Submitted:
23 July 2025
Posted:
24 July 2025
Read the latest preprint version here
Abstract
Focal task-specific dystonia (FTSD) poses a complex interplay of maladaptive neuroplasticity and motor-circuit imbalance. Traditional theories often implicate subcortical nuclei but fail to explain why symptoms remain so tightly bound to a singular, highly practiced skill. Here, we propose that the primary driver of FTSD is a newly formed “dystonic synergy” within the primary motor cortex (M1), in which excitatory circuit synapses are adequate relative to under-strengthened inhibitory circuit synapses, triggering involuntary contractions once the skill’s intensity demands surpass the functional synergy’s excitatory and inhibitory circuit capacity (synaptic strength). In short, we use an extensive single-case observation as the core empirical foundation, we chronicle how a decade of stable piano performance deteriorated following a sudden technical change that forced the finger flexion motor synergy to “overreach”. The patient’s initial phase was dominated by “true weakness,” a condition of task-specific paresis where the motor system is physically unable to generate the required excitatory/inhibitory (E/I) drive to match the attempted movement speed; over repetitive attempts to override that limitation, the excitatory circuit strengthened while the inhibitory circuit lagged, culminating in a fully formed dystonic synergy within three weeks. This maladaptive synergy then manifested in both piano playing and typing—a related digit-based skill—greatly disabling normal function in both tasks. We illustrate that once formed, the dystonic synergy remains stable but not spontaneously progressive, consistent with a saturable excitatory capacity. Moreover, we used a spiking neural network simulation to provide quantitative proof of concept verification of the hypothesis. Other commonly reported structural or electrophysiological alterations—such as basal ganglia and cerebellar changes, sensorimotor smudging in the primary somatosensory cortex (S1), or impaired spinal inhibition—are reframed and proposed as secondary byproducts emerging from chronic hyperexcitation of the M1 synergy. Additionally, we outline a new taxonomy distinguishing (i) “typical” neuroplastic dystonias, including task-specific forms whose primary trigger is repeated overreaching and whose pathophysiology lies in the consequent synergy imbalance; (ii) atypical neuroplastic variants with strong genetic underpinnings but partial plastic compensation; and (iii) non-neuroplastic dystonias resulting from more deterministically causal gene mutations. Finally, we propose and describe a non-invasive motor retraining approach for reversing FTSD: “below or at-threshold retraining” (BATR), wherein the inhibitory circuit of the dystonic synergy is methodically strengthened. This motor strategy, validated in the single-case longitudinal data alongside other published studies using very similar methods, reveals that the dysregulated synergy can be rebalanced to restore fully normal motor function. By integrating these mechanistic and therapeutic insights, we offer a unifying framework for FTSD pathogenesis and highlight a compelling, noninvasive avenue for cure alongside a guided strategy for prevention.
Keywords:
dystonia
; focal dystonia
; musician’s dystonia
; cause
; cure
; mechanism
; plasticity
; rehabilitation
Introduction
Focal task-specific dystonia (FTSD) ranks among the most perplexing motor disorders (Frucht, 2014; Stahl & Frucht, 2017). Affecting individuals who have often achieved a high degree of skill in a specialized movement domain, FTSD typically emerges after years of seemingly normal practice, manifesting in abrupt, involuntary muscle contractions and distortions unique to the targeted task (Rozanski et al., 2015). Although FTSD shares some superficial clinical overlap with other primary dystonias, particularly involuntary twisting or posturing, its distinctly “task-bound” nature (i.e., symptoms triggered almost exclusively by a specific skilled activity) underscores a pivotal role for maladaptive neuroplasticity within the cortical circuitry subserving that motor subtask (Quartarone et al., 2006). Indeed, multiple electrophysiological findings—notably the characteristic reduction of short-interval intracortical inhibition (SICI)—point to an underlying hyperexcitability in the primary motor cortex (M1) (Furuya et al., 2018; Ridding et. al, 1995; Siebner et al., 1999). However, the deep causal chain and precise mechanisms driving these changes have been historically elusive. Most conventional models of dystonia have emphasized basal ganglia (Grossman & Kelly, 1976; Simonyan et al., 2017) or cerebellar dysfunction (Teo et al., 2009), highlighting broad sensorimotor integration deficits. Yet these explanations do not entirely account for why FTSD can remain confined to a single fine motor skill while leaving nearby tasks—or even adjacent digits—spared.
The hallmark transcranial magnetic stimulation (TMS) findings in “typical” forms of dystonia (a term we define later on) have shown that local cortical inhibitory circuits, presumably GABAA-mediated, are functionally impaired (e.g., Levy & Hallett, 2002; Stinear & Byblow, 2004). In task-specific syndromes, these inhibitory deficits appear strongly plasticity-driven: skill repetition at “above-capacity” loads fosters an aberrant reorganization process. Combined, these insights argue for a mechanistic framework wherein the repeated overreaching of the synergy’s excitatory/inhibitory (E/I) capacity gradually sculpts a “dystonic synergy” with excessive excitatory strength relative to its under strengthened inhibitory counterpart. The resulting synergy becomes hyperexcitable, “locking in” involuntary co-contractions or undesired postures whenever that skill is activated. Observations of non-manifesting DYT1 carriers who exhibit partial cortical excitability changes (Edwards et al., 2003), but no clinical dystonia, suggest that genetic predispositions alone are insufficient to produce symptoms for FTSD; environmental triggers or overuse in the relevant synergy appear necessary to amplify the pre-existing E/I imbalance into full-blown dystonic movements (Furuya et al., 2018). Thus, the “cause” of FTSD can be anchored in repeated, maladaptive Hebbian loops that evolve after the synergy’s capacity is reduced by major technique changes and is then relentlessly overreached. In this scenario, any concurrency of “genetic predisposition” further lowers the threshold at which overreaching triggers an entrenched dystonic synergy.
Despite the strong emphasis on cortical plasticity, it is critical to recognize that FTSD, much like other forms of dystonia, can present with broad neural alterations outside M1. Numerous imaging and neurophysiological studies have revealed basal ganglia involvement, potentially abnormal cerebellar feedback loops, and sensorimotor smudging in the primary somatosensory cortex (S1)—collectively reflecting the entire motor system’s capacity to reconfigure in maladaptive ways (Elbert et al. 1998; Kita et al., 2021; Simonyan et al., 2017; Tinazzi et al., 2003). A large body of research, nonetheless, indicates that these changes outside M1 could potentially arise secondarily, through Hebbian adaption from repeated ectopic signals from an M1 circuit locked in chronic hyperexcitability (e.g., Furuya et al., 2018; Hallett, 2011; Tseng et al., 2014). Similarly, spinal inhibitory deficits found in focal hand dystonia may represent downstream changes—persistent pathologic input from the affected synergy reshaping inhibitory interneuron networks (Berardelli et al., 1998). Hence, a unifying explanation posits that once the synergy’s inhibitory dimension fails to strengthen proportionally with excitatory drive, excessive excitatory outflow cascades through cortico-subcortical and corticospinal loops, generating new “byproduct” alterations at many levels of the motor hierarchy.
From a clinical standpoint, FTSD historically has been managed symptomatically: botulinum toxin injections aim to reduce excessive muscle over-activity, medications modulate generalized motor excitability, or surgery intervenes on deeper structures (e.g., globus pallidus) (Grigoriu et al., 2015). While these can provide partial relief, none directly tackle the underlying E/I mismatch at its source, nor restore normal synergy function. Over the past two decades, an emerging recognition that FTSD is a problem of maladaptive plasticity has spurred interest in motor retraining approaches—a set of behavioral protocols designed to reshape disordered synergy circuits by carefully controlling practice conditions (Quartarone & Hallett, 2013). Indeed, case studies of “slow-down exercise” (SDE) have documented complete or near-complete resolution of task-specific dystonias (Yoshie et al., 2015). These rehabilitative successes underline the principle that, if the synergy can re-strengthen its inhibitory circuit without re-engaging the involuntary synergy’s excitatory drive, a rebalancing of synaptic strengths is feasible.
Moreover, throughout this manuscript, we employ the term threshold in two non-interchangeable ways. Symptom-threshold (behavioral threshold) refers to the level of task intensity—whether defined by speed, force, or effort—beyond which performance deteriorates, manifesting as true weakness or as involuntary dystonic contractions; movements executed at or below this intensity remain fully voluntary and symptom-free (Sakai, 2006; Yoshie et al., 2015). In contrast, spike threshold (firing threshold) designates the membrane-potential value at which an individual neuron generates an action potential, with subthreshold denoting excursions that do not reach this firing level. To avoid confusion, we hyphenate symptom-threshold when referring to the behavioral phenomenon and leave threshold unmodified for the cellular “spike-threshold” sense. Where both meanings could plausibly be inferred, the appropriate qualifier is repeated explicitly.
This paper, titled A Proposed Cause, Cure, and Underlying Mechanism for Focal Task-Specific Dystonia, aims to (1) delineate in detail how FTSD can be conceptualized as the direct outcome of repeated overreaching in a synergy whose excitatory and inhibitory capacities have become reduced; (2) show, through firsthand empirical observations, the progression of dystonia and its resolution via “below or at-threshold retraining” (BATR) in a single-case “natural experiment”; and (3) discuss why other documented brain alterations (e.g., in basal ganglia, cerebellum, S1) potentially emerge secondary to the pathologic synergy’s chronic over-excitation and how they remain highly consistent with an M1-centric cause of the dystonia. Ultimately, we propose that rather than attributing FTSD to a purely genetic origin or a patchwork of co-equal network abnormalities, the synergy framework—E/I circuits in M1 overshadowed by maladaptive excitatory growth—provides the most direct mechanistic account for how dystonia forms in skill-specific tasks, as well as how purely behavioral methods (BATR) can eradicate the imbalance. In so doing, we also incorporate an updated taxonomy of dystonia more generally, differentiating “typical” (neuroplastic) forms from “atypical” or “non-neuroplastic” variants whose incomplete penetrance might still hinge on partial compensations but whose core pathology is less reliant on the skill-specific synergy. This reconceptualization clarifies the path from “overreach or technique shift” to “structural cortical changes in inhibitory circuits,” bridging emergent clinical phenomena and clarifying why strategic retraining can, in some patients, re-establish normal motor function without pharmacological or surgical interventions.
In what follows, we present a structured account of these concepts: first, the Empirical Observations, which, in lieu of a conventional Methods/Results section, offers a single-case demonstration of the synergy imbalance model in real time; second, a full exposition of the Mechanism—mapping out how repeated overreaching of skill intensity fosters the formation of a dystonic synergy—and the immediate “Cause,” i.e., the repeated mismatch of excitatory and inhibitory capacities; third, the resulting approach to “Cure” that harnesses BATR to methodically restore balanced synaptic strengths; fourth, an in-silico validation step, in which a spiking neural network simulation provides proof of concept quantitative verification of the hypothesis and pinpoints the input-strength threshold at which the circuit shifts from balanced to hyper-excitable output; fifth, a section on Alterations beyond M1, explaining how basal ganglia, cerebellar, sensory-cortical, and spinal changes can be recast as secondary byproducts of chronic dystonic-synergy activation; and finally, Supplemental Considerations on broader dystonia taxonomy and the generalizability of how the approach extends to other forms. Through this structure, the paper seeks to unify electrophysiological, neuroimaging, computational, and clinical insights with a single synergy-based explanation that is experimentally testable and offers novel rehabilitative possibilities.
Empirical Observations
A single patient (the lead author), a classically trained right-handed male pianist, served as investigator and subject. The patient began learning the piano in 2013 and played for approximately a decade without any neurological or orthopedic issues. During this period, his technique—a systemic approach to playing the piano—was characterized by a general hand shape of predominantly flexed finger posture, achieved through a consistently sustained contraction of the Flexor Digitorum Profundus (FDP) in nearly all circumstances except when playing large intervals (e.g., octaves or larger). In this technique, keystrokes were executed by performing flexion of the metacarpophalangeal (MCP) joint with maintained extension at the proximal and distal interphalangeal (PIP and DIP) joints using the lumbricals and interosseous muscles of the hand. Subsequently, the key release was initiated by relaxing the lumbrical and interossei contractions that maintained MCP flexion (with interphalangeal (IP) joint extension), allowing the finger to return to its initial position before the keystroke (the finger resting on the key). The shapes of the fingers remained constant throughout the playing process, as the IP joint angles were maintained across the stages of pre-keystroke, keystroke, and key release.
On May 27, 2023, seeking a more staccato-oriented (notes to be played in a separated, detached manner) articulation for playing the first movement (“Allegro inquieto”) of Sergei Prokofiev’s “Piano Sonata No. 7, Op. 83,” the patient switched dramatically from the previous technique he had used for a roughly decade to a new “plucking” technique in an attempt to achieve the desired musical articulation. This new technique involved an even more pronounced curvature of the general hand shape compared to the previous technique—by now sustaining an even greater contraction of the FDP across nearly all contexts. Additionally, keystrokes were executed through a "plucking" motion, in contrast with the isolated MCP flexion facilitated by the lumbricals and interosseous muscles in the prior technique. In the new technique, the fingers (already in a flexed posture by FDP activation) undergo further additional flexion at the DIP and PIP joints (using the FDP) to depress the piano keys. The key release occurred the moment the fingertip’s increased flexion caused it to lose contact with the key surface, thereby allowing the key to return to its pre-keystroke position. Immediately thereafter, the fingers returned from their deeper flexed state to the original pre-keystroke curved posture by reducing FDP tension. This method allowed for the desired staccato articulation to be achieved.
Shortly upon adopting this technique, the patient noted a light performance setback. Specifically, any attempt to match the same previously attainable fast performance speeds with the newly adopted plucking technique engendered an acute sense of powerlessness (weakness and partial paralysis-like feelings). He called this sensation “true weakness”. However, the patient observed that the sensation of true weakness was only present when attempting to play speeds above a relatively rapid speed. In contrast, when playing at speeds equal to or below this threshold, the patient reported no true weakness. Undeterred, the patient disregarded the sensation of true weakness and continued practicing at speeds above the true weakness threshold for multiple hours per day, attempting to override or “fight through” the transient difficulty.
By June 16, 2023, the patient’s initial experience of true weakness at higher playing speeds had resolved as he noted that the true weakness threshold gradually became higher and subsequently disappeared. Nonetheless, this was accompanied by the insidious development of involuntary hyperflexion in the right 4th finger during piano playing. Additionally, involuntary hyperflexion manifested in the right 5th finger, a consequence of the musculotendinous interconnections between these two digits, rather than the 5th finger itself contracting involuntarily. Notably, the patient reported a dystonic threshold phenomenon, where dystonic symptoms only manifested when attempting to play at speeds exceeding the speed of the very first initial true weakness threshold that emerged after adopting the plucking technique. Conversely, the patient experienced a complete absence of symptoms when attempting speeds below or at the very first initial true weakness threshold.
Moreover, the patient remarks that the dystonia did not worsen further after this date and that repetitive symptom-triggering no longer increased its severity. Concurrently, during the time frame from May 27 to June 16, the patient observed a spread of the same dystonic symptoms and dystonic threshold phenomenon to typing at the computer keyboard while additionally noting that an abnormal sensation of heaviness or weightedness had developed in the ring and little fingers, causing these digits to feel ‘fused’ at all times—even in the absence of tasks that provoked dystonic symptoms—except during sleep.
The diagnosis of FTSD—in particular, musician’s dystonia—was only recognized by the patient on June 20, 2023, after personal research and officialized after subsequent clinical consultation. Recoiling from common medical interventions (e.g., botulinum toxin injections, oral medications, thalamotomy, deep brain stimulation), the patient sought a neuroplasticity-based motor retraining method after hearing a testimony from a colleague who had noticed improvement in her own musician’s dystonia through such an approach.
After June 20, 2023, the patient, having been newly diagnosed with dystonia and motivated by a colleague’s testimony regarding neuroplastic retraining, searched fervently for a structured protocol that could reverse his symptoms. Despite the popularity of “neuroplasticity-based motor retraining” as a general idea, the patient encountered no clear-cut methodology or guidelines, leaving him in a prolonged trial-and-error phase. Initially, he believed that the dystonic contractions stemmed from “bad technique” (specifically, poor piano ergonomics) and hence attempted repeated technique modifications in rapid succession: he would adopt one technique for a short time, find it inadequate, then abandon it and switch again to a new one—often within days. Such frequent shifts neither alleviated the existing dystonia nor lowered its threshold; in fact, the dystonic threshold speed—and the associated symptom severity—remained identical to what had emerged in mid-June.
While these rapid continual technique changes had no measurable impact on the dystonic threshold speed or the severity of involuntary contractions, the patient reported that they introduced a fresh complication: a new true weakness threshold that emerged precisely from the original dystonic threshold and migrated downward over successive technique changes.
Over the following three weeks, the descending true weakness threshold carved out a middle zone in the patient’s speed range, forcing a performance scenario in which, below or at that new true weakness threshold, no symptoms of any sort were triggered, between that true weakness threshold and the longstanding dystonic threshold only true weakness manifested but in all five digits, and above the original dystonic threshold the familiar involuntary hyperflexion still surfaced in the 4th digit and also the 5th digit due to musculotendinous interconnections, coupled with true weakness in the other digits. Throughout this period, the dystonic threshold remained stable at the higher speed set in mid-June. Yet, every major technique change drove the newly minted true weakness threshold lower, steadily shrinking the “safe” speed zone where neither true weakness nor dystonia emerged. By late October, the patient found himself confined to a multi-tiered scenario—a true weakness threshold at mid-range speeds, an unaltered dystonic threshold at higher speeds, and no improvement in functioning.
In the following days, curious about whether deliberately confining practice to speeds below both the true weakness and dystonic thresholds—speeds at which no symptoms emerged—could restore normal right-hand function, the patient adopted a rigorous motor retraining method he called “below or at-threshold retraining” (BATR). This protocol involved systematically performing a chosen exercise—initially a simple five-finger pattern in C major (C–D–E–F–G–F–E–D–C)—and attempting it at a speed equal to or below the true weakness threshold where no symptoms of any sort occurred. The patient gradually raised the tempo only upon feeling genuine progress: if he sensed the capacity to play faster without triggering symptoms, he would make a small speed increase but still remain decisively under both the true weakness and dystonic thresholds. Should he inadvertently exceed either threshold and feel true weakness or dystonia creeping back, he immediately reverted to the last comfortable speed at which no symptoms arose. Each keystroke was executed at a deliberately tempered pace, often taking as long as ten to twenty seconds per note press—the keystroke was so slow that no audible sound was produced—thereby executing a symptom-free movement. Throughout each daily practice session, he repeated BATR for 4-8 hours with minimal breaks and never practiced the piano in any other way except BATR—thereby never triggering true weakness or dystonia.
By December 6, 2023, after consistently practicing BATR for over a month and observing significant improvement, the patient, driven by curiosity, attempted once again to play at the high speeds that had previously provoked dystonia, in order to see whether involuntary contractions still emerged. On that date, however, no dystonic symptoms emerged in either piano playing or typing no matter how fast the attempted speed was. The patient’s subjective hand sensation had likewise normalized during this period, with the sense of heaviness in the 4th and 5th fingers having disappeared. Despite the complete successful resolution of dystonia, true weakness still persisted at higher speeds above a specific true weakness threshold, even though that threshold had risen considerably over the course of using BATR during this period. Following December 2023, the patient continued exclusively practicing BATR, approaching a near-complete full recovery by late March.
Nevertheless, in late March 2024, the patient’s right hand still had a residual true weakness threshold that had not been fully overcome. Confronted with a looming piano competition in early April 2024, and pressed for time, the patient believed he would be capable of participating in it. He reported that he consciously decided to repeatedly practice at speeds well above the true weakness threshold, consequently experiencing constant true weakness during those practice sessions. Over several days of such practice, a second set of new dystonic symptoms developed, now centered primarily on the third digit. Whenever the patient depressed the third digit at fast speeds, involuntary hyperflexion appeared in both the third and fourth digits, with the fourth digit flexing (but to a smaller degree) due to linked musculotendinous interconnections with the third. Alarmed by this development, the patient immediately reverted to solely practicing BATR again after the competition had passed. Within roughly two weeks—by April 20, 2024—the more recent second dystonia fully resolved, leaving another residual true weakness threshold once again. From April through December 2024, the patient adhered exclusively to practicing BATR. By the end of that year, he reported a complete and lasting 100% recovery, with normal piano playing restored and no true weakness or dystonia at any speed.
Answering the Three Key Questions Regarding Focal Task-Specific Dystonia
The Underlying Mechanism
While synergies are often described at a macro level, using Safavynia et al.’s (2011) definition of motor synergies, we employ the framework of synergy at a micro level in which each “task-specific motor synergy” (TSMS) corresponds to the smallest ensemble of muscles co-activated to produce a unidirectional movement (e.g., finger flexion, extension, abduction, adduction, wrist flexion, wrist extension, wrist radial deviation, wrist ulnar deviation, forearm pronation, forearm supination) at an individual digit or body part. Concretely, rather than treating every single muscle as an entirely independent module, we group all prime movers (intrinsic and/or extrinsic) that jointly create the movement force in that one direction, along with their corresponding alpha motor neurons in the spinal cord and the relevant cortical E/I circuits in the task-specific subregion of M1. Any proximal stabilizers or muscles whose activity merely modulates finger posture indirectly are excluded, leaving only those muscles directly responsible for executing the specific unidirectional motion. In this sense, each synergy can involve multiple muscles (e.g., intrinsic hand muscles plus certain extrinsic forearm muscles) if they routinely co-activate to produce a single direction of movement in that digit. Crucially, a muscle can also belong to more than one synergy if it contributes to multiple directions. By focusing on this tightly defined “unidirectional synergy,” we capture the local population of excitatory and inhibitory neurons that practice together as a functional sub-network for that particular movement. We propose that FTSD can arise in any one of these unidirectional TSMS microcircuits if repeated overreaching selectively strengthens excitatory drive while the corresponding inhibition fails to keep pace.
Moreover, a crucial boundary condition is set by cortical interneuron identity. Across the neocortex, including M1, three non-overlapping molecular families account for virtually all GABAergic cells: parvalbumin-positive (PV), somatostatin-positive (SST), and the ionotropic serotonin receptor 5HT3a (5HT3aR) interneurons (Rudy et al., 2011). Framing FTSD in this canonical tripartite scheme immediately narrows the mechanistic search: any inhibitory deficit in a TSMS must therefore map onto one, or some combination, of these three classes.
Under normal conditions, we propose that a TSMS—encoding a learned unidirectional movement—is represented in the M1 by an ensemble of excitatory (pyramidal) neurons that co-fire to produce the intended motor output. These excitatory neurons rely on local inhibitory interneurons, particularly PV fast-spiking cells, which provide short-latency GABAA-mediated inhibition to keep excitatory drive in check. Whenever pyramidal neurons ramp up their firing, the corresponding PV cells receive strong excitatory input and deliver a timely burst of inhibition, preventing runaway activity (a similar mechanism as the pyramidal-interneuron gamma PING described by Keeley et al., 2017). As skill acquisition proceeds through repeated practice, excitatory and inhibitory synapses within the synergy co-strengthen in parallel via Hebbian-like mechanisms: excitatory-excitatory (E-E) connections among frequently co-activated pyramidal cells become more robust, while excitatory-inhibitory (E-I) connections onto PV interneurons also scale up, ensuring balanced inhibitory feedback. The result under healthy circumstances is a balanced microcircuit, reflected experimentally in normal short-latency intracortical inhibition (SICI) and moderate intracortical facilitation (ICF) when measured by TMS.
In FTSD, we propose that this balance is disrupted by an imbalance of synaptic strengths between pyramidal neurons and local PV interneurons within the TSMS of the affected digit or individual body part in M1. Specifically, the pyramidal cells outpace the inhibitory drive furnished by PV interneurons—the fast-spiking subset of GABAA-mediated cells (Tian & Izumi, 2022) that predominantly govern SICI (Di Lazzaro et al., 2006). In a healthy TSMS, balanced E-I synapses (both E→I and I→E) ensure that pyramidal neurons fire with appropriate spatiotemporal specificity. However, in FTSD, the PV interneuron synapses (E→I and/or I→E) become under-strengthened and disproportionately weak compared to E-E synapses. Regardless of which of those two is more compromised, the net effect is insufficient GABAA current to hyperpolarize or shunt excitatory cells. This weakened short-latency inhibition directly manifests as reduced SICI in TMS studies (Di Lazzaro et al., 2006). Although each TSMS can also contain the two other principal cortical inhibitory interneuron populations (SST and 5HT3aR), across the FTSDs (writer’s cramp, musician’s dystonia, etc.) a substantial body of TMS studies report markedly reduced SICI (e.g., Furuya et al., 2018; Huang et al., 2010; McDonnell et al., 2007; Ridding et al., 1995; Siebner et al., 1999; Stinear & Byblow, 2004). By contrast, results for long-interval intracortical inhibition (LICI) are heterogeneous—normal in some cohorts (Furuya et al., 2018; Meunier et al., 2012), reduced in others (Chen et al., 1997; Espay et al., 2006), occasionally even increased (Caux-Dedeystère et al., 2021). We contend that by itself, the inconsistency of LICI does not imply methodological noise alone; rather, it offers a clue pointing to the specific interneuron subclass that constitutes the principal locus of dysfunction within the TSMS. Compelling support for this inference comes from a direct demonstration of how slow, metabotropic inhibition gates late polysynaptic activity in a study by Shao and Burkhalter (1999). In their study, layer-2/3 stimulation (rat V1) evokes an early glutamatergic EPSP, followed later by a slow GABAB-IPSP that peaks at ~146 ± 13 ms. Bath application of the GABAB antagonist 2-OH-saclofen or CGP 55845 abolishes this IPSP and immediately unveils a large, long-lasting train of reverberant EPSPs, likely suggesting that dendrite-targeting SST neurons normally veto late recurrent excitation. In addition, the underlying circuit—L2/3 pyramids recruiting Martinotti (SST) cells that project to distal dendrites in L1—is highly conserved across the neocortex, including M1 (Jiang et al., 2015). Thus, if one performs the following thought experiment—subtracting SST-mediated GABAB currents while leaving PV-driven GABAA inhibition intact in the context of performing a motor task—the predicted motor phenotype diverges sharply from FTSD. Loss of dendrite-targeting SST cells abolishes the ∼100-200 ms gain-down window that normally vetoes late polysynaptic reverberation. A brief cortical command should then in theory fragment into a series of low-frequency echo-like bursts of polysynaptic activity, causing a cascade of discrete, clonic after-contractions or phasic tremor-like jerks. FTSD, by contrast, is observed clinically to be a sustained, posture-like involuntary contraction that initiates once the task-specific speed or force threshold is crossed and then plateaus for the duration of the action (Sakai, 2006; Yoshie et al., 2015). The absence of involuntary phasic bursting movements in nearly every well-documented case of FTSD therefore argues that dendritic SST gating cannot constitute the primary locus of dysfunction. This empirical result therefore strengthens the inference that PV hypofunction—not SST loss—is the critical failure mode in FTSD.
Repeating the same thought experiment with the variables reversed—weakening PV synapses while sparing SST circuits—recapitulates the clinical picture far more faithfully. As PV basket and chandelier cells clamp the perisomatic membrane within 1-5 ms of each pyramidal spike, their under-strengthening removes the instantaneous brake that normally limits population firing probability. Initial pyramidal discharge therefore rises steeply and, in the face of still-functional SST gain control, settles onto a new, elevated plateau: a hyperexcitable yet largely continuous output drive. Behaviorally, the motor system expresses the excess as a tonic, task-bound spasm, matching the phenomenology of FTSD. That outcome requires no additional failure of SST-mediated inhibition, merely a quantitative mismatch in the E/I ratio at PV synapses. The same deductive framework further argues against a primary role for 5HT3aR interneurons, a significant amount of which express vasoactive-intestinal-peptide (VIP) and serve chiefly to disinhibit SST cells. In healthy cortex, VIP neurons fire in response to cholinergic or serotonergic drive, transiently silencing SST dendrite-targeting interneurons and thereby permitting a momentary increase in pyramidal dendritic excitability. If FTSD were rooted in a loss of VIP/5HT3aR output, SST cells would be chronically over-effective, not under-active; GABAB gain control would strengthen, late polysynaptic reverberation would be further suppressed, and corticospinal output would likely tilt toward bradykinetic or hypometric movements—exactly the opposite of the hyperkinetic, threshold-locked phenotype observed. Conversely, if VIP neurons were pathologically hyper-active, they would disinhibit SST targets so that the functional effect would approximate a direct SST knock-out, again predicting phasic echo bursts rather than the sustained co-contractions that define FTSD. These converging considerations motivate a working hypothesis in which PV hypofunction is necessary—and, at the level of core motor phenomenology, sufficient—for FTSD. Whether SST and/or VIP pathways are spared or impaired remains an open and empirically testable question; current evidence suggests they are not required to be abnormal and their involvement is variable and smaller in magnitude than the PV synaptic strength deficit.
Moreover, when an individual attempts a high-demand or precise motor act—such as writing or playing an instrument—insufficient inhibitory “clamping” of pyramidal populations causes them to fire excessively, leading to the characteristic repetitive involuntary movements of FTSD (Furuya et al., 2018; Ridding et al., 1995). Notably, PV interneuron synapses do still function to some degree; there is not an absolute loss of inhibition. Nonetheless, we propose that their quantitative and qualitative insufficiency adequately explains the loss of surround inhibition that has been repeatedly documented in FTSD (e.g., Beck & Hallett, 2011; Beck et al. 2009; Sohn & Hallett, 2004). Because surround inhibition largely relies on PV interneurons to selectively inhibit neighboring excitatory outputs (Kujirai et al., 1993), weakening these synapses compromises the inhibitory “gating,” resulting in spillover or overflow of excitatory drive into adjacent cortical representations.
Additionally, both the excitatory inputs onto PV interneurons (E→I) and the inhibitory outputs from PV cells to pyramidal neurons (I→E) could be weakened in FTSD. The reduction of SICI in FTSD (e.g., Furuya et al., 2018; Huang et al., 2010; McDonnell et al., 2007; Ridding et al., 1995; Siebner et al., 1999; Stinear & Byblow, 2004) directly reflects a weakened GABAA effect on pyramidal neurons, whether from PV interneurons firing less or having less effective synapses onto excitatory cells. Experimentally, blocking the postsynaptic effect of PV-mediated inhibition in M1 is sufficient to induce dystonic features: in monkey experiments, the focal application of a GABAA antagonist (bicuculline) to motor cortex caused excessive excitatory drive and abnormal co-contraction of agonist/antagonist muscles, thus mimicking dystonic movements (Matsumura et al., 1991). This demonstrates how loss of I→E inhibition alone can critically degrade motor control specificity. In a parallel finding, a peripheral afferent stimulus that normally elicit cortical inhibition instead produced excitation in FTSD patients, a phenomenon attributable to underactive inhibitory interneuron output (Abbruzzese et al., 2001). Collectively, such data strongly implicate deficient I→E synaptic transmission (PV → pyramidal) as a key factor in FTSD pathology. Furthermore, in the healthy motor cortex, a sub-threshold conditioning pulse suppresses late I3-waves but spares the early I1-wave, confirming that I3 activity is gated by intracortical GABA-ergic (likely PV-cell) inhibition (Hanajima et al., 1998). Current-direction studies add a second, complementary probe. Posterior-anterior (PA) stimulation reliably recruits an I1 volley at threshold, with I2/I3 waves emerging only at higher intensities, whereas anterior-posterior (AP) stimulation can, in some individuals, elicit an I3 volley first; in others the initial volley remains I1- or even D-like (Di Lazzaro et al., 2001). Thus, PA currents predominantly interrogate early-wave circuitry, whereas AP currents provide at least partial access to circuits capable of generating later, PV-gated I-waves. This orientation-based “double probe” reveals a distinctive pattern in FTSD. When SICI is tested with PA currents it is markedly reduced, yet the same paradigm with AP currents yields normal inhibition (Hanajima et al., 2008). Because the PA configuration samples early-wave-dominated output, while the AP configuration can still engage late-wave pathways, the selective loss of PA-SICI implies that everyday motor output in FTSD relies disproportionately on the fast, direct I1 route and fails to recruit the PV-interneuron circuitry that shapes later I-waves. Consistently, Stinear and Byblow (2004) showed that higher conditioning intensities are required to elicit SICI in FTSD, indicating that PV interneurons are present but less excitable. Together, these findings support the notion that weakened pyramidal-to-PV synaptic drive leaves the inhibitory network under-recruited.
In summary, we propose that the core mechanism in FTSD is a PV-centered synaptic-strength imbalance within a TSMS, in which PV-mediated inhibitory circuits are insufficiently potentiated relative to excitatory circuits, shifting the synergy into a hyperexcitable regime. The hallmark features—repetitive involuntary movements and a loss of surround inhibition—result directly from reduced inhibitory gating (SICI deficiency) and surplus excitatory drive, ultimately ‘locking’ the cortex into maladaptive activity patterns whenever the learned task is initiated.
The Developmental Cause
We propose that when you switch techniques in a motor skill or at the piano, certain components of your old finger posture or movement pattern overlap with what the new technique needs. Essentially, “overlap” between the old and new techniques arises because certain subpopulations of pyramidal neurons (and their corresponding local inhibitory interneurons acting as modulators) in M1 encode movement features—finger trajectories, velocity profiles, or force levels—that are partly common to both the old and new patterns of piano playing. Even when your new technique changes aspects of hand posture or finger movement, it still relies on muscle activations and joint configurations that substantially resemble fragments of the old pattern—such as maintaining flexion in a particular finger joint, or generating similar wrist alignment. At the level of individual neurons and synapses, this is manifested through a distribution (rather than a one-to-one mapping) of excitatory synapses in M1 that each contribute to subcomponents of the overall movement.
Inside M1, populations of pyramidal cells are organized into partially overlapping ensembles, each broadly tuned to specific movement directions, muscle synergies, or force-speed parameters (Economo et al., 2024 and references therein; Shinotsuka et al., 2023). This organization is not strictly topographic (i.e., not “one neuron, one finger”), but rather a population code: each neuron’s firing reflects a preferred contribution to certain aspects of movement—whether that be flexion of the distal phalanx, extension of the wrist, or stabilizing the thumb. When you adopt a “new technique,” many motor elements do change—different finger angles, distinct wrist orientation, altered timing—but there remains a core set of smaller-scale motion primitives (e.g., the same DIP joint flexion in the index finger) that the new technique still demands.
At a synaptic scale, each pyramidal neuron has thousands of (E-E) connections and receives short-latency inhibitory inputs from local interneurons, especially fast-spiking PV cells. Whenever you execute a particular finger transition—say, depressing a piano key with the index finger while stabilizing adjacent fingers—some subset of pyramidal neurons that formerly participated in the old technique for that same or very similar muscular action will again receive correlated pre- and postsynaptic activity. This is because these neurons were already wired (through prior Hebbian strengthening during your original technique training) to generate precisely that mechanical output. If the new technique retains enough of the old technique’s biomechanical or kinematic subroutines (like a specific angle or force component for pressing a key), those same neurons get reactivated.
Additionally, the local inhibitory circuits recruited alongside these pyramidal neurons reflect the same partial overlap. The PV interneurons that were tuned to provide well-timed inhibitory bursts for controlling the speed or force of that same finger trajectory will still be co-activated. At the synaptic level, this overlapping microcircuit (pyramidal-interneuron-pyramidal loops) is effectively “shared” between the old synergy and the new synergy because it encodes that particular fragment of movement output that both techniques happen to employ.
Thus, the reason certain subcircuits get reused is that the cortical architecture for motor outputs is built around semi-redundant, multifunctional neuronal populations—rather than strictly dedicated “old-technique” vs. “new-technique” neurons. The new technique re-elicits patterns of spiking in those neurons whose preferred movement features match the partial motion components or forces used in both old and new posture. Essentially, any connections that are “useful” for the new synergy still experience synchronous presynaptic and postsynaptic spiking, which is the Hebbian trigger for maintaining (or further potentiating) these synapses.
Meanwhile, the subset of neurons and synapses from the old technique that code for truly unique angles or muscle synergies of the old technique are not recruited by the new technique and are not reliably activated. Crucially, synaptic plasticity in the cortex is governed by spike timing-dependent plasticity (STDP) rules: if a given presynaptic terminal no longer fires in precise synchrony with its target postsynaptic neuron, the relevant connection undergoes long-term depression (LTD) or fails to be reconsolidated during offline phases of protein synthesis. Several mechanisms ensure that unreinforced synapses “fade” over time. These include (1) synaptic tag-and-capture processes, wherein newly reactivated synapses tag themselves for further stabilization proteins, whereas inactive synapses do not (Bin Ibrahim et al., 2024; Frey & Morris, 1997; Redondo & Morris, 2011); (2) competition for plasticity-related proteins (PRPs), meaning that actively firing synapses can “capture” the molecular resources needed to retain high synaptic strength, leaving inactive synapses starved (Govindarajan et al., 2011; Sajikumar et al., 2014); (3) homeostatic mechanisms that prevent indefinite global potentiation by favoring a downregulation of inputs that are not used for the current motor program (Turrigiano et al., 1998; Turrigiano, 2008); and most importantly (4) the principle of occlusion and retrograde interference, wherein a local circuit that has undergone significant long-term potentiation (LTP) from the new technique has diminished capacity for further potentiation, effectively overshadowing or destabilizing old, unreinforced connections (Cantarero et al., 2013). Once the newly formed synergy “dominates,” unique components of the old technique that remain unused are especially prone to LTD or outright pruning through lack of reactivation.
This division of your old technique’s synapses—into those that remain active under the new movement pattern vs. those that do not—creates what we observe behaviorally and propose as a “partial baseline shift”: you retain only that fraction of synaptic strength that the new technique actually calls upon and keeps reactivating. In other words, the new technique’s earliest practice sessions re-potentiate (or at least prevent from decaying and getting downregulated) those old synapses it still needs, while simultaneously allowing the old-technique-unique synapses to undergo LTD. The overlap portion of the old synergy (excitatory neurons plus their matched inhibitory interneurons) is “rescued” and preserved each time you execute the new technique, whereas the segments of the old synergy that are no longer relevant do not receive correlated firing and thus do not keep their former high-potentiation state. We hypothesize that this selective retention is precisely why an individual can notice a performance setback after every technique change: you no longer have 100% of the old synergy’s synaptic capacity but only the part that the new technique still re-activates.
Moreover, a rigorous way to define “intensity” (whether speed, force, volume, etc.) in the nervous system begins by recognizing that the motor output—how fast or forcefully you strike a piano key—arises from the net excitatory minus inhibitory drive to the motor pathway. In piano performance, these motor neurons reside predominantly in layer 5 of M1 for the upper motor component, and in the ventral horn of the spinal cord for the lower motor component. The degree of “intensity” depends on two inter-related mechanisms: (i) recruitment of a larger population of corticospinal neurons and spinal motoneurons, and (ii) rate coding—higher discharge frequencies within those units. Greater recruitment and faster firing raise the descending drive onto spinal interneurons and motoneuron pools; the motoneurons then enlist more motor units and/or elevate their firing rates in the hand muscles, producing quicker and more forceful keystrokes. Thus, in purely neural terms, we define intensity not as a single variable but as an emergent property of how many neurons are active, how extensively they recruit spinal motor circuits, how fast they spike, and—during brief transients—how synchronously they discharge
When you produce a “higher-intensity” keystroke—pressing a key at greater speed or with greater force—the underlying mechanism is that more excitatory synapses onto pyramidal neurons (and onto the downstream spinal circuitry) shift their membrane potentials closer to or beyond threshold in a coherent, time-locked way. Each spike in these upper motor neurons generates descending action potentials along the corticospinal tract. Within the spinal cord, the summation of presynaptic drive onto alpha motor neurons determines how many of those motor neurons discharge, and with what frequency. If you need a lower velocity or gentler force, fewer pyramidal neurons fire, or they do so at lower frequencies, recruiting smaller motor units or firing them sparsely.
To call it “intensity” underscores that the nervous system flexibly scales the net excitatory output in each TSMS. Within M1, a digit-specific TSMS is encoded by ensembles of pyramidal neurons whose firing patterns direct the muscle activity needed to press a piano key with that particular finger. Crucially, the magnitude of pyramidal firing—and the resulting degree of spinal motor neuron recruitment—determines how vigorously the synergy manifests. In other words, the same digit-level TSMS can yield a soft, slow keystroke or a hard, fast keystroke simply by modulating the net excitatory outflow: the same group of prime-mover muscles is activated, but at different intensities of neuronal discharge.
“Attempted intensity” refers to the top-down command specifying how forcefully or how rapidly one intends to move that finger. At a cellular level, premotor and supplementary motor areas, together with M1, generate pre-movement activity reflecting an internal plan for the upcoming velocity or force of the finger TSMS. This plan modifies synaptic input to the relevant pyramidal ensembles, effectively setting a “target excitatory drive.” If you choose to strike a key loudly with, say, your index finger, the cortex mobilizes a stronger excitatory barrage onto the ensemble controlling the finger flexors, while dynamically adjusting local inhibitory interneuron firing to preserve spatiotemporal precision. Once you initiate the movement, spinal and sensory feedback loops refine the ongoing force, but the core mechanism is that the corticospinal command—shaped by basal ganglia gating, cerebellar error correction, and other influences—either ramps up or tapers off the population spike rate of the finger TSMS’s motor neurons to match your “attempted intensity.”
Thus, in the most literal sense, “attempted intensity (e.g., speed, force, volume)” is the set of descending E/I patterns that the cortex generates in anticipation of the required movement amplitude and velocity. It is a forward projection of neural firing rates, shaped by prior learning, that aims to recruit a defined number of spinal motor units at a certain frequency. In this way, “intensity” can be biologically viewed as the net excitatory load placed on the TSMS, and “attempted intensity” is your brain’s command to load the TSMS to a particular level of activity.
Additionally, synaptic strengths in a given TSMS directly shape how much net drive the involved neurons can generate or suppress, because the amount of potentiation or depression at each relevant synapse dictates the amplitude of excitatory or inhibitory postsynaptic potentials (EPSPs or IPSPs), which in turn translates into the population-level firing rates for that TSMS. In the excitatory circuit, synaptic strength governs the amplitude of each EPSP. At every excitatory synapse, the density or conductance of AMPA receptors and the presynaptic release probability determine how much depolarization the postsynaptic neuron receives upon a presynaptic spike. When synapses are strongly potentiated, each incoming spike volley yields a larger total excitatory current, driving the postsynaptic neuron to fire more frequently or recruit additional neurons. Consequently, the population’s overall firing rate in pyramidal cells of the motor cortex can escalate, thereby increasing the descending command that ultimately activates spinal alpha motor neurons. Higher cortical firing rates mean more motor neurons discharge with greater frequency, producing higher muscle force and faster movement of the digit. In this way, robust synaptic strengths in the excitatory circuit allow the TSMS to reach a greater maximum intensity when an individual attempts a high-velocity or high-force action. Conversely, if these synapses are weak, even a strong top-down command fails to generate sufficient postsynaptic spiking, capping the TSMS at a lower maximum speed or force.
Meanwhile, in the inhibitory circuit, synaptic strengths control how effectively interneurons can clamp or limit the TSMS’s excitatory outflow. PV interneurons, among others, receive excitatory inputs (E→I) from pyramidal cells, and the strength of these inputs determines how vigorously those interneurons fire in response to a given excitatory drive. At the same time, the output of these interneurons (I→E) projects back onto the perisomatic region of pyramidal neurons through GABAA-mediated synapses, whose strength dictates how much inhibitory hyperpolarization or shunting each interneuron spike confers on the excitatory cells. If these E→I and I→E connections are robust, a rising excitatory barrage from the pyramidal population will trigger a corresponding burst of interneuron firing, promptly delivering large IPSCs that damp or sculpt the excitatory neurons’ discharge. This mechanism ensures short-latency feedback inhibition, preventing runaway firing and controlling whether only a subset of neurons is active. Alternatively, if inhibitory synapses remain under-strengthened, the TSMS struggles to curb excessive pyramidal activity, risking unregulated hyperexcitability or overshoot behaviors reminiscent of FTSD.
Hence, the amount of excitatory drive that the TSMS can generate at any moment is set by the balance between strong excitatory synapses, which boost EPSPs toward threshold, and sufficiently strong inhibitory synapses, which govern the restraint or timing of that drive. Excitatory strengths fix how large the TSMS’s overall firing can become, thereby defining the upper limit of speed or force. Inhibitory strengths determine how effectively the TSMS can gate or refine that excitatory surge, maintaining precision and mitigating unwanted spillover. When these circuits are well-matched, an individual can translate a chosen “attempted intensity” into real-world movement up to the TSMS’s maximal capacity. If the excitatory side is too weak, no amount of attempted drive will achieve high force or velocity; if the inhibitory side is too weak, the TSMS can overshoot and cause unintended movements.
Furthermore, we propose that the true weakness prominently reported by the patient arises when the excitatory drive required for a given movement exceeds the capacity (synaptic strength) of the TSMS’s excitatory circuit to depolarize and recruit downstream motor neurons. Each synapse in the TSMS’s excitatory pathway (e.g., layer 5 pyramidal cells projecting to spinal motor pools) has a certain degree of potentiation—reflected in features like AMPA receptor density, presynaptic release probability, and dendritic spine morphology—that sets how large an EPSP can become with each incoming spike. When these synaptic strengths are suboptimal or partially depressed, the postsynaptic neurons in the TSMS cannot reach the firing rate or recruitment threshold necessary for generating high levels of force or speed. In other words, the local excitatory circuit is incapable of converting a top-down “attempted intensity” into the corresponding spike output.
At the most immediate scale, true weakness is visible when a strong cortical command fails to produce sufficient spiking in the relevant pyramidal neurons. If synapses have lost potency—through LTD—they simply do not inject enough depolarizing current into the cell bodies and proximal dendrites of the pyramidal neurons. As a result, the neurons saturate at a lower firing frequency and fail to recruit the full range of alpha motor neurons in the spinal cord. The spinal motor neuron pool is responsive to the summation of EPSPs arriving from descending cortical and subcortical pathways; inadequate excitatory amplitude means fewer motor units are activated, and those that are active may not fire at high enough rates to achieve the intended force or velocity. The performer then experiences a pronounced difficulty in generating the expected strength or speed, which feels subjectively like true weakness.
Thus, true weakness arises because the TSMS’s excitatory circuit is below the threshold of synaptic potentiation required to drive the descending corticospinal system at the level mandated by a high-intensity motor command. The result is a clear gap between the intended movement (the performer’s internal sense of how forcefully they want to press) and the TSMS’s actual motor output (the diminished, sluggish or feeble press), precisely because excitatory circuits cannot muster the rapid, high-amplitude depolarizations needed to push the muscle fibers to the desired level of contraction.
When you “overreach" by attempting finger speeds or forces (intensity) beyond your current E/I capacity (synaptic strengths), the cortical and spinal motor networks must still generate repeated motor commands—albeit insufficient ones—to move the fingers at least partially. These repeated presynaptic spikes, even if they fail to match the intended force, reinforce a subset of excitatory synapses that happen to be active during the partial or fragmented movement. We propose that on a population scale, this subset can drift away from the original balanced synergy and form what we call a dystonic synergy/TSMS, specifically above the speed threshold you are trying to surpass. Because the new overreached commands repeatedly push excitatory neurons in the synergy toward a firing pattern aimed at higher intensity—even if the movement is incomplete or weak—the excitatory pyramidal neurons nevertheless still produce some bursts of activity—enough to push their own E-E synapses gradually toward LTP—yet those partial bursts typically lack the amplitude, timing consistency, and synchrony needed to robustly engage the interneurons. For PV interneurons (and other local inhibitory cells) to undergo parallel LTP, they must receive well-timed, sufficiently large excitatory inputs and be able to fire action potentials that coincide with the postsynaptic activity of the pyramidal neurons they are inhibiting. Below are the mechanistic reasons we propose as to why that often fails to happen during overreaching: First, pyramidal-to-inhibitory (E→I) synapses may have higher or more phasic thresholds for effective plasticity, meaning they need a strong, synchronous volley of spikes to trigger the specific intracellular cascades (e.g., adequate calcium influx or NMDA receptor activation) that lead to potentiation. During overreaching, the pyramidal neurons do fire, but not at the robust rates or aligned bursts they would produce if they truly met (or slightly surpassed) their excitatory threshold. The result is bursts that are too sporadic or too brief to reliably boost interneuron spiking to the point of reinforcing E→I synapses. In other words, the partial, “struggling” pyramidal output might be enough to bump the E-E synapses upward (since those synapses can sometimes be potentiated even by submaximal but repeated inputs), but it does not achieve the amplitude or temporal pattern crucial for flipping interneuron synapses into an LTP-supporting mode.
Second, interneurons themselves often have distinctive electrophysiological properties—like fast spiking patterns characterized by short membrane time constants and strong after-hyperpolarizations (typical of hyperpolarization activated currents, .e.g., Ih)—that require a certain threshold of coincident presynaptic drive to stay firing in a synchronized manner. If the incoming excitatory signals arrive in small, uncoordinated “packets” (as happens when you cannot fully achieve the desired speed/force), the interneurons fire a few scattered spikes rather than the coherent, higher-frequency trains that robust inhibitory LTP typically demands. Critically, plasticity in E→I synapses is spike timing-dependent: you need presynaptic pyramidal spikes firing before (after) interneuron spikes for strengthening (weakening). When the excitatory bursts are fragmented and never quite push the interneurons into robust discharges, the spike timing windows for plasticity close, without a stable E→I memory trace forming.
Third, the I→E connections back onto pyramidal cells also require synchronous interneuron firing plus a depolarized postsynaptic target to strengthen. If interneurons only spike fleetingly and the pyramidal cells themselves are not in a well-timed depolarized window, there is no matched pre- and postsynaptic depolarization to anchor LTP. PV interneurons specialize in high-frequency, short-latency bursts that clamp excitatory neurons, but that effect becomes meaningful only if the interneurons are driven strongly enough to run these rapid-fire bursts. During overreaching, the synergy’s excitatory signals remain in a borderline zone, insufficient to coordinate both sides of the circuit at once, and the synergy never “locks in” those reciprocal inhibitory synapses with the proper Hebbian signatures.
Finally, on the molecular side, repeated submaximal firing can still tag and capture plasticity-related proteins (PRPs) for the E-E connections (because at least some portion of the pyramidal ensemble fires consistently), whereas the interneurons—firing too erratically—fail to capture the needed PRPs. In short, the partial bursts feed just enough repeated stimulation to the E-E synapses to accumulate LTP, but not enough to orchestrate the robust co-activation pattern needed for the inhibitory side. Over time, that differential consolidation mechanism explains how the dystonic synergy’s excitatory circuit creeps upward while the inhibitory circuit stalls, never receiving the full co-activation “recipe” required to strengthen in lockstep.
In summary, we propose that a separate dystonic synergy forms under overreaching conditions because the repeated, subthreshold excitatory bursts effectively “peel off” or consolidate a new set of excitatory synapses above the functional synergy’s existing capacity, while simultaneously failing to co-develop the matching inhibitory circuitry. Meanwhile, the original functional synergy/TSMS—the balanced one that could operate comfortably at lower speeds or forces—no longer receives the specific coincident firing needed to maintain or further increase its excitatory and inhibitory synaptic strengths. As a result, those older synapses stall in development while the newly emerging (though imbalanced) dystonic synergy entrenches itself.
Moreover, when the performer consistently tries to move at a speed or force level beyond what the current functional synergy can deliver, the cortical command still generates repeated presynaptic spikes in some pyramidal neurons. Although these bursts are not coherent or fully synchronized enough to reinforce the old synergy’s E/I loops, they do repeatedly tag a new sub-population of E-E synapses. This “tagging” indicates that these partially active synapses are relevant, and they capture plasticity-related proteins (PRPs) such as CaMKII, PKMζ, or BDNF, allowing them to undergo incremental LTP. Over time, this repeated partial activation converges into a subcircuit that resides above the original synergy’s upper threshold—because it is precisely the subcircuit engaged whenever the performer pushes for that higher intensity.
Simultaneously, the older synergy’s excitatory and inhibitory connections cease to experience the correlated presynaptic-postsynaptic activity they need for LTP or even stable reconsolidation. Each time the performer “overreaches,” the descending pattern is specifically geared to exceed the old synergy’s comfortable zone. That means the old synergy’s synapses do not get the consistent spiking patterns that once maintained or increased their strengths. In addition, local plasticity resources (e.g., those PRPs) become partially exhausted or reallocated to the nascent subcircuit, leaving fewer available for the original synergy’s E/I pairing. Without renewed Hebbian co-activation or ample PRPs, the old synergy’s synaptic strengths stagnate.
Essentially, the reason this new subcircuit forms a “dystonic” synergy (i.e., with imbalanced E/I development) is that the partial bursts never robustly recruit the associated inhibitory interneurons. PV-positive interneurons, for instance, require well-synchronized, high-intensity inputs to potentiate their E→I or I→E synapses. Because overreaching yields disjointed, below-threshold excitatory firing, the interneurons never get the consistent, high-amplitude drive crucial for parallel LTP. Hence, these inhibitory synapses stay under-strengthened. The repeated bursts still suffice to incrementally reinforce the excitatory side, but do not align frequently or powerfully enough to entrain the local inhibitory microcircuits. Over time, the subcircuit coalesces into a distinct excitatory-dominant synergy that sits “above” the old functional synergy’s capacity, becoming hyperexcitatory due to the mismatch with its relatively weak or stagnated inhibitory counterpart.
Thus, the older synergy’s E/I network halts in development because it is no longer the primary circuit engaged at these higher intensities, while the emerging circuit gets just enough partial engagement (plus local plasticity resources) to lock in progressively stronger excitatory connections without matching inhibitory gains. This forms a new dystonic synergy that dominates whenever the performer attempts speeds or forces above the old threshold—ultimately manifesting as FTSD at that higher range.
Importantly, when FTSD first develops, its emergent “dystonic synergy” relies partly on the same pyramidal neurons and local circuits that the functional synergy originally employed, especially in the synaptic range below the threshold for symptomatic high-force or high-speed movements. During repeated overreaching above the old synergy’s capacity, excitatory synapses in the new dystonic subcircuit gain incremental LTP, while the matching inhibitory synapses fail to track that potentiation. Initially, this new subcircuit draws on many of the same neuronal pools as the functional synergy at lower intensities, simply adding a further excitatory “extension” above the threshold. Because it is still forming, it partially overlaps with the older synergy’s resources—both excitatory and inhibitory—below that threshold.
However, we hypothesize and propose that metaplastic processes and continued “use” at higher intensities enable the dystonic synergy’s excitatory side to reorganize and add new or re-labeled synapses, effectively building a parallel resource base that was once entirely shared with the functional synergy. Through the tagging and capture of plasticity-related proteins (PRPs) and the recruitment of additional dendritic spines, the dystonic synergy becomes more autonomous. As repeated high-demand episodes reinforce these excitatory connections above the threshold, the subcircuit does not merely borrow old synergy synapses; it stabilizes its own set of strongly potentiated E-E links. At the same time, once again, the old synergy’s E/I loops in the lower domain receive fewer co-activations and fewer PRPs, leading them to stagnate.
Once the dystonic synergy consolidates in this branched-off manner, it no longer relies on the original functional synergy’s “below-symptom threshold” resources. Consequently, when you introduce new technique changes in the lower or moderate intensity range, retrograde interference degrades the old/functional synergy’s unique synapses—because those are the synapses still being partially engaged in normal or subthreshold playing. The dystonic synergy, however, resides at higher intensities (above its threshold) and has already completed its own consolidation. If you are not consistently reactivating that exact high-intensity subcircuit during technique changes—and indeed, you might be deliberately avoiding those speeds to avoid triggering dystonia—its E/I network is not disturbed by the new practice patterns. Moreover, the principle of occlusion indicates that once the dystonic synergy’s excitatory subcircuit saturates, it does not keep accruing more LTP.
Meanwhile, the functional synergy below the symptom threshold remains vulnerable: any time you adopt another new technique, only a fraction of old synergy synapses overlaps, and the unused portion succumbs to downregulation. Because the dystonic synergy no longer meaningfully shares that pool (having “branched off” via metaplastic growth), it is immune to these interference effects, preserving its severity and threshold unaltered. The net result is a three-state scenario: (1) a below-threshold zone of relatively unproblematic movement, albeit weakened; (2) a mid-range zone that prompts true weakness when attempted intensities exceed the functional synergy’s decayed capacity; and (3) a higher zone that triggers the fully formed dystonic synergy, whose imbalance is locked in and unaffected by subsequent changes in the older synergy’s domain. To the best of our knowledge, this accurately reflects the patient’s described circumstance of a developed three-state scenario after repeatedly changing technique without practicing enough using the new technique after the change.
That said, if an individual already with a fully developed dystonic synergy repeatedly adopts new piano techniques without consolidating each one through repetitive practice, the functional synergy’s excitatory and inhibitory synapses progressively degrade due to partial baseline shifts and retrograde interference of the previous technique. This process makes the individual be in a three-state scenario like the example above. This process establishes a true weakness zone, where attempted speeds or forces exceed the newly reduced synaptic capacity but still remain below an established dystonic threshold since a dystonic synergy is already present. Now, if the individual again engages in overreaching within this mid-range zone—pushing repeated below-threshold excitatory bursts—theoretically speaking, we propose that this can gradually form a new 2nd dystonic synergy above that weakened threshold.
Each time you overreach at a newly lowered capacity, certain E-E synapses of pyramidal neurons receive repeated, partial but frequent spikes, enough to nudge them toward incremental LTP. Because these bursts are only partial—never fully synchronized or forceful—they fail to co-activate the local inhibitory interneurons in a matched, Hebbian manner, preventing E→I and I→E connections from similarly strengthening. This mismatch allows an “upper subcircuit” to acquire progressively stronger excitatory synaptic efficacy without the parallel inhibitory feedback that would keep it balanced.
Simultaneously, the existing dystonic synergy remains insulated because it has branched off via metaplastic changes, becoming self-sustaining at an even higher intensity domain. The new synergy under formation in the mid-range zone no longer taps the same pool of synaptic resources that had consolidated in the original dystonic synergy; instead, it reuses or reorganizes the moderately weakened domain’s E-E synapses to form another excitatory-dominant subnetwork. By the principle of occlusion, each new synergy eventually saturates its excitatory side once enough partial bursts have incrementally stabilized those E-E synapses. Its inhibitory half, however, remains underdeveloped due to insufficient synchronous drive.
Theoretically, repeating this cycle could yield multiple discrete dystonic synergies stacked at successively lower intensities: every time you degrade the functional synergy’s capacity through repeated, unreinforced technique changes, then overreach in the new true weakness zone, you carve out a fresh excitatory subcircuit that consolidates into a second or third dystonia. We propose that such an outcome is incredibly rare in practical life because it demands an extreme—and arguably irrational—training pattern: continually changing techniques, never consolidating progress, and persistently pushing above whichever reduced threshold emerges. Nonetheless, from a strict neurological plasticity standpoint, theoretically speaking, there is no fundamental mechanism that categorically prevents an individual from creating multiple dystonic synergies in this layered fashion. Each synergy would simply occupy its own “band” of excitatory intensities above each newly formed true weakness threshold.
Importantly, when a new dystonic synergy first begins to form above the old functional synergy’s threshold, its E-E synapses are only partially potentiated. These subthreshold bursts, although enough to trigger LTP in the newly emerging circuit, have not yet driven all those synapses to their upper capacity as limited by occlusion principles. Consequently, if you keep operating in that same high-intensity range—whether precisely at or somewhat beyond/overreaching the dystonic synergy’s current excitatory capacity—the repeated bursts of presynaptic spiking continue to “tag” and capture plasticity-related proteins (e.g., CaMKII, PKMζ, BDNF) in that same excitatory network. We hypothesize that in the scenario where the individual overreaches repetitively past the capacity of a partially developed excitatory circuit of the dystonic synergy, these repeated activations would simply push the synergy’s E-E connections closer to their maximum potentiation, without creating a separate additional dystonic synergy.
Forming a distinct second dystonic synergy would require going through a different true weakness zone that sits below the newly established dystonic threshold. In other words, one would have to degrade the functional synergy further, establish a new midrange true weakness threshold, and then repeatedly overreach in that range. By contrast, using the existing dystonic synergy—even at intensities that exceed its current excitatory strength—merely keeps reinforcing the same maladaptive subnetwork. The inhibitory subcircuit continues to lag behind because these bursts remain partial or unsynchronized, failing to co-activate PV interneurons robustly. Thus, the result is an incremental climb toward saturation in the existing dystonic synergy rather than the creation of an entirely new one.
In short, once a dystonic synergy has formed in that upper band of intensities, additional attempts to surpass its still-maturing excitatory capacity do not cause a second dystonia; they simply strengthen the ongoing dystonic network until it nears occlusion. A genuinely new dystonic synergy would only emerge if you later carved out another true weakness threshold (through technique changes) and then started overreaching there in the same partial-burst, suboptimal manner that gave rise to the first one.
In addition, “counter-motion,” in our framework, refers to an intentional effort to produce the antagonist movement of a dystonically driven motion. For example, if the dystonic synergy causes involuntary hyperflexion in the right index finger, counter-motion means attempting extension with that same finger while it simultaneously experiences the dystonic symptom of hyperflexion. Critically, we propose that each digit’s representation in M1 consists of multiple partially overlapping TSMS (e.g., flexion, extension, abduction, adduction) with each unidirectional movement pattern qualifying as a separate TSMS at the cortical level. While there is partial overlap in their neuronal populations, each synergy also depends on its own subset of pyramidal cells and PV interneurons.
In this example, the dystonic hyperflexion synergy has developed right above the functional flexion synergy’s existing excitatory and inhibitory capacities with abnormally high E-E synaptic strength with impaired inhibitory feedback (via weakened PV interneuron circuits). As a result, once a patient’s intended movement or speed level crosses the symptom threshold where that dystonic synergy fully engages, its pyramidal neurons outpace competing ensembles for the same finger, generating an involuntary over-firing of flexor-oriented neural output.
When no counter-motion is attempted, we hypothesize that only the dystonic synergy for that digit is significantly active. Because it is hyperexcitable and insufficiently clamped by inhibitory synapses, it saturates large portions of the local pyramidal population that controls the finger’s prime-mover muscles. In other words, we propose that the dystonic ensemble hijacks a large fraction of the available motor cortical neurons for that digit, displacing or overshadowing the functional synergy that would normally execute non-dystonic movement at that force or speed. We propose that this overshadowing arises because many of the same cortical neurons can, in principle, be recruited by either synergy; however, the dystonic synergy’s abnormally strengthened E-E connections and weak E/I regulation make it more likely to discharge robustly and lock the population into a maladaptive pattern of sustained firing. When the patient attempts a counter-motion at the same time as the dystonic symptoms, it means that some subset of pyramidal neurons—those still belonging to the functional synergy for the finger’s antagonist motion—also receive sufficient descending drive to initiate extension. This extension drive, however, remains drastically constrained. We propose that as soon as the dystonic synergy has saturated a large proportion of the digit’s neuronal pool with runaway E-E firing, there are fewer “free” or recruitable pyramidal neurons left to build a robust antagonist command. By analogy, the functional synergy would need to ramp up to a high intensity to push the finger fully into extension, but it cannot fully scale its excitatory output because so many neurons (and so much plasticity “bandwidth”) are already consumed by the dystonic ensemble’s hyperexcitable firing pattern. Consequently, the patient manages only a partial, limited extension—the extension synergy can never ramp to the point of overreaching even if its synaptic strength has not reached the theoretical maximum.
We defined overreaching earlier as a situation in which a functional synergy attempts an intensity beyond its own synaptic capacity, causing the pyramidal neurons to fire in repeated, partial bursts that fail to recruit inhibitory interneurons in a well-timed manner. These repeated submaximal bursts can eventually form a new dystonic subcircuit by selectively potentiating excitatory connections without matching inhibitory strengthening. To create a second dystonia, one would need the functional synergy (now aiming for extension, for example) to chronically operate in that “partial-burst,” suboptimal regime each time the performer tries a force or speed above the synergy’s capacity. However, when the patient is already in a state of active dystonia (i.e., the dystonic synergy in flexion is saturating the cortical motor pool), we hypothesize that the counter-motion synergy cannot approach its upper limit of excitatory drive. Because most excitatory neurons are pinned into the hyperflexion mode (or are at least significantly depolarized in that pattern), the antagonist synergy lacks the neuronal resources to perform repeated subthreshold bursts. Instead, what the patient experiences is a shallow, relatively low-frequency extension command that coexists with the hyperflexion drive. This weak extension command does not represent classical “overreaching,” as the functional extension synergy can never push its excitatory circuit to intensities that outstrip its synaptic strength capacity in isolation; it is simply overshadowed/dwarfed by the dystonic synergy’s robust hyperexcitability and strong E-E feedback loops.
Thus, even though from an external perspective one might assume that trying counter motion against a strong involuntary flexion may potentially seem like overreaching—in the context that the extension synergy’s synaptic strength has not yet reached the theoretical maximum—given the fact that the intensity attempted triggers the dystonic synergy, on a cellular level, the extension synergy is prevented from actually scaling up to that level. Once again, we propose that the active dystonic ensemble hogs resources when the pyramidal neurons that would ordinarily be available to ramp up extension firing are already firing in the flexor-oriented pattern or are so depolarized by the dystonic ensemble’s E-E drive that they cannot respond to another excitatory input with well-timed bursts. Because of this inability to truly overreach, the counter-motion does not generate the repeated subthreshold bursts that are necessary to form a new, second dystonia. On the contrary, it remains a modest extension command whose excitatory drive never enters the unstable zone where excitatory plasticity outstrips inhibitory plasticity. Over time, no new maladaptive synergy is cemented for the antagonist direction. The counter-motion synergy’s presence therefore does not undermine or supersede the dystonic synergy, nor does it form its own pathologically imbalanced subnetwork, as it is physiologically unable to reach the partial-burst, high-intensity regime that can create a second dystonia. Consequently, even as the patient voluntarily tries to extend against the strong involuntary flexion, the net effect is simply a visually observable “tug of war” between a hyperexcitable dystonic synergy and a much weaker, functional synergy.
The Probable Cure
Once FTSD has arisen—manifesting as a TSMS in which PV synapses are under-strengthened relative to the excitatory circuit (E-E) synapses—we propose that the most direct path to recovery is to retrain the deficient inhibitory circuits below or at the threshold that triggers involuntary contractions (or below or at the threshold for the true weakness threshold in a three-state scenario). From discussions above it is reasonable to assume STDP effects can be used as an advantage for retraining. Under these therapeutic conditions, restoring balanced inhibition in the affected dystonic synergy/TSMS hinges on precisely targeted motor retraining protocols that incrementally strengthen the E-E, excitatory-inhibitory (E→I), and inhibitory-excitatory (I→E) synapses within the residual “healthy” functional synergy and, crucially, the inhibitory PV synapses (E→I and I→E) of the dystonic synergy.
The essential principle of below or at-threshold retraining (BATR) is to repeatedly perform the skill—be it piano playing, writing, speaking, or another specialized movement—strictly at intensities that do not elicit involuntary dystonic output (and true weakness if there is a true weakness threshold present too). BATR does in fact closely mirror the “slow-down exercise” (SDE) first detailed by Sakai (2006) and later replicated by Yoshie et al. (2015); however, both studies focused on the protocol’s clinical outcomes and left the underlying neurophysiological mechanisms—such as the synergy-threshold model proposed here—largely unexplored. BATR practice functions by ensuring that every cycle of firing in the functional synergy falls within a range where pyramidal neurons can consistently co-activate PV interneurons in a well-timed, Hebbian manner. When the performer moves at or below the circuits’ current capacity—meaning the speed or force demand does not surpass the synaptic strengths of the excitatory and inhibitory circuits of the functional synergy—the pyramidal neurons of the primary motor cortex enter a stable firing regime. In this stable regime, each burst of excitatory spikes is sufficiently robust to depolarize local interneurons, but not so extreme that it elicits disorganized, partial-burst patterns where interneuron recruitment fails. Because PV interneurons require a properly synchronized volley of excitatory input to achieve LTP at excitatory-to-inhibitory (E→I) synapses (and, in turn, to deliver well-timed GABAA inhibition back onto pyramidal cells at inhibitory-to-excitatory [I→E] synapses), these steady, below-or-at-symptom-threshold bursts provide exactly the coincidence of presynaptic and postsynaptic activity needed to drive inhibitory plasticity.
From a mechanistic standpoint, repeated synaptic co-activation in this zone ensures that the membrane potential of both the pyramidal neuron and the PV interneuron passes through the specific temporal windows conducive to STDP. STDP rules demand that excitatory-inhibitory connections strengthen when the interneuron receives consistent, well-phased (E before I) excitatory postsynaptic potentials (EPSPs) from presynaptic pyramidal cells and, shortly thereafter, fires an action potential that can exert inhibitory postsynaptic currents (IPSCs) back onto those same or closely related excitatory targets. In other words, if the pyramidal cell’s output spike trains are relatively stable rather than erratic or below threshold-fragmented, the PV interneuron experiences well-defined time-locked depolarizations. This triggers intracellular calcium transients via NMDA-type glutamate receptor activation (where present) or high-frequency suprathreshold depolarizations, both of which are mandatory for LTP induction in these local inhibitory circuits. Additionally, each volley of PV interneuron firing that arrives back onto pyramidal cell somas with a short latency helps sculpt a precise postsynaptic potential, reinforcing the timing-based link between the excitatory spike pattern and the inhibitory feedback that follows.
By contrast, if for example, in a three-state scenario where one attempts intensities in the true weakness zone which therefore exceed the functional synergy’s excitatory and inhibitory capacity, pyramidal neurons produce bursts that are neither strong nor consistent enough to recruit interneurons reliably; the partial or highly asynchronous bursts provoke little to no net LTP in E→I synapses. Below-or-at-threshold training avoids that pitfall, because the intensity level is sufficient to elevate pyramidal neurons into well-synchronized discharges—enough to drive the interneuron to spike—yet not so high that the circuitry slips into the maladaptive patterns seen in dystonic overflow. Each successful repetition of a below-threshold movement reaffirms the coincidence of excitatory firing and inhibitory activation, gradually strengthening the crucial E→I and I→E synapses that define SICI.
In addition, each time the pyramidal neurons and PV interneurons fire together with the correct temporal offsets, they engage plasticity-related proteins (e.g., CaMKII, CaMKIV, PKC) in both the presynaptic terminals and postsynaptic compartments. The activated interneuron also undergoes intracellular signaling cascades that upregulate GABAA receptor clustering at the axon terminals projecting back to pyramidal cells, elevating the amplitude of miniature inhibitory postsynaptic currents (mIPSCs) in those neurons. Moreover, the PV interneurons themselves, when receiving repeated patterned input, can exhibit increased presynaptic release probability at their GABAergic terminals, boosting their inhibitory efficacy. In this way, the synergy’s once-compromised inhibitory loops regain the capacity to clamp excitatory surges at higher intensities than before, effectively pushing the threshold for involuntary dystonic firing upward. Meanwhile, because the inhibitory microcircuit in M1 does not exist as a fully segregated set of interneurons for each synergy, we propose that a subset of the PV interneurons that belong to the functional synergy also project to (or receive excitatory input from) the dystonic synergy’s pyramidal population. In other words, these local circuit inhibitory cells are partially shared between neighboring excitatory ensembles encoding movement of the same digit. Whenever the functional synergy recruits those shared PV interneurons in a well-timed, high-fidelity manner, it sends a strong excitatory drive onto them (E→I). The interneurons then fire robustly, and their outputs (I→E) go not only to the functional synergy’s pyramidal neurons but also to overlapping or neighboring pyramidal neurons in the dystonic ensemble. This means that, even though the dystonic synergy’s excitatory subcircuit remains silent at below-threshold speeds, each practice trial with clean inhibition still delivers a coincident burst of excitatory input to the shared PV interneurons and a matched inhibitory volley back onto excitatory cells in that region of M1. The repeated synchronized spiking and inhibitory release foster gradual strengthening of both E→I and I→E synapses in the dystonic synergy’s inhibitory circuits as well.
Thus, the key insight is that the partial overlap in local inhibitory cells serves a dual function: the performer maintains and enhances the functional synergy’s balance of excitation and inhibition while simultaneously giving the dormant side of the dystonic synergy (its under strengthened inhibitory loop) the proper frequency and timing of interneuron co-activation it needs to build up its inhibitory synaptic strength. Each below-threshold repetition avoids driving the dystonic synergy’s E-E connections to any further potentiation—because the performer never surpasses the speed or force threshold that would trigger runaway firing in that maladaptive circuit—but it does keep the local PV interneurons thoroughly engaged in the normal, balanced synergy. Consequently, these interneurons have plenty of well-timed bursts, calcium signaling, and associated PRP capture, enabling their GABAA output synapses onto the dystonic excitatory neurons to slowly “catch up” and strengthen.
Over the course of many practice sessions, this strengthening of inhibitory contacts in the dystonic subnetwork eventually reaches a point at which the dystonic synergy’s excitatory circuit is no longer unopposed, but matched equally in synaptic strength by its inhibitory circuit. Essentially, repeated BATR has tipped the ratio of E-E to E-I back toward equilibrium in that maladaptive synergy. Once the capacity of the local PV interneurons to deliver robust, short-latency inhibition matches the excitatory drive of the dystonic synergy, attempting higher speeds or forces no longer triggers the runaway bursts characteristic of FTSD. Instead, as soon as the dystonic pyramidal cells begin to fire in a large ensemble, the now-strengthened PV interneurons respond with an appropriately timed and magnitude-sufficient inhibitory burst, preventing the cortical hyperexcitability that originally caused involuntary contractions or posturing. The clinical outcome is the gradual disappearance of dystonic symptoms. Practically speaking, this manifests in the performer being able to raise tempo or force beyond the prior dystonic threshold without slipping into the involuntary synergy. Therefore, through BATR, the motor cortex reestablishes the delicate temporal synchrony between excitation and inhibition: the functional synergy’s excitatory and inhibitory circuits strengthen through LTP in lock-step, and—via their shared PV interneurons—the dystonic synergy’s previously weak inhibitory circuit is strengthened through LTP as well, effectively reversing the maladaptive plastic changes that define FTSD and leading to a potentially full 100% recovery.
Clinical Implementation: A Practical Guide for Below or At-Threshold Retraining
BATR relies on strict adherence to performing the affected motor task while not provoking dystonic symptoms (and true weakness if there’s also a true weakness threshold). Whether the individual is a musician, writer, vocalist, athlete, or any other practitioner whose craft hinges on refined motor skills, the same neuroplastic principles apply: the patient must locate and remain below his or her dystonic or true weakness threshold while carefully, gradually expanding that threshold once the inhibitory network has begun to catch up. In a clinical or self-guided setting, the first step is to identify the intensity at which dystonic symptoms vanish. For a pianist, this might mean using a metronome to find the fastest tempo at which no involuntary contractions occur; for a spasmodic dysphonia patient (also known as laryngeal dystonia), it might mean finding the maximum volume for speech at which there are no symptoms (often found to be when whispering or very soft phonation); and for a writer, it might entail discovering the writing speed or pen pressure at which handwriting remains crisp and free of involuntary finger or wrist contraction. Once that threshold point is determined, the recovery journey begins with long daily practice sessions (with ideally minimal breaks and interruptions) of precisely executing movements at this “clean” intensity. The patient should strive for consistently accurate repetitions free of any involuntary muscle firing, all the while cultivating a sharp sensory awareness of each movement. The goal is to ensure repeated, well-timed co-activation of pyramidal cells and PV interneurons, driving the desired adaptive synaptic potentiation discussed previously.
Clinicians should instruct patients to increment speed or intensity in small, carefully measured steps. The attempted intensity should only be increased if improvement is felt during or after a consistent period—often hours, days or weeks—of symptom-free practice. A musician who experiences improvement at a slow tempo may nudge the metronome up a few beats per minute, provided no dystonic signs appear. A writer might gradually reduce the time taken to physically write a sentence, ensuring no dystonic symptoms creep in. If any dystonic symptoms reemerge, the patient should immediately revert to a slightly slower or gentler intensity, ensuring that each successful repetition remains below or at the threshold where no symptoms occur. This cyclical process—discovering the clean range, reinforcing it through repetitive practice, and then gently expanding it—systematically coaxes the inhibitory circuit of the dystonic synergy and both excitatory and inhibitory circuits of the functional synergy to strengthen, a process necessitating weeks to potentially months of continuous daily training (ideally lasting several uninterrupted hours every day).
Clinically, it is crucial to emphasize patience: rebalancing the E/I interplay in FTSD demands high repetition counts, distributed over consistent daily practice. In the early stages, patients may often find that progress remains slow or negligible for days or weeks. It is important that the clinician or therapist helps sustain morale and ensures the therapy adheres to core neuroplastic principles. Relapses into dystonic firing may occur momentarily if the chosen intensity increase after experienced improvement is more than what the recent improvement can provide, serving as a barometer to step back and reconsolidate inhibitory strength at slightly slower intensities. By carefully following this below-threshold protocol, the performer incrementally shrinks the E/I gap that initiated the dystonia. Over time, the thresholds at which dystonic activity or true weakness intrudes move higher, gradually matching or surpassing normal functional demands and eventually resolving.
Addressing Variability in Patient Outcomes
A vital point for both clinicians and patients to understand is that BATR should, in theory, be universally applicable to all focal task-specific dystonias, so long as a below-threshold range can be identified. The fundamental mechanism—properly and regularly engaging shared PV interneurons without reinforcing high-excitability loops—should not vary among different individuals with FTSD. Thus, when a patient reports slow or insufficient progress, it does not indicate that the method “does not work” for that particular case of FTSD. Rather, such variability most often reflects a breakdown in practice protocols or an insufficient duration of uninterrupted training. If the patient increases speed or force too rapidly, inadvertently activating or potentially reinforcing the dystonic synergy, the inhibitory side never engages robustly enough to induce the necessary synaptic strengthening. Similarly, if practice sessions are too brief or too inconsistent, then the neural circuits have not received enough of the consistent E/I co-activation needed to recalibrate its balance.
From the clinician’s perspective, monitoring adherence is paramount. Detailed logs of daily practice—recording the exact speed (e.g., metronome markings), duration of below-threshold performance, and any lapses into dystonic firing—help pinpoint whether the patient is genuinely meeting the criteria for correct execution and implementation of BATR. Crucially, the published SDE protocols (Sakai, 2006; Yoshie et al., 2015) and other below-threshold retraining approaches (e.g., van Vugt et al., 2014, Video S1) have demonstrated objective success in carefully documented cases, indicating that the method is robust across the spectrum of FTSD. The key is that any below-threshold practice must be truly dystonia-free, repeated adequately to induce synaptic potentiation (both E→I and I→E), and carried out with unwavering consistency to allow Hebbian plasticity to accumulate.
In practical terms, clinicians can reassure patients that the BATR protocol works on a solid neurophysiological foundation, backed by empirical data, scientific reasoning, and direct clinical observation. Variations in recovery speed or incomplete symptom resolution typically signify that certain steps of the retraining regimen are being skipped, rushed, or inconsistently implemented. If a patient claims “it doesn’t help,” the immediate clinical response should be to assess whether the proper tempo or force range has ever truly avoided dystonic episodes, whether the practice is performed daily and with adequate repetition, and whether the patient prematurely attempts more challenging speeds or tasks. Only by maintaining a rigorously below-threshold regimen—sometimes for weeks or months—do the PV interneurons have the chance to “catch up” and dampen the excitatory burst patterns underlying FTSD. Hence, persistent use of BATR remains the recommended route to lasting resolution, regardless of individual variation in short-term outcomes.
Hypothesis Verification via Computational Modeling
While much of the evidence for the synergy-based explanation of FTSD comes from clinical observation and electrophysiological findings, computational modeling offers a powerful complementary approach to test and refine these hypotheses. Specifically, by simulating a neural architecture underlying excitatory/inhibitory (E/I) balance, one can systematically manipulate synaptic strengths, plasticity rates, and network connectivity to observe whether, and under what conditions, a “maladaptive synergy” emerges. This strategy allows a more rigorous, iterative cycle of hypothesis generation (from clinical observation to modeling) and hypothesis verification (from modeling back to empirical testing), without actually accessing human patients which is oftentimes not possible. In this section, we present findings from a simulated spiking neural network we used to verify some of the hypotheses described above.
Network Model
We simulated a spiking neural network composed of two interconnected populations: an excitatory population (E) and an inhibitory population (I). Each population consisted of Nex excitatory and Ninh inhibitory neurons, following a leaky integrate-and-fire (LIF) neuron model with synaptic interactions.
The neurons evolved according to the following membrane potential equation:
where V is the membrane potential, Vrest is the resting potential, τm is the membrane time constant, and Isyn represents synaptic input.
Spiking events were generated when V exceeded a threshold Vth, after which the membrane potential was reset to Vreset.
Synaptic Connectivity
Synaptic interactions were implemented with conductance-based synapses, where synaptic current was given by:
where gsyn is the synaptic conductance, and Esyn is the reversal potential of the synapse. Excitatory-to-inhibitory (E→I) and inhibitory-to-excitatory (I→E) connections were defined with a probability of connection p.
Input Drive and External Stimuli
External inputs were applied to neurons via a Poisson-distributed spike train, simulating background noise and task-related input. Input amplitude to the excitatory populations was systematically varied to examine the network’s response to different stimulation intensities. The external drive followed:
where A was the input amplitude and r the baseline firing rate of the Poisson process.
Simulation of Overreaching and Dystonia
To simulate maladaptive plasticity leading to dystonia, we introduced a functional synergy (FS) and a dystonic synergy (DS) within the excitatory population. FS was modeled with balanced excitatory/inhibitory drive (EF, IF), while DS was introduced as an imbalance where excitatory synapses (ED) increased without a corresponding increase in inhibitory synapses (ID). Overreaching was induced by systematically increasing external drive beyond the functional synergy’s limit.
Firing Rate Analysis
Network activity was analyzed by computing population-averaged firing rates for both excitatory populations. The firing rate was calculated as:
For systematic variation of input amplitude to the second population, firing rates of both populations were measured and plotted as a function of the input strength.
Simulation and Implementation
All simulations were implemented in Python using custom-built neuron models. The integration of differential equations was performed using the fourth-order Runge-Kutta method with a time step of dt=0.1ms. The network was simulated for T=2000, and results were analyzed with Matplotlib and Pandas for statistical processing and visualization.
Computational model demonstrates the emergency of dystonic synergy in E/I network
This section analyzes the spiking neural network results under different conditions to understand the transition from normal motor control to dystonia. Three different scenarios are examined: the healthy network representing baseline functional synergy, the mixed functional and dystonic population, and the loss of balance leading to dystonia. The comparison of firing frequencies across these conditions provides insights into how dystonic circuits emerge and disrupt normal motor function.
In Fig 1B we show the spiking activity of a healthy network, where excitatory and inhibitory functional synergies maintain a stable balance. The firing activity remains regular and within a physiological range, ensuring proper motor control. This serves as a baseline reference, illustrating a scenario where neurons fire without abnormal synchrony or instability. Also, the experiment where input strength is increased (simulating speed/intensity), demonstrates that for this case the firing rate follows a linear increase with the input strength. The inset shows the activity of inhibitory cells, typical of PV cells in that brain area that follows a similar linear increase.
This pattern is not always the case, and although in a 2D excitatory/inhibitory (E/I) neuron network model, increasing the amplitude of the external stimulus typically leads to an increase in the firing rate of the neurons a low-pass pattern is expected and could come earlier depending on the balance. This is because a stronger external stimulus can drive the excitatory neurons to fire more frequently but, as the excitatory neurons become more active, they can, in turn, increase the activity of inhibitory neurons due to the network’s connectivity (Fig 1D).
The overall effect on the network’s firing rate can depend on several factors, including the balance between excitation and inhibition, the network’s connectivity, and the specific dynamics of the neurons involved. If the inhibitory feedback is strong enough, it might counteract the increase in excitatory activity, potentially stabilizing the firing rate or even reducing it after an initial increase.
In summary, while you can generally expect an increase in firing rate with a stronger external stimulus, the precise outcome will depend on the specific parameters and dynamics of your E/I network model.
Figure 1.
A. Schematic representation of the experimental setup. Stimulation (depicted by a piano keyboard) leads to motor action through functional synergy involving excitatory (E) and inhibitory (I) neurons. Dystonic synergy is indicated by the overlapping circles. The latter could be present or absent depending on the situation (see text) B. Raster plot showing neuronal firing activity over time for healthy state. Each row represents a different neuron, and each tick mark indicates a spike. C. Graph illustrating the effect of input strength on network firing rate (same as in B). The main plot shows the firing rate increasing with input strength. The inset highlights the inhibitory effect at a specific input strength. D. Graph showing the decrease in firing rate with increasing input strength, indicating a different network response. Different E/I balance and input strengths change the response pattern of the network. E. Comparison of firing rates between dystonic and functional synergies across varying input strengths. A threshold is marked where the firing rate diverges between the two conditions.
Figure 1.
A. Schematic representation of the experimental setup. Stimulation (depicted by a piano keyboard) leads to motor action through functional synergy involving excitatory (E) and inhibitory (I) neurons. Dystonic synergy is indicated by the overlapping circles. The latter could be present or absent depending on the situation (see text) B. Raster plot showing neuronal firing activity over time for healthy state. Each row represents a different neuron, and each tick mark indicates a spike. C. Graph illustrating the effect of input strength on network firing rate (same as in B). The main plot shows the firing rate increasing with input strength. The inset highlights the inhibitory effect at a specific input strength. D. Graph showing the decrease in firing rate with increasing input strength, indicating a different network response. Different E/I balance and input strengths change the response pattern of the network. E. Comparison of firing rates between dystonic and functional synergies across varying input strengths. A threshold is marked where the firing rate diverges between the two conditions.
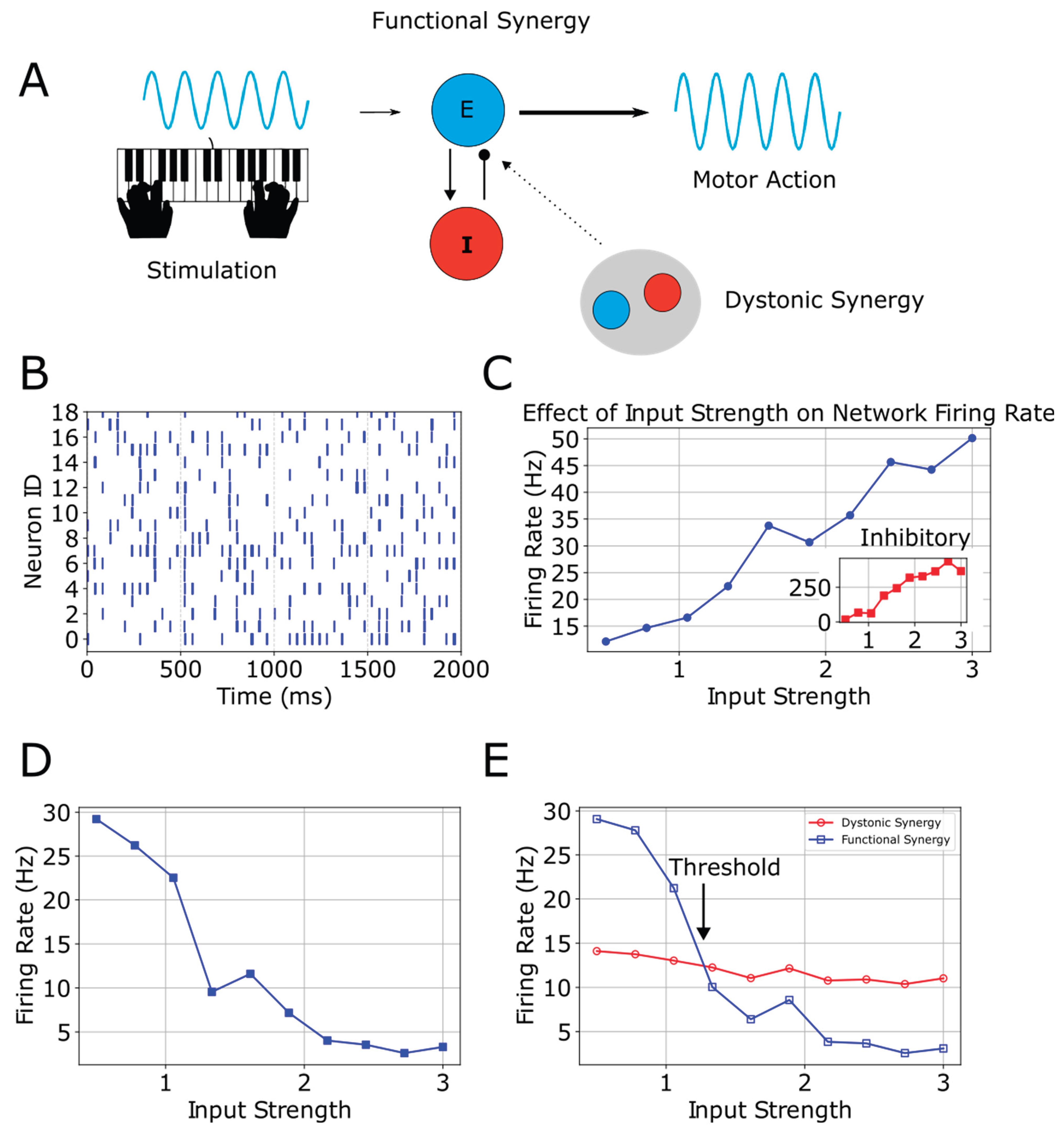
As dystonic synergy emerges alongside the existing functional synergy (Fig 1E), a firing rate competition becomes apparent. Thus, the network consists of two interacting populations. Notice that the dystonic synergy has low firing rate for smaller input strengths and does not affect considerably the functional, but after a certain threshold, it is firing at higher rates than the functional. In other words, the distinct firing rates indicate that both populations coexist and interference is apparent after the threshold, suggesting that dystonic neurons are active but not dominating motor function unless higher input strengths, here associated with speeds, are attempted.
This result is significant because it is associated to unbalanced networks and how dystonia allows excessive synchronization and hyper-excitability in motor circuits. The dystonic synergy overshadows the functional synergy, leading to involuntary contractions due to the loss of independent motor control, pathological co-activation of neurons, and a shift in motor circuit dynamics that reinforces maladaptive plasticity.
In the next section we will discuss alterations beyond M1, explaining how they can be recast as secondary byproducts of repeated dystonic synergy activation.
Alterations
In the sections that follow, we draw on foundational neurobiological principles (Dayan & Abbott, 2001; Kandel et al., 2021; Purves et al., 2018) and the primary studies referenced therein to interpret the involvement of different brain areas in FTSD.
Basal Ganglia
Consistent with the M1-centric framework developed in this paper, we propose that the prominent basal ganglia alterations identified by Simonyan et al. (2017) can be hypothesized as emergent “secondary by-products” of the repeated usage of the maladaptive dystonic synergy. When a dystonic synergy in M1 is repetitively activated for the same specialized movement—such as writing in writer’s cramp or phonation in laryngeal dystonia—the descending signals systematically converge onto the corresponding somatotopic sector of the striatum. This repeated, high-frequency cortical input preferentially engages the medium spiny neurons (MSNs) in that striatal territory.
The essence of “learning” at the level of substantia nigra pars compacta (SNc) dopaminergic neurons boils down to a remodeling of both their afferent synapses—excitatory glutamatergic inputs from the pedunculopontine nucleus (PPN) and subthalamic nucleus (STN) plus inhibitory GABAergic inputs from striatal direct-pathway neurons—and activity-dependent tuning of their intrinsic membrane properties (ion-channel makeup and signaling cascades that determine whether the cell fires tonically, bursts, or stays quiescent). When a movement produces an unexpectedly good outcome—a positive reward-prediction error—those excitatory afferents fire in tight temporal register with the action, driving a precisely timed, high-frequency burst in SNc neurons that delivers a phasic surge of dopamine to the corresponding sector of striatum. However, when the same movement consistently yields erroneous or suboptimal results, a cascade of molecular and synaptic plasticity events within the dopaminergic neurons progressively suppresses that bursting mechanism for that particular movement context. One can break down the “how” of this plastic reconfiguration into three interdependent layers.
First, synaptic plasticity at excitatory glutamate inputs onto SNc dendrites. The dopaminergic neurons in SNc receive glutamatergic inputs from multiple sources, including the STN, PPN, and others. These synapses are capable of both LTP and LTD, much like cortical or striatal synapses, depending on patterns of pre- and postsynaptic firing and the resultant intracellular calcium transients. When a motor action yields a negative or neutral outcome—repeatedly failing to produce the expected sensorimotor match—we hypothesize that various basal ganglia feedback loops (particularly from the striatum via pallidal or nigral reticulata circuits) provide inhibitory or desynchronizing signals that discourage these glutamatergic inputs from firing in tight synchrony with movement onset. At the same time, the dopaminergic neuron itself experiences partial or asynchronous excitatory drive that does not push membrane potentials into the high-threshold bursting range.
Because the dystonic synergy fails to produce a coherent or “successful” motor outcome, we hypothesize that the afferent inputs that typically drive a strong, synchronized excitatory burst in the dopaminergic neurons arrive in an uncoordinated fashion or with insufficient amplitude whenever that synergy is attempted. Normally, if a movement matches internal predictions or yields positive reinforcement, the STN, PPN and other excitatory sources coordinate their firing to coincide tightly with the moment of correct action, pushing the dopaminergic neuron’s membrane potential above a critical threshold and enabling a phasic burst. In the dystonic scenario, however, negative or neutral prediction-error signals flowing through basal ganglia loops disrupt this synchronization. Instead of a single well-timed “surge” of excitatory drive, the midbrain neurons experience smaller or ill-timed bursts from their inputs: sometimes excitatory volleys are too spread out or arrive at off-peak moments, sometimes inhibitory signals from pallidal or nigral reticulata circuits interject. The end result is that each piece of excitatory input by itself is insufficient to overcome the intrinsic potassium currents, calcium channels, and other membrane ionic regulatory mechanisms that demand a tightly clustered, high-amplitude depolarization to elicit a burst. With each repetition of the synergy, these excitatory drives remain fragmented or asynchronous, never coalescing into the strong depolarizing pulse that triggers the characteristic high-frequency dopaminergic discharge. Consequently, the dopaminergic neuron remains only partly activated, entering brief or moderate depolarizations that fall short of the level needed to initiate a full phasic burst. Over time, repeated episodes of subthreshold activation further reinforce this pattern by inducing forms of synaptic depression at excitatory inputs, making it even less likely that future volleys, arriving out of sync, will sum enough to propel the neuron into burst-firing territory.
More specifically, in dopaminergic neurons of the SNc, high-frequency bursts typically require a sufficiently large and synchronous excitatory input to overcome multiple stabilizing currents (e.g., potassium conductances, calcium-dependent after-hyperpolarizations) and push the membrane potential into a plateau or “bursting” voltage range. In a healthy reward-predictive context, excitatory projections from structures like the PPN or STN arrive in tight temporal coordination precisely when the movement outcome is deemed better than expected, producing a robust depolarization that triggers a phasic burst. By contrast, if the excitatory drive is partial, asynchronous, or arrives at the wrong time relative to the motor event, the neuron does not receive the consolidated depolarization needed to cross that bursting threshold. Instead, the neuron experiences multiple subthreshold episodes where each incoming volley of glutamate partially depolarizes the membrane but fails to initiate a full burst.
The significance of these repeated subthreshold depolarizations—and why they lead to synaptic depression rather than potentiation—comes down to the intracellular calcium dynamics and receptor-trafficking rules in dopaminergic neurons. When the neuron depolarizes strongly and synchronously (for example, due to coherent afferent firing), a large calcium influx typically enters through voltage-gated calcium channels and/or NMDA receptors, activating kinases like CaMKII or PKC that phosphorylate key receptor subunits (AMPA or NMDA), stabilizing them in a potentiated state. In a subthreshold scenario, however, the depolarization is weaker or briefer, generating only small or short-lived increases in intracellular calcium. These smaller calcium signals frequently engage phosphatase-dominated pathways—particularly calcineurin (PP2B)—and can also recruit internalization machinery (such as β-arrestin-mediated endocytosis for glutamate receptors). NMDA receptor subunits that remain only partially activated, instead of being fully engaged in a high-calcium “LTP-like” event, end up dephosphorylated, which discourages their synaptic incorporation or stabilizing scaffolds. AMPA receptors also become more susceptible to endocytosis. We propose that repeated occurrences of this partial or poorly synchronized excitatory input—each time the synergy misfires—push the neuron’s glutamatergic synapses toward LTD. The cell’s logic, so to speak, is that “I keep receiving excitatory drive at times that do not align with a meaningful reward or successful action, so I will reduce my sensitivity to these ineffective inputs.” Each such “error repetition” increases the likelihood that these STN/PPN-SNc synapses shed receptor content and reduce synaptic weights, diminishing their ability to deliver enough depolarization for subsequent bursts.
Because the synergy continues to produce the same partially coherent or off-timed excitation during each failed attempt, the dopaminergic neuron is continually subject to subthreshold calcium transients and partial receptor activation, consolidating the LTD-like mechanism. NMDA receptor subunits might be dephosphorylated at certain key residues (like on the NR2B subunit), AMPA receptor subunits (like GluA1) can be internalized, and local scaffolding proteins (e.g., PSD-95) may also be downregulated or displaced. As a result, the neuron’s capacity to generate a strong synchronized burst in subsequent attempts is lowered because the synapses that would normally build the depolarization are now weaker. With repeated occurrences, these depressed inputs no longer arrive with the high-gain, coordinated excitatory potential that would be needed to push the dopaminergic neuron above its threshold for bursting. Consequently, the phasic release of dopamine becomes more and more blunted.
In short, the “why” is that the consistent mismatch of timing and amplitude signals the cell that these excitatory inputs are unproductive or even “erroneous,” triggering a shift toward synaptic depression rather than reinforcement. The “how” is that partial or asynchronous excitatory drive leads to calcium transients too small or too brief to initiate kinase-dependent LTP, favoring phosphatase-heavy LTD processes that dephosphorylate NMDA/AMPA receptor subunits and stimulate receptor endocytosis. Over multiple failed trials, this LTD-like plasticity becomes consolidated, so later volleys from the STN/PPN—even if they occasionally become more synchronous—will find the synapses already depressed and incapable of pushing the neuron into burst mode. We propose that this mechanism thus locks in the reduced phasic dopamine release that characterizes the dystonic state.
Second, homeostatic or adaptive changes in intrinsic membrane conductances. Dopaminergic neurons have distinctive pacemaker properties mediated by voltage-gated calcium channels (particularly Cav1.3 L-type channels), hyperpolarization-activated cyclic nucleotide-gated channels (HCN), potassium channels (SK, K-ATP, etc.), and more. Phasic bursts are usually triggered when afferent excitation converges at the right time to push the membrane into a plateau potential or high-frequency firing mode. However, if repeated sensorimotor mismatches cause negative prediction errors, the dopaminergic neuron experiences perturbations in intracellular calcium that activate a range of phosphatases and other signaling molecules (for instance, calcineurin or specific protein kinases). We propose that over multiple failed attempts, these signals can alter phosphorylation states or expression levels of channels that support bursting. For example, T-type calcium channels in distal dendrites may become less available for activation, or SK channels (which hyperpolarize the cell after bursts) may be upregulated or remain persistently open at lower thresholds. The net effect is to make the neuron more resistant to the high-voltage plateau needed for a strong phasic burst. In other words, the cell “tunes” itself to require more robust excitatory synchrony before it will fire a burst, and such synchrony seldom occurs if the synergy is consistently errant. This adaptation is akin to a negative feedback loop: the cell sees that each time it partially depolarizes, the outcome is not rewarded, so it lowers overall excitatory gain to avoid wasting metabolic resources on fruitless bursts.
Third, negative reward-prediction error signals shaping the mesocircuit. The basal ganglia have a sophisticated architecture in which outputs from the striatum, globus pallidus (GPi/GPe), and substantia nigra pars reticulata (SNr) can modulate the excitatory or inhibitory drive that arrives at SNc. In typical successful motor learning, when the movement is better than expected, striatal signals suppress certain pallidal/nigral outputs that would otherwise inhibit the burst-driving circuits. This suppression allows the STN or PPN excitatory projections to the SNc to align tightly with movement onset, generating a pulse of dopamine release. We propose that when the dystonic synergy keeps generating involuntary contractions and prediction errors, this gating reverses: GPi/SNr inhibition stays high, STN and/or PPN excitation is damped or desynchronized, and additional GABA tone from SNr collaterals to SNc rises. Repetition cements these changes: each unsuccessful attempt slightly retunes the loop so that future STN/PPN volleys are weaker or mistimed. The dopaminergic neuron, as part of this circuit, thus “learns” to withhold bursts when it senses repeated mismatch or negative outcome signals, effectively generating a state of lowered phasic release specific to that maladaptive motor pattern. Hence, the synergy no longer triggers the kind of strong excitatory gating or synchronous conduction that would produce a robust dopamine transient.
Because this plastic reshaping persists over numerous attempts, the synergy never receives large, timely pulses of dopamine. Nonetheless, the striatum can still get low-level or tonic dopamine. From the dopaminergic neurons’ standpoint, continuing to fire large bursts for a motor program that produces consistent errors is a wasteful, undesirable scenario. Through the synaptic and intrinsic changes described, we propose that these neurons reduce the probability of high-frequency firing. The result is a stable (albeit maladaptive) decrease in the amplitude and consistency of phasic dopamine release each time the dystonic synergy is attempted. This is how the dopaminergic system “learns” at a detailed, mechanistic level: repetitive negative prediction errors initiate a chain of synaptic LTD in excitatory inputs, channel phosphorylation state changes that heighten the threshold for bursting, and circuit-level modifications that feed more inhibition or reduced excitatory synchrony onto SNc neurons, effectively locking in a low-bursting regime for that specific motor context.
In addition, despite this reduction in phasic dopamine, there remains at least some residual dopaminergic tone in the striatal compartments where the maladaptive synergy projects—composed of either small or erratically timed pulses and a tonic baseline that never disappears entirely. The cortical drive itself, being abnormally high and repetitive, guarantees that the direct-pathway medium spiny neurons (those expressing D1 receptors) are strongly depolarized and often in an up-state when dopamine arrives, even if that dopaminergic signal is relatively weak or late. D1 receptors couple to Gs/olf proteins, causing an upregulation of cAMP and subsequent activation of protein kinase A (PKA) whenever they bind dopamine during neuronal depolarization. Because the synergy is initiated over and over again, these partial dopamine-plus-glutamate coincidences happen frequently enough to trigger incremental but persistent synaptic and transcriptional changes. PKA phosphorylates key effectors such as DARPP-32 and NMDA receptor subunits, which stabilizes excitatory inputs on D1-expressing cells. Moreover, we propose that repeated partial activation can engage immediate early genes such as c-Fos and ΔFosB that induce long-lasting epigenetic modifications, gradually increasing the number of D1 receptors on the membrane or boosting their downstream signaling potency. Essentially, the repeated large glutamate pulses ensure that D1 neurons remain above threshold for potentiation each time even a trickle of dopamine arrives, so they can accumulate enough intracellular signals to achieve a net strengthening (rather than a net weakening) of the direct pathway. Over many iterations, this process manifests as D1 receptor upregulation, despite the overall depressed phasic dopamine environment. The crux is that D1-MSNs do not require perfectly timed or robust bursts of dopamine so long as the cortical drive consistently pushes them into a depolarized state and there is still some dopamine available to bind D1 receptors repeatedly. Once these neurons begin upregulating D1 receptor density, each subsequent partial dopaminergic pulse has an even greater impact on cAMP and PKA cascades, accelerating the maladaptive reinforcement of the direct pathway within the corresponding somatotopic region. The result is a self-perpetuating cycle of lowered phasic dopamine release for that task alongside a paradoxical but persistent increase in D1 receptor-mediated plasticity, which further cements the dystonic synergy’s dominance over the affected movement.
Furthermore, when we discuss D2 receptor downregulation under conditions of low phasic dopamine plus hyperexcitable cortical (glutamate) input, it helps to distinguish between simple “dopamine deficiency” at the receptor and the dynamic, synaptic plasticity mechanisms triggered by repeated mismatches in timing and amplitude of dopamine relative to excitatory drive. In a typical scenario such as partial denervation (e.g., Parkinson’s disease) where there is an outright dopamine shortfall, we often see a compensatory upregulation of D2 receptors (Blesa et al., 2017; and references therein), but that happens largely when the neurons are starved of dopamine in a relatively quiescent or normal pattern of excitatory input. In focal dystonia, we hypothesize that the difference is that the relevant medium spiny neurons (MSNs) in the indirect pathway are bombarded by powerful, disorganized bursts of glutamate that arrive without well-timed reinforcing dopamine pulses. This mismatch of strong cortical input plus erratic or minimal dopamine triggers a plasticity process that actively suppresses D2 receptor expression at the membrane rather than promoting a compensatory increase.
The principle reason behind this outcome is that iSPNs depend on coherent, instructive dopamine signals to stabilize or adapt their role in suppressing unwanted movements. When the motor cortex is hyperexcitable and repeatedly sends large glutamate volleys to iSPNs, but dopamine release is not only reduced overall but also arrives at the “wrong” times (e.g., too late, too small, or unpredictably absent), the neuron receives recurring “error-laden” activity. Instead of a constructive reinforcement of which synapses should maintain or upregulate D2 receptors, the cell repeatedly experiences partial activation of D2 receptors in a context that does not match a successful or reward-consistent movement. We reason that these partial, non-optimal stimulations can engage intracellular pathways (for instance, involving β-arrestin) that tag the receptor for internalization rather than maintain it. Over many such cycles, the net effect is a genuine downregulation of functional D2 receptor density on the cell surface, because the neuron is effectively “learning” that this pattern of input is not beneficial or is consistently associated with a negative or neutral outcome. It is not a simple homeostatic response to less dopamine—rather, it is an active plasticity process in which large, misaligned excitatory drive arrives in the absence of a coherent dopaminergic reinforcing signal, pushing the neuron toward pruning its receptors.
Mechanistically, the intracellular signaling in iSPNs that leads to downregulation of D2 can be traced to the interplay between glutamate-induced depolarization and low-level, mistimed dopamine. Under normal conditions, well-timed phasic dopamine bursts help iSPNs refine or depress unwanted excitatory inputs and preserve receptor expression in a context that supports balanced inhibition. Here, however, it is conceivable that each time the cortex fires off its excessive burst, iSPNs are hit with too much or erratic glutamate, which drives the cell toward a heightened or prolonged up-state. The minimal or off-phase dopamine then does not confer a clear “LTD” or stabilizing signal but instead can promote partial receptor phosphorylation patterns that favor internalization or degradation. This is sometimes referred to as maladaptive plasticity: it’s not merely that dopamine is low; it’s that the timing is so incompatible with the excitatory input that the iSPNs interpret this as persistent motor “error” rather than a scenario where upregulating D2 would help. As an additional feedback loop, persistent negative reward-prediction error signals propagate through basal ganglia circuits to the substantia nigra, likely further reducing phasic dopamine for that motor context, so each trial of the dystonic movement replays the mismatch. With repetition, these iSPNs systematically shift toward lower receptor availability.
Put simply, the presence of large, frequent, pathologically timed glutamate bursts changes the rules for how a neuron reacts to low dopamine levels. We propose that instead of the typical upregulation that might be seen if dopamine were absent but everything else normal, the iSPNs are forced into repeated partial stimulation under conditions that never match successful or rewarding movement. The cell’s internal signaling machinery responds by internalizing or failing to replace D2 receptors at the membrane, culminating in a real downregulation of D2 function. The “why” is because the mismatch of excitatory input and insufficient, untimely dopamine triggers repeated “negative feedback” states that drive synaptic and receptor-level plasticity away from D2 receptor maintenance. The “how” is through the molecular cascades (like β-arrestin, abnormal calcium transients, or altered phosphorylation patterns) that cause trafficking of D2 away from the membrane rather than upregulating its expression. The end result is that indirect-pathway MSNs lose their receptor population, further undermining their ability to inhibit the maladaptive motor output.
Moreover, we reason that the topological disorganization of D1- and D2-expressing zones, as well as dopaminergic release sites in dystonia, emerges from the same repeated mismatch conditions—strong, poorly timed glutamate from cortex plus reduced and erratic phasic dopamine—that simultaneously drive D1 upregulation, D2 downregulation, and depressed burst firing in SNc. Under healthy conditions, direct and indirect pathway neurons (and their attendant dopaminergic inputs) form partially overlapping fields in the striatum: a single somatotopic zone that controls, for example, finger flexion, will contain interwoven populations of D1-MSNs and D2-MSNs, both of which can be modulated by consistent dopamine bursts arriving in that region. This creates a functional overlap so that any movement—especially a finely tuned action—can be sculpted by a balance of direct-pathway facilitation and indirect-pathway inhibition, all reinforced by well-timed dopaminergic pulses.
In dystonia, prolonged “error-laden” input from the cortex causes large, repeated excitatory signals to converge upon discrete patches of striatum, but the normal, synchronized pulses of dopamine do not arrive. Instead, there is a partial or asynchronous dopaminergic trickle, which has a profoundly different plastic impact on D1- vs. D2-MSNs. Over many training episodes, D1-MSNs in that region get strongly potentiated—both at their glutamatergic synapses and by an upregulation of D1 receptor density—while D2-MSNs in the same territory become downregulated and fail to maintain or expand their receptor population. Crucially, this differential plasticity could begin to carve out sub-territories within the same somatotopic striatal zone in which only the D1-upregulated neurons remain robustly connected to the pathologically active cortical inputs. The D2-MSNs can shrink their dendritic arbors or downregulate synaptic spines, leading to a near “exclusion zone” where the indirect pathway no longer effectively participates. This separation is further enhanced by the fact that the subregions of the striatum where dopamine release typically overlaps with both direct and indirect pathway neurons also undergo reorganization: the misalignment of excitatory bursts and reduced phasic dopamine release fosters localized LTD of excitatory inputs onto dopaminergic neurons (e.g., from STN or PPN) that would otherwise service both D1- and D2-rich compartments.
As a result, we hypothesize that the dopaminergic midbrain no longer provides phasic release in a single, well-shared region of the striatum; instead, the few partial pulses that do occur become channeled or reinforced primarily to the patch of D1-dominated neurons. Because these same D1-MSNs keep receiving robust cortical drive, any dopaminergic pulses that trickle in become relevant almost exclusively to that “winning” D1 subcircuit. Meanwhile, the iSPNs in nearby areas—once part of a shared network—have lost receptor density and synaptic strength, and the net dopaminergic innervation to them is even more attenuated or out of phase. This sets off a vicious cycle of functional segregation: the direct-pathway subregion effectively “grows” in terms of receptor potency and remains tied to the minimal dopaminergic input, whereas the indirect-pathway subregion no longer participates effectively in the same movement domain.
Hence, our model predicts that the previously overlapping topography—where healthy striatal microzones contained a blend of D1- and D2-expressing neurons all subject to the same bursts of dopamine—becomes replaced by more isolated clusters. One cluster is heavily D1-based, hyperresponsive to cortical inputs, and still sees some modicum of dopaminergic effect (leading to upregulation). The other, previously overlapping indirect cluster is diminished, with D2 receptors downregulated or internalized, failing to capture the scarce dopamine that remains. In imaging terms, these appear as distinct or non-overlapping territories for D1 vs. D2 expression, alongside a minimal or missing overlap with dopamine release sites.
An important cell-biological component is that repeated maladaptive motor attempts may drive local microcircuits to reorganize spines and receptor composition in ways that physically confine D1 upregulation to certain dendritic “islands” (and the synapses that feed them). In healthy subjects, microcolumns of direct and indirect pathway neurons interdigitate, each receiving shared dopaminergic terminals. But if the D1 and D2 subpopulations respond differently to the same erroneous input—one fraction being potentiated, the other depressed—they can effectively reorganize their local arbors, losing the shared dopaminergic microenvironments that once overlapped. Meanwhile, the dopaminergic neurons themselves are plastic, reducing or redirecting their excitatory drive inputs in ways that reinforce a specialized, narrower output domain (the subcircuit that still responds). Over time, this combination of structural and synaptic changes could yeild the discrete, topologically segregated D1-dominant vs. D2-deficient zones we see in dystonia, rather than the healthy pattern of partial overlap among D1, D2, and dopaminergic release.
Therefore, we posit that the basal ganglia alterations observed by Simonyan et al.—the reduced phasic dopamine release during the affected task, the upregulation of D1 receptors in the striatum, the downregulation of D2 receptors, and the breakdown of the typical overlap between dopaminergic inputs and direct/indirect pathways—are best interpreted as secondary adaptations of the maladaptive synergy’s repetitive usage.
Cerebellum
Extending the M1-centric view, we suggest that the cerebellar abnormalities reported in FTSD represent adaptive responses to the chronic output of the dystonic synergy rather than a primary lesion within the cerebellum itself. We propose that repeated hyperexcitable signals—originating from a persistent, maladaptive dystonic synergy in the M1 and conveyed indirectly through intermediary pathways—may cause noisy disruptions that gradually reshape cerebellar function. In a healthy system, each climbing fiber (CF) burst that reaches a Purkinje cell triggers a large, transient surge of Ca2+ in the dendrites, facilitated by voltage-gated calcium channels (notably P/Q-type Ca2+ channels in Purkinje somato-dendritic membranes) and the robust depolarization caused by that uniquely powerful synapse. These Ca2+ transients drive downstream second-messenger cascades—most notably involving protein kinase C (PKC), Ca2+/calmodulin-dependent protein kinases (CaMKs), and other signaling intermediaries in the postsynaptic density—that mediate synaptic plasticity (e.g., long-term depression of parallel fiber-Purkinje synapses). Under normal circumstances, CF bursts are relatively infrequent, so they are interpreted as salient “error signals” requiring an adaptive change. However, in FTSD, we hypothesize that if CF barrages become chronically large and frequent—when the cerebellum is continuously exposed to persistent hyperexcitability drive from the dystonic synergy—then intracellular Ca2+ and its downstream signaling elements could enter a state of near-constant upregulation.
When these strong bursts are no longer rare but the new norm, multiple homeostatic and compensatory mechanisms ramp up within Purkinje cells. One prominent and likely response is the biochemical “saturation” of the plasticity machinery: high levels of Ca2+-induced signaling can drive partial inactivation or desensitization of key enzymes such as PKC and CaMKII. Such a phenomenon is inferred from other chronic stimulation paradigms in cerebellar research. Mechanistically, it can happen via diverse routes: for instance, chronic phosphorylation of certain kinase subunits can paradoxically render them less effective, while excessive Ca2+ triggers phosphatase pathways (e.g., calcineurin) that dephosphorylate crucial receptor or enzyme sites. In parallel, negative-feedback loops can alter subunit composition or anchoring of these kinases, further reducing their efficacy over time.
Simultaneously, Purkinje cells undergo homeostatic shifts in intrinsic excitability. Chronically elevated dendritic Ca2+ leads to changes in both the expression and posttranslational modification of voltage-gated Ca2+ channels (including P/Q-type channels), reducing their overall density or modifying subunits to decrease conductance. Parallel adjustments in various K+ channels—such as SK channels, which modulate afterhyperpolarizations, or large-conductance BK channels—and HCN “pacemaker” channels can limit dendritic depolarization triggered by climbing fiber input. Each step curtails the amplitude of subsequent Ca2+ transients and the postsynaptic depolarization that climbing fibers elicit. Over longer time frames, transcriptional regulation (e.g., changes in immediate-early genes) cements these adaptations, ensuring the cell’s baseline biophysical state is reset to resist further CF-induced plasticity surges. Taken together, we propose that Purkinje neurons drift toward an intrinsically “down-scaled” excitability when exposed to persistent, high-gain CF activity.
At the synaptic level, repeated large-amplitude CF events may also disrupt the crucial “coincidence detection” that underpins cerebellar learning, in line with Marr-Albus-Ito theory (Yamazaki & Lennon, 2019). Ordinarily, CF bursts arriving with precisely timed parallel fiber (PF) activity instruct Purkinje cells to strengthen or weaken the relevant PF-Purkinje synapses. This process calibrates which motor commands are flagged as errors. We posit that when CF bursts become near-constant (or so frequent that they coincide with almost all PF firing) no meaningful discrimination occurs. Rather than marking an isolated subset of PF inputs as erroneous, the CF is effectively labeling “everything” as an error, far too often. Consequently, Purkinje neurons can move to a new equilibrium where further parallel fiber-Purkinje synaptic depression (or potentiation) is minimized because their downstream molecular cascades—already saturated or chronically engaged—fail to implement further PF-Purkinje LTD or LTP with the usual specificity. Thus, Purkinje cells no longer produce strong corrective adjustments with each subsequent CF burst; the repeated strong signal is effectively downgraded to what can be considered as “background noise”. The normal CF-driven plasticity events do not initiate as robustly, depriving the system of the carefully targeted corrections that typically refine movement.
In parallel, upstream alterations in the inferior olive (IO) can occur, compounding the problem. Olivary neurons rely on electrotonic coupling (via gap junctions) and subthreshold oscillations to generate highly synchronized “teaching” bursts. Under conditions of uniformly high excitatory input—such as persistent signals from a hyperexcitable M1 synergy—the IO may reduce the degree of synchronous spiking or undergo a shift in its gap-junction conductance, effectively desynchronizing the ensemble that normally fires CF bursts together. Moreover, while direct evidence is limited in FTSD, IO cells can display homeostatic changes in their intrinsic membrane properties (for instance, regulating T-type Ca2+ channels or K+ currents), further degrading the amplitude or timing precision of the CF bursts. As a result, the reliability of the CF “teaching signal” declines. Instead of delivering sharp, well-phased error bursts at carefully timed intervals, the IO provides a duller, less coherent excitatory train. This diminishes the “salience” of each burst and would reinforce the cerebellum’s tendency to accept the elevated CF input as baseline.
Moreover, downstream of Purkinje cells, the deep cerebellar nuclei (DCN) normally integrate the inhibitory output from Purkinje neurons and project excitatory signals back toward the motor thalamus and, ultimately, M1. We speculate that, when Purkinje cells chronically fire in a manner shaped by saturated CF input—meaning they no longer deliver strongly modulated inhibitory commands—DCN neurons can undergo their own homeostatic or plastic adaptations. Over time, and as inferred from studies of cerebellar plasticity in other disorders, DCN cells may reduce the responsiveness to Purkinje inhibition or alter the expression of specific receptors and ion channels that govern how they interpret Purkinje input. For example, persistent mild hyperinhibition (or disorganized inhibitory bursts) can trigger DCN remodeling that blunts further inhibitory influence, effectively ratcheting down the net inhibitory drive traveling up the cerebello-thalamic-cortical pathway. This is the final neural link that would ordinarily “clamp” motor cortical excitability. Hence, once the DCN adapt to a background of excessive or chronically unstructured Purkinje firing, cerebello-cortical inhibition may be functionally weakened.
Altogether, these convergent processes—kinase/phosphatase saturation, channel regulation, altered transcription, local circuit feedback in Purkinje cells, gap-junction changes in the IO, and DCN homeostatic changes—lead the cerebellar circuit to “redefine” what it considers a significant deviation from expectation, though it is important to note that such mechanisms are primarily inferred from animal models and general principles of cerebellar learning. Rather than treating these high-intensity CF bursts as salient error events, the system shifts to an ongoing baseline state wherein the plastic changes that would normally curtail hyperactivity are no longer triggered. Purkinje neurons effectively reset their threshold for responding to the CF, desensitizing to further large inputs so that chronically high CF activity fades into the background.
Consequently, Brighina et al.’s (2009) observation that cerebellar stimulation fails to suppress M1 excitability in dystonia reflects the downstream consequence of this altered plasticity. Under normal conditions, Purkinje cells and DCN together would robustly dampen an overactive M1, but here they are tuned to regard that hyperexcitability as an acceptable baseline. The cerebello-cortical feedforward loop therefore weakens its inhibitory influence on the dystonic synergy. Kita et al.’s (2021) data, showing elevated cerebellar BOLD in pianists with overt dystonia, align with this scenario: despite having “accepted” a high excitatory baseline, the cerebellum remains constantly engaged, performing micro-corrections on local deviations caused by unintended antagonist co-contractions stemming from diminished reciprocal inhibition. Each unanticipated mismatch triggers climbing fiber and granule cell activation, raising the metabolic demand in the cerebellar cortex and DCN. Thus, this system is paradoxically overactive (high BOLD) while simultaneously failing to clamp M1 excitability. Instead of fully suppressing the synergy, it “chases” moment-to-moment motor errors around a chronically heightened setpoint. In healthy individuals, a sudden burst of excessive cortical drive is swiftly recognized and downregulated; in FTSD, we propose that it is processed merely as a modest deviation from an abnormally large baseline. The result is continuous partial corrections, which consume significant cerebellar resources without restoring a normal synergy. Over time, such “locking” of synergy perpetuates the FTSD condition at both the cortical and subcortical levels, with the cerebellum’s “error threshold” effectively reprogrammed to accommodate the persistent hyperexcitability from M1.
Primary Somatosensory Cortex
Cortical “smudging” in FTSD can be viewed as a secondary by-product of maladaptive plasticity within S1. As noted earlier, neocortical inhibition is divided among three molecular classes; of these, PV interneurons—already established as the engines of rapid surround inhibition—are most critical here, while the SST and 5HT3aR lineages supply slower, modulatory braking. Under normal conditions each digit occupies a partly distinct S1 territory. Rodent slice and in-vivo work show that thalamocortical EPSPs in layer-4 principal cells are followed ~1-3 ms later by disynaptic feed-forward IPSCs from PV interneurons, producing an integration window on the order of a single millisecond; this window can broaden to ~10 ms during sustained 10 Hz activity as inhibition depresses (Cruikshank et al., 2007; Gabernet et al., 2005). By extension, a rapid PV discharge in the neighboring digit column is thought to curb spill-over and dampen unwanted co-activation, thereby preserving relatively discrete zones of balanced excitation and inhibition for every finger.
However, in FTSD, repetitive co-activations of two or more digits frequently occur. This can happen when a forceful contraction of a symptomatic digit (say, digit 4) mechanically drags or co-activates its neighbor digit 3—a phenomenon often termed “enslaving” (Zatsiorsky et al., 2000); when the symptomatic finger co-contracts with an unaffected neighboring digit because surround inhibition is inadequate; or when multiple fingers are themselves symptomatic. These simultaneous signals from the co-activation of multiple digits arriving at S1 drive highly correlated presynaptic volleys, including in the horizontal connections (e.g., layer 2/3 pyramidal neuron collaterals) that span digit columns, thereby engaging precisely the STDP that produces LTP among the participating neurons. Over many thousands of repeated co-activations, STDP can begin to strengthen excitatory synapses linking the digit columns, while simultaneously undermining the timed inhibitory connections that once enforced clear demarcation between them. Additionally, we propose that the S1 “learns” that inhibiting the adjacent digit is less effective in this context and begins to synaptically downregulate these inhibitory synapses through either LTD or outright synaptic pruning. On a receptor level, repeatedly failing inhibition may involve reduced GABAA receptor function—e.g., internalization or subunit changes driven by phosphatase-like pathways (calcineurin or β-arrestin) that accelerate receptor endocytosis.
What ensues is a cycle in which strengthening of cross-column excitatory synapses (i.e., “neurons that fire together wire together”) and weakening of time-mismatched inhibition (the “lose their link” phenomenon for ineffective inhibitory firing) combine to degrade the once-robust boundary between digit 3 and digit 4 in S1. Furthermore, this abnormal reorganization is magnified by unmasking previously silent or subthreshold connections: once lateral inhibition wanes through the process described above, any anatomically extant but functionally suppressed connections between the two columns become functionally active. In other words, even before structural LTP fully consolidates the two-digit maps, the sudden “unmasking” of existing synaptic pathways can produce an apparent enlargement or overlap of cortical representation. Such LTP-like changes often involve molecular cascades (e.g., CaMKII, PKA, or CaMKIV activation) that increase AMPA receptor (e.g., GluA1) insertion at the bridging synapses. Repetitive usage cements these connections through Hebbian plasticity, culminating in significantly smudged territories for the affected digits. The well-documented phenomenon in nonhuman primates described by Allard et al. (1991)—where artificially syndactylized fingers received correlated stimulation and ended up with fused cortical territories—provides an elegant parallel to the repetitive co-activations in dystonic hands. Human data from Elbert et al. (1998) reinforce the notion that in focal dystonia (particularly in highly trained musicians), representations of adjacent digits collapse inward and show a reduced distance between peak cortical sites, consistent with losing the carefully timed feedforward inhibition. Furthermore, Yoshie et al. (2015) demonstrate how restoring temporally segregated inputs can reverse or reduce that smudging. Their case study of a pianist who practiced SDE (performing deliberate, slow, and distinct finger movements just like BATR) exemplifies how re-introducing unique, offset patterns of digit activation likely gives interneurons the correct time window to reassert effective inhibition—thus reversing the overlap in the receptive fields and improving two-point discrimination thresholds.
In sum, the process can be conceived of at three levels: (1) at the cellular level, STDP governs synaptic strength: excitatory synapses bridging two digit columns become potent when repeatedly coactivated, while inhibitory synapses fail to curtail excitatory drive (through GABAA receptor downregulation or synaptic pruning) and thus undergo LTD; (2) at the local circuit level, the interneuron timing that was once finely tuned to deliver hyperpolarization a few milliseconds after one digit’s firing is lost, because both digit columns fire too synchronously; the inhibitory bursts arrive in phase with, or even behind, the strong depolarizing events, yielding minimal net suppression. Basket cells (a type of PV interneuron) or chandelier cells may no longer provide a well-timed clamp on excitatory layer 2/3 microcircuits that bridge adjacent digits; and (3) at the larger network level, the representational boundary in S1 dissolves, and previously discrete digit zones fuse into a single smudged territory. This breakdown of surround inhibition plus the unmasking of latent excitatory pathways means the cortex now treats the two digits as a partially unified entity, producing the enlarged or merged receptive fields characteristic of FTSD. Critically, once these maladaptive patterns become entrenched, triggering co-activation of digits reinforces them further. However, as Yoshie et al. (2015) highlighted with SDE, providing each finger with distinct, well-timed sensory input can shift excitatory correlations out of phase and re-enable inhibition at precisely the right offset. Over time, these more physiologically adaptive patterns re-empower the local interneurons that impose digit-specific gating, allowing the cortical columns to regain their separation. In some instances, neuromodulators such as acetylcholine or norepinephrine can further shape synaptic plasticity thresholds by modulating spike timing windows and receptor trafficking, potentially facilitating either more rapid strengthening or faster downregulation, depending on the context. Ultimately, this is why carefully guided retraining can restore digit independence and reverse at least some of the cortical smudging. The system that learned maladaptive timing can, given consistent uncoupled input, learn once more how to keep digit representations distinct.
Moreover, to understand the crux of how the S1 “learns” that inhibiting the adjacent digit is ineffective, we must consider when and how effectively these interneurons are firing relative to the excitatory neurons they are supposed to modulate. Recall that inhibition in the S1 depends crucially on precise timing. Surround inhibition for digit representations is most efficient when a PV interneuron discharges a few milliseconds after the “active” column fires, pre-emptively silencing the adjacent column. That small offset ensures that the excitatory drive coming from one digit’s afferents arrives in the adjacent column at a different phase, allowing the inhibitory burst to arrive in time to diminish or shunt excitatory depolarization there. However, in FTSD—although it is important to note that no single study in humans has directly recorded spike-by-spike timing of PV interneurons in S1 during FTSD (technical and ethical constraints make that impossible)—it can be inferred that inhibitory interneurons are firing “in phase” (i.e., nearly simultaneous) with local excitatory events during the co-activation of digits. For example, data from cortical reorganization studies in both nonhuman primates and human dystonic patients (e.g., Elbert et al. 1998, Yoshie et al. 2015) demonstrate that once adjacent finger zones are chronically co-activated, the representational boundary effectively collapses. A logical explanation for this is that inhibition is not operating in a well-timed “slightly delayed” manner to keep the zones separate, but rather is swamped or arrives when the excitatory wave is too large and too synchronous. This repeated mismatch fosters a scenario in which the network “learns/concludes” that a short-lived or in-phase inhibitory discharge does not reduce spiking—hence synaptic depression or pruning ensues. Essentially, in FTSD when two or more digits simultaneously produce highly correlated sensory volleys that converge onto S1 columns, because the excitatory neurons in each digit’s column are repeatedly firing together and the inhibitory interneurons for columns 3 and 4 are chronically in phase with the excitatory neurons, the well-timed hyperpolarization that actually reduces spiking in the target excitatory cells is never produced. In practice, the biological “credit-assignment” for whether an inhibitory synapse is doing its job relies on feedback signals—calcium transients, membrane potentials, second-messenger cascades (e.g., calcineurin or CaMKIV), and so forth—within both the interneuron and its postsynaptic excitatory targets. If, over thousands of co-activations, excitatory synapses linking digit columns persistently strengthen while the net depolarization of the excitatory neuron is never meaningfully lessened by the inhibitory input, that situation triggers the molecular conditions for LTD. Conceptually, one can imagine each cortical synapse “asking”: “When I fire, does it make any difference?” If the answer is repeatedly “No”—because the excitatory drive from the other column is strong, in sync, and unstoppable—then gene transcription and receptor trafficking gradually shift so that the inhibitory synapses lose synaptic strength, or entire interneuronal branches (dendrites or axon terminals) get pruned as the local circuit has no impetus to preserve an inhibitory connection that never achieves the desired effect (i.e., a measurable drop in the excitatory neuron’s activity). Meanwhile, excitatory synapses bridging the two columns can become positively reinforced through additional AMPA receptor insertion—a classic Hebbian (“neurons that fire together wire together”) rule. Once the bridging inhibitory connections have weakened, the boundary between the two digit representations dissolves, and the incoming afferent signals begin to appear “shared” by both columns—manifesting as enlarged, overlapping receptive fields (i.e., cortical smudging).
Spine
A plausible and detailed mechanistic explanation for how reduced spinal reciprocal inhibition arises as a secondary byproduct in FTSD can be constructed by integrating the concept of the dystonic synergy in M1 once again with well-established principles of Hebbian plasticity in descending motor pathways and spinal interneuron circuits. For example, Walker and Detloff (2021) provide evidence of Hebbian adaptations in spinal networks after injury. In FTSD, a specialized cortical synergy—originally tuned for skilled movement—becomes dominated by excitatory drive and has an insufficient counterbalance from local inhibitory interneurons. Although these cortical phenomena typically receive the most emphasis when explaining task-specific dystonic symptoms, converging evidence from in vivo human studies suggests that spinal circuits, too, adapt over time to the abnormal cortical output, ultimately manifesting as impaired reciprocal inhibition (see Chen et al., 1997; Espay et al., 2006, and references therein).
In clinical terms, the dystonic synergy looks like a single dominant unidirectional movement—often flexion, extension, or twisting—because that subset of cortical excitatory drive encodes that movement and predominates. Nonetheless, the critical detail is that this hyperexcitable ensemble may not be truly unidirectional in an absolute sense: since the antagonist’s representation shares the same finger map in M1 and is not a separate digit zone like adjacent fingers, but rather an opposing subregion of the same digit’s cortical columns (Arbuckle et al., 2020), because surround inhibition is weakened (e.g., Beck & Hallett, 2011; Sohn & Hallett, 2004), a portion of that runaway excitation of the dystonic synergy likely overflows onto the antagonist representation. As a result, whenever the individual attempts a skilled task (e.g., piano playing), there is excessive co-activation of agonist and antagonist muscle representations. Over repeated practice in real life—where the dystonic synergy is activated for hundreds or thousands of attempts at writing or playing an instrument—this aberrant co-contraction signal consistently descends to spinal cord levels.
A useful way to envision the “learning” or adaptation that leads to reduced spinal reciprocal inhibition in FTSD is to track, in detail, what happens at the level of the spinal inhibitory interneuron and its associated circuits when the cortex persistently sends overlapping agonist-antagonist signals. One critical starting point is that the Ia inhibitory interneuron (IaIN) relies on correlated activity to maintain its excitatory synapses.
In a healthy motor system, the IaIN entrusted with reciprocal inhibition is precisely tuned to the idea that whenever the agonist is active (e.g., finger flexors firing strongly), the antagonist (e.g., finger extensors) should be receiving minimal descending excitation. That normal “asymmetry” in descending drive ensures two important conditions. First, the cortical signals and the Ia afferents from the “active” agonist muscle typically reach the inhibitory interneuron in a temporally correlated burst. As a result, the interneuron experiences a robust, clearly timed volley—like a clean on/off switch—and can reliably inhibit the antagonist motoneuron pool. Second, because the antagonist receives minimal or out-of-phase cortical drive, the inhibitory interneuron easily hyperpolarizes those motoneurons, ensuring reciprocal inhibition is swift and obvious.
In FTSD, however, we hypothesize that the motor cortex is co-activating agonist and antagonist representations. This co-activation corrupts those two healthy conditions. First, the interneuron depends on a “pure” or strongly correlated excitatory signal from the agonist side; instead, it sees mixed or partly overlapping descending input. In other words, cortical commands meant for agonist and antagonist arrive at similar times or at slightly asynchronous intervals (disrupting, for example, PING mechanism described in Keeley et al., 2017), and the usual “clear” burst of excitatory input tied only to the agonist is diluted. It is diluted in the sense that the interneuron no longer receives a well-isolated volley that strongly correlates with the agonist alone—some excitatory signals coincide with antagonist firing, which can disrupt or desynchronize the net input pattern the interneuron experiences.
Second, and equally important, the antagonist motoneurons are also likely being driven by excitatory input from cortex. Even if the interneuron manages to fire and release inhibitory neurotransmitters onto those antagonist motoneurons, it is now “firing into” a motoneuron pool that is already partially activated (i.e., partially depolarized). Because excitatory inputs summate with inhibitory inputs in the cell’s membrane potential, a stronger-than-usual excitatory drive can either cancel out the inhibitory effect or at least render it less decisive. In a typical one-agonist-active scenario, the antagonist motoneurons are near resting levels of excitability, and a modest inhibitory push easily keeps them silent. In FTSD, we propose that the same amount of inhibitory release from the interneuron is insufficient to overcome the antagonist’s ongoing excitation, so the net effect of “quelling” antagonist activity is watered down.
When the spinal inhibitory interneuron repeatedly experiences these poorly correlated patterns—meaning that its own output fails to achieve the typical “reciprocal” result, and it receives asynchronous or weaker excitatory commands—we hypothesize that it undergoes synaptic plasticity in the direction of down-regulation. A well-recognized mechanism is LTD at excitatory synapses onto the interneuron (or short-term if timescales are faster). LTD can be triggered by patterns of moderate-frequency presynaptic activity that fail to coincide with robust postsynaptic spiking or by asynchronous activation that does not follow the standard Hebbian “fire together” rule. STDP is an example (Bi & Poo, 1998; Lu et al., 2007; Markram et al., 2012). In the context of FTSD, the interneuron might be partially active (due to some agonist signals), but the antagonist motoneuron remains simultaneously recruited by the cortex, so the net effect is that the interneuron’s success in inhibiting the antagonist is diminished. This mismatch leads to below threshold or erratic depolarizations, precisely the circumstances under which LTD-like processes are known to occur. Over many repetitions—every time the dystonic synergy is activated—it is conceivable that these subtle depressions accumulate, resulting in a net reduction in the strength of excitatory inputs from both corticospinal fibers and peripheral afferents onto the inhibitory interneuron.
In parallel, we speculate that the interneuron may also experience alterations in gene transcription and receptor expression as it remains in an unusual firing regime. For instance, certain neurotrophin or receptor pathways (e.g., BDNF-TrkB signaling) that typically support interneuron health and synaptic maintenance can be down-regulated if the interneuron’s firing pattern does not match normal reciprocal inhibition. Over time, the interneuron’s connectivity may be pruned, either through microglial mechanisms that remove underutilized or “weak” synapses or through an actual decrease in excitatory receptor density on the interneuron’s surface (Andoh & Koyama, 2021). These changes would reduce the interneuron’s responsiveness to incoming drives even further, creating a vicious cycle where each new attempt at a skilled task only reinforces the “co-activation” pattern.
Eventually, the entire circuit arrives at a new, maladaptive equilibrium. The descending corticospinal signals for “agonist contraction” no longer drive robust inhibition of the antagonist, because the interneuron’s synapses have diminished strength (both structurally and functionally). Peripheral afferent input from the agonist muscle also fails to recruit the interneuron effectively, as these synapses have undergone parallel LTD and pruning. The direct result is that reciprocal inhibition measures—like those probed by Chen et al. (1997) and Espay et al. (2006)—are significantly weakened, and antagonist motoneurons do not receive the full inhibitory barrage they do in healthy individuals. In short, the spinal inhibitory circuit has learned through Hebbian-like adaptation that co-contraction is the norm and therefore has no impetus to preserve an inhibitory connection that never achieves the desired effect, thus pruning away the synaptic capacity needed to enforce strict reciprocal inhibition. Additionally, given the observation that LICI is commonly observed to be highly heterogeneous in FTSD, as proposed earlier, we hypothesize that the SST interneurons mediating slower GABAB inhibition could be intact and unaffected in FTSD. In turn, the shortened cortical silent period (CSP) in FTSD could be plausibly explained by the compromised spinal inhibitory mechanisms (e.g., reduced spinal reciprocal inhibition), rather than by dysfunction of the M1 SST-mediated LICI.
Over time, this plasticity at the spinal level becomes a self-sustaining phenomenon, compounding the cortical dysfunction. When one attempts the repetitive skilled movement, the dystonic synergy in M1 continues to send these overlapping signals, the spinal interneuron circuitry fails to robustly silence the antagonist, and the clinical hallmark of co-contraction and involuntary dystonic posturing emerges. It is precisely this cycle—where cortical disinhibition drives repeated abnormal signals, and the spinal inhibitory circuits respond by chronically down-regulating unused or incorrectly timed pathways—that could explain the secondary development of deficient reciprocal inhibition in FTSD.
Proposed Taxonomy, Genetic Framework, and Motor Retraining Implications
A major impetus for this taxonomy stems from the realization that “dystonia” is best understood as an umbrella term that brings together multiple neurophysiological disorders whose superficial clinical similarity (involuntary twisting or abnormal posturing) conceals fundamental mechanistic heterogeneity (Balint et al., 2018; Stephen et al., 2023). While the term “dystonia” can be clinically useful as a clinical descriptive label, lumping all forms of dystonia together—despite often markedly different etiologies—risks obscuring important distinctions and thus can derail research, therapeutic strategies, and even core conceptual progress. The classification system we propose endeavors to disentangle these varied conditions by recognizing that some dystonias are chiefly driven by maladaptive cortical plasticity (the “typical” group), others arise more directly from an intrinsic genetic or metabolic disruption with lesser dependence on environment (“non-neuroplastic” group), and still others (“atypical neuroplastic”) exhibit some hallmark features of plastic reorganization but also have a relatively direct genetic impetus. Our use of typical, atypical, and non-neuroplastic is admittedly simplified, but it highlights their distinct pathophysiological routes while acknowledging some interplay with compensatory neuroplastic processes in each category.
Figure 2.
Taxonomy description.
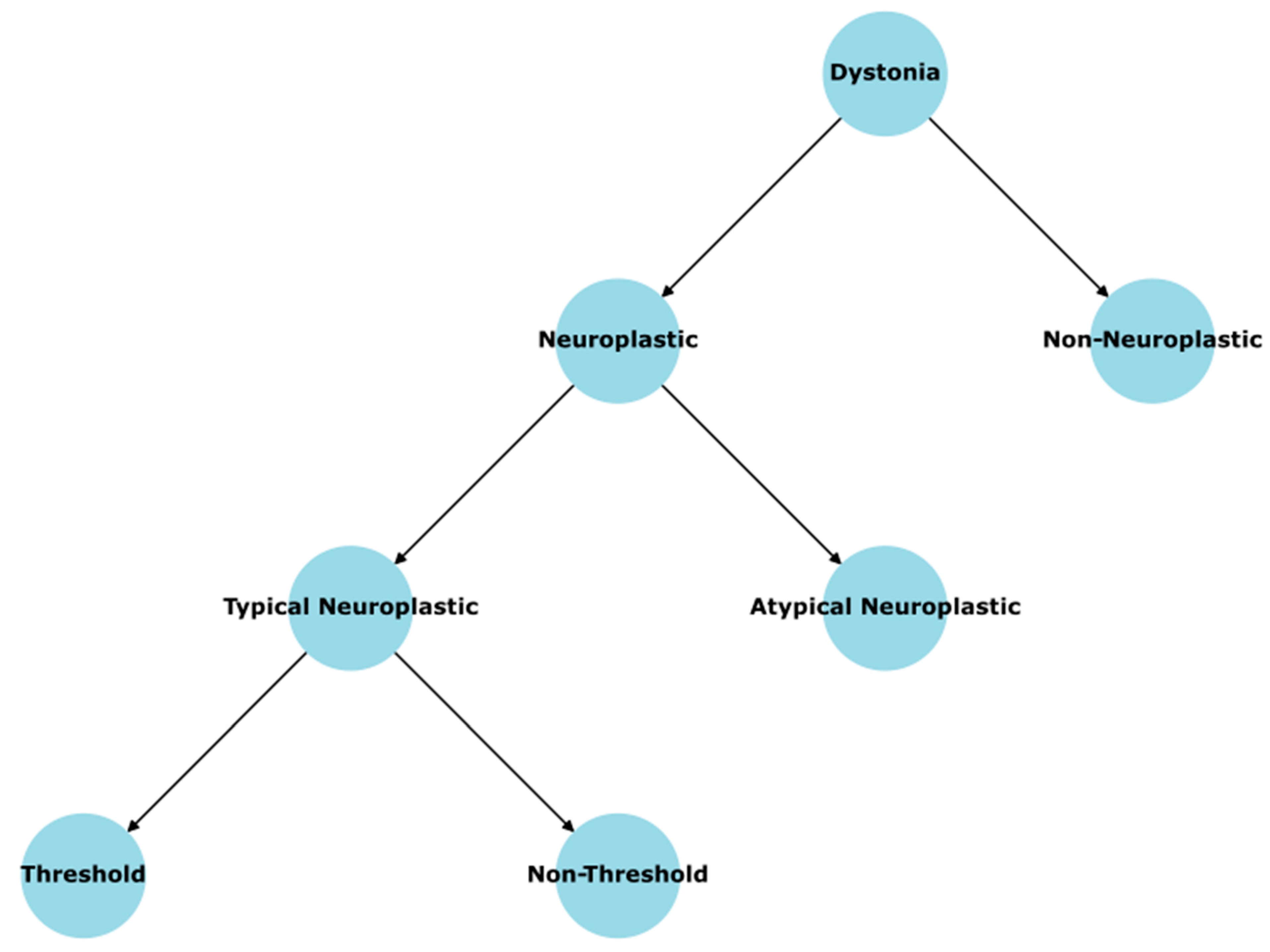
Under this taxonomy, typical dystonias broadly include both threshold (commonly task-specific, such as spasmodic dysphonia and musician’s dystonia because they all have the observed threshold phenomenon) and non-threshold forms (e.g., certain segmental or focal dystonias such as oromandibular dystonia, blepharospasm, and cervical dystonia) that share hallmark TMS findings, particularly reduced SICI (Ginatempo et al., 2023; Kaňovský et al., 2003; Sommer et al., 2002). Numerous electrophysiological studies confirm that in these so-called typical dystonias, the chief pathology involves hyperexcitable cortical circuits and diminished capacity for rapid GABAA-mediated inhibition in M1 (e.g., Furuya et al., 2018; Huang et al., 2010; McDonnell et al., 2007; Ridding et al., 1995; Siebner et al., 1999; Stinear & Byblow, 2004). Critically, these forms display maladaptive cortical plasticity as a central driver of pathogenesis: indeed, when they are task-specific, sustained overreaching in a particular synergy whose excitatory and inhibitory synapses have not yet reached their maximal (occluded) strength triggers abnormal Hebbian loops that yield a dystonic synergy with unbalanced E/I circuits. However, even in non-threshold, typical dystonias, numerous lines of evidence (including the same TMS hallmark deficits; McCambridge & Bradnam, 2021) suggest that these conditions may also arise from maladaptive learning within the motor system, potentially via the same synergy-overreaching framework we propose, thereby locking local M1 circuits into a pathological E/I imbalance. Genes associated with these typical dystonias—like certain DYT1 mutations—act more as predispositions that tilt the neural substrate toward excitatory hyperexcitability. They are rarely fully deterministic. This is exemplified by the phenomenon of “non-manifesting carriers” (e.g., in DYT1) who have partially abnormal cortical inhibition on TMS (reduced SICI) but are asymptomatic until or unless certain environmental or internal stressors amplify the E/I imbalance (Edwards et al., 2003).
By contrast, we distinguish non-neuroplastic dystonias, which tend to have an intrinsic biochemical or degenerative cause, often through single-gene or metabolic mutations that strongly disrupt normal motor pathways from birth. Prototype examples might include dopa-responsive dystonia, various paroxysmal dystonias, or X-linked dystonia-parkinsonism (Gardiner et al., 2015; Ichinose et al., 1999; Reyes et al., 2022; Weissbach et al., 2022). Although these may still display incomplete penetrance, the essential underlying pathology is more direct—the gene exerts a robust effect on, say, dopamine synthesis or on basal ganglia architecture. While incomplete penetrance in these “non-neuroplastic” dystonias could be potentially explained by the idea of neuroplastic compensatory mechanisms like in Parkinson’s disease (Blesa et al., 2017; Petrucci & Valente, 2013), we propose that these conditions are not typically triggered by overreaching or environment-driven learning loops in M1 the same way typical dystonias are. Their baseline pathology is more “deterministic,” in that the mutation itself is fairly severe or functionally disruptive, with individuals’ variable compensatory capacity dictating whether and when it becomes clinically evident.
Finally, atypical neuroplastic dystonias occupy an intermediate or "hybrid" position: these conditions have robust genetic foundations, often involving well-defined monogenic mutations, yet they also display evidence of aberrant plastic remodeling. This is hypothesized from the systematic, stereotyped, and predictable nature of their symptoms, which contrasts with the more sporadic symptom patterns observed in non-neuroplastic dystonia. In such conditions, we hypothesize that the gene triggers a significant structural or molecular defect that the developing brain could partially mask via robust neuroplastic compensation up to a point, thereby explaining why the clinical symptoms are not immediately present at birth. Environmental or maturational factors (e.g., infection, intense stress, mild injuries) could unmask or disturb these compensatory mechanisms, unmasking the underlying genetic defect. Rapid-onset dystonia parkinsonism can exemplify this scenario, wherein patients harbor a well-defined ATP1A3 mutation that likely drives the hyperexcitability, but only after a breakdown of the brain’s typical adaptive responses to the genetic insult after an environmental trigger (de Carvalho Aguiar et al., 2004; Whitlow et al., 2023).
Genetically, then, we propose that the typical forms display genes acting as predispositions, the atypical neuroplastic group displays genes acting more strongly or deterministically but still requiring plastic reorganization to express fully, and the non-neuroplastic group emerges from gene-based or metabolic lesions so profound that they do not need extrinsic maladaptive motor learning to manifest—though they still can be partially suppressed by neuroplastic compensation. This accounts for the wide variation in penetrance across dystonias that seem “mild” in basic science terms (like certain DYT1 carriers who remain healthy) versus severely disruptive degenerative or metabolic forms.
Importantly, this taxonomy informs our therapeutic perspective, particularly in terms of motor retraining. In threshold typical dystonias—paradigmatically the task-specific subtypes—the outlined conceptual model emphasizes how repetitive overreaching fosters a dystonic synergy with excess excitatory capacity and insufficient inhibitory capacity. Because hallmark TMS data indicate the same cortical hyperexcitability and disinhibition in non-threshold typical dystonias (e.g., blepharospasm, cervical dystonia), it is plausible they could share the same primary mechanism of an imbalance of synaptic strengths at the synergy level, even if the “trigger” is not so observable at a discrete intensity threshold. Some form of targeted motor retraining could be adapted here, aiming to fix the hyperexcitable synergy systematically by doing motor exercises in ways that restore adequate inhibitory circuit synaptic strength. The essence is to push the system into repeated, consistent practice of more normal E/I activation patterns so that the gating deficits progressively normalize. For the atypical neuroplastic dystonias, while these forms do not rely as exclusively on repeated skill-based overuse, the underlying circuit disruptions may still revolve around disinhibition or maladaptive plastic changes once the defective gene product disturbs basal ganglia-cortical loops. If the pathology remains partially plastic—i.e., if incomplete penetrance or partial symptom onset suggests that compensatory circuits can keep the dystonia at bay—one can speculate that targeted motor retraining could potentially re-induce those protective compensations. In effect, the therapy tries to “rebuild” or up-regulate the endogenous compensatory mechanisms that were masking the gene’s effects. Lastly, for non-neuroplastic dystonias, the efficacy of solely motor retraining may be limited, as the core pathology—such as a significant metabolic disruption or a degenerative lesion—remains unresolved. These patients typically benefit more from direct biochemical corrections (e.g., L-DOPA in dopa-responsive dystonia) or neurosurgical interventions. Our taxonomy clarifies why motor training alone will rarely fully correct a fundamental dopamine synthetic defect or a catastrophic degenerative pathology, whereas in typical dystonias heavily reliant on plastic maladaptation, such training can in principle reverse the maladaptive synergy.
Laying out this taxonomy—typical, atypical neuroplastic, and non-neuroplastic dystonias—serves not to rigidly pigeonhole disorders, but rather to highlight the dominant pathophysiological driver: maladaptive E/I circuit remodeling (typical), or strongly deterministic gene/metabolic lesions (non-neuroplastic), or some blend of the two (atypical neuroplastic). By extension, it clarifies which forms are most amenable to specialized motor retraining. Overall, this taxonomy aims to refine how we think about “dystonia” as a broad label, preventing the conflation of mechanistically distinct disorders, and paving the way for more effective, mechanism-based therapeutic interventions that reflect each subtype’s underlying physiology rather than relying on a one-size-fits-all approach.
In conclusion, the framework proposed in this paper reinterprets FTSD as a fundamentally cortical disorder in which a specialized synergy in M1 becomes locked in a maladaptive E/I imbalance. Contrary to the longstanding assumption that these dystonias must originate in subcortical structures, we have demonstrated how the hallmark features of FTSD—its task-bound specificity, its abrupt onset when speed or force thresholds are surpassed, its characteristic reduction of SICI and surround inhibition—all follow logically once the synergy’s PV inhibitory networks are under strengthened relative to excitatory synapses. From this perspective, the well-documented alterations in basal ganglia or cerebellum are no longer considered the primary drivers, but rather emerge as secondary manifestations, arising after a prolonged period of heightened and unregulated excitatory output from the dystonic synergy. Indeed, the infiltration of overflow activity into antagonist muscles, the smudging of sensory representations in S1, and the downregulation of reciprocal inhibition in the spinal cord can all be viewed as consequences of repeated aberrant cortical signals rather than the instigating pathology.
Observations from the case reported here—where a decade of stable piano technique gave way to an overreaching of the synergy’s capacity, culminating in a rapid shift from true weakness to an overt dystonic pattern—offer a clear illustration of the proposed mechanism. Each time the performer demanded an intensity beyond the synergy’s current level of E/I balance, excitatory synapses incrementally gained strength while the corresponding inhibitory circuit failed to keep pace. Eventually, the result was a dystonic synergy that hijacked normal movement whenever intensity requirements exceeded a particular threshold, leading to involuntary movements and the classic intensity threshold phenomenon of FTSD. Yet the same observations also revealed that BATR, a method that ensures well-timed co-activation of pyramidal cells and inhibitory interneurons without triggering the dystonic synergy, systematically re-strengthened the inhibitory loop over several months—confirming that the disorder, despite its disabling presentation, can indeed be reversed through neuroplastic means. Importantly, this result exemplifies how repeated practice in a “clean” regime, one that never activates the dystonic synergy, can re-install short-latency inhibition by enabling robust Hebbian potentiation at E→I and I→E synapses.
Beyond clinical descriptions, the synergy imbalance model bridges decades of electrophysiological findings. Reduced CSPs, heterogeneous LICI, and diminished SICI all follow naturally when the PV subset of interneurons is specifically under potentiated in a TSMS. Subcortical changes, such as cerebellar hyperactivity or basal ganglia rewiring, no longer appear random or etiologically primary; rather, they fit a pattern of adaptation and partial compensation in the face of chronically excessive cortical drive. Similarly, the phenomenon of cortical smudging in S1 is recast as a direct outgrowth of repetitive co-activation among digits. By emphasizing that these extra-M1 findings likely emerge only after persistent aberrant cortical output, the synergy imbalance model resolves the fundamental mystery of how dystonia remains so anatomically localized to a single skill (and even a specific finger) for years.
References
- Abbruzzese, G., Marchese, R., Buccolieri, A., Gasparetto, B., & Trompetto, C. (2001). Abnormalities of sensorimotor integration in focal dystonia: a transcranial magnetic stimulation study. Brain: a journal of neurology, 124(3), 537–545.
- Allard, T., Clark, S. A., Jenkins, W. M., & Merzenich, M. M. (1991). Reorganization of somatosensory area 3b representations in adult owl monkeys after digital syndactyly. Journal of neurophysiology, 66(3), 1048-1058. [CrossRef]
- Andoh, M., & Koyama, R. (2021). Microglia regulate synaptic development and plasticity. Developmental Neurobiology, 81(5), 568-590. [CrossRef]
- Arbuckle, S. A. , Weiler, J., Kirk, E. A., Rice, C. L., Schieber, M., Pruszynski, J. A., Ejaz, N., & Diedrichsen, J. (2020). Structure of Population Activity in Primary Motor Cortex for Single Finger Flexion and Extension. The Journal of neuroscience: the official journal of the Society for Neuroscience, 40(48), 9210–9223. [CrossRef]
- Balint, B., Mencacci, N. E., Valente, E. M., Pisani, A., Rothwell, J., Jankovic, J., ... & Bhatia, K. P. (2018). Dystonia. Nature reviews Disease primers, 4(1), 25.
- Beck, S., & Hallett, M. (2011). Surround inhibition in the motor system. Experimental brain research, 210(2), 165–172. [CrossRef]
- Beck, S. , Schubert, M., Richardson, S. P., & Hallett, M. (2009). Surround inhibition depends on the force exerted and is abnormal in focal hand dystonia. Journal of applied physiology (Bethesda, Md.: 1985), 107(5), 1513–1518. [CrossRef]
- Berardelli, A., Rothwell, J. C., Hallett, M., Thompson, P. D., Manfredi, M., & Marsden, C. D. (1998). The pathophysiology of primary dystonia. Brain: a journal of neurology, 121(7), 1195-1212. [CrossRef]
- Bi, G. Q. , & Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. The Journal of neuroscience: the official journal of the Society for Neuroscience, 18(24), 10464–10472. [CrossRef]
- Bin Ibrahim, M. Z. , Wang, Z., & Sajikumar, S. (2024). Synapses tagged, memories kept: synaptic tagging and capture hypothesis in brain health and disease. Philosophical transactions of the Royal Society of London. Series B, Biological sciences, 379(1906), 20230237. [CrossRef]
- Blesa, J. , Trigo-Damas, I., Dileone, M., Del Rey, N. L. G., Hernandez, L. F., & Obeso, J. A. (2017). Compensatory mechanisms in Parkinson’s disease: circuits adaptations and role in disease modification. Experimental neurology, 298, 148-161. [CrossRef]
- Brighina, F. I. L. I. P. P. O., Romano, M., Giglia, G., Saia, V., Puma, A., Giglia, F., & Fierro, B. (2009). Effects of cerebellar TMS on motor cortex of patients with focal dystonia: a preliminary report. Experimental brain research, 192, 651-656. [CrossRef]
- Cantarero, G. , Tang, B., O’Malley, R., Salas, R., & Celnik, P. (2013). Motor learning interference is proportional to occlusion of LTP-like plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience, 33(11), 4634–4641.
- Caux-Dedeystère, A., Allart, E., Morel, P., Kreisler, A., Derambure, P., & Devanne, H. (2021). Late cortical disinhibition in focal hand dystonia. European Journal of Neuroscience, 54(2), 4712-4720. [CrossRef]
- Chen, R., Wassermann, E. M., Caños, M., & Hallett, M. (1997). Impaired inhibition in writer’s cramp during voluntary muscle activation. Neurology, 49(4), 1054-1059. [CrossRef]
- Cruikshank, S. J., Lewis, T. J., & Connors, B. W. (2007). Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nature neuroscience, 10(4), 462–468. [CrossRef]
- Dayan, P., & Abbott, L. F. (2001). Theoretical neuroscience: Computational and mathematical modeling of neural systems. MIT Press.
- de Carvalho Aguiar, P. , Sweadner, K. J., Penniston, J. T., Zaremba, J., Liu, L., Caton, M., Linazasoro, G., Borg, M., Tijssen, M. A., Bressman, S. B., Dobyns, W. B., Brashear, A., & Ozelius, L. J. (2004). Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron, 43(2), 169–175.
- Di Lazzaro, V., Oliviero, A., Saturno, E., Pilato, F., Insola, A., Mazzone, P., … & Rothwell, J. C. (2001). The effect on corticospinal volleys of reversing the direction of current induced in the motor cortex by transcranial magnetic stimulation. Experimental brain research, 138, 268-273. [CrossRef]
- Di Lazzaro, V., Pilato, F., Dileone, M., Ranieri, F., Ricci, V., Profice, P., Bria, P., Tonali, P. A., & Ziemann, U. (2006). GABAA receptor subtype specific enhancement of inhibition in human motor cortex. The Journal of physiology, 575(Pt 3), 721–726. [CrossRef]
- Economo, M. N. , Komiyama, T., Kubota, Y., & Schiller, J. (2024). Learning and Control in Motor Cortex across Cell Types and Scales. The Journal of neuroscience: the official journal of the Society for Neuroscience, 44(40), e1233242024.
- Edwards, M. J. , Huang, Y. Z., Wood, N. W., Rothwell, J. C., & Bhatia, K. P. (2003). Different patterns of electrophysiological deficits in manifesting and non-manifesting carriers of the DYT1 gene mutation. Brain: a journal of neurology, 126(Pt 9), 2074–2080. [CrossRef]
- Elbert, T., Candia, V., Altenmüller, E., Rau, H., Sterr, A., Rockstroh, B., ... & Taub, E. (1998). Alteration of digital representations in somatosensory cortex in focal hand dystonia. Neuroreport, 9(16), 3571-3575. [CrossRef]
- Espay, A. J. , Morgante, F., Purzner, J., Gunraj, C. A., Lang, A. E., & Chen, R. (2006). Cortical and spinal abnormalities in psychogenic dystonia. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society, 59(5), 825-834.
- Frey, U. , & Morris, R. G. (1997). Synaptic tagging and long-term potentiation. Nature, 385(6616), 533–536.
- Frucht S., J. (2014). Focal Task-specific Dystonia-From Early Descriptions to a New, Modern Formulation. Tremor and other hyperkinetic movements (New York, N.Y.), 4, 230.
- Furuya, S. , Uehara, K., Sakamoto, T., & Hanakawa, T. (2018). Aberrant cortical excitability reflects the loss of hand dexterity in musician’s dystonia. The Journal of physiology, 596(12), 2397-2411. [CrossRef]
- Gabernet, L. , Jadhav, S. P., Feldman, D. E., Carandini, M., & Scanziani, M. (2005). Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron, 48(2), 315–327. [CrossRef]
- Gardiner, A. R. , Jaffer, F., Dale, R. C., Labrum, R., Erro, R., Meyer, E., Xiromerisiou, G., Stamelou, M., Walker, M., Kullmann, D., Warner, T., Jarman, P., Hanna, M., Kurian, M. A., Bhatia, K. P., & Houlden, H. (2015). The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain: a journal of neurology, 138(Pt 12), 3567–3580. [CrossRef]
- Ginatempo, F., Manzo, N., Loi, N., Belvisi, D., Cutrona, C., Conte, A., Berardelli, A., & Deriu, F. (2023). Abnormalities in the face primary motor cortex in oromandibular dystonia. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology, 151, 151–160.
- Govindarajan, A. , Israely, I., Huang, S. Y., & Tonegawa, S. (2011). The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron, 69(1), 132–146. [CrossRef]
- Grigoriu, A. I., Dinomais, M., Rémy-Néris, O., & Brochard, S. (2015). Impact of injection-guiding techniques on the effectiveness of botulinum toxin for the treatment of focal spasticity and dystonia: a systematic review. Archives of physical medicine and rehabilitation, 96(11), 2067-2078. [CrossRef]
- Grossman, R. G., & Kelly, P. J. (1976). Physiology of the basal ganglia in relation to dystonia. Advances in neurology, 14, 49-57.
- Hallett, M. (2011). Neurophysiology of dystonia: the role of inhibition. Neurobiology of disease, 42(2), 177-184. [CrossRef]
- Hanajima, R. , Okabe, S., Terao, Y., Furubayashi, T., Arai, N., Inomata-Terada, S., Hamada, M., Yugeta, A., & Ugawa, Y. (2008). Difference in intracortical inhibition of the motor cortex between cortical myoclonus and focal hand dystonia. Clinical neurophysiology: official journal of the International Federation of Clinical Neurophysiology, 119(6), 1400–1407.
- Hanajima, R. , Ugawa, Y., Terao, Y., Sakai, K., Furubayashi, T., Machii, K., & Kanazawa, I. (1998). Paired-pulse magnetic stimulation of the human motor cortex: differences among I waves. The Journal of physiology, 509(2), 607-618.
- Huang, Y. Z. , Rothwell, J. C., Lu, C. S., Wang, J., & Chen, R. S. (2010). Restoration of motor inhibition through an abnormal premotor-motor connection in dystonia. Movement disorders: official journal of the Movement Disorder Society, 25(6), 696–703. [CrossRef]
- Ibanez, V. , Sadato, N., Karp, B., Deiber, M. P., & Hallett, M. (1999). Deficient activation of the motor cortical network in patients with writer’s cramp. Neurology, 53(1), 96-96. [CrossRef]
- Ichinose, H. , Suzuki, T., Inagaki, H., Ohye, T., & Nagatsu, T. (1999). Molecular genetics of dopa-responsive dystonia. [CrossRef]
- Jiang, X., Shen, S., Cadwell, C. R., Berens, P., Sinz, F., Ecker, A. S., Patel, S., & Tolias, A. S. (2015). Principles of connectivity among morphologically defined cell types in adult neocortex. Science (New York, N.Y.), 350(6264), aac9462. [CrossRef]
- Kandel, E. R., Koester, J. D., Mack, S. H., & Siegelbaum, S. A. (2021). Principles of neural science (6th ed.). McGraw-Hill Education.
- Kaňovský, P. , Bareš, M., Streitová, H., Klajblová, H., Daniel, P., & Rektor, I. (2003). Abnormalities of cortical excitability and cortical inhibition in cervical dystonia: evidence from somatosensory evoked potentials and paired transcranial magnetic stimulation recordings. Journal of neurology, 250, 42-50.
- Keeley, S., Fenton, A. A., & Rinzel, J. (2017). Modeling fast and slow gamma oscillations with interneurons of different subtype. Journal of neurophysiology, 117(3), 950-965. [CrossRef]
- Kita, K., Furuya, S., Osu, R., Sakamoto, T., & Hanakawa, T. (2021). Aberrant Cerebello-Cortical Connectivity in Pianists With Focal Task-Specific Dystonia. Cerebral cortex (New York, N.Y.: 1991), 31(10), 4853–4863. [CrossRef]
- Kujirai, T., Caramia, M. D., Rothwell, J. C., Day, B. L., Thompson, P. D., Ferbert, A. A., ... & Marsden, C. D. (1993). Corticocortical inhibition in human motor cortex. The Journal of physiology, 471(1), 501-519. [CrossRef]
- Levy, L. M. , & Hallett, M. (2002). Impaired brain GABA in focal dystonia. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society, 51(1), 93-101.
- Lu, J. T. , Li, C. Y., Zhao, J. P., Poo, M. M., & Zhang, X. H. (2007). Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. The Journal of neuroscience: the official journal of the Society for Neuroscience, 27(36), 9711–9720. [CrossRef]
- Markram, H. , Gerstner, W., & Sjöström, P. J. (2012). Spike-timing-dependent plasticity: a comprehensive overview. Frontiers in synaptic neuroscience, 4, 2. [CrossRef]
- Matsumura, M. , Sawaguchi, T. O. S. H. I. Y. U. K. I., Oishi, T., Ueki, K., & Kubota, K. I. S. O. U. (1991). Behavioral deficits induced by local injection of bicuculline and muscimol into the primate motor and premotor cortex. Journal of neurophysiology, 65(6), 1542-1553. [CrossRef]
- McCambridge, A. B. , & Bradnam, L. V. (2021). Cortical neurophysiology of primary isolated dystonia and non-dystonic adults: A meta-analysis. European Journal of Neuroscience, 53(4), 1300-1323. [CrossRef]
- McDonnell, M. N. , Thompson, P. D., & Ridding, M. C. (2007). The effect of cutaneous input on intracortical inhibition in focal task-specific dystonia. Movement disorders: official journal of the Movement Disorder Society, 22(9), 1286–1292. [CrossRef]
- Meunier, S. , Russmann, H., Shamim, E., Lamy, J. C., & Hallett, M. (2012). Plasticity of cortical inhibition in dystonia is impaired after motor learning and paired-associative stimulation. The European journal of neuroscience, 35(6), 975–986. [CrossRef]
- Mima, T. , Nagamine, T., Nishitani, N., Mikuni, N., Ikeda, A., Fukuyama, H.,... & Shibasaki, H. (1998). Cortical myoclonus: sensorimotor hyperexcitability. Neurology, 50(4), 933-942.
- Petrucci, S., & Valente, E. M. (2013). Genetic issues in the diagnosis of dystonias. Frontiers in neurology, 4, 34. [CrossRef]
- Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A.-S., White, L. E., Mooney, R., & Platt, M. L. (2018). Neuroscience (6th ed.). Sinauer Associates (Oxford University Press).
- Quartarone, A. , & Hallett, M. (2013). Emerging concepts in the physiological basis of dystonia. Movement disorders: official journal of the Movement Disorder Society, 28(7), 958–967.
- Quartarone, A. , Siebner, H. R., & Rothwell, J. C. (2006). Task-specific hand dystonia: can too much plasticity be bad for you?. Trends in neurosciences, 29(4), 192–199. [CrossRef]
- Redondo, R. L. , & Morris, R. G. (2011). Making memories last: the synaptic tagging and capture hypothesis. Nature reviews. Neuroscience, 12(1), 17–30. [CrossRef]
- Reyes, C. J. , Asano, K., Todd, P. K., Klein, C., & Rakovic, A. (2022). Repeat-Associated Non-AUG Translation of AGAGGG Repeats that Cause X-Linked Dystonia-Parkinsonism. Movement disorders: official journal of the Movement Disorder Society, 37(11), 2284–2289.
- Rozanski, V. E. , Rehfuess, E., Bötzel, K., & Nowak, D. (2015). Task-Specific Dystonia in Professional Musicians. A Systematic Review of the Importance of Intensive Playing as a Risk Factor. Deutsches Arzteblatt international, 112(51-52), 871–877.
- Ridding, M. C., Sheean, G., Rothwell, J. C., Inzelberg, R., & Kujirai, T. (1995). Changes in the balance between motor cortical excitation and inhibition in focal, task specific dystonia. Journal of neurology, neurosurgery, and psychiatry, 59(5), 493–498. [CrossRef]
- Rudy, B. , Fishell, G., Lee, S., & Hjerling-Leffler, J. (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Developmental neurobiology, 71(1), 45–61. [CrossRef]
- Safavynia, S., Torres-Oviedo, G., & Ting, L. (2011). Muscle synergies: implications for clinical evaluation and rehabilitation of movement. Topics in spinal cord injury rehabilitation, 17(1), 16-24. [CrossRef]
- Sajikumar, S. , Morris, R. G., & Korte, M. (2014). Competition between recently potentiated synaptic inputs reveals a winner-take-all phase of synaptic tagging and capture. Proceedings of the National Academy of Sciences of the United States of America, 111(33), 12217–12221. [CrossRef]
- Sakai N (2006) Slow-down exercise for the treatment of focal hand dystonia in pianists. Med Probl Perform Art 21: 25-28.
- Shaikh, A. G. , Wong, A. L., Optican, L. M., & Zee, D. S. (2017). Impaired motor learning in a disorder of the inferior olive: is the cerebellum confused?. The Cerebellum, 16, 158-167. [CrossRef]
- Shao, Z. , & Burkhalter, A. (1999). Role of GABAB receptor-mediated inhibition in reciprocal interareal pathways of rat visual cortex. Journal of neurophysiology, 81(3), 1014–1024. [CrossRef]
- Shinotsuka, T. , Tanaka, Y. R., Terada, S. I., Hatano, N., & Matsuzaki, M. (2023). Layer 5 Intratelencephalic Neurons in the Motor Cortex Stably Encode Skilled Movement. The Journal of neuroscience: the official journal of the Society for Neuroscience, 43(43), 7130–7148. [CrossRef]
- Siebner, H. R. , Tormos, J. M., Ceballos-Baumann, A. O., Auer, C., Catala, M. D., Conrad, B., & Pascual-Leone, A. (1999). Low-frequency repetitive transcranial magnetic stimulation of the motor cortex in writer’s cramp. Neurology, 52(3), 529–537. [CrossRef]
- Simonyan, K., Cho, H., Hamzehei Sichani, A., Rubien-Thomas, E., & Hallett, M. (2017). The direct basal ganglia pathway is hyperfunctional in focal dystonia. Brain: a journal of neurology, 140(12), 3179–3190. [CrossRef]
- Sohn, Y. H., & Hallett, M. (2004). Disturbed surround inhibition in focal hand dystonia. Annals of neurology, 56(4), 595–599. [CrossRef]
- Sommer, M. , Ruge, D., Tergau, F., Beuche, W., Altenmüller, E., & Paulus, W. (2002). Intracortical excitability in the hand motor representation in hand dystonia and blepharospasm. Movement disorders: official journal of the Movement Disorder Society, 17(5), 1017-1025. [CrossRef]
- Stahl, C. M., & Frucht, S. J. (2017). Focal task specific dystonia: a review and update. Journal of neurology, 264, 1536-1541. [CrossRef]
- Stephen, C. D. , Simonyan, K., Ozelius, L., Breakefield, X. O., & Sharma, N. (2023). Dystonia. Neurobiology of Brain Disorders, 713-751.
- Stinear, C. M. , & Byblow, W. D. (2004). Elevated threshold for intracortical inhibition in focal hand dystonia. Movement disorders: official journal of the Movement Disorder Society, 19(11), 1312–1317. [CrossRef]
- Teo, J. T. , Van De Warrenburg, B. P. C., Schneider, S. A., Rothwell, J. C., & Bhatia, K. P. (2009). Neurophysiological evidence for cerebellar dysfunction in primary focal dystonia. Journal of Neurology, Neurosurgery & Psychiatry, 80(1), 80-83. [CrossRef]
- Tian, D. , & Izumi, S. I. (2022). Transcranial Magnetic Stimulation and Neocortical Neurons: The Micro-Macro Connection. Frontiers in neuroscience, 16, 866245. [CrossRef]
- Tinazzi, M., Rosso, T., & Fiaschi. (2003). Role of the somatosensory system in primary dystonia. Movement Disorders, 18(6), 605-622. [CrossRef]
- Tseng, Y. J. , Chen, R. S., Hsu, W. Y., Hsiao, F. J., & Lin, Y. Y. (2014). Reduced motor cortex deactivation in individuals who suffer from writer’s cramp. PloS one, 9(5), e97561. [CrossRef]
- Turner, R. S. , & Desmurget, M. (2010). Basal ganglia contributions to motor control: a vigorous tutor. Current opinion in neurobiology, 20(6), 704-716. [CrossRef]
- Turrigiano, G. G. , Leslie, K. R., Desai, N. S., Rutherford, L. C., & Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature, 391(6670), 892–896. [CrossRef]
- Turrigiano G., G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell, 135(3), 422–435. [CrossRef]
- van Vugt, F. T. , Boullet, L., Jabusch, H. C., & Altenmüller, E. (2014). Musician’s dystonia in pianists: long-term evaluation of retraining and other therapies. Parkinsonism & related disorders, 20(1), 8–12. [CrossRef]
- Walker, J. R., & Detloff, M. R. (2021). Plasticity in cervical motor circuits following spinal cord injury and rehabilitation. Biology, 10(10), 976. [CrossRef]
- Weissbach, A. , Pauly, M. G., Herzog, R., Hahn, L., Halmans, S., Hamami, F.,... & Lohmann, K. (2022). Relationship of genotype, phenotype, and treatment in dopa-responsive dystonia: MDSGene review. Movement Disorders, 37(2), 237-252.
- Whitlow, C. T. , Atcheson, K. M., Snively, B. M., Cook, J. F., Kim, J., Haq, I. U., Sweadner, K. J., Ozelius, L. J., & Brashear, A. (2023). Rapid-onset dystonia-parkinsonism is associated with reduced cerebral blood flow without gray matter changes. Frontiers in neurology, 14, 1116723. [CrossRef]
- Yoshie, M., Sakai, N., Ohtsuki, T., & Kudo, K. (2015). Slow-down exercise reverses sensorimotor reorganization in focal hand dystonia: a case study of a pianist. Int J Neurorehabilitation, 2(2), 2376-0281.
- Yamazaki, T., & Lennon, W. (2019). Revisiting a theory of cerebellar cortex. Neuroscience research, 148, 1-8. [CrossRef]
- Zatsiorsky, V. M. , Li, Z. M., & Latash, M. L. (2000). Enslaving effects in multi-finger force production. Experimental brain research, 131(2), 187–195. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



