Submitted:
30 June 2025
Posted:
01 July 2025
You are already at the latest version
Abstract
Mast cells (MCs) are an emerging target in cancer immunotherapy due to their phenotypic plasticity, immunomodulatory capacity, and active recruitment into the tumor microenvironment (TME). While MCs can adopt both pro- and anti-tumor phenotypes, recent evidence suggests that tumors exploit this plasticity by recruiting immature MC progenitors and conditioning them toward tumor-supportive functions. Therapeutic strategies aimed at reprogramming MCs into anti-tumor effectors—rather than depleting them—are gaining traction. However, many of the most promising modulatory agents, including cationic peptides and small-molecule agonists, face significant delivery barriers due to poor systemic stability, off-target effects, and limited tumor penetration. Drug delivery systems (DDSs), particularly nanomedicine platforms, offer a powerful solution by enabling targeted, controlled, and localized delivery of these agents to MCs within the TME. In this review, we highlight the rationale for MC-targeted immunotherapy, evaluate current strategies for phenotypic modulation, and emphasize the critical role of DDSs in overcoming translational barriers. By integrating MC biology with advanced delivery technologies, we outline a path forward for harnessing this underexplored immune population in cancer treatment.
Keywords:
mast cell
; innate immunotherapy
; cancer
; drug delivery systems
1. Introduction
Immunotherapy, including immune checkpoint blockade, chimeric antigen receptor T-cell therapy, and cancer vaccines, is increasingly becoming the frontline treatment for several cancers and has revolutionized cancer care. One emerging target of interest for tumor immunotherapy is the mast cell (MC). MCs are granulocytes that have been implicated in rapid, robust, and sustained inflammatory responses in the body (e.g., anaphylaxis), but primarily have roles in immunity against infectious pathogens [1,2]. The presence of MCs in various tumors has long been recognized and is closely associated with cancer prognosis. While increased MC density correlates with favorable outcomes in some cancers (e.g., gliomas, colorectal, ovarian), it is associated with poor prognosis in others (e.g., melanoma, thyroid, gastric) [3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Additional conflicting results arise in breast and prostate cancer, where outcomes vary depending on MC location within the tumor [17,18,19,20]. Further, MC activation status has corresponded with both improved and worsened prognosis, sometimes within the same disease [7,8,9,10]. These mixed and sometimes even contradictory results highlight the dual role MCs play in cancer and hints at their immunotherapeutic potential (Figure 1).
MCs are often understudied in cancer research, partly because their activation is primarily associated with Type I hypersensitivity reactions, such as anaphylaxis. Anaphylaxis is typically triggered by antigen-specific IgE binding to the high-affinity IgE receptor (FcεRI) receptor on MCs. Upon exposure to an anaphylactic inducing antigen (e.g., peanut Ara h or shellfish tropomyosin), IgE crosslinking activates MCs, leading to rapid systemic inflammation and classic anaphylactic symptoms [21,22]. However, non-IgE activation mechanisms exist, mediated through receptors such as complement receptors (C3a/C5a), the Mas-related G protein receptor-X2 (MRGPRX2), or toll-like receptors (TLR) 2 and 4 [23,24,25]. Upon activation, MCs initiate a triphasic response designed to induce immediate toxicity, signal nearby immune cells, and recruit specialized immune populations. MCs secrete pre-synthesized granule contents, immunomodulatory lipid mediators, and chemokines and cytokines in each phase respectively [22] (Figure 2). The sum contribution of MC secreted factors within the tumor microenvironment (TME) defines whether its phenotype is pro- or anti-tumor.
The systemic dissemination of chemokines and cytokines is critical to MC homeostatic functioning and its immunotherapeutic potential as this ultimately initiates formation of an adaptive immune response [26]. For example, TNF-α released by MCs recruits adaptive immune populations to lymph nodes and enhances T-cell activation [27,28]. This functionality is facilitated by MCs localizing around blood vessels at host-environment interfaces [29]. Their perivascular location allows cytokines and chemokines to rapidly enter systemic circulation. An additional aspect that makes MCs uniquely intriguing for immunotherapy is that they leave the bone marrow as progenitors and mature within their final tissue of residence. MCs are recruited to the airways, intestinal linings, skin, and throughout regions of the brain where they undergo tissue-specific maturation shaped by local cytokines, extracellular matrix (ECM), and matrix metalloproteases (MMPs) [30]. This leads to a remarkably plastic and heterogenetic cellular population with distinct, tissue-dependent transcriptional programs [31] and resulting phenotypes [32,33]. Despite their heterogeneity, MCs are broadly classified in humans by protease content into MCT (tryptase-only) and MCCT (containing tryptase, chymase, carboxypeptidase, and cathepsin G) [34,35,36]. Although the full extent of their phenotypic flexibility is still being explored, MCs are increasingly recognized as dynamic players with therapeutic relevance in modulating the TME.
Strikingly, there is sufficient evidence to suggest that tumors are actively recruiting these immature MC progenitors to the TME. Stem cell factor (SCF) is a key growth factor for MC maturation and differentiation; however, it has been shown to recruit MCs in various cancers, including glioblastoma, hepatocellular carcinoma, and breast cancer [37,38,39]. Further, SCF and MCs have been implicated in promoting metastasis in breast cancer [40]. Several chemokines have been shown in tumor-mediated MC recruitment. CXCL12 colocalizes with CXCR4+ MCs in human glioblastoma [39], and inhibition of CXCR4 prevents MC migration towards pancreatic adenocarcinoma [41]. Similarly, CCL5 colocalizes with MCs in uterine cancer, and lymphoma-secreted CCL5 causes in vitro MC chemotaxis [42,43]. There is significant evidence that MC progenitors are broadly recruited to the TME, where, upon arrival, they can be conditioned to adopt pro- and anti-tumor-supportive functions.
The role of MCs in the TME has been heavily reviewed [36,44,45,46]. Within the context of cancer, MCs are thought to play a dual role – being both pro- and anti-tumor depending on the environment. Their pro-tumor functions can be summarized as promoting angiogenesis, degrading the ECM, and engaging in immunosuppression [44]. These effects are mediated by vasoactive factors such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor-β (PDGF-β), proteases including tryptase or chymase, and matrix metalloproteinases like matrix metalloprotein-9 (MMP-9), as well as inhibitory cytokines such as IL-10 and TGF-β [47]. MC anti-tumor functions are in direct cytotoxicity, immune cell recruitment, and immune cell activation. These effects are mediated by factors like TNF-α, CCL5, and IL-6, respectively. The role of MCs in tumors is complex, and numerous MC modulating agents have been developed and tested for their anti-tumor potential. To improve delivery and efficacy, a few of these compounds have been formulated using drug delivery systems (DDSs). This review will examine MC modulators that have been evaluated for their activity in cancer.
2. Modulating MC Activation as Cancer Immunotherapy
Anti-tumor immunotherapy strategies involving MCs focus on either inhibiting or activating their function. Approaches aimed at inhibition, such as reducing MC numbers or stabilizing them, have been studied as well as MC activation via agonism of receptors like complement receptors or TLRs. Historically, MC activation has been the most studied. Activation of MCs is achieved historically through agents such as compound 48/80 (c48/80), mastoparan, and mastoparan 7 (M7). C48/80, a p-methoxy-N-methyl phenylethylamine crosslinked by formaldehyde, is believed to activate MCs by directly stimulating Gi/o-type G proteins or by activating phospholipase D [57,58,59,60,61]. Mastoparan is an oligopeptide derived from wasp venom and is the parent compound for several second-generation MC agonists such as M7 and third-generation agonists MP12W [55,56,62]. Polymyxins, a class of antibiotics, have also demonstrated MC agonist activity and are considered relatively safe. Aside from mastoparan, many of these compounds have been underexplored for their anti-cancer potential [63,64,65,66,67].
Given that mastoparan has been the most extensively studied, particularly in cancer research, Table 1 highlights a range of wasp venom-derived or -inspired peptides that have been investigated for their MC agonism and anti-cancer potential. These peptides share a characteristic amphipathic structure and are typically used in their amidated form (-NH₂) rather than the carboxylated form (-COOH), as the former has been shown to exhibit greater biological potency [68]. Native mastoparan peptides originate from various wasp species, including Vespa lewisii (Mastoparan-L), V. xanthoptera (Mastoparan-X), V. crabro (Mastoparan-C), and Polybia paulista (Polybia-MPI) [48,49,50,51]. In addition to natural variants, several synthetic analogs have been engineered to enhance efficacy, stability, or selectivity. These include KM8 (a rationally designed analog of Mastoparan-L), [I5, R8] Mastoparan (a modified version with improved immunomodulatory effects), and Mastoparan-3 (a truncated derivative retaining core activity) [51,52,53]. Collectively, these cationic, membrane-penetrating peptides are of significant interest for MC activation and cancer immunotherapy research due to their potential to trigger immune responses or cytotoxic effects in the TME, with the Mas-related G protein-coupled receptor (MRGPR) family identified as their putative target [69,70,71]. These wasp venom-derived peptides represent a promising class of immunomodulatory agents for targeted MC activation and anti-tumor therapy.
2.1. Direct Effects of MC Modulators on Cancer Cells
Inhibition and activation of MCs can be achieved by leveraging agents that selectively induce MCs activation and exhibit cytotoxicity toward tumor cells (Table 2). Table 2 provides a comparative overview of the cytotoxic effects of various MC modulators—both inhibitors and agonists—across a range of cancer and healthy cell lines. Modulator activity is reported at the 50% inhibitory concentration (IC50) as determined through an MTT assay.
Among the MC inhibitors, cromolyn sodium exhibited selective cytotoxicity, showing stronger inhibition of colon cancer cells (HT-29, IC50 = 2.33 µM) compared to healthy breast epithelial cells (MCF-10, IC50 = 7.33 µM), suggesting some degree of tumor selectivity. In contrast, MC agonists such as mastoparan derivatives displayed broad-spectrum cytotoxicity across multiple tumor types. The amidated form, mastoparan-L-NH₂, demonstrated substantially greater potency than its carboxylated counterpart, evidenced by IC50 values for the same breast cancer lines ranged from 20-24 µM for the amidated form compared to 250-432 µM for the carboxylated version. Synthetic analogs such as KM8 exhibited the most potent cytotoxicity, with IC50 values as low as 5–7 µM in breast, lung, and esophageal cancers and significantly higher IC50s in healthy cells (~45 µM), indicating improved therapeutic indices. While natural mastoparans like mastoparan-C and Polybia-MPI show promise in lung, breast, and prostate models, their moderate cytotoxicity toward healthy cells warrants further optimization. The observed variability in IC50 values across cell types and peptide formulations highlights the importance of structure-activity relationships in tuning both efficacy and safety. Future development of MC-targeted therapeutics may benefit from combining potent MC agonists like KM8 with delivery systems that enhance tumor specificity and minimize systemic toxicity. These findings underscore the potential of MC modulators as targeted anti-cancer agents, particularly when modified to enhance potency and selectivity.
2.2. Inhibition of MC Function as Cancer Immunotherapy
In cancer immunotherapy, inhibiting MC function is commonly achieved by reducing their numbers or stabilizing their degranulation. Stabilizing degranulation can result in a reduction in the expression of MC-derived factors such as VEGF and histamine. VEGF plays a central role in angiogenesis, making it a promising target for limiting tumor vascularization and growth [81,82]. Histamine signals through four distinct receptors (H1–H4) that can be expressed on both tumor cells and various immune cells [83,84,85]. However, the complexity of histamine signaling poses a challenge—particularly since individual receptors can have opposing effects. For example, the H2 receptor has been shown to both promote and inhibit tumor progression, depending on the context [86,87].
Historically, reducing MC numbers has been accomplished by targeting c-KIT (CD117) which is the receptor for stem cell factor, an essential growth factor for MC survival and development [88]. One such agent is imatinib, an FDA-approved tyrosine kinase inhibitor used to treat cancers with aberrant c-KIT signaling [89,90]. However, because c-KIT is also expressed on other cell types, such as epithelial cells, imatinib can cause off-target effects. In a prostate cancer model, imatinib reduced adenocarcinoma development but unexpectedly promoted an aggressive neuroendocrine prostate cancer phenotype [91,92]. Similarly, in a triple-negative breast cancer model, imatinib increased tumor growth and induced peripheral tumor-associated clotting [93]. These findings suggest that MC depletion strategies may limiting. Given the variable outcomes of MC depletion, alternative strategies such as stabilizing MC degranulation have also been explored.
With regards to inhibiting MCs through stabilizing degranulation, inhibitory receptors like, Siglec-8 and PD-L1, can be targeted using humanized antibodies, like antolimab (anti-Siglec-8) or infliximab (anti-TNF-α) [28,94]. Blockade of MC-associated PD-L1 was shown to increase T-cell infiltration and activation products in a gastric cancer model [95]. However, these strategies are rarely specific to MCs, as the targeted receptors—such as Siglec-8, which is also expressed on eosinophils and basophils, and PD-L1, which is found on antigen-presenting cells, T cells, B cells, and various other cell types—are broadly expressed. In place of inhibitory antibodies, primarily two small molecules have been used in the context of cancer to stabilize MCs: ketotifen and cromolyn sodium (CS).
Ketotifen is a non-competitive antagonist of H1 receptors that prevents MC degranulation [96,97]. Interestingly, it was able to prolong survival and suppress metastasis formation of neuroendocrine prostate cancer [98]. This is an aggressive prostate cancer subtype promoted by c-KIT targeting, suggesting MCs in prostate cancer can be protective and deleterious depending on the subtype. These findings are supported by contradictory roles of MCs on tumor growth in prostate cancer depending on peri- vs intratumoral localization [17,92]. Cromolyn sodium, an FDA-approved MC stabilizer used for asthma, allergic rhinitis, and mastocytosis, has shown promising preclinical anti-tumor effects in several models, including thyroid and pancreatic cancers (Table 2) [12,99,100,101]. In a thyroid cancer xenograft model, Melillo et al. demonstrated that co-delivery of exogenous MCs with cromolyn significantly reduced tumor growth, accompanied by a decrease in proliferating (Ki67+) cells, compared to delivery of MCs alone [12]. Similarly, in a Myc-driven β-cell carcinoma model, Soucek et al. found that cromolyn halted tumor expansion and induced widespread cell death in established islet tumors, indicating a critical role for MCs in pancreatic tumor maintenance [101]. In contrast, Aliabadi et al. reported only a non-significant reduction in subcutaneous colon tumor mass using a higher cromolyn dose (50 mg/kg) from the previous study, suggesting tumor-type or model specific differences in MC involvement [72]. Together, these findings underscore the nuanced therapeutic role of MCs in tumor treatment and highlight the potential and limitations of both depletion and stabilization strategies.

Table 3.
Relevant preclinical model details and outcomes of soluble delivery of MC-modulating agents. Tumor size was determined via a web-based data digitizer. Daily (qd), every two days (q2d), intraperitoneal (IP), intravenous (IV), not significant (n.s.), and subcutaneous (subQ).
Table 3.
Relevant preclinical model details and outcomes of soluble delivery of MC-modulating agents. Tumor size was determined via a web-based data digitizer. Daily (qd), every two days (q2d), intraperitoneal (IP), intravenous (IV), not significant (n.s.), and subcutaneous (subQ).
| MC AGONIST | CANCER | IN VIVO MODEL | DOSE | DOSING | CONCLUSIONS | REF | |
| Cromolyn sodium | Thyroid | 8505-C subQ in BALB/c-nu | IP 10 mg/kg | qd | Co-injection of MCs with tumor resulted in accelerated tumor growth, treatment with cromolyn reversed this | [12] | |
| Pancreatic | pIns-mycERTAM;RIP7-bcl-xL transgenic | IP 10 mg/kg | qd | Induced apoptosis of pre-existing β-cell tumors; MCs critical for tumor expansion | [101] | ||
| Colon | CT-26 subQ in BALB/c | IP 50 mg/kg | q2d | Non-significant reduction in tumor weight and after survival change | [72] | ||
| Mastoparan | Melanoma | B16F10-Nex2 subQ in C57BL/6 | Peritumoral 5 mg/kg |
qd 5x | 70.29% growth inhibition rate 28.26% prolonged survival ratio |
[73] | |
| Breast | 4T1 orthotopic in BALB/c | IP 6 mg/kg | q2d | Non-significant reduction in tumor growth. significant reduction when combined with gemcitabine |
[68] | ||
| MCF-7/Dox in BALB/c-nu | IV 10 μmol/kg | q2d 7x | 33.6% growth inhibition rate | No signs of damage by histology or blood chemistry | [52] | ||
| KM8 | 81.6% growth inhibition rate | ||||||
2.3. Activation of MCs as Cancer Immunotherapy
While a limited number of cancer therapies focus on inhibiting MCs, a growing number of studies instead aim to activate them. Once activated, MCs have been shown to have direct cytotoxic effects on tumor cells via TNF-α and indirect cytostatic effects through stimulating DC histamine release[87,102]. MCs can also modulate macrophages, eosinophils, dendritic cells, NK cells, and T cells within the TME and recruit additional immune populations [36,81]. For example, activated MCs can release CCL2 and CCL3 that have been shown to recruit plasmacytoid DCs, CD8 T-cells, and NK cells [103,104]. MC-secreted TNF-α then facilitates better CD8 T-cell priming by DCs and can directly activate T cells [28,105]. These agonists predominantly target MRGPR due to non-specificity and anaphylaxis being associated with other MC activating receptors. While activation by these agonists sometimes results in robust degranulation, evidence suggests they can facilitate protective adaptive immune functions [106,107,108].
There is a limited number of in vivo studies regarding cancer and MC agonists, with studies primarily focusing on Mastoparan-L. The greatest reduction in tumor growth was reported by De Azevedo et al., who observed a 70% inhibition in a low-immunogenicity melanoma model following peri-tumoral treatment [73]. Except for this study, all studies used systemic, rather than local, delivery routes (I.V. or I.P.) and evaluated mastoparan and its analogues in breast cancer models. Hilchie et al. saw a non-significant reduction in tumor growth with systemic mastoparan-L treatment, while Zhang et al. observed a 33.6% growth inhibition rate [52,68]. Notably, Hilchie et al. used a murine tumor model whereas Zhang et al. used a xenograft model. Using the same model and route of administration, Zhang et al. showed that mastoparan analog KM8 had an 81.6% inhibition rate. These findings highlight the potential of MC agonists—particularly mastoparan analogs—as anti-tumor agents, though further in vivo studies are needed to optimize delivery methods and clarify their therapeutic efficacy across cancer types and within species.
3. Formulation of MC Modulators in Drug Delivery Systems
To improve stability, bioavailability, and targeted delivery of MC modulators, various advanced DDSs have been applied to modulate MC responses (Table 4). These systems help address key challenges to delivery of MC modulators such as poor solubility, enzymatic degradation, and off-target cytotoxicity [109]. Among the many carriers applied, PEGylated liposomes are noted to enhance circulation time and reduce immune clearance, offering controlled release of the encapsulate. Chitosan nanoparticles are well studied because of their known mucoadhesive properties which can improve mucosal delivery. Similarly, alendronate sodium (e.g. Fosamax) nanoconjugates have been formed with MC modulating compounds for lung delivery. The fluvastatin-mastoparan nanocomplex represents a dual-functional system combining the anti-inflammatory statin properties of fluvastatin with the MC-activating capacity of mastoparan. Soya phosphatidylcholine-based phytosomes enhance the stability and bioavailability of MC modulators by encapsulating them in the phospholipid carriers. Other innovative platforms include outer membrane vesicles (OMVs) from gram-negative bacteria, which serve as natural carriers for immune modulators, and fluorinated polyethylenimine (PEI) nanoparticles, which improve endosomal escape and transfection efficiency. Also, micelles offer a targeted approach for delivering of MC modulators. Collectively, these DDS technologies offer promising strategies for harnessing or inhibiting MC functions in cancer, allergy, and infection, while minimizing systemic toxicity and enhancing therapeutic precision [110,111,112,113,114].
3.1. Cromolyn Formulations for MC Inhibition
Cromolyn has been encapsulated into chitosan nanoparticles and liposomes to facilitate better delivery [115,116,117]. Using ionic gelation techniques, Motwai et al. fabricated 10 formulations of cromolyn-containing chitosan nanoparticles (CCSNPs) [116]. These formulations displayed a wide range of physiochemical characteristics: 100 nm to 2 μm diameter, 0.3-0.9 polydispersity index (PDI), and +5 to +50 mV surface charge (Figure 3A). The formulation “F6” was selected for in vivo interrogation considering output measures of encapsulation efficiency, morphology, and release kinetics (Figure 3A,B). These roughly spherical, positively charged particles displayed a burst release of ~20% in two hours before plateauing at 80% release in 48 hours (Figure 3C). In a chemically induced colorectal cancer model, Motawi et al. showed 33% of mice treated with cromolyn-containing chitosan NPs had tumor lesions compared to 67% for mice receiving soluble cromolyn. Colorectal cancer leads to loss of colon architecture, a phenotype that was rescued only with treatment with cromolyn-containing NPs but not the vehicle or soluble controls. When evaluating colon MCs by toluidine blue staining, they observed that without tumor induction, there were limited MCs present, even with cromolyn treatment. In disease-bearing mice, treatment with cromolyn NPs decreased MC numbers while treatment with soluble cromolyn had no apparent effect on the number of MCs (Figure 3D). Survival was not assessed in these studies. Overall, the study demonstrates that encapsulating the MC inhibitor cromolyn in polymeric chitosan NPs significantly enhances its therapeutic efficacy in a colorectal cancer model.
Kim et al. produced PEGylated liposomes containing cromolyn using a reverse-phase evaporation vesicle method [115]. Liposomes were produced with a fixed dipalmitoylphosphatidylcholine (DPPC), dimyristoylphosphatidylcholine (DMPC), and distearoylphosphatidylcholine (DSPC) molar ratio (4:1:1), while the concentration of a fourth excipient, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-mPEG2000), was optimized by varying its molar ratio from 0.1 to 1.0. A ten-fold increase in DSPE-mPEG2000 resulted in a 3.5x reduction in encapsulation efficiency of cromolyn. The formulation with the lowest ratio of DSPE-mPEG2000 (4:1:1:0.1) was selected for stability evaluations and found to not increase surface-adsorbed serum proteins after 48 hours (Figure 4A). Further the liposomal formulation was able to reduce cell growth of two pancreatic lines in vitro (Figure 4B). In an immunodeficient model of pancreatic cancer, they observed improved tumor growth with their PEGylated liposomal formulation over a soluble cromolyn control, both dosed at 10 mg/kg (Figure 4C). While treatment with a gemcitabine PEGylated liposome outperformed the cromolyn formulation, it was dosed at 80 mg/kg. However, the greatest tumor reduction was observed in mice receiving separate cromolyn and gemcitabine liposomal formulations at 10 and 80 mg/kg respectively. Overall, formulated cromolyn demonstrated potent effects in vivo whether delivered through chitosan particles or liposomes.
3.2. Mastoparan Nanoconjugates
Alhakamy et al. (2021, 2022) developed two distinct systems by conjugating mastoparan to small-molecule drugs, enhancing their therapeutic activity and characterizing their performance in vitro. In 2021 they produced a mastoparan and fluvastatin (a statin) conjugate optimized for a 77.6 nm size [75]. Complexation with mastoparan nearly halved its 24-hour IC₅₀ value against the A549 lung cancer cell line, reducing it from 34.3 to 18.6 μg/mL. They subsequently developed a nanoconjugate composed of mastoparan and alendronate sodium (Fosamax), resulting in a larger particle size of 134.9 nm [118]. When the IC50 assay was conducted over two hours in the same A549 lung cancer cell line, the nanoconjugate demonstrated a markedly enhanced potency, reducing the IC50 from ~14 μM for mastoparan alone to just 1.3 μM. Notably fluvastatin and alendronate sodium have their own anti-tumor effects, and while significance against soluble controls was assessed, synergy was not. Interestingly, mastoparan appears to be more potent at shorter timepoints, which is supported by Hilchie et al.’s finding that cytotoxicity peaked at 8 hours [68]. Together, these studies by Alhakamy et al. demonstrate that conjugating mastoparan to anti-tumor small molecules like fluvastatin and alendronate sodium can significantly enhance cytotoxic potency in vitro, though the potential for synergistic effects remains to be fully explored.
3.3. Phytosome Encapsulation of Mastoparan
Phytosomes are liposome-like vesicular systems formed by complexing bioactive compounds with plant-derived phospholipids, such as soya bean lecithin. Like liposomes, these carriers enhance the stability, bioavailability, and cellular uptake of encapsulated agents. In one study, mastoparan-M was encapsulated into phytosomes to generate positively charged nanoparticles optimized for in vivo delivery (Figure 5A–D) [119]. When applied in a breast cancer xenograft model, Zhao et al. reported that phytosome-encapsulated mastoparan-M led to a significant reduction in tumor volume by day 20, compared to administration of soluble mastoparan-M. Histological analysis using hematoxylin and eosin (HE) staining further demonstrated a marked decrease in proliferative activity and a corresponding increase in apoptosis within tumor tissues treated with the phytosomal formulation (Figure 5E–H). These results suggest that phytosomal delivery enhances the therapeutic efficacy and tumor-targeting potential of mastoparan-based therapies.
3.4. Polybia-MP1 Nanoparticle Formulations
MC agonist Polybia-MP1 (MP1) was formulated into natural and synthetic carrier systems by Ren et al. and Li et al., respectively, to treat bladder cancer [120,121].
Ren et al. genetically engineered gram-negative bacteria to express modified MP1 peptides for improved expression, which were subsequently passively loaded into the bacteria's naturally secreted outer membrane vesicles (OMVs) [121]. Four OMV formulations were made with all found to be ~90 nm with a -17 mV charge (Figure 6A). When taken in vivo in a subcutaneous immunocompetent model of bladder cancer, they identified a lead formulation that both significantly reduced tumor growth and displayed greater apoptosis by TUNEL immunofluorescence staining (Figure 6B,C). They went on to show this formulation improved intratumoral antigen presentation by dendritic cells as well as increasing infiltration by both CD4 and CD8 T-cells (Figure 6D–F). These elevated levels of effector populations correlated with increased IL-6, TNF-α, and IFN-γ within the tumor. Critically, many of these results were shown to be significant compared to the OMV vehicle control, suggesting the heightened inflammation was due to MP1. These findings highlight that MP1-loaded OMVs can enhance anti-tumor immunity through targeted MC activation and modulation of the TME.
Li et al. encapsulated MPI into polyethylenimine (PEI), with or without fluorination (F-PEI), the latter aimed at enhancing transport across the bladder mucosa following intravesical therapy [120]. After successfully producing 260-296 nm sized NPs for F-PEI and PEI, respectively, Li et al. assessed the NP’s mucosal transport in vivo in an orthotopic xenograft bladder cancer model (Figure 7A). Using a fluorescently labeled MP1, they saw an increased penetration of the fluorescent signal into the bladder 60 minutes after infusion (Figure 7B). Initial results in a subcutaneous tumor showed their MP1-containg F-PEI NPs improved tumor control, even when compared to a chemotherapeutic agent, as well as minimal reduction in mouse weights (Figure 7C,D). Finally, they supported their results with an orthotopic bladder cancer model and showed that their F-PEI NPs containing MP1 improved survival at four weeks to 83%, compared to 50% for soluble MPI (Figure 7E). These findings highlight the potential of fluorinated PEI NPs as an effective delivery system for MPI, offering enhanced mucosal penetration, superior tumor control, and improved survival outcomes in bladder cancer models. MCs are thought to promote bladder cancer invasiveness through ECM remodeling [122].
3.5. Micellular Polymyxin E Co-Delivered with Doxorubicin
Lan et al. investigated the use of polymyxin E (PE) co-loaded with doxorubicin into polymeric micelles as a therapeutic strategy for cervical cancer [123]. In a subcutaneous U14 tumor model in Kunming mice, treatment with micelles containing polymyxin E alone showed no significant impact on tumor growth. However, when combined with doxorubicin and administered intravenously at 2.5 mg/kg every other day for a total of seven doses, the co-loaded micelles resulted in a significantly greater reduction in tumor volume compared to either agent alone. These results suggest that while PE may lack direct anti-tumor activity, its combination with chemotherapy can enhance therapeutic efficacy, likely through synergistic mechanisms or improved drug delivery.
4. Conclusions
MCs represent a unique and underexplored target for cancer immunotherapy. Significant evidence suggests MCs play a role in tumor development, with the environmental context dictating whether that role is pro- or anti-tumor. Targeting the process of functional polarization within the TME offers a unique opportunity to potentially reprogram progenitor MCs before they are co-opted by the tumor’s immunosuppressive environment. While more work needs to be done to understand the underlying biology, advances are already being made in potential therapeutics. Approaches to reduce MC numbers and to target their secreted factors are often non-specific and have mixed results across tumor models. MC agonists and stabilizing agents have been shown to decrease tumor growth in vivo in cancer models; however, many of these agents display unfavorable physiochemical properties for systemic administration. Drug delivery systems—particularly those leveraging nanomedicine platforms—offer a promising solution by enabling cell-specific targeting, controlled release kinetics, and co-delivery of synergistic agents. Indeed, some in vivo studies only observed significant reductions in tumor burden upon codelivery of MCs agonists with chemotherapeutic agents [68]. Likely, the success of MC-targeted therapies will be contingent on combination with other immunotherapies[125,126]. As such, the integration of advanced delivery technologies is not merely advantageous but essential for the achieving the precision necessary for effective immunological re-education of MCs in cancer therapy.
Future work should exploit DDS for delivery of MC modulators that affect MC development within the TME. MCs are a distinctive immunotherapeutic target, as many tumors actively recruit progenitor cells and condition them within the TME. Targeting the mechanisms that drive this functional polarization presents a promising strategy to disrupt tumor-mediated immune education—offering the potential to reprogram or eliminate MCs before they are co-opted into supporting tumor growth and immune suppression. In summary, leveraging drug delivery systems to precisely modulate mast cell behavior within the TME may unlock new avenues in cancer immunotherapy, transforming these often-overlooked cells from tumor accomplices into allies in anti-cancer defense.
References
- Flier, J.S.; Underhill, L.H.; Galli, S.J. New Concepts about the Mast Cell. N. Engl. J. Med. 1993, 328, 257–265. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Pang, X.; Letourneau, R.; Sant, G.R. Interstitial Cystitis: A Neuroimmunoendocrine Disordera. Ann. New York Acad. Sci. 1998, 840, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Hedström, G.; Berglund, M.; Molin, D.; Fischer, M.; Nilsson, G.; Thunberg, U.; Book, M.; Sundström, C.; Rosenquist, R.; Roos, G.; et al. Mast cell infiltration is a favourable prognostic factor in diffuse large B-cell lymphoma. Br. J. Haematol. 2007, 138, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Magistris, A.; Loizzi, V.; Lin, F.; Rutgers, J.; Osann, K.; DiSaia, P.J.; Samoszuk, M. Mast cell density, angiogenesis, blood clotting, and prognosis in women with advanced ovarian cancer. Gynecol. Oncol. 2005, 99, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wu, W.; Liu, T.; Zhao, Y.; Wang, Y.; Zhang, H.; Wang, Z.; Dai, Z.; Zhou, X.; Luo, P.; et al. Large-scale bulk RNA-seq analysis defines immune evasion mechanism related to mast cell in gliomas. Front. Immunol. 2022, 13, 914001. [Google Scholar] [CrossRef]
- Gulubova, M.; Vlaykova, T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J. Gastroenterol. Hepatol. 2009, 24, 1265–1275. [Google Scholar] [CrossRef]
- Shen, Y.M.; Zheng, D.B.; Hu, D.M.; Ma, B.M.; Cai, C.M.; Chen, W.M.; Zeng, J.M.; Luo, J.M.; Xiao, D.M.; Zhao, Y.M.; et al. The prognostic value of tumor-associated macrophages in glioma patients. Medicine 2023, 102, e35298. [Google Scholar] [CrossRef]
- Xie, Z.; Niu, L.; Zheng, G.; Du, K.; Dai, S.; Li, R.; Dan, H.; Duan, L.; Wu, H.; Ren, G.; et al. Single-cell analysis unveils activation of mast cells in colorectal cancer microenvironment. Cell Biosci. 2023, 13, 1–17. [Google Scholar] [CrossRef]
- Wang, B.; Li, L.; Liao, Y.; Li, J.; Yu, X.; Zhang, Y.; Xu, J.; Rao, H.; Chen, S.; Zhang, L.; et al. Mast cells expressing interleukin 17 in the muscularis propria predict a favorable prognosis in esophageal squamous cell carcinoma. Cancer Immunol. Immunother. 2013, 62, 1575–1585. [Google Scholar] [CrossRef]
- Guo, X.; Shen, W.; Sun, M.; Lv, J.; Liu, R.; Qin, S. Activated Mast Cells Combined with NRF2 Predict Prognosis for Esophageal Cancer. J. Oncol. 2023, 2023, 1–13. [Google Scholar] [CrossRef]
- Franco, G.; Guarnotta, C.; Frossi, B.; Piccaluga, P.P.; Boveri, E.; Gulino, A.; Fuligni, F.; Rigoni, A.; Porcasi, R.; Buffa, S.; et al. Bone marrow stroma CD40 expression correlates with inflammatory mast cell infiltration and disease progression in splenic marginal zone lymphoma. Blood 2014, 123, 1836–1849. [Google Scholar] [CrossRef]
- Melillo, R.M.; Guarino, V.; Avilla, E.; Galdiero, M.R.; Liotti, F.; Prevete, N.; Rossi, F.W.; Basolo, F.; Ugolini, C.; de Paulis, A.; et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 2010, 29, 6203–6215. [Google Scholar] [CrossRef]
- Molin, D.; Edström, A.; Glimelius, I.; Glimelius, B.; Nilsson, G.; Sundström, C.; Enblad, G. Mast cell infiltration correlates with poor prognosis in Hodgkin's lymphoma. Br. J. Haematol. 2002, 119, 122–124. [Google Scholar] [CrossRef]
- Ribatti, D.; Ennas, M.G.; Vacca, A.; Ferreli, F.; Nico, B.; Orru, S.; Sirigu, P. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur. J. Clin. Investig. 2003, 33, 420–425. [Google Scholar] [CrossRef]
- Taskinen, M.; Karjalainen-Lindsberg, M.-L.; Leppä, S. Prognostic influence of tumor-infiltrating mast cells in patients with follicular lymphoma treated with rituximab and CHOP. Blood 2008, 111, 4664–4667. [Google Scholar] [CrossRef]
- Yano, H.; Kinuta, M.; Tateishi, H.; Nakano, Y.; Matsui, S.; Monden, T.; Okamura, J.; Sakai, M.; Okamoto, S. Mast cell infiltration around gastric cancer cells correlates with tumor angiogenesis and metastasis. Gastric Cancer 1999, 2, 26–32. [Google Scholar] [CrossRef]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikström, P.; et al. Mast Cells Are Novel Independent Prognostic Markers in Prostate Cancer and Represent a Target for Therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef]
- Majorini, M.T.; Colombo, M.P.; Lecis, D. Few, but Efficient: The Role of Mast Cells in Breast Cancer and Other Solid Tumors. Cancer Res. 2022, 82, 1439–1447. [Google Scholar] [CrossRef]
- Reddy, S.M.; Reuben, A.; Barua, S.; Jiang, H.; Zhang, S.; Wang, L.; Gopalakrishnan, V.; Hudgens, C.W.; Tetzlaff, M.T.; Reuben, J.M.; et al. Poor Response to Neoadjuvant Chemotherapy Correlates with Mast Cell Infiltration in Inflammatory Breast Cancer. Cancer Immunol. Res. 2019, 7, 1025–1035. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Tamma, R. Controversial role of mast cells in breast cancer tumor progression and angiogenesis. Clin. Breast Cancer 2021, 21, 486–491. [Google Scholar] [CrossRef]
- Turner, H.; Kinet, J.-P. Signalling through the high-affinity IgE receptor FcεRI. Nature 1999, 402, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Gülen, T.; Akin, C. Anaphylaxis and Mast Cell Disorders. Immunol. Allergy Clin. North Am. 2022, 42, 45–63. [Google Scholar] [CrossRef]
- Stone, K.D. Prussin, and D.D. Metcalfe, IgE, mast cells, basophils, and eosinophils. Journal of Allergy and Clinical Immunology, 2010. 125(2 SUPPL. 2): p. S73-S80.
- Yu, Y.; Blokhuis, B.R.; Garssen, J.; Redegeld, F.A. Non-IgE mediated mast cell activation. Eur. J. Pharmacol. 2016, 778, 33–43. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2016, 6, 620. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Nakae, S.; Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 2005, 6, 135–142. [Google Scholar] [CrossRef]
- McLachlan, J.B.; Hart, J.P.; Pizzo, S.V.; Shelburne, C.P.; Staats, H.F.; Gunn, M.D.; Abraham, S.N. Mast cell–derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat. Immunol. 2003, 4, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Suto, H.; Iikura, M.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast Cells Enhance T Cell Activation: Importance of Mast Cell Costimulatory Molecules and Secreted TNF. J. Immunol. 2006, 176, 2238–2248. [Google Scholar] [CrossRef]
- Nautiyal, K.M.; Ribeiro, A.C.; Pfaff, D.W.; Silver, R. Brain mast cells link the immune system to anxiety-like behavior. Proc. Natl. Acad. Sci. 2008, 105, 18053–18057. [Google Scholar] [CrossRef]
- Gurish, M.F.; Austen, K.F. Developmental Origin and Functional Specialization of Mast Cell Subsets. Immunity 2012, 37, 25–33. [Google Scholar] [CrossRef]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F.; The Immunological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef]
- Katz, H.R.; Stevens, R.L.; Austen, K. Heterogeneity of mammalian mast cells differentiated in vivo and in vitro. J. Allergy Clin. Immunol. 1985, 76, 250–259. [Google Scholar] [CrossRef]
- Xing, W.; Austen, K.F.; Gurish, M.F.; Jones, T.G. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc. Natl. Acad. Sci. 2011, 108, 14210–14215. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, G., O. Ajitawi, and D.S. Chi, The human mast cell: an overview. Mast Cells: Methods and Protocols, 2005: p. 13-34.
- Khazaie, K.; Blatner, N.R.; Khan, M.W.; Gounari, F.; Gounaris, E.; Dennis, K.; Bonertz, A.; Tsai, F.-N.; Strouch, M.J.T.; Cheon, E.; et al. The significant role of mast cells in cancer. Cancer Metastasis Rev. 2011, 30, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Ligan, C. , et al., The regulatory role and mechanism of mast cells in tumor microenvironment. American Journal of Cancer Research, 2024. 14(1): p. 1-1.
- Huang, B.; Lei, Z.; Zhang, G.-M.; Li, D.; Song, C.; Li, B.; Liu, Y.; Yuan, Y.; Unkeless, J.; Xiong, H.; et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood 2008, 112, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Kwok, E.; Everingham, S.; Zhang, S.; Greer, P.A.; Allingham, J.S.; Craig, A.W. FES Kinase Promotes Mast Cell Recruitment to Mammary Tumors via the Stem Cell Factor/KIT Receptor Signaling Axis. Mol. Cancer Res. 2012, 10, 881–891. [Google Scholar] [CrossRef]
- Põlajeva, J.; Sjösten, A.M.; Lager, N.; Kastemar, M.; Waern, I.; Alafuzoff, I.; Smits, A.; Westermark, B.; Pejler, G.; Uhrbom, L.; et al. Mast Cell Accumulation in Glioblastoma with a Potential Role for Stem Cell Factor and Chemokine CXCL12. PLOS ONE 2011, 6, e25222. [Google Scholar] [CrossRef]
- Roy, D.L. , et al., Arthritis augments breast cancer metastasis: role of mast cells and SCF/c-Kit signaling. Breast Cancer Research, 2013. 15(2): p. R32.
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic Mast Cell–Stromal Cell Interactions Promote Growth of Pancreatic Cancer. Cancer Res. 2013, 73, 3927–3937. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Lv, J.-Q.; Lin, Y.; Xiang, M.; Gao, B.-H.; Shi, Y.-F. Expression of chemokines CCL5 and CCL11 by smooth muscle tumor cells of the uterus and its possible role in the recruitment of mast cells. Gynecol. Oncol. 2007, 105, 650–656. [Google Scholar] [CrossRef]
- Fischer, M. , et al., Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. International Journal of Cancer, 2003. 107(2): p. 197-201.
- Oldford, S.A.; Marshall, J.S. Mast cell modulation of the tumor microenvironment. The Tumor Immunoenvironment, 2013: p.
- Varricchi, G. , et al., Future Needs in Mast Cell Biology. International Journal of Molecular Sciences 2019, Vol. 20, Page 4397, 2019. 20(18): p. 4397-4397.
- Varricchi, G. , et al., Are Mast Cells MASTers in Cancer? Frontiers in Immunology, 2017. 8.
- Grützkau, A.; Krüger-Krasagakes, S.; Baumeister, H.; Schwarz, C.; Kögel, H.; Welker, P.; Lippert, U.; Henz, B.M.; Möller, A. Synthesis, Storage, and Release of Vascular Endothelial Growth Factor/Vascular Permeability Factor (VEGF/VPF) by Human Mast Cells: Implications for the Biological Significance of VEGF206. Mol. Biol. Cell 1998, 9, 875–884. [Google Scholar] [CrossRef]
- Silva, O.N.; Torres, M.D.T.; Cao, J.; Alves, E.S.F.; Rodrigues, L.V.; Resende, J.M.; Lião, L.M.; Porto, W.F.; Fensterseifer, I.C.M.; Lu, T.K.; et al. Repurposing a peptide toxin from wasp venom into antiinfectives with dual antimicrobial and immunomodulatory properties. Proc. Natl. Acad. Sci. 2020, 117, 26936–26945. [Google Scholar] [CrossRef]
- Henriksen, R.J. , et al., Side Chain Hydrophobicity Modulates Therapeutic Activity and Membrane Selectivity of Antimicrobial Peptide Mastoparan-X. PLoS ONE, 2014. 9(3): p. e91007.
- Souza, B.M.; Mendes, M.A.; Santos, L.D.; Marques, M.R.; César, L.M.; Almeida, R.N.; Pagnocca, F.C.; Konno, K.; Palma, M.S. Structural and functional characterization of two novel peptide toxins isolated from the venom of the social wasp Polybia paulista. Peptides 2005, 26, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Argiolas, A.; Pisano, J.J. Isolation and characterization of two new peptides, mastoparan C and crabrolin, from the venom of the European hornet, Vespa crabro. J. Biol. Chem. 1984, 259, 10106–10111. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Xing, Z.; Zhong, H.; Yu, D.; Yu, R.; Deng, X. Plasma metabolites-based design of long-acting peptides and their anticancer evaluation. Int. J. Pharm. 2022, 631, 122483. [Google Scholar] [CrossRef] [PubMed]
- Irazazabal, L.N.; Porto, W.F.; Ribeiro, S.M.; Casale, S.; Humblot, V.; Ladram, A.; Franco, O.L. Selective amino acid substitution reduces cytotoxicity of the antimicrobial peptide mastoparan. Biochim. et Biophys. Acta (BBA) - Biomembr. 2016, 1858, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Raynor, R.L.; Kim, Y.-S.; Zheng, B.; Vogler, W.R.; Kuo, J. Membrane interactions of mastoparan analogues related to their differential effects on protein kinase C, Na,K-ATPase and HL60 cells. FEBS Lett. 1992, 307, 275–279. [Google Scholar] [CrossRef]
- Konrad, J.R. , et al., The Heterotrimeric G-protein Gi Is Localized to the Insulin Secretory Granules of β-Cells and Is Involved in Insulin Exocytosis. Journal of Biological Chemistry, 1995. 270(21): p. 12869-12876.
- Ontiveros-Padilla, L.; Hendy, D.A.; Pena, E.S.; Williamson, G.L.; Murphy, C.T.; Lukesh, N.R.; Ashcraft, K.A.; Abraham, M.A.; Landon, C.D.; Staats, H.F.; et al. Broadly active intranasal influenza vaccine with a nanocomplex particulate adjuvant targeting mast cells and toll-like receptor 9. J. Control. Release 2025, 384, 113855. [Google Scholar] [CrossRef]
- McLachlan, J.B.; Shelburne, C.P.; Hart, J.P.; Pizzo, S.V.; Goyal, R.; Brooking-Dixon, R.; Staats, H.F.; Abraham, S.N. Mast cell activators: a new class of highly effective vaccine adjuvants. Nat. Med. 2008, 14, 536–541. [Google Scholar] [CrossRef]
- Stanovnik, L., M. Logonder-Mlinsek, and F. Erjavec, The effect of compound 48/80 and of electrical field stimulation on mast cells in the isolated mouse stomach. Agents Actions, 1988. 23(3-4): p. 300-3.
- Koibuchi, Y. , et al., Binding of active components of compound 48/80 to rat peritoneal mast cells. Eur J Pharmacol, 1985. 115(2-3): p. 171-7.
- Murphy CT, B. EM, and A. KM, Mast Cell Activators as Adjuvants in Mucosal Vaccines. International Journal of Pharmaceutics, In press.
- Schemann, M. , et al., The mast cell degranulator compound 48/80 directly activates neurons. PLoS One, 2012. 7(12): p. e52104.
- John, A.L.S.; Choi, H.W.; Walker, Q.D.; Blough, B.; Kuhn, C.M.; Abraham, S.N.; Staats, H.F. Novel mucosal adjuvant, mastoparan-7, improves cocaine vaccine efficacy. npj Vaccines 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Ellis, H.V.; Johnson, A.R.; Moran, N.C. SELECTIVE RELEASE OF HISTAMINE FROM RAT MAST CELLS BY SEVERAL DRUGS. J. Pharmacol. Exp. Ther. 1970, 175, 627–631. [Google Scholar] [CrossRef]
- Laboratories, B. , Polymyxin B for Injection USP 500,000 units. 2011: Product Insert.
- Yoshino, N.; Endo, M.; Kanno, H.; Matsukawa, N.; Tsutsumi, R.; Takeshita, R.; Sato, S.; Miyaji, E.N. Polymyxins as Novel and Safe Mucosal Adjuvants to Induce Humoral Immune Responses in Mice. PLOS ONE 2013, 8, e61643. [Google Scholar] [CrossRef]
- Berkhout, M.C.; van Velzen, A.J.; Touw, D.J.; de Kok, B.M.; Fokkens, W.J.; Heijerman, H.G.M. Systemic absorption of nasally administered tobramycin and colistin in patients with cystic fibrosis. J. Antimicrob. Chemother. 2014, 69, 3112–3115. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, J.; Liu, H.-X.; Zhu, Y.-G.; Qu, J.-M. Intravenous combined with aerosolised polymyxin versus intravenous polymyxin alone in the treatment of pneumonia caused by multidrug-resistant pathogens: a systematic review and meta-analysis. Int. J. Antimicrob. Agents 2015, 46, 603–609. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Sharon, A.J.; Haney, E.F.; Hoskin, D.W.; Bally, M.B.; Franco, O.L.; Corcoran, J.A.; Hancock, R.E. Mastoparan is a membranolytic anti-cancer peptide that works synergistically with gemcitabine in a mouse model of mammary carcinoma. Biochim. et Biophys. Acta (BBA) - Biomembr. 2016, 1858, 3195–3204. [Google Scholar] [CrossRef]
- Ding, J.; Zhang, S.S.; Fernandopulle, N.A.; Karas, J.A.; Li, J.; Ziogas, J.; Velkov, T.; Mackay, G.A. Differential MRGPRX2-dependent activation of human mast cells by polymyxins and octapeptins. Eur. J. Pharmacol. 2024, 984, 177050. [Google Scholar] [CrossRef] [PubMed]
- Ferry, X.; Brehin, S.; Kamel, R.; Landry, Y. G protein-dependent activation of mast cell by peptides and basic secretagogues. Peptides 2002, 23, 1507–1515. [Google Scholar] [CrossRef]
- Mousli, M.; Bueb, J.-L.; Bronner, C.; Rouot, B.; Landry, Y. G protein activation: a receptor-independent mode of action for cationic amphiphilic neuropeptides and venom peptides. Trends Pharmacol. Sci. 1990, 11, 358–362. [Google Scholar] [CrossRef]
- Aliabadi, A.; Haghshenas, M.R.; Kiani, R.; Panjehshahin, M.R.; Erfani, N. Promising anticancer activity of cromolyn in colon cancer: in vitro and in vivo analysis. J. Cancer Res. Clin. Oncol. 2024, 150, 1–8. [Google Scholar] [CrossRef]
- De Azevedo, R.A. , et al., Mastoparan induces apoptosis in B16F10-Nex2 melanoma cells via the intrinsic mitochondrial pathway and displays antitumor activity in vivo. Peptides, 2015. 68: p. 113-119.
- Alhakamy, N.A.; Okbazghi, S.Z.; Alfaleh, M.A.; Abdulaal, W.H.; Bakhaidar, R.B.; Alselami, M.O.; AL Zahrani, M.; Alqarni, H.M.; Alghaith, A.F.; Alshehri, S.; et al. Wasp venom peptide improves the proapoptotic activity of alendronate sodium in A549 lung cancer cells. PLOS ONE 2022, 17, e0264093. [Google Scholar] [CrossRef] [PubMed]
- Alhakamy, N.A.; Ahmed, O.A.A.; Shadab; Fahmy, U. A. Mastoparan, a Peptide Toxin from Wasp Venom Conjugated Fluvastatin Nanocomplex for Suppression of Lung Cancer Cell Growth. Polymers 2021, 13, 4225. [Google Scholar] [CrossRef]
- Walker, O. , et al., Anti-cancer peptide mastoparan and derivatives kill HCT-116 and HT-29 colon cancer cells by peptide-mediated Lysis | Acadia Scholar. 2020.
- Hilchie, A.L.; Sharon, A.J.; Haney, E.F.; Hoskin, D.W.; Bally, M.B.; Franco, O.L.; Corcoran, J.A.; Hancock, R.E. Mastoparan is a membranolytic anti-cancer peptide that works synergistically with gemcitabine in a mouse model of mammary carcinoma. Biochim. et Biophys. Acta (BBA) - Biomembr. 2016, 1858, 3195–3204. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Wu, Y.; Wang, L.; Ma, C.; Xi, X.; Bininda-Emonds, O.R.; Shaw, C.; Chen, T.; Zhou, M. Evaluation of the bioactivity of a mastoparan peptide from wasp venom and of its analogues designed through targeted engineering. Int. J. Biol. Sci. 2018, 14, 599–607. [Google Scholar] [CrossRef]
- Wang, K.-R. , et al., Antitumor effects, cell selectivity and structure–activity relationship of a novel antimicrobial peptide polybia-MPI. Peptides, 2008. 29(6): p. 963-968.
- da Silva, A.M.B. , et al., Pro-necrotic Activity of Cationic Mastoparan Peptides in Human Glioblastoma Multiforme Cells Via Membranolytic Action. Molecular Neurobiology, 2018. 55(7): p. 5490-5504.
- Maltby, S.; Khazaie, K.; McNagny, K.M. Mast cells in tumor growth: Angiogenesis, tissue remodelling and immune-modulation. Biochim. et Biophys. Acta (BBA) - Rev. Cancer 2009, 1796, 19–26. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Nakata, K.; Oyama, K.; Higashijima, N.; Sagara, A.; Date, S.; Luo, H.; Hayashi, M.; Kubo, A.; Wu, C.; et al. Blockade of histamine receptor H1 augments immune checkpoint therapy by enhancing MHC-I expression in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2024, 43, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Speisky, D. , et al., Histamine H4 Receptor Expression in Triple-negative Breast Cancer: An Exploratory Study. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society, 2022. 70(4): p. 311-322.
- Akdis, C.A.; Blaser, K. Histamine in the immune regulation of allergic inflammation. J. Allergy Clin. Immunol. 2003, 112, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Nakamura, E.; Okabe, S. Histamine regulates growth of malignant melanoma implants via H2 receptors in mice. Inflammopharmacology 2005, 13, 281–289. [Google Scholar] [CrossRef]
- Martner, A.; Wiktorin, H.G.; Lenox, B.; Sander, F.E.; Aydin, E.; Aurelius, J.; Thorén, F.B.; Ståhlberg, A.; Hermodsson, S.; Hellstrand, K. Histamine Promotes the Development of Monocyte-Derived Dendritic Cells and Reduces Tumor Growth by Targeting the Myeloid NADPH Oxidase. J. Immunol. 2015, 194, 5014–5021. [Google Scholar] [CrossRef]
- Jensen, B.M.; Akin, C.; Gilfillan, A.M. Pharmacological targeting of the KIT growth factor receptor: a therapeutic consideration for mast cell disorders. Br. J. Pharmacol. 2008, 154, 1572–1582. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; Von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Hodi, F.S. , et al., Imatinib for melanomas harboring mutationally activated or amplified kit arising on mucosal, acral, and chronically sun-damaged skin. Journal of Clinical Oncology, 2013. 31(26): p. 3182-3190.
- Jachetti, E. , et al., Imatinib spares cKit-expressing prostate neuroendocrine tumors, whereas kills seminal vesicle epithelial-stromal tumors by targeting PDGFR-β. Molecular Cancer Therapeutics, 2017. 16(2): p. 365-375.
- Pittoni, P.; Tripodo, C.; Piconese, S.; Mauri, G.; Parenza, M.; Rigoni, A.; Sangaletti, S.; Colombo, M.P. Mast Cell Targeting Hampers Prostate Adenocarcinoma Development but Promotes the Occurrence of Highly Malignant Neuroendocrine Cancers. Cancer Res. 2011, 71, 5987–5997. [Google Scholar] [CrossRef]
- Samoszuk, M.; Corwin, M.A. Acceleration of tumor growth and peri-tumoral blood clotting by imatinib mesylate (Gleevec™). Int. J. Cancer 2003, 106, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Brock, E.C.; Leung, J.; Falahati, R.; Bochner, B.S.; Rasmussen, H.S.; Peterson, K.; Bebbington, C.; Tomasevic, N. Siglec-8 antibody reduces eosinophils and mast cells in a transgenic mouse model of eosinophilic gastroenteritis. J. Clin. Investig. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhao, Y.; Wang, X.; Chen, N.; Mao, F.; Teng, Y.; Wang, T.; Peng, L.; Zhang, J.; Cheng, P.; et al. Increased intratumoral mast cells foster immune suppression and gastric cancer progression through TNF-α-PD-L1 pathway. J. Immunother. Cancer 2019, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Tachi, M.; Ejima, Y.; Endo, Y.; Toyama, H.; Matsubara, M.; Saito, K.; Yamauchi, M.; Miura, C.; Kazama, I. Anti-Allergic Drugs Tranilast and Ketotifen Dose-Dependently Exert Mast Cell-Stabilizing Properties. Cell. Physiol. Biochem. 2016, 38, 15–27. [Google Scholar] [CrossRef]
- Kidd, M.; McKenzie, S.H.; Steven, I.; Cooper, C.; Lanz, R. Efficacy and safety of ketotifen eye drops in the treatment of seasonal allergic conjunctivitis. Br. J. Ophthalmol. 2003, 87, 1206–1211. [Google Scholar] [CrossRef]
- Ji, Y. , et al., Repurposing ketotifen as a therapeutic strategy for neuroendocrine prostate cancer by targeting the IL-6/STAT3 pathway. Cellular Oncology, 2023. 46(5): p. 1445-1456.
- Hemmati, A.; Nazari, Z.; Motlagh, M.; Goldasteh, S. THE ROLE OF SODIUM CROMOLYN IN TREATMENT OF PARAQUAT-INDUCED PULMONARY FIBROSIS IN RAT. Pharmacol. Res. 2002, 46, 229–234. [Google Scholar] [CrossRef]
- Minutello, K. and V. Gupta, Cromolyn Sodium. StatPearls, 2024.
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Plotkin, J.D.; Elias, M.G.; Fereydouni, M.; Daniels-Wells, T.R.; Dellinger, A.L.; Penichet, M.L.; Kepley, C.L. Human Mast Cells From Adipose Tissue Target and Induce Apoptosis of Breast Cancer Cells. Front. Immunol. 2019, 10, 138. [Google Scholar] [CrossRef]
- Drobits, B.; Holcmann, M.; Amberg, N.; Swiecki, M.; Grundtner, R.; Hammer, M.; Colonna, M.; Sibilia, M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Investig. 2012, 122, 575–585. [Google Scholar] [CrossRef]
- Oldford, S.A.; Haidl, I.D.; Howatt, M.A.; Leiva, C.A.; Johnston, B.; Marshall, J.S. A Critical Role for Mast Cells and Mast Cell-Derived IL-6 in TLR2-Mediated Inhibition of Tumor Growth. J. Immunol. 2010, 185, 7067–7076. [Google Scholar] [CrossRef]
- Dudeck, J.; Ghouse, S.M.; Lehmann, C.H.; Hoppe, A.; Schubert, N.; Nedospasov, S.A.; Dudziak, D.; Dudeck, A. Mast-Cell-Derived TNF Amplifies CD8+ Dendritic Cell Functionality and CD8+ T Cell Priming. Cell Rep. 2015, 13, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Weaver, B.; Choi, H.W.; Abraham, S.N.; Staats, H.F. Mast cell activators as novel immune regulators. Curr. Opin. Pharmacol. 2018, 41, 89–95. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, J.B.; Shelburne, C.P.; Hart, J.P.; Pizzo, S.V.; Goyal, R.; Brooking-Dixon, R.; Staats, H.F.; Abraham, S.N. Mast cell activators: a new class of highly effective vaccine adjuvants. Nat. Med. 2008, 14, 536–541. [Google Scholar] [CrossRef]
- Moulin, D.; Donzé, O.; Talabot-Ayer, D.; Mézin, F.; Palmer, G.; Gabay, C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007, 40, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Berselli, E.; Coccolini, C.; Tosi, G.; Gökçe, E.H.; Oliveira, M.B.P.P.; Fathi, F.; Krambeck, K.; Souto, E.B. Therapeutic Peptides and Proteins: Stabilization Challenges and Biomedical Applications by Means of Nanodelivery Systems. Int. J. Pept. Res. Ther. 2024, 30, 1–15. [Google Scholar] [CrossRef]
- Rische, C.H.; Thames, A.H.; Krier-Burris, R.A.; O’sUllivan, J.A.; Bochner, B.S.; Scott, E.A. Drug delivery targets and strategies to address mast cell diseases. Expert Opin. Drug Deliv. 2023, 20, 205–222. [Google Scholar] [CrossRef]
- Duan, S.; Arlian, B.M.; Nycholat, C.M.; Wei, Y.; Tateno, H.; A Smith, S.; Macauley, M.S.; Zhu, Z.; Bochner, B.S.; Paulson, J.C. Nanoparticles Displaying Allergen and Siglec-8 Ligands Suppress IgE-FcεRI–Mediated Anaphylaxis and Desensitize Mast Cells to Subsequent Antigen Challenge. J. Immunol. 2021, 206, 2290–2300. [Google Scholar] [CrossRef]
- Inoh, Y.; Tadokoro, S.; Tanabe, H.; Inoue, M.; Hirashima, N.; Nakanishi, M.; Furuno, T. Inhibitory effects of a cationic liposome on allergic reaction mediated by mast cell activation. 86, 1731. [Google Scholar] [CrossRef]
- Hendy, D.A.; Johnson-Weaver, B.T.; Batty, C.J.; Bachelder, E.M.; Abraham, S.N.; Staats, H.F.; Ainslie, K.M. Delivery of small molecule mast cell activators for West Nile Virus vaccination using acetalated dextran microparticles. Int. J. Pharm. 2023, 634, 122658–122658. [Google Scholar] [CrossRef]
- Ontiveros-Padilla, L.; Batty, C.J.; Hendy, D.A.; Pena, E.S.; Roque, J.A.; Stiepel, R.T.; Carlock, M.A.; Simpson, S.R.; Ross, T.M.; Abraham, S.N.; et al. Development of a broadly active influenza intranasal vaccine adjuvanted with self-assembled particles composed of mastoparan-7 and CpG. Front. Immunol. 2023, 14. [Google Scholar] [CrossRef]
- Kim, C.-E.; Lim, S.-K.; Kim, J.-S. In vivo antitumor effect of cromolyn in PEGylated liposomes for pancreatic cancer. J. Control. Release 2012, 157, 190–195. [Google Scholar] [CrossRef]
- Motawi, T.K.; El-Maraghy, S.A.; ElMeshad, A.N.; Nady, O.M.; Hammam, O.A. Cromolyn chitosan nanoparticles as a novel protective approach for colorectal cancer. Chem. Interactions 2017, 275, 1–12. [Google Scholar] [CrossRef]
- Motawi, T.K.; El-Maraghy, S.A.; Sabry, D.; Nady, O.M.; Senousy, M.A. Cromolyn chitosan nanoparticles reverse the DNA methylation of RASSF1A and p16 genes and mitigate DNMT1 and METTL3 expression in breast cancer cell line and tumor xenograft model in mice. Chem. Interactions 2022, 365, 110094. [Google Scholar] [CrossRef] [PubMed]
- Alhakamy, N.A.; Okbazghi, S.Z.; Alfaleh, M.A.; Abdulaal, W.H.; Bakhaidar, R.B.; Alselami, M.O.; AL Zahrani, M.; Alqarni, H.M.; Alghaith, A.F.; Alshehri, S.; et al. Wasp venom peptide improves the proapoptotic activity of alendronate sodium in A549 lung cancer cells. PLOS ONE 2022, 17, e0264093. [Google Scholar] [CrossRef]
- Zhao, H.; Bao, S.; Chen, S.; Yang, Q.; Lou, K.; Gai, Y.; Lin, J.; Liu, C.; Liu, H.; Zhang, C.; et al. Phytosomes Loaded with Mastoparan-M Represent a Novel Strategy for Breast Cancer Treatment. Int. J. Nanomed. 2025, ume 20, 109–124. [Google Scholar] [CrossRef]
- Li, G.; Lei, Q.; Wang, F.; Deng, D.; Wang, S.; Tian, L.; Shen, W.; Cheng, Y.; Liu, Z.; Wu, S. Fluorinated Polymer Mediated Transmucosal Peptide Delivery for Intravesical Instillation Therapy of Bladder Cancer. Small 2019, 15, e1900936. [Google Scholar] [CrossRef]
- Ren, C.; Li, Y.; Cong, Z.; Li, Z.; Xie, L.; Wu, S. Bioengineered bacterial outer membrane vesicles encapsulated Polybia–mastoparan I fusion peptide as a promising nanoplatform for bladder cancer immune-modulatory chemotherapy. Front. Immunol. 2023, 14, 1129771. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Chen, Y.; Yeh, C.-R.; Ding, J.; Li, L.; Chang, C.; Yeh, S. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 2015, 7, 7842–7855. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Guo, Q.; Liu, Z.; Liu, K.; He, J.; Li, R.; Sun, H.; Yao, W.; Wang, L. Facile preparation of nanomicelles using polymyxin E for enhanced antitumor effects. J. Biomater. Sci. Polym. Ed. 2021, 33, 329–341. [Google Scholar] [CrossRef]
- Zhao, H.; Bao, S.; Chen, S.; Yang, Q.; Lou, K.; Gai, Y.; Lin, J.; Liu, C.; Liu, H.; Zhang, C.; et al. Phytosomes Loaded with Mastoparan-M Represent a Novel Strategy for Breast Cancer Treatment. Int. J. Nanomed. 2025, ume 20, 109–124. [Google Scholar] [CrossRef]
- Lichterman, J.N.; Reddy, S.M. Mast Cells: A New Frontier for Cancer Immunotherapy. Cells 2021, 10, 1270. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLOS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Varied pro- and anti-tumor effects of mast cells within the tumor microenvironment (TME). The effector molecule is labeled in purple at the bottom of the wedge and the resultant immune effect in grey at the top. Chemokine ligand 5 (CCL5); colony-stimulating factor 2 (CSF2); C-X-C motif chemokine ligand 10 (CXCL10); extracellular matrix (ECM); tumor-associated macrophages (TAM); tumor necrosis factor (TNF); toll-like receptor (TLR).
Figure 1.
Varied pro- and anti-tumor effects of mast cells within the tumor microenvironment (TME). The effector molecule is labeled in purple at the bottom of the wedge and the resultant immune effect in grey at the top. Chemokine ligand 5 (CCL5); colony-stimulating factor 2 (CSF2); C-X-C motif chemokine ligand 10 (CXCL10); extracellular matrix (ECM); tumor-associated macrophages (TAM); tumor necrosis factor (TNF); toll-like receptor (TLR).
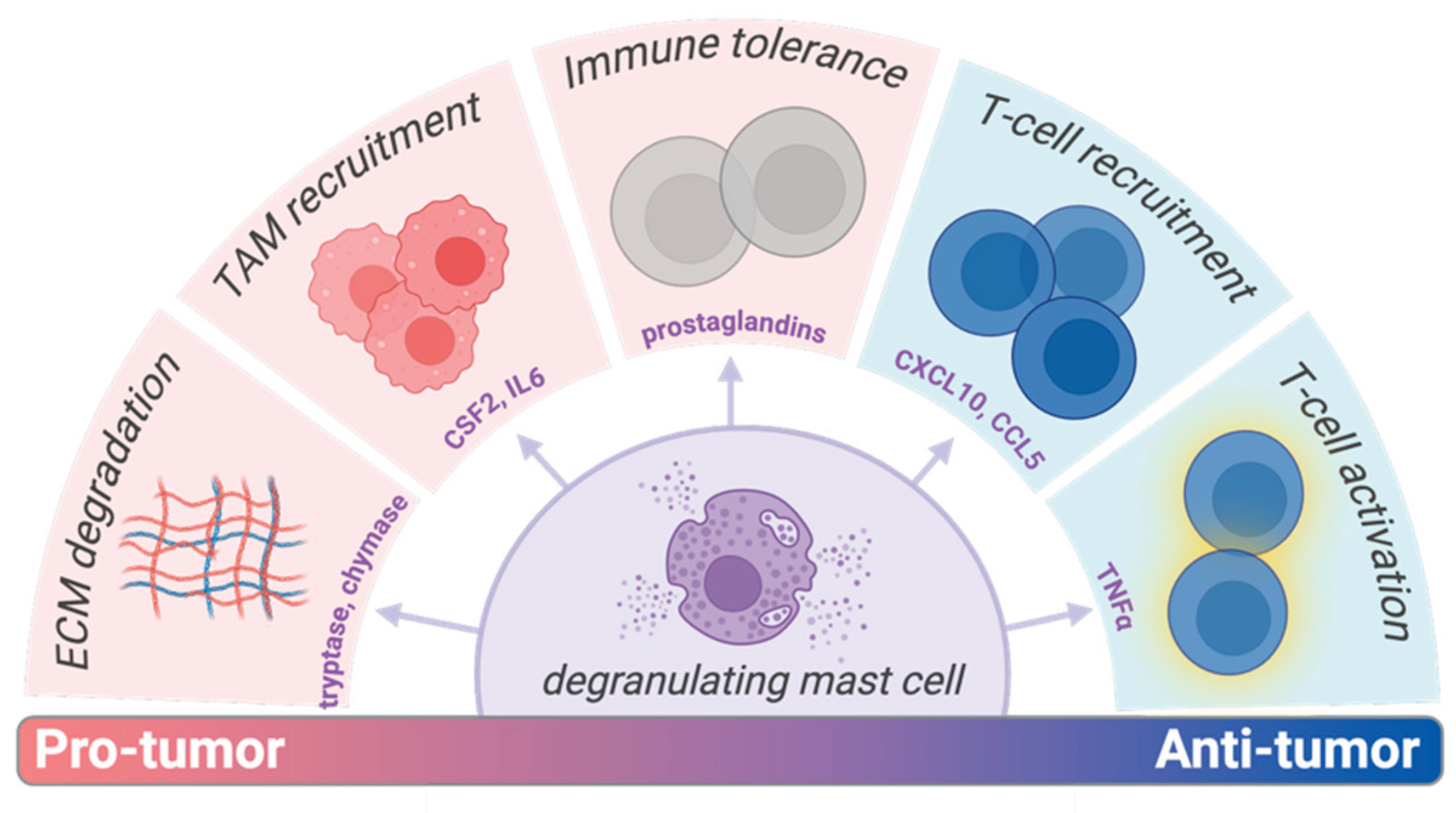
Figure 2.
Sequentially labeled mast cell activation events with their subsequent immune effects. Leukotriene C4 and B4 (LTC4, LTB4); monocyte chemoattractant protein-1 (MCP-1); mas-related G protein receptor-X2 (MRGPRX2); prostaglandin D2 (PGD2); tumor necrosis factor (TNF); toll-like receptor (TLR).
Figure 2.
Sequentially labeled mast cell activation events with their subsequent immune effects. Leukotriene C4 and B4 (LTC4, LTB4); monocyte chemoattractant protein-1 (MCP-1); mas-related G protein receptor-X2 (MRGPRX2); prostaglandin D2 (PGD2); tumor necrosis factor (TNF); toll-like receptor (TLR).
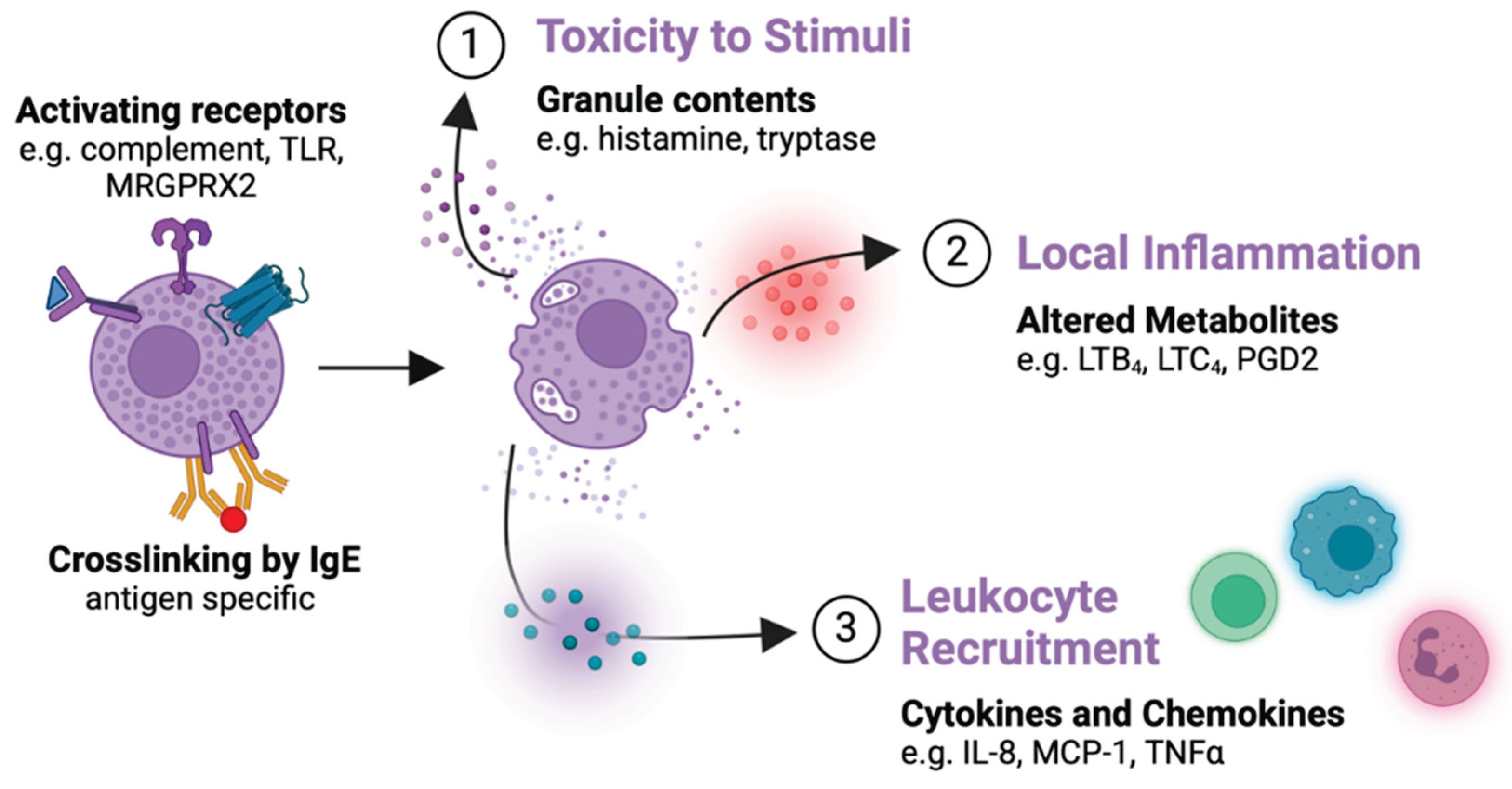
Figure 3.
Characterization and in vivo evaluation of cromolyn chitosan nanoparticles (CCSNP) by Motwai et. al. (A) The physicochemical characteristics of all formulations made with the selected optimized CCSNP “F6” highlighted in blue. (B) TEM images of spherical F6 particles. (C) Release kinetics of five of the ten formulations, F6-F10. (D) MC staining by toluidine blue of the colon of mice with chemically induced tumors including control, soluble cromolyn (without and with tumor) or CCSNP groups. Not determined (ND) [116].
Figure 3.
Characterization and in vivo evaluation of cromolyn chitosan nanoparticles (CCSNP) by Motwai et. al. (A) The physicochemical characteristics of all formulations made with the selected optimized CCSNP “F6” highlighted in blue. (B) TEM images of spherical F6 particles. (C) Release kinetics of five of the ten formulations, F6-F10. (D) MC staining by toluidine blue of the colon of mice with chemically induced tumors including control, soluble cromolyn (without and with tumor) or CCSNP groups. Not determined (ND) [116].
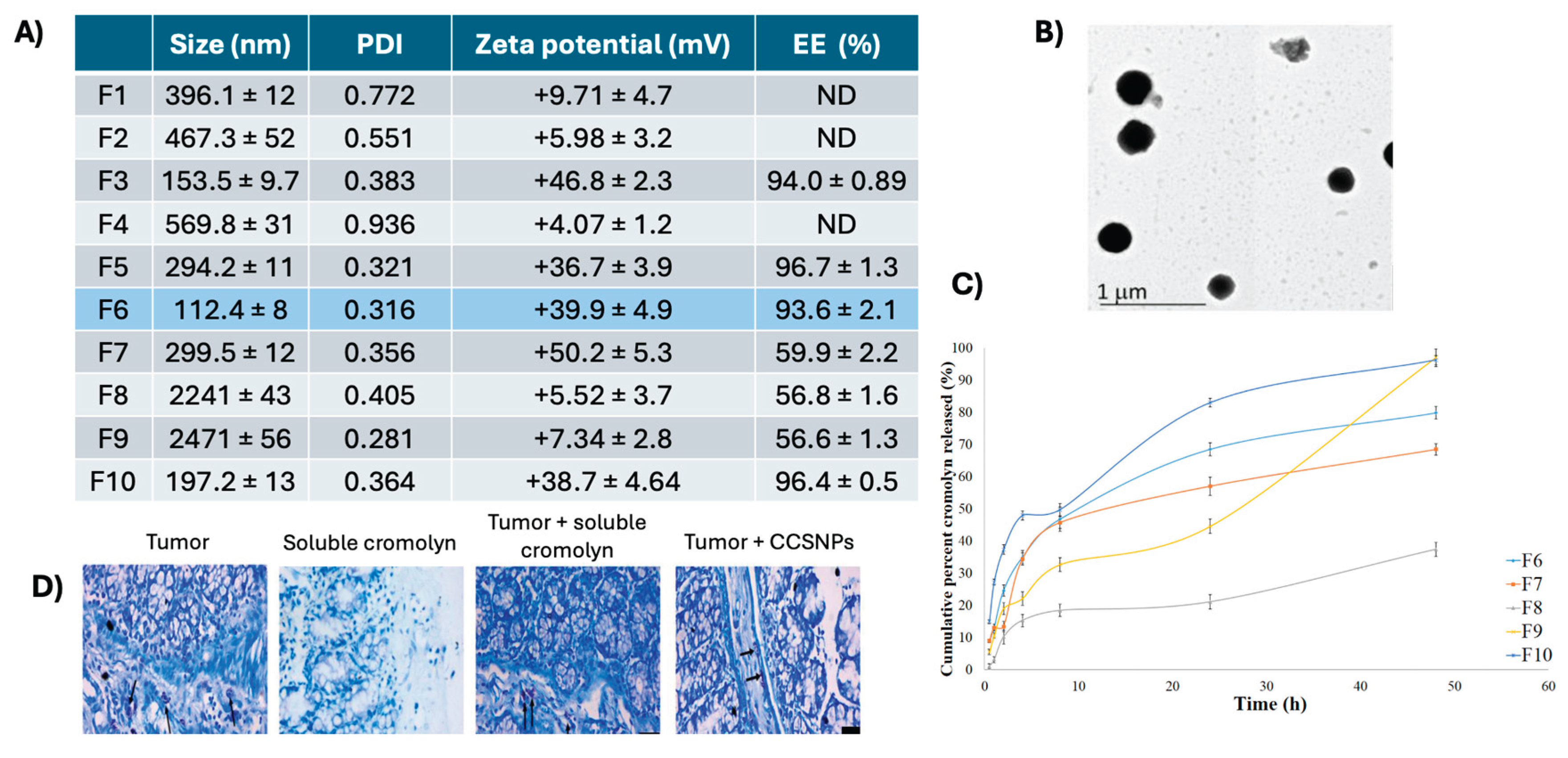
Figure 4.
Stability and efficacy studies of a cromolyn liposomal formulation. (A) Surface adsorbed protein levels after liposome incubation with serum at 30 minutes and 24 hours. Liposomes were either non-PEG (Non-PEG-lipo), PEGylated but blank (PEG-lipo-empty), or PEGylated with cromolyn (PEG-lipo-cro). (B) Cell growth inhibition of the optimized liposomal formulation against two pancreatic cancer lines, Panc-1 and BxPC-3. (C) Tumor growth curves, as measured by calipers, of treatments including PBS, soluble cromolyn, and liposomal formulations of cromolyn, gemcitabine, and a combination of both; ** p<0.01, with significance compared to controls where not clarified. [115].
Figure 4.
Stability and efficacy studies of a cromolyn liposomal formulation. (A) Surface adsorbed protein levels after liposome incubation with serum at 30 minutes and 24 hours. Liposomes were either non-PEG (Non-PEG-lipo), PEGylated but blank (PEG-lipo-empty), or PEGylated with cromolyn (PEG-lipo-cro). (B) Cell growth inhibition of the optimized liposomal formulation against two pancreatic cancer lines, Panc-1 and BxPC-3. (C) Tumor growth curves, as measured by calipers, of treatments including PBS, soluble cromolyn, and liposomal formulations of cromolyn, gemcitabine, and a combination of both; ** p<0.01, with significance compared to controls where not clarified. [115].
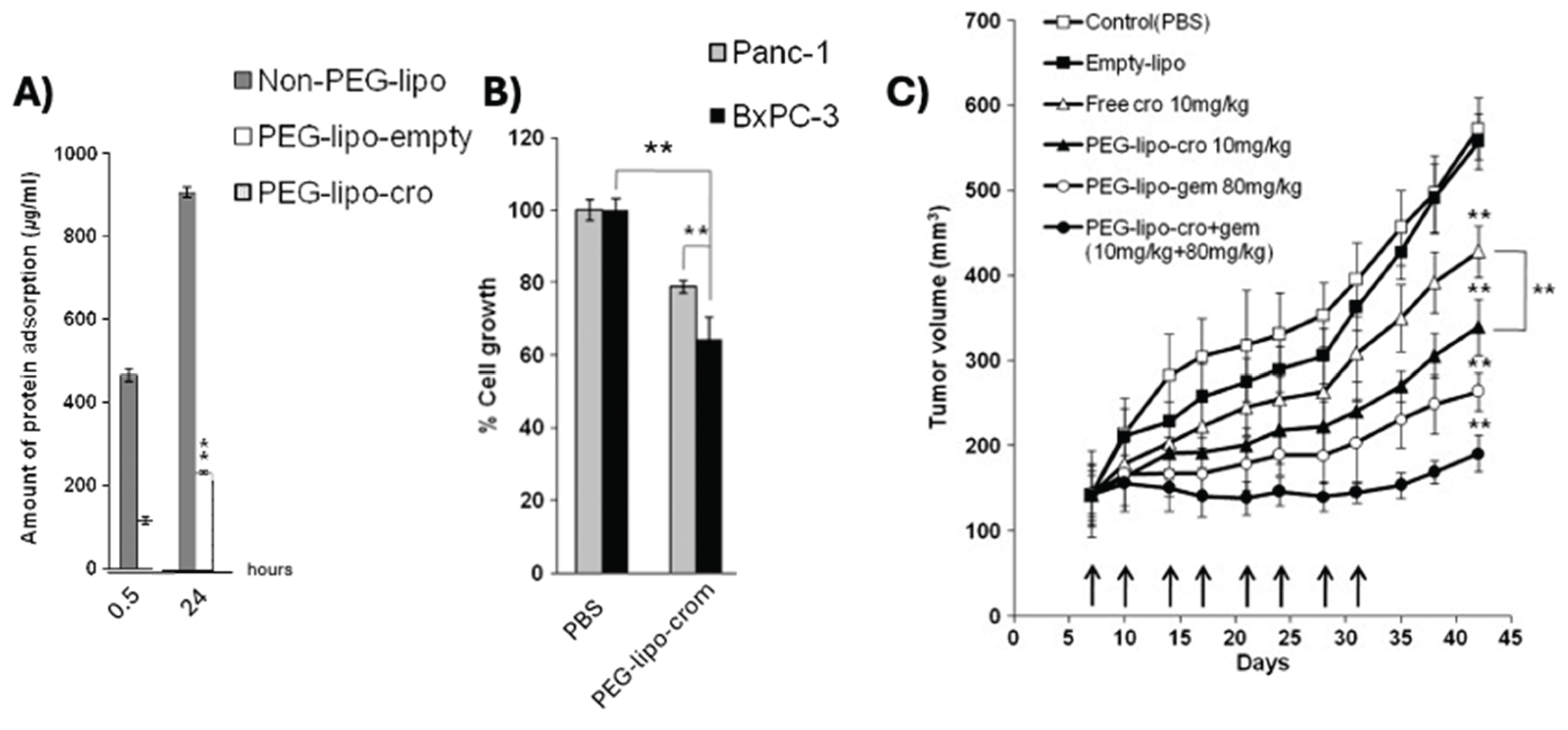
Figure 5.
Characterization of mastoparan-M phytosome formulation. (A) Self-complexation of mastoparan-M with soybean lecithin to create spherical phytosomes. (B) DLS and (C) zeta potential of the final formulation with results being 125 nm and +32 mV respectively. (D) Mouse weight and (E) tumor size shown after the first treatment. Quantitation of HE staining of (F) Ki67+, (G) Bcl-2, and (H) Tunel on excised tumors. [119].
Figure 5.
Characterization of mastoparan-M phytosome formulation. (A) Self-complexation of mastoparan-M with soybean lecithin to create spherical phytosomes. (B) DLS and (C) zeta potential of the final formulation with results being 125 nm and +32 mV respectively. (D) Mouse weight and (E) tumor size shown after the first treatment. Quantitation of HE staining of (F) Ki67+, (G) Bcl-2, and (H) Tunel on excised tumors. [119].
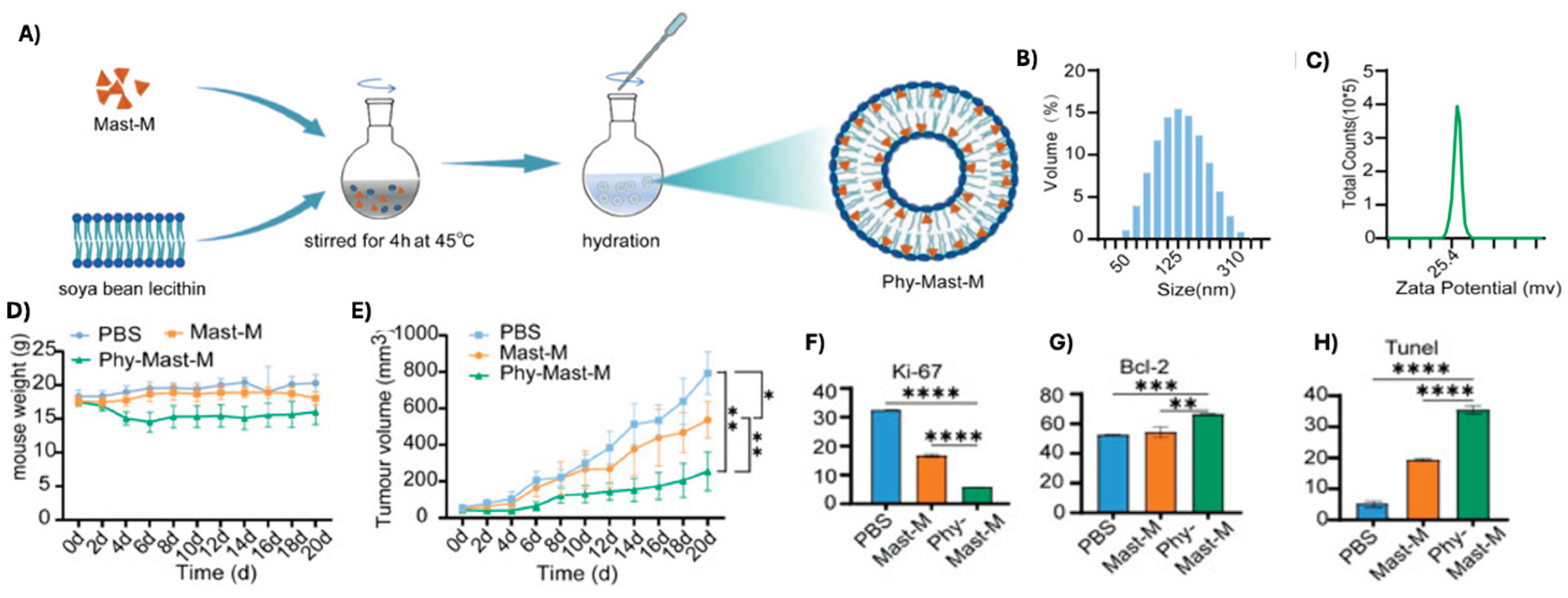
Figure 6.
Results showing successful TME immune modulation after treatment with OMV particles containing MP1. A) size and charge assessed by DLS for four OMV formulations. B) Tumor growth control by caliper measurements and C) apoptosis by immunofluorescent TUNEL assay. Changes in intratumoral immune populations showing increased D) CD80+ CD86+ dendritic cells, E) CD4+ T-cells, and F) CD8+ T-cells. Dendritic cells were defined as CD11c+. [121].
Figure 6.
Results showing successful TME immune modulation after treatment with OMV particles containing MP1. A) size and charge assessed by DLS for four OMV formulations. B) Tumor growth control by caliper measurements and C) apoptosis by immunofluorescent TUNEL assay. Changes in intratumoral immune populations showing increased D) CD80+ CD86+ dendritic cells, E) CD4+ T-cells, and F) CD8+ T-cells. Dendritic cells were defined as CD11c+. [121].
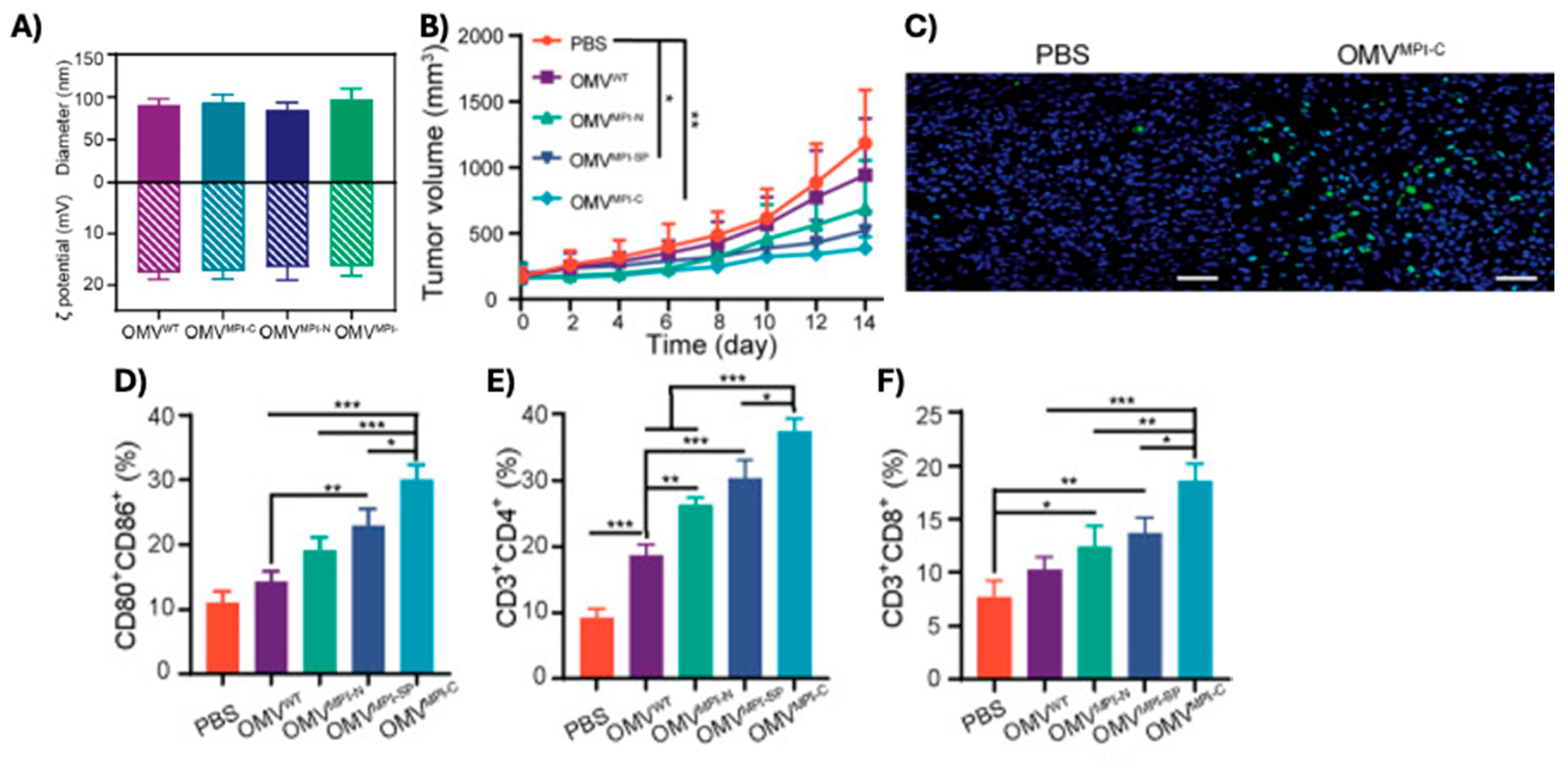
Figure 7.
Successful transmucosal delivery of MP1 for the treatment of bladder cancer. A) schematic of intravesicular therapy. B) Confocal images of resected bladders 60 minutes after treatment with soluble MP1, MP1 encapsulated in PEI NPs, and MP1 encapsulated in F-PEI NPs; MP1 has been fluorescently labeled with Cy5.5 for visualization. C) Tumor growth control in a subQ bladder cancer model and D) mouse weights during time of treatment. E) Survival of mice bearing orthotopic bladder tumors after treatment with MP1 in F-PEI NPs along with several controls. Fluorinated polyethylenimine (F-PEI), mitomycin-c (MMC); polybia-MP1 (MP1); polyethylenimine (PEI) [120].
Figure 7.
Successful transmucosal delivery of MP1 for the treatment of bladder cancer. A) schematic of intravesicular therapy. B) Confocal images of resected bladders 60 minutes after treatment with soluble MP1, MP1 encapsulated in PEI NPs, and MP1 encapsulated in F-PEI NPs; MP1 has been fluorescently labeled with Cy5.5 for visualization. C) Tumor growth control in a subQ bladder cancer model and D) mouse weights during time of treatment. E) Survival of mice bearing orthotopic bladder tumors after treatment with MP1 in F-PEI NPs along with several controls. Fluorinated polyethylenimine (F-PEI), mitomycin-c (MMC); polybia-MP1 (MP1); polyethylenimine (PEI) [120].
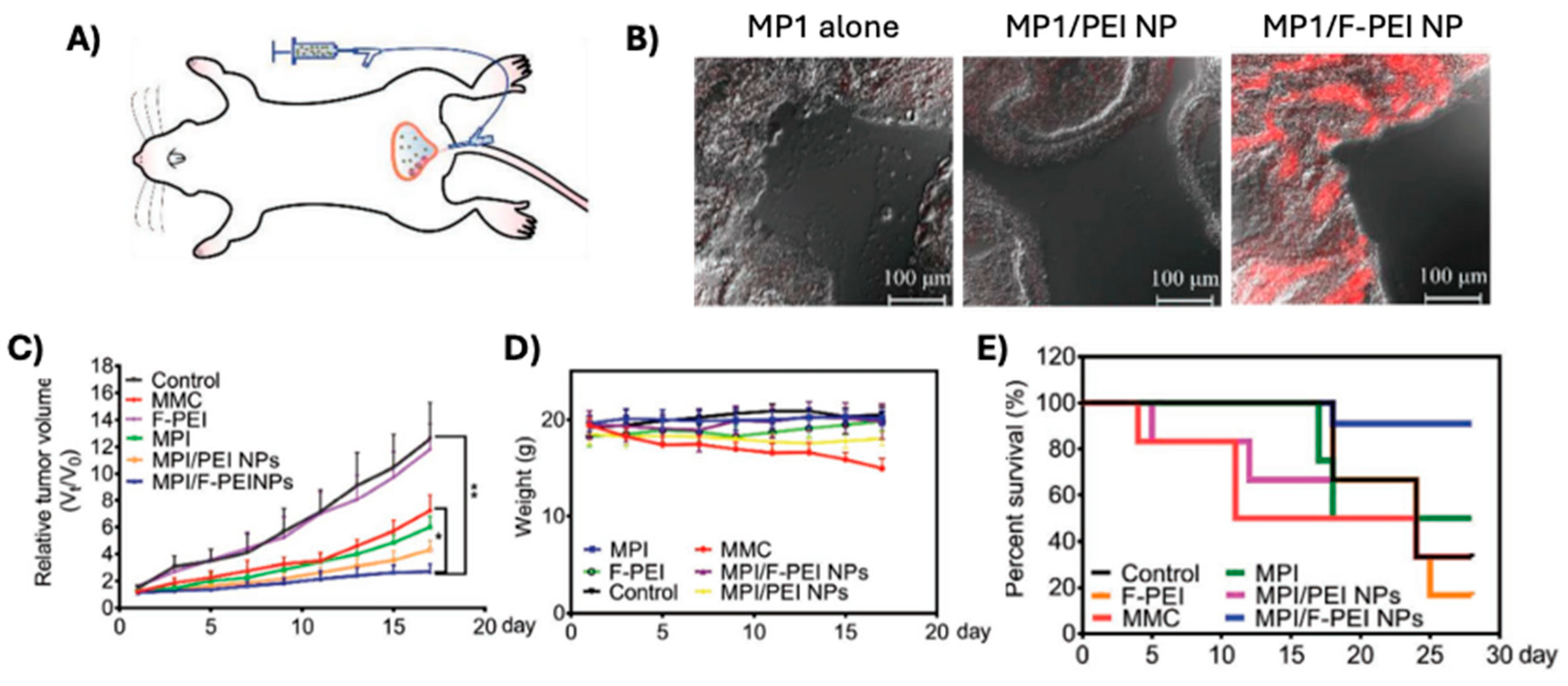
Table 1.
A series of polycationic peptides derived from, or derivatives of wasp venom. Mastoparan-L (M-L).
Table 1.
A series of polycationic peptides derived from, or derivatives of wasp venom. Mastoparan-L (M-L).
| PEPTIDE | SEQUENCE | ORIGIN | REF |
|---|---|---|---|
| Mastoparan-L | INLKALAALAKKIL-NH₂ | Vespa lewisii | [48] |
| Mastoparan-X | INWKGIAAMAKKLL- NH₂ | Vespa xanthoptera | [49] |
| Mastoparan-C | LNLKALLAVAKKIL-NH₂ | Vespa crabro | [50] |
| Polybia-MPI | IDWKKLLDAAKQIL-NH₂ | Polybia paulista | [51] |
| KM8 | KLLKKNLKALAALAKKIL-NH₂ | M-L analog | [52] |
| [I5, R8] Mastoparan | INLKILARLAKALL-NH₂ | M-L analog | [53] |
| Mastoparan-3 | NLKALAALAKKIL-NH₂ | M-L analog | [54] |
| Mastoparan-7 (M7) | INLKALAALAKALL- NH₂ | M-L analog | [55] |
| MP12W | INLKALAALAWALL-NH₂ | M7 analog | [56] |
Table 2.
Cytotoxicity of various peptides and molecules against a range of tumor and healthy cells. Values were identified from MTT assay after incubation for the designated time with respective compound. Glioblastoma (GBM). Time at which in vitro model was evaluated is given in hours. The 50% inhibitory concentration is listed as IC50 in micromolar. NA means not available.
Table 2.
Cytotoxicity of various peptides and molecules against a range of tumor and healthy cells. Values were identified from MTT assay after incubation for the designated time with respective compound. Glioblastoma (GBM). Time at which in vitro model was evaluated is given in hours. The 50% inhibitory concentration is listed as IC50 in micromolar. NA means not available.
| MC Modulator | Cancer Type | In vitro model |
Time (hrs) |
IC50 (μM) |
Ref |
| MC Inhibitor | |||||
| Cromolyn sodium | Colon | HT-29 | 72 | 2.33 | [72] |
| Healthy | MCF-10 | 7.33 | |||
| MC Agonist | |||||
| Mastoparan-L-COOH |
Lymphoma | Jurkat | 24 |
77.9 | [73] |
| GBM | U87 | 311.7 | |||
| Cervical | SiHA | 172.1 | |||
| Breast | MCF-7 | 432.5 | |||
| MDA-MB-231 | 251.25 | ||||
| SK-BR3 | 320.3 | ||||
| Melanoma | A2058 | 140 | |||
| B16F10-Nex2 | 165 | ||||
| Healthy | Melan-a | 411.5 | |||
| HACaT | 428 | ||||
| Mastoparan-L-NH2 |
Lung | A549 | ~14 | [74] | |
| 34.3 | [75] | ||||
| Colon | HCT-116 p53 double knockout | ~30-35 | [76] | ||
| HT-29 | ~40-45 | ||||
| Leukemia | Jurkat | 8-9.2 | [77] |
||
| THP-1 | |||||
| Myeloma | HOPC | 11 | |||
| Breast | MDA-MB-231 | 20–24 |
|||
| T47D | |||||
| MDA-MB-468 | |||||
| 4T1 | |||||
| SKBR3 | |||||
| MCF7 | |||||
| MCF7-TX400 | |||||
| Prostate | PC3 | <50 | |||
| Ovarian | SCOV3 | 25 | |||
| Cervical | HeLa | 10 | |||
| Healthy | PBMCs | 48 | |||
| Leukemia | HL60 | NA | 10 | [54] | |
| Breast | MCF-7 | 48 | 26.6 | [52] | |
| MCF-7/Dox | 27.6 | ||||
| Lung | A549 | 28.3 | |||
| NCI-446 | 28.4 | ||||
| Esophageal | Eca109 | 31.9 | |||
| Healthy | LO2 | 53.2 | |||
| HEK-293 | 51.5 | ||||
| KM8 | Breast | MCF-7 | 5.3 | ||
| MCF-7/Dox | 5.5 | ||||
| Lung | A549 | 6.2 | |||
| NCI-446 | 6.3 | ||||
| Esophageal | Eca109 | 7.4 | |||
| Healthy | LO2 | 45.9 | |||
| HEK-293 | 45.0 | ||||
| Mastoparan-C | Lung | H157 | 24 | 13.57 | [78] |
| Breast | MCF-7 | 25.27 | |||
| Prostate | PC-3 | 6.29 | |||
| GBM | U251-MG | 36.65 | |||
| Healthy | HMEC-1 | 57.15 | |||
| Polybia-MPI |
Prostate | PC-3 | 64.68 | [79] | |
| Bladder | Biu87 | 52.16 | |||
| EJ | 75.51 | ||||
| Healthy | HUVEC | 55.6 | |||
| GBM | T298G | 2 | 32.7 | [80] | |
| Mastoparan-X | 18.05 | ||||
| Leukemia | HL60 | NA | 10 | [54] | |
| Mastoparan-3 | 200 | ||||
| Mastoparan [I5, R8] |
Leukemia | THP-1 | 72 | 24.5 | [53] |
| Healthy | HEK-293 | >200 | |||
Table 4.
Relevant preclinical model details and outcomes of MC-modulating agents in drug delivery system. Tumor size was determined via a web-based data digitizer. Doxorubicin (DOX), dimethylhydrazine (DMH), intraperitoneal (IP), intravenous (IV), mitomycin C (MMC), not significant (n.s.), (fluorinated) polyethylenimine [(F-)PEI], polymyxin E (PE), subcutaneous (subQ), every two days (q2d), every 3 days (q3d), and every 4 days (q4d).
Table 4.
Relevant preclinical model details and outcomes of MC-modulating agents in drug delivery system. Tumor size was determined via a web-based data digitizer. Doxorubicin (DOX), dimethylhydrazine (DMH), intraperitoneal (IP), intravenous (IV), mitomycin C (MMC), not significant (n.s.), (fluorinated) polyethylenimine [(F-)PEI], polymyxin E (PE), subcutaneous (subQ), every two days (q2d), every 3 days (q3d), and every 4 days (q4d).
| MC AGONIST | CANCER | MODEL | VEHICLE | DOSE | SCHEDULE | CONCLUSIONS | REF |
| Cromolyn sodium | Pancreatic | Bx-PC-3 subQ in BALB/c mice | PEGylated liposomes | IV 10mg/kg | Twice weekly for 4 weeks | Encapsulated cromolyn outperformed soluble in tumor growth inhibition, more apoptosis 24 hr after treatment | [115] |
| Colorectal | DMH induced in Wister albino rats | Chitosan nanoparticles | IP 5mg/kg | Twice weekly for 16 weeks | Survival not assessed; maintenance of colon architecture, decreased MC frequency | [116] | |
| Mastoparan | Lung | In vitro |
Alendronate sodium nanoconjugate | A549 24 hr MTT | NA | IC50 = 1.3 μM; vehicle alone 37.6 μM, alone ~13 | [74] |
| Fluvastatin mastoparan nanoconjugate | A549 4hr MTT | More apoptotic proteins present in MAS-FLV-NC; IC50 = 18.6 μM; vehicle alone = 58.4 μM, mas = 34.3 μM | [75] | ||||
| Mastoparan-M | Breast | 4T1 subQ in BALB/c mice | Soya phosphatidylcholines phytosomes | IV 2.7mg/kg | q4d 4x | Encapsulated mastoparan outperformed soluble in tumor growth inhibition with more apoptosis (Bcl-2 and TUNEL) and decreased proliferation (Ki-67) | [124] |
| Polybia-mastoparan I (MPI) | Bladder | MB49 subQ in C57/BL6 mice | Outer membrane vesicles of gram-negative bacteria | IT 100 μg vesicle protein | q3d 4x | DC maturation, macrophage recruitment, CD4+ and CD8+ infiltration | [121] |
| T24 subQ in BALB/c nude mice | Fluorinated PEI nanoparticles | 0.5 mg/kg intravesical | 60min long treatment 1x | NPs resulted in superior tumor control a SOC chemo (MMC), no weight loss | [120] | ||
| Polymyxin E | Cervical | U14 subQ in Kunming mice | PE-Doxorubicin loaded micelles | 2.5mg/kg DOX I.V. | q2d 7x | PE not efficacious alone, but when combined with DOX, better than either alone | [123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



