Submitted:
23 May 2025
Posted:
23 May 2025
You are already at the latest version
Abstract
Obesity, a chronic inflammatory disease, is caused by a positive balance between energy intake and energy expenditure. Adipose tissue (AT) inflammation is the main cause of local and systemic inflammation and oxidative stress and is the link between systemic inflammation and obesity-associated metabolic abnormalities, such as dyslipidemia, hypertension, insulin resistance (IR), fatty liver disease, and dysfunction of pancreatic β-cells. AT macrophages are derived from bone marrow and blood monocytes that, upon arrival and under the pressure of the AT microenvironment, are differentiated into AT-associated macrophages (ATMs). The AT microenvironment in obesity causes the activation of transcription factors that control the expression of a number of inflammatory genes, leading to an ATM M1 phenotype or classically activated ATM. These M1 macrophages express a number of proinflammatory genes and are the main cause of AT inflammation. Herein, we reviewed recently published information on the molecular mechanisms leading to the phenotypic switch of macrophages under the pressure of obese AT. This information is needed to develop novel mechanism-based therapeutics to reduce AT inflammation and thus the metabolic risk associated with obesity.
Keywords:
obesity
; adipose tissue
; metaflammation
; microenvironment
; metabolic risk
; macrophage phenotype
; polarization
Introduction
Macrophages are cells of the innate immune system that are characterized by the detection, phagocytosis and destruction of bacteria and/or other invading microorganisms[1]. These cells are involved in the presentation of antigens for the development of the adaptive immune response and can secrete cytokines and chemokines to initiate the inflammatory process against injury[2].
The phenotypic heterogeneity of macrophages can be divided into two main subtypes: M1-type macrophages and M2-type macrophages[3]. M1 macrophages are described as the proinflammatory type and are important for direct host defense against pathogens, including phagocytosis and the secretion of proinflammatory cytokines and microbicidal molecules[4]. M2 macrophages have opposite functions, regulating the resolution phase of inflammation and repairing damaged tissues[5]. More recent in vitro and larger ex vivo studies have shown that macrophage phenotypes are much more diverse, overlapping with one another in terms of gene expression and function, revealing that these many hybrid states form a continuum of activation states that depend on the microenvironment[6]. There is great diversity in the gene expression profiles of different populations of tissue macrophages, which is why the activation spectrum of macrophages is considered broader and involves a complex regulatory pathway that responds to many different signals from the adipose environment[7,8].
Systemic inflammation plays a crucial role in the development of metabolic risks associated with obesity[9]. Chronic low-grade inflammation, often referred to as "metaflammation", is a hallmark of obesity resulting from the expansion of AT[10]. This expansion leads to hypoxia, mechanical stress on adipocytes, and the release of proinflammatory cytokines[11]. These factors collectively trigger an immune response that recruits inflammatory cells such as macrophages and T cells into the AT, perpetuating the inflammatory state[12]. This systemic inflammation not only exacerbates IR but also contributes to the progression of metabolic disorders such as type 2 diabetes (T2D) and cardiovascular disease (CVD)[13]. Recent research highlights that the interplay between metabolic dysregulation and immune activation is central to the pathogenesis of these obesity-related conditions, underscoring the importance of targeting inflammation in therapeutic strategies for obesity and its associated metabolic risks[14].
In this review, we summarize the most recent findings concerning AT inflammation and obesity, highlighting the physiological, cellular, and molecular mechanisms through which AT inflammation contributes to AT dysfunction, systemic inflammation and obesity-associated metabolic abnormalities.
Macrophage Polarization: M1 and M2 Phenotypes
Circulating monocytes in the blood come from the bone marrow through a process known as hematopoiesis, where hematopoietic stem cells differentiate into myeloid progenitors. In response to growth factors such as GM-CSF or G-CSF, these cells generate granulomonocytic progenitors and then monoblasts, which in turn also give rise to premonocytes in the presence of these growth factors; thus, they ultimately differentiate into mature monocytes, which leave the bone marrow and enter the bloodstream[15]. There are different populations of monocytes in the circulation; classical monocytes represent 85% of circulating monocytes and present CD14++ and CD16- as surface markers; intermediate monocytes represent 5% of monocytes in the blood and have CD14+ and CD16+ surface markers; and nonclassical monocytes present CD14+ and CD16++ surface markers and constitute the remaining 10% of circulating monocytes (Figure 1)[16,17]. These immune cells migrate to peripheral tissues in response to some stimulus; within these tissues, they differentiate into macrophages or dendritic cells and trigger specific responses owing to the tissue microenvironment in which they are found; these responses occur owing to the phenotypic heterogeneity presented by macrophages owing to the previous polarization of monocytes[3,18].
Macrophage polarization is a process by which macrophages produce different functional programs in response to microenvironment signals[5]. Macrophage phenotypes have multiple functions in the body: powerful effector cells of the innate immune system, removal of cellular waste, tissue homeostasis and repair, and embryonic development[19]. Macrophages can be polarized into the M1 and M2 phenotypes, so they perform different functions under certain conditions of the cellular environment; these phenotypes differ in their biological functions owing to their surface markers and secreted cytokines[16].
M1 Macrophage’s Phenotype
The M1 macrophage phenotype is activated by the “classical pathway” through the recognition of pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS); inflammatory cytokines, such as interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα); and reactive oxygen species (ROS) and reactive nitrogen (RNS) species[16]. The surface markers expressed in this macrophage phenotype are CD80 antigen (CD80), CD86 antigen (CD86), Toll-like receptor 2 (TLR-2), Toll-like receptor 4 (TLR-4), major histocompatibility complex class II (HLA-DR) and interferon gamma receptor (IFNγR)[20].
M1-activated macrophages are produced because the activation of signaling pathways leads to the activation of transcription factors; high expression of nuclear factor kappa light polypeptide gene enhancer in B cells (NF-κB) and signal transducer and activator of transcription 1 (STAT1); and the upregulation of signal transducer and activator of transcription 5 (STAT5), interferon regulatory factor 5 (IRF5), interferon regulatory factor 3 (IRF3), activator protein (AP-1), and hypoxia-inducible factor 1 alpha (HIF-1α)[2,8]. The induction of these transcription factors enhances the inflammatory, bactericidal, viricidal and antitumor capacity of these cells via the secretion of high levels of proinflammatory cytokines such as TNFα, interleukin-1 beta (IL-1β), interleukin-6 (IL-6), interleukin-12 (IL-12), interleukin-1 alpha (IL-1α), interleukin-23 (IL-23), interleukin-18 (IL18), and chemokines such as C-X-C motif chemokine ligand 9 (CXCL9), C-X-C motif chemokine ligand 10 (CXCL10), C-X-C motif chemokine ligand 11 (CXCL11), C-C motif chemokine ligand (CCL5), C-C motif chemokine receptor 7 (CCR7), C-X-C motif chemokine ligand 16 (CXCL16), and granulocyte–macrophage colony-stimulating factor (GM-CSF) (Figure 2); these cytokines and chemokines can induce chronic inflammation and lead to disease pathogenesis[21,22].
In addition, to increase their ability to eliminate pathogens, M1 macrophages produce greater amounts of nitric oxide (•NO) and citrulline from arginine, products of inducible nitric oxide synthase (iNOS) pathway metabolism[23]. Owing to their ability to fight pathogens, M1 macrophages are present during acute infectious diseases[24]. Several studies have shown that bacterial infection, the early stage of inflammation and chronic inflammatory processes induce macrophage polarization toward the M1 phenotype, resulting in phagocytosis and the intracellular killing of bacteria as well as the removal of damaged or dead body cells[24,25,26]. Inadequate control of the inflammatory response triggered by M1 macrophages can lead to alterations in tissue homeostasis and prevent injury repair[27].
M2 Macrophage’s Phenotype
The M2 macrophage phenotype is activated by the “alternative or nonclassical pathway”, also known as anti-inflammatory macrophages; activation is produced by high levels of interleukin-4 (IL-4) and interleukin-13 (IL-13) and other ligands, such as interleukin-10 (IL-10), interleukin-33 (IL-33), transforming growth factor beta (TGFβ), interleukin-21 (IL-21) and interleukin-25 (IL-25)[23]. The surface markers expressed in this macrophage phenotype are macrophage-associated antigen (CD163), macrophage scavenger receptor 1 (CD204), mannose receptor type-1 (CD206), CD209 antigen (CD209) and interleukin-4 receptor alpha (IL-4Rα)[2,23].
The activation of M2 macrophages triggers the regulation of signaling pathways that produce the following transcription factors: signal transducer and activator of transcription 6 (STAT6), signal transducer and activator of transcription 3 (STAT3), interferon regulatory factor 4 (IRF4), Krüppel-like factor 4 (KLF4), peroxisome proliferator activated receptor delta (PPARδ) and peroxisome proliferator activated receptor gamma (PPARγ)[22,28]. M2-polarized macrophages secrete interleukins such as IL-10 and IL-4; the chemokines C-C motif chemokine ligand 1 (CCL1), C-C motif chemokine ligand 17 (CCL17), C-C motif chemokine ligand 18 (CCL18), C-C motif chemokine ligand 22 (CCL22), C-C motif chemokine ligand 24 (CCL24), and C-X-C motif chemokine ligand 13 (CXCL13); the growth factors TGFβ and vascular endothelial growth factor (VEGF); and other components involved in humoral immunity, wound healing and tolerance of autoantigens and neoantigens[29,30,31]. Thus, M2 macrophages govern functions at the interfaces of immunity, infection prevention, tissue development, removal and repair, angiogenesis and immunomodulation (Figure 2)[16,21].
M2 macrophages have regulatory or wound-healing functions[2]. Regulatory functions show anti-inflammatory and phagocytic properties, which are important in the resolving phases of inflammation, producing the immunosuppressive cytokine IL-10, and can be triggered by immune complexes, prostaglandins, apoptotic cells, and IL-10[32]. On the other hand, wound-healing functions produce IL-4 and positively regulate the activity of the arginase pathway, obtaining ornithine and urea as products from arginine; this enzyme is involved in the production of polyamines and collagen, thus regenerating damaged tissue[33,34].
Recently, M2 macrophages have been subclassified into subtypes, which leads to more complex systematization: M2a, M2b and M2c[8]. M2a macrophages are activated by IL-4 and IL-13, leading to upregulated expression of arginase-1 (Arg-1) and CD206, impact presentation by the MHC II system, and production of IL-10 and TGFβ, leading to tissue regeneration and the internalization of proinflammatory molecules to prevent the inflammatory response[35]. M2b macrophages produce IL-1, IL-6, IL-10, and TNFα in response to immune complexes or bacterial LPS, leading to Th2 cell activation and anti-inflammatory activity; however, they can inhibit the polarization of naive macrophages into M1 macrophages, thus promoting the survival of microorganisms or tumor cells [36]. M2c macrophages are activated by IL-10, TGFβ, and glucocorticoids[20]. These cells express CD206 and CD163 surface markers and then produce large amounts of IL-10, TGFβ and chemokines, leading to suppression of the inflammatory response. These macrophages are involved in tissue homeostasis due to their high phagocytic capacity[35,37]. Another subtype is M2d macrophages, which respond to IL-6, agonist TLRs and adenosine stimuli[38]. These macrophages are characterized by heterogeneous macrophage populations and pro- and anti-inflammatory functions and are also known as tumor-associated macrophages (TAMs) because of their immunosuppressive capacity and ability to promote angiogenesis in cancer (Table 1)[29].
Although the activation state of M2-type macrophages involves heterogeneous macrophage populations, some markers are shared between subtypes, so the strict division of macrophages into subtypes has not been fully elucidated[36]. Tissues contain a diverse range of stimuli that give rise to mixed populations of M2 macrophages, so these cells are of scientific interest because of their broad spectrum of activation states[6].
Inflammation of the Adipose Tissue in Obesity
Obesity is considered a global epidemic that, as a consequence, reduces quality of life, presents a lower life expectancy and increases health care costs[39]. This disease is a risk factor for the development of IR, T2D, hyperlipidemia, CVD, hyperuricemia and disorders of the immune system[40,41]. Obesity is a multifactorial chronic disease that cannot be considered only the result of an energy imbalance between caloric intake and expenditure; however, a series of metabolic abnormalities, oxidative stress, mitochondrial and immunological dysfunction, and chronic inflammation in obese individuals have also been identified[10,42]. Although AT is well known to generate an immune response to the excessive presence of nutrients, there is still no knowledge about the initial inflammatory trigger involved[43]. Therefore, more attention is currently being devoted to the study of endogenous anti-inflammatory mediators and other important therapeutic targets in AT inflammation.
AT, commonly known as body fat, is a complex loose connective tissue composed of mature adipocytes, which are the cells responsible for fat storage; preadipocytes; fibroblasts; mesenchymal cells; vascular endothelial cells; pericytes; the stromal vascular fraction; immune cells, including macrophages and T cells; and the supportive matrix of extracellular proteins[44,45]. Its functions include storing energy in the form of lipids and protecting and insulating vital organs of the body[46]. This tissue has endocrine activity and secretes various bioactive molecules, including adipokines, cytokines, and hormones, such as leptin, adiponectin, resistin, estradiol and TNFα[47,48]. There are two types of AT, white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is composed of 5–50% of the body weight, stores energy in the form of triglycerides and plays an important role in endocrine signaling by releasing hormones. WAT can be divided into subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT). SAT acts as a thermal insulator and protector, whereas VAT is more related to metabolic processes. BAT is located in the neck and large blood vessels of the thorax, plays an important role in adaptive thermogenesis and decreases with age[49,50].
Adipocytes, the main cells present in the AT, can develop through two processes: hypertrophy, described as an increase in cell size, and hyperplasia, which is the increase in the number of cells from a precursor cell—preadipocyte—that manage to differentiate until its final stage, becoming a mature adipocyte[51]. Both processes of adipocyte development occur under healthy conditions; adipocytes begin to grow at a certain time, increasing their fat volume, and when they reach the critical threshold in size, the hyperplasia process occurs; the hypertrophied cell stimulates a preadipocyte, thus generating a new adipose cell[52]. During the development of adipocytes by hypertrophy, a transient inflammatory state occurs, which is considered necessary to carry out this process[51]. The problem arises when in a state of obesity, this situation is continuous, since a larger size of the adipose cell together with the concomitant inflammation compromises the integrity and functionality of the adipocyte by being excessively hypertrophied, which modifies the metabolic behavior of the cell, generating alterations in the tissue and even triggering the apoptosis of the adipocytes[53]. Under these circumstances, obese AT has an altered secretory profile with greater production of leptin and less adiponectin, lower sensitivity to insulin in tissues, mitochondrial dysfunction and greater stress in the endoplasmic reticulum; these disruptions increase basal lipolysis and decrease de novo lipogenesis, which results in alterations in the cellular structure of adipocytes[54].
In healthy individuals, adipocytes help maintain an adequate energy balance and a normal body temperature[55]. Lipids are predominantly stored in the SAT, but in obese individuals, the SAT can reach its expansion limit or cannot expand adequately to store excess energy [55]. When adipocytes reach their triglyceride storage limit, they begin to be deposited ectopically in other tissues involved in metabolic homeostasis, that is, skeletal muscle, liver and VAT; this is due to the increase in basal lipolysis, a process known as the “overflow hypothesis”, thus altering the metabolic balance, which is associated with the development of chronic inflammation and lipotoxicity[56,57]. The increase in the flow of free fatty acids and the release of inflammatory factors act together as triggers of IR and local inflammation to later cause systemic IR and metaflammation[58]. Furthermore, BAT is often reduced under obesogenic conditions, decreasing the body's ability to burn calories and increasing the risk of metabolic diseases[59]. Overall, the hypertrophy capacity of SAT and fat storage in VAT are critical factors in the development of obesity, IR, and their associated complications[60].
Functions of Macrophages in the Healthy Adipose Tissue
The microenvironment of healthy AT is a complex and dynamic system that plays a crucial role in maintaining energy balance and metabolic homeostasis[61]. In a healthy state, the cells that compose this tissue interact in a balanced manner, promoting an anti-inflammatory and metabolically active environment[62]. The AT microenvironment is characterized by a well-regulated balance in the secretion of adipokines, cytokines, and hormones, which are essential for normal metabolic processes such as glucose and lipid metabolism, insulin sensitivity and immune responses[63]. These adipokines, such as leptin and adiponectin, are in charge of regulating appetite, increasing insulin sensitivity and regulating systemic inflammation[64]. Adiponectin has anti-inflammatory effects and promotes insulin sensitivity, helping to maintain efficient regulation of metabolism[65]. Furthermore, the vascular network within the AT is crucial for nutrient and oxygen supply, supporting the high metabolic activity of adipocytes[66]. In healthy AT, this vasculature is well developed, allowing for efficient transport of lipids and glucose into adipocytes and facilitating the removal of waste products[45]. Additionally, the presence of immune cells, such as macrophages, in healthy AT is tightly regulated by cytokines and transcription factors[67]. These immune cells play a role in tissue remodeling and maintaining a balance between proinflammatory and anti-inflammatory signals, which is critical for preventing chronic inflammation and associated metabolic disorders[67,68]. Anti-inflammatory cytokines such as IL-10 and TGFβ predominate, maintaining controlled immune activity[69]. Macrophages in the AT of healthy individuals are usually of the M2 type, which promotes tissue repair and the resolution of inflammation[70]. Furthermore, transcription factors such as PPARγ and CCAAT/enhancer binding protein α/β (C/EBPα/β) are essential for adipocyte differentiation and lipid metabolism; e.g., PPARγ plays a key role in the formation of new adipocytes and in regulating insulin sensitivity[71,72]. On the other hand, the extracellular matrix (ECM), an important factor in healthy AT, provides structural support and regulates cell signaling, which influences the cell differentiation, proliferation, and survival of resident cells[73]. The ECM components are constantly remodeled to adapt to changes in the size and function required by adipocytes, especially during periods of weight gain or weight loss[74]. A healthy ECM ensures that AT remains flexible and capable of expanding or contracting as needed without triggering fibrosis or other pathological changes[75]. Overall, the microenvironment of healthy AT is finely tuned by a network of cellular and molecular signals that support metabolic health, where the tissue can respond to nutritional changes and interact with other organs through endocrine signaling[76]. Disruptions to this microenvironment, such as excessive adipocyte hypertrophy, chronic inflammation, or impaired angiogenesis, can lead to metabolic diseases or initiate metabolic syndrome[77]. Therefore, maintaining the integrity of the AT microenvironment is essential for overall health and well-being[76].
Macrophages in the Microenvironment of Obese Adipose Tissue
Obesity creates a unique AT microenvironment characterized by chronic inflammation and altered immune responses[77]. This environment plays a key role in metabolic dysfunction and can contribute to several metabolic health issues, including IR, T2D and cancer[78].
AT has a great capacity to adapt to the body's excess energy[63]. AT is a fundamental characteristic of the development of a state of metaflammation due to increases in the size and number of adipocytes[79]. Excess lipid storage leads to adipocyte hypertrophy, resulting in hypoxia, dysfunction and cellular stress. TLR2 is activated by the presence of free fatty acids, which leads to the development of an inflammatory state caused by cellular hypertrophy, causing adipocytes and resident macrophages to secrete proinflammatory cytokines such as TNFα, IL-6, IL-1β and monocyte chemotactic protein-1 (MCP-1); these biomolecules induce the infiltration of immune cells, mainly macrophages[80,81,82]. When macrophages begin to accumulate in the tissue in response to cellular stress, they acquire the M1 phenotype, and together with adipocytes, they secrete a greater amount of proinflammatory cytokines and recruit a greater number of immune cells; the release of these inflammatory factors into the bloodstream can be directed to other tissues, where more alterations are generated at the functional level, which leads to the development of a cycle of perpetual systemic inflammation[10,83,84]. Furthermore, adipokines play a crucial role in the complications associated with obesity[85]. The inflammation produced in AT, known as lipo-inflammation, is also associated with an imbalance in the adipokine-secretory profile, where the relationship between leptin and adiponectin is altered[86,87]. Leptin is known to have an immunomodulatory role, and adiponectin acts as an insulin sensitizer and has an anti-inflammatory role at the systemic level[88]. However, in obese individuals, serum leptin levels are increased, and adiponectin levels are decreased[89]. This abnormal secretory profile of adipokines may be one of the causes of metabolic irregularities associated with obesity[90]. In addition, the structure of the ECM and its angiogenic capacity are altered and seem to be involved as underlying factors in the abnormal expansion capacity of AT (abnormal hypertrophy) in obesity, producing a fibrotic and hypoxic state in AT, which further affects the deregulation of adipocyte function and aggravates the inflammatory state; however, this phenomenon has not been widely studied[74,91].
Adipocytes, which have limited hyperplastic capacity, greater hypertrophy and metaflammation in AT, decrease lipolysis by inhibiting the response to insulin in metabolically active tissues and preventing glucose uptake, contributing to the development of IR and glucose intolerance[92,93]. This is why VAT becomes the main store of triglycerides because the SAT becomes incompetent to continue storing excess energy[94]. Therefore, these alterations in the microenvironment of obese AT and the increase in fat deposition at the visceral level play a central role as significant risk factors because of the increased incidence of metabolic syndrome, IR, T2D, CVD, and certain types of cancer, among others[95,96]. To mitigate these health risks, it is essential to understand the mechanisms that link lipo-inflammation to obesity-associated diseases and thus be able to develop therapies targeting these pathologies[97]. On the other hand, the ease of storing fat in the gluteal‒femoral region in women, compared with the accumulation of fat at the central level in men, explains, to some extent, the lower incidence of cardiovascular events in women than in men[98].
Phenotypic Switch of Macrophages in the “Obese” Adipose Tissue
Studies have shown that a high-fat diet leads to dynamic infiltration of innate and adaptive cells into the AT during the onset of IR and obesity[99]. The immune cells that infiltrate AT are macrophages, lymphocytes, foam cells, mast cells, eosinophils and neutrophils. These immune cells secrete inflammatory factors that alter the adipose microenvironment and may play a key role in metabolic disruption, shifting from an anti-inflammatory to an inflammatory profile[100]. Macrophages play crucial roles in regulating inflammation and metabolic balance in AT; in turn, AT plays an important role in regulating the immune system, particularly in influencing macrophage polarization[101]. The macrophages present in this tissue are referred to as ATMs[49]. ATMs are key modulators of energy metabolism and mitochondrial function in adipocytes; they originate from circulating monocytes that accumulate in ATs via MCP-1 action, differentiate into macrophages in VAT and SAT, and self-renew from ATMs already present in the tissue[49,102].
Impact of Macrophage Polarization in Obesity
The phenotype, quantity and activation state of ATMs have a major impact on the development of obesity-induced metabolic diseases[103]. In lean AT, ATMs acquire the M2 phenotype, promoting an anti-inflammatory response and tissue repair in AT[101]. The main function of M2 macrophages in the tissue is to regulate systemic glucose homeostasis by inhibiting preadipocyte proliferation through the CD206/TGFβ signaling pathway, which induces IL-10 release[104,105]. Furthermore, IL-25 stimulates M2 macrophage polarization and enhances their interaction with adipocytes, promoting energy metabolism, improving mitochondrial functions in adipocytes and regulating lipid accumulation in WAT, VAT and the liver[106]. However, under conditions of energy excess and obesity, the ATM M2 phenotype polarizes to the M1 phenotype, which is the main actor in the initiation and maintenance of the inflammatory state by releasing cytokines that contribute to the infiltration of circulating monocytes attracted by chemoattractants, resulting in greater accumulation of macrophages and the development of chronic low-grade local inflammation; this inflammatory state in AT not only affects its function but also extends to a systemic level[107,108]. It has been shown that inositol-requiring enzyme 1α (IRE1α) is an important protein in the development of obesity and metabolic syndrome because its activation decreases the population of M2 ATMs and increases M1 ATMs through the action of obesogenic factors such as stearic, palmitic and myristic fatty acids and LPS; the IRE1α pathway affects BAT activity and prevents WAT browning[109,110]. Furthermore, the inhibition of factors involved in AT browning and homeostasis, such as fibroblast growth factor 21 (FGF21) and NAD-dependent sirtuin-1 deacetylase (SIRT1), increases miR-34a levels in obese AT[49,111]. miR-34a is released into ATMs by lipid-rich exosomes from hypertrophic adipocytes, which decreases the expression of KLF4, a transcription factor involved in M2 polarization; it also releases miR-155, which regulates M1 macrophage polarization[112,113]. However, preadipocyte-derived SVF exosomes transactivate Arg-1 to drive M2 macrophage polarization by inhibiting miR-34a and miR-155, and these macrophages further promote preadipocyte hyperplasia and AT browning[49,114]. On the other hand, continuous exposure to hyperglycemia activates macrophages through the JNK and ERK pathways, which induce monocyte infiltration in the tissue to differentiate into M1 macrophages[115]. In addition, a high number of macrophages infiltrating AT is positively correlated with the development of obesity in human AT and several mouse models[83,116,117]. This finding indicates that macrophages could be among the most important cells involved in the growth of fat mass, which is why macrophages that infiltrate the AT of obese patients are of greater interest for the study of the interaction between immune cells and metabolism[118].
Hypoxia as the Main Driver of Adipose Tissue Dysfunction
Hypoxia in obesity is a key factor driving metabolic dysfunction and chronic inflammation in AT. As AT expands in response to excessive caloric intake, the increasing distance between adipocytes and the vascular network does not increase proportionally, leading to inadequate oxygen delivery or hypoxia, particularly in hypertrophic adipocytes. The hypoxic environment, tissue hypertrophy, and hyperinsulinemia impede proteasome degradation of HIF-1α, triggering the stabilization and upregulation of this transcription factor in adipocytes and macrophages through the IGF receptor-activated PI3K‒Akt pathway, promoting a cascade of proinflammatory and metabolic changes. In turn, HIF1α, by binding to its coactivator p300/CBP, positively regulates the expression of a wide range of genes involved in glycolysis, angiogenesis and inflammation, including GLUT1 (glucose transporter 1), VEGF and NF-κB. This adaptation aims to support AT survival under low-oxygen conditions, but in obesity, chronic activation of HIF-1α contributes to the deregulation of adipocyte function, promotes IR, inhibits PPARγ and alters adipokine secretion. For example, hypoxia is one of the main causes of an altered adiponectin/leptin balance. Furthermore, early inflammation and hypoxia have been shown to activate NF-κB and induce increased expression of HIF-1α, thereby increasing the production of proinflammatory cytokines, such as TNFα and IL-6, downstream and enhancing macrophage infiltration, driving their polarization toward the proinflammatory M1 phenotype. This exacerbates local and systemic inflammation, worsening IR and metabolic disease. Furthermore, HIF-1α activity impairs adipocyte differentiation and promotes fibrosis in ATs, thereby compromising their ability to store lipids properly, leading to ectopic deposition in the liver and muscle. Hypoxia also increases the expression of ECM components such as collagen, impairing AT flexibility and further exacerbating metabolic dysfunction. Consequently, persistent activation of HIF1α in obese AT is a major contributor to the development of metabolic syndrome, making it a critical area of research for understanding and addressing obesity-related complications.
GLUT1 and GLUT4 are key glucose transporters involved in regulating glucose uptake in various tissues, and their dysregulation plays an important role in the development of glucose intolerance and IR. In obesity, HIF1α upregulates GLUT1 expression in response to hypoxia, enhancing basal glucose uptake in a noninsulin-dependent manner in hypoxic AT. This is part of a metabolic shift known as the “Warburg effect,” where cells rely more on glycolysis (anaerobic metabolism) rather than oxidative phosphorylation to produce energy to maintain their elevated metabolic activity. The upregulation of GLUT1 in the hypoxic environment not only fuels adipocytes but also M1-phenotype macrophages, as these immune cells rely on increased glucose uptake to maintain their proinflammatory activities. However, this shift toward active glucose metabolism often coincides with impaired GLUT4 function due to the resulting IR. As insulin signaling becomes defective by HIF-1α in obese AT and skeletal muscle, the ability of GLUT4 to translocate to the cell membrane in response to insulin is decreased, leading to decreased glucose uptake in these tissues and contributing to hyperglycemia. This creates a vicious cycle: hypoxia promotes HIF1α activation, which supports GLUT1 upregulation and proinflammatory responses, whereas the resulting IR decreases GLUT4 activity, impairing insulin-mediated glucose uptake in AT and skeletal muscle. The contrasting roles of GLUT1 and GLUT4 in obesity highlight the complex metabolic environment in obese individuals. While GLUT1 helps cells survive hypoxia by increasing glucose uptake, it also exacerbates immune cell activation and inflammation; moreover, downregulation or dysfunction of GLUT4 contributes to impaired systemic glucose homeostasis. Therefore, targeting HIF1α to alleviate hypoxia and inflammation, modulating GLUT1 activity, and restoring GLUT4 function in adipose and immune cells could be potential therapeutic strategies to reduce hypoxia-induced inflammation and restore proper insulin sensitivity.
VEGF plays a crucial role in regulating angiogenesis, particularly in response to the increased metabolic demands of AT differentiation and expansion in obesity. As AT expands, the oxygen demand increases due to insufficient vascularization, stimulating VEGF production to promote new blood vessel formation. However, in obese individuals, persistent activation of HIF-1α elevates VEGF levels, leading to excessive and abnormal angiogenesis that fails to adequately oxygenate the tissue. This ongoing hypoxic state exacerbates inflammation, fibrosis, and metabolic dysfunction in obese AT. In addition to HIF-1α, VEGF transcription is controlled by STAT3, PCG-1, and PPARγ. The VEGF-HIF1α pathway contributes to the recruitment of immune cells that further aggravate inflammation. Thus, while VEGF and HIF1α are initially activated to combat hypoxia to maintain healthy AT expansion, their dysregulation in obese patients plays a key role in AT dysfunction, systemic inflammation, and the pathogenesis of metabolic diseases. Targeting VEGF signaling to restore normal vascular function without promoting excessive or abnormal angiogenesis could offer therapeutic strategies to improve tissue oxygenation and reduce inflammation in obese patients.
The important role of platelet-derived growth factor receptor (PDGFR) in regulating AT remodeling and fibrosis in obese patients under hypoxic conditions has recently been revealed. PDGFR exists in two forms, PDGFRα and PDGFRβ, both of which are expressed in adipose progenitor and stromal cells. PDGFRα is crucial for AT expansion, as it regulates the development of new adipocytes from progenitor cells. The PDGFR/HIF-1α relationship is an interesting area of study that highlights the interplay between this growth factor signaling pathway and hypoxia-induced responses in AT. PDGFR and HIF-1α are key players in AT remodeling, inflammation, and fibrosis, which are critical processes in metabolic dysfunction. PDGFR downstream signaling enhanced by HIF-1α drives the proliferation and differentiation of fibroblasts and preadipocytes, contributing to impaired adipogenesis and excessive ECM deposition; these abnormalities lead to reduced flexibility and expandability of AT. As the ability to safely store lipids in adipocytes is impaired, a “spillover effect” is created where excess lipids are deposited in ectopic tissues. Furthermore, increased PDGFR activity under hypoxic conditions has been associated with the recruitment and activation of proinflammatory macrophages, which promote the activation of inflammatory pathways in obese AT. On the other hand, a study revealed that the PDGFRβ+ pathway inhibits PPARγ through the release of HIF-1α-driven factors that limit the adipogenic capacity of adipose cells. Inhibition of HIF-1α in PDGFRβ+ preadipocytes promotes adipogenesis and healthy WAT remodeling and improves glucose tolerance in a PPARγ activation-dependent manner. Thus, the PDGFR/HIF-1α axis plays a pivotal role in pathological WAT remodeling during obesity. Targeting this relationship could provide novel therapeutic approaches to improve metabolic health in obese individuals by mitigating fibrosis, promoting adipogenesis, and reducing WAT inflammation.
The Role of PPARγ in Macrophage Polarization
PPARγ is an important transcription factor in AT that is involved in adipocyte lipid metabolism and anti-inflammatory responses[119,120]. PPARγ-regulated metabolic adaptation leads to subcutaneous and brown fat deposition in adipocytes, reduced circulating lipids, and decreased lipotoxicity[121]. Alterations in PPARγ expression decrease fatty acid uptake and change the metabolic programming of adipocytes and ATMs[122]. The presence of lipid molecules plays a role in macrophage polarization and is influenced by the PPARγ signaling pathway[121,123]. This regulatory pathway promotes the anti-inflammatory M2 metabolic program and inhibits M1 polarization[124]. The metabolic features displayed by PPARγ-regulated ATMs include increased amino acid metabolism by Arg-1, inhibition of glycolysis, increased lipolysis, elevated β-oxidation of fatty acids, and increased mitochondrial biogenesis[124,125]. Studies have shown that elevated IL-4 induces the expression of fatty acid translocase (CD36/FAT), a receptor that transports long-chain fatty acids and oxidizes low-density lipoproteins (ox-LDL); moreover, IL-4 further increases PPARγ expression[126]. Therefore, fatty acid metabolism and PPARγ expression are necessary for anti-inflammatory ATM polarization[122]. The inhibition of PPARγ in ATMs of obese individuals induces an M1 proinflammatory metabolic program via IFNγ, which is characterized by increased aerobic glycolysis and fatty acid synthesis, decreased lipid transport, and amino acid metabolism that is regulated by iNOS and generates a greater amount of ROS[123,127].
In recent years, the role of mammalian target of rapamycin (mTOR) in relation to PPARγ in the metabolic program of adipocytes and macrophages has been studied[128]. Induction of mTOR in AT is associated with adipocyte hypertrophy, but its inhibition results in a decrease in the size and number of adipose cells, resulting in probable protection against obesity[128,129]. However, the inhibition of mTOR, despite decreasing lipid uptake and storage, produces hypertriglyceridemia, which is why mTOR and PPARγ also modulate lipid metabolism in the plasma[130]. In obesity, under conditions of excess energy, mTOR enhances the transcriptional activity of PPARγ and increases the synthesis of proteins involved in lipid metabolism, such as lipoprotein lipase (LPL) and glycerol kinase (GyK)[129]. Therefore, mTOR activation is important for maintaining plasma lipid homeostasis and increasing lipid uptake and storage by activating PPARγ[131]. mTOR acts on PPARγ depending on metabolic and nutritional status to execute metabolic responses (e.g., synthesis of cholesterol, fatty acids and proteins involved in lipid metabolism) and responses associated with inflammation in ATMs[132]. Recently, semaphorin-6D (Sema6D) was shown to link the mTOR and PPARγ pathways in ATM polarization[133]. Sema6D, acting as a receptor (reverse signaling), is activated by mTOR and binds to protein kinase C-Abl; this induces the transcription of PPARγ, allowing the polarization of anti-inflammatory macrophages and the regulation of fatty acid metabolism. c-Abl also activates the differentiation of preadipocytes (hyperplasia)[133,134]. The inhibition of mTOR or Sema6D decreases the expression and activity of PPARγ; as a consequence, the polarization of ATMs toward M2 is affected, their metabolic programming and lipid uptake decrease, the expression of CD36 is also affected, and inflammatory polarization is improved[135].
Adiponectin, a plasma protein secreted by adipocytes, protects the body against CVD and metabolic diseases[136]. There is also a regulatory cycle between PPARγ and adiponectin; the increase in adiponectin available in the blood is associated with greater activation of PPARγ, and this factor further stimulates the synthesis and release of bioavailable adiponectin[137]. A study showed that modified mice that overexpress adiponectin present with hypotriglyceridemia and that lipids are stored in the SAT[138]. On the other hand, this adipokine also exerts a regulatory effect on the polarization of macrophages toward an anti-inflammatory phenotype[139]. Animal studies have revealed that modified mice that do not express adiponectin exhibit increased production of the inflammatory cytokines TNFα, IL-6, and MCP-1 in ATMs, whereas the number of M2 macrophages and their markers, such as Arg-1 and IL-10, decreases[140]. In addition, another study reported that the levels of markers and polarization of M2 macrophages increase and that the level of ROS and polarization toward the M1 phenotype decrease in macrophages treated with recombinant adiponectin[141]. The stimulation of adiponectin expression decreases the progression of obesity and other associated metabolic diseases by promoting the resolution of the low-grade inflammatory state in AT[142].
Studies related to microRNAs have been very important and useful in recent years. One known regulator of inflammation and IR is miR-223[143]. The expression of miR-223 is regulated downstream by PPARγ; consequently, the PPARγ/miR-223 axis regulates the polarization of macrophages toward an anti-inflammatory phenotype to maintain homeostasis in AT and ameliorate insulin sensitivity[144,145]. Investigating the mechanisms by which mTOR-Sema6D and adiponectin modulate PPARγ and by which PPARγ regulates miR-223 expression may be highly important for the development of therapeutic approaches related to PPARγ activity, safe lipid storage, ATM polarization, and metabolic reprogramming of adipocytes and macrophages[146].
Macrophages in the Crown-like Structures (CLS)
ATMs exhibit high proliferative capacity and decreased cell apoptosis in the early stages of WAT hypertrophy following a high-fat diet[147]. However, continuous intake of a high-fat diet leads to the development of glucose intolerance and IR because adipocytes upregulate the endoplasmic reticulum stress pathway C/EBPα/β, resulting in an altered WAT microenvironment[148,149]. Alterations and stress in the microenvironment and elevated lipid storage in adipocytes lead to adipose cell death, and cell debris is removed by ATMs. The large size and volume of apoptotic adipocytes cannot be phagocytosed and engulfed by a single macrophage, and as a result, monocytes infiltrate and subsequently differentiate into macrophages, which aggregate around the adipose cell, forming a crown-shaped structure (CLS), and can eliminate dead adipocytes by lysosomal exocytosis, a hallmark of AT remodeling in obesity. As alterations in adipocyte function and stress progress, lysosomal exocytosis abruptly decreases, and excessive lipid accumulation results in increased tissue damage in AT.
CLS is a primary/key histological feature in the inflammatory and apoptotic process of AT. The dominant macrophages in CLSs are of the M1 phenotype and release proinflammatory factors (NF-κB, iNOS, IL-1β, IL-6, and TNFα) and free fatty acids, which contribute to increased hyperinsulinemia and IR. Furthermore, ATMs of the M1 phenotype activate the transcription factor HIF-1α; as a result, these hypoxic ATMs agglomerate to form CLSs and express elevated iNOS and IL-1β as obesity progresses. These CLSs are the main site of hypoxia and local inflammation in the later stages of obesity. Recently, estrogen receptor beta (ERβ) was shown to be key for the initiation of CLS formation by acting on HIF-1α. A recent study confirmed that CLS formation in SAT and VAT is increased in obese ERβ-/- mice. Obese mice treated with an ERβ agonist show decreased CLS formation in SAT and VAT, as well as decreased HIF-1α activation. Stimulating ERβ in macrophages that compose CLSs and infiltrating macrophages may help reduce the amount of CLSs, decreasing the degree of IR in ATs[150].
Tetraspanin-29 (CD9) macrophages represent a distinct ATM population and have gained attention for their role in metabolic dysfunction. They are cells that, owing to the expression of the surface marker CD9, are able to form CLSs around dead or dying adipocytes, thus playing a role in the removal of apoptotic adipocytes; they also present increased lipid accumulation through exosome formation, are metabolically active and are responsible for amplifying inflammatory signaling in obese ATs. CD9+ ATMs co-express elevated levels of the surface markers CD11c, CD16, and CD206, which express inflammatory transcription factors such as AP-1 and NF-κB, and these induce the release of the proinflammatory mediators IL-6, IL-18, and TNFα, which promote IR and exacerbate AT dysfunction. Research has shown that CD9+ ATMs express genes involved in lipid handling and fibrosis, further implicating them in AT remodeling and metabolic complications.
On the other hand, other subpopulations of CD11c+/CD163+ ATMs are part of the CLS in VAT and SAT and have special relevance because of their dual or transitional role in regulating the local inflammatory and metabolic environment and in processes of remodeling or resolution of inflammation, which adds complexity to the immune response in obesity. CD11c+/CD163+ macrophages actively participate in the response to adipocyte death and in the regulation of inflammation in expanded AT. CD11c expression is related to the proinflammatory M1 profile, while CD163 expression could be involved in the modulation of inflammation and protection against excessive tissue damage due to its anti-inflammatory profile. Targeting CD9+ macrophages as well as hybrid M1/M2 macrophage subtypes could offer a novel therapeutic approach to reduce inflammation and improve metabolic outcomes in obese individuals.
Recently, a subpopulation of macrophages known as lipid-associated macrophages (LAMs) was discovered within the CLS; these macrophages have the main functions of lipid catabolism and modulate the inflammatory response of the AT through the loss of homeostasis and death of hypertrophic adipocytes[151]. The LAM that forms the CLS are the only immune cells in obese AT that express lipid receptor trigger receptor expressed on myeloid cells 2 (Trem2)[152]. Studies have shown that LAM-Trem2- increases hypertrophy in adipocytes and leads to hypercholesterolemia, inflammation, glucose intolerance and IR; therefore, metabolism in obese individuals is further impaired[153,154]. However, LAM-Trem2+ CLSs formed in obese adipocytes can locally contain lipid droplets, decrease AT inflammation and hypertrophy, and prevent the loss of lipid homeostasis at the systemic level, since the transcriptional profile triggered by Trem2 induces the expression of mediators involved in lipid handling, such as CD36 and PLIN2[153,155]. These findings suggest that the downstream pathway of Trem2 is activated by recognition signals, indicating the loss of homeostasis in adipocytes and lipid accumulation in the ECM[156]. Although CLS formation has been reported to be positively correlated with increased severity of obesity and metabolic syndrome, these studies identify Trem2+ CLSs as a new potential therapeutic target in obesity.
Obesity-Associated Metabolic Risk and Macrophage Phenotypic Switch
Obesity-related inflammation is a critical factor in the development of several chronic diseases. One of the primary diseases associated with this condition is T2D. Inflammation impairs insulin signaling pathways, leading to IR, a hallmark of T2DM[157]. Cardiovascular diseases (CVDs) are also closely linked to obesity-induced inflammation. Proinflammatory cytokines contribute to the formation of atherosclerotic plaques and promote endothelial dysfunction, increasing the risk of heart attack and stroke[158]. Nonalcoholic fatty liver disease (NAFLD) is another condition exacerbated by inflammation in obese individuals. The accumulation of fat in liver cells triggers inflammation, which can progress to nonalcoholic steatohepatitis (NASH), fibrosis, and even cirrhosis[159]. Additionally, certain cancers, such as breast, colorectal, and pancreatic cancer, are associated with chronic inflammation stemming from obesity. The inflammatory environment promotes tumor initiation, progression, and metastasis. Understanding the intricate relationships among obesity, inflammation, and these diseases is essential for developing effective prevention and treatment strategies[97].
Interestingly, contrary to studies showing that inflammatory signals mainly exert a negative impact on metabolism, there is new evidence that proinflammatory signaling in the tissue is necessary for proper remodeling and expansion of AT in healthy individuals and could be important in the treatment of obesity[107]. Authors have shown that, in mice, a specific reduction in AT in a proinflammatory state reduces the adipogenic capacity in vivo; this finding is associated with a decrease in the ectopic accumulation of lipids, improved glucose tolerance and decreased systemic inflammation[160]. However, other studies suggest that triggering an inflammatory state of AT can generate a response in SAT that allows safe storage and expansion against excess nutrients, thus decreasing lipid accumulation in VAT, and AT could be protected against metabolic and inflammatory disruptions[161].
Concluding Remarks
The phenotypic switch of macrophages in the obese adipose microenvironment has become a critical therapeutic target for addressing obesity-related inflammation and metabolic dysfunction. Recent advances in understanding the underlying mechanisms of this switch, including the role of hypoxia, lipid signaling, and the interplay of cytokines and adipokines, have paved the way for innovative therapeutic strategies. Emerging approaches include the use of small molecules and biologics aimed at modulating key signaling pathways, such as inhibitors of NF-κB and HIF-1α or activators of PPARγ and STAT6, to promote the anti-inflammatory M2 phenotype. Additionally, nanoparticle-based drug delivery systems are being developed to selectively target macrophages within obese adipose tissue, offering precision in reprogramming their polarization without affecting systemic immunity. Immunotherapy approaches, such as adoptive transfer of engineered macrophages or the use of monoclonal antibodies against proinflammatory cytokines such as TNF-α and IL-6, also show promise. Another exciting avenue is the application of metabolic reprogramming agents, which aim to shift macrophage metabolism from glycolysis (associated with M1 polarization) to oxidative phosphorylation (favoring M2 polarization). Furthermore, therapies targeting the gut‒adipose axis, such as prebiotics and probiotics, are being explored to indirectly influence macrophage function by modulating systemic inflammation and lipid metabolism. While these novel therapies show significant potential, challenges remain in optimizing their safety, efficacy, and delivery, emphasizing the need for continued research and clinical trials to bring these approaches closer to widespread application in managing obesity and its associated diseases.
Acknowledgments
This study was supported by PICT-2021-I-A-0147 from the FONCYT, Agencia Nacional para la Promoción de la Ciencia y la Tecnología, Ministerio de Capital Humano, Gobierno de la República Argentina.
References
- Hirayama D, Iida T, Nakase H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. International Journal of Molecular Sciences. 2018;19(1). [CrossRef]
- Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduction and Targeted Therapy. 2023;8(1). [CrossRef]
- Watanabe S, Alexander M, Misharin A V., Budinger GRS. The role of macrophages in the resolution of inflammation. The Journal of Clinical Investigation. 2019;129(7): 2619–2628. [CrossRef]
- Yousaf H, Khan MIU, Ali I, Munir MU, Lee KY. Emerging role of macrophages in noninfectious diseases: An update. Biomedicine & Pharmacotherapy. 2023;161: 114426. [CrossRef]
- Kadomoto S, Izumi K, Mizokami A. Macrophage Polarity and Disease Control. International Journal of Molecular Sciences. 2022;23(1). [CrossRef]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature reviews. Immunology. 2008;8(12): 958. [CrossRef]
- Ochoa-Carrillo FJ, Bravo-Cuellar A. Los macrófagos, ángeles o demonios. Gaceta Mexicana de Oncología. 2013;12(1): 1–3. https://www.elsevier.es/es-revista-gaceta-mexicana-oncologia-305-articulo-los-macrofagos-angeles-o-demonios-X1665920113933087.
- Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. European journal of pharmacology. 2020;877. [CrossRef]
- Savulescu-Fiedler I, Mihalcea R, Dragosloveanu S, Scheau C, Octavian Baz R, Caruntu A, et al. The Interplay between Obesity and Inflammation. Life 2024, Vol. 14, Page 856. 2024;14(7): 856. [CrossRef]
- Kawai T, Autieri M V., Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. American journal of physiology. Cell physiology. 2021;320(3): C375–C391. [CrossRef]
- Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640): 177–185. [CrossRef]
- Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. The Journal of clinical investigation. 2017;127(1): 1–4. [CrossRef]
- Chait A, den Hartigh LJ. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Frontiers in Cardiovascular Medicine. 2020;7. [CrossRef]
- Ruck L, Wiegand S, Kühnen P. Relevance and consequence of chronic inflammation for obesity development. Molecular and Cellular Pediatrics 2023 10:1. 2023;10(1): 1–18. [CrossRef]
- Chaintreuil P, Kerreneur E, Bourgoin M, Savy C, Favreau C, Robert G, et al. The generation, activation, and polarization of monocyte-derived macrophages in human malignancies. Frontiers in Immunology. 2023;14. [CrossRef]
- Cutolo M, Campitiello R, Gotelli E, Soldano S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Frontiers in Immunology. 2022;13. [CrossRef]
- Hofer TP, van de Loosdrecht AA, Stahl-Hennig C, Cassatella MA, Ziegler-Heitbrock L. 6-Sulfo LacNAc (Slan) as a Marker for Nonclassical Monocytes. Frontiers in Immunology. 2019;10. [CrossRef]
- Hirayama D, Iida T, Nakase H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. International Journal of Molecular Sciences. 2018;19(1). [CrossRef]
- Wu J, Zhang L, Shi J, He R, Yang W, Habtezion A, et al. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. EBioMedicine. 2020;58: 102920. [CrossRef]
- Strizova Z, Benesova I, Bartolini R, Novysedlak R, Cecrdlova E, Foley LK, et al. M1/M2 macrophages and their overlaps – myth or reality? Clinical Science (London, England : 1979). 2023;137(15): 1067. [CrossRef]
- Yao Y, Xu XH, Jin L. Macrophage polarization in physiological and pathological pregnancy. Frontiers in Immunology. 2019;10(MAR): 434399. [CrossRef]
- Duque GA, Descoteaux A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Frontiers in Immunology. 2014;5(OCT). [CrossRef]
- Peng Y, Zhou M, Yang H, Qu R, Qiu Y, Hao J, et al. Regulatory Mechanism of M1/M2 Macrophage Polarization in the Development of Autoimmune Diseases. Mediators of Inflammation. 2023;2023. [CrossRef]
- Mily A, Kalsum S, Loreti MG, Rekha RS, Muvva JR, Lourda M, et al. Polarization of M1 and M2 Human Monocyte-Derived Cells and Analysis with Flow Cytometry upon Mycobacterium tuberculosis Infection. Journal of visualized experiments : JoVE. 2020;2020(163): 1–20. [CrossRef]
- Patel U, Rajasingh S, Samanta S, Cao T, Dawn B, Rajasingh J. Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug discovery today. 2017;22(1): 186. [CrossRef]
- Xue C, Tian J, Cui Z, Liu Y, Sun D, Xiong M, et al. Reactive oxygen species (ROS)-mediated M1 macrophage-dependent nanomedicine remodels inflammatory microenvironment for osteoarthritis recession. Bioactive materials. 2023;33: 545–561. [CrossRef]
- Zhou H cun, Yan X yan Y, Yu W wen, Liang X qin, Du X yan, Liu Z chang, et al. Lactic acid in macrophage polarization: The significant role in inflammation and cancer. International reviews of immunology. 2022;41(1): 4–18. [CrossRef]
- Irey EA, Lassiter CM, Brady NJ, Chuntova P, Wang Y, Knutson TP, et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(25): 12442–12451. [CrossRef]
- Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. International Journal of Molecular Sciences. 2021;22(13). [CrossRef]
- Li X, Mara AB, Musial SC, Kolling FW, Gibbings SL, Gerebtsov N, et al. Coordinated chemokine expression defines macrophage subsets across tissues. Nature immunology. 2024;25(6): 1110–1122. [CrossRef]
- Chen S, Wang M, Lu T, Liu Y, Hong W, He X, et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene. 2023;42(37): 2737–2750. [CrossRef]
- Chandrasekaran P, Izadjoo S, Stimely J, Palaniyandi S, Zhu X, Tafuri W, et al. Regulatory Macrophages Inhibit Alternative Macrophage Activation and Attenuate Pathology Associated with Fibrosis. Journal of immunology (Baltimore, Md. : 1950). 2019;203(8): 2130–2140. [CrossRef]
- Krzyszczyk P, Schloss R, Palmer A, Berthiaume F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Frontiers in Physiology. 2018;9(MAY): 419. [CrossRef]
- Willenborg S, Injarabian L, Eming SA. Role of Macrophages in Wound Healing. Cold Spring Harbor perspectives in biology. 2022;14(12). [CrossRef]
- Gao J, Liang Y, Wang L. Shaping Polarization Of Tumor-Associated Macrophages In Cancer Immunotherapy. Frontiers in Immunology. 2022;13. [CrossRef]
- Liu L, Stokes J V., Tan W, Pruett SB. An optimized flow cytometry panel for classifying macrophage polarization. Journal of immunological methods. 2022;511. [CrossRef]
- Diaz-Jimenez D, Kolb JP, Cidlowski JA. Glucocorticoids as Regulators of Macrophage-Mediated Tissue Homeostasis. Frontiers in Immunology. 2021;12. [CrossRef]
- Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Frontiers in Immunology. 2019;10(JULY): 466337. [CrossRef]
- OECD. The Heavy Burden of Obesity: The economics of prevention. OECD Health Policy Studies. Paris: OECD Publishing; 2019. [CrossRef]
- Ruze R, Liu T, Zou X, Song J, Chen Y, Xu R, et al. Obesity and type 2 diabetes mellitus: connections in epidemiology, pathogenesis, and treatments. Frontiers in Endocrinology. 2023;14. [CrossRef]
- Jin X, Qiu T, Li L, Yu R, Chen X, Li C, et al. Pathophysiology of obesity and its associated diseases. Acta Pharmaceutica Sinica. B. 2023;13(6): 2403. [CrossRef]
- Grundy SM. Multifactorial causation of obesity: implications for prevention. The American journal of clinical nutrition. 1998;67(3 Suppl). [CrossRef]
- Maurizi G, Della Guardia L, Maurizi A, Poloni A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. Journal of cellular physiology. 2018;233(1): 88–97. [CrossRef]
- Esteve Ràfols M. Adipose tissue: Cell heterogeneity and functional diversity. Endocrinología y Nutrición (English Edition). 2014;61(2): 100–112. [CrossRef]
- Richard AJ, White U, Elks CM, Stephens JM. Adipose Tissue: Physiology to Metabolic Dysfunction. Endotext. 2020; https://www.ncbi.nlm.nih.gov/books/NBK555602/.
- Ahmed S, Shah P, Ahmed O. Biochemistry, Lipids. StatPearls. 2023; https://www.ncbi.nlm.nih.gov/books/NBK525952/.
- Maniyadath B, Zhang Q, Gupta RK, Mandrup S. Adipose tissue at single-cell resolution. Cell Metabolism. 2023;35(3): 386–413. [CrossRef]
- Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Archives of Medical Science : AMS. 2013;9(2): 191. [CrossRef]
- Li Y, Yun K, Mu R. A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes. Lipids in Health and Disease. 2020;19(1): 1–9. [CrossRef]
- O’Rourke RW. Adipose tissue and the physiologic underpinnings of metabolic disease. Surgery for obesity and related diseases : official journal of the American Society for Bariatric Surgery. 2018;14(11): 1755–1763. [CrossRef]
- Horwitz A, Birk R. Adipose Tissue Hyperplasia and Hypertrophy in Common and Syndromic Obesity—The Case of BBS Obesity. Nutrients. 2023;15(15). [CrossRef]
- Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. International Journal of Molecular Sciences. 2019;20(9). [CrossRef]
- Röszer T. Adipose Tissue Immunometabolism and Apoptotic Cell Clearance. Cells 2021, Vol. 10, Page 2288. 2021;10(9): 2288. [CrossRef]
- Clemente-Suárez VJ, Redondo-Flórez L, Beltrán-Velasco AI, Martín-Rodríguez A, Martínez-Guardado I, Navarro-Jiménez E, et al. The Role of Adipokines in Health and Disease. Biomedicines. 2023;11(5). [CrossRef]
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444(7121): 847. [CrossRef]
- Goossens GH. The Metabolic Phenotype in Obesity: Fat Mass, Body Fat Distribution, and Adipose Tissue Function. Obesity Facts. 2017;10(3): 207. [CrossRef]
- Carmona WS, Carrillo Álvarez E, Jesús A, Oliver S. Obesity as a Complex Chronic Disease. Current Research in Diabetes & Obesity Journal. 2017;7(1): 1–4. [CrossRef]
- Ziolkowska S, Binienda A, Jablkowski M, Szemraj J, Czarny P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2021;22(20): 11128. [CrossRef]
- Maliszewska K, Kretowski A. Brown Adipose Tissue and Its Role in Insulin and Glucose Homeostasis. International Journal of Molecular Sciences. 2021;22(4): 1–19. [CrossRef]
- Meex RCR, Blaak EE, van Loon LJC. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obesity Reviews. 2019;20(9): 1205. [CrossRef]
- Zhu Q, An YA, Scherer PE. Mitochondrial regulation and white adipose tissue homeostasis. Trends in Cell Biology. 2022;32(4): 351–364. [CrossRef]
- Man K, Kallies A, Vasanthakumar A. Resident and migratory adipose immune cells control systemic metabolism and thermogenesis. Cellular & Molecular Immunology 2021 19:3. 2021;19(3): 421–431. [CrossRef]
- Sakers A, De Siqueira MK, Seale P, Villanueva CJ. Adipose-tissue plasticity in health and disease. Cell. 2022;185(3): 419–446. [CrossRef]
- Nakamura K, Fuster JJ, Walsh K. Adipokines: A link between obesity and cardiovascular disease. Journal of Cardiology. 2014;63(4): 250–259. [CrossRef]
- Luo L, Liu M. Adiponectin: Friend or foe in obesity and inflammation. Medical Review. 2022;2(4): 349–362. [CrossRef]
- Corvera S, Solivan-Rivera J, Yang Loureiro Z. Angiogenesis in adipose tissue and obesity. Angiogenesis. 2022;25(4): 439. [CrossRef]
- Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future lipidology. 2008;3(5): 545. [CrossRef]
- Ross EA, Devitt A, Johnson JR. Macrophages: The Good, the Bad, and the Gluttony. Frontiers in Immunology. 2021;12: 708186. [CrossRef]
- Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117(4): 1162–1172. [CrossRef]
- Li S, Xiong Y, Zhu H, Ma T, Sun X, Xiao J. Microenvironment-responsive nanosystems for osteoarthritis therapy. Engineered Regeneration. 2024;5(1): 92–110. [CrossRef]
- Madsen MS, Siersbæk R, Boergesen M, Nielsen R, Mandrup S. Peroxisome Proliferator-Activated Receptor γ and C/EBPα Synergistically Activate Key Metabolic Adipocyte Genes by Assisted Loading. Molecular and Cellular Biology. 2014;34(6): 939. [CrossRef]
- Chatterjee R, Bhattacharya P, Gavrilova O, Glass K, Moitra J, Myakishev M, et al. Suppression of the C/EBP family of transcription factors in adipose tissue causes lipodystrophy. Journal of molecular endocrinology. 2011;46(3): 175–192. [CrossRef]
- Neto IV de S, Durigan JLQ, da Silva ASR, Marqueti R de C. Adipose Tissue Extracellular Matrix Remodeling in Response to Dietary Patterns and Exercise: Molecular Landscape, Mechanistic Insights, and Therapeutic Approaches. Biology. 2022;11(5): 765. [CrossRef]
- Ruiz-Ojeda FJ, Méndez-Gutiérrez A, Aguilera CM, Plaza-Díaz J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. International Journal of Molecular Sciences. 2019;20(19). [CrossRef]
- Sun K, Li X, Scherer PE. Extracellular Matrix (ECM) and Fibrosis in Adipose Tissue: Overview and Perspectives. Comprehensive Physiology. 2023;13(1): 4387. [CrossRef]
- Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nature reviews. Endocrinology. 2019;15(3): 139. [CrossRef]
- Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Frontiers in Physiology. 2019;10: 1607. [CrossRef]
- Swarup S, Ahmed I, Grigorova Y, Zeltser R. Metabolic Syndrome. StatPearls. 2024; https://www.ncbi.nlm.nih.gov/books/NBK459248/.
- Hildebrandt X, Ibrahim M, Peltzer N. Cell death and inflammation during obesity: “Know my methods, WAT(son)”. Cell Death & Differentiation 2022 30:2. 2022;30(2): 279–292. [CrossRef]
- Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. The Journal of clinical investigation. 2003;112(12): 1785–1788. [CrossRef]
- Fain JN. Release of Inflammatory Mediators by Human Adipose Tissue Is Enhanced in Obesity and Primarily by the Nonfat Cells: A Review. Mediators of Inflammation. 2010;2010(1): 513948. [CrossRef]
- Scarano F, Gliozzi M, Zito MC, Guarnieri L, Carresi C, Macrì R, et al. Potential of Nutraceutical Supplementation in the Modulation of White and Brown Fat Tissues in Obesity-Associated Disorders: Role of Inflammatory Signaling. International journal of molecular sciences. 2021;22(7). [CrossRef]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112(12): 1796–1808. [CrossRef]
- Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6): 7204. [CrossRef]
- Kirichenko T V., Markina Y V., Bogatyreva AI, Tolstik T V., Varaeva YR, Starodubova A V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. International Journal of Molecular Sciences. 2022;23(23). [CrossRef]
- Frühbeck G, Catalán V, Rodríguez A, Ramírez B, Becerril S, Salvador J, et al. Adiponectin-leptin Ratio is a Functional Biomarker of Adipose Tissue Inflammation. Nutrients. 2019;11(2). [CrossRef]
- Guzik TJ, Skiba DS, Touyz RM, Harrison DG. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovascular Research. 2017;113(9): 1009. [CrossRef]
- Luz M, Albarracín G, Yibby A, Torres F, Albarracin G. Adiponectin and Leptin Adipocytokines in Metabolic Syndrome: What Is Its Importance? Dubai Diabetes and Endocrinology Journal. 2020;26(3): 93–102. [CrossRef]
- González-Jurado JA, Suárez-Carmona W, López S, Sánchez-Oliver AJ. Changes in Lipoinflammation Markers in People with Obesity after a Concurrent Training Program: A Comparison between Men and Women. International Journal of Environmental Research and Public Health. 2020;17(17): 1–12. [CrossRef]
- Appari M, Channon KM, Mcneill E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. [CrossRef]
- Crewe C, An YA, Scherer PE. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. The Journal of Clinical Investigation. 2017;127(1): 74. [CrossRef]
- Jocken JWE, Goossens GH, Boon H, Mason RR, Essers Y, Havekes B, et al. Insulin-mediated suppression of lipolysis in adipose tissue and skeletal muscle of obese type 2 diabetic men and men with normal glucose tolerance. Diabetologia. 2013;56(10): 2255. [CrossRef]
- Zoidis E, Papamikos V. Glucose: Glucose Intolerance. Encyclopedia of Food and Health. 2016; 227–232. [CrossRef]
- Kojta I, Chacińska M, Błachnio-Zabielska A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients. 2020;12(5). [CrossRef]
- Ramos-Nino ME. The Role of Chronic Inflammation in Obesity-Associated Cancers. ISRN Oncology. 2013;2013: 1–25. [CrossRef]
- Zhao X, An X, Yang C, Sun W, Ji H, Lian F. The crucial role and mechanism of insulin resistance in metabolic disease. Frontiers in Endocrinology. 2023;14: 1149239. [CrossRef]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006 444:7121. 2006;444(7121): 860–867. [CrossRef]
- Gavin KM, Bessesen DH. Sex Differences in Adipose Tissue Function. Endocrinology and metabolism clinics of North America. 2020;49(2): 215. [CrossRef]
- Shu CJ, Benoist C, Mathis D. The immune system’s involvement in obesity-driven type 2 diabetes. Seminars in Immunology. 2012;24(6): 436–442. [CrossRef]
- Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Frontiers in Endocrinology. 2016;7(APR): 178543. [CrossRef]
- Guha Ray A, Odum OP, Wiseman D, Weinstock A. The diverse roles of macrophages in metabolic inflammation and its resolution. Frontiers in Cell and Developmental Biology. 2023;11. [CrossRef]
- Zheng C, Yang Q, Cao J, Xie N, Liu K, Shou P, et al. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death & Disease 2016 7:3. 2016;7(3): e2167–e2167. [CrossRef]
- Ruggiero AD, Key CCC, Kavanagh K. Adipose Tissue Macrophage Polarization in Healthy and Unhealthy Obesity. Frontiers in Nutrition. 2021;8: 625331. [CrossRef]
- Nawaz A, Aminuddin A, Kado T, Takikawa A, Yamamoto S, Tsuneyama K, et al. CD206+ M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nature Communications. 2017;8(1). [CrossRef]
- Orliaguet L, Dalmas E, Drareni K, Venteclef N, Alzaid F. Mechanisms of Macrophage Polarization in Insulin Signaling and Sensitivity. Frontiers in Endocrinology. 2020;11: 516860. [CrossRef]
- Sun JX, Xu XH, Jin L. Effects of Metabolism on Macrophage Polarization Under Different Disease Backgrounds. Frontiers in Immunology. 2022;13. [CrossRef]
- Michailidou Z, Gomez-Salazar M, Alexaki VI. Innate Immune Cells in the Adipose Tissue in Health and Metabolic Disease. Journal of Innate Immunity. 2022;14(1): 4. [CrossRef]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117(1): 175–184. [CrossRef]
- Huang S, Xing Y, Liu Y. Emerging roles for the ER stress sensor IRE1α in metabolic regulation and disease. The Journal of Biological Chemistry. 2019;294(49): 18726. [CrossRef]
- Meng H, Matthan NR, Wu D, Li L, Rodríguez-Morató J, Cohen R, et al. Comparison of diets enriched in stearic, oleic, and palmitic acids on inflammation, immune response, cardiometabolic risk factors, and fecal bile acid concentrations in mildly hypercholesterolemic postmenopausal women-randomized crossover trial. The American journal of clinical nutrition. 2019;110(2): 305–315. [CrossRef]
- Fu T, Kemper JK. MicroRNA-34a and Impaired FGF19/21 Signaling in Obesity. Vitamins and Hormones. 2016;101: 175–196. [CrossRef]
- Pan Y, Hui X, Chong Hoo RL, Ye D, Cheung Chan CY, Feng T, et al. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. The Journal of Clinical Investigation. 2019;129(2): 834–849. [CrossRef]
- Tryggestad JB, Teague AM, Sparling DP, Jiang S, Chernausek SD. Macrophage-Derived microRNA-155 Increases in Obesity and Influences Adipocyte Metabolism by Targeting Peroxisome Proliferator-Activated Receptor Gamma. Obesity. 2019;27(11): 1856–1864. [CrossRef]
- Zhao H, Shang Q, Pan Z, Bai Y, Li Z, Zhang H, et al. Exosomes From Adipose-Derived Stem Cells Attenuate Adipose Inflammation and Obesity Through Polarizing M2 Macrophages and Beiging in White Adipose Tissue. Diabetes. 2018;67(2): 235–247. [CrossRef]
- Cheng CI, Chen PH, Lin YC, Kao YH. High glucose activates Raw264.7 macrophages through RhoA kinase-mediated signaling pathway. Cellular signaling. 2015;27(2): 283–292. [CrossRef]
- Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of Lipid Research. 2005;46(11): 2347–2355. [CrossRef]
- Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia 2016 59:5. 2016;59(5): 879–894. [CrossRef]
- Chylikova J, Dvorackova J, Tauber Z, Kamarad V. M1/M2 macrophage polarization in human obese adipose tissue. http://biomed.papers.upol.cz/doi/10.5507/bp.2018.015.html. 2018;162(2): 79–82. [CrossRef]
- Luquet S, Gaudel C, Holst D, Lopez-Soriano J, Jehl-Pietri C, Fredenrich A, et al. Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2005;1740(2): 313–317. [CrossRef]
- Göttlicher M, Widmark E, Li Q, Gustafsson JÅ. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10): 4653–4657. [CrossRef]
- Corrales P, Vidal-Puig A, Medina-Gómez G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. International Journal of Molecular Sciences. 2018;19(7). [CrossRef]
- Sun C, Mao S, Chen S, Zhang W, Liu C. PPARs-Orchestrated Metabolic Homeostasis in the Adipose Tissue. International Journal of Molecular Sciences. 2021;22(16). [CrossRef]
- Yu L, Gao Y, Aaron N, Qiang L. A glimpse of the connection between PPARγ and macrophage. Frontiers in Pharmacology. 2023;14. [CrossRef]
- Daniel B, Nagy G, Czimmerer Z, Horvath A, Hammers DW, Cuaranta-Monroy I, et al. The nuclear receptor PPARg controls progressive macrophage polarization as a ligand-insensitive epigenomic ratchet of transcriptional memory. Immunity. 2018;49(4): 615. [CrossRef]
- Kleiboeker B, Lodhi IJ. Peroxisomal regulation of energy homeostasis: Effect on obesity and related metabolic disorders. Molecular Metabolism. 2022;65: 101577. [CrossRef]
- Chen Y, Zhang J, Cui W, Silverstein RL. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. The Journal of Experimental Medicine. 2022;219(6). [CrossRef]
- Wang S, Liu G, Li Y, Pan Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Frontiers in Immunology. 2022;13. [CrossRef]
- Lee PL, Jung SM, Guertin DA. The Complex Roles of mTOR in Adipocytes and Beyond. Trends in endocrinology and metabolism: TEM. 2017;28(5): 319. [CrossRef]
- Blanchard PG, Festuccia WT, Houde VP, St-Pierre P, BrÛlé S, Turcotte V, et al. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. Journal of Lipid Research. 2012;53(6): 1117. [CrossRef]
- Cai H, Dong LQ, Liu F. Recent Advances in Adipose mTOR Signaling and Function: Therapeutic Prospects. Trends in pharmacological sciences. 2016;37(4): 303. [CrossRef]
- Mao Z, Zhang W. Role of mTOR in Glucose and Lipid Metabolism. International Journal of Molecular Sciences 2018, Vol. 19, Page 2043. 2018;19(7): 2043. [CrossRef]
- Hao K, Wang J, Yu H, Chen L, Zeng W, Wang Z, et al. Peroxisome Proliferator-Activated Receptor γ Regulates Lipid Metabolism in Sheep Trophoblast Cells through mTOR Pathway-Mediated Autophagy. PPAR Research. 2023;2023(1): 6422804. [CrossRef]
- Kang S, Nakanishi Y, Kioi Y, Okuzaki D, Kimura T, Takamatsu H, et al. Semaphorin 6D reverse signaling controls macrophage lipid metabolism and anti-inflammatory polarization. Nature immunology. 2018;19(6): 561–570. [CrossRef]
- Wagner W, Ochman B, Wagner W. Semaphorin 6 Family—An Important However, Overlooked Group of Signaling Proteins Involved in Cancerogenesis. Cancers 2023, Vol. 15, Page 5536. 2023;15(23): 5536. [CrossRef]
- Luan D, Dadpey B, Zaid J, Bridge-Comer PE, Deluca JH, Xia W, et al. Adipocyte-Secreted IL-6 Sensitizes Macrophages to IL-4 Signaling. Diabetes. 2023;72(3): 367–374. [CrossRef]
- Lei X, Qiu S, Yang G, Wu Q. Adiponectin and metabolic cardiovascular diseases: Therapeutic opportunities and challenges. Genes & Diseases. 2023;10(4): 1525. [CrossRef]
- Da Silva Rosa SC, Liu M, Sweeney G. Adiponectin Synthesis, Secretion and Extravasation from Circulation to Interstitial Space. Physiology. 2021;36(3): 134. [CrossRef]
- Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Molecular Metabolism. 2017;6(3): 267–275. [CrossRef]
- Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin Promotes Macrophage Polarization toward an Anti-inflammatory Phenotype. The Journal of Biological Chemistry. 2010;285(9): 6153. [CrossRef]
- Salvator H, Grassin-Delyle S, Brollo M, Couderc LJ, Abrial C, Victoni T, et al. Adiponectin Inhibits the Production of TNF-α, IL-6 and Chemokines by Human Lung Macrophages. Frontiers in Pharmacology. 2021;12: 718929. [CrossRef]
- Zhang Y, Aldridge J, Vasileiadis GK, Edebo H, Ekwall AKH, Lundell AC, et al. Recombinant Adiponectin Induces the Production of Pro-Inflammatory Chemokines and Cytokines in Circulating Mononuclear Cells and Fibroblast-Like Synoviocytes From Non-Inflamed Subjects. Frontiers in Immunology. 2021;11. [CrossRef]
- Han Y, Sun Q, Chen W, Gao Y, Ye J, Chen Y, et al. New advances of adiponectin in regulating obesity and related metabolic syndromes. Journal of Pharmaceutical Analysis. 2024;14(5): 100913. [CrossRef]
- Wen D, Qiao P, Wang L. Circulating microRNA-223 as a potential biomarker for obesity. Obesity Research & Clinical Practice. 2015;9(4): 398–404. [CrossRef]
- Jiao P, Wang XP, Luoreng ZM, Yang J, Jia L, Ma Y, et al. miR-223: An Effective Regulator of Immune Cell Differentiation and Inflammation. International Journal of Biological Sciences. 2021;17(9): 2308. [CrossRef]
- Gu J, Xu H, Chen Y, Li N, Hou X. MiR-223 as a Regulator and Therapeutic Target in Liver Diseases. Frontiers in Immunology. 2022;13: 860661. [CrossRef]
- Ying W, Tseng A, Chang RCA, Morin A, Brehm T, Triff K, et al. MicroRNA-223 is a crucial mediator of PPARγ-regulated alternative macrophage activation. The Journal of Clinical Investigation. 2015;125(11): 4149. [CrossRef]
- Muir LA, Kiridena S, Griffin C, DelProposto JB, Geletka L, Martinez-Santibañez G, et al. Rapid adipose tissue expansion triggers unique proliferation and lipid accumulation profiles in adipose tissue macrophages. Journal of leukocyte biology. 2018;103(4): 615. [CrossRef]
- Suzuki T, Gao J, Ishigaki Y, Kondo K, Sawada S, Izumi T, et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell reports. 2017;18(8): 2045–2057. [CrossRef]
- Díaz-Bulnes P, Saiz ML, López-Larrea C, Rodríguez RM. Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Frontiers in Immunology. 2019;10: 2951. [CrossRef]
- Wang L, Zhao R peng, Song X yu, Wu W fu. Targeting ERβ in Macrophage Reduces Crown-like Structures in Adipose Tissue by Inhibiting Osteopontin and HIF-1α. Scientific Reports. 2019;9(1). [CrossRef]
- Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(22): E5096–E5105. [CrossRef]
- Li RY, Qin Q, Yang HC, Wang YY, Mi YX, Yin YS, et al. TREM2 in the pathogenesis of AD: a lipid metabolism regulator and potential metabolic therapeutic target. Molecular Neurodegeneration. 2022;17(1). [CrossRef]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell. 2019;178(3): 686-698.e14. [CrossRef]
- Sharif O, Brunner JS, Korosec A, Martins R, Jais A, Snijder B, et al. Beneficial Metabolic Effects of TREM2 in Obesity Are Uncoupled From Its Expression on Macrophages. Diabetes. 2021;70(9): 2042. [CrossRef]
- Xu R, Vujić N, Bianco V, Reinisch I, Kratky D, Krstic J, et al. Lipid-associated macrophages between aggravation and alleviation of metabolic diseases. Trends in Endocrinology and Metabolism. 2024;0(0). [CrossRef]
- Marinelli S, Basilico B, Marrone MC, Ragozzino D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Seminars in Cell & Developmental Biology. 2019;94: 138–151. [CrossRef]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of clinical investigation. 2006;116(7): 1793–1801. [CrossRef]
- Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444(7121): 875–880. [CrossRef]
- Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology (Baltimore, Md.). 2010;52(5): 1836–1846. [CrossRef]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang Z V., et al. Adipocyte Inflammation Is Essential for Healthy Adipose Tissue Expansion and Remodeling. Cell Metabolism. 2014;20(1): 103–118. [CrossRef]
- Wu H, Ballantyne CM. Metabolic Inflammation and Insulin Resistance in Obesity. Circulation Research. 2020;126(11): 1549–1564. [CrossRef]
- Semenza GL. The Hypoxic Tumor Microenvironment: A Driving Force for Breast Cancer Progression. Biochimica et biophysica acta. 2016;1863(3): 382. [CrossRef]
Figure 1.
Monocyte differentiation and polarization. Differentiation of circulating monocytes into three subsets: classical (CD14++, CD16-), intermediate (CD14+, CD16+), and nonclassical (CD14+, CD16++) monocytes. Classical and intermediate monocytes can differentiate into M1 proinflammatory macrophages, whereas nonclassical monocytes can differentiate into M2 anti-inflammatory macrophages. HLA-DR, major histocompatibility complex class II antigen DR; FCGR1, Fc gamma receptor 1; TNFα, tumor necrosis factor alpha; IL-1β, interleukin-1 beta; IL-6, interleukin-6; CD68, macrophage antigen CD68; CD80, CD80 antigen; TLR2, Toll-like receptor 2; TLR4, Toll-like receptor 4; IL-12, interleukin-12; IL-23, interleukin-23; IL8RA, interleukin-8 receptor A; CXCR4, C-X-C motif chemokine receptor 4; SLAN, selenocysteine insertion sequence-binding protein 2-like; IL-10, interleukin-10.
Figure 1.
Monocyte differentiation and polarization. Differentiation of circulating monocytes into three subsets: classical (CD14++, CD16-), intermediate (CD14+, CD16+), and nonclassical (CD14+, CD16++) monocytes. Classical and intermediate monocytes can differentiate into M1 proinflammatory macrophages, whereas nonclassical monocytes can differentiate into M2 anti-inflammatory macrophages. HLA-DR, major histocompatibility complex class II antigen DR; FCGR1, Fc gamma receptor 1; TNFα, tumor necrosis factor alpha; IL-1β, interleukin-1 beta; IL-6, interleukin-6; CD68, macrophage antigen CD68; CD80, CD80 antigen; TLR2, Toll-like receptor 2; TLR4, Toll-like receptor 4; IL-12, interleukin-12; IL-23, interleukin-23; IL8RA, interleukin-8 receptor A; CXCR4, C-X-C motif chemokine receptor 4; SLAN, selenocysteine insertion sequence-binding protein 2-like; IL-10, interleukin-10.
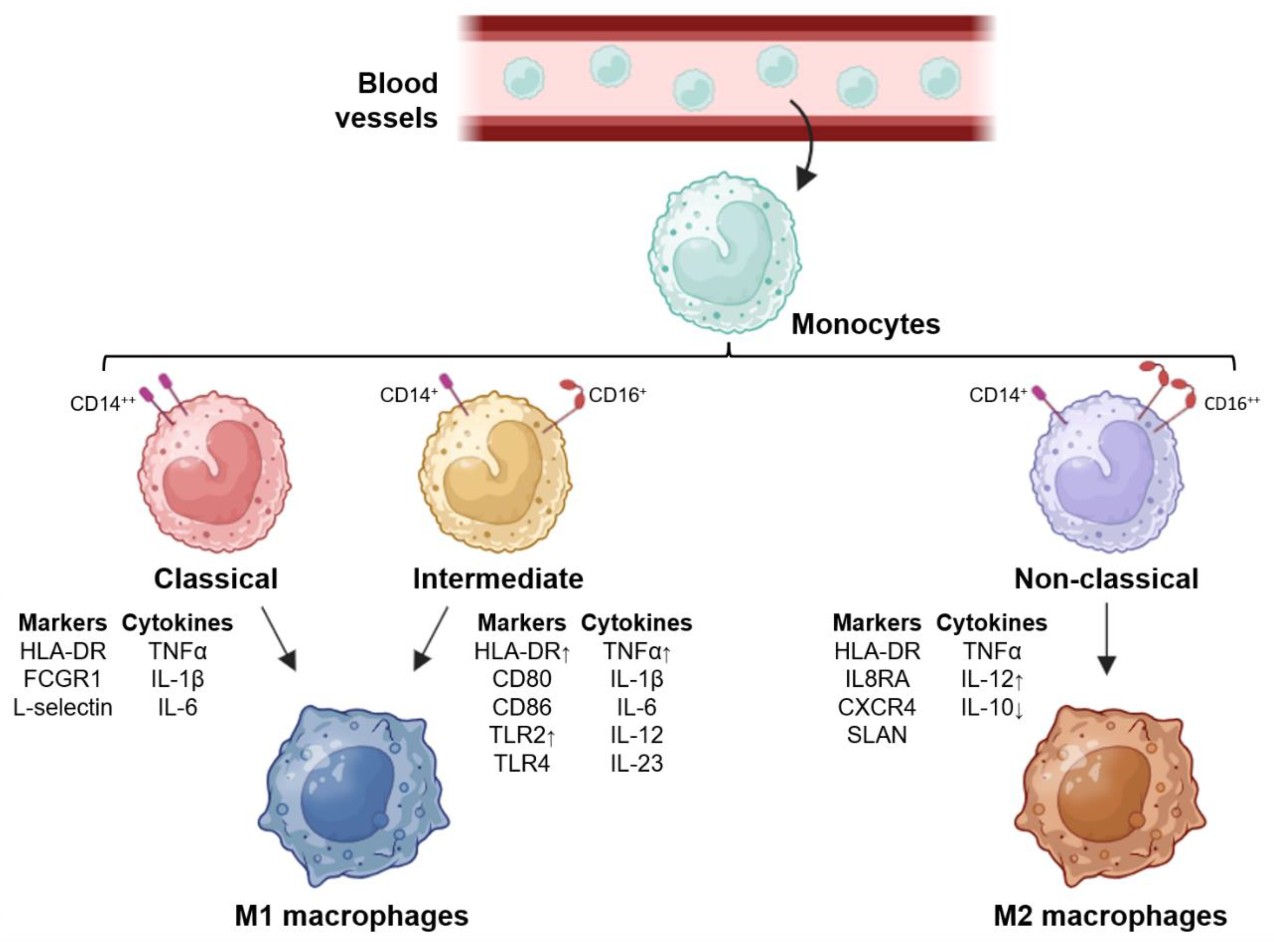
Figure 2.
Mediators secreted during macrophage polarization. The different stimuli or ligands, receptors, transcription factors and metabolism pathways of M1 and M2 macrophages are summarized.
Figure 2.
Mediators secreted during macrophage polarization. The different stimuli or ligands, receptors, transcription factors and metabolism pathways of M1 and M2 macrophages are summarized.
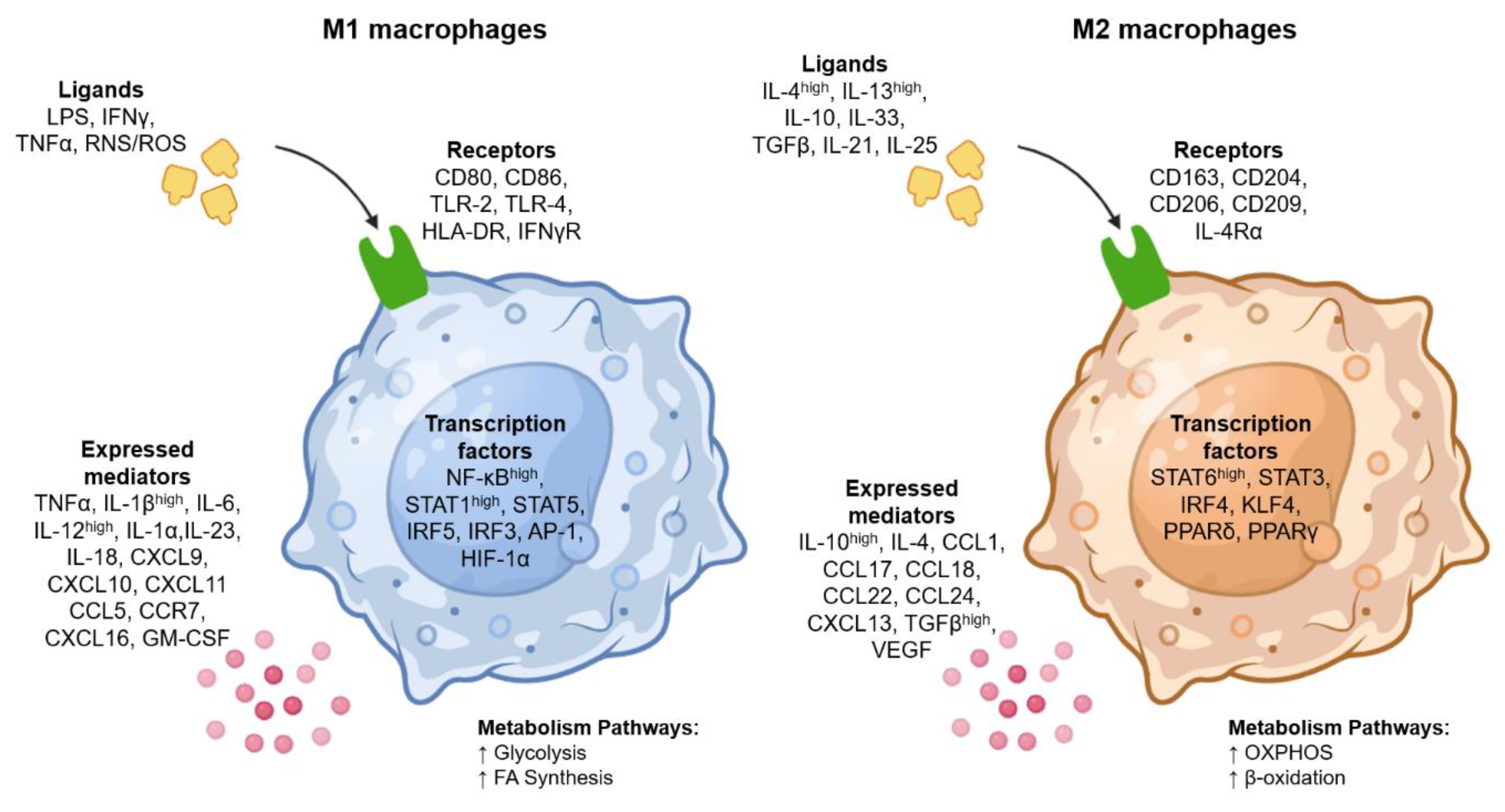

Table 1.
Stimuli, surface markers, secreted cytokines and biological functions of different types of macrophages. Arg-2, arginase type-2; Arg-1, arginase type-1; CSF-1, colony stimulating factor 1; ILT3R, immunoglobulin-like transcript 3.
Table 1.
Stimuli, surface markers, secreted cytokines and biological functions of different types of macrophages. Arg-2, arginase type-2; Arg-1, arginase type-1; CSF-1, colony stimulating factor 1; ILT3R, immunoglobulin-like transcript 3.
| Phenotype | Stimuli | Surface markers | Expressed mediators | Functions | References |
|---|---|---|---|---|---|
| M1 | LPS IFNγ TNFα RNS/ROS |
CD80, CD86, TLR2, TLR4, HLA-DR, IFNγR | TNFα, IL-1βhigh, IL-6, IL-12high, IL-1α, IL-23, IL-18, CXCL9, CXCL10, CXCL11, CCL5, CCR7, CXCL16, GM-CSF Arg-2, iNOS |
Pro-inflammatory Th1 response Anti-tumor capacity |
[2,20,21] |
| M2a | IL-4, IL-13 | CD163, CD206, Arg-1 | IL-10, TGFβ, CCL17, CCL24, CSF-1 |
Anti-inflammatory Tissue remodeling |
[15,35] |
| M2b | IL-1β, TLR ligands, immune complex | CD86 | TNFα, IL-1β, IL-6 IL-10, CCL1 |
Th2 activation, Pro- and anti-inflammatory Immunoregulation |
[15,36] |
| M2c | Glucocorticoids, IL-10, TGFβ | CD206, CD163 Arg-1 |
IL-10, CCL18, CXCL13, TGFβ, CCL16 |
Anti-inflammatory Phagocytic and apoptotic capacity |
[35,37] |
| M2d | IL-6, adenosine receptor ligands, TLR ligands | VEGF, ILT3R |
IL-10, CCL18, CCL22, VEGF |
Pro- and anti-inflammatory Angiogenesis, Immunosuppressive Tumor progression |
[20,29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



