Submitted:
12 July 2025
Posted:
14 July 2025
You are already at the latest version
Abstract
Background/Objectives: Cutaneous T-cell lymphoma (CTCL) is an uncommon diagnosis in the pediatric population, and among its subtypes, subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is exceedingly rare. We present a unique case of pediatric SPTCL initially manifesting as hemophagocytic lymphohistiocytosis (HLH), with diagnosis established only after a skin biopsy. This report aims to highlight diagnostic challenges and therapeutic complexity in such presentations. Methods: We conducted a detailed case review of a pediatric patient who presented with HLH. Extensive in-fectious, autoimmune, and oncologic evaluations were performed prior to diagnostic skin biopsy. Genetic testing was also conducted to assess for germline mutations, including HAVCR2. Results: The patient was ultimately diagnosed with SPTCL following histopathological analysis of skin tissue. His disease course was marked by significant complications, including recurrent HLH episodes. Multiple therapeutic regimens, including immunosuppressants and chemotherapy, were trialed with varying success. Genetic analysis revealed a HAVCR2 mutation, raising important considerations regarding disease pathogenesis and therapeutic direction. Conclusions: This case underscores the importance of considering SPTCL in pediatric HLH of unknown origin and highlights the diagnostic value of skin biopsy. The presence of a HAVCR2 mutation complicates treatment planning and calls into question the appropriateness and timing of hematopoietic stem cell transplantation in this rare population. Greater understanding of the molecular drivers of SPTCL is needed to guide future management strategies.
Keywords:
SPTCL
; HLH
; HAVCR2
1. Introduction
Primary cutaneous lymphomas are defined as non-Hodgkin lymphomas presenting in the skin without evidence of extracutaneous disease. Primary cutaneous lymphomas include two groups: cutaneous T-cell lymphomas (CTCLs) and cutaneous B-cell lymphomas (CBCLs). According to the 2018 WHO guidelines, within CTCL, there is a subtype called subcutaneous panniculitis-like T cell lymphoma (SPTCL) [1]. The overall CTCL incidence is 8.55 per million people [2]. 1% of those diagnosed with primary CTCL have SPTCL; however, this data’s sample primarily consists of adults [1].
The presence of hemophagocytic lymphohistiocytosis (HLH) at SPTCL diagnosis is associated with inferior survival rates [3]. Overall, SPTCL has an 82% 5-year survival rate. However, in a study by Willemze et al., patients with SPTCL complicated by HLH had a statistically significant (P< 0.001) worse 5-year overall survival (46%) compared to patients without HLH (91%) [4].
HLH is a hyperinflammatory disorder initiated by a pathologic activation of cytotoxic T-lymphocytes and macrophages, which leads to a cytokine storm and multi-organ failure. HLH can be either primary due to genetic predisposition or secondary due to malignancy or infection [5].
Results
Written informed consent for this case report was obtained from the patient’s guardian. We report the case of a 17-year-old previously healthy male of Polynesian descent presenting with a three-month history of tachycardia; daily fevers with Tmax of 102F; fatigue; night sweats; anorexia; abdominal pain; emesis; fifty pound weight loss; and multiple 1-4cm, painful, raised, purple-reddish skin lesions on his upper and lower extremities without ulcerations (Figure 1). His medical and family histories were unremarkable except for his father with diabetes. On examination, the patient had upper and lower extremity weakness and was febrile (38.6°C). He had cervical lymphadenopathy, hepatosplenomegaly, and multiple erythematous and tender nodules of variable size.
Laboratory investigations showed normocytic anemia, absolute lymphopenia, and marked transaminitis. Further workup revealed a significantly elevated ferritin (>12,000), and low fibrinogen (127). His broad infectious workup was negative. An extensive rheumatologic work-up proved inconclusive. A liver biopsy displayed steatohepatitis without signs of autoimmune hepatitis. Due to elevated stool calprotectin 2,990 that was suggestive of IBD, an EGD and colonoscopy with biopsies were completed, which were notable for gastritis but were otherwise unremarkable [6].
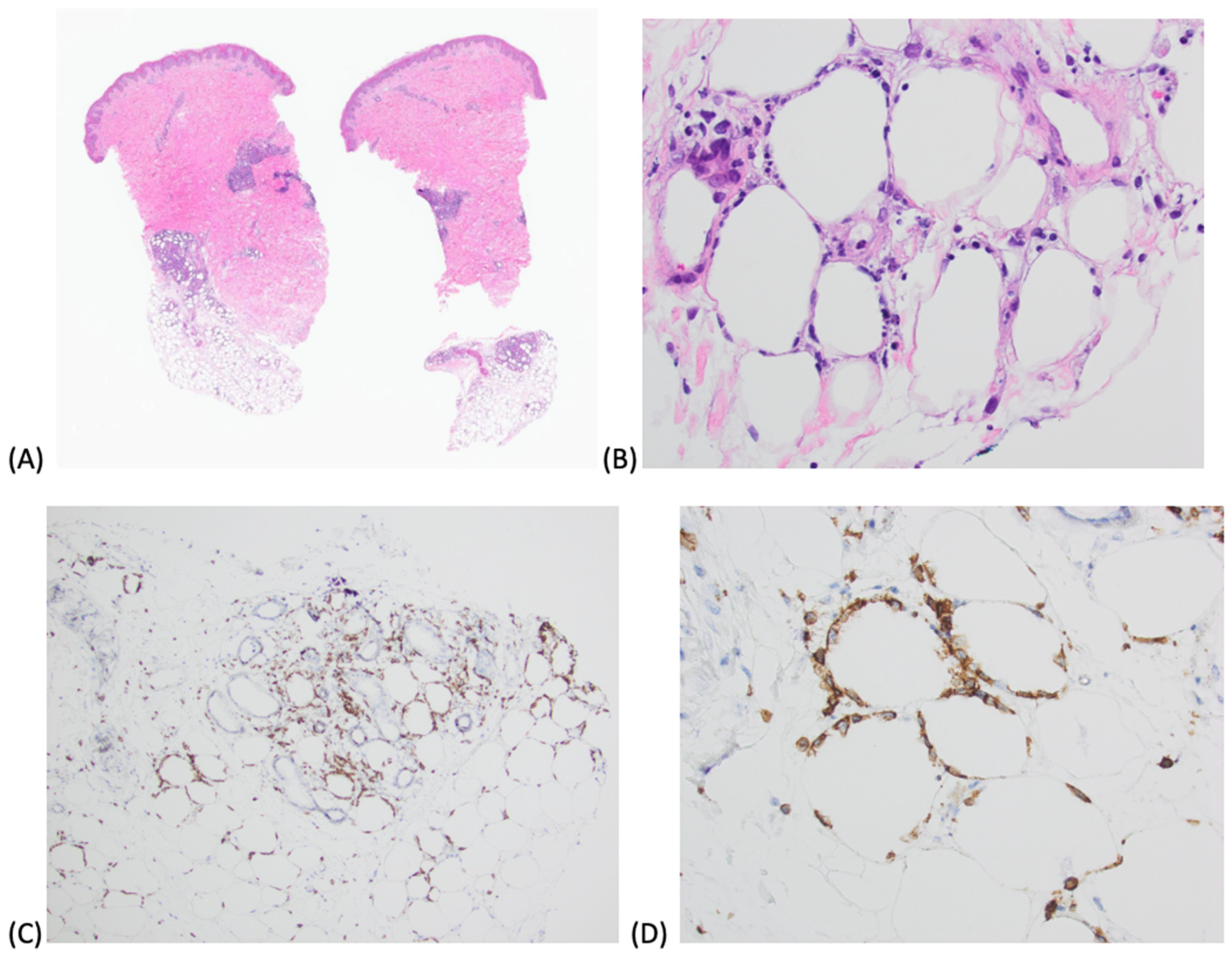
Peripheral blood and bone marrow flow cytometry were unremarkable. Bone marrow biopsy showed no definitive hemophagocytosis. A skin punch biopsy of a nodule was obtained which revealed deep dermal and subcutaneous lymphocytic infiltrate, consistent with SPTCL (Figure 2). Immunohistochemistry supported his SPTCL diagnosis as his lymphocytes were positive for CD3, CD8, and betaF1 (T-cell receptor beta chain), and negative for CD30 and Fite stain.
He met 5 of 8 HLH criteria (fever, splenomegaly, low fibrinogen, elevated ferritin, and elevated soluble CD25) [7]. A Hscore is a validated tool used in adults to estimate the risk of having hemophagocytic syndrome [8]. His Hscore was 254 points with >99% probability of HLH. His HLH genetic panel was sent and was unremarkable. A diagnostic lumbar puncture showed no evidence of HLH and a port-a-cath was placed for long-term access. Per PEDS HLH-94 protocol, he completed an 8-week course of dexamethasone and Etoposide twice weekly for 2 weeks and then weekly for 6 weeks [9]. Following treatment, he had significant improvement in clinical symptoms and laboratory findings.
More superficial forms of CTCL, such as mycosis fungoides and Sézary syndrome, are amenable to phototherapy with ultraviolet A radiation (PUVA) or extracorporeal photochemotherapy (ECP) treatment [10]. However, after consultation with a dermatologist with expertise in CTCL, due to the depth of his lesions, PUVA/ECP were not deemed feasible. Review of literature showed best outcomes with treatment of HLH followed by treatment for T-cell lymphoma [11].
His skin biopsy following induction showed focal lymphocytic infiltrate with limited adipocyte rimming suggestive of minimal residual SPTCL, consistent with response to therapy. His PET scan showed multiple ill-defined soft tissue nodules in the abdomen and pelvis as well as innumerable cutaneous and subcutaneous soft tissue nodules with increased FDG uptake, consistent with his known cutaneous T-cell lymphoma residual disease.
Per T-cell lymphoma Children’s Onocology Group (COG AALL 0434/1231) protocol, he completed interim maintenance I with vincristine, methotrexate, intrathecal methotrexate, and Cal-asparaginase [12,13]. His course was complicated by mucositis, hyperglycemia, and hypovolemic shock with cardiac dysfunction requiring pressors. His ECHO had left ventricular diastolic dysfunction. In addition, he had prolonged QTC up to 550ms, which required adjustments to his anti-emetic regimen. He was discharged after four weeks of hospitalization. He was given leucovorin and reduced methotrexate dosages for subsequent courses.
One month after discharge, he started vorinostat for 3-4 days but was hospitalized for three weeks due to liver dysfunction and dehydration. Vorinostat is a histone deacetylase inhibitor used to treat CTCL in patients with progressive, persistent, or recurrent disease [14]. While in the ICU, he met Baltimore criteria for hepatic veno-occlusive disease with peak total bilirubin 7.4mg/dL, weight gain >5%, ascites, and painful hepatomegaly [15]. His abdominal ultrasound showed dampened monophasic appearance of hepatic venous waveforms, however flow appeared antegrade. His transaminitis and hyperbilirubinemia improved, and he did not start Defibrotide.
A repeat skin biopsy showed no evidence of residual CTCL. Additionally, his PET scan showed a significant decrease in size and FDG activity in his cutaneous and subcutaneous lesions. Following induction per HLH-94, he was anticipated to complete therapy for T-cell lymphoma. However, after presentation at a regional tumor board, due to his severe sensitivity to methotrexate and other agents, it was decided to pursue romidespin monotherapy.
Thus far, he has completed 7 bimonthly cycles of reduced romidespin (7 mg/m2) due to elevated total bilirubin levels and transitioned to full monthly dosaging (14.2 mg/m2) once his bilirubin levels normalized. He has completed Cycle 12 and PET scan shows he is in remission.
Discussion
This case report details a diagnosis of primary SPTCL in a pediatric patient. Its varying presentation makes diagnosis and treatment challenging as there is no standardized SPTCL treatment protocol.
Unfortunately, SPTCL can be complicated by HLH in 15-20% of cases, and often causes mortality [16]. Our patient’s genetic testing was notable for a missense variant in the HAVCR2 gene—c.245A>G (p.Tyr82Cys.)—which has been found to alter T-cell immunoglobulin mucin 3 (TIM-3) [17]. TIM-3 is expressed on some subgroups of T and innate immune cells. In patients with homozygous p.Tyr82Cys mutation, it was found that TIM-3 expression was dramatically decreased which led to the absence of a regulator of innate immunity and inflammatory responses. This variant was also found to be more prevalent in Polynesian and East Asian Populations. From Gayden et al., of the 12 patients he surveyed with altered TIM-3 SPTCL, 6 had complete HLH and the others met criteria for incomplete HLH. This indicates that TIM-3 mutations could be a causative genetic defect in SPTCL-HLH and hold future potential for targeted immunotherapy.
Due to his adverse prognostic factors—presence of HLH and HAVCR2 gene mutation— he is at higher risk for relapse or being refractory to treatment. Typically, patients who relapse or are refractory to treatment proceed towards a stem cell transplant (SCT) with promising results [18]. However, it is difficult to determine if our patient should preemptively proceed towards a SCT versus continuing his current treatment as he is clinically improving. The lack of standard treatment protocols for these patients indicates the need for additional research and clinical trials for SPTCL, especially in the pediatric population.
5. Conclusion
This case underscores the diagnostic and therapeutic complexity of pediatric subcutaneous panniculitis-like T-cell lymphoma (SPTCL), particularly when it presents as hemophagocytic lymphohistiocytosis (HLH) without an identifiable infectious or autoimmune trigger. The diagnostic delay prior to skin biopsy reflects the difficulty of recognizing SPTCL in a pediatric patient, especially when early signs are nonspecific and mimic other systemic inflammatory disorders. Our patient’s clinical course was complicated by treatment-resistant HLH, recurrent cytopenias, and hepatic dysfunction, requiring a multifaceted therapeutic approach including corticosteroids, etoposide, cyclosporine, ruxolitinib, and ultimately hematopoietic stem cell transplantation (HSCT). The identification of a pathogenic HAVCR2 mutation, which is increasingly recognized in association with HLH and SPTCL, offers valuable insight into the underlying pathophysiology and may inform future therapeutic decision-making. However, the role of HSCT in patients with HAVCR2-mutated SPTCL remains poorly defined and warrants further investigation. This case contributes to the limited body of literature on pediatric SPTCL and highlights the urgent need for multicenter collaboration to characterize genotype-phenotype correlations, clarify long-term outcomes, and develop consensus guidelines for optimal management in this rare and often life-threatening disease.
Author Contributions
Meha Krishnareddigari and Jessa Rose A. Li contributed to case analysis, literature review, and manuscript writing. Maxwell A. Fung provided dermatopathology interpretation. Arun Panigrahi supervised clinical care and reviewed the manuscript. All authors approved the final version.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of the University of California, Davis (protocol code IRB #2448257, approved May 2023).
Informed Consent Statement
Written informed consent was obtained from the patient’s legal guardians to publish this case report and any accompanying images.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Acknowledgments
We would like to thank the pediatric hematology-oncology clinical team at UC Davis Health for their continued guidance and support during the patient’s care and follow-up. During the preparation of this manuscript, the authors used ChatGPT-4 (OpenAI, 2025) for editorial suggestions and formatting guidance. The authors have reviewed and edited the output and take full responsibility for the content of this publication. UC Davis Children’s Hospital.
Conflicts of Interest
Arun Panigrahi is a paid speaker for Alexion Pharmaceuticals for Eculizumab/Ravulizumab for atypical HUS.
Abbreviations
The following abbreviations are used in this manuscript:
| Abbreviation | Definition |
| CBCL | Cutaneous B-cell Lymphomas |
| COG | Children’s Onocology Group |
| CTCL | Cutaneous T-cell Lymphoma |
| ECP | Extracorporeal Photochemotherapy |
| HLH | Hemophagocytic Lymphohistiocytosis |
| PUVA | Phototherapy with Ultraviolet A Radiation |
| SCT | Stem Cell Transplant |
| SPTCL | Subcutaneous Panniculitis-like T-cell Lymphoma |
| TIM-3 | T-cell immunoglobulin mucin 3 |
References
- Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, Jaffe ES. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. 2019 Apr 18;133(16):1703-1714. Epub 2019 Jan 11. Erratum in: Blood. 2019 Sep 26;134(13):1112. PMID: 30635287; PMCID: PMC6473500. [CrossRef]
- Cai ZR, Chen ML, Weinstock MA, Kim YH, Novoa RA, Linos E. Incidence Trends of Primary Cutaneous T-Cell Lymphoma in the US From 2000 to 2018: A SEER Population Data Analysis. JAMA Oncol. 2022;8(11):1690–1692. [CrossRef]
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004 Sep 15;101(6):1404-13. PMID: 15368328. [CrossRef]
- Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C, Canninga-van Dijk MR, Carlotti A, Geerts ML, Hahtola S, Hummel M, Jeskanen L, Kempf W, Massone C, Ortiz-Romero PL, Paulli M, Petrella T, Ranki A, Peralto JL, Robson A, Senff NJ, Vermeer MH, Wechsler J, Whittaker S, Meijer CJ; EORTC Cutaneous Lymphoma Group. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008 Jan 15;111(2):838-45. Epub 2007 Oct 12. PMID: 17934071. [CrossRef]
- Knauft, J., Schenk, T., Ernst, T. et al. Lymphoma-associated hemophagocytic lymphohistiocytosis (LA-HLH): a scoping review unveils clinical and diagnostic patterns of a lymphoma subgroup with poor prognosis. Leukemia 38, 235–249 (2024). [CrossRef]
- Turner D, Ricciuto A, Lewis A, Ferdinando D, Dhaliwal J, et al. STRIDE-II: An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology. 160, 1570-1583 (2021). [CrossRef]
- Zhang K, Astigarraga I, Bryceson Y, et al. Familial Hemophagocytic Lymphohistiocytosis. 2006 Mar 22 [Updated 2024 Jun 6]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Table 1. [HLH-2004 Diagnostic Criteria]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1444/table/hlh.T.hlh2004_diagnostic_criteria/.
- Fardet, L., Galicier, L., Lambotte, O., Marzac, C., Aumont, C., Chahwan, D., Coppo, P., & Hejblum, G. (2014). Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis & rheumatology (Hoboken, N.J.), 66(9), 2613–2620. [CrossRef]
- Henter, J. I., Samuelsson-Horne, A., Aricò, M., Egeler, R. M., Elinder, G., Filipovich, A. H., Gadner, H., Imashuku, S., Komp, D., Ladisch, S., Webb, D., Janka, G., & Histocyte Society (2002). Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood, 100(7), 2367–2373. [CrossRef]
- Trautinger, F. Phototherapy of cutaneous T-cell lymphomas. Photochem Photobiol Sci 17, 1904–1912 (2018). [CrossRef]
- Daver, N., McClain, K., Allen, C. E., Parikh, S. A., Otrock, Z., Rojas-Hernandez, C., Blechacz, B., Wang, S., Minkov, M., Jordan, M. B., La Rosée, P., & Kantarjian, H. M. (2017). A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer, 123(17), 3229–3240. [CrossRef]
- Teachey, D. T., Devidas, M., Wood, B. L., Chen, Z., Hayashi, R. J., Hermiston, M. L., Annett, R. D., Archer, J. H., Asselin, B. L., August, K. J., Cho, S. Y., Dunsmore, K. P., Fisher, B. T., Freedman, J. L., Galardy, P. J., Harker-Murray, P., Horton, T. M., Jaju, A. I., Lam, A., Messinger, Y. H., … Raetz, E. A. (2022). Children’s Oncology Group Trial AALL1231: A Phase III Clinical Trial Testing Bortezomib in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia and Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 40(19), 2106–2118. [CrossRef]
- Dunsmore KP, Winter SS, Devidas M, Wood BL, Esiashvili N, Chen Z, Eisenberg N, Briegel N, Hayashi RJ, Gastier-Foster JM, Carroll AJ, Heerema NA, Asselin BL, Rabin KR, Zweidler-Mckay PA, Raetz EA, Loh ML, Schultz KR, Winick NJ, Carroll WL, Hunger SP. Children’s Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J Clin Oncol. 2020 Oct 1;38(28):3282-3293. Epub 2020 Aug 19. PMID: 32813610; PMCID: PMC7526719. [CrossRef]
- Bhupinder S. Mann, John R. Johnson, Martin H. Cohen, Robert Justice, Richard Pazdur, FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma, The Oncologist, Volume 12, Issue 10, October 2007, Pages 1247–1252, . [CrossRef]
- Cairo MS, Cooke KR, Lazarus HM, Chao N. Modified diagnostic criteria, grading classification and newly elucidated pathophysiology of hepatic SOS/VOD after haematopoietic cell transplantation. Br J Haematol. 2020 Sep;190(6):822-836. Epub 2020 Mar 4. PMID: 32133623; PMCID: PMC7483983. [CrossRef]
- Tran NT, Nguyen KT, Le LT, Nguyen KT, Trinh CT, Hoang VT. Subcutaneous Panniculitis-Like T-Cell Lymphoma With Hemophagocytic Lymphohistiocytosis. Journal of Investigative Medicine High Impact Case Reports. 2024;12. [CrossRef]
- Gayden, T., Sepulveda, F., Khuong-Quang, D, et al. Germline HAVC2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nature Genetics. (2018).
- Gibson, J. F., Alpdogan, O., Subtil, A., Girardi, M., Wilson, L. D., Roberts, K., & Foss, F. (2015). Hematopoietic stem cell transplantation for primary cutaneous γδ T-cell lymphoma and refractory subcutaneous panniculitis-like T-cell lymphoma. Journal of the American Academy of Dermatology, 72(6), 1010–5.e5. [CrossRef]
Figure 1.
Cutaneous findings at the initial presentation.
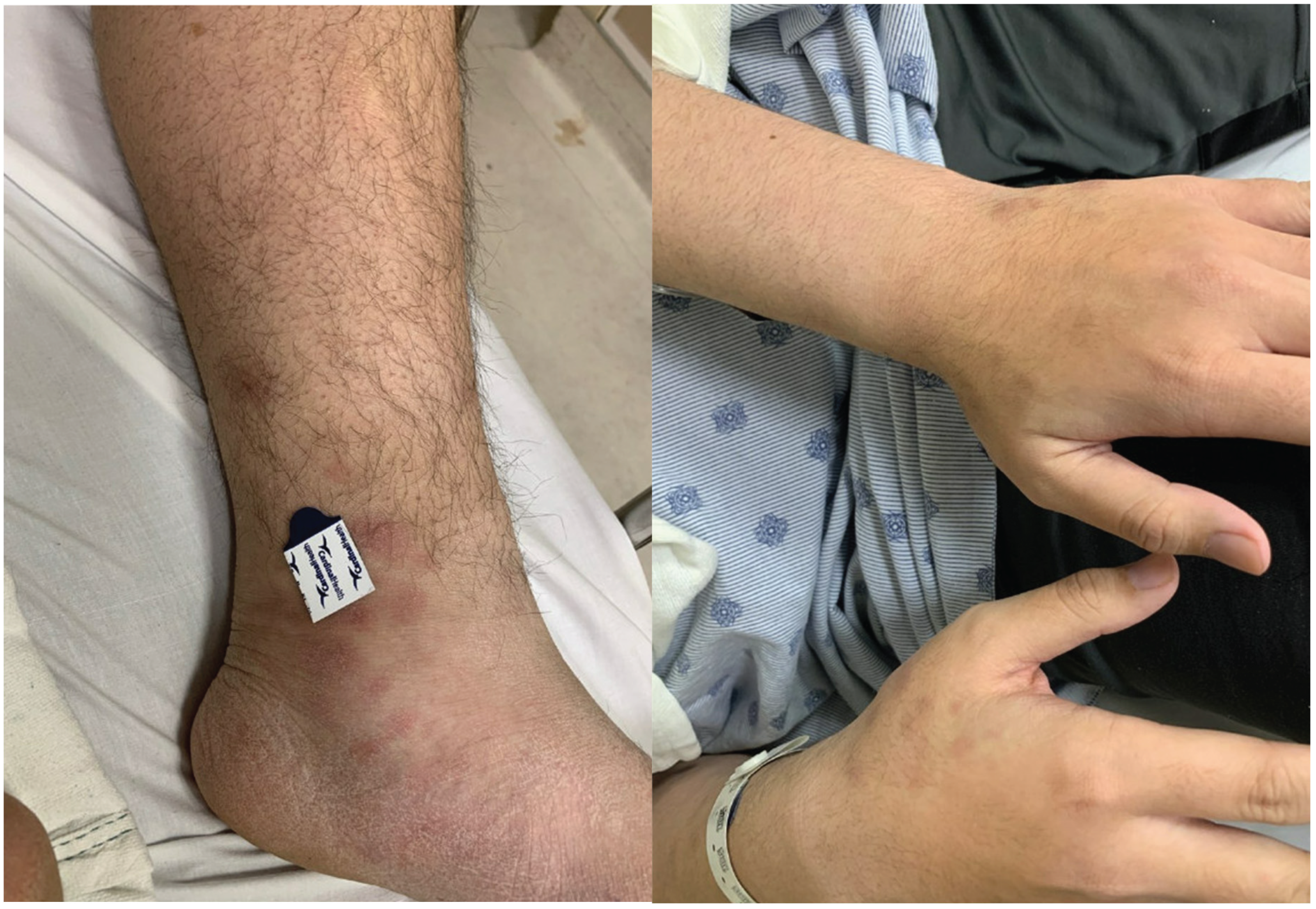
Figure 2.
(A) Subcutaneous panniculitis-like T cell lymphoma (SPTCL). A) Biopsy shows a lobular subcutaneous lymphocytic infiltrate (20x, H&E); B) Adipocyte rimming is characteristic of SPTCL. (400x, H&E); CD8 highlights the lesional T cells (C: 200x, D: 400x)).
Figure 2.
(A) Subcutaneous panniculitis-like T cell lymphoma (SPTCL). A) Biopsy shows a lobular subcutaneous lymphocytic infiltrate (20x, H&E); B) Adipocyte rimming is characteristic of SPTCL. (400x, H&E); CD8 highlights the lesional T cells (C: 200x, D: 400x)).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



