Submitted:
08 December 2025
Posted:
12 December 2025
You are already at the latest version
Abstract
Multiple sclerosis is an immune-driven neurological disease that affect myelinated axons in the central nervous system. However, the trigger of the (dysregulated) immune reactions is not56 known. According to Wilkin’s primary lesion theory, myelin-reactive T cells present in the immune repertoire respond to myelin antigens that are released from idiopathic lesions within the central nervous system. However, neither the cause of the primary lesion, nor the cause of the immune hyper-reactivity are known. We investigated whether these unknown activation signals may be relayed by common herpesviruses. The results highlight human herpesvirus-6A as a potential trigger of primary lesions due to its proven capacity to cause oligodendrogliopathy, cytomegalovirus as a trigger for the formation of effector memory cytotoxic T cells with proven capacity to induce multiple sclerosis pathology in a non-human primate MS model and Epstein Barr Virus due to its capacity to render B cells capable to effectively present a critical myelin antigen to these effector memory cytotoxic T cells. These results lead us to propose the novel paradigm that the immunopathogenesis of multiple sclerosis results from a conspiracy of common herpesviruses.
Keywords:
EBV
; CMV
; HHV-6
; oligodendrogliopathy
; autoimmunity
1. Introduction
Multiple sclerosis (MS) is a chronic neurological disorder causing damage to myelinated axons within the central nervous system (CNS) (Noseworthy et al., 2000). Although broad consensus exists that MS is an immune-mediated disease, the trigger of the immunopathogenic process is unknown.
According to a prevalent outside-in paradigm exposure to a microbial agent elicits the activation of autoreactive T and B cells present in the peripheral immune repertoire. Activated T cells acquire the capacity to transmigrate the blood-brain-barrier (BBB). By interaction with local antigen presenting cells (APC) pathophysiological reactions are elicited that lead to damage of axon-myelin units (Kawakami and Flugel, 2010). The γ1-herpesvirus Epstein Barr Virus (EBV) is the strongest candidate trigger (Soldan and Lieberman, 2023). A concept connecting virus infection with immunity to tissue proteins is ‘molecular mimicry’, being the sharing of immunological ‘look-alike’ motifs between the infectious agent and the target antigen (Libbey et al., 2014a). Examples of mimicry reactions relevant for EBV and MS were published for myelin-specific T cells (Lang et al., 2002) and clonally expanded MS B cells (Lanz et al., 2022).
A more recently developed inside-out paradigm posits that MS is an immunological convolution between primary oligodendrogliopathy and an aberrant immune response of the host (Stys et al., 2012). A mechanistic underpinning is given by Wilkin’s primary lesion theory for autoimmune diseases (’t Hart et al., 2021; Wilkin, 1990), which postulates two integrated processes: 1. Presence of lesions within the target organ from which antigens are released; 2. patients developing an autoimmune disease are high immune responders against released antigens.
Here we discuss whether by integrating published functions of MS-related herpesviruses in Wilkin’s primary lesion theory a viable concept for the initiation and perpetuation of the CNS directed immune attack in MS can be created.
2. Main Players in the Immune Response and Their Interaction in MS Animal Models
The current understanding of immunopathogenic mechanisms in MS is strongly influenced by the mouse experimental autoimmune encephalomyelitis (EAE) model. This model is created by immunization of genetically susceptible strains of inbred strains of immunologically naïve (SPF-bred) laboratory mice with myelin proteins (e.g. MBP, PLP or MOG) formulated with a strong bacterial adjuvant (CFA). This evokes MS-like symptoms and pathology (Baxter, 2007).
The autoimmune attack on the CNS is led by CD4+ T-cells, which are activated through 3 signals (Krovi and Kuchroo, 2022):
Signal 1: An antigenic peptide bound to an major histocompatibility complex (MHC)-II molecule that is recognized by T cells with their specific antigen receptor.
Signal 2: Cognate interaction of co-stimulatory molecules expressed on the T cell and the APC. The expression of co-stimulatory molecules is induced by conserved molecular structures present in viruses and bacteria that signal through evolutionary conserved innate receptors, such as Toll- or Nod-like receptors (Matzinger, 1994). The relay of signal 1 without signal 2 inactivates the CD4+ T cell and abrogates its pathogenic role in the EAE model. The idea that the trigger of MS must be a microbial infection stems from the dependence of EAE induction from signal 2.
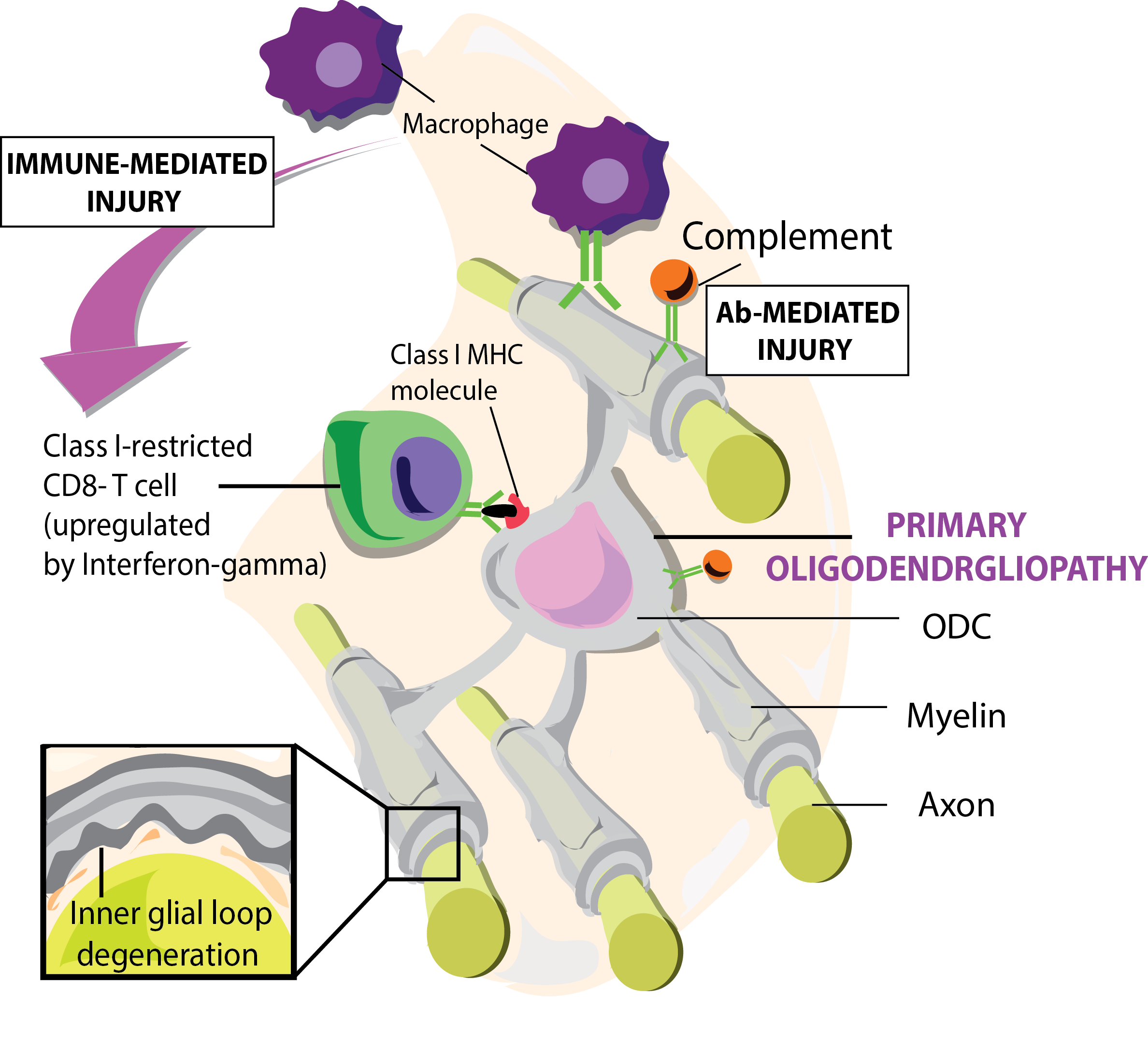
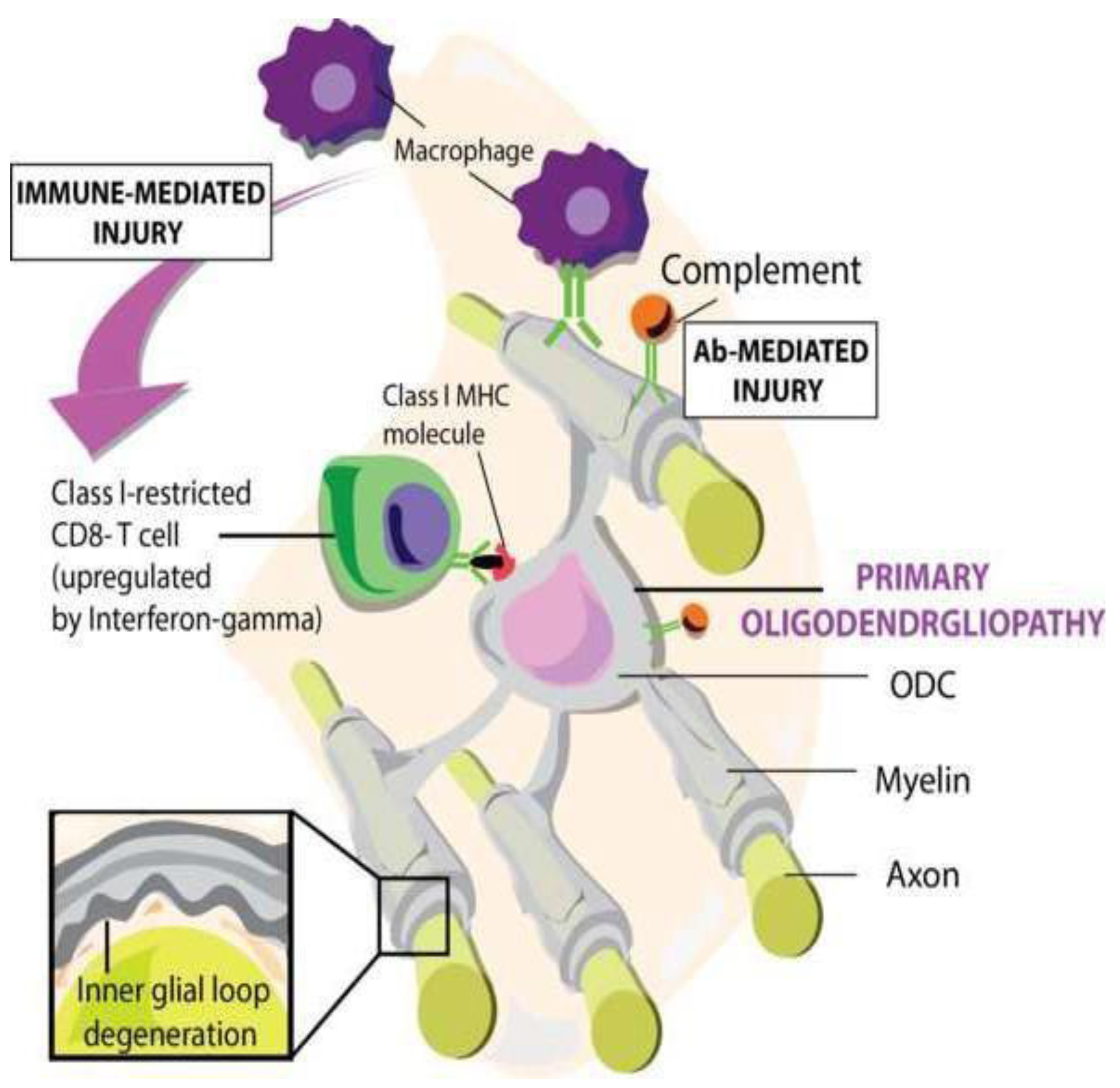
Signal 3: The function of activated T cells is determined by cytokines produced by the APC and tissues cells, which direct activated T cells in pro- or anti-pathogenic direction. The permeabilization of the BBB by CD4+ T-cell-mediated inflammation facilitates secondary infiltration of CD8+ T cells, B cells, macrophages, antibodies and complement factors. Damage to axon-myelin-units (AMU) is mediated by CD8+ T cells attacking oligodendrocytes and/or by antibodies binding to oligodendrocytes and myelin sheaths evoking cytotoxicity of macrophages (ADCC) and /or complement factors (CDC) (Figure 1).
The EAE model is frequently criticized for the artificial manner pathology and symptoms are induced, especially the dependence on CFA (Sriram and Steiner, 2005). Virus-induced MS models, such as with Theiler’s murine encephalomyelitis (TMEV), corroborate the concept that a CNS targeting immune attack can be elicited by a virus (Libbey and Fujinami, 2021). The TMEV SJL/J mouse model is particularly revealing because of the pathological similarities with MS. Immunostaining combined with electron microscopy detected TMEV viral antigens within the inner and outer myelin loops of myelin, indicating primary infection of oligodendrocytes (Rodriguez et al., 1983). Structural studies showed that axonal damage occurred even when the myelin sheath appears intact - with swollen axons, vacuoles, and abnormal spacing between myelin lamellae. These changes weaken the myelin structure, compromise its insulating function and impair axonal signal conduction (Libbey et al., 2014b; Lipton and Dal Canto, 1976).
Difficulties in the translation of scientific findings in mouse EAE models into effective therapies for MS patients raised doubts about the validity of the mouse EAE-based MS pathogenic concept (t Hart et al., 2021). A major discrepancy is that in the EAE model disease can be eliminated by treatment with anti-CD4 antibody (Biasi et al., 1997), whereas this treatment had no or sometimes detrimental effects in MS (van Oosten et al., 1997). A likely explanation is the fundamental differences between the immune systems of genetically homogeneous populations of 10 weeks old SPF-bred laboratory mice and those of a genetically diverse adult population MS patients living in dirty environments (Brodin and Davis, 2017). The human immunological condition is more closely reproduced in captive-bred colonies of non-human primates (‘t Hart et al., 2011). A well-validated EAE model in marmosets revealed a central pathogenic role of MHC-E restricted CD8+ve effector memory cytotoxic T cells (EM- CTL) specific for an epitope (residues 40-48) from the CNS myelin component myelin oligodendrocyte glycoprotein (MOG). These T cells could be directly activated in vivo by immunization with a synthetic MOG peptide (residues 34-56) formulated with a mineral oil (incomplete Freund’s adjuvant, IFA) that lacks the indispensable alarm signals flagging danger to immune cells in mouse EAE models (Jagessar et al., 2010). The requisite APC for these EM- CTL are B cells infected with the EBV-related γ1-herpesvirus Callithricine herpesvirus-3 (CalHV3), which process the epitope and present it via MHC-E molecules (Jagessar et al., 2012). An ex vivo study in EBV-infected B cells revealed that the virus infection is essential for productive processing of the EM-CTL epitope from the immunizing MOG34-56 peptide (Jagessar et al., 2016; Morandi et al., 2017). Thus activated EM-CTL evoke CNS pathology that strikingly reproduces MS pathology, including inflammatory demyelination in the white and grey matter of brain and spinal cord and the disappearance of oligodendrocytes from lesions (‘t Hart et al., 2017).
The MOG34-56 peptide shares a mimicry motif with the major capsid protein of cytomegalovirus (CMV) (Brok et al., 2007). Moreover, a similar population of HLA-E- restricted, CMV-specific EM-CTL has been found in humans (Pietra et al., 2003). We thus hypothesized that the original trigger of the EM-CTL might be a simian CMV. Apparently, the involvement of herpesviruses compensates the lack of signal 2 for pathogenic T cells activation.
3. Herpesviruses Associated with Immune Function
3.1. EBV
EBV (aka human herpesvirus 4; HHV-4), is a γ1-herpesvirus in the genus lymphocryptovirus (LCV) that causes mostly asymptomatic infections in the human population. LCVs are commonly found in both Old and New World primates, where the virus can cause lymphoproliferative disorders, just like EBV does in humans (Muhe and Wang, 2015). Infection occurs mostly by oral transfer of saliva, hence the name kissing disease, and by genital secretions. In Western populations, about half of all five-year-old children and about 90% of adults carry the virus. Infants become susceptible to EBV as soon as maternal antibody protection disappears. Most infected children display no symptoms or symptoms resembling mild childhood illnesses. Infection at adolescent age can cause a more serious clinical condition, infectious mononucleosis. Most infected persons gain adaptive immunity. Once EBV’s initial lytic infection is brought under control, EBV latency persists lifelong in a person’s memory B cells (Thorley-Lawson, 2015).
A strong association of EBV infection with MS, has been suspected for decades but was convincingly proven only in recent years (Bjornevik et al., 2023; Soldan and Lieberman, 2023). The main target of EBV in the immune system is the (memory) B cell (Thorley-Lawson, 2001). The pathogenic role of this cell-type in MS has been underestimated for many years, but gained traction after the discovery that their depletion with a monoclonal antibody against the B- lineage marker CD20 exerts remarkable beneficial effects in relapsing and progressive MS (Hauser et al., 2008; Montalban et al., 2017).
3.2. CMV
CMV (aka human herpesvirus-5, HHV-5) is a β-herpesvirus in the genus Cytomegalovirus that comprises also cytomegaloviruses of non-human primates (Barry and William Chang, 2007). CMV is widely spread throughout the human body. Person to person transmission occurs amongst others via saliva. The infection is usually asymptomatic, but in immunocompromised persons (AIDS, organ transplant recipients) the virus can cause life-threatening clinical problems (Steininger, 2007). Upon infection of healthy persons, the virus remains life-long latent in the body, mainly in myeloid (precursor) cells (Hahn et al., 1998).
CMV engages in complex interactions with the immune system. At increasing age a substantial part of the immune system becomes involved in the maintenance of CMV latency (Sylwester et al., 2005). CMV also exerts a strong influence on the aging of the immune system (immunosenescence) (Goronzy and Weyand, 2013). Immunosenescence implies strong diversity contraction in the naïve and memory T cell compartments contrasting with expansion of CMV-reactive memory T cell clones (Goronzy and Weyand, 2013). Aging diminishes the immune system’s ability to control virus latency (Pawelec, 2022) but increases propensity to maintain low burning systemic inflammation and hyperreactivity of T cells (inflammaging) (Muller and Di Benedetto, 2021).
The relation between MS and CMV is controversial. Some studies report beneficial others detrimental effects (Vanheusden et al., 2015). Thus, the jury is still out. It was found that chronic latent infection with CMV creates a repertoire of MHC-E-restricted effector memory cytotoxic T cells (EM-CTL) expressing a marker of natural killer cells (CD56) that play an important role in the defense against the virus (Moretta et al., 2003; Romagnani et al., 2004). Due to their special features these EM-CTL potentially mediate hyper-reactivity against self- antigens (‘t Hart et al., 2013) and discussion below.
Similar, albeit not 100% identical, types of NK-related CTL have been found in MS lesions (Saikali et al., 2007; Zaguia et al., 2013) and could be implicated in the marmoset EAE model as main driver of MS-like disease (Kap et al., 2008).
4. The Proverbial Primary Lesion
Information of MS pathology prior to diagnosis is limited. Barnett and Prineas reported on extensive oligodendrocyte apoptosis and small clusters of activated microglia in normal- appearing white matter (NAWM) areas with few or no lymphoid or myeloid immune cells present (Barnett and Prineas, 2004). Others reported that these areas contain besides clusters of activated microglia (nodules) with a degenerating axon in the center (Singh et al., 2013) also myelin blisters (Luchicchi et al., 2021). Blisters are particularly prevalent in NAWM areas with increased citrullination of myelin, aka micro-diffusely abnormal white matter (mDAWM) (Luchicchi et al., 2024). Data suggest that blisters are an early stage in the degeneration of axon- myelin units (AMU), making them a candidate early “primary lesion”, from which post- translationally modified (citrullinated) myelin proteins are released.
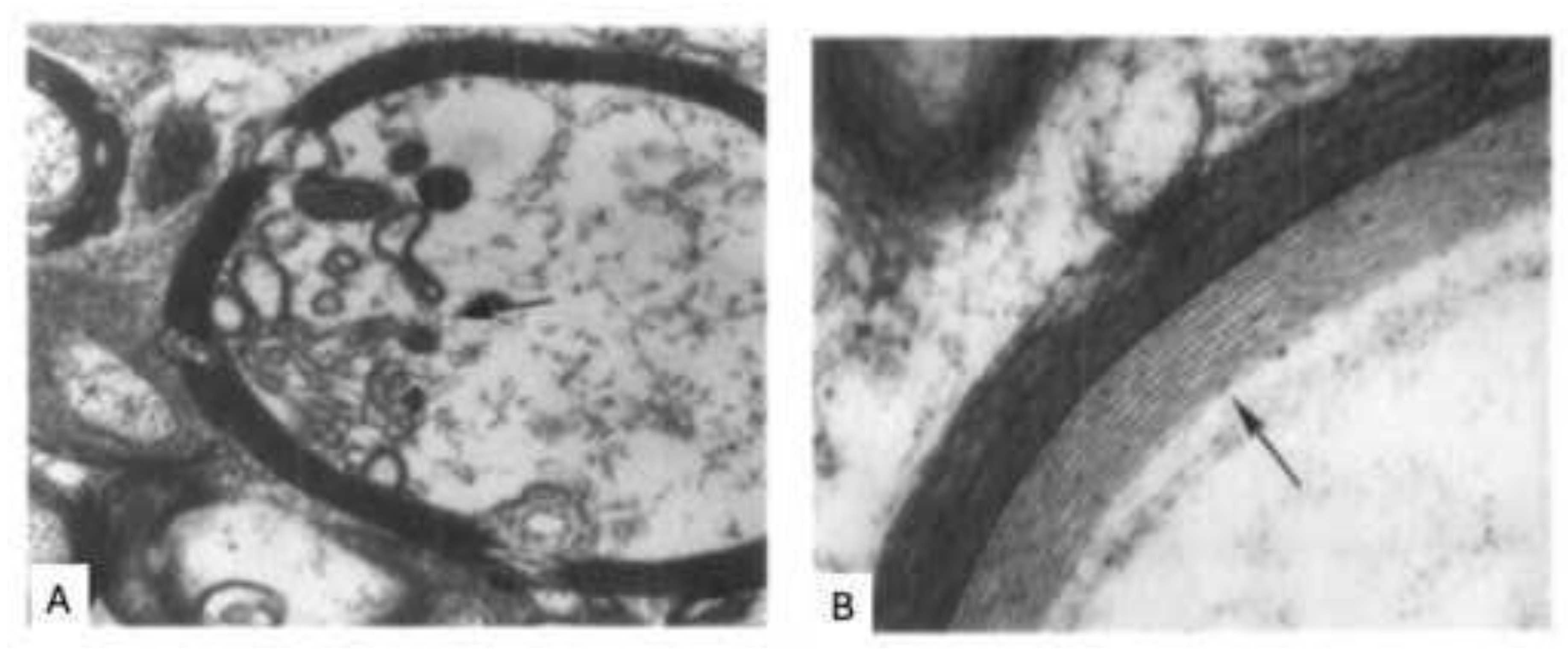
Rodriguez et al. analyzed the ultrastructure of AMU in MS lesions via electron microscopy of 11 brain biopsies (Rodriguez and Scheithauer, 1994). They observed early degenerative changes preceding myelin destruction and immune cell infiltration, including widening of the inner myelin lamellae and disruption of the inner loops while the rest of the sheath remained intact (Figure 2). Different from expectation the first aberration is thus not seen at the surface of myelin sheaths, as in the EAE model, but at the interaction of the myelin sheath’s inner lamellae with the enwrapped axon. Similar observations were reported by Huitinga et al.(van den Bosch et al., 2023). Both studies indicate “dying-back oligodendrogliopathy” as primary event in the pre-immune pathology of MS (Rodriguez, 1985).
4.1. A Putative Viral Cause of Primary Lesions: HHV-6
HHV-6 is a β-herpesvirus in the genus Roseolavirus. HHV-6 infects > 90% of the adult human population, usually at infant age (between 6 months and 2 years) when protective maternal antibodies have disappeared. Person to person transmission occurs via saliva mostly inducing lifelong mostly asymptomatic infections. HHV-6 infection may cause serious clinical complications, especially in immunocompromised individuals. HHV6 can infect a broad range of human cell types including brain glia cells (microglia, astrocytes and oligodendrocytes) but oligodendrocytes are usually the most severely affected (Skuja et al., 2017).
HHV-6 comprises two subtypes, HHV-6A and HHV-6B. HHV-6A has been described as the more neurovirulent and more frequently associated with MS (Alvarez-Lafuente et al., 2006). HHV-6B has been described as the cause of the common childhood disease exanthema subitum. Although in immunocompromised patients, such as transplant recipients, CNS inflammation can be observed (Yoshikawa, 2004).
The first report of a relation between HHV-6 and human demyelinating disease concerned an infected case diagnosed with acute disseminated encephalopathy (Kamei et al., 1997). Elevated levels of myelin basic protein (MBP) in cerebrospinal fluid indicated ongoing demyelination. Subsequent studies corroborated the potential linkage of HHV-6A with MS:
Frequent (immunohistochemical) detection of HHV-6 in MS CNS, particularly associated with active demyelinating disease (Knox et al., 2000).
Patients with CSF IgG oligoclonal bands (OCB) binding HHV-6A were younger and had higher overall OCB levels (Pietilainen-Nicklen et al., 2014).
Serum levels of anti-HHV6 IgM and IgG were higher in MS than in healthy controls. Moreover, HHV-6 levels were higher in in mitogen-stimulated leukocyte cultures from MS patients than healthy controls (Ablashi et al., 2000).
A meta-analysis of the literature revealed a correlation between anti-HHV-6 antibody levels in serum and CSF and the MS clinical course (Voumvourakis et al., 2022).
In a subset of MS patients serum HHV-6A IgG levels increased preceding an increase in serum neurofilament light chain (sNfL), a marker of axon destruction (Grut et al., 2024).
Interrogation of MS brains with PCR and immunohistochemistry detected HHV-6A antigens within oligodendrocytes both inside and adjacent to MS lesions (Challoner et al., 1995; Goodman et al., 2003). The virus latent protein HHV-6A u94A alters the ODC cytoskeleton, contributing to ODC dysfunction (Lyman and Enquist, 2009). HHV-6A infection also worsen endoplasmic reticulum (ER) stress and creates abnormalities in UPR pathways in cells of glial and neural origin (Romeo et al., 2020). Finally, infection of oligodendrocytes with HHV-6A induced similar damage to myelin sheaths as found in MS, such as irregularity of membranes and swelling of lamellae (Skuja et al., 2017).
Collectively, these studies support a role of HHV-6 in the etiopathogenesis of MS. Molecular mimicry seems not to provide a mechanistic explanation for the HHV-6/MS association as no significantly different cross-reactivity with HHV-6 or the myelin antigen MBP could be found in T cell lines isolated from 20 MS patients and 16 controls (Cirone et al., 2002).
In an animal model’s experimental setting, intravenous inoculation of marmoset monkeys with HHV-6A evoked neurological disease (Leibovitch et al., 2013). Using MRI T2 hyperintense small-sized brain lesions were found in some monkeys suggestive of encephalitis. (Immuno)histology revealed multiple microglia nodules and myelin abnormalities.
5. MS Etiopathogenesis, a Conspiracy of Herpesviruses?
This conceptual question was separated into 6 sub-questions: :
5.1. Where Does the Immune Pathogenic Process Start?
Consistent with Wilkin’s primary lesion theory (Wilkin, 1990) we hypothesize that the immune pathogenic process in MS starts with the formation of primary lesions. Studies of the macroscopically nonaffected NAWM of the MS brain revealed oligodendrogliopathy, disturbance of axon-myelin units and microglia nodules as most obvious pathological events. As discussed above, all these pathological features can be associated with HHV-6. There is convincing evidence that serum and CSF levels of neurofilament light (NFL), reporting neuronal damage, are increased in MS patients already in the prodromal phase, i.e. years before clinical diagnosis (Bjornevik et al., 2020). Association of NFL levels with anti-HHV-6 antibody serum levels, reporting virus (re-)activation, has been documented (Grut et al., 2024).
5.2. Where Do EBV-Infected B Cells Pick up Released Myelin Antigens?
In MS patients and MS animal models debris from damaged white matter drain via interstitial and cerebrospinal fluids, ending up in myeloid APC within cervical and lumbar lymph nodes (Engelhardt et al., 2016; Laman and Weller, 2013). This drainage route mainly serves to activate immune cells that can dampen (autoimmune) inflammation within the CNS (Kim et al., 2025). According to Engelhard et al. whether immune tolerance or autoimmunity is induced “is played out at the level of the individual T cell interacting with an individual APC dependent on the reciprocal quantitative and qualitative spectrum of signal 1 (MHC-peptide), signal 2 (panel of costimulatory and co-inhibitory receptors), and signal 3 (spectrum of soluble cytokines)” (Engelhardt et al., 2016). Based on our studies in the marmoset EAE model we posit that involvement of EBV-infected B cells as APC potentially disrupts the tolerance induction cycle and propagates autoimmunity.
EBV-infected B cells have been found localized within cervical lymph nodes (Sarkkinen et al., 2025). It is therefore a reasonable assumption that EBV-infected B cells can pick up draining myelin antigens there. Myelin debris have been detected within myeloid cells both in EAE models (de Vos et al., 2002) and in MS (Fabriek et al., 2005). Whether EBV-infected B cells can directly capture and digest large myelin fragments or whether these must be preprocessed by myeloid APC remains to be established.
5.3. Which Myelin Antigens Are Selected for Presentation to Pathogenic T Cells?
Marmosets (Jagessar et al., 2008) or mice immunized with MOG-deficient (mouse) myelin do not develop chronic EAE. EAE development could be restored by the addition of recombinant MOG (rMOG) (Smith et al., 2005). The critical peptide for chronic EAE development in marmosets is MOG34-56 (Kap et al., 2008).
5.4. Is the Encephalitogenic MOG34-56 Peptide Differentially Processed in LCV-Infected and Non-Infected B Cells.
Immunotherapy studies showed that the requisite APC in rMOG-induced marmoset EAE models is a B cell infected with the EBV-related lymphocryptovirus CalHV3 (‘t Hart and Kap, 2017). The MOG34-56 peptide is instantly destroyed during the processing in B cells but rescued from destructive processing when the B cells are infected with EBV (‘t Hart et al., 2017; ‘t Hart et al., 2016). We propose that this finding provides a mechanistic explanation for the essential role of EBV in the immunopathogenesis of MS.
5.5. Which Pathogenic T Cells Respond to the MOG34-56 Peptide?
Injection of marmosets with synthetic MOG34-56 peptide in IFA, activating mainly memory cells (Croft et al., 1994), induced a neurological disease with MS-like symptoms and pathology (Jagessar et al., 2010). Consistent with the memory status of the marmoset T cell response, immunization of immunologically naïve C57Bl/6 and Biozzi ABH mice with MOG34-56/IFA proved completely inert (Jagessar et al., 2010). Profiling of the T cell response revealed activation of CD3+CD4/8+CD56+ T cells producing IL-17A and TNF-α and displaying cytotoxic activity towards MOG34-56 peptide pulsed autologous EBV-infected B cells. In an earlier study the specific epitope was defined at residues 40 to 48 and the MHC restriction element was defined at Caja-E, being the marmoset equivalent of HLA-E (Jagessar et al., 2012). Based on the sharing of a mimicry motif between the MOG peptide and the UL86 ORF encoded major capsid protein of CMV (Brok et al., 2007) we assume that this virus may be the original inducer of the memory status.
5.6. where Does T Cell Activation by EBV-Infected B Cells Occur?
One possible site of pathogenic T cell activation is inside the CLN (Walsh et al., 2014). Another potential site is inside the CNS, more specific in the meningeal compartment (Magliozzi et al., 2013). A recent study in humanized mice revealed that by EBV-infection human B cells acquire the capacity to infiltrate the CNS parenchyma and to recruit co-infiltration of activated human CD4+ and CD8+ effector memory T cells (Laderach et al., 2025). Predictably, depletion of B cells with rituximab abrogates T cell infiltration into the CNS.
6. Concluding Remarks
We asked whether the two principal aspects of Wilkin’s primary lesion theory might be attributable to herpesviruses:
Formation of a primary lesion from which excess antigen is released. We discussed that HHV-6A is a potential inducer of oligodendrogliopathy that may underlie instability of axon-myelin units. Virus-stressed oligodendrocytes release citrullinated myelin antigens, which render them more immunogenic (Yang et al., 2016).
Hyper-reactivity of the immune system against myelin antigens. We discussed that chronic latent infection with CMV creates a repertoire of potentially auto-aggressive HLA-E-restricted EM-CTL. HLA-E is a non-classical MHC class Ib molecule that is upregulated on cells with reduced or absent expression of classical MHC class Ia molecules to ward off an attack by natural killer cells (Pietra et al., 2009). HLA-E is loaded with processed antigenic peptides in the endoplasmatic reticulum as MHC class Ia molecules, but in autophagosomes (Camilli et al., 2016). The autophagy pathway is virtually inactive in naïve B cells but is strongly upregulated in B cells infected with EBV(Jagessar et al., 2016). Through the association with autophagosomes the pathogenic MOG peptide is not only protected against fast degradation (Morandi et al., 2017) but can also associate with HLA-E.
The concept that the immune attack on axon-myelin units in MS might be caused by the conspiracy of three ubiquitous herpesviruses raises the question why MS is a relatively rare disease. The answer might be found in the specificity of the EBV-infected B cell. B cells are highly efficient APC because they can capture antigens using their highly specific antigen receptors. The clonal organization of the B cell compartment implies that only a small fraction of the B cells (< 1%) bears receptors for the myelin antigen MOG (Elong Ngono et al., 2015). The frequency of B cells that contains EBV is also very low (<0,01%) (Khan et al., 1996). It can thus be envisaged that the chance that a myelin-reactive B cell contains EBV is very low. In mononucleosis infectiosa patients the frequency of EBV-infected B cells can initially increase dramatically, up to 1 per 104 circulating B cells, lowering to 1 per 105 -106 circulating B cells (Kurth et al., 2000). This may explain why the MS risk is about 3-fold higher in people diagnosed with infectious mononucleosis than in the general population (Goldacre, 2024).
Funding
None of the authors received funding for the writing of this publication.
Acknowledgments
The authors declare that they have no competing nor financial interests. The authors thank dr. Antonio Luchicchi (Amsterdam University Medical Center/VUMC) for the artwork, Figure 1 and the graphical abstract.
Declaration of interests
The authors have no interests to declare.
References
- ‘t Hart, B.A.; Chalan, P.; Koopman, G.; Boots, A.M. Chronic autoimmune-mediated inflammation: a senescent immune response to injury. Drug Discov Today 2013, 18(7-8), 372–379. [Google Scholar] [CrossRef]
- ‘t Hart, B.A.; Dunham, J.; Faber, B.W.; Laman, J.D.; van Horssen, J.; Bauer, J.; Kap, Y.S. A B Cell-Driven Autoimmune Pathway Leading to Pathological Hallmarks of Progressive Multiple Sclerosis in the Marmoset Experimental Autoimmune Encephalomyelitis Model. Front Immunol 2017, 8, 804. [Google Scholar] [CrossRef]
- ‘t Hart, B.A.; Gran, B.; Weissert, R. EAE: imperfect but useful models of multiple sclerosis. Trends Mol Med 2011, 17(3), 119–125. [Google Scholar] [CrossRef] [PubMed]
- ‘t Hart, B.A.; Kap, Y.S. An essential role of virus-infected B cells in the marmoset experimental autoimmune encephalomyelitis model. MS journal Exp Translat Clin 2017, 1–6. [Google Scholar]
- ‘t Hart, B.A.; Kap, Y.S.; Morandi, E.; Laman, J.D.; Gran, B. EBV Infection and Multiple Sclerosis: Lessons from a Marmoset Model. Trends Mol Med 2016, 22(12), 1012–1024. [Google Scholar]
- ‘t Hart, B.A.; Luchicchi, A.; Schenk, G.J.; Stys, P.K.; Geurts, J.J.G. Mechanistic underpinning of an inside-out concept for autoimmunity in multiple sclerosis. Annals of clinical and translational neurology 2021, 8(8), 1709–1719. [Google Scholar] [CrossRef]
- Ablashi, D.V.; Eastman, H.B.; Owen, C.B.; Roman, M.M.; Friedman, J.; Zabriskie, J.B.; Peterson, D.L.; Pearson, G.R.; Whitman, J.E. Frequent HHV-6 reactivation in multiple sclerosis (MS) and chronic fatigue syndrome (CFS) patients. J Clin Virol 2000, 16(3), 179–191. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lafuente, R.; Garcia-Montojo, M.; De las Heras, V.; Bartolome, M.; Arroyo, R. Clinical parameters and HHV-6 active replication in relapsing-remitting multiple sclerosis patients. J Clin Virol 2006, 37 Suppl 1, S24–26. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 2004, 55(4), 458–468. [Google Scholar] [CrossRef]
- Barry, P.A.; Chang, William, W.L. Primate betaherpesviruses. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge, 2007. [Google Scholar]
- Baxter, A.G. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol 2007, 7(11), 904–912. [Google Scholar] [CrossRef]
- Biasi, G.; Facchinetti, A.; Monastra, G.; Mezzalira, S.; Sivieri, S.; Tavolato, B.; Gallo, P. Protection from experimental autoimmune encephalomyelitis (EAE): non-depleting anti-CD4 mAb treatment induces peripheral T-cell tolerance to MBP in PL/J mice. J Neuroimmunol 1997, 73(1-2), 117–123. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Munger, K.L.; Cortese, M.; Barro, C.; Healy, B.C.; Niebuhr, D.W.; Scher, A.I.; Kuhle, J.; Ascherio, A. Serum Neurofilament Light Chain Levels in Patients With Presymptomatic Multiple Sclerosis. JAMA neurology 2020, 77(1), 58–64. [Google Scholar] [CrossRef]
- Bjornevik, K.; Munz, C.; Cohen, J.I.; Ascherio, A.; Brodin, P.; Davis, M.M.; Epstein-Barr virus as a leading cause of multiple sclerosis: mechanisms and implications. Human immune system variation. Nat Rev Neurol;Nat Rev Immunol 2023, 19(3) 17(1), 160-171 21-29. [Google Scholar]
- Brok, H.P.; Boven, L.; van Meurs, M.; Kerlero de Rosbo, N.; Celebi-Paul, L.; Kap, Y.S.; Jagessar, A.; Hintzen, R.Q.; Keir, G.; Bajramovic, J.; Ben-Nun, A.; Bauer, J.; Laman, J.D.; Amor, S.; t Hart, B.A. The human CMV-UL86 peptide 981-1003 shares a crossreactive T-cell epitope with the encephalitogenic MOG peptide 34-56, but lacks the capacity to induce EAE in rhesus monkeys. J Neuroimmunol 2007, 182(1-2), 135–152. [Google Scholar] [CrossRef] [PubMed]
- Camilli, G.; Cassotta, A.; Battella, S.; Palmieri, G.; Santoni, A.; Paladini, F.; Fiorillo, M.T.; Sorrentino, R. Regulation and trafficking of the HLA-E molecules during monocyte-macrophage differentiation. J Leukoc Biol 2016, 99(1), 121–130. [Google Scholar] [CrossRef] [PubMed]
- Challoner, P.B.; Smith, K.T.; Parker, J.D.; MacLeod, D.L.; Coulter, S.N.; Rose, T.M.; Schultz, E.R.; Bennett, J.L.; Garber, R.L.; Chang, M. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A 1995, 92(16), 7440–7444. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Cuomo, L.; Zompetta, C.; Ruggieri, S.; Frati, L.; Faggioni, A.; Ragona, G. Human herpesvirus 6 and multiple sclerosis: a study of T cell cross-reactivity to viral and myelin basic protein antigens. J Med Virol 2002, 68(2), 268–272. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Bradley, L.M.; Swain, S.L. Naive versus memory CD4 T cell response to antigen. Memory cells are less dependent on accessory cell costimulation and can respond to many antigen- presenting cell types including resting B cells. J Immunol 1994, 152(6), 2675–2685. [Google Scholar] [CrossRef]
- de Vos, A.F.; van Meurs, M.; Brok, H.P.; Boven, L.A.; Hintzen, R.Q.; van der Valk, P.; Ravid, R.; Rensing, S.; Boon, L.; t Hart, B.A.; Laman, J.D. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol 2002, 169(10), 5415–5423. [Google Scholar] [PubMed]
- Elong Ngono, A.; Lepetit, M.; Reindl, M.; Garcia, A.; Guillot, F.; Genty, A.; Chesneau, M.; Salou, M.; Michel, L.; Lefrere, F.; Schanda, K.; Imbert-Marcille, B.M.; Degauque, N.; Nicot, A.; Brouard, S.; Laplaud, D.A.; Soulillou, J.P. Decreased Frequency of Circulating Myelin Oligodendrocyte Glycoprotein B Lymphocytes in Patients with Relapsing-Remitting Multiple Sclerosis. J Immunol Res 2015, 2015, 673503. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flugel, A.; Laman, J.D.; Weller, R.O. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol 2016, 132(3), 317–338. [Google Scholar] [CrossRef] [PubMed]
- Fabriek, B.O.; Zwemmer, J.N.; Teunissen, C.E.; Dijkstra, C.D.; Polman, C.H.; Laman, J.D.; Castelijns, J.A. In vivo detection of myelin proteins in cervical lymph nodes of MS patients using ultrasound-guided fine-needle aspiration cytology. J Neuroimmunol 2005, 161(1-2), 190–194. [Google Scholar] [CrossRef] [PubMed]
- Goldacre, R. Risk of multiple sclerosis in individuals with infectious mononucleosis: a national population-based cohort study using hospital records in England, 2003-2023. Mult Scler 2024, 30(4-5), 489–495. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.D.; Mock, D.J.; Powers, J.M.; Baker, J.V.; Blumberg, B.M.; Goronzy, J.J.; Weyand, C.M.; Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. Understanding immunosenescence to improve responses to vaccines. J Infect Dis;Nat Immunol 2003, 187(9) 14(5), 1365-1376 428-436. [Google Scholar]
- Grut, V.; Bistrom, M.; Salzer, J.; Stridh, P.; Jons, D.; Gustafsson, R.; Fogdell-Hahn, A.; Huang, J.; Butt, J.; Lindam, A.; Alonso-Magdalena, L.; Bergstrom, T.; Kockum, I.; Waterboer, T.; Olsson, T.; Zetterberg, H.; Blennow, K.; Andersen, O.; Nilsson, S.; Sundstrom, P. Human herpesvirus 6A and axonal injury before the clinical onset of multiple sclerosis. Brain 2024, 147(1), 177–185. [Google Scholar] [CrossRef]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci U S A 1998, 95(7), 3937–3942. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; Langer-Gould, A.; Smith, C.H.; Group, H.T. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008, 358(7), 676–688. [Google Scholar] [CrossRef] [PubMed]
- Jagessar, S.A.; Heijmans, N.; Blezer, E.L.; Bauer, J.; Blokhuis, J.H.; Wubben, J.A.; Drijfhout, J.W.; van den Elsen, P.J.; Laman, J.D.; t Hart, B.A. Unravelling the T-cell-mediated autoimmune attack on CNS myelin in a new primate EAE model induced with MOG34-56 peptide in incomplete adjuvant. Eur J Immunol 2012, 42(1), 217–227. [Google Scholar] [CrossRef] [PubMed]
- Jagessar, S.A.; Holtman, I.R.; Hofman, S.; Morandi, E.; Heijmans, N.; Laman, J.D.; Gran, B.; Faber, B.W.; van Kasteren, S.I.; Eggen, B.J.; t Hart, B.A. Lymphocryptovirus Infection of Nonhuman Primate B Cells Converts Destructive into Productive Processing of the Pathogenic CD8 T Cell Epitope in Myelin Oligodendrocyte Glycoprotein. J Immunol 2016, 197(4), 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Jagessar, S.A.; Kap, Y.S.; Heijmans, N.; van Driel, N.; van Straalen, L.; Bajramovic, J.J.; Brok, H.P.; Blezer, E.L.; Bauer, J.; Laman, J.D.; t Hart, B.A. Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete freund adjuvant. J Neuropathol Exp Neurol 2010, 69(4), 372–385. [Google Scholar] [CrossRef] [PubMed]
- Jagessar, S.A.; Smith, P.A.; Blezer, E.; Delarasse, C.; Pham-Dinh, D.; Laman, J.D.; Bauer, J.; Amor, S.; t Hart, B. Autoimmunity against myelin oligodendrocyte glycoprotein is dispensable for the initiation although essential for the progression of chronic encephalomyelitis in common marmosets. J Neuropathol Exp Neurol 2008, 67(4), 326–340. [Google Scholar] [CrossRef] [PubMed]
- Kamei, A.; Ichinohe, S.; Onuma, R.; Hiraga, S.; Fujiwara, T. Acute disseminated demyelination due to primary human herpesvirus-6 infection. Eur J Pediatr 1997, 156(9), 709–712. [Google Scholar] [CrossRef]
- Kap, Y.S.; Smith, P.; Jagessar, S.A.; Remarque, E.; Blezer, E.; Strijkers, G.J.; Laman, J.D.; Hintzen, R.Q.; Bauer, J.; Brok, H.P.; t Hart, B.A. Fast progression of recombinant human myelin/oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis in marmosets is associated with the activation of MOG34-56-specific cytotoxic T cells. J Immunol 2008, 180(3), 1326–1337. [Google Scholar] [CrossRef]
- Kawakami, N.; Flugel, A. Knocking at the brain’s door: intravital two-photon imaging of autoreactive T cell interactions with CNS structures. Semin Immunopathol 2010, 32(3), 275–287. [Google Scholar] [CrossRef]
- Khan, G.; Miyashita, E.M.; Yang, B.; Babcock, G.J.; Thorley-Lawson, D.A. Is EBV persistence in vivo a model for B cell homeostasis? Immunity 1996, 5(2), 173–179. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Gao, W.; Lichti, C.F.; Gu, X.; Dykstra, T.; Cao, J.; Smirnov, I.; Boskovic, P.; Kleverov, D.; Salvador, A.F.M.; Drieu, A.; Kim, K.; Blackburn, S.; Crewe, C.; Artyomov, M.N.; Unanue, E.R.; Kipnis, J. Endogenous self-peptides guard immune privilege of the central nervous system. Nature 2025, 637(8044), 176–183. [Google Scholar] [CrossRef] [PubMed]
- Knox, K.K.; Brewer, J.H.; Henry, J.M.; Harrington, D.J.; Carrigan, D.R. Human herpesvirus 6 and multiple sclerosis: systemic active infections in patients with early disease. Clin Infect Dis 2000, 31(4), 894–903. [Google Scholar] [CrossRef]
- Krovi, S.H.; Kuchroo, V.K. Activation pathways that drive CD4(+) T cells to break tolerance in autoimmune diseases(). Immunol Rev 2022, 307(1), 161–190. [Google Scholar] [CrossRef]
- Kurth, J.; Spieker, T.; Wustrow, J.; Strickler, G.J.; Hansmann, L.M.; Rajewsky, K.; Kuppers, R. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity 2000, 13(4), 485–495. [Google Scholar] [CrossRef]
- Laderach, F.; Piteros, I.; Fennell, E.; Bremer, E.; Last, M.; Schmid, S.; Rieble, L.; Campbell, C.; Ludwig-Portugall, I.; Bornemann, L.; Gruhl, A.; Eulitz, K.; Gueguen, P.; Mietz, J.; Muller, A.; Pezzino, G.; Schmitz, J.; Ferlazzo, G.; Mautner, J.; Munz, C.; Laman, J.D.; Weller, R.O.; EBV induces CNS homing of B cells attracting inflammatory T cells. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. Nature;J Neuroimmune Pharmacol 2025, 8(4), 840–856. [Google Scholar]
- Lang, H.L.; Jacobsen, H.; Ikemizu, S.; Andersson, C.; Harlos, K.; Madsen, L.; Hjorth, P.; Sondergaard, L.; Svejgaard, A.; Wucherpfennig, K.; Stuart, D.I.; Bell, J.I.; Jones, E.Y.; Fugger, L. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol 2002, 3(10), 940–943. [Google Scholar] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; Schubert, R.D.; Hawes, I.A.; Vazquez, S.E.; Iyer, M.; Zuchero, J.B.; Teegen, B.; Dunn, J.E.; Lock, C.B.; Kipp, L.B.; Cotham, V.C.; Ueberheide, B.M.; Aftab, B.T.; Anderson, M.S.; DeRisi, J.L.; Wilson, M.R.; Bashford-Rogers, R.J.M.; Platten, M.; Garcia, K.C.; Steinman, L.; Robinson, W.H. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603(7900), 321–327. [Google Scholar] [CrossRef] [PubMed]
- Leibovitch, E.; Wohler, J.E.; Cummings Macri, S.M.; Motanic, K.; Harberts, E.; Gaitan, M.I.; Maggi, P.; Ellis, M.; Westmoreland, S.; Silva, A.; Reich, D.S.; Jacobson, S. Novel marmoset (Callithrix jacchus) model of human Herpesvirus 6A and 6B infections: immunologic, virologic and radiologic characterization. PLoS Pathog 2013, 9(1), e1003138. [Google Scholar] [CrossRef]
- Libbey, J.E.; Cusick, M.F.; Fujinami, R.S. Role of pathogens in multiple sclerosis. Int Rev Immunol 2014a, 33(4), 266–283. [Google Scholar] [CrossRef]
- Libbey, J.E.; Fujinami, R.S. Viral mouse models used to study multiple sclerosis: past and present. Arch Virol 2021, 166(4), 1015–1033. [Google Scholar]
- Libbey, J.E.; Lane, T.E.; Fujinami, R.S. Axonal pathology and demyelination in viral models of multiple sclerosis. Discov Med 2014b, 18(97), 79–89. [Google Scholar]
- Lipton, H.L.; Dal Canto, M.C. Chronic neurologic disease in Theiler’s virus infection of SJL/J mice. J Neurol Sci 1976, 30(1), 201–207. [Google Scholar]
- Luchicchi, A.; Hart, B.; Frigerio, I.; van Dam, A.M.; Perna, L.; Offerhaus, H.L.; Stys, P.K.; Schenk, G.J.; Geurts, J.J.G. Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Ann Neurol 2021, 89(4), 711–725. [Google Scholar]
- Luchicchi, A.; Munoz-Gonzalez, G.; Halperin, S.T.; Strijbis, E.; van Dijk, L.H.M.; Foutiadou, C.; Uriac, F.; Bouman, P.M.; Schouten, M.A.N.; Plemel, J.; t Hart, B.A.; Geurts, J.J.G.; Schenk, G.J. Micro-diffusely abnormal white matter: An early multiple sclerosis lesion phase with intensified myelin blistering. Annals of clinical and translational neurology 2024, 11(4), 973–988. [Google Scholar]
- Lyman, M.G.; Enquist, L.W. Herpesvirus interactions with the host cytoskeleton. J Virol 2009, 83(5), 2058–2066. [Google Scholar] [PubMed]
- Magliozzi, R.; Serafini, B.; Rosicarelli, B.; Chiappetta, G.; Veroni, C.; Reynolds, R.; Aloisi, F. B-cell enrichment and Epstein-Barr virus infection in inflammatory cortical lesions in secondary progressive multiple sclerosis. J Neuropathol Exp Neurol 2013, 72(1), 29–41. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu Rev Immunol 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Rammohan, K.W.; Selmaj, K.; Traboulsee, A.; Sauter, A.; Masterman, D.; Fontoura, P.; Belachew, S.; Garren, H.; Mairon, N.; Chin, P.; Wolinsky, J.S.; Investigators, O.C. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med 2017, 376(3), 209–220. [Google Scholar] [CrossRef]
- Morandi, E.; Jagessar, S.A.; t Hart, B.A.; Gran, B. EBV Infection Empowers Human B Cells for Autoimmunity: Role of Autophagy and Relevance to Multiple Sclerosis. J Immunol 2017, 199(2), 435–448. [Google Scholar]
- Moretta, L.; Romagnani, C.; Pietra, G.; Moretta, A.; Mingari, M.C. NK-CTLs, a novel HLA-E- restricted T-cell subset. Trends Immunol 2003, 24(3), 136–143. [Google Scholar]
- Muhe, J.; Wang, F. Non-human Primate Lymphocryptoviruses: Past, Present, and Future. Curr Top Microbiol Immunol 2015, 391, 385–405. [Google Scholar] [PubMed]
- Muller, L.; Di Benedetto, S. How Immunosenescence and Inflammaging May Contribute to Hyperinflammatory Syndrome in COVID-19. Int J Mol Sci 2021, 22(22). [Google Scholar]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N Engl J Med 2000, 343(13), 938–952. [Google Scholar]
- Pawelec, G. Latent CMV makes older adults less naive. EBioMedicine 2022, 77, 103887. [Google Scholar] [CrossRef] [PubMed]
- Pietilainen-Nicklen, J.; Virtanen, J.O.; Uotila, L.; Salonen, O.; Farkkila, M.; Koskiniemi, M. HHV-6-positivity in diseases with demyelination. J Clin Virol 2014, 61(2), 216–219. [Google Scholar] [CrossRef]
- Pietra, G.; Romagnani, C.; Mazzarino, P.; Falco, M.; Millo, E.; Moretta, A.; Moretta, L.; Mingari, M.C. HLA-E-restricted recognition of cytomegalovirus-derived peptides by human CD8+ cytolytic T lymphocytes. Proc Natl Acad Sci U S A 2003, 100(19), 10896–10901. [Google Scholar]
- Pietra, G.; Romagnani, C.; Moretta, L.; Mingari, M.C. HLA-E and HLA-E-bound peptides: recognition by subsets of NK and T cells. Curr Pharm Des 2009, 15(28), 3336–3344. [Google Scholar]
- Rodriguez, M. Virus-induced demyelination in mice: “dying back” of oligodendrocytes. Mayo Clin Proc 1985, 60(7), 433–438. [Google Scholar]
- Rodriguez, M.; Leibowitz, J.L.; Lampert, P.W. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Ann Neurol 1983, 13(4), 426–433. [Google Scholar]
- Rodriguez, M.; Scheithauer, B. Ultrastructure of multiple sclerosis. Ultrastruct Pathol 1994, 18(1-2), 3–13. [Google Scholar] [CrossRef]
- Romagnani, C.; Pietra, G.; Falco, M.; Mazzarino, P.; Moretta, L.; Mingari, M.C. HLA-E- restricted recognition of human cytomegalovirus by a subset of cytolytic T lymphocytes. Hum Immunol 2004, 65(5), 437–445. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Gaeta, A.; D’Orazi, G.; Faggioni, A.; Cirone, M. HHV- 6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis 2020, 1866(3), 165647. [Google Scholar] [PubMed]
- Saikali, P.; Antel, J.P.; Newcombe, J.; Chen, Z.; Freedman, M.; Blain, M.; Cayrol, R.; Prat, A.; Hall, J.A.; Arbour, N. NKG2D-mediated cytotoxicity toward oligodendrocytes suggests a mechanism for tissue injury in multiple sclerosis. J Neurosci 2007, 27(5), 1220–1228. [Google Scholar] [PubMed]
- Sarkkinen, J.; Yohannes, D.A.; Kreivi, N.; Durnsteiner, P.; Elsakova, A.; Huuhtanen, J.; Nowlan, K.; Kurdo, G.; Linden, R.; Saarela, M.; Tienari, P.J.; Kekalainen, E.; Perdomo, M.; Laakso, S.M. Altered immune landscape of cervical lymph nodes reveals Epstein-Barr virus signature in multiple sclerosis. Sci Immunol 2025, 10(104), eadl3604. [Google Scholar]
- Singh, S.; Metz, I.; Amor, S.; van der Valk, P.; Stadelmann, C.; Bruck, W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013, 125(4), 595–608. [Google Scholar]
- Skuja, S.; Zieda, A.; Ravina, K.; Chapenko, S.; Roga, S.; Teteris, O.; Groma, V.; Murovska, M. Structural and Ultrastructural Alterations in Human Olfactory Pathways and Possible Associations with Herpesvirus 6 Infection. PloS one 2017, 12(1), e0170071. [Google Scholar] [CrossRef]
- Smith, P.A.; Heijmans, N.; Ouwerling, B.; Breij, E.C.; Evans, N.; van Noort, J.M.; Plomp, A.C.; Delarasse, C.; t Hart, B.; Pham-Dinh, D.; Amor, S. Native myelin oligodendrocyte glycoprotein promotes severe chronic neurological disease and demyelination in Biozzi ABH mice. Eur J Immunol 2005, 35(4), 1311–1319. [Google Scholar] [CrossRef]
- Soldan, S.S.; Lieberman, P.M. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol 2023, 21(1), 51–64. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Steiner, I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol 2005, 58(6), 939–945. [Google Scholar] [CrossRef]
- Steininger, C. Clinical relevance of cytomegalovirus infection in patients with disorders of the immune system. Clin Microbiol Infect 2007, 13(10), 953–963. [Google Scholar] [CrossRef]
- Stys, P.K.; Zamponi, G.W.; van Minnen, J.; Geurts, J.J. Will the real multiple sclerosis please stand up? Nature reviews. Neuroscience 2012, 13(7), 507–514. [Google Scholar] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; Nelson, J.A.; Picker, L.J. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 2005, 202(5), 673–685. [Google Scholar] [CrossRef] [PubMed]
- t Hart, B.A.; Luchicchi, A.; Schenk, G.J.; Killestein, J.; Geurts, J.J.G. Multiple sclerosis and drug discovery: A work of translation. EBioMedicine 2021, 68, 103392. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 2001, 1(1), 75–82. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence--Introducing the Virus. Curr Top Microbiol Immunol 2015, 390 Pt 1, 151–209. [Google Scholar]
- van den Bosch, A.M.R.; Hummert, S.; Steyer, A.; Ruhwedel, T.; Hamann, J.; Smolders, J.; Nave, K.A.; Stadelmann, C.; Kole, M.H.P.; Mobius, W.; Huitinga, I. Ultrastructural Axon-Myelin Unit Alterations in Multiple Sclerosis Correlate with Inflammation. Ann Neurol 2023, 93(4), 856–870. [Google Scholar] [CrossRef]
- van Oosten, B.W.; Lai, M.; Hodgkinson, S.; Barkhof, F.; Miller, D.H.; Moseley, I.F.; Thompson, A.J.; Rudge, P.; McDougall, A.; McLeod, J.G.; Ader, H.J.; Polman, C.H. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology 1997, 49(2), 351–357. [Google Scholar] [CrossRef]
- Vanheusden, M.; Stinissen, P.; t Hart, B.A.; Hellings, N. Cytomegalovirus: a culprit or protector in multiple sclerosis? Trends Mol Med 2015, 21(1), 16–23. [Google Scholar] [CrossRef] [PubMed]
- Voumvourakis, K.I.; Fragkou, P.C.; Kitsos, D.K.; Foska, K.; Chondrogianni, M.; Tsiodras, S. Human herpesvirus 6 infection as a trigger of multiple sclerosis: an update of recent literature. BMC Neurol 2022, 22(1), 57. [Google Scholar]
- Walsh, J.T.; Zheng, J.; Smirnov, I.; Lorenz, U.; Tung, K.; Kipnis, J.; Wilkin, T.J.; Regulatory T cells in central nervous system injury: a double-edged sword. The primary lesion theory of autoimmunity: a speculative hypothesis. J Immunol;Autoimmunity 2014, 193(10) 7(4), 5013-5022 225-235. [Google Scholar]
- Yang, L.; Tan, D.; Piao, H. Myelin Basic Protein Citrullination in Multiple Sclerosis: A Potential Therapeutic Target for the Pathology. Neurochem Res 2016, 41(8), 1845–1856. [Google Scholar]
- Yoshikawa, T. Human herpesvirus 6 infection in hematopoietic stem cell transplant patients. Br J Haematol 2004, 124(4), 421–432. [Google Scholar] [CrossRef]
- Zaguia, F.; Saikali, P.; Ludwin, S.; Newcombe, J.; Beauseigle, D.; McCrea, E.; Duquette, P.; Prat, A.; Antel, J.P.; Arbour, N. Cytotoxic NKG2C+ CD4 T cells target oligodendrocytes in multiple sclerosis. J Immunol 2013, 190(6), 2510–2518. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



