Submitted:
28 May 2025
Posted:
28 May 2025
You are already at the latest version
Abstract
The cholinergic system is one of the most ancient and widespread signaling systems in the body and is implicated in a variety of pathological conditions, ranging from neuro-degenerative disorders to cancer. Given its broad relevance, there is a growing interest in characterizing this system in diverse cellular models, allowing drug screening and mechanistic studies and assessing further therapeutic avenues. In this work, we investigated four cancer cell lines of neuroblastoma (SH-SY5Y), a lung cancer origin (lung adenocarcinoma line, A549, and two small cell lung carcinoma lines (H69 and H82) for the expression and localization of key components of the cholinergic system, along with their capacity for acetylcholine (ACh) synthesis and release. Whole-cell flow cytometry following membrane permeabilization revealed that all cell lines expressed the ACh-synthesizing enzyme choline acetyltransferase (ChAT). HPLC-MS analysis con-firmed that ChAT was functionally active, as all cell lines synthesized and released ACh into the conditioned media, suggesting the presence of autocrine and/or paracrine ACh signaling circuits in line with previous reports. All cell lines also demonstrated choline uptake, indicative of functional choline and/or organic cation transporters. Additionally, they expressed the ACh-degrading enzymes acetylcholinesterase (AChE) and butyryl-cholinesterase (BChE), along with the nicotinic α7 and muscarinic M1 ACh receptor subtypes. Unexpectedly, flow cytometry of intact SH-SY5Y cells revealed two novel findings: (1) ChAT was localized to the extracellular membrane, which was not evident in the lung cancer cell lines, and (2) BChE, rather than AChE, was the primary mem-brane-bound ACh-degrading enzyme. These results were corroborated by whole-cell and surface-confocal microscopy. In conclusion, our findings suggest that a functional cholinergic phenotype is a shared feature of several carcinoma cell lines, potentially serving as a survival checkpoint. The discovery of extracellular membrane-bound ChAT uniquely in neuroblastoma SH-SY5Y cells points to a novel form of in situ ACh signaling that warrants further investigation.
Keywords:
Cholinergic system
; choline acetyltransferase (ChAT)
; acetylcholine (ACh)
; Cancer
; neuroblastoma
; lung cancer
; flow cytometry
; confocal microscopy
1. Introduction
The cholinergic signalling system, which relies on acetylcholine (ACh), is widespread throughout the body. Cholinergic cells express the enzyme choline acetyltransferase (ChAT), which synthesizes ACh by transferring the acetyl-moiety of an acetyl-coenzyme A molecule (A-CoA) to a choline molecule. The canonical view is that ChAT is a cytosolic enzyme and that ACh biosynthesis occurs in the cytosol. However, recent studies challenge this view, showing that ChAT is released by certain cells into the extracellular fluids, where it maintains an extracellular ACh equilibrium in the presence of fully active ACh-degrading enzymes [1,2].
In addition to ChAT, the cholinergic system includes two primary receptor subtypes: nicotinic receptors (nAChRs), which are ligand-gated ion channels, and muscarinic receptors (mAChRs), which are metabotropic receptors [3]. Two key transporter proteins also contribute to cholinergic function: the vesicular ACh transporter (VAChT), responsible for packaging ACh into vesicles, and the high-affinity choline transporter (hChT), which enables choline reuptake after ACh release and hydrolysis at synapses. Furthermore, two enzymes break down ACh to terminate its action by hydrolyzing it into choline and acetate [3]. Acetylcholinesterase (AChE) is the primary enzyme responsible for ACh degradation, while butyrylcholinesterase (BChE) might act as a decoy, safeguarding AChE’s essential function from inhibitors that may enter the system through dietary sources [4]. While expression of ChAT and synthesis of ACh as a signaling molecule defines the cells as cholinergic, the downstream postsynaptic neurons and glial cells that express one or more subtypes of AChRs are denoted as cholinoceptive cells. Notably, all cholinergic cells can also be considered cholinoceptive given that ACh has autocrine activity, for instance through autoreceptor types of AchRs [3].
There are two main types of cholinergic cells: neuronal and non-neuronal subtypes. Several cholinergic neuronal systems exist. One is the central cholinergic system, originating from the basal forebrain [5]. This system has four main nuclei (Ch1-Ch4) that project across the brain [3]. This network plays an essential role in cognitive function, attention, and memory, and becomes affected in dementia disorders, such as Alzheimer's disease (AD), dementia with Lewy bodies, and Parkinson’s dementia [6,7,8,9,10]. There are also cholinergic interneurons, as part of the nigrostriatal system, especially in regions like the putamen, that are compromised in Parkinson's disease and disorders like corticobasal degeneration and progressive supranuclear palsy [11,12,13,14]. Another neuronal cholinergic system comprises the parasympathetic system, consisting of the 12 cranial nerves which connect directly to organs, muscles, and glands, controlling various bodily functions. These together with cholinergic neuromotor neurons in the brainstem and spinal cord are progressively lost in neuromotor disorders like amyotrophic lateral sclerosis (ALS) [15,16]. Another neuronal cholinergic system consists of the enteric cholinergic circuitries in the enteric nervous system (ENS), controlling motility (muscular) and secretory reflexes within the gastrointestinal tracts [17]. It is suggested that ~64% of neurons in ENS are cholinergic. There is also evidence that cholinergic circuitries in ENS are affected in many neurodegenerative diseases [18].
Non-neuronal cholinergic cells are a variety of ChAT-expressing cell types, including epithelial, mesothelial, endothelial, immune cells [19]. Surprisingly, ChAT expression increases in lymphocytes and macrophages, especially under inflammatory conditions [1]. This expression is thought to be part of a "cholinergic anti-inflammatory pathway," where ACh acts as an immune-regulatory agent, modulating cytokine release and inflammation [19]. In aging and some other diseases, a reduction in this pathway may contribute to excessive inflammatory responses. Accumulating reports also strongly point toward an upregulation of cholinergic machinery in various cancer forms, in particular small cell lung cancer (SCLC) and colon cancer [20,21,22,23]. In this context, ACh may act as an autocrine growth factor [23,24], and use of nicotinic ACh receptor antagonists as anticancer drugs has been proposed [20,25].
While the current view is that ChAT exists mainly as a soluble cytosolic enzyme, there are reports of intracellular membrane-bound forms of ChAT in synaptosomes and cholinergic nerve terminals across several animal species [26,27,28]. As an integral membrane protein, ChAT may behave differently compared to when it is localized in the cytosol [28]. A recent in vitro study indicates that human ChAT protein readily becomes embedded in micellar nanoparticles with a dramatic 10-fold increase in its catalytic rate [29]. ChAT may also retain a nuclear localization signal, allowing its translocation to the nucleus, although the function of ChAT in the nucleus remains elusive [30,31].
In this study, we aimed to investigate the expression of ChAT and related cholinergic markers in four different tumor cell lines to characterize and establish a robust cholinergic model for screening and in vitro characterization of various types of ChAT ligands, such as its inhibitors and/or activators [29,32,33]. In addition, we sought to determine whether ChAT is localized extracellularly as a membrane-bound enzyme, as reported in human spermatozoa [2]. Furthermore, we examined the presence of both cellular cholinergic machinery and compared the surface versus intracellular localization of key components of the cholinergic system across the four different cell lines (a human neuroblastoma cell line, a human adenocarcinoma cell line and two human small cell lung carcinoma cell lines [34]). The results provide novel insights into cholinergic signaling mechanisms involved in neurodegeneration and carcinoma pathology.
2. Results
2.1. Differential ChAT Localization in a Neuroblastoma Cell Line Compared with a Lung Adenocarcinoma and Two Small Cell Lung Carcinoma Cells Lines
The small cell lung carcinoma (SCLC) cell lines H69 and H82, the lung adenocarcinoma cell line A549, and the neuroblastoma cell line SH-SY5Y were evaluated for ChAT cell surface expression (Figure 1). Interestingly, surface ChAT localization was exclusively detected in the SH-SY5Y neuroblastoma cell line. This localization was not detected after blocking the antibody with recombinant ChAT protein, confirming the antibody´s specificity of the staining (Figure 1).
Next, intracellular staining was performed to assess ChAT expression within these cells. Positive intracellular ChAT expression and localization were observed across all four tested cell lines (Figure 2), which is in line with previous reports. ChAT staining with the pre-blocked antibody with recombinant ChAT protein fully eliminated the cell staining, confirming the antibody specificity for ChAT as the target protein. Notably, given that the intracellular staining approach involves cell permeabilization to allow entrance of the antibody into the cells, the resulting staining reflects the total ChAT staining since the extracellularly membrane-bound surface ChAT proteins are still accessible to the antibody. Therefore, a comparison of the number of cells that were positive for surface staining of ChAT (66 ± 1%) to the total number of cells that exhibited ChAT staining (79 ± 3%) indicated that in the neuroblastoma cells a major proportion of ChAT protein was localized extracellularly (compare dot plots in Figure 1 vs Figure 2). In contrast, in the lung carcinoma cell lines H69, H82, and A549, a signal from staining with a ChAT antibody was only evident when cells were permeabilized suggesting that ChAT primarily was only localized intracellularly (compare Figure 1 with Figure 2).
2.2. ChAT Is Functionally Intact in Both Neuroblastoma and Lung Cancer Cells
Measuring ACh and choline in an experiment is crucial for asserting the functionality of the cellular cholinergic machinery, as these two molecules are intimately linked in a functional signalling circuit. As such, their concurrent quantification provides an important measure of the functionality and homeostatic state of the cholinergic system and its auto- and paracrine regulatory mechanisms. In particular, whether ChAT protein is functionally intact, and whether the cells are capable of biosynthesizing and releasing ACh. We therefore measured the biosynthesis and release levels of ACh in the four cancer cell lines using HPLC-MS/MS analyses. The analysis was performed on both cell lysates and the conditioned medium from the cultured cells (to assess ACh release). Complete medium (RPMI) with and without FBS but lacking the cells served as negative controls. To preserve the biosynthesized ACh during preparation of the cell lysate and in the conditioned medium, neostigmine, an inhibitor of both AChE and BChE, was used in parallel. The HPLC results, which are presented in Figure 3, confirmed that all the cell lines were capable of synthesizing ACh, as determined in the cell lysates. To allow normalization and comparison of the levels of ACh and/or choline between the cell lines the total protein levels in the samples were used. The total protein concentration in the cell lysates was 0.58 ± 0.2 mg/mL and statistically did not differ among the lysates of the cell lines (SH-SY5Y = 0.56 ± 0.10; H82 = 0.52 ± 0.16; H69 = 0.59 ± 0.20; and A549 = 0.67 ± 0.28 mg/mL), ascertaining that the data reflects a similar number of cells.
The analyses of the conditioned media indicated that the SH-SY5Y neuroblastoma and the H82 (Figure 3) also released relatively substantial amounts of ACh into the medium as compared to controls. In contrast, only low amounts of ACh were detected in the conditioned medium of H69 and A549 cell lines (Figure 3). In summary, the tested cancer cell lines were all able to store the synthesized ACh intracellularly.
We further measured the levels of choline in SH-SY5Y cells and in the three lung cancer cell lines as well as their conditioned media by HPLC (lower panel of Figure 3). The concentration of choline in the culture medium was 1.8 ± 0.3 μM. The choline levels were generally much higher in the conditioned media than in the cell lysates, indicating, as expected, that the culture media was the source of choline and that the cells were able to take up the choline to use it, for instance, in the biosynthesis of ACh (Figure 3). This also indicates that all the cells express either a specific choline transporter or some general organic cation transporter, or both [35,36]. As expected, the presence or absence of neostigmine had no significant effect on the levels of choline given that choline is not metabolized by AChE or BChE.
2.3. Surface Localization Analysis of ChAT and the Related Cholinergic Markers in Neuroblastoma Cells
In addition to ChAT, we performed flow cytometric analyses with respect to the expression and localization of selected components of the cholinergic machinery, namely the ACh hydrolyzing enzymes, AChE and BChE, and the α7 nicotinic AChR and the M1 muscarinic receptors. The results are shown in Figure 4.
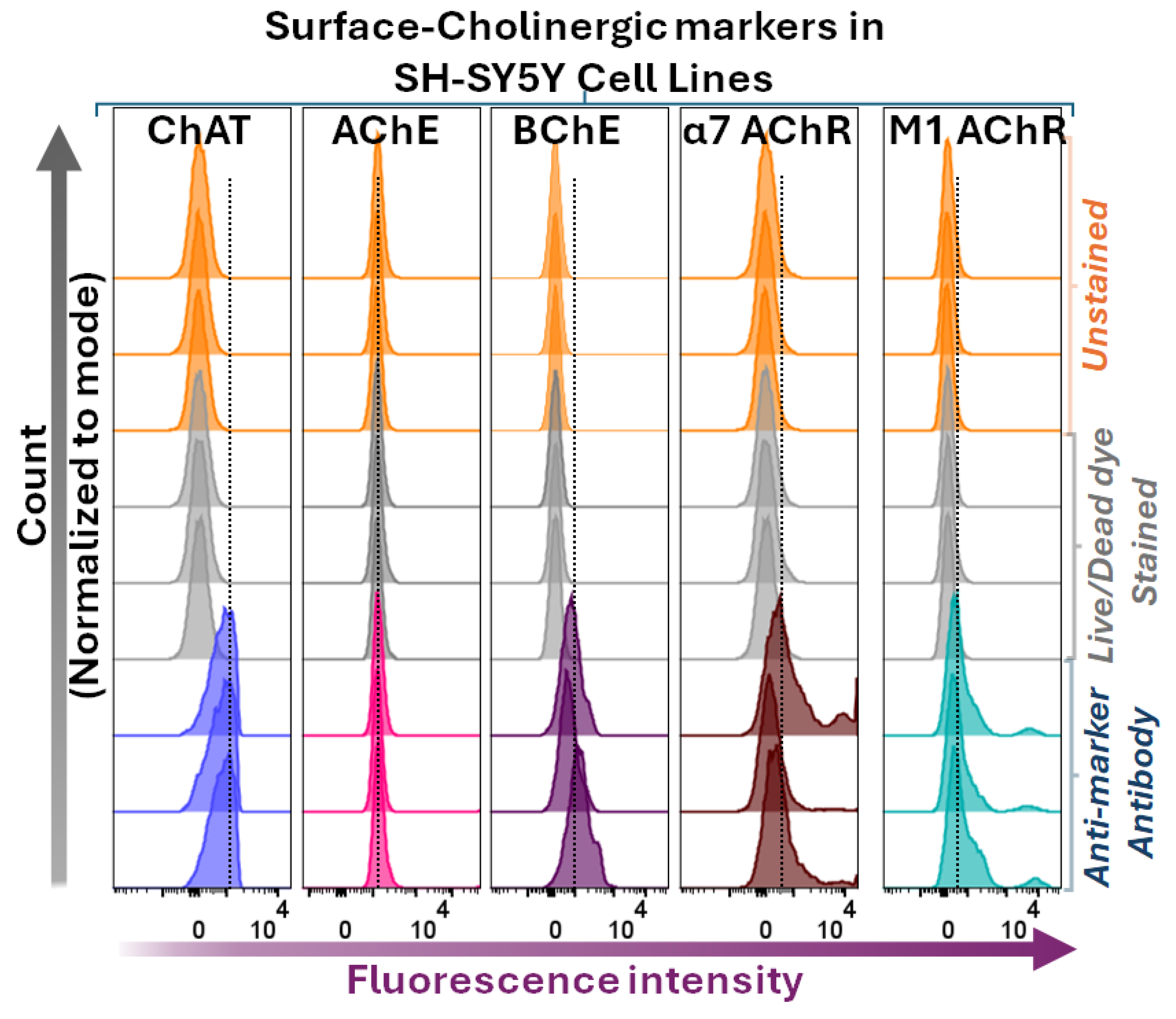
The surface flow cytometric analyses indicated that BChE, α7- and M1-AChRs, but not AChE, were found extracellularly or at the cell surface of SH-SY5Y neuroblastoma cells (Figure 4). These findings are partially unexpected. For instance, the lack of surface/extracellular staining of AChE is in clear contrast to the canonical view of AChE being the main membrane-anchored extracellular enzyme responsible for ACh hydrolysis within the synapses. Secondly, BChE is putatively regarded mainly as a secreted soluble free enzyme, while the current surface flow cytometric data clearly refute this by indicating that BChE is a major surface protein in all four cell lines (Figure 4). Nonetheless, as was expected, all the cells also exhibited positive surface staining for the α7- and M1-AChRs.
Figure 4.
Surface staining and flow cytometric analyses of ChAT in relation to other cholinergic markers in the neuroblastoma cell line, SH-SY5Y. Histograms show surface staining for AChE, BChE, α7-nAChR, and M1-mAChR in comparison with surface ChAT staining in SH-SY5Y neuroblastoma cells. Notably, the cells express all assessed components of the cholinergic machinery at their surface except for AChE. Histograms compare shifts in fluorescence intensities of cells stained with marker-specific antibodies relative to unstained and Live/Dead dye-stained fluorescence intensities. The Live/Dead dye staining was done to ascertain that the membrane of the cells has not been compromised, allowing the antibody to enter the cells. In other words, the staining fluorescence signals originate only from the interaction of the antibody with its target at the cell surface. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
Figure 4.
Surface staining and flow cytometric analyses of ChAT in relation to other cholinergic markers in the neuroblastoma cell line, SH-SY5Y. Histograms show surface staining for AChE, BChE, α7-nAChR, and M1-mAChR in comparison with surface ChAT staining in SH-SY5Y neuroblastoma cells. Notably, the cells express all assessed components of the cholinergic machinery at their surface except for AChE. Histograms compare shifts in fluorescence intensities of cells stained with marker-specific antibodies relative to unstained and Live/Dead dye-stained fluorescence intensities. The Live/Dead dye staining was done to ascertain that the membrane of the cells has not been compromised, allowing the antibody to enter the cells. In other words, the staining fluorescence signals originate only from the interaction of the antibody with its target at the cell surface. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
2.4. Intracellular Expression Analysis of ChAT and Related Cholinergic Markers in Neuroblastoma and Lung Cancer Cells
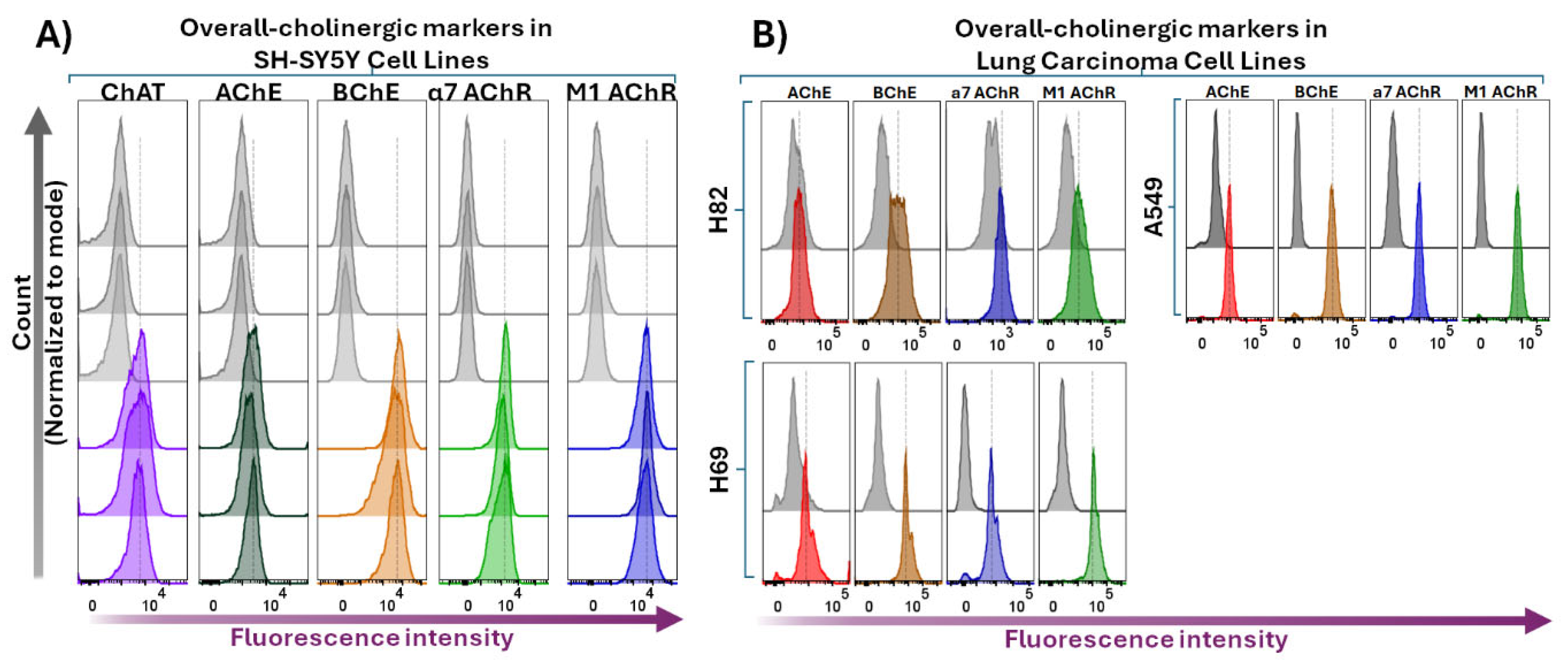
We next performed flow cytometric analyses following membrane permeabilization of the four cell lines. These analyses are expected to detect both surface and intracellular protein expression of the cholinergic markers. The results are presented in Figure 5, which demonstrate that the SH-SY5Y neuroblastoma cells were clearly positive regarding the expression level of all the assessed cholinergic biomarkers (Figure 5A). Essentially, the same finding was observed in the lung cancer cell lines H69, H82, and A549 (Figure 5B). Noteworthy, there was a significant increase in the fluorescence intensities in all cell lines following membrane permeabilization as compared to the surface staining procedure, which was expected given the antibodies had access to all available binding targets. Furthermore, a comparison of the fluorescence intensity profiles for AChE and BChE suggests a much higher expression of BChE relative to AChE (see the corresponding fluorescence intensity shift in the histogram panels in Figure 5), confirming the observations from the flow cytometric surface or extracellular expression analyses of these two enzymes (see Figure 4).
Similarly, the significant increase in intracellular fluorescence signals for the α7- and M1-mAChRs most likely indicates the presence of an intracellular pool of these receptors, comprising ongoing receptor synthesis, intracellular trafficking, or receptor internalization processes.
Figure 5.
Whole-cell-stained flow cytometric analyses of various cholinergic signaling components in the neuroblastoma cell line compared to lung carcinoma cell lines. Panel A shows histogram data depicting overall intracellular and surface targets in SH-SY5Y neuroblastoma cells stained after membrane permeabilization, reflecting whole-cell staining of specific biomolecules (ChAT, AChE, BChE, α7-nAChR, and M1-mAChR). Grey histograms show unstained cells, while color histograms show staining of cholinergic components Noteworthy, the whole-cell staining indicate that upon cell membrane permeabilization the anti-AChE antibody positively stained the cells, suggesting that AChE either was mainly located intracellularly or constituted an isoform that was normally secreted as free extracellular enzyme by the cells. Panel B presents corresponding whole-cell staining data for lung carcinoma cell lines H82, H69, and A549. The data clearly shows that also these cell lines expressed all the tested cholinergic receptors and enzymes, including AChE. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
Figure 5.
Whole-cell-stained flow cytometric analyses of various cholinergic signaling components in the neuroblastoma cell line compared to lung carcinoma cell lines. Panel A shows histogram data depicting overall intracellular and surface targets in SH-SY5Y neuroblastoma cells stained after membrane permeabilization, reflecting whole-cell staining of specific biomolecules (ChAT, AChE, BChE, α7-nAChR, and M1-mAChR). Grey histograms show unstained cells, while color histograms show staining of cholinergic components Noteworthy, the whole-cell staining indicate that upon cell membrane permeabilization the anti-AChE antibody positively stained the cells, suggesting that AChE either was mainly located intracellularly or constituted an isoform that was normally secreted as free extracellular enzyme by the cells. Panel B presents corresponding whole-cell staining data for lung carcinoma cell lines H82, H69, and A549. The data clearly shows that also these cell lines expressed all the tested cholinergic receptors and enzymes, including AChE. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
2.5. Confocal Microscopy Confirms Extracellular Expression of ChAT Cholinergic in Neuroblastoma
The canonical view is that ChAT is a cytosolic enzyme, where it synthesizes ACh. However, we recently reported extracellular localization of ChAT at the surface of human sperm using flow cytometry [2]. Nonetheless, out of the four examined cell lines, only the SH-SY5Y neuroblastoma cells exhibited surface ChAT (Figure 1). To validate this finding, we employed cell surface confocal microscopy of ChAT in the same cells. The cells were stained with fluorescently labelled antibodies under non-permeabilizing conditions to ensure the detection of surface antigens only. In addition, we also performed similar surface microscopy on the other cholinergic markers we assessed by flow cytometry i.e., AChE, BChE, M1-mAChR, and α7-nAChR.
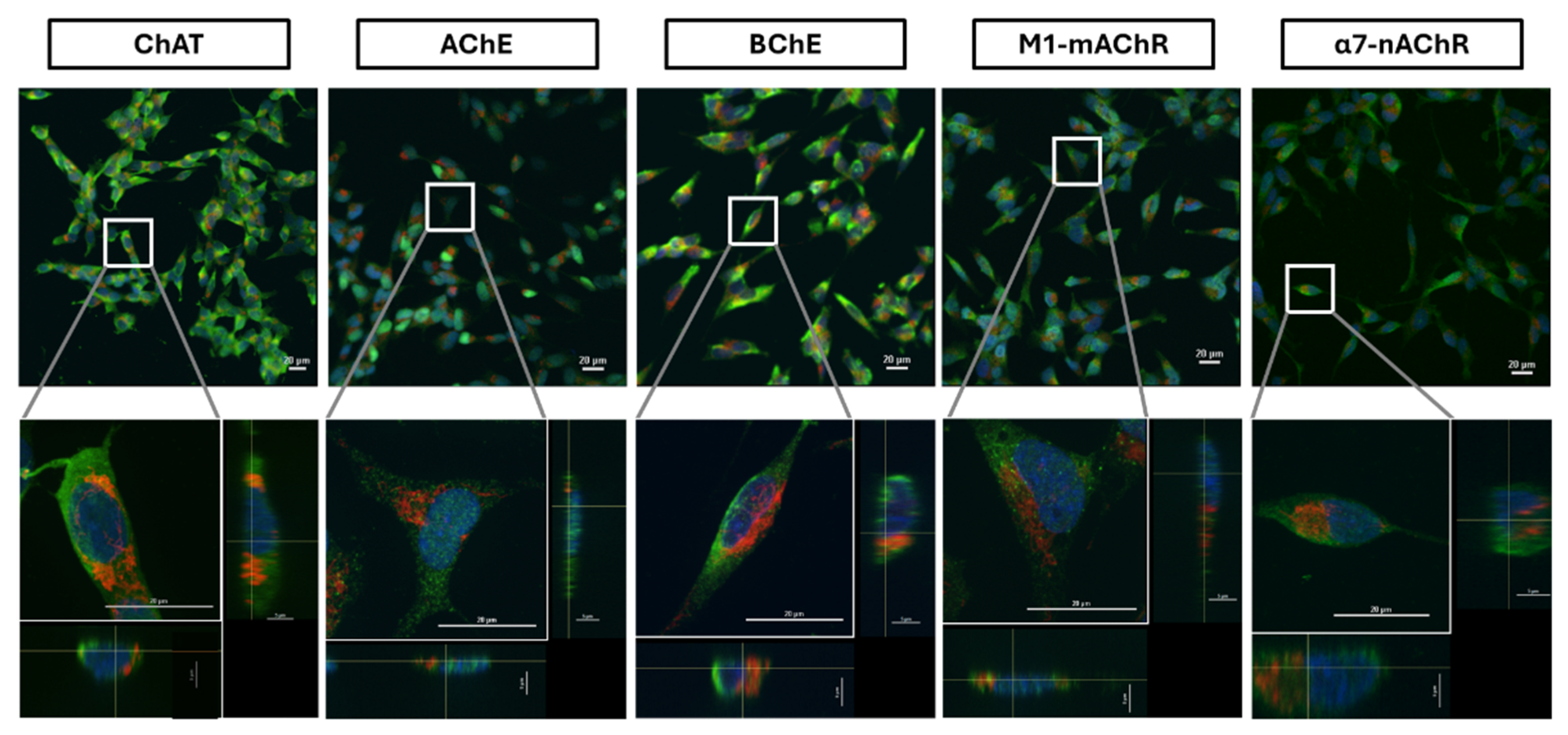
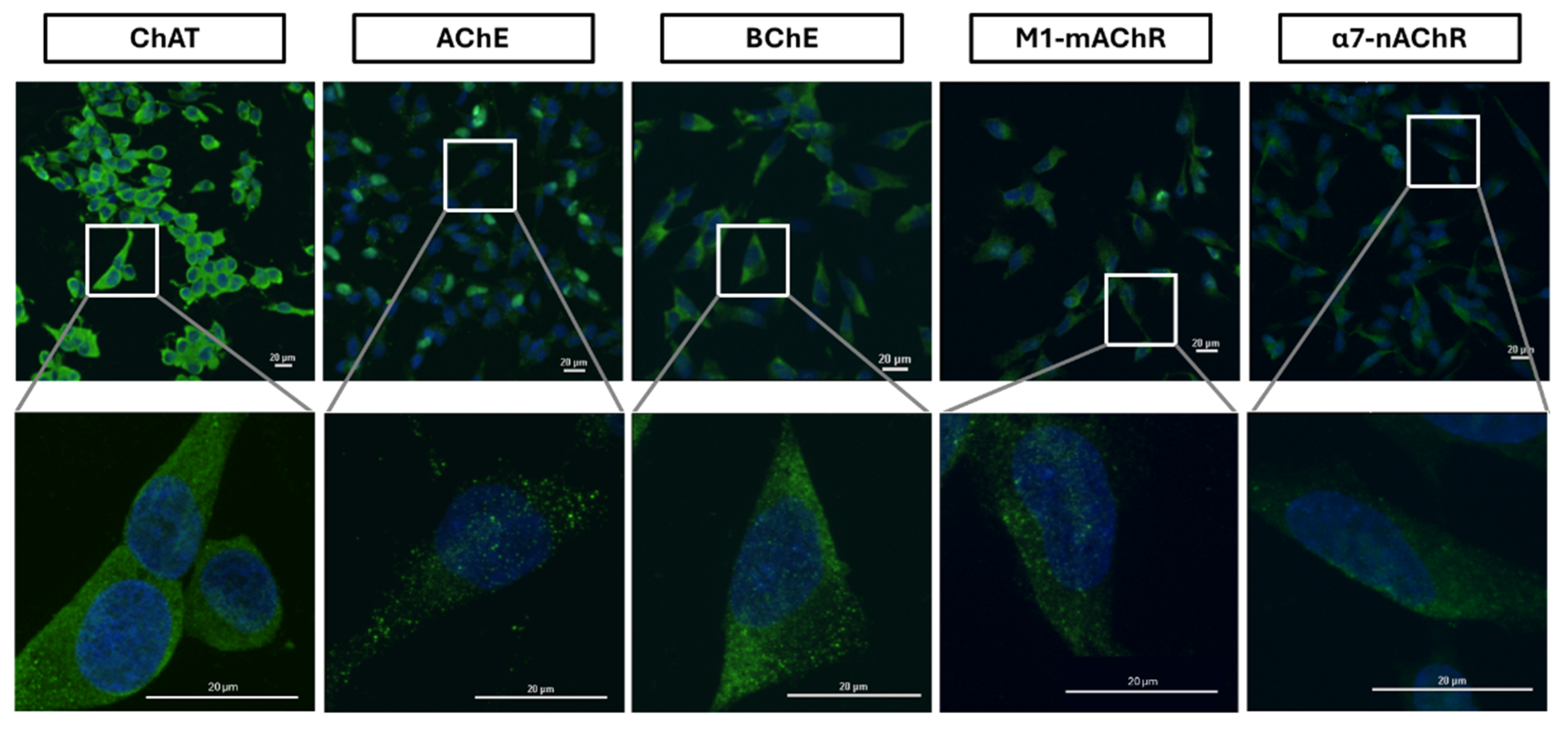
The confocal microscopy investigation of the surface ChAT confirmed the surface flow cytometric finding on surface localization of ChAT in the SH-SY5Y neuroblastoma cells (Figure 6).
Similarly, the surface confocal microscopy analysis also validated the presence of AChE, BChE, M1-mAChR, and α7-nAChR on the surface of the cells (Figure 6). Notably, AChE and α7-nAChR exhibited relatively lower expression intensities as compared to BChE and M1-mAChR. These findings indicate that neuroblastoma SH-SY5Y cells express a functional cholinergic system on their cell surface.
We next did similar analyses, using fluorescent labelled antibodies on the permeabilized cells. Confocal microscopy images are presented in Figure 7. These analyses fully reaffirmed the expression and cellular distribution of ChAT throughout the cells, including in the cytosol. The results of confocal expression of ChAT were also confirmed twice using other ChAT-specific staining antibodies from other vendors (Supplementary Figure S1). Similar staining findings were also evident for the ACh-hydrolyzing enzymes (AChE, BChE) and the ACh-receptors (M1- and α7-AChR). Notably, the intracellular AChE and α7-nAChR expression seemed to exhibit the weakest intensities, with a dot-like expression pattern (Figure 7), as opposed to the intracellular intensities of the other cholinergic markers. In addition, the observed low-level AChE expression is in agreement with the presented flow cytometric data for AChE (see Figure 4 and Figure 5A).
The specificity of secondary antibodies was tested and confirmed by staining cells with secondary antibodies alone as documented in Supplementary Figure S2.
Figure 7.
Extracellular and intracellular staining of cholinergic markers in the neuroblastoma cell line, SH-SY5Y. The cells were subjected to membrane permeabilization followed by incubation with specific antibodies, allowing staining of both surface and intracellular proteins. Micrographs represent images of total extracellular and intracellular staining signals of cholinergic markers (ChAT, AChE, BChE, M1-mAChR, and α7-nAChR) depicted in green, co-labeled with DAPI in blue and mitochondria (mitotracker red) in red. Magnification 20x; Scale bar 20 µm. Zoom-in panels below each set are 60x magnification from the area indicated by the white box. Side views of X-Y cross sections of the Z-stack displayed at the bottom and the right show intra- and extracellular localization of biomarker signals. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
Figure 7.
Extracellular and intracellular staining of cholinergic markers in the neuroblastoma cell line, SH-SY5Y. The cells were subjected to membrane permeabilization followed by incubation with specific antibodies, allowing staining of both surface and intracellular proteins. Micrographs represent images of total extracellular and intracellular staining signals of cholinergic markers (ChAT, AChE, BChE, M1-mAChR, and α7-nAChR) depicted in green, co-labeled with DAPI in blue and mitochondria (mitotracker red) in red. Magnification 20x; Scale bar 20 µm. Zoom-in panels below each set are 60x magnification from the area indicated by the white box. Side views of X-Y cross sections of the Z-stack displayed at the bottom and the right show intra- and extracellular localization of biomarker signals. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
3. Discussion
To the best of our knowledge, this study provides the first compelling evidence for the presence of an extracellularly membrane-bound isoform of ChAT in the neuroblastoma SH-SY5Y cell line. Such extracellularly membrane-bound isoform of ChAT was completely absent in the three lung carcinoma cell lines. This was carefully confirmed with confocal microscopic analyses of the cell surface ChAT protein. Nonetheless, we have previously reported a similar extracellularly membrane-bound ChAT in human spermatozoa [2].
There are reports indicative of the presence of intracellularly membrane-bound ChAT in the synaptic vesicles and synaptosomes in several animal species [26,27,28]. Intriguingly, the synthesized ACh by the membrane-bound ChAT seems to be about forty percent more resistant to hydrolysis by AChE than ACh synthesized by the soluble form of ChAT [26]. A recent study also showed that human ChAT has a great propensity to attach itself to membrane-like micelles, a phenomenon that results in boosting the activity of the enzyme by over 10-fold [29]. Therefore, the distinct extracellular localization of ChAT in the neuroblastoma cell line might indicate a novel physiologically implemented cholinergic enhancing mechanism, involving in a fast in-situ synaptic recycling of ACh, granting ACh a prolonged mode of action in the synapses. This is in line with our previous hypothesis about the extracellularly membrane-bound ChAT in human spermatozoa, where in-situ ACh synthesis was proposed to allow ACh to be synthesized in close proximity to its receptor at the membrane, thereby acting timely as a chemotactic agent in sperm, facilitating or governing their motility [2].
For comparison, we also looked at the expression of ChAT in two SCLC neuroendocrine cell lines i.e., H82 and H69 as well as a NSCLC lung adenocarcinoma cell line, i.e., A549. Although these cells seemed to lack expression of extracellularly membrane-bound ChAT, they clearly had intracellular ChAT staining that was comparable to what was observed in the neuroblastoma SH-SY5Y cell line. Although we just used four cancer cell lines, our results indicate that the extracellular localization of ChAT is not a general phenomenon in cancer cells. To assess whether the expression of ChAT in all these cell lines could have any functional outcome, we measured the production and release levels of ACh. Although the synthesis and release levels seemed to be different in the different cell lines, the results were clear in that all four cell lines did synthesize and release ACh. These findings are in line with previous reports on ACh and choline in human colon cancer cell cultures [22], as well as in SCLCs, where the H82 cell line exhibited like in our study the highest ACh level compared to the H69 cells [20].
The cholinergic machinery also includes, among other components, various forms of nicotinic and muscarinic ACh receptors (nAChRs and mAChRs) as well as ACh-degrading enzymes (AChE and BChE). Many of these components has been shown in SCLCs [20,24,37]. We therefore also looked at some of these components. In the neuroblastoma SH-SY5Y cell line, we first analysed the presence of surface-localized AChE, BChE, M1-mAChR, and α7-nAChR through both flow cytometric analyses and confocal microscopy. Both M1-mAChR and α7-nAChR were detected extracellularly, which is expected given that these are known as plasma membrane-integrated receptors and been reported in various cancer cells [23] as well as these cells [24,38]. The findings on extracellularly membrane-bound AChE and BChE were, however, surprising, since we found that BChE was the main surface cholinesterase rather than AChE in these cells. The putative view is that AChE is the main ACh hydrolyzing enzyme, present extracellularly as membrane-anchored multimeric molecular forms, for example, on the surface of blood cells, within the neuromuscular junctions, and in the synaptic clefts of the cholinergic interfaces in the brain [39]. BChE, on the other hand, has putatively been considered a soluble form. Nonetheless, reports in the recent decade point towards the presence of some membrane-anchored AChE-BChE hybrid molecular forms [40]. Although our findings seem to support this notion, the observation that AChE staining was negligible if not completely absent at the surface of the cells suggests that in the neuroblastoma SH-SY5Y cell line, the membrane-anchored BChE was the dominant form.
Upon membrane permeabilization of the cells, flow cytometric analyses will reflect the overall interaction of the antibody with both the surface and the intracellular target protein expression. A comparison of the overall staining intensity of AChE versus BChE in our flow cytometric data on the neuroblastoma SH-SY5Y cell line indicated that BChE was indeed the much more prevalent enzyme than AChE, supporting the notion that the cells had mainly membrane-anchored BChE. Furthermore, the confocal microscopy results also strongly support this as BChE had an even distribution over the whole cell surface, while AChE presented dotted localized distributions indicative of a discrete focal expression. Nonetheless, this conclusion should be interpreted with caution since two different antibodies against two distinct proteins was used, i.e., given that the antibodies might have different affinity for their target proteins.
Furthermore, we investigated the expression of these cholinergic signaling biomolecules in the lung cancer cell lines, namely the SCLCs H82 and H69 cells, as well as in the lung adenocarcinoma cell line, A549. The results were very similar to that of the neuroblastoma SH-SY5Y cell line, with respect to the expression of not only ChAT and ACh synthesis and release but also the expression pattern of AChE, BChE, M1-mAChR, and α7-nAChR. Noteworthy, even in these three cell lines, the total staining intensity of AChE seemed to be much less than the observed intensity of BChE. Anyway, reports exist for hybrid BChE-AChE molecular forms in human glioma [41].
Intriguingly, given that the examined cell lines are cancerous cell lines, this phenomenon could be a particular feature of cancer pathology. For instance, it is known that BChE has a scavenging property toward cytotoxic agents ingested naturally via e.g., food sources. Therefore, increasing BChE expression may offer tumour cells a protective line of defence against cytotoxic agents. Indeed, a report has indicated that an increase in BChE levels in the serum of patients with cancer of various tissue origins after treatment compared to their baseline or the healthy controls [42]. This hypothesis suggest that a screening of the current cytotoxic agents against BChE is warranted to assess which agents is not metabolized by BChE, i.e. could be more effective to use under such conditions.
Figure 8.
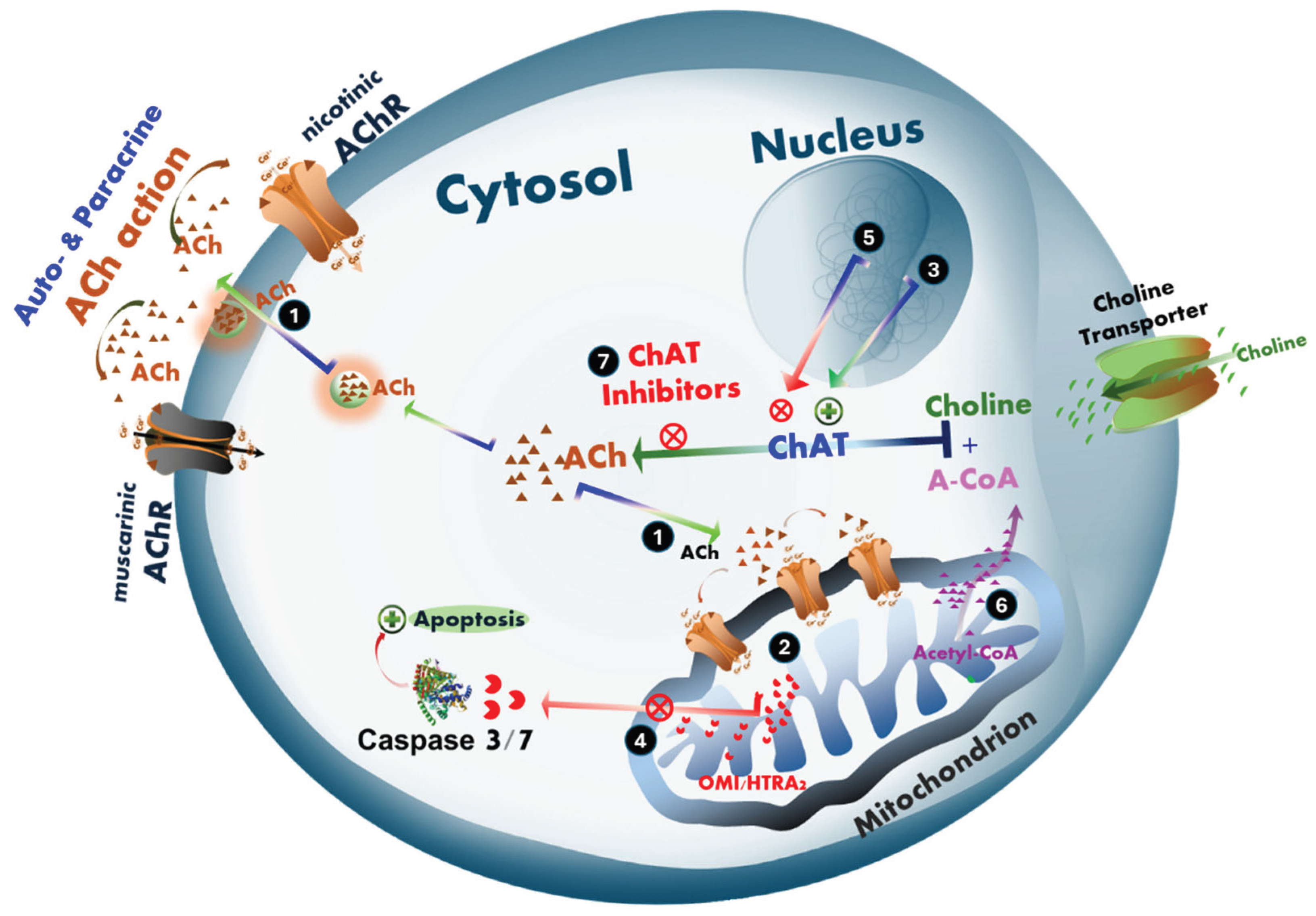
Hypothetical mechanisms of intracellular cholinergic signaling in the action of acetylcholine involved in promoting cell survival and proliferation in cancer in tumor cells or neurodegeneration in Alzheimer’s disease and ALS. (1) ACh promotes cell proliferation and/or survival. We hypothesize that ACh may act through an intracellular signaling mechanism involving the monitoring of mitochondrial bioenergetic function. (2) This mechanism includes regulation of mitochondria-driven apoptosis [43], which may be differentially affected in the progression of cancer and neurodegenerative diseases such as AD and ALS. (3) In cancer, tumor cells upregulate the expression and activity of the ACh-biosynthesizing enzyme, ChAT, resulting in elevated intracellular ACh levels. (4) Tumor cells exploit this pathway as a protective mechanism against cellular damage caused by, for example, radiotherapy or chemotherapeutics, by suppressing mitochondria-driven apoptosis. This provides more time for DNA damage repair, and thereby increases the likelihood of cell survival, allowing metastatic spread and/or recolonization of the tumor site and potentially cancer relapse. (5) In contrast, in cholinergic neurodegenerative disorders such as AD and ALS, a gradual decline in ChAT expression is observed in neurons. This decline may result from aging, reduced stimulation and/or availability of neurotrophic factors such as NGF, BDNF, or both, and/or the inappropriate use of drugs with anticholinergic effects, including proton pump inhibitors (PPIs) and various receptor antagonists. (6) Alternatively or additionally, metabolic and mitochondrial bioenergetic dysfunction may significantly reduce cellular levels of acetyl-Coenzyme A (A-CoA), a co-factor required in equimolar amounts for ACh synthesis by ChAT. A-CoA is primarily produced in mitochondria, where it is partly used in ATP production via the Krebs cycle and partly exported to the cytoplasm for biosynthetic processes including ACh production. Reduced A-CoA production and/or its cytoplasmic availability results in decreased intracellular ACh biosynthesis, compromising the cell’s defense against mitochondria-driven apoptosis. This renders cholinergic neurons more susceptible to degeneration and increasingly sensitive to further anticholinergic insults. (7) This dual-edged mechanistic hypothesis can be tested using selective ChAT inhibitors, such as PPIs [33], and warrants further investigation. The ACh–mitochondria–apoptosis axis may represent a critical pathway both for understanding the selective cholinergic neurodegeneration in dementia and neuromotor disorders, and as a survival strategy exploited by cancer cells.
Figure 8.
Hypothetical mechanisms of intracellular cholinergic signaling in the action of acetylcholine involved in promoting cell survival and proliferation in cancer in tumor cells or neurodegeneration in Alzheimer’s disease and ALS. (1) ACh promotes cell proliferation and/or survival. We hypothesize that ACh may act through an intracellular signaling mechanism involving the monitoring of mitochondrial bioenergetic function. (2) This mechanism includes regulation of mitochondria-driven apoptosis [43], which may be differentially affected in the progression of cancer and neurodegenerative diseases such as AD and ALS. (3) In cancer, tumor cells upregulate the expression and activity of the ACh-biosynthesizing enzyme, ChAT, resulting in elevated intracellular ACh levels. (4) Tumor cells exploit this pathway as a protective mechanism against cellular damage caused by, for example, radiotherapy or chemotherapeutics, by suppressing mitochondria-driven apoptosis. This provides more time for DNA damage repair, and thereby increases the likelihood of cell survival, allowing metastatic spread and/or recolonization of the tumor site and potentially cancer relapse. (5) In contrast, in cholinergic neurodegenerative disorders such as AD and ALS, a gradual decline in ChAT expression is observed in neurons. This decline may result from aging, reduced stimulation and/or availability of neurotrophic factors such as NGF, BDNF, or both, and/or the inappropriate use of drugs with anticholinergic effects, including proton pump inhibitors (PPIs) and various receptor antagonists. (6) Alternatively or additionally, metabolic and mitochondrial bioenergetic dysfunction may significantly reduce cellular levels of acetyl-Coenzyme A (A-CoA), a co-factor required in equimolar amounts for ACh synthesis by ChAT. A-CoA is primarily produced in mitochondria, where it is partly used in ATP production via the Krebs cycle and partly exported to the cytoplasm for biosynthetic processes including ACh production. Reduced A-CoA production and/or its cytoplasmic availability results in decreased intracellular ACh biosynthesis, compromising the cell’s defense against mitochondria-driven apoptosis. This renders cholinergic neurons more susceptible to degeneration and increasingly sensitive to further anticholinergic insults. (7) This dual-edged mechanistic hypothesis can be tested using selective ChAT inhibitors, such as PPIs [33], and warrants further investigation. The ACh–mitochondria–apoptosis axis may represent a critical pathway both for understanding the selective cholinergic neurodegeneration in dementia and neuromotor disorders, and as a survival strategy exploited by cancer cells.
Furthermore, the presence of the major components of cholinergic machinery, including its key enzyme, ChAT, raises the question of a crucial role of cholinergic signalling in cancer cell biology. Our findings are in full agreement with reports that cholinergic signalling is involved in promoting cell survival and proliferation in various cancer, including lung cancer [23,25,35,36,44,45]. Therefore, it seems imperative to boost the research on the role of cholinergic signalling in cancer biology since it may be involved in the development of tolerance against cellular insults by chemo- and/or radiotherapy, and inhibition of this pathway may increase the efficiency of these treatments.
We have hypothesized that ACh may also act as an intracellular signalling pathway involving monitoring mitochondrial bioenergetic function, and thereby regulation of mitochondria-driven apoptosis [43], which could be differentially altered in the course of cancer and neurodegenerative diseases like AD and ALS. In our hypothesis, intracellular ACh is a key player in protecting the cells against mitochondria-driven apoptosis (Figure 8). This might be a key, explaining how two apparently different disorders may have a common key pathological feature. In neurodegenerative disorders (like AD and ALS), a selective neurodegeneration occurs in the central cholinergic neurons and the spinal and peripheral cholinergic motor neurons, as well as the parasympathetic cranial nerves, which is manifested by a severe reduction in ChAT expression [6,7,8,9,10,11,12,13,14,15,16]. Thus, here the expected reduction in intracellular ACh biosynthesis by ChAT will lessen the cellular protection against mitochondria-driven apoptosis and thereby facilitate neuronal degeneration. In tumor cells, the opposite seems to be occurring, i.e., the cancer cells are apparently upregulating a pronounced cholinergic phenotype to protect themselves against apoptosis [20,24,37]. Continuous intracellular ACh biosynthesis for maintaining such a protective mechanism is however a very high energy-demanding bioprocess since it requires equimolar amounts of acetyl-CoA (which otherwise is used to produce ATP). This fact therefore strongly indicates that the upregulated ACh signaling in the tumour cells may have been a worthwhile energy investment adopted by these cells, particularly if we also consider how much energy is required by cancer cells for their proliferation alone. Therefore, the acetylcholine-mitochondria-apoptosis axis is a crucial point of investigation for both a proper understanding of the selective cholinergic neurodegeneration occurring in dementia and neuromotor disorders, as well as a survival checkpoint adopted by tumor cells, including neuroblastoma and lung cancer.
In summary, this study provides compelling evidence for the presence of an extracellularly membrane-bound ChAT, exclusively in the neuroblastoma cells as compared to three lung cancer cell lines tested, although all the studied cell lines expressed ChAT intracellularly. All the examined cells also expressed several other components of the cholinergic machinery, reinforcing that the expression of ChAT had a biological purpose in line with ACh signaling. These findings also underscore the complexity of cholinergic signaling, particularly the roles of membrane-bound and intracellular cholinergic markers. Dysregulation of these components is linked to neurodegenerative diseases such as AD, Parkinson’s disease, and ALS. Membrane and intracellular cholinergic markers influence ACh homeostasis, receptor trafficking, apoptotic pathways, and neuroinflammation, making them critical targets for therapeutic interventions. Further studies are needed to elucidate the molecular pathways governing these markers and their potential as therapeutic targets in both neurodegenerative diseases and cancer. Understanding their interactions and regulatory mechanisms could pave the way for strategies to restore cholinergic system integrity and mitigate disease progression.
4. Materials and Methods
4.1. Reagents and Antibodies
The following reagents and antibodies were used in the study. Cell culture reagents including RPMI-1640, foetal bovine serum (FBS), trypsin, penicillin, and streptomycin, as well as the fixation and permeabilization buffers (cat # 00-8333-56), were all obtained from Thermofisher Scientific. Cell culture containers (T25 and T75 flasks), cell culture plates, and 15 mL falcon tubes were purchased from Corning for growing and maintaining cell lines. Primary antibodies used were APC (allophycocyanin)-conjugated anti-ChAT antibody (cat # AB224001 from Abcam), unconjugated monoclonal mouse anti-ChAT antibody from (MAB-3447 from R&D System), unconjugated monoclonal mouse anti-ChAT antibody (MAB-31383 from Invitrogen), FITC-conjugated anti-AChE antibody (A-11, Sc-373901-FITC from Santa Cruz), PE-conjugated anti-BChE antibody (D-5, Sc-377403-PE from Santa Cruz), AF647-conjugated anti-α7-AChR antibody (319, Sc-58607-AF647 from Santa Cruz), and PE-conjugated anti-M1-AChR antibody (G-9, Sc-365966-PE from Santa Cruz) for flow cytometric assessment of the expression of cholinergic molecules. For surface staining flow cytometric analyses, the cells were also simultaneously incubated for 20 min with a 1:1500 diluted working solution of the LIVE/DEAD™ Cell Stain dye (cat # L23101, Invitrogen), to ascertain the integrity of the cell membrane, as described before [2]. For intracellular staining the cells were treated with a permeabilization buffer (Cat # 88-8824-00, eBioscience™ Intracellular Fixation & Permeabilization Buffer Set, Invitrogen) according to the manufacturer instruction. Secondary antibodies used were FITC-conjugated goat anti-mouse antibody and AF648-conjugated goat anti-rabbit antibody (21244-AF648) from Invitrogen, used in conjunction with the unlabeled primary antibodies. The recombinant human ChAT protein (5.1 mg/mL) was produced and purified by the Protein Science Facility at Karolinska Institutet, as described before [32].
4.2. Cell Lines
In this work, two human small cell lung carcinoma cell lines NCI-H69 (HTB-119 ™, American Type Culture Collection (ATCC)) and NCI-H82 (HTB-175 ™, ATCC)), one lung adenocarcinoma A549 (CRL-185), and one neuroblastoma cell line (SH-SY5Y cells (ATCC, CRL-2266) were used. All cell lines were maintained in RPMI 1640 media supplemented with 10% FBS (Gibco, Invitrogen), 1% penicillin-streptomycin solution (Gibco, Invitrogen), and grown in a humidified cell culture incubator in 5% CO2 atmosphere at 37 °C. The adherent cells (A549 and SH-SY5Y) were detached by trypsination in 0.25% trypsin and 0.02% EDTA when the cells reached 80–90% confluency.
4.3. Assessment of Cholinergic Markers by Flow Cytometry
For the A549 and SH-SY5Y cells, staining was assessed on confluent cell cultures detaching the cells with trypsination followed by washing in PBS and counting. The small cell human lung cell lines (SCLC) H69 and H82 were maintained as suspension cultures and were centrifuged down for the staining experiment. Approximately 1 × 106 cells were used for the staining of each specific protein. The cell staining was performed as follows; the fixation and permeabilization buffers were prepared and used according to the manufacturer’s instructions (cat # 00-8333-56, Thermofisher). Briefly, cells were washed with PBS and fixed in 4% paraformaldehyde solution (30 µl per flow cytometry tube) for 10 minutes at room temperature (RT). The cells were then washed with 300 µl of PBS containing 0.5% BSA (PBS-0.5%BSA) and centrifuged at 2500 rpm for 15 minutes at 4 °C. Next, cells were permeabilized using permeabilization buffer and incubated with primary antibody at indicated concentrations at 4 °C for 30 minutes. Following this, cells were washed with PBS-0.5%BSA, resuspended in 300 µl of PBS, and analyzed by flow cytometry (Beckman Coulter, CytoFLEX S). To confirm the specificity of the ChAT antibody staining, cells were incubated with a 20X concentration of recombinant ChAT protein (~102 μg/mL final concentration) along with the ChAT primary antibody (1/1250 final dilution factor) in the permeabilization solution. These cells were then incubated for 30 minutes at 4 °C, and the staining was performed as above. For the surface staining, the permeabilization step was omitted. A similar staining procedure was applied for the other cholinergic markers, including AChE, BChE, α7-AChR, and M1-AChR with the concentration or dilution of each antibody (1/50 final dilution, all from Santa Cruz). The material details are given in the “Reagents and antibodies” section. All experiments were conducted in triplicate.
4.4. Assessment of ACh Levels by High-Performance Liquid Chromatography (HPLC)
The cells were collected after being in culture for 24-48 hours, and the cell culture medium was transferred into Eppendorf tubes. After washing in PBS, the cells were counted and homogenized using a steel bead with a 25 s ON / 5 s OFF cycle (2 cycles) generating a cell lysate. The cell lysate was then centrifuged at 15,000 rpm for 10 minutes at 4 °C, and the supernatant was transferred to a new Eppendorf tube. Next, 100 µl of pre-chilled acetonitrile (stored at -20 °C) was added to the supernatant, which was vortexed until the solution became cloudy. The sample was centrifuged at 17,000 rpm for 20 minutes at 4 °C. Finally, for the LC-MS/MS determination of ACh and choline concentrations in the samples, 50 µl of each sample was transferred to injection vials, followed by the addition of 10 µl of tuning solution (1 µg/ml solution of Acetylcholine-d9 Chloride) to each vial. As a control, an equivalent amount of complete medium (RPMI + FBS) and RPMI alone (without FBS) without any cells were also collected in separate Eppendorf tubes.
4.5. Immunofluorescence Staining of SH-SY5Y Cells for ChAT and Related Cholinergic Markers
When cells reached approximately confluency, they were trypsinized for 5 minutes, and 50,000–70,000 cells/35 mm culture dish were seeded onto a 20 mm glass well (P35G-1.5-20-C, MatTek Corporation) for a 24-hour incubation. Surface and/or intracellular marker staining was carried out using the two-step optimized method described by Vernay and Cosson [46]. Briefly, cells were fixed in 3.7% paraformaldehyde solution for 10 minutes at 37 °C, followed by the addition of 1% bovine serum albumin (BSA) for 45 minutes to block nonspecific binding, omitting permeabilization. To assess the whole-cell staining of the proteins (i.e., both the surface and intracellular proteins), the cells were subjected to a second fixation and were permeabilized with 0.05% Triton-X for 10 minutes before applying primary and secondary antibodies, as described by Vernay and Cosson [46]. Primary antibodies were applied on ice against mouse monoclonal anti-ChAT (1:70, MAB3447, R&D Systems), rabbit polyclonal anti-ChAT (1:300, PAB14536, Abnova); rabbit polyclonal anti-ChAT (1:70, AB143, Millipore), mouse monoclonal anti-AChE (1:50 or 4 µg/ml, A-11 sc-373901, Santa Cruz), mouse monoclonal anti-BChE (1:50, D-5 sc-377403, Santa Cruz), rat monoclonal anti-α7-nAChR (1:50, sc-58607, Santa Cruz), and mouse monoclonal anti-M1-mAChR (1:50, G-9 sc-365966, Santa Cruz). For visualization, fluorophore-conjugated secondary antibodies were used: anti-mouse IgG (1:375, goat anti-mouse IgG, FITC, Invitrogen), anti-rabbit IgG (1:300, goat anti-rabbit IgG, Alexa Fluor 647, Invitrogen), and anti-rat IgG (1:300, goat anti-rat IgG, Alexa Fluor 647, Invitrogen). Nuclei were counterstained with NucBlue DAPI reagent (R37606, Invitrogen). Multipoint spinning disk confocal images or super-resolution (DeepSIM) 3D Z-stack images were captured by Nikon Ti2 inverted microscope using 405, 477, 545 and 637 nm lasers. To ensure optimal data comparability, microscope settings were kept constant throughout all experiments.
5. Conclusions
In conclusion, our findings, in line with accumulating evidence, suggest that a functional cholinergic phenotype is a shared feature of several carcinoma cell lines, potentially serving as a survival checkpoint. The discovery of extracellular membrane-bound ChAT uniquely in neuroblastoma SH-SY5Y cells points to a novel form of in situ ACh signaling that warrants further investigation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: Extracellular and Intracellular staining of ChAT in SH-SY5Y neuroblastoma cell line with two other different anti-ChAT antibodies; Figure S2: Negative controls for the secondary antibody staining in the SH-SY5Y neuroblastoma cell line.
Author Contributions
Conceptualization, TDS, HB, RL and KV; methodology, TDS, BT, ST and LD; formal analysis, TDS, BT and ST; resources, TDS and HB; data curation, TDS, BT, ST and LD; writing—original draft preparation, TDS and BT; writing—review and editing, TDS, HB, BT, ST, LD, RL and KV; visualization, TDS, BT and ST; supervision, TDS and BH; project administration, TDS; funding acquisition, TDS and HB. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Åhlén-Foundation; Frimurarna Foundation, Ulla-Carin Linquist Foundation for ALS research; Alzheimer´s Association (AARG-24-1310046); Dementia Foundation (Demensfonden); Foundation for Old Servants (Gamla Tjänarinnor); Karolinska Institutet´s Research Foundations; Olle Engkvist Byggmästare Foundation; Magnus Bergvalls Foundation; Gun and Bertil Stohnes Foundation; and grant from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (ALF Med N 20200330), the Swedish Cancer Society (#CAN2021/1469, # 24 3793) and the Stockholm Cancer Society (#221212, #241282 and #221383).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| AChRs | ACh receptors |
| A-CoA | Acetyl-Coenzyme A |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AF647 | Alexa Fluor 647 |
| AF648 | Alexa Fluor 648 |
| APC | Allophycocyanin |
| BChE | Butyrylcholinesterase |
| BDNF | Brain derived neurotrophic factor |
| BSA | bovine serum albumin |
| ChAT | Choline acetyltransferase |
| DMSO | Dimethyl sulfoxide. |
| ENS | Enteric nervous system |
| FBS | Foetal bovine serum |
| FITC | Fluorescein isothiocyanate |
| HPLC-MS/MS | high-performance liquid chromatography-tandem mass spectrometry |
| nAChRs | Nicotinic AChRs |
| NGF | Nerve growth factor |
| mAChRs | Muscarinic AChRs |
| PE | Phycoerythrin |
| PPIs | Proton pump inhibitors |
| SCLCs | Small cell lung cancer cells |
References
- Vijayaraghavan, S.; Karami, A.; Aeinehband, S.; Behbahani, H.; Grandien, A.; Nilsson, B.; Ekdahl, K.N.; Lindblom, R.P.; Piehl, F.; Darreh-Shori, T. Regulated Extracellular Choline Acetyltransferase Activity- The Plausible Missing Link of the Distant Action of Acetylcholine in the Cholinergic Anti-Inflammatory Pathway. PloS one 2013, 8, e65936. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.; Hasooni, L.P.; Gera, R.; Mitra, S.; Bjorndahl, L.; Darreh-Shori, T. Presence of key cholinergic enzymes in human spermatozoa and seminal fluiddagger. Biol Reprod 2024, 110, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer's disease. J Comp Neurol 2013, 521, 4124–4144. [Google Scholar] [CrossRef]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 2003, 4, 131–138. [Google Scholar] [CrossRef]
- Li, X.; Yu, B.; Sun, Q.; Zhang, Y.; Ren, M.; Zhang, X.; Li, A.; Yuan, J.; Madisen, L.; Luo, Q.; Zeng, H.; Gong, H.; Qiu, Z. Generation of a whole-brain atlas for the cholinergic system and mesoscopic projectome analysis of basal forebrain cholinergic neurons. Proc Natl Acad Sci USA 2018, 115, 415–420. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Esiri, M.M.; Bowen, D.M.; Smith, C.C. Alzheimer's disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J Neurol Sci 1982, 57, 407–417. [Google Scholar] [CrossRef]
- Esiri, M.M.; Pearson, R.C.; Steele, J.E.; Bowen, D.M.; Powell, T.P. A quantitative study of the neurofibrillary tangles and the choline acetyltransferase activity in the cerebral cortex and the amygdala in Alzheimer's disease. J Neurol Neurosurg Psychiatry 1990, 53, 161–165. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Ivanco, L.S.; Lopresti, B.; Koeppe, R.A.; Davis, J.G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol 2003, 60, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska, E.B.; Andras, A.; Milne, J.; Abdel-All, Z.; Borr, I.; Jaros, E.; Perry, R.H.; Honer, W.G.; Cleghorn, A.; Doherty, J.; McIntosh, G.; Perry, E.K.; Kalaria, R.N.; McKeith, I.G. Synaptic proteins and choline acetyltransferase loss in visual cortex in dementia with Lewy bodies. J Neuropathol Exp Neurol 2013, 72, 53–60. [Google Scholar] [CrossRef]
- Yates, C.M.; Simpson, J.; Maloney, A.F.; Gordon, A.; Reid, A.H. Alzheimer-like cholinergic deficiency in Down syndrome. Lancet 1980, 2, 979. [Google Scholar] [CrossRef]
- Kanel, P.; Spears, C.C.; Roytman, S.; Koeppe, R.A.; Frey, K.A.; Scott, P.J.H.; Albin, R.L.; Bohnen, N.I. Differential cholinergic systems' changes in progressive supranuclear palsy versus Parkinson's disease: an exploratory analysis. J Neural Transm (Vienna) 2022, 129, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Warren, N.M.; Piggott, M.A.; Perry, E.K.; Burn, D.J. Cholinergic systems in progressive supranuclear palsy. Brain 2005, 128, 239–249. [Google Scholar] [CrossRef]
- Shinotoh, H.; Namba, H.; Yamaguchi, M.; Fukushi, K.; Nagatsuka, S.; Iyo, M.; Asahina, M.; Hattori, T.; Tanada, S.; Irie, T. Positron emission tomographic measurement of acetylcholinesterase activity reveals differential loss of ascending cholinergic systems in Parkinson's disease and progressive supranuclear palsy. Annals of neurology 1999, 46, 62–69. [Google Scholar] [CrossRef]
- Ruberg, M.; Javoy-Agid, F.; Hirsch, E.; Scatton, B.; Lheureux, R.; Hauw, J.J.; Duyckaerts, C.; Gray, F.; Morel-Maroger, A.; Rascol, A.; et al. Dopaminergic and cholinergic lesions in progressive supranuclear palsy. Annals of neurology 1985, 18, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Imai, S.; Nakanishi, I.; Ichikawa, T.; Deguchi, T. Immunohistochemical study on choline acetyltransferase in the spinal cord of patients with amyotrophic lateral sclerosis. Pathology international 1995, 45, 933–939. [Google Scholar] [CrossRef]
- Gillberg, P.G.; Aquilonius, S.M.; Eckernas, S.A.; Lundqvist, G.; Winblad, B. Choline acetyltransferase and substance P-like immuno-reactivity in the human spinal cord: changes in amyotrophic lateral sclerosis. Brain Res 1982, 250, 394–397. [Google Scholar] [CrossRef]
- Harrington, A.M.; Hutson, J.M.; Southwell, B.R. Cholinergic neurotransmission and muscarinic receptors in the enteric nervous system. Progress in histochemistry and cytochemistry 2010, 44, 173–202. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Gershon, M.D. The bowel and beyond: the enteric nervous system in neurological disorders. Nature reviews Gastroenterology & hepatology 2016, 13, 517–528. [Google Scholar] [CrossRef]
- Wessler, I.; Kilbinger, H.; Bittinger, F.; Unger, R.; Kirkpatrick, C.J. The non-neuronal cholinergic system in humans: expression, function and pathophysiology. Life Sci 2003, 72, 2055–2061. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Proskocil, B.; Blusztajn, J.K.; Mark, G.P.; Spindel, E.R. Synthesis of acetylcholine by lung cancer. Life Sci 2003, 72, 2159–2168. [Google Scholar] [CrossRef]
- Chernyavsky, A.I.; Shchepotin, I.B.; Galitovkiy, V.; Grando, S.A. Mechanisms of tumor-promoting activities of nicotine in lung cancer: synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC cancer 2015, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; Davis, R.J.; Nomikos, G.; Raufman, J.P. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation, American journal of physiology. Gastrointestinal and liver physiology 2008, 295, G591–G597. [Google Scholar] [CrossRef]
- Friedman, J.R.; Richbart, S.D.; Merritt, J.C.; Brown, K.C.; Nolan, N.A.; Akers, A.T.; Lau, J.K.; Robateau, Z.R.; Miles, S.L.; Dasgupta, P. Acetylcholine signaling system in progression of lung cancers. Pharmacology & therapeutics 2019, 194, 222–254. [Google Scholar] [CrossRef]
- Song, P.; Sekhon, H.S.; Jia, Y.; Keller, J.A.; Blusztajn, J.K.; Mark, G.P.; Spindel, E.R. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer research 2003, 63, 214–221. [Google Scholar]
- Wang, S.; Hu, Y. alpha7 nicotinic acetylcholine receptors in lung cancer. Oncol Lett 2018, 16, 1375–1382. [Google Scholar] [CrossRef]
- Smith, C.P.; Carroll, P.T. A comparison of solubilized and membrane bound forms of choline-O-acetyltransferase (EC 2.3.1.6) in mouse brain nerve endings. Brain Res 1980, 185, 363–371. [Google Scholar] [CrossRef]
- Carroll, P.T. Membrane-bound choline-O-acetyltransferase in rat hippocampal tissue is associated with synaptic vesicles. Brain Res 1994, 633, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Pahud, G.; Salem, N.; van de Goor, J.; Medilanski, J.; Pellegrinelli, N.; Eder-Colli, L. Study of subcellular localization of membrane-bound choline acetyltransferase in Drosophila central nervous system and its association with membranes. Eur J Neurosci 1998, 10, 1644–1653. [Google Scholar] [CrossRef]
- Dante, D.; Jangra, J.; Baidya, A.T.K.; Kumar, R.; Darreh-Shori, T. Micellar Choline-Acetyltransferase Complexes Exhibit Ultra-Boosted Catalytic Rate for Acetylcholine Synthesis-Mechanistic Insights for Development of Acetylcholine-Enhancing Micellar Nanotherapeutics. Int J Mol Sci 2024, 25, 13602. [Google Scholar] [CrossRef]
- Resendes, M.C.; Dobransky, T.; Ferguson, S.S.; Rylett, R.J. Nuclear localization of the 82-kDa form of human choline acetyltransferase. J Biol Chem 1999, 274, 19417–19421. [Google Scholar] [CrossRef]
- Gill, S.K.; Bhattacharya, M.; Ferguson, S.S.; Rylett, R.J. Identification of a novel nuclear localization signal common to 69- and 82-kDa human choline acetyltransferase. J Biol Chem 2003, 278, 20217–20224. [Google Scholar] [CrossRef]
- Kumar, A.; Lana, E.; Kumar, R.; Lithner, C.U.; Darreh-Shori, T. Soluble Abeta42 Acts as Allosteric Activator of the Core Cholinergic Enzyme Choline Acetyltransferase. Frontiers in molecular neuroscience 2018, 11, 327. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, A.; Nordberg, A.; Langstrom, B.; Darreh-Shori, T. Proton pump inhibitors act with unprecedented potencies as inhibitors of the acetylcholine biosynthesizing enzyme-A plausible missing link for their association with incidence of dementia. Alzheimer's & dementia: the journal of the Alzheimer's Association 2020, 16, 1031–1042. [Google Scholar] [CrossRef]
- Redin, E.; Quintanal-Villalonga, A.; Rudin, C.M. Small cell lung cancer profiling: an updated synthesis of subtypes, vulnerabilities, and plasticity. Trends Cancer 2024, 10, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Inazu, M.; Yamada, T.; Kubota, N.; Yamanaka, T. Functional expression of choline transporter-like protein 1 (CTL1) in small cell lung carcinoma cells: a target molecule for lung cancer therapy. Pharmacological research 2013, 76, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Rui, J.; Li, X.; Meng, X.; Liu, Q. Use of (1)(1)C-Choline positron emission tomography/computed tomography to investigate the mechanism of choline metabolism in lung cancer. Molecular medicine reports 2015, 11, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Sekhon, H.S.; Lu, A.; Arredondo, J.; Sauer, D.; Gravett, C.; Mark, G.P.; Grando, S.A.; Spindel, E.R. M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer research 2007, 67, 3936–3944. [Google Scholar] [CrossRef]
- de Medeiros, L.M.; De Bastiani, M.A.; Rico, E.P.; Schonhofen, P.; Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Grun, L.; Barbe-Tuana, F.; Zimmer, E.R.; Castro, M.A.A.; Parsons, R.B.; Klamt, F. Cholinergic Differentiation of Human Neuroblastoma SH-SY5Y Cell Line and Its Potential Use as an In vitro Model for Alzheimer's Disease Studies. Molecular neurobiology 2019, 56, 7355–7367. [Google Scholar] [CrossRef]
- Grisaru, D.; Sternfeld, M.; Eldor, A.; Glick, D.; Soreq, H. Structural roles of acetylcholinesterase variants in biology and pathology. Eur J Biochem 1999, 264, 672–686. [Google Scholar] [CrossRef]
- Chen, V.P.; Luk, W.K.; Chan, W.K.; Leung, K.W.; Guo, A.J.; Chan, G.K.; Xu, S.L.; Choi, R.C.; Tsim, K.W. Molecular Assembly and Biosynthesis of Acetylcholinesterase in Brain and Muscle: the Roles of t-peptide, FHB Domain, and N-linked Glycosylation. Front Mol Neurosci 2011, 4, 36. [Google Scholar] [CrossRef]
- Garcia-Ayllon, M.S.; Saez-Valero, J.; Munoz-Delgado, E.; Vidal, C.J. Identification of hybrid cholinesterase forms consisting of acetyl- and butyrylcholinesterase subunits in human glioma. Neuroscience 2001, 107, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Zakut, H.; Even, L.; Birkenfeld, S.; Malinger, G.; Zisling, R.; Soreq, H. Modified properties of serum cholinesterases in primary carcinomas. Cancer 1988, 61, 727–737. [Google Scholar] [CrossRef]
- Darreh-Shori, T.; Rezaeianyazdi, S.; Lana, E.; Mitra, S.; Gellerbring, A.; Karami, A.; Bogdanovic, N.; Lithner, C.U.; Winblad, B.; Behbahani, H. Increased Active OMI/HTRA2 Serine Protease Displays a Positive Correlation with Cholinergic Alterations in the Alzheimer's Disease Brain. Molecular neurobiology 2019, 56, 4601–4619. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yao, M.; Xu, J.; Quan, Y.; Zhang, K.; Yang, R.; Gao, W.Q. Autocrine Activation of CHRM3 Promotes Prostate Cancer Growth and Castration Resistance via CaM/CaMKK-Mediated Phosphorylation of Akt. Clinical cancer research: an official journal of the American Association for Cancer Research 2015, 21, 4676–4685. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhi, X.; Zhang, Q.; Wei, S.; Li, Z.; Zhou, J.; Jiang, J.; Zhu, Y.; Yang, L.; Xu, H.; Xu, Z. Muscarinic receptor M3 mediates cell proliferation induced by acetylcholine and contributes to apoptosis in gastric cancer. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine 2016, 37, 2105–2117. [Google Scholar] [CrossRef]
- Vernay, A.; Cosson, P. Immunofluorescence labeling of cell surface antigens in Dictyostelium. BMC Res Notes 2013, 6, 317. [Google Scholar] [CrossRef]
Figure 1.
Surface ChAT staining and flow cytometric analyses in a neuroblastoma cell line compared to three lung cancer cell lines. The left panel shows dot plots of Live/Dead Dye Staining versus ChAT staining in the neuroblastoma SH-SY5Y cell line. The middle dot plot graph shows results after blocking the antibody with recombinant human ChAT protein, and the upper dot plot graph shows unstained cells. The lower dot plot graph indicates the percentage of ChAT staining in SH-SY5Y cells. The digits in the dot plots represent mean ± SD of triplicates. The right panel presents histograms for the small cell lung cancer cells (H82, H69) and the lung adenocarcinoma cell line, A549, stained similarly to the left panel. Abbreviations: ChAT = choline acetyltransferase; APC anti-ChAT Ab = Allophycocyanin-conjugated anti-ChAT antibody.
Figure 1.
Surface ChAT staining and flow cytometric analyses in a neuroblastoma cell line compared to three lung cancer cell lines. The left panel shows dot plots of Live/Dead Dye Staining versus ChAT staining in the neuroblastoma SH-SY5Y cell line. The middle dot plot graph shows results after blocking the antibody with recombinant human ChAT protein, and the upper dot plot graph shows unstained cells. The lower dot plot graph indicates the percentage of ChAT staining in SH-SY5Y cells. The digits in the dot plots represent mean ± SD of triplicates. The right panel presents histograms for the small cell lung cancer cells (H82, H69) and the lung adenocarcinoma cell line, A549, stained similarly to the left panel. Abbreviations: ChAT = choline acetyltransferase; APC anti-ChAT Ab = Allophycocyanin-conjugated anti-ChAT antibody.
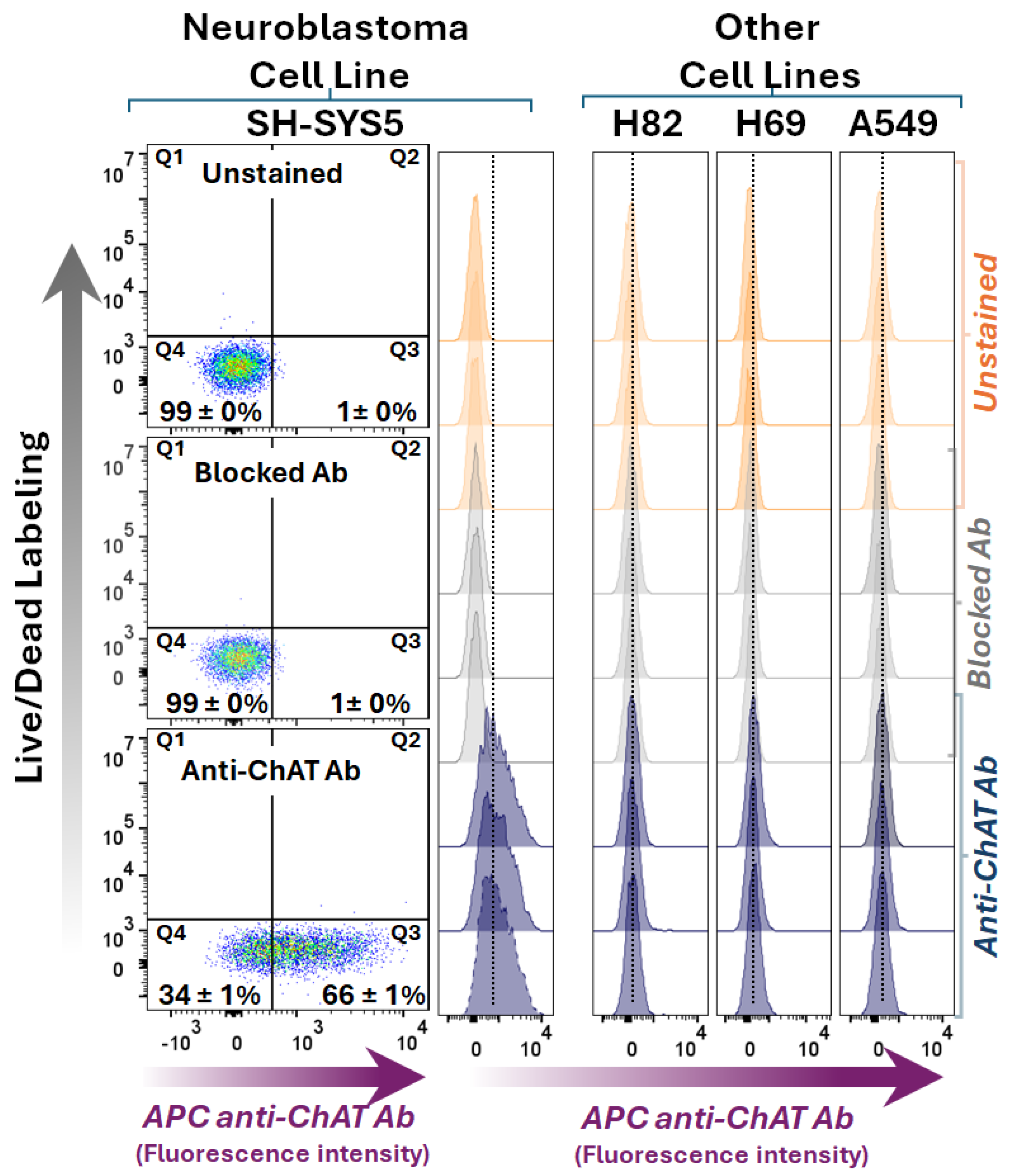
Figure 2.
Whole-cell ChAT staining and flow cytometric analyses in a neuroblastoma cell line compared to three lung cancer cell lines. The dot plots show side scatter area versus ChAT staining in the neuroblastoma SH-SY5Y cell line after membrane permeabilization, allowing the anti-ChAT antibody to access both surface and intracellular ChAT protein, i.e., reflecting the whole-cell ChAT staining. The upper dot plot graph shows unstained cells, the middle dot plot graph shows cells incubated with a pre-blocked anti-ChAT antibody, and the lower dot plot graph indicates the percentage of the neuroblastoma SH-SY-5Y cells were positive with respect to ChAT protein expression. Histograms for the small cell lung carcinoma (H82 and H69) and the lung adenocarcinoma cell line, A549, are shown in comparison with SH-SY5Y neuroblastoma cells. Notably, the ChAT staining in the lung carcinoma cell lines reflect only an intracellularly localized ChAT since these cells lacked surface localized ChAT (as shown in Figure 1). A comparison of the ChAT staining result for the SH-SY-5Y neuroblastoma cells (79 ± 3%) with the extracellular ChAT data in Figure 1 (66 ± 1%), indicate that in contrast to the lung carcinoma cell lines most of the ChAT protein in the neuroblastoma cells were localized extracellularly. The digits in the dot plots represent mean ± SD of triplicates. ChAT = choline-acetyltransferase; APC anti-ChAT Ab = Allophycocyanin-conjugated anti-ChAT antibody.
Figure 2.
Whole-cell ChAT staining and flow cytometric analyses in a neuroblastoma cell line compared to three lung cancer cell lines. The dot plots show side scatter area versus ChAT staining in the neuroblastoma SH-SY5Y cell line after membrane permeabilization, allowing the anti-ChAT antibody to access both surface and intracellular ChAT protein, i.e., reflecting the whole-cell ChAT staining. The upper dot plot graph shows unstained cells, the middle dot plot graph shows cells incubated with a pre-blocked anti-ChAT antibody, and the lower dot plot graph indicates the percentage of the neuroblastoma SH-SY-5Y cells were positive with respect to ChAT protein expression. Histograms for the small cell lung carcinoma (H82 and H69) and the lung adenocarcinoma cell line, A549, are shown in comparison with SH-SY5Y neuroblastoma cells. Notably, the ChAT staining in the lung carcinoma cell lines reflect only an intracellularly localized ChAT since these cells lacked surface localized ChAT (as shown in Figure 1). A comparison of the ChAT staining result for the SH-SY-5Y neuroblastoma cells (79 ± 3%) with the extracellular ChAT data in Figure 1 (66 ± 1%), indicate that in contrast to the lung carcinoma cell lines most of the ChAT protein in the neuroblastoma cells were localized extracellularly. The digits in the dot plots represent mean ± SD of triplicates. ChAT = choline-acetyltransferase; APC anti-ChAT Ab = Allophycocyanin-conjugated anti-ChAT antibody.
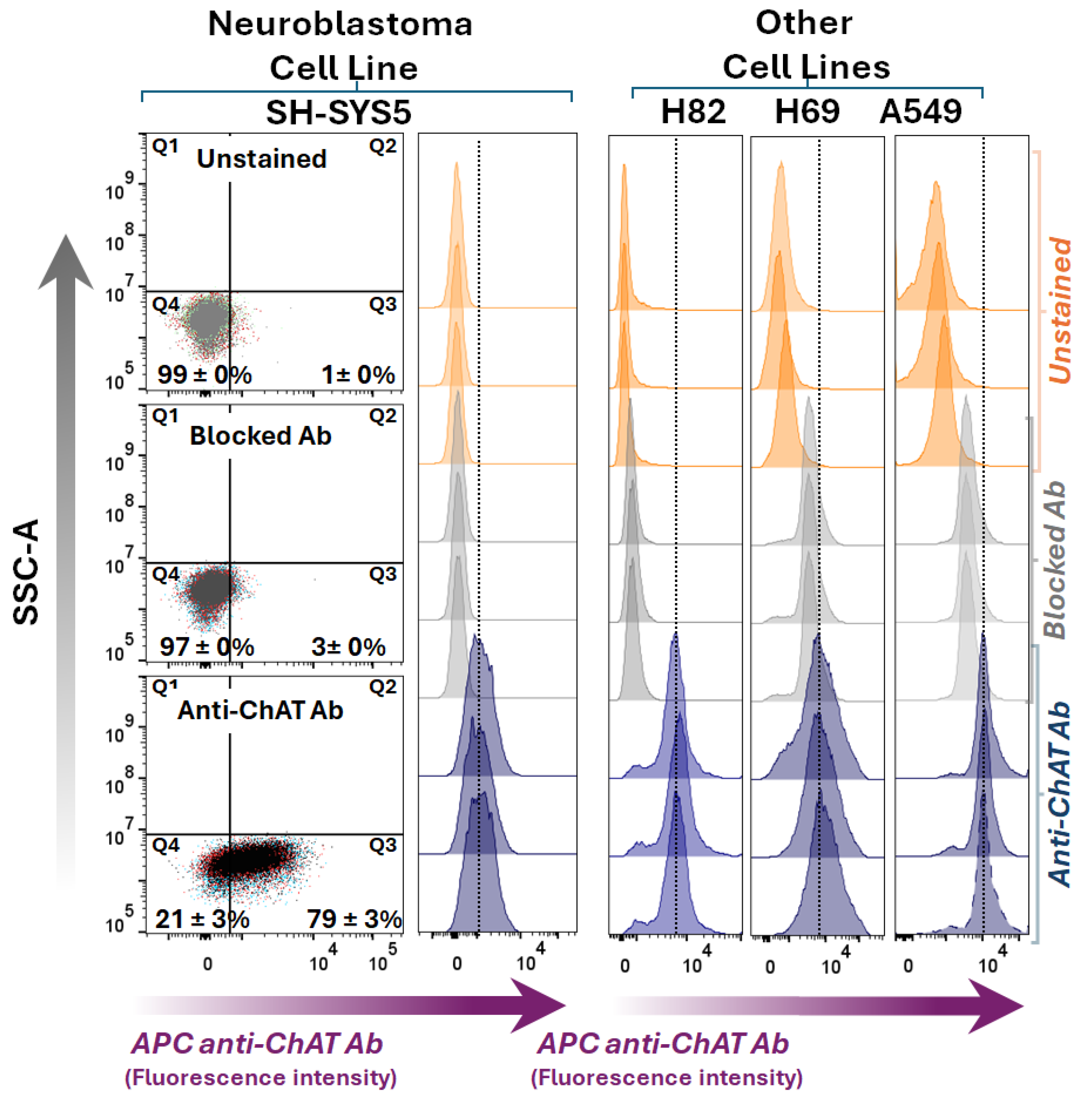
Figure 3.
Synthesis and release of acetylcholine by the neuroblastoma cell line and three small cell lung cancer cell lines. The upper panel graphs show the measured concentration of acetylcholine (ACh) in the cell lysates and conditioned media of the cells, indicating cellular biosynthesis and release of ACh, respectively. Control cells (Ctrl) refer to untreated cells and vehicle (DMSO) treated cells (Veh Ctrl). Neostigmine, a dual inhibitor of the ACh-hydrolysing enzymes, AChE and BChE, was used to preserve ACh against enzymatic hydrolysis, allowing measurement by HPLC-MS technique. Data are given as mean ± SD. AChE = acetylcholinesterase; BChE = butyrylcholinesterase; DMSO = dimethyl sulfoxide.
Figure 3.
Synthesis and release of acetylcholine by the neuroblastoma cell line and three small cell lung cancer cell lines. The upper panel graphs show the measured concentration of acetylcholine (ACh) in the cell lysates and conditioned media of the cells, indicating cellular biosynthesis and release of ACh, respectively. Control cells (Ctrl) refer to untreated cells and vehicle (DMSO) treated cells (Veh Ctrl). Neostigmine, a dual inhibitor of the ACh-hydrolysing enzymes, AChE and BChE, was used to preserve ACh against enzymatic hydrolysis, allowing measurement by HPLC-MS technique. Data are given as mean ± SD. AChE = acetylcholinesterase; BChE = butyrylcholinesterase; DMSO = dimethyl sulfoxide.
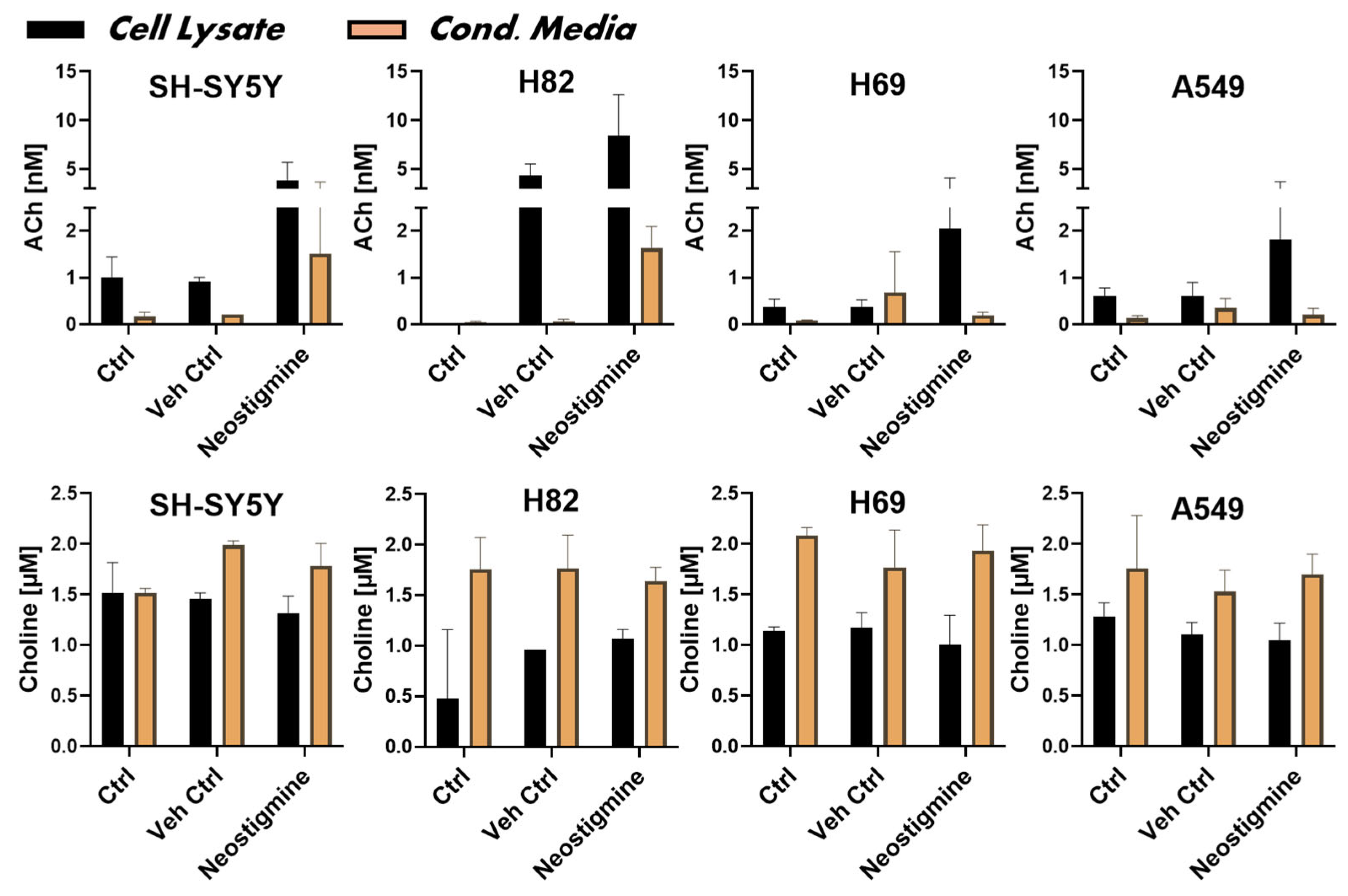
Figure 6.
Surface confocal microscopy of cholinergic markers in the neuroblastoma SH-SY5Y cell line. The neuroblastoma SH-SY5Y cells were incubated with specific antibodies without membrane permeabilization, preventing antibodies from entering the cells and allowing staining of only surface extracellularly bound proteins. Micrographs represent images of surface-stained cholinergic markers (ChAT, AChE, BChE, M1-mAChR, and α7-nAChR) depicted in green and co-labeled with DAPI in blue. Magnification 20x; Scale bar 20 µm. Zoom panels below each set are 60x magnification from the area indicated by the white box. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
Figure 6.
Surface confocal microscopy of cholinergic markers in the neuroblastoma SH-SY5Y cell line. The neuroblastoma SH-SY5Y cells were incubated with specific antibodies without membrane permeabilization, preventing antibodies from entering the cells and allowing staining of only surface extracellularly bound proteins. Micrographs represent images of surface-stained cholinergic markers (ChAT, AChE, BChE, M1-mAChR, and α7-nAChR) depicted in green and co-labeled with DAPI in blue. Magnification 20x; Scale bar 20 µm. Zoom panels below each set are 60x magnification from the area indicated by the white box. ChAT = choline acetyltransferase; AChE = acetylcholinesterase; BChE = butyrylcholinesterase; α7-nAChR = α7-subtype of nicotinic acetylcholine receptor; M1-mAChR = M1-subtype of muscarinic AChR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



