Submitted:
28 July 2025
Posted:
29 July 2025
You are already at the latest version
Abstract
BECLIN-1 is a multidomain protein that through dynamic interaction with a variety of partners controls autophagy and apoptosis, two processes dysregulated in cancer cells, thus playing a crucial role in cell destiny. Mutations of BECN1 gene have not been described, yet its monoallelic deletion leads to insufficient autophagy with an impact on spontaneous cancer development, which identifies it as a haploinsufficient tumor suppressor gene. Epigenetic modulation of its expression, along with alternative splicing, post-translational modifications and alternative partner interactions influence its regulation of autophagy, thus affecting cancer cell behavior in terms of proliferation, motility, and survival under stress. In this review article we describe the structural and functional properties of BECLIN-1 and discuss how its altered expression and interaction with other proteins in cancer cells can be harnessed for diagnostic and therapeutic purposes.
Keywords:
autophagy
; BECN1
; tumor suppressor gene
; apoptosis
; epigenetics
; alternative splicing
; prognosis
1. Introduction
Autophagy is a lysosome-dependent process of disposal of damaged or superfluous cellular structures with recovery of material for resynthesis, essential to maintain homeostasis and cellular survival. This catabolic process was named “autophagy” in 1963 by Christian De Duve, the scholar winner of the Nobel Prize in 1974 for the discovery of lysosomes, since it implies the “eating of self” cellular components [1,2]. The physiological importance of this catabolic process has been neglected for decades till the early 90s, when its underlying molecular mechanisms started to be deciphered in the budding yeast Saccharomyces cerevisiae, primarily due to the pioneering work by Yoshimori Oshumi, who was awarded the Nobel Prize in 2016 for having cloned 15 genes involved in the process, and at the same time also by other researchers including Daniel Klionsky and Michael Thumm [2]. Soon, the orthologs of these genes have been identified in mammalian cells, which made it possible to study and thus better appreciate the pathophysiological relevance of this process also in mammalian organs. Amongst the very first yeast-like mammalian autophagy master regulator, a coiled-coil protein was first identified by the Levine’s Lab as an interactor of the anti-apoptotic BCL-2 and thereby nominated as BECLIN-1 [3]. Dr. Beth Levine (1960-2020) was a highly influential scientist whose groundbreaking research on autophagy and the discovery of BECLIN-1 (and BECLIN-2, see the Appendix) have had significant implications in basic biology and therapeutic applications for various diseases, including cancer and neurodegenerative disorders.
Autophagy is a pro-survival adaptive mechanism that aims to restore cell homeostasis in response to endogenous and environmental stress stimuli, and as such its dysregulation is causally linked to a wide range of diseases [4]. Basal autophagy maintains genomic stability and cell homeostasis by removing damaged organelles and protein aggregates, ensuring macromolecular and organelle turnover [5]. Insufficient autophagy may lead to cumulative oxidative stress and structural damages that eventually impair cell viability (thus leading to cell death) or pave the way to cell transformation as an adaptation to the chronic insults [5,6]. On the opposite side, hyperactivation of autophagy leads to excess eating of vital cellular components (e.g., mitochondria, endoplasmic reticulum, ribosomes), thus causing autophagic cell death [7]. Autophagy has surfaced as a dual-faceted element in cancer biology, oscillating between tumor-suppressive and tumor-promoting functions contingent upon disease stage and microenvironmental settings [8]. For example, in the hypoxic niche, dysregulated autophagy may confer to tumor cells the advantage of surviving in harsh metabolic conditions, leading to the persistence of dormant cells resistant to immune response and chemotherapy [9]. The mechanistic pivot underlying these context-dependent outcomes lies in the molecular composition of the autophagy machinery, particularly the class III PI3K complexes, whose scaffolding protein BECLIN-1 (BECN1) serves as both conductor and choreographer of cellular adaptation or succumbence, representing the switch between cell survival and death [10,11].
Figure 1 schematizes in a simplified model these concepts.
Despite three decades of research since BECN1's discovery, critical gaps persist in harnessing its therapeutic potential. First, the clinical relevance of BECN1 isoforms (e.g., BECN1-Δexon11) remains undefined, with truncated variants demonstrating dominant-negative effects on autophagosome maturation in breast cancer models [12]. Second, post-translational modifications like phosphorylation at Tyr133 by JAK2 and acetylation at Lys430/437, create dynamic "autophagy rheostats" that evade current biomarker assays. Third, the non-autophagic role of BECN1 in STAT3-driven metastasis challenges the paradigm of autophagy-centric targeting.
This review synthesizes emergent insights from structural biology (particularly the domains for post-translational modifications and partner interactions), omics-based stratification, and preclinical models of BECN1-targeted therapy to address these translational roadblocks. By reconciling molecular complexity with clinical realities, we chart a path toward context-aware therapeutic strategies that leverage BECN1's multifaceted biology beyond binary autophagy modulation.
- History of Beclin-1 discovery at glance
Human BECN1 gene, which is an ortholog of Atg6/Vps30 in yeast, is a haploinsufficient tumor suppressor [13]. The gene consists of 11 introns and 12 exons that code for the BECLIN-1 protein composed of 450 amino acids (aa), with a predicted molecular weight of 60 kDa. The seminal works of Levine’s group date to 1999 when the gene BECN1 was precisely mapped on the long arm of chromosome 17 (17q21) localized within 100 kb of the well-known breast cancer susceptibility region that retains BRCA1 and experiences heterozygous deletions in most breast and ovarian cancers [14,15]. After the genomic characterization, Beth Levine’s group set a milestone by identifying the sequence homology of human BECN1 to the yeast Atg6/Vps30. The induction of autophagy was promoted, and tumor cell proliferation was suppressed when BECN1 expression was restored in Atg6/Vps30 defective yeast or in BECN1 mono-allelic deleted MCF7 breast cancer cells [13]. Subsequently, in vivo studies confirmed that Becn1 (+/-) mutant mice suffered from spontaneous tumor development even with a basal expression of the wild-type mRNA and protein [16,17].
These instrumental researches laid the groundwork for further explorations that elaborated on the localization of BECN1 to trans-Golgi networks in the form of a complex with Class III PI3K lipid kinase or VPS34 [18]. Subsequent research continued to characterize the structural construction of BECN1 and its interactome that includes two mutually exclusive Class III PI3K complexes (PI3KC3): C1 (BECN1, VPS34, ATG14, and VPS15) and C2 (BECN1, VPS34, UVRAG, and VPS15), that are involved in the autophagosome formation and maturation, respectively [19,20,21]. Structural investigations elucidating the ECD (Evolutionarily Conserved Domain)/BARA (Beta-Alpha Repeated, Autophagy) domain (residues 244-337) disclose an allosteric switch that regulates the assembly of the PI3KC3-C1/C2 complex, with phosphorylation at Thr388 (ECD) and ubiquitination at Lys117 (BH3) determining membrane recruitment and contact dynamics [22,23].
BECN-1 surpasses its canonical role as an autophagy scaffold, serving as a signaling hub that coordinates endo-lysosomal trafficking, receptor degradation, cell proliferation, and cell death pathways through dynamic partner interchanges [24,25]. For instance, the N-terminal domain of BECN1 (residues 1–144) orchestrates EGFR/HER2 endocytosis via GRB2 coupling, whereas its nuclear pool modulates RB/E2F signaling—mechanisms exploited in HER2+ breast and colorectal cancers to sustain proliferative signaling [26,27].
The discovery of BECLIN-1 and its role in orchestrating the intricate process of autophagy has revolutionized our understanding of cellular homeostasis and disease processes. The findings from Beth Levine’s group not only established BECLIN-1 as a crucial player in autophagy but also stated its significance as a tumor suppressor. As research continues to disentangle the complexity of BECLIN-1 and its regulatory network, we can anticipate significant advancements in our ability to modulate autophagy for therapeutic benefits. The legacy of Dr. Levine’s work on BECLIN-1 will undoubtedly continue to shape the field of autophagy research and impact the development of novel treatment strategies for years to come.
- 2.
- Molecular Architecture and Interaction Landscape of BECLIN-1
The crystal structure of BECLIN-1 has helped identify the various distinct domains involved in protein-protein interactions, asserting its role as a necessary scaffold for complexes with diverse functions [28]. Figure 2 depicts the schematic representation of the different complexes.
An N-terminal BCL-2 homology 3 (BH3) domain (amino acids 108-127) through which it interacts with various BCL-2 family proteins, like BCL-2 and BCL-XL, to inhibit autophagy [29]. Among various BCL-2 homologs, BHRF1 (an Epstein-Barr virus protein structurally similar to BCL-2) has shown that BH3D alone is insufficient for its binding to BECLIN-1 and requires several amino acid residues flanking both sides, specifically from positions 90 to 170 [30]. DIRAS3, a recently found imprinted tumor suppressor, is a 26 kDa GTPase belonging to a unique subgroup of the RAS family. Exceptionally, this epigenetically controlled protein interacts with the N-terminal region of BECLIN-1 (amino acids 1-144), leading to the inhibition of BECLIN-1 homodimerization and the BECLIN-1-BCL-2 interaction. Simultaneously, it enhances the interaction between BECLIN-1 and ATG14L, thereby promoting autophagy [31]. An alternate BECN1-dependent autophagy pathway involves its interaction with the vesicle acidification regulator protein on the endosomal/lysosomal lumen, TMEM9, through the BH3 domain of BECN1, allowing the competitive displacement of BCL-2. This interaction is essential for the activation of Rab9-dependent alternative autophagy [32].
The central coiled-coil domain (CCD) (amino acids 174-266) is an interacting platform important for the autophagic and endocytic cofactors ATG14, AMBRA1, BIF1, UVRAG, and RUBICON [20,33,34]. ATG14 interacts with BECN1 and VPS34 on the primary site of phagophore initiation and in the late stages of autophagosome biogenesis, when autophagosomes are mature and are crucial for autophagy completion [34,35,36]. The BECN1 CCD domain is flanked by a highly conserved flexible helix or coiled-coil 1 (CC1) domain on its left (amino acids 141-171) that is essential for initiating the starvation-induced autophagy by offering autophagy regulators like AMBRA1 to bind [37]. The mutually exclusive competitive interactions between AMBRA1, BECLIN-1, and BCL-2 at the endoplasmic reticulum and at the mitochondrial membrane finely tune the crosstalk between pro-survival autophagy and apoptosis depending on the type and duration of the stress [37,38]. In short, under physiological conditions (when autophagy is regulated at the minimum level necessary to maintain cellular homeostasis) BCL-2 (with NAF-1, nutrient-deprivation autophagy factor-1) at the endoplasmic reticulum binds to BECN1 to down-regulate autophagy while AMBRA1 is largely bound to BCL-2 at mitochondria, whereas upon stress-induction of autophagy AMBRA1 relocates to the endoplasmic reticulum to displace BCL-2 and promoting the formation of the BECN1-PI3KC3 complex to initiate the phagophore biogenesis [37] and at the same time frees BCL-2 to prevent cell death [39].
UVRAG is critical for endosomal trafficking and interacts with BECN1 and VPS34 (PI3KC3) in early and late endosomes [40]. At the same time, RUBICON acts as a negative regulator of autophagy and instead drives alternative endocytic pathways, such as LC3-associated phagocytosis (LAP) and LC3-associated endocytosis (LANDO), utilizing the autophagy machinery in a non-canonical manner [41]. Nutrient deprivation induces BIF1 to act as a sensor, interacting with ATG5 and LC3 on the emerging phagophore to promote autophagosome biogenesis. While the specific interaction site of BIF1 on BECN1 is not explicitly detailed in the literature, it is known that UVRAG mediates their interaction. This BIF1-UVRAG-BECN1 complex is functionally involved in autophagosome formation and, notably, plays a crucial role in receptor degradation and cytokinesis, distinct from the BECN1-ATG14 complex [42,43].
The C-terminal evolutionary conserved domain (ECD) (244-337 aa) is essentially the membrane binding domain for BECN1 that allows its interaction with VPS34, stimulating its lipid kinase activity for producing the lipidic messenger PI3P necessary for autophagosome-related proteins recruitment [44]. This region also comprises three β-sheet-α-helix repeats and is therefore known as β-α repeated, autophagy-specific domain or BARAD [22]. VPS15, a large scaffolding protein (150kDa), plays a crucial role in bridging and stabilizing VPS34 and ATG14 on the BECN1 subcomplex [45]. This complex anchors the catalytic domains of VPS15 and VPS34 onto the autophagosomal membranes for phosphorylation and serves as a scaffold for recruiting other proteins that modulate PI3KC3 activity [46,47]. The interaction between VPS15 and BECN1 occurs via three key BECN1 binding sites: the ECD, which overlaps with the BARA domain; the C-terminal residues 425-450, essential for membrane localization; and an intrinsically disordered N-terminal region that receives the majority of stimulating phosphorylation events. These interactions regulate the PI3KC3-C1 and C2 complexes, with the BECN1-BARAD interacting with the WD40 domain of VPS15 [38,47]. Notably, the 26 amino acids (425-450) at the C-terminal of BECN1 play a fundamental role in its membrane localization and control of autophagosome biogenesis. Deletion of this domain significantly impairs BECN1 membrane localization, highlighting its importance in the complex's function [48]. In addition, GRB2, a well-known adaptor protein for HER2 and other tyrosine kinases, has been identified as a novel BECN1 interacting partner. This interaction occurs specifically through BECN1-ECD, as demonstrated by immunoprecipitation experiments showing that a BECN1 mutant lacking the ECD domain (dECD, d267–450) fails to bind to GRB2. This finding suggests that GRB2 may serve as a molecular bridge between HER2 signaling and autophagy control, potentially explaining how HER2 overexpression can influence autophagy and tumor progression [26].
Lastly, the nuclear export signal (NES) domain of BECN1, corresponding to residues 180-189, has been a subject of evolving research. Initially identified as a leucine-rich motif binding to chromosomal maintenance protein 1 (CRM1) for nuclear export, this domain was thought to be crucial for BECN1's autophagy and tumor suppressor functions [49]. However, recent studies have revealed more complex nuclear functions of BECN1. Intranuclear BECN1 interacts with topoisomerase-II to promote DNA repair independently of autophagy [50]. Furthermore, nuclear BECN1, unable to induce autophagy, can modulate retinoblastoma (Rb) protein expression, thereby regulating cell cycle and colorectal cancer cell growth [27]. While earlier research emphasized the NES-mediated nuclear-cytoplasmic shuttling, current investigations suggest a more nuanced understanding of BECN1's subcellular localization and functional diversity. The domain's significance in promoting specific cellular processes remains an active area of research, challenging previous assumptions about its singular function in nuclear export.
2. Multilayered Regulation of Beclin-1 in Cancer
BECN1 gene mutations are typically not found or very rare in human tumors [51]; however, this gene is monoallelically deleted in at least 75% of ovarian cancer and 50% of breast cancer [14]. Complex mechanisms at the transcriptional and post-transcriptional levels govern BECN-1 expression and function. Epigenetic regulation may also contribute to control transcript levels of this gene through methylation in the promoter region by methyltransferases such as EHMT2/G9a, which in conjunction with DNMT1 can repress the transcription of BECN1 regulating the H3K9me2 [52,53], or other epigenetic regulators such as the histone demethylase LSD1/KDM1A [54], and the acetyltransferases KLF5 and HDAC3 [55]. The transcription regulation in cancers can be orchestrated by multiple mechanisms, including transcription factors adjusting the expression of the BECN1 gene, epigenetic changes affecting chromatin accessibility, and post-transcriptional regulators such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), which are summarized in Figure 3.
These non-coding RNAs have the ability to destabilize the BECN1 mRNA transcript or impede its translation. From a translational perspective, it is interesting to note that many natural products (particularly nutraceuticals such as resveratrol and curcumin) exert antitumor effects also through epigenetic regulation of BECLIN-1-dependent autophagy [56]. Moreover, post-translational modifications like phosphorylation and ubiquitination play a significant role in affecting BECLIN-1 protein stability and function, introducing an additional layer of regulatory intricacy. Recent reports have confirmed the involvement of alternative splicing events that generate unique BECN1 mRNA isoforms, thereby increasing the diversity of BECLIN-1 protein variations [12]. The intricate regulation of this crucial autophagy regulator's expression and functional activity is a testament to the sophisticated control mechanisms that govern it.
2.1. Transcriptional Regulation of BECN1
E2F family of transcription factors (E2F1, E2F2, E2F3) specifically bind within the BECN1 promoter, activating its transcription [57,58,59,60]. Depletion of E2F1-3 in osteosarcoma cells significantly reduces BECN1 mRNA and protein levels, linking E2F activity to autophagy modulation [59].
NF-κB is a major regulator of inflammatory and immune responses which when triggered by stress stimuli such as ROS or starvation, can promote BECN1 transcription through its subunit RelA/p65 that binds to the BECN1 promoter and upregulates its expression in T cells or cancer cells. This marks a potential mechanism by which NF-κB promotes cell survival through autophagy induction [61,62]. The inhibitors of apoptosis proteins (IAPs), X-linked IAP (XIAP), and cellular IAP1 (cIAP1), can further activate NF-κB and induce autophagy by upregulating BECN1 transcription, a mechanism potentially linked to chemotherapy resistance in certain cancers [63]. Conversely, the protein TRIM59 acts as a negative regulator of the NF-κB pathway, attenuating BECN1 transcription and modulating autophagy in non-small cell lung cancer (NSCLC) [64].
GA binding protein or GABP is also known to enhance BECLIN1-dependent autophagy and cell survival in triple-negative breast cancer cells by activating BECN1 transcription and autophagosome formation upon starvation, where forkhead box M1 (FOXM1) directly binds to the BECN1 promoter [65,66]. In contrast, the transcription factor KLF5 (Krüppel-like factor 5), in cooperation with HDAC3 (histone deacetylase 3), can bind to and inhibit the BECN1 promoter, leading to the suppression of BECN1 transcription and autophagy in prostate cancer cells, potentially contributing to docetaxel resistance [67].
2.2. Epigenetic Modifiers of BECN1
The human BECN1 gene contains a 1.5 kb CpG island from its 5' end to intron 2, which can become aberrantly methylated in breast cancer. This highlights DNA methylation in the BECN1 promoter and intronic regions as a silencing mechanism which correlates with its reduced expression in sporadic breast cancers [53]. Epigenetic regulation also plays a pivotal role. EHMT2/G9a, a histone methyltransferase, cooperates with DNMT1 to suppress BECN1 via H3K9me2 deposition at its promoter. Treatment with BIX-01294, an EHMT2 inhibitor, can reverse the suppression of BECN1 by allowing NF-κB and RNA polymerase II recruitment over the promoter region, thereby inducing autophagy in MCF-7 breast cancer cells [52,55]. Alternatively, the well-known signal transducer for tumor growth and progression STAT3 is recognized to epigenetically target BECN1 repression in NSCLC by recruiting HDAC3 to its promoter, reducing acetylation and transcriptional activity [68].
2.3. Post-Transcriptional Control of BECN1 mRNA Translation in Cancer Cells
2.3.1. miRNAs Targeting BECN1 and Their Impact on Cancer Behavior
Micro-RNAs (miRNAs) act as post-transcriptional regulators by binding to complementary sequences in the 3'-untranslated regions (3'-UTR) of target mRNAs, resulting in gene silencing. Numerous miRNAs have been found to modulate BECLIN-1 protein expression by either inhibiting the mRNA translation or degrading its mRNA. Among these, miR-30a and miR-376b are the first identified and the most thoroughly defined as directly inhibiting BECN1 mRNA translation and consequently preventing rapamycin- or starvation-induced autophagy in cancer cells [69,70].
Studies targeting BECN1 mRNA in tumor cells with miRNAs have revealed the dual role (promoting vs. preventing) of BECLIN-1-dependent autophagy in radiotherapy- and chemotherapy-induced cell death, which aligns with the differential role of autophagy in response to toxic stress depending on the tumor context (genetic and epigenetic background; microenvironment) and stage. This is exemplified by the observation that BECN1 mRNA is targeted by both miR-30a and miR-376b, considered tumor suppressor and oncogenic miRNAs, respectively. Additionally, the interpretation of these experiments should also consider the possibility that the same miRNAs can target simultaneously different ATG mRNAs, thus making it difficult to attribute solely to BECLIN-1 downregulation the outcome. For instance, the suppression of BECLIN-1 synthesis by miR-17-5p abrogated autophagy-dependent resistance to irradiation in glioma cells xenografted in nude mice [71]. However, this same microRNA was previously shown to target ATG7 mRNA leading to inhibition of autophagy and concomitant increased sensitivity to low-dose ionizing radiation in human glioblastoma cells [72]. In glioblastoma stem-like cells, miR-93 targeted BECN1 mRNA (along with ATG5, ATG4B, and SQSTM1 mRNAs) to down-regulate autophagy and reduce sphere growth and increasing sensitivity to radiation and temozolomide [73].
Increased radiosensitivity was also induced in pancreatic cancer cells upon inhibition of BECLIN-1-dependent autophagy by miR-216a [74]. In this same line, the inhibition of BECLIN-1 expression (and of autophagy) by over-expression of miR-17-5p restored sensitivity to paclitaxel-induced apoptosis in resistant lung cancer cells [75]. Consistent with the above, the downregulation of miR-30a led to increased BECLIN-1-mediated autophagy and associated chemoresistance in gastric and lung cancer cells [76,77] and viceversa hyper-expression of miR-30a restored sensitivity to cis-platinum in several cancer cell types [78] as well as to imatinib in gastrointestinal stromal cells [79] and in chronic myeloid leukemia cells [80] by inhibiting BECLIN-1-mediated autophagy. A similar sensitization to oxaliplatin was reported in colon cancer cells upon miR-409-3p-mediated suppression of BECLIN-1-dependent autophagy [81]. In non-small lung cancer cells, the sponging effect of the lncRNA PVT1 on miR-216b led to increased BECLIN-1-dependent autophagy and decreased sensitivity to cisplatin [82]. The ectopic expression of miR-216b increased the sensitivity of melanoma cells to vemurafenib (an inhibitor of BRAF (V600E)) by targeting BECLIN-1, UVRAG, and ATG5, and thus suppressing autophagy [83].
Above miRNAs negatively regulating the expression of BECLIN-1 proved to cure the malignant phenotype through inhibition of autophagy. However, there are contexts where BECLIN-1-mediated autophagy elicits anti-tumor effects. For instance, in breast cancer cells, miR-221-mediated down-regulation of BECLIN-1-mediated autophagy resulted in increased aggressiveness and resistance to cell death induced by IL-24 (aka mda-7), a cytokine member of the IL-10 family known to kill specifically cancer cells and able to down-regulate miR-221 [84].
Finally, miR-26a and miR-124-3p are two other microRNAs found to target specifically the UTR of BECN1 mRNA in retinoblastoma and breast cancer cells, respectively [85,86].
2.3.2. LncRNAs Targeting BECN1 and Their Impact on Cancer Biology
Long non-coding RNAs (lncRNAs) have emerged as crucial co-regulators of various biological processes for maintaining cellular homeostasis that when altered can become causative factors in the pathogenesis of several diseases, including cancer [87]. These RNA molecules, although not translated into proteins, play significant roles in regulating gene activity, including the expression of the BECN1 gene, which is central to autophagy.
Several lncRNAs have been identified to regulate BECLIN-1 expression either directly or indirectly. For instance, lncRNA H19 is well known to up-regulate it by inhibiting S-adenosylhomocysteine hydrolase (SAHH), leading to decreased methylation of the BECN1 promoter by DNMT3B. In response to the increased BECN1 expression, breast cancer cells enhanced autophagy and resistance to tamoxifen [88]. LncRNAs also act as competing endogenous RNA (ceRNA), sponging miRNAs to modulate the expression of their targets. For example, lncRNA PVT1, which is overexpressed in cisplatin-resistant lung cancer cells, was found to sponge miR-216b, which essentially targets BECN1 expression, thereby upregulating pro-survival autophagy [89]. Similarly, lncRNA HOTAIR functions as a ceRNA for miR-17-5p, reducing its levels to promote BECN1 expression and enhance autophagy associated with sunitinib resistance in renal cancer cells [90]. Conversely, overexpression of lncRNA PANDAR in lung cancer promoted BECLIN-1 expression at both transcriptional and translational levels through still undefined mechanisms, thereby inhibiting cancer progression [91].
Furthermore, lncRNAs lead inter-regulatory pathways which involve numerous proteins such as the RNA-binding proteins (RBP) that can have a critical impact on the BECN1 mRNA metabolism. These RBPs may influence the mRNA stability, transport, and overall expression dynamics [92]. A significant study by Wang and colleagues conveyed that lncRNA FIRRE interacts and induces the translocation of polypyrimidine tract-binding protein (PTBP1) from the nucleus to the cytoplasm, stabilizing BECN1 mRNA. This stabilization in turn enhances BECN1 protein synthesis ultimately facilitating the autophagic activity in colorectal cancer [93]. More recently, ELAVL1 (embryonic lethal abnormal vision like RNA-binding protein 1) has been identified to exhibit a positive correlation with BECN1 expression levels in a NEAT1-dependent manner. It was shown that ELAVL1 imposes the stabilization of lncRNA NEAT1 in the endometrial cancer cells, thereby modulating BECLIN-1-mediated autophagy and contributing to cancer progression [94].
In summary, the recently found limelight transcriptomic factors or the non-coding RNAs play significant roles in the regulation of BECLIN-1, impacting autophagic activity and cancer progression through complex gene expression mechanisms.
2.3.3. Alternative Splicing of BECN1
Alternative splicing is a process that results in various protein variants being produced from the same mRNA. This mechanism is particularly relevant in tumorigenesis, where it is often deregulated, and it is considered one of the hallmarks of cancer. Recent studies have shown that BECN1 mRNA also undergoes alternative splicing to obtain isoforms with specific functions in autophagy. One first variant, named BECN1s, has been identified in HeLa cells. In vitro experiments displayed that this variant, despite harboring a partial deletion of the ECD and the C-terminal domain, well preserves the binding site for VPS34 and has been shown to drive the PARK2-dependent mitochondria-selective autophagy (mitophagy) process [95]. In our study [12], we corroborated the finding in ovarian cancer cells of splicing variant BECN1-α lacking exon 11, similar to what was observed by another group in human B-cells acute lymphoblastic leukemia cells [96]. The structural prediction of this variant pointed out a truncation (95 aa) in the C-terminal domain comprising the aromatic motif critical for autophagy and the site for VPS15 and VPS34 interaction, that impaired starvation-mediated autophagy. In addition to BECN1-α, two additional splicing variants, BECN1-β and -ɣ have also been identified in our study [12], which are graphically summarized in Figure 4. The first lacks the exons 5, 6, 10, and 11 that cause the deletion of the BH3 domain, losing the inhibitory site for BCL-2 interaction, and partially of the CCD and ECD, which makes the formation of the pro-autophagic complex weaker. Interestingly, the overexpression of this variant is enough to abrogate basal autophagy, suggesting a dominant negative effect on the BECLIN-1 wild-type. Instead, the latter variant lacks the same exons of BECN1- except for exon 10. At the structural level, this variant loses the BH3 domain and 95 aa in the C-terminal domain, but it keeps an intact ECD. At the biological level, this variant is still prone to modulate basal autophagy, even with less efficiency compared to the BECLIN-1 wild-type protein [12]. These findings support further studies to investigate the existence of Beclin-1 variants in other cancer model systems to understand their role in autophagy and endocytosis and explore their potential use as biomarkers in clinical settings.
3. Post-Translational Modifications on BECN1
Beclin-1 and its function in autophagy is extensively regulated by multiple post-translational modifications, including ubiquitination, acetylation, phosphorylation and others, which tune its stability, activity, and interactions with other proteins [38,97].
3.1. Ubiquination of BECN1
The ubiquitin-proteasome system (UPS) is a complex and dynamic regulatory mechanism that modifies proteins through the attachment of ubiquitin molecules to certain lysine residues, forming various connections that determine unique functional outcomes. Importantly, K63-linked ubiquitination often facilitates autophagy by stabilizing BECLIN-1 and augmenting its contacts, whereas K48-linked ubiquitination generally directs BECLIN-1 towards proteasomal destruction, hence impeding autophagy [97]. Moreover, K11-linked ubiquitination has been identified as an alternative degradation signal affecting BECLIN-1 stability [98]. This diversity of ubiquitin chain types provides multiple control mechanisms for BECLIN-1.
Researchers have identified multiple deubiquitinating enzymes (DUBs) that regulate the stability and function of BECLIN-1 and its consequent role in autophagy process. Recently, USP24 was recognized as a pivotal regulator of autophagy-mediated ferroptosis in hepatocellular carcinoma. It was shown that USP24 delays BECLIN-1 degradation by specifically reducing its K48-linked ubiquitination, thereby promoting autophagy and ferroptosis to suppress tumor growth [99]. Deubiquitinase USP5 stabilizes BECLIN-1 and enhances its nuclear accumulation, which inhibits p53-dependent senescence through MDM2-mediated p53 degradation. This USP5-BECLIN-1 axis is critical in overriding p53-dependent senescence in KRAS-driven tumorigenesis [100].
Other deubiquitinating enzymes, such as USP10, directly bind with BECLIN-1 to eliminate K48-linked ubiquitination, predominantly at Lys117 (K117) and Lys263 (K263), thus inhibiting its proteasomal destruction and facilitating autophagy. Conversely, USP13 selectively eliminates K63-linked ubiquitination, particularly at Lys402 (K402), hence influencing BECLIN-1's function in autophagic signaling and lysosomal transport [101]. Moreover, USP11 and USP14, which cleave K48, as well as USP19, which cleaves K437, have established contacts with BECLIN-1, thereby facilitating its stability and enhancing the regulation of autophagy [98,102,103,104]. Nonetheless, USP14, governed by AKT-mediated phosphorylation, inhibits autophagy by cleaving K63-linked ubiquitin chains from BECLIN-1, thus obstructing its association with the class III PI3K complex. USP14 functions as a dual regulator of autophagy and ubiquitin-proteasome system pathways [104]. Therefore, the differential yet complementary actions of these DUBs may highlight their crucial role in autophagic homeostasis, with their dysregulation implicated in cancer progression and other disorders [99,105].
The CUL3 (cullin 3) E3 ubiquitin ligase, in conjunction with the substrate adaptor KLHL38, facilitates K48-linked ubiquitination of BECLIN-1, resulting in its proteasomal destruction. This method inhibits autophagy and promotes tumor advancement in breast and ovarian malignancies, with elevated CUL3 expression associated with unfavorable patient outcomes [106]. Another E3 ligase, RNF216, facilitates K48-linked ubiquitination of BECLIN-1 in TLR-activated macrophages, hindering antimicrobial autophagy and advancing colon cancer progression [107].
The non-proteolytic ubiquitination of BECLIN-1 continues to be an area of active investigation. TRAF6 (E3-ligase tumor necrosis factor receptor (TNFR)-associated factor 6) promotes a non-proteolytic ubiquitination at lysine-117 (K117/Lys117) within the BH3 domain of BECLIN-1, and at lysine-63 (K63/Lys63) that is enhanced by Toll-like receptor-4 (TLR4) activation. On the contrary, the deubiquitinating enzyme A20 disrupts the interaction with BCL-2 promoting the inflammatory-induced autophagy, by deubiquitinating the K63 [108].
AMBRA1 supports, with Rbx/Cul4-ligase, the polyubiquitination of K63, positively sustaining autophagy in response to starvation because of this ubiquitin chains are scaffold for a stronger VPS34 interaction [109]. This mechanism is opposed by WASH protein that directly interacts with BECLIN-1 suppressing ubiquitination and autophagy [109]. The stability of BECLIN-1 is also under the control of ubiquitination machinery, including NEDD4 and HSP90. NEDD4 (E3 ligase neural precursor cell expressed developmentally down-regulated protein 4) polyubiquitinates BECLIN-1 at K11 leading to its proteasomal degradation [110]. On the opposite, the molecular chaperon HSP90 (heat shock protein 90) maintains BECLIN-1 stability preventing the Lys48-linked polyubiquitination chains [111].
3.2. Acetylation
The control of BECLIN-1 by acetylation depends mainly on the regulation of two residues, the lysine-430 (Lys430) and -437 (Lys437), that serve as critical regulatory node for autophagosome maturation having implications spanning neurodegenerative disorders to cancer therapeutics. The acetyltransferase p300 catalyzes acetylation at both these residues to inhibit autophagosome assembly and endocytosis favoring RUBICON recruitment. On the contrary, the NAD+-dependent deacetylase SIRT1 removes these acetyl groups, restoring autophagic flux and promoting autophagosome-lysosome fusion. This regulatory axis has been mechanistically linked to tumor progression, as acetylation-deficient BECLIN-1 mutants (K430R/K437R) enhance autophagosome maturation and suppress tumor growth in xenograft models [112].
The functional implications of BECLIN-1 acetylation are contingent upon the setting. In bladder cancer, SIRT1-mediated deacetylation of BECLIN-1 confers cisplatin resistance by preserving protective autophagy, which enables cancer cells to withstand the stress caused by chemotherapy. Preclinical data indicate that SIRT1 inhibition alleviates this resistance by enhancing BECLIN-1 acetylation, inhibiting autophagy, and reinstating cisplatin sensitivity in xenograft animals [113]. These findings underscore the therapeutic potential of targeting the SIRT1-BECLIN-1 axis to regulate autophagy in cancer therapy.
3.3. Phosphorylation
Most studies have been devoted to understanding phosphorylated sites mainly in the N-terminal and BH3 domain. Several of them have been demonstrated to be regulated in BECLIN-1 domains with pro- or anti-autophagy role.
The downstream target of mTOR, ULK1 phosphorylates BECLIN-1 at serine-15 (Ser15), particularly when bound with ATG14 and VPS34, for initiating autophagy [114]. Moreover, the regulation at the Ser15 is pivotal also for favoring the interaction of BECLIN-1 with PARK2 and its translocation on damaged mitochondria to mediate their specific degradation by mitophagy (see next section) [115]; while the phosphoglycerate kinase-1 (PGK1), critically involved in the production of ATP in the glycolytic pathway, regulates Ser30 in response to glutamine starvation or hypoxic environment [116].
The p38-downstream kinases MK2 and MK3 have as target the serine-90 (Ser90), that is crucial to activate the pro-autophagic complex in response to starvation [117]. Moreover, this same residue is also the target of the other two kinases, DAPK3 and CaMKII (calcium/calmodulin-dependent protein kinase II), respectively demonstrated in cervical cancer and neuroblastoma cell lines [118]. In the latter model system, this phosphorylation enhanced the autophagic-mediated degradation of the protein Id1/2 (inhibitor of differentiation-1/2) to counteract cell differentiation [119].
BECLIN-1 has been found to be under control of the cell cycle checkpoint kinase 2 (CHK2), which is activated by ATM protein in response to DNA damage. It was found that under oxidative stress and hypoxia, CHK2 phosphorylates BECLIN-1 at serine-90 (Ser90) and -93 (Ser93) to detach from BCL-2 and favor the autophagic response (particularly mitophagy) to restore tissue homeostasis, a pathway that is impaired in CHK2-/- knock-out mouse subjected to cerebral ischemia [120]. The center of energy control has in AMPK the main controller. This protein has two specific target sites on BECLIN-1 that consist in the Ser93 and Ser96 and allow autophagy in response to stress stimuli such as glucose starvation [121], when ATP production is compromised. Additionally, it has been shown that the interaction with ATG14 enhances the AMPK-mediated phosphorylation of BECLIN-1 at Ser90 and Ser93 for maximal autophagy in response to starvation [48,121].
A great role in the autophagy is also played by the phosphorylation at threonine-119 (Thr119) [122], in the BH3 domain. This residue is under the control of two kinases, DAPK-1/2, and ROCK-1 (Rho-associated coiled-coil containing protein kinase-1). On the other hand, several phosphorylation abrogates the pro-autophagic function of BECLIN-1. The pro-apoptotic kinase STK4/Mst1 affects the activity of BECLIN-1 through a phosphorylation at threonine-108 (Thr108) in the BH3 domain that allows a BAX-dependent apoptotic cell death [123,124]. Moreover, the kinase JNK phosphorylates the residues threonine-69 (Thr69), serine-70 (Ser70), and serine-97 (Ser97), in the BH3-domain [125] in response to serum starvation to abrogate the BECLIN-1-BCL-2/BCL-XL interaction [122,126,127,128].
Recently, the post-translational regulation of BECLIN-1 has been linked also to tumor microenvironment signals. Interleukin-6 (IL-6), one of the most common cytokines released by inflammatory and cancer cells, was shown to trigger JAK2-mediated phosphorylation of BECLIN-1 at threonine-133 (Thr133), located in the region between BH3 and CCD, which promoted the interaction with VPS34 resulting in enhanced autophagy and chemoresistance in colon cancer [129].
Moreover, in response to glucose withdrawal, AMPK phosphorylates BECLIN-1 also in the C-terminal domain at threonine-388 (Thr388) to facilitate the assembly of the pro-autophagic complex [130]. Recent studies have demonstrated that this is the only known phosphorylation in the ECD/BARA domain that activates rather than inhibits autophagy.
Indeed, several tyrosine phosphorylation events in the ECD domain have inhibitory effects on autophagy. The human epidermal growth factor (EGFR) has been well characterized to interfere with the BECLIN-1-dependent autophagy by phosphorylation at multiple sites in the ECD, namely tyrosine-229 (Y229), -233 (Y233), -352 (Y352). Their role was characterized in non-small cell lung carcinoma in which such phosphorylation inhibits autophagy while enhancing tumorigenesis and cancer progression [131]. Also, in breast cancer, EGFR complexes with HER2 and BECLIN-1 at the plasma membrane, to activate AKT and inhibit autophagy [132]. This inhibition provides an explanation of the link between cell growth and autophagy, and the relevance of this mechanism in tumorigenesis sustained by oncogenic signals [133].
Furtherly, BECLIN-1 is regulated by the casein kinase 1 gamma 2 (CK12) in the ECD at serine-409 (Ser409) that favors the binding with RUBICON to suppress the vesicular transport and autophagic response [112], and by the chimeric tyrosine kinase BCR-ABL, in chronic myeloid leukemia, which both share the same target residue for phosphorylation that is tyrosine-233 (Tyr233), known to impair the interaction with ATG14 [134].
Even the oncoprotein AKT exerts a critical role in the control of the BECLIN-1-dependent autophagy by the phosphorylation at serine-234 (Ser234) and -295 (Ser295). This mechanism sequestered BECLIN-1 to vimentin and intermediate filaments by 14-3-3 protein, preventing the assembly of the pro-autophagic complex [135]. Our group also highlighted the relevance of these phosphorylation sites in controlling autophagy. Its dephosphorylation, mediated by the protein phosphatase PTEN, is pivotal for the autophagosomes complex assembly with ATG14 and VPS34, in a molecular circuitry including AKT itself, in response to starvation and pharmacological approaches affecting the PI3K-mTOR-AKT axis [136].
Figure 5 illustrates the post-translational modifications that regulate BECLIN-1 stability and functions.
4. Functional Role of BECN1 in Cancers
4.1. Pivotal Link Between Autophagy and Apoptosis
BECLIN-1 occupies a pivotal position at the intersection of autophagy and apoptosis, orchestrating cellular responses to stress through its structural and functional plasticity. Notably, the protein's BH3 domain functions as a rheostat that modulates the delicate balance between survival and death by enabling direct interactions with anti-apoptotic BCL-2 family members, like as BCL-2 and BCL-XL [10]. Under nutrient-rich circumstances, BCL-2 interacts with BECN1 and downregulates autophagy by isolating it from the PI3KC3 complex. However, stress-induced phosphorylation of BCL-2 (for example via JNK1) or BECN1 (for example via DAPK) leads to the disruption of the BECN1-BCL-2 complex, facilitating BECN1 heterodimerization with ATG14 and unleashing autophagy to mitigate metabolic stress [137].
BECLIN-1 can also be fragmented through caspase-mediated cleavage. There is no autophagy-inducing capability in the N-terminal region, which contains the BH3 domain. On the other hand, BECLIN-1-C, which is the C-terminal segment, can make cells more sensitive to signals that indicate cell death. The translocation of this BECLIN-1-C fragment to the mitochondrial membrane enhances the BAX oligomerization, hence inducing the release of cytochrome c [138,139,140,141]. Thus, BECLIN-1 functions as a molecular switch, facilitating pro-survival autophagy when intact, but directing cells toward apoptotic death upon cleavage.
Moreover, the anti-oncogenic role of BECLIN-1 in colorectal cancer is demonstrated through autophagy-independent regulation of STAT3 signaling. Without affecting autophagic flux, BECLIN-1 knockdown drives metastasis by increasing STAT3 phosphorylation through stabilizing JAK2-STAT3 connections [142]. Loss of BECLIN-1 in breast cancer stem cells paradoxically hinders carcinogenesis, demonstrating the importance of context-dependent functions. A key factor in treatment resistance, this protein modulates autophagy (via PI3KC3 binding) and apoptosis (through caspase crosstalk) [10]. For instance, O’Donovan and colleagues found that cisplatin-resistant esophageal cancer cells showed elevated BECLIN-1 expression, and that chemotherapy and autophagy suppression worked together to restore apoptosis [134]. The results highlight the possibility of using the BECN1-BCL-2 axis as a therapeutic tool to restore a balance between autophagy and apoptosis in cancer.
4.2. BECLIN-1-Dependent Selective Autophagy
Selective autophagy exerts fine control of damaged organelles turnover in comparison to non-selective autophagy, which randomly degrades damaged materials inside the cells. Each organelle or molecule defines a specific subtype of autophagy, such as mitophagy (mitochondria), lysophagy (lysosomes), ER-phagy (endoplasmic reticulum), ribophagy (ribosomes), aggrephagy (protein aggregates), or xenophagy (pathogens) [135]. For target-specific autophagy, cargo proteins are required to label the materials that are to be degraded. The most widely studied cargo protein is p62 or sequestosome-1, an autophagy receptor that plays a role in the ubiquitin system. As a result of its interaction with LC3, it triggers the degradation of ubiquitinated proteins in the lysosomes after they have been transported to autophagosomes [145].
BECLIN-1 is a cardinal regulator of mitophagy and lysophagy, the two major pathways that have significant consequences for cancer progression and treatment resistance, by interacting dynamically with complexes associated with autophagy and by undergoing post-translational changes.
Mitophagy is a selective form of autophagy that is essential for eliminating depolarized or damaged mitochondria thereby maintaining the quality of mitochondria [146]. Metabolic waste products, including those generated by fatty acid oxidation, Krebs cycle, and oxidative phosphorylation (OXPHOS) [147], can be detrimental to cellular fitness by promoting the oxidation of proteins, nucleic acids, and lipids, and by stimulating cell death pathways [148,149]. BECLIN-1 is indispensable for Parkin-mediated mitophagy. PINK1, a serine/threonine kinase, acts as a sensor for damaged mitochondria by working with the mitochondrial membrane complex TOM20 [150]. Upon activation, Parkin, a ubiquitin ligase, is then recruited to the mitochondria associated membranes (MAMs) and mitophagy can start [151,152,153]. Here, both PINK1 and Parkin can interact directly with BECLIN-1 to trigger autophagosome formation at the mitochondria-ER contact site and shape the omegasome through ULK-1-mediated active phosphorylation of BECLIN-1 at Ser15 [154,155,156,157]. In addition to BECLIN-1, Parkin also interacts with AMBRA1, which facilitates the BECLIN-1-VPS34-dependent assembly of autophagosomes incorporating depolarized mitochondria through the LC3-p62 interaction in the perinuclear clusters, upon prolonged mitochondria depolarization [158]. Notably, BECLIN-1-deficient cells exhibit defective mitochondrial sequestration despite Parkin recruitment, underscoring its non-redundant role in autophagosome-mitochondria tethering [156].
After partial or complete rupture of lysosomes, if repair mechanisms fail, these organelles become ubiquitinated and are degraded through selective autophagy, also known as lysophagy [159]. Lysophagy is typically carried out by a TRIMs-galectin-3-dependent pathway, in which galectin marks damaged lysosomes and ensures their degradation through autophagosome [160], in BECLIN-1-dependent manner [161]. For successful TRIMs interaction, two BECLIN-1 domains are strictly required, one located between BH3 and CCD, and the other overlapping with the ECD [162]. Along with BECLIN-1, ATG16L is recruited to the marked lysosomes, linking the pro-autophagic complex PI3KC3+ with the proteins WIPI2 and LC3 [161]. Recent work demonstrates that BECLIN-1’s interaction with TMEM9, an endolysosomal regulator, activates Rab9-dependent alternative autophagy pathways during lysosomal stress—a process hijacked by cancer cells to evade lysosome-targeted therapies [163].
4.3. BECLIN-1 in Endocytotic Trafficking and Receptor Signaling in Cancer
Endocytosis is an essential cellular process governing the uptake, trafficking, and degradation of molecules, including receptors, that regulate cell signaling and homeostasis [164]. Being the core component of class III phosphatidylinositol 3-kinase complex (PI3KC3), BECLIN-1 participates in endocytic trafficking. Fundamentally, it operates in two distinct complexes: PI3KC3-C1 (with ATG14L) and PI3KC3-C2 (with UVRAG), to maintain cellular homeostasis, primarily through its involvement in autophagy and vesicular trafficking. In particular, PI3KC3-C1 is chiefly engaged in the commencement of autophagosomes, wherein ULK1 phosphorylates BECLIN-1 to activate phagophore nucleation [20,21]. On the contrary, complex PI3KC3-C2 leads the endocytic trafficking, lysosomal maturation, and cytokinesis by generating phosphatidylinositol-3-phosphate (PI3P) on intracellular membranes [43,165,166]. Although the relevance of PI3KC3-C2 in autophagy is contentious, its essential functions in membrane trafficking and endosomal sorting are firmly established. Recent studies have emphasized BECLIN-1's autophagy-independent functions in cancer progression, specifically with its regulation of receptor degradation, maintenance of epithelial integrity, and modulation of ferroptosis.
BECLIN-1 depletion impairs retrograde transport of endosomes to the Golgi compartment, affecting organelle trafficking and neuronal development in C. elegans and mouse models [166,167]. Additionally, BECLIN-1 regulates early endosome maturation in coordination with UVRAG and VPS34, transitioning endosomes from APPL1+/PI3P- to PI3P+ states. These PI3P+ endosomes recruit EEA1 to facilitate fusion and maturation via Rab5 and CMTM7, enabling proper EGF trafficking and receptor degradation [168,169,170,171,172].
PI3P produced by PI3KC3-C2 is recruited by activated Rab5 through VPS15, facilitating the anchoring and fusing of early endosomes [173]. During endosomal maturation, Rab7 supplants Rab5, facilitating the interaction of PI3KC3-C2 with late endosomes through VPS34 and VPS15 [174]. This maturation supports the retromer complex and ESCRT machinery, which recycle cargo proteins like GLUT1 to the plasma membrane or trans-Golgi network and sort ubiquitinated receptors like EGFR into intraluminal vesicles for lysosomal degradation [175,176].
BECLIN-1 tightly regulates the tumor growth and progression, imparting its tumor suppressive effect via endo-lysosomal trafficking, a process that is crucial in controlling the cell surface receptor function. Recently, it was shown that BECLIN-1 promoted the endosomal recruitment of hepatocyte growth factor-regulated tyrosine kinase substrate (HRS), enabling sorting of EGFR and transferrin receptor (TFR1) into intraluminal vesicles for lysosomal degradation. Loss of BECLIN-1 in breast cancer cells reduces HRS recruitment, prolonging EGFR/ERK signaling, and TFR1-driven iron uptake, ultimately supporting the tumor to proliferate while making them more sensitive to EGFR/MEK inhibitors. This pathway is independent of autophagy, as ATG5 or ATG7 deletion does not emulate similar consequences [177]. In congruence, BECLIN-1 loss increased AKT/ERK activation and promoted breast carcinoma invasion by prolonging growth factor receptor retention in signaling compartments enriched with PI3P/APPL1 [170].
Moreover, BECLIN-1 is essential for preserving epithelial structure through the regulation of E-cadherin trafficking. The PI3KC3-C2 complex facilitates the appropriate recycling of E-cadherin to the plasma membrane and inhibits its lysosomal breakdown. The disruption of BECN1 destabilizes E-cadherin localization at adherent junctions, hence activating β-catenin/Wnt signaling and promoting epithelial-mesenchymal transition (EMT) [178,179]. A recent work involving intestinal epithelial cells revealed that BECN1 deletion results in the cytoplasmic mislocalization of E-cadherin and occludin, hence affecting apical F-actin organization and epithelial polarity [179]. In breast cancer models, BECLIN-1 overexpression reinstates plasma membrane E-cadherin levels, inhibiting mesenchymal markers like Snail and Zeb1 [178].
Under starvation or exposure to tyrosine kinase inhibitors (TKI), like erlotinib or gefitinib, EGFR signals are blocked by the detachment of RUBICON from BECLIN-1, mediated by the endosome-associated protein LAPTM4B (lysosomal-associated protein transmembrane 4 beta) [180,181]. Interestingly, several studies point out that contact sites between multivesicular late endosome (MVEs) and ER serve as scaffolds for the endosomal inactive EGFR to undermine the BCL-2-BECLIN-1 interaction, similar to the interaction between inactive EGFR and PTP1B [182,183]. Notably, EGFR internalization via non-clathrin endocytosis (NCE) is critically regulated by the ER-resident protein RTN3 (Reticulon 3), which forms the ER-PM (plasma membrane) contact sites [184]. Surprisingly, this protein enhances endocytic trafficking by promoting the interaction between BECLIN-1 and BCL-2, thereby counteracting autophagy [185]. These mechanisms illustrate BECLIN-1’s multifaceted role in receptor signaling regulation, endocytic maturation, and tumor suppression via trafficking pathways.
4.4. BECLIN-1 Crosstalk with Other Pathways in Cancer
As mentioned above, the most significant interactions of BECLIN-1 occur with apoptotic pathways from the caspase-mediated cleaved fragments of BECLIN-1. In addition to that, recently, the R-BiP/BECLIN-1/p62 complex was identified to play a crucial role in regulating the crosstalk between apoptosis and autophagy. Song and colleagues demonstrated that the disruption of this complex can shift cellular fate from autophagy to apoptosis. Mechanistically, this involved the dephosphorylation of BECLIN-1 at specific residues (Ser 234/295), which resulted in increased cleavage of Beclin-1 and disruption of the R-BiP/BECLIN-1/p62 complex [186].
The PI3K/AKT/mTOR pathway is amongst the major oncogenic cascades for cellular growth and metabolism, which interacts extensively with BECLIN-1. It is established that BECLIN-1 is negatively correlated to this pathway as evidenced by reduced expression levels of PI3K, phosphorylated AKT (p-AKT), and phosphorylated mTOR (p-mTOR) following BECLIN-1 overexpression. Studies have shown that inflammatory cytokines, such as interleukin-7 (IL-7) activate the PI3K/AKT/mTOR signaling pathway thereby downregulating BECLIN-1 expression significantly in the lung cancer cells. This reduction of autophagy creates a tumor-favorable environment by promoting proliferation and survival while suppressing the apoptotic responses [187]. This bidirectional relationship has significant implications for cancer therapy. For instance, LTX-315, a polypeptide that increases BECLIN-1 levels and alters p-AKT and p-mTOR levels, has shown promise in enhancing drug responsiveness in ovarian cancer models [188]. Similarly, Xie-Bai-San (XBS) regulates both mTOR and BECLIN-1, inhibiting gefitinib-induced autophagy in non-small cell lung cancer cells and promoting cell death by enhancing p-mTOR and BCL-2 levels while decreasing BECLIN-1 levels [189]. BECLIN-1’s role in receptor tyrosine kinase (RTK) signaling has been redefined through its novel interactions with HER2/GRB2 and RTN3. HER2 recruits GRB2-BECLIN-1 complexes to the plasma membrane, suppressing autophagy via PI3K/AKT activation. Disrupting this interaction with Tat-BECLIN-1 peptide restores autophagic death in HER2+ breast cancer [26]. Similarly, RTN3, an ER-resident protein, binds BECLIN-1’s N-terminal domain to promote non-clathrin EGFR endocytosis, enhancing EGFR/ERK signaling while inhibiting autophagy which depicts a dual mechanism exploited by tumors for survival [181].
Notable interactions of BECLIN-1 also include activation of STAT3 signaling pathway independently of its known role in autophagy. Mechanistically, BECN1 regulates the interaction between STAT3 and JAK2, which when knocked down promoted STAT3 phosphorylation through enhanced STAT3-JAK2 interaction. Low expressions of BECN1 directly correlated with more aggressive phenotypes of colorectal cancer with high cell motility and invasion [142]. Importantly, this regulatory function of BECLIN-1's suggests additional non-canonical functions of this protein in cancer progression.
The interaction between BECLIN-1 and anti-apoptotic BCL-2 family proteins represents another critical regulatory node, which is modulated by several proteins. One of those proteins is LETM1 which is upregulated in hepatocellular carcinoma and influences both the mechanisms, autophagy and apoptosis. Depletion of LETM1 triggers AMPK-mediated phosphorylation of BCL-2, causing disruption of the BECLIN-1/BCL-2 complex and promoting both apoptosis and autophagy in liver cancer cells [190].
Apparently, recent discoveries have expanded BECLIN-1's role beyond apoptosis and autophagy to include other cell death pathways. Key phosphorylation Ser90, Ser93, and Ser96 on BECLIN-1 by active AMPK leads to the formation of a BECLIN-1-SLC7A11 complex, which directly inhibits system Xc− activity and ultimately promotes ferroptosis, an iron-dependent form of programmed cell death [191]. Paradoxically, BECLIN-1 serves as an inhibitor of necroptosis by associating with phosphorylated MLKL in the necrosome complex, preventing MLKL oligomerization and subsequent necroptotic cell death. Such findings put forth BECLIN-1's complex role in regulating multiple cell death pathways, with significant therapeutic implications [192].
BECLIN-1 interacts extensively with autophagy-related genes to effectively manage autophagy and cancer progression. The CUL3 E3 ligase complex, which includes KLHL38 as a substrate adapter, promotes BECLIN-1 degradation followed by autophagy suppression. Elevated CUL3 levels correlate with poor prognosis in breast and ovarian cancers, suggesting its role in promoting tumor cell growth [106].
The interaction between BECLIN-1 and mitogen-activated protein kinase (MAPK) pathways, including ERK, JNK, and p38, represents another important regulatory network. Studies using Allyl Isothiocyanate (AITC) have shown that it can trigger ERK, AMPK, and JNK signaling pathways while increasing BECLIN-1 expression. Specific knockdown of BECN1 decreases AITC-induced autophagy, suggesting that BECLIN-1 is essential for AITC-induced autophagy through these signaling cascades [193].
5. Clinical Implications and Biomarker Potential
5.1. Prognostic, Predictive, and Diagnostic Value of BECLIN-1 Across Cancer Types
Low expression of BECLIN-1 has been reported in several cancer tissues compared to the normal tissue, which suggests that defective autophagy may favor carcinogenesis and worse prognosis and supports BECLIN-1 as a valuable independent prognostic marker. For instance, decreased BECLIN-1 expression and more aggressive and metastatic phenotype have been reported in breast cancer [194], ovarian cancer [195,196,197], lung cancer [198,199], cholangiocarcinoma [200,201], gastric cancer [202] and colorectal cancer [142,203].
BECLIN-1 has emerged as a valuable prognostic marker across multiple cancer types. High levels of BECN1 are associated with favorable overall survival in salivary gland cancer and cholangiocarcinoma, whereas low levels correlate with poorer survival in breast and gastric cancers [15,200,204,205]. Low levels of BECN1 were found to be associated with the most aggressive pathological traits and linked to poor overall survival in a cohort of estrogen-receptor (ER)-negative subtypes of BC [15], as observed in published datasets in TCGA (n=1033) and METABRIC (n=1929) [51]. Similarly, in Non-Hodgkin Lymphomas and ovarian cancer, elevated BECN1 expression is linked to less aggressive tumors and improved chemotherapy response, contributing to better survival outcomes [206,207]. Further, findings from our group corroborated these observations in diffuse large B-cell lymphoma, where high expression of BECN1 may enhance chemotherapy responsiveness through increased autophagy, making it a valuable prognostic factor correlated with longer survival [208].
However, despite BECN1’s established value as a pro-survival marker and therapeutic target, its role in cancers is more nuanced. Several studies sternly indicate contrasting patterns in breast, gastric, endometrial adenocarcinoma, and colorectal cancer, where the elevated BECN1 levels have been anomalously associated with patient prognosis as well as the aggressive phenotypes, nodal metastasis, and therapy resistance [209,210,211]. These studies signify that the expression and function of BECLIN-1 are tightly interwoven with clinical outcomes across multiple tumor types, and that its role can vary across different tumor types and contexts.
In addition to BECN1, circulating molecular markers, like LC3 and p62, can be integrated into prognostic indexes to further refine patient stratification. For instance, our recent in-silico TCGA analysis of the AML cohort revealed that high expression of BECN1, in conjunction with MAP1LC3B and low p62/SQSTM1 levels (indicative of high autophagy flux) correlated with better prognosis. AML patients with high autophagy–mitophagy signatures driven by BECN1 exhibited the longest overall survival, reinforcing BECN1’s pivotal role in regulating autophagic processes that positively influence clinical outcomes [212].
Moreover, in colorectal cancer, phosphorylation of BECLIN-1 at Y333 by SRC kinase suppresses autophagy, facilitating chemoresistance and establishing it as a predictive marker of therapeutic response [129]. Concomitantly, BECLIN-1 expression in cholangiocarcinoma not only associates with increased autophagy and better patient survival, but its upregulation by the nutraceutical resveratrol also promotes apoptosis, suppresses tumor cell proliferation, and inhibit the pro-tumorigenic IL6/IL6R signaling axis [213].
In hepatocellular carcinoma, serum BECLIN-1 levels distinguish cirrhotic patients with malignancy, while co-evaluation with ATG5 and cachexia scoring improves early detection and risk stratification, highlighting its utility not only as a molecular indicator but also as a potential therapeutic target [214].
Overall, these findings firmly establish BECLIN-1 as a biomarker for prognosis, diagnosis, and therapy response prediction, with clinical implications shaped by tumor type, subtype, and molecular context.
5.2. Therapeutic Targeting of BECLIN-1 Dependent Pathways
Recent advances in understanding the pleiotropic roles of BECN1 have positioned its targeted activation as a promising strategy to overcome therapeutic resistance and reshape immunosuppressive tumor microenvironments. By prioritizing pharmacological and genetic approaches to enhance BECN1-dependent autophagy, researchers are uncovering novel mechanisms to amplify anti-tumor immunity while suppressing pro-metastatic signaling pathways.
Recent advancements have revealed intriguing therapeutic strategies aimed at the BECLIN-1–BCL-2 axis. Emerging peptides like Tat-SP4 and small molecules such as compound 35, which are engineered to selectively disrupt BECLIN-1/BCL-2 interactions without affecting pro-apoptotic pathways, exemplify the potential to precisely modulate autophagy to induce cancer cell death while maintaining normal cellular function [215].
BECN1’s role in immune regulation and tumor suppression underscores its therapeutic potential when strategically induced. Studies have shown that myeloid-specific BECN1 deficiency leads to neutrophilia, aberrant p38 activation, and immunosuppressive interactions between neutrophils and pre-B cells via the CXCL9/CXCR3 chemotaxis pathway. These interactions elevate PD-L1 and IL-10 expression, suppressing CD8+ T cell function and promoting lymphoma. Pharmacological or genetic activation of BECN1 could disrupt these pro-tumorigenic networks by preventing p38 hyperactivation and restoring immune surveillance. Preclinical models indicate that BECN1-inducing agents can reduce PD-L1 expression by 60%, enhancing CD8+ T cell cytotoxicity against tumor cells [216].
Targeting BECN1-mediated autophagy has been shown to disrupt immune suppression in solid tumors. Genetic inhibition of BECN1 in melanoma models enhances NK cell infiltration via CCL5 upregulation, impairing tumor growth [217]. In gastric adenocarcinoma, BECN1 activation correlates with improved outcomes, with high BECN1 expression linked to increased FOXP3+ Treg infiltration, smaller tumors, reduced metastasis, and enhanced CD8+ T cell activity. Nanoparticle systems co-delivering BECN1 mRNA with IL-2 are being explored to regulate Treg recruitment while sustaining anti-tumor immunity [218].
Beyond direct tumor suppression, innovations highlight BECN1’s broader therapeutic relevance. Metabolic reprogramming of CAR T cells using autophagy-inducing agents such as semaglutide and Urolithin A enhances CAR T cell persistence, memory, and anti-tumor efficacy, indirectly leveraging BECN1-mediated autophagy [219].
The evidence that GLP-1R agonists and Urolithin A can collaboratively stimulate autophagy and mitophagy in CAR-T cells provides a foundation for combining metabolic modulation with immunotherapy [219]. These techniques may augment cytotoxic immune responses, especially in refractory or metastatic cancers.
Similarly, the Tat-BECLIN-1 peptide restores autophagic death in HER2+ breast cancer [220], and preclinical studies on Spautin-1, a USP10/13 inhibitor, show promise in stabilizing BECN1 and reducing K48-linked ubiquitination in glioblastoma and NSCLC patients with BECN1 promoter methylation [221,222].
The therapeutic activation of BECN1 also opens avenues for combination strategies with existing modalities. For example, BH3 mimetics such as ABT-737 can liberate BECN1 from inhibitory interactions with anti-apoptotic BCL-2 proteins, promoting autophagy. This induction can sensitize cancer cells to apoptosis when modulated appropriately [223,224,225,226]. BECLIN-1-targeting peptides, such as Tat-SP4, enhance autophagy, triggering autosis—a non-apoptotic cell death form—and demonstrate potent anti-tumor effects by accelerating EGFR degradation [227]. Additionally, agents like metformin bidirectionally regulate autophagy, promoting autophagic cell death in some cancers while suppressing it in others, presenting a dual regulatory role [228,229,230,231].
Combination therapies exploiting autophagy-metabolism crosstalk show promise but also present challenges. For instance, AMPK-mediated BECLIN-1 phosphorylation can trigger ferroptosis in glutamine-deprived tumors but may exacerbate cachexia in advanced patients. Studies emphasize the therapeutic potential of autophagy activation in specific contexts. In PDAC, a cancer type with high basal autophagic activity, BECLIN-1 modulation induces mitochondrial stress and EGFR degradation, bypassing resistance mechanisms associated with apoptotic evasion. These findings position BECLIN-1 as not only a tumor suppressor but also a viable target for designing context-specific autophagy-modulating therapies in PDAC and other malignancies [227].
Figure 6 summarizes the main translational implications of BECLIN-1 described above.
6. Conclusions and Future Prospectives
BECLIN-1 has become a pivotal regulator of autophagy, closely connecting cellular survival, death, and metabolism. Its complex significance is highlighted by several regulatory mechanisms, including genetic and epigenetic control of its transcription, alternative splicing, extensive post-translational modifications, and interactions with essential signaling molecules, which together influence tumor growth and therapeutic response.
Future research should focus on clarifying the functions of BECLIN-1 splice variants in autophagy and endocytosis, enhancing our comprehension of dynamic post-translational modifications to discover new targets for selective autophagy activation, and formulating combination therapies that merge BECLIN-1-targeted agents with current chemotherapeutic, immunotherapeutic, or metabolic modulators.
Overall, a thorough understanding of the regulatory networks governing BECLIN-1 will enhance our strategies for autophagy-based cancer therapies and perhaps facilitate innovative treatments for other pathologies. By coupling structural insights with translational research, BECLIN-1 emerges as a prospective focal point for the advancement of precision therapeutics that harness the full potential of autophagy modulation.
Author Contributions
Conceptualization, C.M., A.C., and C.I.; writing—original draft preparation, C.M., A.C., U.W., C.V., and A.F.; writing—review and editing, D.N.D. and C.I.; figures and literature search, U.W., C.V., and A.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created in this manuscript. Illustrations are original artwork prepared by the authors.
Acknowledgments
U.W. is the recipient of a PhD fellowship granted by Comoli, Ferrari & SpA (Novara, Italy). C.V. was supported with a postdoctoral fellowship from Università degli Studi del Piemonte Orientale “Amedeo Avogadro” (id. 1412) granted by Fondazione Cassa di Risparmio di Torino (CRT, Torino, Italy). A.F. is the recipient of a postdoctoral fellowship awarded by Fondazione Veronesi (FUV 2025) (Milan, Italy). Thanks are due to the Associazione per la Ricerca Medica Ippocrate-Rhazi (Novara, Italy) for supporting the PhD fellowship of C.M.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix Beclin-2
BECLIN-2 was discovered by Beth Levine’s lab in 2003 as a novel autophagy-related protein homologous to BECLIN-1 with a propensity to degrade several G protein-coupled receptors (GPCRs) with GASP1. The BECN2 gene is located on chromosome 1q43 and shares only 57% of sequence homolog with BECN1. In humans, it transcribes a protein with 431 aa and a predicted molecular weight of 49 kDa, with the same functional domains as BECLIN-1 [232]. Unlike BECN1, the deletion of BECN2 (BECN2-/-) does not affect embryonic development and birth in mice, but enhance tissues alterations such as splenomegaly, lymphadenopathy, and diffuse inflammatory state [233,234]. BECLIN-2 has been associated with the inhibition of the inflammatory pathway NF-ΚB through autophagic degradation, independent of BECLIN-1, of MEKK3 and TAK1, which are included in ATG9+-vesicles that fuse with autophagosomes thanks to interactions with STX-5 and -6 [235]. BECLIN-2 also negatively controls inflammasomes activation and directly interacts with component proteins of this complex, such as NLRP3 and NLRP1, AIM2, NLRC4, and CASP1, through its CCD-ECD domain. These proteins are degraded by lysosome with the previously described mechanism involving ATG9+ vesicles [236]. Recent observations confirm the common role for BECLIN-1 and BECLIN-2 in promoting the formation of LC3+-autophagosomes, but a divergent role in mitophagy, where only BECLIN-1 appears necessary, and unexpectedly, the degradation of mitochondria is enhanced in cells lacking BECLIN-2 [237]. Furthermore, BECLIN-2 has been described as unable to interact with RUBICON, and it does not require the detachment of BCL-2 to mediate the starvation-induced autophagy because it lacks the functional residue (Thr119) controlled by DAPK1. However, it displays interactive ability with ATG14, VPS34, and AMBRA1, like BeCLIN-1 [233,238].
References
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against Fatal Sindbis Virus Encephalitis by Beclin, a Novel Bcl-2-Interacting Protein. J. Virol. 1998, 72, 8586–96. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Longo, D.L. Autophagy in Human Diseases. New Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Gómez-Virgilio, L.; Silva-Lucero, M.-D.; Flores-Morelos, D.-S.; Gallardo-Nieto, J.; Lopez-Toledo, G.; Abarca-Fernandez, A.-M.; Zacapala-Gómez, A.-E.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11, 2262. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of cell death. Cell Death Dis. 2023, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2020, 22, 179. [Google Scholar] [CrossRef] [PubMed]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Tian, K.; Ran, Y.; Zhou, H.; Zhou, L.; Ding, Y.; Tang, X. Beclin-1: a therapeutic target at the intersection of autophagy, immunotherapy, and cancer treatment. Front. Immunol. 2024, 15, 1506426. [Google Scholar] [CrossRef]
- Esposito, A.; Ferraresi, A.; Salwa, A.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I. Cancers 2022, 14, 2142. [Google Scholar] [CrossRef]
- Maheshwari, C.; Vidoni, C.; Titone, R.; Castiglioni, A.; Lora, C.; Follo, C.; Isidoro, C. Isolation, Characterization, and Autophagy Function of BECN1-Splicing Isoforms in Cancer Cells. Biomolecules 2022, 12, 1069. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Tang, H.; Sebti, S.; Titone, R.; Zhou, Y.; Isidoro, C.; Ross, T.S.; Hibshoosh, H.; Xiao, G.; Packer, M.; Xie, Y.; et al. Decreased BECN1 mRNA Expression in Human Breast Cancer is Associated With Estrogen Receptor-Negative Subtypes and Poor Prognosis. EBioMedicine 2015, 2, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. Beclin–phosphatidylinositol 3-kinase complex functions at the trans -Golgi network. Embo Rep. 2001, 2, 330–335. [Google Scholar] [CrossRef]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The Evolutionarily Conserved Domain of Beclin 1 is Required for Vps34 Binding, Autophagy, and Tumor Suppressor Function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.-H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–698. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N.; Subramani, S. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Noda, N.N.; Kobayashi, T.; Adachi, W.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of the Novel C-terminal Domain of Vacuolar Protein Sorting 30/Autophagy-related Protein 6 and Its Specific Role in Autophagy. J. Biol. Chem. 2012, 287, 16256–16266. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation–at the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef]
- Peña-Martinez, C.; Rickman, A.D.; Heckmann, B.L. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci. Adv. 2022, 8, eabn1702. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, J.; Zhu, Y.; Wang, L.; Jiang, X.; Liu, B.; He, G. Targeting autophagy and beyond: Deconvoluting the complexity of Beclin-1 from biological function to cancer therapy. Acta Pharm. Sin. B 2023, 13, 4688–4714. [Google Scholar] [CrossRef] [PubMed]
- Montero-Vergara, J.; Plachetta, K.; Kinch, L.; Bernhardt, S.; Kashyap, K.; Levine, B.; Thukral, L.; Vetter, M.; Thomssen, C.; Wiemann, S.; et al. GRB2 is a BECN1 interacting protein that regulates autophagy. Cell Death Dis. 2024, 15, 1–11. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, Z.; Li, J.; Li, J.; Luo, Y.; Li, W.; You, W.; Zhang, Y.; Li, Z.; Yang, J.; et al. Nuclear Beclin 1 Destabilizes Retinoblastoma Protein to Promote Cell Cycle Progression and Colorectal Cancer Growth. Cancers 2022, 14, 4735. [Google Scholar] [CrossRef]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.-L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Wyatt, S.; Glover, K.; Dasanna, S.; Lewison, M.; González-García, M.; Colbert, C.L.; Sinha, S.C. Epstein–Barr Virus Encoded BCL2, BHRF1, Downregulates Autophagy by Noncanonical Binding of BECN1. Biochemistry 2023, 62, 2934–2951. [Google Scholar] [CrossRef]
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.-L.; Bast, R.C., Jr. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy 2014, 10, 1071–1092. [Google Scholar] [CrossRef]
- Baek, S.; Chang, J.-W.; Yoo, S.-M.; Choo, J.; Jung, S.; Nah, J.; Jung, Y.-K. TMEM9 activates Rab9-dependent alternative autophagy through interaction with Beclin1. Cell. Mol. Life Sci. 2024, 81, 1–13. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Liu, R.; Dong, X.; Zhong, Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015, 25, 533–544. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef]
- Strappazzon, F.; Vietri-Rudan, M.; Campello, S.; Nazio, F.; Florenzano, F.; Fimia, G.M.; Piacentini, M.; Levine, B.; Cecconi, F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011, 30, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Fairlie, W.D.; Lee, E.F. BECLIN1: Protein Structure, Function and Regulation. Cells 2021, 10, 1522. [Google Scholar] [CrossRef]
- Levine, B.; Sinha, S.C.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N.; Subramani, S. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Wong, S.; Sil, P.; Martinez, J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J. 2017, 285, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mul, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Thoresen, S.B.; Pedersen, N.M.; Liestøl, K.; Stenmark, H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp. Cell Res. 2010, 316, 3368–3378. [Google Scholar] [CrossRef]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The Evolutionarily Conserved Domain of Beclin 1 is Required for Vps34 Binding, Autophagy, and Tumor Suppressor Function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef]
- Baskaran, S.; Carlson, L.-A.; Stjepanovic, G.; Young, L.N.; Kim, D.J.; Grob, P.; Stanley, R.E.; Nogales, E.; Hurley, J.H. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife 2014, 3, e05115. [Google Scholar] [CrossRef]
- Rostislavleva, K.; Soler, N.; Ohashi, Y.; Zhang, L.; Pardon, E.; Burke, J.E.; Masson, G.R.; Johnson, C.; Steyaert, J.; Ktistakis, N.T.; et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science 2015, 350, aac7365–aac7365. [Google Scholar] [CrossRef]
- Mei, Y.; Glover, K.; Su, M.; Sinha, S.C. Conformational flexibility of BECN1: Essential to its key role in autophagy and beyond. Protein Sci. 2016, 25, 1767–1785. [Google Scholar] [CrossRef]
- Fogel, A.I.; Dlouhy, B.J.; Wang, C.; Ryu, S.-W.; Neutzner, A.; Hasson, S.A.; Sideris, D.P.; Abeliovich, H.; Youle, R.J. Role of Membrane Association and Atg14-Dependent Phosphorylation in Beclin-1-Mediated Autophagy. Mol. Cell. Biol. 2013, 33, 3675–3688. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. . 2001, 61, 3443–9. [Google Scholar]
- Xu, F.; Fang, Y.; Yan, L.; Xu, L.; Zhang, S.; Cao, Y.; Xu, L.; Zhang, X.; Xie, J.; Jiang, G.; et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep. 2017, 7, srep45385. [Google Scholar] [CrossRef] [PubMed]
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational Landscape of the Essential Autophagy Gene BECN1 in Human Cancers. Mol. Cancer Res. 2014, 12, 485–490. [Google Scholar] [CrossRef]
- Park, S.E.; Yi, H.J.; Suh, N.; Park, Y.-Y.; Koh, J.-Y.; Jeong, S.-Y.; Cho, D.-H.; Kim, C.-S.; Hwang, J.J. Inhibition of EHMT2/G9a epigenetically increases the transcription ofBeclin-1via an increase in ROS and activation of NF-κB. Oncotarget 2016, 7, 39796–39808. [Google Scholar] [CrossRef]
- Li, Z.; Chen, B.; Wu, Y.; Jin, F.; Xia, Y.; Liu, X. Genetic and epigenetic silencing of the beclin 1gene in sporadic breast tumors. BMC Cancer 2010, 10, 98–98. [Google Scholar] [CrossRef]
- Ambrosio, S.; Saccà, C.D.; Amente, S.; Paladino, S.; Lania, L.; Majello, B. Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 2017, 36, 6701–6711. [Google Scholar] [CrossRef]
- Shu, F.; Xiao, H.; Li, Q.-N.; Ren, X.-S.; Liu, Z.-G.; Hu, B.-W.; Wang, H.-S.; Wang, H.; Jiang, G.-M. Epigenetic and post-translational modifications in autophagy: biological functions and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 1–23. [Google Scholar] [CrossRef]
- Vidoni, C.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin. Cancer Biol. 2020, 66, 34–44. [Google Scholar] [CrossRef]
- Polager, S.; Ofir, M.; Ginsberg, D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008, 27, 4860–4864. [Google Scholar] [CrossRef]
- Kusama, Y.; Sato, K.; Kimura, N.; Mitamura, J.; Ohdaira, H.; Yoshida, K. Comprehensive analysis of expression pattern and promoter regulation of human autophagy-related genes. Apoptosis 2009, 14, 1165–1175. [Google Scholar] [CrossRef]
- Wang, B.; Ling, S.; Lin, W.-C.; Jin, D.-Y. 14-3-3τ Regulates Beclin 1 and Is Required for Autophagy. PLOS ONE 2010, 5, e10409. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. p65/RelA Modulates BECN1 Transcription and Autophagy. Mol. Cell. Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef]
- Shu, C.-W.; Chang, H.-T.; Wu, C.-S.; Chen, C.-H.; Wu, S.; Chang, H.-W.; Kuo, S.-Y.; Fu, E.; Liu, P.-F.; Hsieh, Y.-D.; et al. RelA-Mediated BECN1 Expression Is Required for Reactive Oxygen Species-Induced Autophagy in Oral Cancer Cells Exposed to Low-Power Laser Irradiation. PLOS ONE 2016, 11, e0160586–e0160586. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Ghislat, G.; Luo, S.; Renna, M.; Siddiqi, F.; Rubinsztein, D.C. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFκB activation. Hum. Mol. Genet. 2015, 24, 2899–2913. [Google Scholar] [CrossRef]
- Han, T.; Guo, M.; Gan, M.; Yu, B.; Tian, X.; Wang, J.-B. TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy 2018, 14, 2035–2048. [Google Scholar] [CrossRef]
- Zhu, W.; Swaminathan, G.; Plowey, E.D. GA binding protein augments autophagy via transcriptional activation ofBECN1-PIK3C3complex genes. Autophagy 2014, 10, 1622–1636. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Delibaşı, N.; Nalbantoglu, U.; Sener, E.F.; Nurdinov, N.; Tascı, B.; Taheri, S.; Özkul, Y.; Donmez-Altuntas, H.; Canatan, H.; et al. FOXM1 plays a role in autophagy by transcriptionally regulating Beclin-1 and LC3 genes in human triple-negative breast cancer cells. J. Mol. Med. 2019, 97, 491–508. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, H.-B.; Shi, Q.; Yang, C.; Ma, J.-B.; Jin, B.; Wang, X.; He, D.; Guo, P. KLF5 downregulation desensitizes castration-resistant prostate cancer cells to docetaxel by increasing BECN1 expression and inducing cell autophagy. Theranostics 2019, 9, 5464–5477. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.-J.; Huang, F.-X.; Sun, Z.-T.; Zhang, R.-X.; Huang, S.-F.; Wang, J. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumor Biol. 2014, 35, 7097–7103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Li, B.; Chen, Y.; Ren, X.; Liu, C.-G.; Yang, J.-M. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009, 5, 816–823. [Google Scholar] [CrossRef]
- Korkmaz, G.; le Sage, C.; Tekirdag, K.A.; Agami, R.; Gozuacik, D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012, 8, 165–176. [Google Scholar] [CrossRef]
- Hou, W.; Song, L.; Zhao, Y.; Liu, Q.; Zhang, S. Inhibition of Beclin-1-Mediated Autophagy by MicroRNA-17-5p Enhanced the Radiosensitivity of Glioma Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 25, 43–53. [Google Scholar] [CrossRef]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtoli, L.; Miracco, C. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef]
- Huang, T.; Wan, X.; Alvarez, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. MIR93(microRNA -93) regulates tumorigenicity and therapy response of glioblastoma by targeting autophagy. Autophagy 2019, 15, 1100–1111. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Lin, S.; Ba, M.; Cui, S. MicroRNA-216a enhances the radiosensitivity of pancreatic cancer cells by inhibiting beclin-1-mediated autophagy. Oncol. Rep. 2015, 34, 1557–1564. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chattopadhyay, D.; Chakrabarti, G.; Mari, B. miR-17-5p Downregulation Contributes to Paclitaxel Resistance of Lung Cancer Cells through Altering Beclin1 Expression. PLOS ONE 2014, 9, e95716. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zou, J.; Zheng, G.; Chu, J. MiR-30a Decreases Multidrug Resistance (MDR) of Gastric Cancer Cells. Med Sci. Monit. 2016, 22, 4509–4515. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Bai, F.; Xu, Y.; Chen, Y.; Chen, L. Intensified Beclin-1 Mediated by Low Expression of Mir-30a-5p Promotes Chemoresistance in Human Small Cell Lung Cancer. Cell. Physiol. Biochem. 2017, 43, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Wu, L.; Ding, H.; Wang, Y.; Zhang, Y.; Chen, X.; Chen, X.; Zhang, C.-Y.; Zhang, Q.; Zen, K. MicroRNA-30a Sensitizes Tumor Cells to cis-Platinum via Suppressing Beclin 1-mediated Autophagy. J. Biol. Chem. 2012, 287, 4148–4156. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.; Liu, H.; Jiang, S.; Wang, G.; Sun, L.; Li, J.; Wang, X.; Yu, S.; Huang, J.; et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, L.; Zhao, M.; Zhu, S.; Kang, R.; Vernon, P.; Tang, D.; Cao, L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012, 26, 1752–1760. [Google Scholar] [CrossRef]
- Tan, S.; Shi, H.; Ba, M.; Lin, S.; Tang, H.; Zeng, X.; Zhang, X. miR-409-3p sensitizes colon cancer cells to oxaliplatin by inhibiting Beclin-1-mediated autophagy. Int. J. Mol. Med. 2016, 37, 1030–1038. [Google Scholar] [CrossRef]
- Chen, L.; Han, X.; Hu, Z.; Chen, L. The PVT1/miR-216b/Beclin-1 regulates cisplatin sensitivity of NSCLC cells via modulating autophagy and apoptosis. Cancer Chemother. Pharmacol. 2019, 83, 921–931. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Wu, S.; O'Connell, D.; Gao, T.; Zhong, H.; Yang, Y. miR-216b enhances the efficacy of vemurafenib by targeting Beclin-1, UVRAG and ATG5 in melanoma. Cell. Signal. 2018, 42, 30–43. [Google Scholar] [CrossRef]
- Pradhan, A.K.; Talukdar, S.; Bhoopathi, P.; Shen, X.-N.; Emdad, L.; Das, S.K.; Sarkar, D.; Fisher, P.B. mda-7/IL-24 Mediates Cancer Cell–Specific Death via Regulation of miR-221 and the Beclin-1 Axis. Cancer Res. 2017, 77, 949–959. [Google Scholar] [CrossRef]
- Li, M.; Chen, X.; Wang, D.; Gan, L.; Qiao, Y. Effects of miR-26a on the expression of Beclin 1 in retinoblastoma cells. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, B.; Long, H.; Yu, J.; Li, F.; Hou, H.; Yang, Q. Decreased miR-124-3p Expression Prompted Breast Cancer Cell Progression Mainly by Targeting Beclin-1. Clin. Lab. 2016, 62, 1139–45. [Google Scholar] [CrossRef] [PubMed]
- Nadhan, R.; Isidoro, C.; Song, Y.S.; Dhanasekaran, D.N. Signaling by LncRNAs: Structure, Cellular Homeostasis, and Disease Pathology. Cells 2022, 11, 2517. [Google Scholar] [CrossRef]
- Wang, J.; Xie, S.; Yang, J.; Xiong, H.; Jia, Y.; Zhou, Y.; Chen, Y.; Ying, X.; Chen, C.; Ye, C.; et al. The long noncoding RNA H19 promotes tamoxifen resistance in breast cancer via autophagy. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Han, X.; Hu, Z.; Chen, L. The PVT1/miR-216b/Beclin-1 regulates cisplatin sensitivity of NSCLC cells via modulating autophagy and apoptosis. Cancer Chemother. Pharmacol. 2019, 83, 921–931. [Google Scholar] [CrossRef]
- Li, D.; Li, C.; Chen, Y.; Teng, L.; Cao, Y.; Wang, W.; Pan, H.; Xu, Y.; Yang, D. LncRNA HOTAIR induces sunitinib resistance in renal cancer by acting as a competing endogenous RNA to regulate autophagy of renal cells. Cancer Cell Int. 2020, 20, 1–14. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Xia, S.; Yang, L.; Wu, D.; Zhou, Y.; Lu, J. Long noncoding RNA PANDAR inhibits the development of lung cancer by regulating autophagy and apoptosis pathways. J. Cancer 2020, 11, 4783–4790. [Google Scholar] [CrossRef]
- Zhang, N.; Wen, K. The role of lncRNA binding to RNA-binding proteins to regulate mRNA stability in cancer progression and drug resistance mechanisms (Review). Oncol. Rep. 2024, 52, 1–12. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Xu, S.; Li, W.; Chen, M.; Jiang, M.; Fan, X. LncRNA FIRRE functions as a tumor promoter by interaction with PTBP1 to stabilize BECN1 mRNA and facilitate autophagy. Cell Death Dis. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Luo, Y.; Zhong, X.; Sun, X.; Fan, J. The RNA-binding protein ELAVL1 promotes Beclin1-mediated cellular autophagy and thus endometrial cancer development by affecting LncRNA-neat stability. Cancer Biol. Ther. 2025, 26, 2469927. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Xu, A.; Qiao, M.; Wu, Q.; Wang, W.; Mei, Y.; Wu, M. BECN1s, a short splice variant of BECN1, functions in mitophagy. Autophagy 2015, 11, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.-N.; Liu, Q.-Q.; Zhang, S.-P.; Yuan, N.; Cao, Y.; Cai, J.-Y.; Lin, W.-W.; Xu, F.; Wang, Z.-J.; Chen, B.; et al. Alternative Messenger RNA Splicing of Autophagic Gene Beclin 1 in Human B-cell Acute Lymphoblastic Leukemia Cells. Asian Pac. J. Cancer Prev. 2014, 15, 2153–2158. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.M.; Wrobel, L.; Rubinsztein, D.C. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 2018, 26, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 2016, 35, 866–880. [Google Scholar] [CrossRef]
- Cao, J.; Wu, S.; Zhao, S.; Wang, L.; Wu, Y.; Song, L.; Sun, C.; Liu, Y.; Liu, Z.; Zhu, R.; et al. USP24 promotes autophagy-dependent ferroptosis in hepatocellular carcinoma by reducing the K48-linked ubiquitination of Beclin1. Commun. Biol. 2024, 7, 1–15. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Luo, Y.; Liu, Y.; Yi, Y.; Li, J.; Pan, Y.; Li, W.; You, W.; Hu, Q.; et al. USP5-Beclin 1 axis overrides p53-dependent senescence and drives Kras-induced tumorigenicity. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef]
- Rong, Y.; Fan, J.; Ji, C.; Wang, Z.; Ge, X.; Wang, J.; Ye, W.; Yin, G.; Cai, W.; Liu, W. USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ. 2021, 29, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rao, S.; Song, C.; Zhu, M.; Zhao, H.; Yuan, S.; Peng, B.; Xu, X. Ubiquitin carboxyl-terminal hydrolase 11 promotes autophagy by de-ubiquitinating and stabilizing Beclin-1. Genome Instab. Dis. 2022, 3, 47–55. [Google Scholar] [CrossRef]
- Xu, D.; Shan, B.; Sun, H.; Xiao, J.; Zhu, K.; Xie, X.; Li, X.; Liang, W.; Lu, X.; Qian, L.; et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016, 30, 1718–1730. [Google Scholar] [CrossRef]
- Tao, L.; Liu, X.; Jiang, X.; Zhang, K.; Wang, Y.; Li, X.; Jiang, S.; Han, T. USP10 as a Potential Therapeutic Target in Human Cancers. Genes 2022, 13, 831. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, K.-B.; Chen, W.; Mai, J.; Wu, X.-Q.; Sun, T.; Wu, R.-Y.; Jiao, L.; Li, D.-D.; Ji, J.; et al. CUL3 (cullin 3)-mediated ubiquitination and degradation of BECN1 (beclin 1) inhibit autophagy and promote tumor progression. Autophagy 2021, 17, 4323–4340. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Feng, K.; Zhao, X.; Huang, S.; Cheng, Y.; Qian, L.; Wang, Y.; Sun, H.; Jin, M.; Chuang, T.-H.; et al. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy 2014, 10, 2239–2250. [Google Scholar] [CrossRef]
- Shi, C.-S.; Kehrl, J.H. TRAF6 and A20 Regulate Lysine 63–Linked Ubiquitination of Beclin-1 to Control TLR4-Induced Autophagy. Sci. Signal. 2010, 3, ra42–ra42. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Du, Y.; Zhao, Z.; Shi, L.; Sun, L.; Huang, G.; Ye, B.; Li, C.; Dai, Z.; et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J. 2013, 32, 2685–2696. [Google Scholar] [CrossRef]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2011, 441, 399–406. [Google Scholar] [CrossRef]
- Xu, C.; Liu, J.; Hsu, L.; Luo, Y.; Xiang, R.; Chuang, T. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J. 2011, 25, 2700–2710. [Google Scholar] [CrossRef]
- Sun, T.; Li, X.; Zhang, P.; Chen, W.-D.; Zhang, H.-L.; Li, D.-D.; Deng, R.; Qian, X.-J.; Jiao, L.; Ji, J.; et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat. Commun. 2015, 6, 7215. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, X.; Tong, H.; Yin, H.; Li, T.; Zhu, J.; Chen, J.; Wu, L.; Zhang, X.; Gou, X.; et al. SIRT1 Promotes Cisplatin Resistance in Bladder Cancer via Beclin1 Deacetylation-Mediated Autophagy. Cancers 2023, 16, 125. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Kumar, A.; Shaha, C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Cai, Q.; Zhang, C.; Yu, Q.; Jiang, Y.; Lee, J.-H.; Hawke, D.; Wang, Y.; Xia, Y.; et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol. Cell 2017, 65, 917–931.e6. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B.; Center, U.S.M.; et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife 2015, 4. [Google Scholar] [CrossRef]
- Fujiwara, N.; Usui, T.; Ohama, T.; Sato, K. Regulation of Beclin 1 Protein Phosphorylation and Autophagy by Protein Phosphatase 2A (PP2A) and Death-associated Protein Kinase 3 (DAPK3). J. Biol. Chem. 2016, 291, 10858–10866. [Google Scholar] [CrossRef]
- Li, X.; Wu, X.-Q.; Deng, R.; Li, D.-D.; Tang, J.; Chen, W.-D.; Chen, J.-H.; Ji, J.; Jiao, L.; Jiang, S.; et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, S.; Zhang, S.; Xu, H.; Li, X.; Guan, Y.; Yi, F.; Zhou, T.; Jiang, B.; Bai, N.; et al. ATM - CHK 2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.-L. Differential Regulation of Distinct Vps34 Complexes by AMPK in Nutrient Stress and Autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. Embo Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.-P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef]
- A Ciechomska, I.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.C.; Levine, B. Dual Role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Eisenstein, M.; Kimchi, A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009, 5, 720–722. [Google Scholar] [CrossRef]
- Shiloh, R.; Gilad, Y.; Ber, Y.; Eisenstein, M.; Aweida, D.; Bialik, S.; Cohen, S.; Kimchi, A. Non-canonical activation of DAPK2 by AMPK constitutes a new pathway linking metabolic stress to autophagy. Nat. Commun. 2018, 9, 1759. [Google Scholar] [CrossRef]
- Gurkar, A.U.; Chu, K.; Raj, L.; Bouley, R.; Lee, S.-H.; Kim, Y.-B.; Dunn, S.E.; Mandinova, A.; Lee, S.W. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Hu, F.; Song, D.; Yan, Y.; Huang, C.; Shen, C.; Lan, J.; Chen, Y.; Liu, A.; Wu, Q.; Sun, L.; et al. IL-6 regulates autophagy and chemotherapy resistance by promoting BECN1 phosphorylation. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, W.; Sun, X.; Xu, D.; Wang, C.; Zhang, Q.; Wang, H.; Luo, W.; Chen, Y.; Chen, H.; et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 2016, 12, 1447–1459. [Google Scholar] [CrossRef]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-Mediated Beclin 1 Phosphorylation in Autophagy Suppression, Tumor Progression, and Tumor Chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef]
- Han, J.; Hou, W.; Lu, C.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. Interaction between Her2 and Beclin-1 Proteins Underlies a New Mechanism of Reciprocal Regulation. J. Biol. Chem. 2013, 288, 20315–20325. [Google Scholar] [CrossRef]
- Levine, B.; Liu, R.; Dong, X.; Zhong, Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015, 25, 533–544. [Google Scholar] [CrossRef]
- Yu, C.; Gorantla, S.P.; Müller-Rudorf, A.; Müller, T.A.; Kreutmair, S.; Albers, C.; Jakob, L.; Lippert, L.J.; Yue, Z.; Engelhardt, M.; et al. Phosphorylation of BECLIN-1 by BCR-ABL suppresses autophagy in chronic myeloid leukemia. Haematologica 2019, 105, 1285–1293. [Google Scholar] [CrossRef]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-Mediated Regulation of Autophagy and Tumorigenesis Through Beclin 1 Phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef]
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7, 84999–85020. [Google Scholar] [CrossRef]
- Prerna, K.; Dubey, V.K. Beclin1-mediated interplay between autophagy and apoptosis: New understanding. Int. J. Biol. Macromol. 2022, 204, 258–273. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef]
- Li, H.; Wang, P.; Yu, J.; Zhang, L. Cleaving Beclin 1 to suppress autophagy in chemotherapy-induced apoptosis. Autophagy 2011, 7, 1239–1241. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Mukherjee, S.; Manivannan, P.; Malathi, K. RNase L Cleavage Products Promote Switch from Autophagy to Apoptosis by Caspase-Mediated Cleavage of Beclin-1. Int. J. Mol. Sci. 2015, 16, 17611–17636. [Google Scholar] [CrossRef]
- Hu, F.; Li, G.; Huang, C.; Hou, Z.; Yang, X.; Luo, X.; Feng, Y.; Wang, G.; Hu, J.; Cao, Z. The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- O’dOnovan, T.R.; O’sUllivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2022, 24, 167–185. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative Phosphorylation at the fin de siècle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Parsons, M.J.; Green, D.R.; Brown, G.C.; Murphy, M.P. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J.; Green, D.R. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLOS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Chen, D.; Gao, F.; Li, B.; Wang, H.; Xu, Y.; Zhu, C.; Wang, G. Parkin Mono-ubiquitinates Bcl-2 and Regulates Autophagy. J. Biol. Chem. 2010, 285, 38214–38223. [Google Scholar] [CrossRef]
- Michiorri, S.; Gelmetti, V.; Giarda, E.; Lombardi, F.; Romano, F.; Marongiu, R.; Nerini-Molteni, S.; Sale, P.; Vago, R.; Arena, G.; et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010, 17, 962–974. [Google Scholar] [CrossRef]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; A Hickey, M.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.-L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Quiles, J.M.; Najor, R.H.; Gonzalez, E.; Jeung, M.; Liang, W.; Burbach, S.M.; Zumaya, E.A.; Diao, R.Y.; Lampert, M.A.; Gustafsson, Å.B. Deciphering functional roles and interplay between Beclin1 and Beclin2 in autophagosome formation and mitophagy. Sci. Signal. 2023, 16, eabo4457. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Vandenberghe, W. Ambra1. Autophagy 2011, 7, 1555–1556. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J. Mol. Biol. 2020, 432, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chauhan, S.; Jain, A.; Ponpuak, M.; Choi, S.W.; Mudd, M.; Peters, R.; Mandell, M.A.; Johansen, T.; Deretic, V. Galectins and TRIMs directly interact and orchestrate autophagic response to endomembrane damage. Autophagy 2017, 13, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.-A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef]
- Baek, S.; Chang, J.-W.; Yoo, S.-M.; Choo, J.; Jung, S.; Nah, J.; Jung, Y.-K. TMEM9 activates Rab9-dependent alternative autophagy through interaction with Beclin1. Cell. Mol. Life Sci. 2024, 81, 1–13. [Google Scholar] [CrossRef]
- Sigismund, S.; Lanzetti, L.; Scita, G.; Di Fiore, P.P. Endocytosis in the context-dependent regulation of individual and collective cell properties. Nat. Rev. Mol. Cell Biol. 2021, 22, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Schink, K.O.; Stenmark, H. Class III phosphatidylinositol 3–kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J. 2013, 280, 2730–2742. [Google Scholar] [CrossRef] [PubMed]
- McKnight, N.C.; Zhong, Y.; Wold, M.S.; Gong, S.; Phillips, G.R.; Dou, Z.; Zhao, Y.; Heintz, N.; Zong, W.-X.; Yue, Z.; et al. Beclin 1 Is Required for Neuron Viability and Regulates Endosome Pathways via the UVRAG-VPS34 Complex. PLOS Genet. 2014, 10, e1004626. [Google Scholar] [CrossRef] [PubMed]
- Ruck, A.; Attonito, J.; Garces, K.T.; Nuñez, L.; Palmisano, N.J.; Rubel, Z.; Bai, Z.; Nguyen, K.C.; Sun, L.; Grant, B.D.; et al. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy inC. elegans. Autophagy 2011, 7, 386–400. [Google Scholar] [CrossRef]
- Lawe, D.C.; Chawla, A.; Merithew, E.; Dumas, J.; Carrington, W.; Fogarty, K.; Lifshitz, L.; Tuft, R.; Lambright, D.; Corvera, S. Sequential Roles for Phosphatidylinositol 3-Phosphate and Rab5 in Tethering and Fusion of Early Endosomes via Their Interaction with EEA1. J. Biol. Chem. 2002, 277, 8611–8617. [Google Scholar] [CrossRef]
- Leonard, D.; Hayakawa, A.; Lawe, D.; Lambright, D.; Bellve, K.D.; Standley, C.; Lifshitz, L.M.; Fogarty, K.E.; Corvera, S. Sorting of EGF and transferrin at the plasma membrane and by cargo-specific signaling to EEA1-enriched endosomes. J. Cell Sci. 2008, 121, 3445–3458. [Google Scholar] [CrossRef]
- A Rohatgi, R.; Janusis, J.; Leonard, D.; Bellvé, K.D.; E Fogarty, K.; Baehrecke, E.H.; Corvera, S.; Shaw, L.M. Beclin 1 regulates growth factor receptor signaling in breast cancer. Oncogene 2015, 34, 5352–5362. [Google Scholar] [CrossRef]
- Liu, B.; Lu, Y.; Zhang, T.; Yu, X.; Wang, Q.; Chi, Y.; Jin, S.; Cheng, G. CMTM7 as a novel molecule of ATG14L-Beclin1-VPS34 complex enhances autophagy by Rab5 to regulate tumorigenicity. Cell Commun. Signal. 2021, 19, 1–16. [Google Scholar] [CrossRef]
- Liu, B.; Su, Y.; Li, T.; Yuan, W.; Mo, X.; Li, H.; He, Q.; Ma, D.; Han, W. CMTM7 knockdown increases tumorigenicity of human non-small cell lung cancer cells and EGFR-AKT signaling by reducing Rab5 activation. Oncotarget 2015, 6, 41092–41107. [Google Scholar] [CrossRef]
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Law, F.; Rocheleau, C.E. Vps34 and the Armus/TBC-2 Rab GAPs: Putting the brakes on the endosomal Rab5 and Rab7 GTPases. Cell. Logist. 2017, 7, e1403530. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Lv, S.; Shi, Y.; Jia, J.; Ma, M.; Han, H.; Zhang, R.; Tan, J.; Zhang, X. RAB21 controls autophagy and cellular energy homeostasis by regulating retromer-mediated recycling of SLC2A1/GLUT1. Autophagy 2022, 19, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey – from activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef]
- Matthew-Onabanjo, A.N.; Janusis, J.; Mercado-Matos, J.; Carlisle, A.E.; Kim, D.; Levine, F.; Cruz-Gordillo, P.; Richards, R.; Lee, M.J.; Shaw, L.M. Beclin 1 Promotes Endosome Recruitment of Hepatocyte Growth Factor Tyrosine Kinase Substrate to Suppress Tumor Proliferation. Cancer Res. 2020, 80, 249–262. [Google Scholar] [CrossRef]
- Wijshake, T.; Zou, Z.; Chen, B.; Zhong, L.; Xiao, G.; Xie, Y.; Doench, J.G.; Bennett, L.; Levine, B. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Tran, S.; Juliani, J.; Harris, T.J.; Evangelista, M.; Ratcliffe, J.; Ellis, S.L.; Baloyan, D.; Reehorst, C.M.; Nightingale, R.; Luk, I.Y.; et al. BECLIN1 is essential for intestinal homeostasis involving autophagy-independent mechanisms through its function in endocytic trafficking. Commun. Biol. 2024, 7, 1–13. [Google Scholar] [CrossRef]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A Kinase-Independent Role for EGF Receptor in Autophagy Initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef]
- Ji, X.; Ma, H.; Du, Y. Role and mechanism of action of LAPTM4B in EGFR-mediated autophagy (Review). Oncol. Lett. 2022, 23, 1–6. [Google Scholar] [CrossRef]
- Eden, E.R.; White, I.J.; Tsapara, A.; Futter, C.E. Membrane contacts between endosomes and ER provide sites for PTP1B–epidermal growth factor receptor interaction. Nat. Cell Biol. 2010, 12, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Thapa, N.; Cryns, V.L.; Anderson, R.A. Regulation of Phosphoinositide Signaling by Scaffolds at Cytoplasmic Membranes. Biomolecules 2023, 13, 1297. [Google Scholar] [CrossRef]
- Caldieri, G.; Barbieri, E.; Nappo, G.; Raimondi, A.; Bonora, M.; Conte, A.; Verhoef, L.G.G.C.; Confalonieri, S.; Malabarba, M.G.; Bianchi, F.; et al. Reticulon 3–dependent ER-PM contact sites control EGFR nonclathrin endocytosis. Science 2017, 356, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Jin, R.; Wu, L.; Ye, X.; Yang, Y.; Luo, K.; Wang, W.; Wu, D.; Ye, X.; Huang, L.; et al. Reticulon 3 attenuates the clearance of cytosolic prion aggregates via inhibiting autophagy. Autophagy 2011, 7, 205–216. [Google Scholar] [CrossRef]
- Song, X.; Lee, D.-H.; Dilly, A.-K.; Lee, Y.-S.; Choudry, H.A.; Kwon, Y.T.; Bartlett, D.L.; Lee, Y.J. Crosstalk Between Apoptosis and Autophagy Is Regulated by the Arginylated BiP/Beclin-1/p62 Complex. Mol. Cancer Res. 2018, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Jian, M.; Yunjia, Z.; Zhiying, D.; Yanduo, J.; Guocheng, J. Interleukin 7 receptor activates PI3K/Akt/mTOR signaling pathway via downregulation of Beclin-1 in lung cancer. Mol. Carcinog. 2018, 58, 358–365. [Google Scholar] [CrossRef]
- Ma, J.; Liu, L.; Ling, Y.; Zheng, J. Polypeptide LTX-315 reverses the cisplatin chemoresistance of ovarian cancer cells via regulating Beclin-1/PI3K/mTOR signaling pathway. J. Biochem. Mol. Toxicol. 2021, 35, e22853. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, X.; Mo, X.; Yu, Y.; Xiao, Z.; Wu, J.; Ding, L.; Lei, C.; Zhu, Y.; Zhang, H. Xie-Bai-San increases NSCLC cells sensitivity to gefitinib by inhibiting Beclin-1 mediated autophagosome formation. Phytomedicine 2024, 125, 155351. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, C.; Yan, X.; Shi, Z.; Xiao, H.; Wei, X.; Jiang, N.; Wu, Z. LETM1 Knockdown Promotes Autophagy and Apoptosis Through AMP-Activated Protein Kinase Phosphorylation-Mediated Beclin-1/Bcl-2 Complex Dissociation in Hepatocellular Carcinoma. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Song, X. , Zhu, S., Chen, P., Hou, W., Wen, Q., Liu, J., Xie, Y., Liu, J., Klionsky, D. J., Kroemer, G., Lotze, M. T., Zeh, H. J., Kang, R., & Tang, D. (2018). AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc- Activity. Current biology : CB, 28(15), 2388–2399.e5. [CrossRef]
- Seo, J.; Seong, D.; Nam, Y.W.; Hwang, C.H.; Lee, S.R.; Lee, C.-S.; Jin, Y.; Lee, H.-W.; Oh, D.-B.; Vandenabeele, P.; et al. Beclin 1 functions as a negative modulator of MLKL oligomerisation by integrating into the necrosome complex. Cell Death Differ. 2020, 27, 3065–3081. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-E.; Lin, J.-F.; Tsai, T.-F.; Lin, Y.-C.; Chou, K.-Y.; Hwang, T.I.-S. Allyl Isothiocyanate Induces Autophagy through the Up-Regulation of Beclin-1 in Human Prostate Cancer Cells. Am. J. Chin. Med. 2018, 46, 1625–1643. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Wan, X.-B.; Yuan, Z.Y.; Wei, L.; Fan, X.J.; Wang, T.-T.; Lv, Y.C.; Li, X.; Chen, Z.-H.; Chen, J.; et al. Low expression of Beclin 1 and elevated expression of HIF-1α refine distant metastasis risk and predict poor prognosis of ER-positive, HER2-negative breast cancer. Med Oncol. 2013, 30, 1–10. [Google Scholar] [CrossRef]
- Minamoto, T.; Nakayama, K.; Nakamura, K.; Katagiri, H.; Sultana, R.; Ishibashi, T.; Ishikawa, M.; Yamashita, H.; Sanuki, K.; Iida, K.; et al. Loss of beclin 1 expression in ovarian cancer: A potential biomarker for predicting unfavorable outcomes. Oncol. Lett. 2017, 15, 1170–1176. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Wang, B.; Wang, H. Prognostic significance of autophagy-related genes Beclin1 and LC3 in ovarian cancer: a meta-analysis. J. Int. Med Res. 2020, 48. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.-D.; Wang, L.-L.; Deng, R.; Zhu, X.-F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef]
- Zhou, W.; Yue, C.; Deng, J.; Hu, R.; Xu, J.; Feng, L.; Lan, Q.; Zhang, W.; Ji, D.; Wu, J.; et al. Autophagic Protein Beclin 1 Serves as an Independent Positive Prognostic Biomarker for Non-Small Cell Lung Cancer. PLOS ONE 2013, 8, e80338. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Chen, L.; Luo, F.; Chen, X.; Li, Y.; Cheng, Q. Beclin-1 expression is associated with prognosis in a Bcl-2-dependent manner in non-small cell lung cancer. Oncol. Lett. 2020, 20, 1–1. [Google Scholar] [CrossRef]
- Dong, L.-W.; Hou, Y.-J.H.; Tan, Y.-X.; Tang, L.; Pan, Y.-F.; Wang, M.; Wang, H.-Y. Prognostic significance of Beclin 1 in intrahepatic cholangiocellular carcinoma. Autophagy 2011, 7, 1222–1229. [Google Scholar] [CrossRef]
- Wang, T.-T.; Cao, Q.-H.; Chen, M.-Y.; Xia, Q.; Fan, X.-J.; Ma, X.-K.; Lin, Q.; Jia, C.-C.; Dong, M.; Ruan, D.-Y.; et al. Beclin 1 Deficiency Correlated with Lymph Node Metastasis, Predicts a Distinct Outcome in Intrahepatic and Extrahepatic Cholangiocarcinoma. PLOS ONE 2013, 8, e80317. [Google Scholar] [CrossRef]
- Zheng, H.-C.; Zhao, S.; Xue, H.; Zhao, E.-H.; Jiang, H.-M.; Hao, C.-L. The Roles of Beclin 1 Expression in Gastric Cancer: A Marker for Carcinogenesis, Aggressive Behaviors and Favorable Prognosis, and a Target of Gene Therapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, M.; Zhao, B. Beclin 1 and LC3 as predictive biomarkers for metastatic colorectal carcinoma. Oncotarget 2017, 8, 59058–59067. [Google Scholar] [CrossRef]
- Liang, L.; Ma, B.; Liang, Y.; Liu, H.; Zheng, G.; Zhang, T.; Chu, M.; Xu, P.; Su, Y.; Liao, G. High expression of the autophagy gene Beclin-1 is associated with favorable prognosis for salivary gland adenoid cystic carcinoma. J. Oral Pathol. Med. 2012, 41, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-H.; Tang, F.; Xu, J.; Wu, X.; Yang, S.-B.; Feng, Z.-Y.; Ding, Y.-G.; Wan, X.-B.; Guan, Z.; Li, H.-G.; et al. Low expression of Beclin 1, associated with high Bcl-xL, predicts a malignant phenotype and poor prognosis of gastric cancer. Autophagy 2012, 8, 389–400. [Google Scholar] [CrossRef]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef]
- Valente, G.; Morani, F.; Nicotra, G.; Fusco, N.; Peracchio, C.; Titone, R.; Alabiso, O.; Arisio, R.; Katsaros, D.; Benedetto, C.; et al. Expression and Clinical Significance of the Autophagy Proteins BECLIN 1 and LC3 in Ovarian Cancer. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Salwa, A.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Moia, R.; Patriarca, A.; Garavaglia, B.; Gaidano, G.; Isidoro, C. High BECN1 Expression Negatively Correlates with BCL2 Expression and Predicts Better Prognosis in Diffuse Large B-Cell Lymphoma: Role of Autophagy. Cells 2023, 12, 1924. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.I.; Koutsopoulos, A.; Chloropoulou, P.; Liberis, V.; Sivridis, E. High Beclin 1 expression defines a poor prognosis in endometrial adenocarcinomas. Gynecol. Oncol. 2011, 123, 147–151. [Google Scholar] [CrossRef]
- I Koukourakis, M.; Giatromanolaki, A.; Sivridis, E.; Pitiakoudis, M.; Gatter, K.C.; Harris, A.L. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Br. J. Cancer 2010, 103, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Huang, S.; Wu, T.-T.; Foster, N.R.; Sinicrope, F.A. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol. Ther. 2013, 14, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Salwa, A.; Ferraresi, A.; Vallino, L.; Maheshwari, C.; Moia, R.; Gaidano, G.; Isidoro, C. High Mitophagy and Low Glycolysis Predict Better Clinical Outcomes in Acute Myeloid Leukemias. Int. J. Mol. Sci. 2024, 25, 11527. [Google Scholar] [CrossRef]
- Thongchot, S.; Ferraresi, A.; Vidoni, C.; Salwa, A.; Vallino, L.; Kittirat, Y.; Loilome, W.; Namwat, N.; Isidoro, C. Preclinical evidence for preventive and curative effects of resveratrol on xenograft cholangiocarcinogenesis. Cancer Lett. 2023, 582, 216589. [Google Scholar] [CrossRef] [PubMed]
- El Shayeb, A.; Deghedy, A.; Bedewy, E.S.; Badawy, S.; Abdeen, N. Serum Beclin 1 and autophagy-related protein-5 and the risk of hepatocellular carcinoma among cirrhotic hepatitis C patients. Egypt. Liver J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Pan, Y.-Z.; Liang, Q.; Tomchick, D.R.; De Brabander, J.K.; Rizo, J. Structural insights for selective disruption of Beclin 1 binding to Bcl-2. Commun. Biol. 2023, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; He, L.; Xing, C.; Mao, J.; Yu, X.; Zhu, M.; Diao, L.; Han, L.; Zhou, Y.; You, J.M.; et al. Myeloid loss of Beclin 1 promotes PD-L1hi precursor B cell lymphoma development. J. Clin. Investig. 2019, 129, 5261–5277. [Google Scholar] [CrossRef]
- Noman, M.Z.; Berchem, G.; Janji, B. Targeting autophagy blocks melanoma growth by bringing natural killer cells to the tumor battlefield. Autophagy 2018, 14, 730–732. [Google Scholar] [CrossRef]
- Song, M.J.; Park, S.; Won, K.Y. Expression of Beclin-1, an autophagy-related protein, is associated with tumoral FOXP3 expression and Tregs in gastric adenocarcinoma: The function of Beclin-1 expression as a favorable prognostic factor in gastric adenocarcinoma. Pathol. - Res. Pr. 2020, 216, 152927. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Shakir; Ansari, M. S.; Divya; Faizan, I.; Chauhan, V.; Singh, A.; Alam, R.; Azmi, I.; Sharma, S.; et al. Bioengineering the metabolic network of CAR T cells with GLP-1 and Urolithin A increases persistence and long-term anti-tumor activity. Cell Rep. Med. 2025, 6, 102021. [Google Scholar] [CrossRef]
- Vega-Rubin-de-Celis, S.; Zou, Z.; Fernandez, A.F.; Ci, B.; Kim, M.; Xiao, G.; Xie, Y.; Levine, B. Increased autophagy blocks HER2-mediated breast tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4176–4181. [Google Scholar] [CrossRef]
- Kona, S.V.; Kalivendi, S.V. The USP10/13 inhibitor, spautin-1, attenuates the progression of glioblastoma by independently regulating RAF-ERK mediated glycolysis and SKP2. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2024, 1870, 167291. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Zhao, C.-J.; Jin, Z.-F.; Zheng, J.; Ma, L.-T. Targeted therapy based on ubiquitin-specific proteases, signalling pathways and E3 ligases in non-small-cell lung cancer. Front. Oncol. 2023, 13, 1120828. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-Only Proteins and BH3 Mimetics Induce Autophagy by Competitively Disrupting the Interaction between Beclin 1 and Bcl-2/Bcl-XL. Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.-C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, X.; Zhang, D.; Xie, Y.; Sun, B.; Li, H.; Sun, L.; Zhang, X. B-cell lymphoma 2 inhibitor ABT-737 induces Beclin1- and reactive oxygen species-dependent autophagy in Adriamycin-resistant human hepatocellular carcinoma cells. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Li, N.; Zhang, X.; Chen, J.; Gao, S.; Wang, L.; Zhao, Y. Perturbation of Autophagy by a Beclin 1-Targeting Stapled Peptide Induces Mitochondria Stress and Inhibits Proliferation of Pancreatic Cancer Cells. Cancers 2023, 15, 953. [Google Scholar] [CrossRef]
- Liu, S.; Yue, C.; Chen, H.; Chen, Y.; Li, G. Metformin Promotes Beclin1-Dependent Autophagy to Inhibit the Progression of Gastric Cancer. OncoTargets Ther. 2020, ume 13, 4445–4455. [Google Scholar] [CrossRef]
- Xia, H.; Tai, X.-J.; Cheng, W.; Wu, Y.; He, D.; Wang, L.-F.; Liu, H.; Zhang, S.-Y.; Sun, Y.-T.; Liu, H.-Z.; et al. Metformin inhibits the growth of SCLC cells by inducing autophagy and apoptosis via the suppression of EGFR and AKT signalling. Sci. Rep. 2025, 15, 1–15. [Google Scholar] [CrossRef]
- Zhuang, A.; Chai, P.; Wang, S.; Zuo, S.; Yu, J.; Jia, S.; Ge, S.; Jia, R.; Zhou, Y.; Shi, W.; et al. Metformin promotes histone deacetylation of optineurin and suppresses tumour growth through autophagy inhibition in ocular melanoma. Clin. Transl. Med. 2022, 12, e660. [Google Scholar] [CrossRef]
- Chen, H.; Lin, C.; Lu, C.; Wang, Y.; Han, R.; Li, L.; Hao, S.; He, Y. Metformin-sensitized NSCLC cells to osimertinib via AMPK-dependent autophagy inhibition. Clin. Respir. J. 2019, 13, 781–790. [Google Scholar] [CrossRef]
- He, C.; Wei, Y.; Sun, K.; Li, B.; Dong, X.; Zou, Z.; Liu, Y.; Kinch, L.N.; Khan, S.; Sinha, S.; et al. Beclin 2 Functions in Autophagy, Degradation of G Protein-Coupled Receptors, and Metabolism. Cell 2013, 154, 1085–1099. [Google Scholar] [CrossRef]
- He, C.; Wei, Y.; Sun, K.; Li, B.; Dong, X.; Zou, Z.; Liu, Y.; Kinch, L.N.; Khan, S.; Sinha, S.; et al. Beclin 2 Functions in Autophagy, Degradation of G Protein-Coupled Receptors, and Metabolism. Cell 2013, 154, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Deng, G.; Tan, P.; Xing, C.; Guan, C.; Jiang, C.; Zhang, Y.; Ning, B.; Li, C.; Yin, B.; et al. Beclin 2 negatively regulates innate immune signaling and tumor development. J. Clin. Investig. 2020, 130, 5349–5369. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Li, C.; Chen, L.; Xing, C.; Fu, C.; Qian, C.; Liu, X.; Wang, H.Y.; Zhu, M.; Wang, R.-F. BECN2 (beclin 2) Negatively Regulates Inflammasome Sensors Through ATG9A-Dependent but ATG16L1- and LC3-Independent Non-Canonical Autophagy. Autophagy 2021, 18, 340–356. [Google Scholar] [CrossRef]
- Quiles, J.M.; Najor, R.H.; Gonzalez, E.; Jeung, M.; Liang, W.; Burbach, S.M.; Zumaya, E.A.; Diao, R.Y.; Lampert, M.A.; Gustafsson, Å.B. Deciphering functional roles and interplay between Beclin1 and Beclin2 in autophagosome formation and mitophagy. Sci. Signal. 2023, 16, eabo4457. [Google Scholar] [CrossRef]
- Su, M.; Li, Y.; Wyborny, S.; Neau, D.; Chakravarthy, S.; Levine, B.; Colbert, C.L.; Sinha, S.C. BECN2 interacts with ATG14 through a metastable coiled-coil to mediate autophagy. Protein Sci. 2017, 26, 972–984. [Google Scholar] [CrossRef]
Figure 1.
BECN1 as a central regulator of cell survival/cell death balance and endocytic trafficking. At the start of autophagy, BECN1 forms a Class III PI3K Complex 1, which is involved in autophagosome formation and the recruitment of other autophagy proteins, allowing the elongation, maturation, and cargo incorporation into the autophagosome. Additionally, BECN1, as part of the Class III PI3K Complex 2, plays a role in endocytic trafficking by regulating the fusion and maturation of early into late endosomes. BECN1 also plays a key role in apoptosis, where it sequesters the anti-apoptotic protein BCL-2 through its BH3 domain, thereby releasing its inhibitory effect on apoptosis.
Figure 1.
BECN1 as a central regulator of cell survival/cell death balance and endocytic trafficking. At the start of autophagy, BECN1 forms a Class III PI3K Complex 1, which is involved in autophagosome formation and the recruitment of other autophagy proteins, allowing the elongation, maturation, and cargo incorporation into the autophagosome. Additionally, BECN1, as part of the Class III PI3K Complex 2, plays a role in endocytic trafficking by regulating the fusion and maturation of early into late endosomes. BECN1 also plays a key role in apoptosis, where it sequesters the anti-apoptotic protein BCL-2 through its BH3 domain, thereby releasing its inhibitory effect on apoptosis.
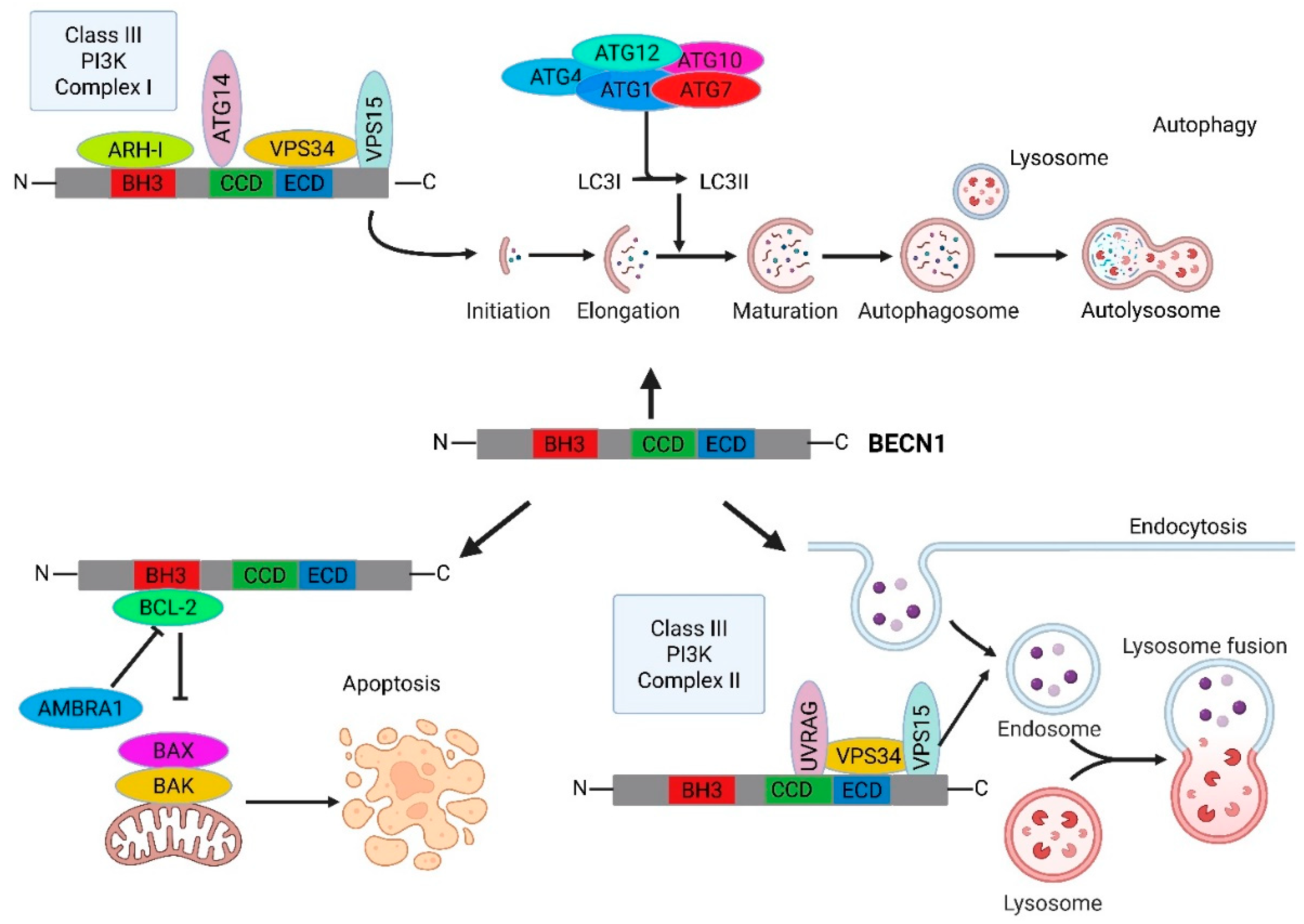
Figure 2.
Domain architecture of BECLIN-1 coding sequence and its interaction landscape. A) Structurally color-coded domains and their respective interactions. BCL-2 homology 3 or BH3 domain (green), coiled-coil domain or CCD (magenta), evolutionary conserved domain or ECD (yellow) and nuclear export signal or NES (orange), B) The CCD can form metastable homodimers of BECLIN-1 that favors its binding to BCL-2/BCL-XL, contributing to the inactivation of BECLIN-1's pro-autophagy functions. This inactivation is reversible, as the metastable homodimers can readily transition to more stable heterodimeric complexes with partners like ATG14L or UVRAG, and C) the two distinct PI3KC3 complexes: Complex I (PI3KC3-C1) — VPS34, VPS15, BECN1, ATG14 and Complex II (PI3KC3-C2) — VPS34, VPS15, BECLIN-1, UVRAG, highlighting the dynamic nature of these complexes and the role of BECLIN-1 as a central scaffold in the initiation and the maturation of the autophagosome, respectively.
Figure 2.
Domain architecture of BECLIN-1 coding sequence and its interaction landscape. A) Structurally color-coded domains and their respective interactions. BCL-2 homology 3 or BH3 domain (green), coiled-coil domain or CCD (magenta), evolutionary conserved domain or ECD (yellow) and nuclear export signal or NES (orange), B) The CCD can form metastable homodimers of BECLIN-1 that favors its binding to BCL-2/BCL-XL, contributing to the inactivation of BECLIN-1's pro-autophagy functions. This inactivation is reversible, as the metastable homodimers can readily transition to more stable heterodimeric complexes with partners like ATG14L or UVRAG, and C) the two distinct PI3KC3 complexes: Complex I (PI3KC3-C1) — VPS34, VPS15, BECN1, ATG14 and Complex II (PI3KC3-C2) — VPS34, VPS15, BECLIN-1, UVRAG, highlighting the dynamic nature of these complexes and the role of BECLIN-1 as a central scaffold in the initiation and the maturation of the autophagosome, respectively.
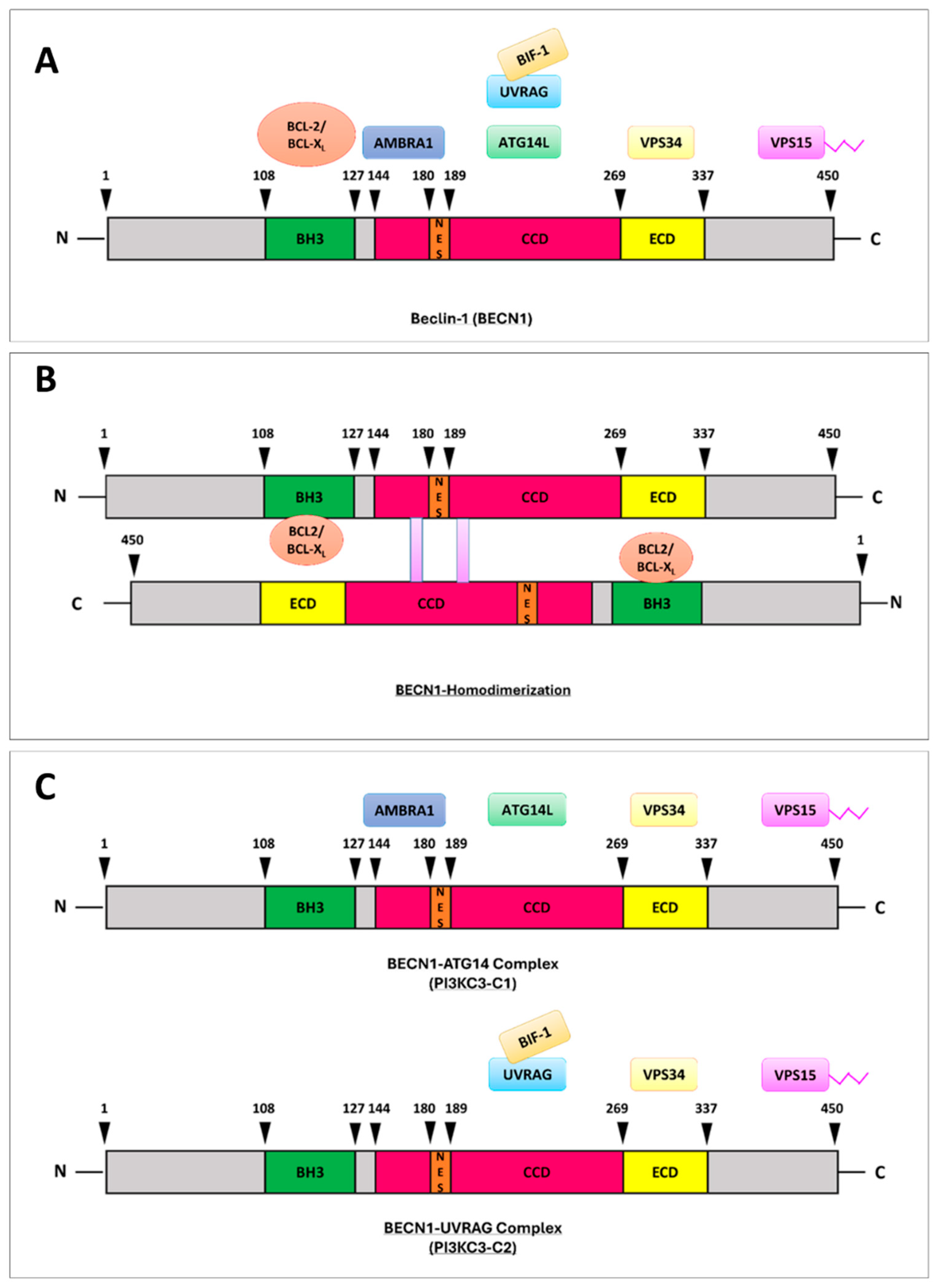
Figure 3.
Epigenetic mechanisms regulating BECN1 expression. Schematic representation of the epigenetic modifiers that control BECN1 mRNA at transcriptional and post-transcriptional levels. The human BECN1 gene (located on chromosome 17q21) undergoes several modifications, including DNA methylation and deacetylation, that result in reduced expression of BECN1. Furthermore, BECN1 mRNA translation is regulated by a subset of lncRNAs and miRNAs. The first ones can act as positive regulators either by stabilizing BECN1 mRNA or by sponging miRNAs targeting BECN1, while the latter ones are responsible for BECN-1 downregulation by eliciting mRNA degradation or translational inhibition.
Figure 3.
Epigenetic mechanisms regulating BECN1 expression. Schematic representation of the epigenetic modifiers that control BECN1 mRNA at transcriptional and post-transcriptional levels. The human BECN1 gene (located on chromosome 17q21) undergoes several modifications, including DNA methylation and deacetylation, that result in reduced expression of BECN1. Furthermore, BECN1 mRNA translation is regulated by a subset of lncRNAs and miRNAs. The first ones can act as positive regulators either by stabilizing BECN1 mRNA or by sponging miRNAs targeting BECN1, while the latter ones are responsible for BECN-1 downregulation by eliciting mRNA degradation or translational inhibition.
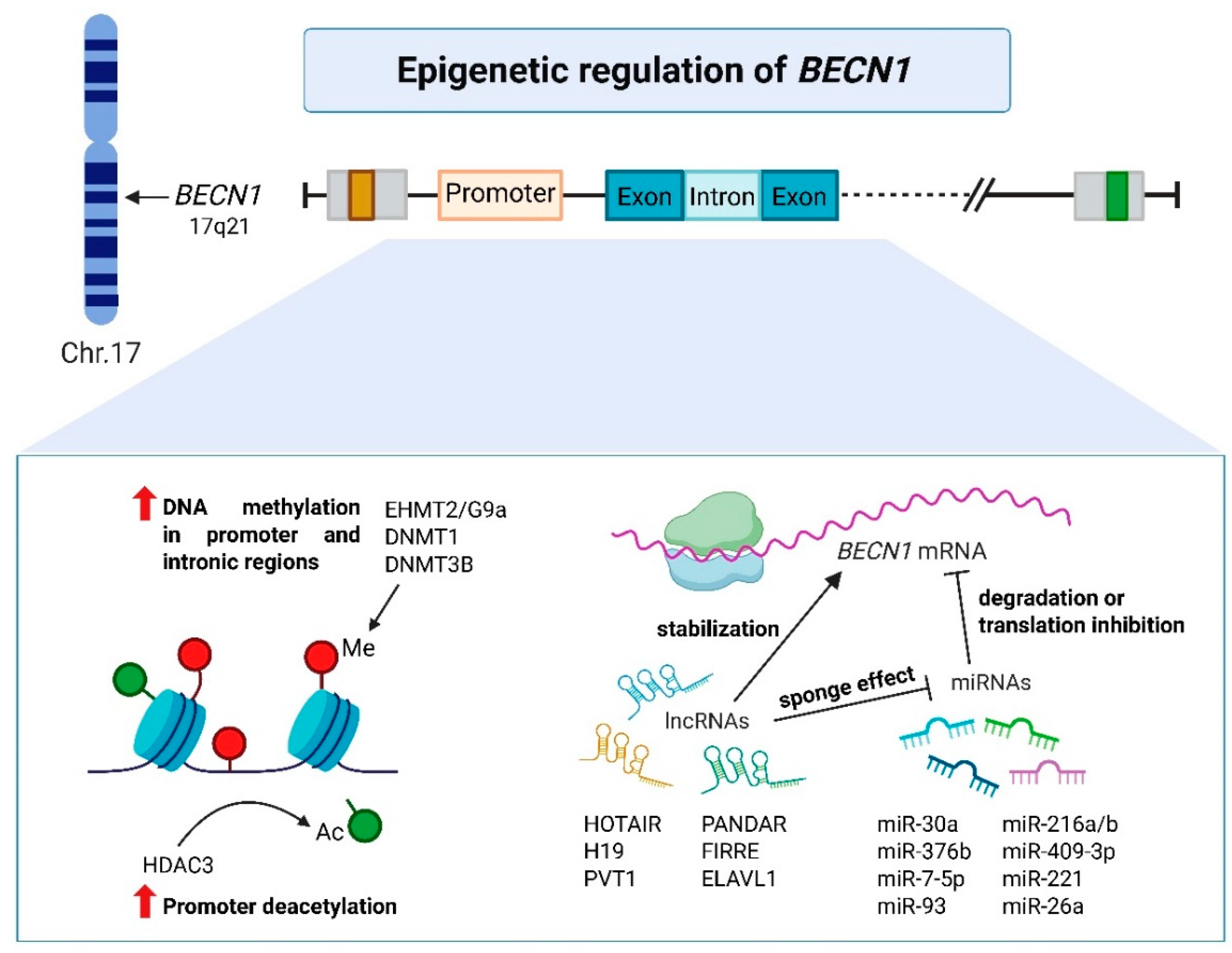
Figure 4.
Alternative splicing of the BECN1 mRNA. The BECN1 mRNA undergoes alternative splicing to produce three BECN1 splice variants (⍺, β, and γ). BECN1-⍺, with a partial deletion in the C-terminal, hampers its interaction with VPS15, affecting its role in starvation-induced autophagy. BECN1-β and -γ cannot interact with BCL-2 and AMBRA1 due to complete and partial deletions of the BH3 domain and CCD. The partial deletion of the ECD in BECN1-β prevents it from interacting with VPS34, which negatively impacts autophagy. BECN1-γ, however, retains its ability to bind VPS34 and only slightly affects autophagy.
Figure 4.
Alternative splicing of the BECN1 mRNA. The BECN1 mRNA undergoes alternative splicing to produce three BECN1 splice variants (⍺, β, and γ). BECN1-⍺, with a partial deletion in the C-terminal, hampers its interaction with VPS15, affecting its role in starvation-induced autophagy. BECN1-β and -γ cannot interact with BCL-2 and AMBRA1 due to complete and partial deletions of the BH3 domain and CCD. The partial deletion of the ECD in BECN1-β prevents it from interacting with VPS34, which negatively impacts autophagy. BECN1-γ, however, retains its ability to bind VPS34 and only slightly affects autophagy.
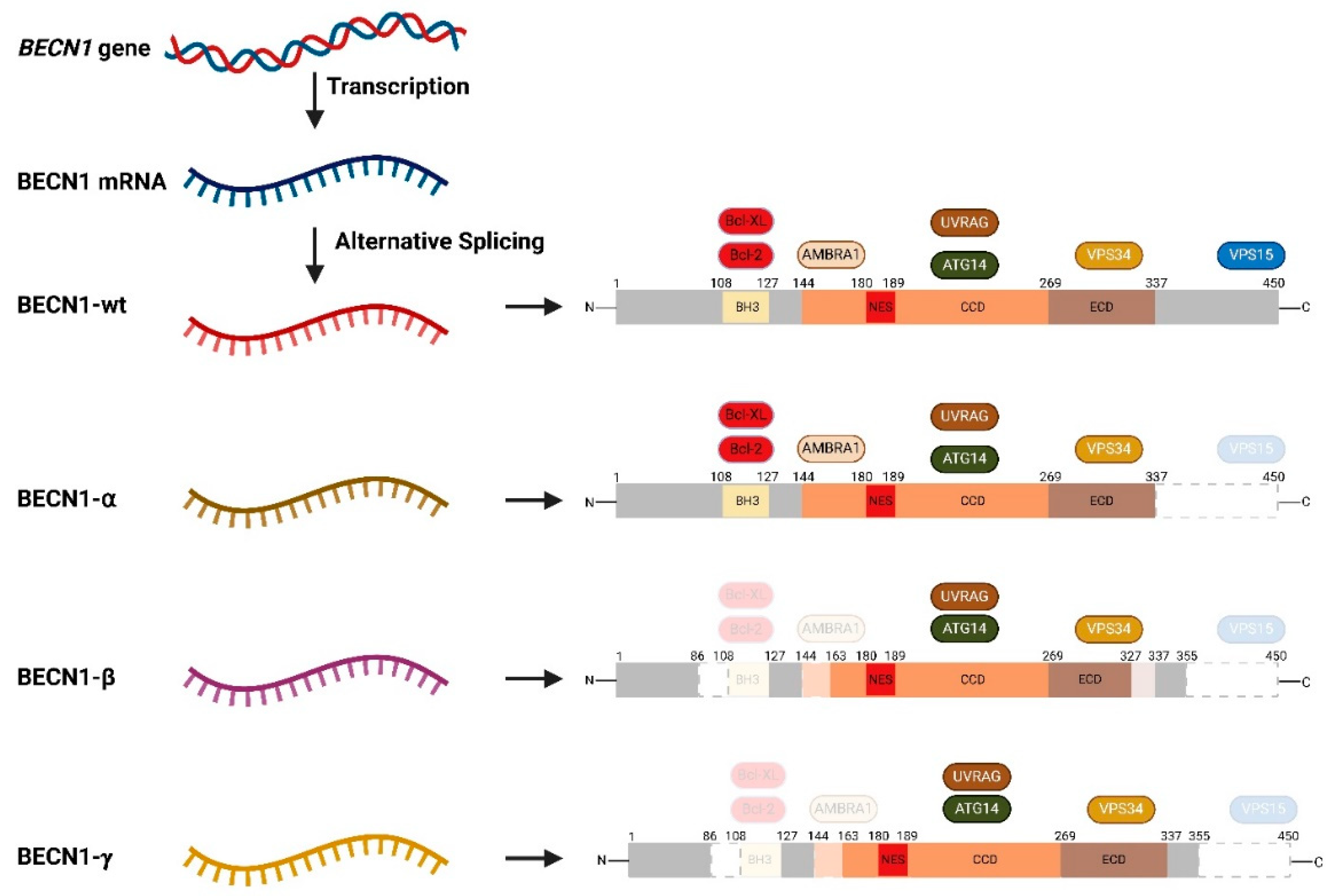
Figure 5.
Site-specific post-translational modification sites on BECLIN-1 amino acid sequence. The picture graphically represents the modifications related to phosphorylation (activating (green color) or inactivating (red color) BECLIN-1 protein function, respectively), ubiquitination, and acetylation of BECLIN-1 operated by the regulators reported in the orange boxes.
Figure 5.
Site-specific post-translational modification sites on BECLIN-1 amino acid sequence. The picture graphically represents the modifications related to phosphorylation (activating (green color) or inactivating (red color) BECLIN-1 protein function, respectively), ubiquitination, and acetylation of BECLIN-1 operated by the regulators reported in the orange boxes.
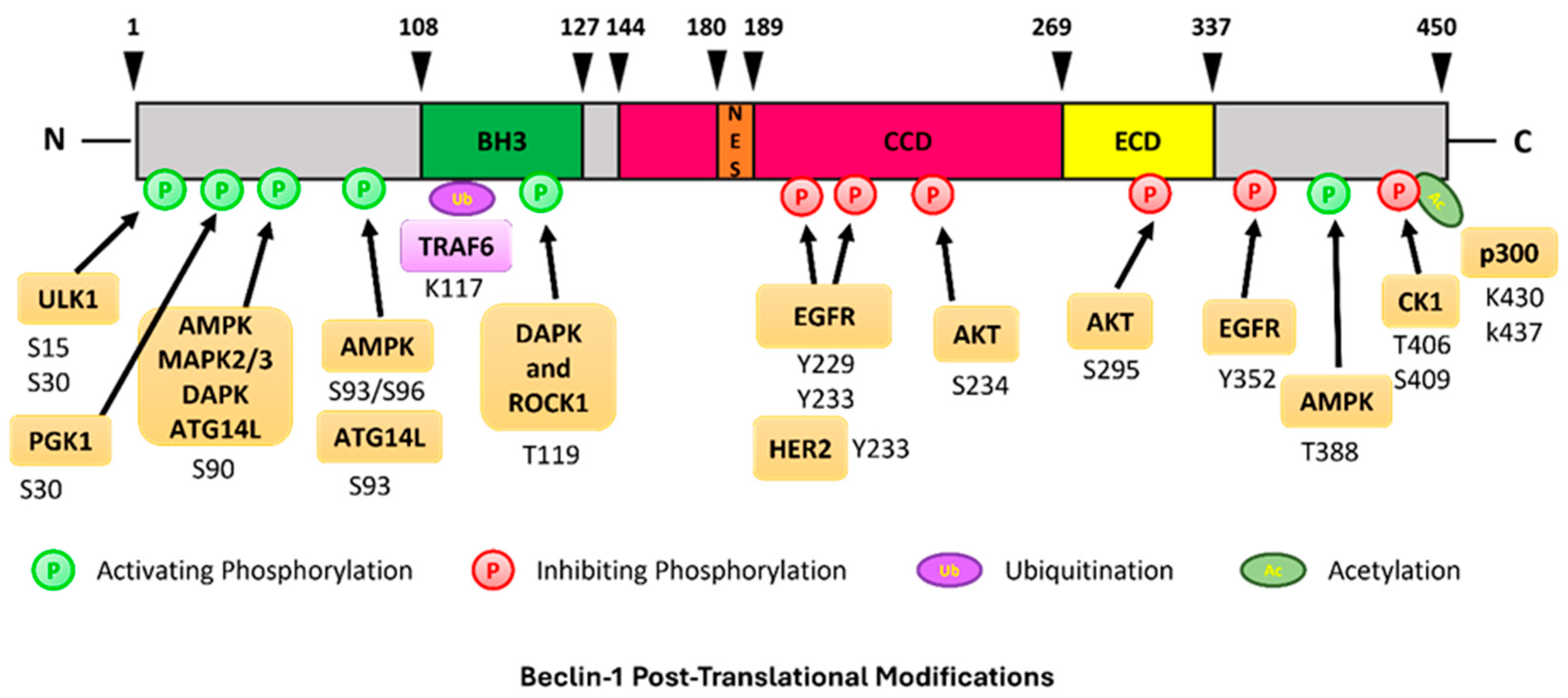
Figure 6.
Clinical relevance of BECN1 expression in cancer. Schematic representation of the main implications of BECN1 in terms of prognostic value and regulation of cancer hallmarks (e.g., metabolic crosstalk in the microenvironment, metastatic behavior, and regulation of anti-cancer immunity). The illustration highlights that BECN1 expression can help in stratifying cancer patients into responder vs. non-responder groups to predict the therapeutic outcomes. This prognostic relevance can be attributed to the functional role of BECN1 in the modulation pro-metastatic, pro-tumorigenic signaling pathways and the immune-suppressive landscape. Abbreviations: N1, type 1 neutrophils (anti-tumor); N2, type 2 neutrophils (pro-tumor); NK, natural killer cells; T CD8+, T-cells; MDSCs, myeloid-derived suppressive cells; T regs, regulatory T-cells.
Figure 6.
Clinical relevance of BECN1 expression in cancer. Schematic representation of the main implications of BECN1 in terms of prognostic value and regulation of cancer hallmarks (e.g., metabolic crosstalk in the microenvironment, metastatic behavior, and regulation of anti-cancer immunity). The illustration highlights that BECN1 expression can help in stratifying cancer patients into responder vs. non-responder groups to predict the therapeutic outcomes. This prognostic relevance can be attributed to the functional role of BECN1 in the modulation pro-metastatic, pro-tumorigenic signaling pathways and the immune-suppressive landscape. Abbreviations: N1, type 1 neutrophils (anti-tumor); N2, type 2 neutrophils (pro-tumor); NK, natural killer cells; T CD8+, T-cells; MDSCs, myeloid-derived suppressive cells; T regs, regulatory T-cells.
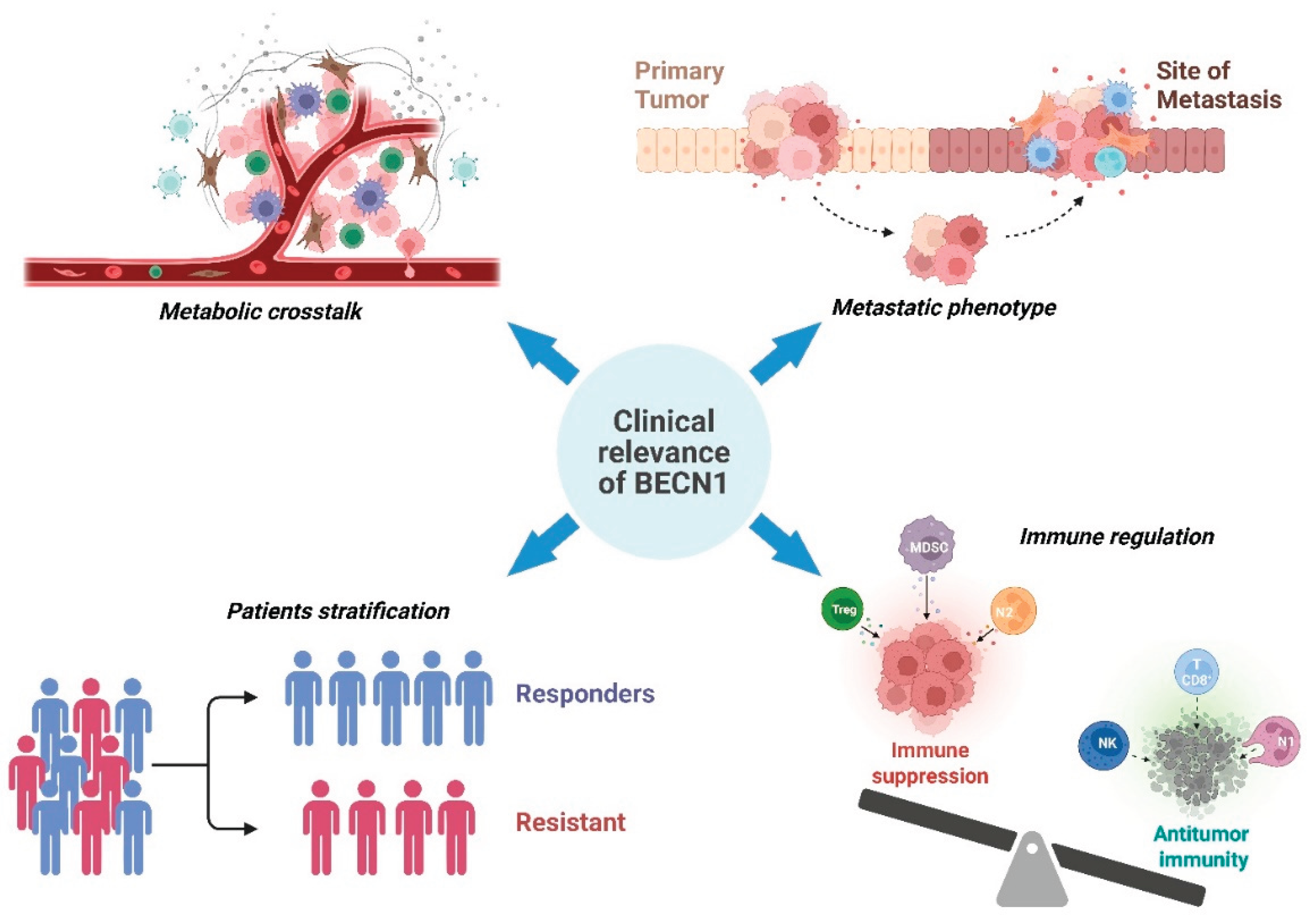
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



