
Submitted:
14 July 2025
Posted:
16 July 2025
You are already at the latest version
Abstract
Central nervous system (CNS) involvement is an extremely rare manifestation in eosinophilic granulomatosis with polyangiitis (EGPA), associated with a poor prognosis.
Here we present a case of 50-year-old female patient previously long-lasting treated with asthma who presented initially with extreme eosinophilia (56%) and severe progressive ascending paresis similar to Guillain-Barré syndrome leading to tetraplegia. After navigating through diagnostic mazes; the diagnosis of EGPA was established based on eosinophilia, anti-neutrophil cytoplasmic antibody positivity (MPO-ANCA), asthma, eosinophil granulomatosis in the gastrointestinal tract, and severe peripheral nervous system involvement complicated with rare central nervous granulomatosis and ischaemia. With combined immunosuppressive and immunomodulatory treatment including high-dose corticosteroids, rituximab and intravenous immunoglobulin along with symptomatic treatment and planned rehabilitation over 6 months our patient recovered gradually from tetraplegia and adverse events such as severe infections and osteoporotic fractures. Now from a 2-year perspective we can conclude a successful treatment leading to decrease in all of her symptoms. Due to persistent eosinophilia after steroid tapering, she is currently on mepolizumab maintaining treatment and demonstating continuous improvement of motor and sensory functions. Thanks to periodically repeated rehabilitation, she became self-sufficient and returned to her previous job.
Our case represents that EGPA patients should be treated in a centre of expertise due to the rarity of disease and complexity of diagnosis and treatment. Careful multidisciplinary cooperation, huge effort of the patient and supportive environment can show a way back from immune-mediated tetraplegia.
Keywords:
eosinophil granulomatosis with polyangiitis
; central nervous system
; tetraplegia
; individualized treatment
1. Introduction
Eosinophilic granulomatosis with polyangiitis (EGPA) previously known as Churg-Straus syndrome is a rare systemic immune-mediated disorder characterized by eosinophilia, ANCA-associated vasculitis (AAV) of small and medium-sized vessels, and pulmonary symptoms [1,2]. Its prevalence ranges from 1.7–13 cases/ million inhabitants. ANCA-positivity ranges from 30–70% of EGPA patients but is usually less frequently observed than in other AAVs [1,2,3,4,5,6,7]. EGPA primarily affects the respiratory tract, lungs, peripheral nervous system, heart, gastrointestinal tract, and skin, but CNS involvement is rarely seen [3]. Some investigators hypothesize an association between EGPA onset and medications such as leukotriene receptor antagonists. However, this is more due to unmasking the underlying disease rather than directly causing EGPA [8]. Several studies considered Human Leukocyte Antigens (HLA-DRB1 and HLA-DRB4) gene as possible susceptibility markers for EGPA [8]. The pathogenesis of EGPA has yet to be completely understood. It is considered primarily T-cell-mediated vascular injury. CD4 positive T lymphocytes secrete gamma interferon, which promotes granulomatous inflammation. Interleukins (IL-4, IL-5, and IL-13) activate eosinophils, thus releasing proteins that cause damage to endothelial cells. This damage leads to the release of eotaxin-3 from endothelial cells, which attracts more eosinophils [8]. Based on previous data EGPA disease course has 3 different phases. The initial prodromal phase typically lasts months to years and includes arthralgia, myalgia, and fever. The eosinophilic phase is characterized by peripheral eosinophilia and the involvement of organs, including the lung, the heart, and the gastrointestinal tract. Finally, the vasculitis phase of EGPA is characterized by sensorimotor peripheral neuropathy and stroke [9]. Anti-myeloperoxidase (MPO)-ANCA positive patients having a more vasculitis phenotype, with peripheral neuropathy, purpura, renal involvement and biopsy-proven vasculitis; and MPO-ANCA negative patients being at higher risk of cardiac involvement [10].
Frequency of CNS manifestations varies from 5% to 13% [4,5]. Ischemic lesions, intracranial hemorrhage, granulomas of the spinal cord and medulla oblongata, cranial nerve palsies, and loss of visual acuity due to optic neuritis have been reported [4,5,6]. Liu et al. reported that more than 50% of patients with CNS involvement also presented gastrointestinal symptoms and fever [4]. Meanwhile peripheral neuropathy considered as a relatively common symptom of EGPA, which occurred in 46-55% of patients [4,5]. Pathophysiology of CNS involvement in EGPA is not completely defined and probably more complex than in other AAV, because the disease frequently associates vasculitis, blood and tissue eosinophilia [5]. Peripheral nerve involvement is caused by axonal degeneration due to ischemia that is secondary to damage of the vasa nervorum. Clinical characteristics are painful paraesthesia, numbness, and later motor impairment and muscular atrophy [9].
During the diagnostic procedures, laboratory and imaging techniques are used to evaluate the organ involvement as follows: computed tomography (CT) for lung involvement, magnetic resonance imaging (MRI) and electroneurography (ENG) for neurological manifestations, endoscopies in case of gastrointestinal symptoms and echocardiography, cardiac MRI for myocardial involvement. Birmingham vasculitis score (BVAS) and other scoring systems like the Five factor score (FFS) are useful to evaluate disease activity and therapeutic response [10,12]. Indirect immunfluorescent techniques and enzyme-linked immunosorbent assay (ELISA) [12] can prove presence of antibodies.
Data regarding therapeutic management and outcome of CNS involvement are scarce. Besides, central nervous system involvement in EGPA represent a huge therapeutic challenge, since it is responsible for long-term sequelae and death, especially in those with intracerebral hemorrhages [5]. Previous treatment guidelines suggested use of corticosteroids combined with cyclophosphamide (CYC) in case of life and/or organ-threatening disease manifestations (i.e., heart, gastrointestinal, central nervous system, severe peripheral neuropathy, severe ocular disease, alveolar hemorrhage and/or glomerulonephritis). Methylprednisolone pulse (0.5–1.0 g/d for 3–5 days), high-dose glucocorticoids (1–2 mg/kg/day) and low-dose regimens also were presented (<0.5 mg/kg/day) in case of mild clinical symptoms. Intravenous immunoglobulin (IVIg), plasma exchange therapy could be considered in therapy refractory cases and intrathecal injections of dexamethasone (10 mg each time) and methotrexate (MTX, 10 mg each time) were used in case of CNS involvement [4,5,6,7,8,11]. Latest guidelines prefer aggressive induction treatment as cyclophosphamide or rituximab in combination with glucocorticoids followed by maintenance therapy (with azathioprine or methotrexate). IVIg remains rescue treatment in case of infective complications [12]. The important role of eosinophils in EGPA and recent development of effective agents to treat other eosinophil-related diseases (e.g., asthma, hypereosinophilic syndrome) have created new therapeutic possibilities as the anti-interleukin-5 agent, mepolizumab. Reslizumab and the IL-5Ra- antagonist benralizumab are both promising as IL-5 is the major cytokine responsible for eosinophil activation, chemo attraction and survival. IL-4 and IL-13 are major cytokines for helper T cells (Th2) profile and eosinophil activation. Agents such as dupilumab, pitakinra, lebrikizumab block IL-4 receptor or the circulating IL-13. Ongoing trials in asthma may open new opportunities for EGPA. Interferon-alpha may be reserved as a third line treatment of therapy refractory cases [11,12].
2. Case Presentation
A 50-year-old female with a history of bronchial asthma previously treated with inhaled beta-2 agonist, antihistamine and inhaled and/or oral low-dose corticosteroids. Patient was referred to our Clinic (a tertiary center in clinical immunology) due to muscle pain, muscle weakness, and numbness in the left upper limb and right lower limb started after an upper-airway infection. MRI of the cervical spine showed protrusion of the intervertebral discs that was not consistent with neurological symptoms. During the first period of diagnostic procedures new symptoms as vomiting and abdominal pain were presented. Chest X-ray and brain CT were negative but further investigations revealed extreme eosinophilia (56%) and a suspicion of cholelithiasis with cholecystitis. Due to diffuse persistent abdominal pain, laparoscopic cholecystectomy was performed, which was converted to exploratory laparotomy due to signs of circulatory disturbance and edema in the wall of the small intestine. Significant findings during laparotomy were edematous small intestinal wall with signs of circulatory disturbance and several small (1 mm) nodules along small mesenteric vessels. Vasculitis was suspected, but unfortunately, histology was partially assessable, inflammatory cellular infiltration was reported. Meanwhile neurological symptoms worsened. ENG examinations revealed peripheral sensory and motor polyneuropathy. Elevated protein, albumin level and elevated leukocyte count was reported after analysis of cerebrospinal fluid. These follows misled the diagnosis to atypical Guillian-Barre syndrome. Laboratory findings as monoclonal gammopathy of unknown significance (MGUS), thrombocytosis, also disturbed the overall picture. Bone marrow biopsy and genetical tests excluded essential thrombocytosis. Neurological symptoms progressed to tetraparesis, areflexia, lack of pyramidal sign as well as tactile and thermal-hypaesthesia at the area of the C4 dermatome. Repeated ENG examination reported severe progressive, axonal dominant sensorimotor polyneuropathy, electromyography (EMG) revealed acute denervation of right anterior tibialis, left vastus lateralis, and right posterior deltoid muscles. Lumbar MRI showed abnormalities consistent with EGPA (spindle-shaped granulomatous lesions along L1-L3) (Figure 1).
Based on asthma, eosinophilia greater than 10% on differential white blood cell count, mononeuropathy (including multiplex), polyneuropathy, mesenterial and spinal cord granulomas and MPO positivity the diagnosis of EGPA was established. After excluding atypical bacterial and viral infections as well as malignancy and parallel to treatment of nosocomial infection, we started pulse steroid (500 mg of methylprednisolone for 3 consecutive days). Beside steroid tapering use of intravenous immunoglobulin (2gr/kg every months) helped the patient through several nosocomial infections (pneumonia, peritonitis, sinusitis and urinary tract infection). Another challenge was the treatment of malabsorption and cessation of weight, specially muscle loss. Intermittent parenteral feeding, vitamin and protein supplementation with personalized exercise program was effective. Although laboratory results improved, extreme eosinophilia seems persistent and repeated ENG showed worsening in axonal and motor neuropathy as well as mononeuritis. We started rituximab (375 mg/m2 per week for 4 weeks) as an induction treatment according to European Alliance of Associations for Rheumatology (EULAR) guideline. Unfortunately, third and fourth dosage need to be delayed due to another nosocomial and severe coronavirus (COVID-19 infection. This viral adverse event reactivated the disease and a new ischaemic/granulomatous lesion was reported in mesencephalon (Figure 2) with increasing MPO titers.
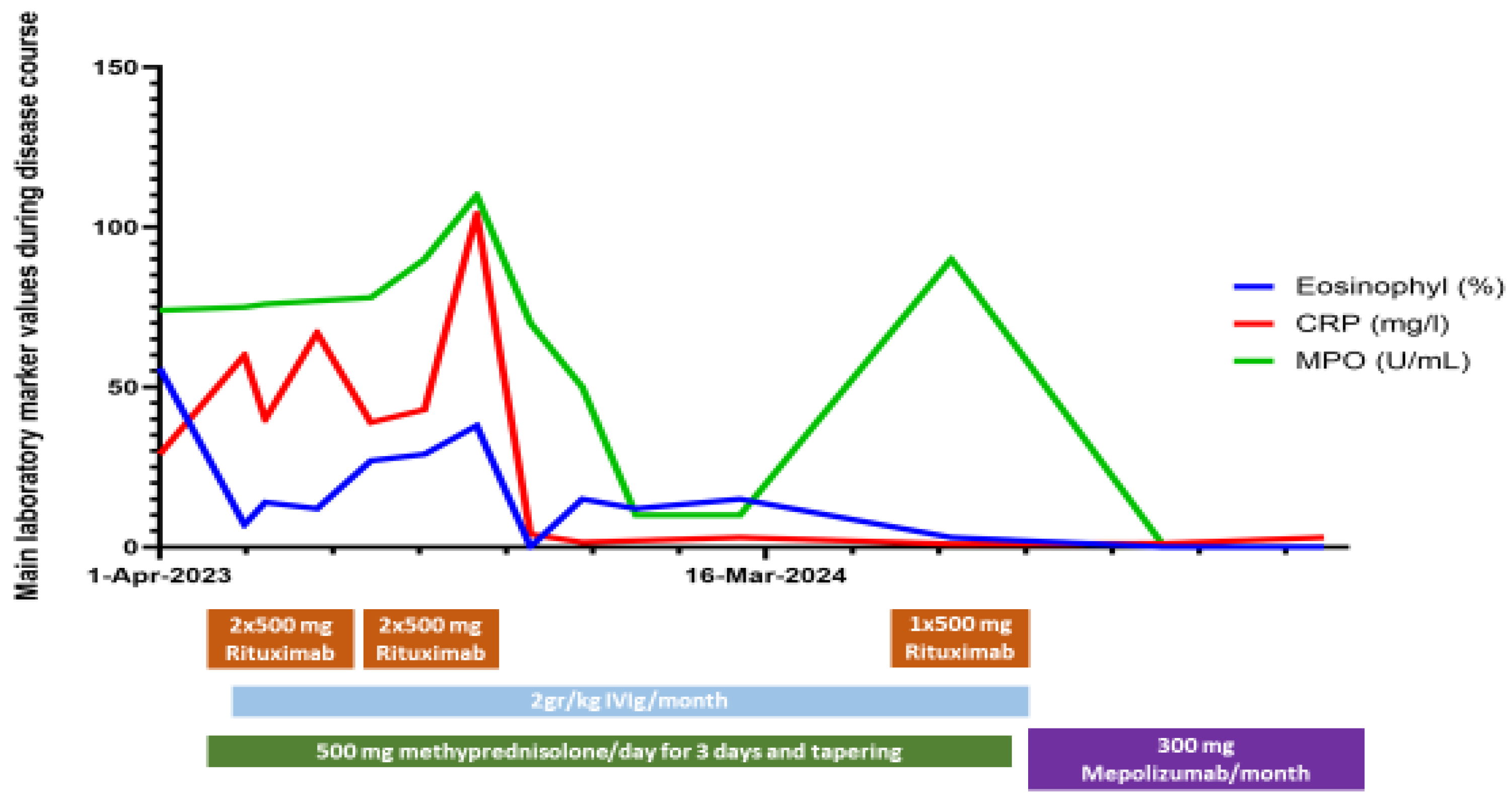
After COVID-19 infection infero-basal hypokinesis appeared on the echocardiography. Careful balance of rituximab and IVIg treatment beside steroid tapering and supportive care partial remission was achieved after 6 months of in-patient treatment. After rehabilitation programs, due to persistent eosinophilia we decided to change the planned rituximab maintaining therapy to 300 mg/months mepolizumab. Due to the high dose of methylprednisolone therapy despite all preventive effort (vitamin D and calcium supplementation, physiotherapy) patient developed osteoporosis and suffered vertebral compressions. Adding teriparatide to her basic treatment and using special auxiliary device, spinal stability improved. Today she can walk without any aid; she is self-sufficient and returned to her previous job. In the last 1,5 years patient improving continuously without relapses. Changes in laboratory markers and therapeutic regimen are presented on Figure 3.
4. Discussion
Our case presented rare and severe combined neurological manifestations of EGPA. Vasculitis-associated manifestations were mesenterial granulomas, mononeuritis, polyneuropathy, CNS ischaemia and granuloma. Eosinophil mediated component was the asthma. Its overlapping features with other vasculitic or eosinophilic diseases, and the wide and heterogeneous range of clinical manifestations, as often in this case also resulted in a delay to diagnosis. Eosinophilic and vasculitic manifestations are often intermingled. Eosinophils may be a component of vascular and perivascular infiltrates and a complete distinction between eosinophilic and vasculitis manifestations cannot be always established. Acute progressive sensory-motor polyneuropathy, which initially mimicked a Guillain-Barre syndrome also meant a diagnostic difficulty. Neuropathy also may have a vasculitic origin and/or an eosinophil-derived neurotoxicity component [13]. Uniqueness of this case the rare granulomatous CNS involvement intertwined with cerebrovascular symptoms, peripherial neuropathy and mononeuritis.
Our case also present a long and hard clinical care with several therapeutic difficulties, which all could have led to fatal outcome. In a French multicentre study, 11 of 26 patients (42%) pateints with CNS involvement died in 3 years from diagnosis [5]. Balance between immunosuppressive treatment and nosocomial infections is usually instable in such a severe case. Availability of IVIg treatment fundamentally determined the end of our case report. Generally, overall survival of EGPA seems good. A French vasculitis study group published outcome data of a cohort of 118 patients with EGPA [14]. Overall survival reached 90% at 7 years, regardless of baseline severity. Older age (≥65 years) was the only factor associated with a higher risk of death. Relapse rate was higher for patients with anti-myeloperoxidase antibody positivity. A larger American cohort with 354 patients and with median follow-up of 7 years showed 4% of mortality [15]. A retrospective cohort study from the US demonstrated that health care costs are approximately 2.5-fold higher in patients with EGPA than in patients with asthma who have similar demographic characteristics. Patients with EGPA also require more health care utilisation and systemic corticosteroid use than patients with asthma. Besides more than one third of those with EGPA experiencing relapses [16]. Chinese cohorts with CNS involvement also reported 34% of relapses during steroid reduction [4]. These results highlight the unmet need of new therapies making possible steroid tapering after induction therapy. New therapies like rituximab, mepolizumab or other biologics also can lead to less common or less severe comorbidities (as osteoporosis, hypertension and diabetes) with a steroid free maintaining treatment. Data from Spanish registry of Systemic Vasculitis (REVAS) also showed improved outcomes with significant decrease in mortality and treatment-related morbidity in patients diagnosed after 2000. Improving outcome data also was related to the implementation of less toxic regimens adapted to the disease activity and stage, and a significant reduction in glucocorticoid dose [17].
5. Conclusions
Our findings indicate that EGPA should be considered as differential diagnosis of rapidly progressing polyneuropathy with or without CNS involvement at a young age especially in the presence of eosinophilia. These patients should be managed in multidisciplinary collaboration with, or in centers with established expertise in the management of small- and medium-sized-vessel vasculitis. Patients with peripheral nerve involvement and motor deficit(s) should also be routinely referred to a physiotherapist. Careful multidisciplinary cooperation, individualized, targeted therapy, huge effort of the patient and supportive environment can show a way back from immune-mediated tetraplegia. New therapeutic options may help achieve remission, steroid sparing, and reduce comorbidities.
Author Contributions
Conceptualization, M.N-V., immunologist, M.N-V, neurologist, T.Á.; writing—original draft preparation, Y. R. and D.Sz.; writing—review and editing, M.N-V.; visualization, E.B.N.; supervision, T.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
All procedures complied with the Declaration of Helsinki. As it is a retrospective case report without any intervention, Institutional Ethical Board doesn’t need to give approval.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to privacy.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AAV | ANCA-associated vasculitis |
| ANCA | anti-neutrophil cytoplasmic antibody |
| BVAS | Birmingham vasculitis score |
| CNS | central nervous system |
| CYC | cyclophosphamide |
| CT | computed tomography |
| COVID-19 | coronavirus |
| ENG | electroneurography |
| EMG | electromyography |
| ELISA | enzyme-linked immunosorbent assay |
| EULAR | European Alliance of Associations for Rheumatology |
| EGPA | eosinophilic granulomatosis with polyangiitis |
| FFS | five factor score |
| IL | lnterleukin |
| IVIg | intravenous immunoglobulin |
| MRI | magnetic resonance imaging |
| MTX | methotrexate |
| MGUS | monoclonal gammopathy of unknown significance |
| MPO | anti-myeloperoxidase |
| REVAS | Spanish registry of Systemic Vasculitis |
| Th | helper T cells |
References
- Trivioli G, Terrier B, Vaglio A. Eosinophilic granulomatosis with polyangiitis: understanding the disease and its management. Rheumatology. 2020 May 1;59(Supplement_3):iii84–94. [CrossRef]
- White J, Dubey S. Eosinophilic granulomatosis with polyangiitis: A review. Autoimmun Rev. 2023 Jan;22(1):103219. [CrossRef]
- Comarmond C, Pagnoux C, Khellaf M, Cordier J, Hamidou M, Viallard J, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum. 2013 Jan 27;65(1):270–81. [CrossRef]
- Liu S, Guo L, Fan X, Zhang Z, Zhou J, Tian X, et al. Clinical features of central nervous system involvement in patients with eosinophilic granulomatosis with polyangiitis: a retrospective cohort study in China. Orphanet J Rare Dis. 2021 Dec 31;16(1):152. [CrossRef]
- André R, Cottin V, Saraux JL, Blaison G, Bienvenu B, Cathebras P, et al. Central nervous system involvement in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Report of 26 patients and review of the literature. Autoimmun Rev. 2017 Sep;16(9):963–9. [CrossRef]
- Rowell J., Lucas S., Dolin P Diverse clinical manifestation of EGPA: a systematic review. Conference abstract. ACR 2024. [CrossRef]
- Samson M, Puechal X, Devilliers H, Ribi C, Cohen P, Stern M, et al. Longterm outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. J Autoimmun. 2013;43:60–9. [CrossRef]
- Menon SG, Hugenberg S, Alkashash AM et al. Colitis as the Initial Presentation of Eosinophilic Granulomatosis with Polyangiitis Case Reports in Rheumatology 2023, Article ID 6620826.
- Yoo I-H, Choi ST, Choi S-H et al. Eosinophilic Granulomatosis with Polyangiitis Presented as Acute Polyneuropathy and Cerebral Vasculitis Exp Neurobiol. 2017 Jun;26(3):168-171.
- Raffray L, Guillevin L Updates for the treatment of EGPA Presse Med 49 (2020) 104036.
- Groh M, Pagnoux C, Baldini C et al. Eosinophilic granulomatosis with polyangiitis (Churg–Strauss) (EGPA) Consensus Task Force recommendations for evaluation andmanagement European Journal of Internal Medicine 26 (2015) 545–553.
- Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 updateAnnals of the Rheumatic Diseases 2024;83:30-47.
- Solans-Laque R, Rúa-Figueroa I., Blanco Aparicio M. et al. Red flags for clinical suspicion of eosinophilic granulomatosis with polyangiitis (EGPA) European Journal of Internal Medicine 128 (2024) 45–52.
- Samson M, Pu’echal X, Devilliers H, Ribi C, Cohen P, Stern M, et al. Long-term outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. J. Autoimmun. 2013 Jun;43:60–9. [CrossRef]
- Doubelt I, Cuthbertson D, Carette S, Chung SA, Forbess LJ, Khalidi NA, et al. Vasculitis clinical research consortium. clinical manifestations and long-term outcomes of eosinophilic granulomatosis with polyangiitis in North America. ACR Open Rheumatol. 2021 Jun;3(6):404–12. [CrossRef]
- Bell CF, Blauer-Peterson C, Mao J. Burden of illness and costs associated with eosinophilic granulomatosis with polyangiitis: evidence from a managed care database in the United States. J. Manag. Care Spec. Pharm. 2021;27(9):1249–59. [CrossRef]
- Solans-Laqu’e R, Fraile G, Rodriguez-Carballeira M, Caminal L, Castillo MJ, Martínez-Valle F, et al. Clinical characteristics and outcome of Spanish patients with ANCA-associated vasculitides: Impact of the vasculitis type, ANCA specificity, and treatment on mortality and morbidity. Medicine (Baltimore) 2017 Feb;96(8):e6083.
Figure 1.
Lumbar spine MRI: (A) T1-weighted sagittal image demonstrate band like intradural lesion with high signal intensity (without contrast) in the level of lumbar vertebrae L1-3. (B) In the same region T2-weighted fat-suppressed sagittal image shows also band like intradural lesion with bright signal, which excludes fat content.
Figure 1.
Lumbar spine MRI: (A) T1-weighted sagittal image demonstrate band like intradural lesion with high signal intensity (without contrast) in the level of lumbar vertebrae L1-3. (B) In the same region T2-weighted fat-suppressed sagittal image shows also band like intradural lesion with bright signal, which excludes fat content.
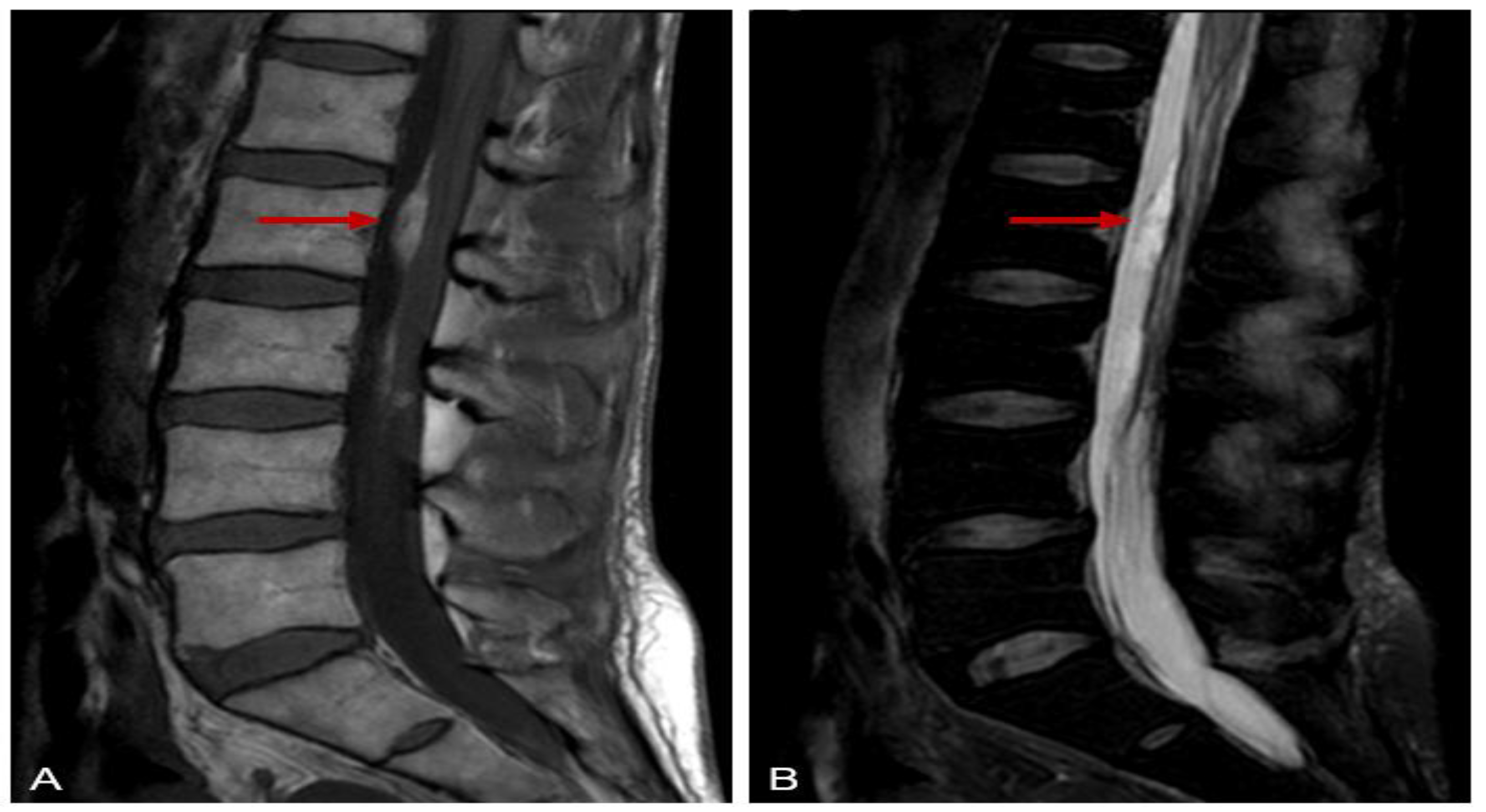
Figure 2.
Brain MRI: (A) Diffusion-weighted axial image and (B) FLAIR (Fluid Attenuated Inversion Recovery) coronal image shows a very bright acute ischaemic lesion in the mesencephalon.
Figure 2.
Brain MRI: (A) Diffusion-weighted axial image and (B) FLAIR (Fluid Attenuated Inversion Recovery) coronal image shows a very bright acute ischaemic lesion in the mesencephalon.
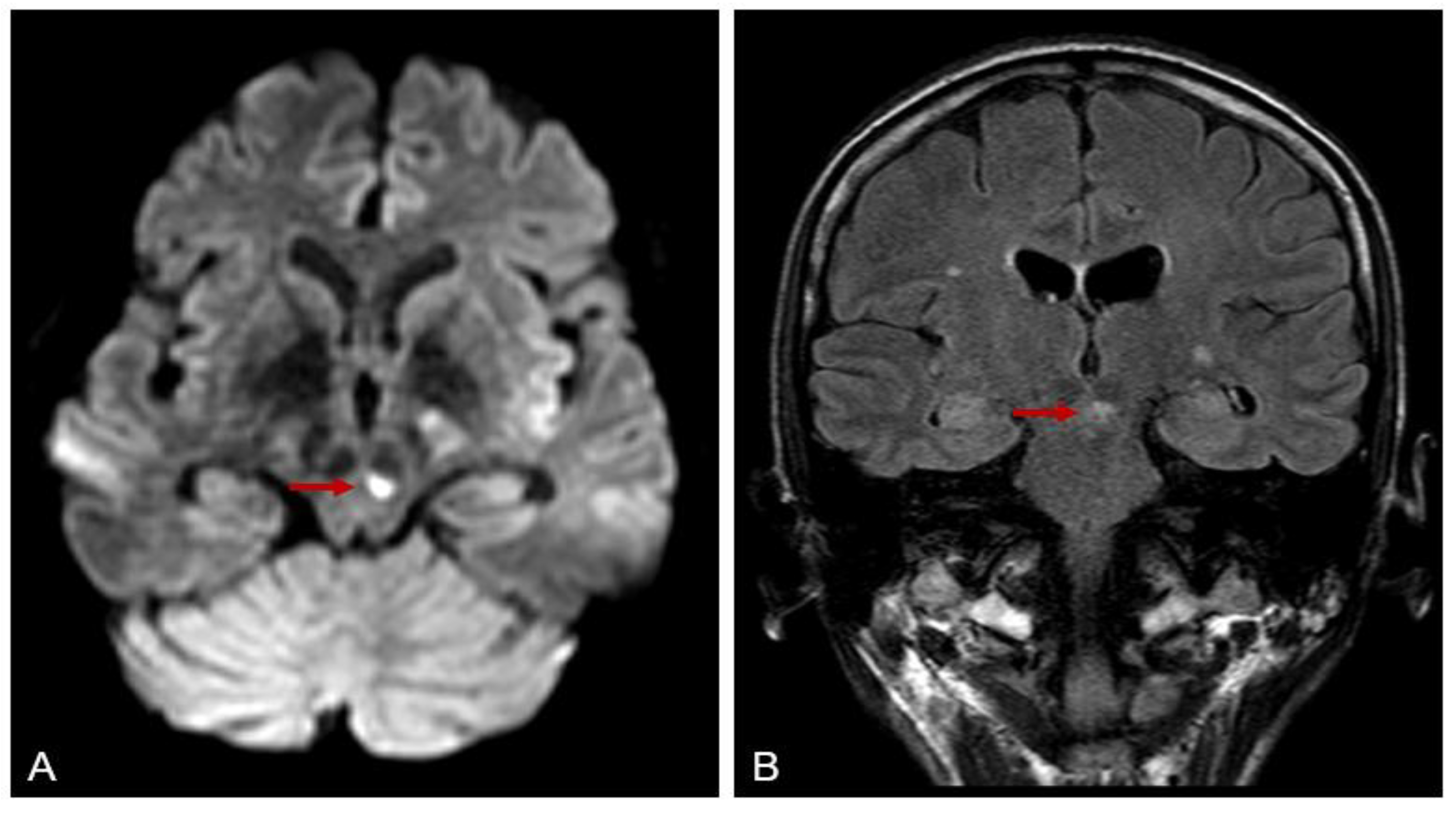
Figure 3.
Changes in most important lab values (eosinophilia (%), C-reactive protein (CRP;U/l) and anti-myeloperoxidase (MPO) antibody titer (U/ml) and treatment regimen.
Figure 3.
Changes in most important lab values (eosinophilia (%), C-reactive protein (CRP;U/l) and anti-myeloperoxidase (MPO) antibody titer (U/ml) and treatment regimen.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



