Submitted:
04 June 2025
Posted:
05 June 2025
You are already at the latest version
Abstract
Cancer is a major threat to human health, and radiotherapy is a key treatment method. However, its effectiveness is often limited by tumor radioresistance. The PI3K/AKT/mTOR signaling pathway is commonly dysregulated in cancers and plays a significant role in radioresistance, though its exact mechanisms remain unclear. This review discusses how this pathway regulates tumor radiosensitivity and highlights recent progress in the development of related inhibitors in preclinical and clinical studies. These findings aim to guide clinical treatment strategies and provide new approaches to overcoming radioresistance.
Keywords:
PI3K
; AKT
; mTOR
; radiosensitivity
1. Introduction
In recent years, the incidence of cancer has been steadily rising, making it one of the leading threats to human health. According to the American Cancer Society's 2024 statistics, cancer has become the primary cause of death for individuals under the age of 85, with over 2 million new cases and 610,000 deaths expected in 2024 [1]. Radiotherapy is a major treatment modality for malignant tumors, with approximately 50% of patients requiring radiotherapy during their treatment [2]. Radiotherapy utilizes high-energy radiation to target tumor tissues, damage cellular DNA, and inhibit cell proliferation, thereby killing tumor cells and controlling tumor growth. However, the resistance of tumor cells to radiotherapy significantly limits its therapeutic efficacy [3,4,5].To address this challenge, research efforts have focused on two main strategies: increasing radiation doses and enhancing tumor radiosensitivity. Advanced technologies such as intensity-modulated radiotherapy (IMRT) and stereotactic body radiotherapy (SBRT) allow higher radiation doses to be delivered while minimizing damage to normal tissues, achieving promising outcomes in diseases like liver and lung cancers [6,7]. Additionally, improving radiosensitivity requires in-depth exploration of the molecular differences between radiosensitive and radioresistant tumor cells, as well as identifying critical targets to achieve radiosensitization by inhibiting relevant signaling pathways [8].
With the advancements in targeted therapy, radiotherapy is no longer limited to local treatment but has evolved into a systemic therapeutic approach by combining with targeted therapies, thereby increasing the treatment efficacy. In addition, occurrence of abscopal effects have been described where radiotherapy-induced regression or remission occurs in unirradiated distant metastatic lesions when a local tumor site is irradiated[9,10]. The PI3K/AKT/mTOR signaling pathway plays a central role in regulating cell proliferation, survival, metabolism, and transcription, and is a critical network in tumor development and progression [11,12,13]. Studies have shown that overactivation of this pathway can reduce tumor radiosensitivity through mechanisms such as promoting DNA damage repair, inhibiting apoptosis, and regulating the cell cycle [14]. In recent years, inhibitors targeting the PI3K/AKT/mTOR pathway have received significant attention, providing new directions to enhance the effectiveness of radiotherapy and demonstrating potential in multiple clinical trials [15]. This review focuses on the role and molecular mechanisms of the PI3K/AKT/mTOR signaling pathway in tumor radioresistance, summarizes the progress in research on related inhibitors, and evaluates their potential applications in clinical radiotherapy. Through this review, we aim to provide new insights and therapeutic strategies for overcoming tumor radioresistance.
2. The Role of the PI3K/AKT/mTOR Pathway in Tumor Radioresistance
The PI3K/AKT/mTOR pathway plays an important role in various processes of irradiated tumor cells including DNA repair, cell ycle regulation, proliferation, apoptosis, autophagy, hypoxia, cancer stem cell slf-renewal, invasion and metastasis as described in the next chapters in more detail (Figure 1).
2.1. DNA Damage Repair
DNA double-strand breaks (DSBs) are among the most severe forms of damage caused by ionizing radiation and can trigger a series of DNA damage responses (DDRs), including damage sensing, signal transduction, cell cycle arrest, and the activation of DNA repair mechanisms [16]. The ability of cells to successfully repair radiation-induced DNA damage largely determines their fate, and dysregulation of DNA repair pathways is closely associated with tumor radioresistance. Ionizing radiation generates approximately 10,000 DNA base lesions, 1,000 single-strand breaks (SSBs), and 40 DSBs per gray of radiation dose [17]. Various types of DNA damage, including adducts, intrastrand and interstrand crosslinks, SSBs, and DSBs, activate distinct repair mechanisms. These primarily include base excision repair, nucleotide excision repair, mismatch repair, homologous recombination (HR), and non-homologous end joining (NHEJ) [18].
The PI3K/AKT/mTOR signaling pathway plays a crucial role in DNA repair by regulating the NHEJ and HR repair pathways. Aberrant activation of this pathway significantly enhances DNA repair capacity, thereby reducing the antitumor effects of radiotherapy. Studies have shown that PI3K/mTOR inhibitors, such as PKI-587 and NVP-BEZ235, can markedly disrupt NHEJ repair efficiency, primarily by modulating the activity of DNA-dependent protein kinase catalytic subunit (DNA-PKcs). The NHEJ process begins with the recognition of DSBs by the Ku70/Ku80 complex, followed by recruitment and activation of DNA-PKcs to facilitate the repair process. Phosphorylation of DNA-PKcs at the Serine 2056 (S2056) site is a critical step in assembling the repair complex. Hyperactivation of the PI3K/mTOR pathway enhances DNA-PKcs phosphorylation, accelerating DSB repair and increasing tumor cell resistance to radiation-induced damage. Conversely, PI3K/mTOR inhibitors significantly reduce DNA-PKcs (S2056) phosphorylation, weaken the binding efficiency of Ku70/Ku80 to broken DNA, and delay repair initiation. This is evidenced by the prolonged dissipation of γ-H2AX foci, indicating reduced NHEJ repair efficiency and increased tumor radiosensitivity [19,20]. Furthermore, Apatinib delays γ-H2AX foci disappearance by suppressing the PI3K/AKT signaling pathway, thereby reducing HR efficiency. It also impairs DDR by inhibiting AKT phosphorylation, leading to decreased recruitment and activation of repair proteins like Rad51 Recombinase(Rad51) [21].
In the HR repair process, mTOR signaling enhances repair efficiency by regulating Fanconi anemia complementation group D2(FANCD2) expression. FANCD2 is a key factor in the Fanconi anemia pathway, and its function depends on the activation of Ataxia telangiectasia mutated(ATM)/Chk2 signaling. Studies have shown that mTOR inhibitors, such as AZD8055, can downregulate FANCD2 expression, reduce ATM/Chk2 activity, and weaken the repair capacity of the Fanconi anemia pathway, thereby exacerbating DNA damage accumulation. Additionally, FANCD2 downregulation further suppresses Ataxia telangiectasia and Rad3-related(ATR)/Chk1 signaling, limiting tumor cells' ability to respond to radiation-induced damage [22].
2.2. Cell Cycle
The cell cycle consists of four phases: the mitotic (M) phase, DNA synthesis (S) phase, and the gap phases G1 and G2 [23]. The radiosensitivity of cells is closely related to their cycle phase, with cells being most radiosensitive during the G2 and M phases, less sensitive during the G1 phase, and least sensitive in the late S phase [24].
In nasopharyngeal carcinoma, overexpression of Chromosome 2 open reading frame 40(C2orf40) regulates the PI3K/AKT/mTOR pathway, significantly reducing the protein expression levels of key cell cycle regulators, including Cyclin-dependent kinase 1(CDK1), Cyclin E1, and Cyclin B1. It also inhibits the phosphorylation of CDK1 and Retinoblastoma protein (Rb), leading to G2/M phase cell cycle arrest. This G2/M arrest not only reduces tumor cell proliferation but also delays the repair of radiation-induced DNA damage [25]. In esophageal squamous cell carcinoma, high Family with sequence similarity 135, member B (FAM135B) expression activates the PI3K/AKT/mTOR pathway, shortening the duration of G2/M arrest. Knockdown of FAM135B significantly decreases p-AKT and p-mTOR levels, reduces CDK1 phosphorylation, and weakens the activities of Chk2 and Cyclin B1, thereby delaying the repair of DSBs [26]. In gastric cancer models, high expression of the adenosine A2a receptor (A2aR) enhances AKT and mTOR activity, accelerating DSB repair and improving tumor cell survival. Additionally, A2aR overexpression induces G1 phase arrest and significantly upregulates stem cell markers such as Nanog, octamer-binding transcription factor 4(OCT-4), and SRY-box transcription factor 2(SOX-2), providing tumor cells with enhanced proliferative and repair capabilities. Treatment with an A2aR-specific antagonist effectively suppresses PI3K/AKT/mTOR signaling activation and reduces the occurrence of G1 phase arrest [27].
2.3. Cell Proliferation and Apoptosis
The proliferation and apoptosis of tumor cells are key characteristics of tumors and important indicators for evaluating tumor malignancy and behavior. In glioblastoma (GBM) models, studies have shown that overexpression of miR-4524b-5p downregulates the PI3K/AKT/mTOR signaling pathway, inhibiting the expression of the proliferation-related factor Ki-67. Furthermore, miR-4524b-5p activates the mitochondrial-mediated apoptosis pathway, characterized by upregulation of pro-apoptotic proteins Bcl-2-associated X protein(Bax) and cleaved caspase-3, and a significant downregulation of the anti-apoptotic protein B-cell lymphoma 2(Bcl-2). These changes markedly enhance radiotherapy-induced apoptosis [28]. Similar molecular alterations have been observed in other tumor models. In prostate cancer, high expression of Apelin Receptor(APLNR) activates the PI3K/AKT/mTOR signaling pathway, increasing levels of p-PI3K, p-AKT, and p-mTOR. This activation further upregulates Bcl-2 expression, inhibits Bax activity, and reduces cleaved caspase-3 levels. These changes diminish tumor cells’ response to radiation-induced apoptosis, thereby increasing radioresistance [29]. In non-small cell lung cancer (NSCLC) models, overexpression of Sirtuin 6(SIRT6) downregulates p-PI3K, p-AKT, and p-mTOR levels, significantly inhibiting the expression of the proliferation marker Ki-67. Additionally, SIRT6 upregulates Bax and cleaved caspase-3 levels while suppressing the anti-apoptotic protein Bcl-2, thereby activating pro-apoptotic pathways. These combined effects substantially enhance tumor cell radiosensitivity [30].
2.4. Cell Invasion and Metastasis
Ionizing radiation can alter tumor cell phenotypes or the tumor microenvironment, increasing the invasiveness of residual tumor cells and thereby raising the likelihood of metastasis [31]. Tumor cells can acquire invasive mesenchymal traits through the epithelial-mesenchymal transition (EMT) process, characterized by the loss of cell polarity, decreased adhesion, and enhanced motility and invasiveness. These features collectively facilitate the metastasis of primary tumors [32].
In radioresistant prostate cancer models, aberrant activation of the PI3K/AKT/mTOR signaling pathway significantly promotes EMT and the formation of cancer stem cell (CSC) phenotypes. This is evident from the downregulation of the epithelial marker E-cadherin and the upregulation of mesenchymal markers such as N-cadherin, Vimentin, OCT3/4, SOX2, and α-smooth muscle actin(SMA). These changes markedly enhance the invasive capability and sphere-forming ability of tumor cells while increasing their resistance to radiotherapy [33].In another prostate cancer study, overexpression of the epithelial cell adhesion molecule (EpCAM) significantly elevated the phosphorylation levels of key PI3K/AKT/mTOR pathway molecules, including p-AKT, p-mTOR, and p-ribosomal protein S6 kinase(S6K), further activating this pathway. High EpCAM expression also promoted the transition of cells from an epithelial state to a mesenchymal state by downregulating E-cadherin and upregulating Vimentin and N-cadherin, thereby enhancing cell migration and invasion [34]. In gastric cancer models, the high expression of caldesmon 1(CALD1) further underscores the pivotal role of the PI3K/AKT/mTOR pathway in EMT induction. CALD1 upregulates PI3K, p-AKT, and p-mTOR expression while inhibiting phosphatase and tensin homolog (PTEN ) expression, significantly activating this pathway. This activation leads to the downregulation of E-cadherin and Claudin-1 and the upregulation of N-cadherin and Vimentin, enhancing tumor cell migration and invasion. Additionally, PI3K/AKT inhibitors can partially reverse the EMT phenotypic changes induced by CALD1, suggesting that CALD1 regulates EMT via the PI3K/AKT/mTOR pathway [35].
2.5. Autophagy and Hypoxia
The hypoxic state of solid tumors is a critical factor contributing to the suboptimal efficacy of radiotherapy. A combination of physical, chemical, and biological processes reduces the sensitivity of hypoxic tumor cells to ionizing radiation and induces hypoxia-related therapeutic resistance [36].
Hypoxia-inducible factor 1-alpha (HIF-1α) is a key transcription factor that supports tumor growth, metabolism, invasion, and metastasis under hypoxic microenvironments [37]. Radiotherapy activates the PI3K/AKT/mTOR signaling pathway, promoting HIF-1α protein synthesis and enhancing its interaction with heat shock protein 90 (Hsp90), thereby stabilizing HIF-1α. Inhibition of Hsp90 or the PI3K/AKT/mTOR pathway significantly reduces radiotherapy-induced HIF-1α expression and diminishes vascular endothelial growth factor (VEGF) levels, weakening angiogenesis and enhancing the radiosensitivity of lung cancer cells. Additionally, HIF-1α collaborates with glucose transporter 1 (Glut-1) to drive hypoxia-induced radioresistance. HIF-1α regulates Glut-1 expression via the PI3K/AKT/mTOR pathway, facilitating glucose uptake and glycolytic metabolism in tumor cells to provide energy support. Knockdown of HIF-1α or Glut-1 genes, or inhibition of the PI3K/AKT/mTOR pathway, reduces hypoxia-induced radioresistance, further validating this mechanism [38].
Under mild hypoxia, autophagy serves as one of the main drivers of tumor cell radioresistance [38]. Studies have shown that cellular Jun proto-oncogene(c-Jun) knockdown suppresses autophagy activation by inhibiting the PI3K/AKT/mTOR signaling pathway, thereby increasing radiation sensitivity. This is evidenced by upregulated expression of p-PI3K, p-AKT, p-mTOR, and Sequestosome-1(P62), alongside downregulated expression of the autophagy-related molecule Microtubule-Associated Protein 1 Light Chain 3 II(LC3-II ) [39]. Furthermore, high expression of Neural precursor cell expressed, developmentally downregulated 8(NEDD8) enhances autophagy activation by inhibiting the PI3K/AKT/mTOR pathway, significantly increasing tumor cell radioresistance. Specifically, NEDD8 overexpression elevates protein levels of autophagy-related molecules Beclin-1, Autophagy-related gene 5 (ATG5), and LC3-II, whereas NEDD8 knockdown reduces these molecules’ expression and markedly improves tumor cell radiosensitivity [40]. In hepatocellular carcinoma models, inhibition of mTOR phosphorylation significantly activates autophagy and enhances radiation-induced apoptosis in tumor cells. Under combined radiotherapy conditions, suppression of mTOR phosphorylation increases autophagosome formation, upregulates the expression of autophagy markers LC3-II and Beclin-1, and downregulates P62 expression, indicating significantly enhanced autophagic activity [41].
2.6. Cancer Stem Cells
Cancer stem cells (CSCs) are a subset of tumor cells with self-renewal capacity and tumor-initiating potential, capable of generating a large population of non-tumorigenic progeny. Because CSCs can initiate new tumors, their incomplete elimination during anticancer treatment may lead to tumor recurrence and metastasis.
The PI3K/AKT/mTOR signaling pathway plays a critical role in CSCs by maintaining stemness, regulating proliferation and differentiation, promoting EMT, facilitating migration, and modulating autophagy [42]. This pathway stabilizes SOX2 within the nucleus by preventing its proteasomal degradation and cytoplasmic retention, thereby maintaining normal stem cell characteristics and functionality [43]. Studies have demonstrated that radiation activates the PI3K/AKT pathway, upregulating SOX2 expression and enhancing the self-renewal and differentiation capacity of colorectal CSC-like cells. Furthermore, inhibition of the PI3K/AKT pathway reduces the expression of SOX2 and CSC markers, such as Cluster of Differentiation 44(CD44), and impairs the migration and invasion capabilities of CSCs [44]. In breast cancer CSC models, HIF-2α upregulates CD44 expression via the PI3K/AKT/mTOR pathway, significantly enhancing CSC survival and radioresistance [45]. Similarly, in radioresistant prostate cancer cell models, activation of the PI3K/AKT/mTOR pathway promotes EMT and reinforces CSC phenotypes. This phenotypic change is characterized by downregulation of E-cadherin and upregulation of mesenchymal markers such as Vimentin and N-cadherin, leading to a marked increase in cell migratory capacity [46].
3. The Impact of Different PI3K/AKT/mTOR Isoforms on Radiosensitivity
3.1. PI3K Isoforms
PI3K is divided into three classes: I, II, and III, each consisting of specific regulatory and catalytic subunits with distinct substrate specificities and functions. These classes regulate cellular signaling and metabolic pathways in diverse ways [47]. Among them, Class I PI3Ks (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ) are the focus of cancer research and the primary subject of this review [48].
PI3Kα regulates key cellular processes such as survival and proliferation, and its hyperactivation is a major contributor to cancer therapy resistance [49]. Danyaei et al. demonstrated that CRISPR/Cas9-mediated knockout of the PI3Kα gene in MDA-MB-231 breast cancer cells, combined with radiotherapy, significantly enhanced apoptosis, inhibited proliferation, and reduced angiogenesis within the tumor microenvironment [50]. In esophageal squamous cell carcinoma (ESCC) models, the PI3Kα-selective inhibitor CYH33 combined with radiotherapy suppressed PI3K/AKT signaling, reduced phosphorylation of p-AKT and Forkhead Box O1 (FOXO1), delayed DSB repair, induced G2/M cell cycle arrest, and enhanced apoptosis while improving the tumor immune microenvironment [51]. Additionally, Korovina et al. found that the PI3Kα inhibitor Alpelisib demonstrated significant radiosensitizing effects in head and neck squamous cell carcinoma (HNSCC) models. However, resistance was observed in some models, which could be overcome by co-inhibiting β1 integrin, further enhancing radiotherapy efficacy [52].
PI3Kβ plays a crucial role in PTEN-deficient tumors and is associated with thrombosis, male fertility, and fragile X syndrome [53]. Regarding DNA damage repair, studies have shown that PI3Kβ recruits Nijmegen Breakage Syndrome 1 protein(Nbs1) protein to activate the MRE11-RAD50-NBS1 complex(MRN) complex and ATM/ATR signaling pathways, significantly enhancing DSB repair capacity [54]. Furthermore, PI3Kβ interacts with Replication Factor C Subunit 1(RFC1) to regulate RFC complex assembly and promotes DNA replication and repair through direct interaction with Proliferating Cell Nuclear Antigen(PCNA) [55,56].
PI3Kγ is expressed in most human tissues, predominantly in tumor-associated macrophages (TAMs), where it is abnormally activated and maintains the immune-suppressive M2 polarization phenotype, thereby weakening anti-tumor immune responses [57]. Inhibition of PI3Kγ can reprogram TAMs into pro-inflammatory M1 phenotypes, enhancing CD8+ T cell-mediated anti-tumor immunity and significantly improving radiotherapy efficacy [58]. Additionally, the PI3Kδ/γ dual inhibitor BR101801 combined with radiotherapy reduces the proportion of regulatory T cells (Tregs) in the tumor microenvironment, enhances CD8+ T cell activity, and significantly increases the occurrence of the abscopal effect [59].
PI3Kδ is predominantly expressed in hematopoietic cells but also plays a role in various tumor cells [60]. PI3Kδ inhibitors combined with radiotherapy can reverse the immunosuppressive state of the tumor microenvironment by reducing Treg and myeloid-derived suppressor cell (MDSC) infiltration, thereby enhancing CD8+ T cell activity [61,62]. Furthermore, PI3Kδ inhibitors such as Idelalisib, when used with radiotherapy, significantly enhance DNA damage response and promote apoptosis. However, in some models, they also increase radiotherapy-induced toxicity [63,64](Figure 2).
3.2. AKT Isoforms
AKT consists of three isoforms: AKT1, AKT2, and AKT3, each playing unique roles in physiological functions and oncogenic mechanisms. AKT1 is essential for placental development, cell proliferation, and growth [65]; AKT2 is critical for maintaining glucose homeostasis [66]; and AKT3 is vital for brain development, as its absence results in brain underdevelopment in mice [67]. Increasing evidence highlights the distinct roles of AKT1, AKT2, and AKT3 in tumor progression and radiation resistance [68].
AKT1 significantly affects radiosensitivity by regulating DNA repair pathways. Studies by Toulany et al. showed that AKT1 activation enhances the activity of DNA-PKcs, promoting NHEJ repair and increasing radiation resistance. Inhibiting AKT1 delays DSB repair and enhances apoptosis through cleaved PARP upregulation [69]. Further research revealed that AKT1 is crucial during both the initiation and completion phases of NHEJ repair, facilitating Ku70/Ku80 binding to DNA. AKT1 inhibition prolongs G2/M phase arrest, thereby enhancing radiotherapy-induced cytotoxicity [70,71]. HER3-dependent nuclear AKT1 activation further promotes NHEJ repair by enhancing DNA-PKcs activity, while HER3 inhibitors reduce AKT1 activity and improve radiosensitivity [72]. Other studies demonstrated that limiting AKT1 phosphorylation delays DSB repair and significantly increases radiation-induced cytotoxicity [73]. In HR, AKT1 enhances repair efficiency by promoting Rad51 focus formation and colocalization with γ-H2AX. Knocking down AKT1 leads to the accumulation of unrepaired DSBs, thereby increasing radiosensitivity [74]. Additionally, activating mutations of AKT1 (e.g., TDSD and E17K) accelerate γ-H2AX focus resolution, improve DSB repair, and enhance radiation resistance [75].
Research by Seiwert et al. revealed that AKT2 regulates radiosensitivity by suppressing autophagy. Radiation-induced DSBs typically activate autophagy as a protective mechanism, but AKT2 suppresses autophagy initiation via mTOR signaling, thereby reducing radiosensitivity [76]. In HR, AKT2 promotes Rad51 focus formation and accelerates radiation-induced DNA repair; conversely, AKT2 knockdown reduces Rad51 recruitment, leading to DNA damage accumulation and increased radiation-induced cell death [77]. AKT2 also interacts with DNA-PKcs and MRE11, participating in alternative NHEJ repair pathways, suggesting its involvement in multiple DNA repair mechanisms [78]. However, studies indicated that in K-RAS mutant cells, AKT1 and AKT3 form stable complexes with DNA-PKcs, while AKT2 does not, suggesting that AKT2’s role in radiosensitivity regulation may be less critical in specific contexts [71].
AKT3 exhibits multifaceted roles in regulating radiation resistance. Research has shown that microRNA-207 directly targets and suppresses AKT3 expression, inhibiting anti-apoptotic signaling and enhancing radiation-induced apoptosis [79]. Moreover, AKT3 phosphorylates the NADPH oxidase subunit p47phox, significantly increasing reactive oxygen species (ROS) levels, which triggers DDR and inhibits tumor cell proliferation. However, in the presence of p53 mutations or deletions, this mechanism facilitates immune evasion and enhances tumor growth [80]. In colorectal cancer models, AKT3 regulates DNA repair through the IL-13/mTOR signaling pathway, showing differential effects in tumors and normal tissues: reduced AKT3 activation in tumors decreases cell survival, while elevated IL-13 levels in normal tissues enhance AKT3 and mTOR signaling, promoting tissue repair [81]. In glioblastoma models, AKT3 gene amplification significantly increases the expression of DNA repair proteins, as evidenced by a significant increase in p-γ-H2AX foci and strong phosphorylation of ATM following γ-irradiation in AKT3-overexpressing cells compared with the empty vector group. This activation confers strong resistance to radiotherapy and chemotherapy (e.g., temozolomide), suggesting that AKT3 amplification may be a key mechanism underlying radiation resistance [82](Figure 3).
3.3. mTOR Isoforms
The mTOR is an evolutionarily conserved serine/threonine protein kinase that forms two multiprotein complexes: mTORC1 and mTORC2. mTORC1 primarily regulates cell growth, while mTORC2 is involved in the regulation of cell survival and proliferation [83].
The mTORC1 complex comprises three core proteins: mTOR, mammalian lethal with SEC13 protein 8(mLST8), and Regulatory-Associated Protein of mTOR(RAPTOR) [84]. Its abnormal activation is closely associated with radiation resistance. At the metabolic level, studies have shown that Fusobacterium nucleatum promotes radiation resistance in nasopharyngeal carcinoma through the Solute Carrier Family 7 Member A5(SLC7A5)/leucine-mTORC1 pathway. Leucine uptake activates mTORC1 signaling, inhibiting radiation-induced oxidative stress and apoptosis. However, a leucine-restricted diet suppresses mTORC1 activity, significantly enhancing the efficacy of radiotherapy, underscoring the critical role of mTORC1 in amino acid metabolism regulation [85]. Additionally, negative regulators of mTORC1, such as DEP domain-containing mTOR-interacting protein(DEPTOR), also play important roles in radiation resistance. Radiation induces DEPTOR degradation, activating mTORC1 signaling to inhibit apoptosis and enhance tumor cell survival. Stabilizing DEPTOR significantly inhibits mTORC1 signaling, increasing the sensitivity of hypopharyngeal cancer cells to radiation therapy [86]. In the context of DNA damage repair, mTORC1 directly regulates HR and NHEJ pathways through its downstream molecules S6K1 and Eukaryotic translation initiation factor 4E-binding protein 1(4E-BP1), enhancing the efficiency of DNA repair and reducing cell death caused by radiation [87]. However, inhibiting mTORC1 alone may trigger compensatory activation of Akt signaling, thereby diminishing the radiosensitization effect. When mTORC1 inhibitors (e.g., rapamycin) are combined with Akt inhibitors (e.g., MK2206), DSB repair is significantly suppressed, further enhancing the efficacy of radiation therapy. This finding highlights the potential of combination targeting strategies [88]. Interestingly, the function of mTORC1 is significantly influenced by the metabolic environment. Murata et al. reported that under nutrient-deficient conditions (e.g., glucose restriction), abnormal activation of mTORC1 increases the radiosensitivity of hepatocellular carcinoma cells. This may be associated with a metabolic stress-induced energy crisis, further indicating that the role of mTORC1 in radiation resistance varies depending on the tumor microenvironment [89].
The mTORC2 complex consists of rictor, mTOR, PPR5, G-protein β-subunit-like protein(GβL), DEPTOR, and stress-activated protein kinase-interacting protein 1(SIN1) [90]. Although studies on mTORC2’s role in radiosensitivity are limited, existing data highlight its critical role in DNA damage repair. Research by Kalpongnukul et al. demonstrated that mTORC2 enhances the activity of DNA repair pathways by phosphorylating BABAM1 (BRISC and BRCA1-A complex member 1), thereby increasing radiation resistance in glioblastoma [91]. Additionally, Tian et al. found that mTORC2 phosphorylates ribonucleotide reductase (RNR), promoting deoxynucleotide synthesis and accelerating DNA replication. This sustained DNA replication capacity plays a crucial role in both gemcitabine resistance and radiation resistance [92]. Currently, specific inhibitors targeting mTORC2 alone are scarce, and most studies focus on dual mTORC1/2 inhibitors. For example, suppressing phosphorylation of mTORC2 downstream target p-Akt has been shown to significantly enhance the efficacy of radiation therapy [93,94,95]. These findings suggest that mTORC2 plays an equally important role in regulating DNA repair and metabolism, and dual targeting of mTORC1 and mTORC2 may be an effective strategy for overcoming radiation resistance in the future.(Figure 4).
4. Preclinical Studies of PI3K/AKT/mTOR Inhibitors Combined with Radiotherapy
4.1. Digestive System Tumors
Digestive system tumors are among the most prevalent malignancies globally, with treatment strategies typically involving surgery, radiotherapy, and systemic chemotherapy [96]. However, the heterogeneity and radiation resistance of these tumors significantly limit the efficacy of radiotherapy.
In colorectal cancer (CRC) models, the combination of the mTOR inhibitor Temsirolimus and the autophagy inhibitor Chloroquine significantly enhanced the antitumor effects of radiotherapy, as evidenced by a marked increase in apoptosis rates. This combination therapy inhibited the phosphorylation of key mTOR signaling proteins, p-S6 and p-4E-BP1, and reduced levels of LC3-II and p62, thereby suppressing autophagy. In vivo experiments demonstrated that this combination therapy effectively inhibited the growth of CRC xenograft tumors without notable toxicity [97]. Another study showed that the dual PI3K/mTOR inhibitor BEZ235 combined with radiotherapy delayed DNA damage repair and significantly reduced the expression of angiogenesis-related factors VEGF-A and HIF-1α. In vivo, the combination therapy substantially inhibited CRC tumor growth, with the maintenance therapy group showing particularly pronounced effects [98].
In hepatocellular carcinoma models, the dual PI3K/mTOR inhibitor PKI-587 and the natural compound Tenacissoside H (TEH) demonstrated radiosensitization potential. PKI-587 interfered with NHEJ and HR repair pathways, delaying the dissipation of γ-H2AX foci induced by radiation and enhancing radiation-induced apoptosis. In vivo, the combination therapy significantly reduced tumor volumes without causing notable toxicity [99]. TEH, on the other hand, enhanced radiotherapy efficacy by downregulating PI3K/Akt/mTOR signaling activity and inducing autophagy. It specifically increased the LC3-II/LC3-I ratio, Beclin-1, and ATG5 expression, while reducing the anti-apoptotic protein Bcl-2 and upregulating the pro-apoptotic protein Bax. The combination of TEH and radiotherapy significantly reduced tumor cell viability and showed enhanced tumor inhibition in in vivo models [100].
The dual PI3K/mTOR inhibitor PF-04691502 also exhibited radiosensitizing effects in gastroenteropancreatic neuroendocrine tumor models. This study compared three administration schedules: simultaneous, pre-radiotherapy, and delayed post-radiotherapy. The group receiving PF-04691502 48 hours after radiotherapy showed the most significant tumor volume reduction without causing notable toxicity to normal tissues [101]. In pancreatic cancer models, the mTOR inhibitor INK128 combined with radiotherapy significantly delayed DNA damage repair and enhanced the cytotoxic effects of radiotherapy by disrupting the formation of the Eukaryotic initiation factor 4F(eIF4F) cap complex, thereby inhibiting protein translation. The radiosensitization effect was most pronounced when INK128 was administered either 1 hour before or within 6 hours after radiotherapy. Delaying administration beyond 6 hours post-radiotherapy markedly reduced its radiosensitization effects, emphasizing the critical role of drug-radiotherapy timing in combination treatments [102].
4.2. Genitourinary System Tumors
Radiotherapy is a crucial therapeutic option for prostate cancer, demonstrating significant efficacy across all stages of the disease [103]. Studies have shown that the dual PI3K/mTOR inhibitor NVP-BEZ235 significantly reduces the survival rate of prostate cancer PC-3 cells post-radiation. Combined treatment not only induces G2/M cell cycle arrest but also markedly enhances apoptosis rates and delays DNA damage repair [104].
In renal cell carcinoma (RCC) models, the combination of the mTOR inhibitor Everolimus and the Survivin inhibitor YM155 (EY-L) significantly potentiates radiosensitization. In vitro experiments demonstrated that EY-L reduces the clonogenic capacity of RCC cells, prolongs the persistence of the DNA damage marker γ-H2AX, and suppresses DNA repair by inhibiting the expression of p-p70S6K and Poly(ADP-ribose)polymerase 1(PARP1). In vivo studies further revealed that EY-L combined with radiotherapy significantly inhibits tumor growth and increases CD8+ T cell infiltration within tumors, thereby amplifying antitumor immune responses [105].
In cervical cancer models, the combination of the PI3K inhibitor Alpelisib and the Aurora kinase inhibitor Alisertib exhibits notable radiosensitizing effects. In vitro data showed that Alpelisib enhances the cytotoxicity of Alisertib, reducing the IC50 of HeLa cells from 19,550 nM to 30.3 nM. Combined treatment significantly prolongs cell division time, induces mitotic failure, and leads to apoptosis or necrosis in 97.9% of cells. Molecular analyses revealed increased levels of cleaved PARP and caspase expression following combination therapy. In vivo experiments further validated that Alpelisib and Alisertib combination therapy significantly suppresses tumor growth in HeLa xenograft mouse models [106].
In bladder cancer models, the mTOR inhibitor Everolimus combined with radiotherapy exhibits significant radiosensitizing effects. In vitro studies revealed that Everolimus (RAD001) markedly reduces the clonogenic survival of bladder cancer cells and enhances radiotherapy efficacy by inhibiting downstream mTOR signaling, such as p-S6 phosphorylation, and regulating the cell cycle. The combination induces dual-phase cell cycle arrest at G0/G1 and G2/M phases, reduces the proportion of S-phase cells, downregulates cyclin D1, and upregulates p21 and p27 expression. In vivo experiments demonstrated that Everolimus combined with radiotherapy significantly reduces tumor weight in bladder cancer xenograft models by approximately 72% and increases p21 expression [107].
4.3. Respiratory System Tumors
Radiotherapy is a key treatment modality for lung cancer, with approximately 77% of patients being eligible for this approach. However, radiotherapy remains underutilized in clinical practice [108]. In small cell lung cancer (SCLC) models, the combination of PI3K/mTOR inhibitors BEZ235 and GSK2126458 with radiotherapy significantly reduced the survival rates of SCLC cells, increased apoptosis rates to 24%-32%, and markedly suppressed clonogenic capacity. The combination therapy enhanced DNA damage, evidenced by significant upregulation of γ-H2AX and p-ATM expression levels [109]. In NSCLC, the combination of MEK inhibitor Trametinib, mTOR inhibitor Temsirolimus, and radiotherapy demonstrated synergistic effects. The combined treatment reduced the survival rates of A549 and NCI-H460 cells under 8 Gy irradiation by approximately 50%, significantly prolonged G2/M phase cell cycle arrest, increased γ-H2AX expression, and elevated radiation-induced apoptosis. In A549 xenograft nude mouse models, tumor growth inhibition in the combination therapy group outperformed both radiotherapy and single-drug treatments [110]. Furthermore, Seol et al. compared the effects of different PI3K inhibitors combined with radiotherapy, finding that both the PI3K-α inhibitor GDC0032 and the pan-PI3K inhibitor GDC0941 reduced A549 cell survival rates to 25%, exhibiting similar radiosensitizing effects. However, GDC0032 demonstrated lower toxicity [111].
Radiotherapy is also a crucial treatment for nasopharyngeal carcinoma (NPC) and certain HNSCC in patients who are inoperable [112]. In NPC models, the mTOR inhibitor Temsirolimus dose-dependently induced caspase-dependent apoptosis and significantly enhanced the radiosensitivity of the resistant cell line C666-1-r. In vivo experiments demonstrated that Temsirolimus delayed tumor growth in C666-1-r models, reducing tumor formation rates from 80% to 20%-55% without notable toxicity [113]. In HNSCC models, the DNA-PK inhibitor AZD7648 combined with radiotherapy significantly delayed the repair of DSBs, induced G2/M phase cell cycle arrest, and suppressed cell proliferation and clonogenic capacity. Additionally, the mTOR inhibitor Sapanisertib exhibited radiosensitizing effects in HPV+ cell lines but showed limited efficacy in HPV− cell lines. In contrast, the dual-function inhibitor CC-115, which targets both DNA-PK and mTOR, significantly enhanced radiotherapy outcomes in both HPV+ and HPV− cell lines, suggesting broader clinical utility [114]. However, the study by Glorieux et al. indicated variability in the radiosensitizing effects of PI3K inhibitors in HNSCC cells. While BKM120 and GDC0980 effectively inhibited p-Akt and p-mTOR expression and weakened DNA repair in some cell lines, their overall radiosensitizing efficacy fell short of expectations. These findings highlight the need for patient-specific selection strategies when employing PI3K inhibitors in clinical applications [115].
4.4. Breast Cancer
Radiotherapy is a critical component of multimodal breast cancer management, widely applied in early-stage, locally advanced, and metastatic cases [116]. In triple-negative breast cancer (TNBC) models with a high risk of brain metastasis, the AKT inhibitor Ipatasertib significantly reduced the survival rates of MDA-MB-231BR cells. However, Ipatasertib did not affect cell migration, whereas the knockout of the AKT1 isoform unexpectedly increased migration potential [117]. Johnson et al. investigated the radiosensitizing effects of various AKT inhibitors and found that MK-2206 reduced AKT1 expression and induced PARP cleavage in MDA-MB-231 cells after 24 hours of treatment. When combined with 4 Gy radiation, MK-2206 significantly enhanced radiation-induced PARP cleavage. In contrast, perifosine reduced AKT1 expression but did not show significant synergistic apoptotic effects, while A-674563 and AZD5363 neither reduced AKT1 levels nor induced apoptosis [118].
Dual PI3K/mTOR inhibitors have shown remarkable radiosensitizing effects across different breast cancer subtypes. Gasimli et al. demonstrated that PKI-402 exhibited strong radiosensitizing effects in Luminal A subtype (MCF-7) and breast CSCs. In MDA-MB-231 cells, PKI-402 combined with radiotherapy significantly increased DSBs levels, but this effect was not observed in MCF-7 or breast CSCs. Mechanistically, PKI-402 reduced phosphorylated Glycogen synthase kinase 3 beta(GSK-3β) and Proline-rich AKT substrate of 40 kDa(PRAS40) levels, thereby modulating mTORC1-related signaling pathways. In MCF-7 cells, PKI-402 further activated apoptotic signals by increasing phosphorylated Bcl-2-associated agonist of cell death(BAD) [119]. Additionally, Masoumi et al. investigated the combined effects of the PI3K/mTOR inhibitor NVP-BEZ235 and the SIRT1 activator SRT1720 under radiotherapy. The study revealed that both NVP-BEZ235 and SRT1720 alone significantly reduced the survival rates of MCF-7 cells, with combined treatment yielding even greater effects. In IL-6 pretreated cells, the combination reduced survival rates to 43% of the control group. Furthermore, combined therapy significantly increased apoptosis and necrosis rates, thereby enhancing the radiosensitizing effect of the treatment [120].
4.5. Central Nervous System Tumors
Although radiotherapy is a key treatment modality for GBM, its efficacy remains limited [121]. Studies have shown that the PI3K-α inhibitor GDC0032, in combination with radiotherapy, significantly reduced the survival rates of LN229 and GL261-luc cells to 27% and 19%, respectively. Combined treatment notably prolonged the persistence of γ-H2AX foci, indicating inhibited DNA damage repair. Furthermore, GDC0032 significantly reduced p-AKT levels, achieving similar effects to the pan-PI3K inhibitor GDC0941 but with lower toxicity. In vivo experiments demonstrated that GDC0032 combined with radiotherapy significantly slowed tumor growth in GL261-luc mouse models and markedly improved survival times [122].
The radiosensitizing effects of the PI3K/mTOR inhibitor PI-103 vary depending on molecular context. In DNA-PK-proficient MO59K cells, PI-103 combined with radiotherapy reduced the clonogenic survival fraction from 0.63 to 0.41, delayed DNA damage repair, and extended the persistence of γ-H2AX and p53-binding protein 1(53BP1) foci. Additionally, PI-103 significantly promoted apoptosis by inducing cleaved PARP and cleaved caspase-3. Conversely, in DNA-PK-deficient MO59J cells, PI-103 unexpectedly accelerated DNA repair, marked by LC3B-II upregulation and p62 downregulation, inducing protective autophagy and increasing the survival fraction from 0.16 to 0.23 [123]. AKT inhibitor MK-2206 also exhibited differential effects across GBM cell lines. It showed no significant radiosensitizing effects in DK-MG cells but paradoxically increased radioresistance in SNB19 cells. Further investigations revealed that while MK-2206 significantly suppressed p-AKT levels, it induced compensatory activation of the mTOR pathway in SNB19 cells, elevating p-mTOR and p-S6 levels and thereby diminishing radiotherapy efficacy. However, when PI-103 was combined with MK-2206, mTOR pathway activity was effectively suppressed, autophagy was enhanced, and DNA repair was delayed, reversing the resistance caused by compensatory activation [124].
5. Clinical Studies on PI3K/AKT/mTOR Inhibitors Combined with Radiotherapy
PI3K inhibitors have emerged as an important strategy in cancer-targeted therapy. These inhibitors, targeting different PI3K isoforms (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ) through selective or pan-inhibition, have shown significant potential in clinical studies. In contrast, most AKT inhibitors are pan-inhibitors, targeting AKT1, AKT2, and AKT3 through ATP-competitive or allosteric mechanisms. However, this broad-spectrum inhibition often results in higher adverse effects, limiting their clinical application. Despite these challenges, the approval of the AKT inhibitor Capivasertib in 2023 for HR+/HER2- advanced metastatic breast cancer marked a significant breakthrough in the clinical translation of this drug class [125]. As the downstream component of the PI3K/AKT/mTOR pathway, mTOR inhibitors mainly target the mTORC1 complex to suppress cell proliferation and metabolism and are widely applied in the treatment of solid and hematologic tumors. Currently, multiple PI3K/AKT/mTOR inhibitors are undergoing clinical trials (Table 1).
Alpelisib, a selective PI3Kα inhibitor, has garnered attention for its radiosensitization effects, particularly in HNSCC. In a phase I trial, Alpelisib combined with cisplatin and concurrent chemoradiotherapy demonstrated good tolerability and achieved local tumor control in most patients with advanced HNSCC. However, some patients experienced distant metastasis, necessitating further phase II trials to evaluate its clinical efficacy [126]. Another study reported that Alpelisib combined with cetuximab and radiotherapy was well-tolerated, though some patients developed severe mucositis and leukopenia, indicating a need for optimized toxicity management [127].Buparlisib, a pan-PI3K inhibitor, significantly reduced hypoxia in tumor regions when combined with low-dose thoracic radiotherapy (20 Gy) in NSCLC patients during a phase I trial. This reduction was closely associated with decreased radioresistance and good tolerability [128]. However, dose-limiting toxicity has restricted its further development. In another phase I trial involving newly diagnosed GBM patients, Buparlisib combined with temozolomide and radiotherapy demonstrated good biomarker suppression but was associated with high rates of hyperglycemia and elevated liver enzymes, limiting dose escalation and failing to establish a maximum tolerated dose (MTD) [129]. Voxtalisib, a dual PI3K/mTOR inhibitor, showed limited radiosensitization effects in high-grade glioma patients, with only a 4% partial response rate in clinical studies. Nevertheless, its safety profile was favorable [130].
Clinical studies on AKT inhibitors are relatively limited. Perifosine, a novel AKT inhibitor [131], was evaluated in a phase I trial combining daily oral doses (50–200 mg) with fractionated radiotherapy in various advanced solid tumors, including NSCLC, bladder cancer, prostate cancer, and esophageal cancer. It showed radiosensitization effects across multiple tumor types, with an infield response rate of 52% (11/21, including 5 partial responses and 6 complete responses) after a median follow-up of 10 months, with an MTD of 150 mg/day [132]. Nelfinavir, initially developed as an HIV protease inhibitor, was later found to downregulate phosphorylated AKT levels, demonstrating potential as a radiosensitizer [133,134]. In a phase I study, Nelfinavir combined with cisplatin and concurrent chemoradiotherapy achieved a remarkable 91% disease-free survival rate (RFS) at a median follow-up of 50 months in cervical cancer patients treated with 1250 mg/day. This regimen was well-tolerated [135]. In another study involving 11 pancreatic cancer patients, Nelfinavir combined with SBRT (40 Gy/5 fractions) showed promising feasibility and safety. Among the 10 patients who received radiotherapy, four underwent surgical resection, and all resected specimens exhibited negative margins. The median overall survival was 13 months despite the premature termination of the study [136]. However, a phase III trial in locally advanced pancreatic cancer failed to demonstrate significant improvements in progression-free survival (PFS) or overall survival (OS), highlighting the need for optimization in specific cancer types [137].
Temsirolimus, an mTOR inhibitor, demonstrated partial responses in patients with locally advanced NSCLC when combined with thoracic radiotherapy in a phase I trial. However, pulmonary toxicity, including pneumonia and hemorrhage, necessitated cautious dose adjustments, with an MTD of 15 mg weekly via intravenous administration [138].In a phase II trial for GBM, Temsirolimus combined with radiotherapy did not significantly improve the 12-month overall survival rate. Nonetheless, patients with high mTOR phosphorylation levels showed better outcomes, suggesting that specific molecular subgroups may benefit from mTOR inhibitors as radiosensitizers [139].Everolimus, another mTOR inhibitor, achieved a 23% pathological complete response rate and a 2-year PFS of 50% when combined with carboplatin and radiotherapy in a phase I study for locally advanced esophageal cancer [140]. In locally advanced cervical cancer, Everolimus demonstrated good tolerability with dose escalation up to 10 mg/day, without irreversible toxicity [141]. However, in newly diagnosed GBM, 10 mg/day of Everolimus combined with temozolomide and radiotherapy failed to significantly improve PFS and was associated with high toxicity [142]. In a study involving patients with biochemical recurrence of prostate cancer after surgery, Everolimus combined with radiotherapy at escalating doses (5, 7.5, and 10 mg/day) resulted in 22% of patients experiencing grade 3 acute toxicity [143].
These studies collectively underscore the potential of PI3K/AKT/mTOR inhibitors as radiosensitizers in various solid tumors. Large-scale randomized controlled trials are essential to validate their efficacy and safety in clinical settings.
6. Future Research Directions
6.1. Molecular Design and Targeted Optimization of Novel Inhibitors
Despite the significant potential of PI3K/AKT/mTOR pathway inhibitors in radiosensitization research, their clinical applications are still limited by systemic toxicity and insufficient target specificity. For instance, PI3K/AKT inhibitors frequently induce adverse reactions such as hypertension, hyperglycemia, and severe pneumonia, posing significant obstacles to their clinical use [144]. A large meta-analysis involving 6,710 patients demonstrated that PI3K/AKT inhibitors are effective in patients with specific genetic mutations, but their safety profiles are relatively poor. Among these, Capivasertib has shown good efficacy and safety in solid tumors like breast cancer, while Idelalisib has exhibited remarkable effects in hematological malignancies such as chronic lymphocytic leukemia. These studies also revealed that AKT inhibitors are generally safer than PI3K inhibitors, which are more prone to causing pneumonia, though the underlying mechanism remains unclear [145].
Balancing efficacy and toxicity control remains a core challenge in clinical research, further driving the development of personalized inhibitors targeting different subtypes of the PI3K/AKT/mTOR pathway. Examples include Alpelisib as a selective PI3Kα inhibitor, GSK2636771 targeting PI3Kβ, Idelalisib and Zandelisib targeting PI3Kδ, and Eganelisib focusing on PI3Kγ, all of which demonstrate strategies for refined targeting. The development of these inhibitors not only enhances efficacy but also significantly reduces damage to normal tissues [146,147,148,149,150]. Additionally, inhibitor development for mTOR complexes has also become more refined. For instance, Everolimus has been developed as an mTORC1-specific inhibitor, while JR-AB2-011 is an mTORC2-specific inhibitor, offering more options for precise pathway regulation [151,152]. Future studies should focus on further optimizing molecular designs, particularly developing specific inhibitors for the three AKT isoforms, to reduce systemic toxicity and improve targeting. This will help ensure therapeutic efficacy while minimizing damage to normal tissues.
6.2. Combination Therapy Strategies
Combination therapy is a critical approach to enhancing the radiosensitization potential of PI3K/AKT/mTOR inhibitors. Studies have shown that the PI3K/AKT/mTOR pathway is often excessively activated after chemotherapy, closely associated with chemotherapy resistance [153]. Combining PI3K/AKT/mTOR inhibitors with standard chemotherapy not only reduces chemotherapy resistance but has also demonstrated significant sensitization effects in preclinical studies on cervical cancer and liver cancer [154]. In radiochemotherapy strategies, chemotherapy enhances the efficacy of radiotherapy through mechanisms such as increasing DNA damage, inhibiting DNA repair, altering the cell cycle, reducing hypoxic cells, and suppressing tumor repopulation [155].
Moreover, combining PI3K/AKT/mTOR inhibitors with radiotherapy and immunotherapy holds great promise. For instance, in preclinical studies on cervical cancer, the mTOR inhibitor RAD001 enhanced radiotherapy-induced PD-L1 expression. When combined with a PD-1 inhibitor, this approach further increased the number and cytotoxicity of CD8+ T cells, demonstrating synergistic effects in activating antitumor immunity [156]. Future research should explore the combination of PI3K/AKT/mTOR inhibitors with other therapeutic modalities, such as DNA damage repair inhibitors and angiogenesis inhibitors, aiming to achieve more precise and effective radiosensitization across various tumor types.
6.3. Identification of Predictive Tumor Biomarkers
With advancements in modern radiotherapy techniques, treatment plans can be highly individualized to match patients' anatomical structures. However, challenges remain in regulating tumor biological characteristics, particularly the lack of biomarkers capable of accurately predicting tumor responses to radiotherapy. Such biomarkers are essential for personalized treatment decisions, dose design, and protocol optimization [157].
Currently, biomarkers such as Phosphoinositide-dependent protein kinase 1(PDK1) and annexin A6(ANXA6) have shown potential in studies on the radiosensitization of PI3K/AKT/mTOR pathway inhibitors, but their universality and predictive value across different tumor types require further validation [158,159]. Moreover, existing research predominantly focuses on single biomarkers, lacking systematic studies that integrate multidimensional analyses, such as molecular expression profiles, phosphorylation states, and liquid biopsy data. Future research should prioritize the development of predictive models incorporating multiple biomarkers, integrating diverse biological datasets to enhance clinical applicability and stability. This approach could enable more precise radiosensitization strategies.
7. Conclusions
The PI3K/AKT/mTOR signaling pathway plays a central role in modulating tumor radiosensitivity by systematically influencing tumor responses to radiotherapy through mechanisms such as DNA damage repair, cell cycle regulation, and the control of cell proliferation and apoptosis. The distinct roles of different PI3K, AKT, and mTOR isoforms in regulating radiosensitivity offer promising directions for precision-targeted therapies.Although preclinical studies demonstrate that targeting specific nodes within this pathway can significantly enhance the effects of radiotherapy, challenges such as limited target selectivity, resistance development, and toxicity continue to hinder clinical applications. Future research should focus on optimizing inhibitor design, developing combination treatment strategies, and identifying predictive biomarkers of sensitivity, aiming to achieve more precise and effective radiosensitization protocols.In summary, the PI3K/AKT/mTOR pathway represents a critical target for understanding tumor radioresistance and developing novel therapeutic strategies. Advances in this field hold the potential to bring new hope to the treatment of cancer.
Author Contributions
J.Z. wrote the original draft and prepared the figures; M.J. critically revised the original draft. All authors have read and agreed to the published version of the manuscript.
Funding
J.Z. was funded by the China Scholarship Council (scholarship number 202508080078).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The figures were created using BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| 53BP1 | p53-binding protein 1 |
| A2aR | Adenosine A2a receptor |
| AKT | Protein Kinase B |
| ANXA6 | Annexin A6 |
| APLNR | Apelin Receptor |
| ATG5 | Autophagy-related gene 5 |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related |
| BABAM1 | BRISC and BRCA1-A complex member 1 |
| BAD | Bcl-2-associated agonist of cell death |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| C2orf40 | Chromosome 2 open reading frame 40 |
| CALD1 | Caldesmon 1 |
| CD44 | Cluster of Differentiation 44 |
| CDK1 | Cyclin-dependent kinase 1 |
| Chk2 | Checkpoint kinase 2 |
| c-Jun | Cellular Jun proto-oncogene |
| CRC | Colorectal cancer |
| CSCs | Cancer stem cells |
| DDRs | DNA damage responses |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DSBs | DNA double-strand breaks |
| EMT | Epithelial-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| ESCC | Esophageal squamous cell carcinoma |
| FA | Fanconi anemia |
| FAM135B | Family with sequence similarity 135, member B |
| FANCD2 | Fanconi anemia complementation group D2 |
| FOXO1 | Forkhead Box O1 |
| GBM | Glioblastoma |
| Glut-1 | Glucose transporter 1 |
| GSK-3β | Glycogen synthase kinase 3 beta |
| GβL | G-protein β-subunit-like protein |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HNSCC | Head and neck squamous cell carcinoma |
| HR | Homologous recombination |
| Hsp90 | Heat shock protein 90 |
| IMRT | Intensity-modulated radiotherapy |
| LC3-II | Microtubule-Associated Protein 1 Light Chain 3 II |
| MDSC | Myeloid-derived suppressor cell |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| MRN | MRE11-RAD50-NBS1 complex |
| mTOR | Mammalian target of rapamycin |
| Nbs1 | Nijmegen Breakage Syndrome 1 protein |
| NEDD8 | Neural precursor cell expressed, developmentally downregulated 8 |
| NHEJ | Non-homologous end joining |
| NPC | Nasopharyngeal carcinoma |
| NSCLC | Non-small cell lung cancer |
| OCT-4 | Octamer-binding transcription factor 4 |
| OS | Overall survival |
| P62 | Sequestosome-1 |
| PARP1 | Poly(ADP-ribose)polymerase 1 |
| PCNA | Proliferating Cell Nuclear Antigen |
| PDK1 | Phosphoinositide-dependent protein kinase 1 |
| PFS | progression-free survival |
| PI3K | Phosphatidylinositol 3-Kinase |
| PRAS40 | Proline-rich AKT substrate of 40 kDa |
| PTEN | phosphatase and tensin homolog |
| RAPTOR | Regulatory-Associated Protein of mTOR |
| Rad51 | RAD51 Recombinase |
| Rb | Retinoblastoma protein |
| RCC | Renal cell carcinoma |
| RFC1 | Replication Factor C Subunit 1 |
| RNR | Phosphorylates ribonucleotide reductase |
| ROS | Reactive oxygen species |
| S2056 | Serine 2056 |
| S6K | Sibosomal protein S6 kinase |
| SBRT | Stereotactic body radiotherapy |
| SCLC | Small cell lung cancer |
| SIN1 | Stress-activated protein kinase-interacting protein 1 |
| SIRT6 | Sirtuin 6 |
| SLC7A5 | Solute Carrier Family 7 Member A5 |
| SMA | Smooth muscle actin |
| SOX2 | SRY-box transcription factor 2 |
| SSBs | Single-strand breaks |
| TAMs | Tumor-associated macrophages |
| TEH | Tenacissoside H |
| VEGF | Vascular endothelial growth factor |
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA. Cancer J. Clin. 2024, 74, 12–49. [CrossRef]
- Lu, Z.; Zheng, X.; Ding, C.; Zou, Z.; Liang, Y.; Zhou, Y.; Li, X. Deciphering the Biological Effects of Radiotherapy in Cancer Cells. Biomolecules 2022, 12, 1167. [CrossRef]
- Xu, Q.; Zhang, P.; Han, X.; Ren, H.; Yu, W.; Hao, W.; Luo, B.; Khan, M.I.; Chen, N. Role of Ionizing Radiation Activated NRF2 in Lung Cancer Radioresistance. Int. J. Biol. Macromol. 2023, 241, 124476. [CrossRef]
- Wang, Q.; Li, Z.; Yang, J.; Peng, S.; Zhou, Q.; Yao, K.; Cai, W.; Xie, Z.; Qin, F.; Li, H.; et al. Loss of NEIL3 Activates Radiotherapy Resistance in the Progression of Prostate Cancer. Cancer Biol. Med. 2021, 19, 1193–1210. [CrossRef]
- Liu, W.; Zheng, M.; Zhang, R.; Jiang, Q.; Du, G.; Wu, Y.; Yang, C.; Li, F.; Li, W.; Wang, L.; et al. RNF126-Mediated MRE11 Ubiquitination Activates the DNA Damage Response and Confers Resistance of Triple-Negative Breast Cancer to Radiotherapy. Adv. Sci. Weinh. Baden-Wurtt. Ger. 2023, 10, e2203884. [CrossRef]
- Kimura, T.; Fujiwara, T.; Kameoka, T.; Adachi, Y.; Kariya, S. The Current Role of Stereotactic Body Radiation Therapy (SBRT) in Hepatocellular Carcinoma (HCC). Cancers 2022, 14, 4383. [CrossRef]
- Shi, Y.; Ma, X.; He, D.; Dong, B.; Qiao, T. Neoadjuvant SBRT Combined with Immunotherapy in NSCLC: From Mechanisms to Therapy. Front. Immunol. 2023, 14, 1213222. [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Nyati, S. Molecular Targets That Sensitize Cancer to Radiation Killing: From the Bench to the Bedside. Biomed. Pharmacother. Biomedecine Pharmacother. 2023, 158, 114126. [CrossRef]
- Meattini, I.; Livi, L.; Lorito, N.; Becherini, C.; Bacci, M.; Visani, L.; Fozza, A.; Belgioia, L.; Loi, M.; Mangoni, M.; et al. Integrating Radiation Therapy with Targeted Treatments for Breast Cancer: From Bench to Bedside. Cancer Treat. Rev. 2022, 108, 102417. [CrossRef]
- Elbanna, M.; Chowdhury, N.N.; Rhome, R.; Fishel, M.L. Clinical and Preclinical Outcomes of Combining Targeted Therapy with Radiotherapy. Front. Oncol. 2021, 11, 749496. [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR Signaling Transduction Pathway and Targeted Therapies in Cancer. Mol. Cancer 2023, 22, 138. [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR Signaling Pathway for Targeted Therapeutic Treatment in Human Cancer. Semin. Cancer Biol. 2022, 85, 69–94. [CrossRef]
- Wanigasooriya, K.; Tyler, R.; Barros-Silva, J.D.; Sinha, Y.; Ismail, T.; Beggs, A.D. Radiosensitising Cancer Using Phosphatidylinositol-3-Kinase (PI3K), Protein Kinase B (AKT) or Mammalian Target of Rapamycin (mTOR) Inhibitors. Cancers 2020, 12, 1278. [CrossRef]
- Mardanshahi, A.; Gharibkandi, N.A.; Vaseghi, S.; Abedi, S.M.; Molavipordanjani, S. The PI3K/AKT/mTOR Signaling Pathway Inhibitors Enhance Radiosensitivity in Cancer Cell Lines. Mol. Biol. Rep. 2021, 48, 1–14. [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [CrossRef]
- Monge-Cadet, J.; Moyal, E.; Supiot, S.; Guimas, V. DNA Repair Inhibitors and Radiotherapy. Cancer Radiother. J. Soc. Francaise Radiother. Oncol. 2022, 26, 947–954. [CrossRef]
- Moosavi, F.; Hassani, B.; Nazari, S.; Saso, L.; Firuzi, O. Targeting DNA Damage Response in Pancreatic Ductal Adenocarcinoma: A Review of Preclinical and Clinical Evidence. Biochim. Biophys. Acta Rev. Cancer 2024, 1879, 189185. [CrossRef]
- Xie, Y.; Liu, C.; Zhang, Y.; Li, A.; Sun, C.; Li, R.; Xing, Y.; Shi, M.; Wang, Q. PKI-587 Enhances Radiosensitization of Hepatocellular Carcinoma by Inhibiting the PI3K/AKT/mTOR Pathways and DNA Damage Repair. PLOS One 2021, 16, e0258817. [CrossRef]
- Mukherjee, B.; Tomimatsu, N.; Amancherla, K.; Camacho, C.V.; Pichamoorthy, N.; Burma, S. The Dual PI3K/mTOR Inhibitor NVP-BEZ235 Is a Potent Inhibitor of ATM- and DNA-PKCs-Mediated DNA Damage Responses. Neoplasia N. Y. N 2012, 14, 34–43. [CrossRef]
- Liao, J.; Jin, H.; Li, S.; Xu, L.; Peng, Z.; Wei, G.; Long, J.; Guo, Y.; Kuang, M.; Zhou, Q.; et al. Apatinib Potentiates Irradiation Effect via Suppressing PI3K/AKT Signaling Pathway in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. CR 2019, 38, 454. [CrossRef]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the mTOR Pathway Contributes to the Resistance of Cancer Cells to DNA Double-Strand Breaks. Cancer Res. 2013, 73, 3393–3401. [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The Cell Cycle in Stem Cell Proliferation, Pluripotency and Differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [CrossRef]
- Pawlik, T.M.; Keyomarsi, K. Role of Cell Cycle in Mediating Sensitivity to Radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [CrossRef]
- Xie, Z.; Li, W.; Ai, J.; Xie, J.; Zhang, X. C2orf40 Inhibits Metastasis and Regulates Chemo-Resistance and Radio-Resistance of Nasopharyngeal Carcinoma Cells by Influencing Cell Cycle and Activating the PI3K/AKT/mTOR Signaling Pathway. J. Transl. Med. 2022, 20, 264. [CrossRef]
- Bi, L.; Wang, H.; Tian, Y. Silencing FAM135B Enhances Radiosensitivity of Esophageal Carcinoma Cell. Gene 2021, 772, 145358. [CrossRef]
- Liu, G.; Yang, S.; Liu, Y.; Xu, Y.; Qiu, H.; Sun, J.; Song, J.; Shi, L. The Adenosine-A2a Receptor Regulates the Radioresistance of Gastric Cancer via PI3K-AKT-mTOR Pathway. Int. J. Clin. Oncol. 2022, 27, 911–920. [CrossRef]
- Yu, H.; Li, X.; Li, Y.; Wang, T.; Wang, M.; Mao, P. MiR-4524b-5p-Targeting ALDH1A3 Attenuates the Proliferation and Radioresistance of Glioblastoma via PI3K/AKT/mTOR Signaling. CNS Neurosci. Ther. 2024, 30, e14396. [CrossRef]
- Li, P.; Cui, Y.; Hu, K.; Wang, X.; Yu, Y. Silencing APLNR Enhances the Radiosensitivity of Prostate Cancer by Modulating the PI3K/AKT/mTOR Signaling Pathway. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2024. [CrossRef]
- Xiong, L.; Tan, B.; Lei, X.; Zhang, B.; Li, W.; Liu, D.; Xia, T. SIRT6 through PI3K/Akt/mTOR Signaling Pathway to Enhance Radiosensitivity of Non-Small Cell Lung Cancer and Inhibit Tumor Progression. IUBMB Life 2021, 73, 1092–1102. [CrossRef]
- Park, C.-M.; Park, M.-J.; Kwak, H.-J.; Lee, H.-C.; Kim, M.-S.; Lee, S.-H.; Park, I.-C.; Rhee, C.H.; Hong, S.-I. Ionizing Radiation Enhances Matrix Metalloproteinase-2 Secretion and Invasion of Glioma Cells through Src/Epidermal Growth Factor Receptor-Mediated P38/Akt and Phosphatidylinositol 3-Kinase/Akt Signaling Pathways. Cancer Res. 2006, 66, 8511–8519. [CrossRef]
- Lan, Q.; Tan, X.; He, P.; Li, W.; Tian, S.; Dong, W. TRIM11 Promotes Proliferation, Migration, Invasion and EMT of Gastric Cancer by Activating β-Catenin Signaling. OncoTargets Ther. 2021, Volume 14, 1429–1440. [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of Epithelial-Mesenchymal Transition and Cancer Stem Cell Phenotypes Is Associated with Activation of the PI3K/Akt/mTOR Pathway in Prostate Cancer Radioresistance. Cell Death Dis. 2013, 4, e875. [CrossRef]
- Ni, J.; Cozzi, P.; Hao, J.; Beretov, J.; Chang, L.; Duan, W.; Shigdar, S.; Delprado, W.; Graham, P.; Bucci, J.; et al. Epithelial Cell Adhesion Molecule (EpCAM) Is Associated with Prostate Cancer Metastasis and Chemo/Radioresistance via the PI3K/Akt/mTOR Signaling Pathway. Int. J. Biochem. Cell Biol. 2013, 45, 2736–2748. [CrossRef]
- Ma, W.-Q.; Miao, M.-C.; Ding, P.-A.; Tan, B.-B.; Liu, W.-B.; Guo, S.; Er, L.-M.; Zhang, Z.-D.; Zhao, Q. CALD1 Facilitates Epithelial-Mesenchymal Transition Progression in Gastric Cancer Cells by Modulating the PI3K-Akt Pathway. World J. Gastrointest. Oncol. 2024, 16, 1029–1045. [CrossRef]
- Beckers, C.; Pruschy, M.; Vetrugno, I. Tumor Hypoxia and Radiotherapy: A Major Driver of Resistance Even for Novel Radiotherapy Modalities. Semin. Cancer Biol. 2024, 98, 19–30. [CrossRef]
- Basheeruddin, M.; Qausain, S. Hypoxia-Inducible Factor 1-Alpha (HIF-1α): An Essential Regulator in Cellular Metabolic Control. Cureus 2024, 16, e63852. [CrossRef]
- Hill, R.M.; Li, C.; Hughes, J.R.; Rocha, S.; Grundy, G.J.; Parsons, J.L. Autophagy Is the Main Driver of Radioresistance of HNSCC Cells in Mild Hypoxia. J. Cell. Mol. Med. 2024, 28, e18482. [CrossRef]
- Sun, Y.; Chen, K.; Lin, G.; Wan, F.; Chen, L.; Zhu, X. Silencing C-Jun Inhibits Autophagy and Abrogates Radioresistance in Nasopharyngeal Carcinoma by Activating the PI3K/AKT/mTOR Pathway. Ann. Transl. Med. 2021, 9, 1085. [CrossRef]
- Yuan, T.-Z.; Lin, H.-Y.; Kuei, C.-H.; Lin, C.-H.; Lee, H.-H.; Lee, H.-L.; Lu, H.-W.; Su, C.-Y.; Chiu, H.-W.; Lin, Y.-F. NEDD8 Promotes Radioresistance via Triggering Autophagy Formation and Serves as a Novel Prognostic Marker in Oral Squamous Cell Carcinoma. Cancer Cell Int. 2023, 23, 41. [CrossRef]
- Wang, B.; Min, W.; Lin, S.; Song, L.; Yang, P.; Ma, Q.; Guo, J. Saikosaponin-d Increases Radiation-Induced Apoptosis of Hepatoma Cells by Promoting Autophagy via Inhibiting mTOR Phosphorylation. Int. J. Med. Sci. 2021, 18, 1465–1473. [CrossRef]
- Karami Fath, M.; Ebrahimi, M.; Nourbakhsh, E.; Zia Hazara, A.; Mirzaei, A.; Shafieyari, S.; Salehi, A.; Hoseinzadeh, M.; Payandeh, Z.; Barati, G. PI3K/Akt/mTOR Signaling Pathway in Cancer Stem Cells. Pathol. Res. Pract. 2022, 237, 154010. [CrossRef]
- Schaefer, T.; Steiner, R.; Lengerke, C. SOX2 and P53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer. Int. J. Mol. Sci. 2020, 21, 4902. [CrossRef]
- Park, J.-H.; Kim, Y.-H.; Shim, S.; Kim, A.; Jang, H.; Lee, S.-J.; Park, S.; Seo, S.; Jang, W.I.; Lee, S.B.; et al. Radiation-Activated PI3K/AKT Pathway Promotes the Induction of Cancer Stem-like Cells via the Upregulation of SOX2 in Colorectal Cancer. Cells 2021, 10, 135. [CrossRef]
- Bai, J.; Chen, W.-B.; Zhang, X.-Y.; Kang, X.-N.; Jin, L.-J.; Zhang, H.; Wang, Z.-Y. HIF-2α Regulates CD44 to Promote Cancer Stem Cell Activation in Triple-Negative Breast Cancer via PI3K/AKT/mTOR Signaling. World J. Stem Cells 2020, 12, 87–99. [CrossRef]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of Epithelial-Mesenchymal Transition and Cancer Stem Cell Phenotypes Is Associated with Activation of the PI3K/Akt/mTOR Pathway in Prostate Cancer Radioresistance. Cell Death Dis. 2013, 4, e875. [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K Isoforms in Cell Signalling and Vesicle Trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [CrossRef]
- Han, B.; Lin, X.; Hu, H. Regulation of PI3K Signaling in Cancer Metabolism and PI3K-Targeting Therapy. Transl. Breast Cancer Res. J. Focus. Transl. Res. Breast Cancer 2024, 5, 33. [CrossRef]
- Vasan, N.; Cantley, L.C. At a Crossroads: How to Translate the Roles of PI3K in Oncogenic and Metabolic Signalling into Improvements in Cancer Therapy. Nat. Rev. Clin. Oncol. 2022, 19, 471–485. [CrossRef]
- Danyaei, A.; Ghanbarnasab-Behbahani, R.; Teimoori, A.; Neisi, N.; Chegeni, N. The Simultaneous Use of CRISPR/Cas9 to Knock out the PI3Kca Gene with Radiation to Enhance Radiosensitivity and Inhibit Tumor Growth in Breast Cancer. Iran. J. Basic Med. Sci. 2024, 27, 1566–1573. [CrossRef]
- Shi, J.-J.; Xing, H.; Wang, Y.-X.; Zhang, X.; Zhan, Q.-M.; Geng, M.-Y.; Ding, J.; Meng, L.-H. PI3Kα Inhibitors Sensitize Esophageal Squamous Cell Carcinoma to Radiation by Abrogating Survival Signals in Tumor Cells and Tumor Microenvironment. Cancer Lett. 2019, 459, 145–155. [CrossRef]
- Korovina, I.; Elser, M.; Borodins, O.; Seifert, M.; Willers, H.; Cordes, N. Β1 Integrin Mediates Unresponsiveness to PI3Kα Inhibition for Radiochemosensitization of 3D HNSCC Models. Biomed. Pharmacother. Biomedecine Pharmacother. 2024, 171, 116217. [CrossRef]
- Yu, Y.; Gu, D.; Cai, L.; Yang, H.; Sheng, R. Development of Small-Molecule Inhibitors That Target PI3Kβ. Drug Discov. Today 2024, 29, 103854. [CrossRef]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear Phosphoinositide 3-Kinase Beta Controls Double-Strand Break DNA Repair. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7491–7496. [CrossRef]
- Redondo-Muñoz, J.; Rodríguez, M.J.; Silió, V.; Pérez-García, V.; Valpuesta, J.M.; Carrera, A.C. Phosphoinositide 3-Kinase Beta Controls Replication Factor C Assembly and Function. Nucleic Acids Res. 2013, 41, 855–868. [CrossRef]
- Marqués, M.; Kumar, A.; Poveda, A.M.; Zuluaga, S.; Hernández, C.; Jackson, S.; Pasero, P.; Carrera, A.C. Specific Function of Phosphoinositide 3-Kinase Beta in the Control of DNA Replication. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7525–7530. [CrossRef]
- Aytenfisu, T.Y.; Campbell, H.M.; Chakrabarti, M.; Amzel, L.M.; Gabelli, S.B. Class I PI3K Biology. Curr. Top. Microbiol. Immunol. 2022, 436, 3–49. [CrossRef]
- Russell, S.N.; Demetriou, C.; Valenzano, G.; Evans, A.; Go, S.; Stanly, T.; Hazini, A.; Willenbrock, F.; Gordon-Weeks, A.N.; Mukherjee, S.; et al. Induction of Macrophage Efferocytosis in Pancreatic Cancer via PI3Kγ Inhibition and Radiotherapy Promotes Tumour Control. Gut 2025, gutjnl-2024-333492. [CrossRef]
- Yoon, Y.N.; Lee, E.; Kwon, Y.-J.; Gim, J.-A.; Kim, T.-J.; Kim, J.-S. PI3Kδ/γ Inhibitor BR101801 Extrinsically Potentiates Effector CD8+ T Cell-Dependent Antitumor Immunity and Abscopal Effect after Local Irradiation. J. Immunother. Cancer 2022, 10, e003762. [CrossRef]
- Xiang, Q.; Dong, S.; Li, X.-H. A Review of Phosphocreatine 3 Kinase δ Subtype (PI3Kδ) and Its Inhibitors in Malignancy. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2021, 27, e932772. [CrossRef]
- Yazdimamaghani, M.; Kolupaev, O.V.; Lim, C.; Hwang, D.; Laurie, S.J.; Perou, C.M.; Kabanov, A.V.; Serody, J.S. Tumor Microenvironment Immunomodulation by Nanoformulated TLR 7/8 Agonist and PI3k Delta Inhibitor Enhances Therapeutic Benefits of Radiotherapy. Biomaterials 2025, 312, 122750. [CrossRef]
- Chang, W.I.; Han, M.G.; Kang, M.H.; Park, J.M.; Kim, E.E.; Bae, J.; Ahn, S.; Kim, I.A. PI3Kαδ Inhibitor Combined with Radiation Enhances the Antitumor Immune Effect of Anti-PD1 in a Syngeneic Murine Triple-Negative Breast Cancer Model. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 845–858. [CrossRef]
- Modi, P.; Balakrishnan, K.; Yang, Q.; Wierda, W.G.; Keating, M.J.; Gandhi, V. Idelalisib and Bendamustine Combination Is Synergistic and Increases DNA Damage Response in Chronic Lymphocytic Leukemia Cells. Oncotarget 2017, 8, 16259–16274. [CrossRef]
- Gryc, T.; Putz, F.; Goerig, N.; Ziegler, S.; Fietkau, R.; Distel, L.V.; Schuster, B. Idelalisib May Have the Potential to Increase Radiotherapy Side Effects. Radiat. Oncol. Lond. Engl. 2017, 12, 109. [CrossRef]
- Chen, W.S.; Xu, P.-Z.; Gottlob, K.; Chen, M.-L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth Retardation and Increased Apoptosis in Mice with Homozygous Disruption of the Akt1 Gene. Genes Dev. 2001, 15, 2203–2208. [CrossRef]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin Resistance and a Diabetes Mellitus-like Syndrome in Mice Lacking the Protein Kinase Akt2 (PKBβ). Science 2001, 292, 1728–1731. [CrossRef]
- Tschopp, O.; Yang, Z.-Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential Role of Protein Kinase Bγ (PKBγ/Akt3) in Postnatal Brain Development but Not in Glucose Homeostasis. Development 2005, 132, 2943–2954. [CrossRef]
- Degan, S.E.; Gelman, I.H. Emerging Roles for AKT Isoform Preference in Cancer Progression Pathways. Mol. Cancer Res. MCR 2021, 19, 1251–1257. [CrossRef]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 Enhances Radiation Toxicity of Human Tumor Cells by Inhibiting DNA-PKcs-Dependent DNA Double-Strand Break Repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [CrossRef]
- Toulany, M.; Lee, K.-J.; Fattah, K.R.; Lin, Y.-F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt Promotes Post-Irradiation Survival of Human Tumor Cells through Initiation, Progression, and Termination of DNA-PKcs-Dependent DNA Double-Strand Break Repair. Mol. Cancer Res. MCR 2012, 10, 945–957. [CrossRef]
- Toulany, M.; Maier, J.; Iida, M.; Rebholz, S.; Holler, M.; Grottke, A.; Jüker, M.; Wheeler, D.L.; Rothbauer, U.; Rodemann, H.P. Akt1 and Akt3 but Not Akt2 through Interaction with DNA-PKcs Stimulate Proliferation and Post-Irradiation Cell Survival of K-RAS-Mutated Cancer Cells. Cell Death Discov. 2017, 3, 17072. [CrossRef]
- Toulany, M.; Iida, M.; Lettau, K.; Coan, J.P.; Rebholz, S.; Khozooei, S.; Harari, P.M.; Wheeler, D.L. Targeting HER3-Dependent Activation of Nuclear AKT Improves Radiotherapy of Non-Small Cell Lung Cancer. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2022, 174, 92–100. [CrossRef]
- Szymonowicz, K.; Oeck, S.; Krysztofiak, A.; van der Linden, J.; Iliakis, G.; Jendrossek, V. Restraining Akt1 Phosphorylation Attenuates the Repair of Radiation-Induced DNA Double-Strand Breaks and Reduces the Survival of Irradiated Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2233. [CrossRef]
- Mueck, K.; Rebholz, S.; Harati, M.D.; Rodemann, H.P.; Toulany, M. Akt1 Stimulates Homologous Recombination Repair of DNA Double-Strand Breaks in a Rad51-Dependent Manner. Int. J. Mol. Sci. 2017, 18, 2473. [CrossRef]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating Akt1 Mutations Alter DNA Double Strand Break Repair and Radiosensitivity. Sci. Rep. 2017, 7, 42700. [CrossRef]
- Seiwert, N.; Neitzel, C.; Stroh, S.; Frisan, T.; Audebert, M.; Toulany, M.; Kaina, B.; Fahrer, J. AKT2 Suppresses Pro-Survival Autophagy Triggered by DNA Double-Strand Breaks in Colorectal Cancer Cells. Cell Death Dis. 2017, 8, e3019. [CrossRef]
- Mohammadian Gol, T.; Rodemann, H.P.; Dittmann, K. Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination. Int. J. Mol. Sci. 2019, 20, 6316. [CrossRef]
- Sahlberg, S.H.; Gustafsson, A.-S.; Pendekanti, P.N.; Glimelius, B.; Stenerlöw, B. The Influence of AKT Isoforms on Radiation Sensitivity and DNA Repair in Colon Cancer Cell Lines. Tumor Biol. 2014, 35, 3525–3534. [CrossRef]
- Tan, P. -x; Du, S. -s; Ren, C.; Yao, Q. -w; Zheng, R.; Li, R.; Yuan, Y. -w MicroRNA-207 Enhances Radiation-Induced Apoptosis by Directly Targeting Akt3 in Cochlea Hair Cells. Cell Death Dis. 2014, 5, e1433. [CrossRef]
- Polytarchou, C.; Hatziapostolou, M.; Yau, T.O.; Christodoulou, N.; Hinds, P.W.; Kottakis, F.; Sanidas, I.; Tsichlis, P.N. Akt3 Induces Oxidative Stress and DNA Damage by Activating the NADPH Oxidase via Phosphorylation of P47phox. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 28806–28815. [CrossRef]
- Li, G.; Wu, A.; Qi, D.; Cui, F.; Zeng, Y.; Xie, F.; Wu, H.; Gu, Y.; Chen, Q.; Zhang, X. Differential Effects of Peptidoglycan on Colorectal Tumors and Intestinal Tissue Post-Pelvic Radiotherapy. Oncotarget 2016, 7, 75685–75697. [CrossRef]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically Amplified Akt3 Activates DNA Repair Pathway and Promotes Glioma Progression. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3421–3426. [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [CrossRef]
- Guo, S.; Xing, S.; Wu, Z.; Chen, F.; Pan, X.; Li, Q.; Liu, W.; Zhang, G. Leucine Restriction Ameliorates Fusobacterium Nucleatum-Driven Malignant Progression and Radioresistance in Nasopharyngeal Carcinoma. Cell Rep. Med. 2024, 5, 101753. [CrossRef]
- Wang, X.; Cao, Z.; Yue, X.; Liu, T.; Wen, G.; Jiang, D.; Wu, W.; Le, L.; Wang, Y.; Wang, C.; et al. Stabilization of DEPTOR Sensitizes Hypopharyngeal Cancer to Radiotherapy via Targeting Degradation. Mol. Ther. - Oncolytics 2022, 26, 330–346. [CrossRef]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. mTORC1 Pathway in DNA Damage Response. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1293–1311. [CrossRef]
- Holler, M.; Grottke, A.; Mueck, K.; Manes, J.; Jücker, M.; Rodemann, H.P.; Toulany, M. Dual Targeting of Akt and mTORC1 Impairs Repair of DNA Double-Strand Breaks and Increases Radiation Sensitivity of Human Tumor Cells. PLOS One 2016, 11, e0154745. [CrossRef]
- Murata, Y.; Uehara, Y.; Hosoi, Y. Activation of mTORC1 under Nutrient Starvation Conditions Increases Cellular Radiosensitivity in Human Liver Cancer Cell Lines, HepG2 and HuH6. Biochem. Biophys. Res. Commun. 2015, 468, 684–690. [CrossRef]
- Mir, S.A.; Dar, A.; Alshehri, S.A.; Wahab, S.; Hamid, L.; Almoyad, M.A.A.; Ali, T.; Bader, G.N. Exploring the mTOR Signalling Pathway and Its Inhibitory Scope in Cancer. Pharm. Basel Switz. 2023, 16, 1004. [CrossRef]
- Kalpongnukul, N.; Bootsri, R.; Wongkongkathep, P.; Kaewsapsak, P.; Ariyachet, C.; Pisitkun, T.; Chantaravisoot, N. Phosphoproteomic Analysis Defines BABAM1 as mTORC2 Downstream Effector Promoting DNA Damage Response in Glioblastoma Cells. J. Proteome Res. 2022, 21, 2893–2904. [CrossRef]
- Tian, L.; Chen, C.; Guo, Y.; Zhang, F.; Mi, J.; Feng, Q.; Lin, S.; Xi, N.; Tian, J.; Yu, L.; et al. mTORC2 Regulates Ribonucleotide Reductase to Promote DNA Replication and Gemcitabine Resistance in Non-Small Cell Lung Cancer. Neoplasia N. Y. N 2021, 23, 643–652. [CrossRef]
- Miyahara, H.; Yadavilli, S.; Natsumeda, M.; Rubens, J.A.; Rodgers, L.; Kambhampati, M.; Taylor, I.C.; Kaur, H.; Asnaghi, L.; Eberhart, C.G.; et al. The Dual mTOR Kinase Inhibitor TAK228 Inhibits Tumorigenicity and Enhances Radiosensitization in Diffuse Intrinsic Pontine Glioma. Cancer Lett. 2017, 400, 110–116. [CrossRef]
- Liu, Z.-G.; Tang, J.; Chen, Z.; Zhang, H.; Wang, H.; Yang, J.; Zhang, H. The Novel mTORC1/2 Dual Inhibitor INK128 Enhances Radiosensitivity of Breast Cancer Cell Line MCF-7. Int. J. Oncol. 2016, 49, 1039–1045. [CrossRef]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mTORC1/mTORC2 Inhibitor AZD2014 Enhances the Radiosensitivity of Glioblastoma Stem-like Cells. Neuro-Oncol. 2014, 16, 29–37. [CrossRef]
- Wang, H.; An, J.; He, S.; Liao, C.; Wang, J.; Tuo, B. Chloride Intracellular Channels as Novel Biomarkers for Digestive System Tumors (Review). Mol. Med. Rep. 2021, 24, 630. [CrossRef]
- Shiratori, H.; Kawai, K.; Hata, K.; Tanaka, T.; Nishikawa, T.; Otani, K.; Sasaki, K.; Kaneko, M.; Murono, K.; Emoto, S.; et al. The Combination of Temsirolimus and Chloroquine Increases Radiosensitivity in Colorectal Cancer Cells. Oncol. Rep. 2019, 42, 377–385. [CrossRef]
- Chen, Y.-H.; Wang, C.-W.; Wei, M.-F.; Tzeng, Y.-S.; Lan, K.-H.; Cheng, A.-L.; Kuo, S.-H. Maintenance BEZ235 Treatment Prolongs the Therapeutic Effect of the Combination of BEZ235 and Radiotherapy for Colorectal Cancer. Cancers 2019, 11, 1204. [CrossRef]
- Xie, Y.; Liu, C.; Zhang, Y.; Li, A.; Sun, C.; Li, R.; Xing, Y.; Shi, M.; Wang, Q. PKI-587 Enhances Radiosensitization of Hepatocellular Carcinoma by Inhibiting the PI3K/AKT/mTOR Pathways and DNA Damage Repair. PLOS One 2021, 16, e0258817. [CrossRef]
- Lin, J.; Ruan, J.; Zhu, H.; Chen, Z.; Chen, J.; Yu, H. Tenacissoside H Induces Autophagy and Radiosensitivity of Hepatocellular Carcinoma Cells by PI3K/Akt/mTOR Signaling Pathway. Dose-Response Publ. Int. Hormesis Soc. 2021, 19, 15593258211011023. [CrossRef]
- Chow, Z.; Johnson, J.; Chauhan, A.; Izumi, T.; Cavnar, M.; Weiss, H.; Townsend, C.M.; Anthony, L.; Wasilchenko, C.; Melton, M.L.; et al. PI3K/mTOR Dual Inhibitor PF-04691502 Is a Schedule-Dependent Radiosensitizer for Gastroenteropancreatic Neuroendocrine Tumors. Cells 2021, 10, 1261. [CrossRef]
- Hayman, T.J.; Wahba, A.; Rath, B.H.; Bae, H.; Kramp, T.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. The ATP-Competitive mTOR Inhibitor INK128 Enhances in Vitro and in Vivo Radiosensitivity of Pancreatic Carcinoma Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 110–119. [CrossRef]
- Murgić, J.; Fröbe, A.; Kiang Chua, M.L. RECENT ADVANCES IN RADIOTHERAPY MODALITIES FOR PROSTATE CANCER. Acta Clin. Croat. 2022, 61, 57–64. [CrossRef]
- Zhu, W.; Fu, W.; Hu, L. NVP-BEZ235, Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor, Prominently Enhances Radiosensitivity of Prostate Cancer Cell Line PC-3. Cancer Biother. Radiopharm. 2013, 28, 665–673. [CrossRef]
- Rachamala, H.K.; Madamsetty, V.S.; Angom, R.S.; Nakka, N.M.; Dutta, S.K.; Wang, E.; Mukhopadhyay, D.; Pal, K. Targeting mTOR and Survivin Concurrently Potentiates Radiation Therapy in Renal Cell Carcinoma by Suppressing DNA Damage Repair and Amplifying Mitotic Catastrophe. J. Exp. Clin. Cancer Res. CR 2024, 43, 159. [CrossRef]
- Tayyar, Y.; Idris, A.; Vidimce, J.; Ferreira, D.A.; McMillan, N.A. Alpelisib and Radiotherapy Treatment Enhances Alisertib-Mediated Cervical Cancer Tumor Killing. Am. J. Cancer Res. 2021, 11, 3240–3251.
- Nassim, R.; Mansure, J.J.; Chevalier, S.; Cury, F.; Kassouf, W. Combining mTOR Inhibition with Radiation Improves Antitumor Activity in Bladder Cancer Cells in Vitro and in Vivo: A Novel Strategy for Treatment. PLOS One 2013, 8, e65257. [CrossRef]
- Vinod, S.K.; Hau, E. Radiotherapy Treatment for Lung Cancer: Current Status and Future Directions. Respirol. Carlton Vic 2020, 25 Suppl 2, 61–71. [CrossRef]
- Deng, H.; Chen, Y.; Wang, L.; Zhang, Y.; Hang, Q.; Li, P.; Zhang, P.; Ji, J.; Song, H.; Chen, M.; et al. PI3K/mTOR Inhibitors Promote G6PD Autophagic Degradation and Exacerbate Oxidative Stress Damage to Radiosensitize Small Cell Lung Cancer. Cell Death Dis. 2023, 14, 652. [CrossRef]
- Kim, S.Y.; Jeong, E.-H.; Lee, T.-G.; Kim, H.-R.; Kim, C.H. The Combination of Trametinib, a MEK Inhibitor, and Temsirolimus, an mTOR Inhibitor, Radiosensitizes Lung Cancer Cells. Anticancer Res. 2021, 41, 2885–2894. [CrossRef]
- Seol, M.Y.; Choi, S.H.; Yoon, H.I. Combining Radiation with PI3K Isoform-Selective Inhibitor Administration Increases Radiosensitivity and Suppresses Tumor Growth in Non-Small Cell Lung Cancer. J. Radiat. Res. (Tokyo) 2022, 63, 591–601. [CrossRef]
- Su, Y.-C.; Lee, W.-C.; Wang, C.-C.; Yeh, S.-A.; Chen, W.-H.; Chen, P.-J. Targeting PI3K/AKT/mTOR Signaling Pathway as a Radiosensitization in Head and Neck Squamous Cell Carcinomas. Int. J. Mol. Sci. 2022, 23, 15749. [CrossRef]
- Huang, S.; Du, K.; Liu, Z.; Li, J. Inhibition of mTOR by Temsirolimus Overcomes Radio-Resistance in Nasopharyngeal Carcinoma. Clin. Exp. Pharmacol. Physiol. 2022, 49, 703–709. [CrossRef]
- Klieber, N.; Hildebrand, L.S.; Faulhaber, E.; Symank, J.; Häck, N.; Härtl, A.; Fietkau, R.; Distel, L.V. Different Impacts of DNA-PK and mTOR Kinase Inhibitors in Combination with Ionizing Radiation on HNSCC and Normal Tissue Cells. Cells 2024, 13, 304. [CrossRef]
- Glorieux, M.; Dok, R.; Nuyts, S. The Influence of PI3K Inhibition on the Radiotherapy Response of Head and Neck Cancer Cells. Sci. Rep. 2020, 10, 16208. [CrossRef]
- Upadhyay, R.; Bazan, J.G. Advances in Radiotherapy for Breast Cancer. Surg. Oncol. Clin. N. Am. 2023, 32, 515–536. [CrossRef]
- Kempska, J.; Oliveira-Ferrer, L.; Grottke, A.; Qi, M.; Alawi, M.; Meyer, F.; Borgmann, K.; Hamester, F.; Eylmann, K.; Rossberg, M.; et al. Impact of AKT1 on Cell Invasion and Radiosensitivity in a Triple Negative Breast Cancer Cell Line Developing Brain Metastasis. Front. Oncol. 2023, 13, 1129682. [CrossRef]
- Johnson, J.; Chow, Z.; Napier, D.; Lee, E.; Weiss, H.L.; Evers, B.M.; Rychahou, P. Targeting PI3K and AMPKα Signaling Alone or in Combination to Enhance Radiosensitivity of Triple Negative Breast Cancer. Cells 2020, 9, 1253. [CrossRef]
- Gasimli, R.; Kayabasi, C.; Ozmen Yelken, B.; Asik, A.; Sogutlu, F.; Celebi, C.; Yilmaz Susluer, S.; Kamer, S.; Biray Avci, C.; Haydaroglu, A.; et al. The Effects of PKI-402 on Breast Tumor Models’ Radiosensitivity via Dual Inhibition of PI3K/mTOR. Int. J. Radiat. Biol. 2023, 99, 1961–1970. [CrossRef]
- Masoumi, H.; Soltani, A.; Ghatrehsamani, M. The Beneficial Role of SIRT1 Activator on Chemo- and Radiosensitization of Breast Cancer Cells in Response to IL-6. Mol. Biol. Rep. 2020, 47, 129–139. [CrossRef]
- Janjua, T.I.; Rewatkar, P.; Ahmed-Cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Kumeria, T.; Ziegler, D.S.; Kavallaris, M.; Mazzieri, R.; et al. Frontiers in the Treatment of Glioblastoma: Past, Present and Emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138. [CrossRef]
- Seol, M.Y.; Choi, S.H.; Lee, I.J.; Park, H.S.; Kim, H.R.; Kim, S.K.; Yoon, H.I. Selective Inhibition of PI3K Isoforms in Brain Tumors Suppresses Tumor Growth by Increasing Radiosensitivity. Yonsei Med. J. 2023, 64, 139–147. [CrossRef]
- Djuzenova, C.S.; Fischer, T.; Katzer, A.; Sisario, D.; Korsa, T.; Steussloff, G.; Sukhorukov, V.L.; Flentje, M. Opposite Effects of the Triple Target (DNA-PK/PI3K/mTOR) Inhibitor PI-103 on the Radiation Sensitivity of Glioblastoma Cell Lines Proficient and Deficient in DNA-PKcs. BMC Cancer 2021, 21, 1201. [CrossRef]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Sisario, D.; Brosch, P.K.; Göhrung, A.; Frister, S.; Zimmermann, H.; Flentje, M.; et al. Differential Effects of the Akt Inhibitor MK-2206 on Migration and Radiation Sensitivity of Glioblastoma Cells. BMC Cancer 2019, 19, 299. [CrossRef]
- Luboff, A.J.; DeRemer, D.L. Capivasertib: A Novel AKT Inhibitor Approved for Hormone-Receptor-Positive, HER-2-Negative Metastatic Breast Cancer. Ann. Pharmacother. 2024, 58, 1229–1237. [CrossRef]
- Day, D.; Prawira, A.; Spreafico, A.; Waldron, J.; Karithanam, R.; Giuliani, M.; Weinreb, I.; Kim, J.; Cho, J.; Hope, A.; et al. Phase I Trial of Alpelisib in Combination with Concurrent Cisplatin-Based Chemoradiotherapy in Patients with Locoregionally Advanced Squamous Cell Carcinoma of the Head and Neck. Oral Oncol. 2020, 108, 104753. [CrossRef]
- Dunn, L.A.; Riaz, N.; Fury, M.G.; McBride, S.M.; Michel, L.; Lee, N.Y.; Sherman, E.J.; Baxi, S.S.; Haque, S.S.; Katabi, N.; et al. A Phase 1b Study of Cetuximab and BYL719 (Alpelisib) Concurrent with Intensity Modulated Radiation Therapy in Stage III-IVB Head and Neck Squamous Cell Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2020, 106, 564–570. [CrossRef]
- McGowan, D.R.; Skwarski, M.; Bradley, K.M.; Campo, L.; Fenwick, J.D.; Gleeson, F.V.; Green, M.; Horne, A.; Maughan, T.S.; McCole, M.G.; et al. Buparlisib with Thoracic Radiotherapy and Its Effect on Tumour Hypoxia: A Phase I Study in Patients with Advanced Non-Small Cell Lung Carcinoma. Eur. J. Cancer Oxf. Engl. 1990 2019, 113, 87–95. [CrossRef]
- Wen, P.Y.; Rodon, J.A.; Mason, W.; Beck, J.T.; DeGroot, J.; Donnet, V.; Mills, D.; El-Hashimy, M.; Rosenthal, M. Phase I, Open-Label, Multicentre Study of Buparlisib in Combination with Temozolomide or with Concomitant Radiation Therapy and Temozolomide in Patients with Newly Diagnosed Glioblastoma. ESMO Open 2020, 5, e000673. [CrossRef]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C.; et al. Phase I Dose-Escalation Study of the PI3K/mTOR Inhibitor Voxtalisib (SAR245409, XL765) plus Temozolomide with or without Radiotherapy in Patients with High-Grade Glioma. Neuro-Oncol. 2015, 17, 1275–1283. [CrossRef]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a Novel Akt Inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [CrossRef]
- Vink, S.R.; Schellens, J.H.M.; Beijnen, J.H.; Sindermann, H.; Engel, J.; Dubbelman, R.; Moppi, G.; Hillebrand, M.J.X.; Bartelink, H.; Verheij, M. Phase I and Pharmacokinetic Study of Combined Treatment with Perifosine and Radiation in Patients with Advanced Solid Tumours. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2006, 80, 207–213. [CrossRef]
- Goda, J.; Pachpor, T.; Basu, T.; Chopra, S.; Gota, V. Targeting the AKT Pathway: Repositioning HIV Protease Inhibitors as Radiosensitizers. Indian J. Med. Res. 2016, 143, 145. [CrossRef]
- Buijsen, J.; Lammering, G.; Jansen, R.L.H.; Beets, G.L.; Wals, J.; Sosef, M.; Den Boer, M.O.; Leijtens, J.; Riedl, R.G.; Theys, J.; et al. Phase I Trial of the Combination of the Akt Inhibitor Nelfinavir and Chemoradiation for Locally Advanced Rectal Cancer. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 107, 184–188. [CrossRef]
- Garcia-Soto, A.E.; McKenzie, N.D.; Whicker, M.E.; Pearson, J.M.; Jimenez, E.A.; Portelance, L.; Hu, J.J.; Lucci, J.A.; Qureshi, R.; Kossenkov, A.; et al. Phase 1 Trial of Nelfinavir Added to Standard Cisplatin Chemotherapy with Concurrent Pelvic Radiation for Locally Advanced Cervical Cancer. Cancer 2021, 127, 2279–2293. [CrossRef]
- Lin, C.; Verma, V.; Lazenby, A.; Ly, Q.P.; Berim, L.D.; Schwarz, J.K.; Madiyalakan, M.; Nicodemus, C.F.; Hollingsworth, M.A.; Meza, J.L.; et al. Phase I/II Trial of Neoadjuvant Oregovomab-Based Chemoimmunotherapy Followed by Stereotactic Body Radiotherapy and Nelfinavir for Locally Advanced Pancreatic Adenocarcinoma. Am. J. Clin. Oncol. 2019, 42, 755–760. [CrossRef]
- Mukherjee, S.; Qi, C.; Shaw, R.; Jones, C.M.; Bridgewater, J.A.; Radhakrishna, G.; Patel, N.; Holmes, J.; Virdee, P.S.; Tranter, B.; et al. Standard or High Dose Chemoradiotherapy, with or without the Protease Inhibitor Nelfinavir, in Patients with Locally Advanced Pancreatic Cancer: The Phase 1/Randomised Phase 2 SCALOP-2 Trial. Eur. J. Cancer Oxf. Engl. 1990 2024, 209, 114236. [CrossRef]
- Waqar, S.N.; Robinson, C.; Bradley, J.; Goodgame, B.; Rooney, M.; Williams, K.; Gao, F.; Govindan, R. A Phase I Study of Temsirolimus and Thoracic Radiation in Non--Small-Cell Lung Cancer. Clin. Lung Cancer 2014, 15, 119–123. [CrossRef]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; van den Bent, M.J.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4797–4806. [CrossRef]
- Saba, N.F.; Force, S.; Staley, C.; Fernandez, F.; Willingham, F.; Pickens, A.; Cardona, K.; Chen, Z.; Goff, L.; Cardin, D.; et al. Phase IB Study of Induction Chemotherapy with XELOX, Followed by Radiation Therapy, Carboplatin, and Everolimus in Patients with Locally Advanced Esophageal Cancer. Am. J. Clin. Oncol. 2019, 42, 331–336. [CrossRef]
- de Melo, A.C.; Grazziotin-Reisner, R.; Erlich, F.; Fontes Dias, M.S.; Moralez, G.; Carneiro, M.; Ingles Garces, Á.H.; Guerra Alves, F.V.; Novaes Neto, B.; Fuchshuber-Moraes, M.; et al. A Phase I Study of mTOR Inhibitor Everolimus in Association with Cisplatin and Radiotherapy for the Treatment of Locally Advanced Cervix Cancer: PHOENIX I. Cancer Chemother. Pharmacol. 2016, 78, 101–109. [CrossRef]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.-H.; Stieber, V.W.; Malone, S.C.; et al. A Randomized Phase II Study of Everolimus in Combination with Chemoradiation in Newly Diagnosed Glioblastoma: Results of NRG Oncology RTOG 0913. Neuro-Oncol. 2018, 20, 666–673. [CrossRef]
- Narayan, V.; Vapiwala, N.; Mick, R.; Subramanian, P.; Christodouleas, J.P.; Bekelman, J.E.; Deville, C.; Rajendran, R.; Haas, N.B. Phase 1 Trial of Everolimus and Radiation Therapy for Salvage Treatment of Biochemical Recurrence in Prostate Cancer Patients Following Prostatectomy. Int. J. Radiat. Oncol. Biol. Phys. 2017, 97, 355–361. [CrossRef]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [CrossRef]
- Zhang, Y.; Xu, X.; Yang, K.; Wang, S.; Zhang, T.; Hui, F.; Zheng, F.; Geng, H.; Xu, C.; Xun, F.; et al. The Efficacy and Safety of PI3K and AKT Inhibitors for Patients with Cancer: A Systematic Review and Network Meta-Analysis. Eur. J. Pharmacol. 2024, 983, 176952. [CrossRef]
- Savas, P.; Lo, L.L.; Luen, S.J.; Blackley, E.F.; Callahan, J.; Moodie, K.; van Geelen, C.T.; Ko, Y.-A.; Weng, C.-F.; Wein, L.; et al. Alpelisib Monotherapy for PI3K-Altered, Pretreated Advanced Breast Cancer: A Phase II Study. Cancer Discov. 2022, 12, 2058–2073. [CrossRef]
- Sarker, D.; Dawson, N.A.; Aparicio, A.M.; Dorff, T.B.; Pantuck, A.J.; Vaishampayan, U.N.; Henson, L.; Vasist, L.; Roy-Ghanta, S.; Gorczyca, M.; et al. A Phase I, Open-Label, Dose-Finding Study of GSK2636771, a PI3Kβ Inhibitor, Administered with Enzalutamide in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5248–5257. [CrossRef]
- Shah, A.; Mangaonkar, A. Idelalisib: A Novel PI3Kδ Inhibitor for Chronic Lymphocytic Leukemia. Ann. Pharmacother. 2015, 49, 1162–1170. [CrossRef]
- Goto, H.; Izutsu, K.; Ennishi, D.; Mishima, Y.; Makita, S.; Kato, K.; Hanaya, M.; Hirano, S.; Narushima, K.; Teshima, T.; et al. Zandelisib (ME-401) in Japanese Patients with Relapsed or Refractory Indolent Non-Hodgkin’s Lymphoma: An Open-Label, Multicenter, Dose-Escalation Phase 1 Study. Int. J. Hematol. 2022, 116, 911–921. [CrossRef]
- Hong, D.S.; Postow, M.; Chmielowski, B.; Sullivan, R.; Patnaik, A.; Cohen, E.E.W.; Shapiro, G.; Steuer, C.; Gutierrez, M.; Yeckes-Rodin, H.; et al. Eganelisib, a First-in-Class PI3Kγ Inhibitor, in Patients with Advanced Solid Tumors: Results of the Phase 1/1b MARIO-1 Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023, 29, 2210–2219. [CrossRef]
- Moein, A.; Jin, J.Y.; Wright, M.R.; Wong, H. Quantitative Assessment of Drug Efficacy and Emergence of Resistance in Patients with Metastatic Renal Cell Carcinoma Using a Longitudinal Exposure-Tumor Growth Inhibition Model: Apitolisib (Dual PI3K/mTORC1/2 Inhibitor) versus Everolimus (mTORC1 Inhibitor). J. Clin. Pharmacol. 2024, 64, 1101–1111. [CrossRef]
- Kořánová, T.; Dvořáček, L.; Grebeňová, D.; Kuželová, K. JR-AB2-011 Induces Fast Metabolic Changes Independent of mTOR Complex 2 Inhibition in Human Leukemia Cells. Pharmacol. Rep. PR 2024, 76, 1390–1402. [CrossRef]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like Growth Factor (IGF) Signaling in Tumorigenesis and the Development of Cancer Drug Resistance. Genes Dis. 2015, 2, 13–25. [CrossRef]
- De Vera, A.A.; Reznik, S.E. Combining PI3K/Akt/mTOR Inhibition with Chemotherapy. In Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy; Elsevier, 2019; pp. 229–242 ISBN 978-0-12-816435-8.
- Rallis, K.S.; Lai Yau, T.H.; Sideris, M. Chemoradiotherapy in Cancer Treatment: Rationale and Clinical Applications. Anticancer Res. 2021, 41, 1–7. [CrossRef]
- Song, L.; Liu, S.; Zhao, S. Everolimus (RAD001) Combined with Programmed Death-1 (PD-1) Blockade Enhances Radiosensitivity of Cervical Cancer and Programmed Death-Ligand 1 (PD-L1) Expression by Blocking the Phosphoinositide 3-Kinase (PI3K)/Protein Kinase B (AKT)/Mammalian Target of Rapamycin (mTOR)/S6 Kinase 1 (S6K1) Pathway. Bioengineered 2022, 13, 11240–11257. [CrossRef]
- Bleaney, C.W.; Abdelaal, H.; Reardon, M.; Anandadas, C.; Hoskin, P.; Choudhury, A.; Forker, L. Clinical Biomarkers of Tumour Radiosensitivity and Predicting Benefit from Radiotherapy: A Systematic Review. Cancers 2024, 16, 1942. [CrossRef]
- Bamodu, O.A.; Chang, H.-L.; Ong, J.-R.; Lee, W.-H.; Yeh, C.-T.; Tsai, J.-T. Elevated PDK1 Expression Drives PI3K/AKT/MTOR Signaling Promotes Radiation-Resistant and Dedifferentiated Phenotype of Hepatocellular Carcinoma. Cells 2020, 9, 746. [CrossRef]
- Chen, Q.; Zheng, W.; Zhu, L.; Yao, D.; Wang, C.; Song, Y.; Hu, S.; Liu, H.; Bai, Y.; Pan, Y.; et al. ANXA6 Contributes to Radioresistance by Promoting Autophagy via Inhibiting the PI3K/AKT/mTOR Signaling Pathway in Nasopharyngeal Carcinoma. Front. Cell Dev. Biol. 2020, 8, 232. [CrossRef]
Figure 1.
Figure 1. The overview of the Influence of the PI3K/AKT/mTOR signaling pathway on the radiosensitivity of tumor Cells.
Figure 1.
Figure 1. The overview of the Influence of the PI3K/AKT/mTOR signaling pathway on the radiosensitivity of tumor Cells.
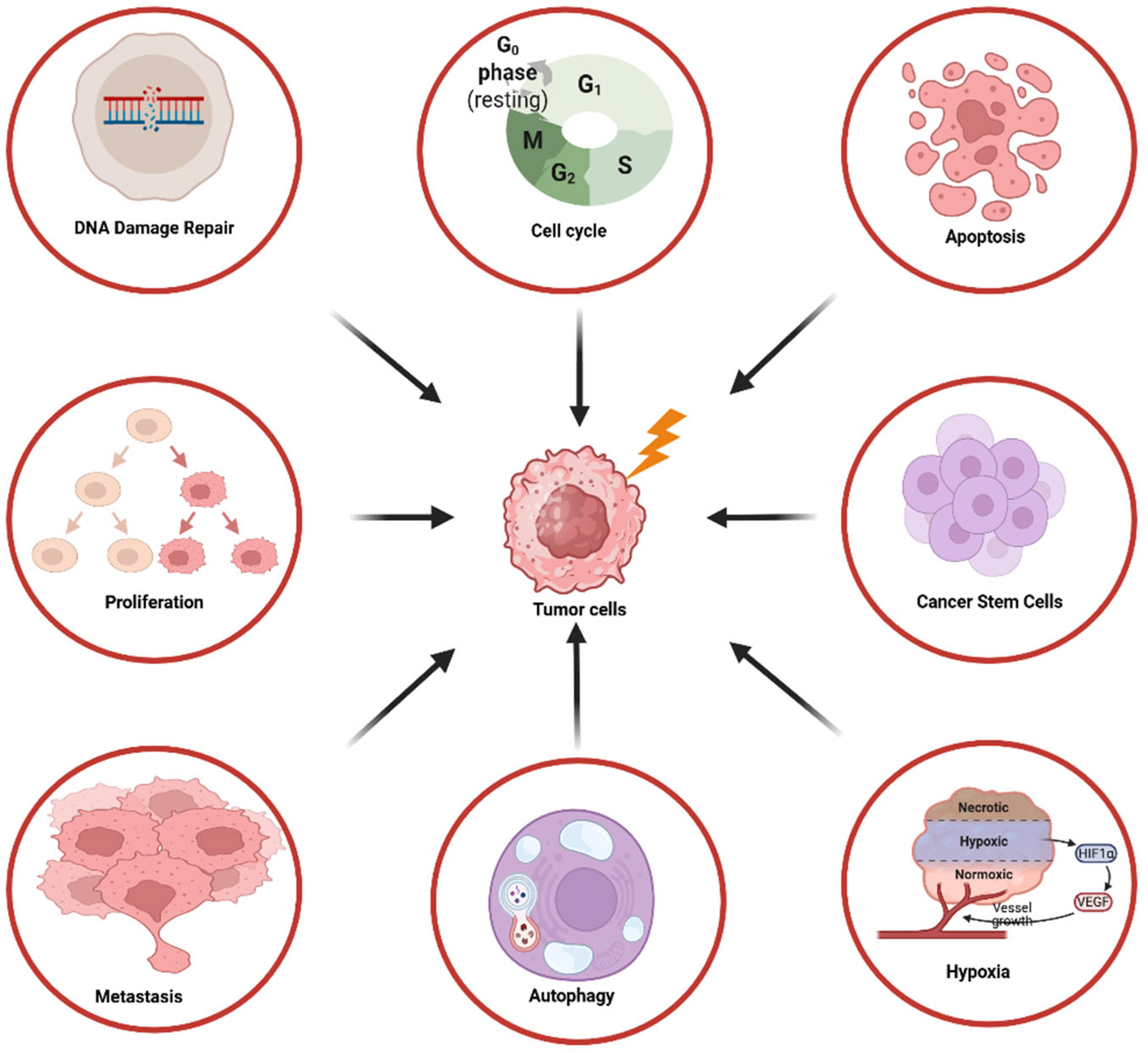
Figure 2.
The influence of PI3K isoform specificity on tumor radiosensitivity.
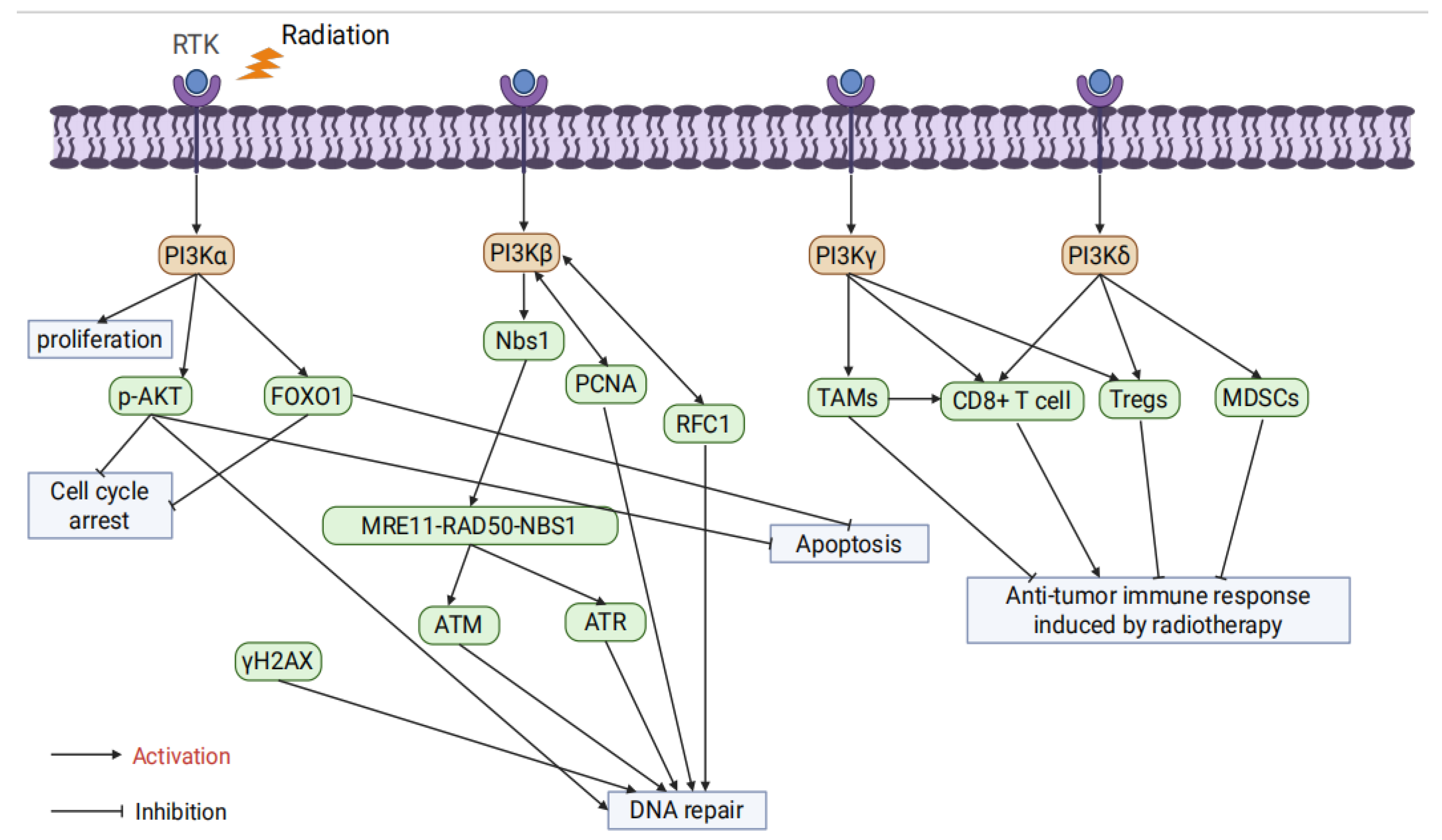
Figure 3.
The influence of AKT isoform specificity on tumor radiosensitivity.
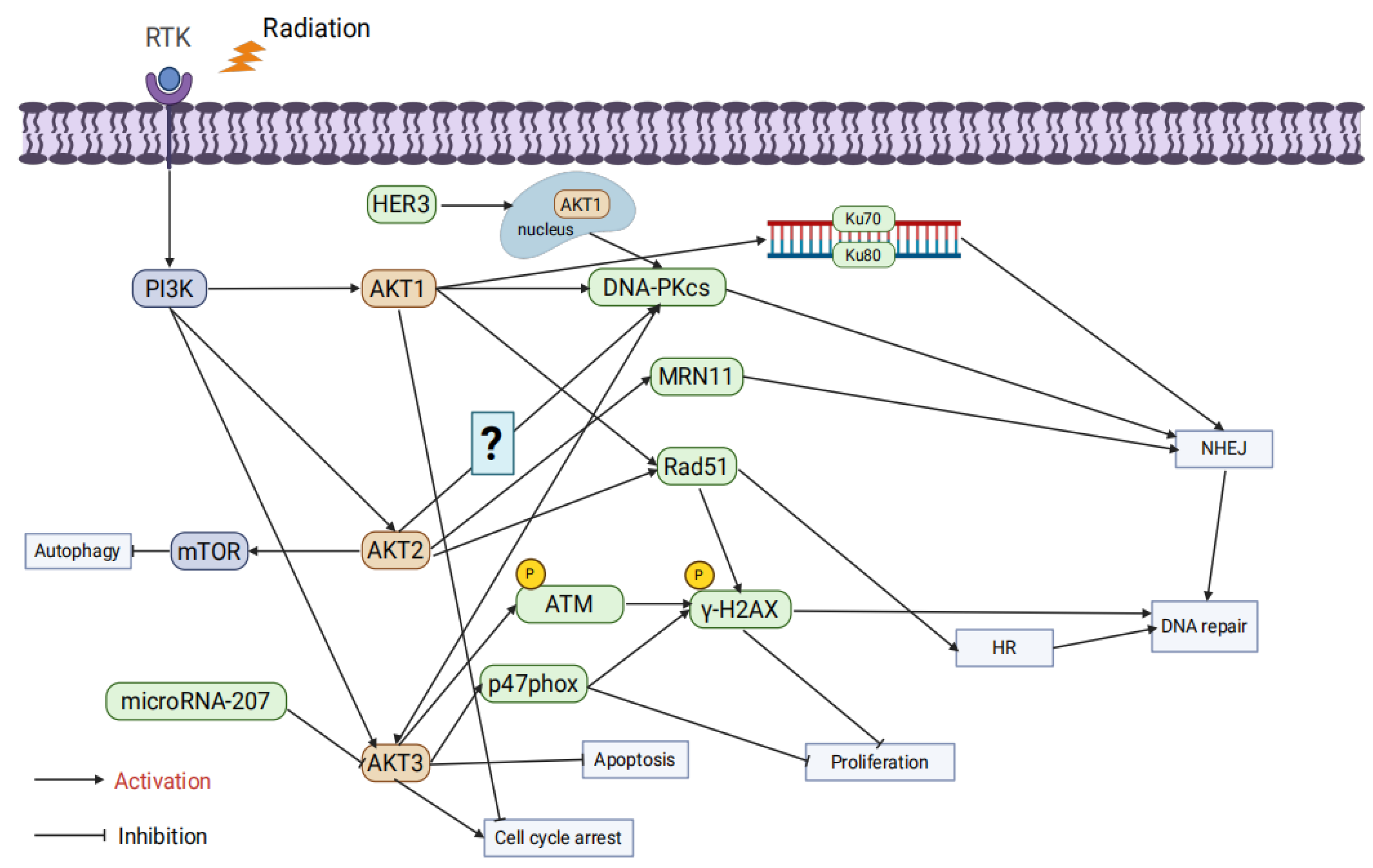
Figure 4.
The influence of mTOR on tumor radiosensitivity.
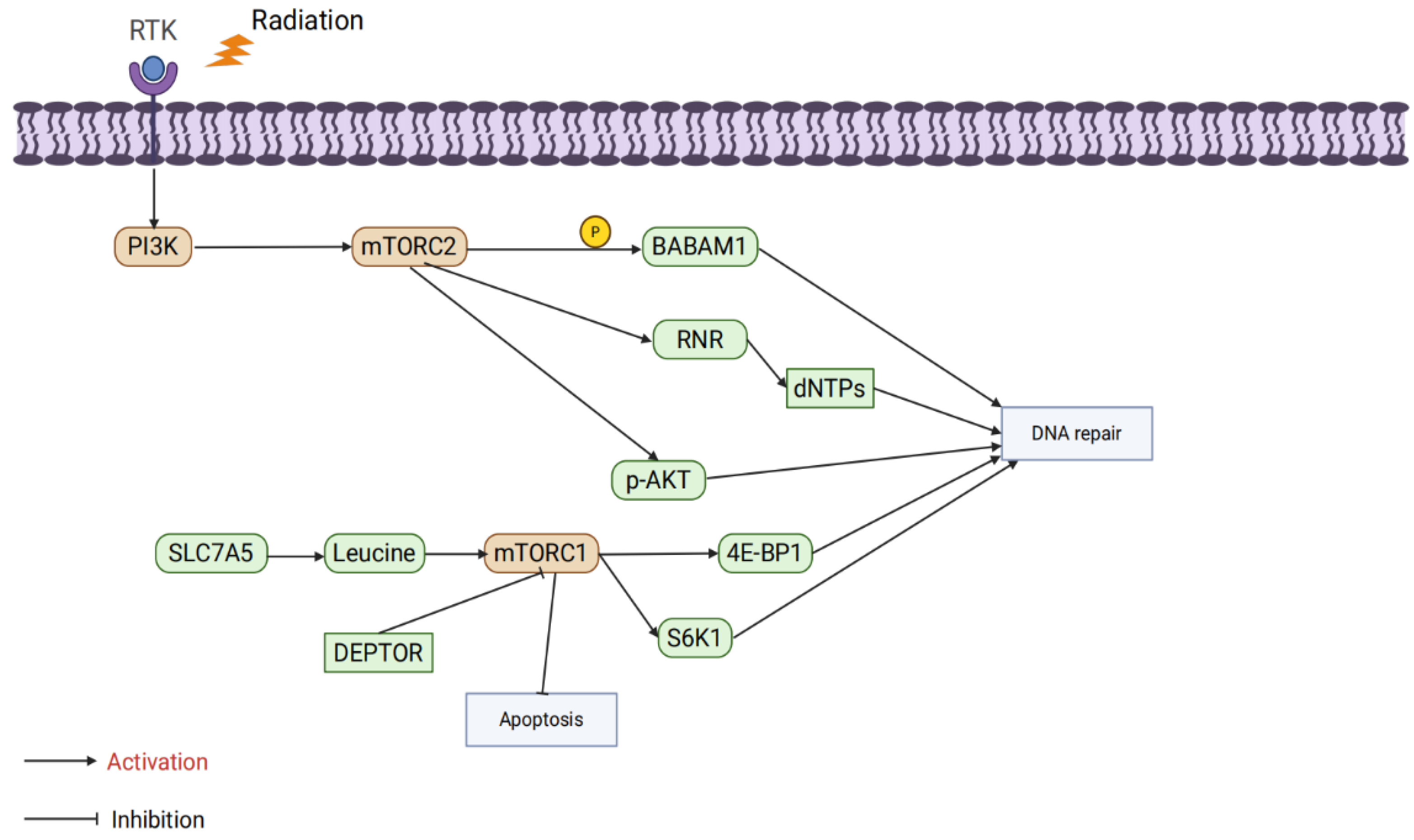

Table 1.
PI3K/AKT/mTOR pathway inhibitors combined with radiotherapy in clinical trials
| Inhibitor | Target | Population | Phase | ClinicalTrials.Gov Identifier |
| Alpelisib | PI3K | Head and Neck Squamous Cell Cancer | 1 | NCT02282371 Start date: October 2014 Completion date: October 2021 |
| PI3K | Head and Neck Cancer | 1 | NCT02537223 Start date: September 2015 Completion date: February 2020 | |
| Buparlisib | PI3K | Carcinoma, Non-Small-Cell Lung | 1 | NCT02128724 Start date: April 2013 Completion date: October 2017 |
| PI3K | Head and Neck Cancer | 1 | NCT02113878 Start date: September 2014 Completion date: January 2022 | |
| PI3K | Glioblastoma | 1 | NCT01473901 Start date: December 2011 Completion date: May 2017 | |
| Voxtalisib | PI3K/mTOR | Glioblastoma | 1 | NCT00704080 Start date: August 2008 Completion date: February 2013 |
| Paxalisib | PI3K/mTOR | Brain Metastases | 1 | NCT04192981 Start date: December 2019 Completion date: December 2025 |
| PI3K/mTOR | Diffuse Midline Gliomas | 2 | NCT05009992 Start date: October 2021 Completion date: June 2029 | |
| PI3K/mTOR | Glioblastoma | 2/3 | NCT03970447 Start date: July 2019 Completion date: June 2028 |
|
| Capivasertib | AKT | Breast Cancer | 2 | NCT06607757 Start date: December 2024 Completion date: August 2026 |
| Ipatasertib | AKT | Head and Neck Cancer | 1 | NCT05172245 Start date: September 2022 Completion date: June 2026 |
| Nelfinavir | AKT | Glioblastoma | 1 | NCT00694837 Start date: March 2009 Completion date: January 2013 |
| AKT | Oligometastases | 2 | NCT01728779 Start date: January 2014 Completion date: December 2020 | |
| AKT | Cervical Cancer | 1 | NCT01485731 Start date: January 2012 Completion date: February 2015 | |
| AKT | Pancreatic Cancer | 1 | NCT01068327 Start date: November 2007 Completion date: February 2015 | |
| AKT | Pancreatic Cancer | 2 | NCT01959672 Start date: September 2013 Completion date: December 2018 | |
| AKT | Glioblastoma | 1 | NCT01020292 Start date: April 2009 Completion date: December 2017 | |
| AKT | Non-Small Cell Lung Cancer | 1/2 | NCT00589056 Start date: June 2007 Completion date: March 2012 | |
| AKT | Pancreatic Cancer | 1/2 | NCT00589056 Start date: March 2016 Completion date: June 2021 | |
| AKT | Cervical Cancer | 1 | NCT02363829 Start date: February 2015 Completion date: February 2020 | |
| AKT | Head and Neck Squamous Cell Cancer | 2 | NCT02207439 Start date: July 2014 Completion date: May 2022 | |
| Everolimus | mTOR | High Grade Glioma | 2 | NCT05843253 Start date: August 2024 Completion date: August 2034 |
| mTOR | Neuroendocrine Tumors | 3 | NCT05918302 Start date: October 2023 Completion date: July 2028 | |
| mTOR | Diffuse Intrinsic Pontine Glioma | 3 | NCT05476939 Start date: September 2022 Completion date: September 2031 | |
| mTOR | Glioblastoma | 1/2 | NCT00553150 Start date: March 2009 Completion date: November 2019 | |
| mTOR | Head and Neck Cancer | 1 | NCT00858663 Start date: March 2009 Completion date: July 2013 | |
| mTOR | Glioblastoma | 1/2 | NCT01062399 Start date: December 2010 Completion date: May 2022 | |
| mTOR | Cervix Cancer | 1 | NCT01217177 Start date: December 2011 Completion date: April 2014 | |
| Temsirolimus | mTOR | Rhabdomyosarcoma | 3 | NCT02567435 Start date: June 2016 Completion date: October 2025 |
| mTOR | Diffuse Intrinsic Pontine Glioma | 1 | NCT02420613 Start date: October 2015 Completion date: October 2024 | |
| mTOR | Non-Small Cell Lung Cancer | 1 | NCT00796796 Start date: March 2009 Completion date: July 2011 | |
| mTOR | Glioblastoma | 1 | NCT00316849 Start date: May 2006 Completion date: November 2010 | |
| mTOR | Glioblastoma | 2 | NCT01019434 Start date: October 2009 Completion date: March 2014 | |
Clinical trial data was obtained from clinicaltrials.gov in May 2025.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



