
Submitted:
16 June 2025
Posted:
17 June 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Post-COVID-19 syndrome (PCS) is an escalating global health concern, marked by persistent cognitive, neurological, and psychiatric symptoms following acute SARS-CoV-2 infection. Although its underlying mechanisms remain incompletely understood, mounting evidence implicates chronic neuroinflammation as a key driver. Sustained microglial and astrocyte activation, blood-brain barrier disruption, and aberrant cytokine signaling contribute to prolonged immune dysregulation within the central nervous system, promoting long-term brain dysfunction. In this expert review, we synthesize emerging insights into how neuroimmune processes impair brain function in PCS. We explore novel mechanistic pathways - including local sleep intrusions, impaired memory reconsolidation, and astrocyte-mediated destabilization of functional networks - that may underlie the syndrome's fluctuating and heterogeneous presentation. We evaluate fluid biomarkers of neuroinflammation, including glial fibrillary acidic protein (GFAP), soluble TREM2, S100β, and pro-inflammatory cytokines such as interleukin-6 and tumor necrosis factor-a. In parallel, we highlight converging neuroimaging biomarkers derived from PET and MRI studies. These include increased TSPO-PET binding in limbic and frontal regions, alterations in cerebral blood flow and oxygen metabolism, neurometabolic changes detected via MR spectroscopy (e.g., elevated myo-inositol and choline), and increased free water content on diffusion imaging - each suggestive of glial activation and network-level dysfunction. We propose a multiscale, longitudinal framework that integrates molecular, neuroimaging, and behavioral data to link immune dysregulation with brain network instability and symptom emergence. Such integrative approaches are critical for advancing precision diagnostics and informing the development of targeted, mechanism-based treatments for individuals affected by PCS.
Keywords:
Post-COVID19 syndrome
; neuroinflammation
; biomarkers
1. Introduction
The global spread of SARS-CoV-2 has precipitated not only an acute public health emergency but also a sustained wave of long-term morbidity. While early public and scientific focus centered on the respiratory manifestations of COVID-19, it has become increasingly clear that a substantial proportion of individuals experience persistent symptoms long after the resolution of the initial infection[1,2,3]. This condition - variously termed post-COVID-19 syndrome (PCS), Long COVID, or post-acute sequelae of SARS-CoV-2 infection (PASC) - encompasses a constellation of enduring symptoms including fatigue, cognitive impairment (“brain fog”), mood disturbances, headaches, and sleep disruptions. In many cases, these symptoms follow a fluctuating or relapsing-remitting course[2,3]. Such chronic post-infectious manifestations have profound implications for quality of life, productivity, and mental health and represent an urgent yet under-addressed global health concern[1,2,3]. Throughout this review, we adopt the term post-COVID-19 syndrome (PCS) to describe persistent neuropsychiatric symptoms following acute SARS-CoV-2 infection, in alignment with case definitions established by the World Health Organization (WHO) and the UK National Institute for Health and Care Excellence (NICE)[4,5]. While alternative terms such as Long COVID and PASC are widely used in the literature, they encompass overlapping but non-identical diagnostic criteria. For clarity and consistency, we use PCS to emphasize the syndrome’s neurocognitive and affective dimensions, which are central to its psychiatric relevance.
Despite the heterogeneity of its clinical presentation, accumulating evidence suggests that chronic neuroinflammation is a core mechanism underlying PCS[2,6,7]. Immune dysregulation in the central nervous system (CNS) - characterized by sustained glial reactivity, enhanced pro-inflammatory cytokine signaling, disruption of the blood-brain barrier (BBB), and altered neurovascular and neurometabolic dynamics - may destabilize neural circuits, contributing to the persistence and unpredictability of symptoms. Critically, these neuroimmune processes not only emerge during acute infection but may also remain active for months or even years, often without a clear correlation to initial COVID-19 severity.
This expert review synthesizes emerging evidence and advances a multiscale model of neuroinflammation in PCS, linking molecular and cellular immune events to systems-level brain dysfunction and clinical symptomatology. We first outline the key biological pathways -including viral neuroinvasion, glial reactivity, autoimmunity, BBB compromise, mast cell activation, and vagus nerve signaling - that may sustain chronic inflammation in the CNS. We then examine converging neuroimaging and fluid biomarker findings that support the presence of distributed, low-grade neuroinflammation. Importantly, we propose a hypothesis-generating framework that integrates these mechanisms with recent insights into local sleep intrusions, impaired memory reconsolidation, and fluctuating symptom expression. Finally, we discuss therapeutic implications, highlighting novel targets such as astrocyte modulators, histaminergic agents, vagus nerve stimulation, and interventions targeting the skull–marrow–meninges immune axis.
2. Mechanisms of Neuroinflammation in Post-COVID-19 Syndrome
Neuroinflammation in post-COVID-19 syndrome (PCS) likely arises from several interrelated and self-reinforcing mechanisms. These include direct viral invasion of the CNS, persistent glial activation, BBB dysfunction, autoimmune phenomena, latent viral reactivation, neurovascular injury, vagal dysregulation, and mast cell activation. Elucidating these mechanisms is critical for identifying reliable biomarkers of disease activity and for informing the development of targeted therapeutic strategies. In the sections below, we synthesize current evidence on how each mechanism may contribute to chronic CNS inflammation in PCS.
2.1. Direct Viral Invasion of Brain Parenchyma
The perception of the brain as an immune-privileged organ protected by the BBB is increasingly being challenged by evidence of viral neuroinvasion. Like other human coronaviruses, SARS-CoV-2 has demonstrated neurotropic potential[8], and growing evidence suggests that it can directly invade neural tissues, causing neuronal injury and localized inflammation[9,10].
Multiple entry routes into the CNS have been described for respiratory viruses, including SARS-CoV-2. These include transcribrial invasion via the olfactory nerve, retrograde transport through cranial nerves innervating the respiratory tract, hematogenous spread across a compromised BBB, and trafficking via infected immune cells[11]. Within the CNS, SARS-CoV-2 can infect neurons, astrocytes, microglia, and oligodendrocytes[12]. Its spike protein structure plays a key role in mediating axonal transport and determining neurovirulence Since the onset of the COVID-19 pandemic, the direct effects of SARS-CoV-2 on the nervous system have been increasingly documented[13,14]. Autopsy studies have revealed the presence of spike protein not only in cortical tissues and meninges but also in skull bone marrow - even in individuals who died from non-COVID-19-related causes - suggesting long-term persistence of viral proteins in reservoirs that can bidirectionally communicate with the brain through recently discovered osseous microchannels[15,16]. This chronic presence could act as a persistent trigger for neuroinflammation and potentially contribute to autoimmune mechanisms implicated in PCS.
Given the high degree of sequence homology between SARS-CoV-2 and other neurotropic coronaviruses such as SARS-CoV-1 and MERS-CoV[17,18], there is a pressing need to clarify the extent and consequences of direct CNS infection. Understanding the role of viral persistence in sustaining inflammatory cascades and neuroimmune dysregulation may be critical to explaining long-term neurological sequelae in PCS.
2.2. Glial Reactivity
Microglia and astrocytes play central roles in the neuroinflammatory processes underlying PCS. Both glial cell types adopt reactive phenotypes in response to viral infection, systemic inflammation, or neural injury, thereby sustaining a pro-inflammatory environment and impairing neural function[12]. Microglia, the brain’s resident immune cells, become chronically activated in PCS due to prolonged exposure to cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-alpha (TNF-α) or to residual viral components[19]. In this activated state, they release neurotoxic mediators, disrupt glutamate homeostasis, and may eliminate functional synapses via complement-mediated pruning—mechanisms thought to contribute to cognitive deficits and affective symptoms[14,20,21].
Importantly, neuroimaging and post-mortem studies suggest that this activation is not limited to phenotypic change but may also reflect increased glial cell density. In vivo PET imaging using TSPO ligands has revealed elevated binding in PCS patients, particularly in the anterior cingulate, striatum, and thalamus, a pattern interpreted as reflecting increased glial density based on recent post-mortem validation studies[6,22]. Supporting this, SARS-CoV-2-infected non-human primates exhibit microglial proliferation and clustering in the olfactory cortex and limbic areas[19], while human autopsy studies have identified persistent Iba1⁺ and CD68⁺ cell aggregates near spike protein reservoirs in the meninges and cortex[15]. These findings converge on the interpretation that microglial abundance is increased in PCS, potentially sustaining chronic inflammation.
Astrocytes are also key immunocompetent glial cells and represent direct cellular targets of SARS-CoV-2. Viral entry - facilitated by ACE2 and cofactors such as neuropilin-1 - disrupts astrocytic metabolic homeostasis, impairs neuronal support, and promotes pro-inflammatory cytokine release[23,24]. These changes are particularly deleterious in brain regions linked to memory, interoception, fatigue, and olfaction[24]. Moreover, astrocytes, which contribute to the structural and functional integrity of the BBB, can adopt neurotoxic A1-like phenotypes when exposed to microglial cytokines[24,25,26]. Such reactive astrocytes may further compromise BBB integrity and amplify inflammation. Astrocytic activation has also been observed via PET imaging using MAO-B ligands, which show increased signal in PCS patients - potentially reflecting a regional expansion of reactive astrocytes.
The reciprocal crosstalk between activated microglia and astrocytes forms a self-sustaining inflammatory loop that destabilizes local neural circuits and may underlie the relapsing-remitting symptom profile observed in PCS[27,28]. In sum, glial activation—amplified by both direct viral tropism and persistent immune signaling - represents a central mechanistic contributor to ongoing brain dysfunction. Targeting glial reactivity and neuron-glia–immune interactions may offer promising therapeutic avenues to interrupt this maladaptive cascade.
2.3. Blood Brain Barrier and Blood-CSF Barrier
Persistent BBB dysfunction is also a feature of neuroinflammation in PCS. Systemic inflammation and endothelial injury during acute infection can compromise BBB integrity, allowing peripheral cytokines and immune cells to access the CNS[28]. Dynamic contrast-enhanced MRI has revealed sustained BBB permeability in PCS patients, especially those with cognitive symptoms, and elevated serum markers such as GFAP and S100β support ongoing barrier disruption[29]. SARS-CoV-2 spike protein may further exacerbate endothelial inflammation, reinforcing a cycle of vascular injury and immune infiltration[30]. Autopsy and animal studies confirm associated cerebrovascular pathology, including fibrinogen leakage and endothelial loss[31,32].
In parallel, the choroid plexus - critical for CSF regulation and a pivotal hub commanding brain-body communication during inflammation[33] - also shows immune activation post-COVID[34]. Inflammatory signaling in this context can promote cytokine and immune cell trafficking into the brain, thereby amplifying neuroinflammation[35]. Together, BBB and choroid plexus dysfunction creates a conduit for chronic peripheral-to-central immune signaling, contributing to ongoing glial activation, neuronal damage, and cognitive impairment in PCS.
2.4. Meningeal Immunity and Skull Bone Marrow
The meninges and adjacent skull bone marrow form an active immunological interface capable of sustaining neuroinflammation[16]. In PCS, recent studies have shown persistent SARS-CoV-2 spike protein in the skull–meninges–brain axis months after infection[15]. Using tissue clearing and 3D imaging, viral antigens were detected in skull marrow, meningeal layers, and even perivascular spaces of the cortex—often co-localized with Iba1⁺ immune cells and near NeuN⁺ neurons[15]. These findings suggest that viral remnants may persist in peripheral immune niches, serving as a chronic stimulus for neuroimmune activation.
The skull bone marrow connects directly to the meninges via microscopic vascular channels, through which it supplies monocytes, neutrophils, and B cells[16,36]. In PCS, this pathway may allow sustained immune cell trafficking into the CNS, driven by ongoing antigenic stimulation. Spike persistence has been linked to elevated CSF levels of tau, neurofilament light, and GFAP, indicating ongoing neuronal and glial injury[15]. Meningeal immune cells - including macrophages, dendritic cells, and mast cells - likely remain in a pro-inflammatory state, releasing cytokines that can diffuse into CSF and brain parenchyma. This may contribute to cortical dysfunction and symptoms, including cognitive slowing, sensory hypersensitivity, and headaches.
2.5. Autoimmunity
Autoimmunity is increasingly recognized as a potential mechanism contributing to sustained neuroinflammation in PCS[2]. SARS-CoV-2 infection can trigger autoimmune responses through molecular mimicry, whereby viral proteins share structural similarities with host antigens, leading to the immune system’s misrecognition and subsequent attack on host tissues[37]. Elevated levels of autoantibodies targeting neural and vascular tissues have been observed in PCS patients, potentially mediating ongoing inflammatory and neurological damage[38,39], as well as autonomic instability when autoantibodies target vasoregulatory and autonomic nervous system (ANS) receptors[40]. These autoimmune responses may exacerbate existing neuroinflammation, further impairing neurological function and contribute to chronic symptomatology. Identifying specific autoantibodies and understanding their pathogenic roles could facilitate the development of targeted therapeutic strategies, such as immunosuppressive or immunomodulatory treatments, aimed at mitigating autoimmune-driven neuroinflammation and improving patient outcomes in PCS.
2.6. Reactivation of Endogenous Retroviruses and Latent Viral Elements
PCS is increasingly recognized as a condition of sustained immune dysregulation, and growing evidence suggests that this immune imbalance may lead to the reactivation of endogenous human retroviruses (HERVs) and latent neurotropic viruses[41,42]. These reactivated viral elements may act as chronic immune stimuli, further propagating neuroinflammation and symptom persistence. HERVs, particularly the HERV-W and HERV-K families, are normally silenced by epigenetic mechanisms but can be re-expressed under inflammatory or oxidative stress conditions[43]. SARS-CoV-2 infection - through its associated cytokine surge, mitochondrial dysfunction, and histone modification pathways - can derepress these elements. Reactivated HERV-W ENV protein[44] has been shown to activate microglia via TLR4, increase pro-inflammatory cytokine production, and induce astroglial dysfunction, contributing to synaptic destabilization and white matter pathology[45,46,47]. This mechanism is implicated in both multiple sclerosis and myalgic encephalomyelitis, and early data suggest similar patterns in PCS[48].
In parallel, reactivation of latent viruses such as Epstein-Barr virus (EBV) and human herpesvirus 6 (HHV-6) has been reported in individuals with PCS[49]. These viruses can persist in CNS-supporting cells such as microglia, astrocytes, and oligodendrocytes, and their reactivation may contribute to episodic flares of inflammation, BBB compromise, and cognitive or affective symptoms[48]. EBV, in particular, has been associated with elevated B cell activation, autoimmune responses, and glial reactivity - features also seen in PCS. These viral reactivations are not merely epiphenomena but may represent active drivers of chronic CNS immune activation, creating a self-sustaining loop between peripheral immune dysfunction, glial sensitization, and neural network instability. Understanding these mechanisms offers a compelling rationale for targeted antiviral or immunomodulatory interventions in select PCS subgroups.
2.7. Neurovascular Unit and Oxygen Metabolism
The neurovascular unit (NVU) - comprising neurons, glial cells, endothelial cells, pericytes, and the extracellular matrix - is critical for maintaining cerebral homeostasis, including oxygen delivery and metabolic support to the brain[50]. In PCS, emerging evidence suggests substantial disruption of the NVU, contributing significantly to persistent neuroinflammation and neurological dysfunction. SARS-CoV-2 infection induces endothelial injury and is accompanied by the formation of fibrin-amyloid microclots[51], resulting in impaired cerebral blood flow regulation, microvascular dysfunction, and diminished oxygen delivery to neural tissues[52,53]. Chronic hypo and delayed perfusion and associated hypoxic conditions within the brain may exacerbate neuronal injury and glial activation, perpetuating a cycle of inflammation and neurodegeneration[54,55]. Moreover, impaired oxygen metabolism and NVU dysfunction are evidenced by elevated markers of endothelial damage and metabolic stress[56,57]. Persistent endothelial inflammation further compromises the integrity of the BBB, facilitating inflammatory mediator infiltration, thus intensifying neuroimmune interactions[29,58].
2.8. Vagus Nerve Pathway
The vagus nerve, a core component of the parasympathetic nervous system, regulates inflammation via the cholinergic anti-inflammatory pathway[59,60]. In PCS, impaired vagal function due to SARS-CoV-2-induced systemic inflammation could, therefore, contribute significantly to sustained neuroinflammation, glial activation, and related symptoms, including cognitive dysfunction, fatigue, mood disorders, and autonomic imbalance[61,62]. Beyond its anti-inflammatory efferent signals, vagal afferent pathways transmit peripheral inflammatory signals directly to brain regions involved in immune regulation, emotional processing, and autonomic control[63,64]. Persistent inflammation in PCS could chronically activate these afferent pathways, forming a feedback loop that perpetuates neuroinflammation, amplifying symptom severity in PCS patients.
2.9. Mast Cell Activation and the Histamine System
Mast cells, strategically positioned at immunological interfaces such as mucosal surfaces and the BBB, have emerged as potential drivers of chronic neuroinflammation in PCS[65]. Increasing clinical and molecular evidence suggests persistent mast cell activation in PCS, with significant symptom overlap with mast cell activation syndrome (MCAS), including fatigue, brain fog, headaches, and dysautonomia[66,67]. Upon activation, mast cells release histamine along with cytokines, chemokines, and proteases - potent mediators of neuroimmune signaling[68]. Elevated histamine levels may increase BBB permeability (including increased permeability of endothelial cells to viral particles[69]), promoting immune cell infiltration and glial activation[65,68]. The histamine H4 receptor (H4R), enriched on mast cells, dendritic cells, eosinophils, and microglia, amplifies cytokine release and leukocyte recruitment in inflamed tissues[70]. In PCS, sustained H4R activation may perpetuate neuroinflammation by promoting microglial reactivity and inflammatory signaling within the brain.
Notably, recent studies have identified the histamine H1 receptor (HRH1) as a potential alternative receptor for SARS-CoV-2, capable of binding the viral spike protein and facilitating viral entry independently of ACE2[71]. HRH1 also appears to enhance ACE2-mediated infection by promoting co-localization of viral and host entry factors. HRH1 is highly expressed in the brain[72]. These findings suggest that histaminergic pathways may not only modulate immune responses but also participate directly in viral pathogenesis, potentially amplifying neuroinflammation and tissue injury in PCS.
Beyond immune functions, histamine also plays key roles as a central neuromodulator, influencing cognition, sleep-wake regulation, mood, and autonomic control[72,73]. Dysregulated histaminergic signaling, likely driven by chronic mast cell activity, can disrupt the balance of other neurotransmitter systems, including serotonin, dopamine, and norepinephrine[74]. As histamine dysregulation affects both emotional and cognitive functions, it might exacerbate symptoms associated with neuroinflammation and undermine overall neural stability, leading to a compounded burden on affected individuals. Supporting this notion, emerging evidence suggests that antihistamines may alleviate symptoms of PCS, particularly those associated with mast cell activation and histamine dysregulation[75].
3. Biomarker Evidence
3.1. Neuroimaging Biomarkers
Although no single neuroimaging biomarker can yet definitively capture neuroinflammation specifically, converging evidence from multiple modalities supports the presence of persistent, low-grade brain inflammation in PCS. Positron Emission Tomography (PET) using second-generation TSPO tracers (e.g., [¹⁸F]FEPPA, [¹¹C]PBR28) has revealed widespread glial reactivity in PCS patients, particularly in the anterior cingulate, striatum, thalamus, and basal ganglia[6,76]. These signals correlate with cognitive and neuropsychiatric symptoms, including psychomotor slowing, anhedonia, and memory impairment, positioning PET-derived neuroinflammatory markers as promising tools for future clinical stratification[77]. Based on the recent understanding of the 18 kDa translocator protein (TSPO) expression in human post-mortem studies[22,78], these studies may provide evidence of increased glia/immune cell density in patients with PCS. Similarly, elevated astrocyte activity has specifically been observed via MAO-B PET ligands (e.g., [¹¹C]SL25.1188)[79]. However, in this case, the links between PET and symptoms seem negative, hinting that increased astrocyte reactivity, as assessed by MAO-B binding might somehow be protective.
MRI-based methods offer complementary insights into neurovascular dysfunction. Dynamic contrast-enhanced MRI (DCE-MRI) has demonstrated BBB disruption in patients with “brain fog,” with increased permeability in frontal and temporal regions correlating with symptom severity and gray matter atrophy[29]. Reductions in glymphatic clearance, as measured by diffusion tensor imaging along perivascular spaces (DTI-ALPS) have similarly been reported[80].
Magnetic Resonance Spectroscopy (MRS) studies further reveal decreased N-acetylaspartate (NAA), decreased glutamate/glutamine (Glx), and increased choline levels - indicative of neuronal dysfunction and possibly inflammatory responses - associated with poorer cognitive and physical performance[81,82,83]. Notably, one recent study using magnetic resonance spectroscopic imaging thermometry has shown increased brain temperature - particularly in the left olfactory tubercle - following mild COVID-19 infection, supporting the presence of subtle neuroinflammatory changes[84].
Advanced diffusion MRI studies indicate widespread microstructural and network-level disruptions. Diffusion imaging consistently shows increased extracellular water and reduced neurite density in limbic, striatal, and frontoparietal regions, suggestive of inflammatory edema and axonal injury[85,86,87,88]. Finally, recent structural neuroimaging studies have identified enlargement of the choroid plexus in individuals with PCS, suggesting a potential role in ongoing neuroinflammatory processes[34,35]. Together, these modalities reveal the presence of distributed, yet subtle, neuroinflammatory processes in PCS, supporting the notion of a persistent, low-grade brain immune response with clinically meaningful consequences.
While neuroimaging studies provide converging support for ongoing brain inflammation in PCS, limitations remain. TSPO-PET tracers, while widely used, lack cell-type specificity and may not distinguish microglial activation from other glial or endothelial responses[22,78]. Furthermore, variations in binding affinity across TSPO genotypes complicate group comparisons. Similarly, advanced MRI methods such as ALPS, free-water imaging, and MRS lack standardized protocols, and sample sizes remain small in many studies. Harmonized acquisition protocols and multicenter replication are essential to validate these neuroimaging biomarkers for clinical translation. Notably, not all studies have detected evidence of neuroinflammation in PCS. Some cohorts show minimal changes in TSPO-PET or MRS indices, and findings often vary by clinical phenotype or time since infection[77,80,88,89]. This heterogeneity underscores the need for phenotypically stratified analyses, larger sample sizes, and careful matching for confounders such as age, sex, baseline psychiatric risk, and severity of acute illness.
3.2. Soluble Biomarkers
Analyses of soluble biomarkers provide additional evidence implicating inflammation—including within the central nervous system— in the pathophysiology of PCS. Indeed, studies have shown elevated levels of interferon (IFN)-γ, IFN-β, C-X-C motif chemokine ligand 9 (CXCL9), and CXCL10 in the serum of PCS patients[90,91]. Similarly, a review revealed patients with PCS can be differentiated from asymptomatic controls with high accuracy based on cytokine levels, including IFN-β, pentraxin 3 - PTX3, IFN-β, IFN-γ, IFN-λ2/3, interleukin-6 - IL-6, CXCL9, CXCL10, IL-5, IL-8, IL-9, IL-12, IL-13, IL-33, and tumor necrosis factor-alpha (TNF-α)[92].
These results were confirmed by a recent meta-analysis exploring individual levels of 58 cytokines and chemokines[93]. Overall results showed an increase in IL-1β, IL-6, IL-8, TNF-α, CXCL1, CXCL10, and chemokine (C-C motif) ligand 3 in patients with PCS when compared to controls. Interestingly, the authors also found the presence of higher levels of vascular mediators, including vascular endothelial growth factor (VEGF), vascular cell adhesion protein-1 (VCAM-1), and epidermal growth factor (EGF) in these patients. These alterations in the vascular environment might be a consequence of the increased inflammation and can lead to the promotion of cell adhesion and pro-coagulation, further contributing to neuroinflammatory processes. This idea is supported by one recent study identifying positive links between peripheral concentrations of many of these vascular factors (including fibrinogen, α2-macroglobulin, orosomucoid (alpha-1-acid glycoprotein or AGP), fetuin-A, sL-selectin (soluble leukocyte selectin, or sCD62L), pentraxin-2 (serum amyloid P component, or SAP) and haptoglobin) and increased TSPO binding in the brain of patients[6].
Additionally, markers indicating glial activation, such as glial fibrillary acidic protein (GFAP) and soluble triggering receptor expressed on myeloid cells 2 (sTREM2), are frequently elevated in PCS patients, reflecting glial cell involvement in sustained inflammatory processes[89,94,95]. In particular, the higher levels of GFAP detected in these patients provide further support for the hypothesis suggesting the intricate involvement of the CNS in the pathophysiology of the syndrome[94]. Of interest, these findings are in line with previous research that has also observed increased GFAP levels in PCS patients, without a concurrent increase in other neural injury biomarkers, therefore reinforcing the notion that GFAP may serve as a possible indicator of CNS involvement in the context of PCS[96]. Furthermore, significant associations between elevated GFAP levels and cognitive decline have been reported in these patients[97]. Considering the above, it is possible to conclude that identifying and validating these biomarkers is essential for diagnosing and monitoring disease progression, as well as for therapeutic response in PCS patients.
4. Neuroinflammation, Brain Dysfunction, and the Non-linear Relapse-Remitting Time-Course of Symptoms: A Speculative Biologically Grounded Mechanistic Model
In this section, we propose a speculative yet biologically grounded framework that aims to guide hypothesis-driven investigation of how neuroinflammation may contribute to the fluctuating and heterogeneous symptoms of PCS. While empirical evidence continues to accumulate, the mechanisms linking immune dysregulation to persistent cognitive, affective, and neurological disturbances remain incompletely understood[98,99]. Here, we synthesize findings across molecular, cellular, and systems neuroscience to outline a speculative model that may guide future research.
Chronic low-grade neuroinflammation is increasingly implicated in PCS and may underpin symptom persistence and non-linear progression. Clinical reports often describe a relapsing-remitting course, with episodes of apparent improvement punctuated by flares following infection, stress, exertion, or disrupted sleep. We hypothesize that such flares reflect transient reactivations of systemic or neuroinflammatory cascades, which destabilize already vulnerable neural circuits - particularly those with altered excitatory–inhibitory (E/I) balance and constrained metabolic resilience. Repeated episodes may entrench maladaptive brain states, increasing the likelihood of chronicity.
At the molecular level, elevated levels of pro-inflammatory cytokines - especially IL-6, IL-1β, and TNF-α - interact with microglia and astrocytes to drive neuroimmune activation[98,99,100]. These processes perturb neuromodulatory systems, including glutamate, GABA, dopamine, serotonin, and histamine, thereby disrupting neurotransmission and plasticity[101]. The resulting imbalance may impair synaptic remodeling, dendritic stability, and long-term potentiation - processes essential for adaptive cognitive and emotional function.
A critical, though often overlooked, downstream effect of this neuroimmune environment may be the induction of “local sleep”: a phenomenon in which small populations of cortical neurons enter a sleep-like, silent state during wakefulness[102,103]. This has been observed in response to metabolic stress and adenosine accumulation - both of which are potentiated by inflammatory cytokines[103,104]. In PCS, such local sleep intrusions may occur preferentially in high-demand regions involved in attention, working memory, interoception, and emotional regulation, giving rise to “brain fog,” attentional lapses, and slowed thought. Moreover, these local offline states may impair neural replay and memory reconsolidation, a neuroplastic process that updates stored memories upon retrieval[105]. If task-relevant neuronal ensembles are intermittently silenced or inhibited by inflammation, this may result in the persistence of outdated or emotionally dysregulated memories - contributing to anxiety, emotional lability, and distorted self-perception. Over time, local disruptions may propagate to large-scale neural networks, particularly those involving the medial prefrontal cortex, anterior cingulate cortex, insula, and the default mode network[106,107]. These regions are integral to integrating sensory, cognitive, and emotional signals[108]. Their disconnection - possibly exacerbated by altered glial-neuronal coupling and neurotransmitter imbalance - may explain the complex symptom constellation seen in PCS, including chronic fatigue, anhedonia, dissociative experiences, and affective instability[109].
Each flare-up might weaken network coherence further, lowering the threshold for subsequent exacerbations. Through recurrent inflammation, cumulative adenosine burden, and impaired reconsolidation, a self-reinforcing loop may emerge in which recovery becomes increasingly difficult - pushing the brain toward a state of sustained dysfunction. This model remains speculative but integrates converging findings from basic and clinical research to generate testable hypotheses, which we discuss below in turn.
5. Therapeutic Implications and Opportunities for Drug Development/Repurposing
Given the central role of chronic neuroinflammation in PCS, targeted strategies to modulate immune pathways offer promising avenues for symptom relief and functional recovery. While general immunosuppressants - such as corticosteroids, IL-6 inhibitors, and JAK-STAT pathway blockers - are used in selected cases, emerging mechanistic insights point toward more precise interventions[110].
Persistent viral antigens in immune-privileged sites such as the meninges and skull bone marrow may drive ongoing inflammation. Approaches under investigation include antiviral agents[111,112], immunotherapies aimed at viral clearance[113], and techniques to enhance meningeal lymphatic drainage to promote immune clearance from the CNS[114]. The skull–marrow–meninges axis, in particular, represents a novel therapeutic target in post-infectious neuroinflammation[114]. Stabilizing the BBB and protecting the NVU are also promising strategies. Endothelial-protective therapies (i.e., defibotide), antioxidants (i.e., flavonoids), and agents that restore tight junction integrity could reduce peripheral immune cell infiltration and mitigate neurovascular inflammation[28,53]. Astrocyte modulators such as MAO-B inhibitors (e.g., selegiline)[115], fingolimod[116], ibudilast[117], and palmitoylethanolamide (PEA)[118] may reduce glial reactivity and cytokine release while preserving synaptic function. Targeting astroglial dysfunction offers a direct way to counteract central neuroinflammation and excitotoxicity. Given growing evidence implicating mast cells and histamine dysregulation, interventions like H1/H2 blockers, mast cell stabilizers (e.g., cromolyn[119], ketotifen[120]), and selective H3/H4 receptor antagonists hold promise[121]. Notably, H4R antagonists reduce microglial activation and neuroinflammation in preclinical models and merit exploration in PCS-specific trials[122].
Non-pharmacological neuromodulatory strategies, particularly vagal nerve stimulation (VNS), have shown the potential to dampen systemic and central immune responses via the cholinergic anti-inflammatory reflex[123,124]. VNS has a strong safety profile and may be especially useful in reducing the frequency and severity of PCS flares[124]. These approaches are most effective when integrated into multidisciplinary rehabilitation programs that combine physical therapy, cognitive-behavioral interventions, autonomic regulation, and lifestyle adjustments. Tailoring such programs to the biology of neuroinflammation and the fluctuating symptom profile of PCS can help restore cognitive function, support memory reconsolidation, and alleviate fatigue.
6. Outstanding Questions and Directions for Further Research
Despite recent efforts, several outstanding questions remain concerning the role of neuroinflammation in PCS, requiring focused, multidisciplinary research. Our proposed mechanistic model - linking chronic inflammation to local sleep intrusions, reconsolidation impairments, adenosine dysregulation, and cyclical symptom flares - highlights novel and largely unexplored domains that deserve urgent attention. These additions complement established research directions and collectively form a comprehensive roadmap for advancing the field. Key areas include elucidating the specific neuroimmune pathways activated during persistent inflammation, particularly those that influence regional brain activity and contribute to phenomena such as local sleep. Although cytokines such as IL-1β and TNF-α are known to modulate sleep pressure and adenosine signaling, how they trigger focal, sleep-like silencing of cortical microcircuits during wakefulness remains unclear. Targeted neurophysiological and imaging studies could help characterize this phenomenon in PCS and establish its relevance to cognitive dysfunction.
The influence of initial COVID-19 disease severity on long-term neuroinflammatory profiles is another area warranting clarification. Stratified, longitudinal biomarker and imaging studies are needed to determine whether more severe acute illness leads to greater chronic neuroimmune activation and a higher risk of PCS[6]. Moreover, the temporal dynamics of inflammation - from acute infection through long-term recovery - are still not well-defined. A better understanding of the time course of cytokine and glial responses, along with their interaction with metabolic stress and neuromodulatory tone, could identify windows of vulnerability where intervention may be most effective[125]. Genetic and epigenetic contributions to individual susceptibility remain underexplored. Investigating whether certain host factors increase the likelihood of persistent neuroinflammation could help explain the heterogeneity in PCS presentations[126]. These efforts will benefit from large-scale genomic and epigenomic datasets linked to longitudinal phenotyping. Identifying reliable neuroinflammatory biomarkers that predict long-term neurological and psychiatric symptoms, and mapping them onto distinct symptom clusters using advanced neuroimaging, remains an essential priority to refine diagnosis and personalize care.
Therapeutically, there is a critical need for clinical trials evaluating immunomodulatory interventions, especially those targeting microglial and astrocytic activation in vulnerable cortical regions. These trials should also consider agents that modulate adenosine signaling or support metabolic resilience, as these may mitigate the emergence of local sleep intrusions and restore circuit stability. In parallel, more research is needed to understand how flare cycles - defined by transient worsening of symptoms - are biologically triggered and whether they reflect surges in neuroinflammatory or purinergic activity[104]. Time-resolved biomarkers, wearable sensors, and digital phenotyping may be useful tools for detecting these flare states in real-time. Furthermore, evaluating rehabilitation strategies tailored to neuroinflammatory symptoms remains vital. Interventions that take into account the fluctuating nature of PCS symptoms - linked to dynamic changes in brain state - could improve recovery and reduce relapses. Cognitive and behavioral therapies should be tested for their capacity to enhance reconsolidation, restore attentional control, and regulate interoceptive awareness[127].
Ultimately, the development of integrated, multidisciplinary care models bringing together neurology, psychiatry, immunology, sleep medicine, and rehabilitation sciences will be essential to optimizing patient outcomes. These models must be informed by evolving mechanistic insights and grounded in personalized, adaptive care approaches[128].
Figure 1.
Microscopic evidence supports multiple mechanisms of CNS involvement in post-COVID-19 syndrome (PCS). SARS-CoV-2 can invade the brain via olfactory pathways, with viral proteins (spike, nucleocapsid) detected in neurons (a) and glial cells. Markers of viral replication (dsRNA) have been observed in both in vitro and in vivo models. COVID-19 also induces microglial proliferation and activation, particularly in the olfactory cortex (b). Elevated peripheral cytokines may disrupt the BBB, as shown by astrocytic markers in the neurovascular unit (c), increasing vulnerability to neuroinflammation. Micrographs are from a non-human primate model developed at the California National Primate Research Center[19]. We note that similar evidene of glial reactivity exist in humans but the detection of direct invasion of human brain tissue might be challenging mostly because the postmortem brain interval is often too long for keeping viral proteins viable for detection[9,10].
Figure 1.
Microscopic evidence supports multiple mechanisms of CNS involvement in post-COVID-19 syndrome (PCS). SARS-CoV-2 can invade the brain via olfactory pathways, with viral proteins (spike, nucleocapsid) detected in neurons (a) and glial cells. Markers of viral replication (dsRNA) have been observed in both in vitro and in vivo models. COVID-19 also induces microglial proliferation and activation, particularly in the olfactory cortex (b). Elevated peripheral cytokines may disrupt the BBB, as shown by astrocytic markers in the neurovascular unit (c), increasing vulnerability to neuroinflammation. Micrographs are from a non-human primate model developed at the California National Primate Research Center[19]. We note that similar evidene of glial reactivity exist in humans but the detection of direct invasion of human brain tissue might be challenging mostly because the postmortem brain interval is often too long for keeping viral proteins viable for detection[9,10].
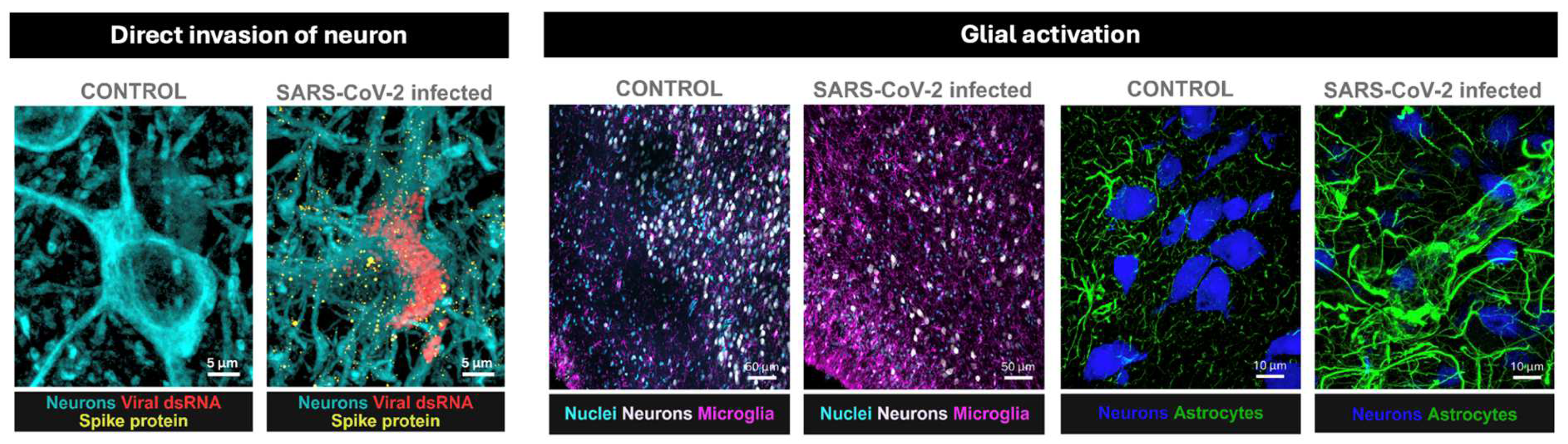
Figure 2.
Multilevel pathways of neuroinflammation in post-COVID-19 syndrome (PCS). This schematic illustrates the key anatomical and cellular routes likely contributing to neuroinflammation in PCS. The left panels show how peripheral immune activation—including vagus nerve signaling and cytokine release—impacts brain structures such as the choroid plexus and meninges. The top-left inset highlights immune traffic through skull–meninges channels. The central panel emphasizes the role of blood–brain and blood–CSF barrier dysfunction, enabling immune cell infiltration, cytokine diffusion, and autoantibody entry into the CNS. Right panels illustrate microglial and astrocyte activation, neuronal degeneration, and the transition from healthy to diseased states (orange to grey). Together, these mechanisms converge to sustain a chronic pro-inflammatory environment in the brain.
Figure 2.
Multilevel pathways of neuroinflammation in post-COVID-19 syndrome (PCS). This schematic illustrates the key anatomical and cellular routes likely contributing to neuroinflammation in PCS. The left panels show how peripheral immune activation—including vagus nerve signaling and cytokine release—impacts brain structures such as the choroid plexus and meninges. The top-left inset highlights immune traffic through skull–meninges channels. The central panel emphasizes the role of blood–brain and blood–CSF barrier dysfunction, enabling immune cell infiltration, cytokine diffusion, and autoantibody entry into the CNS. Right panels illustrate microglial and astrocyte activation, neuronal degeneration, and the transition from healthy to diseased states (orange to grey). Together, these mechanisms converge to sustain a chronic pro-inflammatory environment in the brain.
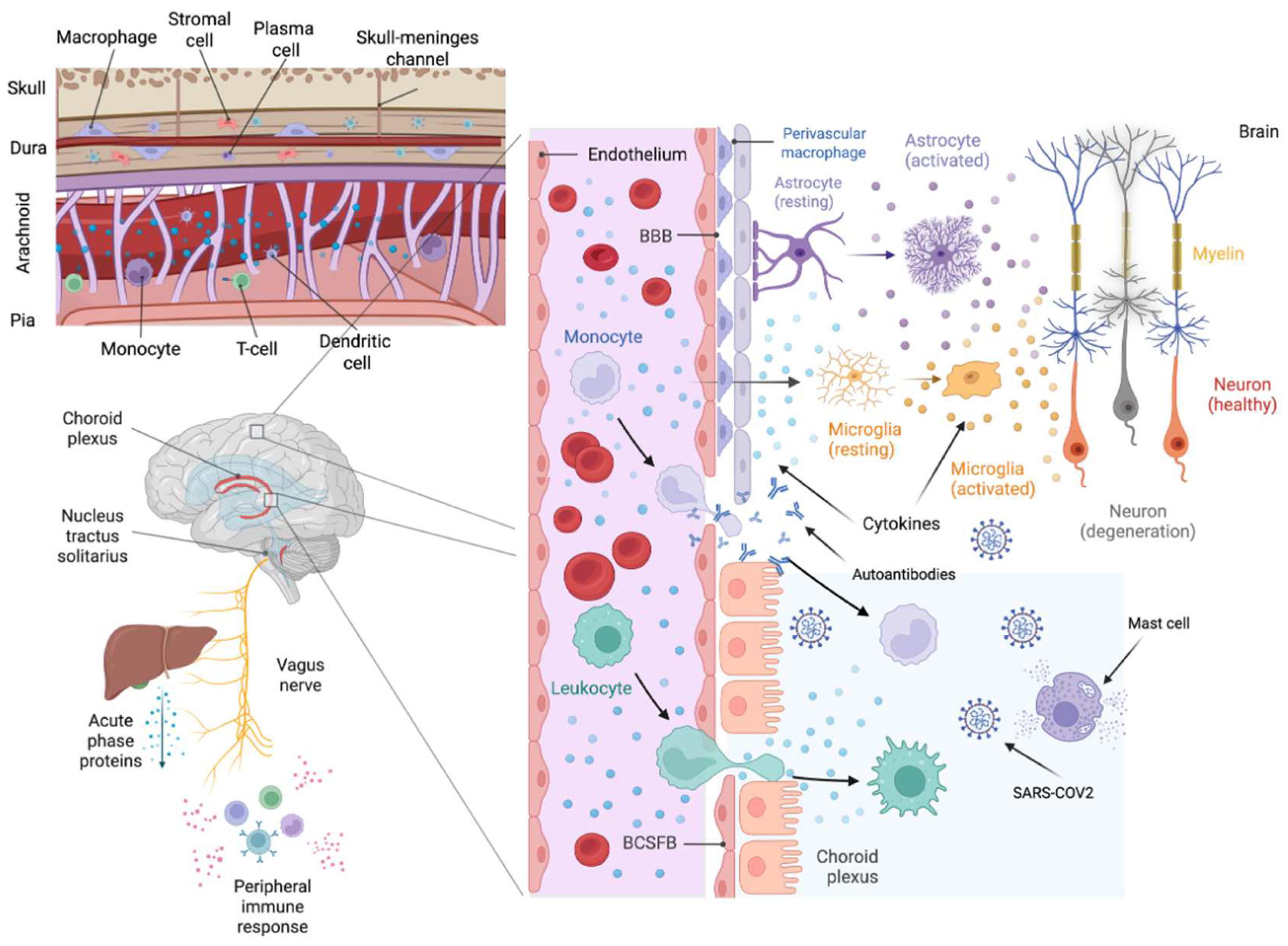
Figure 3.
A dynamic multiscale model of neuroinflammation and symptom exacerbation in PCS. This conceptual model proposes how systemic triggers (e.g., infection, exertion, stress, sleep disruption) reactivate neuroinflammatory cascades in vulnerable individuals. Pro-inflammatory cytokines (e.g., IL-1β, TNF-α) and glial activation contribute to local neuronal silencing (offline activity) during wakefulness, which disrupts replay and memory reconsolidation processes. These localized disturbances impair global functional connectivity across brain networks, ultimately manifesting as fluctuating neuropsychiatric symptoms, including cognitive lapses, emotional dysregulation, and fatigue. The model highlights recursive interactions between inflammation, brain network instability, and symptom re-emergence in PCS.
Figure 3.
A dynamic multiscale model of neuroinflammation and symptom exacerbation in PCS. This conceptual model proposes how systemic triggers (e.g., infection, exertion, stress, sleep disruption) reactivate neuroinflammatory cascades in vulnerable individuals. Pro-inflammatory cytokines (e.g., IL-1β, TNF-α) and glial activation contribute to local neuronal silencing (offline activity) during wakefulness, which disrupts replay and memory reconsolidation processes. These localized disturbances impair global functional connectivity across brain networks, ultimately manifesting as fluctuating neuropsychiatric symptoms, including cognitive lapses, emotional dysregulation, and fatigue. The model highlights recursive interactions between inflammation, brain network instability, and symptom re-emergence in PCS.
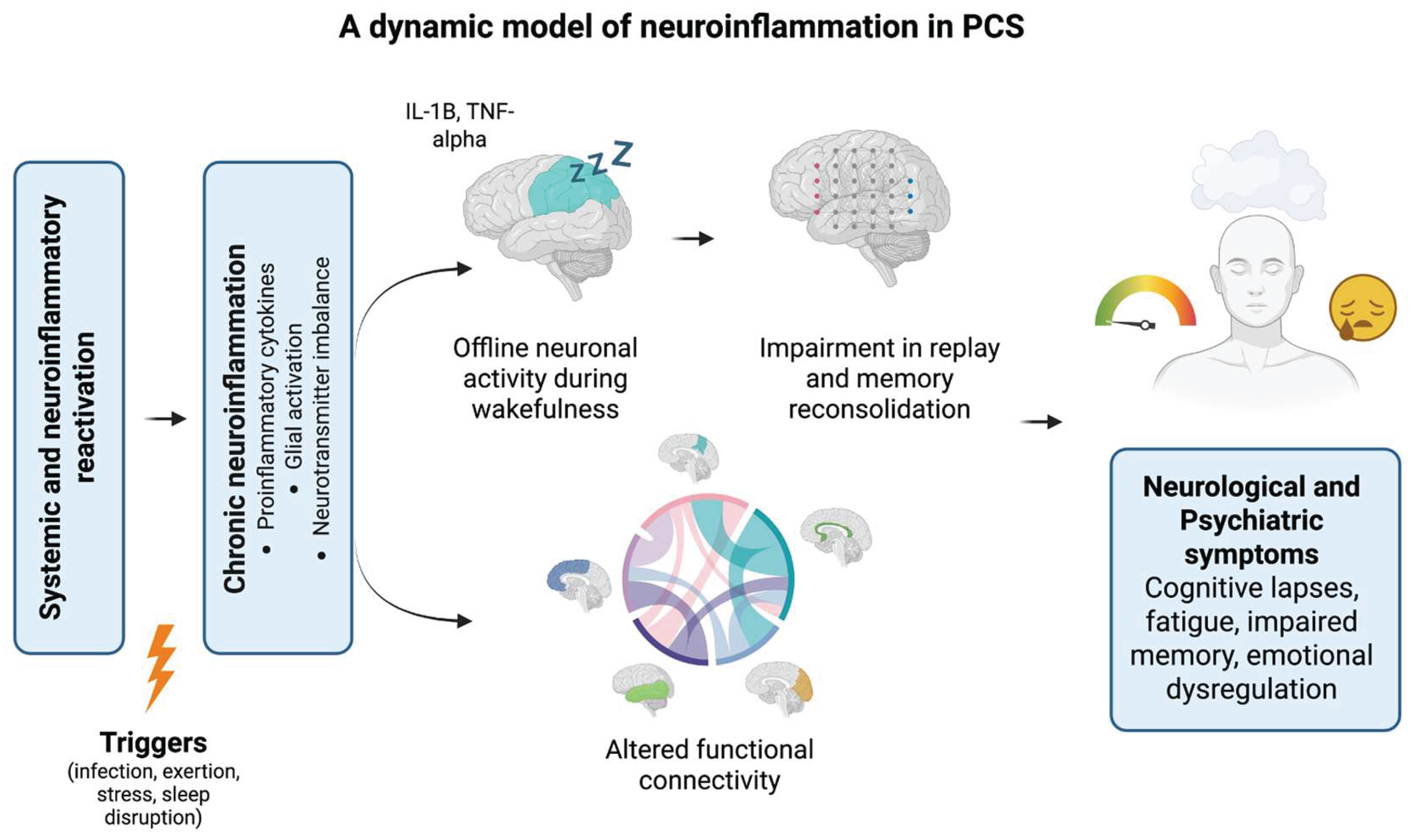

Table 1.
Ten outstanding questions on neuroinflammation in PCS and suggested research approaches.
|
Outstanding Questions on Neuroinflammation in PCS |
Suggested Research Approaches |
|---|---|
| 1. What specific neuroimmune pathways are activated during persistent neuroinflammation in post-COVID-19 syndrome, and do they induce local sleep in cortical microcircuits? | Multi-omics analyses; animal models; EEG-fMRI studies of local sleep under inflammation |
| 2. How do neuroinflammatory profiles differ based on initial COVID-19 severity? | Stratified longitudinal studies using biomarkers and neuroimaging |
| 3. What is the progression and resolution timeline of neuroinflammatory markers post-infection, and how does this relate to symptom fluctuation and flare cycles? | Longitudinal observational cohorts with time-resolved biomarker sampling and digital phenotyping |
| 4. Which neuroinflammatory biomarkers predict persistent neurological and cognitive symptoms, including those linked to reconsolidation failure or fatigue? | Prospective cohort studies and biomarker validation trials |
| 5. Are genetic or epigenetic markers associated with susceptibility to neuroinflammation, local sleep phenomena, or reconsolidation impairments in PCS? | Genome-wide association studies (GWAS); epigenetic analyses linked to cognitive profiles |
| 6. Can neuroimaging reliably differentiate neuroinflammatory patterns and local sleep signatures linked to specific symptom clusters? | Functional and structural neuroimaging studies (MRI, PET, mobile EEG) |
| 7. Which immunomodulatory therapies most effectively reduce neuroinflammatory markers, support metabolic resilience, and stabilize network function? | Randomized controlled clinical trials; mechanistically informed pharmacological studies |
| 8. Can targeting glial activation and adenosine signaling reduce sustained neuroinflammation and associated cognitive or emotional symptoms? | Preclinical experiments; clinical trials of astrocyte/microglia and purinergic-targeted therapies |
| 9. Which rehabilitation strategies most effectively support circuit reintegration, reconsolidation processes, and resilience to flare cycles? | Controlled trials of cognitive-behavioral and neuroplasticity-based rehabilitation approaches |
| 10. How can integrated multidisciplinary care models dynamically respond to fluctuating brain states and optimize long-term recovery in PCS? | Clinical pathway optimization involving neurology, psychiatry, immunology, and rehabilitation |
7. Conclusions
Post-COVID-19 syndrome (PCS) presents a complex and evolving public health challenge, characterized by persistent neurological, cognitive, and psychiatric symptoms. Growing evidence implicates chronic neuroinflammation as a central driver of these long-term sequelae. Our proposed multiscale model suggests that sustained immune activation disrupts neurotransmitter balance, impairs synaptic and network plasticity, and gives rise to phenomena such as local sleep intrusions, memory reconsolidation failure, and episodic symptom flares - offering a novel framework for understanding the fluctuating and heterogeneous nature of PCS. Future research must focus on clarifying the interplay between inflammation, energy metabolism, and brain network stability. Translating these insights into reliable biomarkers and targeted therapies - particularly those addressing glial activation, purinergic signaling, and reconsolidation mechanisms - will be essential. Personalized rehabilitation strategies tailored to the dynamic neurobiological state of each patient may also enhance recovery. Meeting this challenge will require sustained, cross-disciplinary collaboration among neurologists, psychiatrists, immunologists, sleep researchers, and rehabilitation specialists. Only through such integrated efforts can we advance diagnostic precision, therapeutic innovation, and long-term outcomes for individuals living with PCS.
Conflict of Interests
Nothing to declare.
Author Contributions
Daniel Martins: Conceptualization, Writing—Original Draft, Supervision, Funding acquisition. Danielle Beckman: Writing—Review & Editing, Visualization. Marco Loggia: Writing—Review & Editing. Alessandra Costanza: Writing—Review & Editing. Alessandra Borsini: Writing—Review & Editing.
Acknowledgments
DM is supported by the NIHR Maudsley Biomedical Research Centre, South London, and Maudsley NHS Foundation Trust. MLL is supported by 3R01DA047088–05S1.
References
- Li, J.; Zhou, Y.; Ma, J.; Zhang, Q.; Shao, J.; Liang, S.; Yu, Y.; Li, W.; Wang, C. The long-term health outcomes, pathophysiological mechanisms and multidisciplinary management of long COVID. Signal Transduct Target Ther 2023, 8, 416. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Su, S.; Zhao, Y.; Zeng, N.; Liu, X.; Zheng, Y.; Sun, J.; Zhong, Y.; Wu, S.; Ni, S.; Gong, Y. , et al. Epidemiology, clinical presentation, pathophysiology, and management of long COVID: an update. Mol Psychiatry 2023, 28, 4056–4069. [Google Scholar] [CrossRef]
- Shah, W.; Hillman, T.; Playford, E.D.; Hishmeh, L. Managing the long term effects of covid-19: summary of NICE, SIGN, and RCGP rapid guideline. BMJ 2021, 372, n136. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.B.; Murthy, S.; Marshall, J.C.; Relan, P.; Diaz, J.V.; Condition, W.H.O.C.C.D.W.G.o.P.-C.-. . A clinical case definition of post-COVID-19 condition by a Delphi consensus. Lancet Infect Dis 2022, 22, e102–e107. [Google Scholar] [CrossRef]
- VanElzakker, M.B.; Bues, H.F.; Brusaferri, L.; Kim, M.; Saadi, D.; Ratai, E.M.; Dougherty, D.D.; Loggia, M.L. Neuroinflammation in post-acute sequelae of COVID-19 (PASC) as assessed by [(11)C]PBR28 PET correlates with vascular disease measures. Brain Behav Immun 2024, 119, 713–723. [Google Scholar] [CrossRef]
- Dos Reis, R.S.; Selvam, S.; Ayyavoo, V. Neuroinflammation in Post COVID-19 Sequelae: Neuroinvasion and Neuroimmune Crosstalk. Rev Med Virol 2024, 34, e70009. [Google Scholar] [CrossRef]
- Bergmann, C.C.; Lane, T.E.; Stohlman, S.A. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol 2006, 4, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S. , et al. Multiorgan and Renal Tropism of SARS-CoV-2. N Engl J Med 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S. , et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci 2021, 24, 168–175. [Google Scholar] [CrossRef]
- Dube, M.; Le Coupanec, A.; Wong, A.H.M.; Rini, J.M.; Desforges, M.; Talbot, P.J. Axonal Transport Enables Neuron-to-Neuron Propagation of Human Coronavirus OC43. J Virol 2018, 92. [Google Scholar] [CrossRef]
- Awogbindin, I.O.; Ben-Azu, B.; Olusola, B.A.; Akinluyi, E.T.; Adeniyi, P.A.; Di Paolo, T.; Tremblay, M.E. Microglial Implications in SARS-CoV-2 Infection and COVID-19: Lessons From Viral RNA Neurotropism and Possible Relevance to Parkinson's Disease. Front Cell Neurosci 2021, 15, 670298. [Google Scholar] [CrossRef]
- Guerrero, J.I.; Barragan, L.A.; Martinez, J.D.; Montoya, J.P.; Pena, A.; Sobrino, F.E.; Tovar-Spinoza, Z.; Ghotme, K.A. Central and peripheral nervous system involvement by COVID-19: a systematic review of the pathophysiology, clinical manifestations, neuropathology, neuroimaging, electrophysiology, and cerebrospinal fluid findings. BMC Infect Dis 2021, 21, 515. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Streit, S.; Dittmayer, C.; Manitius, R.V.; Radbruch, H.; Heppner, F.L. The neurobiology of SARS-CoV-2 infection. Nat Rev Neurosci 2024, 25, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Mai, H.; Ebert, G.; Kapoor, S.; Puelles, V.G.; Czogalla, J.; Hu, S.; Su, J.; Prtvar, D.; Singh, I. , et al. Persistence of spike protein at the skull-meninges-brain axis may contribute to the neurological sequelae of COVID-19. Cell Host Microbe 2024, 32, 2112–2130. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, X.; Chai, Y.; Zhang, J.; Deng, Q.; Chen, X. Skull bone marrow and skull meninges channels: redefining the landscape of central nervous system immune surveillance. Cell Death Dis 2025, 16, 53. [Google Scholar] [CrossRef]
- Fodoulian, L.; Tuberosa, J.; Rossier, D.; Boillat, M.; Kan, C.; Pauli, V.; Egervari, K.; Lobrinus, J.A.; Landis, B.N.; Carleton, A. , et al. SARS-CoV-2 Receptors and Entry Genes Are Expressed in the Human Olfactory Neuroepithelium and Brain. iScience 2020, 23, 101839. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019: A Review. JAMA Neurol 2020, 77, 1018–1027. [Google Scholar] [CrossRef]
- Beckman, D.; Bonillas, A.; Diniz, G.B.; Ott, S.; Roh, J.W.; Elizaldi, S.R.; Schmidt, B.A.; Sammak, R.L.; Van Rompay, K.K.A.; Iyer, S.S. , et al. SARS-CoV-2 infects neurons and induces neuroinflammation in a non-human primate model of COVID-19. Cell Rep 2022, 41, 111573. [Google Scholar] [CrossRef]
- Samudyata; Oliveira, A. O.; Malwade, S.; Rufino de Sousa, N.; Goparaju, S.K.; Gracias, J.; Orhan, F.; Steponaviciute, L.; Schalling, M.; Sheridan, S.D., et al. SARS-CoV-2 promotes microglial synapse elimination in human brain organoids. Mol Psychiatry 2022, 27, 3939–3950. [Google Scholar] [CrossRef]
- Jeong, G.U.; Lyu, J.; Kim, K.D.; Chung, Y.C.; Yoon, G.Y.; Lee, S.; Hwang, I.; Shin, W.H.; Ko, J.; Lee, J.Y. , et al. SARS-CoV-2 Infection of Microglia Elicits Proinflammatory Activation and Apoptotic Cell Death. Microbiol Spectr 2022, 10, e0109122. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, S.S.; Rowe, J.B.; Mason, H.D.; Allinson, K.S.J.; Thomas, R.; Vontobel, D.S.; Fryer, T.D.; Hong, Y.T.; Bacioglu, M.; Spillantini, M.G. , et al. Post-mortem validation of in vivo TSPO PET as a microglial biomarker. Brain, 1093. [Google Scholar]
- Tavcar, P.; Potokar, M.; Kolenc, M.; Korva, M.; Avsic-Zupanc, T.; Zorec, R.; Jorgacevski, J. Neurotropic Viruses, Astrocytes, and COVID-19. Front Cell Neurosci 2021, 15, 662578. [Google Scholar] [CrossRef] [PubMed]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.; Vendramini, P.H.; Valenca, A.G.F.; Brandao-Teles, C.; Zuccoli, G.D.S. , et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc Natl Acad Sci U S A 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Andrews, M.G.; Mukhtar, T.; Eze, U.C.; Simoneau, C.R.; Ross, J.; Parikshak, N.; Wang, S.; Zhou, L.; Koontz, M.; Velmeshev, D. , et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc Natl Acad Sci U S A 2022, 119, e2122236119. [Google Scholar] [CrossRef]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R. , et al. Neuropilin-1 Mediates SARS-CoV-2 Infection of Astrocytes in Brain Organoids, Inducing Inflammation Leading to Dysfunction and Death of Neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef] [PubMed]
- Schwabenland, M.; Salie, H.; Tanevski, J.; Killmer, S.; Lago, M.S.; Schlaak, A.E.; Mayer, L.; Matschke, J.; Puschel, K.; Fitzek, A. , et al. Deep spatial profiling of human COVID-19 brains reveals neuroinflammation with distinct microanatomical microglia-T-cell interactions. Immunity 2021, 54, 1594–1610. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell Mol Immunol 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Greene, C.; Connolly, R.; Brennan, D.; Laffan, A.; O'Keeffe, E.; Zaporojan, L.; O'Callaghan, J.; Thomson, B.; Connolly, E.; Argue, R. , et al. Blood-brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat Neurosci 2024, 27, 421–432. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q. , et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther 2021, 6, 337. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Muller-Fielitz, H.; Schuster, R.; Zille, M.; Muller, K.; Krohn, M.; Korbelin, J.; Zhang, L.; Ozorhan, U. , et al. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Steiner, J.; Pasternack, N.; Li, W.; Maric, D.; Safavi, F.; Horkayne-Szakaly, I.; Jones, R.; Stram, M.N. , et al. Neurovascular injury with complement activation and inflammation in COVID-19. Brain 2022, 145, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hehnly, C.; Lehtinen, M.K. The choroid plexus: a command center for brain-body communication during inflammation. Curr Opin Immunol 2025, 93, 102540. [Google Scholar] [CrossRef]
- Rau, A.; Gonzalez-Escamilla, G.; Schroeter, N.; Othman, A.; Dressing, A.; Weiller, C.; Urbach, H.; Reisert, M.; Groppa, S.; Hosp, J.A. Inflammation-Triggered Enlargement of Choroid Plexus in Subacute COVID-19 Patients with Neurological Symptoms. Ann Neurol 2024, 96, 715–725. [Google Scholar] [CrossRef]
- Diez-Cirarda, M.; Yus-Fuertes, M.; Delgado-Alonso, C.; Gil-Martinez, L.; Jimenez-Garcia, C.; Gil-Moreno, M.J.; Gomez-Ruiz, N.; Oliver-Mas, S.; Polidura, C.; Jorquera, M. , et al. Choroid plexus volume is enlarged in long COVID and associated with cognitive and brain changes. Mol Psychiatry, 1038. [Google Scholar]
- Mazzitelli, J.A.; Pulous, F.E.; Smyth, L.C.D.; Kaya, Z.; Rustenhoven, J.; Moskowitz, M.A.; Kipnis, J.; Nahrendorf, M. Skull bone marrow channels as immune gateways to the central nervous system. Nat Neurosci 2023, 26, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Castilla, J.; Stebliankin, V.; Baral, P.; Balbin, C.A.; Sobhan, M.; Cickovski, T.; Mondal, A.M.; Narasimhan, G.; Chapagain, P.; Mathee, K. , et al. Potential Autoimmunity Resulting from Molecular Mimicry between SARS-CoV-2 Spike and Human Proteins. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Santos Guedes de Sa, K.; Silva, J.; Bayarri-Olmos, R.; Brinda, R.; Alec Rath Constable, R.; Colom Diaz, P.A.; Kwon, D.I.; Rodrigues, G.; Wenxue, L.; Baker, C. , et al. A causal link between autoantibodies and neurological symptoms in long COVID. medRxiv, 1101. [Google Scholar]
- Hansen, N. Psychiatric Symptoms in Acute and Persisting Forms of COVID-19 Associated with Neural Autoantibodies. Antibodies (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Seibert, F.S.; Stervbo, U.; Wiemers, L.; Skrzypczyk, S.; Hogeweg, M.; Bertram, S.; Kurek, J.; Anft, M.; Westhoff, T.H.; Babel, N. Severity of neurological Long-COVID symptoms correlates with increased level of autoantibodies targeting vasoregulatory and autonomic nervous system receptors. Autoimmun Rev 2023, 22, 103445. [Google Scholar] [CrossRef] [PubMed]
- Kitsou, K.; Kotanidou, A.; Paraskevis, D.; Karamitros, T.; Katzourakis, A.; Tedder, R.; Hurst, T.; Sapounas, S.; Kotsinas, A.; Gorgoulis, V. , et al. Upregulation of Human Endogenous Retroviruses in Bronchoalveolar Lavage Fluid of COVID-19 Patients. Microbiol Spectr 2021, 9, e0126021. [Google Scholar] [CrossRef]
- Gimenez-Orenga, K.; Pierquin, J.; Brunel, J.; Charvet, B.; Martin-Martinez, E.; Perron, H.; Oltra, E. HERV-W ENV antigenemia and correlation of increased anti-SARS-CoV-2 immunoglobulin levels with post-COVID-19 symptoms. Front Immunol 2022, 13, 1020064. [Google Scholar] [CrossRef]
- Douville, R.N.; Nath, A. Human endogenous retroviruses and the nervous system. Handb Clin Neurol 2014, 123, 465–485. [Google Scholar]
- Brunel, J.; Paganini, J.; Galloux, M.; Charvet, B.; Perron, H. HERV-W ENV transcription in B cells predicting symptomatic COVID-19 and risk for long COVID can express a full-length protein despite stop codon in mRNA from chromosome X via a ribosome readthrough. Microbes Infect, 1016. [Google Scholar]
- Gruchot, J.; Reiche, L.; Werner, L.; Herrero, F.; Schira-Heinen, J.; Meyer, U.; Kury, P. Molecular dissection of HERV-W dependent microglial- and astroglial cell polarization. Microbes Infect, 1016. [Google Scholar]
- Gruchot, J.; Lewen, I.; Dietrich, M.; Reiche, L.; Sindi, M.; Hecker, C.; Herrero, F.; Charvet, B.; Weber-Stadlbauer, U.; Hartung, H.P. , et al. Transgenic expression of the HERV-W envelope protein leads to polarized glial cell populations and a neurodegenerative environment. Proc Natl Acad Sci U S A 2023, 120, e2308187120. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Brunel, J.; Pierquin, J.; Iampietro, M.; Decimo, D.; Queruel, N.; Lucas, A.; Encabo-Berzosa, M.D.M.; Arenaz, I.; Marmolejo, T.P. , et al. SARS-CoV-2 awakens ancient retroviral genes and the expression of proinflammatory HERV-W envelope protein in COVID-19 patients. iScience 2023, 26, 106604. [Google Scholar] [CrossRef] [PubMed]
- Josephs, S.F.; Henry, B.; Balachandran, N.; Strayer, D.; Peterson, D.; Komaroff, A.L.; Ablashi, D.V. HHV-6 reactivation in chronic fatigue syndrome. Lancet 1991, 337, 1346–1347. [Google Scholar] [CrossRef]
- Rousseau, B.A.; Bhaduri-McIntosh, S. Inflammation and Epstein-Barr Virus at the Crossroads of Multiple Sclerosis and Post-Acute Sequelae of COVID-19 Infection. Viruses 2023, 15. [Google Scholar] [CrossRef]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat Neurosci 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Kotze, M.J.; Oladejo, S.O.; Watson, L.R.; Rajaratnam, K.; Watson, B.W.; Kell, D.B. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc Diabetol 2022, 21, 148. [Google Scholar] [CrossRef]
- Fekete, M.; Lehoczki, A.; Szappanos, A.; Toth, A.; Mahdi, M.; Sotonyi, P.; Benyo, Z.; Yabluchanskiy, A.; Tarantini, S.; Ungvari, Z. Cerebromicrovascular mechanisms contributing to long COVID: implications for neurocognitive health. Geroscience 2025, 47, 745–779. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Endothelial inflammation in COVID-19. Science 2024, 386, 972–973. [Google Scholar] [CrossRef]
- Ikemizu, Y.; Oda, Y.; Hirose, Y.; Sasaki, T.; Iyo, M. Cerebellar and Occipital Alterations in Brain Perfusion in a Patient With Post-acute COVID-19 Encephalopathy Misdiagnosed As Primary Psychotic Disorder. Cureus 2024, 16, e52953. [Google Scholar] [CrossRef]
- Chien, C.; Heine, J.; Khalil, A.; Schlenker, L.; Hartung, T.J.; Boesl, F.; Schwichtenberg, K.; Rust, R.; Bellmann-Strobl, J.; Franke, C. , et al. Altered brain perfusion and oxygen levels relate to sleepiness and attention in post-COVID syndrome. Ann Clin Transl Neurol 2024, 11, 2016–2029. [Google Scholar] [CrossRef]
- Adingupu, D.D.; Soroush, A.; Hansen, A.; Twomey, R.; Dunn, J.F. Brain hypoxia, neurocognitive impairment, and quality of life in people post-COVID-19. J Neurol 2023, 270, 3303–3314. [Google Scholar] [CrossRef] [PubMed]
- Szogi, T.; Borsos, B.N.; Masic, D.; Radics, B.; Bella, Z.; Banfi, A.; Ordog, N.; Zsiros, C.; Kiricsi, A.; Pankotai-Bodo, G. , et al. Novel biomarkers of mitochondrial dysfunction in Long COVID patients. Geroscience 2025, 47, 2245–2261. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hashita, T.; Yanagida, S.; Sato, H.; Yasuhiko, Y.; Okabe, K.; Noda, T.; Nishida, M.; Matsunaga, T.; Kanda, Y. SARS-CoV-2 causes dysfunction in human iPSC-derived brain microvascular endothelial cells potentially by modulating the Wnt signaling pathway. Fluids Barriers CNS 2024, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Gallowitsch-Puerta, M.; Pavlov, V.A. Neuro-immune interactions via the cholinergic anti-inflammatory pathway. Life Sci 2007, 80, 2325–2329. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Targeting the cholinergic anti-inflammatory pathway with vagus nerve stimulation in patients with Covid-19? Bioelectron Med 2020, 6, 15. [Google Scholar] [CrossRef]
- Llados, G.; Massanella, M.; Coll-Fernandez, R.; Rodriguez, R.; Hernandez, E.; Lucente, G.; Lopez, C.; Loste, C.; Santos, J.R.; Espana-Cueto, S. , et al. Vagus nerve dysfunction in the post-COVID-19 condition: a pilot cross-sectional study. Clin Microbiol Infect 2024, 30, 515–521. [Google Scholar] [CrossRef]
- Komegae, E.N.; Farmer, D.G.S.; Brooks, V.L.; McKinley, M.J.; McAllen, R.M.; Martelli, D. Vagal afferent activation suppresses systemic inflammation via the splanchnic anti-inflammatory pathway. Brain Behav Immun 2018, 73, 441–449. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex--linking immunity and metabolism. Nat Rev Endocrinol 2012, 8, 743–754. [Google Scholar] [CrossRef]
- Wu, M.L.; Xie, C.; Li, X.; Sun, J.; Zhao, J.; Wang, J.H. Mast cell activation triggered by SARS-CoV-2 causes inflammation in brain microvascular endothelial cells and microglia. Front Cell Infect Microbiol 2024, 14, 1358873. [Google Scholar] [CrossRef]
- Sumantri, S.; Rengganis, I. Immunological dysfunction and mast cell activation syndrome in long COVID. Asia Pac Allergy 2023, 13, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, L.B.; Brook, J.B.; Walters, A.S.; Goris, A.; Afrin, L.B.; Molderings, G.J. Mast cell activation symptoms are prevalent in Long-COVID. Int J Infect Dis 2021, 112, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; An, Q.; Zhang, W.; Li, Y.; Zhang, Q.; Yan, H. Histamine and receptors in neuroinflammation: Their roles on neurodegenerative diseases. Behav Brain Res 2024, 465, 114964. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Leo, M.D. Histamine Potentiates SARS-CoV-2 Spike Protein Entry Into Endothelial Cells. Front Pharmacol 2022, 13, 872736. [Google Scholar] [CrossRef]
- de Esch, I.J.; Thurmond, R.L.; Jongejan, A.; Leurs, R. The histamine H4 receptor as a new therapeutic target for inflammation. Trends Pharmacol Sci 2005, 26, 462–469. [Google Scholar]
- Yu, F.; Liu, X.; Ou, H.; Li, X.; Liu, R.; Lv, X.; Xiao, S.; Hu, M.; Liang, T.; Chen, T. , et al. The histamine receptor H1 acts as an alternative receptor for SARS-CoV-2. mBio 2024, 15, e0108824. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the nervous system. Physiol Rev 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Yamada, Y.; Yoshikawa, T.; Naganuma, F.; Kikkawa, T.; Osumi, N.; Yanai, K. Chronic brain histamine depletion in adult mice induced depression-like behaviours and impaired sleep-wake cycle. Neuropharmacology 2020, 175, 108179. [Google Scholar] [CrossRef]
- Flik, G.; Folgering, J.H.; Cremers, T.I.; Westerink, B.H.; Dremencov, E. Interaction Between Brain Histamine and Serotonin, Norepinephrine, and Dopamine Systems: In Vivo Microdialysis and Electrophysiology Study. J Mol Neurosci 2015, 56, 320–328. [Google Scholar] [CrossRef]
- Salvucci, F.; Codella, R.; Coppola, A.; Zacchei, I.; Grassi, G.; Anti, M.L.; Nitisoara, N.; Luzi, L.; Gazzaruso, C. Antihistamines improve cardiovascular manifestations and other symptoms of long-COVID attributed to mast cell activation. Front Cardiovasc Med 2023, 10, 1202696. [Google Scholar] [CrossRef]
- Braga, J.; Lepra, M.; Kish, S.J.; Rusjan, P.M.; Nasser, Z.; Verhoeff, N.; Vasdev, N.; Bagby, M.; Boileau, I.; Husain, M.I. , et al. Neuroinflammation After COVID-19 With Persistent Depressive and Cognitive Symptoms. JAMA Psychiatry 2023, 80, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Clouston, S.A.P.; Vaska, P.; Babalola, T.; Gardus, J., 3rd; Huang, C.; Soriolo, N.; Fontana, A.; DeLorenzo, C.; Parsey, R.; Luft, B.J. Glial activation among individuals with neurological post-acute sequelae of coronavirus disease 2019: A positron emission tomography study of brain fog using [(18)F]-FEPPA. Brain Behav Immun Health 2025, 44, 100945. [Google Scholar] [CrossRef]
- Nutma, E.; Fancy, N.; Weinert, M.; Tsartsalis, S.; Marzin, M.C.; Muirhead, R.C.J.; Falk, I.; Breur, M.; de Bruin, J.; Hollaus, D. , et al. Translocator protein is a marker of activated microglia in rodent models but not human neurodegenerative diseases. Nat Commun 2023, 14, 5247. [Google Scholar] [CrossRef] [PubMed]
- Braga, J.; Kuik, E.J.Y.; Lepra, M.; Rusjan, P.M.; Kish, S.J.; Vieira, E.L.; Nasser, Z.; Verhoeff, N.; Vasdev, N.; Chao, T. , et al. Astrogliosis Marker [(11)C]SL25.1188 After COVID-19 With Ongoing Depressive and Cognitive Symptoms. Biol Psychiatry 2025, 97, 816–824. [Google Scholar] [CrossRef]
- Chaganti, J.R.; Talekar, T.K.; Brew, B.J. Asymmetrical glymphatic dysfunction in patients with long Covid associated neurocognitive impairment- correlation with BBB disruption. BMC Neurol 2025, 25, 112. [Google Scholar] [CrossRef]
- Ernst, T.; Ryan, M.C.; Liang, H.J.; Wang, J.P.; Cunningham, E.; Saleh, M.G.; Kottilil, S.; Chang, L. Neuronal and Glial Metabolite Abnormalities in Participants With Persistent Neuropsychiatric Symptoms After COVID-19: A Brain Proton Magnetic Resonance Spectroscopy Study. J Infect Dis 2023, 228, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo, D.; Dezortova, M.; Hajek, M.; Ibrahimova, M.; Ibrahim, I. Metabolic changes assessed by 1H MR spectroscopy in the corpus callosum of post-COVID patients. MAGMA 2024, 37, 937–946. [Google Scholar] [CrossRef]
- Chaganti, J.; Poudel, G.; Cysique, L.A.; Dore, G.J.; Kelleher, A.; Matthews, G.; Darley, D.; Byrne, A.; Jakabek, D.; Zhang, X. , et al. Blood brain barrier disruption and glutamatergic excitotoxicity in post-acute sequelae of SARS COV-2 infection cognitive impairment: potential biomarkers and a window into pathogenesis. Front Neurol 2024, 15, 1350848. [Google Scholar] [CrossRef]
- Sharma, A.A.; Nenert, R.; Goodman, A.M.; Szaflarski, J.P. Brain temperature and free water increases after mild COVID-19 infection. Sci Rep 2024, 14, 7450. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E. , et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Hosp, J.A.; Reisert, M.; Dressing, A.; Gotz, V.; Kellner, E.; Mast, H.; Arndt, S.; Waller, C.F.; Wagner, D.; Rieg, S. , et al. Cerebral microstructural alterations in Post-COVID-condition are related to cognitive impairment, olfactory dysfunction and fatigue. Nat Commun 2024, 15, 4256. [Google Scholar] [CrossRef] [PubMed]
- Kenji Sudo, F.; Pinto, T.P.; F, G.Q.B.-A.; Bramati, I.; Marins, T.F.; Monteiro, M.; Meireles, F.; Soares, R.; Erthal, P.; Calil, V. , et al. Cognitive, behavioral, neuroimaging and inflammatory biomarkers after hospitalization for covid-19 in Brazil. Brain Behav Immun, 1016. [Google Scholar]
- Serrano Del Pueblo, V.M.; Serrano-Heras, G.; Romero Sanchez, C.M.; Landete, P.P.; Rojas-Bartolome, L.; Feria, I.; Morris, R.G.M.; Strange, B.; Mansilla, F.; Zhang, L. , et al. Brain and cognitive changes in patients with long COVID compared with infection-recovered control subjects. Brain 2024, 147, 3611–3623. [Google Scholar] [CrossRef]
- Besteher, B.; Rocktaschel, T.; Garza, A.P.; Machnik, M.; Ballez, J.; Helbing, D.L.; Finke, K.; Reuken, P.; Gullmar, D.; Gaser, C. , et al. Cortical thickness alterations and systemic inflammation define long-COVID patients with cognitive impairment. Brain Behav Immun 2024, 116, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Deitchman, A.N.; Torres, L.; Iyer, N.S.; Munter, S.E.; Nixon, C.C.; Donatelli, J.; Thanh, C.; Takahashi, S.; Hakim, J. , et al. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep 2021, 36, 109518. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J. , et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Espin, E.; Yang, C.; Shannon, C.P.; Assadian, S.; He, D.; Tebbutt, S.J. Cellular and molecular biomarkers of long COVID: a scoping review. EBioMedicine 2023, 91, 104552. [Google Scholar] [CrossRef]
- Almulla, A.F.; Thipakorn, Y.; Zhou, B.; Vojdani, A.; Maes, M. Immune activation and immune-associated neurotoxicity in Long-COVID: A systematic review and meta-analysis of 103 studies comprising 58 cytokines/chemokines/growth factors. Brain Behav Immun 2024, 122, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Vrettou, C.S.; Vassiliou, A.G.; Keskinidou, C.; Mourelatos, P.; Asimakos, A.; Spetsioti, S.; Diamantopoulos, A.; Jahaj, E.; Antonoglou, A.; Katsaounou, P. , et al. A Prospective Study on Neural Biomarkers in Patients with Long-COVID Symptoms. J Pers Med 2024, 14. [Google Scholar] [CrossRef]
- Pacheco-Jaime, L.; Garcia-Vicente, C.; Ariza, M.; Cano, N.; Garolera, M.; Carreras-Vidal, L.; Roura, I.; Capdevila-Lacasa, C.; Oltra, J.; Pardo, J. , et al. Structural brain changes in post-COVID condition and its relationship with cognitive impairment. Brain Commun 2025, 7, fcaf070. [Google Scholar] [CrossRef]
- Telser, J.; Grossmann, K.; Weideli, O.C.; Hillmann, D.; Aeschbacher, S.; Wohlwend, N.; Velez, L.; Kuhle, J.; Maleska, A.; Benkert, P. , et al. Concentrations of Serum Brain Injury Biomarkers Following SARS-CoV-2 Infection in Individuals with and without Long-COVID-Results from the Prospective Population-Based COVI-GAPP Study. Diagnostics (Basel) 2023, 13. [Google Scholar]
- Bark, L.; Larsson, I.M.; Wallin, E.; Simren, J.; Zetterberg, H.; Lipcsey, M.; Frithiof, R.; Rostami, E.; Hultstrom, M. Central nervous system biomarkers GFAp and NfL associate with post-acute cognitive impairment and fatigue following critical COVID-19. Sci Rep 2023, 13, 13144. [Google Scholar] [CrossRef] [PubMed]
- Lempriere, S. Single-cell transcriptomics reveals neuroinflammation in severe COVID-19. Nat Rev Neurol 2021, 17, 461. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, M.M.; Sivandzade, F.; Albekairi, T.H.; Alqahtani, F.; Cucullo, L. Neuroinflammation and Its Impact on the Pathogenesis of COVID-19. Front Med (Lausanne) 2021, 8, 745789. [Google Scholar] [CrossRef]
- Fekete, R.; Simats, A.; Biro, E.; Posfai, B.; Cserep, C.; Schwarcz, A.D.; Szabadits, E.; Kornyei, Z.; Toth, K.; Ficho, E. , et al. Microglia dysfunction, neurovascular inflammation and focal neuropathologies are linked to IL-1- and IL-6-related systemic inflammation in COVID-19. Nat Neurosci 2025, 28, 558–576. [Google Scholar] [CrossRef]
- Vitkovic, L.; Bockaert, J.; Jacque, C. "Inflammatory" cytokines: neuromodulators in normal brain? J Neurochem 2000, 74, 457–471. [Google Scholar] [CrossRef]
- Siclari, F.; Tononi, G. Local aspects of sleep and wakefulness. Curr Opin Neurobiol 2017, 44, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.M.; Nguyen, J.T.; Dykstra-Aiello, C.J.; Taishi, P. Local sleep. Sleep Med Rev 2019, 43, 14–21. [Google Scholar] [CrossRef]
- Pietrobon, A.J.; Andrejew, R.; Custodio, R.W.A.; Oliveira, L.M.; Scholl, J.N.; Teixeira, F.M.E.; de Brito, C.A.; Glaser, T.; Kazmierski, J.; Goffinet, C. , et al. Dysfunctional purinergic signaling correlates with disease severity in COVID-19 patients. Front Immunol 2022, 13, 1012027. [Google Scholar] [CrossRef]
- Olafsdottir, H.F.; Bush, D.; Barry, C. The Role of Hippocampal Replay in Memory and Planning. Curr Biol 2018, 28, R37–R50. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, G.; Zeng, W.; Zhu, R.; Li, C.; Zhu, Y.; Li, D.; Liu, J.; Wang, W.; Li, P. , et al. Segregation and integration of resting-state brain networks in a longitudinal long COVID cohort. iScience 2025, 28, 112237. [Google Scholar] [CrossRef]
- Verger, A.; Doyen, M.; Heyer, S.; Goehringer, F.; Bruyere, A.; Kaphan, E.; Chine, M.; Menard, A.; Horowitz, T.; Guedj, E. Reorganization of brain connectivity in post-COVID condition: a (18)F-FDG PET study. EJNMMI Res 2025, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, J.; Bernhardt, B.C.; Leech, R.; Bzdok, D.; Jefferies, E.; Margulies, D.S. The default mode network in cognition: a topographical perspective. Nat Rev Neurosci 2021, 22, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Madden, D.; Stephens, T.M.; Scott, J.; O'Neal Swann, C.; Prather, K.; Hoffmeister, J.; Ding, L.; Dunn, I.F.; Conner, A.K.; Yuan, H. Functional connectivity of default mode network in non-hospitalized patients with post-COVID cognitive complaints. Front Neurosci 2025, 19, 1576393. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, C.; Deng, J.; Zhou, J. JAK inhibition as a new treatment strategy for patients with COVID-19. Biochem Pharmacol 2022, 202, 115162. [Google Scholar] [CrossRef] [PubMed]
- Wee, L.E.; Lim, J.T.; Tay, A.T.; Chiew, C.J.; Young, B.E.; Wong, B.; Lim, R.; Lee, C.L.; Tan, J.; Vasoo, S. , et al. Nirmatrelvir/ritonavir treatment and risk for post-acute sequelae of COVID-19 in older Singaporeans: author's response. Clin Microbiol Infect 2025, 31, 139–140. [Google Scholar] [CrossRef]
- Al-Aly, Z. SARS-CoV-2 antivirals and post-COVID-19 condition. Lancet Infect Dis 2025, 25, 6–8. [Google Scholar] [CrossRef]
- AminJafari, A.; Ghasemi, S. The possible of immunotherapy for COVID-19: A systematic review. Int Immunopharmacol 2020, 83, 106455. [Google Scholar] [CrossRef]
- Zhang, Q.; Niu, Y.; Li, Y.; Xia, C.; Chen, Z.; Chen, Y.; Feng, H. Meningeal lymphatic drainage: novel insights into central nervous system disease. Signal Transduct Target Ther 2025, 10, 142. [Google Scholar] [CrossRef]
- Nam, M.H.; Sa, M.; Ju, Y.H.; Park, M.G.; Lee, C.J. Revisiting the Role of Astrocytic MAOB in Parkinson's Disease. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Lee, D.H.; Seubert, S.; Huhn, K.; Brecht, L.; Rotger, C.; Waschbisch, A.; Schlachetzki, J.; Klausmeyer, A.; Melms, A.; Wiese, S. , et al. Fingolimod effects in neuroinflammation: Regulation of astroglial glutamate transporters? PLoS One 2017, 12, e0171552. [Google Scholar]
- Oliveros, G.; Wallace, C.H.; Chaudry, O.; Liu, Q.; Qiu, Y.; Xie, L.; Rockwell, P.; Figueiredo-Pereira, M.E.; Serrano, P.A. Repurposing ibudilast to mitigate Alzheimer's disease by targeting inflammation. Brain 2023, 146, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Toti, A.; Micheli, L.; Lucarini, E.; Ferrara, V.; Ciampi, C.; Margiotta, F.; Failli, P.; Gomiero, C.; Pallecchi, M.; Bartolucci, G. , et al. Ultramicronized N-Palmitoylethanolamine Regulates Mast Cell-Astrocyte Crosstalk: A New Potential Mechanism Underlying the Inhibition of Morphine Tolerance. Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Dong, H.; Wang, Y.; Zhang, X.; Zhang, X.; Qian, Y.; Ding, H.; Zhang, S. Stabilization of Brain Mast Cells Alleviates LPS-Induced Neuroinflammation by Inhibiting Microglia Activation. Front Cell Neurosci 2019, 13, 191. [Google Scholar] [CrossRef]
- Ramirez-Ponce, M.P.; Flores, J.A.; Barrella, L.; Ales, E. Ketotifen is a microglial stabilizer by inhibiting secretory vesicle acidification. Life Sci 2023, 319, 121537. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front Immunol 2018, 9, 1873. [Google Scholar] [CrossRef]
- Aldossari, A.A.; Assiri, M.A.; Ansari, M.A.; Nadeem, A.; Attia, S.M.; Bakheet, S.A.; Albekairi, T.H.; Alomar, H.A.; Al-Mazroua, H.A.; Almanaa, T.N. , et al. Histamine H4 Receptor Antagonist Ameliorates the Progression of Experimental Autoimmune Encephalomyelitis via Regulation of T-Cell Imbalance. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Mehranfard, D.; Speth, R.C. Cholinergic anti-inflammatory pathway and COVID-19. Bioimpacts 2022, 12, 171–174. [Google Scholar]
- Khan, M.W.Z.; Ahmad, M.; Qudrat, S.; Afridi, F.; Khan, N.A.; Afridi, Z.; Fahad; Azeem, T. ; Ikram, J. Vagal nerve stimulation for the management of long COVID symptoms. Infect Med (Beijing) 2024, 3, 100149. [Google Scholar] [CrossRef] [PubMed]
- Wood, G.K.; Sargent, B.F.; Ahmad, Z.U.; Tharmaratnam, K.; Dunai, C.; Egbe, F.N.; Martin, N.H.; Facer, B.; Pendered, S.L.; Rogers, H.C. , et al. Posthospitalization COVID-19 cognitive deficits at 1 year are global and associated with elevated brain injury markers and gray matter volume reduction. Nat Med 2025, 31, 245–257. [Google Scholar] [CrossRef]
- Calzari, L.; Dragani, D.F.; Zanotti, L.; Inglese, E.; Danesi, R.; Cavagnola, R.; Brusati, A.; Ranucci, F.; Di Blasio, A.M.; Persani, L. , et al. Epigenetic patterns, accelerated biological aging, and enhanced epigenetic drift detected 6 months following COVID-19 infection: insights from a genome-wide DNA methylation study. Clin Epigenetics 2024, 16, 112. [Google Scholar] [CrossRef]
- Saunders, E.G.; Pouliopoulou, D.V.; Miller, E.; Billias, N.; MacDermid, J.C.; Brunton, L.; Pereira, T.V.; Quinn, K.L.; Bobos, P. Rehabilitation interventions and outcomes for post-COVID condition: a scoping review. BMJ Public Health 2025, 3, e001827. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.; Lavelle, B.; Miller, J.; Jimenez, M.; Lim, P.H.; Orban, Z.S.; Clark, J.R.; Tomar, R.; Ludwig, A.; Ali, S.T. , et al. Multidisciplinary Center Care for Long COVID Syndrome-A Retrospective Cohort Study. Am J Med 2025, 138, 108–120. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



