Submitted:
11 July 2025
Posted:
15 July 2025
You are already at the latest version
Abstract
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a debilitating, multi-system disease characterized by profound fatigue, post-exertional malaise (PEM), and a constellation of immune, neurological, and autonomic symptoms. Despite its global prevalence, the pathophysiology of ME/CFS remains elusive, and there are no FDA-approved treatments targeting the underlying mechanisms. Symptom-based pharmacologic management is often complicated by hypersensitivity reactions and mitochondrial toxicity. Non-pharmacologic interventions, such as energy conservation, autonomic regulation, and nutritional strategies, are frequently employed to mitigate symptom burden. Emerging research points to mitochondrial dysfunction as a core contributor to ME/CFS pathology, marked by impaired ATP production, oxidative stress, and bioenergetic failure. Mitochondrial-derived peptides (MDPs), particularly MOTS-c, offer a novel therapeutic avenue by enhancing mitochondrial biogenesis, reducing oxidative damage, and modulating inflammatory responses through AMPK and NRF2 activation. Preclinical evidence suggests that MOTS-c improves glucose metabolism, increases mitochondrial density, and enhances fatigue resistance. However, safety and efficacy data in humans are lacking. Future investigations are needed to evaluate MOTS-c's potential as a disease-modifying therapy in ME/CFS.
Keywords:
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
; Mitochondrial-derived peptides (MDPs)
; MOTS-c
; AMP-activated protein kinase (AMPK)
1. Introduction
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a complex, multi-system disease that severely disrupts the lives of those affected. It's characterized by a range of persistent symptoms, including extreme fatigue, muscle and joint pain, immune system dysfunction, mitochondrial issues, cognitive impairments (also known as "brain fog”), poor sleep quality, autonomic nervous system disturbances, and a key feature called post-exertional malaise (PEM)—a worsening of symptoms after minimal physical or mental activity that can last days or even weeks. To be diagnosed, symptoms must last for more than six months. In many cases, fatigue is so severe that individuals become housebound or bedridden for extended periods. It's estimated that 17 to 24 million people worldwide live with ME/CFS [1]. Despite its widespread impact, the underlying pathogenesis of ME/CFS remains poorly understood, and there are currently no U.S. Food and Drug Administration (FDA)-approved pharmaceutical treatments specifically for the condition. Clinical management is therefore aimed at symptom relief, often requiring a highly individualized and cautious approach.
1.1. Pharmacologic Management
Pharmacologic treatments in ME/CFS are symptom-targeted and must be used with exceptional care. The U.S. ME/CFS Clinician Coalition advises clinicians to proceed with caution, noting that patients often experience heightened drug sensitivities and paradoxical reactions that can worsen symptoms [2]. Common pharmacologic strategies include:
- Antidepressants such as tricyclic antidepressants (TCAs) or serotonin-norepinephrine reuptake inhibitors (SNRIs) for sleep disturbances, chronic pain, and mood stabilization.
- Sleep aids, including alpha-blockers or sedative-hypnotics, to manage persistent insomnia.
- Neuropathic pain medications, like low-dose anticonvulsants or selective serotonin reuptake inhibitors (SSRIs), manage nerve-related pain.
- Stimulants, such as modafinil, are used with caution to alleviate severe fatigue.
- Immunomodulators, including intravenous immunoglobulin (IVIG), in cases with significant immune dysfunction.
- Gastrointestinal antibiotics or antimicrobials to treat gut infections or dysbiosis.
However, the frequent need for multiple medications (polypharmacy) increases the risk of adverse effects. Many ME/CFS patients report drug hypersensitivity, worsening existing symptoms, or even the development of new complications such as gastrointestinal upset, heightened inflammation, or worsened autonomic dysfunction. Importantly, many medications have been shown to negatively impact mitochondrial function in ME/CFS by impairing respiration, depleting ATP production, and increasing oxidative stress [5,6,7,8]. Additionally, long-term use of certain drugs, especially benzodiazepines or opioids, may lead to tolerance or dependency [9].
1.2. Non-Pharmacologic Management
The U.S. ME/CFS Clinician Coalition and numerous clinical experts advocate for a cautious, low-dose approach to pharmacologic treatment, given the high incidence of drug hypersensitivity and paradoxical reactions in ME/CFS patients [2,10,11]. These sensitivities are thought to stem from underlying mitochondrial dysfunction, altered detoxification pathways, and potential polymorphisms in cytochrome P450 enzymes, which may contribute to abnormal pharmacokinetics and heightened adverse drug reactions [9,12,13,14]. As such, pharmacologic interventions are best complemented by comprehensive, patient-centered lifestyle strategies aimed at symptom mitigation and quality-of-life improvement.
Key non-pharmacologic interventions include activity pacing and energy conservation, which are vital in managing post-exertional malaise (PEM), a hallmark feature of ME/CFS characterized by delayed symptom exacerbation following minimal exertion. Pacing strategies, such as the “energy envelope” theory, encourage patients to remain within their energy limits to prevent crashes and allow for more predictable functioning [14,15]. Evidence suggests that PEM may be driven by redox imbalance, impaired ATP synthesis, and mitochondrial energy deficits, which may be worsened by physical or cognitive overexertion [14,16]. Sleep hygiene techniques, including minimizing blue light exposure in the evening, using earplugs or white noise, and maintaining consistent bedtime routines, are commonly recommended to address non-restorative sleep [17]. These approaches may help regulate circadian rhythms and attenuate autonomic arousal, which are often disrupted in ME/CFS due to HPA-axis dysfunction and abnormal melatonin secretion [17,18]. Stress management and autonomic regulation through mindfulness, meditation, breathing exercises, and biofeedback may offer benefits for managing neuroendocrine symptoms and mental fatigue. Techniques such as heart rate variability (HRV) training and vagal stimulation aim to rebalance autonomic tone by reducing sympathetic overactivity and enhancing parasympathetic regulation [19]. However, psychological interventions should be applied with care to avoid the misattribution of ME/CFS as psychosomatic [20]. For orthostatic intolerance (OI), often presenting as neurally mediated hypotension (NMH) or postural orthostatic tachycardia syndrome (POTS), a range of non-pharmacologic strategies have demonstrated benefit. These include increased fluid intake (2–3 liters/day), sodium supplementation (up to 8–10 grams/day), use of compression garments, IV saline or electrolyte infusions, magnesium supplementation, and regular posture changes [21,22,23]. Importantly, increasing sodium intake necessitates careful monitoring of potassium status, as excessive sodium may lead to hypokalemia and exacerbate fatigue, muscle weakness, or cardiac arrhythmias [21,22,23]. Potassium-rich foods such as bananas, avocados, and coconut water are recommended to help maintain electrolyte balance. Low-intensity, recumbent physical therapy programs, including supine stretching, resistance bands, or aquatic therapy, may improve vascular tone and blood volume regulation without triggering PEM [24]. These programs must be carefully individualized, as even modest overexertion can lead to significant deterioration in some patients.
In addition, nutritional strategies targeting metabolic and inflammatory pathways are increasingly utilized. These include:
- Low-oxalate diets, useful for individuals with oxalate sensitivity or nephrolithiasis, reduce intake of foods such as spinach, beets, and almonds to prevent calcium oxalate accumulation and associated inflammation [27].
- Low-glutamate diets, which avoid foods high in free glutamate (e.g., MSG, soy sauce, aged cheeses), may help reduce excitotoxicity and neuroinflammation mediated by NMDA receptor overactivation [28].
Additionally, some patients utilize targeted dietary supplements in efforts to improve mitochondrial function, enhance energy metabolism, and reduce systemic inflammation. Among these, coenzyme Q10 (CoQ10) plays a critical role in the electron transport chain and cellular ATP production, with small clinical studies reporting improvements in fatigue symptoms in ME/CFS [29]. Acetyl-L-carnitine facilitates the transport of long-chain fatty acids into mitochondria for β-oxidation and has been associated with benefits in mental clarity and mood regulation [30]. D-ribose, a naturally occurring sugar and essential component of ATP, may support energy generation in metabolically stressed tissues and has shown promise in improving energy and well-being in preliminary studies [33]. NAD⁺ precursors, including nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), are increasingly used to support redox balance, sirtuin activation, and mitochondrial biogenesis, with emerging evidence suggesting potential therapeutic value in disorders of energy metabolism [34]. Omega-3 fatty acids, particularly EPA and DHA, exert anti-inflammatory effects via modulation of cytokine production and resolution pathways, offering support for neuroimmune regulation [35]. Finally, plant-derived polyphenols such as curcumin and resveratrol may help mitigate oxidative stress and neuroinflammation through inhibition of NF-κB and activation of Nrf2 signaling cascades, key regulators of the cellular antioxidant response [36,37].
While many of these supplements demonstrate mechanistic plausibility and favorable safety profiles, the overall evidence remains preliminary. Larger, well-controlled clinical trials are needed to establish their efficacy and optimal use in ME/CFS populations. Taken together, these non-pharmacologic strategies form a crucial pillar of ME/CFS care. They target key physiological disruptions, including mitochondrial dysfunction, neuroinflammation, and autonomic imbalance, while minimizing the risks associated with pharmacologic therapy. A flexible, multidisciplinary approach, emphasizing personalized pacing, dietary optimization, electrolyte monitoring, and mitochondrial support, remains essential to meet the complex and evolving needs of this patient population.
2. Mitochondrial Function in Health and Its Role in ME/CFS Pathophysiology
2.1. Role of Mitochondria in Normal Biochemical Functioning
Mitochondria are essential organelles that play a pivotal role in maintaining cellular homeostasis and energy metabolism. Their primary function is the production of adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS) within the electron transport chain (ETC), which is critical for cellular energy demands, including muscle contraction, neurotransmission, and the biosynthesis of proteins, nucleotides, and lipids [38]. Beyond ATP generation, mitochondria regulate intracellular calcium homeostasis, which is vital for signal transduction, muscle function, and synaptic activity [39]. Mitochondria are also central to the regulation of oxidative stress. They produce reactive oxygen species (ROS) as natural byproducts of respiration; while low levels of ROS serve signaling functions, excessive ROS leads to oxidative damage and contributes to cellular aging and disease progression [39,40]. Furthermore, mitochondria play a key role in apoptosis through cytochrome c release, triggering caspase activation and the removal of damaged or dysfunctional cells [41]. Mitochondria are also responsible for the biosynthesis of critical molecules such as heme, steroid hormones, and certain amino acids [42,43,44]. Given these multifaceted roles, mitochondrial dysfunction can precipitate widespread metabolic and immune disturbances—a feature increasingly recognized in ME/CFS.
2.2. Mitochondrial Dysfunction in ME/CFS
Although research on glycolytic flux and respiratory chain function in ME/CFS remains inconsistent, there is a growing consensus that mitochondrial dysfunction lies at the heart of the disease's pathophysiology. This dysfunction manifests as impaired ATP production, chronic oxidative stress, neuroinflammation, and broader metabolic imbalance [8,40]. Patients with ME/CFS frequently exhibit clinical features consistent with mitochondrial bioenergetic failure—such as profound fatigue, post-exertional malaise (PEM), and orthostatic intolerance—which are suggestive of poor energy recovery and cellular stress under low-grade exertion. Mitochondrial ultrastructural abnormalities have been reported in ME/CFS patients, including cristae branching, mitochondrial swelling, vacuolation, and the appearance of myelin figures, all of which indicate degenerative changes [45,46,47]. However, these findings remain inconsistent and have not been uniformly verified across studies. Despite this, functional studies—many of which assess metabolic responses to stressors or post-exertion—reveal patterns of impaired oxidative phosphorylation, altered redox status, and diminished energy generation [45,46,47].
A systematic review by Holden et al. examined studies comparing mitochondrial profiles of ME/CFS patients to healthy controls, using various case definitions (Fukuda, Canadian Consensus, International Consensus, and IOM)[45]. The review highlighted consistent findings of reduced mitochondrial enzyme activity, impaired ETC performance, diminished ATP synthesis, and altered mitochondrial density and morphology. Genetic contributions to these dysfunctions are also proposed, suggesting a possible heritable mitochondrial vulnerability in some patients [50]. Moreover, mitochondrial dysfunction is likely interlinked with neuroinflammation, which has been visualized via PET imaging in patients with ME/CFS and is believed to contribute to the fatigue, cognitive dysfunction, and sensory hypersensitivity reported in both ME/CFS and Long COVID [51,52].
3. Mitochondrial-Derived Peptides and the Therapeutic Potential of MOTS-c in ME/CFS
Mitochondrial dysfunction plays a central role in the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS), contributing to impaired ATP production, oxidative stress, redox imbalance, and immune dysregulation [45,46,47]. These molecular deficits underline many of the clinical features of ME/CFS, including post-exertional malaise (PEM), fatigue, cognitive dysfunction, and orthostatic intolerance [14,45,46,47]. Given the limitations of traditional pharmacologic treatments, mitochondria-targeted therapeutics have gained interest as a potential disease-modifying strategy. One promising candidate in this domain is MOTS-c, a 16-amino acid mitochondrial-derived peptide (MDP) encoded within the 12S rRNA gene of the mitochondrial genome [53].
3.1. Biochemical Mechanisms of MOTS-c
MOTS-c is unique in that it is mitochondrial-encoded but functions as a cytoplasmic and nuclear signaling peptide. In response to metabolic stress, it translocates to the nucleus and regulates transcription of genes involved in energy metabolism and antioxidant defense. One of its primary actions is activation of the AMP-activated protein kinase (AMPK) pathway, a key regulator of cellular energy balance. MOTS-c accomplishes this by inhibiting the methionine–folate cycle, reducing purine synthesis, and promoting the accumulation of AICAR, a known AMPK activator [53]. Upon AMPK activation, the downstream phosphorylation of PGC-1α enhances mitochondrial biogenesis, fatty acid oxidation, and oxidative phosphorylation [38]. MOTS-c also activates NRF2 (nuclear factor erythroid 2–2-related factor 2), a master transcription factor that upregulates antioxidant enzymes such as glutathione S-transferase and NAD(P)H quinone dehydrogenase. This helps reduce mitochondrial ROS accumulation and protects mitochondrial DNA and membranes from oxidative damage [53,54]. In muscle and adipose tissue, MOTS-c improves glucose metabolism by promoting GLUT4 translocation and enhancing insulin sensitivity, even under conditions of insulin resistance [54,55,56]. These effects occur without inducing hypoglycemia and mimic many of the metabolic benefits of exercise, an especially valuable trait for ME/CFS patients who experience worsening symptoms following exertion.
3.2. Preclinical and Clinical Relevance to ME/CFS
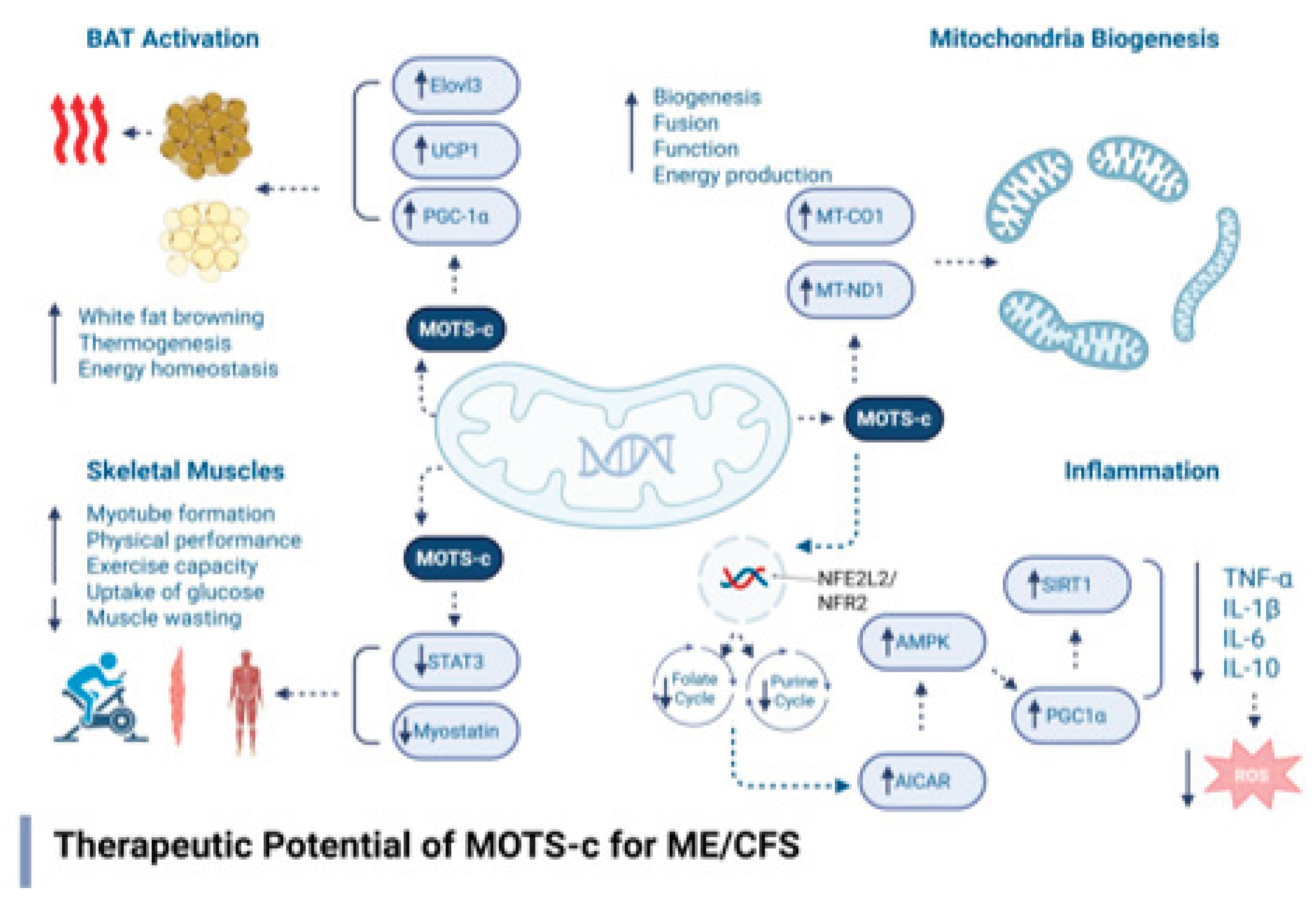
Preclinical research has identified several key mechanisms through which MOTS-c may offer therapeutic benefit in the context of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). One prominent mechanism is its ability to enhance insulin sensitivity and promote glucose uptake in skeletal muscle. These actions help restore metabolic flexibility and counteract the hypometabolic state observed in many ME/CFS patients, where cells exhibit impaired glucose utilization and energy generation [57,58]. In addition to its metabolic effects, MOTS-c has demonstrated anti-inflammatory properties, notably reducing levels of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) in high-fat diet-induced obesity models [59,60]. These cytokines have been implicated in the neuroinflammatory processes observed in ME/CFS, including in neuroimaging studies that reveal microglial activation in affected brain regions [51]. MOTS-c also promotes increased energy expenditure and mitochondrial density through its activation of brown adipose tissue (BAT) and the browning of white adipose tissue, which enhances thermogenesis and lipid oxidation [56]. These effects are particularly relevant to ME/CFS, as impaired mitochondrial function and inefficient lipid metabolism are frequently reported in patients [14,48]. Furthermore, in aging models, MOTS-c administration led to improvements in physical performance, muscle strength, and endurance, outcomes that closely parallel the functional impairments experienced by individuals with ME/CFS [55]. These findings suggest that MOTS-c may also alleviate fatigue and exercise intolerance, two of the most debilitating symptoms in this population. Figure 1 describes the core biochemical and therapeutic mechanisms of MOTS-c.
3.3. Pharmacological Profile and Delivery Considerations
MOTS-c exhibits several pharmacological characteristics that make it a compelling candidate for therapeutic development, particularly in conditions like ME/CFS that involve widespread metabolic and mitochondrial dysregulation. In terms of pharmacodynamics, MOTS-c has been shown to have a relatively short plasma half-life of approximately 30 minutes. However, despite this rapid clearance, it exerts long-lasting biological effects by activating key transcriptional pathways, most notably the AMP-activated protein kinase (AMPK) and nuclear factor erythroid 2–2-related factor 2 (NRF2) pathways. These pathways drive sustained improvements in mitochondrial biogenesis, antioxidant defense, and cellular energy homeostasis [53,54,55]. Regarding tissue distribution, preclinical studies indicate that MOTS-c preferentially accumulates in skeletal muscle, liver, and adipose tissue—all critical sites of metabolic regulation and notably affected in ME/CFS. These tissues are involved in glucose uptake, lipid metabolism, and systemic energy production, functions that are often impaired in patients with ME/CFS [14,48]. This targeted distribution profile supports the relevance of MOTS-c as a potential treatment for correcting the bioenergetic and metabolic disturbances characteristic of the illness.
3.4. Safety Considerations of MOTS-c Therapy
Although MOTS-c shows strong therapeutic promise in preclinical studies, especially for conditions involving mitochondrial dysfunction such as ME/CFS, its safety in humans remains to be fully established. While it is an endogenously encoded mitochondrial-derived peptide, exogenous administration, particularly at pharmacologic doses, introduces a range of potential safety concerns that warrant careful consideration in translational research and early-phase clinical trials.
1. Immunogenicity: Despite being a naturally occurring peptide, synthetic or recombinant MOTS-c may be perceived as foreign by the immune system, particularly with chronic or high-dose administration. There is a potential for the development of neutralizing antibodies, allergic reactions, or even autoimmune phenomena, as has been observed with other peptide therapeutics [61,62]. Strategies such as PEGylation or encapsulation in biocompatible delivery vehicles may reduce immunogenicity but would require additional safety testing.
2. Off-Target Effects and Cellular Signaling: The complete spectrum of MOTS-c’s receptor interactions and tissue-specific signaling remains incompletely mapped. Its principal mechanism—activation of AMP-activated protein kinase (AMPK)can affect a wide array of downstream targets, including those regulating lipid metabolism, muscle protein synthesis, and autophagy [53,54,55,56,57,58,59,60]. Chronic or excessive AMPK activation in tissues without energy deficits could potentially disrupt normal anabolic processes, impair growth, or lead to undesired catabolism.
3. Metabolic Dysregulation: MOTS-c has demonstrated beneficial effects on glucose metabolism and insulin sensitivity in skeletal muscle and adipose tissue, making it attractive for treating metabolic disorders [54,60]. However, in individuals with normal glucose metabolism or in combination with other glucose-lowering agents, there is a theoretical risk of hypoglycemia. Similarly, its impact on lipid mobilization and thermogenesis may contribute to unintended weight loss, especially in patients with low BMI or energy intake.
4. Mitochondrial Overactivation and Oxidative Stress: By stimulating mitochondrial biogenesis through the AMPK–PGC-1α pathway, MOTS-c may increase mitochondrial workload and oxygen consumption [53]. While this is typically beneficial, in the context of preexisting redox imbalance—as often seen in ME/CFS—it could exacerbate reactive oxygen species (ROS) production if not coupled with adequate antioxidant responses. Although MOTS-c activates the NRF2 antioxidant pathway, the capacity of this compensatory mechanism may vary between individuals.
5. Cardiovascular and Autonomic Effects: AMPK activation and enhanced mitochondrial activity influence vascular tone, heart rate, and thermoregulation. In ME/CFS patients, many of whom experience orthostatic intolerance (OI) and autonomic dysfunction, careful monitoring is needed to assess whether MOTS-c impacts blood pressure regulation or exacerbates dysautonomia. Its effects on brown adipose tissue (BAT) activation and body temperature regulation [56] also necessitate caution in individuals with abnormal thermoregulatory responses.
6. Long-Term and Proliferative Risks: While AMPK generally exerts tumor-suppressive effects by inhibiting cell growth under low-energy conditions, prolonged metabolic stimulation through pathways such as PGC-1α or mitochondrial biogenesis could, in theory, influence proliferation in pre-malignant cells or interfere with normal apoptosis pathways [63]. No evidence to date links MOTS-c to carcinogenesis, but long-term safety data in humans are lacking.
7. Pharmacokinetics and Administration Route: In animal studies, MOTS-c is commonly delivered via subcutaneous or intraperitoneal injection, with a short plasma half-life (~30 minutes) but sustained biological effects due to transcriptional activation [65]. In human application, degradation by serum peptidases and variable bioavailability present challenges. Optimizing delivery methods—such as sustained-release formulations or alternative routes (e.g., intranasal or transdermal)—will be critical to achieving safe and consistent therapeutic exposure.
8. Patient-Specific Variables and Comorbidities: Safety profiles may vary significantly based on the metabolic, immune, and autonomic profiles of individuals. For instance, ME/CFS patients with mitochondrial disease, mast cell activation syndrome (MCAS), or postural orthostatic tachycardia syndrome (POTS) may exhibit heightened sensitivity to metabolic modulators. Additionally, interactions with supplements commonly used in ME/CFS (e.g., CoQ10, NAD⁺ precursors) or pharmacologic agents (e.g., beta blockers, antidepressants) must be considered to avoid additive or antagonistic effects. Additionally, MOTS-c may synergize with existing mitochondrial therapies such as NAD⁺ precursors (e.g., nicotinamide riboside), CoQ10, acetyl-L-carnitine, and omega-3 fatty acids. These combinations could amplify mitochondrial biogenesis, improve ATP synthesis, and reduce oxidative stress more effectively than monotherapy.
3.5. Regulatory and FDA Guidance for Peptide Therapies
MOTS-c is currently classified as an investigational peptide and is not approved by the U.S. Food and Drug Administration (FDA) for therapeutic use. As a biologic agent, it is regulated under the FDA’s Investigational New Drug (IND) framework. This regulatory pathway mandates comprehensive preclinical evaluation—including studies on pharmacokinetics, toxicology, and immunogenicity—prior to progressing through phased clinical trials designed to establish safety and efficacy in humans [65]. Special considerations for peptide therapies include formulation stability (e.g., PEGylation, encapsulation), manufacturing consistency, and methods for enhancing bioavailability. Given its endogenous origin and physiological activity, MOTS-c is well-positioned for translation pending successful completion of early-phase trials.
4. Conclusions
ME/CFS presents a major clinical challenge due to its complex symptomatology, lack of targeted therapies, and high patient heterogeneity. While current treatment focuses on symptom relief, an increasing body of research implicates mitochondrial dysfunction as a central driver of ME/CFS pathology. This has spurred interest in mitochondria-targeted therapies, particularly mitochondrial-derived peptides like MOTS-c. With its ability to modulate energy metabolism, antioxidant defenses, and immune responses, MOTS-c represents a compelling therapeutic candidate. Yet, careful assessment of its pharmacodynamics, safety profile, and individual variability is essential. The integration of mitochondrial-based interventions with individualized, multidisciplinary care holds promise for improving outcomes in ME/CFS.
Author Contributions
Conceptualization, A.C.; Writing—original draft preparation, A.C.; Writing—review and editing, A.C., I.R., V.R., A.F., K.D., and N.K.; Literature review, L.T. and S.P.; Visualization, I.R. and V.R.; Supervision, A.C.; Critical review and feedback, I.R., V.R., A.F., K.D., and N.K. All authors have read and approved the final version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing does not apply to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ME/CFS | Myalgic Encephalomyelitis/Chronic Fatigue Syndrome |
| PEM | Post-Exertional Malaise |
| MDPs | Mitochondrial-Derived Peptides |
| MOTS-c | Mitochondrial Open Reading Frame of the 12S rRNA-c |
| AMPK | AMP-Activated Protein Kinase |
| PGC-1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha |
| NRF2 | Nuclear Factor Erythroid 2–Related Factor 2 |
| OXPHOS | Oxidative Phosphorylation |
| ETC | Electron Transport Chain |
| ROS | Reactive Oxygen Species |
| ATP | Adenosine Triphosphate |
| FDA | U.S. Food and Drug Administration |
| IND | Investigational New Drug |
| OI | Orthostatic Intolerance |
| POTS | Postural Orthostatic Tachycardia Syndrome |
| BAT | Brown Adipose Tissue |
| NMN | Nicotinamide Mononucleotide |
| NR | Nicotinamide Riboside |
| CoQ10 | Coenzyme Q10 |
References
- Institute of Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Washington, DC: The National Academies Press; 2015.
- ME/CFS Clinician Coalition. Treatment recommendations for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). 2021. Available from: https://mecfscliniciancoalition.org/.
- DePace, N.L.; Vinik, A.I.; Acosta, C.; Santos, L.; Murray, G.L.; Colombo, J. ORAL VASOACTIVE MEDICATIONS.
- Miller, A.J.; Raj, S.R. Pharmacotherapy for postural tachycardia syndrome. Autonomic Neuroscience. 2018, 215, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Cortes Rivera, M.; Mastronardi, C.; Silva-Aldana, C.T.; Arcos-Burgos, M.; Lidbury, B.A. Myalgic encephalomyelitis/chronic fatigue syndrome: a comprehensive review. Diagnostics. 2019, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Neustadt, J.; Pieczenik, S.R. Medication-induced mitochondrial damage and disease. Molecular nutrition & food research. 2008, 52, 780–8. [Google Scholar]
- Cohen, B.H. Pharmacologic effects on mitochondrial function. Developmental disabilities research reviews. 2010, 16, 189–99. [Google Scholar] [CrossRef] [PubMed]
- Vuda, M.; Kamath, A. Drug induced mitochondrial dysfunction: Mechanisms and adverse clinical consequences. Mitochondrion. 2016, 31, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Janhsen, K.; Roser, P.; Hoffmann, K. The problems of long-term treatment with benzodiazepines and related substances: Prescribing practice, epidemiology, and the treatment of withdrawal. Deutsches Ärzteblatt International. 2015, 112, 1. [Google Scholar] [PubMed]
- Rowe, P.C.; Underhill, R.A.; Friedman, K.J.; Gurwitt, A.; Medow, M.S.; Schwartz, M.S.; Speight, N.; Stewart, J.M.; Vallings, R.; Rowe, K.S. Myalgic encephalomyelitis/chronic fatigue syndrome diagnosis and management in young people: a primer. Frontiers in pediatrics. 2017, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Grach, S.L.; Seltzer, J.; Chon, T.Y.; Ganesh, R. Diagnosis and management of myalgic encephalomyelitis/chronic fatigue syndrome. InMayo Clinic Proceedings 2023 Oct 1 (Vol. 98, No. 10, pp. 1544-1551). Elsevier.
- Evrard, A.; Mbatchi, L. Genetic polymorphisms of drug metabolizing enzymes and transporters: the long way from bench to bedside. Current Topics in Medicinal Chemistry. 2012, 12, 1720–9. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab Brain Dis. 2014, 29, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Naviaux, J.C.; Li, K.; et al. Metabolic features of chronic fatigue syndrome. Proc Natl Acad Sci USA. 2016, 113, E5472–E5480. [Google Scholar] [CrossRef] [PubMed]
- Jason, L. The Energy Envelope Theory and myalgic encephalomyelitis/chronic fatigue syndrome. Aaohn Journal. 2008, 56, 189–95. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, T.; Cagnin, S.; Bondi, D.; Santangelo, C.; Marramiero, L.; Purcaro, C.; Bonadio, R.S.; Di Filippo, E.S.; Mancinelli, R.; Fulle, S.; Verratti, V. Myalgic encephalomyelitis/chronic fatigue syndrome from current evidence to new diagnostic perspectives through skeletal muscle and metabolic disturbances. Acta Physiologica. 2024, 240, e14122. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.M. Sleep Hygiene. Integrative Sleep Medicine. 2021, 113. [Google Scholar]
- Baranwal, N.; Phoebe, K.Y.; Siegel, N.S. Sleep physiology, pathophysiology, and sleep hygiene. Progress in cardiovascular diseases. 2023, 77, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Manresa-Rocamora, A.; Sarabia, J.M.; Javaloyes, A.; Flatt, A.A.; Moya-Ramon, M. Heart rate variability-guided training for enhancing cardiac-vagal modulation, aerobic fitness, and endurance performance: A methodological systematic review with meta-analysis. International journal of environmental research and public health. 2021, 18, 10299. [Google Scholar] [CrossRef] [PubMed]
- Sloan, M.; Bosley, M.; Gordon, C.; Pollak, T.A.; Mann, F.; Massou, E.; Morris, S.; Holloway, L.; Harwood, R.; Middleton, K.; Diment, W. ‘I still can’t forget those words’: mixed methods study of the persisting impact on patients reporting psychosomatic and psychiatric misdiagnoses. Rheumatology. 2025, 64, 3842–53. [Google Scholar] [CrossRef] [PubMed]
- Terlou, A.; Ruble, K.; Stapert, A.F.; Chang, H.C.; Rowe, P.C.; Schwartz, C.L. Orthostatic intolerance in survivors of childhood cancer. European Journal of Cancer. 2007, 43, 2685–90. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.P. General information brochure on orthostatic intolerance and its treatment. Chronic Fatigue Clinic Johns Hopkins Children’s Center. 2014.
- Rowe, P.C.; Barron, D.F.; Calkins, H.; Maumenee, I.H.; Tong, P.Y.; Geraghty, M.T. Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. Am J Med Genet A. 1999, 82, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.F.; Ellmore, M.; Middleton, G.; Murgatroyd, P.M.; Gee, T.I. Effects of resistance band exercise on vascular activity and fitness in older adults. International journal of sports medicine. 2017, 38, 184–92. [Google Scholar] [CrossRef] [PubMed]
- Jakše, B.; Jakše, B.; Pajek, M.; Pajek, J. Uric acid and plant-based nutrition. Nutrients. 2019, 11, 1736. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xue, X.; Ma, L.; Zhou, S.; Li, K.; Wang, C.; Sun, W.; Li, C.; Chen, Y. Effect of low-purine diet on the serum uric acid of gout patients in different clinical subtypes: a prospective cohort study. European Journal of Medical Research. 2024, 29, 449. [Google Scholar] [CrossRef] [PubMed]
- Siener, R.; Hesse, A. Influence of dietary oxalate intake on urinary oxalate excretion. World J Urol. 2006, 24, 305–309. [Google Scholar]
- Holton, K. The potential role of dietary intervention for the treatment of neuroinflammation. Translational Neuroimmunology 2023, 7, 239–66. [Google Scholar]
- Castro-Marrero, J.; Cordero, M.D.; Sáez-Francas, N.; et al. Effect of coenzyme Q10 plus NADH supplementation on maximum heart rate after exercise testing in chronic fatigue syndrome–a randomized, controlled, double-blind trial. Clin Nutr. 2016, 35, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Vacante, M.; Giordano, M.; et al. Oral acetyl-l-carnitine therapy reduces fatigue in older patients with chronic fatigue syndrome. Arch Gerontol Geriatr. 2008, 46, 181–190. [Google Scholar] [CrossRef] [PubMed]
- .
- .
- Teitelbaum, J.E.; Johnson, C.; St Cyr, J. The use of D-ribose in chronic fatigue syndrome and fibromyalgia: a pilot study. J Altern Complement Med. 2006, 12, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, H.; Kawanami, T.; Ogasawara, K.; et al. Effects of nicotinamide riboside and NMN on cardiovascular health and metabolism. Nutrients. 2021, 13, 1171. [Google Scholar]
- Bent, S.; Bertoglio, K.; Ashwood, P.; et al. A pilot randomized controlled trial of omega-3 fatty acids for autism spectrum disorder. J Autism Dev Disord. 2020, 50, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Daverey, A.; Agrawal, S.K. Curcumin protects against white matter injury through NF-κB and Nrf2 cross talk. Journal of Neurotrauma. 2020, 37, 1255–65. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomedicine & Pharmacotherapy. 2020, 127, 110234. [Google Scholar]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best practice & research Clinical endocrinology & metabolism. 2012, 26, 711–23. [Google Scholar]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox biology. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radical Biology and Medicine. 2009, 47, 333–43. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell death & differentiation. 1999, 6, 99–104. [Google Scholar]
- Papadopoulos, V.; Miller, W.L. Role of mitochondria in steroidogenesis. Best practice & research Clinical endocrinology & metabolism. 2012, 26, 771–90. [Google Scholar]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: more than a structural role. haematologica. 2014, 99, 973. [Google Scholar] [CrossRef] [PubMed]
- Kalf, G.F. Deoxyribonucleic acid in mitochondria and its role in protein synthesis. Biochemistry. 1964, 3, 1702–6. [Google Scholar] [CrossRef] [PubMed]
- Holden, S. Mitochondrial dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): A systematic review and quality assessment of the evidence. J Transl Med. 2020, 18, 290. [Google Scholar] [CrossRef] [PubMed]
- Behan, W.M.; More, I.A.; Behan, P.O. Mitochondrial abnormalities in the postviral fatigue syndrome. Acta Neuropathol. 1991, 83, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. International journal of clinical and experimental medicine. 2009, 2, 1. [Google Scholar] [PubMed]
- Tomas, C.; Brown, A.E.; Newton, J.L. Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS One. 2017, 13, e0192817. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, E.; Ryan, M.; Edgar, C.; MacKay, A.; Vallings, R.; Tate, W. Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome. Int J Immunopathol Pharmacol. 2020, 34, 2058738420933686. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.; Light, K.; Light, A.; Bateman, L.; Cannon-Albright, L.A. Evidence for a heritable predisposition to Chronic Fatigue Syndrome. BMC neurology. 2011, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nakatomi, Y.; Mizuno, K.; Ishii, A.; et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: An ¹¹C-(R)-PK11195 PET study. J Nucl Med. 2014, 55, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.J.; Camacho, A.; Martin, C.; et al. Neuroimaging findings in Long COVID and ME/CFS: An overlapping pattern? Front Neurol. 2020, 11, 1026. [Google Scholar]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018, 28, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.C.; Lai, R.W.; Woodhead, J.S.T.; et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat Commun. 2021, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Nguyen, A.; Lee, C. Mitochondrial peptide MOTS-c promotes thermogenesis and browning of white fat. Proc Natl Acad Sci USA. 2021, 118, e2023993118. [Google Scholar]
- Hoel, F.; Hoel, A.; Pettersen, I.K.; Rekeland, I.G.; Risa, K.; Alme, K.; Sørland, K.; Fosså, A.; Lien, K.; Herder, I.; Thürmer, H.L. A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. JCI insight. 2021, 6, e149217. [Google Scholar] [CrossRef] [PubMed]
- Anastasia, M. Metabolic Dysregulation in ME/CFS: The Role of Hypometabolism and Mitochondrial Dysfunction in Post-Exertional Malaise (Doctoral dissertation).
- Tang, M.; Su, Q.; Duan, Y.; Fu, Y.; Liang, M.; Pan, Y.; Yuan, J.; Wang, M.; Pang, X.; Ma, J.; Laher, I. The role of MOTS-c-mediated antioxidant defense in aerobic exercise alleviating diabetic myocardial injury. Scientific Reports. 2023, 13, 19781. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wei, X.; Wei, P.; Lu, H.; Zhong, L.; Tan, J.; Liu, H.; Liu, Z. MOTS-c functionally prevents metabolic disorders. Metabolites. 2023, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C. Overcoming the shortcomings of peptide-based therapeutics. Future Drug Discovery. 2022, 4, FDD75. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nature reviews Drug discovery. 2021, 20, 309–25. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nature reviews Molecular cell biology. 2023, 24, 255–72. [Google Scholar] [PubMed]
- Miller, B.; Kim, S.J.; Kumagai, H.; Yen, K.; Cohen, P. Mitochondria-derived peptides in aging and healthspan. The Journal of clinical investigation. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Guidance for Industry: Peptide Drug Products. 2022. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/peptide-drug-products.
Figure 1.
Mechanistic pathways through which MOTS-c may ameliorate ME/CFS pathology. MOTS-c promotes AMPK activation, mitochondrial biogenesis, anti-inflammatory signaling, skeletal muscle function, and brown adipose tissue activation—addressing core pathophysiologic features of ME/CFS. Image was created using BioRender.
Figure 1.
Mechanistic pathways through which MOTS-c may ameliorate ME/CFS pathology. MOTS-c promotes AMPK activation, mitochondrial biogenesis, anti-inflammatory signaling, skeletal muscle function, and brown adipose tissue activation—addressing core pathophysiologic features of ME/CFS. Image was created using BioRender.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



