Submitted:
11 June 2025
Posted:
12 June 2025
You are already at the latest version
Abstract
Clinically used imaging modalities such as positron emission tomography (PET) and single photon emission computed tomography (SPECT) are highly sensitive non-invasive imaging modalities utilized in combination with magnetic resonance imaging (MRI) or computed tomography (CT) for the diagnosis of a multitude of disorders. Efficient and robust radiolabeling methods are needed to accommodate for the increasing demand for PET and SPECT tracer development. Copper-mediated radiohalogenation (CMRH) reactions have been reported for the rapid late-stage preparation of radiolabeled arenes, yet synthetic challenges and instability of corresponding radiolabeling precursors can limit the applications of CMRH radiolabeling approaches. In this work, a general protocol for the preparation of 18F-labeled and 123I-labeled arenes applying CMRH of stable aryl boronate substrates is reported. The method utilizes aryl boronic acid 1,1,2,2-tetraethylethylene glycol esters [ArB(Epin)s] and aryl boronic acid 1,1,2,2-tetrapropylethylene glycol esters [ArB(Ppin)s] as stable and versatile precursor building blocks for radiolabeling via CMRH. Both ArB(Epin)s and ArB(Ppin)s based radiolabeling precursors were prepared in a one-step synthesis from the corresponding aryl boronic acids with chemical yields of 49-99%. Radiolabeling of the aryl boronic esters with fluorine-18 or iodine-123 via CMRH furnished the corresponding radiolabeled arenes with RCC of 7-99% and 10-99%, respectively. Notably, a radiohalogenated prosthetic group containing a vinyl sulfone motif was obtained with an activity yield (AY) of 18 ± 3%, and applied towards the preparation of two clinically-relevant PET tracers.
Keywords:
Copper-mediated radiohalogenation (CMRH)
; 18F-fluorination
; 123I-iodination
; positron emission tomography (PET)
; radiohalogenation
; single photon emission computed tomography (SPECT)
; prosthetic group
1. Introduction
Positron emission tomography (PET) and single photon emission computed tomography (SPECT) are diagnostic non-invasive imaging modalities typically combined with magnetic resonance imaging (MRI) or computed tomography (CT) imaging modalities in nuclear medicine for the real-time hybrid visualization of metabolic disorders and malignancies with high precision [1,2,3,4,5,6]. In addition to radiometal-based radiopharmaceuticals, the many PET and SPECT tracers are radiosynthesized as radiohalogenated molecules. Radiohalogenated compounds are also routinely used to obtain biodistribution and pharmacokinetic information in drug discovery and development [7,8,9,10].
Fluorine-18, the most predominant diagnostic radionuclide for PET imaging, has a number of advantageous physicochemical properties including a suitable half-life (t1/2= 109.7 min) for multi-step radiosynthesis and PET tracer transport between institutions [11].
Additionally, the high positron decay ratio (>97% β+) and low positron energy of fluorine-18 (0.635 MeV) affords high resolution PET images. As of June 2025, out of 19 FDA-approved PET radiopharmaceuticals, 12 contain fluorine-18 [12,13]. The most clinically prevalent PET tracer for oncological applications, 2-deoxy-2-[18F]fluoro-d-glucose or commonly named [18F]fluorodeoxyglucose ([18F]FDG), is a glucose analogue where fluorine-18 replaces a hydroxyl substituent in the two-position as a bioisostere [14].
In contrast to fluorine-18, which functions solely as positron-emitting radionuclide, compounds containing radioiodine offer more functional versatility, and can be used as PET or SPECT tracers, and for therapeutic applications. Radioiodinated compounds, irrespective of the radionuclide (e.g., iodine-123, iodine-124 or iodine-131) employed, are chemically indistinguishable and therefore exhibit identical pharmacological properties [15]. Iodine-123, (t1/2 = 13.2 h), decays via electron capture (EC) and is used primarily for SPECT imaging. Notable clinical applications of 123I-labeled SPECT tracers include [123I]NaI for thyroid disease imaging [16], meta-[123I]iodobenzylguanidine ([123I]MIBG) for cardiac imaging [17,18], and [123I]Ioflupane for Parkinson’s disease diagnosis [19]. Further, 123I-Labeled PARP inhibitors have also been used for Auger-Meitner electron based radiotherapeutic applications [20]. Iodine-124 serves as a long-lived β+-emitting radionuclide (t1/2 = 4.2 d), which is particularly useful for long term clinical and small animal studies [21]. Iodine-125 (t1/2 = 59.4 d) is typically used as an Auger-Meitner electron emitter for brachytherapy, and for preclinical radioligand assays [15,22]. Iodine-131 decays via EC and β–-emission (t1/2 = 8.02 days) and is considered the first example of a true theranostic radionuclide, due to its imaging and radiotherapeutic applications for thyroid diseases, such as thyrotoxicosis and thyroid cancer [23,24,25]. Currently, eight FDA-approved radiopharmaceuticals contain radioiodine [26].
Due to their multi-faceted utility in nuclear medicine applications, radiohalogenation reactions have been the focus of a plethora of radiosynthetic procedures [11,27,28,29,30,31,32,33]. However, adaptation of halogenation strategies from larger scale (e.g., 0.1-1 M) concentrations in organic syntheses cannot be easily translated towards radiohalogenation approaches due to the divergent reactivity of radiohalogens at low concentrations (1-10 mM and below). Therefore, novel and robust radiohalogenation techniques that may facilitate the development of future radiopharmaceuticals are highly sought after.
Radiolabeled peptides are vital tools for PET and SPECT imaging, owing to their high selectivity for cell surface receptors that are typically overexpressed in various cancers. Moreover, radiolabeled peptides are powerful biovectors for targeted radiotherapeutic applications (e.g., theranostic approach) [34,35,36,37]. Consequently, robust radiolabeling protocols for the efficient development of radiohalogenated peptides hold significant potential for clinical applications. Despite the advancements in radiohalogenation techniques, the majority of radiohalogenated peptide preparations are performed through conjugation with prosthetic groups [32,38,39,40,41,42]. Indirect radiolabeling approaches towards selective bioconjugation of peptides is typically achieved via bioconjugation of a radiolabeled prosthetic group with lysine and cysteine residues [43,44]. Cysteine-selective thiol-based conjugation strategies are generally favored over lysine modification, due to increased regioselectivity and minimization of potential side-product formation [45,46]. Recently, Murphy et al. developed a promising prosthetic group containing a vinyl sulfone motif, which offers a number of radiosynthetic advantages compared to cysteine-specific 18F-labeling of peptides via maleimide-based synthons [32,47,48,49].
Cu-mediated radiohalogenation (CMRH) protocols, which focus on the preparation of radiohalogenated arenes from aryl-stannanes or aryl-boronates have proven to be efficient, robust and practical for radiopharmaceutical preparations [50,51,52,53,54,55,56]. Gouverneur et al. applied the Cu-mediated radiofluorination (CMRF) approach towards the preparation of 18F-labeled arenes using electron-rich, electron-neutral and electron-poor aryl-boronate ester substrates (Scheme 1A) [57]. The use of aryl-boronic acids by Scott et al. further expanded the scope of CMRF reactions (Scheme 1B) [51]. Radioiodination, radiobromination and radioastatination via CMRH was also demonstrated to be feasible (Scheme 1C,D) [58,59,60]. Despite the progress of CMRH protocols, radiolabeling precursor synthesis can be challenging due to the inherent toxicity of stannyl compounds or to the relative instability of aryl boronic esters groups, which may decompose during silica gel chromatography purification or in aqueous conditions. Oka et al. reported the use of aryl boronic acid 1,1,2,2-tetraethylethylene glycol esters [ArB(Epin)s] as more stable substrates for Suzuki-Miyaura coupling reactions compared to the respective aryl boronic acid and aryl boronic acid 1,1,2,2-tetramethylethylene glycol esters [ArB(pin)s] counterparts [61]. The work attributed the increased stability to the four ethyl groups within ArB(Epin) compounds, which could dynamically protect the otherwise exposed empty orbital of the boron atom. The elevated stability presents practical synthetic advantages, as purification of the ArB(Epin) substrates using silica-based chromatographic techniques is facilitated, compound decomposition under alkaline conditions is significantly reduced, and the reactivity of the ArB(Epin) towards Suzuki-Miyaura coupling with aryl bromides was increased [62,63].
2. Materials and Methods
2.1. Materials
Unless stated otherwise, all solvents and reagents were obtained from commercial vendors, and utilized without additional purification. All solvents used in experiments were of HPLC or analytical grade, with the exception of water which was ultrapure (>18.2 MΩ cm−1).
2.2. General Information
Unless otherwise stated, all moisture and/or oxygen sensitive reactions were carried out using the Schlenk technique under an argon atmosphere. For this, glass reaction vessels were heated several times under high vacuum (10–3 bar) with a heat gun and filled with argon. Solvents or liquid chemicals were added via septa using stainless steel cannulas. Thin layer chromatography (TLC) was carried out using silica coated Carl Roth plates (TLC plates ROTI®ChromaPlate Alu 60 UV, Art. No.: 1A18.1) and visualization took place under UV light (254 nm and 366 nm) or with vanillin staining reagent. Crude products were purified via flash column chromatography at the Biotage® Selekt System using BÜCHI FlashPure EcoFlex chromatography cartridges. Beforehand, the crude products were dissolved in CH2Cl2, silica was added, and the mixture was evaporated under reduced pressure and the crude product adsorbed on silica was dry loaded via a pre-column onto the system. The products were collected by tracking the UV activity in the range of 200 to 400 nm.
All NMR spectra were recorded at 25 °C using a Bruker Avance III 400 MHz/Agilent DD2-400 MHz (1H: 400 MHz, 13C: 101 MHz, 19F: 376 MHz) or Bruker Avance 600 Hz/Agilent DD2–600 MHz (1H: 600 MHz, 13C: 151 MHz). 13C-NMR spectra were recorded decoupled from 1H-NMR spectra. The evaluation of the NMR spectra was carried out using the program Mestrelab MestReNova (version 15.0.1).
High-resolution mass spectra were obtained on a Q-TOF MS using electrospray ionization: Agilent 1260 Infinity II HPLC (Santa Clara, California, USA; pump G7104C, autosampler G7129C, column oven G7116A, DAD detector G7117C) coupled to γ detector Gabi Star (Raytest Isotopenmeßgeräte GmbH, Straubenhardt, Germany) followed by accurate mass Revident Q-TOF LC/Q-TOF G6575A. Unless otherwise stated, the measurements were performed in bypass mode using an eluent consisting of (A): MeCN and (B): 0.1% formic acid in H2O; flow rate 0.2 mL/min). A reference mass solution containing hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazene, and purine was continuously co-injected via dual AJS ESI source. The system was operated using Agilent Masshunter Workstation 3.6 – LC/MS data aquisition software (Version 12.0) and data evaluation was performed using Agilent Masshunter Workstation 3.6 Qualitative Analysis software (Version 12.0 Update 1).
An Acquity I-Class UPLC system (binary gradient pump BSM, autosampler FTN, column manager CM, and diode array detector PDAeλ) with a Waters Xevo TQ-S mass spectrometer (Waters, Milford, Massachusetts, USA), was used to acquire low-resolution mass spectra and assess the purity of the prepared substances. Unless otherwise stated, mass spectra were obtained by positive electrospray ionization (ESI+) in bypass mode and purity analysis was performed by UPLC analysis (ACQUITY UPLC BEH C18 column (1.7 μm, 130 Å, 100x2.1 mm with respective VanGuard precolumn 5x2.1 mm) at a flow rate of 0.4 ml/min and gradient elution (mobile phase: A: 0.1% acetic acid in water (v/v) B: CH3OH/CH3CN/acetic acid 50/50/0.1 (v/v/v), t0 min 95/5 - t0.5 min 95/5 - t5.5 min 5/95 –t7.0 min 5/95 - t8.0 min 95/5 – t8.5 min 95/5)). The UPLC chromatograms and mass spectra were analyzed using MassLynx 4.1 software.
All radiochemistry experiments were performed at the Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf (HZDR). [18F]Fluoride was produced via the (p,n) reaction using the in-house TR-Flex (Advanced Cyclotron Systems Inc., ACSI, Canada) cyclotron by irradiating [18O]H2O with 18 MeV protons [64]. No-carrier-added sodium [123I]iodide (Na [123I]I) was produced using the in-house TR-Flex cyclotron (ACSI) and the gas target KIPROS 200 from ZAG Zyklotron AG (Eggenstein-Leopoldshafen, Germany) by bombardment of highly enriched 124Xe gas with 30 MeV protons via, amongst others, the nuclear reaction 124Xe(p, pn)123Xe → 123I. Concentration of crude [123I]iodide and formulation in 0.02 M aqueous NaOH was performed by ROTOP Pharmaka GmbH at the HZDR campus. Aliquots containing [123I]iodide in an activity concentration of ~10 MBq/µL were used for further experiments and diluted accordingly with NaOH (0.02 M). Semi-preparative HPLC for determination of isolated radiochemical yield for radioiodinated compounds was performed on the following system: Jasco HPLC (interface LC-Net II/ADC, pump PU-2080Plus, gradient mixer LG-980-02; degasser DG-980-50, UV detector UV-2075Plus; γ detector Gabi Star (Raytest Isotopenmeßgeräte GmbH, Straubenhardt, Germany) with a 6-way column switching valve BESTA and fractionation valve BESTA; column Onyx Monolithic Semi-PREP C18, LC Column 100 x 10 mm); eluent: (A): 0.1% trifluoroacetic acid in H2O, (B): 0.1% trifluoroacetic acid in MeCN. flow rate 5 mL/min, gradient (eluent A/B): t0 min 95/5 - t2 min 95/5 – t27 min 5/95 – t30 min 5/95 – t31 min 95/5 – t40 min 95/5.
Analytical Radio-(U)HPLC was performed on the following system: Shimadzu Nexera X2 UHPLC system (Shimadzu Corporation, Kyoto, Japan; degasser DGU-20A3R and DGU-20A5R, pump LC-30AD, autosampler SIL-30AC, column oven CTO-20AC with two column switching valves FCV-14AH, diode array detector SPD-M30A, fluorescence detector RF-20A, γ detector Gabi Star (Raytest Isotopenmeßgeräte GmbH, Straubenhardt, Germany), communication bus module CBM-20A; UPLC column Kinetex C-18 (Phenomenex Inc., Torrance, United States of America; 50 × 2.1 mm, 1.7 µm, 100 Å with Security Guard precolumn) or HPLC column Kinetex C-18 (Phenomenex Inc., Torrance, United States of America; 250 × 4.6 mm, 5 µm, 100 Å with Security Guard precolumn), Gradient A Radio-UHPLC: flow rate 0.5 mL/min, eluent (A): 0.1% trifluoroacetic acid in H2O, (B): MeCN. (eluent A/B): t0 min 95/5 - t0.3 min 95/5 - t4.5 min 5/95 – t5.5 min 5/95 - t6.0 min 95/5 – t7.5 min 95/5. Gradient B: Radio-HPLC 40 min: flow rate 1.0 mL/min, gradient (eluent A/B): t0 min 95/5 – t3.0 min 95/5- t28.0 min 5/95 – t34.0 min 5/95 – t35.0 min 95/5 – t40.0 min 95/5. Gradient C: Radio-HPLC 30 min: flow rate 1.0 mL/min, gradient (eluent A/B): t0 min 95/5 – t1.0 min 95/5- t21.0 min 5/95 – t23.5 min 5/95 – t25.0 min 95/5 – t30.0 min 95/5.
Semi-preparative HPLC for determination of isolated radiochemical yield for radiofluorinated compounds was performed on the following system: Agilent Series 1100 HPLC (interface Hewlett Packard 35900, pump G1311A, thermostatted column compartment G1316A; degasser G1322A, Variable Wavelength Detector (VWD) G1314A; γ detector Gabi Star (Raytest Isotopenmeßgeräte GmbH, Straubenhardt, Germany) with an external 4 port column switching valve VICI (AG); column Agilent ZORBAX SB-C18, 5 µm, 9.4 x 250 mm; eluent: (A): 0.1% trifluoroacetic acid in H2O, (B): 0.1% trifluoroacetic acid in MeCN. Flow rate 4 mL/min, gradient (eluent A/B): t0 min 90/10 – t3 min 90/10 – t23 min 5/95 – t27 min 5/95 – t28 min 90/10 – t33 min 90/10.
The radiochemical conversion (RCC) based on radio-(U)HPLC was determined by analyzing an aliquot after dilution of the crude radiofluorination reaction mixture (MeCN) with MeCN/H2O (1:1) and are based on relative peak areas of the γ detector channel of the chromatogram. The identity of the radiolabeled products was determined by associating the UV-(U)HPLC traces of the suitable unlabeled reference compounds with the radio-(U)HPLC traces of the radiolabeled products. The radiochemical purity (RCP) of radiolabeled products was determined based on the integration of the peaks in the chromatogram in the radio-(U)HPLC. The isolated activity yield (AY) of a radiolabeled product is the non-decay-corrected (n.d.c.) yield given as a percentage value, which is determined by dividing the product activity at the end of radiosynthesis (E.O.S.) by the initial starting activity and multiplying by one hundred. The molar activity (Am) was determined by HPLC analysis based on injection of a known activity amount followed by determination of the injected amount of substance. For that, the UV peak area corresponding to the radiolabeled product was determined and concentration of the non-radiolabeled “carrier” compound was calculated based on a calibration curve (See supporting Information Figure S300).
2.3. General Procedure for the Synthesis of B(Epin) and B(Ppin) Compounds Starting from the Corresponding Boronic Acids
In a 10 mL one-neck flask 2 mL of CH2Cl2 were placed under an argon atmosphere. 3,4-Diethylhexane-3,4-diol (2, Epin, 209 mg, 1.20 mmol, 1.2 eq.) or 4,5-dipropyloctane-4,5-diol (4, Ppin, 276 mg, 1.20 mmol, 1.2 eq.), the corresponding boronic acid (1.00 mmol, 1.0 eq.), and Na2SO4 (284 mg, 2.00 mmol, 2.0 eq.) were successively added (Scheme 3). The suspension was warmed and refluxed for 16 h. After cooling to room temperature, the reaction mixture was transferred to a separatory funnel with EtOAc and washed with water. The phases were separated and the aqueous phase was washed three times with EtOAc. The combined organic phases were dried over Na2SO4, filtered, and the solvent was removed in vacuo. After adsorption on silica gel, the crude product was purified by column chromatography on silica using a Biotage Select system (mobile phase gradient: 0-20% EtOAc in cyclohexane).
2.4. General Procedure for the Preparation of Radiolabeled Arenes via CMRF
The radiosynthetic procedure towards [18F]33 and [18F]56-74 commenced with the adsorption of [18F]fluoride (50-10,000 MBq) on an anion exchange resin (QMA light carbonate cartridge, Waters Corp.) followed by washing twice with dry MeOH (2 x 0.7 mL) from the male side and elution of 18F with a solution of 3.0 mg (9.6 µmol) BnBu3NCl in 0.7 mL MeOH from the female side (Scheme 5). The methanol was removed under vacuum at 70 °C for 5 min. The radiolabeling precursor (10 µmol) in 1.2 mL DMI/n-BuOH (2:1, v/v) was added to the cooled reaction vessel and allowed to react for 30 min at 120 °C with stirring. The reaction mixture containing the crude radiofluorinated products was quenched with 2 mL H2O and analyzed using analytical radio-(U)HPLC for product identification and RCC determination. The RCC determination was carried out following the quenching of the reaction mixture, taking an aliquot (5–20 µL) of the crude reaction mixture and adding the mixture to a mixture of (50-200 µL) H2O/MeCN (1:1, v/v) and analyzed using a radio-UHPLC. The RCC value was determined by integrating the radiolabeled product peak area, and dividing this value by the total integrated peak area values, and multiplying the value by one hundred. Unless noted otherwise, each experiment was performed in triplicate.
For the purification of selected radiofluorinated compounds, dilution of the crude reaction mixture with 5 mL H2O was performed before loading the mixture onto a HLB cartridge solid-phase extraction (SPE) cartridge (hydrophilic lipophilic balance, OasisTM HLB Plus LP extraction cartridge, Waters Corp). The HLB cartridge was washed with 10 mL H2O. Thereafter, elution of the radiolabeled product with 2 mL EtOH was performed prior to semi-preparative HPLC purification (mobile phase gradient: 10-95% MeCN in H2O, system Agilent 1100).
2.5. General Procedure for the Preparation of Radiolabeled Arenes via CMRI
The CMRI was performed according to a previously reported method [60,65]. Screening of substrate scope was performed for each substrate in duplicate by adding 0.5-2 MBq [123I]iodide in 0.02 M NaOH (1 µL) to a solution of the radiolabeling precursor in MeCN (10 µL, final concentration 0.8 mM, (U)HPLC grade) and [Cu(OTf)2(py)4] in MeOH (40 µL, final concentration: 4 mM, (U)HPLC grade). Without stirring, the reaction mixture was allowed to react for 10 min at room temperature. Afterwards the solution was diluted with 200 µL H2O/MeCN (1:1, v/v). The reaction mixture was analyzed by radio-HPLC (System Shimadzu UPLC Gradient C) and identification was achieved by coinjection with the authentic non-radioactive reference at a concentration of 100 µg/mL.
EPin-based radiolabeling precursor 36 was radioiodinated applying the general procedure at a total volume of 306 µL (~73 MBq [123I]iodide in 6 µL 0.02 M NaOH; 300 µL 0.8 mM 36 and 4 mM [Cu(OTf)2(py)4] in MeOH/MeCN, 4:1) followed by dilution with 1.5 mL MeCN/H2O and semipreparative HPLC (system JASCO HPLC, tR ~15 min). Isolated RCY was determined based on activity of product obtained after semi-preparative HPLC related to the starting activity.
3. Results
3.1. Organic Synthesis of Radiolabeling Precursors and Reference Compounds
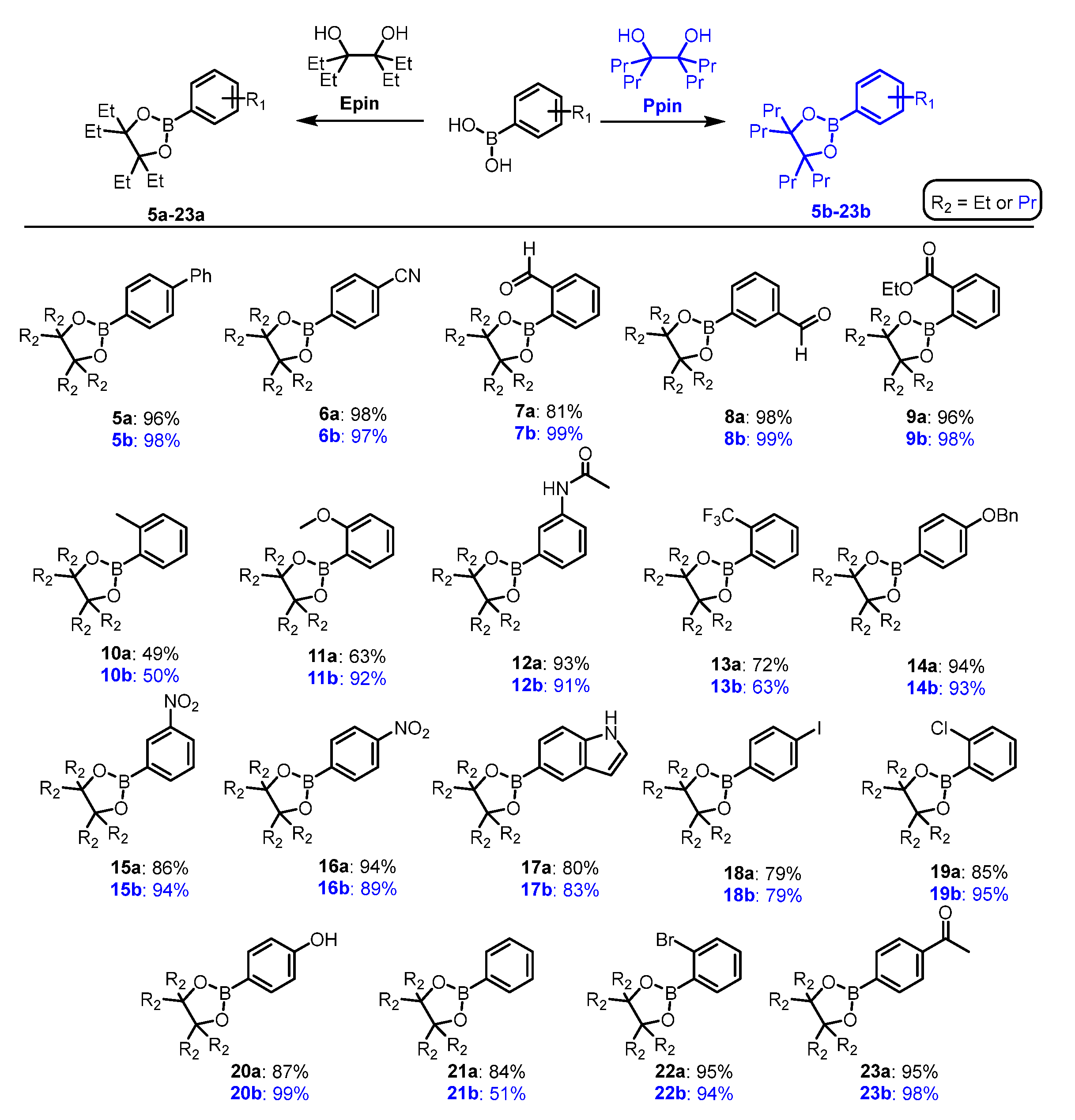
The synthetic routes towards ArB(Epin) (5a-23a) and ArB(Ppin) (5b-23b) radiolabeling precursors started with the preparation of Epin (2) and Ppin (4) using a McMurry reaction following a slightly modified literature procedure (Scheme 2) [66]. The radiolabeling precursors were obtained following the dehydrative esterification of commercially available aryl boronic acids with yields ranging from 49-99% (Scheme 3).
The synthesis of the prosthetic group radiolabeling precursor 36, for cysteine-specific bioconjugation of peptides, and the reference compounds 33 and 35 started from the thiophenols 24-26, which were first alkylated using 2-bromoethanol (Scheme 4). Oxidation of 27-29 to the sulfones with mCPBA followed by dehydration provided the reference substances 33 and 35 with overall yields of 47% and 15% respectively. Thereafter, 34 was subsequently borylated with bis(ethylpinacolato)diboron to give the radiolabeling precursor 36 with an overall yield of 33%.
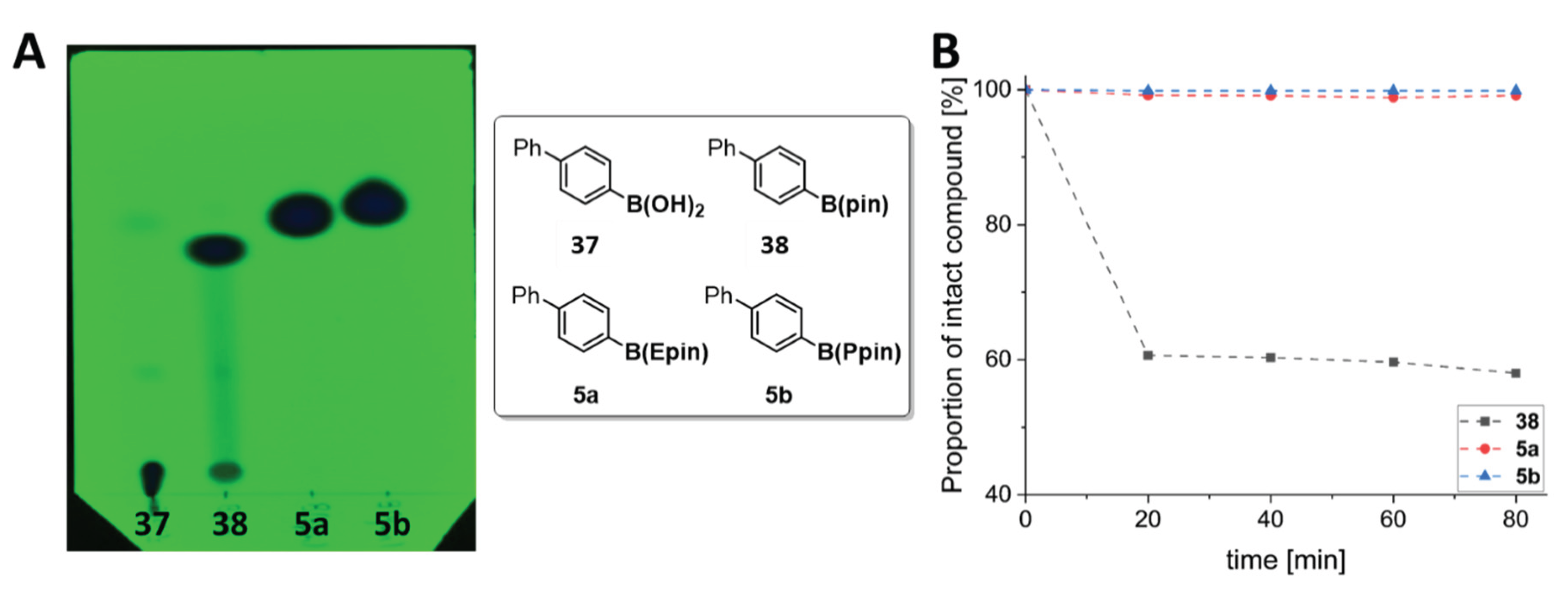
To investigate the stability of the ArB(Epin) and ArB(Ppin) compounds compared to their aryl boronic acid and ArBpin counterparts, we evaluated the chromatographic behavior on TLC plates and the stability in aqueous acetonitrile (Figure 1). The stability of the four biphenyl compounds on silica-based thin-layer chromatography (TLC) plates was observed using UV-visualization (Figure 1 A). The boronic acid 37 was not eluted by the mobile phase as expected, showing a Rf value of ~0. This finding suggests that purification of aryl-boronic acids by normal phase chromatography (using this mobile phase system) could prove challenging. For Bpin 38, a clear decomposition was observed as indicated by multiple spots and smearing on the TLC plate, with partial formation of 37 from 38 likely following deesterification. The ArB(Epin) (5a) and ArB(Ppin) (5b) derivatives were observed to be stable on TLC. Similarly, the radiolabeling precursor compounds 5a-23a and 5b-23b were observed to be stable on silica following TLC analysis (See Supporting Information Figure S216). Moreover, the stability of 38, 5a and 5c was evaluated when incubated in MeCN/H2O (1:1), a standard mobile phase system for reversed-phase chromatographic separations and for semi-preparative HPLC purifications (Figure 2B). Within only 20 min, approximately 40% compound degradation of the ArBpin derivative (38) to the corresponding aryl boronic acid (37) was observed. This finding underlines that semi-preparative HPLC purification of radiolabeling precursors containing an ArBpin may prove challenging due to decomposition [54]. From our experiments, we have observed that both ArB(Epin) and ArB(Ppin) compounds both have a similarly high stability on silica and in aqueous acetonitrile.
3.2. Optimization of Radiofluorination and Substrate Scope of CMRF and CMRI
The optimization of the CMRF was performed by modifying one variable at a time using 6a (NC-ArB(Epin) as a model substrate. The optimal molar equivalents of the [Cu(OTf)2(py)4] mediator was found to be 25 µmol with respect to 10 µmol 6a (Figure 2A) for reactions in 1.2 mL. The highest RCC values were found when using the DMI/n-BuOH solvent (2:1) system (Figure 2B). Additionally, the optimal reaction temperature for the CMRF was found to be 120 °C for 30 min (See Supporting Information Table S8). The phase transfer agent (PTA) BnBu3NCl (3 mg, 13.1 µmol) provided slightly higher RCC with smaller deviations compared to conventional PTAs Et4NHCO3 (1.8 mg, 9.6 µmol) and Et4NOTf (2.68 mg, 9.6 µmol), although the overall values hardly differed (See Supporting Information Table S2) [11]. Using the optimized conditions, for both ArB(Epin) and ArB(Ppin), substituents in the para-position were generally well tolerated and resulted in RCC values of >70%, independent of the electron-withdrawing groups -CN ([18F]59), -NO2 ([18F]69), -I ([18F]71) or –COMe ([18F]76) or the electron-donating -Ph ([18F]58) or -OH([18F]73). The only exception was [18F]67 with a benzyloxy group in para-position to the -B(Epin) and -B(Ppin) groups, which gave lower RCC values of 64% and 62%, respectively. Ortho-substituted ArB(Epin) and ArB(Ppin) compounds with stronger electron-withdrawing groups such as -CHO ([18F]60), -CO2Et ([18F]62) or -CF3 ([18F]66) were not well accepted, as shown by RCC values of 10-37 %. The weakly electron-withdrawing group -Cl ([18F]72) and the electron-donating group -OMe ([18F]64) also showed only moderate RCCs of 53-64%. Only the simple -Me derivative ([18F]63) showed very good RCCs with 96% and 93% from the Epin and the Ppin precursor, respectively. Among the meta-substituted compounds, the electron-donating group -NHCOMe ([18F]65) was clearly better accepted than the electron-withdrawing groups -CHO ([18F]61) and -NO2 ([18F]68). The only bicyclic heterocycle indole [18F]70 in this substrate set gave only moderate RCC values of 64% each from Epin and the Ppin precursor. Generally, the RCCs differed only marginally when the Epin or the Ppin precursor of the same derivative were used.
CMRI was performed as recently described to screen substrate scope of ArB(Epin) and ArB(Ppin) precursors [60,65,67]. RCC was determined by radio-HPLC and product identification was performed by coinjection of the authentic non-radioactive reference. Generally, radioiodination furnished the anticipated 123I-labeled products in RCC between 11-99%, while for ArB(Epin) substituted compounds the RCC were typically comparable or higher compared to the respective ArE(Ppin) substituted analogs (Scheme 5). Interestingly, the unsubstituted arenes phenyl-B(Epin) (21a) and phenyl-B(Ppin) (21b) afforded [123I]55 only in low RCC values of 17% and 26%, respectively. For both ArB(Epin) and ArB(Ppin), substituents in ortho-position were generally well-tolerated and afforded >80% RCC values, irrespective of the electron-withdrawing -CHO ([123I]41), -CO2Et ([123I]43), -CF3 ([123I]47), -Cl ([123I]53), or -Br ([123I]56) groups or of electron-donating -OMe ([123I]45). The only exception was [123I]44 having a methyl group in ortho-position to the -B(Epin) and -B(Ppin) groups which afforded lower RCC values of 57% and 13%, respectively. Para-substituted ArB(Epin) and ArB(Ppin) compounds with electron donating groups like Ph ([123I]39), BnO ([123I]48) or OH ([123I]54) as well as electron withdrawing groups like -I ([123I]52), -COMe ([123I]57), -CN ([123I]40) or NO2 ([123I]50) gave mostly lower but still moderate RCC values of 26-81%. For meta-substituted compounds only few examples have been employed here that gave no obvious trend. Meta-substitution with -CHO ([123I]42) or -NHCOMe ([123I]46) was not well tolerated, as indicated by RCC values of 19-37% while meta-NO2 substituted compound [123I]49 gave high RCC of 86% from the Epin and 61% from the Ppin precursor. The only bicyclic heterocycle indole [123I]51 in this substrate set was obtained in only low RCC of 11% from the Epin and 18% from the Ppin precursor. Of note, for radioiodination of ArB(Ppin) substituted compounds in some cases an unidentified but common radioiodinated byproduct was formed in varying amounts which gives rise to the hypothesis that a stabilized [123I]IB(Ppin) species [68] forms in concurrence to the CMRI product (See Supporting Information Figure S218A–D).
In conclusion, in CMRI both ArB(Epin) and ArB(Ppin) showed high tolerance for a broad aromatic substitution pattern. No aryl substituent completely prohibited reactivity in CMRI. However, ortho-substitution seems favored over para-substitution at this stage of the explored substrate scope.
3.3. Preparation of Radiohalogenated Prosthetic Groups Towards Peptide Radiolabeling
Utilizing the optimized CMRH conditions for manual radiosynthesis, [18F]fluoro-4-(vinylsulfonyl)benzene ([18F]33) and [123I]iodo-4-(vinylsulfonyl)benzene ([123I]35) could be obtained from the radiolabeling precursor 36 with RCC values of 83 ± 2% and 26 ± 7% respectively (Scheme 6A). Semi-preparative purification of [18F]33 afforded the isolated product with 18 ± 3% AY. Similarly, following semi-preparative HPLC purification the radioiodinated prosthetic group [123I]33 was obtained with 48.7 ± 2.6% RCY. The bioconjugation using [18F]33 was evaluated using three thiol-containing substrates in a similar manner as previously reported (Scheme 6B–D) [47] N-Boc-cysteine and the cyclic cysteine-containing RGD analogue peptide (RADfC, 79) were applied as commercially available model substances and showed high RCC of 98% and 99% respectively. Of note, RGD displays high affinity for the biomarker integrin αvβ3 and radiolabeled RGD analogues have been used in PET for angiogenesis imaging [69,70]. Further, a cysteine-containing TATE analogue (81) was designed and used for bioconjugation with [18F]33, allowing [18F]82 as potential 18F-labeled SSTR2 ligand in RCC of 99%. Currently, there exists no FDA or EMA-approved clinical 18F-labeled PET tracer for neuroendocrine tumor (NET) imaging as an alternative to the existing radiometal-based counterparts (e.g., [68Ga]Ga-DOTA-TATE, [68Ga]Ga-DOTA-TOC, or [64Cu]Cu-DOTA-TATE). However, [18F]SiTATE and [18F]AlF-NOTA-octreotide have both been under clinical investigation and early access programs in specific centers [71,72].
4. Discussion
The physicochemical properties of radiohalogens enable broad functional capabilities suited for diagnostic and therapeutic applications in nuclear medicine. Thus, radiohalogenation strategies play a significant role in the production and development of radiopharmaceuticals. Despite the advantages of CMRH protocols, the utility of the radiolabeling methodology can be hindered by the necessity to prepare and handle potentially instable ArB(OH)2 or ArBpin and/or toxic aryl-stannane substrates in the suitable quantities assuming 10-60 µmol of radiolabeling precursor needed per radiolabeling experiment [51,55,57,73,74]. For example, the carbon−boron (C−B) bond in radiolabeling precursor compounds containing ArB(OH)2 may cleave during functional group transformations [61]. Unfortunately, also purification of aryl-boronic acids or their ArBpin derivatives can in certain cases cause problems. Under acidic conditions or during chromatographic purification hydrolysis of ArBpin esters to aryl-boronic acids as well as the formation of proto-deborylated or hydroxy-deborylated side products is frequently observed [62,63]. Moreover, the purification of ArB(OH)2 is non-trivial due to the strong absorption onto the silica gel stationary phase. However, reversed phase purification of ArB(OH)2 may be employed to circumvent this limitation. Despite the relatively enhanced additional stability provided by ArB(pin) substrates, the purification of radiolabeling precursors containing ArB(pin) on silica gel should be performed rapidly in order to prevent significant losses of overall yield. Isobe et al. have developed a robust method for the purification of ArB(pin) derivatives on silica gel preconditioned with boric acid (B-silica gel) [75]; however, the resolution efficiency of B-silica gel is considerably lower than the standard silica gel chromatography making purification using this technique a challenge [61].
The preparation of the ArB(Epin/Ppin)-substituted arenes (5a-23a and 5b-23b) was achieved via one-step dehydrative esterification of commercially available aryl-boronic acids with either Epin (2) or Ppin (4). Oka et al. reported numerous methods of furnishing ArB(Epin)s via dehydrative esterification using various temperatures and solvents [61]. Pd-catalyzed Miyaura borylation of aryl-bromides using a B2(Epin2) reagent, and metalation of aryl-bromides also reportedly afforded ArB(Epin)s in high yields. Importantly, the stability of ArB(Epin) and ArB(Ppin) radiolabeling precursors facilitated purification via silica gel column chromatography, and the compounds provided clear defined spots on silica gel thin layer chromatography (TLC) compared to ArBpin compounds (Figure 2 A).
The application of stable ArB(Epin) and novel ArB(Ppin) substrates allowed the facile preparation of radiohalogenated arenes via CMRH. From the corresponding aryl-boronic ester substrates, a small library of 19 radiofluorinated compounds [18F]58-76, and 19 radioiodinated compounds [123I]39-57 containing various substituents and diverse electronic properties were obtained with RCC values of 7-99%, indicating excellent functional group tolerability. The RCC values obtained were comparable with the values previously reported in the literature. The optimization of the radiofluorination was performed by varying the reaction temperature, the molar equivalents of [Cu(OTf)2(py)4] with respect to the radiolabeling precursor, the PTA used for 18F-elution and the reaction solvent. In nearly all cases, longer reaction times resulted in higher RCC values, likely due to formation of the strong aryl-18F bond, which remained stable. Our findings identified the optimal CMRF conditions to include: 30 min reaction time, reaction temperature of 120 °C, reaction solvent of 1200 µL DMI/n-BuOH (2:1), BnBu3NCl as the optimal PTA, and 10 µmol radiolabeling precursor (10 µmol) and 25 µmol [Cu(OTf)2(py)4].
Aside from the enhanced stability of the radiolabeling precursors, the capacity of either ArB(Epin) and ArB(Ppin) radiolabeling precursors to be subjected to either CMRF or CMRI, allows either substrate to be used for the preparation of either a PET or SPECT radiotracer depending on the desired application [60]. Noteworthily, due to the chemically indistinguishable nature of the various radioiodides, CMRH of ArB(Epin) and ArB(Ppin) substrates using iodine-124/-125/-131 remains a distinct prospect, and may be the focus of a future study. Furthermore, the CMRH protocol may also be applied towards radiobromination and astatination of ArB(Epin)s and ArB(Ppin)s in a future study [76,77].
Using the optimized radiolabeling conditions, we have furnished both an 18F-labeled and 123I-labeled prosthetic groups ([18F]33 and [123I]35) from the starting radiolabeling precursor 36 (Scheme 6A) in high RCP. The optimized site-specific bionconjugation of [18F]33 upon clinically-relevant Cys-containing peptides (79 and 81) allows rapid access to both radiolabeled RGD and TATE analogues ([18F]80 and [18F]82, respectively).
5. Conclusions
Our findings broaden the applicability of CMRH by introducing two new substrate classes, ArB(Epin)s and ArB(Ppin)s, to the radiochemist’s toolkit. Key advantages include improved precursor stability and moderate-to-high RCC values achieved through CMRH. This protocol provides a robust method for preparing radiohalogenated molecules, for both diagnostic and potential therapeutic applications. Importantly, the enhanced stability of the B(Epin) and B(Ppin) groups offers greater flexibility in preparing radiolabeling precursors, which can improve PET and SPECT radiotracer development.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Experimental Procedures for Organic Syntheses, Copies of radio-(U)HPLC, HRMS and NMR data are provided within the supporting information.
Author Contributions
Conceptualization, A.C., F.S. and M.L.; methodology, A.C., F.S., and M.L.; investigation, A.C., F.S., F.B. and M.L.; resources, K.K. and S.S.; writing—original draft preparation, A.C.; writing—review and editing, F.S. and M.L.; visualization, A.C.; supervision, K.K. and S.S.; project administration, A.C., K.K. and S.S.; funding acquisition, A.C., K.K. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Helmholtz Enterprise funding (associated with Helmholtz-Gemeinschaft Deutscher Forschungszentren e.V., Impuls- und Vernetzungsfonds), project code HE-2023-08.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Material.
Acknowledgements
The authors are grateful for the support of the Dr. Martin Kreller and cyclotron team for the provision of fluorine-18 and production iodine-123 required for this work. We thank Rotop Radiopharmacy GmbH for providing [123I]iodide for research purposes. We also wish to thank Jan Opitz and Ian Moore Gilpin for their support in the preparation and analysis of the radiolabeling precursors.
Conflicts of Interest
Austin Craig, Frederik Sachse and Markus Laube are co-inventors for a patent application based on this manuscript’s findings submitted to the German Patent Office (PCT/DE2025/100397).
References
- Alavi, A.; Werner, T. J.; Stępień, E. Ł.; Moskal, P. Unparalleled and Revolutionary Impact of PET Imaging on Research and Day to Day Practice of Medicine. Bio-Algorithms and Med-Systems 2022, 17, 203–212. [Google Scholar] [CrossRef]
- Alavi, A.; Basu, S. Planar and SPECT Imaging in the Era of PET and PET–CT: Can It Survive the Test of Time? Eur J Nucl Med Mol Imaging 2008, 35, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Israel, O.; Pellet, O.; Biassoni, L.; De Palma, D.; Estrada-Lobato, E.; Gnanasegaran, G.; Kuwert, T.; La Fougère, C.; Mariani, G.; Massalha, S.; Paez, D.; Giammarile, F. Two Decades of SPECT/CT – the Coming of Age of a Technology: An Updated Review of Literature Evidence. Eur J Nucl Med Mol Imaging 2019, 46, 1990–2012. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Lee, D.; Shim, H. Metabolic Positron Emission Tomography Imaging in Cancer Detection and Therapy Response. Seminars in Oncology 2011, 38, 55–69. [Google Scholar] [CrossRef]
- Phelps, M. E. Positron Emission Tomography Provides Molecular Imaging of Biological Processes. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 9226–9233. [Google Scholar] [CrossRef]
- Ametamey, S. M.; Honer, M.; Schubiger, P. A. Molecular Imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef]
- Donnelly, D. J. Small Molecule PET Tracers in Drug Discovery. Seminars in Nuclear Medicine 2017, 47, 454–460. [Google Scholar] [CrossRef]
- Donnelly, D. J. PET Imaging in Drug Discovery and Development. In Handbook of Radiopharmaceuticals; Scott, P., Kilbourn, M., Eds.; Wiley, 2020; pp 703–725. [CrossRef]
- Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Kunos, C. A.; Mankoff, D. A.; Schultz, M. K.; Graves, S. A.; Pryma, D. A. Radiopharmaceutical Chemistry and Drug Development—What’s Changed? Seminars in Radiation Oncology 2021, 31, 3–11. [Google Scholar] [CrossRef]
- Craig, A.; Kogler, J.; Laube, M.; Ullrich, M.; Donat, C. K.; Wodtke, R.; Kopka, K.; Stadlbauer, S. Preparation of 18F-Labeled Tracers Targeting Fibroblast Activation Protein via Sulfur [18F]Fluoride Exchange Reaction. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- FDA-Approved PET Radiopharmaceuticals: http://www.radiopharmaceuticals.info/pet-radiopharmaceuticals.html (accessed 2024-08-04).
- Clarke, B. N. PET Radiopharmaceuticals: What’s New, What’s Reimbursed, and What’s Next? J Nucl Med Technol 2018, jnmt.117.205021. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H. H.; Elsinga, P. H.; Iwata, R.; Kilbourn, M. R.; Pillai, M. R. A.; Rajan, M. G. R.; Wagner, H. N.; Zaknun, J. J. Fluorine-18 Radiopharmaceuticals beyond [18F]FDG for Use in Oncology and Neurosciences. Nuclear Medicine and Biology 2010, 37, 727–740. [Google Scholar] [CrossRef]
- Bowden, G. D.; Scott, P. J. H.; Boros, E. Radiochemistry: A Hot Field with Opportunities for Cool Chemistry. ACS Cent. Sci. 2023, 9, 2183–2195. [Google Scholar] [CrossRef]
- Park, H.-M. 123I: Almost a Designer Radioiodine for Thyroid Scanning. J Nucl Med 2002, 43, 77. [Google Scholar] [PubMed]
- Snay, E. R.; Treves, S. T.; Fahey, F. H. Improved Quality of Pediatric 123I-MIBG Images with Medium-Energy Collimators. Journal of Nuclear Medicine Technology 2011, 39, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Gross, M. D. Radiochemistry, Biochemistry, and Kinetics of 131 I-metaiodobenzylguanidine (MIBG) and 123 I-MIBG: Clinical Implications of the Use of 123 I-MIBG. Med. Pediatr. Oncol. 1987, 15, 170–177. [Google Scholar] [CrossRef]
- Nuvoli, S.; Spanu, A.; Piras, M. R.; Nieddu, A.; Mulas, A.; Rocchitta, G.; Galleri, G.; Serra, P. A.; Madeddu, G. 123I-Ioflupane Brain SPECT and 123I-MIBG Cardiac Planar Scintigraphy Combined Use in Uncertain Parkinsonian Disorders. Medicine 2017, 96, e6967. [Google Scholar] [CrossRef] [PubMed]
- Chan, C. Y.; Chen, Z.; Guibbal, F.; Dias, G.; Destro, G.; O’Neill, E.; Veal, M.; Lau, D.; Mosley, M.; Wilson, T. C.; Gouverneur, V.; Cornelissen, B. [ 123 I]CC1: A PARP-Targeting, Auger Electron–Emitting Radiopharmaceutical for Radionuclide Therapy of Cancer. J Nucl Med 2023, jnumed.123.265429. [Google Scholar] [CrossRef]
- Koehler, L.; Gagnon, K.; McQuarrie, S.; Wuest, F. Iodine-124: A Promising Positron Emitter for Organic PET Chemistry. Molecules 2010, 15, 2686–2718. [Google Scholar] [CrossRef]
- Adam, M. J.; Wilbur, D. S. Radiohalogens for Imaging and Therapy. Chem. Soc. Rev. 2005, 34, 153. [Google Scholar] [CrossRef]
- Ehrhardt Jr, J. D.; Güleç, S. A Review of the History of Radioactive Iodine Theranostics: The Origin of Nuclear Ontology. Mirt 2020, 29, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, E. B. Radioiodine: The Classic Theranostic Agent. Seminars in Nuclear Medicine 2012, 42, 164–170. [Google Scholar] [CrossRef]
- Anger, H. O. Scintillation Camera. Review of Scientific Instruments 1958, 29, 27–33. [Google Scholar] [CrossRef]
- FDA-Approved Radiopharmaceuticals: http://www.radiopharmaceuticals.info/radiopharmaceuticals.html (accessed 2024-08-04).
- Antuganov, D.; Zykov, M.; Timofeev, V.; Timofeeva, K.; Antuganova, Y.; Orlovskaya, V.; Fedorova, O.; Krasikova, R. Copper-Mediated Radiofluorination of Aryl Pinacolboronate Esters: A Straightforward Protocol by Using Pyridinium Sulfonates. Eur J Org Chem 2019, 2019, 918–922. [Google Scholar] [CrossRef]
- Jeon, M. H.; Kwon, Y.-D.; Kim, M. P.; Torres, G. B.; Seo, J. K.; Son, J.; Ryu, Y. H.; Hong, S. Y.; Chun, J.-H. Late-Stage 18 F/ 19 F Isotopic Exchange for the Synthesis of 18 F-Labeled Sulfamoyl Fluorides. Org. Lett. 2021, 23, 2766–2771. [Google Scholar] [CrossRef]
- Orlovskaya, V. V.; Craig, A. S.; Fedorova, O. S.; Kuznetsova, O. F.; Neumaier, B.; Krasikova, R. N.; Zlatopolskiy, B. D. Production of 6-l-[18F]Fluoro-m-Tyrosine in an Automated Synthesis Module for 11C-Labeling. Molecules 2021, 26, 5550. [Google Scholar] [CrossRef]
- Lee, E.; Kamlet, A. S.; Powers, D. C.; Neumann, C. N.; Boursalian, G. B.; Furuya, T.; Choi, D. C.; Hooker, J. M.; Ritter, T. A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 2011, 334, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, B. H.; Stephenson, N. A.; Vasdev, N.; Liang, S. H. Spirocyclic Hypervalent Iodine(III)-Mediated Radiofluorination of Non-Activated and Hindered Aromatics. Nat Commun 2014, 5. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D. O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Dubost, E.; McErlain, H.; Babin, V.; Sutherland, A.; Cailly, T. Recent Advances in Synthetic Methods for Radioiodination. J. Org. Chem. 2020, 85, 8300–8310. [Google Scholar] [CrossRef]
- Ambrosini, V.; Zanoni, L.; Filice, A.; Lamberti, G.; Argalia, G.; Fortunati, E.; Campana, D.; Versari, A.; Fanti, S. Radiolabeled Somatostatin Analogues for Diagnosis and Treatment of Neuroendocrine Tumors. Cancers 2022, 14, 1055. [Google Scholar] [CrossRef] [PubMed]
- Okarvi, S. M. Peptide-based Radiopharmaceuticals: Future Tools for Diagnostic Imaging of Cancers and Other Diseases. Medicinal Research Reviews 2004, 24, 357–397. [Google Scholar] [CrossRef] [PubMed]
- Neels, O. C.; Kopka, K.; Liolios, C.; Afshar-Oromieh, A. Radiolabeled PSMA Inhibitors. Cancers 2021, 13, 6255. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Maecke, H. R.; Okarvi, S. M. Radiolabeled Peptides: Valuable Tools for the Detection and Treatment of Cancer. Theranostics 2012, 2, 481–501. [Google Scholar] [CrossRef]
- Gröner, B.; Willmann, M.; Donnerstag, L.; Urusova, E. A.; Neumaier, F.; Humpert, S.; Endepols, H.; Neumaier, B.; Zlatopolskiy, B. D. 7-[18 F]Fluoro-8-Azaisatoic Anhydrides: Versatile Prosthetic Groups for the Preparation of PET Tracers. J. Med. Chem. 2023, 66, 12629–12644. [Google Scholar] [CrossRef]
- Marik, J.; Sutcliffe, J. L. Click for PET: Rapid Preparation of [18F]Fluoropeptides Using CuI Catalyzed 1,3-Dipolar Cycloaddition. Tetrahedron Letters 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Glaser, M.; Årstad, E. “Click Labeling” with 2-[18 F]Fluoroethylazide for Positron Emission Tomography. Bioconjugate Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Richter, S.; Wuest, F. 18F-Labeled Peptides: The Future Is Bright. Molecules 2014, 19, 20536–20556. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Wängler, B.; Bailey, J.; Bernard-Gauthier, V.; Schirrmacher, E.; Wängler, C. Small Prosthetic Groups in 18 F-Radiochemistry: Useful Auxiliaries for the Design of 18 F-PET Tracers. Seminars in Nuclear Medicine 2017, 47, 474–492. [Google Scholar] [CrossRef]
- Chalker, J. M.; Bernardes, G. J. L.; Lin, Y. A.; Davis, B. G. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chemistry An Asian Journal 2009, 4, 630–640. [Google Scholar] [CrossRef]
- Gundam, S. R.; Callstrom, M. R.; Pandey, M. K. Synthesis and Application of 1-[18 F]Fluoro-4-Isothiocyanatobenzene for Radiofluorination of Peptides in Aqueous Medium. J. Org. Chem. 2025, 90, 458–470. [Google Scholar] [CrossRef]
- Spicer, C. D.; Davis, B. G. Selective Chemical Protein Modification. Nat Commun 2014, 5, 4740. [Google Scholar] [CrossRef]
- Moore, T. M.; Akula, M. R.; Kabalka, G. W. Fluorine-18 Radiochemistry: A Novel Thiol-Reactive Prosthetic Group, [18F]FBAMPy. NS 2016, 08, 1–7. [Google Scholar] [CrossRef]
- Ma, G.; McDaniel, J. W.; Murphy, J. M. One-Step Synthesis of [18 F]Fluoro-4-(Vinylsulfonyl)Benzene: A Thiol Reactive Synthon for Selective Radiofluorination of Peptides. Org. Lett. 2021, 23, 530–534. [Google Scholar] [CrossRef]
- Failla, M.; Floresta, G.; Abbate, V. Peptide-Based Positron Emission Tomography Probes: Current Strategies for Synthesis and Radiolabelling. RSC Med. Chem. 2023, 14, 592–623. [Google Scholar] [CrossRef]
- Kniess, T.; Kuchar, M.; Pietzsch, J. Automated Radiosynthesis of the Thiol-Reactive Labeling Agent N-[6-(4-[18F]Fluorobenzylidene)Aminooxyhexyl]Maleimide ([18F]FBAM). Applied Radiation and Isotopes 2011, 69, 1226–1230. [Google Scholar] [CrossRef]
- Ichiishi, N.; Brooks, A. F.; Topczewski, J. J.; Rodnick, M. E.; Sanford, M. S.; Scott, P. J. H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(Aryl)Iodonium Salts. Org Lett 2014, 16, 3224–3227. [Google Scholar] [CrossRef]
- Mossine, A. V.; Brooks, A. F.; Makaravage, K. J.; Miller, J. M.; Ichiishi, N.; Sanford, M. S.; Scott, P. J. H. Synthesis of [ 18 F]Arenes via the Copper-Mediated [ 18 F]Fluorination of Boronic Acids. Org. Lett. 2015, 17, 5780–5783. [Google Scholar] [CrossRef]
- Makaravage, K. J.; Brooks, A. F.; Mossine, A. V.; Sanford, M. S.; Scott, P. J. H. Copper-Mediated Radiofluorination of Arylstannanes with [ 18 F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- Lee, S. J.; Makaravage, K. J.; Brooks, A. F.; Scott, P. J. H.; Sanford, M. S. Copper-Mediated Aminoquinoline-Directed Radiofluorination of Aromatic C−H Bonds with K 18 F. Angew. Chem. Int. Ed. 2019, 58, 3119–3122. [Google Scholar] [CrossRef]
- Craig, A.; Kolks, N.; Urusova, E. A.; Zischler, J.; Brugger, M.; Endepols, H.; Neumaier, B.; Zlatopolskiy, B. D. Preparation of Labeled Aromatic Amino Acids via Late-Stage 18 F-Fluorination of Chiral Nickel and Copper Complexes. Chem. Commun. 2020, 56, 9505–9508. [Google Scholar] [CrossRef]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B. D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chemistry 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Bolik, K.-V.; Hellmann, J.; Maschauer, S.; Neu, E.; Einsiedel, J.; Riss, P.; Vogg, N.; König, J.; Fromm, M. F.; Hübner, H.; Gmeiner, P.; Prante, O. Heteroaryl Derivatives of Suvorexant as OX1R Selective PET Ligand Candidates: Cu-Mediated 18F-Fluorination of Boroxines, in Vitro and Initial in Vivo Evaluation. EJNMMI Res 2024, 14, 80. [Google Scholar] [CrossRef]
- Tredwell, M.; Preshlock, S. M.; Taylor, N. J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18 F Fluorination of Arenes. Angew Chem Int Ed 2014, 53, 7751–7755. [Google Scholar] [CrossRef]
- Mixdorf, J. C.; Hoffman, S. L. V.; Aluicio-Sarduy, E.; Barnhart, T. E.; Engle, J. W.; Ellison, P. A. Copper-Mediated Radiobromination of (Hetero)Aryl Boronic Pinacol Esters. J. Org. Chem. 2023, 88, 2089–2094. [Google Scholar] [CrossRef]
- Reilly, S. W.; Makvandi, M.; Xu, K.; Mach, R. H. Rapid Cu-Catalyzed [ 211 At]Astatination and [ 125 I]Iodination of Boronic Esters at Room Temperature. Org. Lett. 2018, 20, 1752–1755. [Google Scholar] [CrossRef]
- Wilson, T. C.; McSweeney, G.; Preshlock, S.; Verhoog, S.; Tredwell, M.; Cailly, T.; Gouverneur, V. Radiosynthesis of SPECT Tracers via a Copper Mediated 123 I Iodination of (Hetero)Aryl Boron Reagents. Chem. Commun. 2016, 52, 13277–13280. [Google Scholar] [CrossRef]
- Oka, N.; Yamada, T.; Sajiki, H.; Akai, S.; Ikawa, T. Aryl Boronic Esters Are Stable on Silica Gel and Reactive under Suzuki–Miyaura Coupling Conditions. Org. Lett. 2022, 24, 3510–3514. [Google Scholar] [CrossRef]
- Gaube, G.; Miller, D.; McCallum, R.; Pipaon Fernandez, N.; Leitch, D. Base-Free Palladium-Catalyzed Borylation of Enol Carboxylates and Further Reactivity toward Deboronation and Cross-Coupling. September 19, 2024. [CrossRef]
- Geaneotes, P. J.; Janosko, C. P.; Afeke, C.; Deiters, A.; Floreancig, P. E. Potent and Selective Oxidatively Labile Ether-Based Prodrugs through Late-Stage Boronate Incorporation. Angew Chem Int Ed 2024, e202409229. [Google Scholar] [CrossRef]
- Kreller, M.; Pietzsch, H.; Walther, M.; Tietze, H.; Kaever, P.; Knieß, T.; Füchtner, F.; Steinbach, J.; Preusche, S. Introduction of the New Center for Radiopharmaceutical Cancer Research at Helmholtz-Zentrum Dresden-Rossendorf. Instruments 2019, 3, 9. [Google Scholar] [CrossRef]
- Laube, M.; Brandt, F.; Kopka, K.; Pietzsch, H.-J.; Pietzsch, J.; Loeser, R.; Wodtke, R. Development of 123I-Labelled Acrylamides as Radiotracer Candidates for Transglutaminase 2. Nuclear Medicine and Biology 2021, 96–97, S79–S80. [Google Scholar] [CrossRef]
- Rubio-Presa, R.; Suárez-Pantiga, S.; Pedrosa, M. R.; Sanz, R. Molybdenum-Catalyzed Sustainable Friedländer Synthesis of Quinolines. Adv Synth Catal 2018, 360, 2216–2220. [Google Scholar] [CrossRef]
- Kanagasundaram, T.; Laube, M.; Wodtke, J.; Kramer, C. S.; Stadlbauer, S.; Pietzsch, J.; Kopka, K. Radiolabeled Silicon-Rhodamines as Bimodal PET/SPECT-NIR Imaging Agents. Pharmaceuticals 2021, 14, 1155. [Google Scholar] [CrossRef]
- Murphy, C. L.; Hall, A.; Roberts, E. J.; Ryan, M. D.; Clary, J. W.; Singaram, B. Preparation and Reactions of 4-Iodobutyl Pinacolborate. Synthesis of Substituted Alkyl and Aryl Pinacolboronates via 4-Iodobutyl Pinacolborate Utilizing Tetrahydrofuran as the Leaving Group. Tetrahedron Letters 2015, 56, 3032–3033. [Google Scholar] [CrossRef]
- Cai, H.; Conti, P. S. RGD-based PET Tracers for Imaging Receptor Integrin αv β3 Expression. Labelled Comp Radiopharmac 2013, 56, 264–279. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled Cyclic RGD Peptides as Integrin αv β3 -Targeted Radiotracers: Maximizing Binding Affinity via Bivalency. Bioconjugate Chem. 2009, 20, 2199–2213. [Google Scholar] [CrossRef]
- Beyer, L.; Gosewisch, A.; Lindner, S.; Völter, F.; Mittlmeier, L. M.; Tiling, R.; Brendel, M.; Cyran, C. C.; Unterrainer, M.; Rübenthaler, J.; Auernhammer, C. J.; Spitzweg, C.; Böning, G.; Gildehaus, F. J.; Jurkschat, K.; Wängler, C.; Wängler, B.; Schirrmacher, R.; Wenter, V.; Todica, A.; Bartenstein, P.; Ilhan, H. Dosimetry and Optimal Scan Time of [18F]SiTATE-PET/CT in Patients with Neuroendocrine Tumours. Eur J Nucl Med Mol Imaging 2021, 48, 3571–3581. [Google Scholar] [CrossRef]
- Pauwels, E.; Cleeren, F.; Tshibangu, T.; Koole, M.; Serdons, K.; Boeckxstaens, L.; Dekervel, J.; Vandamme, T.; Lybaert, W.; Den Broeck, B. V.; Laenen, A.; Clement, P. M.; Geboes, K.; Cutsem, E. V.; Stroobants, S.; Verslype, C.; Bormans, G.; Deroose, C. M. 18 F-AlF-NOTA-Octreotide Outperforms68 Ga-DOTATATE/NOC PET in Neuroendocrine Tumor Patients: Results from a Prospective, Multicenter Study. J Nucl Med 2023, 64, 632–638. [Google Scholar] [CrossRef]
- Taylor, N. J.; Emer, E.; Preshlock, S.; Schedler, M.; Tredwell, M.; Verhoog, S.; Mercier, J.; Genicot, C.; Gouverneur, V. Derisking the Cu-Mediated 18 F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc. 2017, 139, 8267–8276. [Google Scholar] [CrossRef]
- Wright, J. S.; Sharninghausen, L. S.; Preshlock, S.; Brooks, A. F.; Sanford, M. S.; Scott, P. J. H. Sequential Ir/Cu-Mediated Method for the Meta -Selective C–H Radiofluorination of (Hetero)Arenes. J. Am. Chem. Soc. 2021, 143, 6915–6921. [Google Scholar] [CrossRef]
- Hitosugi, S.; Tanimoto, D.; Nakanishi, W.; Isobe, H. A Facile Chromatographic Method for Purification of Pinacol Boronic Esters. Chemistry Letters 2012, 41, 972–973. [Google Scholar] [CrossRef]
- Ellison, P. A.; Olson, A. P.; Barnhart, T. E.; Hoffman, S. L. V.; Reilly, S. W.; Makvandi, M.; Bartels, J. L.; Murali, D.; DeJesus, O. T.; Lapi, S. E.; Bednarz, B.; Nickles, R. J.; Mach, R. H.; Engle, J. W. Improved Production of 76Br, 77Br and 80mBr via CoSe Cyclotron Targets and Vertical Dry Distillation. Nuclear Medicine and Biology 2020, 80–81, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Samanta, M.; Martorano, P.; Lee, H.; Gitto, S. B.; Patel, K.; Groff, D.; Pogoriler, J.; Martinez, D.; Riad, A.; Dabagian, H.; Zaleski, M.; Taghvaee, T.; Xu, K.; Lee, J. Y.; Hou, C.; Farrel, A.; Batra, V.; Carlin, S. D.; Powell, D. J.; Mach, R. H.; Pryma, D. A.; Maris, J. M. Pre-Clinical Investigation of Astatine-211-Parthanatine for High-Risk Neuroblastoma. Commun Biol 2022, 5, 1260. [Google Scholar] [CrossRef]
Scheme 1.
(A–D) Previous CMRH preparations of radiolabeled arenes. (E) This work: Preparation of radiolabeled arenes via CMRH of divergent ArB(Epin/Ppin) substrates.
Scheme 1.
(A–D) Previous CMRH preparations of radiolabeled arenes. (E) This work: Preparation of radiolabeled arenes via CMRH of divergent ArB(Epin/Ppin) substrates.
Figure 1.
A) UV-visualized TLC experiment using radiolabeling precursors 37, 38, 5a and 5b, mobile phase: cyclohexane/EtOAc (4:1); B) Stability studies using aliquots of 38, 5a and 5b in MeCN/H2O (1:1).
Figure 1.
A) UV-visualized TLC experiment using radiolabeling precursors 37, 38, 5a and 5b, mobile phase: cyclohexane/EtOAc (4:1); B) Stability studies using aliquots of 38, 5a and 5b in MeCN/H2O (1:1).
Scheme 2.
Preparation of A) Epin (2) and B) Ppin (4) from commercially available reagents.
Scheme 3.
Preparation of ArB(Epin) and ArB(Ppin) radiolabeling precursors 5a-23a and 5b-23b, respectively from aryl boronic acids. Conditions: ArB(OH)2 1 (1.0 equiv), Epin (1.2 equiv) and Na2SO4 (2.0 equiv.) in CH2Cl2 (0.50 M) at 40 °C for 16 h.
Scheme 3.
Preparation of ArB(Epin) and ArB(Ppin) radiolabeling precursors 5a-23a and 5b-23b, respectively from aryl boronic acids. Conditions: ArB(OH)2 1 (1.0 equiv), Epin (1.2 equiv) and Na2SO4 (2.0 equiv.) in CH2Cl2 (0.50 M) at 40 °C for 16 h.
Scheme 4.
Preparation of divergent radiolabeling precursor 36 and reference compounds 33 & 35.
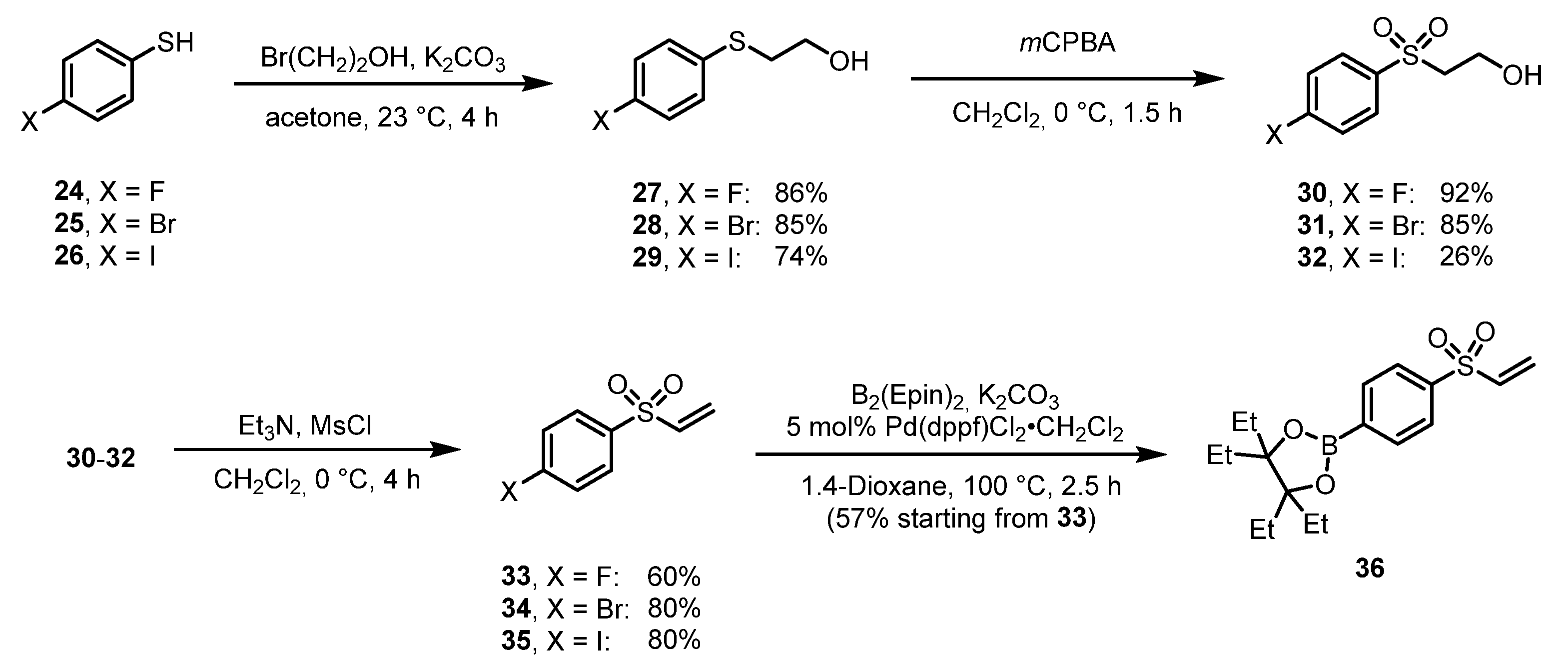
Figure 2.
A) 18F-Fluorination optimization with varying Cu mediator equivalents; and B) CMRF optimization with various reaction solvents; and C) 18F-Elution with various PTAs.
Figure 2.
A) 18F-Fluorination optimization with varying Cu mediator equivalents; and B) CMRF optimization with various reaction solvents; and C) 18F-Elution with various PTAs.
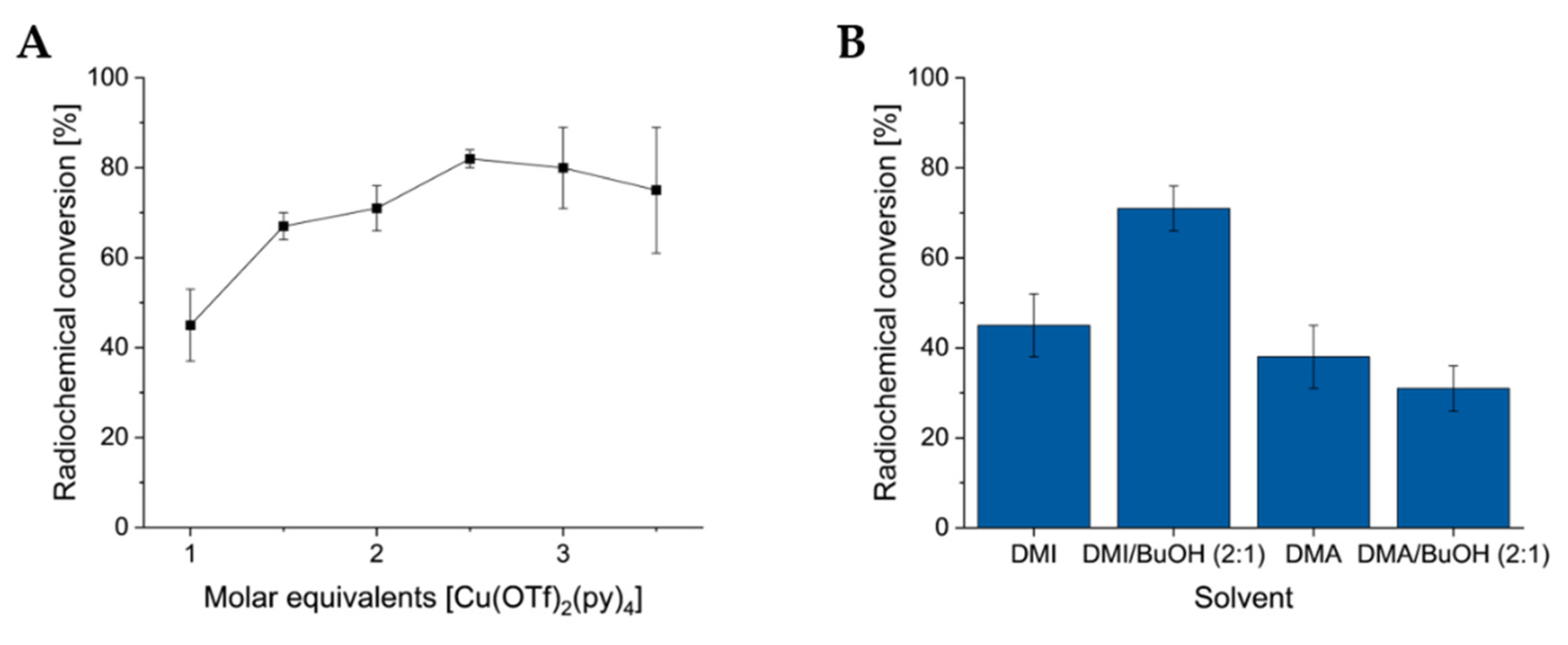
Scheme 5.
Substrate scope for the CMRH of ArB(Epin)s and ArB(Ppin)s. Radiolabeled products were prepared from the respective [a] (hetero)ArB(Epin), or [b] (hetero)ArB(Ppin) precursors. If not otherwise stated, mean (±) standard deviation values for RCC are provided. All experiments were performed at least in duplicate for radioiodinations, and triplicate for radiofluorinations.
Scheme 5.
Substrate scope for the CMRH of ArB(Epin)s and ArB(Ppin)s. Radiolabeled products were prepared from the respective [a] (hetero)ArB(Epin), or [b] (hetero)ArB(Ppin) precursors. If not otherwise stated, mean (±) standard deviation values for RCC are provided. All experiments were performed at least in duplicate for radioiodinations, and triplicate for radiofluorinations.
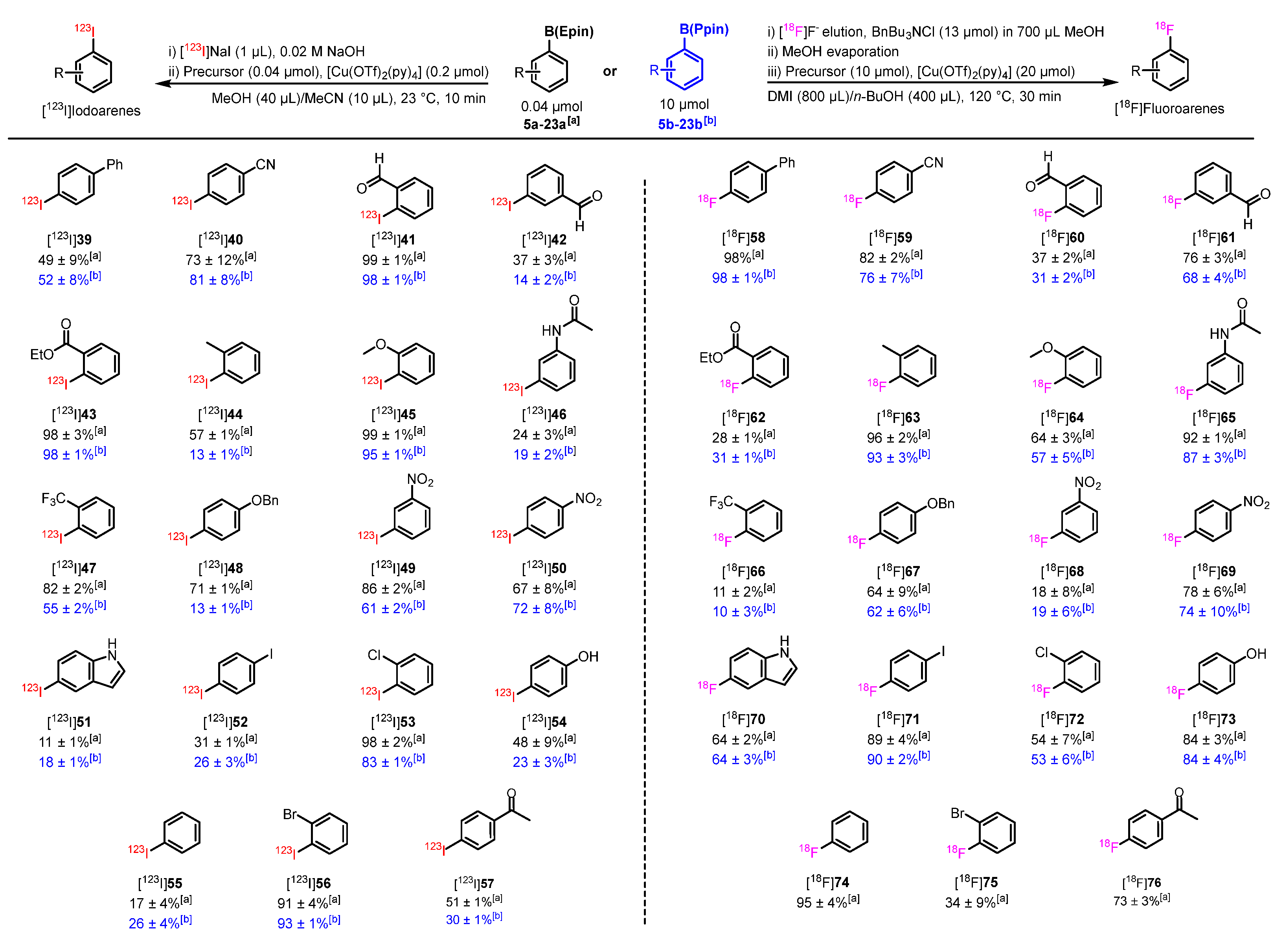
Scheme 6.
A) Preparation of [18F]33; B-D) Regioselective 18F-fluorination compounds via cysteine bioconjugation using [18F]33. Reaction conditions: Cys-containing compound (0.5-3.0 mg), [18F]33 (50-500 MBq), sodium borate buffer pH 8.5/MeOH (1:1, 1-2 mL), 35 °C, 30 min. Radio-HPLC was used to determine the non-isolated radiochemical yields (RCYHPLC) of the radiolabeled products. The RCYHPLC and was calculated by the division of the integrated product peak area by the total integrated 18F-labeled peaks. The identity of the 18F-labeled products was confirmed by coinjection with the “cold” 19F-reference standard.
Scheme 6.
A) Preparation of [18F]33; B-D) Regioselective 18F-fluorination compounds via cysteine bioconjugation using [18F]33. Reaction conditions: Cys-containing compound (0.5-3.0 mg), [18F]33 (50-500 MBq), sodium borate buffer pH 8.5/MeOH (1:1, 1-2 mL), 35 °C, 30 min. Radio-HPLC was used to determine the non-isolated radiochemical yields (RCYHPLC) of the radiolabeled products. The RCYHPLC and was calculated by the division of the integrated product peak area by the total integrated 18F-labeled peaks. The identity of the 18F-labeled products was confirmed by coinjection with the “cold” 19F-reference standard.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



