Submitted:
28 May 2025
Posted:
29 May 2025
You are already at the latest version
Abstract
α-Hydroxyphosphonates the most prominent class within phosphonates remain of utmost importance because of their potential and real biological activity as pharmaceutical or pesticide agenst that is the consequence of their enzyme inhibitory properties. After optimizing the synthesis, 23 members of a new family, α-hydroxy-α-(benzothiophen-2-yl)-methylphosphonates were synthesized by the Pudovik reaction of benzo[b]thiophene-2-carboxaldehydes and diethyl phosphite performed at 26 °C in the presence of triethylamine as the catalyst. One of the products was also characterized by single-crystal X-ray analysis. The cytotoxic effect of the α-hydroxy-α-benzothiophenyl-methylphosphonates was tested on U266 myeloma, A2058 melanoma, HT-29 colon and EBC-1 lung cancer cell lines. Most of the molecules showed significant activity; the greatest effects were seen after treatment with hydroxyphosphonates with a trifluoromethyl group in the benzene ring.
Keywords:
α-Hydroxyphosphonate
; Pudovik reaction
; X-ray structure
; Biological activity
; Cytostatic effect
1. Introduction
The large family of α-hydroxyphosphonates and their derivatives is still in the focus these days due to their biological activity [1,2]. A broad scale of the activity has been described, including pesticidal [3], insecticidal [4,5] and herbicidal [3,6] effects. As regards the pharmaceutical relevancies, antibacterial [7,8], antimicrobial [7,8,9,10,11], antifungal [7,8,11] and antioxidant [12,13] activities were described. An actual challenge is the applicability of hydroxyphosphonic derivatives in the treatment of cancer. There are encouraging data on their cytotoxic activity [14,15,16,17,18]. The above biological effects are all connected with the enzyme inhibitory properties of the α-hydroxyphosphonic derivatives [19]. The species under discussion may act as farnesyl protein transferase inhibitors [20], HIV protease- [21] and viral cysteine protease inhibitors [22], inhibitors of CD45 tyrosine phosphatase [23] and undecaprenyl diphosphate phosphatase [24], as well as an influencer of the P5C reductase [25].
The practical method for the synthesis of α-hydroxyphosphonates is the Pudovik reaction involving the addition of dialkyl phosphites to the carbonyl carbon atom of an oxo compound that may be an aldehyde or ketone [26,27]. A wide variety of methods were described [28]. Special efforts were made to elaborate green synthetic methods including catalyst-free and/or microwave-assisted accomplishments [29]. Solvent-free additions on the surface of solid catalysts (e.g. on aluminum oxide/KF) were also developed, however, these methods required solvents during the work-up comprising extraction, chromatography and recrystallization [30]. The group of the senior author of this paper described an indeed green procedure for the synthesis of α-hydroxy-benzylphosphonates applying triethylamine as the catalyst, and only a small quantity of acetone as the medium. The arylaldehyde – dialkyl phosphite adducts precipitated in a crystalline form from the reaction mixtures on cooling [31].
In this article, we describe the synthesis of α-benzothiophenyl-α-hydroxymethylphosphonates, a brand new family of compounds alloying two scaffolds bearing potential activity: the hydroxyphosphonate skeleton and the benzothiophene heterocyclic ring.
2. Results and Discussion
2.1. Synthesis of the new α-benzothiophenyl-α-hydroxy-ethylphosphonates
Our plan was to prepare a family of new α-hydroxyphosphonates (2) by the Pudovik reaction of a series of benzo[b]thiophene-2-carboxaldehydes (1) with diethyl phosphite. For the optimization, the addition of the phosphite to the 5-fluorobenzothiophene-2-carboxaldehyde (1a) served as the model reaction. It was found that carrying out the addition in dichloromethane at room temperature (26 °C), there was need for 4 equivalents of triethylamine, and 4 equivalents of diethyl phosphite. No complete conversions were attained by applying smaller amounts of the base and the P-reagent. Then, the reaction was extended to benzothiophene-carboxaldehydes (2b-w) containing methyl, ethyl, halogen and trifluoromethyl substituents in different positions of the benzothiophene ring. The aldehyde - diethyl phosphite adducts (2a-w) were purified by flash column chromatography, and their structures were characterized by spectroscopic methods, such as 31P, 13C and 1H NMR, as well as HRMS. In this way, 23 new α-hydroxyphosphonate derivatives (2a-w) were prepared in good to excellent yields. The preparative results are summarized in Table 1.
2.2. Spectroscopic Characterization
The structural elucidation of the synthesized α-hydroxyphosphonates was primarily supported by multinuclear NMR and IR spectroscopic data, which provided consistent and highly informative datasets across the compound library.
2.2.1. 13C NMR Spectroscopy
The 13C spectra confirmed structural features through diagnostic 13C-31P and, where applicable, 13C-19F couplings. Universally, a strong 1-bond 13C-31P coupling (1JC-P ≈ 165–168 Hz) was observed for the carbon atoms α to the phosphorus atom, typically appearing around δ 66-67 ppm. For the O-ethyl moieties, the ethyl CH2 and CH3 carbons exhibited 2- and 3-bond 13C-31P couplings (2JC-P ≈ 7 Hz and 3JC-P 5–6 Hz, respectively). In 3-methyl-substituted compounds, a weak 4-bond 13C-31P coupling (4JC-P ≈ 1-1.5 Hz) was observable in the methyl carbon resonance. In contrast, non-methylated analogues displayed a 3-bond coupling on the aromatic C-3 carbon (3JC-P = 8–10 Hz). The 31P coupling influence extended across the aromatic system, with 4- and even 5-bond couplings (4,5JC-P = 1–3 Hz) detectable in certain ring carbons of the benzothiophene moiety.
2.2.2. 1H NMR Spectroscopy
In the 1H NMR spectra, the aromatic protons of the benzothiophene ring appeared downfield, typically in the range of δ 8.0–6.8 ppm, with splitting patterns reflecting the substitution pattern of the aromatic ring. Fluorine substituents introduced additional splitting due to 1H-19F couplings, increasing spectral complexity. In most cases, a broad or multiplet resonance attributable to the hydroxyl proton was also observed, which lacked the corresponding 1H-13C HSQC cross-peak, consistent with assignment as a hydroxyl proton.
The methine proton adjacent to the hydroxyl group typically appeared as a doublet or doublet of doublets, depending on whether the coupling with the OH proton was resolved. Notably, a 2-bond 1H-31P coupling (2JH-P ≈ 10–11 Hz) was consistently observed. For O-ethyl substituents, the methylene protons of the ethyl groups showed complex multiplets, while the terminal methyl protons appeared as triplets with the most upfield chemical shifts. In compounds with a 3-methyl substituent, a methyl doublet was seen around δ 2.5 ppm, showing a 5-bond 1H-31P coupling (5JH-P ≈ 3 Hz). In non-methylated analogues, the aromatic proton in position 3 exhibited a 4-bond 1H-31P coupling (4JH-P ≈ 3.5 Hz).
2.2.3. 31P NMR Spectroscopy
A sharp singlet resonance was consistently detected around δ 19–20 ppm in the 31P NMR spectra, confirming the presence of the α-hydroxyphosphonate moiety.
Typical values of the coupling constants 1H or 13C nuclei with 31P nuclei are summarized in Figure 1, indicated on the general structural formula.
2.2.4. IR Spectroscopy
The IR spectra exhibited characteristic absorption bands associated with phosphonate functional groups. Strong P=O stretching bands were seen in the range of 1220–1260 cm-1, while P–O–C stretching vibrations occurred around 1020–1060 cm-1. Additional bands observed between 700–800 cm-1 were assigned to P–C stretching modes. These vibrational features provide further verification of the core phosphonate structure.
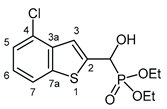
2.3. Single Crystal X-ray analysis of diethyl α-(7-chlorobenzothiophenyl-)α-hydroxy-methylphosphonate (2f)
The molecular structure of α-(7-chlorobenzo[b]thiophen-2-yl)-α-hydroxy-methylphosphonate 2f was determined by single crystal X-ray diffraction that is shown in Figure 2. The P(O)(OEt)2 group of the molecule is disordered at least over two, most probably over more than two positions. Only a splitting over two positions was considered. Further splitting did not result in an improvement of the structure model. Due to the strong disorder, the C-O and C-C distances of the OEt groups were fixed. Only one of the two positions of the P(O)(OEt)2 group is shown for clarity. In addition, the OH group is also disordered over two positions, most probably resulting from the two enantiomers occupying the same crystallographic position. Also, in this case, only one of the positions is shown for clarity. Atom distances and bond angles are within the expected ranges (Table SI 1-3). Nevertheless, in addition to proving the structure of the compound, some interesting structural features can be observed. The heterocyclic part of the molecule is, as expected planar, and the carbon atom C1A of the -CH(OH)- group is also lying in the plane of the heterocycle. This arrangement results in a flat molecule except the -P(O)(OEt)2 unit, which is rotated out of the molecular plane with a dihedral angle P1A-C1A-C2-S1 of 117.8(2)°. This particular structural feature is anticipated to have an impact on the biological activity of benzothiophenyl α-hydroxyphosphonates.
2.4. Bioactivity study of the α-benzothiophenyl-α-hydroxy-methylphosphonates (2a-w)
A series of 23 different α-hydroxy-α-benzothiophenyl-methylphosphonates (2a-w) were screened for their effects on cell viability in four human tumor cell lines: U266 (multiple myeloma), EBC-1 (lung cancer), A2058 (melanoma), and HT-29 (colon cancer). Based on the cell viability results shown in Table 2, most compounds, regardless of the substituents in positions R1-R5, exhibited at least a mild viability-reducing effect across the tested cell lines with normalized cell viability values of 0.8 or lower.
Among the compounds with R1 = H, the appearance of a floro-substituent as R3-R5 in the benzothiophene ring (as in 2a, 2b and 2c) proved effective against the U266 myeloma cell line. In case of R3 = Br (as in 2g), only a moderate decrease in the cell viability of the A2058 melanoma and the HT-29 colon cell lines was observed, while the EBC-1 lung and U266 myeloma cell lines were clearly impacted. Furthermore, the appearance of a chloro- or iodo substituent as R2 or R5 had mostly favorable effects (as in 2d, 2e, 2f and 2h), similarly to the presence of a trifluoromethyl group as R2 (2j). Similar results were observed following the treatment of the cell lines with compound 2w (R² = F, R⁵ = I).
Interestingly, when R1 = H was replaced with a methyl group (as in 2i), long-term treatments had no significant effects on the cell viability of any cell line. Furthermore, the substitution of the benzene ring of species 2i with one or two fluorine atoms (as in 2k, 2l, 2m, 2n, 2o, and 2p) did not markedly alter the antiproliferative effects as compared to homologue 2i. Nevertheless, hydroxophosphonates 2o and 2p exhibited some degree of effectiveness on the U266 cell line. This result aligns with earlier observations from treatments using derivatives 2a, 2b or 2c. The two chlorinated species, 2q and 2r displayed profiles similar to those obtained for compounds 2d, 2e and 2f. Hydroxyphosphonates 2s, 2t, and 2u containing a trifluoromethyl group as the R3, R4, and R5 group, respectively, showed varying effectiveness in the cell viability assays. The para-substituted hydroxyphosphonate (2u) exhibited the weakest activity, while the meta-substituted species (2t) showed the strongest activity on three cell lines (U266, EBC-1 and HT-29) out of the four. It should be pointed out that, on the U266 cell line, compound 2t reduced cell viability to 0.09 ± 0.02. Changing R1 = Me to Et (as in 2v) resulted in reduced activity according. Overall, U266 cells appeared to be the most sensitive to the hydroxyphosphonates applied in the tests. However, the effect of the vehicle control DMSO cannot be overlooked, as it had a profound impact on the viability of U266 cells, but not on A2058, EBC-1, or HT-29 cells.
3. Experimental
3.1. General
All NMR spectra were recorded in CDCl3 or DMSO-d6 solution in standard 5 mm NMR tubes at 295 K, on a Bruker Avance III HD 600 spectrometer (1H: 600.00 MHz, 13C: 150.89 MHz, 31P: 242.87 MHz). The 31P, 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) relative to the internal standard H3PO4, TMS and the NMR solvent (CDCl3), respectively. Coupling constants (J) are given in hertz (Hz). The following abbreviations were used to designate multiplicities: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, br = broad. IR spectra were obtained on a Bruker Alpha FT-IR spectrometer in transmission mode in KBr pellets. High-resolution mass spectra (HRMS) were recorded on an Agilent 7250 Q-TOF mass spectrometer coupled to an Agilent 8890 gas chromatographic system. All melting points were determined on a Büchi B-540 capillary melting point apparatus and are uncorrected. Reactions were monitored by thin-layer chromatography (TLC) carried out on silica gel plates (60 F254) using UV light as a visualising agent. Purifications by column chromatography were carried out using Merck 107736 silica gel 60 H with a CH2Cl2–MeOH solvent system. All reagents were purchased from commercial sources, and were used without further purification. Benzo[b]thiophene-2-carboxaldehydes (2) were prepared according to literature methods [33,34,35].
3.2. General procedure for the synthesis of diethyl [(1-benzothiopen-2-yl)(hydroxy)methyl]phosphonates 2
The appropriate aldehyde (1, 1.0 mmol) was dissolved in dichloromethane (5 mL), then triethylamine (0.61 mL, 0.445 g, 4.4 mmol) and diethyl phosphite (0.57 mL, 0.608 g, 4.4 mmol) were added to this solution. The reaction mixture was stirred at room temperature for 24 hours, diluted with dichloromethane (10 mL) and washed with water (2 × 10 mL). The organic layer was dried (Na2SO4), filtered and the solvent removed under reduced pressure. The residue was purified by flash column chromatography on silica gel (CH2Cl2 → CH2Cl2/MeOH 9:1) to afford products 2. Analytical samples were obtained by recrystallization from the solvents or solvent mixtures given below in parentheses after the melting point.
The following products were thus prepared:
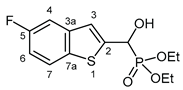
3.2.1. Diethyl [(5-fluoro-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2a)
Yield: 240 mg (75%). White crystals. Mp 109‒111 °C (hexane‒EtOAc). IR (KBr): 3424, 3205, 1445, 1226, 1204, 1044, 870 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.72 (dd, J1 = 8.8 Hz, J2 = 4.8 Hz 1H, H-7), 7.37 (dd, J1 = 9.4 Hz, J2 = 2.4 Hz, 1H, H-4), 7.34 (d, J = 3.4 Hz, 1H, H-3), 7.07 (dt, Jd = 2.5 Hz, Jt = 8.8 Hz, 1H, H-6), 5.31 (d, J = 11.9 Hz, 1H, CH), 4.93 (br s, 1H, OH), 4.20‒4.11 (m, 4H, 2×CH2), 1.32 (t, J = 7.1 Hz, 3H, CH3), 1.28 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 160.7 (d, J = 241.5 Hz, C-5), 143.3 (d, J = 1.6 Hz, C-2), 140.3 (dd, J1 = 9.5 Hz, J2 = 2.7 Hz, C-3a), 135.1 (t, J = 1.5 Hz, C-7a), 123.3 (d, J = 9.4 Hz, C-7), 121.9 (dd, J1 = 8.1 Hz, J2 = 4.3 Hz, C-3), 113.1 (dd, J1 = 25.3, J2 = 1.0 Hz, C-6), 109.0 (dd, J1 = 22.9 Hz, J2 = 0.7 Hz, C-4), 67.5 (d, J = 165.4 Hz, CH), 64.0 (d, J = 7.1 Hz, CH2), 63.5 (d, J = 7.4 Hz, CH2), 16.4 (d, J = 5.6 Hz, 2×CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.0 ppm. HRMS: [M]+ Calcd for C13H16FO4PS 318.0491; Found 318.0474.
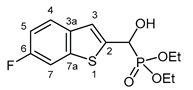
3.2.2. Diethyl [(6-fluoro-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2b)
Yield: 277 mg (87%). White crystals. Mp 114‒116 °C (EtOAc). IR (KBr): 3196, 2985, 1601, 1469, 1223, 1058, 1023, 568 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.64 (dd, J1 = 8.7 Hz, J2 = 5.1 Hz 1H, H-4), 7.48 (dd, J1 = 8.7 Hz, J2 = 2.0 Hz, 1H, H-7), 7.33 (d, J = 3.4 Hz, 1H, H-3), 7.08 (td, Jt = 8.9 Hz, Jd = 2.3 Hz, 1H, H-5), 5.29 (d, J = 11.6 Hz, 1H, CH), 5.22 (br s, 1H, OH), 4.20‒4.10 (m, 4H, 2×CH2), 1.31 (t, J = 7.1 Hz, 3H, CH3), 1.28 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 160.4 (dd, J1 = 244.3 Hz, J2 = 1.2 Hz, C-6), 140.7 (dd, J1 = 10.3 Hz, J2 = 1.5 Hz, C-7a), 140.3 (dd, J1 = 3.7 Hz, J2 = 1.4 Hz, C-2), 135.8 (m, C-3a), 124.6 (dd, J1 = 9.1 Hz, J2 = 0.8 Hz, C-4), 121.8 (d, J = 8.3 Hz, C-3), 113.3 (d, J = 24.2 Hz, C-5), 108.3 (d, J = 25.4 Hz, C-7), 67.3 (d, J = 166.2 Hz, CH), 63.9 (d, J = 7.1 Hz, CH2), 63.5 (d, J = 7.3 Hz, CH2), 16.39 (d, J = 5.5 Hz, CH3), 16.38 (d, J = 5.8 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.2 ppm. HRMS: [M]+ Calcd for C13H16FO4PS 318.0491; Found 318.0486.
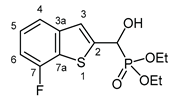
3.2.3. Diethyl [(7-fluoro-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2c)
Yield: 299 mg (94%). Off-white crystals. Mp 125‒127 °C (EtOAc). IR (KBr): 3405, 3230, 1470, 1228, 1058, 1016, 978, 784 cm-1. 1H NMR (CDCl3, 600 MHz): δ 6.52‒6.49 (m, 1H, H-4), 7.42‒7.40 (m, 1H, H-3), 7.31‒7.27 (m, 1H, H-5), 7.03‒6.99 (m, 1H, H-6), 5.33 (d, J = 11.9 Hz, 1H, CH), 5.03 (br s, 1H, OH), 4.22‒4.13 (m, 4H, 2×CH2), 1.32 (t, J = 7.1 Hz, 3H, CH3), 1.29 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 157.5 (dd, J1 = 246.8 Hz, J2 = 1.0 Hz, C-7), 142.7 (dd, J1 = 4.6 Hz, J2 = 2.6 Hz, C-3a), 142.1 (t, J = 1.5 Hz, C-2), 126.6 (dd, J1 = 18.1 Hz, J2 = 1.5 Hz, C-7a), 125.6 (d, J = 6.8 Hz, C-5), 122.4 (dd, J1 = 8.0 Hz, J2 = 2.2 Hz, C-3), 119.4 (dd, J1 = 3.3 Hz, J2 = 0.8 Hz, C-4), 109.3 (dd, J1 = 18.5 Hz, J2 = 0.8 Hz, C-6), 67.4 (d, J = 165.2 Hz, CH), 64.0 (d, J = 7.1 Hz, CH2), 63.4 (d, J = 7.4 Hz, CH2), 16.42 (d, J = 5.4 Hz, CH3), 16.40 (d, J = 5.7 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 18.9 ppm. HRMS: [M]+ Calcd for C13H16FO4PS 318.0491; Found 318.0483.
3.2.4. Diethyl [(4-chloro-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2d)
Yield: 241 mg (72%). White crystals. Mp 90‒92 °C (hexane–EtOAc). IR (KBr): 3239, 1414, 1253, 1203, 1036, 761, 542 cm-1. 1H NMR (DMSO-d6, 600 MHz): δ 7.96 (d, J = 8.0 Hz, 1H, H-7), 7.47 (d, J = 3.6 Hz, 1H, H-3), 7.46–7.45 (m, 1H, H-5), 7.34 (t, J = 7.9 Hz, 1H, H-6), 6.91 (dd, J1 = 14.5 Hz, J2 = 6.0 Hz, 1H, OH), 5.45 (dd, J1 = 14.5 Hz, J2 = 5.4 Hz, 1H, CH), 4.10–4.06 (m, 2H, CH2), 4.06–4.01 (m, 2H, CH2), 1.22 (t, J = 7.1 Hz, 3H, CH3), 1.21 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (DMSO-d6, 150 MHz): δ 145.9 (C-7a), 140.5 (d, J = 1.4 Hz, C-2), 137.1 (d, J = 2.5 Hz, C-3a), 127.0 (d, J = 1.1 Hz, C-4), 125.4 (C-6), 124.4 (C-5), 121.8 (C-7), 119.2 (d, J = 8.4 Hz, C-3), 65.7 (d, J = 168.1 Hz, CH), 63.0 (d, J = 7.0 Hz, CH2), 62.6 (d, J = 6.8 Hz, CH2), 16.54 (d, J = 5.6 Hz, CH3), 16.51 (d, J = 5.5 Hz, CH3) ppm. 31P NMR (DMSO-d6, 242 MHz): δ 19.4 ppm. HRMS: [M]+ Calcd for C13H16ClO4PS 334.0195; Found 334.0186.
3.2.5. Diethyl [(5-chloro-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2e)
Yield: 207 mg (62%). Off-white crystals. Mp 104‒105 °C (hexane–EtOAc). IR (KBr): 3425, 3169, 1440, 1224, 1067, 1052, 1027, 545 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.70 (d, J = 8.6 Hz, 1H, H-7), 7.67 (br s, 1H, H-4), 7.30 (d, J = 3.2 Hz, 1H, H-3), 7.28–7.25 (m, 1 H, H-6), 5.32 (br s, 1H, OH), 5.31 (d, J = 11.0 Hz, 1H, CH), 4.20–4.15 (m, 2H, CH2), 4.15–4.10 (m, 2H, CH2), 1.31 (t, J = 7.0 Hz, 3H, CH3), 1.27 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 143.0 (d, J = 1.5 Hz, C-2), 140.5 (d, J = 2.6 Hz, C-3a), 137.8 (d, J = 1.6 Hz, C-7a), 130.4 (C-5), 124.7 (C-6), 123.2 (C-7), 123.0 (C-4), 121.4 (d, J = 7.9 Hz, C-3), 67.4 (d, J = 165.8 Hz, CH), 64.0 (d, J = 7.0 Hz, CH2), 63.5 (d, J = 7.4 Hz, CH2), 16.39 (d, J = 5.4 Hz, CH3), 16.38 (d, J = 5.7 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.0 ppm. HRMS: [M]+ Calcd for C13H16ClO4PS 334.0202; Found 334.0194.
3.2.6. Diethyl [(7-chloro-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2f)
Yield: 287 mg (86%). White crystals. Mp 119‒120 °C (EtOAc). IR (KBr): 3324, 2980, 1453, 1198, 1045, 974, 766 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.62 (d, J = 7.3 Hz, 1H, H-4), 7.41 (d, J = 3.4 Hz, 1H, H-3), 7.32–7.29 (m, 1H, H-6), 7.29–7.26 (m, 1H, H-5), 5.34 (d, J = 11.9 Hz, 1H, CH), 5.23 (br s, 1H, OH), 4.21–4.13 (m, 4H, 2×CH2), 1.32 (t, J = 7.1 Hz, 3H, CH3), 1.29 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 142.1 (d, J = 1.4 Hz, C-2), 140.7 (d, J = 2.6 Hz, C-3a), 138.9 (d, J = 1.6 Hz, C-7a), 127.6 (C-7), 125.4 (C-5), 123.8 (C-6), 122.7 (d, J = 8.0 Hz, C-3), 122.0 (d, J = 0.8 Hz, C-4), 67.4 (d, J = 165.5 Hz, CH), 64.0 (d, J = 7.0 Hz, CH2), 63.6 (d, J = 7.3 Hz, CH2), 16.43 (d, J = 5.4 Hz, CH3), 16.40 (d, J = 5.6 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 18.9 ppm. HRMS: [M]+ Calcd for C13H16ClO4PS 334.0195; Found 334.0190.
3.2.7. Diethyl [(5-bromo-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2g)
Yield: 277 mg (73%). White crystals. Mp 96‒100 °C (EtOAc). IR (KBr): 3166, 2982, 1580, 1437, 1224, 1122, 1061, 1026, 980 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.85 (d, J = 1.7 Hz, 1H, H-4), 7.66 (d, J = 8.5 Hz, 1H, H-7), 7.41 (dd, J1 = 8.6 Hz, J2 = 1.4 Hz, 1H, H-6), 7.31 (d, J = 3.4 Hz, 1H, H-3), 5.31 (dd, J1 = 11.8 Hz, J2 = 4.7 Hz, 1H, CH), 4.85 (br s, 1H, OH), 4.20–4.10 (m, 4H, 2×CH2), 1.32 (t, J = 7.1 Hz, 3H, CH3), 1.28 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 142.7 (d, J = 1.7 Hz, C-2), 140.9 (d, J = 2.6 Hz, C-3a), 138.3 (d, J = 1.5 Hz, C-7a), 127.3 (d, J = 0.9 Hz, C-6), 126.2 (d, J = 0.9 Hz, C-4), 123.6 (C-7), 121.4 (d, J = 8.1 Hz, C-3), 118.2 (C-5), 67.5 (d, J = 165.1 Hz, CH), 64.0 (d, J = 7.0 Hz, CH2), 63.6 (d, J = 7.3 Hz, CH2), 16.42 (d, J = 5.8 Hz, CH3), 16.41 (d, J = 5.8 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 18.9 ppm. HRMS: [M]+ Calcd for C13H16BrO4PS 377.9690; Found 377.9687.
3.2.8. Diethyl [hydroxy(4-iodo-1-benzothiophen-2-yl]methyl]phosphonate (2h)Yield: 283 mg (66%). White crystals. Mp 94‒95 °C (hexane–EtOAc). IR (KBr): 3188, 2979, 1522, 1441, 1404, 1255, 1238, 1196, 1022, 976 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.77 (d, J = 8.0 Hz, 1H, H-7), 7.75 (dd, J1 = 7.6 Hz, J2 = 0.7 Hz, 1H, H-5), 7.46 (d, J = 3.5 Hz, 1H, H-3), 7.02 (t, J = 8.0 Hz, 1H, H-6), 5.35 (d, J = 11.7 Hz, 1H, CH), 4.24–4.14 (m, 5H, 2×CH2, OH), 1.35 (t, J = 7.1 Hz, 3H, CH3), 1.32 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 142.3 (d, J = 2.6 Hz, C-3a), 141.2 (d, J = 2.5 Hz, C-2), 138.9 (d, J = 1.5 Hz, C-7a),134.3 (C-5), 126.3 (d, J = 8.3 Hz, C-3), 125.5 (d, J = 0.8 Hz, C-6), 122.3 (C-7), 90.1 (d, J = 1.5 Hz, C-4), 67.7 (d, J = 164.8 Hz, CH), 64.0 (d, J = 7.0 Hz, CH2), 63.6 (d, J = 7.3 Hz, CH2), 16.5 (d, J = 5.4 Hz, CH3), 16.4 (d, J = 5.8 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 18.7 ppm. HRMS: [M]+ Calcd for C13H16IO4PS 425.9552; Found 425.9553.
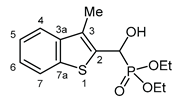
3.2.9. Diethyl [(hydroxy(3-methyl-1-benzothiophen-2-yl)methyl]phosphonate (2i)
Yield: 193 mg (61%). White crystals. Mp 128‒130 °C (hexane‒EtOAc). IR (KBr): 3216, 2974, 1438, 1230, 1196, 1051, 1019, 964, 751 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.81 (d, J = 7.9 Hz, 1H, H-7), 7.68 (d, J = 7.9 Hz, 1H, H-4), 7.38 (t, J = 7.3 Hz, 1H, H-5 or H-6), 7.33 (t, J = 7.6 Hz, 1H, H-5 or H-6), 5.52 (dd, J1 = 10.7 Hz, J2 = 3.1 Hz, 1H, CH), 2.40 (d, J = 3.0 Hz, 3H, CH3), 1.30 (t, J = 6.6 Hz, 3H, CH2CH3), 1.28 (t, J = 7.2 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 140.1 (d, J = 2.9 Hz, C-3a), 139.2 (d, J = 1.6 Hz, C-7a), 134.2 (d, J = 5.2 Hz, C-2), 129.7 (d, J = 10.1 Hz, C-3), 124.4 (d, J = 1.1 Hz, C-5 or C-6), 123.9 (C-5 or C-6), 122.4 (d, J = 1.0 Hz, C-4), 121.8 (d, J = 1.3 Hz. C-7), 66.4 (d, J = 167.6 Hz, CH), 63.9 (d, J = 6.9 Hz, CH2), 63.2 (d, J = 7.3 Hz, CH2), 16.4 (t, J = 5.9 Hz, 2 × OCH2CH3), 12.3 (d, J = 1.3 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 20.0 ppm. HRMS: [M]+ Calcd for C14H19O4PS 314.0742; Found 314.0740.
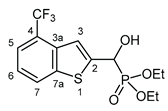
3.2.10. Diethyl {hydroxy [4-(trifluoromethyl)-1-benzothiophen-2-yl]methyl}phosphonate (2j)
Yield: 289 mg (79%). White crystals. Mp 86‒89 °C (hexane–EtOAc). IR (KBr): 3444, 3240, 1426, 1328, 1221, 1120, 1057, 1031 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.99‒7.96 (m, 1H, H-7), 7.65‒7.63 (m, 1H, H-5), 7.58 (br s, 1H, H-3), 7.39‒7.35 (m, 1H, H-6), 5.43 (d, J = 11.5 Hz, 1H, CH), 4.91 (br s, 1H, OH), 4.22‒4.12 (m, 4H, 2×CH2), 1.32 (t, J = 7.1 Hz, 3H, CH3), 1.29 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 143.9 (C-2), 141.2 (d, J = 1.6 Hz, C-7a), 135.3 (C-3a), 126.0 (C-7), 124.42 (q, J = 32.2 Hz, C-4), 124.35 (q, J = 273.1 Hz, CF3), 123.2 (C-6), 121.9 (q, J = 5.0 Hz, C-5), 119.9 (qd, Jq = 1.6 Hz, Jd = 8.1 Hz, C-3), 67.5 (d, J = 165.6 Hz, CH), 64.4 (d, J = 7.0 Hz, CH2), 63.6 (d, J = 7.6 Hz, CH2), 16.3 (d, J = 5.6 Hz, CH3), 16.2 (d, J = 6.0 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 18.6 ppm. HRMS: [M]+ Calcd for C14H16F3O4PS 368.0459; Found 368.0456.
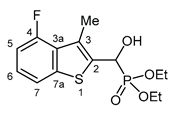
3.2.11. Diethyl [(4-fluoro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2k)
Yield: 236 mg (71%). White crystals. Mp 105‒107 °C (EtOAc). IR (KBr): 3216, 2988, 1467, 1230, 1058, 1024, 973 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.54 (d, J = 7.9 Hz, 1H, H-7), 7.24‒7.20 (m, 1H, H-6), 6.97 (dd, J1 = 7.9 Hz, J2 = 11.8 Hz, 1H, H-5), 5.50 (d, J = 10.4 Hz, 1H, CH), 4.22‒4.15 (m, 2H, CH2), 4.15‒4.11 (m, 1H, CH2), 4.11‒4.07 (m, 1H, CH2), 2.53 (dd, J1 = 2.5 Hz, J2 = 3.0 Hz, 1H, CH3), 1.30 (t, J = 7.1 Hz, 6H, 2×CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 158.8 (dd, J1 = 1.5 Hz, J2 = 251.7 Hz, C-4), 141.7 (dd, J1 = 1.6 Hz, J2 = 6.6 Hz, C-7a), 134.8 (dd, J1 = 1.1 Hz, J2 = 4.9 Hz, C-2), 128.7 (dd, J1 = 3.0 Hz, J2 = 15.1 Hz, C-3a), 128.1 (dd, J1 = 4.5 Hz, J2 = 10.3 Hz, C-3), 125.0 (dd, J1 = 1.0 Hz, J2 = 7.7 Hz, C-6), 118.3 (dd, J1 = 1.1 Hz, J2 = 3.9 Hz, C-7), 109.8 (d, J = 20.6 Hz, C-5), 65.9 (d, J = 168.2 Hz, CH), 64.1 (d, J = 7.0 Hz, CH2), 63.2 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.5 Hz, CH2CH3), 16.4 (d, J = 5.9 Hz, CH2CH3), 14.5 (dd, J1 = 6.6 Hz, J2 = 1.1 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.9 ppm. HRMS: [M]+ Calcd for C14H18FO4PS 332.0648; Found 332.0649.

3.2.12. Diethyl [(5-fluoro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2l)
Yield: 264 mg (79%). White crystals. Mp 125‒126 °C (EtOAc). IR (KBr): 3211, 1605, 1445, 1230, 1194, 1051, 1019, 801 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.71 (dd, J1 = 4.9 Hz, J2 = 8.7 Hz, 1H, H-7), 7.31 (dd, J1 = 2.4 Hz, J2 = 9.7 Hz, 1H, H-4), 7.08 (dt, Jd = 2.4 Hz, Jt = 8.8 Hz, 1H, H-6), 5.51 (dd, J1 = 4.3 Hz, J2 = 10.6 Hz, 1H, CH), 4.90 (br s, 1H, OH), 4.19–4.14 (m, 2H, CH2), 4.14–4.06 (m, 2H, CH2), 2.33 (d, J = 2.9 Hz, 3H, CH3), 1.29 (t, J = 7.2 Hz, 3H, CH2CH3), 1.28 (t, J = 7.1 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 160.7 (d, J = 241.3 Hz, C-5), 141.3 (dd, J1 = 2.9 Hz, J2 = 8.7 Hz, C-3a), 137.3 (d, J = 5.0 Hz, C-2), 134.4 (C-7a), 129.1 (dd, J1 = 4.3 Hz, J2 = 10.1 Hz, C-3), 123.5 (d, J = 9.2 Hz, C-7), 113.1 (d, J = 25.1 Hz, C-6), 107.4 (d, J = 22.9 Hz, C-4), 66.3 (d, J = 167.9 Hz, CH) 64.0 (d, J = 7.0 Hz, CH2), 63.2 (d, J = 7.5 Hz, CH2), 16.4 (d, J = 5.5 Hz, CH2CH3), 16.3 (d, J = 5.8 Hz, CH2CH3), 12.3 (d, J = 1.1 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.9 ppm. HRMS: [M]+ Calcd for C14H18FO4PS 332.0648; Found 332.0637.
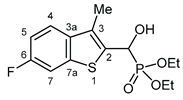
3.2.13. Diethyl [(6-fluoro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2m)
Yield: 169 mg (51%). White crystals. Mp 139‒140 °C (EtOAc). IR (KBr): 3212, 2925, 1602, 1469, 1230, 1194, 1053, 1021, 965, 854 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.60 (dd, J1 = 8.7 Hz, J2 = 5.1 Hz, 1H, H-4), 7.49 (dd, J1 = 8.7, J2 = 1.9 Hz, 1H, H-7), 7.12 (td, J1 = 8.7 Hz, J2 = 1.9 Hz, 1H, H-5), 5.47 (dd, J1 = 10.5 Hz, J2 = 3.5 Hz, 1H, CH), 4.23‒4.21 (m, 1H, OH), 4.21‒4.14 (m, 2H, CH2), 4.14‒4.07 (m, 2H, CH2), 2.38 (d, J = 3.0 Hz, 3H, CH3), 1.30 (t, J = 7.1 Hz, 6H, 2×CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 160.6 (dd, J1 = 244.3 Hz, J2 = 1.3 Hz, C-6), 140.1 (dd, J1 = 10.2 Hz, J2 = 1.6 Hz, C-7a), 136.6 (dd, J1 = 3.1 Hz, J2 = 1.3 Hz, C-3a), 133.8 (dd, J1 = 5.2 Hz, J2 = 3.7 Hz, C-2), 129.1 (d, J = 10.1 Hz, C-3), 122.8 (dd, J1 = 9.2 Hz, J2 = 1.3 Hz, C-4), 112.8 (d, J = 24.1 Hz, C-5), 108.5 (dd, J1 = 25.2 Hz, J2 = 1.0 Hz, C-7), 66.3 (d, J = 7.0 Hz, CH), 63.9 (d, J = 7.0 Hz, CH2), 63.2 (d, J = 7.4 Hz, CH2), 16.5 (d, J = 5.6 Hz, CH2CH3), 16.4 (d, J = 5.9 Hz, CH2CH3), 12.4 (d, J = 1.3 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.9 ppm. HRMS: [M]+ Calcd for C14H18FO4PS 332.0648; Found 332.0647.
3.2.14. Diethyl [(7-fluoro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2n)
Yield: 264 mg (79%). White crystals. Mp 134‒136 °C (EtOAc). IR (KBr): 3216, 1551, 1473, 1229, 1196, 1058, 1025, 779, 713 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.45 (d, J = 8.0 Hz, H-4), 7.34‒7.30 (m, 1H, H-5), 7.03 (t, J = 8.8 Hz, H-6), 5.54 (dd, J1 = 4.4 Hz, J2 = 10.6 Hz, CH), 5.08 (br s, 1H, OH), 4.23‒4.20 (m, 2H, CH2), 4.16‒4.12 (m, 1H, CH2), 4.10‒4.02 (m, 1H, CH2), 2.38 (d, J = 2.38 Hz, CH3), 1.31 (t, J = 7.1 Hz, CH2CH3), 1.28 (t, J = 7.1 Hz, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 157.6 (dd, J1 = 1.3 Hz, J2 = 246.7 Hz, C-7), 143.6 (dd, J1 = 3.1 Hz, J2 = 4.5 Hz, C-3a), 131.6 (dd, J1 = 1.3 Hz, J2 = 5.0 Hz, C-2), 129.7 (dd, J1 = 2.1 Hz, J2 = 10.0 Hz, C-3), 125.8 (dd, J1 = 1.5 Hz, J2 = 18.3 Hz, C-7a), 125.2 (d, J = 6.8 Hz, C-5), 117.5 (dd, J1 = 1.7 Hz, J2 = 3.0 Hz, C-4), 109.5 (d, J = 18.6 Hz, C-6), 66.2 (d, J = 167.9 Hz, CH), 64.1 (d, J = 7.0 Hz, CH2), 63.2 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.5 Hz, CH2CH3), 16.4 (d, J = 5.8 Hz, CH2CH3), 12.6 (CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.8 ppm. HRMS: [M]+ Calcd for C14H18FO4PS 332.0648; Found 332.0638.

3.2.15. Diethyl [(5,7-difluoro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2o)
Yield: 262 mg (75%). White crystals. Mp 150‒153 °C (EtOAc). IR (KBr): 3205, 2977, 1620, 1578, 1423, 1228, 1193, 1136, 1059, 1019 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.15 (dd, J1 = 9.1 Hz, J2 = 2.0 Hz, 1H, H-4), 6.85 (td, Jt = 9.3 Hz, Jd = 1.5 Hz, 1H, H-6), 5.51 (dd, J1 = 10.7 Hz, J2 = 4.0 Hz, 1H, CH), 5.27‒5.21 (m, 1H, OH), 4.25‒4.20 (m, 2H, CH2), 4.19‒4.11 (m, 1H, CH2), 4.11‒4.05 (m, 1H, CH2), 2.34 (d, J = 3.2 Hz, 3H, CH3), 1.32 (t, J = 7.1 Hz, 3H, CH2CH3), 1.29 (t, J = 7.1 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 160.7 (dd, J1 = 243.5 Hz, J2 = 10.5 Hz, C-5), 157.1 (ddd, J1 = 248.7, J2 = 13.6 Hz, J3 = 1.3 Hz, C-7), 143.0 (m, C-3a), 138.4 (dd, J1 = 4.9 Hz, J2 = 1.2 Hz, C-2), 129.3 (ddd, J1 = 9.8 Hz, J2 = 4.7 Hz, J3 = 2.3 Hz, C-3), 121.6 (d, J = 18.5 Hz, C-7a), 103.4 (dd, J1 = 23.0, J2 = 2.5 Hz, C-4), 99.8 (dd, J1 = 28.9 Hz, J2 = 22.6 Hz, C-6), 66.2 (d, J = 167.8 Hz, CH), 64.3 (d, J = 7.0 Hz, CH2), 63.3 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.4 Hz, CH2CH3), 16.4 (d, J = 5.9 Hz, CH2CH3), 12.5 (CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.5 ppm. HRMS: [M]+ Calcd for C14H17F2O4PS 350.0553; Found 350.0553.
3.2.16. Diethyl [(6,7-difluoro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2p)
Yield: 197 mg (56%). White crystals. Mp 176‒179 °C (EtOAc). IR (KBr): 3226, 2984, 1623, 1497, 1440, 1268, 1205, 1018 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.36 (dd, J1 = 8.7 Hz, J2 = 3.9 Hz, H-4), 7.22‒7.18 (m, 1H, H-5), 5.49 (dd, J1 = 10.4 Hz, J2 = 3.7 Hz, 1H, CH), 4.91‒4.85 (m, 1H, OH), 4.25‒4.18 (m, 2H, CH2), 4.18‒4.12 (m, 1H, CH2), 4.12‒4.07 (m, 1H, CH2), 2.36 (d, J = 3.3 Hz, 3H, CH3), 1.32 (t, J = 7.1 Hz, 3H, CH2CH3), 1.30 (t, J = 7.1 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 147.1 (ddd, J1 = 245.2 Hz, J2 = 11.1 Hz, J3 = 1.3 Hz, C-6), 145.1 (ddd, J1 = 250.0 Hz, J2 = 15.5 Hz, J3 = 1.3 Hz, C-7), 138.8 (m, C-3a), 135.7 (t, J = 3.7 Hz, C-2), 129.3 (d, J = 10.0 Hz, C-3), 127.7 (d, J = 14.5 Hz, C-7a), 117.2 (m, C-4), 114.6 (d, J = 19.9 Hz, C-5), 66.2 (d, J = 168.0 Hz, CH), 64.2 (d, J = 7.0 Hz, CH2), 63.3 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.4 Hz, CH2CH3), 16.4 (d, J = 5.8 Hz, CH2CH3), 12.5(CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.6 ppm. HRMS: [M]+ Calcd for C14H17F2O4PS 350.0553; Found 350.0556.
3.2.17. Diethyl [(5-chloro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2q)
Yield: 276 mg (79%). White crystals. Mp 130‒131 °C (EtOAc). IR (KBr): 3279, 2982, 1444, 1231, 1050, 977, 857, 617 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.70 (d, J = 8.5 Hz, 1H, H-7), 7.63 (s, 1H, H-4), 7.30‒7.27 (m, 1H, H-6), 5.50 (d, J = 10.7 Hz, 1H, CH), 4.85 (br s, 1H, OH), 4.20‒4.14 (m, 2H, CH2), 4.14‒4.10 (m, 1H, CH2), 4.10‒4.03 (m, 1H, CH2), 2.34 (d, J = 3.1 Hz, 3H, CH3), 1.28 (t, J = 7.0 Hz, 6H, 2 × CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 141.3 (d, J = 3.0 Hz, C-4a), 137.4 (d, J = 1.6 Hz, C-7a), 136.9 (d, J = 5.0 Hz, C-2), 130.2 (C-5), 128.7 (d, J = 10.0 Hz, C-3), 124.7 (C-6), 123.4 (d, J = 0.7 Hz, C-7), 121.4 (d, J = 1.2 Hz, C-4), 66.2 (d, J = 167.8 Hz, CH), 64.1 (d, J = 6.9 Hz, CH2), 63.2 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.6 Hz, CH2CH3), 16.4 (d, J = 5.7 Hz, CH2CH3), 12.3 (d, J = 1.2 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.8 ppm. HRMS: [M]+ Calcd for C14H18ClO4PS 348.0352; Found 348.0339.
3.2.18. Diethyl [(6-chloro-3-methyl-1-benzothiophen-2-yl)(hydroxy)methyl]phosphonate (2r)
Yield: 229 mg (66%). White crystals. Mp 133‒135 °C (MeCN). IR (KBr): 3213, 1453, 1228, 1061, 1020, 809, 758 cm-1. 1H NMR (DMSO-d6, 600 MHz): δ 8.08 (d, J = 1.7 Hz, 1H, H-7), 7.74 (d, J = 8.6 Hz, 1H, H-4), 7.41 (dd, J1 = 1.8 Hz, J2 = 8.6 Hz, 1H, H-5), 6.76 (dd, J1 = 4.5 Hz, J2 = 17.6 Hz, 1H, OH), 5.41 (dd, J1 = 4.5 Hz, J2 = 17.6 Hz, 1H, CH), 4.07–4.02 (m, 2H, CH2), 4.02–3.96 (m, 2H, CH2), 2.34 (d, J = 3.0 Hz, 3H, CH3), 1.21 (t, J = 7.1 Hz, 3H, CH2CH3), 1.18 (t, J = 7.0 Hz, 3H, CH2CH3) ppm. 13C NMR (DMSO-d6, 150 MHz): δ 139.9 (C-7a), 138.9 (d, J = 2.7 Hz, C-3a), 138.8 (d, J = 3.3 Hz, C-2), 129.3 (C-6), 128.0 (d, J = 9.9 Hz, C-3), 124.6 (C-5), 123.3 (C-4), 122.1 (C-7), 65.0 (d, J = 172.1 Hz, CH), 62.8 (d, J = 7.2 Hz, CH2), 62.4 (d, J = 6.7 Hz, CH2), 16.6 (d, J = 5.5 Hz, CH2CH3), 16.5 (d, J = 5.5 Hz, CH2CH3), 12.2 (CH3) ppm. 31P NMR (DMSO-d6, 242 MHz): δ 19.9 ppm. HRMS: [M]+ Calcd for C14H18ClO4PS 348.0352; Found 348.0341.
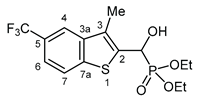
3.2.19. Diethyl {hydroxy [3-methyl-5-(trifluoromethyl)-1-benzothiophen-2-yl]methyl}phosphonate (2s)
Yield: 253 mg (66%). White crystals. Mp 138‒139 °C (EtOAc). IR (KBr): 3241, 2987, 1446, 1350, 1330, 1295, 1203, 1104, 1023, 809 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.92 (s, 1H, H-4), 7.90 (d, J = 8.4 Hz, 1H, H-7), 7.55 (d, J = 8.3 Hz, 1H, H-6), 5.54 (d, J = 10.4 Hz, 1H, CH), 4.23‒4.18 (m, 2H, CH2), 4.16‒4.12 (m, 1H, CH2), 4.12‒4.05 (m, 1H, CH2), 2.42 (d, J = 3.0 Hz, 3H, CH3), 1.29 (t, J = 7.4 Hz, 3H, OCH2CH3), 1.30 (t, J = 7.3 Hz, 3H, OCH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 142.3 (C-7a), 139.7 (d, J = 3.1 Hz, C-3a), 137.0 (d, J = 5.3 Hz, C-2), 129.5 (d, J = 9.9 Hz, C-3), 126.5 (q, J = 32.3 Hz, C-5), 124.7 (q, J = 272.0 Hz, CF3), 122.9 (C-7), 120.6 (d, J = 2.5 Hz, C-6), 118.9 (d, J = 2.9 Hz, C-4), 66.2 (d, J = 167.5 Hz, CH), 64.2 (d, J = 7.0 Hz, CH2), 63.3 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.4 Hz, OCH2CH3), 16.4 (d, J = 5.8 Hz, OCH2CH3), 12.3 (d, J = 1.2 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.7 ppm. HRMS: [M]+ Calcd for C15H18F3O4PS 382.0616; Found 382.0609.
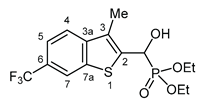
3.2.20. Diethyl {hydroxy [3-methyl-6-(trifluoromethyl)-1-benzothiophen-2-yl]methyl}phosphonate (2t)
Yield: 265 mg (69%). White crystals. Mp 141‒143 °C (EtOAc). IR (KBr): 3229, 2991, 1327, 1230, 1159, 1128, 1056, 1021, 822 cm-1. 1H NMR (CDCl3, 600 MHz): δ 8.10 (s, 1H, H-7), 7.76 (d, J = 8.4 Hz, 1H, H-4), 7.60 (dd, J1 = 8.4 Hz, J2 = 0.9 Hz, H-5), 5.53 (d, J = 10.6 Hz, 1H, CH), 4.37 (br s, 1H, OH), 4.23‒4.17 (m, 2H, CH2), 4.16‒4.05 (m, 2H, CH2), 2.43 (d, J = 3.2 Hz, 3H, CH3), 1.31 (t, J = 7.3 Hz, 3H, CH2CH3), 1.30 (t, J = 7.3 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 142.4 (C-7a), 138.8 (d, J = 1.6 Hz, C-3a), 138.2 (d, J = 5.3 Hz, C-2), 129.2 (d, J = 9.7 Hz, C-3), 126.5 (q, J = 32.3 Hz, C-6), 124.5 (q, J = 272.1 Hz, CF3), 122.1 (d, J = 1.4 Hz, C-4), 120.7 (d, J = 3.0 Hz, C-5), 119.8 (d, J = 3.4 Hz, C-7), 65.9 (d, J = 166.7 Hz, CH), 64.1 (d, J = 7.0 Hz, CH2), 63.3 (d, J = 7.5 Hz, CH2), 16.5 (d, J = 5.5 Hz, CH2CH3), 16.4 (d, J = 5.8 Hz, CH2CH3), 12.4 (d, J = 1.3 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.5 ppm. HRMS: [M]+ Calcd for C15H18F3O4PS 382.0616; Found 382.0618.
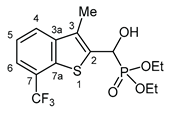
3.2.21. Diethyl {hydroxy [3-methyl-7-(trifluoromethyl)-1-benzothiophen-2-yl]methyl}phosphonate (2u)
Yield: 299 mg (78%). White crystals. Mp 153‒157 °C (EtOAc). IR (KBr): 3441, 3237, 2985, 1629, 1446, 1312, 1246, 1213, 1155, 1113, 1026 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.82 (d, J = 8.0 Hz, 1H, H-4), 7.63 (d, J = 7.4 Hz, 1H, H-6), 7.46 (t, J = 7.7 Hz, 1H, H-5), 5.55 (d, J = 10.3 Hz, 1H, CH), 4.27‒4.20 (m, 2H, CH2), 4.18‒4.11 (m, 1H, CH2), 4.11‒4.04 (m, 2H, CH2, OH), 2.40 (d, J = 3.2 Hz, 3H, CH3), 1.23 (t, J = 7.1 Hz, 3H, CH2CH3), 1.29 (t, J = 7.1 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 141.8 (d, J = 3.1 Hz, C-3a), 136.5 (d, J = 5.3 Hz, C-2), 135.6 (m, C-7a), 128.8 (d, J = 9.8 Hz, C-3), 125.2 (C-4), 124.6 (qd, J1 = 32.7 Hz, J2 = 1.2 Hz, C-7), 124.2 (q, J = 273.4 Hz, CF3), 123.7 (C-5), 122.1 (q, J = 4.5 Hz, C-6), 66.3 (d, J = 167.4 Hz, CH), 64.3 (d, J = 7.0 Hz, CH2), 63.3 (d, J = 7.5 Hz, CH2), 16.4 (d, J = 5.5 Hz, CH2CH3), 16.3 (d, J = 5.9 Hz, CH2CH3), 12.4 (d, J = 1.3 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.6 ppm. HRMS: [M]+ Calcd for C15H18F3O4PS 382.0616; Found 382.0607.
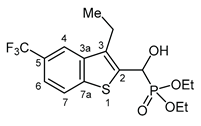
3.2.22. Diethyl [(3-ethyl-5-trifluoromethyl-1-benzothiophen-2-yl)](hydroxy)methyl]phosphonate (2v)
Yield: 306 mg (77%). White crystals. Mp 156‒157 °C (EtOAc). IR (KBr): 3258, 2975, 1756, 1610, 1567, 1439, 1394, 1354, 1328, 1235, 1221, 1116, 978 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.95 (s, 1H, H-4), 7.91 (d, J = 8.4 Hz, 1H, H-7), 7.55 (d, J = 8.4 Hz, 1H, H-6), 5.53 (d, J = 10.9 Hz, 1H, CH), 4.20‒4.17 (m, 2H, OCH2CH3), 4.16‒4.11 (m, 2H, OCH2CH3), 2.97‒2.95 (m, 1H, CH2CH3), 2.91‒2.89 (m, 1H, CH2CH3), 1.29 (m, 6H, OCH2CH3), 1.28 (m, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 142.9 (C-7a), 138.7 (d, J = 2.6 Hz, C-3a), 136.6 (d, J = 4.6 Hz, C-2), 136.3 (d, J = 10.4 Hz, C-3), 126.5 (q, J = 32.2 Hz, C-5), 124.6 (q, J = 272.0 Hz, CF3), 123.1 (C-7), 120.6 (m, C-6), 118.9 (m, C-4), 65.8 (d, J = 168.0 Hz, CH), 64.1 (d, J = 7.0 Hz, OCH2CH3), 63.3 (d, J = 7.5 Hz, OCH2CH3), 20.1 (CH2CH3), 16.5 (d, J = 5.5 Hz, OCH2CH3), 16.4 (d, J = 5.7 Hz, OCH2CH3), 14.4 (d, J = 2.6 Hz, CH2CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 19.6 ppm. HRMS: [M+H]+ Calcd for C16H21F3O4PS 397.0850; Found 397.0842.
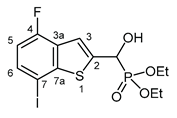
3.2.23. Diethyl [(4-fluoro-7-iodo-1-benzothiopen-2-yl)(hydroxy)methyl]phosphonate (2w)
Yield: 300 mg (68%). White crystals. Mp 145‒146 °C (EtOAc). IR (KBr): 3225, 2983, 1458, 1251, 1203, 1042, 977 cm-1. 1H NMR (CDCl3, 600 MHz): δ 7.72 (d, J = 3.5 Hz, 1H, H-3), 7.57 (dd, J1 = 8.3 Hz, J2 = 4.6 Hz, 1H, H-6), 6.81 (dd, J1 = 9.7 Hz, J2 = 8.3 Hz, 1H, H-5), 5.35 (d, J = 12.1 Hz, 2H, CH, OH), 4.25‒4.15 (m, 4H, 2 × CH2), 1.35 (t, J = 7.1 Hz, 3H, CH3), 1.32 (t, J = 7.1 Hz, 3H, CH3) ppm. 13C NMR (CDCl3, 150 MHz): δ 157.8 (dd, J1 = 252.8 Hz, J2 = 1.1 Hz, C-4), 148.3 (dd, J1 = 6.4 Hz, J2 = 1.5 Hz, C-7a), 142.1 (d, J = 1.7 Hz, C-2), 134.1 (d, J = 6.4 Hz, C-6), 128.0 (dd, J1 = 20.0 Hz, J2 = 2.6 Hz, C-3a), 118.2 (d, J = 8.0 Hz, C-3), 111.4 (d, J = 20.1 Hz, C-5), 80.0 (d, J = 3.0 Hz, C-7), 67.5 (d, J = 165.5 Hz, CH), 64.2 (d, J = 7.1 Hz, CH2), 63.3 (d, J = 7.4 Hz, CH2), 16.5 (d, J = 5.3 Hz, CH3), 16.4 (d, J = 5.3 Hz, CH3) ppm. 31P NMR (CDCl3, 242 MHz): δ 18.6 ppm. HRMS: [M+1]+ Calcd for C13H16FIO4PS 444.9536; Found 444.9531.
3.3. X-Ray Experimental
3.3.1. Single Crystal X-Ray Diffraction Studies
Single crystals of compound 2f, suitable for X-ray diffraction, were obtained by slow evaporation of ethyl acetate solution. The crystals were introduced into perfluorinated oil and a suitable single crystal was carefully mounted on the top of a thin glass wire. Data collection was performed with an Oxford Xcalibur 3 diffractometer equipped with a Spellman generator (50 kV, 40 mA) and a Kappa CCD detector, operating with Mo-Kα radiation (λ = 0.71071 Ǻ).
Data collection and data reduction were performed with the CrysAlisPro software [36].) Absorption correction using the multiscan method [36] was applied. The structures were solved with SHELXS-97 [37], refined with SHELXL-97 [38] and finally checked using PLATON [39]. Details for data collection and structure refinement are summarized in Table 1.
CCDC- 2440279 contains supplementary crystallographic data for this compound. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.

Table 3.
Details for X-ray data collection and structure refinement for compound 2f.
| 2f | |
|---|---|
| Empirical formula | C13H16ClO4PS |
| Formula mass | 334.74 |
| T[K] | 123(2) |
| Crystal size [mm] | 0.40 × 0.25 × 0.20 |
| Crystal description | colorless block |
| Crystal system | triclinic |
| Space group | P-1 |
| a [Ǻ] | 7.6579(4) |
| b [Ǻ] | 7.8950(4) |
| c [Ǻ] | 13.9491(6) |
| α [°] | 78.234(4) |
| β [°] | 85.603(4) |
| γ [°] | 65.219(5) |
| V [Ǻ3] | 749.57(7) |
| Z | 2 |
| ρcalcd. [g cm-3] | 1.483 |
| μ [mm-1] | 0.509 |
| F(000) | 348 |
| Θ range [°] | 2.89 – 25.24 |
| Index ranges | -10 ≤ h ≤ 10 |
| -10 ≤ k ≤ 10 | |
| -18 ≤ l ≤ 18 | |
| Reflns. collected | 12988 |
| Reflns. obsd. | 3270 |
| Reflns. unique | 3718 (Rint = 0.0204) |
| R1, wR2 (2σ data) | 0.0405, 0.1050 |
| R1, wR2 (all data) | 0.0471, 0.1111 |
| GOOF on F2 | 1.029 |
| Peak/hole [e Ǻ-3] | 0.754 / -0.457 |
3.4. Bioactivity Experimental
3.4.1. Cell Culturing
In our study, four different human cell lines were tested to screen the antitumor effect of the α-hydroxyphosphonates. The myeloma (U266), colon adenocarcinoma cell line (HT-29) and the metastatic melanoma (A2058) were obtained from the European Collection of Authenticated Cell Cultures (ECACC, Salisbury, UK). The lung squamous carcinoma cells (EBC-1) were purchased from the Japanese Collection of Research Bioresources Cell Bank (JCRB Cell Bank, Osaka, Japan). The EBC-1, A2058 and HT-29 cell lines have adherent growth properties, but the U266 cells (85051003 ECACC) grow in suspension.
EBC-1 cells were cultured in DMEM medium (Sigma Ltd., St. Louis, MO, USA), while HT-29, A2058 and U266 cells were cultured in RPMI 1640 (Sigma Ltd., St. Louis, MO, USA). Both media were supplemented with 10% fetal bovine serum (Invitrogen Corporation, New York, NY, USA), 1% L-glutamine (Invitrogen Corporation, New York, NY, USA), and 1% penicillin/streptomycin (Invitrogen Corporation, New York, NY, USA). The medium for EBC-1 was further completed with 1% non-essential amino acids (Sigma Ltd., St. Louis, MO, USA) and 1% sodium pyruvate (Sigma Ltd., St. Louis, MO, USA).
3.4.2. Cell Viability Assays
Compounds were dissolved in dimethyl sulfoxide (DMSO; AppliChem GmbH, Darmstadt, Germany) to prepare stock solutions at a concentration of 10⁻¹ M. During experiments, the final DMSO concentration was kept below 1% (v/v). Stock solutions were stored at −80 °C and working solutions were freshly prepared prior to each experiment. Adherent EBC-1, A2058, and HT-29 cells were seeded in transparent 96-well plates (Sarstedt AG, Nümbrecht, Germany) at a density of 1 × 10⁵ cells/mL. After overnight incubation, cells were treated with the test compounds at a final concentration of 100 μM, alongside appropriate controls (medium only and vehicle control with DMSO). Following a 72-hour incubation period with the compounds, 11 μL of alamarBlue reagent (Thermo Scientific, Waltham, MA, USA) was added to each well, and plates were incubated for an additional 3 hours. Fluorescence was measured using a Fluoroskan FL Microplate Fluorometer and Luminometer (Thermo Scientific, Waltham, MA, USA) at excitation/emission wavelengths of 530–560 nm and 590 nm, respectively.
Due to technical difficulties in assessing U266 cell viability using the alamarBlue assay, the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) was employed instead, following the protocol described in our previous publication [40]. All experiments were conducted in triplicate, and results were normalized to the DMSO vehicle control using OriginPro 8 software (OriginLab Corporation, Northampton, MA, USA) and reported as mean ± standard deviation (SD).
4. Conclusions
Encouraged by our earlier experiences that the modification of α-hydroxyphosphonates may lead to derivatives displaying significant biological activity including cytotoxic effects, 23 members of a new α-hydroxyphosphonate family with the benzothiophene scaffold were synthesized by the Pudovik reaction of benzo[b]thiophene-2-carboxaldehydes and diethyl phosphite performed at 26 °C in the presence of triethylamine as the catalyst. All compounds were fully characterized including one single crystal X-ray analysis. With a few exceptions, most of the compounds affected the viability of the tumor cell lines, such as the U266 (multiple myeloma), the EBC-1 (lung cancer), the A2058 (melanoma) and the HT-29 (colon cancer) cell cultures - with U266 cells displaying the highest sensitivity. Single “halogenation” generally had weaker effects as compared to double “halogenation” or substitution with a trifluoromethyl group. Notably, the meta-substituted hydroxyphosphonate bearing a trifluoromethyl group showed the strongest antiproliferative effect on three of the four cell lines, underscoring its potential as a promising lead compound.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org, X-ray data for 7-chlorobenzothiophenyl-α-hydroxy-methylphosphonate (2f); 31P, 13C 1H NMR and IR spectra for compounds 2a-w synthesized; HSQC, HMBC and COSY NMR spectra for the compounds synthesized; SI Table 1. Selected bond lengths (Å) of hydroxy-methylphosphonate 2f; SI Table 2. Selected bond angles (°) of hydroxy-methylphosphonate 2f; SI Table 3. Selected torsion angles (°) of hydroxy-methylphosphonate 2f.
Author Contributions
Conceptualization, M.M, G.K. and L.K.; methodology, M.M., Z.G. and A.T..; formal analysis, Z.G., A.T. and K.K.; investigation, A.S.K, T.J. and A.T; resources, M.M. and G.K.; data curation, K.K., A.T. and Z.S.; writing—original draft preparation, M.M, G.K, Z.G., K.K, L.K and A,T.; writing—review and editing, M.M. G.K., L.K. and A.T.; visualization, Z.S.; supervision, M.M., G.K., and L.K.; project administration, M.M., G.K. and L.K; funding acquisition, G.K. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the National Research, Development and Innovation Office (NKKP-ADVANCED 149447).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, X.B.; Shi, D.Q. Synthesis and biological activity of novel phosphonate derivatives containing of pyridyl and 1,2,3-triazole rings. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 1134–1144. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Chiral hydroxyl phosphonates: Synthesis, configuration and biological properties. Russ. Chem. Rev. 2006, 75, 227–253. [Google Scholar] [CrossRef]
- Song, H.; Mao, H.; Shi, D. Synthesis and Herbicidal Activity of α-Hydroxy Phosphonate Derivatives Containing Pyrimidine Moiety. Chin. J. Chem. 2010, 28, 2020–2024. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L. P.; Ning, B. K.; Mao, M. Z.; Xue, C.; Wang, H. Y. Synthesis and insecticidal activities of O,O-dialkyl-2-[3-bromo-1-(3-chloropyridin-2-yl)-1H-pyrazole-5-carbonyloxy] (aryl)methylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1362–1367. [Google Scholar] [CrossRef]
- Lorenz, W.; Henglein, A.; Schrader, G. The New Insecticide O,O-Dimethyl 2,2,2-Trichloro-1-hydroxyethylphosphonate. J. Am. Chem. Soc. 1955, 77, 2554–2556. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Y.; Peng, H.; He, H. W.; Lu, X. T. Synthesis and herbicidal activity of α-[(substituted phenoxybutyryloxy or valeryoxy)]alkylphosphonatesand 2-(substituted phenoxybutyryloxy)alkyl-5,5-dimethyl-1,3,2-dioxaphosphinan-2-one containing fluorine. J. Fluorine Chem. 2017, 193, 8–16. [Google Scholar] [CrossRef]
- Pokalwar, R.U.; Hangarge, R.V.; Maske, P.V.; Shingare, M.S. Synthesis and antibacterial activities of α-hydroxyphosphonates and α-acetyloxyphosphonates derived from 2-chloroquinoline-3-carbaldehyde. Arkivoc 2006, 11, 196–204. [Google Scholar] [CrossRef]
- Kategaonkar, A.H.; Pokalwar, R.U.; Sonar, S.S.; Gawali, V.U.; Shingate, B.B.; Shingare, M.S. Synthesis, in vitro antibacterial and antifungal evaluations of new α-hydroxyphosphonate and new α-acetoxyphosphonate derivatives of tetrazolo [1, 5-a] quinoline. Eur. J. Med. Chem. 2010, 45, 1128–1132. [Google Scholar] [CrossRef]
- Reddy, G. S.; Sundar, C.S.; Prasad, S.S.; Dadapeer, E.D.; Raju, C.N.; Reddy, C.S. Synthesis, spectral characterization and antimicrobial activity of α-hydroxyphosphonates. Der Pharma Chemica 2012, 4, 2208–2213, http://derpharmachemica.com/vol4-iss6/DPC-2012-4-6-2208-2213.pdf. [Google Scholar]
- Sampath, S.C.; Raju, N.C.; Rao, V. An efficient synthesis, spectral characterization, antimicrobial, and antioxidant activities of novel α-hydroxyphosphonates and α-hydroxyphosphinates. Phosphorus Sulfur Silicon 2016, 191, 95–99. [Google Scholar] [CrossRef]
- Patil, N.S.; Deshmukh, G.B.; Patil, S.V.; Bholay, A.D.; Gaikwad, N.D. Synthesis and biological evaluation of novel N-aryl maleimide derivatives clubbed with α-hydroxyphosphonates. Eur. J. Med. Chem. 2014, 83, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.U.M.; Sundar, C.S.; Prasad, S.S.; Rani, C.R.; Reddy, C.S. Neat Synthesis and Anti-oxidant Activity of α-Hydroxyphosphonates. Bull. Korean Chem. Soc. 2011, 32, 3343–3347. [Google Scholar] [CrossRef]
- Naidu, K.R.M.; Kumar, K.S.; Arulselvan, P.; Reddy, C.B.; Lasekan, O. Synthesis of α-Hydroxyphosphonates and Their Antioxidant Properties. Arch. Pharm. Chem. Life Sci. 2012, 345, 957–963. [Google Scholar] [CrossRef]
- Yang, J.; Ma, J.; Che,W.; Li, M.; Li, G.; Song, B. Microwave-assisted synthesis and antitumor activity of salicyl acyloxy phosphonate derivatives. Chin. J. Org. Chem. 2014, 34, 2566–2571. [CrossRef]
- Bagchi, S.; Rathee, P.; Jayaprakash, V. Banerjee, S. Farnesyl Transferase Inhibitors as Potential Anticancer Agents. Mini-Rev. Med. Chem. 2018, 18, 1611–1623. [Google Scholar] [CrossRef]
- Al-Kali, A.; Gandhi, V.; Ayoubi, M.; Keating, M.; Ravandi, F. Forodesine: Review of Preclinical and Clinical Data. Future Oncol. 2010, 6, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Yokomatsu, T.; Abe, H.; Sato, M. Suemune, K.; Kihara, T.; Soeda, S.; Shimeno, H.; Shibuya, S. Synthesis of 1,1-difluoro-5-(1H-9-purinyl)-2-pentenylphosphonic acids and the related methano analogues. Remarkable effect of the nucleobases and the cyclopropane rings on inhibitory activity toward purine nucleoside phosphorylase. Bioorg. Med. Chem. 1998, 6, 2495–2505. [CrossRef]
- Kalla, R.M.N.; Lee, H.R.; Cao, J.; Yoo, J.W.; Kim, I. Phospho sulfonic acid: an efficient and recyclable solid acid catalyst for the solvent-free synthesis of α-hydroxyphosphonates and their anticancer properties. New J. Chem. 2015, 39, 3916–3922. [Google Scholar] [CrossRef]
- Patel, D.V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C.A.; Rogers, W.L.; Smith, S.A.; DeForrest, J.M.; Oehl, R.S.; Petrillo Jr., E. W..alpha.-Hydroxy Phosphinyl-Based Inhibitors of Human Renin. J. Med. Chem. 1995, 38, 4557–4569. [Google Scholar] [CrossRef]
- Stowasser, B.; Budt, K.-H.; Jian-Qi, L.; Peyman, A.; Ruppert, D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett. 1992, 33, 6625–6628. [Google Scholar] [CrossRef]
- Prior, A.M.; Kim, Y.; Weerasekara, S.; Moroze, M.; Alliston, K.R.; Uy, R.A.; Groutas, W.C.; Chang, K.O.; Hua, D.H. Design, synthesis, and bioevaluation of viral 3C and 3C-like protease inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6317–6320. [Google Scholar] [CrossRef] [PubMed]
- Pompliano, D.L.; Rands, E.; Schaber, M.D.; Mosser, S.D.; Anthony, N.J.; Gibbs, J.B. Steady-state kinetic mechanism of ras farnesyl:protein transferase. Biochemistry 1992, 31, 3800–3807. [Google Scholar] [CrossRef] [PubMed]
- Frechette, R.F.; Ackerman, C.; Beers, S.; Look, R.; Moore, J. Novel hydroxyphosphonate inhibitors of CD-45 tyrosine phosphatase. Bioorg. Med. Chem. Lett. 1997, 7, 2169–2172. [Google Scholar] [CrossRef]
- Desai, J.; Wang, Y.; Wang, K.; Malwal, S.R.; Oldfield, E. Isoprenoid Biosynthesis Inhibitors Targeting Bacterial Cell Growth. Chem. Med. Chem. 2016, 11, 2205–2215. [Google Scholar] [CrossRef]
- Forlani, G.; Occhipinti, A.; Berlicki, Ł.; Dziedzioła, G.; Wieczorek, A.; Kafarski, P. Tailoring the Structure of Aminobisphosphonates To Target Plant P5C Reductase. J. Agric. Food Chem. 2008, 56, 3193–3199. [Google Scholar] [CrossRef] [PubMed]
- Pudovik, A.N.; Zametaeva, G.A. New synthesis of esters of phosphonic and thiophosphonic acids. XIII. Addition of diethyl thiophosphite to ketones and aldehydes. Izv. Akad. Nauk SSSR Ser. Khim. 1952, 1952, 932–939. [Google Scholar]
- Pudovik, A.N.; Konovalova, I.V. Addition reactions of esters of phosphorus(III) acids with unsaturated systems. Synthesis 1979, 2, 81–96. [Google Scholar] [CrossRef]
- Rádai, Z.; Keglevich, G. Synthesis and Reactions of α-Hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef]
- Keglevich, G.; Tóth, V.R.; Drahos, L. Microwave-Assisted Synthesis of α-Hydroxybenzylphosphonates and -benzylphosphine Oxides. Heteroatom Chem. 2011, 22, 15–17. [Google Scholar] [CrossRef]
- Texier-Boullet, F.; Foucaud, A. Synthesis of 1-Hydroxyalkanephosphonic Esters on Alumina. Synthesis 1982, 916, 25. [Google Scholar] [CrossRef]
- Keglevich, G.; Rádai, Z.; Kiss, N.Z. To date the greenest method for the preparation of α-hydroxyphosphonates from substituted benzaldehydes and dialkyl phosphites. Green Process Synth. 2017, 6, 197–201. [Google Scholar] [CrossRef]
- DIAMOND, Crystal Impact GbR. Version 3.2i; Bonn, Germany. 2014.
- Matsunaga, N,; Kaku, T.; Itoh, F.; Tanaka, T.; Hara, T.; Miki, H.; Iwasaki, M.; Aono, T.; Yamaoka, M.; Kusaka, M.; Tasaka, A. C17,20-Lyase inhibitors I. Structure-based de novo design and SAR study of C17,20-lyase inhibitors. Bioorg. Med. Chem. 2004, 12, 2251–2273. [CrossRef] [PubMed]
- Hur, W.; Rosen, H.; Gray, N.S. A benzo[b]thiophene-based selective type 4 S1P receptor agonist. Bioorg. Med. Chem. Lett. 2017, 27, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shigeno, M.; Fujii, Y.; Kajima, A.; Nozawa-Kumada, K.; Kondo, Y. Catalytic Deprotonative α-Formylation of Heteroarenes by an Amide Base Generated in Situ from Tetramethylammonium Fluoride and Tris(trimethylsilyl)amine. Org. Process. Res. Dev. 2019, 23, 443–451. [Google Scholar] [CrossRef]
- Program package ‘CrysAlisPro 1.171.40.82a (Rigaku OD, 2020)’.
- Sheldrick, G. M. (1997) SHELXS-97: Program for Crystal Structure Solution, University of Göttingen, Germany.
- Sheldrick, G. M. (1997) SHELXL-97: Program for the Refinement of Crystal Structures, University of Göttingen, Germany.
- Spek, A. L. (1999) PLATON: A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands.
- Szalai, Z.; Bednárik, J.; Tóth, BS.; Takács, A.; Tekula, S.; Kőhidai, L.; Karaghiosoff, K.; Drahos, L.; Keglevich, G. Cytotoxic Activity of Bisphosphonic Derivatives Obtained by the Michaelis-Arbuzov or the Pudovik Reaction. Pharmaceuticals (Basel). 2025, 18, 91. [Google Scholar] [CrossRef]
Figure 1.
Typical 1H–31P (blue) and 13C–31P (red) coupling constants (in Hz) observed in the synthesized α-hydroxyphosphonates, shown on the general structural formula.
Figure 1.
Typical 1H–31P (blue) and 13C–31P (red) coupling constants (in Hz) observed in the synthesized α-hydroxyphosphonates, shown on the general structural formula.
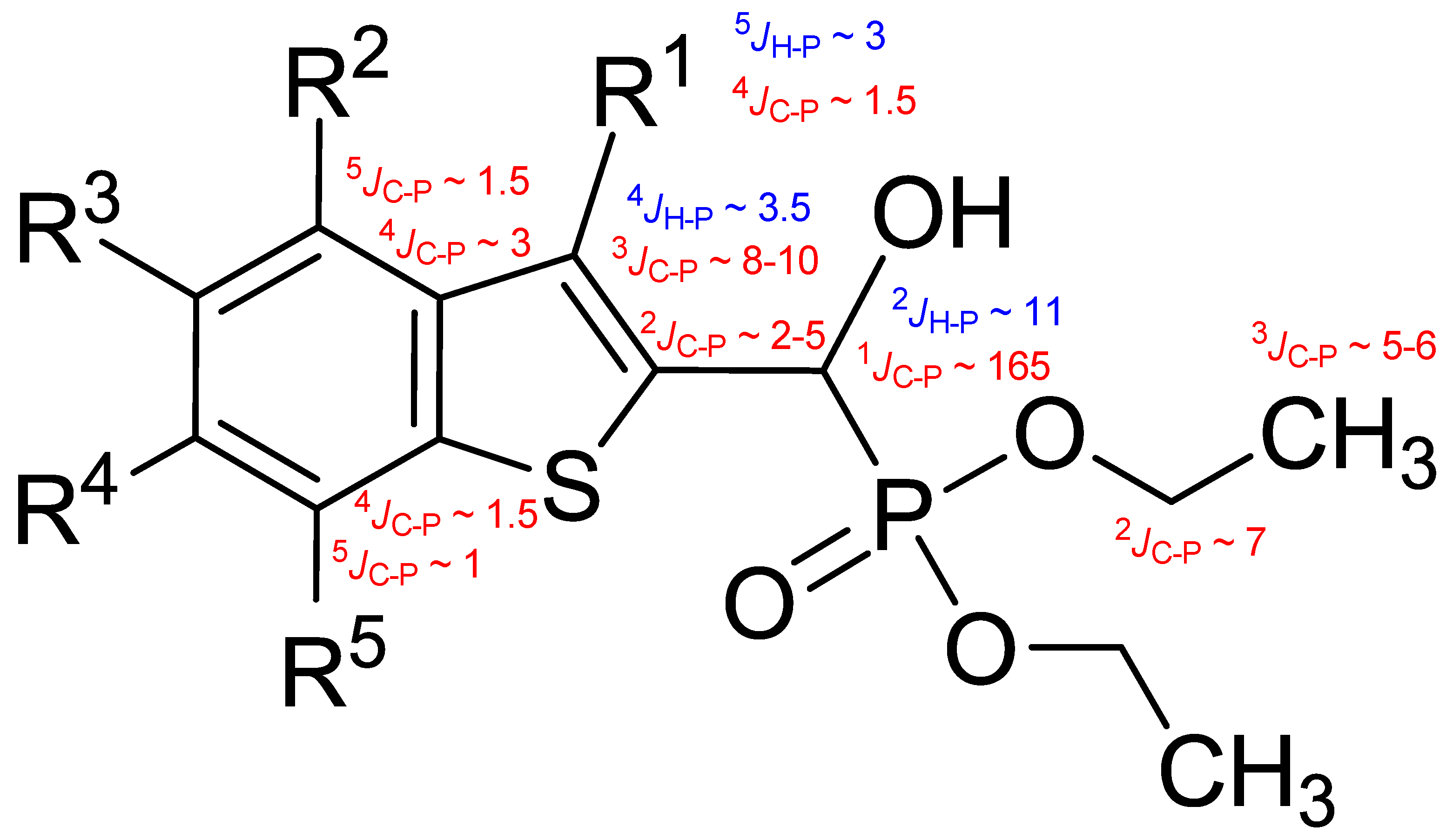
Figure 2.
Molecular structure of 7-chlorobenzothiophenyl-α-hydroxy-methylphosphonate (2f) in the crystal. DIAMOND [32] representation; thermal ellipsoids are drawn at 50 % probability level.
Figure 2.
Molecular structure of 7-chlorobenzothiophenyl-α-hydroxy-methylphosphonate (2f) in the crystal. DIAMOND [32] representation; thermal ellipsoids are drawn at 50 % probability level.
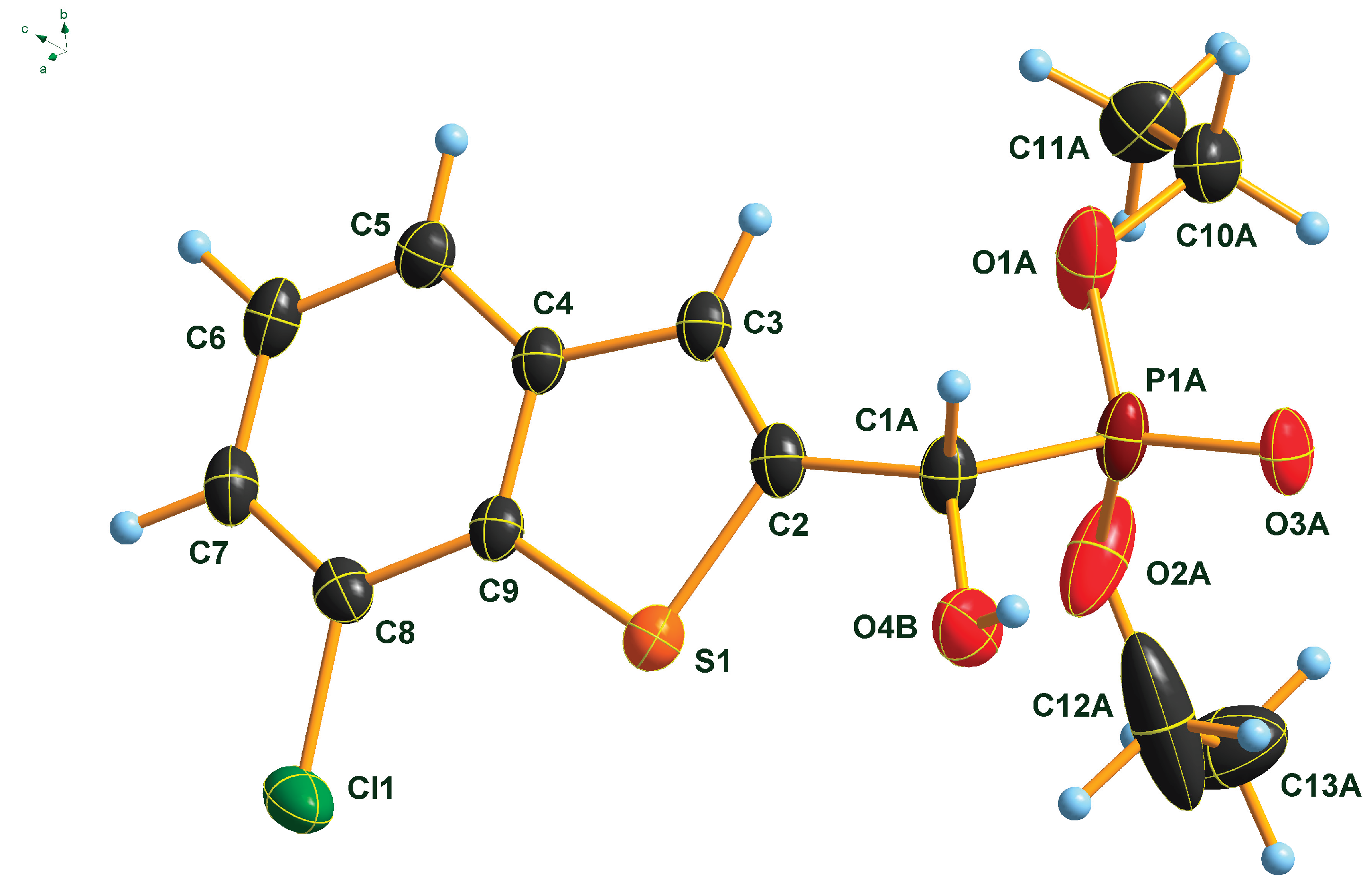
Table 1.
Preparative data for the new α-benzothiophenyl-α-hydroxy-ethylphosphonates (2a-w)
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | R3 | R4 | R5 | Yield (%) |
| 1 | 2a | H | H | F | H | H | 75 |
| 2 | 2b | H | H | H | F | H | 87 |
| 3 | 2c | H | H | H | H | F | 94 |
| 4 | 2d | H | Cl | H | H | H | 72 |
| 5 | 2e | H | H | Cl | H | H | 62 |
| 6 | 2f | H | H | H | H | Cl | 86 |
| 7 | 2g | H | H | Br | H | H | 73 |
| 8 | 2h | H | I | H | H | H | 66 |
| 9 | 2i | Me | H | H | H | H | 61 |
| 10 | 2j | H | CF3 | H | H | H | 79 |
| 11 | 2k | Me | F | H | H | H | 71 |
| 12 | 2l | Me | H | F | H | H | 79 |
| 13 | 2m | Me | H | H | F | H | 51 |
| 14 | 2n | Me | H | H | H | F | 79 |
| 15 | 2o | Me | H | F | H | F | 75 |
| 16 | 2p | Me | H | H | F | F | 56 |
| 17 | 2q | Me | H | Cl | H | H | 79 |
| 18 | 2r | Me | H | H | Cl | H | 66 |
| 19 | 2s | Me | H | CF3 | H | H | 66 |
| 20 | 2t | Me | H | H | CF3 | H | 69 |
| 21 | 2u | Me | H | H | H | CF3 | 78 |
| 22 | 2v | Et | H | CF3 | H | H | 77 |
| 23 | 2w | H | F | H | H | I | 68 |
Table 2.
Cell viability of U266 myeloma, EBC-1 lung, A2058 melanoma and HT-29 colon cell lines following long-term treatment (72 h) with α-benzothiophenyl-α-hydroxy-methylphosphonate derivatives (2a-w) at 100 μM. The data are expressed as mean ± standard deviation (SD), with a sample size of n = 3. Statistical analysis was performed using a one-way ANOVA followed by Fisher’s LSD post hoc test. Significance levels are denoted as follows: *: p < 0.05, **: p < 0.01 or ***: p < 0.001.
Table 2.
Cell viability of U266 myeloma, EBC-1 lung, A2058 melanoma and HT-29 colon cell lines following long-term treatment (72 h) with α-benzothiophenyl-α-hydroxy-methylphosphonate derivatives (2a-w) at 100 μM. The data are expressed as mean ± standard deviation (SD), with a sample size of n = 3. Statistical analysis was performed using a one-way ANOVA followed by Fisher’s LSD post hoc test. Significance levels are denoted as follows: *: p < 0.05, **: p < 0.01 or ***: p < 0.001.
| U266 | EBC-1 | A2058 | HT-29 | |
|---|---|---|---|---|
| Compounds | 100 µM | |||
| Medium | 2.05 ± 0.21*** | 0.92 ± 0.04*** | 1.03 ± 0.04 | 0.87 ± 0.02*** |
| DMSO | 1.00 ± 0.05 | 1.00 ± 0.02 | 1.00 ± 0.07 | 1.00 ± 0.04 |
| 2a | 0.85 ± 0.07*** | 0.93 ± 0.02** | 0.96 ± 0.06** | 0.90 ± 0.02* |
| 2b | 0.76 ± 0.04*** | 0.97 ± 0.01 | 0.97 ± 0.06*** | 0.97 ± 0.02 |
| 2c | 0.78 ± 0.06*** | 0.89 ± 0.02*** | 0.93 ± 0.05** | 0.90 ± 0.01** |
| 2d | 0.55 ± 0.07*** | 0.77 ± 0.01*** | 0.82 ± 0.03*** | 0.83 ± 0.03*** |
| 2e | 0.52 ± 0.05*** | 0.71 ± 0.02*** | 0.90 ± 0.02*** | 0.86 ± 0.02*** |
| 2f | 0.51 ± 0.03*** | 0.66 ± 0.03*** | 0.78 ± 0.11*** | 0.79 ± 0.03*** |
| 2g | 0.46 ± 0.01*** | 0.71 ± 0.01*** | 0.92 ± 0.01*** | 0.91 ± 0.06** |
| 2h | 0.51 ± 0.01*** | 0.66 ± 0.04*** | 0.76 ± 0.01*** | 0.81 ± 0.04*** |
| 2i | 0.94 ± 0.01 | 0.99 ± 0.03* | 1.06 ± 0.01 | 0.98 ± 0.01 |
| 2j | 0.61 ± 0.02*** | 0.76 ± 0.02*** | 0.80 ± 0.02*** | 0.78 ± 0.03*** |
| 2k | 1.08 ± 0.05 | 0.78 ± 0.03*** | 0.83 ± 0.03*** | 0.83 ± 0.02*** |
| 2l | 0.75 ± 0.08*** | 0.93 ± 0.01** | 0.97 ± 0.05* | 0.92 ± 0.03* |
| 2m | 0.87 ± 0.01** | 0.89 ± 0.02*** | 0.90 ± 0.04*** | 0.95 ± 0.04* |
| 2n | 0.81 ± 0.08*** | 0.86 ± 0.03*** | 0.90 ± 0.01*** | 0.91 ± 0.02* |
| 2o | 0.63 ± 0.02*** | 0.93 ± 0.05* | 0.98 ± 0.04* | 0.96 ± 0.02 |
| 2p | 0.69 ± 0.02*** | 0.85 ± 0.01*** | 0.82 ± 0.01*** | 0.91 ± 0.02** |
| 2q | 0.52 ± 0.06*** | 0.91 ± 0.04*** | 0.92 ± 0.02*** | 0.92 ± 0.02** |
| 2r | 0.51 ± 0.05*** | 0.68 ± 0.02*** | 0.89 ± 0.06*** | 0.84 ± 0.04*** |
| 2s | 0.30 ± 0.01*** | 0.71 ± 0.01*** | 1.02 ± 0.01 | 0.86 ± 0.03*** |
| 2t | 0.09 ± 0.02*** | 0.65 ± 0.01*** | 0.95 ± 0.05** | 0.58 ± 0.14*** |
| 2u | 0.55 ± 0.05*** | 0.85 ± 0.01*** | 0.82 ± 0.04*** | 0.87 ± 0.02*** |
| 2v | 0.77 ± 0.02*** | 1.03 ± 0.01 | 1.05 ± 0.01 | 0.99 ± 0.01 |
| 2w | 0.33 ± 0.01*** | 0.69 ± 0.01*** | 0.71 ± 0.01*** | 0.75 ± 0.01*** |
Blue indicates a reduction in cell viability to 80–65%, pale orange to 65–50%, and pale pink to 50% or less.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



