Submitted:
01 July 2025
Posted:
02 July 2025
You are already at the latest version
Abstract
x.FASTQ is a suite of modular Bash scripts designed to streamline RNA-Seq data analysis by enabling remote, command-line execution of standardized pipelines. Targeting research environments where bioinformatics expertise and infrastructures are limited, x.FASTQ is designed with remote access in mind: the tool, once installed on a remote machine, can be easily accessed via SSH and used even without advanced computer skills, only requiring basic shell knowledge. The suite provides a complete transcriptomic workflow from raw read acquisition to expression matrix generation, leveraging trusted third-party tools such as FastQC, BBDuk, STAR, RSEM, and MultiQC. Each module runs by default persistently in the background, allowing users to work asynchronously, but it also supports optional chaining into fully automated pipelines. A Python script, Moliere, enables single-command execution of end-to-end analyses. With minimal configuration, consistent file naming conventions, detailed logging, and version tracking, x.FASTQ offers a reproducible, scalable, and user-friendly platform for transcriptome (meta)analyses across distributed research teams.
Keywords:
RNA-Seq data analysis
; pipeline automation
; command-line bioinformatics
; Bash wrappers
; reproducibility
1. Introduction
1.1. Background and Motivations
RNA Sequencing (RNA-Seq) has become a fundamental technique in transcriptomics, enabling quantitative and qualitative assessments of transcript abundance across diverse biological conditions. Despite rapid advancements in sequencing technologies and bioinformatics methodologies, practical implementation remains challenging for many research groups, particularly those lacking dedicated computational expertise or resources. Standard RNA-Seq workflows involve multiple steps—from raw read acquisition and quality control to alignment, quantification, and data visualization—which require substantial bioinformatics proficiency, computational infrastructure, and robust pipeline management skills [1].
While several bioinformatics frameworks exist to streamline RNA-Seq analyses, many require familiarity with scripting languages (Bash, Python, R) and/or containerization (Docker, Singularity) [2,3,4,5,6,7]. Furthermore, most of these analyses require considerable computational resources to be accomplished, limiting their utility for laboratories without local hardware capacity or specialized personnel.
In this regard Galaxy (https://usegalaxy.org/) [8] represents a common answer to these issues and a valuable entry point for RNA-Seq analysis, offering a user-friendly graphical interface, support for reproducibility, integration of numerous tools, and deployment on both cloud and local servers.
While we acknowledge Galaxy as a fully viable solution, upholding high standards of quality and reproducibility, we also recognize several critical limitations:
- resource constraints on the Galaxy server hosting the analysis may limit data uploading speed, available storage space, and/or computational capacity, potentially slowing down workflows during periods of high usage;
- the availability of dozens of tools for similar tasks (e.g., multiple aligners, trimmers, and differential expression packages) can be overwhelming—particularly for users who prefer a more streamlined and minimal environment;
- tool selection is not guided and terminology (e.g., GTF vs GFF3, strandedness, normalization methods) assumes bioinformatics background;
- the interface, while point-and-click, becomes dense and overwhelming quickly;
- as a result, despite its apparent simplicity, gaining proficiency in Galaxy’s internal logic (histories, datasets, job statuses, and parameter settings) can require a significant learning curve;
- on the other hand, for experienced users, Galaxy can feel less transparent, flexible, and customizable than command-line alternatives;
- integrating Galaxy tools into custom command-line pipelines can also be challenging;
- error reporting is often opaque, especially for tools that rely on complex stacks of dependencies and configurations.
In synthesis, we feel that Galaxy—as well as other similar web-based tools such as BaseSpace Sequence Hub by Illumina (https://basespace.illumina.com/) or Broad Institute’s GenePattern (https://www.genepattern.org/)—often sits awkwardly between being too complex for novices and too restrictive for experts. For this reason, when, as bioinformaticians, we were asked for advice by “wet-lab” researchers interested in performing gene expression analysis, we opted for a model of supervised autonomy and trained them in the use of x.FASTQ, a suite of Bash-based wrappers of both original and third-party software for RNA-Seq data analysis that features an extremely fast learning curve.
The design philosophy of x.FASTQ stems from a pragmatic observation about collaborative research: in many real-world settings, a limited number of bioinformaticians are called upon to support a broader network of wet-lab researchers, often dispersed across different institutions. In this scenario, x.FASTQ promotes a model of distributed autonomy under expert supervision. The bioinformatician acts as the technical facilitator, responsible for installing x.FASTQ on a remote workstation, configuring its dependencies and environment, and ensuring secure access via SSH. Wet-lab collaborators, in turn, require only minimal computational skills (basic familiarity with the command line and access to an SSH client, typically preinstalled on most systems) to independently perform comprehensive RNA-Seq (meta)analyses. This model empowers experimental researchers to run complex workflows without being hindered by technical barriers, while still maintaining a close, guided, and reassuring connection to bioinformatics expertise. For the bioinformatician, x.FASTQ offers a structured and reproducible framework that abstracts and automates common RNA-Seq operations, making it feasible to support many concurrent projects with minimal overhead.
1.2. Key Features
Based on these considerations, the key features of x.FASTQ include:
- Remote operability
Due to the typical hardware requirements for read alignment and subsequent transcript abundance quantification, x.FASTQ has been designed to be installed on one (or a few) remote Linux systems and accessed by multiple users via SSH. Accordingly, each x.FASTQ module runs by default in the background and persistently (i.e., ignoring the HUP hangup signal), so that the user is not forced to keep the connection active for the entire duration of the analysis, but only for job scheduling.
- Standardization
Most x.FASTQ scripts are wrappers of lower-level applications that are commonly used as standard tools in RNA-Seq data analysis and widely appreciated for their performance (e.g., FastQC [9], BBDuk [10], MultiQC [11], STAR [3], RSEM [4]).
- Simplification
Scripts expose a limited number of options by making extensive use of default settings (suitable for the majority of standard RNA-Seq analyses) and by taking over the management of input and output data formats and their organization. Crucially, however, they allow for high parameter customization if needed.
- Automation
All scripts are designed to loop over sets of target files properly stored in the same directory. In addition, although designed to run independently, each module can optionally be chained to the next in a single pipeline to automate the entire analysis workflow (-w | --workflow option).
- Completeness
The tools provided by x.FASTQ allow for a complete RNA-Seq workflow, from raw read retrieval to count matrix generation.
- No bioinformatics skills required
The only requirement for the end-user is a basic knowledge of the Unix shell and an SSH client installed on the local machine. Each x.FASTQ module is a CLI-executable Bash command with a --help option that provides extensive documentation.
- Reproducibility
Each x.FASTQ module is tightly versioned and designed to save detailed log files at each run [12]. Utilities are also available to print full version reports on x.FASTQ modules and dependencies (i.e., x.fastq -r and -d options, respectively). Containerization of the individual tools is planned for the future.
2. From Reads to Counts
2.1. x.FASTQ Modules
Core Modules. x.FASTQ consists of seven core modules, designed to be run directly by the end user, each corresponding to a distinct analytical step of a general RNA-Seq data analysis pipeline, from raw read acquisition to expression matrix generation.
- getFASTQ allows the user to download NGS raw data in FASTQ format from the ENA database (https://www.ebi.ac.uk/ena/browser/home) to the machine hosting x.FASTQ;
- trimFASTQ uses BBDuk, from the BBTools suite [10] (https://archive.jgi.doe.gov/data-and-tools/software-tools/bbtools/), to remove adapter sequences and perform quality trimming;
- anqFASTQ uses STAR [3] (https://github.com/alexdobin/STAR) and RSEM [4] (https://github.com/deweylab/RSEM) to align reads and quantify transcript abundance, respectively, supporting both gene-level and isoform-level analyses;
- qcFASTQ is an interface for multiple quality-control tools, including FastQC [9] (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), MultiQC [11] (https:// multiqc.info/), and PCA analysis;
- tabFASTQ merges counts from multiple samples into a single TSV expression table, choosing among multiple metrics (TPM, FPKM, RSEM expected counts) and levels (gene or isoform). Optionally, it inserts experimental design information into the matrix header and appends annotations regarding gene symbol, gene name, and gene type (Ensembl gene/transcript IDs are required for annotation);
- metaharvest fetches Sample and Study metadata from GEO (https://www.ncbi.nlm.nih.gov/geo/) and/or ENA (https://www.ebi.ac.uk/ena/browser/home) databases, then it parses the retrieved metadata and saves a local copy of them as a CSV-formatted table;
- x.FASTQ is a cover-script that performs a number of common tasks of general utility, such as dependency checking, symlink creation, version monitoring, and disk usage reporting.
In addition, a Python script called Moliere is included to easily run a full RNA-Seq analysis pipeline with a single command.
Auxiliary Scripts. x.FASTQ also contains a number of complementary scripts (written in Bash, R, or Python) that are not intended to be run directly by the end user, but are called by the main modules. Most of them are found in the workers subfolder.
- x.funx.sh contains variables and functions that must be shared (i.e., sourced) by all other x.FASTQ modules;
- progress_funx.sh collects all the functions for tracking the progress of the different modules (see the -p option below);
- trimmer.sh is the actual BBDuk wrapper, called by trimFASTQ;
- starsem.sh is the actual STAR/RSEM wrapper, called by trimFASTQ;
- assembler.R implements the matrix assembly procedure required by tabFASTQ;
- pca_hc.R implements Principal Component Analysis and Hierarchical Clustering of samples as required by the qcfastq --tool=PCA ... option;
- fuse_csv.R is called by metaharvest to merge the cross-referenced metadata down- loaded from both GEO and ENA databases;
- parse_series.R is called by metaharvest to extract metadata from a GEO-retrieved SOFT formatted family file;
- re_uniq.py is used to reduce redundancy when STAR and RSEM logs are displayed in the console as anqFASTQ progress reports.
Common Features and Options. All suite modules enjoy some internal consistency:
- upon running x.fastq.sh -l <target_path> from the local x.FASTQ repository directory, each x.FASTQ module can be invoked from any location on the remote machine using its fully lowercase name (provided that <target_path> is already included in $PATH);
- by default, each script launches in the background a persistent job (or a queue of jobs) by using a custom re-implementation of the nohup command (namely the _hold_on function from x.funx.sh);
- each module (except x.FASTQ and metaharvest) saves its own log file inside the project directory using the filename pattern
Z_<ModuleName>_<ID>_<DateStamp>.log
-
some common flags keep the same meaning across all modules (even if not all of them are always available):
- ‣
- -h | --help to display the script-specific help;
- ‣
- -v | --version to display the script-specific version;
- ‣
- -q | --quiet to run the script silently;
- ‣
- -w | --workflow to make processes run in the foreground, useful when used in pipelines;
- ‣
- -p | --progress to see the progress of possibly ongoing processes;
- ‣
- -k | --kill to gracefully terminate possibly ongoing processes;
- ‣
- -a | --keep-all not to delete intermediate files upon script execution;
- all core modules are versioned according to the three-number Semantic Versioning system (https://semver.org/). x.fastq -r can be used to get a version report of all scripts along with the summary version of the whole x.FASTQ suite;
- if -p is followed by no other arguments, the script will search the current directory for log files from which to infer the progress of the last namesake task;
- with the -q option, scripts do not print anything to the screen except for possible error messages that stop execution (i.e., fatal errors); however, logging is never disabled.
2.2. Usage and Workflow Examples
Practical usability is here illustrated with clear workflow examples. Assuming that you have identified a study of interest from GEO (e.g., GSE138309), have already created a project folder somewhere (mkdir <any_path>/GSE138309), and have moved into it (cd <any_path>/GSE138309), here are some possible examples of workflows.
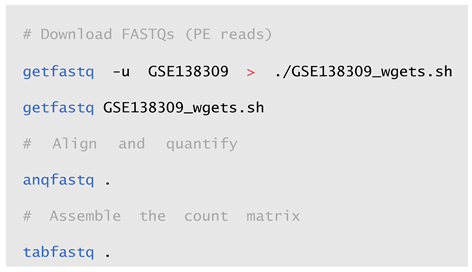
Minimal Step-by-Step Workflow. As an example of a minimal workflow, we can think of the following set of commands to (sequentially) retrieve the FASTQ files of interest, align and quantify them, and generate the gene-level count matrix (in TPMs).
Complete Step-by-Step Workflow. A more robust workflow might include the parallel download of FASTQ files along with their metadata, read trimming, multiple quality control steps, and the inclusion of gene annotations and experimental design information in the count matrix, here computed at isoform-level and saved as RSEM expected counts for possible subsequent Differential Expression Analysis (DEA).
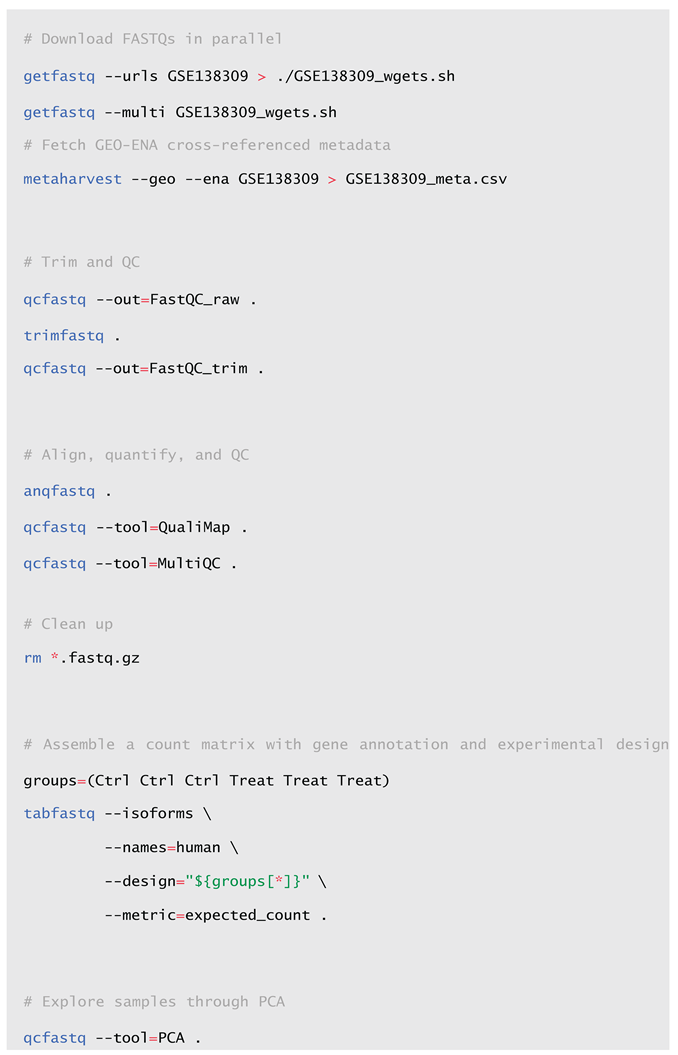
Please note that the study chosen here as an example has non-interleaved (i.e., dual- file) paired-end (PE) reads, but x.FASTQ also supports single-ended (SE) and interleaved PE formats. In those cases, you must use the [-s | --single-end] or [-i | -- interleaved] options, respectively, when both trimming and aligning.
Complete Workflow in Batch Mode. As already mentioned in Section 1.2, each x.FASTQ module is designed to run by default in the background and persistently. In this way, each x.FASTQ module is meant to be run independently as a single analysis step. Alternatively, multiple modules can be chained together in a single pipeline to automate the entire analysis workflow by using the -w | --workflow option for foreground execution. Here is the batched version of the previous workflow
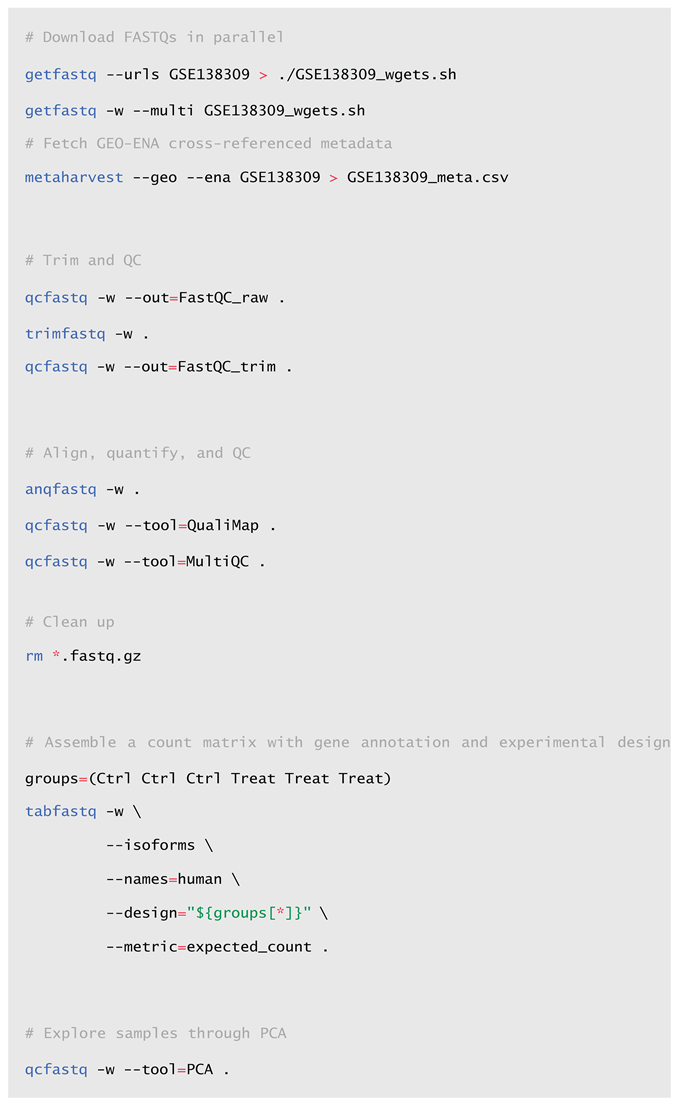
Just save this pipeline as a single script file (e.g., pipeline.xfastq) and run the entire workflow with nohup and in the background

Complete Workflow with Moliere. Alternatively, a similar workflow can be performed in a single command using Moliere, a “precasted” Python script that runs, in order, getfastq, qcfastq, trimfastq, qcfastq (again), anqfastq, and tabfastq, covering the whole analysis process with sensible defaults.

The final output is the “expected counts” matrix in the ./GSE138309/Counts/ folder. If you need other metrics, you may call tabFASTQ manually: Moliere does not delete the quantifications.
Moliere batches the job to only download a set of FASTQ files at once per cycle, so that your hard drive does not get full with downloaded files. It will handle this transparently for you, by default downloading 20 files at once. Of course, the final tabfastq call will be performed only after all files in all batches are processed correctly.
In the spirit of other x.FASTQ scripts, you can check where Moliere is in the analysis (i.e., read the .moliere file) by issuing the command
from the project directory. This will give you an overall view of the analysis, plus the outputs (if relevant) of the x.FASTQ scripts when called with the --progress option.
If an error occurs in the analysis, you can fix it and then resume the process where it was left off with moliere resume (run from the project directory).
Moliere renders routine RNA-Seq retrieval, alignment and quantification extremely easy and convenient.
2.3. Installation and Deployment
As already noted in Section 1.2, an SSH client is the only local software requirement for the end-user to interact with x.FASTQ, provided it has already been installed on some remote workstation by the system administrator. Installation details are precisely doc- umented on the official x.FASTQ GitHuib page (https://github.com/TCP-Lab/x.FASTQ/tree/foreground/docs), encompassing dependency management and system-specific configurations via the install.paths file, also including the procedure to generate a new STAR genome index for each alternative model of interest, as well as the corresponding reference for RSEM.
All the dependencies required by x.FASTQ for its proper functioning are listed in Appendix A.2
2.4. Practical Considerations and Advanced Hints
On Trimming. In its current implementation, trimFASTQ wraps BBDuk to perform a quite conservative trimming of the reads, based on three steps:
- Adapter trimming: adapters are automatically detected based on BBDuk’s adapters.fa database and then right-trimmed using 23-to-11 base-long kmers allowing for one mismatch (i.e., Hamming distance =1). See the KTrimmed stat in the log file.
- Quality trimming: it is performed on both sides of each read using a quality score threshold trimq=10. See the QTrimmed stat in the log file.
- Length filtering: all reads shorter than 25 bases are discarded. See the Total Removed stat in the log file.
Adapter trimming always precedes quality trimming to avoid partial adapter removal affecting downstream alignments [10]. On top of that, it should be noted that, in case you are sequencing for counting applications (like DEA, ChIP-seq, ATAC-seq) read trimming is generally not required anymore when using modern aligners. Modern alignment tools—like STAR—inherently handle untrimmed reads by soft-clipping, minimizing the necessity for aggressive trimming in expression analyses [14]. Only when reads are used for variant analyses, genome annotation or assembly purposes, read trimming is still recommended, including both adapter and quality trimming.
Library Strandedness. Currently x.FASTQ allows analyzing unstranded libraries only, meaning that even possible stranded libraries are analyzed as unstranded. STAR does not use library strandedness information for mapping, while RSEM can be configured to work with stranded libraries with the --strandedness <none|forward|reverse> option, with the recommendation of using reverse for Illumina TruSeq Stranded protocols. Thus, in case of need, it should be straightforward to add a new option to anqFASTQ for selecting the correct RSEM’s strandedness value. An issue for this is already open here: https://github.com/TCP-Lab/x.FASTQ/issues/30.
On RSEM Quantification. Among quantification approaches, a fundamental distinction exists between methods that estimate transcript abundance using generative probabilistic models (e.g., RSEM, BitSeq, salmon) and those that rely on straightforward read counting (e.g., HTSeq, featureCounts). The former typically offer superior accuracy at the gene level and also enable transcript-level resolution. However, generative models require prior knowledge of the fragment length distribution—the length of the physical molecule of DNA/RNA captured in the library prep stage and (partially) sequenced by the sequencer. This distribution is essential for computing effective transcript lengths, which influence fragment-to-transcript assignment probabilities. In paired-end sequencing, the fragment length distribution can be inferred from either the FASTQ files or the alignment data. In contrast, this is not possible for single-end reads, and RSEM users are therefore strongly encouraged to provide empirical fragment length statistics using the –fragment-length-mean and -fragment-length-sd options. Incorporating fragment length specifications substantially improves the accuracy of transcript quantification from single-end RNA-Seq data [4], however, if these parameters (i.e., the BioAnalyzer output) cannot be obtained or recovered, RSEM will not take a fragment length distribution into consideration. Nonetheless, the inference process remains relatively robust to such omissions: although maximum likelihood estimates may exhibit minor shifts, the use of identical distributional assumptions across all samples ensures that most effects of misspecification are cancelled out in downstream differential analyses.
3. Conclusions
The increasing availability of RNA-Seq data in public repositories, combined with the growing demand for transcriptomic analysis in life sciences, calls for tools that are at once robust, reproducible, and accessible to non-specialist users. x.FASTQ—initially developed at the Turin Cell Physiology Lab (TCP-Lab) to standardize and accelerate RNA-Seq data analyses—addresses this need by offering a lightweight, modular, and fully command-line-based solution that bridges the gap between usability and control. By abstracting the complexity of RNA-Seq workflows into background-executing Bash scripts, x.FASTQ empowers wet-lab researchers to autonomously perform comprehensive transcriptomic analyses without requiring in-depth bioinformatics skills.
x.FASTQ design enables scalable deployment across collaborative research environments, facilitating distributed analysis while minimizing the bioinformatician’s supervision load. At the same time, by relying on well-established tools, version-controlled scripting, and log-rich execution, it guarantees high standards of transparency and reproducibility.
Overall, this framework empowers the bioinformatician with clearly structured tools to automate RNA-Seq pipelines, while simultaneously equips collaborators who lack formal training in bioinformatics with a set of simple command-line tools that can be executed independently via SSH.
Looking forward, future developments will focus on containerization for enhanced portability, improved support for stranded protocols, and the addition of downstream modules for statistical analysis and visualization. In its current form, however, x.FASTQ already represents a pragmatic and effective compromise between flexibility, performance, and user autonomy in transcriptome data processing.
4. Software Availability
The x.FASTQ suite is freely available under the permissive MIT license and can be accessed via its public GitHub repository at https://github.com/TCP-Lab/x.FASTQ. The repository includes the full source code for all Bash modules, auxiliary R and Python scripts, and the Moliere orchestration utility. A dedicated test/ directory is provided to validate the functionality of all core features in a local environment; this folder contains example input data and a separate README.md file with detailed instructions for executing the complete pipeline in test mode. Installation guidelines, dependency specifications, and usage examples are available within the docs/ subdirectory. All components follow semantic versioning and implement integrated version reporting and logging to support reproducibility. Users and system administrators are encouraged to adapt the install.paths configuration file to their specific environments. Contributions and issue reports can be submitted through the GitHub issue tracker.
Appendix A
Appendix A.1. File Naming
In the current implementation of x.FASTQ, filenames are very meaningful. Each FASTQ file is required to have a name matching the regex pattern
i.e., beginning with an alphanumeric ID (usually an ENA Run ID of the type (E|D|S)RR[0-9]{6,}), immediately followed by the extension .fastq.gz, or separated from the rest of the filename by an underscore or other characters other than [a-zA-Z0-9]. Valid examples are GSM34636.fastq.gz, SRR19592966_1.fastq.gz, etc. This leading ID is propagated to the names of the log files printed by the getFASTQ module and BBDuk (stored in the Trim_stats subdirectory), as well as all output files from FastQC, STAR, and RSEM (stored in the FastQC_* and Counts subfolders, respectively). tabFASTQ will then assume each RSEM output file being saved into a sample- or run-specific subdirectory, whose name will be used for count matrix heading. Similarly, but at a lower level, even MultiQC needs each STAR and RSEM output to be properly prefixed with a suitable sample or run ID to be correctly accounted for. Note, however, that all of this should come naturally when FASTQs are downloaded from the ENA database using the getFASTQ module, since, as part of the INSDC [13], ENA guarantees a high degree of uniformity in its archive-generated FASTQs files, including file naming. In contrast, it is recommended that the user to manually name each project folder (i.e., each directory containing the entire set of FASTQ files from a single experiment) with a meaningful study ID (typically the associated GEO Series ID GSE[0-9]+ or the ENA BioProject accession PRJ(E|D|N)[A-Z][0-9]+). The name of the project directory is used as the project ID by most x.FASTQ modules to name their log files. The same applies to the MultiQC HTML global report, the PCA results and the name of the final expression matrix.
^[a-zA-Z0-9]+([^a-zA-Z0-9]*.*)?\.fastq\.gz$
Appendix A.2. Dependencies
The following software is required for x.FASTQ to function properly. You can always use
x.fastq -d to get a complete report about the current dependency status.
| • Development Environments | https://www.java.com/ |
| ► Java | https://www.python.org/ |
| ► Python | https://www.r-project.org/ |
| ► R | https://www.bioconductor.org/ |
| ► Bioconductor Packages | |
| - BiocManager | |
| - PCAtools | |
| - org.Hs.eg.db | |
| - org.Mm.eg.db | |
| ► CRAN Packages | https://cran.r-project.org/ |
| - gtools | |
| - stringi | |
| • Linux Tools | |
| ► hostname | |
| ► jq | https://jqlang.org/ |
| ► figlet (optional) | |
| • QC Tools | |
| ► FastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| ► MultiQC | https://seqera.io/multiqc/ |
| ► QualiMap | http://qualimap.conesalab.org/ |
| • NGS Software | |
| ► BBDuk | https://archive.jgi.doe.gov/data-and-tools/software-tools/bbtools/ |
| ► STAR | https://github.com/alexdobin/STAR |
| ► RSEM | https://github.com/deweylab/RSEM |
References
- Conesa, A.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Patro, R. , Duggal, G., Love, M.I., Irizarry, R.A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Dobin, A.; et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B. & Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I. , Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kulkarni, N.; et al. Reproducible bioinformatics project: a community for reproducible bioinformatics analysis pipelines. BMC Bioinformatics 2018, 19. [Google Scholar] [CrossRef]
- Beccuti, M.; et al. SeqBox: RNAseq/ChIPseq reproducible analysis on a consumer game computer. Bioinformatics 2018, 34, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; et al. Babraham Bioinformatics - FastQC: A Quality Control tool for High Throughput Sequence Data. (2012). at https://www.bioinformatics.babraham. ac.uk/projects/fastqc/.
- Bushnell, B. , Rood, J. & Singer, E. BBMerge – Accurate paired shotgun read merging via overlap. PLOS ONE 2017, 12, e185056. [Google Scholar] [CrossRef]
- Ewels, P. , Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinform. (Oxf. Engl. ) 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Sandve, G.K. , Nekrutenko, A., Taylor, J. & Hovig, E. Ten Simple Rules for Reproducible Computational Research. PLOS Comput. Biol. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Arita, M. , Karsch-Mizrachi, I. & Cochrane, G. The international nucleotide sequence database collaboration. Nucleic Acids Res. 2021, 49, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.R. , Baccarella, A., Parrish, J.Z. & Kim, C.C. Trimming of sequence reads alters RNA-Seq gene expression estimates. BMC Bioinform. 2016, 17, 103. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



