers contain carboxylate groups with optimal hydrocarbon chain length, resulting in more favorable solvation free energy. | [152] |
| Tacrine | Chitosan, PBCA | Molecular dynamics | LAMMPS | PCFF | Interaction between tacrine and polymeric nanoparticles increased with the length of the polymer chain. | [157] |
| Indomethacin | PEG, PLA | Molecular dynamics | Material Studio 8.0 | COMPASS | Indomethacin exhibited significant miscibility with both PEG and PLA as carriers, resulting in high encapsulation efficiency. | [153] |
| Clonazepam, ibuprofen, fenofibrate, alprazolam | PVP–VA64, HPMC, Eudragit® EPO | Molecular dynamics | Materials Studio 7.0 | COMPASS | Ibuprofen/PVP–VA64 and ibuprofen/Eudragit® EPO formed strong hydrogen bonds, resulting in stable solid dispersions. | [145] |
| Felodipine | HPMC | Molecular dynamics | AMBER | GLYCAM | Miscibility of HPMC at various concentrations was supported by observation of single Tg values from DSC. | [150] |
| Aspirin, caffeine, carbamazepine, finasteride, flufenamic acid, flutamide, mefenamic acid, salicylamide, theophylline | PVP–VA64, poly (glycerol adipate) and derivatives | Molecular dynamics | GROMACS | CHARMM | Solubility and interaction parameters did not correlate with miscibility; six of nine API–PGA polymers were miscible. | [154] |
| Ibuprofen, carbamazepine | Soluplus®, PEG | Molecular docking | AutoDock Vina | CHARMM | Miscible blends were formed for ibuprofen–Soluplus®/PEG and carbamazepine–Soluplus®/PEG, with the latter showing stronger interactions. | [147] |
| 6–Mercaptopurine | PLA, PEG–modified PLA | Molecular docking | XenoView v.3.7.9.0 | PCFFD | The degree of polymerization was found to be optimal for solubility of 6–mercaptopurine in PLA and PEG polymers. | [155] |
| Olmesartan medoxomil | PVP–VA64, Soluplus® | Molecular dynamics | Maestro Schrodinger | OPLS | Strong hydrogen bonding between the carbonyl residues of pyrrolidone and acetate monomers in PVP–VA64 and the tetrazole and aromatic rings of olmesartan medoxomil inhibited recrystallization. | [156] |
| Simvastatin | PVP | Molecular dynamics | XenoView v.3.7.9.0 | PCFF | Simvastatin contains hydrogen bond donor and acceptor groups, while the PVP chain contains hydrogen bond acceptors, resulting in intermolecular interactions that stabilized the amorphous solid dispersion. | [194] |
| Rivaroxaban | Soluplus® | Molecular dynamics | XenoView | PCFF | Strong specific homo molecular interactions and Soluplus® chain shrinkage led to recrystallization under high relative humidity. | [158] |
| Naproxen, indomethacin | PVP, PVA | Quantum mechanical | COSMO–SAC | GAUSSIAN | Drug solubility in the polymer and thermodynamic compatibility of drug and polymer was investigated. | [118] |
| Ritonavir | Lutrol® | Molecular dynamics | GROMACS | AMBER 99SB-ILDN, AMBER (GAFF) | Strong intermolecular interactions suppressed molecular mobility, supported the amorphous state, and prevented recrystallization. | [142] |
| Erlotinib HCl | PEG, PVP | Molecular dynamics | Material Studio 7 | COMPASS | Erlotinib HCl formed weak hydrogen bonds with PEG and PVP individually, while the composite polymer enhanced molecular interactions through hydrogen bonding. | [159] |
Table 5.
Recent machine learning prediction for amorphous solid dispersion properties, stability, and formulation.
Table 5.
Recent machine learning prediction for amorphous solid dispersion properties, stability, and formulation.
| Year | Target feature | Input feature | Algorithm | Dataset | Reference |
|---|---|---|---|---|---|
| 2011 | Dispersion potential of drug–polymer (miscible dispersion) | Molecular descriptors and 3D structure derived from molecular structure, topology, and atomic properties | LR | Twelve compounds solidified with PVP–VA64 | [166] |
| 2011 | Percentage drug release at 60 min, time to 90 % drug dissolution, floating properties, physical stability | Proportions of drug, polymer, and effervescent agents | ANN/GP | Twenty-five mixture proportions | [167] |
| 2013 | The percentage of Tibolone dissolved in 30 min (Y30min) | Molecular weight of PEG, mixing temperature, drug amount, and total mixing time | ANN | Thirty-six experiments with four independent factors | [168] |
| 2015 | Enhanced dissolution rate | Optimization of ternary solid dispersions of carbamazepine, Soluplus®, and Lutrol® F68 | ANN | Twenty-two using D-optimal mixture experimental design and three for predictive modeling | [151] |
| 2019 | Physical stability of solid dispersions at 3 months and 6 months | Drug loading ratio, polymer molecular weight, drug properties, environmental conditions, preparation method, and temperature | ANN, SVM, RF, DT, Light GBM, kNN, NB, DNN | Fifty drug compounds with ten molecular descriptors | [169] |
| 2020 | Quantification and differentiation of amorphous solid dispersion systems | Crystalline and amorphous drug content of rivaroxaban with Soluplus® | ANN, PLS, PCR | Thirty sample formulations | [158] |
| 2022 | Dissolution percentage after 10 min (Q10) and 30 min (Q30) | Percentages of carbamazepine, Neusilin, and Kollidon® VA64. | GRNN, MLP | Twenty drug compounds | [170] |
| 2023 | Amorphization and chemical stability of ASD via HME | Proportions of drug and polymer, extruder configuration, barrel temperature, screw speed, and feed rate | XGBoost, Light GBM, RF, SVM, SHAP, IG | Forty-nine drug molecules | [171] |
| 2024 | Glass transition temperature determination (Tg) | Hydrophilic backbone methylation, hydrophilic feed fraction, hydrophobic backbone methylation | RF | Fifty unique copolymers with probucol | [172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



