Submitted:
25 June 2025
Posted:
26 June 2025
You are already at the latest version
Abstract
With the rise in immunocompromised individuals and patients with immune-related comorbidities such as COVID-19, the rate of fungal infections is growing. This increase, along with the current plateau in anti-fungal drug development, has made understanding the pathogenesis and dissemination of these organisms more pertinent than ever. The mouse model of fungal infection, while informative on a basic science level, has severe limitations in terms of translation to the human disease. Here we present data supporting the implementation of the human cerebral organoid model, which is generated from human embryonic stem cells and accurately recapitulates relevant brain cell types and structures, to study fungal infection and dissemination to the central nervous system (CNS). This approach provides direct insight into the relevant pathogenesis of specific fungal organisms in human tissues where in vivo models are impossible. With this model system we assessed the specific brain tropisms and cellular effects of fungal pathogens known to cross the blood brain barrier (BBB) such as Cryptococcus neoformans. We determined the effects of this fungal pathogen has on the overall gross morphology, cellular architecture, and cytokine release from these model organoids. Furthermore, we demonstrated that C. neoformans penetrates and invades the organoid tissue and remains present throughout the course of infection. These results demonstrate the utility of this new model to the field and highlight the potential for this system to elucidate fungal pathogenesis to new therapeutic strategies to prevent and treat the disseminated stages of fungal diseases such as cryptococcal meningitis.
Keywords:
cerebral organoid
; fungal pathogen
; Cryptococcus neoformans
1. Introduction
Invasive fungal pathogens pose an increasing risk to the growing population of immunocompromised individuals and patients worldwide, especially ubiquitous environmental yeasts like Cryptococcus neoformans [1,2]. This fungal pathogen has made its environmental reservoir in tree soil and pigeon guano, making it an extreme and ever-present risk to those with compromised immune systems. Furthermore, C. neoformans is a professional pathogen that can adapt to the various environments of the host to establish a severe and life-threatening infection killing 600,000 people annually [3]. One of these environments is that of the human brain. Following inhalation into the lungs, C. neoformans traverses the blood brain barrier (BBB) and persists in the CNS. This brain infection, or meningococcal encephalitis, is extremely difficult to treat due to the lack of new or affordable fungicidal drugs and the inherent resistance of C. neoformans to one of the three classes of available antifungals, echinocandins. Annually, it is estimated that one million people develop cryptococcal meningitis and that over 60% of these individuals succumb to the disease within a few months of diagnosis [4]. Considering the extremely high mortality rate of these infections, it is vital that we further study the interactions between this pathogenic fungus and the human brain in order to enhance patient outcomes following this lethal stage of Cryptococcal disease.
Understanding the mechanisms by which this pathogen traverses the BBB has intrigued the field and many groups have established in vitro models and cell lines for elucidating how in fact C. neoformans yeast enter the brain parenchyma from the blood vessel lumen. From these studies, it is thought that C. neoformans cells usurp macrophages via a trojan horse mechanism to cross through the tight junctions and subsequently escape from the macrophage once safely within the brain environment [5]. Others propose that the yeast cells undergo transcytosis themselves to enter the central nervous system without the need for other host cells [6,7]. While this research has been very informative, our understanding of what occurs after traversal that leads to the establishment of infection is severely limited. While mouse models of C. neoformans brain infections have yielded some insights, some have highlighted the limitations and often lack of translation of animal models to the clinic [8]. To date, the only human models of brain infections remain the use of human cadavers from individuals infected with cryptococcomas, which provide excellent insights into late-stage fatal disease, but do not aid in our understanding of infection initiation or progression [9].
To address these shortcomings, we present this the first use of cerebral organoids derived from human-embryonic stem cells (hESCs) as a model for studying the development of fungal disease in the brain. hESCs are a useful platform to generate models of human tissues of interest, including lung, liver, kidney, intestine, and, importantly for this study, the brain. Organoid models derived from human primary cells have been shown to reliably reproduce findings that either match patient data or correlate with clinical trials [10]. The data presented here provide evidence for successful infection of human brain organoids with multiple pathogenic and neurotropic fungi (i.e., C. neoformans and Candida auris), while also revealing a lack of infection and functional consequence of exposing these organoids to non-pathogenic fungi (i.e., Saccharomyces cerevisiae). Furthermore, the tissue-penetrating fungal infection caused by C. neoformans not only affects gross morphology and cellular organization/viability, but also generates an immune response evident through increased cytokine production and an inflammatory transcript response indicative of infection. Taken together, this work demonstrates the potential utility of human organoid models as a strategy for studying fungal-induced pathogenesis.
2. Results
2.1. Organoids Display Growth Defect in Response to Pathogenic Fungi
To determine if human cerebral organoids can be infected by the neurotropic pathogenic fungus C. neoformans, we co-cultured 21-day old cerebral organoids with C. neoformans at two different ratios of fungal cell: organoid cell: 0.1:2 (low) and 1:2 (high). On days 7, 14, and 21 we imaged the organoids and measured size changes by diameter length over time compared to the uninfected control (Figure 1A). By day 7, regardless of the amount of C. neoformans added, the infected organoids are significantly smaller compared to the control. This size reduction is similar to that of organoids infected with neurotropic viruses, often indicating a strong disruption of cellular organization and the induction of cytopathology [11]. Unlike viruses, however, fungal organisms can rapidly grow in cell culture media, consuming the nutrients that cerebral organoids need and potentially inducing growth defects via nutrient deprivation. To ensure our observed phenotype is a function of pathogenicity and not simply the presence of a fast-growing fungal organism, we also examined organoid growth over time in the presence of another pathogenic fungus known to infect the brain, Candida auris [12], as well as a not traditionally pathogenic fungal organism, Saccharomyces cerevisiae, the Baker’s yeast. Excitingly, the pathogenic C. auris strain also induced a dramatic reduction in organoid growth, similar to C. neoformans, while the growth rate of organoids co-cultured with the non-pathogenic S. cerevisiae was not significantly different from uninfected controls at the end of the infection (day 21) despite an early growth deficit (Figure 1B). These infection experiments demonstrate that human fungal pathogens like C. neoformans and C. auris can infect human brain organoids and cause significant growth defects by day 7 post infection as opposed to non-pathogenic strains like S. cerevisiae, which do not induce the same growth defects.
2.2. C. neoformans Effectively Penetrates Organoid Tissues
While the growth defect these organoids present in response to the presence of C. neoformans is striking, we next needed to determine if this fungus was in fact penetrating the organoid tissues to establish infection. Using a periodic-acid Schiff (PAS) stain to detect polysaccharides in tissues, we determined that after 7 days of a C. neoformans challenge, organoids have large amounts of polysaccharides present within their tissue layers compared to an uninfected control (Figure 2A (magenta)). To assess this fungal penetration at the species level, we also imaged these organoids using an antibody against the main component of the C. neoformans polysaccharide capsule, glucuronoxylomannan (GXM). Using confocal microscopy, we imaged C. neoformans infected organoids at days 7, 14, and 21 and visualized a time-dependent infiltration of GXM-positive fungal organisms into the organoid tissue (Figure 2B (yellow)). This penetration and staining pattern were not seen in the uninfected control, confirming that C. neoformans is capable of penetrating into the structure of cerebral organoids without the presence of vasculature. This finding is crucial as it supports our assertion that cerebral organoids could be a useful model for neuroinvasion of fungal pathogens.
2.3. C. neoformans Infection Disrupts Organoid Cellular Architecture
Having identified both growth defects and invasion of C. neoformans into cerebral organoids following infection, we next examined the impacts on organoid cytoarchitecture. During growth, cerebral organoids display a very consistent architectural pattern wherein neural progenitor cells (NPCs) arrange into ventricular patterns and asymmetrically divide outwards to produce neurons and other neural cell types of the brain. Examining the earliest timepoint in which we saw growth defects, 7 days post infection, we stained organoid sections with antibodies against the transcription factor SOX2 and mitogen-associated protein 2 (MAP2), to image NPCs and neurons, respectively. Excitingly, at this early timepoint (day 7), we were able to see significant differences in the organization of both NPCs and neurons, likely suggesting that our observed growth defects were due to a significant disruption of the standard function and division of both neurons and NPCs. Following a C. neoformans infection, both MAP2 and SOX2 positive cells are very sparse and dispersed and do not resemble the organization patterns of the uninfected organoid (Figure 3). Furthermore, in an uninfected organoid, the MAP2 positive neuronal cells surround the developing ventricle, but in the infected organoid, these cells are dispersed throughout and do not establish any ventricle-associated pattern. Therefore, this C. neoformans infection is completely disrupting the typical organization and neuronal structure of these organoids.
2.4. Cryptococcal Infection Induces Cytokine Induction in Brain Organoids
A key outcome of cryptococcal CNS infection is the subsequent induction of a wide array of cytokines [13,14,15,16,17,18,19,20,21,22], which are a major contributor to disease progression. Importantly, however, not all cells can produce every cytokine. The major cells identified in human cerebral organoids are NPCs, neurons, oligodendrocyte precursor cells, and astrocytes [23]. When comparing the cytokines that these cells are capable of producing with the cytokines known to be induced by cryptococcal infections in the brain, a small list is generated: CXCL10, IL6, CCL5, and IFN-γ. Therefore, we decided to assess the induction of these cytokines in the lysates of our infected and uninfected organoids. Following a 7-day infection with both low and high doses of C. neoformans, we were able to detect increased levels of all four cytokines by immunoblot analyses (Figure 4). These same cytokines were not found in the lysates of uninfected organoids providing further evidence that these fungal pathogens are not only infecting these tissues but are also triggering an inflammatory response in a physiologically relevant manner. These proteins were not detected in the lysates of organoids infected with C. neoformans on Day 3 (data not shown) indicating that the induction of these cytokines is time dependent. We did not assess protein induction past day 7 as we found the protein of these infected organoids to be completely degraded and unstable for analysis likely due to the severe disorganization of the organoids themselves as highlighted in Figure 3.
2.5. Organoid Transcriptional Response to Fungal Infection
While understanding the ability for this persisting cryptococcal infection to stimulate the release of specific inflammatory cytokines is important, it does not reveal the full immune response elicited. Therefore, we performed bulk RNA-sequencing on the RNA extracted from organoids infected at days 3 and 7 with both our organism of interest, C. neoformans (both high and low dose), and another neurotropic fungal pathogen that we found to affect organoid growth over time, C. auris (Figure 1B). Importantly, when we attempted this sequencing for day 14 or 21 post infection, the RNA was determined unstable and degraded for organoids that had been infected with both C. neoformans and C. auris.
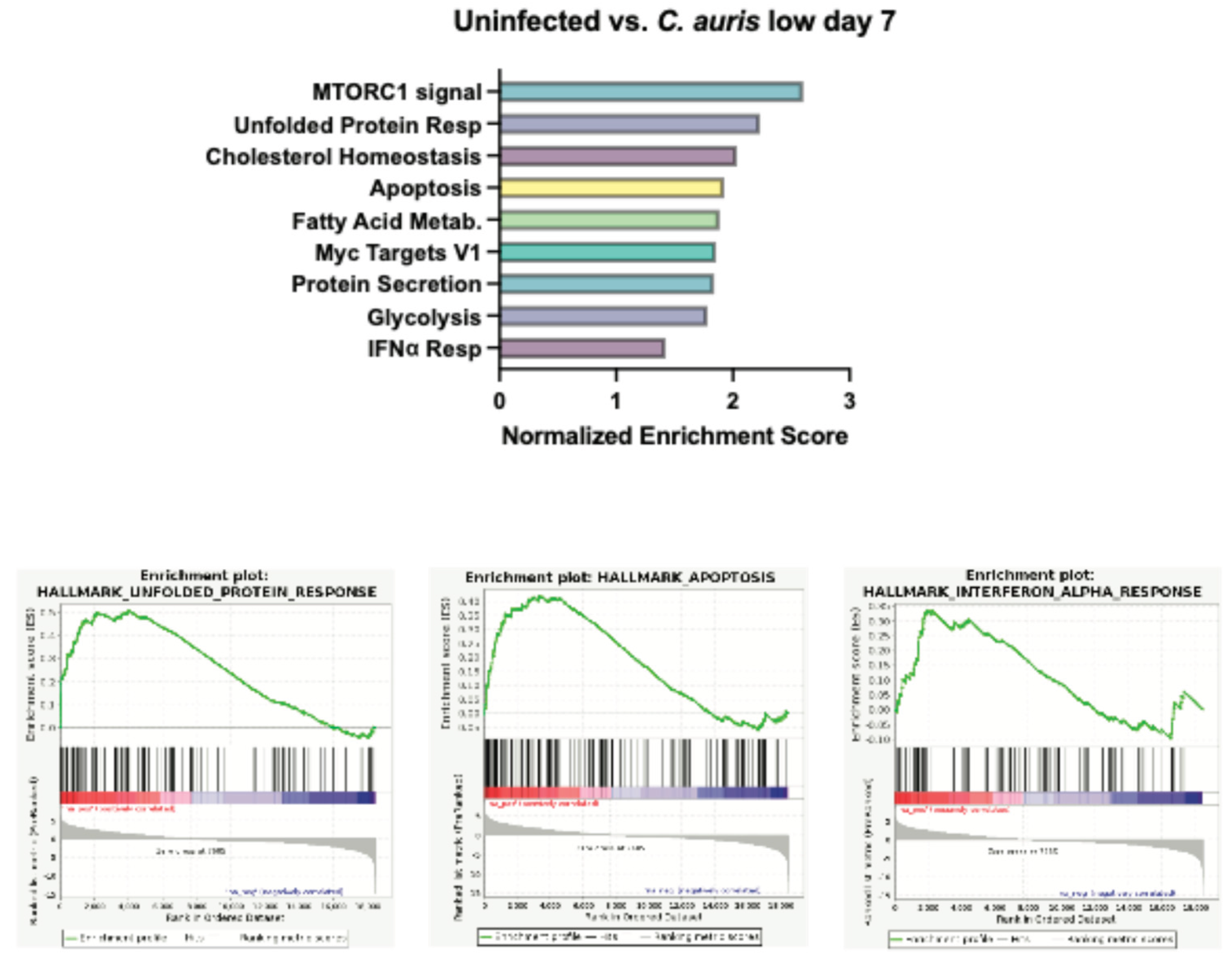
To evaluate our results in an unbiased manner, we turned to gene set enrichment analysis (GSEA). Compared to uninfected controls at day 3, a low dose of C. neoformans led to increased expression of genes involved in pancreas beta cell regulation and genes that are targets of MYC (Figure 5A). Neurons and beta cells share many of the transcription factors involved in differentiations as they both originate from the ectoderm, which might explain the increased expression of pancreas beta cell regulatory genes we observe at day 3 as cells are undergoing stress from infection [24]. MYC targets are expressed early during brain development and demonstrate increased expression later in the context of traumatic brain injury or cognitive disease [25]. Studies have shown that MYC and MYC target expression is tightly regulated under normal brain development, but overexpression can sensitize cells to apoptosis and cell cycle activation [26,27]. At a high dose of C. neoformans at the same timepoint, we did not observe MYC targets, however there was increased expression of genes involved in protein secretion, potentially upregulating cytokine release in response to infection. Perhaps most important was the shared induction of the unfolded protein response (UPR) pathway, a hallmark of cellular stress and often a precursor to both apoptosis and inflammation (Fig 5A-B). Interestingly, at day 7 in organoids exposed to both low and high dose C. neoformans, we observed an increase in transcriptional and cell cycle regulator pathways (E2F targets and G2M checkpoint) as well as a strong upregulation of the type I interferon (IFN) response (Figure 5C-D). We also noted upregulation of the IFN response pathway in response to C. auris infected organoids at day 7, however, at a slightly weaker level than those seen following C. neoformans infection (Supp Figure S1). C. auris appeared to induce the UPR and apoptosis pathways more than type I IFN response, perhaps suggesting that C. auris infection in the brain is less immunostimulatory and instead characterized by a stronger induction of cellular stress. Given this upregulation of inflammatory signatures at day 7 across our pathogenic fungal infected organoids, we also examined the transcriptional state of organoids exposed to S. cerevisiae at this same time point. Excitingly, we did not observe induction of IFN response pathways in our S. cerevisiae infected organoids, indicating that this inflammatory response is pathogen-dependent (Table 1). We did however, observe an increase in the E2F targets and G2M checkpoint pathways in response to S. cerevisiae, indicating that although this is not a traditional pathogen, the presence of this yeast in the brain still affects the transcription and cell cycle progression of these neurons and astrocytes. Interestingly, the C. auris infected organoids at day 3 and 7 had approximately 3-fold more pathways turned on in response to infection than C. neoformans at either dose. These pathways included the unfolded protein response, cholesterol synthesis, apoptosis, and other inflammatory responses (Table 1).
Figure 5.
Gene set enrichment analysis of cerebral organoids following infection with C. neoformans. A-B Graphed normalized enrichment scores of the Hallmark pathways that were significantly enriched (FDR < 0.05) and a set of enrichment plots for select pathways for organoids infected with a low (A) and high (B) dose of C. neoformans 3 days post infection. C-D Graphed normalized enrichment scores of the Hallmark pathways that were significantly enriched (FDR < 0.05) and a set of enrichment plots for select pathways for organoids infected with a low (A) and high (B) dose of C. neoformans 7 days post infection.
Figure 5.
Gene set enrichment analysis of cerebral organoids following infection with C. neoformans. A-B Graphed normalized enrichment scores of the Hallmark pathways that were significantly enriched (FDR < 0.05) and a set of enrichment plots for select pathways for organoids infected with a low (A) and high (B) dose of C. neoformans 3 days post infection. C-D Graphed normalized enrichment scores of the Hallmark pathways that were significantly enriched (FDR < 0.05) and a set of enrichment plots for select pathways for organoids infected with a low (A) and high (B) dose of C. neoformans 7 days post infection.
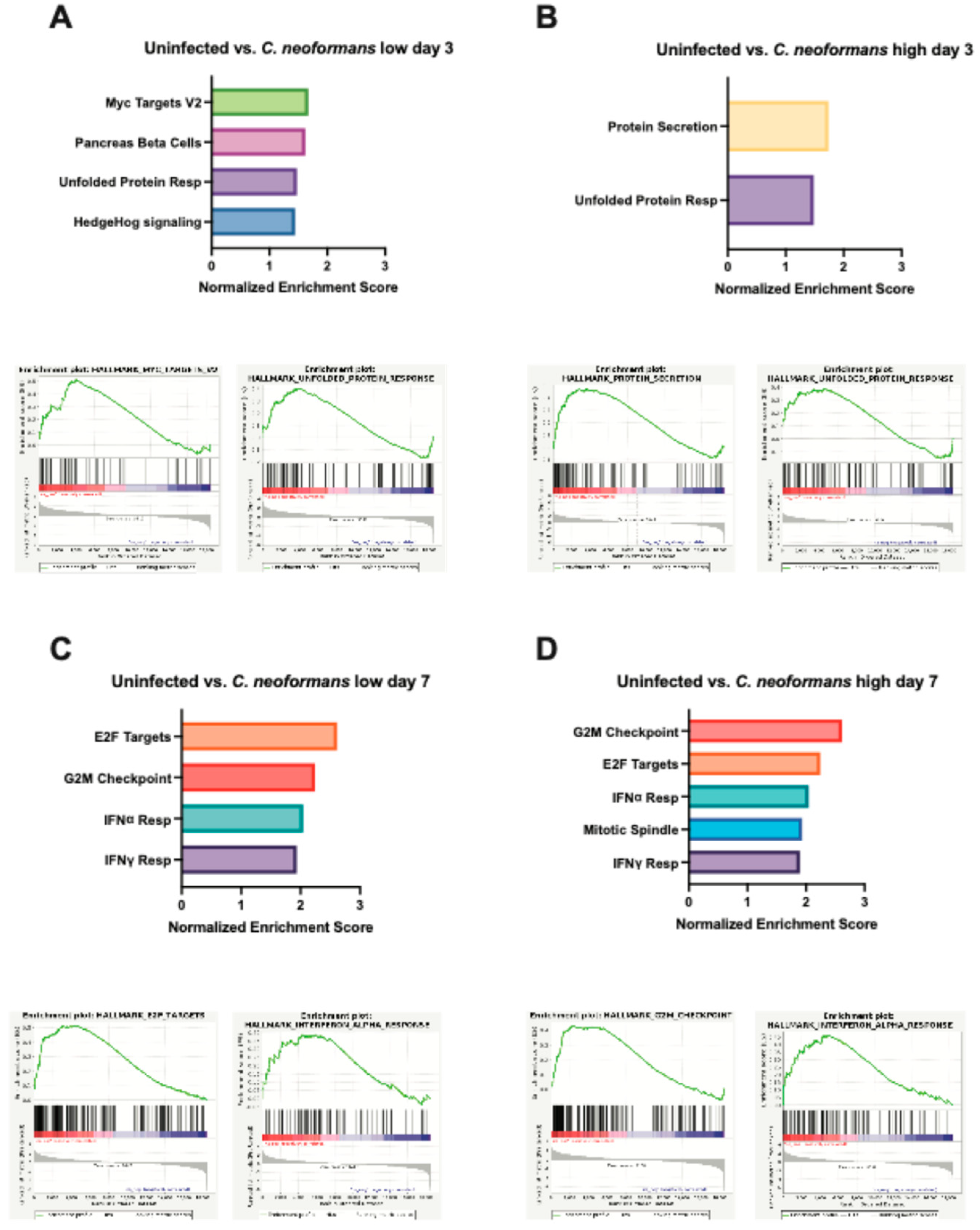
Figure S1.
Gene Set enrichment analysis of cerebral organoids following infection with C. auris. Graphed normalized enrichment scores of the Hallmark pathways that were significantly enriched (FDR < 0.05) and a set of enrichment plots for select pathways for organoids infected with C. auris seven days post infection.
Figure S1.
Gene Set enrichment analysis of cerebral organoids following infection with C. auris. Graphed normalized enrichment scores of the Hallmark pathways that were significantly enriched (FDR < 0.05) and a set of enrichment plots for select pathways for organoids infected with C. auris seven days post infection.
3. Discussion
The results presented here are the first to describe a C. neoformans infection in human cerebral organoids. We have demonstrated that organoids infected with two different doses of C. neoformans have significant growth defects over time compared to uninfected controls. Additionally, we have displayed, through two different methods of fungal detection, a time-dependent infiltration of fungal organism into the tissue of the organoid. Furthermore, these infected organoids display defects in cyto-architecture, increased cytokine secretion, and a dose-and time-dependent upregulation of inflammatory pathways. We also note that through the addition of S. cerevisiae and C. auris as non-pathogenic/negative and positive controls, respectively, we discovered that cerebral organoids respond differently to different fungal organisms both regarding growth and the transcriptional response. Therefore, these findings present a novel and relevant model system for studying the initial interaction and disease progression when fungal cells reach the human cerebral environment causing life-threatening disease.
Understanding the interaction between neurotropic fungal pathogens like C. neoformans and the brain is essential to improving diagnoses, treatment, and patient outcome in patients that develop these lethal brain infections. In vitro cell lines and various in vivo animal models for studying cryptococcal meningitis have existed for decades and have uncovered important details about the mechanisms of fungal entry into the CNS, however they are still lacking in terms of translational potential. Many investigators use human brain endothelial cell lines (i.e., hCMEC/D3) to uncover cryptococcal transmigration across the BBB [28,29,30]. These studies have exposed the complex and multifaceted ways C. neoformans reaches the CNS through the BBB. There is evidence to suggest both a passive transcytosis across the endothelial cell layer and an active “trojan horse” phagocyte-dependent crossing of engulfed C. neoformans yeast in macrophages through tight junctions between endothelial cells [28,31]. These detailed studies are made possible by the highly controlled environment of working with in vitro cell lines and the field has progressed due to these findings, however they only tell part of the pathogenesis story and reveal little about what occurs once the yeast interact with the complex brain environment. Therefore, other research groups use the mouse, rat, rabbit, or zebrafish vertebrate models to investigative the disease progression of C. neoformans to the brain. The transparency of zebrafish larvae, tractable nature of rabbit CNS infections, natural susceptibility of the rat to Cryptococcus, and the genetic flexibility of mouse models are just a few of the positives that using these vertebrates provide [8]. However, the lack of clinical translatability and high cost of maintenance has interfered with research progress specifically in regard to elucidating the dissemination of C. neoformans to the CNS. Additionally, some investigators attempting to study the pathogenesis of this infection using a clinically relevant model for studying cryptococcal brain infection dissect cadavers of human patients who have succumb to this disease. While this approach has revealed interesting aspects about the pathology of this late and terminal stage of cryptococcal disease in actual human patient brain sections [32], this does not investigate the early interactions between fungal cells and the host brain environment. Therefore, a similarly relevant, but less invasive method for studying cryptococcal meningitis is essential. By using human derived cerebral organoids as a model to study C. neoformans infection, we are not only able to study disease initiation and progression amongst clinically relevant cell types, but also quickly process meaningful quantities of samples to reveal key insights into the cellular, cytokine, and transcriptional responses this infection evokes in the human brain environment at various timepoints throughout pathogenesis.
The cytokine response uncovered in organoids infected with both a low and high dose of C. neoformans reveals interesting insights into the behavior of astrocytes, glial cells, neurons, and endothelial cells in response to fungal pathogen. The increased secretion of IL-6 and CXCL10 are particularly exciting because of previous studies connecting these cytokines to not only neuroinflammation, but also fungal disease. IL-6 is repeatedly described as highly induced in the CNS during times of neuroinflammation such as viral meningitis, murine cerebral malaria, systemic lupus, and HIV-1 [33,34,35,36]. The cell types that secrete IL-6 (neurons, astrocytes, and endothelial cells) are all present in the human cerebral organoids in our model system making the detection of this cytokine highly likely during neuro-stress and infection [33]. Furthermore, cryptococcal researchers have demonstrated that without IL-6, there is an increased permeability of the BBB during fungal meningitis indicating that this C. neoformans-induced IL-6 is integral to preventing fungal migration from the periphery to the CNS [13,14]. Similarly, the proinflammatory chemokine CXCL10 has been consistently reported as upregulated in the CNS in response to infection with neurotropic viruses like herpes, HIV-1, hepatitis, and West Nile [37]. CXCL10 is also induced in response to cerebral malaria and trypanosome infection [38]. It is believed that this increased chemokine response is essential for mediating the influx of inflammatory leukocytes into the CNS during neuroinflammation through binding its receptor, CXCR3. In the context of cryptococcal meningitis, CXCR3 is essential for lethal brain pathology but dispensable for pathogen clearance [22]. This study examined mice infected with C. neoformans and identified the CXCL10-CXCR3 axis as important for the recruitment of T-cells to the CNS, similar to what has been demonstrated with viral infection [22]. Furthermore, CXCL10-producing cell types like neurons, astrocytes, and endothelial cells are present in our C. neoformans-infected organoids. We further discovered upregulation of IFN-γ in response to C. neoformans infection. CXCL10 is also known as IFN-γ–induced protein 10 and therefore, this induction could be related to the CXCR3 axis response. Importantly, IFN-γ has been linked to increased fungal cell clearance in a patient population with HIV-associated cryptococcal meningitis and in a murine model of infection [39,40]. CCL5, which was also induced in our infected organoids, is an important chemokine produced by astrocytes in response to inflammation, especially in diseases such as multiple sclerosis and intracerebral hemorrhage [41,42]. However much of this induction is essential for microglial recruitment [43], which is something our organoid model currently lacks. Therefore, it could be that C. neoformans infection induces astrocyte secretion of CCL5 to then interact with receptor CCR5 and recruit various inflammatory cell types like microglia and monocytes. Future studies that incorporate these cell types into our existing organoids will reveal a role for increased CCL5 in the response to C. neoformans-induced cerebral damage.
The transcriptional upregulation of specific pathways in response to varied fungi in a dose- and time-dependent manner as revealed by bulk RNA-sequencing is especially intriguing because it not only exposes the cellular reaction to infection, but also reveals a fungal pathogen-specific regulatory response. What is particularly interesting, is the upregulation of type I IFN responses in the C. neoformans and C. auris-infected organoids at day 7 that was not present in the S. cerevisiae-infected group. This fungal pathogen-specific type I IFN response has excited the field lately as many have connected diverse fungal infections to type I IFN signaling pathways and functional responses [44,45,46,47,48,49,50,51]. A recent finding even identified this IFN response as fungal pathogen-specific and connected the degree of type I IFN signaling to the ability for host cells to endocytose diverse fungal extracellular vesicles (EVs) [52]. The fact that we have identified an upregulation of type I IFN signaling in response to specific fungi in this model, but not others begs the question: might fungal EVs be involved in eliciting this specific response? Future studies investigating not only EVs, but other fungal ligands (B-glucan, mannan, capsule, melanin, etc.) as the stimuli that activates these neuronal cell types will elucidate the physical interaction that results in this proinflammatory phenotype of fungal-infected cerebral organoids.
Overall, these data establish the use of a novel model system for studying the neuro-pathogenesis of fungal pathogens. This approach provides direct insight into the relevant pathogenesis of specific fungal organisms in human tissues where in vivo models are impossible and in vitro models are lacking. Human organoid models have been used to study a variety of both viral and bacterial pathogens and represent a promising avenue for research of fungal pathogens that disseminate and cause disease in the brain. Our findings support the ability to use this model to assess the specific brain tropisms and cellular effects of fungal pathogens that are known to cross the BBB such as Cryptococcus neoformans. These insights will allow us to better understand the effects of fungal organisms on the function of the human brain and help us develop treatment strategies for the terminal stages of these fungal diseases such as cryptococcal meningitis.
4. Methods
4.1. Organoid Generation from Human Embryonic Stem Cells (hESCs)
Human embryonic stem cells were generated as previously described [53]. Briefly, WIBR3 cells were plated into ultra-low attachment round bottom 96-well plates to generate single embryoid bodies (EBs). EBs were maintained in these plates for approximately 6-days before being exposed to neural induction medium. Between 4 and 6 days after the addition of neural induction medium, EBs were embedded in droplets of matrigel and cultured neural maturation media in stationary dishes for 4 more days. Organoids were then transferred to an orbital shaker and rotated continuously at 80 rpm for the remainder of experimentation. Approximately 35-days after formation, organoids were considered mature and exposed to fungal pathogens or maintained as untreated controls.
4.2. Fungal Infection of Organoids
Following organoid growth and maturation for 21 days, fungal organisms were added to the wells containing the growing organoids. Overnight cultures of wildtype C. neoformans (H99), Saccharomyces cerevisiae (S288C), and Candida auris (AR387) were washed 3 times with 1x PBS, counted on a LUNA cytometer device, and added at specific ratio to the organoids: 0.1:2 fungal cell: organoid cell for ‘low’ C. neoformans and 1:2 fungal cell: organoid cell for ‘high’ C. neoformans and for controls S. cerevisiae and C. auris. Organoids were maintained shaking at 150 rpm in low adherent 12 well plates at 37 °C and 5% CO2 and media was changed every 3 days. Uninfected organoids were given fresh maturation media on Day 0 and refreshed every 3 days in the same way.
4.3. Growth Measurements
Prior to infection and at pre-determined timepoints (7, 14, 21), images of each organoid were taken on an epifluorescence scope at 4x magnification to ensure entirety of organoid and diameter was visible. Pictures were processed and diameters were measured in fiji (ImageJ2 version 2.3.0/1.53q). Graphs and statistical analysis (2-way ANOVA with multiple Dunnet’s comparisons test between infected and uninfected at each timepoint) for growth over time was done in Prism 10. * = 0.0314, *** = 0.0005, **** < 0.0001.
4.4. Organoid Collection for Staining, Embedding and Sectioning
On day of collection, isolate individua organoid into sterile tube. Aspirate media and incubate in 4% PFA for 30 minutes at RT to fix the cells. Aspirate PFA and wash 5 times with 1x PBS. After the final wash, add 15% sucrose in PBS (weight/volume) incubate overnight at 4°C. Aspirate 15% sucrose and add 30% sucrose in PBS (weight/volume), parafilm the tube to prevent evaporation and store at 4°C until all samples are collected. Organoids were then embedded in OCT compound and frozen in liquid nitrogen cooled 2-methyl-butane. Organoid blocks were stored at -80C and cryosectioned using a Leica CM1850 Cryostat. Sections were placed on microscope slides and allowed to dry at room temp before storing at -80C until staining.
4.3. Immunofluorescence Staining
Organoid sections were warmed to room temperature and then washed in PBS before permeabilization (0.1% Triton X-100 in PBS) and blocking (3% normal goat serum in PBS) for one hour. Primary antibodies used included a rabbit monoclonal anti-SOX2 (1:500, Cell Signaling Technology), chicken polyclonal anti-MAP2A/B (1:10,000, EnCor Biotechnology), and mouse anti-GXM clone 18B7 (1:1,000, Millipore Sigma) in blocking buffer at 4C overnight. Samples were then washed three times in PBST (0.25% Triton-X). Secondary Alexa fluor conjugated goat anti-mouse, anti-chicken, and anti-rabbit antibodies (Invitrogen) were diluted 1:1000 and added to blocking buffer with DAPI (1 ug/mL, Invitrogen) and incubated at room temp for 1 hr. Samples were then washed three times with PBST before being mounted with #1.5 thickness coverslips using ProLong Diamond antifade mounting reagent. Images were captured on a Nikon Inverted Microscope Eclipse Ti-E equipped with a CSU-X1 confocal spinning disk head (Yokogawa), and a Coherent 4 W continuous-wave laser excited the sample. A 20× high-numerical aperture objective was used. Images were obtained using an EMCCD camera (Hamamatsu Photonics). Image processing was performed using FIJI 2 (ImageJ version 2.3.0/1.53q).
4.4. Fungal, Periodic Acid Schiff Stain
Periodic Acid Schiff staining was done following the standard kit procedure (abcam). Cryosection samples and washed in PBS and tap water before being immersed in the Periodic Acid Solution for 10 minutes. Slides were then washed with distilled water 4 times before being immersed in the Schiff’s solution for 30 minutes. Slides were then rinsed with hot water and distilled water before staining in light green solution for 2 minutes. Slides were rinsed with absolute alcohol and then dehydrated with two more rinses before being mounted with #1.5 thickness coverslips. Images taken at 10x or 40x on Nikon eclipse TS100 epifluorescence microscope and obtained using an Excelis camera and processed using FIJI 2 (ImageJ version 2.3.0/1.53q).
4.5. Immunoblot Analysis
Lysates were collected after 7 days co-cultured incubation at 37 °C and 5% CO2 into sterile tube. Growth media was aspirated, accutase was used to break up structure and then incubated for 10 mins at 37 °C and 5% CO2. Cells were broken up into single cell suspension via pipetting and collected via centrifugation at 1000g for 5 mins. Lysates were collected using mammalian protein extraction reagent lysis buffer (Thermo Scientific, no. 78501) with sodium orthovanadate and protease inhibitors. Lysates were spun down (14,000g for 5 min at 4 °C), transferred to a fresh tube and subsequently mixed with 4× NuPage lithium dodecyl sulfate loading buffer and 10× NuPage reducing agent. Western blot assays were performed using a 4–12% NuPage gel, with 2-[N-morpholino]ethanesulfonic acid running buffer (NuPage gels, Thermo Fisher Scientific), and transferred to methanol activated polyvinylidene difluoride membrane (Perkin Elmer, Waltham, MA) using transfer buffer (0.025 M Tris, 0.192 M glycine and 20% methanol) and electrophoretic transfer at 100 V for 1 h. For detection of proteins, polyvinylidene difluoride membranes were blocked for 1 h at RT in 5% milk in TBS 0.01% Tween 20 (TBST). To detect CXCL10 (Cell Signaling 14969), CCL5 (Cell Signaling 2988), IL-6 (Cell Signaling 12153) proteins, blots were incubated for overnight at 4°C in PBST, 1% BSA and primary antibody (1:1,000). Following incubation with primary antibody, blots were subsequently washed in either PBST or TBST 3× and incubated with secondary swine anti-rabbit horseradish peroxidase conjugated antibody at 1:2,000 (Agilent DAKO, P0399) ( Jackson ImmunoResearch) at 1:2,000 in 1% milk in PBST/TBST for 1 h at RT. To detect total protein via actin, blocked blots were incubated for 1 h at RT with simultaneous probing for actin (Cell Signaling 1:2,500) and secondary swine anti-rabbit horseradish peroxidase conjugated antibody. Membranes were washed 3× and then visualized using Western Lightning Plus ECL chemiluminescent substrate (Perkin Elmer) on Kodak BioMax XAR film (MilliporeSigma). Films were then scanned and processed using Adobe Illustrator. Any contrast adjustments were applied evenly to the entire image and adheres to standards set forth by the scientific community.
4.6. RNA Isolation and Sequencing
Collected individual organoids into sterile tube. Aspirated media, added 200 uL accutase, incubated in incubator for 10 mins at 37 °C and 5% CO2, pipette up and down to break up into single cell suspension, spin at 1000g for 5 mins. Resuspended in RLT buffer + BME. RNA was then further extracted from organoid cells using the Qiagen RNeasy minikit according to standard protocols. Samples were then submitted to the MIT BioMicrocenter and total RNA was sequenced using the paired end Illumina NovaSeq 6000 platform. Raw reads were then run through the RNA-seq Nextflow pipeline (revision 3.16.0) using the hg38 assembly to generate differential expression data (Deseq2) and Gene set enrichment analysis (GSEA) for the described comparisons.
Acknowledgments
The Gehrke Lab is supported by NCI grant: U19AI131135 (LG). The work in the Vyas Lab is supported by grants from the NIH: 1R01AI150181-01A1 and 5R21AI109303-02 (JMV). HBH is supported by the BroadIgnite Fellowship. RNA-sequencing experiments were completed through the Barbara K. Ostrom (1978) Bioinformatics and Computing Core Facility of the Swanson Biotechnology Center at MIT P30-CA14051. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 2017;17: 873–881. [CrossRef]
- Bratton EW, El Husseini N, Chastain CA, Lee MS, Poole C, Stürmer T, et al. Comparison and Temporal Trends of Three Groups with Cryptococcosis: HIV-Infected, Solid Organ Transplant, and HIV-Negative/Non-Transplant. Scheurer M, editor. PLoS One. 2012;7: e43582. [CrossRef]
- Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 2017;17: 873–881. [CrossRef]
- Sabiiti W, May RC. Mechanisms of Infection by the Human Fungal Pathogen Cryptococcus Neoformans. Future Microbiol. 2012;7: 1297–1313. [CrossRef]
- Casadevall A. Cryptococci at the brain gate: break and enter or use a Trojan horse? J Clin Invest. 2010;120: 1389. [CrossRef]
- Chang YC, Stins MF, McCaffery MJ, Miller GF, Pare DR, Dam T, et al. Cryptococcal Yeast Cells Invade the Central Nervous System via Transcellular Penetration of the Blood-Brain Barrier. Infect Immun. 2004;72: 4985. [CrossRef]
- Santiago-Tirado FH, Klein RS, Doering TL. An In Vitro Brain Endothelial Model for Studies of Cryptococcal Transmigration into the Central Nervous System. Curr Protoc Microbiol. 2019;53: e78. [CrossRef]
- Normile TG, Bryan AM, Del Poeta M. Animal Models of Cryptococcus neoformans in Identifying Immune Parameters Associated With Primary Infection and Reactivation of Latent Infection. Front Immunol. 2020;11: 581750. [CrossRef]
- Lee SC, Dickson DW, Casadevall A. Pathology of cryptococcal meningoencephalitis: Analysis of 27 patients with pathogenetic implications. Hum Pathol. 1996;27: 839–847. [CrossRef]
- Wensink GE, Elias SG, Mullenders J, Koopman M, Boj SF, Kranenburg OW, et al. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis Oncol. 2021;5: 1–13. https://doi.org/10.1038/S41698-021-00168-1;SUBJMETA=1059,2423,308,4028,53,575,67,692;KWRD=CANCER+THERAPY,PREDICTIVE+MARKERS,TRANSLATIONAL+RESEARCH.
- Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, et al. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell. 2016;19: 258. [CrossRef]
- Halioti A, Vrettou CS, Neromyliotis E, Gavrielatou E, Sarri A, Psaroudaki Z, et al. Cerebrospinal Drain Infection by Candida auris: A Case Report and Review of the Literature. Journal of Fungi. 2024;10: 859. [CrossRef]
- Reguera-Gomez M, Munzen ME, Hamed MF, Charles-Niño CL, Martinez LR. IL-6 deficiency accelerates cerebral cryptococcosis and alters glial cell responses. J Neuroinflammation. 2024;21: 242. [CrossRef]
- Li X, Liu G, Ma J, Zhou L, Zhang Q, Gao L. Lack of IL-6 increases blood–brain barrier permeability in fungal meningitis. J Biosci. 2015;40: 7–12. [CrossRef]
- Jarvis JN, Meintjes G, Bicanic T, Buffa V, Hogan L, Mo S, et al. Cerebrospinal Fluid Cytokine Profiles Predict Risk of Early Mortality and Immune Reconstitution Inflammatory Syndrome in HIV-Associated Cryptococcal Meningitis. PLoS Pathog. 2015;11. [CrossRef]
- Huffnagle GB, McNeil LK. Dissemination of C. neoformans to the central nervous system: Role of chemokines, Th1 immunity and leukocyte recruitment. J Neurovirol. 1999;5: 76–81. [CrossRef]
- Hansakon A, Jeerawattanawart S, Pattanapanyasat K, Angkasekwinai P. IL-25 Receptor Signaling Modulates Host Defense against Cryptococcus neoformans Infection. The Journal of Immunology. 2020;205: 674–685. [CrossRef]
- Zhou Q, Gault RA, Kozel TR, Murphy WJ. Protection from Direct Cerebral Cryptococcus Infection by Interferon-γ-Dependent Activation of Microglial Cells. The Journal of Immunology. 2007;178: 5753–5761. [CrossRef]
- Lee SC, Dickson DW, Brosnan CF, Casadevall A. Human astrocytes inhibit Cryptococcus neoformans growth by a nitric oxide-mediated mechanism. Journal of Experimental Medicine. 1994;180: 365–369. [CrossRef]
- Uicker WC, Doyle HA, McCracken JP, Langlois M, Buchanan KL. Cytokine and chemokine expression in the central nervous system associated with protective cell-mediated immunity against Cryptococcus neoformans. Med Mycol. 2005;43: 27–38. [CrossRef]
- Neal LM, Xing E, Xu J, Kolbe JL, Osterholzer JJ, Segal BM, et al. Cd4+ T cells orchestrate lethal immune pathology despite fungal clearance during cryptococcus neoformans meningoencephalitis. mBio. 2017;8. [CrossRef]
- Xu J, Neal LM, Ganguly A, Kolbe JL, Hargarten JC, Elsegeiny W, et al. Chemokine receptor CXCR3 is required for lethal brain pathology but not pathogen clearance during cryptococcal meningoencephalitis. Sci Adv. 2020;6: eaba2502. [CrossRef]
- Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Yang SM, Berger DR, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545: 48. [CrossRef]
- Juan-Mateu J, Rech TH, Villate O, Lizarraga-Mollinedo E, Wendt A, Turatsinze JV, et al. Neuron-enriched RNA-binding Proteins Regulate Pancreatic Beta Cell Function and Survival. Journal of Biological Chemistry. 2017;292: 3466–3480. [CrossRef]
- Ferrer I, Blanco R. N-myc and c-myc expression in Alzheimer disease, Huntington disease and Parkinson disease. Molecular Brain Research. 2000;77: 270–276. [CrossRef]
- Lee HG, Casadesus G, Nunomura A, Zhu X, Castellani RJ, Richardson SL, et al. The Neuronal Expression of MYC Causes a Neurodegenerative Phenotype in a Novel Transgenic Mouse. Am J Pathol. 2009;174: 891–897. [CrossRef]
- Lee HP, Kudo W, Zhu X, Smith MA, Lee H gon. Early induction of c-Myc is associated with neuronal cell death. Neurosci Lett. 2011;505: 124–127. [CrossRef]
- Sorrell TC, Juillard PG, Djordjevic JT, Kaufman-Francis K, Dietmann A, Milonig A, et al. Cryptococcal transmigration across a model brain blood-barrier: evidence of the Trojan horse mechanism and differences between Cryptococcus neoformans var. grubii strain H99 and Cryptococcus gattii strain R265. Microbes Infect. 2016;18: 57–67. [CrossRef]
- Santiago-Tirado FH, Klein RS, Doering TL. An In Vitro Brain Endothelial Model for Studies of Cryptococcal Transmigration into the Central Nervous System. Curr Protoc Microbiol. 2019;53. [CrossRef]
- Vu K, Weksler B, Romero I, Couraud PO, Gelli A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by cryptococcus neoformans. Eukaryot Cell. 2009;8: 1803–1807. [CrossRef]
- Casadevall A. Cryptococci at the brain gate: break and enter or use a Trojan horse? J Clin Invest. 2010;120: 1389. [CrossRef]
- Lee SC, Dickson DW, Casadevall A. Pathology of cryptococcal meningoencephalitis: Analysis of 27 patients with pathogenetic implications. Hum Pathol. 1996;27: 839–847. [CrossRef]
- Erta M, Quintana A, Hidalgo J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int J Biol Sci. 2012;8: 1254. [CrossRef]
- Metcalfe RD, Putoczki TL, Griffin MDW. Structural Understanding of Interleukin 6 Family Cytokine Signaling and Targeted Therapies: Focus on Interleukin 11. Front Immunol. 2020;11: 1424. [CrossRef]
- Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system invol vement. Arthritis Rheum. 1990;33: 644–649. [CrossRef]
- Laurenzi MA, Sidén Å, Persson MAA, Norkrans G, Hagberg L, Chiodi F. Cerebrospinal fluid interleukin-6 activity in HIV infection and inflammatory and noninflammatory diseases of the nervous system. Clin Immunol Immunopathol. 1990;57: 233–241. [CrossRef]
- Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, et al. Neuronal CXCL10 Directs CD8+ T-Cell Recruitment and Control of West Nile Virus Encephalitis. J Virol. 2005;79: 11457. [CrossRef]
- Petrisko TJ, Bloemer J, Pinky PD, Srinivas S, Heslin RT, Du Y, et al. Neuronal CXCL10/CXCR3 Axis Mediates the Induction of Cerebral Hyperexcitability by Peripheral Viral Challenge. Front Neurosci. 2020;14. [CrossRef]
- Chen Y, Strickland A, Shi M. IFN-γ signaling is essential for the clearance of C. neoformans in the brain and survival of the infected mice. The Journal of Immunology. 2020;204: 231.2-231.2. [CrossRef]
- Siddiqui AA, Brouwer AE, Wuthiekanun V, Jaffar S, Shattock R, Irving D, et al. IFN-γ at the Site of Infection Determines Rate of Clearance of Infection in Cryptococcal Meningitis. The Journal of Immunology. 2005;174: 1746–1750. [CrossRef]
- Lin J, Xu Y, Guo P, Chen YJ, Zhou J, Xia M, et al. CCL5/CCR5-mediated peripheral inflammation exacerbates blood‒brain barrier disruption after intracerebral hemorrhage in mice. J Transl Med. 2023;21: 1–19. [CrossRef]
- Pittaluga A. CCL5-glutamate cross-talk in astrocyte-neuron communication in multiple sclerosis. Front Immunol. 2017;8: 285787. [CrossRef]
- Lin J, Xu Y, Guo P, Chen YJ, Zhou J, Xia M, et al. CCL5/CCR5-mediated peripheral inflammation exacerbates blood‒brain barrier disruption after intracerebral hemorrhage in mice. J Transl Med. 2023;21: 1–19. [CrossRef]
- Stawowczyk M, Naseem S, Montoya V, Baker DP, Konopka J, Reich NC. Pathogenic effects of IFIT2 and interferon-β during fatal systemic Candida albicans infection. mBio. 2018;9. [CrossRef]
- delFresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, et al. Interferon-β Production via Dectin-1-Syk-IRF5 Signaling in Dendritic Cells Is Crucial for Immunity to C. albicans. Immunity. 2013;38: 1176–1186. [CrossRef]
- Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, et al. Type I Interferons Promote Fatal Immunopathology by Regulating Inflammatory Monocytes and Neutrophils during Candida Infections. PLoS Pathog. 2012;8: 10. [CrossRef]
- Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol. 2017;2. [CrossRef]
- Smeekens SP, Ng A, Kumar V, Johnson MD, Plantinga TS, Van Diemen C, et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat Commun. 2013;4. [CrossRef]
- Pekmezovic M, Hovhannisyan H, Gresnigt MS, Iracane E, Oliveira-Pacheco J, Siscar-Lewin S, et al. Candida pathogens induce protective mitochondria-associated type I interferon signalling and a damage-driven response in vaginal epithelial cells. Nature Microbiology 2021 6:5. 2021;6: 643–657. [CrossRef]
- Brown Harding H, Kwaku G, Reardon C, Khan N, Zamith-Miranda D, Zarnowski R, et al. Candida albicans Extracellular Vesicles Trigger Type I IFN Signaling via cGAS and STING. Nat Microbiol. 2024;9: 95–107. [CrossRef]
- Han F, Guo H, Wang L, Zhang Y, Sun L, Dai C, et al. The cGAS-STING signaling pathway contributes to the inflammatory response and autophagy in Aspergillus fumigatus keratitis. Exp Eye Res. 2021;202: 108366. [CrossRef]
- Kwaku GN, Jensen KN, Simaku P, Floyd DJ, Saelens JW, Reardon CM, et al. Extracellular vesicles from diverse fungal pathogens induce species-specific and endocytosis-dependent immunomodulation. PLoS Pathog. 2025;21: e1012879. [CrossRef]
- Li Y, Muffat J, Omer A, Bosch I, Lancaster MA, Sur M, et al. Induction of expansion and folding in human cerebral organoids. Cell Stem Cell. 2016;20: 385. [CrossRef]
Figure 1.
Organoids infected with fungal pathogens decrease in size. 1A: Diameters of mature organoids infected with a high and low dose of C. neoformans were measured at the largest cross section on day 0, 7, 14 and 21. These diameters were compared to an uninfected control at the same time points and the fold change in size is plotted with the mean and SD. A minimum of n = 4 organoids were measured for each time point. Significance assessed by an ordinary one-way ANOVA and Dunnet’s multiple comparisons test, p* = 0.0314, p*** = .0005, and p**** <0.0001. 1B: Diameters of mature organoids infected with S. cerevisiae and C. auris were measured at the largest cross section on day 0, 7, 14 and 21. These diameters were compared to an uninfected control at the same time points the fold change in size is plotted with the mean and SD. A minimum of n = 4 organoids were measured for each time point. Significance assessed by an ordinary one-way ANOVA and Dunnet’s multiple comparisons test, p* = 0.0128, p** = .0079, and p**** <0.0001.
Figure 1.
Organoids infected with fungal pathogens decrease in size. 1A: Diameters of mature organoids infected with a high and low dose of C. neoformans were measured at the largest cross section on day 0, 7, 14 and 21. These diameters were compared to an uninfected control at the same time points and the fold change in size is plotted with the mean and SD. A minimum of n = 4 organoids were measured for each time point. Significance assessed by an ordinary one-way ANOVA and Dunnet’s multiple comparisons test, p* = 0.0314, p*** = .0005, and p**** <0.0001. 1B: Diameters of mature organoids infected with S. cerevisiae and C. auris were measured at the largest cross section on day 0, 7, 14 and 21. These diameters were compared to an uninfected control at the same time points the fold change in size is plotted with the mean and SD. A minimum of n = 4 organoids were measured for each time point. Significance assessed by an ordinary one-way ANOVA and Dunnet’s multiple comparisons test, p* = 0.0128, p** = .0079, and p**** <0.0001.
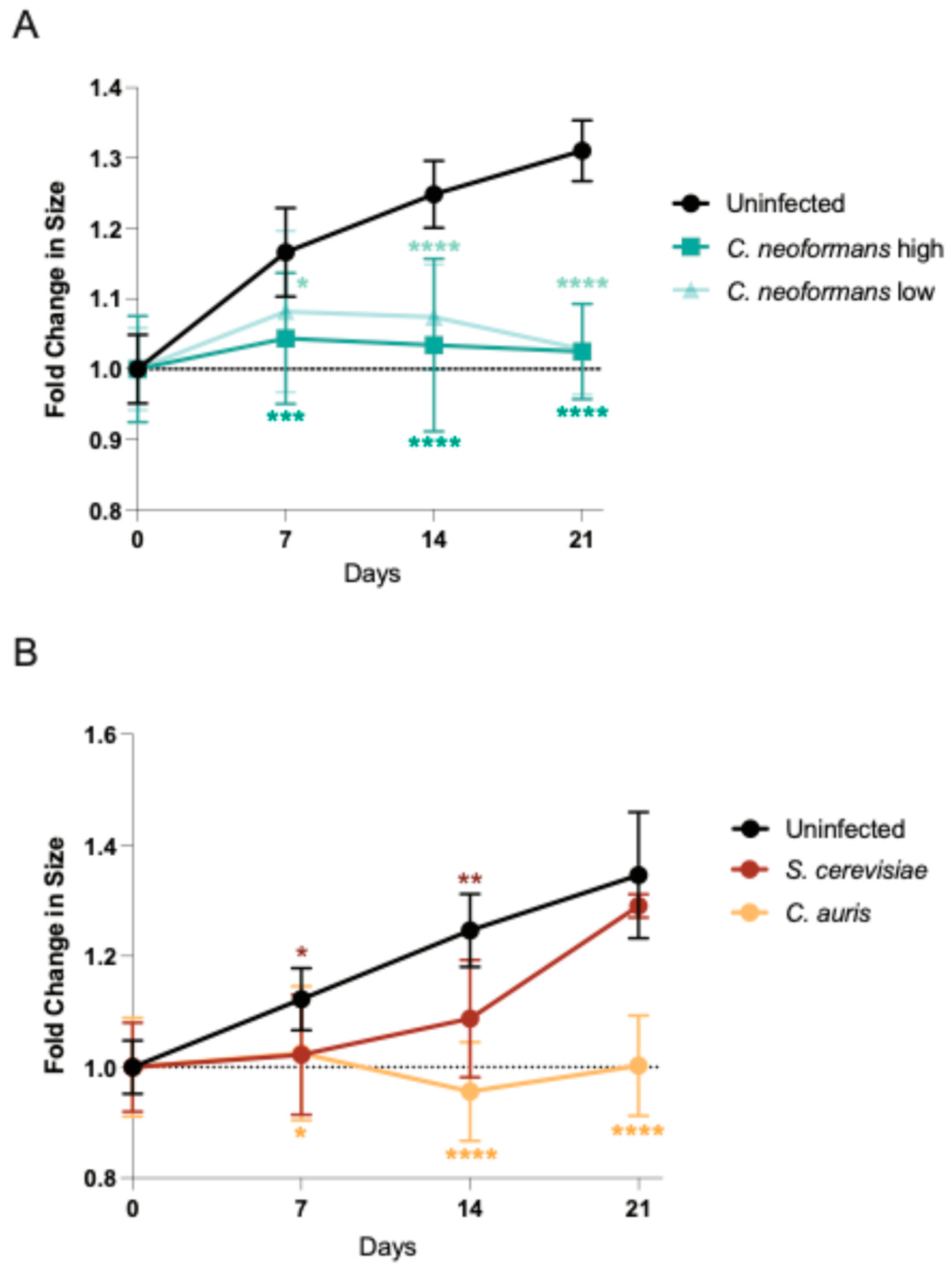
Figure 2.
Organoids have C. neoformans present throughout infection. 2A: Periodic-acid Schiff (PAS) stain to detect polysaccharides in organoid sections infected with a high dose of C. neoformans compared to an uninfected control at day 7. The blue-stained sections at both 10x and 40x represent tissue, magenta-stained sections represent fungal cell walls (polysaccharides and glycogen). Scale bar for 10x = 100 μm, scale bar for 40x = 25 μm. 2B: Confocal microscopy images of sections of organoids infected with a high dose of C. neoformans compared to an uninfected control at day 7, 14, and 21. Sections were incubated with anti-GXM clone 18B7 and subsequently the secondary Alexa fluor conjugated goat anti-mouse with DAPI. The blue represents nuclei that are positive for DAPI and the yellow indicates areas that are positive for GXM (fungal capsule polysaccharide). Scale bar = 50 μm.
Figure 2.
Organoids have C. neoformans present throughout infection. 2A: Periodic-acid Schiff (PAS) stain to detect polysaccharides in organoid sections infected with a high dose of C. neoformans compared to an uninfected control at day 7. The blue-stained sections at both 10x and 40x represent tissue, magenta-stained sections represent fungal cell walls (polysaccharides and glycogen). Scale bar for 10x = 100 μm, scale bar for 40x = 25 μm. 2B: Confocal microscopy images of sections of organoids infected with a high dose of C. neoformans compared to an uninfected control at day 7, 14, and 21. Sections were incubated with anti-GXM clone 18B7 and subsequently the secondary Alexa fluor conjugated goat anti-mouse with DAPI. The blue represents nuclei that are positive for DAPI and the yellow indicates areas that are positive for GXM (fungal capsule polysaccharide). Scale bar = 50 μm.

Figure 3.
Organoids have decreased Neural Progenitor Cells and neurons after infection. Confocal microscopy panels of organoid sections infected with a high dose of C. neoformans at day 7 compared to an uninfected control. Sections were stained with antibodies against the NPC transcription factor SOX2 (red), the neuronal marker MAP2 (green), and the nuclear marker (DAPI). The merged images reveal organoid architecture and organization in uninfected mature organoids and a disordered state in the infected organoids. Scale bar = 50 μm.
Figure 3.
Organoids have decreased Neural Progenitor Cells and neurons after infection. Confocal microscopy panels of organoid sections infected with a high dose of C. neoformans at day 7 compared to an uninfected control. Sections were stained with antibodies against the NPC transcription factor SOX2 (red), the neuronal marker MAP2 (green), and the nuclear marker (DAPI). The merged images reveal organoid architecture and organization in uninfected mature organoids and a disordered state in the infected organoids. Scale bar = 50 μm.
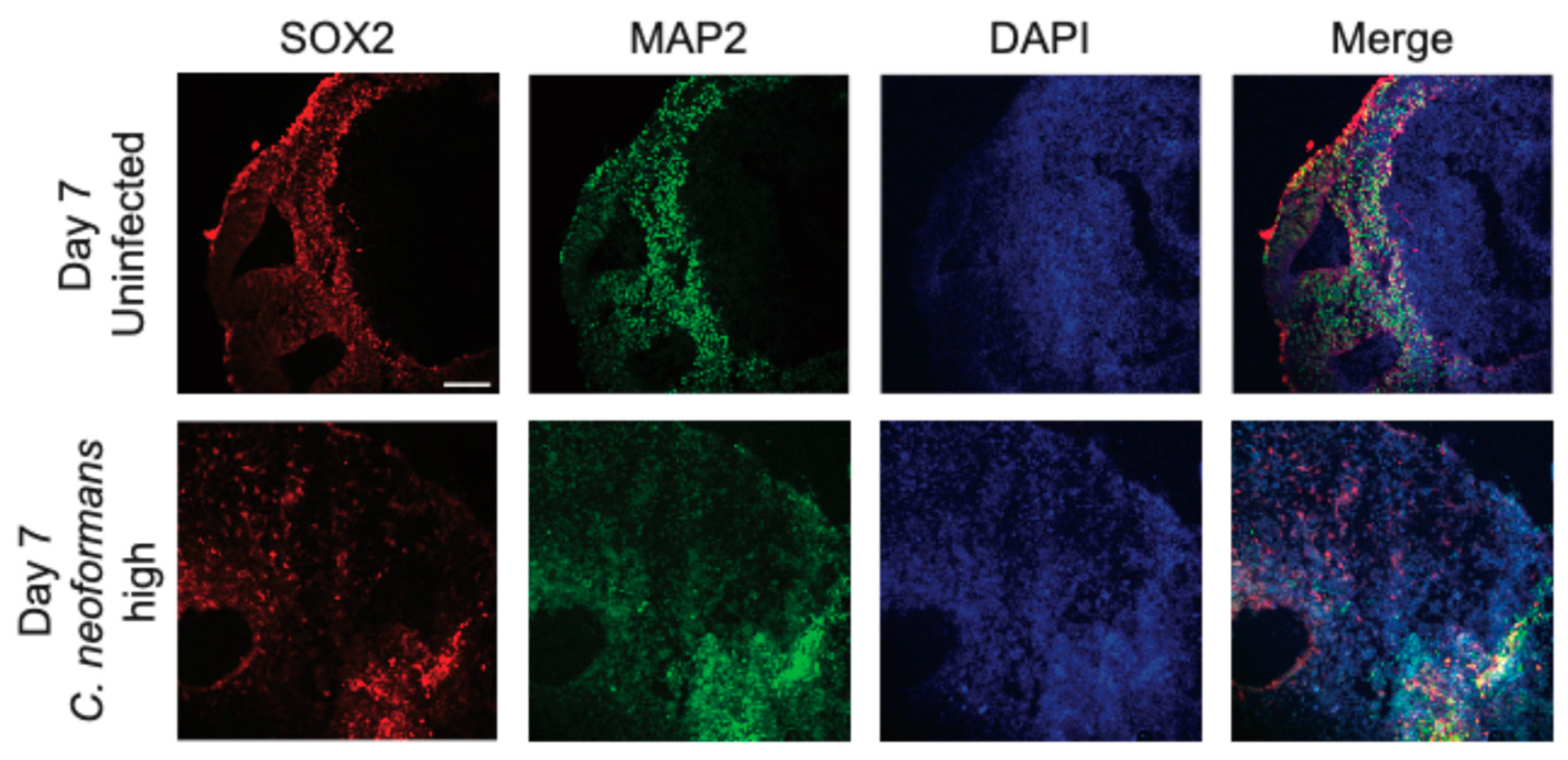
Figure 4.
C. neoformans infected organoids have increased cytokine expression and upregulated proinflammatory pathways. Immunoblot of IL-6, CXCL10, CCL5, IFN-γ and actin (loading control) from the lysate of uninfected and C. neoformans infected organoids at both low and high doses at day 7.
Figure 4.
C. neoformans infected organoids have increased cytokine expression and upregulated proinflammatory pathways. Immunoblot of IL-6, CXCL10, CCL5, IFN-γ and actin (loading control) from the lysate of uninfected and C. neoformans infected organoids at both low and high doses at day 7.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



