Submitted:
04 June 2025
Posted:
05 June 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Respiratory infections caused by severe acute respiratory syndrome coronavirus 2, influenza virus, and respiratory syncytial virus pose significant global health challenges, leading to high morbidity and mortality, particularly in vulnerable populations. Despite their distinct virological characteristics, these viruses exploit host cellular metabolism to support replication, modulate immune responses, and promote disease progression. Emerging evidence shows that they induce metabolic reprogramming, shifting cellular energy production toward glycolysis to meet the bioenergetic demands of viral replication. Additionally, alterations in lipid metabolism, including enhanced fatty acid synthesis and disrupted cholesterol homeostasis, facilitate viral entry, replication, and immune evasion. Dysregulation of mitochondrial function and oxidative stress pathways also contributes to disease severity and long-term complications, such as persistent inflammation and immune exhaustion. Understanding these metabolic shifts is crucial for identifying new therapeutic targets and novel biomarkers for early disease detection, prognosis, and patient stratification. This review provides an overview of the metabolic alterations induced by severe acute respiratory syndrome coronavirus 2, influenza virus, and respiratory syncytial virus, highlighting shared and virus-specific mechanisms and potential therapeutic interventions.
Keywords:
infectious diseases
; influenza
; metabolism
; respiratory infections
; respiratory syncytial virus
; severe acute respiratory syndrome coronavirus 2
; viral infections
1. Introduction
Respiratory viral infections remain a significant global health burden, contributing to substantial morbidity and mortality, particularly among vulnerable populations [1,2]. Among these, infections caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), influenza virus, and respiratory syncytial virus (RSV) represent the most clinically relevant threats, leading to pandemics, seasonal outbreaks, and severe respiratory complications [3]. Despite their distinct virological characteristics, these viruses share a common strategy of hijacking host cellular metabolism to facilitate their replication, modulate immune responses, and drive pathogenesis.
Emerging evidence indicates that viral infections induce profound metabolic reprogramming, shifting cellular energy production, lipid metabolism, and amino acid utilization to favor viral propagation. SARS-CoV-2, influenza virus, and RSV all promote a metabolic switch toward glycolysis (resembling the Warburg effect observed in cancer cells) to meet the bioenergetic and biosynthetic demands of viral replication. Additionally, alterations in lipid metabolism, including increased fatty acid synthesis and disrupted cholesterol homeostasis, play a pivotal role in viral entry, replication, and immune evasion. Furthermore, dysregulation of mitochondrial function and oxidative stress pathways contributes to disease severity and long-term complications, such as persistent inflammation and immune exhaustion [4,5,6].
Understanding these metabolic alterations is crucial for identifying novel therapeutic targets and optimizing treatment strategies. While current antiviral therapies primarily focus on direct inhibition of viral replication, targeting host metabolic pathways offers a promising complementary approach to limit viral spread and mitigate severe disease outcomes [7,8]. Moreover, investigating virus-induced metabolic reprogramming has the potential to reveal novel diagnostic and prognostic biomarkers, enabling earlier detection of disease progression and stratification of patients based on metabolic signatures [9]. This review aims to provide a comprehensive overview of the metabolic alterations induced by SARS-CoV-2, influenza virus, and RSV, highlighting shared and virus-specific mechanisms, their implications for disease pathogenesis, and potential metabolic interventions for future therapeutic development.
2. Lipid Metabolism
Viruses utilize lipid rafts for cell entry, manipulate fatty acid synthesis and β-oxidation to fuel replication, and disrupt cholesterol homeostasis, impairing the innate immune response. Lipid droplets also act as platforms for viral assembly and immune modulation. The interaction between viral infections and mitochondria is essential in lipid metabolism during infectious diseases. In catabolism, mitochondria are the primary site for lipid β-oxidation, where fatty acids are broken down into acetyl-CoA, which enters the Krebs cycle to generate adenosine triphosphate (ATP). In anabolism, mitochondria produce citrate, which is transported to the cytosol and converted into acetyl-CoA for fatty acid synthesis. Thus, mitochondria regulate the balance between lipid degradation and synthesis, adjusting metabolic processes in response to cellular energy demands during infection.
2.1. Role of Lipid Rafts in Viral Entry
In animal cells, phospholipids comprise most of the lipids in cell membranes. However, the plasma membrane also incorporates glycolipids and cholesterol, essential for maintaining membrane fluidity [10]. While lipids typically move freely within the lipid bilayer, components such as cholesterol, glycosphingolipids, glycosylphosphatidylinositol (GPI)-anchored proteins, and some transmembrane proteins can cluster into specialized domains called lipid rafts [11,12]. The 2006 Keystone Symposium of Lipid Rafts and Cell Function defined these rafts as: “Small (10–200 nm), heterogenous, highly dynamic, sterol- and sphingolipid-enriched domains that compartmentalize cellular processes. Small rafts can sometimes be stabilized to form larger platforms through protein-protein and protein-lipid interactions” [13] (Figure 1).
Numerous viruses utilize these lipid rafts as key elements in the fusion process with the host cell. This complex process first requires virus binding to the cell surface, often via a cell receptor. These interactions induce conformational changes that promote viral entry. Cholesterol, an essential component of rafts, is fundamental in stabilizing these complexes, promoting membrane fluidity and facilitating fusion with the viral particles [14]. Before entry, many viruses establish weak ionic interactions with cell surface glycosaminoglycans, such as heparan and chondroitin sulfate, or with glycosphingolipids [15]. These interactions aid the virion's adhesion to the membrane, allowing it to move across the cell surface until it finds the specific receptor needed for internalization [16]. Viral trafficking on the cell surface or within the endosomal network facilitates its arrival at the unmasking site while providing signals to activate its fusion machinery.
The location of viral receptors within lipid rafts affects the efficiency of the fusion process between the viral particle and the cell membrane. Some receptors are constitutively located in lipid rafts, while others are recruited to rafts after virus binding [17,18,19,20,21]. This dynamics highlights the complexity of the fusion process and underscores the regulatory role of lipid microdomains in infection. Different viruses may employ various routes to enter host cells, with some relying entirely on lipid rafts while others exploit alternative pathways. Raft-mediated entry mechanisms often involve the concentration of viral receptors within these domains, optimizing viral adhesion and fusion. In other cases, the virus attaches to regions outside the rafts before being directed to these structures to complete its entry [14,22,23,24].
In addition to direct fusion with the cell membrane [25,26,27], viruses may exploit endocytic pathways related to lipid rafts. Endocytosis is a cellular process that allows the internalization of substances from the extracellular environment. It can be divided into phagocytosis and pinocytosis. Phagocytosis is primarily employed by specialized cells, such as macrophages, to digest bacteria and large particles. Pinocytosis, conversely, is a more generalized, non-specific process for the uptake of liquids and macromolecules through endocytic vesicles [28]. Within this process, two primary mechanisms are distinguished: clathrin-mediated endocytosis and clathrin-independent endocytosis. Clathrin-mediated endocytosis [29] is the most widely studied pathway for internalizing small and medium-sized viruses. Virions, after moving laterally across the cell surface, are incorporated into clathrin-coated invaginations, which then detach from the plasma membrane to form endocytic vesicles. These vesicles lose their clathrin coat before fusing with endosomes. This mechanism depends on dynamin-II, a guanosine-5'-triphosphatase essential for separating clathrin-coated vesicles from the plasma membrane.
On the other side, clathrin-independent endocytosis [30,31,32] encompasses various cholesterol-sensitive pathways, including caveolae-mediated and other pathways that are neither clathrin- nor caveolae-dependent. Caveolae are specialized microdomains of lipid rafts that form invaginations in the plasma membrane. They are coated internally by caveolins, proteins that regulate cholesterol organization and caveola biogenesis. Additionally, cavins, which stabilize the caveola structure, play a role in membrane curvature and the formation of endocytic vesicles [33,34,35].
The central role of lipids in regulating these events has been recognized in the past decade. Although phosphatidylinositol (PI) is the least abundant phospholipid in the cell membrane, its signaling capacity is crucial for endosomal trafficking and maturation [10]. The differential phosphorylation of PI, regulated by specific kinases and phosphatases, generates various phosphorylated species that control cellular compartmentalization and associated signaling [36,37]. Many viruses have developed strategies to exploit PI-mediated signaling at different stages of infection, especially to coordinate their entry and reprogram the host cell.
The phosphoinositide 3-kinase (PI3K) pathway is one of the major signaling pathways activated during viral entry. The activation of PI3K and the production of PI triphosphate serve as anchoring platforms for proteins with lipid-binding domains, including Akt, a key regulator of the PI3K pathway [38]. This signaling pathway is involved in multiple forms of endocytosis, though its role has been best characterized in macropinocytosis, where it regulates cytoskeletal rearrangement and membrane dynamics during macropinosome formation [39]. Various viruses, such as influenza [40,41], activate PI3K signaling to facilitate their internalization. Additionally, some viruses use this pathway to regulate post-internalization events, such as viral replication and assembly.
Different viruses rely on lipid rafts to varying degrees. SARS-CoV-2 primarily enters cells through the binding of spike protein to the angiotensin-converting enzyme 2 (ACE2) receptor, a process facilitated by lipid rafts [42]. These cholesterol-rich microdomains stabilize the receptor and enhance viral fusion, and their disruption has been shown to reduce SARS-CoV-2 entry into epithelial cells [43], highlighting their importance in infection [44]. However, although lipid rafts are a major platform for SARS-CoV-2 entry, it has recently been reported that the virus can also bind to ACE2 outside of these microdomains and be internalized via fusion [45], suggesting that lipid rafts are not strictly required for viral entry. The influenza virus utilizes multiple entry mechanisms, including clathrin-mediated endocytosis and macropinocytosis [44,46,47], which may or may not involve lipid rafts. However, it has been suggested that rafts are attachment points, aiding multivalent binding and increasing infection efficiency [48]. While lipid rafts play a role in influenza virus attachment, their membrane fusion necessity is less pronounced than SARS-CoV-2. RSV has a less well-defined relationship with lipid rafts. While certain studies suggest that its fusion proteins are associated with raft-like domains [49], others indicate that infection can occur independently of these structures [50].
In summary, viruses may employ lipid rafts as platforms for viral attachment, fusion, and entry. Many viruses, including SARS-CoV-2, influenza, and RSV, exploit these microdomains to enhance infection efficiency. However, the degree of dependence on lipid rafts varies among viruses. While SARS-CoV-2 primarily utilizes these structures for entry, influenza virus and RSV exhibit greater flexibility, often engaging alternative pathways to facilitate infection. Beyond their role in viral entry, lipid metabolism influences downstream processes critical to viral replication and assembly. In particular, many viruses fine-tune their infection cycle by hijacking phosphoinositide-mediated pathways, ensuring efficient propagation within the host.
2.2. Fatty Acid Synthesis, Lipid Droplets and β-Oxidation Dysregulation
The relationship between lipid synthesis and glycolysis is schematized in Figure 2. Fatty acid synthesis is a highly regulated anabolic pathway that converts acetyl-CoA into long-chain fatty acids and usually occurs in the liver and adipose tissues. This process is primarily controlled by acetyl-CoA carboxylase, which converts acetyl-CoA to malonyl-CoA, and fatty acid synthase (FASN), which catalyzes the sequential elongation of fatty acid chains. The sterol regulatory element-binding proteins (SREBPs), particularly SREBP-1c, play a key role in regulating the expression of these enzymes in response to metabolic cues. Additionally, the activity of ATP-citrate lyase, which provides cytosolic acetyl-CoA, and elongases that extend fatty acid chains further modulate lipid biosynthesis [51].
Viruses exploit fatty acid synthesis to support their replication. SARS-CoV-2 [52,53], influenza [54], and RSV [55] upregulate FASN expression to promote lipid biosynthesis. Increased fatty acid synthesis provides a lipid-rich environment for forming viral replication complexes and membranous vesicles that serve as viral replication platforms. Pharmacological inhibition of FASN has been shown to reduce viral replication in multiple models, highlighting its potential as an antiviral target [56].
Lipid droplets (LD) are dynamic organelles of a neutral lipid core surrounded by a phospholipid monolayer. They store triacylglycerols and cholesterol esters, which can be mobilized through lipolysis under metabolic stress or increased cellular demand [57]. LD formation is regulated by diacylglycerol O-acyltransferase 1 and 2 (DGAT1 and DGAT2), which mediate triacylglycerol synthesis and packaging within the endoplasmic reticulum membrane [58]. The release of LD into the cytoplasm is influenced by SREBP-1 and perilipin, a droplet membrane protein [59].
SARS-CoV-2, influenza A, and RSV manipulate LD within host cells through distinct mechanisms to enhance their replication. SARS-CoV-2 promotes their synthesis, as evidenced by increased LD accumulation in monocytes from coronavirus disease (COVID)-19 patients compared to uninfected individuals. In vitro studies have shown that SARS-CoV-2 modulates SREBP-1 and DGAT-1 expression, triggering LD formation [60]. In contrast, influenza A virus activates the mechanistic target of rapamycin (mTOR) complexes 1 and 2, inducing autophagy and LD degradation to support viral replication [61,62,63]. Although RSV’s interaction with LD is less understood, animal studies indicate that they disperse and degrade LD, contributing to oxidative stress, inflammation, and airway hyperresponsiveness [64].
Fatty acid oxidation (β-oxidation) breaks down fatty acids into acetyl-CoA for ATP production in mitochondria and peroxisomes and is regulated by carnitine palmitoyltransferases (CPT) [65,66]. Because β-oxidation is a significant energy source in times of high demand, viruses have evolved mechanisms to manipulate this pathway to optimize their replication, either by downregulating it to accumulate free fatty acids for membrane synthesis or upregulating it to boost ATP production [67,68]. For instance, SARS-CoV-2 has been shown to impair fatty acid oxidation in alveolar epithelial cells, leading to decreased expression of CPT1A and peroxisome proliferator-activated receptor-γ coactivator 1- α (PGC-1α), both critical for mitochondrial function [69]. Influenza virus can suppress β-oxidation by downregulating carnitine palmitoyltransferase II (CPT II) and altering mitochondrial dynamics, contributing to metabolic reprogramming in infected cells [70]. RSV infection has also been linked to metabolic dysfunction, including disruptions in mitochondrial fatty acid oxidation [71] though evidence remains limited and requires further confirmation.
2.3. Disrupted Cholesterol Homeostasis and Innate Immune Evasion
Cholesterol is essential to eukaryotic cell membranes, ensuring structural integrity, regulating membrane fluidity, and facilitating key cellular functions such as endocytosis, vesicular transport, and immune response modulation. Cellular cholesterol homeostasis is tightly controlled through endogenous synthesis, receptor-mediated uptake of low-density lipoproteins (LDL), and high-density lipoprotein (HDL)-dependent efflux, governed by SREBP and liver X receptors. [72].
HDL particles are considered to be a part of the innate immune system. Under normal conditions, HDL exhibit anti-inflammatory, antioxidant, and protective properties [73]. They facilitate reverse cholesterol transport, prevent lipid oxidation, and modulate immune cell activity, thereby reducing oxidative stress and inflammation. Additionally, HDL help neutralize bacterial toxins and support defense mechanisms [74].
However, during infection and inflammation, the host initiates a cascade of responses known as the acute-phase response, which profoundly disrupts lipid metabolism, particularly HDL function. During the acute-phase response, the levels of key proteins involved in HDL-mediated reverse cholesterol transport decline, including lecithin:cholesterol acyltransferase, cholesterol ester transfer protein, phospholipid transfer protein, apolipoprotein A-I, and paraoxonase 1 [75]. These alterations impair HDL's ability to remove excess cholesterol and neutralize oxidative damage. Simultaneously, HDL composition shifts, characterized by cholesterol ester depletion and enrichment in free cholesterol, triglycerides, and free fatty acids. Furthermore, apolipoprotein J and serum amyloid A levels increase substantially, replacing apolipoprotein A-I and transforming HDL into a pro-atherogenic and pro-inflammatory particle [76,77]. As a result, HDL loses its protective functions, instead promoting endothelial dysfunction, oxidative stress, and chronic inflammation, which may contribute to immune evasion by pathogens (Figure 3).
In addition to alterations in HDL composition, infectious diseases induce changes in other lipoproteins. The enhanced secretion of very low-density lipoproteins (VLDL) is attributed to increased lipolysis in adipose tissue, augmented hepatic fatty acid synthesis, and suppressed fatty acid oxidation. Impaired lipoprotein lipase and apolipoprotein E activity further contribute to reduced VLDL clearance. LDL are also affected, with their levels varying depending on the type and severity of infection. In some viral infections, LDL concentrations decrease due to increased catabolism or reduced hepatic synthesis, whereas in others, LDL oxidation contributes to foam cell formation, exacerbating inflammation and atherosclerosis. These disruptions in cholesterol homeostasis not only impair lipid transport but may also facilitate immune evasion by pathogens, highlighting the intricate interplay between lipid metabolism and host defense mechanisms [75,77,78].
Recently, significant advances have been made in understanding lipoprotein alterations in COVID-19 using proton nuclear magnetic resonance (1H-NMR). This technique offers the unique advantage of simultaneously measuring the quantity, size, and composition of lipoprotein particles with exceptional accuracy, providing a more comprehensive understanding of the metabolic changes associated with the disease [79]. These studies have indicated that COVID-19 patients have larger and more abundant VLDL particles than healthy individuals, along with elevated VLDL-cholesterol and VLDL-triglyceride concentrations. In contrast, LDL-cholesterol concentrations were lower, with LDL particles being larger and fewer. HDL particles showed reduced cholesterol content and increased triglycerides, with a notable decrease in small HDL particles [80,81,82,83,84].
Similar lipoprotein alterations have been observed in influenza, though the pattern varies, and analyses have been done by methods other than 1H-NMR. HDL cholesterol levels are often reduced in this disease, while LDL cholesterol levels tend to increase during the acute phase [85]. However, these changes are generally less pronounced than in SARS-CoV-2 infection and do not appear to be major drivers of disease progression. The exception is low HDL levels at hospital admission, which have been associated with higher inflammation and increased mortality, particularly in males [86]. Information on lipoprotein alterations in RSV infection is scarce. Recent in vitro studies [87] suggested that RSV infection activates SREBP2 and low-density lipoprotein receptor (SREBP2-LDLR) pathway, which is a key regulatory mechanism in cholesterol uptake. Activating this pathway increases the expression of LDL receptors and promotes cholesterol uptake into cells, contributing to cholesterol accumulation in lysosomes, which supports viral replication. This mechanism suggests that lipoprotein changes during RSV infection may involve increased LDL uptake, although specific alterations in lipoprotein profile (such as changes in HDL or LDL levels) have not yet been characterized.
Respiratory virus infections not only alter circulating lipoprotein concentrations but also modify intracellular cholesterol metabolism to enhance viral replication and release. A recent preprint reported that SARS-CoV-2 disrupts host lipid metabolism by inducing lysosomal cholesterol sequestration, a process facilitated by the interaction between the viral protein ORF3a and the host protein VPS39, which impairs cholesterol trafficking and contributes to viral pathogenesis [88]. In contrast, influenza A virus exploits cholesterol through distinct mechanisms, primarily relying on cholesterol-rich lipid rafts to facilitate receptor clustering and endocytosis via its hemagglutinin glycoprotein. Disrupting these lipid rafts with cholesterol-depleting agents, such as methyl-β-cyclodextrin, impairs viral entry and reduces infectivity [89]. This virus primarily manipulates intracellular cholesterol to promote viral ribonucleoprotein export, virion assembly at the plasma membrane, and efficient budding. Cholesterol-rich microdomains are essential for releasing infectious virions, and cholesterol depletion has been shown to result in malformed particles with diminished infectivity [90]. Unlike SARS-CoV-2 and influenza A virus, RSV does not appear to induce widespread systemic cholesterol dysregulation. However, cholesterol depletion in host cells has been shown to impair RSV fusion and reduce viral replication [87], highlighting its dependence on lipid rafts. Additionally, RSV infection seems to modulate cholesterol metabolism to support viral assembly and budding, although the exact molecular mechanisms remain less well characterized than other respiratory viruses [50].
Alterations of respiratory virus infections on the host lipid metabolism are summarized in Table 1.
3. Energy Metabolism and Mitochondrial Dynamics
The metabolic response to viral infections is characterized by alterations in energy demand and mitochondrial function [71]. Cellular energy metabolism is primarily driven by three interconnected pathways: glycolysis, the Krebs cycle, and oxidative phosphorylation (OXPHOS). Glycolysis occurs in the cytosol, breaking down glucose into pyruvate while generating ATP and reduced nicotinamide adenine dinucleotide (NADH). Under aerobic conditions, pyruvate is transported into mitochondria, where it enters the Krebs cycle, producing NADH and flavin adenine dinucleotide, which fuel the electron transport chain for ATP synthesis via OXPHOS. The balance between these metabolic pathways is crucial for cellular homeostasis, immune responses, and host defense mechanisms against viral infections [91].
Mitochondria play a central role in cellular bioenergetics, integrating energy production with immune signaling. Viral infections frequently disrupt mitochondrial function, leading to altered ATP generation, shifts in metabolic fluxes, and modulation of host defense mechanisms [71,92].
3.1. Glycolysis and the Warburg-like Effect in Viral Infections
Viruses extensively reprogram host cell metabolism to facilitate their replication and persistence. One of the most well-documented metabolic alterations induced by viral infections is the shift towards increased glycolysis, a phenomenon reminiscent of the Warburg effect observed in cancer cells (Figure 4). This metabolic adaptation, often called a "Warburg-like effect," is characterized by enhanced glycolytic flux despite oxygen availability, leading to reduced mitochondrial respiration [93,94]. This strategy provides infected cells with a rapid supply of ATP and metabolic intermediates essential for viral genome replication, protein synthesis, and lipid membrane production. The Warburg-like effect in viral infections is driven by multiple host and viral factors, including the activation of hypoxia-inducible factor 1-α (HIF-1α), OXPHOS suppression, and modulation of key metabolic enzymes [95,96]. Respiratory viruses exhibit profound metabolic reprogramming that favors glycolysis over mitochondrial respiration.
One of the key mechanisms by which SARS-CoV-2 enhances glycolysis is through the upregulation of enzymes, including hexokinase 2, phosphofructokinase, and lactate dehydrogenase [97]. This metabolic shift ensures a continuous supply of ATP and biosynthetic precursors, supporting viral replication and assembly. Moreover, SARS-CoV-2 has been shown to suppress OXPHOS by disrupting mitochondrial function, leading to metabolic dependence on glycolysis [98]. In addition to direct metabolic reprogramming, inflammatory responses associated with COVID-19 further reinforce glycolytic metabolism. Infected immune cells, particularly monocytes and macrophages, exhibit an increased glycolytic phenotype, contributing to a hyperinflammatory state [96]. This excessive metabolic shift may exacerbate disease severity, promoting lung tissue damage and cytokine storm.
Influenza virus reprograms host cell metabolism by promoting a shift toward glycolysis, primarily through the activation of HIF-1α, a key regulator of cellular metabolism under hypoxic conditions [99,100]. During infection, HIF-1α activation has been shown to upregulate glucose transporters (GLUT1, GLUT3) and key glycolytic enzymes, enhancing glucose uptake and metabolic flux [101,102]. This metabolic adaptation ensures a rapid supply of energy and biosynthetic intermediates essential for viral protein synthesis and replication while modulating the host immune response. However, excessive glycolytic activity may impair antiviral defense mechanisms, increasing host cell susceptibility to severe infection. In addition to enhancing glycolysis, influenza virus disrupts mitochondrial function by interfering with mitochondrial dynamics. Specifically, it has been reported to induce mitochondrial fission, resulting in fragmented mitochondria with diminished OXPHOS efficiency. This mitochondrial disruption further reinforces the reliance on glycolysis, creating a metabolic environment that facilitates viral replication and propagation [103].
RSV is another important pathogen known to rewire host cell metabolism towards glycolysis. Like SARS-CoV-2 and influenza, this virus enhances glycolytic flux to sustain viral replication. RSV infection has been shown to activate the insulin receptor-PI3K-Akt axis, upregulated the translation and activity of HIF-1α, increased the expression of GLUT1, GLUT3, and GLUT4, hexokinase 1 and 2, and platelet-type phosphofructokinase, and promoted glucose uptake and glycolysis. In addition, mitochondrial damage induced by RSV resulted in the generation of large amounts of reactive oxygen species (ROS) in infected cells, which contributed to stabilizing HIF-1α [104].
The shift towards glycolysis in virally infected cells has profound implications for the host immune response. Activated immune cells, including macrophages, dendritic cells, and T cells, rely on glycolysis for rapid energy production and effector function. However, excessive metabolic reprogramming can harm immune function and disease progression. SARS-CoV-2-infected macrophages exhibit a hyperglycolytic state, leading to increased production of pro-inflammatory cytokines such as interleukin (IL)-6, tumor necrosis factor-α, (TNF-α) and IL-1β [96]. This effect contributes to the hyperinflammatory response observed in severe COVID-19 cases. T cell activation depends on glycolysis, but persistent viral infections can lead to metabolic exhaustion, reducing T cell proliferation and cytotoxic function [105]. Metabolic reprogramming in dendritic cells affects their ability to present antigens and stimulate adaptive immune responses, potentially impairing viral clearance [106].
The Warburg-like effect in viral infections highlights the intricate relationship between host metabolism and viral pathogenesis. By shifting cellular energy production towards glycolysis while suppressing mitochondrial function, viruses create an environment conducive to their replication. This metabolic reprogramming sustains viral growth and impacts immune function, influencing disease severity and outcomes. Understanding these metabolic alterations provides new insights into antiviral strategies that target host cell metabolism, potentially leading to novel therapeutic interventions for respiratory viral infections.
3.2. Mitochondrial Dysfunction and Bioenergetic Failure
Mitochondria are essential organelles that serve as the cell's powerhouse and key regulators of innate immunity. Their ability to generate ATP through OXPHOS is crucial for cellular homeostasis, while their role in immune signaling makes them a primary target for viral manipulation. Numerous viruses have evolved sophisticated mechanisms to disrupt mitochondrial function [107], leading to bioenergetic failure and immune evasion. This mitochondrial dysfunction contributes to viral persistence, increased pathogenicity, and exacerbation of disease severity [108].
One of the major consequences of viral infections on mitochondrial function is the suppression of OXPHOS, which leads to decreased ATP production [109]. Many viruses disrupt electron transport chain activity, impairing efficient ATP synthesis [110]. For example, research indicates that SARS-CoV-2 can suppress the expression of both nuclear-encoded and mitochondrial-encoded mitochondrial genes, impairing mitochondrial function. This downregulation affects electron transport chain components, including complexes I and III, thereby disrupting oxidative phosphorylation and ATP production [111,112].
Mitochondria are highly dynamic organelles that continuously undergo fission (division) and fusion (joining) to maintain their function and adapt to cellular demands [113]. Viral infections often hijack these processes to optimize viral replication and evade host immune responses. Fission is primarily mediated by dynamin-related protein 1 (Drp1), which translocates to the mitochondria upon activation, causing mitochondrial fragmentation [114]. SARS-CoV-2, influenza, and Dengue, exploit Drp1-mediated fission to disrupt mitochondrial homeostasis [115]. SARS-CoV-2 has been shown to upregulate Drp1 and increase mitochondrial fission, decreasing ATP production in cultured cells [116]. This mitochondrial fragmentation also impairs immune cell function, dampening antiviral responses and facilitating viral persistence. Moreover, lymphocytes from patients recovered from severe COVID-19 were reported to have high Drp1 expression, a disruption in mitochondrial dynamics, as well as a lack of structural integrity in the electron transport chain, and altered circulating levels of IL-1β, interferon (IFN)-α2, and IL-27 [117]. Conversely, fusion is controlled by mitofusins 1 and 2 and optic atrophy 1, which help maintain mitochondrial integrity and bioenergetic efficiency [118]. Some viruses actively suppress fusion to hinder mitochondrial repair and prolong cellular dysfunction. However, influenza virus has been reported to exert a dual effect in cultured cells, inducing mitochondrial fragmentation under serum starvation while promoting fusion and mitochondrial elongation under optimal culture conditions [119].
A crucial component of mitochondrial antiviral defense is the mitochondrial antiviral-signaling protein (MAVS) [120]. MAVS is localized on the outer mitochondrial membrane and acts as a signaling hub for type I IFN responses upon viral detection. Respiratory viruses have evolved strategies to inhibit MAVS function [121]. SARS-CoV-2 protein ORF degrades MAVS, thereby blocking downstream IFN signaling and allowing the virus to evade immune detection [122]. In addition to MAVS, other mitochondrial proteins play roles in antiviral defense. The NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, a key component of innate immunity, is activated in response to mitochondrial stress [123]. Studies in cultured cells showed that SARS-CoV-2 and influenza virus exploit mitochondrial damage to overactivate NLRP3. Instead of effectively fighting the infection, excessive NLRP3 activation triggers uncontrolled inflammation, which can be harmful to the host and may even lead to organ failure and death [98,124,125].
Mitochondria possess quality control mechanisms, such as mitophagy (mitochondrial autophagy), to eliminate damaged mitochondria and maintain cellular homeostasis. Some viruses hijack these pathways to their advantage. For example, the Zika virus activates mitophagy in cultured trophoblasts to degrade mitochondria, suppressing immune responses and prolonging infection [126]. Conversely, SARS-CoV-2 has been reported to impair mitophagy, accumulating dysfunctional mitochondria and excessive inflammation. The inhibition of mitophagy can result in prolonged mitochondrial dysfunction, contributing to disease pathology and immune dysregulation [127].
Mitochondrial dysfunction is a common feature of many viral infections, contributing to metabolic reprogramming, immune evasion, and disease progression. By suppressing OXPHOS, promoting mitochondrial fragmentation, disrupting MAVS signaling, and manipulating mitochondrial quality control, many viruses, including SARS-CoV-2, IV, and RSV, create an environment conducive to favor their replication and impair the host defense mechanisms.
3.3. Cellular Metabolic Sensors and Viral Manipulation
As discussed in the previous sections, mitochondrial dysfunction and the reprogramming of energy metabolism are hallmarks of viral infections, driving a shift from OXPHOS to glycolysis to support viral replication. Beyond these mitochondrial alterations, viruses also target key cellular metabolic sensors, including AMP-activated protein kinase (AMPK) and mTOR, to further manipulate host cell metabolism in their favor. These pathways are critical in determining whether a cell enters a catabolic or anabolic state, thereby influencing viral replication and immune responses [128,130] (Figure 5).
AMPK serves as a central energy sensor that becomes activated under conditions of energy stress, such as viral infection. When ATP levels drop, AMPK is phosphorylated and activated to restore energy homeostasis by promoting catabolic pathways, including fatty acid oxidation and autophagy, while simultaneously inhibiting anabolic processes such as protein and lipid synthesis [130]. By reducing cellular biosynthetic activity, AMPK activation creates a less favorable environment for viral replication, as many viruses rely on an abundance of host-derived macromolecules. Consequently, some viruses have evolved mechanisms to inhibit AMPK signaling to prevent host cells from entering an energy-saving state. For example, hepatitis B and C viruses have been reported to suppress AMPK activity to ensure sufficient lipid availability for viral envelope formation [129,131]. In contrast, certain viruses may activate AMPK to promote selective autophagy (mitophagy), removing damaged mitochondria that could trigger antiviral immune responses [132]. RSV, for instance, has been shown to induce mitophagy in infected cells, which may contribute to immune evasion by reducing mitochondrial ROS production and dampening innate immune activation [133].
While AMPK functions as a metabolic checkpoint that limits viral replication, mTOR plays an opposing role by promoting anabolic metabolism and cellular growth [129]. mTOR is a key regulator of protein and lipid synthesis, which viruses often hijack to facilitate their replication cycle. Many viruses, including influenza virus, RSV, hepatitis C virus, and human cytomegalovirus, activate mTOR signaling to enhance the production of viral proteins and structural components [134,135]. The mTOR pathway is particularly important for regulating translation through its downstream effectors, such as ribosomal protein S6 kinase and eukaryotic initiation factor 4E-binding protein 1, which govern ribosome biogenesis and mRNA translation [136]. Additionally, mTOR activation can suppress autophagy and autophagy-induced catabolism by phosphorylating unc-51-like kinase 1, autophagy-related gene 13, and other molecules [137]. Since autophagy can degrade viral components, some viruses actively evade this process. Conversely, in certain scenarios, viruses may also exploit autophagy to generate intracellular membranes necessary for viral replication compartments, as observed in SARS-CoV-2 infection [138].
Another key metabolic signaling pathway frequently manipulated by viruses is the PI3K-Akt pathway. This pathway is a major upstream activator of mTOR, promoting cell growth and survival by integrating signals from growth factors and nutrients. Activation of PI3K leads to Akt phosphorylation, which activates mTOR complex 1, thereby regulating processes such as protein synthesis and autophagy suppression [139]. PI3K-Akt signaling plays a central role in cell survival, proliferation, and glucose metabolism, making it an attractive target for viral interference. Activation of this pathway promotes glycolysis by upregulating glucose transporters and key glycolytic enzymes, ensuring a continuous supply of biosynthetic precursors required for viral assembly [137,138,139,140]. Influenza virus, RSV, and SARS-CoV-2, have been reported to modulate the PI3K-Akt pathway to create a metabolic environment conducive to replication [140]. This pro-survival signaling facilitates sustained viral replication and contributes to immune evasion by preventing premature cell death and subsequent antigen presentation.
The interplay between AMPK, mTOR, and PI3K-Akt highlights the intricate metabolic rewiring during viral infections. While AMPK activation generally acts as a barrier to viral replication by inducing catabolic pathways and inhibiting biosynthesis, mTOR and PI3K-Akt activation facilitate viral propagation by driving anabolic metabolism and cell survival. However, the context-dependent effects of these metabolic regulators must be carefully considered, as their inhibition could also impact immune cell function and host defense mechanisms.
Overall, viruses have evolved sophisticated strategies to hijack metabolic signaling pathways to optimize their replication and evade immune responses. Understanding the interplay between AMPK, mTOR, and PI3K-Akt in the context of viral infections provides valuable insights into host-virus interactions and potential therapeutic targets to restore metabolic balance and enhance antiviral immunity.
Table 2 provides a comparative overview of the main characteristics of how SARS-CoV-2, influenza virus, and RSV affect glycolysis, mitochondrial function, and cellular metabolic sensors in host cells.
4. Metabolic Alterations in Amino Acid and Nucleotide Pathways
Respiratory viral infections induce profound disruptions in amino acid and nucleotide metabolism, which are crucial in disease progression and influence viral replication, immune function, and cellular stress responses. Understanding these metabolic shifts provides insight into the host-pathogen interaction and the consequences of viral infections on cellular homeostasis.
4.1. Amino Acid Metabolism
Glutamine is a critical amino acid involved in energy production, nitrogen balance, and the biosynthesis of nucleotides and other amino acids. During viral infections, glutamine metabolism is frequently upregulated to support increased energy demands and nucleotide synthesis required for viral replication. SARS-CoV-2, influenza virus, and RSV infections have been shown to enhance glutaminolysis, contributing to elevated levels of glutamate and downstream metabolites, which provide metabolic intermediates that support viral protein synthesis [141,142,143]. Additionally, glutamine is crucial in maintaining cellular redox balance by serving as a precursor for glutathione synthesis, a major antioxidant [144]. Therefore, while direct evidence linking glutamine depletion to oxidative stress in respiratory virus infections is limited, it is plausible that such depletion could contribute to increased oxidative stress by impairing glutathione production.
In addition to supporting viral replication, glutamine metabolism influences immune cell function. Activated lymphocytes, macrophages, and dendritic cells rely heavily on glutamine for proliferation and cytokine production [145]. Decreased glutamine availability during infection can impair adaptive immune responses, leading to suboptimal viral clearance. Moreover, metabolic competition between the host and the virus for glutamine may exacerbate disease severity by inducing metabolic stress in host cells.
Arginine plays a pivotal role in NO production, which is essential for immune defense [146]. SARS-CoV-2, influenza virus, and RSV infections, lead to decreased arginine availability due to increased activity of arginase enzymes [147,148]. This reduction in arginine levels suppresses NO-mediated antiviral responses and promotes an immunosuppressive environment. Studies on patients with severe COVID-19 have shown that arginine depletion is associated with T-cell dysfunction and impaired antiviral immunity [149]. Similarly, influenza and RSV infections trigger a comparable metabolic shift, exacerbating disease severity through immune evasion [150,151].
Arginine metabolism also influences polyamine synthesis, which plays a role in viral RNA stabilization and replication [152]. Some viruses, including SARS-CoV-2, exploit polyamine metabolism to enhance their replication efficiency [153]. Depletion of arginine may, therefore, serve a dual purpose: impairing host immune responses while simultaneously facilitating viral proliferation [154]. Understanding the regulation of arginine metabolism during infection could provide insights into host-virus interactions and potential metabolic vulnerabilities.
Tryptophan metabolism is significantly altered during respiratory viral infections, primarily through activation of the kynurenine pathway by IFN-stimulated enzymes such as indoleamine 2,3-dioxygenase. Elevated serum kynurenine levels have been reported in SARS-CoV-2 infections, correlating with immune suppression and increased inflammatory markers [155,156,157]. Similar findings have been observed in influenza and RSV infections, where tryptophan catabolism reduces serotonin synthesis, impacting mood and immune function. The modulation of this pathway may contribute to the prolonged symptoms observed in post-viral syndromes, including psychological disturbances [158].
Tryptophan metabolism also plays a key role in regulatory T-cell function and immune tolerance. Kynurenine and its downstream metabolites can suppress effector T-cell responses while promoting an anti-inflammatory environment. While beneficial in preventing excessive immune activation, viruses may exploit this mechanism to evade immune detection [159]. Therefore, the balance between tryptophan catabolism and immune activation is critical to disease outcomes in respiratory viral infections.
Cysteine availability is crucial for synthesizing glutathione (GSH), a major antioxidant that protects cells from oxidative stress. GSH synthesis is limited by the availability of cysteine, which serves as a rate-limiting substrate in this process [160]. Respiratory viral infections often lead to GSH depletion, exacerbating oxidative damage and inflammation. For instance, studies have shown that SARS-CoV-2 infection downregulates the nuclear factor erythroid 2–related factor 2 (NRF2) levels and NRF2-dependent gene expression in human airway epithelial cells and in the lungs of mice, leading to reduced antioxidant responses [161]. Similarly, increased ROS production can activate the nuclear factor κB pathway during influenza infections, leading to lung damage. RSV infections have also been associated with oxidative stress and ROS-mediated cellular events [162,163]. Thus, the depletion of cysteine and glutathione plays a critical role in the pathophysiology of these infections. Moreover, GSH influences immune signaling and cytokine production beyond its role in mitigating oxidative stress. Specifically, GSH regulates the activity of transcription factors modulating inflammatory responses [164]. A decline in GSH levels can lead to exaggerated cytokine release, contributing to the hyperinflammatory state observed in severe COVID-19 and other viral pneumonia.
In summary, maintaining adequate cysteine levels is essential for GSH synthesis, which in turn plays a pivotal role in protecting against oxidative stress, regulating inflammation, and ensuring proper immune function during respiratory viral infections.
4.2. Nucleotide Metabolism
Respiratory viruses induce significant alterations in the host’s nucleotide metabolism to ensure sufficient nucleotide availability for viral replication and transcription. They upregulate purine and pyrimidine biosynthesis pathways, facilitating rapid viral genome replication. The increased nucleotide demand imposes metabolic stress on host cells, often leading to nucleotide depletion and cellular dysfunction. In response, host cells enhance nucleotide salvage pathways to compensate. However, the competition between host and viral replication can result in nucleotide shortages, impairing dna acid repair mechanisms and immune cell proliferation. This metabolic bottleneck and impaired immune function may contribute to the prolonged recovery observed in severe viral infections [165,166,167,168].
SARS-CoV-2, influenza virus, and RSV exploit pyrimidine metabolism, increasing uridine triphosphate and cytidine triphosphate synthesis to support viral genome replication while depleting precursors essential for the host’s adaptive immunity. This metabolic competition can impair T-cell proliferation and antibody production [143,166,169,170]. Similarly, purine metabolism is disrupted, affecting energy balance and immune signaling. ATP and guanosine triphosphate are energy carriers, while purine metabolites such as adenosine modulate immune responses. Altered adenosine metabolism in COVID-19 has been associated with excessive inflammation, while influenza and RSV dysregulate purine pathways, contributing to cytokine storms and prolonged inflammation [171,172]. By modulating de novo biosynthesis and salvage pathways, these viruses create nucleotide imbalances that compromise cellular repair and immune responses, ultimately exacerbating disease severity [173].
Table 3 summarizes the main characteristics of how SARS-CoV-2, influenza virus, and RSV alter amino acid and nucleotide metabolism, contributing to viral replication, immune modulation, and systemic inflammation.
5. Oxidative Stress and the Inflammatory Reaction
5.1. Intracellular Factors Linking Oxidative Stress and Inflammation
Respiratory viruses trigger increased ROS production through mitochondrial dysfunction, activation of NADPH oxidase (NOX), and inhibition of endogenous antioxidant systems [174,175,176]. Excessive ROS levels produce lipid peroxidation, protein oxidation, and DNA damage, leading to cellular dysfunction. ROS also activate nuclear factor kappa B (NF-κB) and activator protein-1 [177,178], promoting the transcription of pro-inflammatory cytokines, while inflammatory mediators like TNF-α and IL-1β further stimulate ROS production, creating a self-perpetuating cycle of oxidative stress and inflammation [179].
One proposed way virus-induced oxidative stress can enhance the inflammatory response is by activating inflammasomes, which are multiprotein complexes that initiate an immune response [180]. Inflammasomes, such as NLRP3, play a key role in the inflammatory response. ROS activate NLRP3 in macrophages, triggering their assembly and the synthesis of compounds like the chemokine (C-C motif) ligand 2 (CCL2, formerly termed monocyte chemoattractant protein-1, MCP-1), which recruits immune cells to the infection site [181,182]. This chemokine is upregulated after tissue injury and can induce endoplasmic reticulum stress and autophagy and regulate NF-kB expression [183].
Pattern recognition receptors are proteins primarily expressed by cells of the innate immune system that detect molecules characteristic of pathogens. They recognize two main classes of molecular patterns: pathogen-associated molecular patterns, which are derived from microbial pathogens, and damage-associated molecular patterns, which originate from host cell components released during cellular damage or death. Recognition of these patterns leads to the activation of NF-κB and the subsequent production of adhesion molecules and chemokines [184,185,186,187]. Additional pathways, such as PI3K, mitogen-activated protein kinase MAPK, and Janus kinase/signal transducers and activators of transcription, further regulate inflammatory responses and cellular stress [188,189].
The unfolded protein response, activated through inositol-requiring enzyme 1, protein kinase R-like endoplasmic reticulum kinase, and activating transcription factor 6, links oxidative stress with endoplasmic reticulum stress and inflammation [190,191,192]. Endoplasmic reticulum stress, driven by CCL2, can modulate inflammation by upregulating C–C chemokine receptor type 2 expression and is linked to cell death and autophagy [193]. Autophagy plays a crucial role in maintaining mitochondrial integrity, and its dysregulation contributes to inflammasome activation and age-related disease susceptibility [194]. Therefore, mitochondrial dysfunction, autophagy impairment, and metabolic alterations are interconnected in the pathology of respiratory viral infections.
5.2. Alterations in Endogenous Antioxidant Systems
While inflammation is essential for viral clearance, excessive immune activation can cause severe complications, such as cytokine storms, leading to vascular leakage and multiorgan dysfunction, as seen in severe COVID-19 and influenza cases. Chronic inflammation also depletes antioxidant defenses, increasing cellular vulnerability to oxidative damage.
The organism relies on multiple antioxidant systems to defend against oxidative stress. Respiratory virus infections can profoundly disrupt these protective mechanisms. Among the key components involved in maintaining redox homeostasis and regulating inflammatory responses are GSH, superoxide dismutase (SOD), catalase (CAT), and paraoxonase 1 (PON1). Emerging evidence suggests that although respiratory viruses share common pathogenic pathways, they exert distinct and virus-specific effects on the antioxidant systems they impair.
GSH, the most abundant intracellular non-enzymatic antioxidant, protects cells from oxidative damage by directly scavenging ROS and as a cofactor for glutathione peroxidase. In COVID-19, a marked depletion of GSH has been observed in patients with moderate to severe disease [195,196]. This depletion correlates with elevated oxidative stress markers and pro-inflammatory cytokines, suggesting that inadequate GSH levels may contribute to cytokine storm and tissue injury. Similarly, in influenza virus infection, GSH levels drop significantly in the respiratory tract epithelium [197]. However, in contrast to COVID-19, the depletion appears transient and less systemically profound. In infants, RSV infection also leads to GSH oxidation and disruption of the GSH/GSSG balance [198], exacerbating inflammation and mucus hypersecretion.
SOD, which catalyzes the dismutation of superoxide radicals into hydrogen peroxide and oxygen, is another critical enzymatic defense. There are three isoforms of SOD: cytosolic (SOD1), mitochondrial (SOD2), and extracellular (SOD3) [199]. In SARS-CoV-2 infection, decreased activity of SOD, especially SOD2, has been reported in severe cases [200,201], likely due to mitochondrial dysfunction due to persistent ROS production. In influenza, initial upregulation of SOD occurs as a compensatory response, but this is not sustained in advanced stages of infection [202,203]. RSV also induces oxidative stress with high superoxide levels, and although SOD expression increases early in infection, this upregulation is often insufficient to neutralize the oxidative burden [198].
CAT, responsible for converting hydrogen peroxide into water and oxygen, prevents the accumulation of this toxic intermediate and the subsequent formation of highly reactive hydroxyl radicals. Reduced CAT activity has been reported in SARS-CoV-2, influenza vitus, and RSV infections, contributing to redox imbalance and alveolar damage [198,202,204].
PON1, an esterase associated with HDL, plays a multifaceted antioxidant and anti-inflammatory role. It degrades oxidized lipids, modulates macrophage activation, and helps preserve endothelial function (Figure 6) [76,77,78]. Serum PON1 activity is markedly decreased in COVID-19, especially in patients with comorbidities such as diabetes or cardiovascular disease [205,206,207], and the measurement of the serum levels of this enzyme has been proposed as a diagnostic marker [206]. For influenza virus and RSV, the available literature is limited. However, given their known ability to increase oxidative stress and alter HDL structure and composition [75], it is plausible that these infections also lead to changes in PON1 activity similar to those observed in COVID-19, albeit possibly to a lesser extent.
In comparison, and considering the overall evidence on respiratory viral infections and antioxidant systems, SARS-CoV-2 infection appears to induce a more systemic and persistent oxidative imbalance, likely driven by prolonged viral shedding, endothelial involvement, and dysregulated immune responses. The impact on GSH and PON1 is particularly pronounced, suggesting that these components may be crucial in modulating the severity of COVID-19. Influenza infections, while capable of triggering oxidative stress, often provoke a more localized and transient antioxidant response, with early compensatory increases that may protect against severe tissue damage in immunocompetent hosts. RSV, predominantly affecting infants and older people, leads to intense localized oxidative stress in the lower airways. Depleting GSH and suppressing CAT activity are especially relevant to its pathogenesis. In conclusion, respiratory viruses such as SARS-CoV-2, influenza, and RSV disrupt the host's antioxidant defense systems through distinct but overlapping mechanisms. The depletion of GSH, reduced activity of SOD and CAT, and suppression of PON1 are key features that contribute to disease severity and tissue damage.
Table 4 summarizes the main changes in endogenous antioxidant systems in infections caused by SARS-CoV-2, influenza virus, and RSV.
6. Analysis of Peripheral Circulating Metabolites and Search for Metabolic Biomarkers
6.1. Omics Disciplines and the Rationale for Metabolic Biomarkers in Viral Infections
In the previous sections, we have reviewed how respiratory viruses induce profound alterations in host cell metabolism, reflecting both viral replication strategies and the host immune response. These metabolic disruptions converge on key pathways, including glycolysis, lipid metabolism, mitochondrial function, amino acid turnover, and redox homeostasis. As a result, metabolites and enzymes involved in these pathways represent promising candidates for biomarkers of disease severity, prognosis, and therapeutic response [96,208]. Early stratification of patients based on metabolic profiles could inform treatment decisions, identify individuals requiring hospitalization, and anticipate complications such as acute respiratory distress or post-viral syndromes [209].
Modern science provides powerful analytical tools for exploring the potential of metabolic parameters as disease biomarkers. In addition to traditional techniques such as spectrophotometry and immunoassays, recent decades have witnessed the emergence of advanced approaches, notably multi-omics. Multi-omics refers to the integrative analysis of multiple layers of biological data to comprehensively understand biological systems. This approach reveals complex interactions among genes, proteins, metabolites, and other biomolecules, offering more profound insights into the mechanisms of health, disease progression, and treatment response [210].
Omics disciplines can be broadly classified into molecular and phenotypic/clinical categories. Molecular omics encompasses genomics, transcriptomics, proteomics, metabolomics, and epigenomics, each focusing on specific molecular components—DNA, RNA, proteins, metabolites, and epigenetic modifications. Phenotypic or clinical omics includes data derived from clinical observations and diagnostic technologies, such as radiomics, pathomics, and hematological omics. Radiomics extracts quantitative features from medical imaging; pathomics integrates molecular profiles with digital pathology and histological data; and hematological omics involves the detailed molecular and cellular analysis of blood to elucidate the complex biology of hematologic conditions [211].
Among these, metabolomics plays a particularly important role [212]. One of its main advantages in clinical research is the relative ease and minimal invasiveness of sample collection, often requiring only a simple blood draw. This practicality facilitates large-scale studies and longitudinal monitoring, making metabolomics a powerful tool in the search for clinically relevant biomarkers in viral infections.
Metabolomics relies on various analytical techniques, each with distinct advantages regarding sensitivity, specificity, and metabolome coverage. The two most widely used platforms are mass spectrometry (MS), often coupled with chromatographic separation methods such as gas chromatography (GC) or liquid chromatography (LC), and 1H-NMR spectroscopy [79,213,214]. The latter offers high reproducibility and non-destructive analysis with minimal sample preparation, making it particularly suitable for longitudinal and comparative studies. However, its relatively lower sensitivity limits the detection of low-abundance metabolites. In contrast, MS-based approaches, especially when combined with GC or LC, provide high sensitivity and broad metabolite coverage, allowing for detecting and quantifying a wide range of compounds at low concentrations [215,216]. Targeted metabolomics focuses on predefined sets of metabolites, often associated with specific pathways or conditions, offering high accuracy and quantitative precision. Untargeted metabolomics, on the other hand, aims to profile as many metabolites as possible without prior bias, facilitating the discovery of novel biomarkers or previously unrecognized metabolic alterations [217,218,219]. Together, these complementary approaches provide a robust framework for elucidating metabolic signatures associated with viral infections and identifying potential diagnostic or prognostic biomarkers.
The term biomarker is frequently and, at times, indiscriminately used in the biomedical literature. However, the mere presence of a statistically significant difference in the levels of a given parameter between two clinical conditions is not sufficient to classify that parameter as a biomarker. An accurate biomarker is a measurable indicator that distinguishes between two well-defined biological states, such as health versus disease, favorable versus poor prognosis, or responsiveness versus resistance to a specific treatment [220,221]. For a parameter to fulfill this role, it must enable unambiguous discrimination between these conditions, ideally with minimal or no overlap in their respective distributions. This characteristic implies exceptionally high diagnostic performance, typically characterized by sensitivity and specificity values approaching 100% and an area under the receiver operating characteristic (ROC) curve (AUC) nearing 1.0 [222]. These stringent criteria are rarely met, particularly in the context of metabolic biomarkers.
This section reviews the most relevant studies identifying candidate metabolites as biomarkers of respiratory viral infections. We also critically evaluate their diagnostic accuracy and practical limitations.
6.2. Lipid Signatures and the Lipidome
A comparative lipidomic analysis revealed significant alterations in serum lipid mediators between COVID-19-positive patients and healthy individuals, with elevated levels of O-octanoyl-L-carnitine (CAR 8:0) and lysophosphatidylethanolamine (LPE), and decreased levels of arachidonic acid and oxylipins like 9/13-HODE and 15-HETE [223]. Elevated CAR 8:0 suggests mitochondrial dysfunction, a known feature of viral infections including COVID-19, reflecting impaired fatty acid β-oxidation and potential lung injury through surfactant inhibition [224,225,226]. However, similar lipid disturbances were observed in COVID-19-negative patients with bacterial infections, indicating a general inflammatory response rather than a SARS-CoV-2 effect. To refine specificity, comparisons with COVID-19-negative patients with bacterial infections highlighted an increased phosphatidylcholine/lysophosphatidylcholine (PC/LPC) ratio in COVID-19 patients, showing strong diagnostic potential (AUC = 0.950) [223]. Nonetheless, the generalizability of this marker is limited by inconsistent findings across studies, likely due to differences in disease severity, patient comorbidities, and the choice of control groups. Indeed, prior studies have reported varying patterns, including decreased PC with increased LPC, the reverse, or simultaneous reductions in both lipid classes [227,228,229,230,231,232,233].
Lipidomic profiles of lung cells further underscore disease-specific patterns. COVID-19-positive patients exhibited elevated long-chain triglycerides, particularly in immune cells, which could modulate inflammatory signaling. Similar serum triglycerides, VLDL, and polyunsaturated fatty acid elevations have been noted in Ebola and COVID-19 infections [234,235,236,237]. Importantly, some lipid species—like long-chain triglycerides, PC 36:5, LPC 22:6-sn2, PC 36:1, and secondary bile acids—were distinctly altered between COVID-19-positive and -negative groups. These may contribute to long-term cardiovascular risk and point to potential post-discharge interventions, such as dietary or lipid-lowering strategies [223].
One of the most promising findings was the significant reduction of arachidonic acid in COVID-19 patients, a pattern also observed in cases of influenza A and RSV infections [238]. This result holds practical relevance, as arachidonic acid benefits from the availability of specific calibrators and antibodies, enabling the development of simple, cost-effective assays suitable for routine use in general clinical laboratories—unlike more complex lipids such as LPC or PC. Arachidonic acid plays a central role in both the synthesis of inflammatory mediators and the formation of viral membranes. Notably, in vitro studies have shown that its supplementation can inhibit viral replication [239]. Its depletion in patients likely reflects increased utilization for host immune responses or viral replication requirements and has been associated with disease severity [223,240].
Recently, a novel biosensing platform has been developed using a competitive immunoassay integrated with magnetic microbeads and screen-printed carbon electrodes to quantify arachidonic acid in serum. This system demonstrated high sensitivity, reproducibility, and practicality for clinical settings, with strong concordance with conventional assays [241]. Although still at the prototype stage and not commercially available, the platform holds significant promise for point-of-care diagnostics and the personalized assessment of vaccine responses.
1H-NMR spectroscopy has provided valuable insights into serum lipoprotein alterations associated with respiratory viral infections. These studies have highlighted significant changes in lipoprotein profiles and have investigated their potential as biomarkers for disease severity and progression. In COVID-19 patients, 1H-NMR-based analyses have consistently revealed notable disruptions in lipoprotein profiles. Specifically, there is a marked reduction in HDL particle numbers, particularly small HDL particles, alongside elevated levels of triglyceride-rich lipoproteins, especially very small and small subfractions. These alterations are often accompanied by increased levels of systemic inflammation and elevated concentrations of branched-chain amino acids and beta-hydroxybutyrate, indicating hepatic dysfunction and altered energy metabolism [80,84].
Further studies have identified correlations between specific cytokine clusters and lipoprotein alterations in COVID-19 patients. For instance, cytokines such as IL-6, IL-18, and IFN-γ show strong positive correlations with LDL subfractions and negative correlations with HDL subfractions, suggesting an interplay between immune responses and lipid metabolism [81]. Moreover, 1H-NMR-based metabolomic profiling has demonstrated that alterations in lipoprotein composition can serve as prognostic markers for COVID-19 severity. In a multicentric Spanish cohort, machine learning models utilizing 1H-NMR-derived lipoprotein parameters, such as increased triglyceride-rich particles and altered HDL composition, accurately classified disease severity [242]. Interestingly, even months after recovery, some COVID-19 patients exhibit persistent metabolic abnormalities, including modified lipoprotein profiles, despite being asymptomatic. This finding suggests that SARS-CoV-2 infection can have lasting effects on lipid metabolism, potentially contributing to long-term health risks [237,243,244].
In contrast, data on 1H-NMR-measured lipoprotein alterations in influenza A and RSV infections are less extensive. However, available studies indicate that these infections also impact lipid metabolism, albeit with differing patterns. For instance, influenza A virus infection has been associated with decreased HDL cholesterol levels and increased triglyceride levels, reflecting an inflammatory response similar to that observed in COVID-19. A study demonstrated that low HDL concentrations predict poor outcomes and correlate with increased inflammation parameters in hospitalized influenza patients [86].
Regarding RSV infection, studies have identified changes in lipid profiles, including alterations in HDL and LDL levels, which may influence disease severity and outcomes. Researchers analyzed nasopharyngeal lipidome data in a multicenter prospective study of 800 infants hospitalized for RSV or rhinovirus bronchiolitis. They found that specific lipid species, including PC, dihydroceramide, and several fatty acids, were significantly associated with the risk of requiring positive pressure ventilation, indicating a link between lipid alterations and disease severity [245].
While 1H-NMR spectroscopy has proven to be a valuable tool in characterizing lipoprotein alterations induced by respiratory viral infections, several factors must be considered when evaluating these parameters as biomarkers. Pre-analytical variables, such as sample handling and storage conditions, can significantly impact 1H-NMR measurements. For instance, heat inactivation of samples, a common practice for viral decontamination, has been shown to induce substantial changes in lipoprotein and metabolite profiles, potentially leading to artifactual pseudo-biomarkers and the destruction of genuine biomarkers [246]. The specificity of lipoprotein alterations to particular viral infections is also critical. Many of the observed changes in lipoprotein profiles, such as reduced HDL levels and increased triglycerides, are common responses to systemic inflammation [84].
Furthermore, the clinical applicability of lipoprotein profiling requires careful standardization and validation across diverse populations and clinical settings. Comorbidities, medications, and demographic variables can influence lipoprotein metabolism, confounding data interpretation [247,248,249]. Thus, large-scale, longitudinal studies are necessary to establish the reliability and generalizability of these potential biomarkers.
6.3. Altered Carbohydrate Flux, Energy Imbalance, and Perturbations in Amino Acid and Nucleotide Metabolism
Comparative studies analyzing the serum metabolomic profiles of healthy individuals, COVID-19-positive patients, and COVID-19-negative patients with infections have revealed pronounced differences in pentose glucuronate interconversion, ascorbate and fructose metabolism, the nucleotide sugar biosynthetic route, as well as nucleotide and amino acid metabolic processes, all of which are related to carbohydrate and energy metabolism [250]. Among these routes, pentose and glucuronate interconversion emerged as the most characteristic alteration observed in COVID-19-positive patients, with notably elevated activity compared to COVID-19-negative individuals and healthy controls. This pathway is crucial for detoxification, wherein d-glucuronic acid conjugates with hydroxyl or amino groups of toxic compounds under the catalysis of UDP-glucuronosyltransferase, enhancing their water solubility and facilitating excretion through bile or urine [251]. Although investigations into this metabolic alteration in COVID-19 are still limited, recent reports have associated enhanced pentose and glucuronate interconversion with microbiome disruptions in patients suffering from oral infections [252,253]. Additionally, pharmacological studies have indicated that the modulation of this pathway could be a mechanism through which certain anti-inflammatory drugs exert their effects in both human subjects and animal models [254,255].
During viral transcription, the energy requirements and precursor molecules for building viral structural components are primarily supplied by an upregulation of aerobic glycolysis and activation of the pentose phosphate pathway [256,257]. Enhanced aerobic glycolysis increases the function of hexokinase, the enzyme that controls the pace of glycolytic flux, consequently supporting the activation of the pentose phosphate pathway. Hexokinase initiates this process by phosphorylating glucose to form glucose-6-phosphate, which is then oxidized by glucose-6-phosphate dehydrogenase within the pentose phosphate pathway to produce ribose-5-phosphate. Ribose-5-phosphate is essential for nucleotide biosynthesis, while sugar-phosphate intermediates derived from this pathway are also critical for producing amino acids and NADPH [258,259]. Several viruses, such as the influenza virus, SARS-CoV-2, hepatitis C virus, and HIV-1, have been shown to enhance the activity of the pentose phosphate pathway [260,261,262]. Overall, these findings highlight significant disruptions in pathways linked to energy production, nucleotide synthesis, and amino acid metabolism, which are highly interconnected and exert mutual influences.
Researchers have also explored whether metabolic profiles differed among COVID-19 patients according to comorbidities and disease severity. Particularly notable were the observations related to the severity of illness. Volcano plot analyses revealed that individuals who required intensive care unit ICU admission or who succumbed to the disease exhibited elevated serum levels of lauric acid [250]. When ingested from oils, lauric acid is metabolized into laurate-monoglyceride, a compound capable of inactivating enveloped viruses by disrupting the viral membrane and preventing viral entry into host cells [263,264]. Additional studies have described that laurate-monoglyceride can disintegrate viral envelopes, leading to viral inactivation [265]. At first glance, these findings may seem paradoxical, as higher lauric acid levels were associated with worse clinical outcomes. A potential explanation proposed is that free lauric acid may not display the same virucidal activity as its monoglyceride form; thus, elevated levels of free lauric acid might correspond with reduced amounts of the active monoglyceride form. Alternatively, an upsurge in lauric acid might reflect an attempt by the body to boost monoglyceride synthesis in response to viral infection. These hypotheses point to a promising area of future investigation into the relationship between lauric acid dynamics and COVID-19 severity.
Furthermore, lower xylitol concentrations were documented in patients requiring intensive care compared to less severe cases. Xylitol, a metabolite derived from the pentose and glucuronate interconversion pathway, has been recognized for its anti-inflammatory, antiglycemic, antiviral, and antibacterial actions in the context of pulmonary infections [266]. It has been shown that xylitol can reduce salt concentrations in the airway surface liquid of the lungs, thereby enhancing antibody function [267]. In vitro research has further demonstrated that xylitol-treated macrophages show a tenfold reduction in adhesion capacity relative to untreated controls, alongside diminished expression of adhesion molecules — a key process in modulating pulmonary inflammatory responses [268]. Additional preclinical studies have reported that dietary xylitol supplementation reduces viral loads in mice infected with human RSV or influenza A virus [267,269].
Moreover, machine learning-based analyses [250] identified maltose, glyceric acid, mannonic acid, xylitol, and erythronic acid as the most discriminative metabolites for distinguishing COVID-19-positive patients from healthy controls (AUC = 0.98). In contrast, the combination of succinic, phosphoric, and malic acids, hypoxanthine, and S-adenosylhomocysteine most effectively differentiated COVID-19-positive from COVID-19-negative individuals (AUC = 0.85).
The biological significance of these observations is not straightforward. Since maltose, mannonic acid, and erythronic acid are predominantly plant-derived compounds and are not endogenously synthesized in large amounts by humans, it has been hypothesized that changes in their serum concentrations may be influenced by alterations in gut microbiota secondary to infection. Indeed, the concept of a gut–lung axis has been proposed, suggesting that respiratory infections can impact the gut microbiota and vice versa, ultimately manifesting in altered circulating metabolite profiles [270]. Dysbiosis of the gut microbiome has been implicated in respiratory diseases and infections [271], and antibiotic-induced microbiome disruptions have been shown to influence lung disease outcomes [272]. Moreover, modifications of the lung microbiota are known to affect gut microbial communities reciprocally [273]. Several studies have connected alterations in maltose, mannose, succinate, and erythronic acid serum levels with changes in the gastrointestinal microbiome [274,275,276,277,278]. Recently, a multiomics investigation further detailed complex networks linking gut microbes, metabolites, and cytokines in the context of COVID-19 [279]. Of particular interest, shifts in the gut microbiota have been associated with changes in metabolites belonging to the pentose and glucuronate interconversion pathway [280,281,282].
Although reprogramming of carbohydrate and energy metabolism is a shared feature among respiratory viral infections, the specific signatures of SARS-CoV-2 differ in essential ways and suggest the presence of unique metabolic biomarkers. Influenza virus, for instance, also enhances glycolysis and pentose phosphate pathway (PPP) activity to support viral replication [70,142,283], but does not appear to induce the same degree of alteration in detoxification-related pathways, such as the pentose and glucuronate interconversion route, which is markedly affected in COVID-19. Studies exploring the utility of such detoxification pathway metabolites as biomarkers in influenza are limited or nonexistent.
RSV infection, on the other hand, promotes mitochondrial fragmentation and significantly alters amino acid metabolism, especially glutamine and arginine pathways [284]. While these findings are mechanistically relevant, systematic analyses to identify consistent metabolic biomarkers of RSV infection remain scarce.
6.4. Redox Imbalance and the Antioxidant Response
GSH is the most abundant intracellular antioxidant and a critical regulator of redox homeostasis. GSH depletion and a decreased GSH/GSSG ratio have been consistently reported in viral infections. However, the severity and persistence of these changes appear greater in COVID-19 compared to influenza and RSV. A mechanistic hypothesis [195] posits that endogenous GSH deficiency may be a central factor in developing severe COVID-19 manifestations, due to unchecked oxidative stress and a weakened immune response. Clinical studies have since confirmed that patients hospitalized with COVID-19 present significantly lower plasma GSH levels, which correlate with elevated markers of systemic inflammation [285,286]. Although no robust ROC-based metrics have been reported for GSH alone, its measurement contributes valuable prognostic information when incorporated into broader biomarker panels.
SOD and CAT are key enzymatic antioxidants that neutralize superoxide radicals and hydrogen peroxide, respectively. Respiratory virus infections usually elicit a short-lived increase in their activities, followed by suppression or depletion in advanced stages of the disease [287]. This dynamic shift is indicative of sustained oxidative pressure and impaired mitochondrial function. While SOD and CAT have been included in some prognostic models, their individual diagnostic accuracy, measured by sensitivity, specificity, or AUC, remains insufficiently documented.
PON1 activity is reduced during viral infections, but the decrease is especially pronounced in COVID-19 compared to influenza and RSV. Hospitalized COVID-19 patients showed significantly reduced PON1 arylesterase activity [205]. In a model designed to differentiate COVID-19 from other acute respiratory syndromes, PON1 activity demonstrated a sensitivity of 84%, specificity of 79%, and an AUC of 0.87, making it a promising biomarker for differential diagnosis [206].
Although not classical antioxidants, the tryptophan–kynurenine pathway metabolites are modulated by oxidative stress and systemic inflammation. In COVID-19, there is a sustained increase in the kynurenine/tryptophan (Kyn/Trp) ratio, associated with immune activation and disease severity. A metabolomic study reported a high discriminatory performance (AUC = 0.95) for distinguishing COVID-19-positive from COVID-19-negative patients; however, the authors cautioned that these findings should be interpreted carefully due to the limited sample size [156].
While redox imbalance is a shared hallmark of respiratory viral infections, accumulating evidence suggests that distinct plasma antioxidant and metabolic signatures may serve as virus-specific biomarkers. In the case of COVID-19, several alterations stand out: a marked reduction in GSH and PON1, a consistently elevated kynurenine-to-tryptophan ratio, persistent SOD downregulation, and increased levels of oxidative lipid byproducts such as 4-hydroxynonenal and malondialdehyde. In contrast, influenza virus infection is typically associated with transient oxidative stress, featuring moderate GSH depletion, variable upregulation of enzymatic antioxidants such as SOD and CAT, and subsequent normalization of redox markers during the convalescent phase. RSV infection presents a different profile, characterized by early mitochondrial oxidative injury, depletion of GSH during the acute phase, and elevated lipid peroxidation. However, data on enzymatic antioxidant responses remain limited.
These findings suggest that the combined presence of low GSH and PON1 activity, along with elevated kynurenine levels and oxidative lipid metabolites, may constitute a distinctive redox-based biosignature for COVID-19. Importantly, this biosignature not only differentiates COVID-19 from healthy controls, but results indicate that it may also help to distinguish it from other respiratory viral infections.
7. Identification of Potential Therapeutic Targets
The metabolic reprogramming induced by respiratory viral infections creates vulnerabilities that can be exploited for therapeutic purposes. Beyond the canonical antiviral approaches, targeting host metabolic pathways altered during infection provides an opportunity to modulate viral replication, control inflammation, and mitigate long-term sequelae. This section identifies promising therapeutic targets emerging from preclinical and clinical studies on SARS-CoV-2, influenza, and RSV, focusing on host metabolic and redox pathways.
7.1. Inhibiting Viral Entry
Several host factors crucial for viral entry are modulated by cellular metabolic pathways, offering indirect avenues for antiviral intervention. For instance, the expression of the transmembrane protease, serine 2 (TMPRSS2) and ACE2, key entry factors for SARS-CoV-2, can be influenced by metabolic status and hormonal regulation linked to insulin signaling and energy-sensing pathways such as AMPK. Similarly, the fusion machinery of RSV depends on host membrane lipid composition and cholesterol content, both shaped by metabolic activity. These insights have prompted the exploration of drugs that modulate host metabolism to interfere with viral entry. Camostat mesylate, a TMPRSS2 inhibitor, has shown promise in preclinical models by blocking SARS-CoV-2 spike protein priming, thus preventing cell entry [19]. By altering cholesterol synthesis and lipid raft composition, statins may reduce viral attachment and assembly, with some observational studies reporting improved outcomes in viral pneumonia [89]. Chloroquine and hydroxychloroquine, despite their controversial clinical use, were found to interfere with endosomal pH and glycosylation of ACE2, thereby affecting SARS-CoV-2 entry in vitro [288].
7.2. Targeting Lipid Metabolism
Various studies have highlighted the reliance of respiratory viruses on host lipid metabolism, including de novo lipogenesis and cholesterol trafficking. FASN inhibitors, such as orlistat, disrupt viral envelope formation and replication in enveloped viruses by interfering with host lipid biosynthesis, and have shown antiviral activity in vitro against SARS-Cov-2, influenza, and RSV [52,289].
Statins, which inhibit HMG-CoA reductase and reduce cholesterol biosynthesis, have demonstrated anti-inflammatory and potential antiviral effects in observational studies of COVID-19, with meta-analyses suggesting a protective effect on mortality [290,291,292,293].
Omega-3 fatty acids and their specialized pro-resolving mediators, including resolvins and protectins, have also emerged as modulators of inflammation in viral infections and may counteract the persistent inflammatory response observed in long COVID [294].
7.3. Inhibiting Glycolysis
The enhanced glycolytic flux activ[195–298ation observed in infected cells reflects a shift toward anabolism. Inhibiting key glycolytic enzymes such as hexokinase 2 or 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) can reduce viral replication and inflammation. For instance, 2-deoxy-D-glucose has shown efficacy in reducing SARS-CoV-2 replication in vitro and in clinical trials in India, where it shortened oxygen dependency and hospital stay in moderately ill patients [195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295,296,297,298]. Similarly, inhibition of PFKFB3 has shown potential in reducing viral replication and inflammatory responses in preclinical models of influenza. Recent studies, such as those using the selective inhibitor KAN0438757 [299], demonstrated significant suppression of H1N1 influenza virus replication and cytokine production in human epithelial cells [300]. In addition, although the compound 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) has also been reported to attenuate influenza-related inflammation, its specificity as a PFKFB3 inhibitor is debated, suggesting that its observed effects may involve off-target mechanisms [301,302,303]. Therefore, while targeting PFKFB3 is a promising therapeutic strategy, further studies using more selective inhibitors are needed to validate its efficacy and mechanism of action.
7.4. Modulating Mitochondrial Dynamics and Bioenergetics
One promising therapeutic avenue is the modulation of mitochondrial dynamics, especially the inhibition of excessive mitochondrial fission, which is associated with inflammation and cell death. Mitochondrial division inhibitor 1 (Mdivi-1), a selective inhibitor of Drp1, prevents mitochondrial fragmentation by blocking Drp1-mediated fission. Studies in experimental animals showed that Mdivi-1 reduces viral-induced mitochondrial fragmentation, restores mitochondrial membrane potential, and attenuates inflammatory cytokine release [304,305].
Moreover, mitochondrial protectants such as melatonin, a potent antioxidant and regulator of mitochondrial homeostasis, have demonstrated anti-inflammatory and antiviral properties in COVID-19 models. Melatonin reduces oxidative stress, supports mitochondrial biogenesis via PGC-1α activation, and modulates immune responses [306]. Coenzyme Q10, another mitochondrial antioxidant, may similarly improve mitochondrial respiration and reduce oxidative injury [307,308], although clinical data in viral infections remain limited.
Other pharmacological candidates targeting mitochondrial metabolism include metformin, which supports mitochondrial function via AMPK activation and reduces proinflammatory signaling in viral pneumonia [309,310], and Szeto-Schiller (SS) peptides, such as SS-31, which selectively target the inner mitochondrial membrane, reduce mitochondrial ROS, and improve mitochondrial efficiency in models of oxidative stress [311]. Despite their promise in models of ischemia, neurodegeneration, and metabolic dysfunction [312,313,314,315], no published evidence supports the use of SS peptides in viral infections. Nevertheless, their mechanisms of action align well with the known mitochondrial disturbances induced by respiratory viruses, making them candidates for future investigation.
Preserving mitochondrial function and preventing mitochondrial fragmentation represent rational strategies to limit inflammation, promote cellular survival, and improve outcomes in respiratory viral infections. Drugs such as Mdivi-1, melatonin, and SS-31 illustrate the potential of this approach, which warrants further investigation in clinical settings. Agents enhancing OXPHOS, such as dichloroacetate, may also counteract the Warburg-like phenotype in infected cells and restore energy balance [316]. Moreover, supplementation with mitochondrial cofactors like coenzyme Q10 and nicotinamide riboside has been proposed to alleviate COVID-19-related mitochondrial dysfunction [317,318].
7.5. Antioxidant System Modulation
N-acetylcysteine (NAC), a well-known precursor of cysteine and GSH, has been widely studied among antioxidant therapies. In patients with COVID-19, NAC administration has been associated with improved oxygenation, reduction in inflammatory markers, and lower risk of mechanical ventilation in observational studies [319,320]. Studies in cultured A549 lung cancer cells showed that NAC inhibited oxidant-sensitive pathways, including NF-κB and mitogen-activated protein kinase p38. Moreover, it reduced viral replication and production of pro-inflammatory molecules [321], and prophylactic benefits by NAC were observed in elderly individuals during influenza seasons [322].
Melatonin, an endogenous molecule with antioxidant and anti-inflammatory properties, upregulates enzymatic defenses such as SOD and CAT, and downregulates NF-κB-mediated cytokine production. In preclinical models of viral infections, including RSV and influenza, melatonin treatment has reduced pulmonary inflammation and oxidative stress [323,324,325]. Recent reclinical and clinical studies are evaluating its efficacy as an adjuvant therapy in COVID-19 [306,326,327,328]
Tempol, a membrane-permeable nitroxide that mimics SOD activity, has shown efficacy in preclinical models by neutralizing superoxide radicals, reducing lipid peroxidation, and attenuating inflammation [329]. Tempol treatment significantly reduced viral replication and lung pathology in Syrian hamsters infected with SARS-CoV-2. The treated animals showed decreased viral RNA levels in the lungs, reduced infectious virus titers, and less severe histopathological changes compared to control groups [330], though human studies are lacking.
Ebselen, a glutathione peroxidase mimetic, has shown dual action in COVID-19in an in silico study. This compound possesses direct antiviral activity through inhibition of the SARS-Cov-2 main protease (Mpro) and has antioxidant effects [331].
Despite the growing interest in these antioxidant strategies, the therapeutic modulation of PON1 in viral infections remains largely unexplored. Although reduced PON1 activity has been consistently reported in COVID-19 and associated with disease severity, no pharmacological interventions have been developed to restore PON1 function. Experimental approaches to increase PON1 activity, such as dietary polyphenols, HDL-targeted therapies, or gene modulation, have been proposed in cardiovascular and metabolic diseases [332,333]. However, no clinical or preclinical studies have evaluated their efficacy in respiratory viral infections. This gap highlights a potential opportunity for future research, particularly given PON1’s role in neutralizing lipid peroxides and modulating inflammatory responses. Whether increasing PON1 activity could mitigate oxidative stress or reduce complications in viral infections such as COVID-19 or influenza remains an open question.
7.6. Modulation of the Kynurenine Pathway
The kynurenine pathway is the principal route of tryptophan catabolism, and its activation plays a central role in immune regulation, oxidative stress, and metabolic control during viral infections. One of its key regulatory enzymes, indoleamine 2,3-dioxygenase 1 (IDO1), is upregulated in response to inflammatory cytokines, particularly IFN-γ, leading to increased conversion of tryptophan into kynurenine and other downstream metabolites. In COVID-19, a persistently elevated Kyn/Trp ratio has been consistently associated with disease severity, lymphopenia, and systemic immune dysregulation [156,230]. This metabolic shift serves both antiviral and immunoregulatory purposes. On the one hand, tryptophan depletion can limit viral replication, but on the other, kynurenine and its metabolites exert immunosuppressive effects by inducing regulatory T cells (Tregs), inhibiting effector T cell responses, and modulating dendritic cell function [334]. Kynurenine also acts as an endogenous ligand of the aryl hydrocarbon receptor (AhR), a transcription factor that regulates mucosal immunity, barrier function, and inflammatory cytokine production [335]. Chronic activation of AhR by elevated kynurenine levels may contribute to persistent inflammation and immune exhaustion in severe COVID-19 and possibly other viral infections.
Given its role in immune escape and homeostasis, the kynurenine pathway has emerged as a potential therapeutic target, particularly in diseases marked by chronic inflammation and immune dysfunction. Inhibitors of IDO1, such as epacadostat and navoximod, have been tested extensively in oncology for their ability to reverse immune suppression and restore antitumor immunity [336,337,338]. Although these agents have not yet been tested in large trials for viral infections, their mechanism of action may be relevant for conditions such as severe COVID-19, where elevated kynurenine activity correlates with worse outcomes [230].
In addition to IDO1 blockade, targeting downstream components of the pathway may offer therapeutic opportunities. For instance, kynurenic acid and quinolinic acid exert neuroactive and pro-oxidant effects, and the possibility exists that they are implicated in the pathophysiology of viral encephalopathy [339]. Another promising approach is modulating the AhR, which is activated by kynurenine and related metabolites. Selective AhR antagonists or modulators, such as CH223191, have been shown to reduce the traffic-derived particulate matter-induced cytokine release in human macrophages and bronchial epithelial cells [340].
Nutritional modulation of the kynurenine pathway is also under investigation. Niacin (vitamin B3), a downstream product of the pathway, may act as a feedback regulator of tryptophan metabolism and has been proposed as an adjunctive therapy to buffer oxidative stress and inflammation [341]. Furthermore, gut microbiota composition influences tryptophan availability and kynurenine pathway activity, suggesting that probiotic or microbiome-directed interventions might indirectly affect this immunometabolic axis [342].
Despite these promising avenues, no therapeutic interventions targeting the kynurenine axis have yet been approved or validated in acute viral respiratory infections. Given the strong correlation between the kynurenine pathway and disease severity in COVID-19, this biochemical route represents a compelling target for future translational research.
Table 5 provides an overview of the principal drugs investigated, their associated metabolic pathways, and the viral infections in which they have been proposed as therapeutic agents.
7.7. Multi-Omics Guided Therapies and Personalized Interventions
Multi-omics approaches provide a comprehensive, systems-level view of disease pathophysiology, enabling the identification of biosignatures that serve as robust diagnostic or prognostic biomarkers and reveal actionable therapeutic targets. Rather than isolated molecular readouts, multi-omics facilitates the construction of dynamic interaction networks that map the cascade of host responses, from gene expression to metabolite accumulation, offering unprecedented precision for therapeutic intervention.
In the context of respiratory viral infections, this holistic approach allows for the stratification of patients based on their individual metabolic and immune profiles. For instance, redox status, lipidomic alterations, and amino acid imbalances can guide the personalized use of antioxidant regimens, lipid-modulating agents, amino acid supplementation, or targeted metabolic inhibitors. Notably, integrating metabolic and immune data can help identify patient subgroups more likely to benefit from specific interventions, minimizing ineffective or potentially harmful treatments [343,344].
One prominent example is the recognition of arginine deficiency in severe COVID-19, derived from metabolomic analyses showing significant depletion of circulating arginine levels. This finding has led to the hypothesis that therapeutic arginine depletion or modulation might influence immune responses and viral replication. Pegylated arginase (PEG-Arg1), which depletes extracellular arginine, has been proposed as a potential therapy to modulate hyperinflammation in COVID-19 [345]. Similarly, glutamine metabolism, which is altered in both influenza and SARS-CoV-2 infections, has been proposed as a therapeutic target, with inhibitors of glutaminolysis showing antiviral effects in preclinical models [346].
Furthermore, machine learning–enhanced metabolomics has demonstrated that combinations of metabolites (e.g., maltose, xylitol, glyceric acid) can differentiate COVID-19 patients from healthy controls and patients without COVID-19 with high accuracy. These panels offer diagnostic utility and may assist in therapeutic stratification, identifying patients with specific metabolic phenotypes that respond better to tailored interventions [250].
Another key application of multi-omics is in the monitoring of therapeutic response. Changes in metabolic or lipidomic profile during treatment can serve as dynamic biomarkers, providing real-time feedback on drug efficacy or therapeutic adjustment [223,250]. While integrating multi-omics data holds promise for personalizing pharmacological or nutritional interventions in respiratory viral infections, current clinical trials have not yet adopted this approach. Existing studies on omega-3 supplementation provide valuable insights but do not utilize patient-specific omics data to tailor interventions. Further research is needed to develop and assess personalized nutritional strategies guided by comprehensive omics profiling. [347,348].
Ultimately, the multi-omics framework paves the way for a multi-therapeutic approach, combining pharmacological and nutritional strategies adapted to the individual's molecular profile. For example, a patient showing GSH depletion, dysregulated kynurenine metabolism, and altered mitochondrial lipid composition could be managed with a tailored regimen combining NAC, AhR modulators, and mitochondrial stabilizers. This personalized, multi-targeted intervention model represents a shift from disease-based to mechanism-based precision medicine, especially relevant for complex viral syndromes like COVID-19 or severe influenza.
However, translating multi-omics findings into clinical practice poses significant challenges, including data standardization, analytical complexity, cost, and interdisciplinary coordination. Nonetheless, ongoing systems biology and computational medicine efforts are progressively overcoming these barriers, bringing personalized multi-therapeutic strategies closer to clinical application [349,350].
8. Conclusions and Future Perspectives
Respiratory viral infections such as those caused by SARS-CoV-2, influenza virus, and RSV continue to exert a significant global health burden. Although distinct in structure and host tropism, these viruses share a common feature: the ability to hijack and reprogram host metabolic pathways to facilitate their replication, evade immune responses, and modulate disease progression. This review has detailed how respiratory viruses manipulate lipid metabolism, energy production, amino acid and nucleotide pathways, and oxidative stress responses, often driving the host cell into a pro-viral metabolic state.
A key advance in recent years has been integrating multi-omics technologies, allowing for a more comprehensive understanding of the host response to infection. These tools facilitate the discovery of biosignatures with diagnostic and prognostic value and highlight actionable metabolic targets. In particular, personalized therapeutic approaches, such as antioxidant regimens tailored to redox imbalance, or amino acid and lipid-based interventions guided by metabolomic signatures, have emerged as promising strategies. Furthermore, targeting virus-induced mitochondrial dysfunction, glycolysis, or pathways like kynurenine metabolism holds translational potential.
While these conceptual and technical advances have been largely propelled by the intense global research efforts on COVID-19, there is a growing recognition that similar efforts are urgently needed for influenza and RSV. Despite the unprecedented depth of metabolic and immunological profiling conducted for SARS-CoV-2, comparable multi-omics datasets and therapeutic trials remain limited for other respiratory viruses. This disparity is especially concerning given recent epidemiological trends. For example, during the 2023–2024 season in the United States, influenza was responsible for approximately 40 million illnesses, 470,000 hospitalizations, and 28,000 deaths, which now rival or exceed those associated with COVID-19 in the same period [351]. Similarly, RSV is increasingly recognized as a serious pathogen in older adults, causing 60,000–160,000 hospitalizations and 6,000–10,000 deaths annually in individuals over 65 [352].
Given this context, future research should build upon the methodologies and insights developed during the COVID-19 pandemic to study influenza and RSV with equivalent rigor. Priority areas include: expanding the application of multi-omics to uncover metabolic vulnerabilities; conducting randomized clinical trials to validate metabolically guided therapies; and integrating machine learning models for patient stratification and prediction of disease trajectories. Additionally, systems biology approaches that combine omics data with clinical phenotyping will be essential to unravel the complex interplay between viral infection, host metabolism, and immune response.
In conclusion, metabolic reprogramming is a central and dynamic aspect of respiratory viral pathogenesis. Leveraging this knowledge deepens our mechanistic understanding and opens new avenues for diagnosis, risk stratification, and therapeutic intervention. Bridging the current research gap between SARS-CoV-2 and other respiratory viruses like influenza and RSV is essential to ensure that all patients benefit from the advances in precision medicine that are now within reach.
Author Contributions
Conceptualization, J.C.; S.I.; A.C. and J.J.; investigation, J.C., S.I. and A.J.F.; validation, J.C.; visualization, J.C. and S.I.; writing—original draft preparation, J.C. and S.I.; writing—review and editing, J.C., S.I., A.C. and J.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
During the preparation of this work, the authors used CHAT GPT 4.0 (developed by OpenAI) to improve the grammar, syntax, and clarity of the text and GRAMMARLY (from Grammarly Inc.) for orthographic corrections. After using these tools, the authors reviewed and edited the content as needed and took full responsibility for the content of the published article. The content, ideas, and scientific conclusions presented in this manuscript are solely the authors' work and have not been generated by AI. The AI tools were utilized exclusively to enhance the readability and presentation of the text.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACE2 | Angiotensin-converting enzyme 2 |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| AUC | Area under the curve |
| AhR | Aryl hydrocarbon receptor |
| BCAA | Branched-chain amino acids |
| CAR 8:0 | O-octanoyl-L-carnitine |
| CAT | Catalase |
| COVID-19 | Coronavirus disease 2019 |
| CPT | Carnitine palmitoyltransferases |
| CCL2 | Chemokine (C-C motif) ligand 2 |
| DGAT | Diacylglycerol O-acyltransferase |
| Drp1 | Dynamin-related protein 1 |
| FASN | Fatty acid synthase |
| GC | Gas chromatography |
| GLUT | Glucose transporter |
| GSH | Glutathione |
| HDL | High-density lipoproteins |
| HETE | Hydroxyeicosatetraenoic acid |
| HIF-1α | Hypoxia-inducible factor 1-α |
| HODE | Hydroxyoctadecadienoic acid |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| IFN | Interferon |
| IL | Interleukin |
| Kyn/Trp | Kynurenine/tryptophan |
| LC | Liquid chromatography |
| LD | Lipid droplets |
| LDL | Low-density lipoproteins |
| LPC | Lysophosphatidylcholine |
| LPE | Lysophosphatidylethanolamine |
| MAVS | Mitochondrial antiviral-signaling protein |
| MCP-1 | Monocyte chemoattractant protein-1 |
| Mdivi-1 | Mitochondrial division inhibitor 1 |
| MS | Mass spectrometry |
| mTOR | Mechanistic target of rapamycin |
| MTP | Mitochondrial trifunctional protein |
| NAC | N-acetylcysteine |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa B |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NMR | Nuclear magnetic resonance |
| NOX | NADPH oxidase |
| NRF2 | Nuclear factor erythroid 2–related factor 2 |
| OXPHOS | Oxidative phosphorylation |
| PGC-1α | Peroxisome proliferator-activated receptor-γ coactivator 1-α |
| PC | Phosphatidylcholine |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PI | Phosphatidylinositol |
| PI3K | Phosphoinositide 3-kinase |
| 3PO | 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one |
| PON1 | Paraoxonase-1 |
| ROC | Receiver operating characteristic |
| ROS | Reactive oxygen species |
| RLR | Retinoic acid-inducible gene-I-like receptors, |
| RSV | Respiratory syncytial virus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SOD | Superoxide dismutase |
| SREBP | Sterol regulatory element-binding proteins |
| SREBP2-LDLR | Sterol regulatory element-binding protein 2 and low-density lipoprotein receptor |
| TLR | Toll-like receptors |
| TMPRSS2 | Transmembrane protease, serine 2 |
| TNF-α | Tumor necrosis factor-α |
| VLDL | Very low-density lipoproteins |
References
- Safiri, S.; Mahmoodpoor, A.; Kolahi, A.A.; Nejadghaderi, S.A.; Sullman, M.J.M.; Mansournia, M.A.; Ansarin, K.; Collins, G.S.; Kaufman, J.S.; Abdollahi, M. Global burden of lower respiratory infections during the last three decades. Front. Public Health. 2023, 10, 1028525. [Google Scholar] [CrossRef] [PubMed]
- Gravenstein, S. Foreword: Prevention of COVID-19, influenza, and respiratory syncytial virus in at-risk populations. Infect. Dis. Ther. 2025, 14 (Suppl 1), 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hanage, W.P.; Schaffner, W. Burden of acute respiratory infections caused by influenza virus, respiratory syncytial virus, and SARS-CoV-2 with consideration of older adults: A narrative review. Infect. Dis. Ther. 2025, 14 (Suppl 1), 5–37. [Google Scholar] [CrossRef] [PubMed]
- Kleinehr, J.; Wilden, J.J.; Boergeling, Y.; Ludwig, S.; Hrincius, E.R. Metabolic modifications by common respiratory viruses and their potential as new antiviral targets. Viruses 2021, 13, 2068. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.N.S.; Arjona, S.P.; Santerre, M.; Sawaya, B.E. Hallmarks of metabolic reprogramming and their role in viral pathogenesis. Viruses 2022, 14, 602. [Google Scholar] [CrossRef]
- Shahpar, A.; Sofiani, V.H.; Nezhad, N.Z.; Charostad, M.; Ghaderi, R.; Farsiu, N.; Kiskani, A.K.; Pezeshki, S.; Nakhaie, M. A narrative review: exploring viral-induced malignancies through the lens of dysregulated cellular metabolism and glucose transporters. BMC Cancer 2024, 24, 1329. [Google Scholar] [CrossRef]
- Konaklieva, M.I.; Plotkin, B.J. Targeting host-specific metabolic pathways-opportunities and challenges for anti-infective therapy. Front. Mol. Biosci. 2024, 11, 1338567. [Google Scholar] [CrossRef]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; Azhar, E.; Hoelscher, M.; Maeurer, M; Host-Directed Therapies Network consortium. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect. Dis. 2016, 16, e47–e63. [CrossRef]
- Palmer, C.S. Innate metabolic responses against viral infections. Nat. Metab. 2022, 4, 1245–1259. [Google Scholar] [CrossRef]
- Mazzon, M.; Mercer, J. Lipid interactions during virus entry and infection. Cell. Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef]
- Waheed, A.A.; Freed, E.O. The role of lipids in retrovirus replication. Viruses 2010, 2, 1146–1180. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Ripa, I., Andreu, S.; López-Guerrero, J.A.; Bello-Morales, R. Membrane rafts: Portals for viral entry. Front. Microbiol. 2021, 12, 631274. [CrossRef]
- Lorizate, M.; Kräusslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Virus entry: open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef]
- Glende, J.; Schwegmann-Wessels, C.; Al-Falah, M.; Pfefferle, S.; Qu, X.; Deng, H.; Drosten, C.; Naim, H.Y.; Herrler, G. Importance of cholesterol-rich membrane microdomains in the interaction of the S protein of SARS-coronavirus with the cellular receptor angiotensin-converting enzyme 2. Virology 2008, 381, 215–221. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; Müller, M.A.; Drosten, C.; Pöhlmann, S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [CrossRef]
- Chakraborty, S.; Veettil, M.V.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 7146–7147. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, S.; Shen, S.; Guo, H.; Huang, H.; Wei, W. Methyl-β-cyclodextrin inhibits EV-D68 virus entry by perturbing the accumulation of virus particles and ICAM-5 in lipid rafts. Antiviral Res. 2020, 176, 104752. [CrossRef]
- Roncato, R.; Angelini, J.; Pani, A.; Talotta, R. Lipid rafts as viral entry routes and immune platforms: A double-edged sword in SARS-CoV-2 infection? Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 2022, 1867, 159140. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Mukhamedova, N.; Sviridov, D. Lipid rafts and pathogens: the art of deception and exploitation. J. Lipid Res. 2020, 61, 601–610. [Google Scholar] [CrossRef]
- Song, M.S.; Lee, D.K.; Lee, C.Y.; Park, S.C.; Yang, J. Host subcellular organelles: Targets of viral manipulation. Int. J. Mol. Sci. 2024, 25, 1638. [Google Scholar] [CrossRef]
- Rawat, S.S.; Viard, M.; Gallo, S.A.; Rein, A.; Blumenthal, R.; Puri, A. Modulation of entry of enveloped viruses by cholesterol and sphingolipids. Mol. Membr. Biol. 2003, 20, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [CrossRef]
- Barrett, C.T.; Dutch, R.E. Viral membrane fusion and the transmembrane domain. Viruses 2020, 12, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Basturea, G. Endocytosis Mater. Methods 2019, 9, 2752. [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef]
- Sandvig, K.; Kavaliauskiene, S.; Skotland, T. Clathrin-independent endocytosis: an increasing degree of complexity. Histochem. Cell Biol. 2018, 150, 107–118. [Google Scholar] [CrossRef]
- Shafaq-Zadah, M.; Dransart, E.; Johannes, L. Clathrin-independent endocytosis, retrograde trafficking, and cell polarity. Curr. Opin. Cell Biol. 2020, 65, 112–121. [Google Scholar] [CrossRef]
- Ruzzi, F.; Cappello, C.; Semprini, M.S.; Scalambra, L.; Angelicola, S.; Pittino, O.M.; Landuzzi, L.; Palladini, A.; Nanni, P.; Lollini, P.L. Lipid rafts, caveolae, and epidermal growth factor receptor family: friends or foes? Cell. Commun. Signal. 2024, 22, 489. [Google Scholar] [CrossRef]
- McMahon, K.A.; Zajicek, H.; Li, W.P.; Peyton, M.J.; Minna, J.D.; Hernandez, V.J.; Luby-Phelps, K.; Anderson, R.G. SRBC/cavin-3 is a caveolin adapter protein that regulates caveolae function. EMBO J. 2009, 28, 1001–1015. [Google Scholar] [CrossRef]
- Kovtun, O.; Tillu, V.A.; Ariotti, N.; Parton, R.G.; Collins, B.M. Cavin family proteins and the assembly of caveolae. J. Cell Sci. 2015, 128, 1269–1278. [CrossRef]
- Vicinanza, M.; D’angelo, G.; Di Campli, A.; De Matteis, M.A. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008, 27, 2457–2470. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell. Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chakraborty, S.; Basu, A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J. Neurochem. 2010, 115, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Bohdanowicz, M.; Grinstein, S. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol. Rev. 2013, 93, 69–106. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell. Microbiol. 2011, 13, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Tsuda, M.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. The Ras-PI3K signaling pathway is involved in clathrin-independent endocytosis and the internalization of influenza viruses. PLoS ONE 2011, 6, e16324. [Google Scholar] [CrossRef]
- Palacios-Rápalo, S.N.; De Jesús-González, L.A.; Cordero-Rivera, C.D.; Farfan-Morales, C.N.; Osuna-Ramos, J.F.; Martínez-Mier, G.; Quistián-Galván, J.; Muñoz-Pérez, A.; Bernal-Dolores, V.; Del Ángel, R.M.; Reyes-Ruiz, J.M. Cholesterol-rich lipid rafts asplatforms for SARS-CoV-2 entry. Front. Immunol. 2021, 12, 796855. [Google Scholar] [CrossRef]
- El Khoury, M.; Naim, H.Y. Lipid rafts disruption by statins negatively impacts the interaction between SARS-CoV-2 S1 subunit and ACE2 in intestinal epithelial cells. Front. Microbiol. 2024, 14, 1335458. [Google Scholar] [CrossRef]
- Zhang, Y.; Whittaker, G.R. Influenza entry pathways in polarized MDCK cells. Biochem. Biophys. Res. Commun. 2014, 450, 234–239. [Google Scholar] [CrossRef]
- Bolland, W.; Marechal, I.; Petiot, C.; Porrot, F.; Guivel-Benhassine, F.; Brelot, A.; Casartelli, N.; Schwartz, O.; Buchrieser, J. SARS-CoV-2 entry and fusion are independent of ACE2 localization to lipid rafts. J. Virol. 2025, 99, e0182324. [Google Scholar] [CrossRef]
- Rossman, J.S.; Leser, G.P.; Lamb, R.A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 2012, 86, 10950–10960. [Google Scholar] [CrossRef]
- de Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jiménez, V.; Scholte, F.; García-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.K.; Gupta, D.; Lal, S.K. Host lipid rafts play a major role in binding and endocytosis of Influenza A virus. Viruses 2018, 10, 650. [Google Scholar] [CrossRef]
- Fleming, E.H.; Kolokoltsov, A.A.; Davey, R.A.; Nichols, J.E.; Roberts, N.J. Jr. Respiratory syncytial virus F envelope protein associates with lipid rafts without a requirement for other virus proteins. J. Virol. 2006, 80, 12160–12170. [Google Scholar] [CrossRef]
- Shaikh, F.Y.; Crowe, J.E. Jr. Molecular mechanisms driving respiratory syncytial virus assembly. Future Microbiol. 2013, 8, 123–131. [Google Scholar] [CrossRef]
- Chandel, N.S. Lipid metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040576. [Google Scholar] [CrossRef]
- Chu, J.; Xing, C.; Du, Y.; Duan, T.; Liu, S.; Zhang, P.; Cheng, C.; Henley, J.; Liu, X.; Qian, C.; Yin, B.; Wang, H.Y.; Wang, RF. Pharmacological inhibition of fatty acid synthesis blocks SARS-CoV-2 replication. Nat. Metab. 2021, 3, 1466–1475. [Google Scholar] [CrossRef]
- Williams, C.G.; Jureka, A.S.; Silvas, J.A.; Nicolini, A.M.; Chvatal, S.A.; Carlson-Stevermer, J.; Oki, J.; Holden, K.; Basler, C.F. Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-CoV-2 replication. Cell. Rep. 2021, 36, 109479. [Google Scholar] [CrossRef]
- Limsuwat, N.; Boonarkart, C.; Phakaratsakul, S.; Suptawiwat, O.; Auewarakul, P. Influence of cellular lipid content on influenza A virus replication. Arch. Virol. 2020, 165, 1151–1161. [Google Scholar] [CrossRef]
- Ohol, Y.M.; Wang, Z.; Kemble, G.; Duke, G. Direct inhibition of cellular fatty acid synthase impairs replication of respiratory syncytial virus and other respiratory viruses. PLoS One 2015, 10, e0144648. [Google Scholar] [CrossRef]
- Aliyari, S.R.; Ghaffari, A.A.; Pernet, O.; Parvatiyar, K.; Wang, Y.; Gerami, H.; Tong, A.J.; Vergnes, L.; Takallou, A.; Zhang, A.; Wei, X.; Chilin, L.D.; Wu, Y.; Semenkovich, C.F.; Reue, K.; Smale, S.T.; Lee, B.; Cheng, G. Suppressing fatty acid synthase by type I interferon and chemical inhibitors as a broad spectrum anti-viral strategy against SARS-CoV-2. Acta Pharm. Sin. B. 2022, 12, 1624–1635. [Google Scholar] [CrossRef]
- Wölk, M.; Fedorova, M. The lipid droplet lipidome. FEBS Lett. 2024, 598, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mak, H.Y.; Lukmantara, I.; Li, Y.E.; Hoehn, K.L.; Huang, X.; Du, X.; Yang, H. CDP-DAG synthase 1 and 2 regulate lipid droplet growth through distinct mechanisms. J. Biol. Chem. 2019, 294, 16740–16755. [Google Scholar] [CrossRef] [PubMed]
- Monks, J.; Orlicky, D.J.; Libby, A.E.; Dzieciatkowska, M.; Ladinsky, M.S.; McManaman, J.L. Perilipin-2 promotes lipid droplet-plasma membrane interactions that facilitate apocrine lipid secretion in secretory epithelial cells of the mouse mammary gland. Front. Cell. Dev. Biol. 2022, 10, 958566. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.S.G.; Soares, V.C.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; Nunes da Silva, M.A.; Barreto, E.; Mattos, M.; de Freitas, C.S.; Azevedo-Quintanilha, I.G.; Manso, P.P.A.; Miranda, M.D.; Siqueira, M.M.; Hottz, E.D.; Pão, C.R.R.; Bou-Habib, D.C.; Barreto-Vieira, D.F.; Bozza, F.A.; Souza, T.M.L.; Bozza, P.T. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar] [CrossRef]
- Chawla, K.; Subramanian, G.; Rahman, T.; Fan, S.; Chakravarty, S.; Gujja, S.; Demchak, H.; Chakravarti, R.; Chattopadhyay, S. Autophagy in virus infection: A race between host immune response and viral antagonism. Immuno. 2022, 2, 153–169. [Google Scholar] [CrossRef]
- Herrera-Moro Huitron, L.; De Jesús-González, L.A.; Martínez-Castillo, M.; Ulloa-Aguilar, J.M.; Cabello-Gutierrez, C.; Helguera-Repetto, C.; Garcia-Cordero, J.; León Juárez, M. Multifaceted nature of lipid droplets in viral interactions and pathogenesis. Microorganisms 2023, 11, 1851. [Google Scholar] [CrossRef]
- Kuss-Duerkop, S.K.; Wang, J.; Mena, I.; White, K.; Metreveli, G.; Sakthivel, R.; Mata, M.A.; Muñoz-Moreno, R.; Chen, X.; Krammer, F.; Diamond, M.S.; Chen, Z.J.; García-Sastre, A.; Fontoura, B.M.A. Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathog. 2017, 13, e1006635. [Google Scholar] [CrossRef]
- Dai, P.; Tang, Z.; Qi, M.; Liu, D.; Bajinka, O.; Tan, Y. Dispersion and utilization of lipid droplets mediates respiratory syncytial virus-induced airway hyperresponsiveness. Pediatr. Allergy Immunol. 2022, 33, e13651. [Google Scholar] [CrossRef]
- Cheung, W.; Gill, M.; Esposito, A.; Kaminski, C.F.; Courousse, N.; Chwetzoff, S.; Trugnan, G.; Keshavan, N.; Lever, A.; Desselberger, U. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J. Virol. 2010, 84, 6782–6798. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Talley, J.T.; Mohiuddin, SS. Biochemistry, Fatty Acid Oxidation; StatPearls [Internet]. StatPearls Publishing: Treasure Island (FL), USA, 2025. [PubMed]
- Tanner, L.B.; Chng, C.; Guan, X.L.; Lei, Z.; Rozen, S.G.; Wenk, M.R. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res. 2014, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Andrade Silva, M.; da Silva, A.R.P.A.; do Amaral, M.A.; Fragas, M.G.; Câmara, N.O.S. Metabolic alterations in SARS-CoV-2 infection and its implication in kidney dysfunction. Front. Physiol. 2021, 12, 624698. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Solaymani-Mohammadi, F.; Namdari, H.; Arjeini, Y.; Mousavi, M.J.; Rezaei, F. Metabolic host response and therapeutic approaches to influenza infection. Cell. Mol. Biol. Lett. 2020, 25, 15. [Google Scholar] [CrossRef]
- Pérez, S.E.; Gooz, M.; Maldonado, E.N. Mitochondrial dysfunction and metabolic disturbances induced by viral infections. Cells 2024, 13, 1789. [Google Scholar] [CrossRef]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., Kalra, S., Kaltsas, G., Kapoor, N., Koch, C., Kopp, P., Korbonits, M., Kovacs, C.S., Kuohung, W., Laferrère, B., Levy, M., McGee, E.A., McLachlan, R., New, M., Purnell, J., Sahay, R., Shah, A.S., Singer, F., Sperling, M.A., Stratakis, C.A., Trence, D.L., Wilson, D. Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2024. Bookshelf ID: NBK305896. [PubMed]
- Navab, M.; Reddy, S.T.; Van Lenten, B.J.; Fogelman, A.M. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 2011, 8, 222–232. [Google Scholar] [CrossRef]
- Catapano, A.L.; Pirillo, A.; Bonacina, F.; Norata, G.D. HDL in innate and adaptive immunity. Cardiovasc. Res. 2014, 103, 372–383. [Google Scholar] [CrossRef]
- Camps, J.; Iftimie, S.; García-Heredia, A.; Castro, A.; Joven, J. Paraoxonases and infectious diseases. Clin. Biochem. 2017, 50, 804–811. [Google Scholar] [CrossRef]
- G, H.B.; Rao, V.S.; Kakkar, V.V. Friend turns foe: Transformation of anti-inflammatory HDL to proinflammatory HDL during acute-phase response. Cholesterol 2011, 2011, 274629. [Google Scholar] [CrossRef]
- Camps, J.; Castañé, H.; Rodríguez-Tomàs, E.; Baiges-Gaya, G.; Hernández-Aguilera, A.; Arenas, M.; Iftimie, S.; Joven, J. On the role of paraoxonase-1 and chemokine ligand 2 (C-C motif) in metabolic alterations linked to inflammation and disease. A 2021 update. Biomolecules 2021, 11, 971. [Google Scholar] [CrossRef]
- Camps, J.; Iftimie, S.; Arenas, M.; Castañé, H.; Jiménez-Franco, A.; Castro, A.; Joven, J. Paraoxonase-1: How a xenobiotic detoxifying enzyme has become an actor in the pathophysiology of infectious diseases and cancer. Chem. Biol. Interact. 2023, 380, 110553. [Google Scholar] [CrossRef]
- Mallol, R.; Amigó, N.; Rodríguez, M.A.; Heras, M.; Vinaixa, M.; Plana, N.; Rock, E.; Ribalta, J.; Yanes, O.; Masana, L.; Correig, X. Liposcale: a novel advanced lipoprotein test based on 2D diffusion-ordered 1H NMR spectroscopy. J. Lipid Res. 2015, 56, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Ballout, R.A.; Kong, H.; Sampson, M.; Otvos, J.D.; Cox, A.L.; Agbor-Enoh, S.; Remaley, AT. The NIH lipo-COVID study: A pilot NMR investigation of lipoprotein subfractions and other metabolites in patients with severe COVID-19. Biomedicines 2021, 9, 1090. [Google Scholar] [CrossRef] [PubMed]
- Lodge, S.; Nitschke, P.; Kimhofer, T.; Coudert, J.D.; Begum, S.; Bong, S.H.; Richards, T.; Edgar, D.; Raby, E.; Spraul, M.; Schaefer, H.; Lindon, J.C.; Loo, R.L.; Holmes, E.; Nicholson, J.K. NMR spectroscopic windows on the systemic effects of SARS-CoV-2 infection on plasma lipoproteins and metabolites in relation to circulating cytokines. J. Proteome Res. 2021, 20, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Schmelter, F.; Föh, B.; Mallagaray, A.; Rahmöller, J.; Ehlers, M.; Lehrian, S.; von Kopylow, V.; Künsting, I.; Lixenfeld, A.S.; Martin, E.; Ragab, M.; Meyer-Saraei, R.; Kreutzmann, F.; Eitel, I.; Taube, S.; Käding, N.; Jantzen, E.; Graf, T.; Sina, C.; Günther, U.L. Metabolic and lipidomic markers differentiate COVID-19 from non-hospitalized and other intensive care patients. Front. Mol. Biosci. 2021, 8, 737039. [Google Scholar] [CrossRef]
- Rössler, T.; Berezhnoy, G.; Singh, Y.; Cannet, C.; Reinsperger, T.; Schäfer, H.; Spraul, M.; Kneilling, M.; Merle, U.; Trautwein, C. Quantitative serum NMR spectroscopy stratifies COVID-19 patients and sheds light on interfaces of host metabolism and the immune response with cytokines and clinical parameters. Metabolites 2022, 12, 1277. [Google Scholar] [CrossRef]
- Iftimie, S.; Amigó, N.; Martínez-Micaelo, N.; López-Azcona, A.F.; Martínez-Navidad, C.; Castañé, H.; Jiménez-Franco, A.; Ribalta, J.; Parra, S.; Castro, A.; Camps, J.; Joven, J. Differential analysis of lipoprotein and glycoprotein profiles in bacterial infections and COVID-19 using proton nuclear magnetic resonance and machine learning. Heliyon 2024, 10, e37115. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Wagner, A.C.; Anantharamaiah, G.M.; Garber, D.W.; Fishbein, M.C.; Adhikary, L.; Nayak, D.P.; Hama, S.; Navab, M.; Fogelman, A.M. Influenza infection promotes macrophage traffic into arteries of mice that is prevented by D-4F, an apolipoprotein A-I mimetic peptide. Circulation 2002, 106, 1127–1132. [Google Scholar] [CrossRef]
- Heinzl, M.W.; Freudenthaler, M.; Fellinger, P.; Kolenchery, L.; Resl, M.; Klammer, C.; Obendorf, F.; Schinagl, L.; Berger, T.; Egger, M.; Dieplinger, B.; Clodi, M. High-density lipoprotein predicts intrahospital mortality in influenza. J. Clin. Med. 2024, 13, 7242. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Xu, W.; Chen, J.; Tang, Y.; Xiong, S.; Li, Y.; Zhang, H.; Li, M.; Liu, Z. Cholesterol-rich lysosomes induced by respiratory syncytial virus promote viral replication by blocking autophagy flux. Nat. Commun. 2024, 15, 6311. [Google Scholar] [CrossRef]
- Doyle, A.; Goodson, B.A.; Kolaczkowski, O.M.; Liu, R.; Jia, J.; Wang, H.; Han, X.; Ye, C.; Bradfute, S.B.; Kell, A.M.; Lemus, M.R.; Pu. J. Manipulation of host cholesterol by SARS-CoV-2. bioRxiv [Preprint]. 2024, 2024.11.13.623299. [CrossRef]
- Li, Y.J.; Chen, C.Y.; Yang, J.H.; Chiu, Y.F. Modulating cholesterol-rich lipid rafts to disrupt influenza A virus infection. Front. Immunol. 2022, 13, 982264. [Google Scholar] [CrossRef]
- Carter, T.; Iqbal, M. The influenza A virus replication cycle: A comprehensive review. Viruses 2024, 16, 316. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.; Wang, J.; Guo, X.; Song, Y.; Fu, K.; Gao, Z.; Liu, D.; He, W.; Yang, L.L. Energy metabolism in health and diseases. Signal Transduct. Target. Ther. 2025, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Amar, S.; Gehlot, P.; Patra, S.K.; Kanwar, N.; Kanwal, A. Mitochondrial modulations, autophagy pathways shifts in viral infections: Consequences of COVID-19. Int. J. Mol. Sci. 2021, 22, 8180. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [CrossRef]
- Singh, S.; Singh, P.K.; Suhail, H.; Arumugaswami, V.; Pellett, P.E.; Giri, S.; Kumar, A. AMP-activated protein kinase restricts Zika virus replication in endothelial cells by potentiating innate antiviral responses and inhibiting glycolysis. J. Immunol. Baltim. Md. 2020, 1950, 1810–1824. [Google Scholar] [CrossRef]
- Pouysségur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Ždralević, M.; Vucetic, M. 'Warburg effect' controls tumor growth, bacterial, viral infections and immunity-Genetic deconstruction and therapeutic perspectives. Semin. Cancer Biol. 2022, 86, 334–346. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; Jimenez Restrepo, J.L.; Vendramini, P.H.; Reis-de-Oliveira, G.; Bispo Dos Santos, K.; Toledo-Teixeira, D.A.; Parise, P.L.; Martini, M.C.; Marques, R.E.; Carmo, H.R.; Borin, A.; Coimbra, L.D.; Boldrini, V.O.; Brunetti, N.S.; Vieira, A.S.; Mansour, E.; Ulaf, R.G.; Bernardes, A.F.; Nunes, T.A.; Ribeiro, L.C.; Palma, A.C.; Agrela, M.V.; Moretti, M.L.; Sposito, A.C.; Pereira, F.B.; Velloso, L.A.; Vinolo, M.A.R.; Damasio, A.; Proença-Módena, J.L.; Carvalho, R.F.; Mori, M.A.; Martins-de-Souza, D.; Nakaya, H.I.; Farias, A.S.; Moraes-Vieira, P.M. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent Axis. Cell. Metab. 2020, 32, 437-446.e5. [CrossRef]
- Santos, A.F.; Póvoa, P.; Paixão, P.; Mendonça, A.; Taborda-Barata, L. Changes in glycolytic pathway in SARS-COV 2 infection and their importance in understanding the severity of COVID-19. Front. Chem. 2021, 9, 685196. [Google Scholar] [CrossRef]
- Guarnieri, J.W.; Lie, T.; Albrecht, Y.E.S.; Hewin, P.; Jurado, K.A.; Widjaja, G.A.; Zhu, Y.; McManus, M.J.; Kilbaugh, T.J.; Keith, K.; Potluri, P.; Taylor, D.; Angelin, A.; Murdock, D.G.; Wallace, D.C. Mitochondrial antioxidants abate SARS-COV-2 pathology in mice. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2321972121. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Z.; Zhang, W.; Meng, X.; Zhu, Y.; Han, P.; Zhou, X.; Hu, Y.; Wang, R. Nuclear translocation of HIF-1α induced by influenza A (H1N1) infection is critical to the production of proinflammatory cytokines. Emerg. Microbes Infect. 2017, 6, e39. [Google Scholar] [CrossRef]
- Meng, X.; Zhu, Y.; Yang, W.; Zhang, J.; Jin, W.; Tian, R.; Yang, Z.; Wang, R. HIF-1α promotes virus replication and cytokine storm in H1N1 virus-induced severe pneumonia through cellular metabolic reprogramming. Virol. Sin. 2024, 39, 81–96. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, J.; Cheng, L.; Xu, K.; Yang, Y.; Su, X. Deficiency of HIF-1α enhances influenza A virus replication by promoting autophagy in alveolar type II epithelial cells. Emerg. Microbes Infect. 2020, 9, 691–706. [Google Scholar] [CrossRef]
- Reyes, A.; Duarte, L.F.; Farías, M.A.; Tognarelli, E.; Kalergis, A.M.; Bueno, S.M.; González, P.A. Impact of hypoxia over human viral infections and key cellular processes. Int. J. Mol. Sci. 2021, 22, 7954. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Ren, C:; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [CrossRef]
- Chen, L.F.; Cai, J.X.; Zhang, J.J.; Tang. Y.J.; Chen, J.Y.; Xiong, S.; Li, Y.L.; Zhang, H.; Liu, Z.; Li, M.M. Respiratory syncytial virus co-opts hypoxia-inducible factor-1α-mediated glycolysis to favor the production of infectious virus. mBio. 2023, 14, e0211023. [CrossRef]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479-480, 180–193. [CrossRef]
- Wu, L.; Yan, Z.; Jiang, Y.; Chen, Y.; Du, J.; Guo, L.; Xu, J.; Luo, Z.; Liu, Y. Metabolic regulation of dendritic cell activation and immune function during inflammation. Front. Immunol. 2023, 14, 1140749. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Weissenberger, S.; Ptacek, R.; Anders, M.; Raboch, J.; Büttiker, P. Viruses and mitochondrial dysfunction in neurodegeneration and cognition: An evolutionary perspective. Cell. Mol. Neurobiol. 2024, 44, 68. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.; Desquiret-Dumas, V.; Nagot, N.; Rapenne, C.; Van de Perre, P.; Reynier, P.; Molès, J.P. Long-term persistence of mitochondrial dysfunctions after viral infections and antiviral therapies: A review of mechanisms involved. J. Med. Virol. 2024, 96, e29886. [Google Scholar] [CrossRef] [PubMed]
- Elesela, S.; Lukacs, N.W. Role of mitochondria in viral infections. Life (Basel) 2021, 11, 232. [Google Scholar] [CrossRef]
- Purandare, N.; Ghosalkar, E.; Grossman, L.I.; Aras, S. Mitochondrial oxidative phosphorylation in viral infections. Viruses 2023, 15, 2380. [Google Scholar] [CrossRef]
- Guarnieri, J.W.; Dybas, J.M.; Fazelinia, H.; Kim, M.S.; Frere, J.; Zhang, Y.; Soto Albrecht, Y.; Murdock, D.G.; Angelin, A.; Singh, L.N.; Weiss, S.L.; Best, S.M.; Lott, M.T.; Zhang, S.; Cope, H.; Zaksas, V.; Saravia-Butler, A.; Meydan, C.; Foox, J.; Mozsary, C.; Bram, Y.; Kidane, Y.; Priebe, W.; Emmett, M.R.; Meller, R.; Demharter, S.; Stentoft-Hansen, V.; Salvatore, M.; Galeano, D.; Enguita, F.J.; Grabham, P.; Trovao, N.S.; Singh, U.; Haltom, J.; Heise, M.T.; Moorman, N.J.; Baxter, V.K.; Madden, E.A.; Taft-Benz, S.A.; Anderson, E.J.; Sanders, W.A.; Dickmander, R.J.; Baylin, S.B.; Wurtele, E.S.; Moraes-Vieir, P.M.; Taylor, D.; Mason, C.E.; Schisler, J.C.; Schwartz, R.; Beheshti, A.; Wallace, D.C. Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts. Sci. Transl. Med. 2023, 15, eabq1533. [Google Scholar] [CrossRef]
- Miller, B.; Silverstein, A.; Flores, M.; Cao, K.; Kumagai, H.; Mehta, H.H.; Yen, K.; Kim, S.J.; Cohen, P. Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Sci. Rep. 2021, 11, 3. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell. Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Mehmood, T.; Nasir, Q.; Younis, I.; Muanprasat, C. Inhibition of mitochondrial dynamics by mitochondrial division inhibitor-1 suppresses cell migration and metastatic markers in colorectal cancer HCT116 cells. J. Exp. Pharmacol. 2025, 17, 143–157. [Google Scholar] [CrossRef]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Dasgupta, A.; Chen, K.H.; Wu, D.; Baid, K.; Mamatis, J.E.; Gonzalez, V.; Read, A.; Bentley, R.E.; Martin, A.Y.; Mewburn, J.D.; Dunham-Snary, K.J.; Evans, G.A.; Levy, G.; Jones, O.; Al-Qazazi, R.; Ring, B.; Alizadeh, E.; Hindmarch, C.C.; Rossi, J.; Lima, P.D.; Falzarano, D.; Banerjee, A.; Colpitts, C.C. SARS-CoV-2 mitochondriopathy in COVID-19 pneumonia exacerbates hypoxemia. Redox Biol. 2022, 58, 102508. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Delgado, I.; López-Pastor, A.R.; González-Jiménez, A.; Ramos-Acosta, C.; Hernández-Garate, Y.; Martínez-Micaelo, N.; Amigó, N.; Espino-Paisán, L.; Anguita, E.; Urcelay, E. Long-term mitochondrial and metabolic impairment in lymphocytes of subjects who recovered after severe COVID-19. Cell. Biol. Toxicol. 2025, 41, 27. [Google Scholar] [CrossRef]
- Tábara, L.C.; Morris, J.L.; Prudent, J. The complex dance of organelles during mitochondrial division. Trends Cell. Biol. 2021, 31, 241–253. [Google Scholar] [CrossRef]
- Duan, X.; Liu, R.; Lan, W.; Liu, S. The essential role of mitochondrial dynamics in viral infections. Int. J. Mol. Sci. 2025, 26, 1955. [Google Scholar] [CrossRef]
- Vazquez, C.; Horner, S.M. MAVS coordination of antiviral innate immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef]
- Qi, Y.; Yin, J.; Xia, W.; Yang, S. Exploring the role of mitochondrial antiviral signaling protein in cardiac diseases. Front. Immunol. 2025, 16, 1540774. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; Wang, W.; Zhao, Y.; Zhao, Y.; Xu, C.; Ma, X.; Gao, Y.; He, H. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell. Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Hsieh, L.L.; Looney, M.; Figueroa, A.; Massaccesi, G.; Stavrakis, G.; Anaya, E.U.; D'Alessio, F.R.; Ordonez, A.A.; Pekosz, A.S.; DeFilippis, V.R.; Karakousis, P.C.; Karaba, A.H.; Cox, AL. Bystander monocytic cells drive infection-independent NLRP3 inflammasome response to SARS-CoV-2. mBio. 2024, 15, e0081024. [Google Scholar] [CrossRef]
- Pandey, K.P.; Zhou, Y. Influenza A virus infection activates NLRP3 inflammasome through trans-Golgi network dispersion. Viruses 2022, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Shin, O.S. Zika virus modulates mitochondrial dynamics, mitophagy, and mitochondria-derived vesicles to facilitate viral replication in trophoblast cells. Front. Immunol. 2023, 14, 1203645. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; Li, X. SARS-CoV-2 causes mitochondrial dysfunction and mitophagy impairment. Front. Microbiol. 2022, 12, 780768. [Google Scholar] [CrossRef] [PubMed]
- Bhutta, M.S.; Gallo, E.S.; Borenstein, R. Multifaceted role of AMPK in viral infections. Cells 2021, 10, 1118. [Google Scholar] [CrossRef]
- Camps, J.; Rodríguez-Gallego, E.; García-Heredia, A.; Triguero, I.; Riera-Borrull, M.; Hernández-Aguilera, A.; Luciano-Mateo, F.; Fernández-Arroyo, S.; Joven, J. Paraoxonases and chemokine (C-C motif) ligand-2 in noncommunicable diseases. Adv. Clin. Chem. 2014, 63, 247–308. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell. Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Mankouri, J.; Tedbury, P.R.; Gretton, S.; Hughes, M.E.; Griffin, S.D.; Dallas, M.L.; Green, K.A.; Hardie, D.G.; Peers, C.; Harris, M. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 11549–11554. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, Y.; Yin, L.; Liang, W.; Zhang, J.; Ma, C.; Zhang, Y.; Liu, B.; Wang, J.; Zhao, W.; Li, M.; Wei, L. The nonstructural protein 1 of respiratory syncytial virus hijacks host mitophagy as a novel mitophagy receptor to evade the type I IFN response in HEp-2 cells. mBio. 2023, 14, e0148023. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the stress response: Viral subversion of the mTOR signaling pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef]
- Vincent, H.A.; Ziehr, B.; Moorman, N.J. Human cytomegalovirus strategies to maintain and promote mRNA translation. Viruses 2016, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Majeed, S.T.; Batool, A.; Majeed, R.; Bhat, N.N.; Zargar, M.A.; Andrabi, K.I. mTORC1 induces eukaryotic translation initiation factor 4E interaction with TOS-S6 kinase 1 and its activation. Cell Cycle 2021, 20, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lei, X.; Zhao, G.; Guo, R.; Cui, N. mTOR in programmed cell death and its therapeutic implications. Cytokine Growth Factor Rev. 2023, 71–72, 66–81. [CrossRef]
- Twu, W.I.; Lee, J.Y.; Kim, H.; Prasad, V.; Cerikan, B.; Haselmann, U.; Tabata, K.; Bartenschlager, R. Contribution of autophagy machinery factors to HCV and SARS-CoV-2 replication organelle formation. Cell. Rep. 2021, 37, 110049. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Dunn, E.F.; Connor, J.H. HijAkt: The PI3K/Akt pathway in virus replication and pathogenesis. Prog. Mol. Biol. Transl. Sci. 2012, 106, 223–250. [Google Scholar] [CrossRef]
- Greene, K. S:; Choi, A.; Yang, N.; Chen, M.; Li, R.; Qiu, Y.; Ezzatpour, S.; Rojas, K.S.; Shen, J.; Wilson, K.F.; Katt, W.P.; Aguilar, H.C.; Lukey, M.J.; Whittaker, G.R.; Cerione, R.A. Glutamine metabolism is essential for coronavirus replication in host cells and in mice. J. Biol. Chem. 2025, 301, 108063. [Google Scholar] [CrossRef]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; Roussel, M.F.; Green, D.R.; Pasa-Tolic, L.; Thomas, P.G. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell. Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, S.; Sun, H.; Shan, J.; Shen, C.; Ji, J.; Lin, L.; Xu, J.; Peng, L.; Dai, C.; Xie, T. Analysis of temporal metabolic rewiring for human respiratory syncytial virus infection by integrating metabolomics and proteomics. Metabolomics 2023, 19, 30. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and immune function, supplementation and clinical translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef]
- Wu, G.; Meininger, C.J.; McNeal, C.J.; Bazer, F.W.; Rhoads, J.M. Role of L-arginine in nitric oxide synthesis and health in humans. Adv. Exp. Med. Biol. 2021, 1332, 167–187. [Google Scholar] [CrossRef] [PubMed]
- Moraes, T.J. Arginase and respiratory viral infections. The Open Nitric Oxide Journal, 2010, 2, 64–68. [CrossRef]
- van den Berg, M.P.; Meurs, H.; Gosens, R. Targeting arginase and nitric oxide metabolism in chronic airway diseases and their co-morbidities. Curr. Opin. Pharmacol. 2018, 40, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.D.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Baiamonte, L.B.; Philbrook, P.; Majumder, R.; Vander Heide, R.S.; Dunkenberger, L.; Thylur, R.P.; Nossaman, B.; Roberts, W.M.; Chapple, A.G.; Wu, J.; Hicks, C.; Collins, J.; Luke, B.; Johnson, R.; Koul, H.K.; Rees, C.A.; Morris, C.R.; Garcia-Diaz, J.; Ochoa, A.C. Severe COVID-19 is characterized by an impaired type I interferon response and elevated levels of arginase producing granulocytic myeloid derived suppressor cells. Front. Immunol. 2021, 12, 695972. [Google Scholar] [CrossRef] [PubMed]
- Churiso, G.; Husen, G.; Bulbula, D.; Abebe, L. Immunity cell responses to RSV and the role of antiviral inhibitors: A systematic review. Infect. Drug Resist. 2022, 15, 7413–7430. [Google Scholar] [CrossRef]
- West, E.E.; Merle, N.S.; Kamiński, M.M.; Palacios, G.; Kumar, D.; Wang, L.; Bibby. J.A.; Overdahl, K.; Jarmusch, A.K.; Freeley, S.; Lee, D.Y.; Thompson, J.W.; Yu, Z.X.; Taylor, N.; Sitbon, M.; Green, D.R.; Bohrer, A.; Mayer-Barber, K.D.; Afzali, B.; Kazemian, M.; Scholl-Buergi, S.; Karall, D.; Huemer, M.; Kemper, C. Loss of CD4+ T cell-intrinsic arginase 1 accelerates Th1 response kinetics and reduces lung pathology during influenza infection. Immunity 2023, 56, 2036–2053.e12. [CrossRef]
- Mounce, B.C.; Olsen, M.E.; Vignuzzi, M.; Connor, J.H. Polyamines and their role in virus infection. Microbiol. Mol. Biol. Rev. 2017, 81, e00029–17. [Google Scholar] [CrossRef]
- Firpo, M.R.; Mastrodomenico, V.; Hawkins, G.M.; Prot, M.; Levillayer, L.; Gallagher, T.; Simon-Loriere, E.; Mounce, B.C. Targeting polyamines inhibits coronavirus infection by reducing cellular attachment and entry. ACS Infect. Dis. 2021, 7, 1423–1432. [Google Scholar] [CrossRef]
- Cruz-Pulido, Y.E.; Mounce, B.C. Good cop, bad cop: Polyamines play both sides in host immunity and viral replication. Semin. Cell. Dev. Biol. 2023, 146, 70–79. [Google Scholar] [CrossRef]
- Lionetto, L.; Ulivieri, M.; Capi, M.; De Bernardini, D.; Fazio, F.; Petrucca, A.; Pomes, L.M.; De Luca, O.; Gentile, G.; Casolla, B.; Curto, M.; Salerno, G.; Schillizzi, S.; Torre, M.S.; Santino, I.; Rocco, M.; Marchetti, P.; Aceti, A.; Ricci, A.; Bonfini, R.; Nicoletti, F.; Simmaco, M.; Borro, M. Increased kynurenine-to-tryptophan ratio in the serum of patients infected with SARS-CoV2: An observational cohort study. Biochim. Biophys. Acta Mol. Basis. Dis. 2021, 1867, 166042. [Google Scholar] [CrossRef]
- Thomas, T.; Stefanoni, D.; Reisz, J.A.; Nemkov, T.; Bertolone, L.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hansen, K.C.; Hod, E.A.; Spitalnik, S.L.; D'Alessandro, A. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5, e140327. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Heydari, M.; Panahi, H.K.S.; Lewin, S.R.; Heng, B.; Brew, B.J.; Guillemin, G.J. The roles of the kynurenine pathway in COVID-19 neuropathogenesis. Infection 2024, 52, 2043–2059. [Google Scholar] [CrossRef]
- Karimi, Z.; Chenari, M.; Rezaie, F.; Karimi, S.; Parhizgari, N.; Mokhtari-Azad, T. Proposed pathway linking respiratory infections with depression. Clin. Psychopharmacol. Neurosci. 2022, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Williams, R.O. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol. Sci. 2023, 44, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Haas de Mello, A.; Morris, D.R.; Jones-Hall, Y.L.; Ivanciuc, T.; Sattler, R.A.; Paessler, S.; Menachery, V.D.; Garofalo, R.P.; Casola, A. SARS-CoV-2 inhibits NRF2-mediated antioxidant responses in airway epithelial cells and in the lung of a murine model of infection. Microbiol. Spectr. 2023, 11, e0037823. [Google Scholar] [CrossRef]
- Fernandes, I.G.; de Brito, C.A.; Dos Reis, V.M.S.; Sato, M.N.; Pereira, N.Z. SARS-CoV-2 and other respiratory viruses: What does oxidative stress have to do with it? Oxid. Med. Cell. Longev. 2020, 2020, 8844280. [Google Scholar] [CrossRef]
- Yang, X.; Liu, X.; Nie, Y.; Zhan, F.; Zhu, B. Oxidative stress and ROS-mediated cellular events in RSV infection: potential protective roles of antioxidants. Virol. J. 2023, 20, 224. [Google Scholar] [CrossRef]
- Jones, J.T.; Qian, X.; van der Velden, J.L.; Chia, S.B.; McMillan, D.H.; Flemer, S.; Hoffman, S.M.; Lahue, K.G.; Schneider, R.W.; Nolin, J.D.; Anathy, V.; van der Vliet, A.; Townsend, D.M.; Tew, K.D.; Janssen-Heininger, Y.M. Glutathione S-transferase pi modulates NF-κB activation and pro-inflammatory responses in lung epithelial cells. Redox Biol. 2016, 8, 375–382. [Google Scholar] [CrossRef]
- Mazzarino, R.C. Targeting future pandemics, a case for de novo purine synthesis and basic research. Front. Immunol. 2021, 12, 694300. [Google Scholar] [CrossRef]
- Qin, C.; Rao, Y.; Yuan, H.; Wang, T.Y.; Zhao, J.; Espinosa, B.; Liu, Y.; Zhang, S.; Savas, A.C.; Liu, Q.; Zarinfar, M.; Rice, S.; Henley, J.; Comai, L.; Graham, N.A.; Chen, C.; Zhang, C.; Feng, P. SARS-CoV-2 couples evasion of inflammatory response to activated nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2122897119. [Google Scholar] [CrossRef]
- Diehl, F.F.; Miettinen, T.P.; Elbashir, R.; Nabel, C.S.; Darnell, A.M.; Do, B.T.; Manalis, S.R.; Lewis, C.A.; Vander Heiden, M.G. Nucleotide imbalance decouples cell growth from cell proliferation. Nat. Cell. Biol. 2022, 24, 1252–1264. [Google Scholar] [CrossRef]
- Sahan, A.Z.; Hazra, T.K.; Das, S. The pivotal role of DNA repair in infection mediated-inflammation and cancer. Front. Microbiol. 2018, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Okesli, A.; Khosla, C.; Bassik, M.C. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr. Opin. Biotechnol. 2017, 48, 127–134. [Google Scholar] [CrossRef]
- Luganini, A.; Boschi, D.; Lolli, M.L.; Gribaudo, G. DHODH inhibitors: What will it take to get them into the clinic as antivirals? Antiviral Res. 2025, 236, 106099. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Rubini, M.; Burns, J.S. The potential of purinergic signaling to thwart viruses including SARS-CoV-2. Front. Immunol. 2022, 13, 904419. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Hernández, L.M.; Ramírez-Noyola, J.A.; Gómez-García, I.A.; Ignacio-Cortés, S.; Zúñiga, J.; Choreño-Parra, J.A. Comparing the cytokine storms of COVID-19 and pandemic influenza. J. Interferon Cytokine Res. 2022, 42, 369–392. [Google Scholar] [CrossRef]
- Ali, E.S.; Ben-Sahra, I. Regulation of nucleotide metabolism in cancers and immune disorders. Trends Cell. Biol. 2023, 33, 950–966. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox biology of respiratory viral infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell. Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef]
- Yang, W.S.; Yi, Y.S.; Kim, D.; Kim, M.H.; Park, J.G.; Kim, E.; Lee, S.Y.; Yoon, K.; Kim, J.H.; Park, J.; Cho, J.Y. Nuclear factor kappa-B- and activator protein-1-mediated immunostimulatory activity of compound K in monocytes and macrophages. J. Ginseng Res. 2017, 41, 298–306. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive oxygen species signaling and oxidative stress: Transcriptional regulation and evolution. Antioxidants (Basel) 2024, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef] [PubMed]
- Naiditch, H.; Betts, M.R.; Larman, H.B.; Levi, M.; Rosenberg, A.Z. Immunologic and inflammatory consequences of SARS-CoV-2 infection and its implications in renal disease. Front. Immunol. 2025, 15, 1376654. [Google Scholar] [CrossRef] [PubMed]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Ziehr, B.K.; MacDonald, J.A. Regulation of NLRPs by reactive oxygen species: A story of crosstalk. Biochim. Biophys. Acta Mol. Cell. Res. 2024, 1871, 119823. [Google Scholar] [CrossRef]
- Kolattukudy, P.E.; Niu, J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ Res. 2012, 110, 174–189. [Google Scholar] [CrossRef]
- Pandey, E.; Nour, A.S.; Harris, E.N. Prominent receptors of liver sinusoidal endothelial cells in liver homeostasis and disease. Front. Physiol. 2020, 11, 873. [Google Scholar] [CrossRef]
- Andersson, U.; Ottestad, W.; Tracey, K.J. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol. Med. 2020, 26, 42. [Google Scholar] [CrossRef]
- Shamilov, R.; Ackley, T.W.; Aneskievich, B.J. Enhanced wound healing -and inflammasome- associated gene expression in TNFAIP3-interacting protein 1-(TNIP1-) deficient HaCaT keratinocytes parallels reduced reepithelialization. Mediators Inflamm. 2020, 2020, 5919150. [Google Scholar] [CrossRef]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma. Emerg. Surg. 2020, 46, 751–775. [Google Scholar] [CrossRef]
- Afrose, S.S.; Junaid, M.; Akter, Y.; Tania, M.; Zheng, M.; Khan, M.A. Targeting kinases with thymoquinone: a molecular approach to cancer therapeutics. Drug Discov. Today 2020, 25, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Dantonio, P.M.; Klein, M.O.; Freire, M.R.V.B.; Araujo, C.N.; Chiacetti, A.C.; Correa, R.G. Exploring major signaling cascades in melanomagenesis: a rationale route for targetted skin cancer therapy. Biosci. Rep. 2018, 38, BSR20180511. [Google Scholar] [CrossRef] [PubMed]
- Slaine, P.D.; Kleer, M.; Duguay, B.A.; Pringle, E.S.; Kadijk, E.; Ying, S. , Balgi, A.; Roberge, M.; McCormick, C.; Khaperskyy, D.A. Thiopurines activate an antiviral unfolded protein response that blocks influenza A virus glycoprotein accumulation. J. Virol. 2021, 95, e00453–21. [Google Scholar] [CrossRef] [PubMed]
- Féral, K.; Jaud, M.; Philippe, C.; Di Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER stress and unfolded protein response in leukemia: Friend, Foe, or Both? Biomolecules 2021, 11, 199. [Google Scholar] [CrossRef]
- Robinson, C.M.; Talty, A.; Logue, S.E.; Mnich, K.; Gorman, A.M.; Samali, A. An emerging role for the unfolded protein response in pancreatic cancer. Cancers (Basel) 2021, 13, 261. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Dymkowska, D. The involvement of autophagy in the maintenance of endothelial homeostasis: The role of mitochondria. Mitochondrion 2021, 57, 131–147. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous deficiency of glutathione as the most likely cause of serious manifestations and death in COVID-19 patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The role of glutathione in protecting against the severe inflammatory response triggered by COVID-19. Antioxidants (Basel) 2020, 9, 624. [Google Scholar] [CrossRef]
- De Angelis, M.; Amatore, D.; Checconi, P.; Zevini, A.; Fraternale, A.; Magnani, M.; Hiscott, J.; De Chiara, G.; Palamara, A.T.; Nencioni, L. Influenza virus down-modulates G6PD expression and activity to induce oxidative stress and promote its replication. Front. Cell. Infect. Microbiol. 2022, 11, 804976. [Google Scholar] [CrossRef]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Hua, L.; Liu, X.; Xiong, H.; Jiang, F.; Zhou, W.; Wang, L.; Xue, G. Superoxide dismutase alterations in COVID-19: implications for disease severity and mortality prediction in the context of omicron variant infection. Front. Immunol. 2024, 15, 1362102. [Google Scholar] [CrossRef] [PubMed]
- Tavassolifar, M.J.; Aghdaei, H.A.; Sadatpour, O.; Maleknia, S.; Fayazzadeh, S.; Mohebbi, S.R.; Montazer, F.; Rabbani, A.; Zali, M.R.; Izad, M.; Meyfour, A. New insights into extracellular and intracellular redox status in COVID-19 patients. Redox Biol. 2023, 59, 102563. [Google Scholar] [CrossRef]
- Choi, A.M.; Knobil, K.; Otterbein, S.L.; Eastman, D.A.; Jacoby, D.B. Oxidant stress responses in influenza virus pneumonia: gene expression and transcription factor activation. Am. J. Physiol. 1996, 271, L383–L391. [Google Scholar] [CrossRef]
- Chen, F.; Chen, L.; Liang, J.; Chen, Z.; Zhang, C.; Zhang, Z.; Yang, J. Potential role of superoxide dismutase 3 (SOD3) in resistance to Influenza A virus infection. Antioxidants (Basel) 2023, 12, 354. [Google Scholar] [CrossRef]
- Hasan Anber, Z.N.; Oied Saleh, B.; Hassan Majed, R. Assessment of oxidative stress parameters in Iraqi male patients with Covid-19; A case control study. Rep. Biochem. Mol. Biol. 2024, 13, 167–173. [Google Scholar] [CrossRef]
- Rodríguez-Tomàs, E.; Iftimie, S.; Castañé, H.; Baiges-Gaya, G.; Hernández-Aguilera, A.; González-Viñas, M.; Castro, A.; Camps, J.; Joven, J. Clinical performance of paraoxonase-1-related variables and novel markers of inflammation in coronavirus disease-19. A machine learning approach. Antioxidants (Basel) 2021, 10, 991. [Google Scholar] [CrossRef]
- Gabaldó, X.; Juanpere, M.; Castañé, H.; Rodríguez-Tomàs, E.; López-Azcona, A.F.; Baiges-Gaya, G.; Castro, L.; Valverde-Díaz, E.; Muñoz-Blázquez, A.; Giménez-Cuenca, L.; Felipo-Balada, L.; Ballester, F.; Pujol, I.; Simó, J.M.; Castro, A.; Iftimie, S.; Camps, J.; Joven, J. Usefulness of the measurement of serum paraoxonase-1 arylesterase activity in the diagnoses of COVID-19. Biomolecules 2022, 12, 879. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, J.R.; Lee, I.C.; Kwon, H.J. Native high-density lipoproteins (HDL) with higher paraoxonase exerts a potent antiviral effect against SARS-CoV-2 (COVID-19), while glycated HDL lost the antiviral activity. Antioxidants (Basel) 2021, 10, 209. [Google Scholar] [CrossRef]
- Thaker, S.K.; Ch'ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, J.; Liang, Q.; Zheng, H.; Ye, Y.; Nan, W.; Zhang, X.; Gao, H.; Li, Y. Metabolomics profile in acute respiratory distress syndrome by nuclear magnetic resonance spectroscopy in patients with community-acquired pneumonia. Respir. Res. 2022, 23, 172. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Jiménez-Franco, A.; García-Pablo, R.; Joven, J.; Arenas, M. Artificial intelligence-driven integration of multi-omics and radiomics: A new hope for precision cancer diagnosis and prognosis. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167841. [Google Scholar] [CrossRef]
- Hu, L.; Liu, J.; Zhang, W.; Wang, T.; Zhang, N.; Lee, Y.H.; Lu, H. Functional metabolomics decipher biochemical functions and associated mechanisms underlie small-molecule metabolism. Mass Spectrom. Rev. 2020, 39, 417––433. [Google Scholar] [CrossRef]
- Fuertes-Martín, R.; Taverner, D.; Vallvé, J.; Paredes, S.; Masana, L.; Correig Blanchar, X. Characterization of 1H NMR plasma glycoproteins as a new strategy to identify inflammatory patterns in rheumatoid arthritis. J. Proteome Res. 2018, 17, 3730–3739. [Google Scholar] [CrossRef]
- Fuertes-Martin, R.; Moncayo, S.; Insenser, M.; Martínez-García, M.A.; Luque-Ramírez, M.; Grau, N.A.; Blanchar, X.C.; Escobar-Morreale, H.F. Glycoprotein A and B height-to-width ratios as obesity-independent novel biomarkers of low-grade chronic inflammation in women with polycystic ovary syndrome (PCOS) J. Proteome Res. 2019, 18, 4038–4045. [Google Scholar] [CrossRef]
- Riera-Borrull, M.; Rodríguez-Gallego, E.; Hernández-Aguilera, A.; Luciano, F.; Ras, R.; Cuyàs, E.; Camps, J.; Segura-Carretero, A.; Menendez, J.A.; Joven, J.; Fernández-Arroyo, S. Exploring the process of energy generation in pathophysiology by targeted metabolomics: Performance of a simple and quantitative method. J. Am. Soc. Mass Spectrom. 2016, 27, 168–177. [Google Scholar] [CrossRef]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; Neuzil, J.; Menendez, J.A. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef]
- Nikolskiy, I.; Siuzdak, G.; Patti, G.J. Discriminating precursors of common fragments for large-scale metabolite profiling by triple quadrupole mass spectrometry. Bioinformatics 2015, 31, 2017–2023. [Google Scholar] [CrossRef]
- Patti, G.J. Separation strategies for untargeted metabolomics. J. Sep. Sci. 2011, 34, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Ivanisevic, J.; Want, E.J. From samples to insights into metabolism: uncovering biologically relevant information in LC-HRMS metabolomics data. Meta 2019, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Strimbu K, Tavel JA. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [CrossRef]
- FDA-NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and other Tools) Resource [Internet]; Silver Spring: Maryland, USA, 2016; Bookshelf ID: NBK326791. [PubMed]
- Pepe, M.S.; Janes, H.; Longton, G.; Leisenring, W.; Newcomb, P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am. J. Epidemiol. 2004, 159, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Castañé, H.; Iftimie, S.; Baiges-Gaya, G.; Rodríguez-Tomàs, E.; Jiménez-Franco, A.; López-Azcona, A.F.; Garrido, P.; Castro, A.; Camps, J.; Joven, J. Machine learning and semi-targeted lipidomics identify distinct serum lipid signatures in hospitalized COVID-19-positive and COVID-19-negative patients. Metabolism 2022, 131, 155197. [Google Scholar] [CrossRef]
- Mai, M.; Tönjes, A.; Kovacs, P.; Stumvoll, M.; Fiedler, G.M.; Leichtle, A.B. Serum levels of acylcarnitines are altered in prediabetic conditions. PLoS One 2013, 8, e82459. [Google Scholar] [CrossRef]
- Ayres, J.S. A metabolic handbook for the COVID-19 pandemic. Nat. Metab. 2020, 2, 572–585. [Google Scholar] [CrossRef]
- Otsubo, C.; Bharathi, S.; Uppala, R.; Ilkayeva, O.R.; Wang, D.; McHugh, K.; Zou, Y.; Wang, J.; Alcorn, J.F.; Zuo, Y.Y.; Hirschey, M.D.; Goetzman, E.S. Long-chain acylcarnitines reduce lung function by inhibiting pulmonary surfactant. J. Biol. Chem. 2015, 290, 23897–23904. [Google Scholar] [CrossRef]
- Wu, D.; Shu, T.; Yang, X.; Song, J.X.; Zhang, M.; Yao, C.; Liu, W.; Huang, M.; Yu, Y.; Yang, Q.; Zhu, T.; Xu, J.; Mu, J.; Wang, Y.; Wang, H.; Tang, T.; Ren, Y.; Wu, Y.; Lin, S.H.; Qiu, Y.; Zhang, D.Y.; Shang, Y.; Zhou, X. Plasma metabolomic and lipidomic alterations associated with COVID-19. Natl. Sci. Rev. 2020, 7, 1157–1168. [Google Scholar] [CrossRef]
- Song, J.W.; Lam, S.M.; Fan, X.; Cao, W.J.; Wang, S.Y.; Tian, H.; Chua, G.H.; Zhang, C.; Meng, F.P.; Xu, Z.; Fu, J.L.; Huang, L.; Xia, P.; Yang, T.; Zhang, S.; Li, B.; Jiang, T.J.; Wang, R.; Wang, Z.; Shi, M.; Zhang, J.Y.; Wang, F.S.; Shui, G. Omics-driven systems interrogation of metabolic dysregulation in COVID-19 pathogenesis. Cell. Metab. 2020, 32, 188–202. [Google Scholar] [CrossRef]
- Barberis, E.; Timo, S.; Amede, E.; Vanella, V.V.; Puricelli, C.; Cappellano, G.; Raineri, D.; Cittone, M.G.; Rizzi, E.; Pedrinelli, A.R.; Vassia, V.; Casciaro, F.G.; Priora, S.; Nerici, I.; Galbiati, A.; Hayden, E.; Falasca, M.; Vaschetto, R.; Sainaghi, P.P.; Dianzani, U.; Rolla, R.; Chiocchetti, A.; Baldanzi, G.; Marengo, E.; Manfredi, M. Large-scale plasma analysis revealed new mechanisms and molecules associated with the host response to SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 8623. [Google Scholar] [CrossRef]
- Fraser, D.D.; Slessarev, M.; Martin, C.M.; Daley, M.; Patel, M.A.; Miller, M.R.; Patterson, E.K.; O'Gorman, D.B.; Gill, S.E.; Wishart, D.S.; Mandal, R.; Cepinskas, G. Metabolomics profiling of critically ill coronavirus disease 2019 patients: identification of diagnostic and prognostic biomarkers. Crit. Care Explor. 2020, 2, e0272. [Google Scholar] [CrossRef] [PubMed]
- Delafiori, J.; Navarro, L.C.; Siciliano, R.F.; de Melo, G.C.; Busanello, E.N.B.; Nicolau, J.C.; Sales, G.M.; de Oliveira, A.N.; Val, F.F.A.; de Oliveira, D.N.; Eguti, A.; Dos Santos, L.A.; Dalçóquio, T.F.; Bertolin, A.J.; Abreu-Netto, R.L.; Salsoso, R.; Baía-da-Silva, D.; Marcondes-Braga, F.G.; Sampaio, V.S.; Judice, C.C.; Costa, F.T.M.; Durán, N.; Perroud, M.W.; Sabino, E.C.; Lacerda, M.V.G.; Reis, L.O.; Fávaro, W.J.; Monteiro, W.M.; Rocha, A.R.; Catharino, R.R. Covid-19 automated diagnosis and risk assessment through metabolomics and machine learning. Anal. Chem. 2021, 93, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Zhang, Z.; Feng, G.; Chen, M.; Wan, Q.; Lin, J.; Wu, L.; Nie, W.; Chen, S. Distinct lipid metabolic dysregulation in asymptomatic COVID-19. iScience 2021, 24, 102974. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.M.; Zhang, C.; Wang, Z.; Ni, Z.; Zhang, S.; Yang, S.; Huang, X.; Mo, L.; Li, J.; Lee, B.; Mei, M.; Huang, L.; Shi, M.; Xu, Z.; Meng, F.P.; Cao, W.J.; Zhou, M.J.; Shi, L.; Chua, G.H.; Li, B.; Cao, J.; Wang, J.; Bao, S.; Wang, Y.; Song, J.W.; Zhang, F.; Wang, F.S.; Shui, G. A multi-omics investigation of the composition and function of extracellular vesicles along the temporal trajectory of COVID-19. Nat. Metab. 2021, 3, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.E.; Burnum-Johnson, K.E.; Wendler, J.P.; Eisfeld, A.J.; Halfmann, P.J.; Watanabe, T.; Sahr, F.; Smith, R.D.; Kawaoka, Y.; Waters, K.M.; Metz, T.O. Plasma lipidome reveals critical illness and recovery from human Ebola virus disease. Proc Natl Acad Sci U S A. 2019, 116, 3919–3928. [Google Scholar] [CrossRef]
- Nguyen, M.; Bourredjem, A.; Piroth, L.; Bouhemad, B.; Jalil, A.; Pallot, G.; Le Guern, N.; Thomas, C.; Pilot, T.; Bergas, V.; Choubley, H.; Quenot, J.P.; Charles, P.E.; Lagrost, L.; Deckert, V.; de Barros, J.P.; Guinot, P.G.; Masson, D.; Binquet, C.; Gautier, T.; Blot, M; Lymphonie study group. High plasma concentration of non-esterified polyunsaturated fatty acids is a specific feature of severe COVID-19 pneumonia. Sci. Rep. 2021, 11, 10824. [CrossRef]
- Bizkarguenaga, M.; Bruzzone, C.; Gil-Redondo, R.; SanJuan, I.; Martin-Ruiz, I.; Barriales, D.; Palacios, A.; Pasco, S.T.; González-Valle, B.; Laín, A.; Herrera, L.; Azkarate, A.; Vesga, M.A.; Eguizabal, C.; Anguita, J.; Embade, N.; Mato, J.M.; Millet, O. Uneven metabolic and lipidomic profiles in recovered COVID-19 patients as investigated by plasma NMR metabolomics. NMR Biomed. 2022, 35, e4637. [Google Scholar] [CrossRef]
- Bruzzone, C.; Bizkarguenaga, M.; Gil-Redondo, R.; Diercks, T.; Arana, E.; García de Vicuña, A.; Seco, M.; Bosch, A.; Palazón, A.; San Juan, I.; Laín, A.; Gil-Martínez, J.; Bernardo-Seisdedos, G.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Embade, N.; Lu, S.; Mato, J.M.; Millet, O. SARS-CoV-2 infection dysregulates the metabolomic and lipidomic profiles of serum. iScience 2020, 23, 101645. [Google Scholar] [CrossRef]
- Iftimie, S.; Gabaldó-Barrios, X., Penadés-Nadal, J.; Canela-Capdevila, M.; Piñana, R.; Jiménez-Franco, A.; López-Azcona, A.F.; Castañé, H.; Cárcel, M.; Camps, J.; Castro, A.; Joven, J. Serum levels of arachidonic acid, interleukin-6, and C-reactive protein as potential indicators of pulmonary viral infections: Comparative analysis of influenza A, respiratory syncytial virus infection, and COVID-19. Viruses 2024, 16, 1065. [CrossRef]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.; Yuen, K.Y. Characterization of the lipidomic profile of human coronavirus-infected cells: Implications for lipid metabolism remodeling upon coronavirus replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang. C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; Ge, W.; Liu, W.; Liang, S.; Chen, H.; Zhang, Y.; Li, J.; Xu, J.; He, Z.; Chen, B.; Wang, J.; Yan, H.; Zheng, Y.; Wang, D.; Zhu, J.; Kong, Z.; Kang, Z.; Liang, X.; Ding, X.; Ruan, G.; Xiang, N.; Cai, X.; Gao, H.; Li, L.; Li, S.; Xiao, Q.; Lu, T.; Zhu, Y.; Liu, H.; Chen, H.; Guo, T. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 2020, 182, 59–72. [CrossRef]
- Torrente-Rodríguez, R.M., Ruiz-Valdepeñas Montiel, V.; Iftimie, S.; Montero-Calle, A.; Pingarrón, J.M.; Castro, A.; Camps, J.; Barderas, R.; Campuzano, S.; Joven, J. Contributing to the management of viral infections through simple immunosensing of the arachidonic acid serum level. Mikrochim. Acta 2024, 191, 369. [CrossRef]
- Amigó, N.; Martínez-Micaelo, N.; Velasco, M.; Casas, M.L.; Correig, X.; Guijarro, C. Identification of a molecular signature associated with covid-19 severity using a comprehensive 1H-NMR serum metabolomics profiling strategy. Atherosclerosis 2024, 395 (Suppl.1), 117627. [Google Scholar] [CrossRef]
- López-Hernández, Y.; Oropeza-Valdez, J.J.; García Lopez, D.A.; Borrego, J.C.; Murgu, M.; Valdez, J.; López, J.A.; Monárrez-Espino, J. Untargeted analysis in post-COVID-19 patients reveals dysregulated lipid pathways two years after recovery. Front. Mol. Biosci. 2023, 10, 1100486. [Google Scholar] [CrossRef]
- Washirasaksiri, C.; Sayabovorn, N.; Ariyakunaphan, P.; Kositamongkol, C.; Chaisathaphol, T.; Sitasuwan, T.; Tinmanee, R.; Auesomwang, C.; Nimitpunya, P.; Woradetsittichai, D.; Chayakulkeeree, M.; Phoompoung, P.; Mayurasakorn, K.; Sookrung, N.; Tungtrongchitr, A.; Wanitphakdeedecha, R.; Muangman, S.; Senawong, S.; Tangjittipokin, W.; Sanpawitayakul, G.; Nopmaneejumruslers, C.; Vamvanij, V.; Phisalprapa, P.; Srivanichakorn, W. Long-term multiple metabolic abnormalities among healthy and high-risk people following nonsevere COVID-19. Sci. Rep. 2023, 13, 14336. [Google Scholar] [CrossRef] [PubMed]
- Kyo, M.; Zhu, Z.; Shibata, R.; Fujiogi, M.; Mansbach, J.M.; Camargo, C.A.; Hasegawa, K. Respiratory virus-specific nasopharyngeal lipidome signatures and severity in infants with bronchiolitis: A prospective multicenter study. J. Infect. Dis. 2023, 228, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Loo, R.L.; Lodge, S.; Kimhofer, T.; Bong, S.H.; Begum, S.; Whiley, L.; Gray, N.; Lindon, J.C.; Nitschke, P.; Lawler, N.G.; Schäfer, H.; Spraul, M.; Richards, T.; Nicholson, J.K.; Holmes, E. Quantitative in-vitro diagnostic NMR spectroscopy for lipoprotein and metabolite measurements in plasma and serum: Recommendations for analytical artifact minimization with special reference to COVID-19/SARS-CoV-2 samples. J. Proteome Res. 2020, 19, 4428–4441. [Google Scholar] [CrossRef] [PubMed]
- Corn, G.; Lund, M.; Andersson, N.W.; Dohlmann, T.L.; Hlatky, M.A.; Wohlfahrt, J.; Melbye, M. Low-density lipoprotein cholesterol response to statins according to comorbidities and co-medications: A population-based study. Am. Heart J. 2024, 274, 102–112. [Google Scholar] [CrossRef]
- Zimodro, J.M.; Mucha, M.; Berthold, H.K.; Gouni-Berthold, I. Lipoprotein metabolism, dyslipidemia, and lipid-lowering therapy in women: A comprehensive review. Pharmaceuticals (Basel) 2024, 17, 913. [Google Scholar] [CrossRef]
- Berta, E.; Zsíros, N.; Bodor, M.; Balogh, I.; Lőrincz, H.; Paragh, G.; Harangi, M. Clinical aspects of genetic and non-genetic cardiovascular risk factors in familial hypercholesterolemia. Genes (Basel) 2022, 13, 1158. [Google Scholar] [CrossRef]
- Baiges-Gaya, G.; Iftimie, S.; Castañé, H.; Rodríguez-Tomàs, E.; Jiménez-Franco, A.; López-Azcona, A.F.; Castro, A.; Camps, J.; Joven, J. Combining semi-targeted metabolomics and machine learning to identify metabolic alterations in the serum and urine of hospitalized patients with COVID-19. Biomolecules 2023, 13, 163. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, A.H.; Song, Q.; Fang, H.; Liu, X.Y.; Su, J.; Yang, L.; Yu, M.D.; Wang, X.J. Functional metabolomics discover pentose and glucuronate interconversion pathways as promising targets for Yang Huang syndrome treatment with Yinchenhao Tang. RSC Adv. 2018, 8, 36831–36839. [Google Scholar] [CrossRef]
- Chen, S.; Niu, C.; Lv, W. Multi-omics insights reveal the remodeling of gut mycobiome with P. gingivalis. Front. Cell. Infect. Microbiol. 2022, 12, 937725. [Google Scholar] [CrossRef]
- Lu, X.; Liu, T.; Zhou, J.; Liu, J.; Yuan, Z.; Guo, L. Subgingival microbiome in periodontitis and type 2 diabetes mellitus: An exploratory study using metagenomic sequencing. J. Periodontal Implant. Sci. 2022, 52, 282–297. [Google Scholar] [CrossRef]
- Xiong, H.; Li, N.; Zhao, L.; Li, Z.; Yu, Y.; Cui, X.; Liu, Q.; Zhao, C. Integrated serum pharmacochemistry, metabolomics, and network pharmacology to reveal the material basis and mechanism of Danggui Shaoyao San in the treatment of primary dysmenorrhea. Front. Pharmacol. 2022, 13, 942955. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, K.; Zeng, M.; Qiao, B.; Zhou, B. Serum metabolomics analysis of the anti-inflammatory effects of gallic acid on rats with acute inflammation. Front. Pharmacol. 2022, 13, 830439. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, D.; Gao, X.; Wang, X.; Zhang, L. 2’- and 3’-ribose modifications of nucleotide analogues establish the structural basis to inhibit the viral replication of SARS-CoV-2. J. Phys. Chem. Lett. 2022, 13, 4111–4118. [Google Scholar] [CrossRef]
- Guo, X.; Wu, S.; Li, N.; Lin, Q.; Liu, L.; Liang, H.; Niu, Y.; Huang, Z.; Fu, X. Accelerated metabolite levels of aerobic glycolysis and the pentose phosphate pathway are required for efficient replication of infectious spleen and kidney necrosis virus in Chinese perch brain cells. Biomolecules 2019, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kaminiski, R.; Deshmane, S.; Langford, D.; Khalili, K.; Amini, S.; Datta, P.K. Role of hexokinase-1 in the survival of HIV-1-infected macrophages. Cell Cycle 2015, 14, 980–989. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; Keller, M.A.; Breitenbach, M.; Brindle, K.M.; Rabinowitz, J.D.; Ralser, M. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Chen, I.T.; Aoki, T.; Huang, Y.T.; Hirono, I.; Chen, T.C.; Huang, J.Y. White spot Syndrome virus induces metabolic changes resembling the Warburg effect in shrimp hemocytes in the early stage of infection. J. Virol. 2011, 85, 12919–12928. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Soto, M.E.; Guarner-Lans, V..; Manzano-Pech, L.; Soria-Castro, E. The possible role of glucose-6-phosphate dehydrogenase in the SARS-CoV-2 infection. Cells 2022, 11, 1982. [CrossRef]
- Bojkova, D.; Costa, R.; Reus, P.; Bechtel, M.; Jaboreck, M.C.; Olmer, R.; Martin, U.; Ciesek, S.; Michaelis, M.; Cinatl, J., Jr. Targeting the pentose phosphate pathway for SARS-CoV-2 therapy. Metabolites 2021, 11, 699. [Google Scholar] [CrossRef]
- Isaacs, C.E.; Kim, K.S.; Thormar, H. Inactivation of enveloped viruses in human bodily fluids by purified lipids. Ann. N. Y. Acad. Sci. 1994, 724, 457–464. [Google Scholar] [CrossRef]
- Nefedova, E.; Koptev, V.; Bobikova, A.S; Cherepushkina, V.; Mironova, T.; Afonyushkin, V.; Shkil, N.; Donchenko, N.; Kozlova, Y.; Sigareva, N.; Davidova, N.; Bogdanchikova, N.; Pestryakov, A.; Toledano-Magaña, Y. The infectious bronchitis coronavirus pneumonia model presenting a novel insight for the SARS-CoV-2 dissemination route. Vet. Sci. 2021, 8, 239. [Google Scholar] [CrossRef]
- Thormar, H.; Isaacs, C.E.; Brown, H.R.; Barshatzky, M.R.; Pessolano, T. Inactivation of enveloped viruses and killing of cells by fatty acids and monoglycerides. Antimicrob. Agents Chemother. 1987, 31, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Cheudjeu, A. Correlation of D-xylose with severity and morbidity-related factors of COVID-19 and possible therapeutic use of D-xylose and antibiotics for COVID-19. Life Sci. 2020, 260, 118335. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.S.; Ad Souza, M.; Raposo, N.R.B.; Ferreira, A.P.; Silva, S.S. Xylitol inhibits J774A.1 macrophage adhesion in vitro. Braz. Arch. Biol. Technol. 2011, 54, 1211–1216. [Google Scholar] [CrossRef]
- Xu, M.L.; Wi, G.; Kim, H.J.; Kim, H.J. Ameliorating effect of dietary xylitol on human respiratory syncytial virus (hRSV) infection. Biol. Pharm. Bull. 2016, 39, 540–546. [Google Scholar] [CrossRef]
- Yin, S.Y.; Kim, H.J.; Kim, H.J. Protective effect of dietary xylitol on influenza A virus infection. PLoS ONE 2014, 9, e84633. [Google Scholar] [CrossRef]
- Anand, S.; Mande, S.S. Diet, microbiota and gut-lung connection. Front. Microbiol. 2018, 9, 2147. [Google Scholar] [CrossRef]
- Shukla, S.D.; Budden, K.F.; Neal, R.; Hansbro, P.M. Microbiome effects on immunity, health and disease in the lung. Clin. Transl. Immunol. 2017, 6, e133. [Google Scholar] [CrossRef]
- Russell, S.L.; Gold, M.J.; Willing, B.P.; Thorson, L.; Mcnagny, K.M.; Finlay, B.B. Perinatal antibiotic treatment affects murine microbiota, immune responses and allergic asthma. Gut Microbes 2013, 4, 158–164. [Google Scholar] [CrossRef]
- Looft, T.; Allen, H.K. Collateral effects of antibiotics on mammalian gut microbiomes. Gut Microbes 2012, 3, 463–467. [Google Scholar] [CrossRef]
- Xie, J.; Cho, H.; Lin, B.M.; Pillai, M.; Heimisdottir, L.H.; Bandyopadhyay, D.; Zou, F.; Roach, J.; Divaris, K.; Wu, D. Improved metabolite prediction using microbiome data-based elastic net models. Front. Cell. Infect. Microbiol. 2021, 11, 734416. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, Y.; He, W.; Tian, Z.; Lin, J.; Liu, Z.; Li, Y.; Chen, M.; Han, S.; Liang, J.; Shi, Y.; Wang, X.; Zhou, L.; Cao, Y.; Liu, J.; Wu, K. Gut microbiota and metabolite changes in patients with ulcerative colitis and Clostridioides difficile infection. Front. Microbiol. 2022, 13, 802823. [Google Scholar] [CrossRef] [PubMed]
- Colonetti, K.; de Carvalho, E.L.; Rangel, D.L.; Pinto, P.M.; Roesch, L.F.W.; Pinheiro, F.C.; Schwartz, I.V.D. Are the bacteria and their metabolites contributing for gut inflammation on GSD-Ia patients? Metabolites 2022, 12, 873. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Ma, T.; Xu, N.; Jin, H.; Zhao, F.; Kwok, L.Y.; Zhang, H.; Zhang, S.; Sun, Z. Adjunctive probiotics alleviates asthmatic symptoms via modulating the gut microbiome and serum metabolome. Microbiol. Spectr. 2021, 9, e0085921. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Hannou, S.A.; Wang, Y.; Astapova, I.; Sargsyan, A.; Monn, R.; Thiriveedi, V.; Li, D.; McCann, J.R.; Rawls, J.F.; Roper, J.; Zhang, G.F.; Herman, M.A. . The intestine is a major contributor to circulating succinate in mice. FASEB J. 2022, 36, e22546. [Google Scholar] [CrossRef]
- Nagata, N.; Takeuchi, T.; Masuoka, H.; Aoki, R.; Ishikane, M.; Iwamoto, N.; Sugiyama, M.; Suda, W.; Nakanishi, Y.; Terada-Hirashima, J.; Kimura, M.; Nishijima, T.; Inooka, H.; Miyoshi-Akiyama, T.; Kojima, Y.; Shimokawa, C.; Hisaeda, H.; Zhang, F.; Yeoh, Y.K.; Ng, S.C.; Uemura, N.; Itoi, T.; Mizokami, M.; Kawai, T.; Sugiyama, H.; Ohmagari, N.; Ohno, H. Human gut microbiota and its metabolites impact immune responses in COVID-19 and its complications. Gastroenterology 2023, 164, 272–288. [Google Scholar] [CrossRef]
- Liao, J.; Li, Q.; Lei, C.; Yu, W.; Deng, J.; Guo, J.; Han, Q.; Hu, L.; Li, Y.; Pan, J.; Zhang, H.; Chang, Y.F.; Tang, Z. Toxic effects of copper on the jejunum and colon of pigs: Mechanisms related to gut barrier dysfunction and inflammation influenced by the gut microbiota. Food Funct. 2021, 12, 9642–9657. [Google Scholar] [CrossRef]
- Yu, W.; Shang, J.; Guo, R.; Zhang, F.; Zhang, W.; Zhang, Y.; Wu, F.; Ren, H.; Liu, C.; Xiao, J.; Zhao, Z. The gut microbiome in differential diagnosis of diabetic kidney disease and membranous nephropathy. Ren. Fail. 2020, 42, 1100–1110. [Google Scholar] [CrossRef]
- Yin, J.; Li, Y.; Han, H.; Liu, Z.; Zeng, X.; Li, T.; Yin, Y. Long-term effects of lysine concentration on growth performance, intestinal microbiome, and metabolic profiles in a pig model. Food Funct. 2018, 9, 4153–4163. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, W.; Zhang, J.; Zhang, J.; Zhang, H.; Zhu, Y.; Meng, X.; Yi, Z.; Wang, R. Influenza A virus (H1N1) infection induces glycolysis to facilitate viral replication. Virol. Sin. 2021, 36, 1532–1542. [Google Scholar] [CrossRef]
- Martín-Vicente, M.; González-Riaño, C.; Barbas, C.; Jiménez-Sousa, M.Á.; Brochado-Kith, O.; Resino, S.; Martínez, I. Metabolic changes during respiratory syncytial virus infection of epithelial cells. PLoS One 2020, 15, e0230844. [Google Scholar] [CrossRef]
- Fratta Pasini, A.M.; Stranieri, C.; Girelli, D.; Busti, F.; Cominacini, L. Is ferroptosis a key component of the process leading to multiorgan damage in COVID-19? Antioxidants (Basel) 2021, 10, 1677. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Olive, L.; Marx, W.; O'Neil, A.; Athan, E.; Carvalho, A.F.; Maes, M.; Walder, K.; Berk, M. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 258, 118166. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses. 2020, 143, 110102. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell. Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Ammer, E.; Nietzsche, S.; Rien, C.; Kühnl, A.; Mader, T.; Heller, R.; Sauerbrei, A.; Henke, A. The anti-obesity drug orlistat reveals anti-viral activity. Med. Microbiol. Immunol. 2015, 204, 635–645. [Google Scholar] [CrossRef]
- Kow, C.S.; Hasan, S.S. Meta-analysis of effect of statins in patients with COVID-19. Am. J. Cardiol. 2020, 134, 153–155. [Google Scholar] [CrossRef]
- Diaz-Arocutipa, C.; Melgar-Talavera, B.; Alvarado-Yarasca, Á.; Saravia-Bartra, M.M.; Cazorla, P.; Belzusarri, I.; Hernandez, A.V. Statins reduce mortality in patients with COVID-19: an updated meta-analysis of 147 824 patients. Int. J. Infect. Dis. 2021, 110, 374–381. [Google Scholar] [CrossRef]
- Onorato, D.; Pucci, M.; Carpene, G.; Henry, B.M.; Sanchis-Gomar, F.; Lippi, G. Protective effects of statins administration in European and North American patients infected with COVID-19: A meta-analysis. Semin. Thromb. Hemost. 2021, 47, 392–399. [Google Scholar] [CrossRef]
- Florêncio de Mesquita, C.; Rivera, A.; Araújo, B.; Durães, V.L.; Queiroz, I.; Carvalho, V.H.; Haque, T.; Bes, T.M. Adjunctive statin therapy in patients with Covid-19: A systematic review and meta-analysis of randomized controlled trials. Am. J. Med. 2024, 137, 966–973.e11. [CrossRef]
- Arnardottir, H.; Pawelzik, S.C.; Öhlund Wistbacka. U.; Artiach, G.; Hofmann, R.; Reinholdsson, I.; Braunschweig, F.; Tornvall, P.; Religa, D.; Bäck, M. Stimulating the resolution of inflammation through omega-3 polyunsaturated fatty acids in COVID-19: Rationale for the COVID-Omega-F trial. Front. Physiol. 2021, 11, 624657. [CrossRef]
- Huang, Z.; Chavda, V.P.; Vora, L.K.; Gajjar, N.; Apostolopoulos, V.; Shah, N.; Chen, Z.S. 2-deoxy-D-glucose and its derivatives for the COVID-19 treatment: An update. Front. Pharmacol. 2022, 13, 899633. [Google Scholar] [CrossRef]
- Bhatt, A.N.; Shenoy, S.; Munjal, S.; Chinnadurai, V.; Agarwal, A.; Vinoth Kumar, A.; Shanavas, A.; Kanwar, R.; Chandna, S. 2-deoxy-D-glucose as an adjunct to standard of care in the medical management of COVID-19: a proof-of-concept and dose-ranging randomised phase II clinical trial. BMC Infect. Dis. 2022, 22, 669. [Google Scholar] [CrossRef]
- Verma, A.; Adhikary, A.; Woloschak, G.; Dwarakanath, B.S.; Papineni, R.V.L. A combinatorial approach of a polypharmacological adjuvant 2-deoxy-D-glucose with low dose radiation therapy to quell the cytokine storm in COVID-19 management. Int. J. Radiat. Biol. 2020, 96, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Sandepogu, T.S.; Dara, C.; Mallamgunta, S.; Jogi, S.; Sree Podila, K.; Chandrasekhar, J.; N, V.; Sivakumar, S. Role of 2-deoxy-D-glucose in enhancing the efficacy of standard of care for moderate to severe COVID-19: A comparative analysis of clinical outcomes. Cureus 2024, 16, e73993. [Google Scholar] [CrossRef] [PubMed]
- Ergashev, A.; Shi, F.; Liu, Z.; Pan, Z.; Xie, H.; Kong, L.; Wu, L.; Sun, H.; Jin, Y.; Kong, H.; Geng, D.; Ibrohimov, A.; Obeng, E.; Wang, Y.; Ma, F.; Chen, G.; Zhang, T. KAN0438757, a novel PFKFB3 inhibitor, prevent the progression of severe acute pancreatitis via the Nrf2/HO-1 pathway in infiltrated macrophage. Free Radic. Biol. Med. 2024, 10, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Ye, Z.W.; Chu, N. Host PFKFB3-dependent glycolytic reprogramming as a broad-spectrum antiviral strategy. Open Forum Infect. Dis. 2023, 10 Suppl. 2, ofad500.976.
- Klarer, A.C.; O'Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014, 2, 2. [Google Scholar] [CrossRef]
- Clem, B.F.; O'Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A. 2nd.; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; Telang, S.; Chesney, J. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [CrossRef]
- Boyd, S.; Brookfield, J.L.; Critchlow, S.E.; Cumming, I.A.; Curtis, N.J.; Debreczeni, J.; Degorce, S.L.; Donald, C.; Evans, N.J.; Groombridge, S.; Hopcroft, P.; Jones, N.P.; Kettle, J.G.; Lamont, S.; Lewis, H.J.; MacFaull, P.; McLoughlin, S.B.; Rigoreau, L.J.; Smith, J.M.; St-Gallay, S.; Stock, J.K.; Turnbull, A.P.; Wheatley, E.R.; Winter, J.; Wingfield, J. Structure-based design of potent and selective inhibitors of the metabolic kinase PFKFB3. J. Med. Chem. 2015, 58, 3611–3625. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, Y.; Zhu, Z.; Wen, W.; Li, X. Mitofusin-mediated mitochondrial fusion inhibits Pseudorabies virus infection in porcine cells. Vet. Sci. 2025, 12, 368. [Google Scholar] [CrossRef]
- Gregorczyk-Zboroch, K.; Szulc-Dąbrowska, L.; Pruchniak, P.; Gieryńska, M.; Mielcarska, M.B.; Biernacka, Z.; Wyżewski, Z.; Lasocka, I.; Świtlik, W.; Szepietowska, A.; Kukier, P.; Kwiecień-Dębska, A.; Kłęk, J. Modifications of mitochondrial network morphology affect the MAVS-dependent immune response in L929 murine fibroblasts during Ectromelia virus infection. Pathogens 2024, 13, 717. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. COVID-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Arenas-de Larriva, A.P.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 supplementation for the reduction of oxidative stress: Clinical implications in the treatment of chronic diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gutierrez-Mariscal, F.M.; Yubero-Serrano, E.M.; Villalba, J.M.; Lopez-Miranda, J. Coenzyme Q10: From bench to clinic in aging diseases, a translational review. Crit. Rev. Food Sci. Nutr. 2019, 59, 2240–2257. [Google Scholar] [CrossRef]
- Shikama, Y.; Otsuka, K.; Shikama, Y.; Furukawa, M.; Ishimaru, N.; Matsushita, K. Involvement of metformin and aging in salivary expression of ACE2 and TMPRSS2. Biofactors 2025, 51, e2154. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, S.; Sun, H.; Zhou, L.; Wang, H.; Li, Y.; Gilson, E.; Lu, Y.; Hu, L.; Ye, J. Metformin alleviates inflammatory response and severity rate of COVID-19 infection in elderly individuals. Sci. Rep. 2025, 15, 11340. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Hernandez-Mijares, A.; Garcia-Malpartida, K.; Bañuls, C.; Bellod, L.; Victor, V.M. Mitochondria-targeted antioxidant peptides. Curr. Pharm. Des. 2010, 16, 3124–3131. [Google Scholar] [CrossRef]
- Gasanoff, E.S.; Yaguzhinsky, L. .; Garab, G. Cardiolipin, non-bilayer structures and mitochondrial bioenergetics: Relevance to cardiovascular disease. Cells 2021, 10, 1721. [Google Scholar] [CrossRef]
- Ding, X.W.; Robinson, M.; Li, R.; Aldhowayan, H.; Geetha, T.; Babu, J.R. Mitochondrial dysfunction and beneficial effects of mitochondria-targeted small peptide SS-31 in Diabetes Mellitus and Alzheimer's disease. Pharmacol. Res. 2021, 171, 105783. [Google Scholar] [CrossRef]
- Li, X.; Ma, L.; Fu, P. The mitochondrion-targeted antioxidants in kidney disease. Curr. Med. Chem. 2021, 28, 4190–4206. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer's disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef]
- Jantz-Naeem, N.; Guvencli, N.; Böttcher-Loschinski, R.; Böttcher, M.; Mougiakakos, D.; Kahlfuss, S. Metabolic T-cell phenotypes: from bioenergetics to function. Am. J. Physiol. Cell Physiol. 2025, 328, C1062–C1075. [Google Scholar] [CrossRef]
- Official Study Title: NIRVANA: NIcotinamide Riboside in SARSCoV-2 pAtients for reNAl protection. Available online: https://cdn.clinicaltrials.gov/large-docs/16/NCT04818216/Prot_SAP_002.pdf (accessed on 15 May 2025).
- Naidu, A.S.; Wang, C.K.; Rao, P.; Mancini, F.; Clemens, R.A.; Wirakartakusumah, A.; Chiu, H.F.; Yen, C.H.; Porretta, S.; Mathai, I.; Naidu, S.A.G. Precision nutrition to reset virus-induced human metabolic reprogramming and dysregulation (HMRD) in long-COVID. NPJ Sci. Food. 2024, 8, 19. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Aretha, D.; Komninos, D.; Dimitropoulou, D.; Lagadinou, M.; Leonidou, L.; Oikonomou, I.; Mouzaki, A.; Marangos, M. N-acetyl-cysteine reduces the risk for mechanical ventilation and mortality in patients with COVID-19 pneumonia: a two-center retrospective cohort study. Infect. Dis. (Lond). 2021, 53, 847–854. [Google Scholar] [CrossRef]
- Ibrahim, H.; Perl, A.; Smith, D.; Lewis, T.; Kon, Z.; Goldenberg, R.; Yarta, K.; Staniloae, C.; Williams, M. Therapeutic blockade of inflammation in severe COVID-19 infection with intravenous N-acetylcysteine. Clin. Immunol. 2020, 219, 108544. [Google Scholar] [CrossRef]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.W.; Cinatl, J. Jr. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem. Pharmacol. 2010, 79, 413–420. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Grassi, C.; Carati, L. Attenuation of influenza-like symptomatology and improvement of cell-mediated immunity with long-term N-acetylcysteine treatment. Eur. Respir. J. 1997, 10, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.Y.; Ye, J.J.; Zhang, D.W.; Hu, L.; Wu, H.M.; Fei, G.H. Melatonin rescues Influenza A virus-induced cellular energy exhaustion via OMA1-OPA1-S in acute exacerbation of COPD. J. Pineal Res. 2024, 76, e12991. [Google Scholar] [CrossRef]
- Xu, M.M.; Kang, J.Y.; Wang, Q.Y.; Zuo, X.; Tan, Y.Y.; Wei, Y.Y.; Zhang, D.W.; Zhang, L.; Wu, H.M.; Fei, G.H. Melatonin improves influenza virus infection-induced acute exacerbation of COPD by suppressing macrophage M1 polarization and apoptosis. Respir. Res. 2024, 25, 186. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, C.; Liu, X.; Tang, W.; Gui, H.; Sun, T.; Xu, D.; He, M.; Han, M.; Qiu, H.; Chen, M.; Huang, S. Melatonin suppresses TLR4-mediated RSV infection in the central nervous cells by inhibiting NLRP3 inflammasome formation and autophagy. J. Cell. Mol. Med. 2024, 28, e18338. [Google Scholar] [CrossRef]
- Qin, J.; Wang, G.; Han, D. Benefits of melatonin on mortality in severe-to-critical COVID-19 patients: A systematic review and meta-analysis of randomized controlled trials. Clinics (Sao Paulo) 2025, 80, 100638. [CrossRef]
- Kakad, U.U.; Khopkar-Kale, P.S.; Tripathy, S.P.; Bhawalkar, J.S. Potential of melatonin as a treatment option for long COVID: A call for research. Br. J. Clin. Pharmacol. 2025, 91, 493–494. [Google Scholar] [CrossRef]
- Tirkan, A.; Eskandari, D.; Roham, M.; Aloosh, O.; Ramim, T.; Afshar, H. Investigating the effectiveness of melatonin in the treatment of critically ill patients with COVID-19 hospitalized in the Intensive Care Unit: A double-blind randomized clinical trial. Med. J. Islam. Repub. Iran 2024, 38, 41. [Google Scholar] [CrossRef]
- Bahmyari, R.; Zare, M.; Sharma, R.; Agarwal, A.; Halvaei, I. The efficacy of antioxidants in sperm parameters and production of reactive oxygen species levels during the freeze-thaw process: A systematic review and meta-analysis. Andrologia 2020, 52, e13514. [Google Scholar] [CrossRef]
- Maio, N.; Cherry, S.; Schultz, D.C.; Hurst, B.L.; Linehan, W.M.; Rouault, T.A. TEMPOL inhibits SARS-CoV-2 replication and development of lung disease in the Syrian hamster model. iScience 2022, 25, 105074. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; Duan, Y.; Yu, J.; Wang, L.; Yang, K.; Liu, F.; Jiang, R.; Yang, X.; You, T.; Liu, X.; Yang, X.; Bai, F.; Liu, H.; Liu, X.; Guddat, L.W.; Xu, W.; Xiao, G.; Qin, C.; Shi, Z.; Jiang, H.; Rao, Z.; Yang, H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Gouédard, C.; Barouki, R.; Morel, Y. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism. Mol. Cell. Biol. 2004, 24, 5209–5222. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Marsillach, J.; Joven, J. Pharmacological and lifestyle factors modulating serum paraoxonase-1 activity. Mini Rev. Med. Chem. 2009, 9, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Cicin, I.; Plimack, E.R.; Gurney, H.; Leibowitz, R.; Alekseev, B.Y.; Parnis, F.X.; Peer, A.; Necchi, A.; Bellmunt, J.; Nishiyam, H.; Clark, J.; Munteanu, M.; Kataria, R.; Jia, C.; Powles, T.; Sternberg, C.N. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab for advanced urothelial carcinoma: results from the randomized phase III ECHO-303/KEYNOTE-698 study. BMC Cancer 2024, 23 (Suppl 1), 1256. [Google Scholar] [CrossRef]
- Lara, P.N. Jr.; Villanueva, L.; Ibanez, C.; Erman, M.; Lee, J.L.; Heinrich, D.; Lipatov, O.N.; Gedye, C.; Gokmen, E.; Acevedo, A.; Semenov, A.; Park, S.H.; Gafanov, R.A.; Kose, F.; Jones, M.; Du, X.; Munteanu, M.; Perini, R.; Choueiri, T.K.; Motzer, R.J. A randomized, open-label, phase 3 trial of pembrolizumab plus epacadostat versus sunitinib or pazopanib as first-line treatment for metastatic renal cell carcinoma (KEYNOTE-679/ECHO-302). BMC Cancer 2024, 23 (Suppl 1), 1253. [Google Scholar] [CrossRef]
- Chen, F.; Zhao, D.; Huang, Y.; Wen, X.; Feng, S. Synergetic impact of combined navoximod with cisplatin mitigates chemo-immune resistance via blockading IDO1+ CAFs-secreted Kyn/AhR/IL-6 and pol ζ-prevented CIN in human oral squamous cell carcinoma. Life Sci. 2023, 335, 122239. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J. Implications of the kynurenine pathway and quinolinic acid in Alzheimer's disease. Redox Rep. 2002, 7, 199–206. [Google Scholar] [CrossRef]
- Låg, M.; Skuland, T.; Ballangby, J.; Grytting, V.S.; Jørgensen, R.B.; Snilsberg, B.; Øvrevik, J.; Holme, J.A.; Refsnes, M. Mechanisms involved in pro-inflammatory responses to traffic-derived particulate matter (PM) in THP-1 macrophages compared to HBEC3-KT bronchial epithelial cells. Toxicology 2025, 516, 154174. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; He, Y.; Chopra, A.; Chieng, H.C.; Tiwari, A.; Judson, M.A.; Paulson, B.; Brademan, D.R.; Zhu, Y.; Serrano, L.R.; Linke, V.; Drake, L.A.; Adam, A.P.; Schwartz, B.S.; Singer, H.A.; Swanson, S.; Mosher, D.F.; Stewart, R.; Coon, J.J.; Jaitovich, A. Large-scale multi-omic analysis of COVID-19 severity. Cell. Syst. 2021, 12, 23–40.e7. [CrossRef]
- Grimes, J.M.; Khan, S.; Badeaux, M.; Rao, R.M.; Rowlinson, S.W.; Carvajal, R.D. Arginine depletion as a therapeutic approach for patients with COVID-19. Int. J. Infect. Dis. 2021, 102, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Greene, K.S.; Choi, A.; Chen, M.; Yang, N.; Li, R.; Qiu, Y.; Lukey, M.J.; Rojas, K.S.; Shen, J.; Wilson, K.F.; Katt, W.P.; Whittaker, G.R.; Cerione, R.A. Inhibiting glutamine metabolism blocks coronavirus replication in mammalian cells. bioRxiv [Preprint], 2023, 28, 2023.09.27.559756. [CrossRef]
- Rodríguez-Vera, D.; Salazar, J.R.; Soriano-Ursúa, M.A.; Guzmán-Pérez, J.; Vergara-Castañeda, A.; Muñoz-Durán, H.; Ramírez-Velez, G.L.; Vivar-Sierra, A.; Naranjo-Navarro, C.R.; Meza-Meneses, P.A.; Loza-Mejía, M.A.; Pinto-Almazán, R. Effectiveness of omega-3 fatty acid supplementation in improving the metabolic and inflammatory profiles of Mexican adults hospitalized with COVID-19. Diseases 2024, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Safaei Ardestani, S.Z.; Rahideh, S.T. The effect of omega-3 fatty acid supplementation on clinical and biochemical parameters of critically ill patients with COVID-19: a randomized clinical trial. J. Transl. Med. 2022, 20, 32. [Google Scholar] [CrossRef]
- Wang, K.; Khoramjoo, M.; Srinivasan, K.; Gordon, P.M.K.; Mandal, R.; Jackson, D.; Sligl, W.; Grant, M.B.; Penninger, J.M.; Borchers, C.H.; Wishart, D.S.; Prasad, V.; Oudit, G.Y. Sequential multi-omics analysis identifies clinical phenotypes and predictive biomarkers for long COVID. Cell. Rep. Med. 2023, 4, 101254. [Google Scholar] [CrossRef]
- Pinero, S.; Li, X.; Liu, L.; Li, J.; Lee, S.H.; Winter, M.; Nguyen, T.; Zhang, J.; Le, T.D. Integrative multi-omics framework for causal gene discovery in long COVID. medRxiv, 2025, 2025.02.09.25321751. [CrossRef]
- Centers for Disease Control and Prevention. Influenza severity assessment and estimated influenza illnesses, medical visits, hospitalizations, and deaths that occurred and those that were prevented by vaccination in the United States – 2023-2024 influenza season. https://www.cdc.gov/flu/whats-new/flu-summary-addendum-2023-2024.html (accessed 16 May 2025).
- Havers, F.P.; Whitaker, M.; Melgar, M.; Chatwani, B.; Chai, S.J.; Alden, N.B.; Meek, J.; Openo, K.P.; Ryan, P.A.; Kim, S.; Lynfield, R.; Shaw, Y.P.; Barney, G.; Tesini, B.L.; Sutton, M.; Talbot, H.K.; Olsen, K.P.; Patton, M.E.; RSV-NET Surveillance Team. Characteristics and outcomes among adults aged ≥60 years hospitalized with laboratory-confirmed respiratory syncytial virus - RSV-NET, 12 States, July 2022-June 2023. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 1075–1082. [CrossRef]
Figure 1.
Conceptual illustration of lipid raft organization within cellular membranes. Cholesterol (yellow) and phospholipids (blue and brown) are present in both leaflets of the bilayer, while sphingolipids (violet) are predominantly localized in the outer leaflet. Lipids in raft regions typically feature long, saturated acyl chains (violet and brown), in contrast to the shorter and more unsaturated acyl chains (blue) found in non-raft areas. Raft microdomains are enriched in proteins with dual acylation (green) and glycosylphosphatidylinositol (GPI) anchors (brown), whereas transmembrane (blue) and prenylated (green) proteins are more commonly located in non-raft regions. GPLs: Glycerophospholipids; GSLs: Glycerosphingolipids; SM: Sphingomyelin. Reproduced from [11]. Copyright by the authors under a CC BY 4.0 license.
Figure 1.
Conceptual illustration of lipid raft organization within cellular membranes. Cholesterol (yellow) and phospholipids (blue and brown) are present in both leaflets of the bilayer, while sphingolipids (violet) are predominantly localized in the outer leaflet. Lipids in raft regions typically feature long, saturated acyl chains (violet and brown), in contrast to the shorter and more unsaturated acyl chains (blue) found in non-raft areas. Raft microdomains are enriched in proteins with dual acylation (green) and glycosylphosphatidylinositol (GPI) anchors (brown), whereas transmembrane (blue) and prenylated (green) proteins are more commonly located in non-raft regions. GPLs: Glycerophospholipids; GSLs: Glycerosphingolipids; SM: Sphingomyelin. Reproduced from [11]. Copyright by the authors under a CC BY 4.0 license.
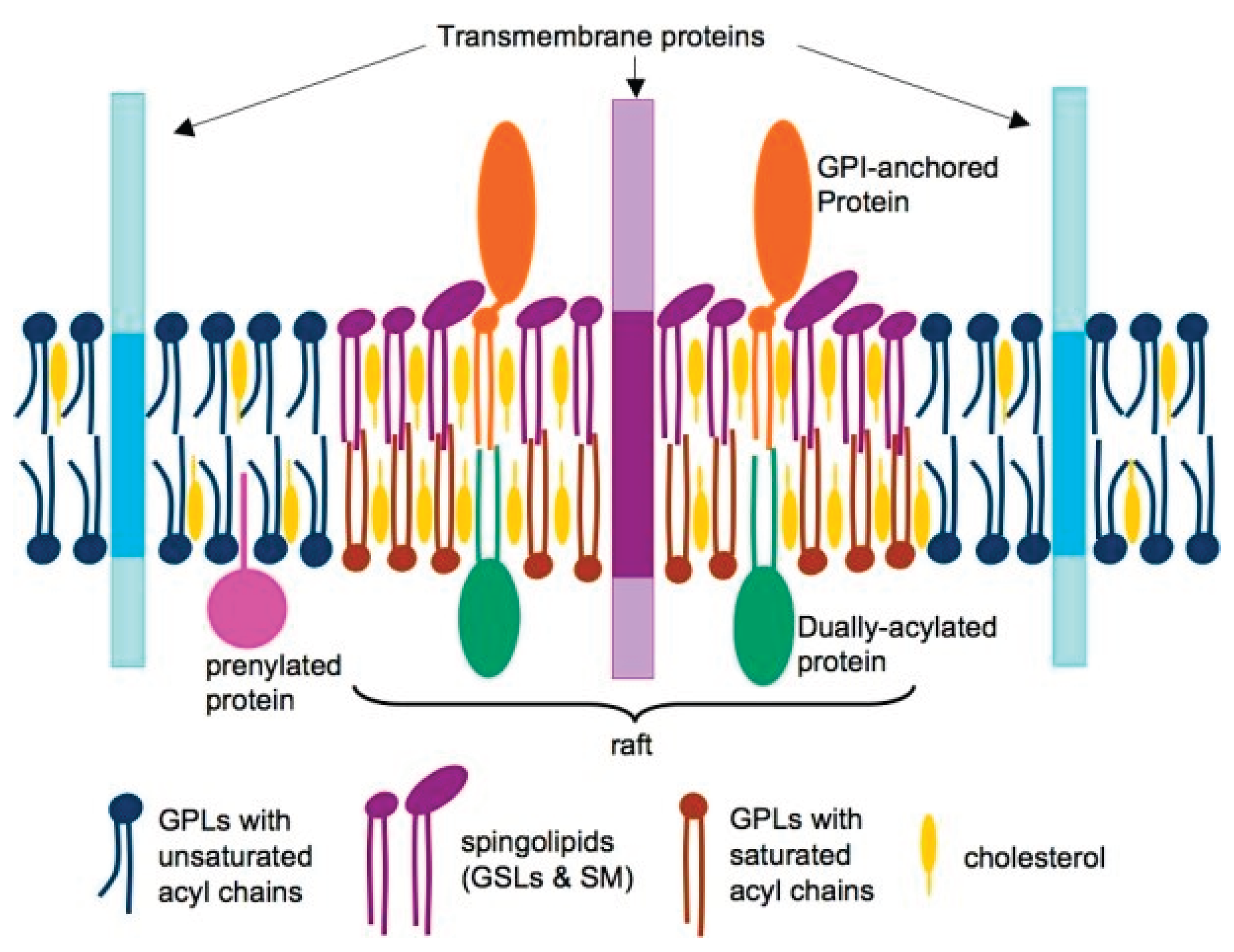
Figure 2.
Schematic representation of the pathways of lipolysis (blue) and lipogenesis (red) in relation to glycolysis and the tricarboxylic acid (TCA) cycle.
Figure 2.
Schematic representation of the pathways of lipolysis (blue) and lipogenesis (red) in relation to glycolysis and the tricarboxylic acid (TCA) cycle.
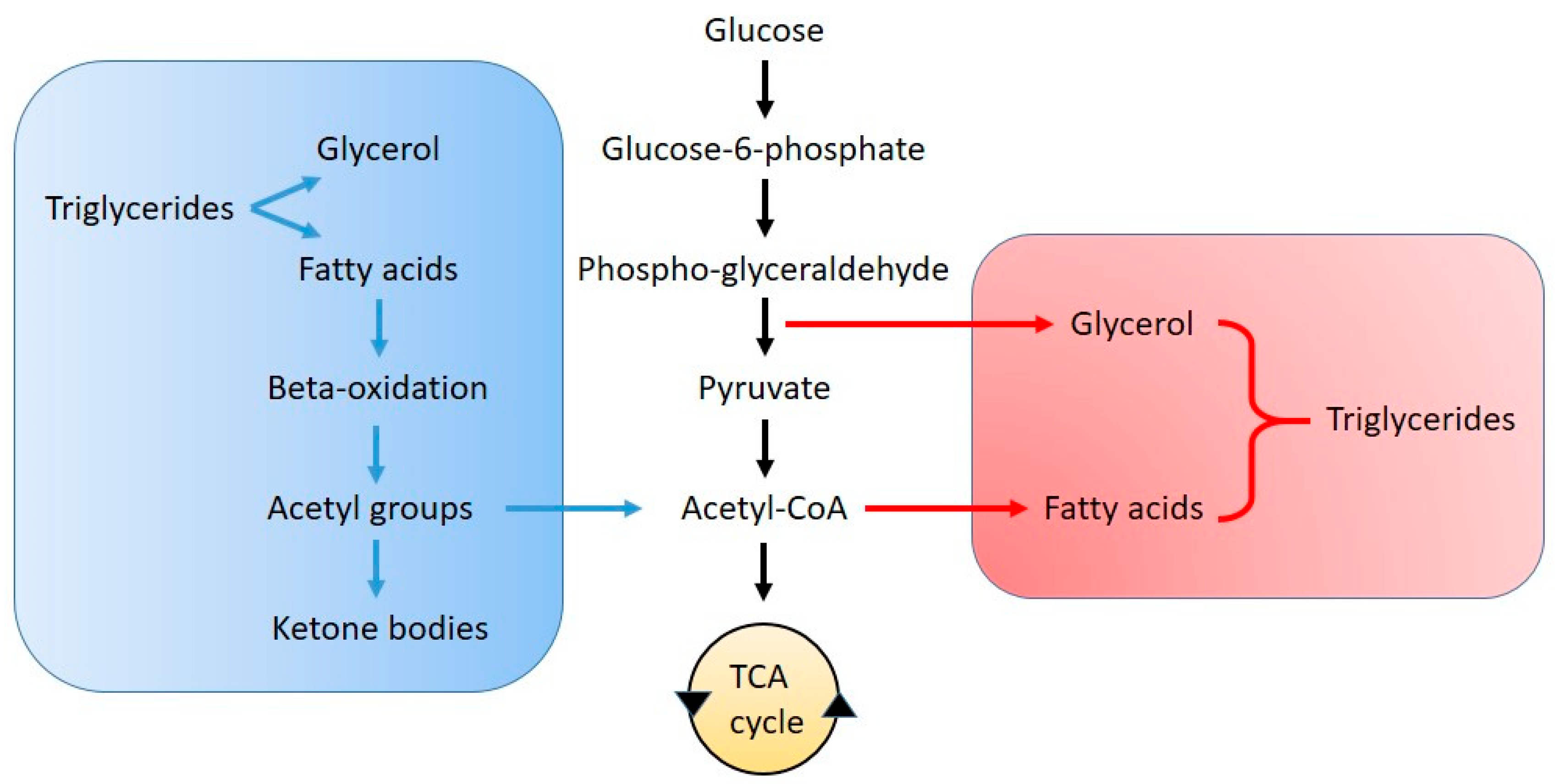
Figure 3.
Changes in the structure of high-density lipoproteins (HDL) produced by inflammation. Chronic inflammatory processes cause a decrease in the content of paraoxnase-1 (PON1), apolipoprotein AI (Apo AI), lecithin:cholesterol acyltransferase (LCAT), and cholesterol ester transfer protein (CETP), and an increase in the concentration of serum amyloid A (SAA). Reproduced from [77]. Copyright by the authors under a CC BY 4.0 license.
Figure 3.
Changes in the structure of high-density lipoproteins (HDL) produced by inflammation. Chronic inflammatory processes cause a decrease in the content of paraoxnase-1 (PON1), apolipoprotein AI (Apo AI), lecithin:cholesterol acyltransferase (LCAT), and cholesterol ester transfer protein (CETP), and an increase in the concentration of serum amyloid A (SAA). Reproduced from [77]. Copyright by the authors under a CC BY 4.0 license.
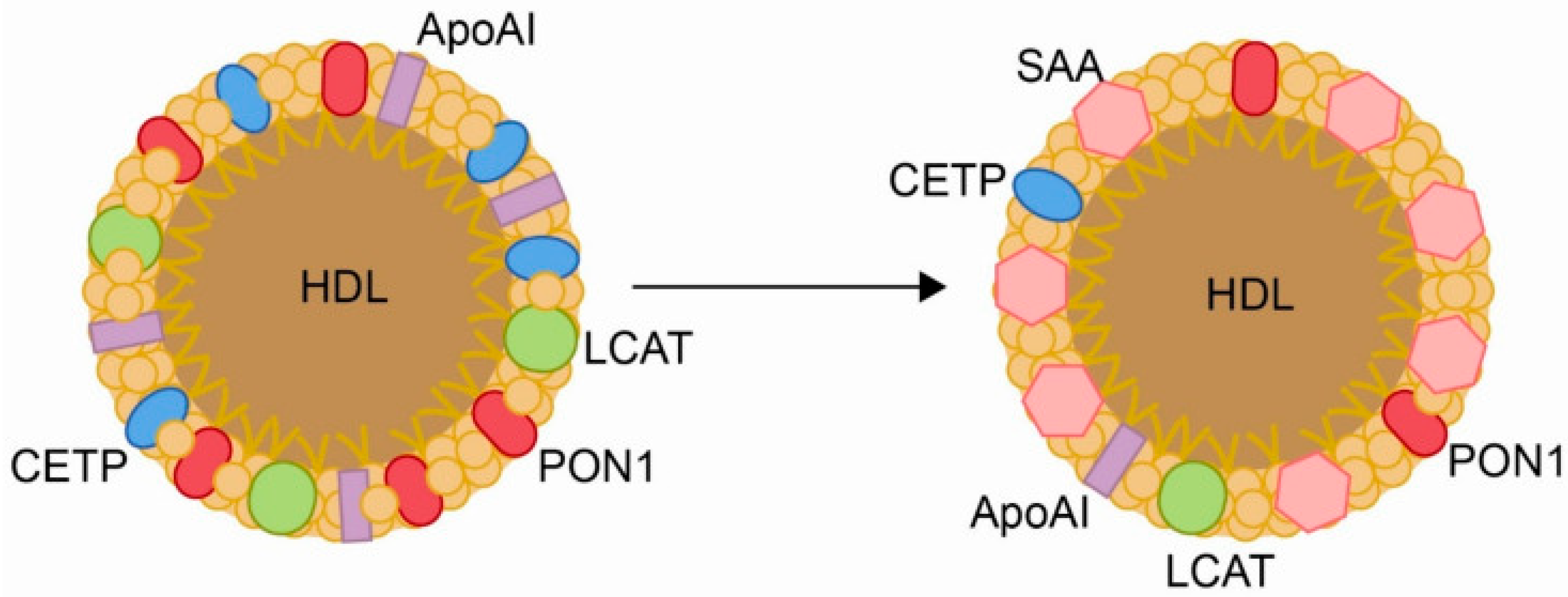
Figure 4.
Schematic representation of the “Warburg-like effect” in viral infections.
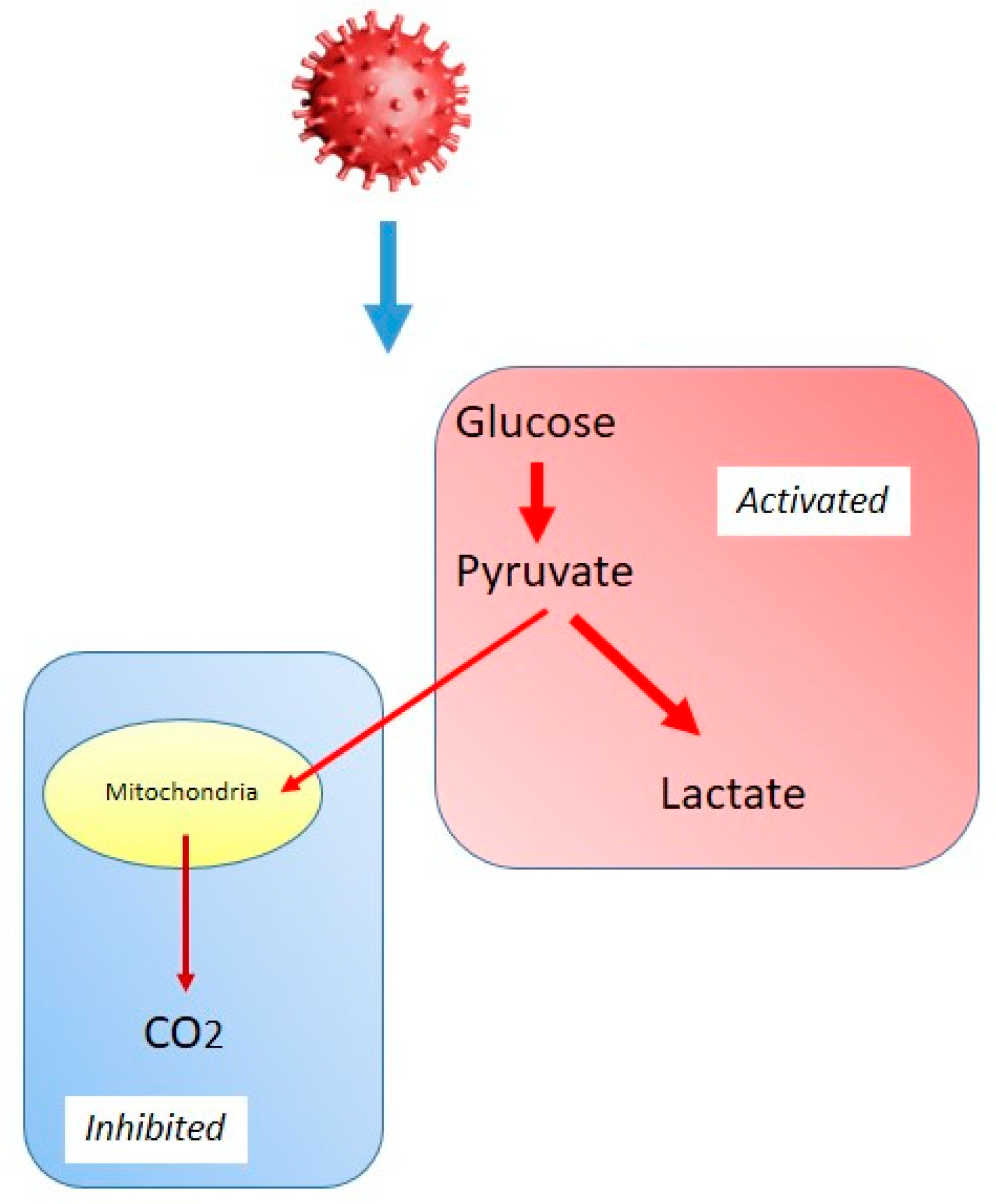
Figure 5.
The mechanistic target of rapamycin (MTOR) belongs to the phosphatidylinositol 3-kinase-related kinase protein family. It is a serine/threonine protein kinase that regulates autophagy, cell growth, cell proliferation, cell motility, cell survival, protein synthesis, and transcription. MTOR integrates the input from upstream pathways that are involved in noncommunicable diseases, including insulin, growth factors, and the status of cellular nutrient, oxygen, and energy. Of note, other pathways unrelated to energy metabolism may also be activated. For instance, recombinationactivating gene (RAG) protein is a substrate of AMPK (A). MTOR is the catalytic subunit of two molecular complexes: MTORC1 and MTORC2. The role of MTORC1 is to activate translation of proteins and is composed of MTOR, regulatory-associated protein of MTOR (Raptor), mammalian lethal with SEC13 protein 8 (MLST8), and other partners such as PRAS40 and DEPTOR (B). MTORC2 functions as regulator of the cytoskeleton and phosphorylates the serine/threonine protein kinase Akt/PKB at the serine residue S473. Phosphorylation of the serine stimulates Akt phosphorylation at the threonine Thr-308 residue via PDK1 and leads to full Akt activation. MTORC2 is composed of MTOR, rapamycin-insensitive companion of MTOR (RICTOR), GbL, and mammalian stressactivated protein kinase-interacting protein 1 (mSIN1) (C). Other abbreviations: ATG-13, autophagy-related protein 13; ERK, extracellular signal-regulated kinase; HIF-1, hypoxia-inducible factor 1; IRS, insulin receptor substrate; LEBP, lung epithelial binding peptide; PGC-1a, peroxisome proliferators-activated receptor g coactivator-1a; PI3K, phosphatidylinositol 3-kinase; PPARg, peroxisome proliferators-activated receptor g; PTEN, phosphatase and tensin homolog deleted on chromosome TEN; pTOS6k, p70 ribosomal S6 kinase; Rag, Ras-related GTPase; REDD, regulated in development and DNA damage responses; Rheb, Ras homologue enriched in brain; TSC2, tuberous sclerosis protein 2. Reproduced from [129] with permission. Copyright by Elsevier.
Figure 5.
The mechanistic target of rapamycin (MTOR) belongs to the phosphatidylinositol 3-kinase-related kinase protein family. It is a serine/threonine protein kinase that regulates autophagy, cell growth, cell proliferation, cell motility, cell survival, protein synthesis, and transcription. MTOR integrates the input from upstream pathways that are involved in noncommunicable diseases, including insulin, growth factors, and the status of cellular nutrient, oxygen, and energy. Of note, other pathways unrelated to energy metabolism may also be activated. For instance, recombinationactivating gene (RAG) protein is a substrate of AMPK (A). MTOR is the catalytic subunit of two molecular complexes: MTORC1 and MTORC2. The role of MTORC1 is to activate translation of proteins and is composed of MTOR, regulatory-associated protein of MTOR (Raptor), mammalian lethal with SEC13 protein 8 (MLST8), and other partners such as PRAS40 and DEPTOR (B). MTORC2 functions as regulator of the cytoskeleton and phosphorylates the serine/threonine protein kinase Akt/PKB at the serine residue S473. Phosphorylation of the serine stimulates Akt phosphorylation at the threonine Thr-308 residue via PDK1 and leads to full Akt activation. MTORC2 is composed of MTOR, rapamycin-insensitive companion of MTOR (RICTOR), GbL, and mammalian stressactivated protein kinase-interacting protein 1 (mSIN1) (C). Other abbreviations: ATG-13, autophagy-related protein 13; ERK, extracellular signal-regulated kinase; HIF-1, hypoxia-inducible factor 1; IRS, insulin receptor substrate; LEBP, lung epithelial binding peptide; PGC-1a, peroxisome proliferators-activated receptor g coactivator-1a; PI3K, phosphatidylinositol 3-kinase; PPARg, peroxisome proliferators-activated receptor g; PTEN, phosphatase and tensin homolog deleted on chromosome TEN; pTOS6k, p70 ribosomal S6 kinase; Rag, Ras-related GTPase; REDD, regulated in development and DNA damage responses; Rheb, Ras homologue enriched in brain; TSC2, tuberous sclerosis protein 2. Reproduced from [129] with permission. Copyright by Elsevier.
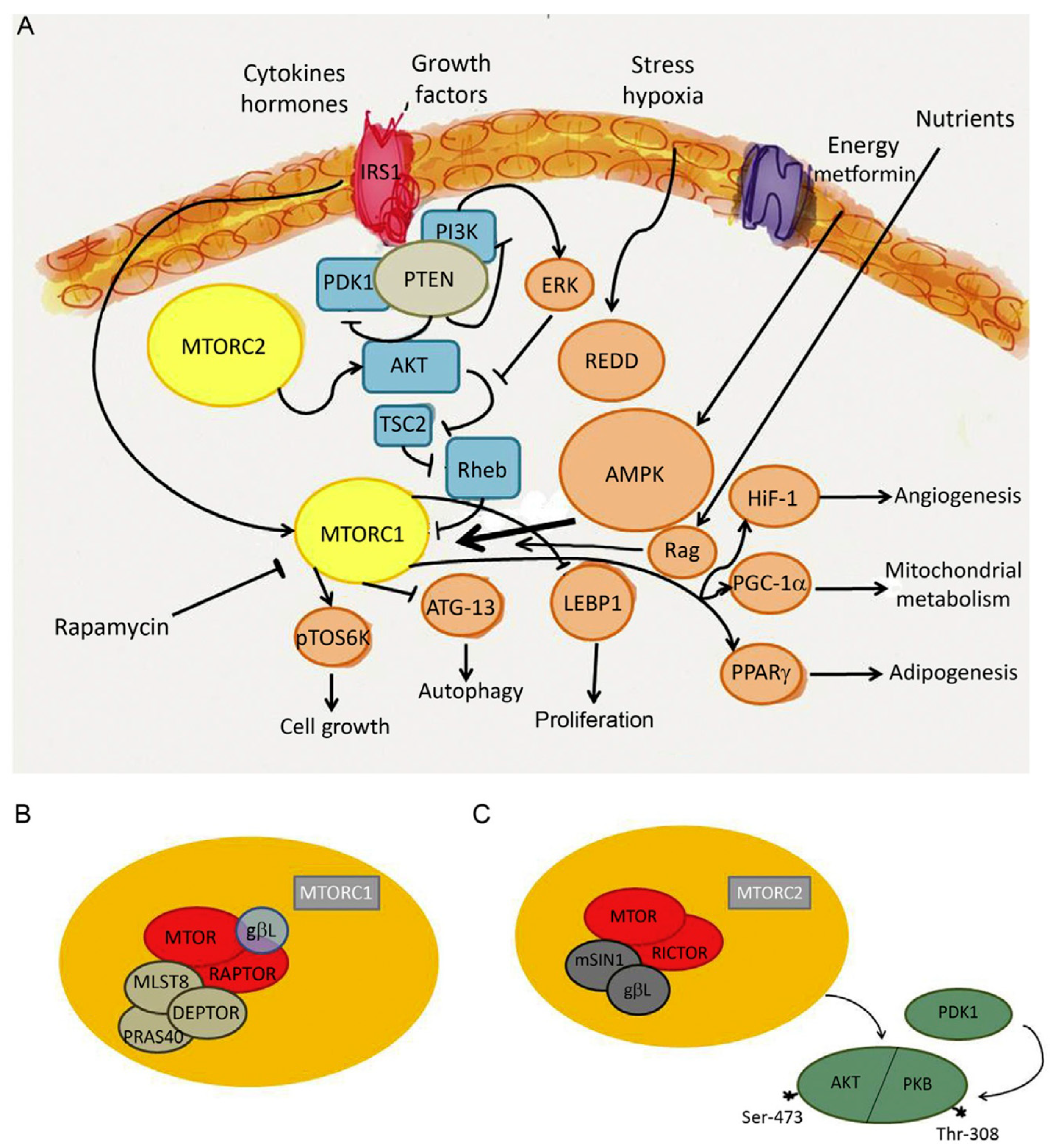
Figure 6.
Oxidation, inflammation, and disturbances in energy metabolism are closely related. To date, the evidence reported suggests that excessive production of reactive oxygen species (ROS) would inhibit paraoxonase-1 (PON1) activity in high-density lipoprotein (HDL) particles and in the mitochondrial membranes of somatic cells. At the same time, it would stimulate the synthesis of chemokine (C-C motif) ligand 2 (CCL2) through several pathways, notably that of pathogen-associated molecular patterns/damage-associated molecular patterns/pattern-recognition receptors (PAMP/DAMP/PRR). The decrease in PON1 activity and the increase in CCl2 would cause alterations in mitochondrial metabolism and an inhibition of autophagy. At the same time, CCL2 would interact with its receptor (CRR2) and present on monocytes, promoting their migration to sites of injury, their differentiation to macrophages, and their synthesis of new ROS, producing a vicious circle that would trigger and aggravate the disease. Reproduced from [77]. Copyright by the authors under a CC BY 4.0 license.
Figure 6.
Oxidation, inflammation, and disturbances in energy metabolism are closely related. To date, the evidence reported suggests that excessive production of reactive oxygen species (ROS) would inhibit paraoxonase-1 (PON1) activity in high-density lipoprotein (HDL) particles and in the mitochondrial membranes of somatic cells. At the same time, it would stimulate the synthesis of chemokine (C-C motif) ligand 2 (CCL2) through several pathways, notably that of pathogen-associated molecular patterns/damage-associated molecular patterns/pattern-recognition receptors (PAMP/DAMP/PRR). The decrease in PON1 activity and the increase in CCl2 would cause alterations in mitochondrial metabolism and an inhibition of autophagy. At the same time, CCL2 would interact with its receptor (CRR2) and present on monocytes, promoting their migration to sites of injury, their differentiation to macrophages, and their synthesis of new ROS, producing a vicious circle that would trigger and aggravate the disease. Reproduced from [77]. Copyright by the authors under a CC BY 4.0 license.
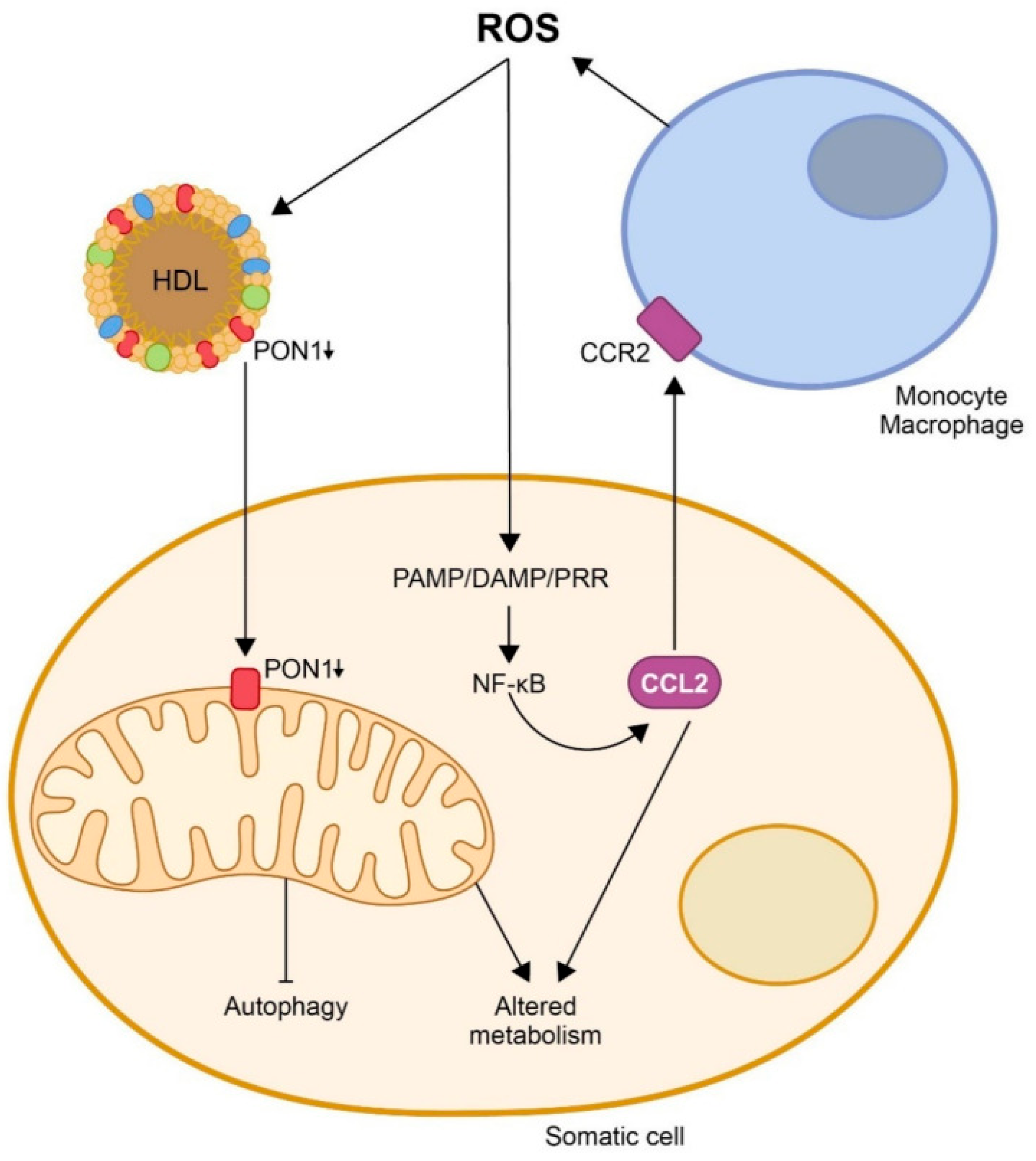

Table 1.
Comparison of lipid metabolism in SARS-CoV-2, influenza virus, and RSV infections.
| Feature | SARS-CoV-2 | Influenza virus | RSV |
| Lipid rafts in viral entry | Spike protein binding to ACE2 and entry. | Mainly clathrin-dependent endocytosis involving lipid rafts. | May bind to heparan sulfate and use rafts for attachment and fusion. |
| Fatty acid synthesis | Increased FASN | Increased FASN | Increased FASN |
| LD involvement | Induces LD accumulation for immune evasion and viral assembly. | Depletes or degrades LD via mTOR activation to fuel viral replication; inhibits LD biogenesis. | Disperses and reduces LD content; mechanisms less well characterized, but likely interferes with LD-associated immune signaling. |
| β-oxidation alterations | Inhibits β-oxidation, causing lipid accumulation. | Suppresses β-oxidation; shifts toward lipogenesis. | Mitochondrial dysfunction affects β-oxidation; less studied. |
| Cholesterol dependency | Cholesterol metabolism dysregulation. | Cholesterol metabolism dysregulation. | Not well characterized. |
| Lipoprotein metabolism | Decreased HDL; increased VLDL and LDL. | Transient decreased HDL and increased LDL. | Not well characterized. |
FASN: Fatty acid synthase; HDL: High-density lipoproteins; LD: Lipid droplets; LDL: Low-density lipoproteins; mTOR: Mechanistic target of rapamycin; RSV: Respiratory syncytial virus; VLDL: Very low-density lipoproteins.
Table 2.
Effects of respiratory viruses on energy metabolism and mitochondrial dynamics.
| Feature | SARS-CoV-2 | Influenza virus | RSV |
| Glycolysis and OXPHOS | Increased glycolysis (Warburg-like shift); decreased OXPHOS. |
Increased glycolysis (Warburg-like shift); decreased OXPHOS. |
Increased glycolysis (Warburg-like shift); decreased OXPHOS. |
| Mitochondrial dysfunction |
Altered morphology, decreased function; increased ROS; inhibited MAVS; activated NLRP3. |
Altered morphology, decreased function; increased ROS; activated NLRP3. |
Altered morphology, decreased function; increased ROS. |
| Bioenergetic failure | Decreased ATP production; impaired mitochondrial respiration; shift to anaerobic metabolism. | Decreased ATP production; impaired mitochondrial respiration; shift to anaerobic metabolism. | Decreased ATP production; impaired mitochondrial respiration; shift to anaerobic metabolism. |
| Cellular metabolic sensors |
HIF-1α activation; AMPK inhibition; altered mTOR and PI3K–Akt signaling. | HIF-1α and PI3K–Akt–mTOR axis activation; energy reprogramming. | mTOR and PI3K–Akt upregulation. |
AMPK: AMP-activated protein kinase; ATP: Adenosine triphosphate; HIF-1α: Hypoxia-inducible factor 1α; MAVS: Mitochondrial antiviral-signaling protein; mTOR: Mechanistic target of rapamycin; NLRP3: NOD-, LRR and pyrin domain-containing protein 3; PI3K–Akt: Phosphoinositide 3 kinase-Akt; ROS: Reactive oxygen species; RSV: Respiratory syncytial virus.
Table 3.
Main alterations in amino acid and nucleotide metabolism in SARS-CoV-2, influenza virus, and RSV infections.
Table 3.
Main alterations in amino acid and nucleotide metabolism in SARS-CoV-2, influenza virus, and RSV infections.
| Pathway | SARS-CoV-2 | Influenza virus | RSV |
| Glutamine/glutamate | Increased glutaminolysis | Increased glutaminolysis | Increased glutaminolysis |
| Arginine metabolism | Arginine depletion; arginase upregulation; decreased NO production. | Arginine depletion; arginase upregulation; decreased NO production. | Arginine depletion; arginase upregulation; decreased NO production. |
| Tryptophan–kynurenine | Strongly activated; increased IDO1 activity; correlates with severity | Moderate activation; role in immune modulation | Upregulated IDO1 in severe cases; less characterized |
| Cysteine metabolism | Decreased cysteine availability. | Decreased cysteine availability. | Decreased cysteine availability. |
| Nucleotide metabolism | Increased biosynthesis and salvage pathways | Increased biosynthesis and salvage pathways | Increased biosynthesis and salvage pathways |
IDO1: Indoleamine 2,3-dioxygenase 1; GSH: Glutathione; RSV: Respiratory syncytial virus.
Table 4.
Main alterations in endogenous antioxidant systems during SARS-CoV-2, influenza virus, and RSV infections.
Table 4.
Main alterations in endogenous antioxidant systems during SARS-CoV-2, influenza virus, and RSV infections.
| Pathway | SARS-CoV-2 | Influenza virus | RSV |
| Glutathione | Increased glutahione oxidation. | Transient increased glutahione oxidation. | Increased glutahione oxidation. |
| Superoxide dismutase | Decreased activity in plasma and tissues; associated with disease severity. | Variable: Decreased activity in severe disease; oxidative burden overwhelms the enzyme. | Variable: Decreased activity in severe disease; oxidative burden overwhelms the enzyme. |
| Catalase | Decreased activity. | Decreased activity. | Decreased activity. |
| Paraoxonase 1 | Markedly decreased activity. | Less characterized. | Less characterized. |
RSV: Respiratory syncytial virus.
Table 5.
Candidate drugs targeting virus-induced metabolic alterations in respiratory infections.
| Metabolic pathway | Proposed drug | Targeted virus |
| Viral entry | Camostat mesylate Statins Chloroquine |
SARS-CoV-2 Influenza SARS-CoV-2 |
| Lipid metabolism | Orlistat Statins Omega-3 fatty acids |
SARS-CoV-2, Influenza, RSV SARS-CoV-2 SARS-CoV-2 |
| Glycolysis | 2-deoxy-D-glucose KAN0438757 3PO |
SARS-CoV-2 Influenza Influenza |
| Mitochondrial dynamics Bioenergetics |
Mdivi 1 Melatonin Coenzyme Q10 Metformin Szeto-Schiller peptides |
Other viruses SARS-CoV-2, Influenza, RSV Not tested on viruses SARS-CoV-2 Not tested on viruses |
| Endogenous antioxidants | N-acetylcysteine Melatonin Tempol Ebselen |
SARS-CoV-2, Influenza SARS-CoV-2, Influenza, RSV SARS-CoV-2 SARS-CoV-2 |
| Kynurenine pathway | Epacadostat, navoximod | Not tested on viruses |
3PO: 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one; Mdivi 1: Mitochondrial division inhibitor 1; RSV: Respiratory syncytial virus.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



