Submitted:
31 July 2025
Posted:
01 August 2025
You are already at the latest version
Abstract
Obesity is a complex, heterogeneous, chronic and progressive disease, which correlates with an augmented risk to develop several comorbidities with an enhanced risk of death. In addition to metabolic and cardiovascular diseases, obesity is related to depressive disorders and neurodegenerative diseases, in which gut microbiota dysbiosis plays a central role in triggering the meta-neuroinflammation through the gut-brain axis. In obese patients, the release of inflammatory mediators by dysfunctional adipocytes and the oxidative stress induces meta-neuroinflammation, which have been identified as the main physio-pathological pathway underlying chronic pain syndromes, particularly osteoar-thritis, low back pain, fibromyalgia, headache, and painful diabetic peripheral neu-ropathy. Both the peripheral and the central nervous system are involved in neuroin-flammatory processes, leading to central sensitization and pain chronification. Me-ta-neuroinflammation is also a potential peripheral target of treatment in degenerative joint disease, in order to minimize the traditional pharmacological approaches use. The ultramicronized palmitoylethanolamide is able to control the body weight, to exert neuroprotective, anti-neuroinflammatory and analgesic actions, and to restore intestinal eubiosis, with beneficial effects on mental disorders via the gut-brain axis. Finally, adelmidrol, as a PEA congener, available for intra-articular injection associated with hyaluronic acid, has been shown to modulate meta-neuroinflammation in knee osteo-arthritis.
Keywords:
obesity
; chronic pain
; meta-neuroinflammation
; low back pain
; osteoarthritis
; oxidative stress
; palmitoylethanolamide
; gut microbiota
; gut dysbiosis
; cognitive impairment
1. Epidemiology of Obesity and Chronic Pain Syndromes
1.1. Obesity and Its Comorbidities
Obesity is a complex, heterogeneous, chronic, and progressive disease, which substantially affects more than 890 million (13%) adults [1]. According to the World Health Organization (WHO), the diagnosis of overweight is made by measuring a Body Mass Index (BMI) greater than or equal to 25 kg/m2 and obesity by a BMI greater than or equal to 30 kg/m2. Central obesity is defined by a waist circumference greater than 102 cm in men and 88 cm in women. In 2022, 2.5 billion adults worldwide were overweight and about 16% of population were obese [2].
Worldwide, obesity is a major public health problem associated with increased morbidity and mortality for all-causes [3]. Although obesity is recognized as a high-risk condition for the development of other chronic degenerative non-communicable diseases, this pathology has been declared as a disease per se that leads to a reduction in the quality and expectancy of life [4]. In obesity patients, the adipocyte hypertrophy, visceral and ectopic adiposity, increased production of adipokines with anorexigenic function, such as leptin, and of a plethora of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL) -6, IL-1β, and resistin, predisposes the organism to the adiposopathy (or “sick fat”) [5]. The latter is defined as a “pathologic adipose tissue (AT) anatomic/functional disturbances induced by positive caloric balance in genetically susceptible individuals that results in adverse endocrine and immune responses that may cause or worsen metabolic dysfunction” [3]. This alteration is also sustained by dysfunctional adipocytes, resulting in an imbalance between the production of pro-inflammatory and anti-inflammatory cytokines, in favor of the first. This condition predisposes the body to the chronic low-grade inflammation or meta-inflammation, which is observed in all tissues involved in energy homeostasis [6]. The low-grade inflammation is also sustained by infiltration of bone marrow-derived immune cells that signal via the production of cytokines and chemokines. Despite its low-grade nature, meta-inflammation negatively impacts remote organ function, a phenomenon that is considered causative of the complications of obesity. The visceral and ectopic fat, either in the liver, muscle or heart, can increase the risk of developing insulin resistance, type 2 diabetes mellitus (DM), and cardiovascular diseases (CVDs) [7]. In fact, the obesity correlates with an augmented risk of several comorbidities that exponentially increase the risk of death. In this regard, the risk of all-cause mortality increases as the number of years lived with obesity increases, regardless of the current BMI [8]. Notably, two-thirds of obesity-related mortality is attributable to CVDs, including atherosclerotic disease, heart failure, thromboembolic disease, arrhythmias, and sudden cardiac death. In fact, for every additional 2 years lived with obesity, the risk of cardio-vascular mortality rises significantly by 7% [8]. CVDs are followed by type 2 DM, cancer (especially esophagus, colon, rectum and liver), and chronic kidney disease (CKD) [9,10]. Obesity contributes to the development of CKD among 15–30% of patients, though direct and indirect mechanisms [11]. Among the direct mechanisms, the altered secretion of adipokines and the lipotoxicity lead to the accumulation of perirenal AT and of fatty acids in the renal parenchyma. The result is a tubule-interstitial damage, that involves either the proximal tubular epithelial cells and endothelial cells [12]. The hemodynamic changes, such as glomerular hyperfiltration and microvascular stretching, and the hyperactivation of renin-angiotensin-aldosterone system are the other two direct mechanisms contributing to inflammation, oxidative stress and fibrosis [13,14]. This triad is exacerbated if the patient presents type 2 DM, arterial hypertension and atherosclerosis, namely comorbidities and indirect mechanisms of obesity itself, that are responsible for the development and progression of CKD [15].
Obesity is also related to structural and functional abnormalities that reduced patients quality of life (QoL). These include gastrointestinal reflux disease, gallbladder disease, osteoarthritis (OA), obstructive sleep apnea/obesity hypoventilation syndrome, psychological and eating behavior disorders, anxiety and depression and impairment of the physical performance [16]. In fact, obese patients often have impaired cognitive functioning and present a major depressive disorder, with negative effects on measures of processing speed and executive function, as evidenced by mood assessment questionnaires and neuropsychological tests [17]. Furthermore, obesity appears to be associated with greater emotional dysregulation, compared to normal-weight condition. In fact, obesity shares many psychological features with eating disorders, especially with binge eating disorder and alexithymia [18].
1.2. Obesity and Chronic Pain
Chronic pain is a widespread health issue, which affects over 20% of adult population [19]. In obese patients, chronic pain syndromes are among the most common observed comorbidities, with their relevant psychosocial consequences. These two phenomena are closely related, with each condition adversely impacting the other, because of limited mobility, mood disorders, and common chemical mediators [20].
Numerous studies investigated the relationship between OA-related pain and obesity, which is the focus of this review. Among older people, long-term obesity has been identified as a significant predictor of pain, particularly with an increased risk of back, hip, and knee pain [21], which are the most common sites of OA. Among OA-related pain syndromes, low back pain (LBP) has the highest prevalence in general population, affecting over 600 million people globally. LBP is supposed to be a major determinant of age-standardized disability-adjusted life-years (DALYs) in the next 25 years, while improvement of BMI has been proposed as one of the strategies for improving life expectancy [22]. Overweight and obesity have been recognized as risk factors for LBP. Obesity increased the incidence of LBP, with an odd ratio of 1.36 and 1.4, respectively in men and women [23]. In a recent cohort study conducted in Norway, higher values of BMI have been associated with higher incidence of LBP, particularly among very obese women [24], however the exact magnitude of this phenomenon is still under investigation.
Advancing age and adiposity also contribute to musculoskeletal degenerative diseases, which lead to sarcopenic obesity (SO), a condition that links osteopenia/osteoporosis, muscle loss, and obesity [25]. SO is associated with reduced physical activity, loss of independence among older adults and is a determinant risk factor for frailty [26]. In postmenopausal women, SO had greater effect on knee OA compared to obesity without sarcopenia and to sarcopenia without obesity [27].
Obesity has been also associated also with other pain syndromes, such as painful diabetic peripheral neuropathy [28], headache [29], and fibromyalgia [30].
Diabetic neuropathy is the most common complication of DM, affecting about 50% of diabetes patients and about 70% of patients with diabetic neuropathies receive medications for neuropathic pain [31]. Visceral obesity is an independent risk factor for diabetic peripheral neuropathy [32].
Different studies investigated obesity as a risk factor for headache/migraine. Although migraine frequency was not associated with BMI, obese patients seem to have an higher prevalence of frequent and severe migraine headaches [33]. The exact relationship between these two comorbidities is not fully understood, however, there is evidence that obesity could be a consequence of migraine, through the effect of inflammatory mediators, adipokines, and alterations of gut microbiota [34].
Finally, 35% of adults with fibromyalgia are obese and obesity impacts most of the clinical features of fibromyalgia, such as tenderness and stiffness, fatigue, physical functioning, sleep, and cognitive function, leading to a reduced QoL [35]. Among women suffering from fibromyalgia, obese patients displayed higher levels of anxiety and depression, compared with the normal-weight subjects [36]. Even in this case, it is not possible to understand whether obesity is a cause or a consequence of fibromyalgia [30].
2. Mechanisms Underlying Obesity and Osteoarthritis: Role of Oxidative Stress and Meta-Neuroinflammation
2.1. Obesity and Neuroinflammation
AT is an endocrine organ distributed throughout the body and is characterized by high metabolic and dynamic activity [37]. AT regulates several physiological mechanisms through the secretion of adipocytokines (also called adipokines) into the bloodstream, creating a communication with other tissues and organs [38]. In adult mammals, AT is classified in two different types: white and beige. White AT (WAT) accounts for the largest percentage of AT in the human body and is localized around the viscera, subcutis, and perivascular. WAT stores excess energy in the form of triglycerides and secretes adipokines and vasoactive factors. Its phenotype changes in patients affected by obesity, becoming hyperplasic and hypertrophic, suffering the infiltration of the immune cells and secreting vasoconstrictor factors. Beige AT (BAT) mainly surrounds the thoracic aorta. It possesses anti-inflammatory and cardioprotective properties and is involved in the thermogenesis, dissipating energy as heat. For this reason, BAT has anti-obesogenic and anti-diabetic properties, ensuring cardio-metabolic health [39]. Lean individuals with normo-metabolic function present an increased production of anti-inflammatory ILs (like IL-10, IL-5, IL-4, IL-13, IL-25, IL-33) and anti-inflammatory adipokines (such as adiponectin, omentin, apelin and secreted frizzled-related protein-Sfrp-5) [40]. Moreover, in a healthy AT, macrophages constitute 5-10% of the cells, of which only a small part is in a pro-inflammatory state (M1), because the remainder of the resident macrophages is in an anti-inflammatory state (M2) [41]. During chronic low-grade inflammation, which characterizes patients with obesity, AT is mainly characterized by hypertrophic adipocytes that accumulate lipid droplets, secrete pro-inflammatory adipokines (like leptin, resistin, and visfatin), and amplify the infiltration into AT itself of pro-inflammatory cells (such as M1 macrophages, T helper 1 cells, natural killer cells, CD8+ T cells, neutrophils, and mast cells).
The dysbiosis of the gut microbiota also plays a central role in exacerbating AT inflammation, negatively impacting on distant organ function, such as the brain. In fact, despite its low-grade nature, chronic inflammation is the leitmotif that links obesity with neuroinflammation. The central nervous system (CNS) requires a highly controlled microenvironment to support its physiological functioning. This is possible thanks to the presence of three biological barrier at the blood-brain interface, that effectively separates the brain from the rest of the body [42]. These include the blood–brain barrier (BBB), the blood–cerebrospinal fluid barrier and the arachnoid barrier [43]. Brain microvascular endothelial cells display distinctive morphological, structural and functional characteristics that differentiate them from other vascular endothelia. In detail, these cells express: (i) the tight junctions (TJs), namely intercellular protein complexes that preserve tissue homeostasis and integrity through the control of paracellular pathways between adjacent endothelial cells, thus preventing the unregulated passage of polar molecules between the blood and the brain [44]; (ii) the absence of fenestrations [45]; (iii) the lack of pinocytic vesicles [46]; (iv) the active transport mechanisms, specifically expressed by the endothelial cells of the cerebral capillaries. These mechanisms ensure the transport of nutrients and essential amino acids into the CNS and the blockade of endogenous and xenobiotics molecules, that could be harmful to the ideal milieu for neural transmission [47]. For this reason, the BBB is an anatomo-functional structure that protects the CNS from systemic circulation, not allowing the pro-inflammatory factors, toxins, immune cells and pathogens to be translocated into the brain [48]. The integrity of the BBB is compromised in patients with obesity. Moreover, obese patients have as a comorbidity type 2 DM that downregulates the TJs proteins, leading to the destruction of claudin-5, zonula occludens-1 (ZO-1), occludin and caveolin [49]. The upregulation of the matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs) is observed in obese subjects, and it is associated with some obesity-related parameters, including BMI, waist circumference, blood pressure and endothelial-dependent response [50]. In addition, hyperglycemia leads to the amplification of oxidative respiration and the production of reactive oxygen species (ROS). ROS react with nitric oxide to produce peroxynitrite, which mediates MMPs activation and TIMPs inhibition, causing basement membrane (BM) degradation [51] (namely the second barrier of entry for the immune system) [52]. The BM regeneration is unable to compensate the protease activity of the MMPs. In fact, the increase in fibronectin, collagen IV and laminin compromises the attachment of cells to the BM and the downregulation of heparin-sulfated proteoglycans removes anionic protein binding sites, destabilizing the BM [53] and allowing the extravasation of leukocytes. Leukocytes express highly glycosylated molecules on their surface, namely the P-selectin glycoprotein ligand-1 (PSGL-1), consenting selectins adhesion receptors to bind them and triggering the neuroinflammation response through the microglia activation. The interaction between PSGL-1 and P-selectin and E-selectin mediate the initial capture and the rolling of leukocytes on the vascular endothelium in search of a point for extravasation, which can occur by paracellular and transcellular diapedesis. Most transmigration into the perivascular space occurs via a paracellular mechanism. The immune cells extend pseudopods and pass through the endothelium, thanks to the interaction with platelet endothelial cell adhesion molecule (PECAM) and junctional adhesion molecule-A (JAM-A). When leucocytes cannot find an endothelial junction, transcellular diapedesis occurs [54,55]. The leukocyte extravasation into the brain parenchyma is also permitted by MMP-9, which remove away BM filaments [56]. The impaired BM also becomes thicker, leading to increased vascular permeability [57]. This process is favored by the activation of protein kinase C (PKC), advanced glycation end-products (AGEs), transforming growth factor -β (TGF-β) and connective tissue growth factor [52]. AGEs act on AGE receptor (RAGE) to intensify nuclear factor kappa β activation (NF-κβ), increasing pro-inflammatory gene expression, including RAGE itself and pro-inflammatory cytokines, like TNF-α [58]. Astrocyte endfeet wrap around the entire CNS vascular tree and perform important functions in regulating the BBB, through the cerebral blood flow, nutrient uptake, and waste elimination [59]. During neuroinflammation, astrocytes produce and secrete a wide range of molecules and chemokines to attract circulating peripheral immune cells, including CD8+ T cells, B cells, NK cells, monocytes and macrophages into the CNS [60]. Conversely, astrocytes can boost effector functions of peripheral immune cells, including NK cells and CD8+ T cells, through the production of IL-15. TH 17 cells promote pathogenic activities of astrocytes by expressing receptor activator of nuclear factor-kappa β (RANK) ligand and granulocyte-macrophage colony-stimulating factor (GM-CSF). The RANK activation by TH 17 cell–expressed RANK ligand triggers the production of C-C motif chemokine ligand 20 - CCL20, triggering the recruitment of effector T cells in the CNS. In astrocytes, GM-CSF induces the expression of pro-inflammatory genes [61], creating a cytotoxic state mediated by the production of IL-1β, IL-6, TNF-α, prostaglandins and vascular endothelial growth factor (VEGF) that migrate into the perivascular space through the destroyed BM [52]. Chronic overexposure of VEGF also increases the expression of intercellular adhesion molecule-1 (ICAM-1) and major histocompatibility complex (MHC) class I and II expression, modulating immune responses in the CNS through opening of the BBB and allowing contacts between CNS antigens and blood-borne immune mediators [62]. Activated microglia migrates to the injured area and release proinflammatory cytokines, NO, ROS, prostaglandins, and chemokines, resulting in the additional chemoattraction of circulating leukocytes [63]. Moreover, leptin leads to the activation of the mechanistic target of rapamycin (mTOR) and hypoxia-inducible factor 1 (HIF-1) in the endothelial cells of the CNS, leading to VEGF production. VEGF activates PKC-β and Rho-kinase (ROCK), exacerbating neuroinflammation. Activation of PKC-β is also due to increased diacylglycerol concentrations, typically observed in hyperglycemic conditions [64,65]. This pathway increases the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, producing O2-. O2- mediates the phosphorylation of the inhibitor of kappa β kinase (IKK) and induces downstream degradation of Iκβα, leading to the nuclear localization and transcriptional activation of NF-κβ [66]. The activation of NF-κβ regulates the synthesis, the release and the recruitment of various inflammatory mediators capable of disrupting the BBB through the reorganization of the actin cytoskeleton in brain microvascular endothelial cells, the disruption of TJs formation, the inhibition of the proliferative and migratory capacity of endothelial cells and astrocytes, and the apoptosis [67]. On the other hand, ROCK-mediated cellular pathway inhibits the expression of endothelial nitric oxide synthase (eNOS), which reduces the availability of NO, inducing endothelial dysfunction [68]. This is due to increased endothelial Na2+ channels (EnNaCs) activity, leading to polymerization of cortical actin fibers. Subsequently, it is possible to observe a reduction of eNOS activity and a decrease of nitric oxide (NO) production, resulting in increased vascular stiffness [39]. Mediators of obesity-induced endothelial dysfunction also include altered sirtuin 1 expression, oxidative stress, autophagy machinery and endoplasmic reticulum stress [68]. Inactivation of eNOS causes the activation of microglia, promoting a pro-inflammatory phenotype in the brain, downregulating the claudin-5 and occludin and increasing the BBB permeability caused by VEGF signaling from astrocytes [69]. Moreover, eNOS-deficient mice exhibit impaired cognitive performance, suggesting that loss of endothelial NO has a detrimental effect on the functions of neuronal cells [70]. At the same time, an increased NO production in the CNS is associated with the pathogenesis of neurodegenerative diseases, such as Parkinson’s disease, and Alzheimer’s disease (AD) [71]. In fact, the pathological manifestations of AD include not only the accumulation of amyloidbeta-protein (Aβ) and hyperphosphorylated tau (pTau) in the brain, but also microgliosis, astrocytosis, and neurodegeneration mediated by metabolic dysregulation and neuroinflammation [72]. Aβ also increases ROCK-1 activity in neurons [73] and, in turn, ROCK-1 enhances cleavage of the amyloid precursor protein (APP), producing increased Aβ formation [74]. Neuroinflammation is characterized by a dysfunction of the influx transporters that impedes the supply of glucose and nutrient molecules, leading to hypometabolism that is detrimental to neuronal activity. Downregulation or decreased activity of efflux transporters fails to eliminate neurotoxic molecules, particularly Aβ, aggravating Aβ deposition in the parenchyma and brain [75]. Moreover, the downregulation of P-glycoprotein, amino acids and leptin transporters is observed [52]. In individuals with obesity, decreased leptin signalling in the CNS is also caused by downregulation of the leptin receptor (LepR) and by deficiency of leptin signalling at the LepR [76]. Leptin resistance is characterized by reduced satiety, over-consumption of foods, and increased total fat mass [77]. The leptin plasma levels may be protective against the development or progression of AD. Lower leptin plasma levels have a significant association with higher Aβ deposition in the brain, although there is not a significant association between leptin plasma levels and tau deposition [78]. High leptin levels were independently associated with a reduced risk of cognitive decline in elderly Italians [79]. In a cohort of older adults with mild cognitive impairment, plasma leptin levels were not associated with cognitive function, nor did they predict risk of dementia. Although leptin levels increase in obesity, leptin resistance prevents to leptin to exert its functions [80]. N-acetylaspartate (NAA) is synthesized by neurons and is involved in neuronal metabolism and axonal myelination. The concentration of NAA has been associated with cognitive dysfunction in neurodegenerative and metabolic diseases. NAA levels depend on age, BMI, and glucose levels [81]. In the study conducted by Coplan et al., higher BMI (≥ 25 kg/m2) has been associated with decreased levels of neural integrity and with neuronal injury, expressed by reduced concentrations of NAA in the hippocampus [82]. Irisin is an adipo-myokine hormone produced during physical exercise, through the expression of the peroxisome proliferative activated receptor-γ coactivator-1 α (PGC-1α). Expressed PGC-1α causes the production of the fibronectin type III domain containing 5 (FNDC5) protein, which is cleaved in skeletal muscle fibers by proteases to produce irisin [83]. Irisin binds to its integrin αV/β5 receptor with these consequences i) WAT browning; ii) improving of insulin sensitivity and metabolic balance, by enhancing mitochondrial functions and by reducing oxidative stress; iii) promoting osteogenesis and mitigating the bone loss; iv) attenuating the cognitive dysfunction, by decreasing Aβ toxicity, neuroinflammation, and oxidative stress and by improving brain-derived neurotrophic factor (BDNF) signaling, which rescues cognition and synaptic health; v) regulating dopamine pathways, alleviating neuropsychiatric symptoms like depression and apathy; and vi) mitigating cardiac injury [84,85]. The levels of irisin are significantly lower in patients with obesity, osteoporosis, sarcopenia, AD, and CVDs [85]. The dysfunctional phenotype caused by low levels of irisin is exacerbated in patients with SO. In fact, SO compromises mitochondrial oxidative capacity and lipid oxidation in skeletal muscle and suppresses sarcolipin-induced sarcoplasmic reticulum calcium ATPase (SERCA) activation, impairing the ability to switch between glucose and lipid metabolism in response to nutrients and physical exercise and resulting in reduced oxidative capacity, diminished energy expenditure, and increased adiposity [86]. SO patients display a smaller total gray matter volume [87] and show higher serum levels of IL-6, IL-18, TNF-α, TNF-like weak inducer of apoptosis (TWEAK) and leptin compared to non-sarcopenic patients; in contrast, the levels of insulin growth factor 1, insulin, and adiponectin are significantly lower [88]. For these reasons, Irisin may represent a therapeutic potential biomarker for metabolic diseases, osteoporosis, sarcopenia, and neurodegenerative diseases [84].
2.2. Obesity, Oxidative Stress and Osteoarthritis
In obese patients, many pro-inflammatory molecules and mediators are express during acute and chronic high-intensity loading [89], namely extracellular matrix (ECM) components and remodelers, joint cell- and AT cell-derived mediators (cytokines, adipokines), AGEs, and ROS [90]. Besides predisposing to OA for loading mio-mechanical reasons, obesity increases the risk of OA development also in non-weight-bearing joints, such as hands, through the activity of systemic inflammatory mediators, such as adipokines, free fatty acids (FFAs), and ROS released by dysfunctional abdominal AT [91]. End-stage OA patients showed increased levels of ROS and decreased antioxidant capacity [92]. Many of these inflammatory mediators have been linked to cartilage matrix synthesis and degradation, and synovial tissue inflammation. For instance, leptin is a 16-kd polypeptide hormone secreted by adipocytes, regulating adipose mass and body weight. Impaired leptin signalling was related to protection against obesity-induced OA, even in absence of elevated body fat, thus corroborating the pro-inflammatory effects of leptin in OA [93]. Leptin secretion from the infrapatellar fat induces MMPs 1 and 13 gene expression in OA chondrocytes [94]. Recent literature supported the hypothesis that underlying mechanisms for OA go further than mechanical load and stress, since a correlation with CVDs, metabolic syndrome (MetS), and especially factors such as arterial hypertension, DM, dyslipidemia, has been observed [91], with a positive correlation with radiological OA severity [92]. In animal studies, omega-3 polyunsaturated fatty acids reduced the expression of inflammatory markers, cartilage degradation and oxidative stress in chondrocytes, while the opposite was true after omega-6 polyunsaturated fatty acid and saturated fatty acid administration, both in animals and in humans; hence, omega-3 polyunsaturated fatty acid supplementation may have a beneficial effect on pain and functionality through a reduction in structural damage [95].
2.3. Osteoarthritis
2.3.1. Osteoarthritis and Neuroinflammation
Chronic painful conditions, including OA, are accompanied and sustained by inflammatory responses in peripheral tissues, e.g. joints, as well as in the peripheral and central nervous system: such phenomena are described as “neuroinflammation”, and they rely on a bidirectional signalling between nervous structures and cells and the peripheral damaged tissues. Animal models of OA showed that joint neurons, especially high-threshold C and Aδ afferents, undergo plastic changes [96] and develop an important sensitization, hence mechanical stimuli are perceived as painful in behavioural tests [97,98]. For instance, after induction of knee OA via intra-articular monosodium iodoacetate (MIA) injection, destabilization of the medial meniscus (DMM), or partial meniscectomy, animals display mechanical hyperalgesia, with pain evoked by simple gentle pressure or by normal-range joint mobilization [99]. Nociceptors that are initially considered as responsive to other stimuli, namely cold, heat, or chemicals, and silent to mechanical stimuli, become mechanosensitive too, taking on a polymodal phenotype [100]. Such peripheral findings are correlated with a higher response to the same stimuli in dorsal root ganglia (DRG) neurons, where nociceptors are activated via paracrine mechanisms [101]. Many mediators derived from degradation of inflamed cartilage are implicated in the sensitization and excitation of DRG neurons [102]. Under physiological conditions, several cytotypes in the joints, namely chondrocytes, fibroblast-like synoviocytes (FLS), synovial macrophages, and mast cells (MCs) are represented in a quiet state, as “sentinels” against pathogens and possible injuries [103,104]. When joint inflammation occurs, the disruption of the ECM allows for the release of damage-associated molecular patterns to the joint cavity, and consequent activation of pattern recognition receptors, namely Toll-like receptors (TLRs), on such sentinel cells. These phenomena lead to the production and release of inflammatory molecules, catabolic factors, and the activation of the complement cascade [105]. Particularly, MCs secrete granules containing proinflammatory substances, such as histamine, proteinases (tryptases and chymases) [106], as well as chemokines and cytokines (TNF-α, IL-1β, IL-6, IL-8, CCL2, PG2, VEGF, and others), thus leading to vasodilatation, angiogenesis, as well as recruitment of other inflammatory cells from the bloodstream [107]. MCs also produce neurotrophin nerve growth factor (NGF) [108] which, after being secreted by MCs, binds to neuroptrophin p75 and tropomyosin-related kinase (TRK)-A receptors on several inflammatory cells, including other MCs, promoting their degranulation. The increase of NGF has been correlated with sprouting of pain fibres in vitro [109], microglia activation (e.g. “microgliosis”) in the dorsal horn [110], and other structural changes leading to neuroinflammation and pain chronification [111]. In the wake of this, clinical trials with humanized neutralizing monoclonal antibodies against NGF are ongoing for OA patients, as they have already been reported to have analgesic effects in animal models, both as prophylaxis and in therapeutic protocols. Accordingly, TRK-A inhibition showed analgesic results in animals [112,113]. Other immune cells, including circulating macrophages, infiltrate the DRG in OA rodent models [114], especially after HFDs administration, possibly hinting at a correlation between the latter and chronic pain [115]. Macrophage infiltration in the DRG in OA animals was positively correlated with persistence of pain [116]. Most of recent preclinical research has focused on the role of glial cells, such as microglia, in the DRG and the dorsal horn, which are activated during neuroinflammatory processes [117,118]. Microglia is actually the resident macrophages of the CNS: its activation in the DRG occurs in animal models after OA induction, with different timing based on the OA inducer: for instance, microglia activation is detectable as soon as one week after MIA injection, while it takes DMM models 8 to 16 weeks to display such activation [110]. These findings may correlate with the difficulty of treating OA in late stages, where drugs targeting the CNS are often necessary, such as opioids, while others, namely nonsteroidal anti-inflammatory drugs, are insufficient for pain control [100]. Microglial cells produce cytokines, such as TNFα, IL-1, and NGF, and release other molecules, namely substance P, which may further activate receptors expressed by macrophages in a paracrine manner, and sustain a shift in their phenotype to a M1 proinflammatory one [119]. Microglia activation seems to be accompanied by overexpression of stress markers in the DRG, and with allodynia; the latter is reversible via glia inhibition through monicycline and fluorocitrate [120]. For example, NF-κB/p65 is overexpressed in astrocytes in the dorsal horn in rat MIA OA model, alongside cytokines like IL-1β, TNF-α and IL-33; moreover, astrocytes themselves are heightened in number and mechanical hyperalgesia is advisable, albeit reversible through spinal inhibition of NF-κB/p65 [121]. A predominant role for NF-κB was also confirmed recently by Sun et al., who showed that inhibition of bromodomain-containing protein 4 (Brd4), a bromodomain and extra-terminal epigenetic reader protein that usually promotes gene transcription, attenuated MIA-induced pain behaviours in rats with OA through a reduced activation of inflammatory genes. Brd4 inhibition also promoted antioxidant responses in the DRG and in the spinal cord [122]. Oxidative stress has been linked to various inflammatory and degenerative conditions of the CNS [123] and may be a contributing factor for arthritic pain: overexpression of anti-inflammatory molecules, namely sestrin2 (Sesn2) [124], and inhibition of pro-inflammatory ones, such as GSK-3β [125,126], reduced ROS and cytokines levels in the spinal cord in MIA- and complete Freund’s adjuvant (CFA)-induced OA, respectively, with analgesic effects on OA pain. Interestingly, OA symptom burden seems to be more severe in women than men in clinical practice [127,128], probably due to higher inflammatory responses in female than males [129,130], or, possibly, to differences in neuroimmune signalling. Kosek et al. showed that female subjects scheduled for total knee replacement displayed higher levels of IL-6 and IL-8 in synovial fluid (SF) and cartilage, correlated with more severe pain and higher pain sensitivity. Moreover, when considering the relative presence of different cytokines in the CSF, serum, SF, and cartilage, discrepancies between men and women were found, suggesting sex-related peculiarities in the cross-talk among peripheral damaged tissue and the CNS. Such communications rely, among other factors, on the presence of monocyte chemoattractant protein 1 (MCP1), which is responsible for increased BBB permeability, as well as spinal infiltration of monocytes and their eventual differentiation into activated microglial cells. Besides being produced by monocytes/macrophages, synovial fibroblasts, and chondrocytes, MCP1 is also detectable in nociceptive afferent fibers in peripheral tissues with possible implication in neuropathic manifestation, especially mechanical allodynia in animal models. Interestingly, higher concentrations of MCP1 and IL-8 in the CSF were correlated with lower pain levels, whereas their presence in joints led to higher pain sensitivity. Hence, presence of proinflammatory markers in the CNS may be part of a compensatory response against peripheral disease, such as OA [131]. This may be corroborated by the higher levels of both myo-inositol, a glial-derived neuroinflammation marker, and choline, a cell membrane metabolism and cellular turnover marker, in patients with knee OA, with positive correlation with pain, stiffness, and disability scores. On the other hand, concentrations of NAA, a marker for neuronal integrity, were reduced compared to healthy controls, but returned to normal-range levels after total knee arthroplasty, thus suggesting that surgical treatment may have a role in counteracting the maladaptive mechanisms leading to pain sensitization and chronification in subjects with OA. More specifically, since NAA is produced in oligodendrocytic and neuronal mitochondria, the restoration of its physiological levels after surgery may be the consequence of ameliorated mitochondrial function [132], further confirming the role of oxidative processes in neuroinflammation. Age, too, is correlated with higher levels of inflammatory markers in the DRG and the spinal cord, as well as peripherally, e.g. the sciatic nerve, in mice with MIA-induced OA: in particular, overexpression of CD68, CD11b, activating transcription factor 3, and TNF as neuroinflammatory markers were higher in old animals, and they were counteracted by subcutaneous morphine administration 7 days after OA induction [133]. Opioids are still pivotal in chronic pain management; however, they are burdened by important adverse effects, and tolerance may develop after long-term administration [134]. Research has focused on possible mechanisms and molecules involved in opioid tolerance, in order to counteract, or even prevent it. For instance, sigma-1 (σ1) receptors are expressed in CNS areas implicated in pain perception, namely DRG, dorsal horns, and periaqueductal grey: given their cross-talk with the opioid system, their antagonism allowed for prevention of both pain sensitization and opioid tolerance in MIA-induced OA mice, hinting at a possible role for σ1 receptors antagonists as analgesics [135].
A tight relationship between joint disease and CNS pathology is conveyed by the effect of collagen-derived AGEs on neuronal structure and functionality. AGEs derive from glycation of the ECM (ECMGC): this phenomenon is widely described in osteoarthritic joints, and recent data points out that it may hinder neuronal cell attachment and neurite formation, and elicit neuronal excitation, neuropeptide and neurotransmitters release, and eventually peripheral sensitization. Such effects are counteracted by morphine administration, thus suggesting that ECMGC may represent a new target for chronic pain treatment [136]. Moreover, a connection between peripheral damaged tissues and the CNS is suggested by the elevated levels of chemokines prokineticin (PK)-1 and PK-2, which act via interaction with two receptors, namely PK receptor (PKR)-1 and PKR2. This system was found to be upregulated in OA mice 28 days after MIA injection, with synovial fibroblasts and macrophages producing such chemokines, alongside elevated levels of several cytokines in the spinal cord. Concurrent development of allodynia, motor deficits, and fatigue was assessed, as well as mood disorders, particularly anxiety and depression. In fact, PK upregulation also occurred in brain areas that are typically related to mood control, such as the prefrontal cortex and the hippocampus. Administration of PK antagonist PC1 reverted such findings, possibly by both anti-inflammatory effects in damaged joints and in brain regions [137]. Similarly, heightened levels of proinflammatory cytokines and an anxio-depressive state were found in MIA-driven OA mice, with morphine administration contrasting both phenomena [138].
Preclinical research also pointed out that pain alters genetic expression [139], and genes expressed through early to chronic stages of pain are different [140]. A role may be played by long non-coding RNAs (lncRNAs), which act at transcriptional and post-transcriptional levels, via interaction with the DNA and regulation of mRNA translation into proteins, up to epigenetic modifications. lncRNAs play a role in neuronal functionality, immune responses, and inflammatory diseases, such as OA: in fact, different lncRNAs may be up- or downregulated, with modulating effects on inflammatory and apoptotic signalling pathways, and consequent impact on chondrocyte viability and functionality [141]. Among others, lncRNAs are responsible for the regulation and genetic expression of several inflammatory molecules, as is the case for member of the Krüppel-like transcription factors (KLFs) family, KLF4, which is implicated in inflammatory responses in various pathological condition, ranging from IBD, renal inflammation, pneumonia, neuroinflammatory conditions, and OA. In fact, KLF4 inhibits ECM-degrading enzymes in human chondrocytes, synovial, and meniscus cells via an anti-inflammatory effect [142]. Hence, lncRNAs may represent a novel therapeutic target for OA management.
2.3.2. Osteoarthritis and Oxidative Stress
Besides being triggered and aggravated by biomechanical trauma, OA is known to be an inflammatory chronic condition, in which the oxidative stress probably plays a pivotal role in its pathogenesis and progression, as demonstrated by recent literature, both in vitro and in vivo [143]. Basically, an imbalance between the production of ROS and antioxidant defensive mechanisms occurs, with negative impact on joint structure and related pain [144]. Since OA pain is still largely undertreated, thus infecting functionality and QoL, the reduction of oxidative processes may be a new target for its treatment [145]. The role of inflammatory mediators, such as cytokines and chemokines, has been widely assessed in OA [146,147]. Such mediators may derive from lipid peroxidation processes, which are found to be enhanced in OA synovial cells compared to rheumatoid arthritis and healthy controls [148]. Many lipid peroxidation products, such as 8-isoprostane F2α [149], malondialdehyde, 4-Hydroxy-2-nonenal [150], and so on, are in fact elevated in synovial biopsies from patients with OA compared to healthy subjects. The NO production by inducible nitric oxide synthase (NOS2) was also correlated with the pathogenesis of OA [151], and NO itself secreted by chondrocytes has been correlated with the induction of lipid peroxidation [152], as well as ECM degradation by MMPs [153] and inhibition of proteoglycans and collagen synthesis [154]. On the other hand, a growing body of evidence suggests a role for NO as an inhibitor of the NF-κB pathway [155] and a stimulator of collagen synthesis in vitro [156]. It is possible for NO to have pro-inflammatory effects only when in the form of redox derivative peroxynitrite, which is a known inducer of lipid peroxidation [157]. Mitochondria are key players in the oxidation pathways. Damaged mitochondria are continuously replaced by new ones, in order to preserve mitochondrial function: this process is known as mitochondrial biogenesis, and recent literature shows that it may be an indicator of OA [158]. Several transcription factors regulate mitochondrial renewal, namely PGC-1α [159] and nuclear respiratory factor (Nrf)-1 and Nrf-2, the latter further upregulating the expression of other factors, such as mitochondrial transcription factor A and nuclear-encoded mitochondrial proteins [160]. PGC-1α is deacetylated by NAD-dependent deacetylase sirtuin 1 (SIRT1), which is activated by AMP-activated protein kinase [161]. Nrf-2 was shown to positively modulate the expression of many endogenous antioxidants, namely NAD(P)H oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), superoxide dismutase (SOD), glutathione (GSH), and glutathione peroxidase (GPx) [162,163]. Nrf-2 was also shown to inhibit pro-inflammatory pathways, such as NF-κB [164], hence reducing levels of inflammatory cytokines, namely IL-1β and TNF-α [165]. Nrf-2 promotes macrophage differentiation to M2 [166], it modulates osteoclastogenesis [167], and it inhibits the activation of inflammatory synovial fibroblasts [168]. The latter are responsible for the production of metabolic degradation factors, such as MMPs and disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), resulting in synovial and ECM degradation [169,170]. Furtherly, mitochondrial dynamics imply their continuous mixing with each other and dividing into daughters [171], alongside with removal of damaged ones through autophagy [172]. When such mechanisms are impaired, a reduction in ATP generation occurs, as well as augmented ROS production, mtDNA mutations, and mitochondrial membrane dysfunction [173]: such phenomena are linked to a wide range of degenerative and inflammatory diseases, including OA [174]. Dysfunctional mitochondrial activity has been increasingly linked to OA onset and progression [175], as assessed in animal models [176] and human chondrocytes [158]. Nrf2 induction in models of surgically-induced OA prevented OA progression via inhibition of NLR family pyrin domain containing 3 (NLRP3) inflammasome [177], a complex playing a pivotal in triggering inflammatory responses in several pathological conditions [178]. The latter is upregulated in the synovial tissue of mice with collagen-induced arthritis [179], as well as in sensory neurons in the DRG in MIA-induced OA. Accordingly, inhibition of NLRP3 inflammasome prevented the transition from an acute to a chronic painful condition in such animal models [180]. GSH is a well-known antioxidant that plays a key role against oxidative stress, with pleiotropic effects, including activation of Nrf2 [181]. Reduced GSH and its precursor molecule, N-acetylcysteine, have demonstrated a specific role in oxidative stress resistance [182], and efficacy in reducing inflammation markers and cartilage degradation, as well as significant amelioration in pain control and functionality [183]. One of the ways to increase GSH levels is through hydrogen sulfide [184]. Administration of slow-releasing hydrogen sulfide donors phenyl isothiocyanate [185], allyl isothiocyanate [186], diallyl disulfide [187] and morpholin-4-ium 4-methoxyphenyl(morpholino) phosphinodithioate dichloromethane complex (GYY4137) [188] alleviated the mechanical allodynia, the grip strength and memory deficits, as well as the depressive-like behaviors accompanying OA through inhibition of activated microglia, downregulation of NOS2, CD11b/c, phosphatidylinositol-3-kinase-PI3K, and phosphorylated Akt- p-Akt all while maintaining high levels of antioxidant/detoxificant molecules in central regions such as the hippocampus, the amygdala, periaqueductal gray matter, and infralimbic cortex, thus demonstrating the antinociceptive potential of NOS2 inhibition [189].
3. Therapeutic Perspectives
3.1. Palmitoylethanolamide
Palmitoylethanolamide (PEA, N-hexadecanoylethanolamide) is a N-acylethanolamine (NAE), and it was first discovered in the late 1950’s in soybean, peanut oil, and egg yolk [190]; later on, it was also identified in mammalian tissues [191]. PEA is a highly lipophilic molecule, which may hinder its absorption: in fact, the micronization was found to be useful, since this process makes the original compound smaller [192,193]. After absorption, PEA is hydrolysed to ethanolamine and palmitic acid by fatty acid amide hydrolase (FAAH), which is a membrane-bound enzyme with a heterodimeric structure. It is located in the endoplasmic reticulum and it acts on multiple substrates, including N-acylamines, NAEs, and N-acyltaurines [194]. PEA is also metabolized by FAAH-2, which is localised in lipid droplets [195], and by lysosomal enzyme NAE acid amidase [196]. Such enzymes act on either endogenous [197,198,199] or exogenous [200,201] PEA, and are found in different tissues and cytotypes, ranging from the gastrointestinal tract [199] [202], to joints [203], to the brain [204,205,206]. Various attempts to bypass its presystemic metabolism were made, either using prodrugs [207,208] or PEA analogues [209,210,211,212] however, such compounds need further investigation and regulation. Since it is almost insoluble in water, the bioavailability of PEA per se is poor [213]. Data on its distribution is overall quite scarce. Nonetheless, given its lipophilic nature, the volume of distribution for PEA is high [214], and way greater than its plasma volume [214]. As shown by early studies in rats, after intraperitoneal administration PEA tends to distribute in several organs and tissues, according to the following order: adrenal > diaphragm > spleen > kidney > testis > lung > liver > heart > brain > plasma > erythrocytes [215]. When orally administrated, PEA is able to cross the BBB, albeit in small amounts [216]. PEA is now recognized as a lipid mediator, with anti-inflammatory properties [217] via interaction with several receptors and mediators. Among the latter, peroxisome proliferator-activated receptor (PPAR) is a nuclear transcription factor, with several ligands [218], including arachidonic acid and fibrates, the latter being used to treat dyslipidaemias [219]. In fact, PPAR activation leads to the transcription of genes involved in lipid metabolism [220], as well as inflammatory responses [221], with consequent enhanced expression of pro-inflammatory molecules, such as NF-κB. In fact, highly bioavailable formulations of PEA, e.g. micronized (m-PEA) and ultramicronized (um-PEA), were found to promote white-to-beige AT conversion [222], reducing fat mass; and counteracting lipid and glucose dysmetabolism [223]. Recently, comicronized PEA with rutin (m-PEA– rutin), a plant-derived polyphenol compound, well-known for its antioxidant and anti-inflammatory properties, have been investigated in a murine model of obesity-induced metabolic alterations, supporting their effect in counteracting glucose and lipid dysmetabolism associated with diabesity [223]. Clinical results against adiposopathy have been found when m-PEA– rutin and hydroxytyrosol (HTyr), a phenolic compound found in olive leaves, fruits and extra-virgin olive oil, have been added to the Mediterranean diet as a potential conservative treatment for patients with MetS. HTyr has been shown to reduce oxidative stress, and modulate inflammatory pathways [224].
3.1.1. Palmitoylethanolamide and Osteoarthritis
PEA is present in high concentrations in healthy joints and acts as a MCs modulator [225]. Physiologically, MCs, alongside macrophages, represent about 3% of resident cellular cell population and act as sentinels for possible pathogens and injuries. In arthritic joints, the number of MCs increases via maturation and proliferation of resident cells, as well as recruitment of progenitors from the blood stream, via paracrine mechanisms [107]. PEA reduces the degranulation of MCs in vitro [226] and in vivo [227].
3.1.2. Palmitoylethanolamide and Neuroinflammation
PEA is also among the many substances produced, released, and hydrolyzed by microglia [228]. The involvement of PEA in neuroinflammatory processes was thoroughly studied, and it may be mediated by its direct and indirect interaction with various receptors, namely PPAR [229], cannabinoid receptors (CB)-1 and CB2 [230], as well as non-CB1 and non-CB2 [231], with paracrine effects on microglia itself, furtherly boosting neuroinflammation [232], even more so considering that recent literature has highlighted the existing link between MCs and glial activation [225]. Neuroinflammatory responses may be driven by the effects of PEA on TRPV1 channels [233], which are knowingly implicated in such processes [234], possible via modulation of cannabinoid pathways [235]. Such cross-communications between these systems may give reason to the possible role for PEA in modulating intestinal inflammatory conditions [236,237]. PEA levels increase in several animal models of neurological disorders, and in human models, e.g. of migraine [238]. Administration of exogenous PEA was found to be efficacious in experimental models of mast cell-mediated acute and neurogenic inflammation [239].
Moreover, it showed promising results in reverting amyloid β-peptide-induced astrogliosis [240,241] and consequent learning and memory impairment in mice [242], hinting at a possible role for PEA as adjuvant and add-on therapy for human neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases [243,244,245].
PEA was found to reverse histopathological changes, reduce joint swelling, and reduce serum levels of NO, IL-1β, leukotriene B4, TNF-α, and prostaglandin E2, as well as cartilage-degrading MMPs in MIA-induced knee OA rats, hence preserving cartilage structure [246]. PEA administration decreased neuropathic pain, especially mechanical hyperalgesia, in Sprague-Dawley rats that were injured via L5 and L6 spinal nerves transection: interestingly, intrarticular injection of PEA was effective at lower doses compared to intraperitoneal PEA [247]. PEA administration had beneficial effects in dogs with chronic OA and lameness, with the latter having significant improvement, alongside better functioning and pain control [248].
3.1.3. Palmitoylethanolamide and Low Back Pain
m-PEA administration was found to be useful for pain control in patients with LBP, with efficacy against neuropathic features. Although m-PEA may not have a role on functional improvement when administrated alone at low doses (600 mg/die) [249], um-PEA showed promising results when administrated at higher dosage (600 mg twice a day), at least in an early stage, as a support therapy during rehabilitation, with improvement in mental and physical components of QoL, as well as disability scores [250]. Moreover, add-on therapy with um-PEA was correlated with reduced intake of opioids in patients with chronic LBP, with a significant reduction in pain perception and neuropathic manifestations, overall maintaining a good tolerability and safety profile [251,252], which makes um-PEA a good option even in older patients [253]. In addition, um-PEA was found to ameliorate pain control in patients with failed back surgery syndrome as an add-on therapy, together with dual opioid tapentadol and anticonvulsant pregabalin [254]. The modulatory properties of PEA on neuropathic pain are potentially driven by both its effects on CB1, Transient Receptor Potential Vanilloid 1- TRPV1, and PPAR-γ, and a reduction of proinflammatory cytokines, namely NGF and TNF-α [255].
3.1.4. Palmitoylethanolamide and Gut Dysbiosis
Gut microbiota exerts a significant influence on both human physical and mental health [256]. The bidirectional communication between the gastrointestinal tract and the CNS, defined as “gut microbiota-brain axis”, occurs through the neuroendocrine system, the activation of immune system and the production of bacterial metabolites by the gut microbiota (Figure 1) [257]. In detail, the neuroendocrine system refers to the hypothalamus-pituitary-adrenal- HPA axis, while the immune system to microbial- and pathogen-associated molecular patterns (such as lipopolysaccharide-LPS pathway), and finally the bacterial metabolites to the production of short-chain fatty acids (SCFAs), neurotransmitters (dopamine, norepinephrine, serotonin- 5-HT, histamine and gamma-aminobutyric acid- GABA) and tryptophan (Trp) [257]. The communication between the gut microbiota and the HPA axis is closely interconnected with the immune system, microbial metabolites, gut hormones, BBB, and sensory and autonomic nervous system [258]. Gut dysbiosis is defined as an alteration in the gut microbiota composition, detected by an increase in the number of pathobionts and a decrease in symbionts [259], a consequence of high-fat diets (HFDs) and Western diet [260], drugs, immune system dysfunction, stress conditions [261] and diseases (including those inflammatory, autoimmune, metabolic, neoplastic, neurodegenerative and CKD) [262,263,264,265,266,267]. In chronic pain patients, gut microbiota may be affected by opioid administration [268] and drugs used for managing opioid induced constipation [269]. The HPA axis is part of the limbic system and is the main regulator of the stress response [270]. Signals generated by the hypothalamus reach the pituitary and adrenal glands and communicate with entero-epithelial cells via the HPA axis. The signals influence the gut microbiota through the enteric nervous system (ENS), located in the submucosa and myenteric plexus of the gut wall [271]. However, the relationship between the gut dysbiosis and the HPA axis is bidirectional [272]. In fact, stress conditions can alter the gut microbiota composition [273]. Gut microbiota dysbiosis, in turn, influence HPA axis activity, resulting in excess circulating cortisol production through the synthesis of corticotropin-releasing hormone (CRH) by the hypothalamus [274]. CRH stimulates the anterior pituitary gland to produce adrenocorticotropic hormone- ACTH, resulting in the release of an excess cortisol by the adrenal cortex [275]. Cortisol overproduction is implicated in neuroinflammation and impairments of the hippocampus and prefrontal cortex, namely brain regions involved in emotional regulation and memory [276]. While cortisol is necessary for an adaptive stress response, its chronic and excessive production can have neurotoxic effects [277], manifesting with depressive symptoms, cognitive deficits, neuropsychiatric disorders and impaired adaptation to stress, and exacerbating gut dysbiosis [278]. In fact, impairment of the hippocampus and prefrontal cortex, in response to cortisol overproduction, damages the CNS, inducing a reduced neuronal plasticity, a decreased level of BDNF, and atrophy of hippocampus and prefrontal cortex [279]. The overactivity of the HPA axis contributes to the alteration of the composition of the gut microbiota, with an increase in the abundance of Firmicutes phyla and a decrease in Bacteroidetes one [280]. Both phyla are responsible for the production of SCFAs (acetate, propionate and butyrate). The latter are metabolites produced by bacterial fermentation from indigestible carbohydrates (dietary fibre) in the gastrointestinal tract [281]. They perform beneficial functions for the gut microbiota, regulating positively its composition, increasing the amount of TJs proteins and improving the function of intestinal epithelial barrier. SCFAs produced adhere to FFA receptors on the surface of intestinal epithelial cells and interact with neurons or enter the bloodstream [271]. SCFAs modulate TJs protein expression also in the BBB. Indeed, fecal transfer of gut microbiota to germ-free mice decreases BBB permeability and regulates the expression of claudin-5 and ZO-1 in the CNS, reducing BBB permeability [282]. The reduction of SCFAs is closely associated with the onset and development of metabolic and inflammatory diseases (such as type 2 DM, obesity, CKD, arterial hypertension, inflammatory bowel disease-IBD and colorectal cancer) [283]. Firmicutes are responsible for the production of butyrate, while Bacteroidetes of acetate and propionate. However, decreased Bacteroidetes abundance results in reduced acetate and propionate production. Rats treated with HFD are associated with increased Firmicutes phyla and decreased Bacteroidetes phyla abundance. This leads to a change in SCFAs production, with an increase in butyrate levels and a decrease in propionate levels. The globally decrease in SCFAs production induces to a loss of enteric neurons and thus a weakening of the ENS [271]. Gut dysbiosis is also associated with increased Gram-negative bacteria, which activate the immune system and neuroendocrine system via LPS [257,260], the major component of the outer membrane of Gram-negative bacteria [284]. The increase of Gram-negative bacteria in the gastrointestinal tract allows LPS to act locally and systemically, after crossing the impaired gut barrier and entering in the systemic circulation. In fact, the LPS can enter in the bloodstream thanks to increased gut permeability, as pointed out by decreased levels of TJs proteins, zonulin-1 and occluding [285]. High concentrations of circulating LPS are defined as metabolic endotoxemia, which is responsible for triggering chronic-metabolic diseases [286]. Therefore, once LPS has crossed the intestinal barrier to enter in the systemic circulation, it binds to TLR4 and leads to systemic inflammation by stimulating the production of pro-inflammatory cytokines and activating the innate immune system response [287]. Pro-inflammatory cytokines, such as IL-6 and TNF-α, further activate HPA axis, stimulating the release of cortisol [288], exacerbating depression and contributing to symptoms such as apathy, demotivation, and fatigue [274]. Restoring the eubiosis of gut microbiota could be beneficial to nervous peripheral and central disorders related to gut dysbiosis [289]. Over the years, the role of PEA on the gut microbiota has been studied in the literature. For the first time, Couch and co-Authors have investigated the effects of PEA on the permeability of the human gastrointestinal tract in vitro, ex vivo and in vivo. In vitro and ex vivo studies, PEA prevented inflammation-induced permeability of dextran. In the in vivo study, the aspirin-induced increase in intestinal permeability, detected by an increase in urinary lactulose-to-mannitol ratio, was rescued by PEA administration at the dose of 600 mg. These data suggest how PEA is able to reduce the permeability in the human colon [290]. The effects of um-PEA on the gut microbiota have been better elucidated by Pirozzi and co-Authors on HFD-fed mice. The Authors showed, in this animal model, that um-PEA, administered at the dose of 30 mg/kg/die per os for 7 weeks, has been able to reduce the inflammatory response in the gut, restore Trp metabolism and remodel the gut microbiota of mice. The reduction of the inflammatory response is supported by the immunomodulatory effects exerted by um-PEA. Indeed, its administration was able to limit immune cell recruitment and activation of intestinal MCs and macrophages, leading to a reduction in the expression of intestinal pro-inflammatory factors, namely IL-1β, TNF-α, cyclooxygenase-2, and NOS2. The effects on Trp metabolism were evidenced by the ability of the um-PEA to restore 5-HT/kynurenine (KYN) levels in the mice colon, rescuing altered 5-HT turnover, decreasing gastrointestinal indoleamine-2,3-dioxygenase (IDO) expression and resulting in reduced Trp oxidation in KYN [291]. Trp metabolism connects the gut to the CNS, as the enzyme IDO oxidizes Trp in KYN, decreasing circulating levels of 5-HT. KYN reaches the systemic circulation, crosses the BBB and acts at the CNS level, causing depression, anhedonia, anxiety, and metabolic dysfunction related to obesity and insulin resistance [292]. The same Authors pointed out that the administration of um-PEA in HFD-fed mice was able to reduce the Firmicutes/Bacteroidetes ratio and to increase the Bifidobacterium genera and the Oscillospiraceae family, both butyrate producers. Confirming this, the Authors have highlighted an increased expression of monocarboxylate transporters-1- MCT-1, a gene related to butyrate activity that mediates its transport in the colon mucosa, and of G-protein-coupled receptors-43- GPR43, which is involved in the control of the gut inflammation. Finally, the increase of SCFAs-producing bacterial species, such as the genera Turicibacter sanguinis, has been highlighted in the gut [291]. To date, only a randomized, placebo controlled, double-blind study conducted by Batacan and co-Authors has investigated the role of PEA on the gut microbiome of overweight adults (BMI 30-40 kg/m2), at the dose of 700 mg/day for 12 weeks. At the end of the study, the Authors demonstrated the ability of PEA in reducing triglycerides and IL-2 levels. The in vivo effects of PEA remain unclear. In fact, no significant differences in overall microbiota composition were found after PEA administration. Moreover, the microbiota richness and diversity remained constant for both groups [293]. In light of this, further studies are needed to better determine the action of PEA on gut dysbiosis.
3.2. Adelmidrol
Adelmidrol (ADM) is a synthetic derivate of azelaic acid and a member of the Autacoid Local Injury Antagonist Amides (ALIAmides) family, with both amphipathic and amphiphilic properties, which make it particularly suitable for topical and intra-articular administration [294]. A gel combining 2% ADM with 0.1% hyaluronic acid (HA) showed antioxidant effects in the inflamed gut in a mouse model of colitis after intrarectal administration, with protective effects on the epithelial barrier [295]. Moreover, aerosol administration of 2% ADM reduced inflammatory damage in a LPS-induced mice model of acute lung injury, particularly through a reduction in MC activation [296]. Topical ADM (mucoadhesive gel) showed anti-inflammatory effects in small animals with inflammatory pathologies [297,298,299]. A combination of 2% ADM with 1% high molecular weight HA has been approved for intra-articular injections in knee OA, with a significant improvement in analgesia and functionality [300]. In OA joints, ADM acts as a PEA enhancer, leading to higher PEA levels [301]. Moreover, ADM leads to a reduction in inflammatory cytokines and cartilage degradation, through its effects on MCs [211]. Activated MCs release lytic enzymes that accelerate degradation of HA in the joints. Therefore, ADM, by normalizing MCs activity, increase the bioavailability of HA. Moreover, it supports the phenotypical switch of MCs, from an hyperactivated to a physiological state, with consequent restoration of their function of heparin secretion. Since heparin is a precursor of HA, ADM displayed a visco-inductive effect in OA joints in preclinical models [300]. The maintenance of results at follow-up after intra-articular administration of ADM-HA 2%/1% were affected mainly by the BMI of patients and dysmetabolic disorders [300]. Finally, a clinical investigation comparing ADM-HA 2%/1% versus HA alone, showed that at 2-years follow-up, ADM improved all components of the WOMAC scale in the treatment of knee OA [302]. These data suggest a possible combined used of Adelmidrol-HA in obese patients, together with other systemic strategies for targeting meta-neuroinflammations, such as um-PEA and the association m-PEA– rutin and HTyr, with the aim of optimizing body mass index, reducing inflammation biomarkers, and preventing pain chronification (Figure 2).
4. Conclusions
Globally, obesity is a serious public health problem associated with increased morbidity and mortality from all causes, leading to a reduction in patients’ quality of life and life expectancy. Suffering from obesity, the adipocyte hypertrophy, visceral and ectopic adiposity predispose the organism to the meta-inflammation. The dysbiosis of the gut microbiota is typically of obesity, contributing to the meta-neuroinflammation. In fact, the gut microbiota and the CNS are in close communication, defining the gut microbiota-brain axis. Moreover, the integrity of the BBB is compromised in obesity. In light of this innovative point of view, the um-PEA and the m-PEA– rutin and HTyr seem to be able to restore the eubiosis of the gut microbiota and to reduce body weight, body mass index, fat mass, and inflammation biomarkers, to such an extent that they are considered a new adjuvant therapy against adiposopathy. This evidence supports a modern and innovative approach to many comorbidities associated with obesity, particularly for chronic pain syndromes. Osteoarthrosis is the most commonly observed painful disease in obese patients and targeting meta-neuroinflammation is the challenge of the next future. Clinicians should be aware that a modern approach to OA in obese patients require the ability to “think outside the box”, by focusing not only on the single organ or disease, but on the close interconnection between systems, sustained by meta-neuroinflammation. Nowadays, in clinical practice, both central and peripheral mechanisms of neuroinflammation can be managed by using natural bioactive compounds with antioxidant and anti-neuroinflammatory properties. In particular, clinical evidence is available for um-PEA, through systemic administration, and for adelmidrol, as intra-articular injections for knee OA. Further studies are warranted to support these data and open new perspectives for the future of obese patients suffering from chronic pain in OA.
Author Contributions
Conceptualization, F.C. and A.N.; writing—original draft preparation, K.C and M.S.S. ; writing—review and editing, F.C. and A.N.; visualization, F.C., K.C., M.S.S., A.N.; supervision, F.C. and A.N. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by Epitech Group SpA, Via Leonardo da Vinci, 3 35030 Saccolongo (PD) Italy.
Conflicts of Interest
This paper was supported by Epitech Group S.p.A, which co-funded the PhD Program of Dr. Kevin Cornali. The Professor Flaminia Coluzzi and the Professor Annalisa Noce served as speakers and consultants for Epitech Group S.p.A.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Alzheimer’s disease |
| ADAMTS | Disintegrin and Metalloproteinase with Thrombospondin Motifs |
| ADM | Adelmidrol |
| AGEs | Advanced glycation end-products |
| APP | Amyloid Precursor Protein |
| AT | Adipose tissue |
| Aβ | Amyloidbeta-protein |
| BAT | Beige adipose tissue |
| BBB | Blood–brain barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| BM | Basement membrane |
| BMI | Body Mass Index |
| Brd4 | Bromodomain-containing protein 4 |
| CB | Cannabinoid Receptors |
| CFA | Complete Freund’s Adjuvant |
| CKD | Chronic kidney disease |
| CNS | Central nervous system |
| CRH | Corticotropin-Releasing Hormone |
| CVDs | Cardiovascular diseases |
| DALYs | Disability-Adjusted Life-Years |
| DM | Diabetes mellitus |
| DMM | Medial meniscus |
| DRG | Dorsal root ganglia |
| ECM | Extracellular matrix |
| ECMGC | Glycation of the extracellular matrix |
| EnNaCs | Endothelial Na2+ Channels |
| eNOS | Endothelial Nitric Oxide Synthase |
| ENS | Enteric nervous system |
| FAAH | Fatty Acid Amide Hydrolase |
| FFAs | Free fatty acids |
| FLS | Fibroblast-Like Synoviocytes |
| FNDC5 | Fibronectin type III domain containing 5 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| GPx | Glutathione Peroxidase |
| GSH | Glutathione |
| HA | Hyaluronic acid |
| HFDs | High-fat diets |
| HIF-1 | Hypoxia-Inducible Factor 1 |
| HO-1 | Heme Oxygenase-1 |
| HTyr | Hydroxytyrosol |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| IDO | Indoleamine-2,3-Dioxygenase |
| IKK | Inhibitor of Kappa β Kinase |
| IL | Interleukin |
| JAM-A | Junctional Adhesion Molecule-A |
| KLFs | Krüppel-Like Transcription Factors |
| KYN | Kynurenine |
| LBP | low back pain |
| LepR | Leptin receptor |
| lncRNAs | Long non-coding RNAs |
| LPS | Lipopolysaccharide |
| m-PEA | Micronized palmitoylethanolamide |
| M1 | Pro-inflammatory macrophages |
| M2 | Anti-inflammatory macrophages |
| MCP1 | Monocyte Chemoattractant Protein 1 |
| MCs | Mast cells |
| MetS | Metabolic syndrome |
| MHC | Major Histocompatibility Complex |
| MIA | Monosodium iodoacetate |
| MMPs | Matrix metalloproteinases |
| m-PEA– rutin | Comicronized palmitoylethanolamide with rutin |
| mTOR | Mechanistic Target of Rapamycin |
| NAA | N-acetylaspartate |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NAE | N-acylethanolamine |
| NF-κβ | Nuclear Factor kappa β |
| NGF | Neurotrophin Nerve Growth Factor |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| NO | Nitric oxide |
| NOS2 | Inducible nitric oxide synthase |
| NQO1 | NAD(P)H Oxidoreductase 1 |
| Nrf | Nuclear Respiratory Factor |
| OA | Osteoarthritis |
| PEA | Palmitoylethanolamide |
| PECAM | Platelet Endothelial Cell Adhesion Molecule |
| PGC-1α | Peroxisome Proliferative Activated Receptor-γ Coactivator-1α |
| PK | Prokineticin |
| PKC | Protein Kinase C |
| PKR | Prokineticin receptor |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| pTau | Hyperphosphorylated Tau |
| QoL | Quality of Life |
| RAGE | Advanced glycation end-products receptor |
| RANK | Receptor Activator of Nuclear Factor-kappa β |
| ROCK | Rho-kinase |
| ROS | Reactive Oxygen Species |
| SCFAs | Short-Chain Fatty Acids |
| SERCA | Sarcoplasmic Reticulum Calcium ATPase |
| Sesn2 | Sestrin2 |
| SF | Synovial fluid |
| SIRT1 | NAD-Dependent Deacetylase Sirtuin 1 |
| SO | Sarcopenic obesity |
| SOD | Superoxide Dismutase |
| TGF-β | Transforming Growth Factor-β |
| TIMPs | Tissue Inhibitor of Metalloproteinases |
| TJs | Tight junctions |
| TLRs | Toll-Like Receptors |
| TNF-α | Tumor Necrosis Factor-α |
| TRK | Tropomyosin-Related Kinase |
| Trp | Tryptophan |
| TWEAK | Tumour Necrosis Factor-Like Weak Inducer of Apoptosis |
| um-PEA | Ultramicronized palmitoylethanolamide |
| VEGF | Vascular Endothelial Growth Factor |
| WAT | White adipose tissue |
| WHO | World Health Organization |
| ZO-1 | Zonula occludens-1 |
| σ1 | Sigma-1 |
References
- Lingvay, I.; Cohen, R.V.; Roux, C.W.L.; Sumithran, P. Obesity in adults. Lancet 2024, 404, 972–987. [Google Scholar] [CrossRef]
- Islam, A.; Sultana, H.; Nazmul Hassan Refat, M.; Farhana, Z.; Abdulbasah Kamil, A.; Meshbahur Rahman, M. The global burden of overweight-obesity and its association with economic status, benefiting from STEPs survey of WHO member states: A meta-analysis. Prev Med Rep 2024, 46, 102882. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Gratteri, S.; Gualtieri, P.; Cammarano, A.; Bertucci, P.; Di Renzo, L. Why primary obesity is a disease? J Transl Med 2019, 17, 169. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Van Craenenbroeck, E.M.; Antoniades, C.; Bluher, M.; Gorter, T.M.; Hanssen, H.; Marx, N.; McDonagh, T.A.; Mingrone, G.; Rosengren, A.; et al. Obesity and cardiovascular disease: an ESC clinical consensus statement. Eur Heart J 2024, 45, 4063–4098. [Google Scholar] [CrossRef]
- Elagizi, A.; Kachur, S.; Carbone, S.; Lavie, C.J.; Blair, S.N. A Review of Obesity, Physical Activity, and Cardiovascular Disease. Curr Obes Rep 2020, 9, 571–581. [Google Scholar] [CrossRef]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front Immunol 2021, 12, 746151. [Google Scholar] [CrossRef] [PubMed]
- Favaretto, F.; Bettini, S.; Busetto, L.; Milan, G.; Vettor, R. Adipogenic progenitors in different organs: Pathophysiological implications. Rev Endocr Metab Disord 2022, 23, 71–85. [Google Scholar] [CrossRef]
- Abdullah, A.; Wolfe, R.; Stoelwinder, J.U.; de Courten, M.; Stevenson, C.; Walls, H.L.; Peeters, A. The number of years lived with obesity and the risk of all-cause and cause-specific mortality. Int J Epidemiol 2011, 40, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Kloock, S.; Ziegler, C.G.; Dischinger, U. Obesity and its comorbidities, current treatment options and future perspectives: Challenging bariatric surgery? Pharmacol Ther 2023, 251, 108549. [Google Scholar] [CrossRef] [PubMed]
- Basilicata, M.; Pieri, M.; Marrone, G.; Nicolai, E.; Di Lauro, M.; Paolino, V.; Tomassetti, F.; Vivarini, I.; Bollero, P.; Bernardini, S.; et al. Saliva as Biomarker for Oral and Chronic Degenerative Non-Communicable Diseases. Metabolites 2023, 13. [Google Scholar] [CrossRef]
- Rhee, C.M.; Ahmadi, S.F.; Kalantar-Zadeh, K. The dual roles of obesity in chronic kidney disease: a review of the current literature. Curr Opin Nephrol Hypertens 2016, 25, 208–216. [Google Scholar] [CrossRef]
- Czaja-Stolc, S.; Potrykus, M.; Stankiewicz, M.; Kaska, L.; Malgorzewicz, S. Pro-Inflammatory Profile of Adipokines in Obesity Contributes to Pathogenesis, Nutritional Disorders, and Cardiovascular Risk in Chronic Kidney Disease. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Martin-Taboada, M.; Vila-Bedmar, R.; Medina-Gomez, G. From Obesity to Chronic Kidney Disease: How Can Adipose Tissue Affect Renal Function? Nephron 2021, 145, 609–613. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Chen, Y.; Dong, Y. Kidney Damage Caused by Obesity and Its Feasible Treatment Drugs. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Taccone-Gallucci, M.; Noce, A.; Bertucci, P.; Fabbri, C.; Manca-di-Villahermosa, S.; Della-Rovere, F.R.; De Francesco, M.; Lonzi, M.; Federici, G.; Scaccia, F.; et al. Chronic treatment with statins increases the availability of selenium in the antioxidant defence systems of hemodialysis patients. J Trace Elem Med Biol 2010, 24, 27–30. [Google Scholar] [CrossRef]
- Jastreboff, A.M.; Kotz, C.M.; Kahan, S.; Kelly, A.S.; Heymsfield, S.B. Obesity as a Disease: The Obesity Society 2018 Position Statement. Obesity (Silver Spring) 2019, 27, 7–9. [Google Scholar] [CrossRef]
- Restivo, M.R.; McKinnon, M.C.; Frey, B.N.; Hall, G.B.; Syed, W.; Taylor, V.H. The impact of obesity on neuropsychological functioning in adults with and without major depressive disorder. PLoS One 2017, 12, e0176898. [Google Scholar] [CrossRef] [PubMed]
- Amianto, F.; Martini, M.; Olandese, F.; Davico, C.; Abbate-Daga, G.; Fassino, S.; Vitiello, B. Affectionless control: A parenting style associated with obesity and binge eating disorder in adulthood. Eur Eat Disord Rev 2021, 29, 178–192. [Google Scholar] [CrossRef]
- Zimmer, Z.; Fraser, K.; Grol-Prokopczyk, H.; Zajacova, A. A global study of pain prevalence across 52 countries: examining the role of country-level contextual factors. Pain 2022, 163, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Okifuji, A.; Hare, B.D. The association between chronic pain and obesity. J Pain Res 2015, 8, 399–408. [Google Scholar] [CrossRef]
- Chen, S.; Min, M.; Du, L.; Gao, Y.; Xie, L.; Gao, J.; Li, L.; Zhong, Z. Trajectories of obesity indices and their association with pain in community-dwelling older adults: Findings from the English longitudinal study of ageing. Arch Gerontol Geriatr 2025, 129, 105690. [Google Scholar] [CrossRef]
- Disease, G.U.B.o.; Forecasting, C. Burden of disease scenarios by state in the USA, 2022–2050: a forecasting analysis for the Global Burden of Disease Study 2021. Lancet 2024, 404, 2341–2370. [Google Scholar] [CrossRef]
- Zhang, T.T.; Liu, Z.; Liu, Y.L.; Zhao, J.J.; Liu, D.W.; Tian, Q.B. Obesity as a Risk Factor for Low Back Pain: A Meta-Analysis. Clin Spine Surg 2018, 31, 22–27. [Google Scholar] [CrossRef]
- Heuch, I.; Heuch, I.; Hagen, K.; Zwart, J.A. Overweight and obesity as risk factors for chronic low back pain: a new follow-up in the HUNT Study. BMC Public Health 2024, 24, 2618. [Google Scholar] [CrossRef]
- Vincent, H.K.; Raiser, S.N.; Vincent, K.R. The aging musculoskeletal system and obesity-related considerations with exercise. Ageing Res Rev 2012, 11, 361–373. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, Q.; Zhou, J.; Wu, J. A comprehensive meta-analysis of risk factors associated with osteosarcopenic obesity: a closer look at gender, lifestyle and comorbidities. Osteoporos Int 2024, 35, 759–773. [Google Scholar] [CrossRef]
- Kim, H.I.; Ahn, S.H.; Kim, Y.; Lee, J.E.; Choi, E.; Seo, S.K. Effects of sarcopenia and sarcopenic obesity on joint pain and degenerative osteoarthritis in postmenopausal women. Sci Rep 2022, 12, 13543. [Google Scholar] [CrossRef]
- Lawal, Y.; Mshelia-Reng, R.; Omonua, S.O.; Odumodu, K.; Shuaibu, R.; Itanyi, U.D.; Abubakar, A.I.; Kolade-Yunusa, H.O.; David, Z.S.; Ogunlana, B.; et al. Predictors of Peripheral Neuropathy Among Persons with Diabetes Mellitus: A Multicenter Cross-Sectional Study. J Am Podiatr Med Assoc 2025, 115. [Google Scholar] [CrossRef] [PubMed]
- Chai, N.C.; Scher, A.I.; Moghekar, A.; Bond, D.S.; Peterlin, B.L. Obesity and headache: part I--a systematic review of the epidemiology of obesity and headache. Headache 2014, 54, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Naty, S.; Grembiale, R.D. Fibromyalgia and obesity: the hidden link. Rheumatol Int 2011, 31, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.X.; Yazdi, C.; Islam, R.K.; Anwar, A.I.; Alvares-Amado, A.; Townsend, H.; Allen, K.E.; Plakotaris, E.; Hirsch, J.D.; Rieger, R.G.; et al. Diabetic Neuropathy: A Guide to Pain Management. Curr Pain Headache Rep 2024, 28, 1067–1072. [Google Scholar] [CrossRef]
- Wu, R.L.; Chen, N.; Chen, Y.; Wu, X.; Ko, C.Y.; Chen, X.Y. Visceral Adiposity as an Independent Risk Factor for Diabetic Peripheral Neuropathy in Type 2 Diabetes Mellitus: A Retrospective Study. J Diabetes Res 2024, 2024, 9912907. [Google Scholar] [CrossRef]
- Bigal, M.E.; Liberman, J.N.; Lipton, R.B. Obesity and migraine: a population study. Neurology 2006, 66, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Jahromi, S.R.; Martami, F.; Morad Soltani, K.; Togha, M. Migraine and obesity: what is the real direction of their association? Expert Rev Neurother 2023, 23, 75–84. [Google Scholar] [CrossRef] [PubMed]
- D’Onghia, M.; Ciaffi, J.; Lisi, L.; Mancarella, L.; Ricci, S.; Stefanelli, N.; Meliconi, R.; Ursini, F. Fibromyalgia and obesity: A comprehensive systematic review and meta-analysis. Semin Arthritis Rheum 2021, 51, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, V.A.; Ortega, F.B.; Carbonell-Baeza, A.; Camiletti, D.; Ruiz, J.R.; Delgado-Fernandez, M. Relationship of weight status with mental and physical health in female fibromyalgia patients. Obes Facts 2011, 4, 443–448. [Google Scholar] [CrossRef]
- Fruhbeck, G.; Gomez-Ambrosi, J.; Muruzabal, F.J.; Burrell, M.A. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am J Physiol Endocrinol Metab 2001, 280, E827–847. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J Mol Cell Biol 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ Res 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Nuszkiewicz, J.; Kukulska-Pawluczuk, B.; Piec, K.; Jarek, D.J.; Motolko, K.; Szewczyk-Golec, K.; Wozniak, A. Intersecting Pathways: The Role of Metabolic Dysregulation, Gastrointestinal Microbiome, and Inflammation in Acute Ischemic Stroke Pathogenesis and Outcomes. J Clin Med 2024, 13. [Google Scholar] [CrossRef]
- Xu, S.; Lu, F.; Gao, J.; Yuan, Y. Inflammation-mediated metabolic regulation in adipose tissue. Obes Rev 2024, 25, e13724. [Google Scholar] [CrossRef]
- Liebner, S.; Czupalla, C.J.; Wolburg, H. Current concepts of blood-brain barrier development. Int J Dev Biol 2011, 55, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol Dis 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Nehme, Z.; Roehlen, N.; Dhawan, P.; Baumert, T.F. Tight Junction Protein Signaling and Cancer Biology. Cells 2023, 12. [Google Scholar] [CrossRef]
- Anthony, D.P.; Hegde, M.; Shetty, S.S.; Rafic, T.; Mutalik, S.; Rao, B.S.S. Targeting receptor-ligand chemistry for drug delivery across blood-brain barrier in brain diseases. Life Sci 2021, 274, 119326. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxid Redox Signal 2001, 3, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Sorokin, L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 2009, 31, 497–511. [Google Scholar] [CrossRef]
- Mou, Y.; Du, Y.; Zhou, L.; Yue, J.; Hu, X.; Liu, Y.; Chen, S.; Lin, X.; Zhang, G.; Xiao, H.; et al. Gut Microbiota Interact With the Brain Through Systemic Chronic Inflammation: Implications on Neuroinflammation, Neurodegeneration, and Aging. Front Immunol 2022, 13, 796288. [Google Scholar] [CrossRef]
- Sheikh, M.H.; Errede, M.; d’Amati, A.; Khan, N.Q.; Fanti, S.; Loiola, R.A.; McArthur, S.; Purvis, G.S.D.; O’Riordan, C.E.; Ferorelli, D.; et al. Impact of metabolic disorders on the structural, functional, and immunological integrity of the blood-brain barrier: Therapeutic avenues. FASEB J 2022, 36, e22107. [Google Scholar] [CrossRef]
- Boumiza, S.; Chahed, K.; Tabka, Z.; Jacob, M.P.; Norel, X.; Ozen, G. MMPs and TIMPs levels are correlated with anthropometric parameters, blood pressure, and endothelial function in obesity. Sci Rep 2021, 11, 20052. [Google Scholar] [CrossRef]
- Chen, H.; Guan, B.; Chen, S.; Yang, D.; Shen, J. Peroxynitrite activates NLRP3 inflammasome and contributes to hemorrhagic transformation and poor outcome in ischemic stroke with hyperglycemia. Free Radic Biol Med 2021, 165, 171–183. [Google Scholar] [CrossRef]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front Neurosci 2018, 12, 930. [Google Scholar] [CrossRef]
- Turksen, K.; Aubin, J.E.; Sodek, J.; Kalnins, V.I. Localization of laminin, type IV collagen, fibronectin, and heparan sulfate proteoglycan in chick retinal pigment epithelium basement membrane during embryonic development. J Histochem Cytochem 1985, 33, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Angiari, S.; Constantin, G. Selectins and their ligands as potential immunotherapeutic targets in neurological diseases. Immunotherapy 2013, 5, 1207–1220. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front Cell Neurosci 2021, 15, 661838. [Google Scholar] [CrossRef]
- Song, J.; Wu, C.; Korpos, E.; Zhang, X.; Agrawal, S.M.; Wang, Y.; Faber, C.; Schafers, M.; Korner, H.; Opdenakker, G.; et al. Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep 2015, 10, 1040–1054. [Google Scholar] [CrossRef]
- Junker, U.; Jaggi, C.; Bestetti, G.; Rossi, G.L. Basement membrane of hypothalamus and cortex capillaries from normotensive and spontaneously hypertensive rats with streptozotocin-induced diabetes. Acta Neuropathol 1985, 65, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Cross, K.; Vetter, S.W.; Alam, Y.; Hasan, M.Z.; Nath, A.D.; Leclerc, E. Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses. Biomolecules 2024, 14. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Robel, S.; Mishra, A. Astrocyte Endfeet in Brain Function and Pathology: Open Questions. Annu Rev Neurosci 2023, 46, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Lee, J.H.; Flausino, L.E.; Quintana, F.J. Neuroinflammation: An astrocyte perspective. Sci Transl Med 2023, 15, eadi7828. [Google Scholar] [CrossRef] [PubMed]
- Sanmarco, L.M.; Wheeler, M.A.; Gutierrez-Vazquez, C.; Polonio, C.M.; Linnerbauer, M.; Pinho-Ribeiro, F.A.; Li, Z.; Giovannoni, F.; Batterman, K.V.; Scalisi, G.; et al. Gut-licensed IFNgamma(+) NK cells drive LAMP1(+)TRAIL(+) anti-inflammatory astrocytes. Nature 2021, 590, 473–479. [Google Scholar] [CrossRef]
- Proescholdt, M.A.; Heiss, J.D.; Walbridge, S.; Muhlhauser, J.; Capogrossi, M.C.; Oldfield, E.H.; Merrill, M.J. Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J Neuropathol Exp Neurol 1999, 58, 613–627. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Avignon, A.; Sultan, A. PKC-B inhibition: a new therapeutic approach for diabetic complications? Diabetes Metab 2006, 32, 205–213. [Google Scholar] [CrossRef]
- Goto, K.; Kondo, H. Diacylglycerol kinase in the central nervous system--molecular heterogeneity and gene expression. Chem Phys Lipids 1999, 98, 109–117. [Google Scholar] [CrossRef]
- Yee, Y.H.; Chong, S.J.F.; Kong, L.R.; Goh, B.C.; Pervaiz, S. Sustained IKKbeta phosphorylation and NF-kappaB activation by superoxide-induced peroxynitrite-mediated nitrotyrosine modification of B56gamma3 and PP2A inactivation. Redox Biol 2021, 41, 101834. [Google Scholar] [CrossRef] [PubMed]
- Kadir, R.R.A.; Alwjwaj, M.; McCarthy, Z.; Bayraktutan, U. Therapeutic hypothermia augments the restorative effects of PKC-beta and Nox2 inhibition on an in vitro model of human blood-brain barrier. Metab Brain Dis 2021, 36, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, K.; Nguyen, Q.M.; Gabani, M.; Kassan, A.; Kumar, S.; Choi, S.K.; Gonzalez, A.A.; Khataei, T.; Sahyoun, A.M.; Chen, C.; et al. MicroRNAs and obesity-induced endothelial dysfunction: key paradigms in molecular therapy. Cardiovasc Diabetol 2020, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest 2012, 122, 2454–2468. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Austin, S.A. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J 2014, 35, 888–894. [Google Scholar] [CrossRef]
- Iova, O.M.; Marin, G.E.; Lazar, I.; Stanescu, I.; Dogaru, G.; Nicula, C.A.; Bulboaca, A.E. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders-An Overview. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Abeta and Tau in the Pathogenesis of Alzheimer’s Disease. Int J Biol Sci 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-beta levels in brain. J Neurochem 2016, 138, 525–531. [Google Scholar] [CrossRef]
- Hu, Y.B.; Ren, R.J.; Zhang, Y.F.; Huang, Y.; Cui, H.L.; Ma, C.; Qiu, W.Y.; Wang, H.; Cui, P.J.; Chen, H.Z.; et al. Rho-associated coiled-coil kinase 1 activation mediates amyloid precursor protein site-specific Ser655 phosphorylation and triggers amyloid pathology. Aging Cell 2019, 18, e13001. [Google Scholar] [CrossRef]
- Yue, Q.; Leng, X.; Xie, N.; Zhang, Z.; Yang, D.; Hoi, M.P.M. Endothelial Dysfunctions in Blood-Brain Barrier Breakdown in Alzheimer’s Disease: From Mechanisms to Potential Therapies. CNS Neurosci Ther 2024, 30, e70079. [Google Scholar] [CrossRef]
- Dragano, N.R.; Haddad-Tovolli, R.; Velloso, L.A. Leptin, Neuroinflammation and Obesity. Front Horm Res 2017, 48, 84–96. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front Endocrinol (Lausanne) 2021, 12, 585887. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Byun, M.S.; Yi, D.; Ahn, H.; Jung, G.; Jung, J.H.; Chang, Y.Y.; Kim, K.; Choi, H.; Choi, J.; et al. Plasma Leptin and Alzheimer Protein Pathologies Among Older Adults. JAMA Netw Open 2024, 7, e249539. [Google Scholar] [CrossRef]
- Littlejohns, T.J.; Kos, K.; Henley, W.E.; Cherubini, A.; Ferrucci, L.; Lang, I.A.; Langa, K.M.; Melzer, D.; Llewellyn, D.J. Serum leptin and risk of cognitive decline in elderly italians. J Alzheimers Dis 2015, 44, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Tezapsidis, N.; Johnston, J.M.; Smith, M.A.; Ashford, J.W.; Casadesus, G.; Robakis, N.K.; Wolozin, B.; Perry, G.; Zhu, X.; Greco, S.J.; et al. Leptin: a novel therapeutic strategy for Alzheimer’s disease. J Alzheimers Dis 2009, 16, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Daniele, G.; Campi, B.; Saba, A.; Codini, S.; Ciccarone, A.; Giusti, L.; Del Prato, S.; Esterline, R.L.; Ferrannini, E. Plasma N-Acetylaspartate Is Related to Age, Obesity, and Glucose Metabolism: Effects of Antidiabetic Treatment and Bariatric Surgery. Front Endocrinol (Lausanne) 2020, 11, 216. [Google Scholar] [CrossRef]
- Coplan, J.D.; Fathy, H.M.; Abdallah, C.G.; Ragab, S.A.; Kral, J.G.; Mao, X.; Shungu, D.C.; Mathew, S.J. Reduced hippocampal N-acetyl-aspartate (NAA) as a biomarker for overweight. Neuroimage Clin 2014, 4, 326–335. [Google Scholar] [CrossRef]
- De Sousa, R.A.L. Exercise-produced irisin effects on brain-related pathological conditions. Metab Brain Dis 2024, 39, 1679–1687. [Google Scholar] [CrossRef]
- Paoletti, I.; Coccurello, R. Irisin: A Multifaceted Hormone Bridging Exercise and Disease Pathophysiology. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Liu, S.; Cui, F.; Ning, K.; Wang, Z.; Fu, P.; Wang, D.; Xu, H. Role of irisin in physiology and pathology. Front Endocrinol (Lausanne) 2022, 13, 962968. [Google Scholar] [CrossRef]
- Della Guardia, L.; Shin, A.C. Obesity-induced tissue alterations resist weight loss: A mechanistic review. Diabetes Obes Metab 2024, 26, 3045–3057. [Google Scholar] [CrossRef]
- Vints, W.A.J.; Kusleikiene, S.; Sheoran, S.; Valatkeviciene, K.; Gleizniene, R.; Himmelreich, U.; Paasuke, M.; Cesnaitiene, V.J.; Levin, O.; Verbunt, J.; et al. Body fat and components of sarcopenia relate to inflammation, brain volume, and neurometabolism in older adults. Neurobiol Aging 2023, 127, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Yu, K.; Shyh-Chang, N.; Li, G.X.; Jiang, L.J.; Yu, S.L.; Xu, L.Y.; Liu, R.J.; Guo, Z.J.; Xie, H.Y.; et al. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J Cachexia Sarcopenia Muscle 2019, 10, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Senol, O.; Gundogdu, G.; Gundogdu, K.; Miloglu, F.D. Investigation of the relationships between knee osteoarthritis and obesity via untargeted metabolomics analysis. Clin Rheumatol 2019, 38, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Bonet, M.L.; Granados, N.; Palou, A. Molecular players at the intersection of obesity and osteoarthritis. Curr Drug Targets 2011, 12, 2103–2128. [Google Scholar] [CrossRef]
- Courties, A.; Gualillo, O.; Berenbaum, F.; Sellam, J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthritis Cartilage 2015, 23, 1955–1965. [Google Scholar] [CrossRef]
- Tootsi, K.; Martson, A.; Kals, J.; Paapstel, K.; Zilmer, M. Metabolic factors and oxidative stress in osteoarthritis: a case-control study. Scand J Clin Lab Invest 2017, 77, 520–526. [Google Scholar] [CrossRef]
- Liu, Z.; Xie, W.; Li, H.; Liu, X.; Lu, Y.; Lu, B.; Deng, Z.; Li, Y. Novel perspectives on leptin in osteoarthritis: Focus on aging. Genes Dis 2024, 11, 101159. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; Litherland, G.J.; Elias, M.S.; Kitson, G.I.; Cawston, T.E.; Rowan, A.D.; Young, D.A. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. Ann Rheum Dis 2012, 71, 455–462. [Google Scholar] [CrossRef]
- Loef, M.; Schoones, J.W.; Kloppenburg, M.; Ioan-Facsinay, A. Fatty acids and osteoarthritis: different types, different effects. Joint Bone Spine 2019, 86, 451–458. [Google Scholar] [CrossRef]
- Obeidat, A.M.; Miller, R.E.; Miller, R.J.; Malfait, A.M. The nociceptive innervation of the normal and osteoarthritic mouse knee. Osteoarthritis Cartilage 2019, 27, 1669–1679. [Google Scholar] [CrossRef]
- Zhang, L.; Li, M.; Li, X.; Liao, T.; Ma, Z.; Zhang, L.; Xing, R.; Wang, P.; Mao, J. Characteristics of sensory innervation in synovium of rats within different knee osteoarthritis models and the correlation between synovial fibrosis and hyperalgesia. J Adv Res 2022, 35, 141–151. [Google Scholar] [CrossRef]
- Shin, Y.; Cho, D.; Kim, S.K.; Chun, J.S. STING mediates experimental osteoarthritis and mechanical allodynia in mouse. Arthritis Res Ther 2023, 25, 90. [Google Scholar] [CrossRef] [PubMed]
- Kuyinu, E.L.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Animal models of osteoarthritis: classification, update, and measurement of outcomes. J Orthop Surg Res 2016, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Malfait, A.M. Osteoarthritis pain: What are we learning from animal models? Best Pract Res Clin Rheumatol 2017, 31, 676–687. [Google Scholar] [CrossRef]
- Miller, R.E.; Tran, P.B.; Ishihara, S.; Syx, D.; Ren, D.; Miller, R.J.; Valdes, A.M.; Malfait, A.M. Microarray analyses of the dorsal root ganglia support a role for innate neuro-immune pathways in persistent pain in experimental osteoarthritis. Osteoarthritis Cartilage 2020, 28, 581–592. [Google Scholar] [CrossRef]
- Tonello, R.; Silveira Prudente, A.; Hoon Lee, S.; Faith Cohen, C.; Xie, W.; Paranjpe, A.; Roh, J.; Park, C.K.; Chung, G.; Strong, J.A.; et al. Single-cell analysis of dorsal root ganglia reveals metalloproteinase signaling in satellite glial cells and pain. Brain Behav Immun 2023, 113, 401–414. [Google Scholar] [CrossRef]
- Nigrovic, P.A.; Lee, D.M. Mast cells in inflammatory arthritis. Arthritis Res Ther 2005, 7, 1–11. [Google Scholar] [CrossRef]
- Magarinos, N.J.; Bryant, K.J.; Fosang, A.J.; Adachi, R.; Stevens, R.L.; McNeil, H.P. Mast cell-restricted, tetramer-forming tryptases induce aggrecanolysis in articular cartilage by activating matrix metalloproteinase-3 and -13 zymogens. J Immunol 2013, 191, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Zappia, J.; Sanchez, C.; Florin, A.; Dubuc, J.E.; Henrotin, Y. The Damage-Associated Molecular Patterns (DAMPs) as Potential Targets to Treat Osteoarthritis: Perspectives From a Review of the Literature. Front Med (Lausanne) 2020, 7, 607186. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, A.; Filice, E.; Caffarelli, C.; Ricci, G.; Pession, A. Mast Cell Activation Disorders. Medicina (Kaunas) 2021, 57. [Google Scholar] [CrossRef]
- Loucks, A.; Maerz, T.; Hankenson, K.; Moeser, A.; Colbath, A. The multifaceted role of mast cells in joint inflammation and arthritis. Osteoarthritis Cartilage 2023, 31, 567–575. [Google Scholar] [CrossRef]
- Sousa-Valente, J.; Calvo, L.; Vacca, V.; Simeoli, R.; Arevalo, J.C.; Malcangio, M. Role of TrkA signalling and mast cells in the initiation of osteoarthritis pain in the monoiodoacetate model. Osteoarthritis Cartilage 2018, 26, 84–94. [Google Scholar] [CrossRef]
- Barker, P.A.; Mantyh, P.; Arendt-Nielsen, L.; Viktrup, L.; Tive, L. Nerve Growth Factor Signaling and Its Contribution to Pain. J Pain Res 2020, 13, 1223–1241. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.B.; Miller, R.E.; Ishihara, S.; Miller, R.J.; Malfait, A.M. Spinal microglial activation in a murine surgical model of knee osteoarthritis. Osteoarthritis Cartilage 2017, 25, 718–726. [Google Scholar] [CrossRef]
- Ohashi, Y.; Uchida, K.; Fukushima, K.; Satoh, M.; Koyama, T.; Tsuchiya, M.; Saito, H.; Takahira, N.; Inoue, G.; Takaso, M. NGF Expression and Elevation in Hip Osteoarthritis Patients with Pain and Central Sensitization. Biomed Res Int 2021, 2021, 9212585. [Google Scholar] [CrossRef]
- Obeidat, A.M.; Donner, A.; Miller, R.E. An update on targets for treating osteoarthritis pain: NGF and TRPV1. Curr Treatm Opt Rheumatol 2020, 6, 129–145. [Google Scholar] [CrossRef]
- I, O.S.; Kc, R.; Singh, G.; Das, V.; Ma, K.; Li, X.; Mwale, F.; Votta-Velis, G.; Bruce, B.; Natarajan Anbazhagan, A.; et al. Sensory Neuron-Specific Deletion of Tropomyosin Receptor Kinase A (TrkA) in Mice Abolishes Osteoarthritis (OA) Pain via NGF/TrkA Intervention of Peripheral Sensitization. Int J Mol Sci 2022, 23. [CrossRef]
- Raoof, R.; Martin Gil, C.; Lafeber, F.; de Visser, H.; Prado, J.; Versteeg, S.; Pascha, M.N.; Heinemans, A.L.P.; Adolfs, Y.; Pasterkamp, J.; et al. Dorsal Root Ganglia Macrophages Maintain Osteoarthritis Pain. J Neurosci 2021, 41, 8249–8261. [Google Scholar] [CrossRef]
- Song, Z.; Xie, W.; Chen, S.; Strong, J.A.; Print, M.S.; Wang, J.I.; Shareef, A.F.; Ulrich-Lai, Y.M.; Zhang, J.M. High-fat diet increases pain behaviors in rats with or without obesity. Sci Rep 2017, 7, 10350. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, T.; Winter, D.R.; Miller, R.J.; Miller, R.E.; Malfait, A.M. Neuroimmune interactions and osteoarthritis pain: focus on macrophages. Pain Rep 2021, 6, e892. [Google Scholar] [CrossRef]
- Adaes, S.; Almeida, L.; Potes, C.S.; Ferreira, A.R.; Castro-Lopes, J.M.; Ferreira-Gomes, J.; Neto, F.L. Glial activation in the collagenase model of nociception associated with osteoarthritis. Mol Pain 2017, 13, 1744806916688219. [Google Scholar] [CrossRef]
- Ogbonna, A.C.; Clark, A.K.; Malcangio, M. Development of monosodium acetate-induced osteoarthritis and inflammatory pain in ageing mice. Age (Dordr) 2015, 37, 9792. [Google Scholar] [CrossRef] [PubMed]
- Martin Gil, C.; Raoof, R.; Versteeg, S.; Willemen, H.; Lafeber, F.; Mastbergen, S.C.; Eijkelkamp, N. Myostatin and CXCL11 promote nervous tissue macrophages to maintain osteoarthritis pain. Brain Behav Immun 2024, 116, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, V.; Deamond, H.; Yousefpour, N.; Fitzcharles, M.A.; Ribeiro-da-Silva, A. Pain-related behavior is associated with increased joint innervation, ipsilateral dorsal horn gliosis, and dorsal root ganglia activating transcription factor 3 expression in a rat ankle joint model of osteoarthritis. Pain Rep 2020, 5, e846. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Guo, J.; Guo, X.; Feng, Z.; Zhao, X. Spinal NF-kB upregulation contributes to hyperalgesia in a rat model of advanced osteoarthritis. Mol Pain 2020, 16, 1744806920905691. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, X.H.; Song, F.H.; Li, D.Y.; Gao, S.J.; Zhang, L.Q.; Wu, J.Y.; Liu, D.Q.; Wang, L.W.; Zhou, Y.Q.; et al. Inhibition of Brd4 alleviates osteoarthritis pain via suppression of neuroinflammation and activation of Nrf2-mediated antioxidant signalling. Br J Pharmacol 2023, 180, 3194–3214. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, II.; Radu, C.I.; Vladacenco, O.; Roza, E.; Costachescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Song, F.H.; Wu, J.Y.; Zhang, L.Q.; Li, D.Y.; Gao, S.J.; Liu, D.Q.; Zhou, Y.Q.; Mei, W. Sestrin2 overexpression attenuates osteoarthritis pain via induction of AMPK/PGC-1alpha-mediated mitochondrial biogenesis and suppression of neuroinflammation. Brain Behav Immun 2022, 102, 53–70. [Google Scholar] [CrossRef]
- Hu, Y.D.; Yue, Y.F.; Chen, T.; Wang, Z.D.; Ding, J.Q.; Xie, M.; Li, D.; Zhu, H.L.; Cheng, M.L. Alleviating effect of lycorine on CFA-induced arthritic pain via inhibition of spinal inflammation and oxidative stress. Exp Ther Med 2023, 25, 241. [Google Scholar] [CrossRef]
- Yang, H.Y.; Sun, X.; Zhen, S.Q.; Yu, L.Z.; Ding, J.Q.; Liu, L.; Xie, M.; Zhu, H.L. GSK-3beta inhibition alleviates arthritis pain via reducing spinal mitochondrial reactive oxygen species level and inflammation. PLoS One 2023, 18, e0284332. [Google Scholar] [CrossRef]
- Laitner, M.H.; Erickson, L.C.; Ortman, E. Understanding the Impact of Sex and Gender in Osteoarthritis: Assessing Research Gaps and Unmet Needs. J Womens Health (Larchmt) 2021, 30, 634–641. [Google Scholar] [CrossRef]
- Tschon, M.; Contartese, D.; Pagani, S.; Borsari, V.; Fini, M. Gender and Sex Are Key Determinants in Osteoarthritis Not Only Confounding Variables. A Systematic Review of Clinical Data. J Clin Med 2021, 10. [Google Scholar] [CrossRef]
- Mun, C.J.; Letzen, J.E.; Nance, S.; Smith, M.T.; Khanuja, H.S.; Sterling, R.S.; Bicket, M.C.; Haythornthwaite, J.A.; Jamison, R.N.; Edwards, R.R.; et al. Sex Differences in Interleukin-6 Responses Over Time Following Laboratory Pain Testing Among Patients With Knee Osteoarthritis. J Pain 2020, 21, 731–741. [Google Scholar] [CrossRef]
- Perruccio, A.V.; Badley, E.M.; Power, J.D.; Canizares, M.; Kapoor, M.; Rockel, J.; Chandran, V.; Gandhi, R.; Mahomed, N.M.; Davey, J.R.; et al. Sex differences in the relationship between individual systemic markers of inflammation and pain in knee osteoarthritis. Osteoarthr Cartil Open 2019, 1, 100004. [Google Scholar] [CrossRef] [PubMed]
- Kosek, E.; Finn, A.; Ultenius, C.; Hugo, A.; Svensson, C.; Ahmed, A.S. Differences in neuroimmune signalling between male and female patients suffering from knee osteoarthritis. J Neuroimmunol 2018, 321, 49–60. [Google Scholar] [CrossRef]
- Weerasekera, A.; Morrissey, E.; Kim, M.; Saha, A.; Lin, Y.; Alshelh, Z.; Torrado-Carvajal, A.; Albrecht, D.; Akeju, O.; Kwon, Y.M.; et al. Thalamic neurometabolite alterations in patients with knee osteoarthritis before and after total knee replacement. Pain 2021, 162, 2014–2023. [Google Scholar] [CrossRef]
- Amodeo, G.; Franchi, S.; Galimberti, G.; Comi, L.; D’Agnelli, S.; Baciarello, M.; Bignami, E.G.; Sacerdote, P. Osteoarthritis Pain in Old Mice Aggravates Neuroinflammation and Frailty: The Positive Effect of Morphine Treatment. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Muchhala, K.H.; Jacob, J.C.; Dewey, W.L.; Akbarali, H.I. Role of beta-arrestin-2 in short- and long-term opioid tolerance in the dorsal root ganglia. Eur J Pharmacol 2021, 899, 174007. [Google Scholar] [CrossRef] [PubMed]
- Carcole, M.; Kummer, S.; Goncalves, L.; Zamanillo, D.; Merlos, M.; Dickenson, A.H.; Fernandez-Pastor, B.; Cabanero, D.; Maldonado, R. Sigma-1 receptor modulates neuroinflammation associated with mechanical hypersensitivity and opioid tolerance in a mouse model of osteoarthritis pain. Br J Pharmacol 2019, 176, 3939–3955. [Google Scholar] [CrossRef] [PubMed]
- Bufalo, M.C.; Almeida, M.E.S.; Jensen, J.R.; DeOcesano-Pereira, C.; Lichtenstein, F.; Picolo, G.; Chudzinski-Tavassi, A.M.; Sampaio, S.C.; Cury, Y.; Zambelli, V.O. Human Sensory Neuron-like Cells and Glycated Collagen Matrix as a Model for the Screening of Analgesic Compounds. Cells 2022, 11. [Google Scholar] [CrossRef]
- Galimberti, G.; Amodeo, G.; Magni, G.; Riboldi, B.; Balboni, G.; Onnis, V.; Ceruti, S.; Sacerdote, P.; Franchi, S. Prokineticin System Is a Pharmacological Target to Counteract Pain and Its Comorbid Mood Alterations in an Osteoarthritis Murine Model. Cells 2023, 12. [Google Scholar] [CrossRef]
- Amodeo, G.; Franchi, S.; D’Agnelli, S.; Galimberti, G.; Baciarello, M.; Bignami, E.G.; Sacerdote, P. Supraspinal neuroinflammation and anxio-depressive-like behaviors in young- and older- adult mice with osteoarthritis pain: the effect of morphine. Psychopharmacology (Berl) 2023, 240, 2131–2146. [Google Scholar] [CrossRef]
- Wistrom, E.; Chase, R.; Smith, P.R.; Campbell, Z.T. A compendium of validated pain genes. WIREs Mech Dis 2022, 14, e1570. [Google Scholar] [CrossRef]
- Schumacher, M.A. Peripheral Neuroinflammation and Pain: How Acute Pain Becomes Chronic. Curr Neuropharmacol 2024, 22, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Jiao, P.; Wang, J.; Li, Y.; Bao, B.; Luoreng, Z.; Wang, X. Role of Long Noncoding RNAs in the Regulation of Cellular Immune Response and Inflammatory Diseases. Cells 2022, 11. [Google Scholar] [CrossRef]
- Liang, Y.; Zhao, J.; Dai, T.; Li, X.; Chen, L.; He, Z.; Guo, M.; Zhao, J.; Xu, L. A review of KLF4 and inflammatory disease: Current status and future perspective. Pharmacol Res 2024, 207, 107345. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Y.; Ahmad, N.; Haqqi, T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed Pharmacother 2020, 129, 110452. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Molnar, V.; Matisic, V.; Kodvanj, I.; Bjelica, R.; Jelec, Z.; Hudetz, D.; Rod, E.; Cukelj, F.; Vrdoljak, T.; Vidovic, D.; et al. Cytokines and Chemokines Involved in Osteoarthritis Pathogenesis. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Grigolo, B.; Roseti, L.; Fiorini, M.; Facchini, A. Enhanced lipid peroxidation in synoviocytes from patients with osteoarthritis. J Rheumatol 2003, 30, 345–347. [Google Scholar] [PubMed]
- Franz, A.; Joseph, L.; Mayer, C.; Harmsen, J.F.; Schrumpf, H.; Frobel, J.; Ostapczuk, M.S.; Krauspe, R.; Zilkens, C. The role of oxidative and nitrosative stress in the pathology of osteoarthritis: Novel candidate biomarkers for quantification of degenerative changes in the knee joint. Orthop Rev (Pavia) 2018, 10, 7460. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, L.; Guo, Z.; Wang, G.; Xu, J.; Zheng, Z.; Sun, K.; Guo, F. Lipid peroxidation in osteoarthritis: focusing on 4-hydroxynonenal, malondialdehyde, and ferroptosis. Cell Death Discov 2023, 9, 320. [Google Scholar] [CrossRef]
- Jiang, H.; Ji, P.; Shang, X.; Zhou, Y. Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Bentz, M.; Zaouter, C.; Shi, Q.; Fahmi, H.; Moldovan, F.; Fernandes, J.C.; Benderdour, M. Inhibition of inducible nitric oxide synthase prevents lipid peroxidation in osteoarthritic chondrocytes. J Cell Biochem 2012, 113, 2256–2267. [Google Scholar] [CrossRef]
- Akool el, S.; Kleinert, H.; Hamada, F.M.; Abdelwahab, M.H.; Forstermann, U.; Pfeilschifter, J.; Eberhardt, W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol Cell Biol 2003, 23, 4901–4916. [Google Scholar] [CrossRef]
- Mastbergen, S.C.; Bijlsma, J.W.; Lafeber, F.P. Synthesis and release of human cartilage matrix proteoglycans are differently regulated by nitric oxide and prostaglandin-E2. Ann Rheum Dis 2008, 67, 52–58. [Google Scholar] [CrossRef]
- Katsuyama, K.; Shichiri, M.; Marumo, F.; Hirata, Y. NO inhibits cytokine-induced iNOS expression and NF-kappaB activation by interfering with phosphorylation and degradation of IkappaB-alpha. Arterioscler Thromb Vasc Biol 1998, 18, 1796–1802. [Google Scholar] [CrossRef]
- Xia, W.; Szomor, Z.; Wang, Y.; Murrell, G.A. Nitric oxide enhances collagen synthesis in cultured human tendon cells. J Orthop Res 2006, 24, 159–172. [Google Scholar] [CrossRef]
- Rubbo, H. Nitric oxide and peroxynitrite in lipid peroxidation. Medicina (B Aires) 1998, 58, 361–366. [Google Scholar]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol 2015, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1alpha Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic Biol Med 2022, 189, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Noce, A.; Ferrannini, M.; Fabrini, R.; Bocedi, A.; Dessi, M.; Galli, F.; Federici, G.; Palumbo, R.; Di Daniele, N.; Ricci, G. Erythrocyte glutathione transferase: a new biomarker for hemodialysis adequacy, overcoming the Kt/V(urea) dogma? Cell Death Dis 2012, 3, e377. [Google Scholar] [CrossRef]
- Sheng, W.; Yue, Y.; Qi, T.; Qin, H.; Liu, P.; Wang, D.; Zeng, H.; Yu, F. The Multifaceted Protective Role of Nuclear Factor Erythroid 2-Related Factor 2 in Osteoarthritis: Regulation of Oxidative Stress and Inflammation. J Inflamm Res 2024, 17, 6619–6633. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, C. Nrf2-mediated anti-inflammatory polarization of macrophages as therapeutic targets for osteoarthritis. Front Immunol 2022, 13, 967193. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, Y.; Zhou, X.; Tian, Y.; Miao, Z.; Ko, C.C.; Hu, X. Nrf2 differentially regulates osteoclast and osteoblast differentiation for bone homeostasis. Biochem Biophys Res Commun 2023, 674, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Jie, P.; Wu, Y.; Song, C.; Cheng, Y.; Liu, Y.; Chen, K. Mechanism of Nrf2/miR338-3p/TRAP-1 pathway involved in hyperactivation of synovial fibroblasts in patients with osteoarthritis. Heliyon 2023, 9, e21412. [Google Scholar] [CrossRef]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Zhang, L.; Xing, R.; Huang, Z.; Ding, L.; Zhang, L.; Li, M.; Li, X.; Wang, P.; Mao, J. Synovial Fibrosis Involvement in Osteoarthritis. Front Med (Lausanne) 2021, 8, 684389. [Google Scholar] [CrossRef]
- Green, A.; Hossain, T.; Eckmann, D.M. Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission. Front Cell Dev Biol 2022, 10, 1010232. [Google Scholar] [CrossRef]
- Wang, K.; Klionsky, D.J. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Osko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczynska, K. Mitochondrial Oxidative Stress-A Causative Factor and Therapeutic Target in Many Diseases. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Qi, Z.; Zhu, J.; Cai, W.; Lou, C.; Li, Z. The role and intervention of mitochondrial metabolism in osteoarthritis. Mol Cell Biochem 2024, 479, 1513–1524. [Google Scholar] [CrossRef]
- Cheung, C.; Tu, S.; Feng, Y.; Wan, C.; Ai, H.; Chen, Z. Mitochondrial quality control dysfunction in osteoarthritis: Mechanisms, therapeutic strategies & future prospects. Arch Gerontol Geriatr 2024, 125, 105522. [Google Scholar] [CrossRef]
- Shen, K.; Zhou, H.; Zuo, Q.; Gu, Y.; Cheng, J.; Yan, K.; Zhang, H.; Song, H.; Liang, W.; Zhou, J.; et al. GATD3A-deficiency-induced mitochondrial dysfunction facilitates senescence of fibroblast-like synoviocytes and osteoarthritis progression. Nat Commun 2024, 15, 10923. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Qi, W.; Zhan, J.; Lin, Z.; Lin, J.; Xue, X.; Pan, X.; Zhou, Y. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J Cell Mol Med 2020, 24, 13046–13057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid Med Cell Longev 2020, 2020, 4063562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, Y.; Li, H. NLRP3 Inflammasome Plays an Important Role in the Pathogenesis of Collagen-Induced Arthritis. Mediators Inflamm 2016, 2016, 9656270. [Google Scholar] [CrossRef]
- Silva Santos Ribeiro, P.; Willemen, H.; Versteeg, S.; Martin Gil, C.; Eijkelkamp, N. NLRP3 inflammasome activation in sensory neurons promotes chronic inflammatory and osteoarthritis pain. Immunother Adv 2023, 3, ltad022. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.J.; Lee, H.; Hong, S.H.; Park, C.; Park, S.H.; Kim, G.Y.; Kim, S.; Kim, H.S.; Hwang, H.J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Makosa, D.; Miller, B.; Griffin, T.M. Glutathione as a mediator of cartilage oxidative stress resistance and resilience during aging and osteoarthritis. Connect Tissue Res 2020, 61, 34–47. [Google Scholar] [CrossRef]
- Setti, T.; Arab, M.G.L.; Santos, G.S.; Alkass, N.; Andrade, M.A.P.; Lana, J. The protective role of glutathione in osteoarthritis. J Clin Orthop Trauma 2021, 15, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Lucarini, E.; Micheli, L.; Martelli, A.; Testai, L.; Calderone, V.; Ghelardini, C.; Di Cesare Mannelli, L. Efficacy of isothiocyanate-based compounds on different forms of persistent pain. J Pain Res 2018, 11, 2905–2913. [Google Scholar] [CrossRef]
- Che, H.; Shao, Z.; Ding, J.; Gao, H.; Liu, X.; Chen, H.; Cai, S.; Ge, J.; Wang, C.; Wu, J.; et al. The effect of allyl isothiocyanate on chondrocyte phenotype is matrix stiffness-dependent: Possible involvement of TRPA1 activation. Front Mol Biosci 2023, 10, 1112653. [Google Scholar] [CrossRef]
- Chen, Y.; Xue, R.; Jin, X.; Tan, X. Antiarthritic Activity of Diallyl Disulfide against Freund’s Adjuvant-Induced Arthritic Rat Model. J Environ Pathol Toxicol Oncol 2018, 37, 291–303. [Google Scholar] [CrossRef]
- Ma, J.; Yang, P.; Zhou, Z.; Song, T.; Jia, L.; Ye, X.; Yan, W.; Sun, J.; Ye, T.; Zhu, L. GYY4137-induced p65 sulfhydration protects synovial macrophages against pyroptosis by improving mitochondrial function in osteoarthritis development. J Adv Res 2025, 71, 173–188. [Google Scholar] [CrossRef]
- Batalle, G.; Cabarga, L.; Pol, O. The Inhibitory Effects of Slow-Releasing Hydrogen Sulfide Donors in the Mechanical Allodynia, Grip Strength Deficits, and Depressive-Like Behaviors Associated with Chronic Osteoarthritis Pain. Antioxidants (Basel) 2019, 9. [Google Scholar] [CrossRef]
- Keppel Hesselink, J.M.; de Boer, T.; Witkamp, R.F. Palmitoylethanolamide: A Natural Body-Own Anti-Inflammatory Agent, Effective and Safe against Influenza and Common Cold. Int J Inflam 2013, 2013, 151028. [Google Scholar] [CrossRef] [PubMed]
- Bachur, N.R.; Masek, K.; Melmon, K.L.; Udenfriend, S. Fatty Acid Amides of Ethanolamine in Mammalian Tissues. J Biol Chem 1965, 240, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Fusco, R.; Cordaro, M.; Peritore, A.F.; D’Amico, R.; Gugliandolo, E.; Crupi, R.; Genovese, T.; Evangelista, M.; Di Paola, R.; et al. The Protective Effects of Pre- and Post-Administration of Micronized Palmitoylethanolamide Formulation on Postoperative Pain in Rats. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Cordaro, M.; Verde, R.; Schiano Moriello, A.; Marcolongo, G.; Schievano, C.; Siracusa, R.; Piscitelli, F.; Peritore, A.F.; Crupi, R.; et al. Oral Ultramicronized Palmitoylethanolamide: Plasma and Tissue Levels and Spinal Anti-hyperalgesic Effect. Front Pharmacol 2018, 9, 249. [Google Scholar] [CrossRef]
- Fowler, C.J.; Jonsson, K.O.; Tiger, G. Fatty acid amide hydrolase: biochemistry, pharmacology, and therapeutic possibilities for an enzyme hydrolyzing anandamide, 2-arachidonoylglycerol, palmitoylethanolamide, and oleamide. Biochem Pharmacol 2001, 62, 517–526. [Google Scholar] [CrossRef]
- Kaczocha, M.; Glaser, S.T.; Chae, J.; Brown, D.A.; Deutsch, D.G. Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J Biol Chem 2010, 285, 2796–2806. [Google Scholar] [CrossRef]
- Piomelli, D.; Scalvini, L.; Fotio, Y.; Lodola, A.; Spadoni, G.; Tarzia, G.; Mor, M. N-Acylethanolamine Acid Amidase (NAAA): Structure, Function, and Inhibition. J Med Chem 2020, 63, 7475–7490. [Google Scholar] [CrossRef] [PubMed]
- Congiu, M.; Micheli, L.; Santoni, M.; Sagheddu, C.; Muntoni, A.L.; Makriyannis, A.; Malamas, M.S.; Ghelardini, C.; Di Cesare Mannelli, L.; Pistis, M. N-Acylethanolamine Acid Amidase Inhibition Potentiates Morphine Analgesia and Delays the Development of Tolerance. Neurotherapeutics 2021, 18, 2722–2736. [Google Scholar] [CrossRef]
- Takizawa, M.; Hatta, T.; Iitsuka, H.; Katashima, M.; Sato, Y.; Kuroishi, K.; Nagashima, H. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ASP3652, a Reversible Fatty Acid Amide Hydrolase Inhibitor, in Healthy, Nonelderly, Japanese Men and Elderly, Japanese Men and Women: A Randomized, Double-blind, Placebo-controlled, Single and Multiple Oral Dose, Phase I Study. Clin Ther 2020, 42, 906–923. [Google Scholar] [CrossRef]
- Alhouayek, M.; Bottemanne, P.; Subramanian, K.V.; Lambert, D.M.; Makriyannis, A.; Cani, P.D.; Muccioli, G.G. N-Acylethanolamine-hydrolyzing acid amidase inhibition increases colon N-palmitoylethanolamine levels and counteracts murine colitis. FASEB J 2015, 29, 650–661. [Google Scholar] [CrossRef]
- Bonezzi, F.T.; Sasso, O.; Pontis, S.; Realini, N.; Romeo, E.; Ponzano, S.; Nuzzi, A.; Fiasella, A.; Bertozzi, F.; Piomelli, D. An Important Role for N-Acylethanolamine Acid Amidase in the Complete Freund’s Adjuvant Rat Model of Arthritis. J Pharmacol Exp Ther 2016, 356, 656–663. [Google Scholar] [CrossRef]
- Alhouayek, M.; Bottemanne, P.; Makriyannis, A.; Muccioli, G.G. N-acylethanolamine-hydrolyzing acid amidase and fatty acid amide hydrolase inhibition differentially affect N-acylethanolamine levels and macrophage activation. Biochim Biophys Acta Mol Cell Biol Lipids 2017, 1862, 474–484. [Google Scholar] [CrossRef]
- Larauche, M.; Mulak, A.; Ha, C.; Million, M.; Arnett, S.; Germano, P.; Pearson, J.P.; Currie, M.G.; Tache, Y. FAAH inhibitor URB597 shows anti-hyperalgesic action and increases brain and intestinal tissues fatty acid amides in a model of CRF(1) agonist mediated visceral hypersensitivity in male rats. Neurogastroenterol Motil 2024, 36, e14927. [Google Scholar] [CrossRef]
- Zhou, P.; Xiang, L.; Yang, Y.; Wu, Y.; Hu, T.; Liu, X.; Lin, F.; Xiu, Y.; Wu, K.; Lu, C.; et al. N-Acylethanolamine acid amidase (NAAA) inhibitor F215 as a novel therapeutic agent for osteoarthritis. Pharmacol Res 2019, 145, 104264. [Google Scholar] [CrossRef]
- Sagheddu, C.; Scherma, M.; Congiu, M.; Fadda, P.; Carta, G.; Banni, S.; Wood, J.T.; Makriyannis, A.; Malamas, M.S.; Pistis, M. Inhibition of N-acylethanolamine acid amidase reduces nicotine-induced dopamine activation and reward. Neuropharmacology 2019, 144, 327–336. [Google Scholar] [CrossRef]
- Bottemanne, P.; Guillemot-Legris, O.; Paquot, A.; Masquelier, J.; Malamas, M.; Makriyannis, A.; Alhouayek, M.; Muccioli, G.G. N-Acylethanolamine-Hydrolyzing Acid Amidase Inhibition, but Not Fatty Acid Amide Hydrolase Inhibition, Prevents the Development of Experimental Autoimmune Encephalomyelitis in Mice. Neurotherapeutics 2021, 18, 1815–1833. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Demartini, C.; Zanaboni, A.; Casini, I.; De Icco, R.; Reggiani, A.; Misto, A.; Piomelli, D.; Tassorelli, C. Characterization of the peripheral FAAH inhibitor, URB937, in animal models of acute and chronic migraine. Neurobiol Dis 2021, 147, 105157. [Google Scholar] [CrossRef]
- Vacondio, F.; Bassi, M.; Silva, C.; Castelli, R.; Carmi, C.; Scalvini, L.; Lodola, A.; Vivo, V.; Flammini, L.; Barocelli, E.; et al. Amino Acid Derivatives as Palmitoylethanolamide Prodrugs: Synthesis, In Vitro Metabolism and In Vivo Plasma Profile in Rats. PLoS One 2015, 10, e0128699. [Google Scholar] [CrossRef]
- Manzo, E.; Schiano Moriello, A.; Tinto, F.; Verde, R.; Allara, M.; De Petrocellis, L.; Pagano, E.; Izzo, A.A.; Di Marzo, V.; Petrosino, S. A Glucuronic Acid-Palmitoylethanolamide Conjugate (GLUPEA) Is an Innovative Drug Delivery System and a Potential Bioregulator. Cells 2021, 10. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Peritore, A.F.; Piras, C.; Cuzzocrea, S.; Crupi, R. Palmitoylethanolamide and Related ALIAmides: Prohomeostatic Lipid Compounds for Animal Health and Wellbeing. Vet Sci 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Roa-Coria, J.E.; Navarrete-Vazquez, G.; Fowler, C.J.; Flores-Murrieta, F.J.; Deciga-Campos, M.; Granados-Soto, V. N-(4-Methoxy-2-nitrophenyl)hexadecanamide, a palmitoylethanolamide analogue, reduces formalin-induced nociception. Life Sci 2012, 91, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Di Paola, R.; Cordaro, M.; Gugliandolo, E.; Casili, G.; Morittu, V.M.; Britti, D.; Esposito, E.; Cuzzocrea, S. Adelmidrol, a palmitoylethanolamide analogue, as a new pharmacological treatment for the management of acute and chronic inflammation. Biochem Pharmacol 2016, 119, 27–41. [Google Scholar] [CrossRef]
- Wallace, V.C.; Segerdahl, A.R.; Lambert, D.M.; Vandevoorde, S.; Blackbeard, J.; Pheby, T.; Hasnie, F.; Rice, A.S. The effect of the palmitoylethanolamide analogue, palmitoylallylamide (L-29) on pain behaviour in rodent models of neuropathy. Br J Pharmacol 2007, 151, 1117–1128. [Google Scholar] [CrossRef]
- Clayton, P.; Hill, M.; Bogoda, N.; Subah, S.; Venkatesh, R. Palmitoylethanolamide: A Natural Compound for Health Management. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, L.; Mattsson, S.; Fowler, C.J. Palmitoylethanolamide for the treatment of pain: pharmacokinetics, safety and efficacy. Br J Clin Pharmacol 2016, 82, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Zhukov, O.D. [Distribution of N-([1-14C]-palmitoyl)ethanolamine in rat tissues]. Ukr Biokhim Zh (1999) 1999, 71, 124–125. [Google Scholar]
- Artamonov, M.; Zhukov, O.; Shuba, I.; Storozhuk, L.; Khmel, T.; Klimashevsky, V.; Mikosha, A.; Gula, N. Incorporation of labelled N-acylethanolamine (NAE) into rat brain regions in vivo and adaptive properties of saturated NAE under x-ray irradiation. Ukr Biokhim Zh (1999) 2005, 77, 51–62. [Google Scholar] [PubMed]
- di Marzo, V.; Skaper, S.D. Palmitoylethanolamide: biochemistry, pharmacology and therapeutic use of a pleiotropic anti-inflammatory lipid mediator. CNS Neurol Disord Drug Targets 2013, 12, 4–6. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: a patent review (2014-present). Expert Opin Ther Pat 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Katsiki, N.; Nikolic, D.; Montalto, G.; Banach, M.; Mikhailidis, D.P.; Rizzo, M. The role of fibrate treatment in dyslipidemia: an overview. Curr Pharm Des 2013, 19, 3124–3131. [Google Scholar] [CrossRef]
- Keller, H.; Mahfoudi, A.; Dreyer, C.; Hihi, A.K.; Medin, J.; Ozato, K.; Wahli, W. Peroxisome proliferator-activated receptors and lipid metabolism. Ann N Y Acad Sci 1993, 684, 157–173. [Google Scholar] [CrossRef]
- Youssef, J.; Badr, M. Role of Peroxisome Proliferator-Activated Receptors in Inflammation Control. J Biomed Biotechnol 2004, 2004, 156–166. [Google Scholar] [CrossRef]
- Annunziata, C.; Pirozzi, C.; Lama, A.; Senzacqua, M.; Comella, F.; Bordin, A.; Monnolo, A.; Pelagalli, A.; Ferrante, M.C.; Mollica, M.P.; et al. Palmitoylethanolamide Promotes White-to-Beige Conversion and Metabolic Reprogramming of Adipocytes: Contribution of PPAR-alpha. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Melini, S.; Pirozzi, C.; Lama, A.; Comella, F.; Opallo, N.; Del Piano, F.; Di Napoli, E.; Mollica, M.P.; Paciello, O.; Ferrante, M.C.; et al. Co-Micronized Palmitoylethanolamide and Rutin Associated With Hydroxytyrosol Recover Diabesity-Induced Hepatic Dysfunction in Mice: In Vitro Insights Into the Synergistic Effect. Phytother Res 2024, 38, 6035–6047. [Google Scholar] [CrossRef]
- Cornali, K.; Di Lauro, M.; Marrone, G.; Masci, C.; Montalto, G.; Giovannelli, A.; Schievano, C.; Tesauro, M.; Pieri, M.; Bernardini, S.; et al. The Effects of a Food Supplement, Based on Co-Micronized Palmitoylethanolamide (PEA)-Rutin and Hydroxytyrosol, in Metabolic Syndrome Patients: Preliminary Results. Nutrients 2025, 17. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L. Mast cell-glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos Trans R Soc Lond B Biol Sci 2012, 367, 3312–3325. [Google Scholar] [CrossRef]
- Petrosino, S.; Schiano Moriello, A.; Verde, R.; Allara, M.; Imperatore, R.; Ligresti, A.; Mahmoud, A.M.; Peritore, A.F.; Iannotti, F.A.; Di Marzo, V. Palmitoylethanolamide counteracts substance P-induced mast cell activation in vitro by stimulating diacylglycerol lipase activity. J Neuroinflammation 2019, 16, 274. [Google Scholar] [CrossRef]
- De Filippis, D.; Luongo, L.; Cipriano, M.; Palazzo, E.; Cinelli, M.P.; de Novellis, V.; Maione, S.; Iuvone, T. Palmitoylethanolamide reduces granuloma-induced hyperalgesia by modulation of mast cell activation in rats. Mol Pain 2011, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, G.G.; Stella, N. Microglia produce and hydrolyze palmitoylethanolamide. Neuropharmacology 2008, 54, 16–22. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, I.; Rostampour, M.; Shabani, M.; Naderi, N.; Motamedi, F.; Babaei, P.; Khakpour-Taleghani, B. Palmitoylethanolamide attenuates PTZ-induced seizures through CB1 and CB2 receptors. Epilepsy Res 2015, 117, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Im, D.S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: involvement of the CB2 receptor. Sci Rep 2017, 7, 375. [Google Scholar] [CrossRef]
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARalpha agonist palmitoylethanolamide. Br J Pharmacol 2013, 168, 1430–1444. [Google Scholar] [CrossRef]
- Coluzzi, F.; Scerpa, M.S.; Alessandri, E.; Romualdi, P.; Rocco, M. Role of TRP Channels in Cancer-Induced Bone Pain. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Petrosino, S.; Schiano Moriello, A.; Cerrato, S.; Fusco, M.; Puigdemont, A.; De Petrocellis, L.; Di Marzo, V. The anti-inflammatory mediator palmitoylethanolamide enhances the levels of 2-arachidonoyl-glycerol and potentiates its actions at TRPV1 cation channels. Br J Pharmacol 2016, 173, 1154–1162. [Google Scholar] [CrossRef]
- Capasso, R.; Orlando, P.; Pagano, E.; Aveta, T.; Buono, L.; Borrelli, F.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide normalizes intestinal motility in a model of post-inflammatory accelerated transit: involvement of CB(1) receptors and TRPV1 channels. Br J Pharmacol 2014, 171, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Karwad, M.A.; Macpherson, T.; Wang, B.; Theophilidou, E.; Sarmad, S.; Barrett, D.A.; Larvin, M.; Wright, K.L.; Lund, J.N.; O’Sullivan, S.E. Oleoylethanolamine and palmitoylethanolamine modulate intestinal permeability in vitro via TRPV1 and PPARalpha. FASEB J 2017, 31, 469–481. [Google Scholar] [CrossRef] [PubMed]
- De Icco, R.; Greco, R.; Demartini, C.; Vergobbi, P.; Zanaboni, A.; Tumelero, E.; Reggiani, A.; Realini, N.; Sances, G.; Grillo, V.; et al. Spinal nociceptive sensitization and plasma palmitoylethanolamide levels during experimentally induced migraine attacks. Pain 2021, 162, 2376–2385. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Glia and mast cells as targets for palmitoylethanolamide, an anti-inflammatory and neuroprotective lipid mediator. Mol Neurobiol 2013, 48, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by beta-amyloid peptide. J Cell Mol Med 2011, 15, 2664–2674. [Google Scholar] [CrossRef]
- Beggiato, S.; Borelli, A.C.; Ferraro, L.; Tanganelli, S.; Antonelli, T.; Tomasini, M.C. Palmitoylethanolamide Blunts Amyloid-beta42-Induced Astrocyte Activation and Improves Neuronal Survival in Primary Mouse Cortical Astrocyte-Neuron Co-Cultures. J Alzheimers Dis 2018, 61, 389–399. [Google Scholar] [CrossRef]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-beta25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef]
- Kiani, A.K.; Miggiano, G.A.D.; Aquilanti, B.; Velluti, V.; Matera, G.; Gagliardi, L.; Bertelli, M. Food supplements based on palmitoylethanolamide plus hydroxytyrosol from olive tree or Bacopa monnieri extracts for neurological diseases. Acta Biomed 2020, 91, e2020007. [Google Scholar] [CrossRef]
- Facchinetti, R.; Valenza, M.; Bronzuoli, M.R.; Menegoni, G.; Ratano, P.; Steardo, L.; Campolongo, P.; Scuderi, C. Looking for a Treatment for the Early Stage of Alzheimer’s Disease: Preclinical Evidence with Co-Ultramicronized Palmitoylethanolamide and Luteolin. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Brotini, S.; Schievano, C.; Guidi, L. Ultra-micronized Palmitoylethanolamide: An Efficacious Adjuvant Therapy for Parkinson’s Disease. CNS Neurol Disord Drug Targets 2017, 16, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.I.; Lee, H.S.; Jeon, Y.E.; Kim, S.M.; Hong, S.H.; Moon, J.M.; Lim, C.Y.; Kim, Y.H.; Kim, E.J. Anti-inflammatory activity of palmitoylethanolamide ameliorates osteoarthritis induced by monosodium iodoacetate in Sprague-Dawley rats. Inflammopharmacology 2021, 29, 1475–1486. [Google Scholar] [CrossRef]
- Seol, T.K.; Lee, W.; Park, S.; Kim, K.N.; Kim, T.Y.; Oh, Y.N.; Jun, J.H. Effect of palmitoylethanolamide on inflammatory and neuropathic pain in rats. Korean J Anesthesiol 2017, 70, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Della Rocca, G.; Re, G. Palmitoylethanolamide and Related ALIAmides for Small Animal Health: State of the Art. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Cruccu, G.; Stefano, G.D.; Marchettini, P.; Truini, A. Micronized Palmitoylethanolamide: A Post Hoc Analysis of a Controlled Study in Patients with Low Back Pain - Sciatica. CNS Neurol Disord Drug Targets 2019, 18, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, D.; Asaro, C.; Lauricella, L.; Tomasello, S.; Varrassi, G.; Letizia Mauro, G. Combination of Rehabilitative Therapy with Ultramicronized Palmitoylethanolamide for Chronic Low Back Pain: An Observational Study. Pain Ther 2020, 9, 319–326. [Google Scholar] [CrossRef]
- Passavanti, M.B.; Fiore, M.; Sansone, P.; Aurilio, C.; Pota, V.; Barbarisi, M.; Fierro, D.; Pace, M.C. The beneficial use of ultramicronized palmitoylethanolamide as add-on therapy to Tapentadol in the treatment of low back pain: a pilot study comparing prospective and retrospective observational arms. BMC Anesthesiol 2017, 17, 171. [Google Scholar] [CrossRef]
- Chirchiglia, D.; Paventi, S.; Seminara, P.; Cione, E.; Gallelli, L. N-Palmitoyl Ethanol Amide Pharmacological Treatment in Patients With Nonsurgical Lumbar Radiculopathy. J Clin Pharmacol 2018, 58, 733–739. [Google Scholar] [CrossRef]
- Germini, F.; Coerezza, A.; Andreinetti, L.; Nobili, A.; Rossi, P.D.; Mari, D.; Guyatt, G.; Marcucci, M. N-of-1 Randomized Trials of Ultra-Micronized Palmitoylethanolamide in Older Patients with Chronic Pain. Drugs Aging 2017, 34, 941–952. [Google Scholar] [CrossRef]
- Paladini, A.; Varrassi, G.; Bentivegna, G.; Carletti, S.; Piroli, A.; Coaccioli, S. Palmitoylethanolamide in the Treatment of Failed Back Surgery Syndrome. Pain Res Treat 2017, 2017, 1486010. [Google Scholar] [CrossRef]
- Costa, B.; Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain 2008, 139, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell Mol Life Sci 2019, 76, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Asadi, A.; Shadab Mehr, N.; Mohamadi, M.H.; Shokri, F.; Heidary, M.; Sadeghifard, N.; Khoshnood, S. Obesity and gut-microbiota-brain axis: A narrative review. J Clin Lab Anal 2022, 36, e24420. [Google Scholar] [CrossRef]
- Farzi, A.; Frohlich, E.E.; Holzer, P. Gut Microbiota and the Neuroendocrine System. Neurotherapeutics 2018, 15, 5–22. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Essa, M.M.; Rathipriya, A.G.; Bishir, M.; Ray, B.; Mahalakshmi, A.M.; Tousif, A.H.; Sakharkar, M.K.; Kashyap, R.S.; Friedland, R.P.; et al. Gut dysbiosis, defective autophagy and altered immune responses in neurodegenerative diseases: Tales of a vicious cycle. Pharmacol Ther 2022, 231, 107988. [Google Scholar] [CrossRef]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Madry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol Life Sci 2017, 74, 2959–2977. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat Rev Immunol 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Di Lauro, M.; Guerriero, C.; Cornali, K.; Albanese, M.; Costacurta, M.; Mercuri, N.B.; Di Daniele, N.; Noce, A. Linking Migraine to Gut Dysbiosis and Chronic Non-Communicable Diseases. Nutrients 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, L.; Gualtieri, P.; Romano, L.; Marrone, G.; Noce, A.; Pujia, A.; Perrone, M.A.; Aiello, V.; Colica, C.; De Lorenzo, A. Role of Personalized Nutrition in Chronic-Degenerative Diseases. Nutrients 2019, 11. [Google Scholar] [CrossRef]
- Noce, A.; Marrone, G.; Di Daniele, F.; Ottaviani, E.; Wilson Jones, G.; Bernini, R.; Romani, A.; Rovella, V. Impact of Gut Microbiota Composition on Onset and Progression of Chronic Non-Communicable Diseases. Nutrients 2019, 11. [Google Scholar] [CrossRef]
- Noce, A.; Marchetti, M.; Marrone, G.; Di Renzo, L.; Di Lauro, M.; Di Daniele, F.; Albanese, M.; Di Daniele, N.; De Lorenzo, A. Link between gut microbiota dysbiosis and chronic kidney disease. Eur Rev Med Pharmacol Sci 2022, 26, 2057–2074. [Google Scholar] [CrossRef]
- Canale, M.P.; Noce, A.; Di Lauro, M.; Marrone, G.; Cantelmo, M.; Cardillo, C.; Federici, M.; Di Daniele, N.; Tesauro, M. Gut Dysbiosis and Western Diet in the Pathogenesis of Essential Arterial Hypertension: A Narrative Review. Nutrients 2021, 13. [Google Scholar] [CrossRef]
- Coluzzi, F.; Scerpa, M.S.; Loffredo, C.; Borro, M.; Pergolizzi, J.V.; LeQuang, J.A.; Alessandri, E.; Simmaco, M.; Rocco, M. Opioid Use and Gut Dysbiosis in Cancer Pain Patients. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Alvaro, D.; Caraceni, A.T.; Coluzzi, F.; Gianni, W.; Lugoboni, F.; Marinangeli, F.; Massazza, G.; Pinto, C.; Varrassi, G. What to Do and What Not to Do in the Management of Opioid-Induced Constipation: A Choosing Wisely Report. Pain Ther 2020, 9, 657–667. [Google Scholar] [CrossRef]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr Physiol 2016, 6, 603–621. [Google Scholar] [CrossRef]
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10. [Google Scholar] [CrossRef]
- Rusch, J.A.; Layden, B.T.; Dugas, L.R. Signalling cognition: the gut microbiota and hypothalamic-pituitary-adrenal axis. Front Endocrinol (Lausanne) 2023, 14, 1130689. [Google Scholar] [CrossRef]
- Geng, S.; Yang, L.; Cheng, F.; Zhang, Z.; Li, J.; Liu, W.; Li, Y.; Chen, Y.; Bao, Y.; Chen, L.; et al. Gut Microbiota Are Associated With Psychological Stress-Induced Defections in Intestinal and Blood-Brain Barriers. Front Microbiol 2019, 10, 3067. [Google Scholar] [CrossRef]
- Bertollo, A.G.; Santos, C.F.; Bagatini, M.D.; Ignacio, Z.M. Hypothalamus-pituitary-adrenal and gut-brain axes in biological interaction pathway of the depression. Front Neurosci 2025, 19, 1541075. [Google Scholar] [CrossRef]
- Suda, T. Adrenocorticotropic hormone (ACTH). Jpn J Med 1989, 28, 789–791. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- P. , S.; Vellapandian, C. Hypothalamic-Pituitary-Adrenal (HPA) Axis: Unveiling the Potential Mechanisms Involved in Stress-Induced Alzheimer’s Disease and Depression. Cureus 2024, 16, e67595. [Google Scholar] [CrossRef]
- Anand, N.; Gorantla, V.R.; Chidambaram, S.B. The Role of Gut Dysbiosis in the Pathophysiology of Neuropsychiatric Disorders. Cells 2022, 12. [Google Scholar] [CrossRef]
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front Mol Neurosci 2023, 16, 1247422. [Google Scholar] [CrossRef] [PubMed]
- Hamamah, S.; Aghazarian, A.; Nazaryan, A.; Hajnal, A.; Covasa, M. Role of Microbiota-Gut-Brain Axis in Regulating Dopaminergic Signaling. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Zhang, D.; Jian, Y.P.; Zhang, Y.N.; Li, Y.; Gu, L.T.; Sun, H.H.; Liu, M.D.; Zhou, H.L.; Wang, Y.S.; Xu, Z.X. Short-chain fatty acids in diseases. Cell Commun Signal 2023, 21, 212. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Gamage, H.; Laird, A.S. Butyrate as a potential therapeutic agent for neurodegenerative disorders. Neurochem Int 2024, 176, 105745. [Google Scholar] [CrossRef]
- Maldonado, R.F.; Sa-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol Rev 2016, 40, 480–493. [Google Scholar] [CrossRef]
- Basnet, T.B.; Gc, S.; Basnet, R.; Fatima, S.; Safdar, M.; Sehar, B.; Alsubaie, A.S.R.; Zeb, F. Interaction between gut microbiota metabolites and dietary components in lipid metabolism and metabolic diseases. Access Microbiol 2023, 5, acmi000403. [Google Scholar] [CrossRef]
- Mohr, A.E.; Crawford, M.; Jasbi, P.; Fessler, S.; Sweazea, K.L. Lipopolysaccharide and the gut microbiota: considering structural variation. FEBS Lett 2022, 596, 849–875. [Google Scholar] [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front Immunol 2020, 11, 594150. [Google Scholar] [CrossRef]
- Lu, S.; Zhao, Q.; Guan, Y.; Sun, Z.; Li, W.; Guo, S.; Zhang, A. The communication mechanism of the gut-brain axis and its effect on central nervous system diseases: A systematic review. Biomed Pharmacother 2024, 178, 117207. [Google Scholar] [CrossRef]
- Russo, R.; Cristiano, C.; Avagliano, C.; De Caro, C.; La Rana, G.; Raso, G.M.; Canani, R.B.; Meli, R.; Calignano, A. Gut-brain Axis: Role of Lipids in the Regulation of Inflammation, Pain and CNS Diseases. Curr Med Chem 2018, 25, 3930–3952. [Google Scholar] [CrossRef] [PubMed]
- Couch, D.G.; Cook, H.; Ortori, C.; Barrett, D.; Lund, J.N.; O’Sullivan, S.E. Palmitoylethanolamide and Cannabidiol Prevent Inflammation-induced Hyperpermeability of the Human Gut In Vitro and In Vivo-A Randomized, Placebo-controlled, Double-blind Controlled Trial. Inflamm Bowel Dis 2019, 25, 1006–1018. [Google Scholar] [CrossRef]
- Pirozzi, C.; Coretti, L.; Opallo, N.; Bove, M.; Annunziata, C.; Comella, F.; Turco, L.; Lama, A.; Trabace, L.; Meli, R.; et al. Palmitoylethanolamide counteracts high-fat diet-induced gut dysfunction by reprogramming microbiota composition and affecting tryptophan metabolism. Front Nutr 2023, 10, 1143004. [Google Scholar] [CrossRef]
- Correia, A.S.; Vale, N. Tryptophan Metabolism in Depression: A Narrative Review with a Focus on Serotonin and Kynurenine Pathways. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Batacan, R., Jr.; Briskey, D.; Bajagai, Y.S.; Smith, C.; Stanley, D.; Rao, A. Effect of Palmitoylethanolamide Compared to a Placebo on the Gut Microbiome and Biochemistry in an Overweight Adult Population: A Randomised, Placebo Controlled, Double-Blind Study. Biomedicines 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, D.; D’Amico, A.; Cinelli, M.P.; Esposito, G.; Di Marzo, V.; Iuvone, T. Adelmidrol, a palmitoylethanolamide analogue, reduces chronic inflammation in a carrageenin-granuloma model in rats. J Cell Mol Med 2009, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Palenca, I.; Seguella, L.; Zilli, A.; Basili Franzin, S.; Del Re, A.; Pepi, F.; Troiani, A.; Pesce, M.; Rurgo, S.; De Palma, F.D.E.; et al. Intrarectal Administration of Adelmidrol plus Hyaluronic Acid Gel Ameliorates Experimental Colitis in Mice and Inhibits Pro-Inflammatory Response in Ex Vivo Cultured Biopsies Derived from Ulcerative Colitis-Affected Patients. Int J Mol Sci 2023, 25. [Google Scholar] [CrossRef]
- Interdonato, L.; D’Amico, R.; Cordaro, M.; Siracusa, R.; Fusco, R.; Peritore, A.F.; Gugliandolo, E.; Crupi, R.; Coaccioli, S.; Genovese, T.; et al. Aerosol-Administered Adelmidrol Attenuates Lung Inflammation in a Murine Model of Acute Lung Injury. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Cerrato, S.; Brazis, P.; Della Valle, M.F.; Miolo, A.; Puigdemont, A. Inhibitory effect of topical adelmidrol on antigen-induced skin wheal and mast cell behavior in a canine model of allergic dermatitis. BMC Vet Res 2012, 8, 230. [Google Scholar] [CrossRef]
- Bonello, D.; Squarzoni, P. Effect of a mucoadhesive gel and dental scaling on gingivitis in dogs. J Vet Dent 2008, 25, 28–32. [Google Scholar] [CrossRef]
- Abramo, F.; Salluzzi, D.; Leotta, R.; Auxilia, S.; Noli, C.; Miolo, A.; Mantis, P.; Lloyd, D.H. Mast cell morphometry and densitometry in experimental skin wounds treated with a gel containing adelmidrol: a placebo controlled study. Wounds 2008, 20, 149–157. [Google Scholar]
- Guida, F.; Rocco, M.; Luongo, L.; Persiani, P.; Vulpiani, M.C.; Nusca, S.M.; Maione, S.; Coluzzi, F. Targeting Neuroinflammation in Osteoarthritis with Intra-Articular Adelmidrol. Biomolecules 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Del Re, A.; Palenca, I.; Seguella, L.; Pesce, M.; Corpetti, C.; Steardo, L.; Rurgo, S.; Sarnelli, G.; Esposito, G. Oral Adelmidrol Administration Up-Regulates Palmitoylethanolamide Production in Mice Colon and Duodenum through a PPAR-gamma Independent Action. Metabolites 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Vulpiani MC, Nusca SM, Latini E, Santoboni F, Musa F, Trischitta D, Coluzzi F, Lardo D, Ippolito, E. Hyaluronic acid alone versus hyaluronic acid associated with adelmidrol for Intra-articular treatment of knee osteoarthritis: A long-term follow-up. International Journal of Physiotherapy and Research 2023, 11, 4453–4460. [CrossRef]
Figure 1.
Meta-neuroinflammation and gut-brain axis. The communication between central nervous system and gut occurs through the hypothalamus-pituitary-adrenal axis, the activation of immune system that involved the lipopolysaccharide (LPS) pathway, and through the production of bacterial metabolites by the gut microbiota, such as short-chain fatty acids (SCFAs) and neurotransmitters. The hyperactivation of HPA axis, as consequence of stress conditions and metabolic diseases leads to an overproduction of cortisol, influencing the gut microbiota composition by the enteric nervous system, located in the submucosa and in the myenteric plexus of the gut wall. This HPA axis hyperactivation is implicated in neuroinflammation, impairments of the hippocampus and prefrontal cortex and gut eubiosis. In the dysbiotic gut microbiota, the lower production of SCFAs, contributes to increase the permeability of the intestinal barrier, as shown by a reduction in zonula occludens-1 and occludin. Moreover, the translocation of LPS in the bloodstream is responsible of bacterial endotoxemia, that dysregulates the immune system. The ultramicronized-palmitoylethanolamine (um-PEA) can restore the eubiosis of the gut microbiota, through the SCFAs-producing bacterial species, such as Turicibacter sanguinis, Bifidobacterium, Oscillospiraceae family. In fact, the enhancement release of butyrate increases the production of tight junctions and the expression of monocarboxylate transporters-1. Finally, um-PEA is able to limit immune cells recruitment and activation of intestinal mast cells and macrophages and to restore serotonin/kynurenine levels. Abbreviations: ACTH, adrenocorticotropic hormone; BBB, blood–brain barrier; BDNF, brain-derived neurotrophic factor; COX-2, cyclooxygenase-2; CRH, corticotropin-releasing hormone; GABA, gamma-aminobutyric acid; HFD, high-fat diet; HPA, hypothalamus-pituitary-adrenal axis; IDO, indoleamine-2,3-dioxygenase; IL-1β, Interleukin-1β; iNOS, inducible nitric oxide synthase; KYN, kynurenine; LPS, lipopolysaccharide; MCT-1, monocarboxylate transporters-1; TJs, tight junctions; TLR-4, toll-like receptors-4; TNF-α, tumor necrosis factor-α; TRP, tryptophan; um-PEA, ultramicronized palmitoylethanolamide; ZO-1, zonula occludens-1; 5-HT, serotonin.
Figure 1.
Meta-neuroinflammation and gut-brain axis. The communication between central nervous system and gut occurs through the hypothalamus-pituitary-adrenal axis, the activation of immune system that involved the lipopolysaccharide (LPS) pathway, and through the production of bacterial metabolites by the gut microbiota, such as short-chain fatty acids (SCFAs) and neurotransmitters. The hyperactivation of HPA axis, as consequence of stress conditions and metabolic diseases leads to an overproduction of cortisol, influencing the gut microbiota composition by the enteric nervous system, located in the submucosa and in the myenteric plexus of the gut wall. This HPA axis hyperactivation is implicated in neuroinflammation, impairments of the hippocampus and prefrontal cortex and gut eubiosis. In the dysbiotic gut microbiota, the lower production of SCFAs, contributes to increase the permeability of the intestinal barrier, as shown by a reduction in zonula occludens-1 and occludin. Moreover, the translocation of LPS in the bloodstream is responsible of bacterial endotoxemia, that dysregulates the immune system. The ultramicronized-palmitoylethanolamine (um-PEA) can restore the eubiosis of the gut microbiota, through the SCFAs-producing bacterial species, such as Turicibacter sanguinis, Bifidobacterium, Oscillospiraceae family. In fact, the enhancement release of butyrate increases the production of tight junctions and the expression of monocarboxylate transporters-1. Finally, um-PEA is able to limit immune cells recruitment and activation of intestinal mast cells and macrophages and to restore serotonin/kynurenine levels. Abbreviations: ACTH, adrenocorticotropic hormone; BBB, blood–brain barrier; BDNF, brain-derived neurotrophic factor; COX-2, cyclooxygenase-2; CRH, corticotropin-releasing hormone; GABA, gamma-aminobutyric acid; HFD, high-fat diet; HPA, hypothalamus-pituitary-adrenal axis; IDO, indoleamine-2,3-dioxygenase; IL-1β, Interleukin-1β; iNOS, inducible nitric oxide synthase; KYN, kynurenine; LPS, lipopolysaccharide; MCT-1, monocarboxylate transporters-1; TJs, tight junctions; TLR-4, toll-like receptors-4; TNF-α, tumor necrosis factor-α; TRP, tryptophan; um-PEA, ultramicronized palmitoylethanolamide; ZO-1, zonula occludens-1; 5-HT, serotonin.
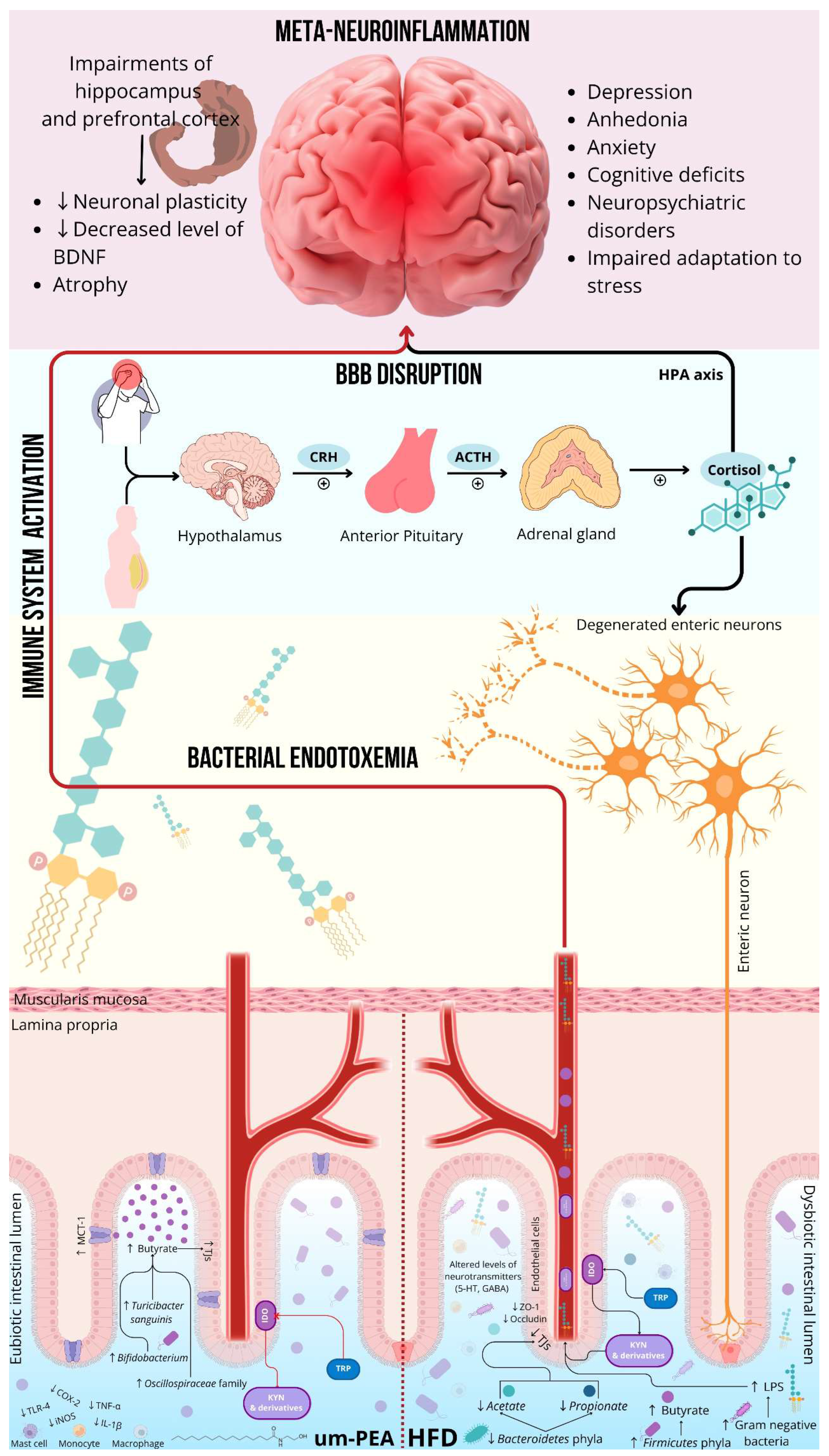
Figure 2.
Mechanisms of meta-neuroinflammation underlying OA. Possible targets of treatment for managing meta-neuroinflammation in obese patients suffering from OA-related chronic pain include: the pro-inflammatory state sustained by the adipose tissue, the microglial activation in the CNS, and peripheral hyperactivation of mast cells in the peripheral joints. The association m-PEA– rutin and HTyr has been shown to restore the eubiosis of the gut microbiota and to reduce body weight, body mass index, fat mass, and inflammation biomarkers. Um-PEA is proven to restore physiological role of microglia and to prevent central sensitization, which is a critical factor for pain chronification. Adelmidrol, a PEA congener, facilitates the switch of mast cells from the hyperactivated to the physiological state, therefore it reduces the degranulation of lytic enzymes and prevent HA degradation. Moreover, by normalizing mast cells production of heparin, it further increases the bioavailability of HA, supporting a visco-induction in the overloaded joints of obese patients. Abbreviations: HA, hyaluronic acid; HTyr, hydroxytyrosol; IL-1β, interleukin-1β; IL-6, interleukin-6; SP, substance P; TGF, transforming growth factor; TNF-α, tumor necrosis factor-α; TRKA, tropomyosin receptor kinase A; TRPV-1, transient receptor potential vanilloid; um-PEA, ultramicronized palmitoylethanolamide.
Figure 2.
Mechanisms of meta-neuroinflammation underlying OA. Possible targets of treatment for managing meta-neuroinflammation in obese patients suffering from OA-related chronic pain include: the pro-inflammatory state sustained by the adipose tissue, the microglial activation in the CNS, and peripheral hyperactivation of mast cells in the peripheral joints. The association m-PEA– rutin and HTyr has been shown to restore the eubiosis of the gut microbiota and to reduce body weight, body mass index, fat mass, and inflammation biomarkers. Um-PEA is proven to restore physiological role of microglia and to prevent central sensitization, which is a critical factor for pain chronification. Adelmidrol, a PEA congener, facilitates the switch of mast cells from the hyperactivated to the physiological state, therefore it reduces the degranulation of lytic enzymes and prevent HA degradation. Moreover, by normalizing mast cells production of heparin, it further increases the bioavailability of HA, supporting a visco-induction in the overloaded joints of obese patients. Abbreviations: HA, hyaluronic acid; HTyr, hydroxytyrosol; IL-1β, interleukin-1β; IL-6, interleukin-6; SP, substance P; TGF, transforming growth factor; TNF-α, tumor necrosis factor-α; TRKA, tropomyosin receptor kinase A; TRPV-1, transient receptor potential vanilloid; um-PEA, ultramicronized palmitoylethanolamide.
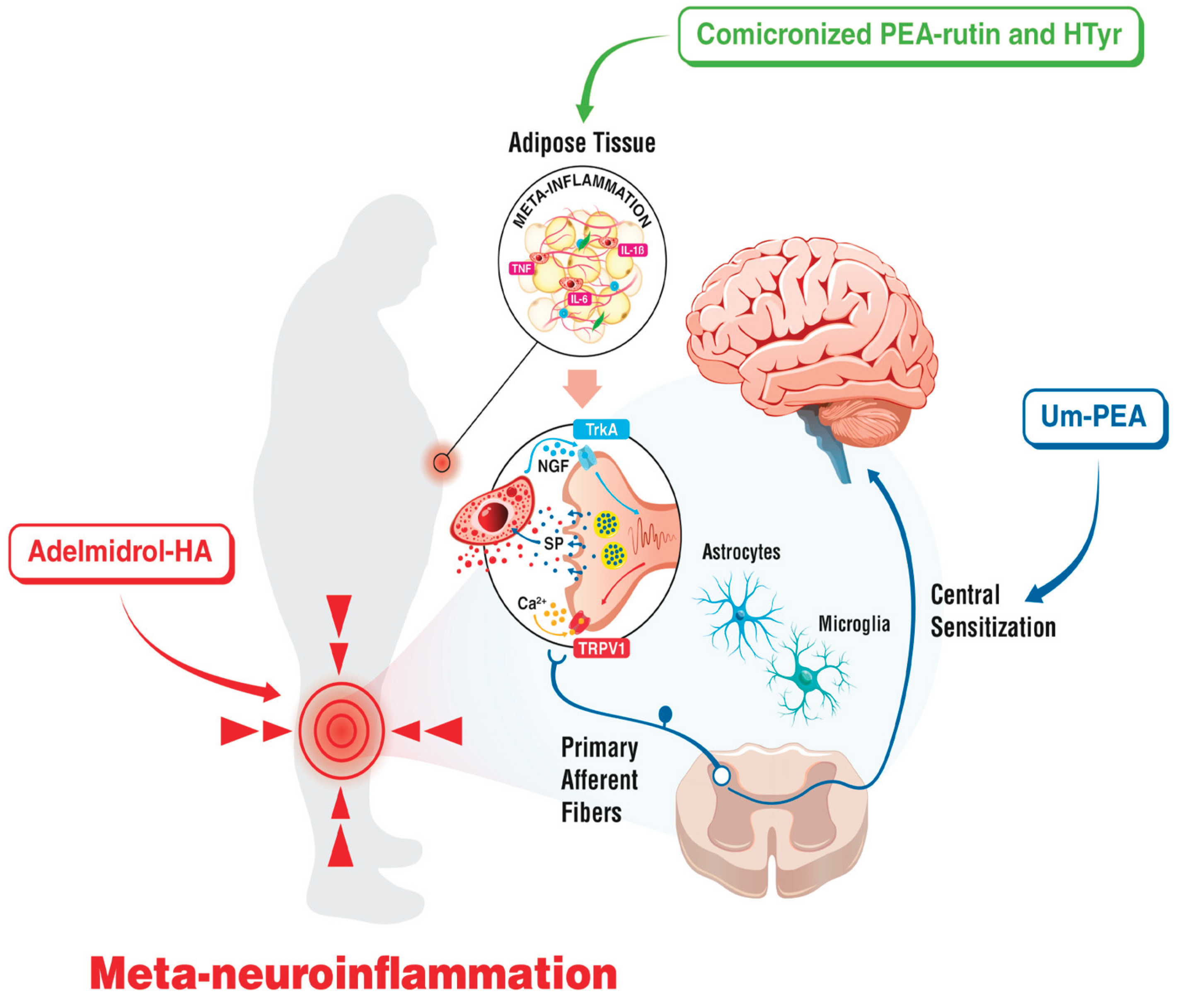
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



