
Submitted:
28 July 2025
Posted:
28 July 2025
You are already at the latest version
Abstract
Methyl groups are either obtained from the diet (labile methyl groups) or produced endogenously (methylneogenesis) via the one-carbon (C1-) metabolism as S-adenosylmethionine (SAM). The essential nutrients, folate and choline (through betaine) are metabolically entwined to feed their methyl groups into the C1-metabolism. A choline deficient diet in rats produces a 31-40% reduction in liver folate content, 50% lower hepatic SAM levels and doubling of plasma homocysteine. Similarly, folate deficiency results in decreased total hepatic choline. Thus, sufficient intakes of both folate and choline (or betaine) contribute to safeguarding the methyl balance in the body. A significant amount of choline (as phosphatidylcholine) is produced in the liver via the SAM-dependent phosphatidylethanolamine methyltransferase. Experimental studies using a diet deficient in several methyl donors have shown that supplemental betaine was able to rescue not only plasma betaine, but also plasma folate.Fasting plasma homocysteine concentrations are mainly determined by folate intake or status, while the effect of choline or betaine on fasting plasma homocysteine is minor. This appears to contradict the finding that approximately 50% of cellular SAM is provided via the betaine homocysteine methyltransferase (BHMT) pathway that uses dietary choline (after oxidation to betaine) or betaine to convert homocysteine to methionine and then SAM. However, it has been shown that the relative contribution of choline and betaine to the cellular methylation is better reflected by measuring plasma homocysteine after a methionine load test. Choline or betaine supplementation significantly lowers post methionine load homocysteine. Whereas, folate supplementation has a minor effect on post methionine load homocysteine concentrations. This review highlights the interactions between folate and choline and the essentiality of choline as a key player in C1-metabolism. We further address some areas of interest for future work.
Keywords:
betaine
; choline
; folate
; homocysteine
; methionine-load test
; metabolism
; methyl donor
; S-adenosylmethionine
1. Introduction
Methyl groups are used by numerous methyltransferases that play a role in cellular processes such as DNA-methylation or synthesis of neurotransmitters and amino acids [1;2]. Methyl groups can be obtained either from the diet (labile methyl groups) or produced endogenously (methylneogenesis) via one-carbon (C1-) metabolism [3]. Folate, choline (or lecithin), betaine and methionine are examples of the dietary sources of labile methyl groups (Figure 1).
The possibility to derive methyl groups from multiple nutrients allows the body a buffer in safeguarding the methylation reservoir. The requirements for each of the dietary methyl donors are partly determined by the amount of other methyl donors in the diet and additionally depend on factors such as genetic polymorphisms in C1-metabolism and life stage (i.e., during pregnancy and lactation). Conservation of methyl groups in the body is necessary to optimize C1-metabolism [10], meet body requirements and excrete methylated substrates in the urine. This narrative review highlights the essentiality of folate and choline as key players in C1-metabolism and their interactions and discusses some of the translational aspects of these functional interactions.
2. Health Effects of Sufficient Folate and Choline Intakes
Studies in humans have established causal associations between insufficient folate intake and status and several disease conditions. There is a high-grade evidence that supplementation of folate beyond the usual dietary intake can reduce the risk of serious birth defects [11] or improve the course of neurodevelopmental [12] or neuropsychiatric disorders [13]. These effects may be explained by the role of folate in DNA-synthesis and stabilization, enhancing cellular methylation and lowering plasma fasting homocysteine concentrations.
Besides being a source of methyl groups, choline has several other functions in the body such building cell membranes (as phosphatidylcholine) and neurotransmission (as acetylcholine). Depletion of dietary choline in adults (50 mg/day) causes fatty liver within 3 weeks, while choline supplementation (i.e., 500 mg/day) normalized liver function [14;15]. Depletion of dietary choline during pregnancy in rodents caused fatty liver to develop towards the end of the pregnancy not only in the mother, but also in the fetus [16]. Therefore, a sufficient choline intake during all life stages, especially during pregnancy and lactation is necessary for normal function of the liver. A recent systematic review and meta-analysis of observational studies has shown that a lower maternal choline intake is associated with a higher risk of neural tube defects [17]. This association suggests that choline and folate intakes during pregnancy may have additive or even synergistic preventative effects.
Depletion-studies in animals have shown that on the one hand folate deficiency causes depletion of liver choline [18] and exaggeration of hepatic steatosis [19]. On the other hand, a diet depleted of choline (or choline and methionine) causes depletion of liver folate [7;8;20]. Therefore, the ability to switch between folate and choline as sources of methyl groups can secure cellular methylation and underscores the necessity of supply redundancy to safeguard this critically important methyl pool. The essential nutrients folate and choline are metabolically entwined to feed their methyl groups into the C1-metabolism (Figure 1). People carrying polymorphisms in genes involved in folate or choline metabolism may have higher requirements for both nutrients [21]. Isolated deficiencies of folate and choline show similar (e.g., elevated homocysteine, lowered methylation potential), but also distinct phenotypes. Based on this, it is tempting to assume that higher dietary folate and choline intakes can be translated into more effective disease-prevention on a population level.
3. Formation of Methyl Groups in C1-Metabolism
C1-metabolism ensures disposition of CO2 from different sources such as glucose metabolism, glycine decarboxylation and 10-formyltetrahydrofolate dehydrogenase into the folate pathway. Additionally, it regenerates adenine triphosphate (ATP) from adenine diphosphate (ADP) through conversion of 5-formyltetrahydrofolate (5-formyl-THF) to tetrahydrofolate (THF). C1-metabolism contributes to synthesis of glutathione from cysteine, and the de novo synthesis of methionine, adenosine, guanosine and thymidylate. Seventy percent of methionine (a protein-forming amino acid) is obtained from dietary protein degradation. The remaining 30% is formed from homocysteine in a remethylation pathway that contributes to the body methyl balance [22]. Serine, formate, glycine, dimethylglycine and sarcosine introduce C1-units into the 5,10-methylenetetrahydrofolate pool.
3.1 Homocysteine Methylation to Methionine
Homocysteine is remethylated to methionine via methionine synthase (MS) and betaine homocysteine methyltransferase (BHMT). Both enzymes are highly expressed in the liver and the kidney [23;24]. Homocysteine is converted to methionine via MS using the methyl group of 5-methyltetrahydrofolate (5-methyl-THF) in the presence of vitamin B12 (as methylcobalamin). In a subsequent step, methionine adenosyltransferase, an ATP-dependent enzyme, converts methionine into S-adenosylmethionine (SAM). SAM is a universal methyl donor for numerous methyltransferase-dependent cell reactions. S-adenosylhomocysteine (SAH) is formed after transferring a methyl-group from SAM to a methyl acceptor (Figure 1). SAH is converted to homocysteine in an irreversible reaction and homocysteine is either recycled back to methionine to further produce methyl groups or is converted to cystathionine, and finally glutathione [25].
The methyl groups of betaine are used to convert homocysteine to methionine via the BHMT pathway thus generating SAM [26]. Betaine is obtained either directly from the diet or via an irreversible mitochondrial oxidation of choline mediated by choline dehydrogenase (CHDH) (Figure 1).
Hyperhomocystinuria is an inherited disorder due to the deficiency of either MS or cystathionine beta synthase (converting homocysteine to cystathionine and thereby cysteine) resulting in accumulation of homocysteine. Intervention with betaine has been used to lower plasma homocysteine in these patients [27], where folic acid supplementation may not be sufficient and homocysteine can be mobilized via the BHMT pathway. In healthy humans, the BHMT-pathway is naturally upregulated to remove homocysteine after ingesting a methionine-rich diet, which is the basis of the methionine load test. Massive disruption of the BHMT pathway such as in Bhmt-/- mice causes elevated homocysteine that cannot be normalized by supplementing folate (i.e., through the MS pathway) [28]. In addition to hyperhomocysteinemia, Bhmt-/- mice show 48% lower liver SAM content, 21-fold higher liver betaine and histological evidence of liver steatosis compared to the Bhmt+/+ mice [4]. The majority of the knockout animals also developed liver tumors later in life [4]. Failure to export triacylglycerol from the liver of the Bhmt-/- mice can be explained by markedly decreased levels of choline derivatives such as phosphocholine, phosphatidylcholine (PtdCho), and sphingomyelin [4]. The liver synthesis of choline derivatives relies on availability of sufficient methyl groups. This might explain the role of adequate betaine and choline intake in removing the triglycerides from liver cells [15]. The knockout animal models provided quantitative estimate on the relative contribution of the BHMT in C1-metabolism, although we acknowledge the limitations of a direct extrapolation of the results to humans.
C1-metabolism is subject to feedback mechanisms and it is regulated by the relative availability of dietary methyl donors. Inadequate methyl groups due to prolonged fasting or short term limited intake of proteins, serine, folate or choline causes upregulation of the re-methylation pathways to convert more homocysteine to methionine. While under these conditions, the flow of homocysteine via the transsulfuration pathway to cystathionine is downregulated [22]. Excess SAM inhibits both the BHMT and methylenetetrahydrofolate reductase (MTHFR) enzymes in order to slow down the production of SAM.
Experimental dietary folate deficiency in humans (50 µg total folate/day for 6 weeks) leads to 115% increase of fasting plasma homocysteine compared to people on a control diet with sufficient folate [9]. The 115% increase of fasting plasma homocysteine under the folate deficient diet was associated with only 37% lower remethylation rate of homocysteine to methionine compared to the control diet [9]. Thus, the rise in fasting plasma homocysteine in folate deficiency is not explained by a corresponding decline in folate-mediated homocysteine remethylation. The question is, where could this additional homocysteine come from? The authors of this study argued that under the folate depletion model in the study, the BHMT-mediated remethylation of homocysteine to methionine was not stimulated to compensate for folate deficiency [9]. Instead it was posited, folate deficiency may cause a reduction in homocysteine flow through the MS pathway, and cause the one-carbon unit of 5,10-methylenetetrahydrofolate to be directed toward serine synthesis which may save the C1-units for important cellular reactions [9]. However, the study did not control for betaine and choline intakes. It is theoretically possible that the BHMT pathway was upregulated in the liver of folate deficient people as an alternative source of SAM. Formation of SAH from SAM after the methyl group transfer may explain the 115% increase in plasma homocysteine.
Therefore, the BHMT and MS pathways have distinct roles in C1-metabolism and folate and betaine/choline roles in this pathway are not fully exchangeable. Normal function of the BHMT pathway (including sufficient intake of choline) contributes to normal homocysteine metabolism, provision of methyl groups, lipid transport and normal liver function.
4. Determinants and Indicators of C1-Metabolism
Elevated fasting plasma homocysteine concentrations may indicate disturbed C1-metabolism due to folate deficiency [29]. Concentrations of fasting plasma homocysteine and plasma folate are routinely used to diagnose folate deficiency. The diagnostic utility of SAM, SAH, choline, betaine or any methylated substrates (such as DNA-methylation) has not been established due to inherent analytical and biological challenges.
Increasing folate intake is the most effective measure of lowering fasting plasma homocysteine. Betaine and choline can also lower plasma homocysteine, albeit to a lesser extent. A 2-fold higher choline and betaine intake (383 mg vs. 689 mg/day) was associated with approx. 1 µmol/L lower fasting plasma homocysteine (-8% versus the low intake category) in adults [30]. The association between choline and betaine intake on the one hand and fasting homocysteine on the other hand was most pronounced among women with a low folate intake [30], suggesting enhanced homocysteine remethylation via the BHMT pathway in women with low folate. Supplementing choline (2.6g per day as phosphatidylcholine for 2 weeks) [31] or betaine (1.5g, 3g and 6 g per day for 6 weeks) [32] caused a dose-response, although moderate, reduction of fasting homocysteine compared to the placebo (-1.3 µmol/L to -2.2 µmol/L or -12% to -20%). Supplementing folic acid for 6 weeks lowered fasting homocysteine to a higher extent than betaine (-18% for folic acid vs. -11% for betaine; both versus placebo) [5]. These results show that folate has a dominant homocysteine-lowering effect compared to betaine and choline.
5. The Methionine Load Test: A Functional Test of the BHMT Pathway
This is a functional test that uses the accumulation of homocysteine in the blood after the administration of a bolus load of methionine to determine the capacity of C1-metabolism by the degree of homocysteine clearance. The methionine load test can therefore detect disorders in C1-metabolism not captured by measuring fasting homocysteine. In this test, 75-100 mg methionine per kg body weight (5.6g to 7.5g methionine for a 75 kg person) is ingested after a sample for fasting plasma homocysteine is drawn and then measured after 4, 6, 8, or 12 hours post methionine load (PML) [33]. Roughly 50% of people with hyperhomocysteinemia after methionine load may have normal fasting plasma homocysteine results [34,35,36].
Van der Griend et al., defined PML-hyperhomocysteinemia as an increase in homocysteine concentration after methionine load (6 hour minus fasting concentration) of > 42.3 µmol/L (the 95th percentile of control subjects) [34]. The definition of PML hyperhomocysteinemia varies between studies. Direct comparison of the results of the test between studies is not possible due to lack of standardization with regard to the time point of measuring homocysteine after methionine dose or the need to adjust for baseline homocysteine [33]. Furthermore, the PML-homocysteine concentrations can be influenced by nutritional and genetic factors (Table 1). Neveretheless, a positive PML test can indicate where choline or betaine supplementation could be used to enhance homocysteine removal via the BHMT pathway (Table 1).
Both acute and chronic intakes of either betaine or choline cause a significant reduction of PML homocysteine response. A single dose of 1.5g, 3g or 6g betaine attenuated the increase of plasma homocysteine after methionine load (by 16%, 23%, and 35%, respectively compared to the placebo) [32]. Similarly, a single dose of 1.5g choline (from phosphatidylcholine) lowered the PML homocysteine by 15% (-4.8 µmol/L; 95% CI: -6.8, -2.8 µmol/L) compared to the placebo [31], suggesting that choline was converted to betaine and used as an immediate source of methyl groups. A 32% reduction of PML-homocysteine was also reported after 6 hours of a meal that was rich either in betaine or in choline compared to a control meal that was depleted of the two nutrients [6]. Therefore, the PML-homocysteine concentration seems to be a sensitive marker of recent intake of choline and betaine. This test may be used to define the intakes of betaine or choline at the inflection point of the homocysteine curve (i.e., when higher intake of betaine or choline does not lead to a further reduction in PML-homocysteine).
It has been shown that concentrations of plasma betaine correlate with PML-homocysteine concentrations in general [38;39]. The correlation remained significant when people received supplements containing folic acid [38] or when plasma folate concentrations were accounted for [39]. Thus, PML-homocysteine is a sensitive marker of low plasma betaine (and possibly choline). This suggestion is strengthened by results from a randomized placebo controlled trial measuring PML-homocysteine levels before and after supplementing either 400 µg x 2 per day folic acid or 3g betaine x 2 per day for a duration of 6 weeks (both interventions were tested against a placebo) (Table 2) [5]. Betaine supplementation for 6 weeks caused a strong reduction of the PML-homocysteine concentrations compared to the placebo group [mean difference (95%CI) at 6 hours = -17.7 (-31.8, -5.3) µmol/L]. Folic acid supplementation caused a non-significant change of the PML-homocysteine compared to the placebo [mean difference (95%CI) at 6 hours = 1.1 (-13.2, 15.4) µmol/L] [5] (Table 2). Similarly, supplementation of 2.6g choline per day (as phosphatidylcholine) for 2 weeks caused 29% lower PML-homocysteine concentrations compared to the placebo [31] (Table 2).
At present, the methionine load test is not widely used in clinical practice due to its tedious nature. In the absence of recognized markers of choline and betaine status and intake, the PML-homocysteine can be considered a functional marker of the BHMT pathway. This marker shows the relative contribution of betaine and choline as methyl donors. Available evidence suggests that PML-hyperhomocysteinemia is a functional marker of disturbed C1-metabolism. Combined supplementation of folate and choline or betaine can normalize C1-metabolism, although this effect is not mirrored by lowering fasting plasma homocysteine concentrations but well reflected by PML-homocysteine. Some gaps in knowledge about potential use of PML-homocysteine in diagnostic and research areas are shown in Table 3.
6. Safeguarding the Methyl Balance Through Diet or Methylneogenesis
The diet provides a wide intake range of methyl sources. People achieving the current dietary intake recommendations of methyl donors are unlikely to have impaired methylation. However, the methylation equilibrium could be disrupted under conditions of high requirement, increased loss or low intake of one or more methyl donors. De-novo formation of new methyl groups (methylneogenesis) can maintain the net amount of methyl groups in the body when the dietary intake of methyl donors is temporarily limited [50] and under prolonged fasting conditions.
The body methylation balance is influenced by renal excretion of SAM and other methylated compounds such as creatine, creatinine, N-methyl nicotinic acid, carnitine, methylated amino acids, and the terminal oxidation of methyl groups, including that of sarcosine (or N-methylglycine) [51]. The relative contribution from the loss of methyl groups in the bile and stool to body methyl balance remains unknown. In addition, biliary excretion of PtdCho was estimated to consume 5 mmol SAM per day, but it is unknown whether this PtdCho is reabsorbed.
There have been some attempts to qualify body methyl flux that reflects overall body dynamics of methyl groups in µmol.kg-1.h-1. Using stable isotope infusion technique, the daily methyl flux in healthy subjects was estimated to be between 16.7 and 23.4 mmol/L (for a 70 kg person) combining fasting and fed situations within 24 hours [50]. Approximately 40% of the daily formed homocysteine is remethylated to methionine and 54% of the methyl groups required for this remethylation was generated via the methylneogenic pathway [52]. Homocysteine remethylation using the methyl group of 5-methyl-THF has been estimated to be between 2 and 8 µmol.kg-1.h-1 [52], suggesting that homocysteine remethylation via methionine synthase constitutes a small fraction of human one-carbon metabolic flux. Considering that Bhmt knockout mice had 48% lower liver SAM compared to the controls, it can be inferred that roughly 50% of SAM is coming from the BHMT pathway [4].
Folate and choline are also key sources of one carbon units that feed again into the folate cycle [50]. Sarcosine can be synthesized from SAM-dependent methylation of glycine or from oxidation of choline to betaine that is converted to dimethylglycine and then to sarcosine. A substantial amount of one carbon units originates from glycine and serine [53]. Methionine load (such as after a protein rich meal) stimulates activities of some methyltransferases such as guanidoacetic methyltransferase [54] and glycine N-methyltransferase [51] that can lead to forming creatine and sarcosine, respectively.
7. Factors affecting methylneogenesis
C1-metabolism is influenced by the availability of one carbon moieties in the diet, sex, age, genetic polymorphisms, and life stage (e.g., pregnancy, lactation and early life). Some of these factors have been extensively investigated.
The activities of several enzymes involved in C1-metabolism differ by sex [48]. The expression of the phosphatidylethanolamine methyl transferase (PEMT) gene is responsive to estrogen [55;56]. Therefore, the reliance of PEMT on methyl groups is high in premenopausal women and highest during the third trimester of pregnancy. Whereas, PEMT activity may be lowest in postmenopausal women and men, suggesting that choline requirements could be higher in those subgroups. Moreover, people with a homozygous PEMT SNP [57;58] are more sensitive to develop symptoms of choline deficiency (e.g., fatty liver or muscle damage) than people without this SNP, especially under restricted choline and or folate intake.
The presence of homozygous variant of the common polymorphism in the MTHFR gene (C677T: TT genotype) causes lower activity of the enzyme and results in lower circulating concentrations of folate. Mthfr-/- mice have high incidence of postnatal death [59], hyperhomocysteinemia, hypomethlyation and are prone to develop fatty liver under choline deficient diet conditions [60]. Betaine supplementation to pregnant mice throughout the pregnancy and until weaning of the pups at 3 weeks of age decreased the mortality of Mthfr-/- mice from 83% to 26%, lowered plasma homocysteine and increased methionine and SAM concentrations in the liver and the brain [59], showing that the BHMT pathway can rescue at least part of severe metabolic and phenotypic consequences of MTHFR deficiency.
In humans, the MTHFR677TT genotype is associated with elevated fasting plasma homocysteine, low serum and blood folate concentrations [61;62] and attenuated response to folate supplementation [61]. Yan et al., showed that the demand for betaine to generate methionine from homocysteine is likely higher in individuals with the 677TT genotype [62]. In a 12-week study of 60 men with known genotype for MTHFRC677T, a daily 550 mg or 1100 mg choline was supplemented for 9 weeks followed by d-9 labeled choline for another 3 weeks [62]. The higher ratio of d-9-betaine to d-9-PtdCho in the 1100 mg choline group vs the 550 mg group indicated greater shunting away from the CDP-pathway and down the PEMT pathway [62]. The higher plasma betaine/CDP-choline ratio in subjects with the TT genotype is consistent with the increased demand for choline and therefore, the imperative to channel a higher proportion of choline into betaine under limited folate status.
The above discussed factors that influence methylneogenesis can significantly impact personalized intake of methyl donor nutrients in some populations or population subgroups. So far, population-intake recommendations for choline and folate have taken some of these considerations into account. For example, a higher intake of folate and choline are recommended during pregnancy and lactation [63;64]. The United States National Academy of Medicine (NAM) has upgraded choline intake recommendations for men compared to women (+ 50 mg/day higher recommendation for men) [65]. The European Food and Safety Authority have upgraded population intake recommendations of folate by 15% due to the prevalent MTHFR C677T polymorphism [63]. In contrast, polymorphisms in PEMT and BHMT genes (e.g., rs1316753 and rs1915706) were not taken into consideration while setting the intake recommendations of folate and choline, but evidence suggests that this issue deserves re-assessment.
8. Phosphatidylethanolamine methyltransferase role in methylneogenesis
The enzyme PEMT is a SAM-dependent enzyme that is responsible for synthesis of 30% of PtdCho in the liver [66;67]. PEMT utilizes three SAM molecules to convert one molecule of phosphatidylethanolamine into PtdCho [68] and as a result, 3 molecules SAH and consequently homocysteine are produced. One of the three methyl groups of PtdCho originates from homocysteine remethylation via the BHMT pathway.
When deuterium-labeled choline is used and the methyl groups are tracked, it was found that, the choline diphosphate (CDP) deuterated methyl groups are detected as d-9-PtdCho, showing that the labelled methyl group is channeled down the Kennedy pathway. When choline is driven down the PEMT pathway via methionine, then only one deuterated methyl group can be detected as d-3- PtdCho (Figure 2). PtdCho derived from the PEMT pathway requires three sequential methylation reactions of phosphotidylethanolamine (PE) that will generate d-3-PtdCho, d-6-PtdCho (if it has two methyl groups from the originally labeled choline or betaine) or rarely d-9 (three deutereum labeled methyl groups).
Metabolites can also be identified by this method (d-6-dimethylglycine, d-3-sarcosine, d-3-methionine and d-3-SAM). Figure 2 illustrates the metabolic fate of choline's methyl groups as demonstrated by compartmentalization studies [65].
The PEMT pathway is a significant contributor to methyl group homeostasis [50]. It has been estimated that PEMT-mediated PtdCho synthesis may consume 5 mmol SAM per day [69], thus implying that an equivalent amount of SAH and homocysteine is produced. Stead et al., reported that mice lacking PEMT activity have 50% lower plasma homocysteine and suggested that the PEMT pathway is a significant source of homocysteine and therefor SAM in the body [69]. Independent experiments have shown that hepatocytes isolated from PEMT-/- mice have lower PEMT activity and secrete less homocysteine than hepatocytes from control mice [70]. In contrast, studies on CTP: phosphocholine cytidylyltransferase-(CT) knockout mice where higher PEMT flux leads to higher PtdCho production, the concentrations of plasma homocysteine were 20-40% higher than in the control mice [71]. Seventy percent of liver PtdCho is produced via the CDP-choline pathway. In CTP: phosphocholine cytidylyltransferase-(CT) knockout mice [71], hepatic CDP-choline pathway is 80% lower than in the control mice while PEMT mRNA, protein, and activity are upregulated possibly to compensate for the absence of the CDP-pathway and produce more PtdCho under the same standard diet [72]. Also the BHMT activity was increased by 80% in the CT knockout mice, thus securing a source of methyl groups needed for the PEMT activity through oxidation of choline to betaine. Therefore, the need to maintain sufficient amount of PtdCho in this genetic mice model caused compensatory induction of PEMT and BHMT pathways of the same magnitude [71]. Although BHMT-induction does not lead to normalizing plasma homocysteine, it may maintain SAM at control levels [71]. The compensatory induction of BHMT is similar to the situation in Mthfr-/- and Cbs-/- mice models where homocysteine accumulates [73;74]. On the other hand, deletion of the BHMT gene in mice has been shown to cause 8-fold increase in plasma homocysteine, a massive disturbance in hepatic methylation potential (low SAM and high SAH) and accumulation of fats in the liver due to inability to synthesize sufficient PtdCho to export the triglycerides from the hepatic cells [4]. Therefore, the BHMT pathway that relies on adequate intake of choline and betaine is a significant source of methyl groups needed to produce PtdCho via PEMT. The PEMT pathway consumes SAM, but also provides SAH and thereby homocysteine that is recycled to methionine and new methyl groups.
PtdCho that is produced via the PEMT pathway using methyl groups of folate, betaine and choline transports long chain polyunsaturated fatty acids (PUFA) such as docosahexaenoic acid (DHA) (22:6n−3) [75], the fatty acid highly enriched in brain and retina. In contrast, phospholipids derived through the CDP pathway carry medium chain, monounsaturated fatty acids such as oleic acid [76].
9. Tracking the Methyl Groups of Betaine and Choline
The transmethylation reaction via BHMT provides one methyl group to convert homocysteine to methionine and retains the remaining 2 methyl groups of betaine that forms dimethylglycine. One methyl group of dimethylglycine provides C1-unit that is used to convert THF to 5,10-methylene-THF and sarcosine is produced. Sarcosine or N-methylglycine carries the last methyl group of dimethylglycine which is used to convert THF to 5,10-methylene-THF and producing glycine [77]. Thus, one of choline’s methyl groups is used to produce SAM and the remaining 2 methyl groups reenter the C1-metabolim pool as 5,10-methylene-THF via formaldehyde. Choline enriches the folate pathway with 2 carbon units that can join C1-metabolism as labile methyl groups via 5-methyl-THF (Figure 3).
The role of choline in enrichment of cellular folate has been shown in a study in newborn pigs [78]. The animals were fed a methyl deficient diet (without folate, choline and betaine) at the age of 4-8 days for 7 days and then received a rescue with either folate or betaine between days 7 and 10 [78]. The folic acid group showed a normalization of plasma folate, a decline of plasma homocysteine and no change in plasma betaine [78]. Whereas, in the betaine rescue group, besides showing correction of plasma betaine, there was a significant rise of plasma folate, demonstrating that folate was endogenously produced after conversion of betaine to sarcosine (Figure 3).
10. Interdependency of Folate and Choline
It has also been previously reported that 60% of choline is converted to betaine in the liver and this was confirmed in an in-vitro study by DeLong et al., in which the levels of betaine were found to be three times as high as choline in normal hepatocyte culture [67]. When hepatocytes were exposed to isotope labelled choline (d9-choline), the ratio of d9-labeled betaine to choline was the same as the unlabeled betaine to choline. When choline is absent from the cell system, no betaine can be detected, demonstrating that oxidation of choline is an obligatory source of betaine in hepatocytes [67]. Liver cancer cells (RH7777 Hepatoma) do not have the enzyme system to convert choline to betaine and hence the 3:1 ratio of betaine to choline is extinguished, thus confirming the significant contribution of choline’s methyl groups to betaine in the liver [67].
In the presence of a choline/betaine deficiency, there is greater dependence on 5-methyl-THF to generate PtdCho via the PEMT pathway (Figure 2). A choline deficient diet for 2 weeks in rats caused a 31 – 40% reduction in liver folate content, which was reversible when choline refeeding occurred [7;8]. Rats fed diets deficient in both methionine and choline for 5 weeks had hepatic folate concentrations that were 50% of those in controls [8]. Tetrahydrofolate deficiency, induced by methotrexate [79,80,81,82,83] or by dietary folate deficiency [18], resulted in decreased hepatic total choline, with the greatest decrease occurring in hepatic phosphocholine concentrations. During choline deficiency, hepatic SAM concentrations also decreased by as much as 50% [84,85,86,87].
These results suggest that temporary low intake of one of these nutrients such as due to seasonal food availability or food choice can be compensated for by other nutrients or methylneogenesis in the C1-metabolism. Whereas one sided nutrition or lack of methyl donors in critical stages of the life cycle such as during pregnancy, lactation and early life can have serious consequences on health.
11. Conclusions
The essential nutrients, folate and choline are metabolically entwined to provide SAM for functional methylation reactions. A folate deficient diet depletes liver choline and a choline deficient diet depletes liver folate by up to 40%, suggesting that long-term depletion of one of these nutrients can have devastating effects on cellular methylation capacity. Dietary folate intake or plasma concentrations of 5-Methyl-THF play a dominant role in determining fasting plasma homocysteine concentrations, but this role is not exclusive. Intakes of choline and betaine show weak associations with fasting plasma homocysteine. Studies on Bhmt-/- mice suggest that this choline- or betaine-dependent enzyme contributes to 50% of cellular SAM, but its role in methylation is not reflected by measuring fasting plasma homocysteine, nor by the effect of supplemental betaine or choline on lowering fasting homocysteine. Instead, the BHMT pathway’s major contribution to removing homocysteine can only be appreciated after a standard methionine load test (100 mg methionine/kg body weight), while folate has a limited effect on post methionine hyperhomocysteinemia. Measuring post methionine load (PML) homocysteine concentrations can be used as a sensitive marker of the contribution of choline and betaine to cellular methylation. The multiple scientific studies reviewed here support a significant and so-far underestimated role of choline and betaine as sources of methyl groups.
Author Contributions
JB wrote the first draft of the paper. RO provided input and revisions to all parts of the paper.
Funding
no funding was received for this article.
Institutional Review Board Statement
not applicable.
Informed Consent Statement
not applicable.
Data Availability Statement
not applicable.
Conflicts of Interest
JB is employed by Balchem Corporation. RO received honoraria for lectures and served as advisory board member for P&G Health GmbH, Wörwag Pharma GmbH, Balchem Corporation, Merck & Cie, and HIPP GmbH.
Abbreviations
The following abbreviations are used in this manuscript: ADP, adenine diphosphate; ATP, adenine triphosphate; BHMT, betaine homocysteine methyltransferase; CHDH, choline dehydrogenase; C1-metabolism, one carbon metabolism; 5-formyl-THF, 5-formyltetrahydrofolate; PML, post methionine load; PtdCho, phosphatidylcholine; 5-methyl-THF, 5-Methyltetrahydrofolate; MS, methionine synthase; PEMT, phosphatidylethanolamine methyl transferase; MTS, methionine adenosyltransferase; MTHFR, methylenetetrahydrofolate reductase; NAM, The United States National Academy of Medicine; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; THF, tetrahydrofolate.
References
- Falnes, P.Ø. Closing in on human methylation—the versatile family of seven-β-strand (METTL) methyltransferases. Nucleic Acids Res. 2024, 52, 11423–11441. [CrossRef]
- da Mota, J.C.N.; Ribeiro, A.A.; Carvalho, L.M.; Esteves, G.P.; Sieczkowska, S.M.; Goessler, K.F.; Gualano, B.; Nicoletti, C.F. Impact of Methyl-Donor Micronutrient Supplementation on DNA Methylation Patterns: A Systematic Review and Meta-Analysis of in vitro, Animal, and Human Studies. Lifestyle Genom. 2023, 16, 192–213. [CrossRef]
- Finkelstein, J.D.; Kyle, W.E.; Harris, B.J. Methionine metabolism in mammals: Regulatory effects of S-adenosylhomocysteine. Arch. Biochem. Biophys. 1974, 165, 774–779. [CrossRef]
- Teng, Y.-W.; Mehedint, M.G.; Garrow, T.A.; Zeisel, S.H. Deletion of Betaine-Homocysteine S-Methyltransferase in Mice Perturbs Choline and 1-Carbon Metabolism, Resulting in Fatty Liver and Hepatocellular Carcinomas. J. Biol. Chem. 2011, 286, 36258–36267. [CrossRef]
- Steenge, G.R.; Verhoef, P.; Katan, M.B. Betaine Supplementation Lowers Plasma Homocysteine in Healthy Men and Women. J. Nutr. 2003, 133, 1291–1295. [CrossRef]
- Atkinson, W.; Elmslie, J.; Lever, M.; Chambers, S.T.; George, P.M. Dietary and supplementary betaine: acute effects on plasma betaine and homocysteine concentrations under standard and postmethionine load conditions in healthy male subjects. Am. J. Clin. Nutr. 2008, 87, 577–585. [CrossRef]
- Selhub, J.; Seyoum, E.; A Pomfret, E.; Zeisel, S.H. Effects of choline deficiency and methotrexate treatment upon liver folate content and distribution.. 1991, 51, 16–21.
- Horne DW, Cook RJ, Wagner C. Effect of dietary methyl group deficiency on folate metabolism in rats. J Nutr 1989;119:618-21.
- Cuskelly GJ, Stacpoole PW, Williamson J, Baumgartner TG, Gregory JF, III. Deficiencies of folate and vitamin B(6) exert distinct effects on homocysteine, serine, and methionine kinetics. Am J Physiol Endocrinol Metab 2001;281:E1182-E1190.
- Finkelstein, J.D.; Martin, J.J. Methionine metabolism in mammals. Distribution of homocysteine between competing pathways.. J. Biol. Chem. 1984, 259, 9508–9513. [CrossRef]
- Barry MJ, Nicholson WK, Silverstein M, Chelmow D, Coker TR, Davis EM et al. Folic Acid Supplementation to Prevent Neural Tube Defects: US Preventive Services Task Force Reaffirmation Recommendation Statement. JAMA 2023;330:454-9.
- Zhang, C.; Chen, Y.; Hou, F.; Li, Y.; Wang, W.; Guo, L.; Zhang, C.; Li, L.; Lu, C. Safety and Efficacy of High-Dose Folinic Acid in Children with Autism: The Impact of Folate Metabolism Gene Polymorphisms. Nutrients 2025, 17, 1602. [CrossRef]
- Maruf AA, Poweleit EA, Brown LC, Strawn JR, Bousman CA. Systematic Review and Meta-Analysis of L-Methylfolate Augmentation in Depressive Disorders. Pharmacopsychiatry 2022;55:139-47.
- Zeisel SH, da Costa KA, Franklin PD, Alexander EA, Lamont JT, Sheard NF et al. Choline, an essential nutrient for humans. FASEB J 1991;5:2093-8.
- Buchman, A.L.; Dubin, M.; Jenden, D.; Moukarzel, A.; Roch, M.H.; Rice, K.; Gornbein, J.; E Ament, M.; Eckhert, C.D. Lecithin increases plasma free choline and decreases hepatic steatosis in long-term total parenteral nutrition patients.. 1992, 102, 1363–70.
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food allergens. Choline and contribution to normal liver function of the foetus and exclusively breastfed infants: evaluation of a health claim pursuant to Article 14 of Regulation (EC) No 1924/2006. EFSA Journal 2023;21: 1-12.
- Obeid R, Derbyshire E, Schon C. Association between maternal choline, foetal brain development and child neurocognition; systematic review and meta-analysis of human studies. Adv Nutr 2022; 13:2445-57.
- Kim, Y.-I.; Miller, J.W.; da Costa, K.-A.; Nadeau, M.; Smith, D.; Selhub, J.; Zeisel, S.H.; Mason, J.B. Severe Folate Deficiency Causes Secondary Depletion of Choline and Phosphocholine in Rat Liver. J. Nutr. 1994, 124, 2197–2203. [CrossRef]
- Johnson, B.C.; James, M.F. Choline Deficiency in the Baby Pig. J. Nutr. 1948, 36, 339–349. [CrossRef]
- Varela-Moreiras, G.; Ragel, C.; de Miguelsanz, J.P. Choline deficiency and methotrexate treatment induces marked but reversible changes in hepatic folate concentrations, serum homocysteine and DNA methylation rates in rats.. J. Am. Coll. Nutr. 1995, 14, 480–485. [CrossRef]
- Chmurzynska, A.; Seremak-Mrozikiewicz, A.; Malinowska, A.M.; Różycka, A.; Radziejewska, A.; KurzawiŃska, G.; Barlik, M.; Wolski, H.; Drews, K. Associations between folate and choline intake, homocysteine metabolism, and genetic polymorphism of MTHFR, BHMT and PEMT in healthy pregnant Polish women. Nutr. Diet. 2019, 77, 368–372. [CrossRef]
- Mudd, S.; Poole, J.R. Labile methyl balances for normal humans on various dietary regimens. Metabolism 1975, 24, 721–735. [CrossRef]
- Chen LH, Liu ML, Hwang HY, Chen LS, Korenberg J, Shane B. Human methionine synthase. cDNA cloning, gene localization, and expression. J Biol Chem 1997;272:3628-34.
- Finkelstein, J.A.M.E.S.D. Pathways and Regulation of Homocysteine Metabolism in Mammals. Semin. Thromb. Hemost. 2000, ume 26, 219–226. [CrossRef]
- Stipanuk MH. Metabolism of sulfur-containing amino acids. Annu Rev Nutr 1986;6:179-209.
- Wilcken, D.E.L.; Wilcken, B.; Dudman, N.P.B.; Tyrrell, P.A. Homocystinuria — The Effects of Betaine in the Treatment of Patients Not Responsive to Pyridoxine. New Engl. J. Med. 1983, 309, 448–453. [CrossRef]
- Wilcken, D.E.; Dudman, N.P.; Tyrrell, P.A. Homocystinuria due to cystathionine β-synthase deficiency—The effects of betaine treatment in pyridoxine-responsive patients. Metabolism 1985, 34, 1115–1121. [CrossRef]
- Teng, Y.-W.; Cerdena, I.; Zeisel, S.H. Homocysteinemia in Mice with Genetic Betaine HomocysteineS -Methyltransferase Deficiency Is Independent of Dietary Folate Intake. J. Nutr. 2012, 142, 1964–1967. [CrossRef]
- Selhub, J.; Jacques, P.F.; Wilson, P.W.F.; Rush, D.; Rosenberg, I.H. Vitamin Status and Intake as Primary Determinants of Homocysteinemia in an Elderly Population. JAMA 1993, 270, 2693–2698. [CrossRef]
- E Chiuve, S.; Giovannucci, E.L.; E Hankinson, S.; Zeisel, S.H.; Dougherty, L.W.; Willett, W.C.; Rimm, E.B. The association between betaine and choline intakes and the plasma concentrations of homocysteine in women. Am. J. Clin. Nutr. 2007, 86, 1073–1081. [CrossRef]
- Olthof MR, Brink EJ, Katan MB, Verhoef P. Choline supplemented as phosphatidylcholine decreases fasting and postmethionine-loading plasma homocysteine concentrations in healthy men. Am J Clin Nutr 2005;82:111-7.
- Olthof, M.R.; Verhoef, P.; van Vliet, T.; Boelsma, E. Low Dose Betaine Supplementation Leads to Immediate and Long Term Lowering of Plasma Homocysteine in Healthy Men and Women. J. Nutr. 2003, 133, 4135–4138. [CrossRef]
- Ubbink, J.B.; Becker, P.J.; Delport, R.; Bester, M.; Riezler, R.; Vermaak, W. Variability of post-methionine load plasma homocysteine assays. Clin. Chim. Acta 2003, 330, 111–119. [CrossRef]
- Van Der Griend, R.; Haas, F.J.; Duran, M.; Biesma, D.H.; Meuwissen, O.J.; Banga, J.-D. Methionine loading test is necessary for detection of hyperhomocysteinemia. J. Lab. Clin. Med. 1998, 132, 67–72. [CrossRef]
- van der Griend, R.; Biesma, D.H.; Banga, J.-D. Postmethionine-load homocysteine determination for the diagnosis hyperhomocysteinaemia and efficacy of homocysteine lowering treatment regimens. Vasc. Med. 2002, 7, 29–33. [CrossRef]
- Bostom, A.G.; Jacques, P.F.; Nadeau, M.R.; Williams, R.R.; Ellison, R.; Selhub, J. Post-methionine load hyperhomocysteinemia in persons with normal fasting total plasma homocysteine: initial results from The NHLBI Family Heart Study. Atherosclerosis 1995, 116, 147–151. [CrossRef]
- Lever, M.; Slow, S.; O McGregor, D.; Dellow, W.J.; George, P.M.; Chambers, S.T. Variability of plasma and urine betaine in diabetes mellitus and its relationship to methionine load test responses: an observational study. Cardiovasc. Diabetol. 2012, 11, 34–34. [CrossRef]
- Holm, P.I.; Bleie, Ø.; Ueland, P.M.; Lien, E.A.; Refsum, H.; Nordrehaug, J.E.; NygårD, O. Betaine as a Determinant of Postmethionine Load Total Plasma Homocysteine Before and After B-Vitamin Supplementation. Arter. Thromb. Vasc. Biol. 2004, 24, 301–307. [CrossRef]
- Holm, P.I.; Ueland, P.M.; Vollset, S.E.; Midttun, Ø.; Blom, H.J.; Keijzer, M.B.; Heijer, M.D. Betaine and Folate Status as Cooperative Determinants of Plasma Homocysteine in Humans. Arter. Thromb. Vasc. Biol. 2005, 25, 379–385. [CrossRef]
- Lee JE, Jacques PF, Dougherty L, Selhub J, Giovannucci E, Zeisel SH et al. Are dietary choline and betaine intakes determinants of total homocysteine concentration? Am J Clin Nutr 2010;91:1303-10.
- da Costa, K.-A.; E Gaffney, C.; Fischer, L.M.; Zeisel, S.H. Choline deficiency in mice and humans is associated with increased plasma homocysteine concentration after a methionine load. Am. J. Clin. Nutr. 2005, 81, 440–444. [CrossRef]
- Verhoef, P.; Steenge, G.R.; Boelsma, E.; van Vliet, T.; Olthof, M.R.; Katan, M.B. Dietary serine and cystine attenuate the homocysteine-raising effect of dietary methionine: a randomized crossover trial in humans. Am. J. Clin. Nutr. 2004, 80, 674–679. [CrossRef]
- Figueroa-Torres, A.G.; Matias-Aguilar, L.O.; Coria-Ramirez, E.; Bonilla-Gonzalez, E.; Gonzalez-Marquez, H.; Ibarra-Gonzalez, I.; Hernandez-Lopez, J.R.; Hernandez-Juarez, J.; Dominguez-Reyes, V.M.; Isordia-Salas, I.; et al. Cystathionine β-synthase and methylenetetrahydrofolate reductase mutations in Mexican individuals with hyperhomocysteinemia. SAGE Open Med. 2020, 8. [CrossRef]
- A Lievers, K.J.; Kluijtmans, L.A.J.; Heil, S.G.; Boers, G.H.J.; Verhoef, P.; Heijer, M.D.; Trijbels, F.J.M.; Blom, H.J. Cystathionine β-synthase polymorphisms and hyperhomocysteinaemia: an association study. Eur. J. Hum. Genet. 2003, 11, 23–29. [CrossRef]
- Bhat, D.S.; Gruca, L.L.; Bennett, C.D.; Katre, P.; Kurpad, A.V.; Yajnik, C.S.; Kalhan, S.C.; Loor, J.J. Evaluation of tracer labelled methionine load test in vitamin B-12 deficient adolescent women. PLOS ONE 2018, 13, e0196970. [CrossRef]
- I Chiang, E.-P.; Selhub, J.; Bagley, P.J.; Dallal, G.; Roubenoff, R. Pyridoxine supplementation corrects vitamin B6 deficiency but does not improve inflammation in patients with rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1404–11. [CrossRef]
- de JR, Griffioen PH, van ZB, Brouns RM, Visser W, Lindemans J. Evaluation of a shorter methionine loading test. Clin Chem Lab Med 2004;42:1027-31.
- Sadre-Marandi, F.; Dahdoul, T.; Reed, M.C.; Nijhout, H.F. Sex differences in hepatic one-carbon metabolism. BMC Syst. Biol. 2018, 12, 1–13. [CrossRef]
- Nelen, W.L.; Blom, H.J.; Thomas, C.M.G.; Steegers, E.A.; Boers, G.H.J.; Eskes, T.K.; Steegers Methylenetetrahydrofolate Reductase Polymorphism Affects the Change in Homocysteine and Folate Concentrations Resulting from Low Dose Folic Acid Supplementation in Women with Unexplained Recurrent Miscarriages. J. Nutr. 1998, 128, 1336–1341. [CrossRef]
- Mudd, S.H.; Brosnan, J.T.; Brosnan, M.E.; Jacobs, R.L.; Stabler, S.P.; Allen, R.H.; Vance, D.E.; Wagner, C. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007, 85, 19–25. [CrossRef]
- Mudd, S.; Ebert, M.H.; Scriver, C.R. Labile methyl group balances in the human: The role of sarcosine. Metabolism 1980, 29, 707–720. [CrossRef]
- Storch, K.J.; Wagner, D.A.; Burke, J.F.; Young, V.R. Quantitative study in vivo of methionine cycle in humans using [methyl-2H3]- and [1-13C]methionine. Am. J. Physiol. Metab. 1988, 255, E322–E331. [CrossRef]
- Lamers, Y.; Williamson, J.; Gilbert, L.R.; Stacpoole, P.W.; Gregory, J.F. Glycine Turnover and Decarboxylation Rate Quantified in Healthy Men and Women Using Primed, Constant Infusions of [1,2-13C2]Glycine and [2H3]Leucine ,. J. Nutr. 2007, 137, 2647–2652. [CrossRef]
- Im YS, Chiang PK, Cantoni GL. Guanidoacetate methyltransferase. Purification and molecular properties. J Biol Chem 1979;254:11047-50.
- Resseguie, M.; Song, J.; Niculescu, M.D.; Costa, K.-A.; Randall, T.A.; Zeisel, S.H. PhosphatidylethanolamineN-methyltransferase(PEMT)gene expression is induced by estrogen in human and mouse primary hepatocytes. FASEB J. 2007, 21, 2622–2632. [CrossRef]
- Resseguie, M.E.; da Costa, K.-A.; Galanko, J.A.; Patel, M.; Davis, I.J.; Zeisel, S.H. Aberrant Estrogen Regulation of PEMT Results in Choline Deficiency-associated Liver Dysfunction. J. Biol. Chem. 2011, 286, 1649–1658. [CrossRef]
- Costa, K.-A.; Kozyreva, O.G.; Song, J.; Galanko, J.A.; Fischer, L.M.; Zeisel, S.H. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J. 2006, 20, 1336–1344. [CrossRef]
- Fischer, L.M.; Dacosta, K.A.; Kwock, L.; Stewart, P.W.; Lu, T.-S.; Stabler, S.P.; Allen, R.H.; Zeisel, S.H. Sex and menopausal status influence human dietary requirements for the nutrient choline. Am. J. Clin. Nutr. 2007, 85, 1275–1285. [CrossRef]
- Schwahn BC, Laryea MD, Chen Z, Melnyk S, Pogribny I, Garrow T et al. Betaine rescue of an animal model with methylenetetrahydrofolate reductase deficiency. Biochem J 2004;382:831-40.
- Chen, Z.; Karaplis, A.C.; Ackerman, S.L.; Pogribny, I.P.; Melnyk, S.; Lussier-Cacan, S.; Chen, M.F.; Pai, A.; John, S.W.; Smith, R.S.; et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001, 10, 433–443. [CrossRef]
- Colson, N.J.; Naug, H.L.; Nikbakht, E.; Zhang, P.; McCormack, J. The impact of MTHFR 677 C/T genotypes on folate status markers: a meta-analysis of folic acid intervention studies. Eur. J. Nutr. 2015, 56, 247–260. [CrossRef]
- Yan, J.; Wang, W.; Gregory, J.F.; Malysheva, O.; Brenna, J.T.; Stabler, S.P.; Allen, R.H.; A Caudill, M. MTHFR C677T genotype influences the isotopic enrichment of one-carbon metabolites in folate-compromised men consuming d9-choline. Am. J. Clin. Nutr. 2011, 93, 348–355. [CrossRef]
- EFSA Panel on Dietetic Products, Nutrition and Allergies NDA. Scientific opinion on dietary reference values for folate. EFSA Journal 2014;12: 3893.
- EFSA Panel on Dietetic Products, Nutrition and Allergies NDA. Dietary reference values for choline. EFSA Journal 2016;14: 4484.
- Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; The National Academies Press: Washington, DC, USA, 1998; pp. 306–356.
- Vance DE, Ridgway ND. The methylation of phosphatidylethanolamine. Prog Lipid Res 1988;27:61-79.
- DeLong, C.J.; Hicks, A.M.; Cui, Z. Disruption of Choline Methyl Group Donation for Phosphatidylethanolamine Methylation in Hepatocarcinoma Cells. J. Biol. Chem. 2002, 277, 17217–17225. [CrossRef]
- E Vance, D.; Walkey, C.J.; Cui, Z. Phosphatidylethanolamine N-methyltransferase from liver. Biochim. et Biophys. Acta (BBA) - Lipids Lipid Metab. 1997, 1348, 142–150. [CrossRef]
- Stead LM, Brosnan JT, Brosnan ME, Vance DE, Jacobs RL. Is it time to reevaluate methyl balance in humans? Am J Clin Nutr 2006;83:5-10.
- Shields, D.J.; Lingrell, S.; Agellon, L.B.; Brosnan, J.T.; Vance, D.E. Localization-independent Regulation of Homocysteine Secretion by Phosphatidylethanolamine N-Methyltransferase. J. Biol. Chem. 2005, 280, 27339–27344. [CrossRef]
- Jacobs, R.L.; Stead, L.M.; Devlin, C.; Tabas, I.; Brosnan, M.E.; Brosnan, J.T.; Vance, D.E. Physiological Regulation of Phospholipid Methylation Alters Plasma Homocysteine in Mice. J. Biol. Chem. 2005, 280, 28299–28305. [CrossRef]
- Jacobs, R.L.; Devlin, C.; Tabas, I.; Vance, D.E. Targeted Deletion of Hepatic CTP:phosphocholine Cytidylyltransferase α in Mice Decreases Plasma High Density and Very Low Density Lipoproteins. J. Biol. Chem. 2004, 279, 47402–47410. [CrossRef]
- Schwahn, B.C.; Wendel, U.; Lussier-Cacan, S.; Mar, M.-H.; Zeisel, S.H.; Leclerc, D.; Castro, C.; A Garrow, T.; Rozen, R. Effects of betaine in a murine model of mild cystathionine-β-synthase deficiency. Metabolism 2004, 53, 594–599. [CrossRef]
- Schwahn BC, Chen Z, Laryea MD, Wendel U, Lussier-Cacan S, Genest J, Jr. et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J 2003;17:512-4.
- Watkins, S.M.; Zhu, X.; Zeisel, S.H. Phosphatidylethanolamine-N-methyltransferase Activity and Dietary Choline Regulate Liver-Plasma Lipid Flux and Essential Fatty Acid Metabolism in Mice. J. Nutr. 2003, 133, 3386–3391. [CrossRef]
- DeLong, C.J.; Shen, Y.-J.; Thomas, M.J.; Cui, Z. Molecular Distinction of Phosphatidylcholine Synthesis between the CDP-Choline Pathway and Phosphatidylethanolamine Methylation Pathway. J. Biol. Chem. 1999, 274, 29683–29688. [CrossRef]
- Augustin, P.; Hromic, A.; Pavkov-Keller, T.; Gruber, K.; Macheroux, P. Structure and biochemical properties of recombinant human dimethylglycine dehydrogenase and comparison to the disease-related H109R variant. FEBS J. 2016, 283, 3587–3603. [CrossRef]
- Robinson, J.L.; McBreairty, L.E.; Randell, E.W.; Harding, S.V.; Bartlett, R.K.; Brunton, J.A.; Bertolo, R.F. Betaine or folate can equally furnish remethylation to methionine and increase transmethylation in methionine-restricted neonates. J. Nutr. Biochem. 2018, 59, 129–135. [CrossRef]
- Barak, A.J.; Kemmy, R.J. Methotrexate effects on hepatic betaine levels in choline-supplemented and choline-deficient rats.. 1982, 1, 275–8.
- Barak AJ, Tuma DJ, Beckenhauer HC. Methotrexate hepatotoxicity. J Am Coll Nutr 1984;3:93-6.
- Freeman-Narrod, M.; Narrod, S.A.; Custer, R.P. Chronic Toxicity of Methotrexate in Rats: Partial to Complete Protection of the Liver by Choline: Brief Communication23. JNCI J. Natl. Cancer Inst. 1977, 59, 1013–1017. [CrossRef]
- Pomfret, E.A.; Dacosta, K.-A.; Zeisel, S.H. Effects of choline deficiency and methotrexate treatment upon rat liver. J. Nutr. Biochem. 1990, 1, 533–541. [CrossRef]
- Ueland, P.M.; Berge, R.K.; Aarsland, A.; Aarsaether, N.; Refsum, H.; Svardal, A.M.; Lønning, P.E. Effect of methotrexate on homocysteine and other sulfur compounds in tissues of rats fed a normal or a defined, choline-deficient diet. Cancer Chemother. Pharmacol. 1988, 21, 313–318. [CrossRef]
- POIRIER, L.; GRANTHAM, P.; ROGERS, A. EFFECTS OF A MARGINALLY LIPOTROPE-DEFICIENT DIET ON HEPATIC LEVELS OF S-ADENOSYLMETHIONINE AND ON URINARY METABOLITES OF 2-ACETYLAMINOFLUORENE IN RATS. 1977, 37, 744–748.
- Barak, A.J.; Kemmy, R.J.; Tuma, D.J. The effect of methotrexate on homocysteine methylating agents in rat liver.. 1982, 1, 303–6.
- Shivapurkar, N.; Poirier, L.A. Tissue levels of S-adenosylmethionine and S-adenosylhomocysteine in rats fed methyl-deficient, amino acid-defined diets for one to five weeks. Carcinog. 1983, 4, 1051–1057. [CrossRef]
- Zeisel, S.H.; Zola, T.; A Dacosta, K.; A Pomfret, E. Effect of choline deficiency on S-adenosylmethionine and methionine concentrations in rat liver. Biochem. J. 1989, 259, 725–729. [CrossRef]
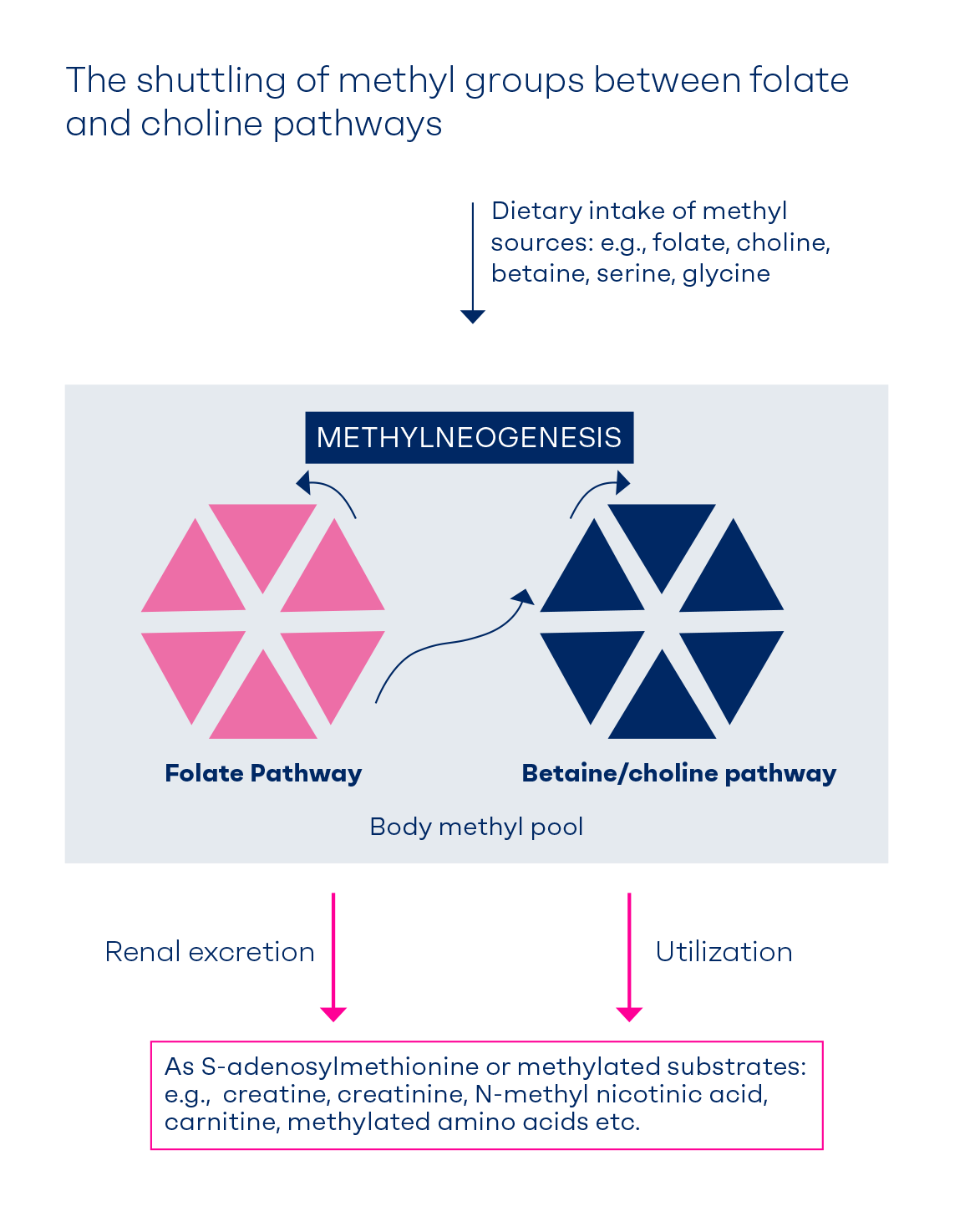
Figure 1.
Flux of methyl groups of folate and choline. Coordination between the betaine homocysteine methyl transferase (BHMT) and folate pathways (methionine synthase, MS) ensures balanced methylation. The relative contribution of the BHMT pathway versus folate pathway to the methylation balance may vary according to the marker used to capture cellular methylation. The BHMT pathway provides 50% of the methionine or SAM [4] and is a major contributor to removing homocysteine after a methionine load [5;6]. A choline deficient diet lowers liver folate by 31 – 40% [7;8]. Folate deficiency causes 37% lower methylation of homocysteine to methionine (thus elevated fasting homocysteine) [9], but has a limited effect on post methionine hyperhomocysteinemia [5;9]. CHDH, choline dehydrogenase; MTHFR, methylenetetrahydrofolate reductase; PEMT, phosphatidylethanolamine methyl transferase.
Figure 1.
Flux of methyl groups of folate and choline. Coordination between the betaine homocysteine methyl transferase (BHMT) and folate pathways (methionine synthase, MS) ensures balanced methylation. The relative contribution of the BHMT pathway versus folate pathway to the methylation balance may vary according to the marker used to capture cellular methylation. The BHMT pathway provides 50% of the methionine or SAM [4] and is a major contributor to removing homocysteine after a methionine load [5;6]. A choline deficient diet lowers liver folate by 31 – 40% [7;8]. Folate deficiency causes 37% lower methylation of homocysteine to methionine (thus elevated fasting homocysteine) [9], but has a limited effect on post methionine hyperhomocysteinemia [5;9]. CHDH, choline dehydrogenase; MTHFR, methylenetetrahydrofolate reductase; PEMT, phosphatidylethanolamine methyl transferase.
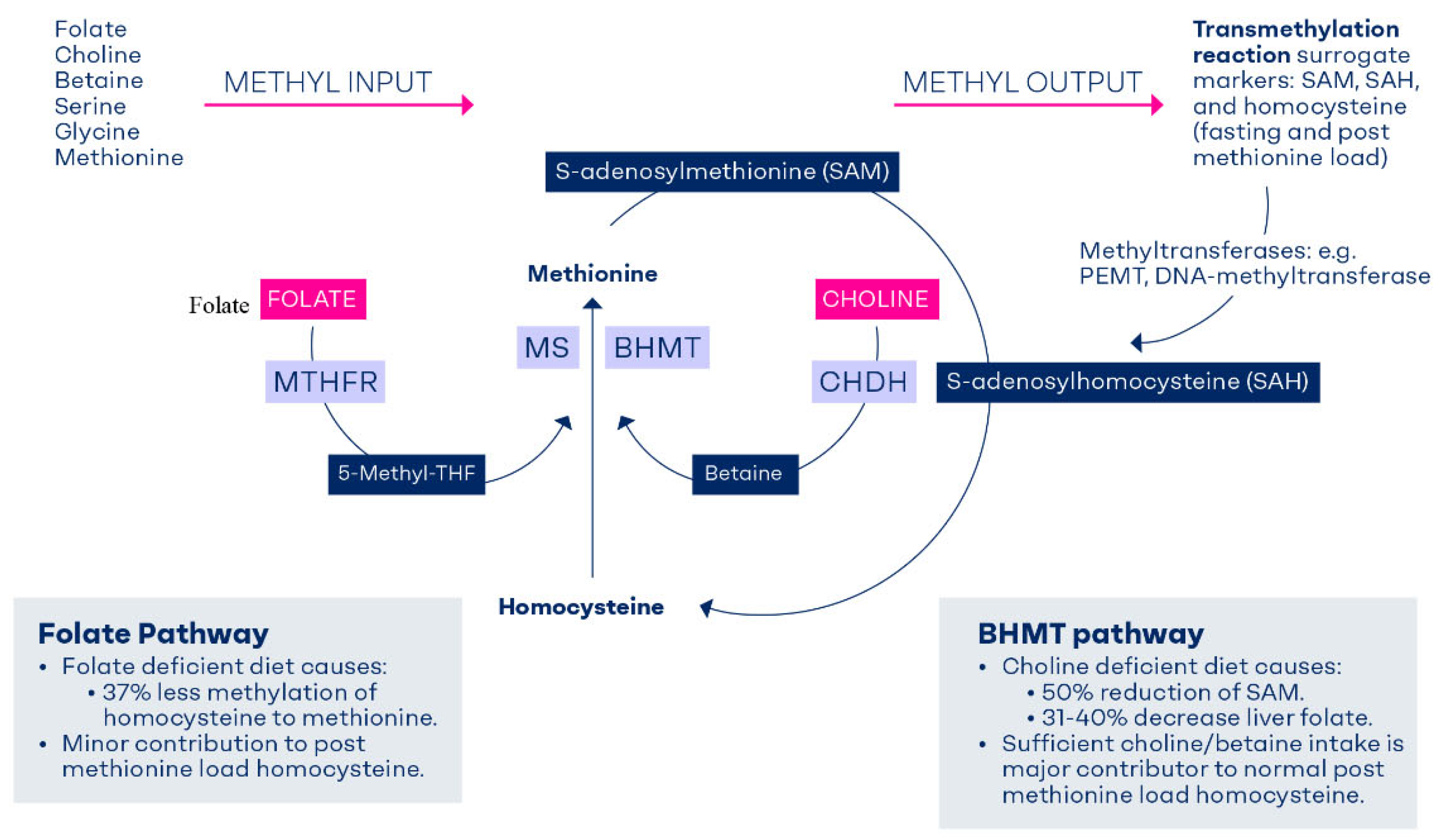
Figure 2.
Metabolic fate of the orally consumed deuterium-labelled choline. The d-9-choline tracer contained three deuterium-labelled methyl groups. After irreversible oxidation of choline to betaine by choline dehydrogenase (CHDH) and betaine aldehyde dehydrogenases (BADH), one of the three labelled methyl group is used to convert homocysteine to methionine and appears in S-adenosylmethionine molecule. The second labelled methyl group (now on dimethylglycine) is traced in 5,10-methylene-tetrahydrofolate. The third methyl group of choline is channeled via sarcosine and then transferred to tetrahydrofolate and is traced in 5,10-methylene-tetrahydrofolate. Thus, each choline molecule that becomes oxidized to betaine contributes to the methyl reservoir by three methyl groups. Synthesis of phosphatidylcholine from phosphatidylethanolamine via phosphatidylethanolamine methyl transferase (PEMT) utilizes three methyl groups from the methylation pool, but it also generates S-adenosylhomocysteine that feeds back into the homocysteine remethylation pathway.
Figure 2.
Metabolic fate of the orally consumed deuterium-labelled choline. The d-9-choline tracer contained three deuterium-labelled methyl groups. After irreversible oxidation of choline to betaine by choline dehydrogenase (CHDH) and betaine aldehyde dehydrogenases (BADH), one of the three labelled methyl group is used to convert homocysteine to methionine and appears in S-adenosylmethionine molecule. The second labelled methyl group (now on dimethylglycine) is traced in 5,10-methylene-tetrahydrofolate. The third methyl group of choline is channeled via sarcosine and then transferred to tetrahydrofolate and is traced in 5,10-methylene-tetrahydrofolate. Thus, each choline molecule that becomes oxidized to betaine contributes to the methyl reservoir by three methyl groups. Synthesis of phosphatidylcholine from phosphatidylethanolamine via phosphatidylethanolamine methyl transferase (PEMT) utilizes three methyl groups from the methylation pool, but it also generates S-adenosylhomocysteine that feeds back into the homocysteine remethylation pathway.
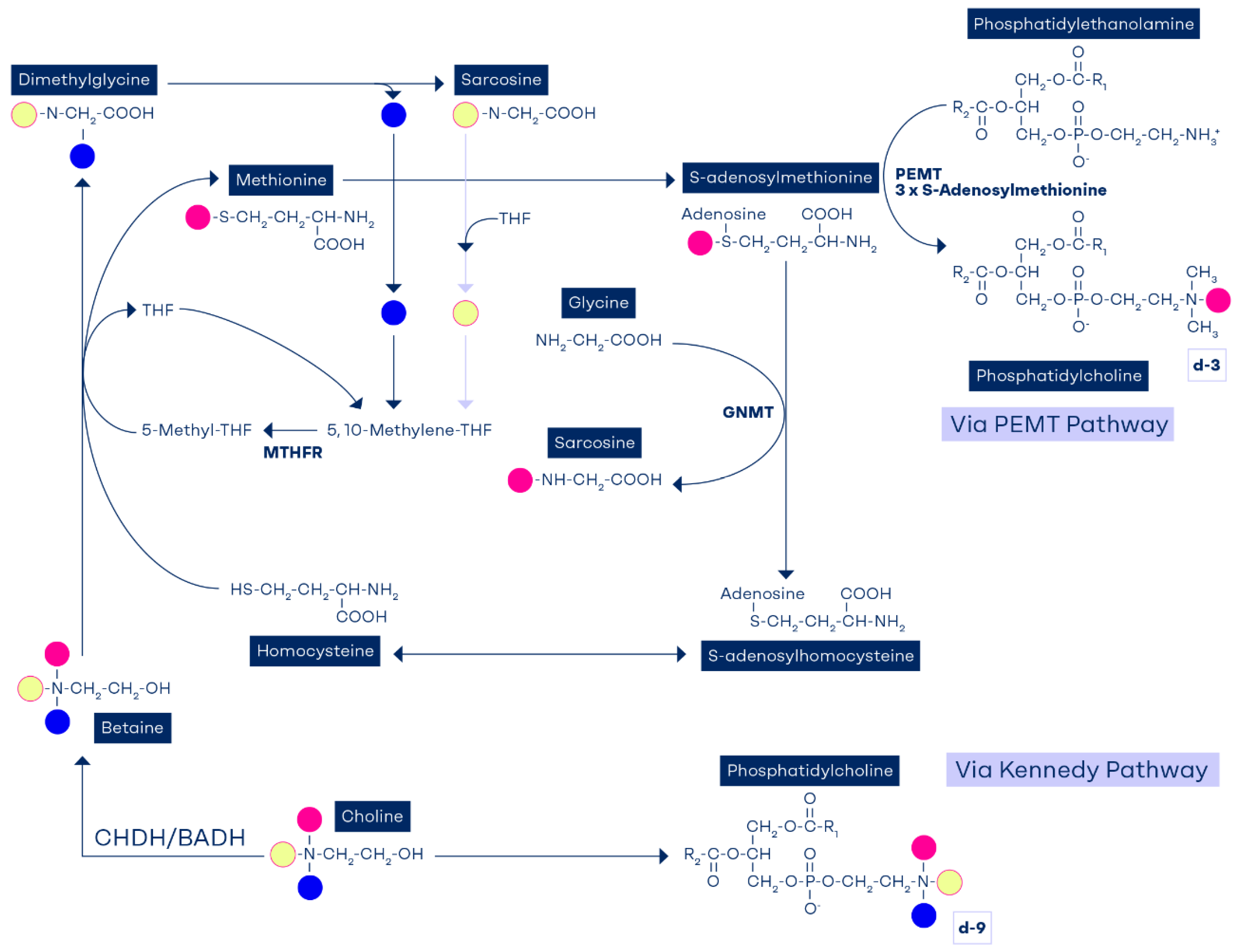
Figure 3.
The methyl groups of choline and betaine are highly conserved and used to replenish the methyl reservoir. Each choline molecule that becomes oxidized to betaine can theoretically convert three homocysteine molecules to form three S-adenosylmethionine (SAM) molecules; one SAM is formed directly from betaine via the BHMT pathway; the other two SAMs are synthesized through formation of 5-methyl-THF (one from dimethylglycine and the other one from sarcosine). The two 5-methyl-THF molecules then augment SAM via the methionine synthase pathway. This could explain why betaine supplementation causes a rise in plasma folate in a methyl deficient animal model [78]. .
Figure 3.
The methyl groups of choline and betaine are highly conserved and used to replenish the methyl reservoir. Each choline molecule that becomes oxidized to betaine can theoretically convert three homocysteine molecules to form three S-adenosylmethionine (SAM) molecules; one SAM is formed directly from betaine via the BHMT pathway; the other two SAMs are synthesized through formation of 5-methyl-THF (one from dimethylglycine and the other one from sarcosine). The two 5-methyl-THF molecules then augment SAM via the methionine synthase pathway. This could explain why betaine supplementation causes a rise in plasma folate in a methyl deficient animal model [78]. .
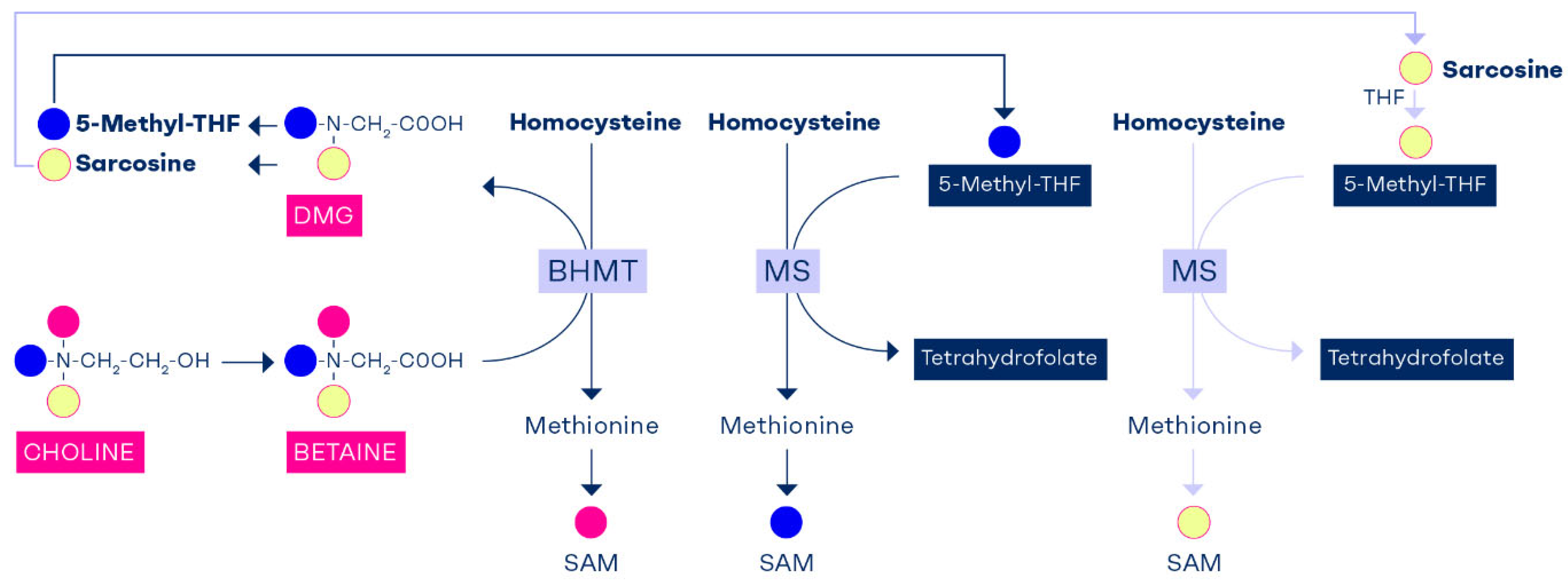

Table 1.
Determinants and Conditions of homocysteine concentrations after methionine load.
| Determinants of homocysteine concentrations after methionine load | Direction |
| Elevated fasting plasma homocysteine [37;38]. | ↑↑ |
| Higher plasma betaine [37,38,39]; betaine intake (diet or supplements) [5;6;40]; or choline intake (diet or supplements) [6;40;41]. | ↓ ↓ ↓ |
| Acute intake of choline/betaine (single dose studies or after a meal) [31;32]. | ↓ ↓ ↓ |
| Higher intake of serine or cysteine [42]; higher folate status [39]; or folate intake [5]. | ↓ |
| Polymorphisms in the transsulfuration pathway (e.g., cystathionine β-synthase) [43;44]. | (↓↑) |
| Low vitamin B12 status [45]; vitamin B6 supplementation [38;46]. | (↑); (↓) |
| Conditions where the post-methionine load test may be used to detect hyperhomocysteinemia | |
| Insufficient choline intake or status. | |
| Carriers of polymorphisms in methylenetetrahydrofolate reductase (MTHFR) [47], phosphatidylethanolamine methyl transferase (PEMT) or BHMT genes. | |
| Anti-folate drugs (e.g., methotrexate, antimalarial) or drugs interfering with folate absorption/metabolism. | |
| Pregnant and lactating women and children with high choline requirements not met through diet. | |
| B12 deficiency (e.g., vegan, elderly). | |
| Mild to moderate fasting hyperhomocysteinemia not explained by low folate, B6 or B12 concentrations. | |
| ↓ lower, (↑) slightly higher, (↓) slightly lower, (↓↑) no clear effect. | |
Table 2.
Relationship between post methionine load (PML) and fasting (F)-homocysteine levels under different intake conditions.
Table 2.
Relationship between post methionine load (PML) and fasting (F)-homocysteine levels under different intake conditions.
| PML-homocysteine | F-homocysteine | PML- minus F-homocysteine | %change (PML vs. F-homocysteine) | Prediction of PML-homocysteine from F-homocysteine under different intake conditions |
| 1- Native condition - no supplement (Olthof et al., [31] and Steenge et al., [5]) | ||||
| 32.6 µmol/L | 15.6 µmol/L | 17.0 µmol/L | +109.0% | PML-homocysteine = F-homocysteine * 2.09. |
| 34.8 µmol/L | 12.2 µmol/L | 22.6 µmol/L | +185.2% | PML-homocysteine = F-homocysteine * 2.85. |
| 31.6 µmol/L | 13.0 µmol/L | 18.6 µmol/L | +143.1% | PML-homocysteine = F-homocysteine * 2.43. |
| Mean = 33.0 µmol/L | 13.6 µmol/L | 19.4 µmol/L | +145.8% | PML-homocysteine = F-homocysteine * 2.46. |
| 2- Supplemented with 2.6 g/d choline for 2 weeks (Olthof et al., [31]) | ||||
| 22.3 µmol/L | 13.6 µmol/L | 8.7 µmol/L | +64.0% | PML-homocysteine = F-homocysteine * 1.64. Methionine load test in choline intake-optimized persons led to roughly 55% lower PML-homocysteine compared to non-supplemented people (8.7 vs. 19.4 µmol/L) |
| 3- Supplemented with 3*2 g/d betaine for 6 weeks (Steenge et al., [5]) | ||||
| 17.6 µmol/L | 10.9 µmol/L | 6.7 µmol/L | +61.5% | PML-homocysteine = F-homocysteine * 1.62. Methionine load test in betaine intake optimized persons led to roughly 65% lower PML-homocysteine compared to non-supplemented people (6.7 vs. 19.4 µmol/L) |
| 4- Supplemented with 400 µg* 2/d folic acid for 6 weeks (Steenge et al., [5]) | ||||
| 33.0 µmol/L | 10.7 µmol/L | 22.3 µmol/L | +208.4% | PML-homocysteine = F-homocysteine * 3.1. Optimization of folate status has no lowering effect on PML-homocysteine compared to non-supplemented people (22.3 vs. 19.4 µmol/L) |
Table 3.
Gaps in knowledge surrounding use of PML-homocysteine to capture disorders in methyl group flux through the BHMT pathway (or alternatively choline and betaine).
Table 3.
Gaps in knowledge surrounding use of PML-homocysteine to capture disorders in methyl group flux through the BHMT pathway (or alternatively choline and betaine).
| Question | Elaboration |
| Sex differences: Is the methylation flux higher in men than in women? | The expressions of several enzymes in C1-metabolism show sex-differences [48]. For example, men have higher plasma homocysteine and betaine than pre-menopausal women because the PEMT gene is upregulated by estrogen. |
| Is there a dose-response relationship between choline intake and PML-homocysteine? | A dose response relationship between betaine intake and PML-homocysteine has been demonstrated [32]. Does the same apply for choline and what is the intake level of choline to achieve a maximal reduction of PML-homocysteine? |
| Could high dose betaine or choline compensate for folate deficiency in terms of lowering PML-homocysteine? | Addressing metabolic capacity to upregulate methyl group flow via betaine/choline in people with folate deficiency or MTHFRC677T TT genotype. |
| Can PML-homocysteine be used to define the optimal intake of choline or betaine in pregnant and lactating women? | Homocysteine concentrations after a methionine load test can be tested before and after loading the ‘gap’ of choline or betaine intakes. |
| Can PML-homocysteine test be used to identify women at high risk of neural tube defects or other pregnancy complications such as recurrent pregnancy loss, gestational diabetes or preeclampsia? | In one study among women with a history of recurrent pregnancy loss, folic acid supplementation (0.5 mg/d for 2 months) did not lower PML-homocysteine in 53% of the women [49]. In theory, the PML-homocysteine test may identify women who could benefit from choline/betaine supplements through increasing methyl group flux via the BHMT pathway and normalizing PML-homocysteine. This may influence disease risk. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



