Submitted:
04 August 2025
Posted:
05 August 2025
You are already at the latest version
Abstract
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease, characterized by progressive degeneration of upper and lower motor neurons. Clinically heterogeneous, ALS presents with varying patterns of muscle weakness, spasticity, and atrophy, ultimately impairing mobility, communication, swallowing, and respiration. The pathogenesis of ALS involves a complex interplay of genetic and environmental factors. Despite extensive research, ALS remains incurable, with only modestly effective disease-modifying therapies currently available. This review summarizes current understanding of ALS pathophysiology, clinical phenotypes, diagnosis, and ongoing therapeutic developments.
Keywords:
amyotrophic lateral sclerosis
; genes
; diagnosis
; preclinical models
; clinical trials
; treatments
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disorder, and it is the most common form of motor neuron disease (MND) [1]. The global prevalence of ALS is estimated at 4 to 6 cases per 100,000 people, classifying it as a rare disease. Regional differences in prevalence have been observed, with higher rates reported in Europe and North America compared to Asia and Africa [2]. This disparity may reflect underdiagnosis or limited access to neurological care, but genetic and environmental modifiers likely also contribute [3].
ALS is characterized by progressive muscle weakness, spasticity, and atrophy, ultimately impairing mobility, communication, swallowing, and breathing [4]. “Amyotrophy”, which is muscular weakness, is caused by lower motor neuron (LMN) degeneration, whereas “lateral sclerosis” refers to corticospinal tract degeneration in the lateral columns of the spinal cord due to upper motor neuron (UMN) degeneration [5]. Clinical findings associated with LMN degeneration include muscle weakness and fasciculations while, clinical findings associated with UMN degeneration include hyperreflexia, incoordination, and spasticity. Importantly, sensory nerves are typically spared in ALS [4].
ALS differs in age of onset and initially affected areas. Early-onset and late-onset ALS refer to the age at which symptoms of ALS first appear. Bulbar-onset and limb-onset ALS, describe different clinical presentations of the same disease, distinguished by the initial region of symptom manifestation and progression patterns [4,6]. The average survival time following symptom onset ranges from 2 to 5 years. Early-onset ALS tends to show a male predominance (male:female, 1.5:1) [7], whereas bulbar-onset displays a female predominance [8]. The sex difference in ALS incidence diminishes with increasing age, and in late-onset ALS, men and women are affected almost equally [9].
2. Genetics
2.1. ALS Genes
ALS is characterized by considerable genetic heterogeneity and complex genetic architecture featuring monogenic, oligogenic, polygenic inheritance and varying penetrance [10]. The disease also demonstrates significant phenotypic variability and clinical overlap with other neurodegenerative disorders, such as frontotemporal dementia (FTD), adding diagnostic complexity. Approximately 10% of ALS cases are familial (fALS) with at least another affected family member, whereas the remaining 90% are considered sporadic (sALS) without any known family history [11]. Although there is a clear environmental component influencing disease development, twin studies support an estimated heritability of ~60%, indicating a strong genetic component.
The first major breakthrough in ALS genetics came in 1991 when a study reported linkage of fALS to chromosome 21q [12], followed by the identification of mutations in the SOD1 gene in 1993 [13]. SOD1 gene encodes superoxide dismutase 1, a cytoplasmic antioxidant enzyme protecting cells from oxidative stress by metabolizing superoxide radicals. Since then, more than 200 mutations in SOD1 have been described, the majority of which are missense mutations inherited in an autosomal dominant manner. SOD1 mutations account for approximately 12% of fALS and 1-2% of sALS [14].
In 2008, mutations in the TARDBP gene, encoding transactive response DNA-binding protein 43 (TDP-43), were identified as a cause of autosomal dominant ALS [15]. Although TARDBP mutations are relatively rare, accounting for approximately 4% of fALS and 1% of sALS, TDP-43 proteinopathy, marked by cytoplasmic aggregates and nuclear loss, is observed in ~97% of ALS cases, making it a major pathological hallmark of the disease.
The following year, in 2009, FUS (Fused in Sarcoma) was discovered as another ALS-related gene, encoding a multifunctional RNA-binding protein [16]. Mutations in FUS are especially clustered in the C-terminal domain responsible for nuclear localization. Like TDP-43, FUS mutations result in toxic cytoplasmic inclusions and are more common in juvenile and early-onset ALS, especially in certain populations.
A major advance occurred in 2011 with the discovery of the GGGGCC (G4C2) hexanucleotide repeat expansion in the first intron of C9orf72 gene, which became recognized as the most common genetic cause of both ALS and FTD [17,18]. This finding resolved a long-standing mystery surrounding the chromosome 9p locus, which had shown strong linkage in fALS/FTD [19,20,21] and had also been replicated by genome-wide association studies (GWAS) in sALS/FTD [22,23,24,25], but lacked a defined causal gene due to technical limitations in detecting repeat expansions. The breakthrough was enabled by next-generation sequencing combined with repeat-primed long-read PCR and changed the landscape of ALD/FTD genetics by providing an explanation for a big proportion of the missing heritability. The example of C9orf72 discovery highlights how technical limitations and structural variants may contribute to the missing heritability in ALS. C9orf72 expansions now account for up to 40% of fALS and 7-10% of sALS, although frequencies vary geographically.
Overall, pathogenic variants in C9orf72, SOD1, TARDBP and FUS genes account for approximately 60% of fALS and 11% of sALS. The frequencies of several ALS mutations differ between populations of different geographical regions due to founder effects. For instance, the C9orf72 hexanucleotide repeat expansion is the most common genetic cause of fALS in Europe [26] whereas it is rare in Japan [27,28]. Similarly, mutations in SOD1 and FUS are more frequently seen in fALS cases in Japan [29] while mutations in TARDBP are more common in Europe [30].
Recent technological advances, particularly in GWAS, whole-exome sequencing, and whole-genome sequencing, have led to the discovery of additional ALS genes, bringing the total number of ALS-associated genes to over 40, and expanding our understanding of pathogenic pathways. These discoveries have not only clarified disease mechanisms but also provided a genetic explanation for approximately 75% of fALS and 25% of sALS cases [31]. Nevertheless, a significant proportion of cases remain genetically unexplained, suggesting a role for structural variants, non-coding regulatory mutations, epigenetic factors, and gene-environment interactions.
2.2. Genetic Diagnosis and Counseling Challenges
Diagnosing ALS and providing effective genetic counseling is particularly challenging due to the disease’s genetic complexity and phenotypic overlap with other neurodegenerative conditions. Moreover, distinguishing between familial and sporadic forms of ALS is increasingly challenging, as pathogenic variants often occur in individuals with no known family history due to factors such as reduced penetrance, variable expressivity, de novo mutations, or unrecognized phenotypic variability in relatives (e.g., FTD). This complicates the interpretation of genetic test results and limits the ability to provide clear prognostic information. Furthermore, the emergence of variants of uncertain significance adds to diagnostic ambiguity, requiring careful assessment and reclassification over time. The growing amount of genetic information must be carefully integrated into clinical practice, balancing the benefit of early ALS diagnosis with the management of the complex ethical and psychological implications due to the devastating nature of the disease and the lack of therapy.
3. Pathophysiology
Histopathologically, ALS is characterized by the degeneration of pyramidal Betz cells in the motor cortex, as well as motor neurons in the spinal cord and brainstem, occurring in the context of widespread neuroinflammation [32,33]. The hallmark feature is the presence of cytoplasmic aggregation of phosphorylated (p), ubiquitinated, and cleaved at the C-terminus TDP-43, which is present across most ALS subtypes, except in patients with SOD1 or FUS mutations [34,35]. This hallmark has gained particular attention due to its prion-like propagation along defined neuroanatomical circuits [36,37]. Myelinated fibers and long-projecting motor neurons that innervate spinal regions appear especially vulnerable, perhaps reflecting their high metabolic demands [38,39]. However, pTDP-43 inclusions are not confined to motor regions but also appear in areas such as the basal ganglia, thalamus, and cerebral cortex, suggesting widespread involvement beyond classical motor systems. pTDP-43 inclusions are not restricted to motor neurons; non-motor neurons such as the pyramidal layer II or III cells of large areas of the neocortex [40], and glial cells have also been found to contain cytoplasmic TDP-43-positive inclusions [41]. Morphologically, pTDP-43 inclusions can be ‘skein-like’, filamentous, or rounded [42], and are accompanied by nuclear clearance of TDP-43, disrupting its essential functions in RNA metabolism and splicing [43]. The origins of this TDP-43 proteinopathy remain unclear, but multiple hypotheses, ranging from disrupted RNA homeostasis to impaired autophagy, have been proposed [44].
In fALS, pathological presentation often varies significantly depending on the underlying genetic mutation, leading to distinct patterns of neurodegeneration. In FUS-related ALS, mutations disrupt the nuclear localization of FUS, resulting in loss of its regulatory functions in transcription and RNA maturation, and the formation of toxic FUS aggregates in the cytoplasm [45,46]. In SOD1-related ALS, mutations cause motor neuron damage through conformational changes which may lead to excitotoxicity, ER stress, oxidative stress, mitochondrial dysfunction, and prion-like spread [47]. In C9orf72-related ALS, the most common genetic form of the disease, pTDP-43 aggregates are commonly observed [48]. In addition, the expanded G4C2 repeat in C9orf72 generates RNA foci that sequester RNA-binding proteins. These foci are predominantly found in neuron cytoplasm but are also present in glial cells [34,37,49]. G4C2 repeat undergoes repeat-associated non-ATG (RAN) translation, resulting in the production of DPRs [50]. DPRs are neurotoxic and accumulate in regions like the cerebellum and hippocampus, often co-localizing with p62, a marker of impaired autophagic degradation [51].
Toxic accumulation of aggregates triggers neuroinflammatory responses which contribute to ALS progression. Microglial activation was linked to protein aggregates [52], leading to synapse loss [53], potentially mediated by complement activation products [54]. Astrocytes, integral to tripartite synapses, lose their supportive functions, acquire detrimental roles, and exhibit impaired interactions with motor neurons [55]. Notably, recent evidence suggests that neuroinflammation in ALS might be neuron-initiated. Activation of the stimulator of interferon genes (STING) pathway, central to innate immunity, has been observed in large layer V Betz cells, among the earliest to degenerate in ALS [56].
4. Preclinical Studies on ALS
4.1. In Vivo Studies
Both naturally occurring and experimentally induced models have advanced our understanding of ALS. Among the latter, transgenic rodents and invertebrates including zebrafish, Drosophila, and C. elegans, expressing mutant ALS genes, have been utilized for recapitulating disease mechanisms and identifying therapeutic targets (Table 1). The SOD1^G93A transgenic mouse was the first widely studied ALS model. This model overexpresses human SOD1 with a glycine-to-alanine mutation at codon 93 [57]. It replicates core ALS features, including motor neuron loss, mitochondrial dysfunction, oxidative stress, and glutamate excitotoxicity, leading to paralysis and death. SOD1^G93A supported the development of FDA-approved drugs like riluzole and edaravone [58]. Other SOD1 mutations, such as G37R and G85R, offer alternative disease trajectories and mechanistic insights [59]. Furthermore, rodent models of TDP-43, the main component of ALS-linked protein inclusions, show motor neuron loss and RNA processing defects, with cytoplasmic mislocalization and have been utilized to explore the contribution of gain versus loss of TDP-43 function [60]. FUS mutation rodent models display cytoplasmic mislocalization, motor neuron degeneration, and variable disease severity depending on mutation type, promoter, and dosage [59,61]. C9orf72 repeat expansion rodent models reveal hallmark features like RNA foci, DPR protein accumulation, and TDP-43 pathology, though outcomes vary based on construct and background [62].
4.2. In Vitro Studies
4.2.1. Human iPSC-Derived Organoids
Induced pluripotent stem cell (iPSC)-derived brain and spinal organoids have emerged as transformative tools for modeling early-stage ALS within a human neurodevelopmental context. These 3D culture systems preserve cytoarchitecture and cellular diversity, enabling the recapitulation of complex molecular and cellular phenotypes that are difficult to capture in traditional 2D cultures or animal models.
Notably, the C9orf72-ALS/FTD cerebral organoids reproduce the full triad of hallmark molecular pathologies, TDP-43 mislocalization, DPR accumulation, and p62-positive inclusions, during a presumptive presymptomatic stage [115]. These organoids also exhibited key neurodevelopmental abnormalities, including reduced numbers of deep-layer neurons, disorganized glial architecture, and impaired glutamatergic synaptic function, all of which closely mirror changes observed in post-mortem ALS brains. Cerebral organoids exposed to pathological extracts from ALS tissue developed robust TDP-43 pathology, astrocyte proliferation, and signs of DNA double-strand break-associated apoptosis [116]. These findings not only support a mechanistic link between proteinopathy and cell death but also highlight the utility of organoids in modeling both endogenous and exogenous pathological triggers. Moreover, C9orf72-ALS cerebral organoid slice cultures revealed DPR accumulation, particularly poly(GA), in deep-layer neurons, increased p62 expression in astrocytes, and signs of nuclear pyknosis [117]. Remarkably, these pathological changes were reversed by treatment with GSK2606414, a PERK inhibitor, underscoring the therapeutic potential of targeting integrated stress response pathways.
In parallel, organoid models have been extended to the neuromuscular axis. Sensorimotor organoids harboring ALS mutations in TARDBP, SOD1, and Profilin 1 (PFN1), identified early cellular impairments at the neuromuscular junction [118]. ALS neuromuscular organoids (NMOs) could faithfully model peripheral pathology, including reduced contractile force, neural denervation, Schwann cell loss, and glutamatergic dysfunction [119]. These NMOs also recapitulated hallmark features such as RNA foci and DPR protein aggregation in neurons and astrocytes. Acute treatment with GSK2606414 enhanced muscular contraction two-fold and reduced DPR aggregation and autophagic stress, reinforcing the value of NMOs for therapeutic screening.
Collectively, these studies validate iPSC-derived brain organoids as a versatile and physiologically relevant platform for modeling both cell-autonomous and non-cell-autonomous mechanisms of ALS.
4.2.2. Human iPSC-Derived Neurons and Glia
Recent in vitro studies in iPSC-derived neurons and glial subtypes have built upon earlier research and provided novel insights into ALS molecular pathogenesis. From a cellular pathology perspective, deficits in synaptic structure and axonal function are central features of ALS. iPSC-derived motor neurons provide high-resolution access to these vulnerable compartments. In a recent study, C9orf72-mutant motor neurons exhibited impaired autophagy, reduced lifespan, and cytoplasmic TDP-43 inclusions [120]. Moreover TBK1 autophosphorylation was detected within those aggregates, linking proteinopathy to defective stress responses. Interestingly, C9orf72 knockout did not directly affect TDP-43 aggregation or axonal transport, but led to a reduction in mature lysosome numbers, indicating selective impairment of autophagic flux.
Further insight into neuron-intrinsic stress responses was provided by a study demonstrating that both TDP-43 depletion and DPR expression induce DNA damage and activate the STING innate immune pathway in neurons [56]. These results converge on a model in which synaptic and axonal degeneration emerge as early, central events in ALS, driven by a combination of RNA dysregulation, impaired organellar function, and aberrant immune signaling.
Astrocytes derived from iPSCs have proven essential in elucidating non-cell-autonomous drivers of motor neuron degeneration. Insights from iPSC studies indicate that while astrocytes are relatively resistant to TDP-43 propagation compared to motor neurons, they play an active role in modulating proteinopathy spread [121]. Rather than acting as passive bystanders, astrocytes function as a dynamic buffer, one that may limit or facilitate pathological transmission depending on the context. Further supporting this dual role, organoids exposed to ALS-derived extracts develop reactive astrogliosis [116], indicating that astrocytes contribute both to the local tissue response and potentially to disease propagation. These observations reinforce the concept of astroglia as both potential protectors and facilitators of neurodegeneration, depending on disease stage and microenvironmental cues.
iPSC-derived microglia models have added an additional layer of insight into neuroinflammatory pathways in ALS. C9orf72-ALS microglia displayed a pro-inflammatory transcriptional profile and secreted matrix metalloproteinase-9 (MMP-9) in response to lipopolysaccharide, promoting motor neuron apoptosis [122]. Although direct toxicity was not observed in baseline co-culture conditions, C9 microglia altered the extracellular milieu and increased dipeptidyl peptidase 4 (DPP4) expression, suggesting early immune priming and subtle glia-neuron crosstalk. These findings align with a growing body of evidence supporting the idea that glial cells, particularly microglia, contribute to the creation of a permissive, or even toxic, environment that accelerates neuronal loss in ALS.
5. Clinical Presentation
ALS typically presents with an insidious onset and follows a relentlessly progressive course [6]. Although ALS is a distinct clinical entity, the relative involvement of UMNs and LMNs varies significantly between individuals, influencing both the clinical presentation and prognosis. ALS shares overlapping clinical features with other MNDs, including progressive muscular atrophy (PMA), progressive bulbar palsy (PBP), and primary lateral sclerosis (PLS), which can complicate the diagnostic process [5]. Furthermore, multiple disease phenotypes exist, defined by the site of onset and the subsequent pattern of clinical progression [123]. In most cases, the initial presentation dictates the trajectory of disease evolution [5].
5.1. Classical ALS
5.1.1. Typical Clinical Features
Classical ALS typically begins with weakness in the distal part of one extremity, either upper or lower [4]. As the disease progresses, it spreads to adjacent regions of the same limb, followed by involvement of the distal part of the contralateral limb, resulting in an asymmetrical pattern of progression. Eventually, bulbar muscles become affected, and later, the trunk and respiratory muscles are involved. Death usually occurs from respiratory failure within 2 to 5 years of symptom onset. Notably, sphincter and ocular muscles are generally spared throughout the disease course [124].
Lower extremity-onset classical ALS is characterized by distal muscle atrophy and spasticity, often manifesting as foot drop. Weakness gradually ascends to involve the calf and thigh muscles, followed by involvement of the contralateral limb in a similar pattern. On examination, patients typically present with painless weakness, preserved sensory function, and exaggerated tendon reflexes. A positive Babinski sign may also be observed. Early presentation with spastic weakness in the lower limbs can resemble PLS, with a definitive ALS diagnosis becoming apparent only as LMN signs emerge over time [4].
Similarly, upper extremity-onset ALS presents with distal hand weakness and spasticity in the fingers, sometimes appearing as wrist drop. While the finger extensors, abductors, and adductors are affected, the long finger extensors are often spared, allowing preservation of grip strength in the early stages. As the disease progresses, weakness extends to the forearm and upper arm, accompanied by fasciculations, hand atrophy, increased spasticity, and generalized hyperreflexia. A positive Hoffmann sign may be present [4].
While bulbar muscle involvement typically follows limb involvement in ALS, bulbar-onset ALS occurs in approximately 25% of cases. The most common initial symptoms are dysarthria and dysphagia, often followed by sialorrhea. Physical examination frequently reveals tongue atrophy and fasciculations. The trunk and respiratory muscles, such as the abdominal, thoracic, posterior neck muscles, and the diaphragm, are usually affected in the later stages of the disease. However, rare presentations of thoracic-onset or respiratory-onset ALS do occur. These forms are characterized by symptoms such as camptocormia (forward flexion of the trunk) and head drop, and are associated with early respiratory failure [6].
Based on the site of symptom onset, several clinical ALS phenotypes have been described, including flail arm syndrome, flail leg syndrome, spinal-onset, bulbar-onset, hemiplegic, pseudo-polyneuritic, thoracic-onset, respiratory-onset, and mixed-onset ALS [123,125]. Flail arm syndrome is defined by symmetric proximal weakness in the upper limbs, whereas flail leg syndrome involves distal-onset weakness in the lower limbs. These syndromes can be difficult to distinguish from classical ALS with limb-onset due to overlapping features. However, flail arm and flail leg syndromes generally have a slower progression, with neurological deficits remaining confined to the limbs for a longer duration. Accurate differentiation of these phenotypes is crucial for establishing an appropriate prognosis and guiding management [126].
5.1.2. Additional Clinical Features
ALS may be accompanied by pseudobulbar palsy, which is characterized by pathological crying, laughing, and yawning due to involvement of the frontopontine pathways. Although less common, autonomic symptoms such as constipation and sensory disturbances can also occur, a presentation referred to as ALS-plus syndrome [127]. Cognitive impairment is increasingly recognized as a significant non-motor feature of ALS. Approximately 30-50% of individuals with ALS exhibit some degree of cognitive dysfunction, while 15-20% meet the criteria for FTD. Patients with ALS-FTD spectrum disorder often display impairments in executive functioning and verbal fluency, along with behavioral changes such as apathy, disinhibition, and agitation [5,123].
5.2. Other Motor Neuron Disease Subtypes
5.2.1. Progressive Muscular Atrophy (PMA)
Progressive muscular atrophy (PMA) occurs more commonly in males and is characterized primarily by the solely involvement of LMN [128]. The most frequent presentation is distal upper extremity-onset, marked by hand weakness and atrophy, with progression to the forearm and upper arm muscles. Less commonly, the disease may begin in the proximal upper limbs or the lower extremities. On physical examination, patients typically exhibit reduced or absent tendon reflexes, consistent with LMN dysfunction. Over time, UMN signs may develop in a significant proportion of individuals, leading to a reclassification as LMN-onset ALS [129,130]. Interestingly, even in patients without clinical UMN involvement during life, postmortem histopathological studies have revealed corticospinal tract degeneration in approximately 50% of cases [131]. Although PMA is a progressive disease, its course is generally slower than that of classical ALS, especially in individuals with early-onset PMA (<50 years old) [128,132].
5.2.2. Progressive Bulbar Palsy (PBP)
Individuals with PBP typically present with dysarthria and dysphagia, which progressively worsen over time. Common symptoms include impaired articulation, nasal speech, food retention in the mouth, coughing, and choking due to dysphagia. In the early stages, tongue atrophy and fasciculations are often observed. As the disease advances, physical examination may reveal loss of the pharyngeal reflex and an exaggerated jaw jerk, reflecting increased weakness of the muscles of mastication. Some patients initially exhibit pseudobulbar signs, such as pathological laughing, crying, or yawning, due to spastic bulbar weakness in the absence of atrophy. PBP gradually progresses to involve the respiratory muscles, ultimately leading to death. Notably, early-onset PBP is associated with a poorer prognosis [4,133].
5.2.3. Primary Lateral Sclerosis (PLS)
PLS is a rare motor neuron disease, accounting for approximately 2-4% of all ALS cases. It is characterized by isolated UMN degeneration, without clinical or electrophysiological evidence of LMN involvement. The typical presentation involves asymmetric stiffness in the lower extremities, which gradually progresses to spastic paraparesis. Over time, spasticity may extend to the upper extremities and bulbar muscles, leading to dysarthria and pseudobulbar palsy. Less common initial presentations include bulbar-onset PLS, which is marked by early pseudobulbar signs, and unilateral spasticity (also known as Mills’ hemiparetic pattern). In some cases, patients may present with pure spastic paraparesis, which subsequently spreads to involve other regions. Although PLS has a slower progression and more favorable prognosis than ALS, accurate diagnosis is essential, as early-stage ALS can mimic PLS before LMN signs appear [4,134].
6. Disease Diagnosis
In the absence of definitive biomarkers, ALS remains a clinical diagnosis of exclusion, which can pose significant challenges for physicians. However, the development of standardized diagnostic criteria, alongside the use of electrophysiological and neuroimaging techniques, has greatly enhanced diagnostic accuracy [123].
The El Escorial criteria, initially established to identify individuals eligible for ALS clinical trials, have undergone several revisions. The currently used version is the 2000 revision, also known as the Awaji criteria [135,136]. Under these criteria, electromyographic (EMG) findings are considered equivalent to clinical evidence of LMN involvement. To establish a diagnosis of clinically definite ALS, the following criteria must be met: a. evidence of both UMN and LMN degeneration, b. disease progression, either within a single anatomical region or to other regions, and c. the exclusion of alternative diagnoses through clinical evaluation, laboratory testing, electrophysiology, and neuroimaging. The anatomical regions used for disease classification include bulbar, cervical, thoracic, and lumbar (Table 2).
All individuals suspected of having ALS should undergo nerve conduction studies (NCS) and electromyography (EMG) [6,137]. NCS may assist in diagnosis by detecting reduced action potential amplitudes, which can precede clinically apparent weakness. EMG is essential for the diagnosis of nearly all ALS cases [4]. Characteristic EMG findings include: a. acute denervation, evidenced by fibrillation potentials and positive sharp waves, b. chronic denervation, shown by long-duration, complex motor unit action potentials (cMUAPs), and c. chronic reinnervation, identified by large-amplitude MUAPs [138]. According to EMG criteria for ALS diagnosis, there must be evidence of either acute or chronic denervation in at least three anatomical regions (bulbar, cervical, thoracic, lumbosacral), or in three extremities involving at least two muscles per limb, each innervated by different roots [139]. Importantly, EMG should not show signs of demyelination or sensory nerve involvement, as these are not typical in ALS.
Magnetic resonance imaging (MRI) is the imaging modality of choice, primarily used to exclude alternative diagnoses such as structural or demyelinating diseases. While MRI is often normal in ALS [4,124], subtle findings, such as motor cortex atrophy and T2-weighted or FLAIR hyperintensities in the posterior limb of the internal capsule, descending corticospinal tracts, or spinal cord, may suggest corticospinal tract Wallerian degeneration [6,140]. These imaging findings can be particularly valuable in cases where prominent LMN signs obscure UMN involvement.
Additional laboratory testing is necessary to exclude other potential conditions that may mimic ALS [124]. In some cases, individuals with ALS may exhibit mildly elevated serum creatine kinase and normal or slightly increased cerebrospinal fluid protein levels [4,124]. Muscle biopsy is not required for diagnosis but can support it by demonstrating neurogenic denervation in affected muscle tissue [4]. The diagnostic approach to ALS is summarized in Figure 1.
7. Treatment
To date, there is no cure for ALS. Management primarily focuses on symptomatic treatment, although four drugs have been approved to delay disease progression [141]. Numerous other agents are currently under investigation in ongoing clinical trials (Table 3).
The four approved therapies are riluzole, edaravone, relyvrio, and tofersen. Riluzole, a glutamate antagonist, was the first drug approved for ALS in 1995 [142]. It has neuroprotective effects, delays the need for ventilatory support, and extends survival by approximately three months. Riluzole can be administered orally or intravenously. Common side effects include nausea and hepatotoxicity [143]. Edaravone, an antioxidant and free radical scavenger, was approved by the FDA in 2017 [144]. It has been shown to slow disease progression, particularly in early-stage ALS. Initially available only as an intravenous formulation, an oral suspension is now also offered. Side effects may include skin erythema, headache, and gait disturbances. Notably, the efficacy of edaravone remains inconclusive and inconsistent, as clinical trials have reported conflicting results [145,146]. Due to the limited evidence of effectiveness, edaravone has not been approved for use in Europe. Relyvrio (sodium phenylbutyrate-taurursodiol), an anti-apoptotic neuroprotective agent, received FDA approval in 2022 [147]. It delays disease progression and improves survival, and is administered orally as a powder. Common adverse effects include headache, nausea, and fatigue. Tofersen, an antisense oligonucleotide targeting SOD1 gene mutations, was approved in 2023 for patients carrying SOD1 mutations [148]. While tofersen’s impact on disease progression has been supported by reductions in neurofilament light chain (NFL) levels, a biomarker of neuronal injury, its clinical efficacy is still being evaluated. The drug is administered intrathecally and may cause side effects such as myalgia, arthralgia, and fatigue [148,149].
The treatment of ALS largely depends on symptomatic therapy for the appropriate management of symptoms [4]. For the treatment of spasticity, baclofen and tizanidine are widely used [160]. For the treatment of sialorrhea, glycopyrrolate and amitriptyline are usually prescribed. Botulinum toxin injections in the parotid and submandibular glands are also effective. In refractory cases, radiation therapy to salivary glands is an option [161,162]. In the presence of dysphagia, special diets need to be administered. However, in patients with severe dysphagia, percutaneous endoscopic gastrostomy can be performed, in order to avoid the risk of aspiration [162]. In patients with dyspnea, cough-assist devices, opioids, and non-invasive ventilation can be used [163]. For the treatment of cognitive, behavioral, and mood disturbances antidepressants and antipsychotics should be administered accordingly [164]. Additionally, dextromethorphan/quinidine has been approved for the treatment of pseudobulbar symptoms [165]. Depending on the cause and severity of pain, patients may receive either non-steroidal anti-inflammatory drugs, opioids, or gabapentin in the case of neuropathic pain [166]. Overall, for the appropriate and holistic management of patients with ALS a multidisciplinary team comprising of neurologists, psychiatrists, internists, speech and language therapists, physiotherapists, and palliative care therapists is essential.
8. Concluding Remarks
Early diagnosis, the development of reliable biomarkers, and stratification of patients based on genetic and phenotypic subtypes may enable precision medicine approaches in ALS management. Continued investment in translational research, particularly the development of novel complex models that more accurately recapitulate disease pathology, including models with multiple genetic mutations or variations, alongside well-designed clinical trials, may accelerate the emergence of disease-modifying therapies for ALS.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.P., I.M.; writing—original draft preparation, I.S., S.S.de L., A.S.; writing—review and editing I.S., S.S. de L., A.T., M.T., E.P., I.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Park, J.; Kim, J.-E.; Song, T.-J. The Global Burden of Motor Neuron Disease: An Analysis of the 2019 Global Burden of Disease Study. Front Neurol 2022, 13, 864339. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef]
- Verma, A. Clinical Manifestation and Management of Amyotrophic Lateral Sclerosis. In Amyotrophic Lateral Sclerosis; Araki, T., Ed.; Exon Publications: Brisbane (AU), 2021 ISBN 978-0-6450017-7-8.
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb Perspect Med 2017, 7, a024117. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N Engl J Med 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. ; EURALS Incidence of Amyotrophic Lateral Sclerosis in Europe. J Neurol Neurosurg Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef]
- Burrell, J.R.; Vucic, S.; Kiernan, M.C. Isolated Bulbar Phenotype of Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler 2011, 12, 283–289. [Google Scholar] [CrossRef]
- Grassano, M.; Moglia, C.; Palumbo, F.; Koumantakis, E.; Cugnasco, P.; Callegaro, S.; Canosa, A.; Manera, U.; Vasta, R.; De Mattei, F.; et al. Sex Differences in Amyotrophic Lateral Sclerosis Survival and Progression: A Multidimensional Analysis. Annals of Neurology 2024, 96, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging Insights into the Complex Genetics and Pathophysiology of ALS. Lancet Neurol 2022, 21, 465–479. [Google Scholar] [CrossRef]
- Barberio, J.; Lally, C.; Kupelian, V.; Hardiman, O.; Flanders, W.D. Estimated Familial Amyotrophic Lateral Sclerosis Proportion: A Literature Review and Meta-Analysis. Neurol Genet 2023, 9, e200109. [Google Scholar] [CrossRef] [PubMed]
- Siddique, T.; Figlewicz, D.A.; Pericak-Vance, M.A.; Haines, J.L.; Rouleau, G.; Jeffers, A.J.; Sapp, P.; Hung, W.Y.; Bebout, J.; McKenna-Yasek, D. Linkage of a Gene Causing Familial Amyotrophic Lateral Sclerosis to Chromosome 21 and Evidence of Genetic-Locus Heterogeneity. N Engl J Med 1991, 324, 1381–1384. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Chiò, A.; Traynor, B.J.; Lombardo, F.; Fimognari, M.; Calvo, A.; Ghiglione, P.; Mutani, R.; Restagno, G. Prevalence of SOD1 Mutations in the Italian ALS Population. Neurology 2008, 70, 533–537. [Google Scholar] [CrossRef]
- Kühnlein, P.; Sperfeld, A.-D.; Vanmassenhove, B.; Van Deerlin, V.; Lee, V.M.-Y.; Trojanowski, J.Q.; Kretzschmar, H.A.; Ludolph, A.C.; Neumann, M. Two German Kindreds With Familial Amyotrophic Lateral Sclerosis Due to TARDBP Mutations. Archives of Neurology 2008, 65, 1185–1189. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Al-Chalabi, A.; Andersen, P.M.; Hosler, B.; Sapp, P.; Englund, E.; Mitchell, J.E.; Habgood, J.J.; de Belleroche, J.; Xi, J.; et al. A Locus on Chromosome 9p Confers Susceptibility to ALS and Frontotemporal Dementia. Neurology 2006, 66, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Al-Chalabi, A.; Ruddy, D.; Smith, B.N.; Hu, X.; Sreedharan, J.; Siddique, T.; Schelhaas, H.J.; Kusters, B.; Troost, D.; et al. Familial Amyotrophic Lateral Sclerosis with Frontotemporal Dementia Is Linked to a Locus on Chromosome 9p13. 2-21.3. Brain 2006, 129, 868–876. [Google Scholar] [CrossRef]
- Boxer, A.L.; Mackenzie, I.R.; Boeve, B.F.; Baker, M.; Seeley, W.W.; Crook, R.; Feldman, H.; Hsiung, G.-Y.R.; Rutherford, N.; Laluz, V.; et al. Clinical, Neuroimaging and Neuropathological Features of a New Chromosome 9p-Linked FTD-ALS Family. J Neurol Neurosurg Psychiatry 2011, 82, 196–203. [Google Scholar] [CrossRef]
- van Es, M.A.; Veldink, J.H.; Saris, C.G.J.; Blauw, H.M.; van Vught, P.W.J.; Birve, A.; Lemmens, R.; Schelhaas, H.J.; Groen, E.J.N.; Huisman, M.H.B.; et al. Genome-Wide Association Study Identifies 19p13. 3 (UNC13A) and 9p21.2 as Susceptibility Loci for Sporadic Amyotrophic Lateral Sclerosis. Nat Genet 2009, 41, 1083–1087. [Google Scholar] [CrossRef]
- Laaksovirta, H.; Peuralinna, T.; Schymick, J.C.; Scholz, S.W.; Lai, S.-L.; Myllykangas, L.; Sulkava, R.; Jansson, L.; Hernandez, D.G.; Gibbs, J.R.; et al. Chromosome 9p21 in Amyotrophic Lateral Sclerosis in Finland: A Genome-Wide Association Study. The Lancet Neurology 2010, 9, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Shatunov, A.; Mok, K.; Newhouse, S.; Weale, M.E.; Smith, B.; Vance, C.; Johnson, L.; Veldink, J.H.; van Es, M.A.; van den Berg, L.H.; et al. Chromosome 9p21 in Sporadic Amyotrophic Lateral Sclerosis in the UK and Seven Other Countries: A Genome-Wide Association Study. Lancet Neurol 2010, 9, 986–994. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Sleiman, P.M.A.; Martinez-Lage, M.; Chen-Plotkin, A.; Wang, L.-S.; Graff-Radford, N.R.; Dickson, D.W.; Rademakers, R.; Boeve, B.F.; Grossman, M.; et al. Common Variants at 7p21 Are Associated with Frontotemporal Lobar Degeneration with TDP-43 Inclusions. Nat Genet 2010, 42, 234–239. [Google Scholar] [CrossRef]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 Expansion Mutation Is a Common Cause of ALS+/−FTD in Europe and Has a Single Founder. Eur J Hum Genet 2013, 21, 102–108. [Google Scholar] [CrossRef]
- Ogaki, K.; Li, Y.; Atsuta, N.; Tomiyama, H.; Funayama, M.; Watanabe, H.; Nakamura, R.; Yoshino, H.; Yato, S.; Tamura, A.; et al. Analysis of C9orf72 Repeat Expansion in 563 Japanese Patients with Amyotrophic Lateral Sclerosis. Neurobiol Aging 2012, 33, 2527.e11-16. [Google Scholar] [CrossRef]
- Nishiyama, A.; Niihori, T.; Warita, H.; Izumi, R.; Akiyama, T.; Kato, M.; Suzuki, N.; Aoki, Y.; Aoki, M. Comprehensive Targeted Next-Generation Sequencing in Japanese Familial Amyotrophic Lateral Sclerosis. Neurobiol Aging 2017, 53, 194.e1-194.e8. [Google Scholar] [CrossRef]
- Nishiyama, A.; Niihori, T.; Warita, H.; Izumi, R.; Akiyama, T.; Kato, M.; Suzuki, N.; Aoki, Y.; Aoki, M. Comprehensive Targeted Next-Generation Sequencing in Japanese Familial Amyotrophic Lateral Sclerosis. Neurobiol Aging 2017, 53, 194.e1-194.e8. [Google Scholar] [CrossRef]
- Borghero, G.; Pugliatti, M.; Marrosu, F.; Marrosu, M.G.; Murru, M.R.; Floris, G.; Cannas, A.; Parish, L.D.; Occhineri, P.; Cau, T.B.; et al. Genetic Architecture of ALS in Sardinia. Neurobiology of Aging 2014, 35, 2882.e7-2882.e12. [Google Scholar] [CrossRef] [PubMed]
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A.; et al. Systematic Evaluation of Genetic Mutations in ALS: A Population-Based Study. J Neurol Neurosurg Psychiatry 2022, 93, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N Engl J Med 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Piao, Y.-S.; Wakabayashi, K.; Kakita, A.; Yamada, M.; Hayashi, S.; Morita, T.; Ikuta, F.; Oyanagi, K.; Takahashi, H. Neuropathology with Clinical Correlations of Sporadic Amyotrophic Lateral Sclerosis: 102 Autopsy Cases Examined between 1962 and 2000. Brain Pathol 2003, 13, 10–22. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem Biophys Res Commun 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 Control in Neurodegeneration: Autoregulation, Localization and Aggregation. Nat Rev Neurosci 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.A.; Akiyama, H.; et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains. Cell Rep 2013, 4, 124–134. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Riva, N.; Gentile, F.; Cerri, F.; Gallia, F.; Podini, P.; Dina, G.; Falzone, Y.M.; Fazio, R.; Lunetta, C.; Calvo, A.; et al. Phosphorylated TDP-43 Aggregates in Peripheral Motor Nerves of Patients with Amyotrophic Lateral Sclerosis. Brain 2022, 145, 276–284. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 Pathology in Amyotrophic Lateral Sclerosis. Ann Neurol 2013, 74, 20–38. [Google Scholar] [CrossRef]
- Tan, C.-F.; Eguchi, H.; Tagawa, A.; Onodera, O.; Iwasaki, T.; Tsujino, A.; Nishizawa, M.; Kakita, A.; Takahashi, H. TDP-43 Immunoreactivity in Neuronal Inclusions in Familial Amyotrophic Lateral Sclerosis with or without SOD1 Gene Mutation. Acta Neuropathol 2007, 113, 535–542. [Google Scholar] [CrossRef]
- Robinson, J.L.; Geser, F.; Stieber, A.; Umoh, M.; Kwong, L.K.; Van Deerlin, V.M.; Lee, V.M.-Y.; Trojanowski, J.Q. TDP-43 Skeins Show Properties of Amyloid in a Subset of ALS Cases. Acta Neuropathol 2013, 125, 121–131. [Google Scholar] [CrossRef]
- Koike, Y. Abnormal Splicing Events Due to Loss of Nuclear Function of TDP-43: Pathophysiology and Perspectives. JMA J 2024, 7, 313–318. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Takanashi, K.; Yamaguchi, A. Aggregation of ALS-Linked FUS Mutant Sequesters RNA Binding Proteins and Impairs RNA Granules Formation. Biochemical and Biophysical Research Communications 2014, 452, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic Gain of Function from Mutant FUS Protein Is Crucial to Trigger Cell Autonomous Motor Neuron Loss. The EMBO Journal 2016, 35, 1077–1097. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat Rev Neurol 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Mizielinska, S.; Lashley, T.; Norona, F.E.; Clayton, E.L.; Ridler, C.E.; Fratta, P.; Isaacs, A.M. C9orf72 Frontotemporal Lobar Degeneration Is Characterised by Frequent Neuronal Sense and Antisense RNA Foci. Acta Neuropathol 2013, 126, 845–857. [Google Scholar] [CrossRef]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN Translation at C9orf72-Associated Repeat Expansions Is Selectively Enhanced by the Integrated Stress Response. Nat Commun 2017, 8, 2005. [Google Scholar] [CrossRef]
- Mann, D.M.A.; Rollinson, S.; Robinson, A.; Bennion Callister, J.; Thompson, J.C.; Snowden, J.S.; Gendron, T.; Petrucelli, L.; Masuda-Suzukake, M.; Hasegawa, M.; et al. Dipeptide Repeat Proteins Are Present in the P62 Positive Inclusions in Patients with Frontotemporal Lobar Degeneration and Motor Neurone Disease Associated with Expansions in C9ORF72. Acta Neuropathol Commun 2013, 1, 68. [Google Scholar] [CrossRef]
- Swanson, M.E.V.; Mrkela, M.; Murray, H.C.; Cao, M.C.; Turner, C.; Curtis, M.A.; Faull, R.L.M.; Walker, A.K.; Scotter, E.L. Microglial CD68 and L-Ferritin Upregulation in Response to Phosphorylated-TDP-43 Pathology in the Amyotrophic Lateral Sclerosis Brain. Acta Neuropathol Commun 2023, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.-Y.; et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017, 95, 297–308.e6. [Google Scholar] [CrossRef] [PubMed]
- Bahia El Idrissi, N.; Bosch, S.; Ramaglia, V.; Aronica, E.; Baas, F.; Troost, D. Complement Activation at the Motor End-Plates in Amyotrophic Lateral Sclerosis. J Neuroinflammation 2016, 13, 72. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The Multi-Dimensional Roles of Astrocytes in ALS. Neurosci Res 2018, 126, 31–38. [Google Scholar] [CrossRef]
- Marques, C.; Held, A.; Dorfman, K.; Sung, J.; Song, C.; Kavuturu, A.S.; Aguilar, C.; Russo, T.; Oakley, D.H.; Albers, M.W.; et al. Neuronal STING Activation in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Acta Neuropathol 2024, 147, 56. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor Neuron Degeneration in Mice That Express a Human Cu,Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Genç, B.; Gautam, M.; Helmold, B.R.; Koçak, N.; Günay, A.; Goshu, G.M.; Silverman, R.B.; Hande Ozdinler, P. NU-9 Improves Health of hSOD1G93A Mouse Upper Motor Neurons in Vitro, Especially in Combination with Riluzole or Edaravone. Sci Rep 2022, 12, 5383. [Google Scholar] [CrossRef]
- Zhu, L.; Li, S.; Li, X.-J.; Yin, P. Pathological Insights from Amyotrophic Lateral Sclerosis Animal Models: Comparisons, Limitations, and Challenges. Translational Neurodegeneration 2023, 12, 46. [Google Scholar] [CrossRef]
- Tsao, W.; Jeong, Y.H.; Lin, S.; Ling, J.; Price, D.L.; Chiang, P.-M.; Wong, P.C. Rodent Models of TDP-43: Recent Advances. Brain Res 2012, 1462, 26–39. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-Associated ALS and FTD: Insights from Rodent Models. Acta Neuropathologica Communications 2016, 4, 99. [Google Scholar] [CrossRef]
- Batra, R.; Lee, C.W. Mouse Models of C9orf72 Hexanucleotide Repeat Expansion in Amyotrophic Lateral Sclerosis/ Frontotemporal Dementia. Front Cell Neurosci 2017, 11, 196. [Google Scholar] [CrossRef]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal Loss of the Glutamate Transporter EAAT2 in a Transgenic Rat Model of SOD1 Mutant-Mediated Amyotrophic Lateral Sclerosis (ALS). Proceedings of the National Academy of Sciences 2002, 99, 1604–1609. [Google Scholar] [CrossRef]
- Harrison, J.M.; Rafuse, V.F. Muscle Fiber-Type Specific Terminal Schwann Cell Pathology Leads to Sprouting Deficits Following Partial Denervation in SOD1G93A Mice. Neurobiology of Disease 2020, 145, 105052. [Google Scholar] [CrossRef]
- Cai, M.; Lee, K.-W.; Choi, S.-M.; Yang, E.J. TDP-43 Modification in the hSOD1(G93A) Amyotrophic Lateral Sclerosis Mouse Model. Neurol Res 2015, 37, 253–262. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic Mice Expressing an Altered Murine Superoxide Dismutase Gene Provide an Animal Model of Amyotrophic Lateral Sclerosis. Proc Natl Acad Sci U S A 1995, 92, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An Adverse Property of a Familial ALS-Linked SOD1 Mutation Causes Motor Neuron Disease Characterized by Vacuolar Degeneration of Mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 Misplacing and Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis Pathogenesis. Front Cell Neurosci 2015, 9, 336. [Google Scholar] [CrossRef]
- Quarta, E.; Bravi, R.; Scambi, I.; Mariotti, R.; Minciacchi, D. Increased Anxiety-like Behavior and Selective Learning Impairments Are Concomitant to Loss of Hippocampal Interneurons in the Presymptomatic SOD1(G93A) ALS Mouse Model. J Comp Neurol 2015, 523, 1622–1638. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 Mutant Transgenic Mice Develop Features of ALS and Frontotemporal Lobar Degeneration. Proc Natl Acad Sci U S A 2009, 106, 18809–18814. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-Linked TDP-43 Mutations Produce Aberrant RNA Splicing and Adult-Onset Motor Neuron Disease without Aggregation or Loss of Nuclear TDP-43. Proc Natl Acad Sci U S A 2013, 110, E736–E745. [Google Scholar] [CrossRef] [PubMed]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive Motor Weakness in Transgenic Mice Expressing Human TDP-43. Neurobiol Dis 2010, 40, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Tong, J.; Bi, F.; Zhou, H.; Xia, X.-G. Mutant TDP-43 in Motor Neurons Promotes the Onset and Progression of ALS in Rats. J Clin Invest 2012, 122, 107–118. [Google Scholar] [CrossRef]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 Transgenic Mice Develop Spastic Paralysis and Neuronal Inclusions Characteristic of ALS and Frontotemporal Lobar Degeneration. Proc Natl Acad Sci U S A 2010, 107, 3858–3863. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J Neurosci 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, C.; Chen, H.; Wang, D.; Landel, C.P.; Xia, P.Y.; Bowser, R.; Liu, Y.-J.; Xia, X.G. Transgenic Rat Model of Neurodegeneration Caused by Mutation in the TDP Gene. PLoS Genet 2010, 6, e1000887. [Google Scholar] [CrossRef]
- Huang, C.; Tong, J.; Bi, F.; Wu, Q.; Huang, B.; Zhou, H.; Xia, X.-G. Entorhinal Cortical Neurons Are the Primary Targets of FUS Mislocalization and Ubiquitin Aggregation in FUS Transgenic Rats. Hum Mol Genet 2012, 21, 4602–4614. [Google Scholar] [CrossRef]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.-J.; Wang, D.; Wei, X.; Xia, X.-G. FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. PLoS Genet 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-Associated Mutant FUS Induces Selective Motor Neuron Degeneration through Toxic Gain of Function. Nat Commun 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.K.; Deykin, A.V.; Bronovitsky, E.V.; Ovchinnikov, R.K.; Ustyugov, A.A.; Shelkovnikova, T.A.; Kukharsky, M.S.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Early Lethality and Neuronal Proteinopathy in Mice Expressing Cytoplasm-Targeted FUS That Lacks the RNA Recognition Motif. Amyotroph Lateral Scler Frontotemporal Degener 2015, 16, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.J.; et al. Overexpression of Human Wild-Type FUS Causes Progressive Motor Neuron Degeneration in an Age- and Dose-Dependent Fashion. Acta Neuropathol 2013, 125, 273–288. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.-Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-Associated Mutation FUS-R521C Causes DNA Damage and RNA Splicing Defects. J Clin Invest 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Sephton, C.F.; Tang, A.A.; Kulkarni, A.; West, J.; Brooks, M.; Stubblefield, J.J.; Liu, Y.; Zhang, M.Q.; Green, C.B.; Huber, K.M.; et al. Activity-Dependent FUS Dysregulation Disrupts Synaptic Homeostasis. Proc Natl Acad Sci U S A 2014, 111, E4769–E4778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.-F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 Poly(GA) Aggregates Sequester and Impair HR23 and Nucleocytoplasmic Transport Proteins. Nat Neurosci 2016, 19, 668–677. [Google Scholar] [CrossRef]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. Neurodegeneration. C9ORF72 Repeat Expansions in Mice Cause TDP-43 Pathology, Neuronal Loss, and Behavioral Deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef]
- Herranz-Martin, S.; Chandran, J.; Lewis, K.; Mulcahy, P.; Higginbottom, A.; Walker, C.; Valenzuela, I.M.-P.Y.; Jones, R.A.; Coldicott, I.; Iannitti, T.; et al. Viral Delivery of C9orf72 Hexanucleotide Repeat Expansions in Mice Leads to Repeat-Length-Dependent Neuropathology and Behavioural Deficits. Dis Model Mech 2017, 10, 859–868. [Google Scholar] [CrossRef]
- Yanovsky-Dagan, S.; Mor-Shaked, H.; Eiges, R. Modeling Diseases of Noncoding Unstable Repeat Expansions Using Mutant Pluripotent Stem Cells. World J Stem Cells 2015, 7, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Vande Velde, C.; et al. Gain and Loss of Function of ALS-Related Mutations of TARDBP (TDP-43) Cause Motor Deficits in Vivo. Hum Mol Genet 2010, 19, 671–683. [Google Scholar] [CrossRef]
- Lemmens, R.; Van Hoecke, A.; Hersmus, N.; Geelen, V.; D’Hollander, I.; Thijs, V.; Van Den Bosch, L.; Carmeliet, P.; Robberecht, W. Overexpression of Mutant Superoxide Dismutase 1 Causes a Motor Axonopathy in the Zebrafish. Human Molecular Genetics 2007, 16, 2359–2365. [Google Scholar] [CrossRef]
- Hewamadduma, C.A.A.; Grierson, A.J.; Ma, T.P.; Pan, L.; Moens, C.B.; Ingham, P.W.; Ramesh, T.; Shaw, P.J. Tardbpl Splicing Rescues Motor Neuron and Axonal Development in a Mutant Tardbp Zebrafish. Hum Mol Genet 2013, 22, 2376–2386. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, L.; Ghilardi, A.; Rottoli, E.; De Maglie, M.; Prosperi, L.; Perego, C.; Baruscotti, M.; Bucchi, A.; Del Giacco, L.; Francolini, M. INaP Selective Inhibition Reverts Precocious Inter- and Motorneurons Hyperexcitability in the Sod1-G93R Zebrafish ALS Model. Sci Rep 2016, 6, 24515. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of Function of C9orf72 Causes Motor Deficits in a Zebrafish Model of Amyotrophic Lateral Sclerosis. Ann Neurol 2013, 74, 180–187. [Google Scholar] [CrossRef]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.H.M.; Beattie, C.E. A Genetic Model of Amyotrophic Lateral Sclerosis in Zebrafish Displays Phenotypic Hallmarks of Motoneuron Disease. Dis Model Mech 2010, 3, 652–662. [Google Scholar] [CrossRef]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal Transport Defects Are a Common Phenotype in Drosophila Models of ALS. Hum Mol Genet 2016, 25, 2378–2392. [Google Scholar] [CrossRef]
- Yuva-Aydemir, Y.; Almeida, S.; Gao, F.-B. Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci 2018, 41, 457–469. [Google Scholar] [CrossRef]
- Xia, R.; Liu, Y.; Yang, L.; Gal, J.; Zhu, H.; Jia, J. Motor Neuron Apoptosis and Neuromuscular Junction Perturbation Are Prominent Features in a Drosophila Model of Fus-Mediated ALS. Mol Neurodegener 2012, 7, 10. [Google Scholar] [CrossRef]
- Wang, J.-W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-Associated Proteins FUS and TDP-43 Function Together to Affect Drosophila Locomotion and Life Span. J Clin Invest 2011, 121, 4118–4126. [Google Scholar] [CrossRef]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila Fused in Sarcoma (FUS) Homologue Causes Deficient Locomotive Behavior and Shortening of Motoneuron Terminal Branches. PLoS One 2012, 7, e39483. [Google Scholar] [CrossRef]
- Mockett, R.J.; Radyuk, S.N.; Benes, J.J.; Orr, W.C.; Sohal, R.S. Phenotypic Effects of Familial Amyotrophic Lateral Sclerosis Mutant Sod Alleles in Transgenic Drosophila. Proc Natl Acad Sci U S A 2003, 100, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Staveley, B.E.; Phillips, J.P.; Hilliker, A.J. Phenotypic Consequences of Copper-Zinc Superoxide Dismutase Overexpression in Drosophila Melanogaster. Genome 1990, 33, 867–872. [Google Scholar] [CrossRef]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 Affects Drosophila Motoneurons Terminal Synapsis and Locomotive Behavior. FEBS Lett 2009, 583, 1586–1592. [Google Scholar] [CrossRef]
- Chang, J.-C.; Morton, D.B. Drosophila Lines with Mutant and Wild Type Human TDP-43 Replacing the Endogenous Gene Reveals Phosphorylation and Ubiquitination in Mutant Lines in the Absence of Viability or Lifespan Defects. PLoS One 2017, 12, e0180828. [Google Scholar] [CrossRef]
- Chang, J.-C.; Hazelett, D.J.; Stewart, J.A.; Morton, D.B. Motor Neuron Expression of the Voltage-Gated Calcium Channel Cacophony Restores Locomotion Defects in a Drosophila, TDP-43 Loss of Function Model of ALS. Brain Res 2014, 1584, 39–51. [Google Scholar] [CrossRef]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 Results in Motor Neuron Degeneration and Stress Sensitivity in C. Elegans. PLoS One 2013, 8, e83450. [Google Scholar] [CrossRef]
- Murakami, T.; Yang, S.-P.; Xie, L.; Kawano, T.; Fu, D.; Mukai, A.; Bohm, C.; Chen, F.; Robertson, J.; Suzuki, H.; et al. ALS Mutations in FUS Cause Neuronal Dysfunction and Death in Caenorhabditis Elegans by a Dominant Gain-of-Function Mechanism. Hum Mol Genet 2012, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Zhang, Y.-J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic Effects of TDP-43 Overexpression in C. Elegans. Hum Mol Genet 2010, 19, 3206–3218. [Google Scholar] [CrossRef] [PubMed]
- Baskoylu, S.N.; Yersak, J.; O’Hern, P.; Grosser, S.; Simon, J.; Kim, S.; Schuch, K.; Dimitriadi, M.; Yanagi, K.S.; Lins, J.; et al. Single Copy/Knock-in Models of ALS SOD1 in C. Elegans Suggest Loss and Gain of Function Have Different Contributions to Cholinergic and Glutamatergic Neurodegeneration. PLoS Genet 2018, 14, e1007682. [Google Scholar] [CrossRef]
- Li, J.; Li, T.; Zhang, X.; Tang, Y.; Yang, J.; Le, W. Human Superoxide Dismutase 1 Overexpression in Motor Neurons of Caenorhabditis Elegans Causes Axon Guidance Defect and Neurodegeneration. Neurobiol Aging 2014, 35, 837–846. [Google Scholar] [CrossRef]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-Linked Mutant SOD1 Produces a Locomotor Defect Associated with Aggregation and Synaptic Dysfunction When Expressed in Neurons of Caenorhabditis Elegans. PLoS Genet 2009, 5, e1000350. [Google Scholar] [CrossRef]
- van der Geest, A.T.; Jakobs, C.E.; Ljubikj, T.; Huffels, C.F.M.; Cañizares Luna, M.; Vieira de Sá, R.; Adolfs, Y.; de Wit, M.; Rutten, D.H.; Kaal, M.; et al. Molecular Pathology, Developmental Changes and Synaptic Dysfunction in (Pre-) Symptomatic Human C9ORF72-ALS/FTD Cerebral Organoids. Acta Neuropathologica Communications 2024, 12, 152. [Google Scholar] [CrossRef]
- Tamaki, Y.; Ross, J.P.; Alipour, P.; Castonguay, C.-É.; Li, B.; Catoire, H.; Rochefort, D.; Urushitani, M.; Takahashi, R.; Sonnen, J.A.; et al. Spinal Cord Extracts of Amyotrophic Lateral Sclerosis Spread TDP-43 Pathology in Cerebral Organoids. PLoS Genet 2023, 19, e1010606. [Google Scholar] [CrossRef] [PubMed]
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD Brain Organoid Slice Cultures Display Distinct Early Astrocyte and Targetable Neuronal Pathology. Nat Neurosci 2021, 24, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.-C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human Sensorimotor Organoids Derived from Healthy and Amyotrophic Lateral Sclerosis Stem Cells Form Neuromuscular Junctions. Nat Commun 2021, 12, 4744. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Shi, Q.; Pan, X.; Chen, J.; Zhang, Y.; Lang, J.; Wen, S.; Liu, X.; Cheng, T.-L.; Lei, K. Neuromuscular Organoids Model Spinal Neuromuscular Pathologies in C9orf72 Amyotrophic Lateral Sclerosis. Cell Rep 2024, 43, 113892. [Google Scholar] [CrossRef]
- Beckers, J.; Tharkeshwar, A.K.; Fumagalli, L.; Contardo, M.; Van Schoor, E.; Fazal, R.; Thal, D.R.; Chandran, S.; Mancuso, R.; Van Den Bosch, L.; et al. A Toxic Gain-of-Function Mechanism in C9orf72 ALS Impairs the Autophagy-Lysosome Pathway in Neurons. Acta Neuropathol Commun 2023, 11, 151. [Google Scholar] [CrossRef]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.-R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct Responses of Neurons and Astrocytes to TDP-43 Proteinopathy in Amyotrophic Lateral Sclerosis. Brain 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Vahsen, B.F.; Nalluru, S.; Morgan, G.R.; Farrimond, L.; Carroll, E.; Xu, Y.; Cramb, K.M.L.; Amein, B.; Scaber, J.; Katsikoudi, A.; et al. C9orf72-ALS Human iPSC Microglia Are pro-Inflammatory and Toxic to Co-Cultured Motor Neurons via MMP9. Nat Commun 2023, 14, 5898. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G. ; PARALS study group Phenotypic Heterogeneity of Amyotrophic Lateral Sclerosis: A Population Based Study. J Neurol Neurosurg Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Tian, J.; Fan, D. Differentiating Slowly Progressive Subtype of Lower Limb Onset ALS From Typical ALS Depends on the Time of Disease Progression and Phenotype. Front Neurol 2022, 13, 872500. [Google Scholar] [CrossRef]
- McCluskey, L.; Vandriel, S.; Elman, L.; Van Deerlin, V.M.; Powers, J.; Boller, A.; Wood, E.M.; Woo, J.; McMillan, C.T.; Rascovsky, K.; et al. ALS-Plus Syndrome: Non-Pyramidal Features in a Large ALS Cohort. J Neurol Sci 2014, 345, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. ; On Behalf of the Eurals Consortium Prognostic Factors in ALS: A Critical Review. Amyotrophic Lateral Sclerosis 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Kim, W.-K.; Liu, X.; Sandner, J.; Pasmantier, M.; Andrews, J.; Rowland, L.P.; Mitsumoto, H. Study of 962 Patients Indicates Progressive Muscular Atrophy Is a Form of ALS. Neurology 2009, 73, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y.; Atsuta, N.; Yoshida, M.; Tatsumi, S.; Iwasaki, Y.; Mimuro, M.; Watanabe, H.; Ito, M.; Senda, J.; Nakamura, R.; et al. Differential Motor Neuron Involvement in Progressive Muscular Atrophy: A Comparative Study with Amyotrophic Lateral Sclerosis. BMJ Open 2014, 4, e005213. [Google Scholar] [CrossRef]
- Ince, P.G.; Evans, J.; Knopp, M.; Forster, G.; Hamdalla, H.H.M.; Wharton, S.B.; Shaw, P.J. Corticospinal Tract Degeneration in the Progressive Muscular Atrophy Variant of ALS. Neurology 2003, 60, 1252–1258. [Google Scholar] [CrossRef]
- Chiò, A.; Brignolio, F.; Leone, M.; Mortara, P.; Rosso, M.G.; Tribolo, A.; Schiffer, D. A Survival Analysis of 155 Cases of Progressive Muscular Atrophy. Acta Neurologica Scandinavica 1985, 72, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Karam, C.; Scelsa, S.N.; Macgowan, D.J.L. The Clinical Course of Progressive Bulbar Palsy. Amyotroph Lateral Scler 2010, 11, 364–368. [Google Scholar] [CrossRef] [PubMed]
- PRINGLE, C.E.; HUDSON, A.J.; MUNOZ, D.G.; KIERNAN, J.A.; BROWN, W.F.; EBERS, G.C. PRIMARY LATERAL SCLEROSIS: CLINICAL FEATURES, NEUROPATHOLOGY AND DIAGNOSTIC CRITERIA. Brain 1992, 115, 495–520. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. ; World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial Revisited: Revised Criteria for the Diagnosis of Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000, 1, 293–299. [Google Scholar] [CrossRef]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. The Awaji Criteria for Diagnosis of ALS. Muscle Nerve 2011, 44, 456–457, author reply 457. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N Engl J Med 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Behnia, M.; Kelly, J.J. Role of Electromyography in Amyotrophic Lateral Sclerosis. Muscle Nerve 1991, 14, 1236–1241. [Google Scholar] [CrossRef]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic Criteria for Diagnosis of ALS. Clin Neurophysiol 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat Rev Dis Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Wong, W. Managed Care Considerations to Improve Health Care Utilization for Patients with ALS. Am J Manag Care 2023, 29, S120–S126. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and Edaravone: A Tale of Two Amyotrophic Lateral Sclerosis Drugs. Med Res Rev 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol 2017, 16, 505–512. [CrossRef]
- Lunetta, C.; Moglia, C.; Lizio, A.; Caponnetto, C.; Dubbioso, R.; Giannini, F.; Matà, S.; Mazzini, L.; Sabatelli, M.; Siciliano, G.; et al. The Italian Multicenter Experience with Edaravone in Amyotrophic Lateral Sclerosis. J Neurol 2020, 267, 3258–3267. [Google Scholar] [CrossRef]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Günther, R.; Wolf, J.; Hermann, A.; et al. Safety and Effectiveness of Long-Term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol 2022, 79, 121–130. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. New England Journal of Medicine 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Jiang, L.; Wang, L.; Pan, Y.; Wang, X.; Qu, H.; Liao, X.; Zhou, X.; Zhang, S.; Kang, M.; et al. RAG-17, a Novel siRNA Therapy for SOD1-ALS: Safety and Preliminary Efficacy from a First-in-Human Trial (S5. 003). Neurology 2025, 104, 5451. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; Defor, T.; Levine, B.L.; June, C.H.; Rubinstein, P.; et al. Infusion of Ex Vivo Expanded T Regulatory Cells in Adults Transplanted with Umbilical Cord Blood: Safety Profile and Detection Kinetics. Blood 2011, 117, 1061–1070. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Miller, J.S.; McKenna, D.H.; Hippen, K.L.; DeFor, T.E.; Sumstad, D.; Curtsinger, J.; Verneris, M.R.; MacMillan, M.L.; Levine, B.L.; et al. Umbilical Cord Blood-Derived T Regulatory Cells to Prevent GVHD: Kinetics, Toxicity Profile, and Clinical Effect. Blood 2016, 127, 1044–1051. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; Lindborg, S.R.; Goyal, N.A.; Miller, R.G.; Burford, M.J.; Berry, J.D.; Nicholson, K.A.; Mozaffar, T.; Katz, J.S.; Jenkins, L.J.; et al. A Randomized Placebo-Controlled Phase 3 Study of Mesenchymal Stem Cells Induced to Secrete High Levels of Neurotrophic Factors in Amyotrophic Lateral Sclerosis. Muscle Nerve 2022, 65, 291–302. [Google Scholar] [CrossRef]
- Kim, G.; Nakayama, L.; Blum, J.A.; Akiyama, T.; Boeynaems, S.; Chakraborty, M.; Couthouis, J.; Tassoni-Tsuchida, E.; Rodriguez, C.M.; Bassik, M.C.; et al. Genome-Wide CRISPR Screen Reveals v-ATPase as a Drug Target to Lower Levels of ALS Protein Ataxin-2. Cell Rep 2022, 41, 111508. [Google Scholar] [CrossRef]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy Induction Enhances TDP43 Turnover and Survival in Neuronal ALS Models. Nat Chem Biol 2014, 10, 677–685. [Google Scholar] [CrossRef]
- Parmar, D.V.; Kansagra, K.A.; Momin, T.; Patel, H.B.; Jansari, G.A.; Bhavsar, J.; Shah, C.; Patel, J.M.; Ghoghari, A.; Barot, A.; et al. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Oral NLRP3 Inflammasome Inhibitor ZYIL1: First-in-Human Phase 1 Studies (Single Ascending Dose and Multiple Ascending Dose). Clin Pharmacol Drug Dev 2023, 12, 202–211. [Google Scholar] [CrossRef]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-Term Survival Analysis of Masitinib in Amyotrophic Lateral Sclerosis. Ther Adv Neurol Disord 2021, 14, 17562864211030365. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022, 19, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.D.; Cudkowicz, M.E.; Windebank, A.J.; Staff, N.P.; Owegi, M.; Nicholson, K.; McKenna-Yasek, D.; Levy, Y.S.; Abramov, N.; Kaspi, H.; et al. NurOwn, Phase 2, Randomized, Clinical Trial in Patients with ALS: Safety, Clinical, and Biomarker Results. Neurology 2019, 93, e2294–e2305. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.; Peterson, K.; Helfand, M. Comparative Efficacy and Safety of Skeletal Muscle Relaxants for Spasticity and Musculoskeletal Conditions: A Systematic Review. J Pain Symptom Manage 2004, 28, 140–175. [Google Scholar] [CrossRef] [PubMed]
- Garuti, G.; Rao, F.; Ribuffo, V.; Sansone, V.A. Sialorrhea in Patients with ALS: Current Treatment Options. Degener Neurol Neuromuscul Dis 2019, 9, 19–26. [Google Scholar] [CrossRef]
- Bjelica, B.; Petri, S. Narrative Review of Diagnosis, Management and Treatment of Dysphagia and Sialorrhea in Amyotrophic Lateral Sclerosis. J Neurol 2024, 271, 6508–6513. [Google Scholar] [CrossRef] [PubMed]
- Filipe, C.B.; Carreira, N.R.; Reis-Pina, P. Optimizing Breathlessness Management in Amyotrophic Lateral Sclerosis: Insights from a Comprehensive Systematic Review. BMC Palliat Care 2024, 23, 100. [Google Scholar] [CrossRef]
- Kurt, A.; Nijboer, F.; Matuz, T.; Kübler, A. Depression and Anxiety in Individuals with Amyotrophic Lateral Sclerosis: Epidemiology and Management. CNS Drugs 2007, 21, 279–291. [Google Scholar] [CrossRef]
- Fralick, M.; Sacks, C.A.; Kesselheim, A.S. Assessment of Use of Combined Dextromethorphan and Quinidine in Patients With Dementia or Parkinson Disease After US Food and Drug Administration Approval for Pseudobulbar Affect. JAMA Internal Medicine 2019, 179, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S. Pain in Amyotrophic Lateral Sclerosis: A Narrative Review. JYMS 2022, 39, 181–189. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
ALS diagnostic algorithm. Created using Biorender.com.
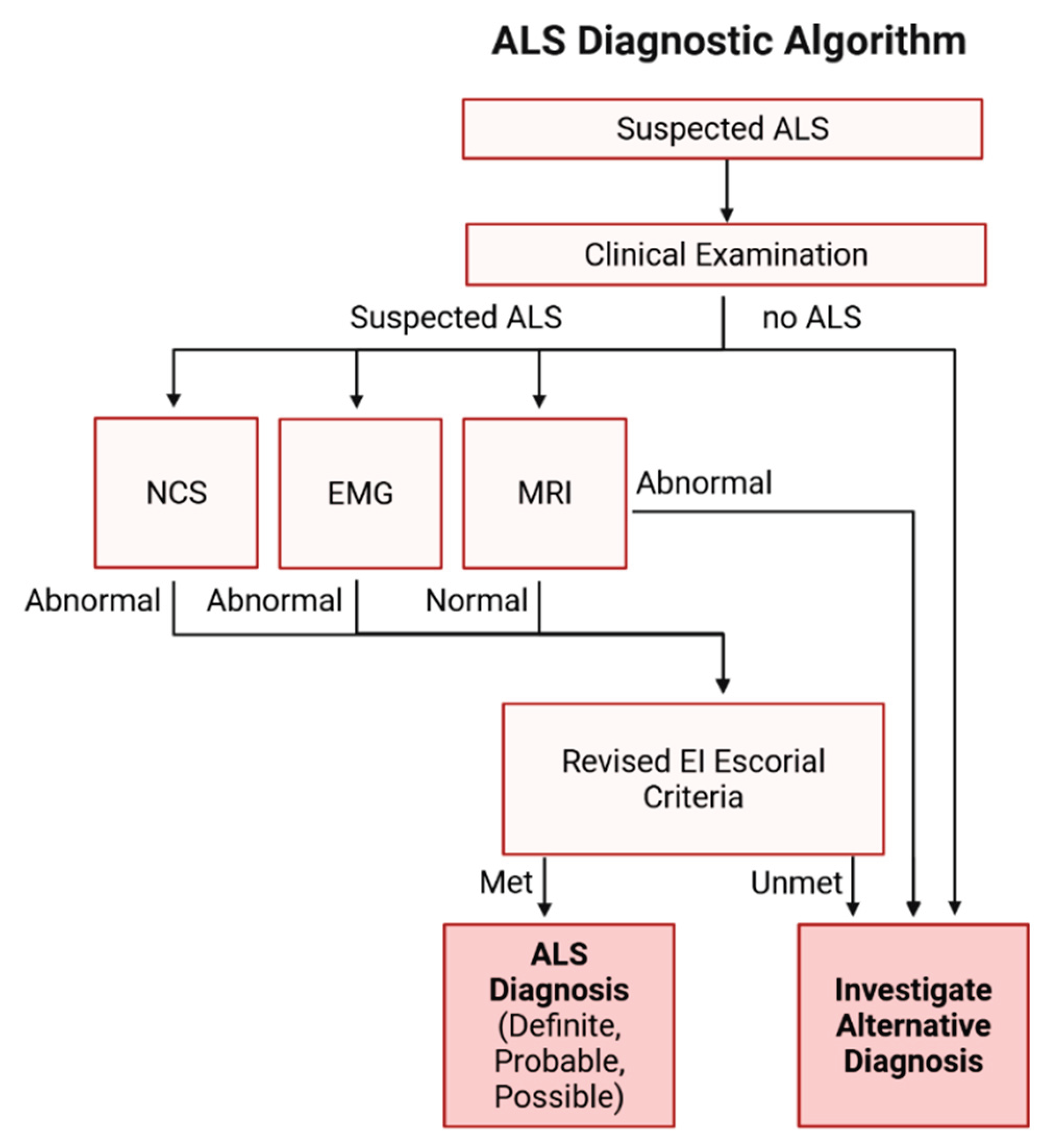

Table 1.
Transgenic models for ALS common mutations.
| Gene | Promoter(s)/ Model |
Species | Pathology | Relevance | References |
|---|---|---|---|---|---|
| Vertebrates | |||||
|
SOD1 G93A, G86R, G37R, A4V, D90A |
Human SOD1 | Mouse Rat |
Motor neuron loss, iron accumulation, gliosis, axon degeneration, mitochondrial damage | Model for fALS, well-characterized | [63,64,65,66,67,68,69,70] |
|
TDP-43 Knock-in, A315T, M337V, Q331K, G348C |
BAC, mPrp, mThy1 | Mouse Rat |
Cytoplasmic TDP-43 aggregates, motor deficits, neuroinflammation | Models TDP-43 pathology | [71,72,73,74,75,76,77] |
|
FUS Knock-out, overexpression, R521C, R521G, P525L |
Tau, mPrp, CAG | Mouse Rat |
Cytoplasmic FUS aggregates, nuclear clearance, motor neuron loss | Found in early-onset ALS, shares features with TDP-43 | [78,79,80,81,82,83,84] |
|
C9ORF72 [100-1000]n [500]n [64]n [147]n [450]n |
BAC, AAV | Mouse | RNA foci, DPRs, nucleocytoplasmic transport defects, TDP-43 pathology, p62 in aggregates | Most common genetic cause of ALS and FTD | [85,86,87,88,89,90,91,92] |
| Invertebrates | |||||
|
Sod1, tdp-43, fus, c9orf72 |
Morpholino knockdown or gene overexpression; CRISPR/Cas9; injections of human mRNA | Zebrafish | Motor axon length decrease, swimming deficits, synaptic abnormalities | Transparent embryos, live imaging, high-throughput | [93,94,95,96,97,98] |
| TDP-43, FUS, SOD1, DPRs | Gene mutants or overexpression; UAS-Gal4 system for expression of human transgenes | Drosophila melanogaster | Motor neuron degeneration, locomotor defects, inclusion formation, axonal transport defects | Fast generation, conserved pathways |
[99,100,101,102,103,104,105,106,107,108] |
| TDP-43, SOD1, DPRs | Knockdown; human transgene expression | Caenorhabditis elegans | Locomotor defects, neurodegeneration, cytosolic aggregates, axonal abnormalities | Genetic screens and modifier identification | [109,110,111,112,113,114] |
Table 2.
Revised El Escorial criteria [135].
Table 2.
Revised El Escorial criteria [135].
| DIAGNOSIS | CRITERIA |
|---|---|
| Definite ALS | Presence of both UMN and LMN signs in 3 anatomical regions. |
| Probable ALS | Presence of both UMN and LMN signs in ≥2 anatomical regions, with UMN signs observed rostral to LMN signs, is suggestive of ALS. |
| Probable ALS, supported by laboratory results | Presence of both UMN and LMN signs in 1 region, with EMG evidence of LMN involvement in at least 1 other region. |
| Possible ALS | Presence of UMN and LMN signs in a single region, or UMN signs in 2 or 3 regions without LMN involvement. |
| Anatomical regions used for disease stratification: Bulbar, cervical, thoracic, lumbar. ALS, Amyotrophic lateral sclerosis; UMN, upper motor neuron; LMN, lower motor neuron. | |
Table 3.
Therapeutic agents currently under evaluation for the treatment of ALS through clinical trials.
Table 3.
Therapeutic agents currently under evaluation for the treatment of ALS through clinical trials.
| NCT NUMBER | THERAPY | MECHANISM/ TARGET | PHASE | STATUS | TYPE | REFERENCES |
|---|---|---|---|---|---|---|
| NCT05903690 | RAG-17 | siRNA targeting the SOD1 gene | Early Phase I | Completed | Interventional | [150] |
| NCT06849609 | VGN-R13 | AAV vector | Early Phase I | Recruiting | Interventional | N/A |
| NCT06645197 | SNUG01 | AAV vector | Early Phase I | Not yet Recruiting | Interventional | N/A |
| NCT06665165 | AMX0114 | AMX0114 ASO | Phase I | Recruiting | Interventional | N/A |
| NCT06307301 | Abatacept & IL-2 | Reduction of Treg numbers & suppression | Phase I | Active, not recruiting | Interventional | N/A |
| NCT05695521 | CK0803 | Allogeneic Tregs (cord blood) | Phase I | Active, not recruiting | Interventional | [151,152,153] |
| NCT04948645 | Fosigotifator | ISR modulator (eIF2B activator) | Phase I | Active, not recruiting | Interventional | N/A |
| NCT05633459 | QRL-201 | STMN2 ASO | Phase I | Recruiting | Interventional | N/A |
| NCT03626012 | BIIB-078 | C9orf72 ASO | Phase I | Completed | Interventional | N/A |
| NCT04494256 | BIIB-105 | ATXN2 ASO (TDP-43) | Phase I/II | Terminated | Interventional | [154] |
| NCT02238626 | MN-166 | Anti-inflammatory (TLR4 inhibitor) | Phase II | Completed | Interventional | N/A |
| NCT03456882 | RNS60 | Mitochondrial metabolic enhancer | Phase II | Completed | Interventional | N/A |
| NCT04569435 | ANX005 | Anti-C1q monoclonal antibody | Phase II | Completed | Interventional | N/A |
| NCT05039099 | AP-101 | Anti-misfolded SOD1 antibody | Phase II | Active, not recruiting | Interventional | N/A |
| NCT03359538 | Rapamycin | Autophagy enhancers | Phase II | Completed | Interventional | [155] |
| NCT05981040 | ZYIL1 (Usnoflast) | NLRP3 inhibitor | Phase II | Completed | Interventional | [156] |
| NCT04615923 | Pridopidine | Sigma-1 receptor agonist | Phase II/III | Completed | Interventional | N/A |
| NCT04297683 | DNL343 | ISR modulator | Phase II/III | Active, not recruiting | Interventional | N/A |
| NCT04220190 | RAPA-501 | Regulatory T-cell immunotherapy | Phase II/III | Recruiting | Interventional | N/A |
| NCT04414345 | CNM-Au8 | Nascent nanocrystal therapy | Phase II/III | Completed | Interventional | N/A |
| NCT02588677 | Masitinib in combination with riluzole | Tyrosine kinase inhibitor | Phase II/III | Completed | Interventional | [157] |
| NCT04856982 | Tofersen | SOD1 ASO | Phase III | Active, not recruiting | Interventional | [158] |
| NCT04944784 | Reldesemtiv | Troponin activator | Phase III | Terminated | Interventional | N/A |
| NCT03280056 | NurOwn | MSC-NTF cell therapy | Phase III | Completed | Interventional | [159] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



