Submitted:
22 May 2025
Posted:
23 May 2025
You are already at the latest version
Abstract
Hemolytic uremic syndrome (HUS) is characterized by the triad of mechanical micro-angiopathic hemolytic anemia, thrombocytopenia and renal impairment. Atypical Hemolytic Uremic Syndrome (aHUS); non Shiga-toxin-HUS, represents 5 -10% of HUS in children and is associated with a mortality rate of 20–25% and a morbidity of 48%, since pediatric patients typically progress to end stage renal disease (ESRD)1 It’s either caused by inherited pathogenic variants in complement gene variants, which account for 50% to 60% or by autoantibodies directed against complement factor H We report the first aHUS case, confirmed by Whole Exome Sequencing (WES), following two injections of purified Vero cell culture rabies vaccine (PCEC), which was satisfactorily managed using a combination of plasma exchange (PEX) and immunosuppressive therapy. Our case highlights the possible association between aHUS and rabies vaccination in genetically susceptible individuals.
Keywords:
Immune dysregulation
; Atypical HUS
; Rabies vaccine
Background:
Endotheliopathy is increasingly recognized as the root of several disorders, including micro- angiopathic hemolytic anemia (MAHA)
Endothelial inflammation can be induced by several mechanisms, including viral or bacterial components, and genetically determined complement dysregulation.
A study by Liuba et al. has pointed out that endothelial dysfunction was detected in recipients after influenza vaccination, which might be a possible explanation for increased coronary syndromes risk after influenza vaccination [2].
Another “trendy” hypothesis suggests that long COVID-19 manifestations are due to persistent endothelial dysfunction after COVID-19 infection or vaccination. [3]
It is currently believed that the vaccine-induced thrombocytopenia seen after Vaxzevira (ChAdOx1-S); the recombinant COVID-19 vaccine, is related to endothelial involvement. [4]
The clinical spectrum of Endotheliopathy can range from increased arterial stiffness leading to increased cardiovascular risk, strokes, to severe hemolysis and platelet damage, as known as MAHA.
We report a case of MAHA following Rabies vaccination and with a concomitant recent history of COVID-19 infection, in a genetically predisposed individual, leaving multiple questions about whether endotheliopathy in this case has been triggered by the vaccination or infection or an intermingling of both.
Case report:
a 10-year old male presented to the emergency room on the 17th of September 2023 with manifestations of acute hemolysis in terms of; fever, jaundice and dark urine of two days duration.
The condition was of acute onset and progressive course. Fever was low-grade, intermittent, responsive to antipyretics and started six days earlier than the associated symptoms.
The condition was not associated with diarrhea, change in colour of stools nor bleeding from any orifice, yet was preceded by a dog bite in his little finger, after which he received 2 doses of intramuscular injection of purified Vero cell culture rabies vaccine (PCEC) , under the trade name RabAvert ,on the 6th and 10th of September 2023.
On physical examination, he was vitally stable, had yellow skin and sclera, but no hepatosplenomegaly, yet palpable deep cervical lymph nodes.
Laboratory investigations on admission showed Haemoglobin (Hb) 6.9 gm/dL, MCV 77 fl, Corrected reticulocytic count 15%, Platelets (Plt) 56,000/mm³, Total leukocytic count 19x10³/mm³, Negative direct and indirect Coomb’s test, Alanine transaminase (ALT) 24 U/L (Normal range 0-40 U/L), Aspartate aminotransferase (AST) U/L 81 U/L (Normal range 0-38 U/L), Lactate dehydrogenase (LDH) 1950 U/L (Normal range 140-280 U/L) , total bilirubin 2.4 mg/dL, direct bilirubin 0.47 mg/dL, Creatinine 0.6 mg/dL and Blood urea nitrogen 70.
The patient then received 1.5 Units of filtered irradiated packed RBCs due to severe decompensation, with post-transfusion Haemoglobin (Hb) level of 9.6 gm/dL. Follow up investigations two days later (on day 3 of admission) showed sudden elevation of kidney functions (Creatinine 2.9 mg/dL, Urea 200) , non-oliguric AKI, grade 2 according to KDIGO.
The condition was then classified as Micro-angiopathic haemolytic anaemia (MAHA) for differential diagnosis.
Haemolytic uremic syndrome (HUS) workup was done; Fragmented RBCs 1.1%, Haptoglobin 0.1 g/L (normal reference range 0.3-2) and shiga toxin in stools was negative, done by PCR method.
C3 was consumed; 47.8 mg/dL (reference range 90-180 mg/dl) , yet normal C4 level;14 mg/dL (reference range 10-40 mg/dl) ANA was negative.
Urine anaylsis showed granular casts and urine Albumin 500 mg/dL, with an Albumin/Creatinine ratio of 12029 mg/g creat (Normal <30) D-dimer 5600 ng/mL. Unfortunately, ADAMTS13 autoantibodies and Anti-Factor H antibody kits were not available. COVID IgM was negative, yet IgG positive. Pelvi-abdominal Ultrasonography showed normal site and size of both kidneys, with increased echogenicity grade I (right more than the left) and increased bilateral renal resistance indices in the main & segmental arteries.
On Day 7 of admission, there was still ongoing hemolysis with persistent thrombocytopenia, albuminuria and elevated kidney functions; Hb dropped to 7.5 gm/dL, PLT count was 74,000/ mm³, corrected reticulocytic count 10% and a creatinine level of 2.75 mg/dL
Due to severe pallor and tachycardia, 1 Unit of packed RBCs was transfused again. Based on the clinical and laboratory findings, Intravenous Methylprednisolone (MP) was initiated at a dose of 30 mg/kg/day for 3 days followed by oral prednisone at a dose of 2 mg/kg/day.
Follow up investigations after three days of IV Methyl Prednisolone showed Hb level 8 gm/dL, PLT count 79,000/mm³, corrected reticulocytic count 20% and creatinine level 2.4 mg/dL, and after four days of oral steroids Hb level 11 gm/dL, PLT count 30,000/mm³ and creatinine level 1.4 mg/dL
While labs on the thirteenth day of steroids intake collectively showed Hb level 8.5 gm/dL, PLT count 27,000/mm³, corrected reticulocytic count 7% and Creatinine level 1 mg/dL (Figure 1.)
Figure 1.
.
Mycophenolate mofetil (MMF) was then prescribed at a dose of 600 mg/m²/12 hrs, still with very poor response and Whole Exome Sequencing (WES) was requested for the patient.
Renal biopsy showed findings compatible with Thrombotic microangiopathy and Focal Segmental Glomerulosclerosis, possibly secondary.
By Light microscopy: Seventeen glomeruli were seen, out of which none was globally sclerosed. Six glomeruli showed marked endothelial swelling, mesangiolysis and segmental peripheral duplication. Three glomeruli showed segmental tuft sclerosis.
Tubules showed mild injury. Interstitium was within normal limits. Arteries and arterioles were unremarkable.
Plasma exchange was then initiated, a total of eleven sessions, each about 1500-2000 ml (50 ml/kg) Plasma.
The patient is currently in hematological remission, with normal Haemoglobin and platelet values in all his follow up investigations (Figure 2), (Figure 3) yet still has persistent mild renal insufficiency, in terms of elevated Creatinine level and persistent proteinuria. (Table 1.)
He has discontinued steroids therapy after gradual tapering, and MMF is currently being tapered to 500mg/day
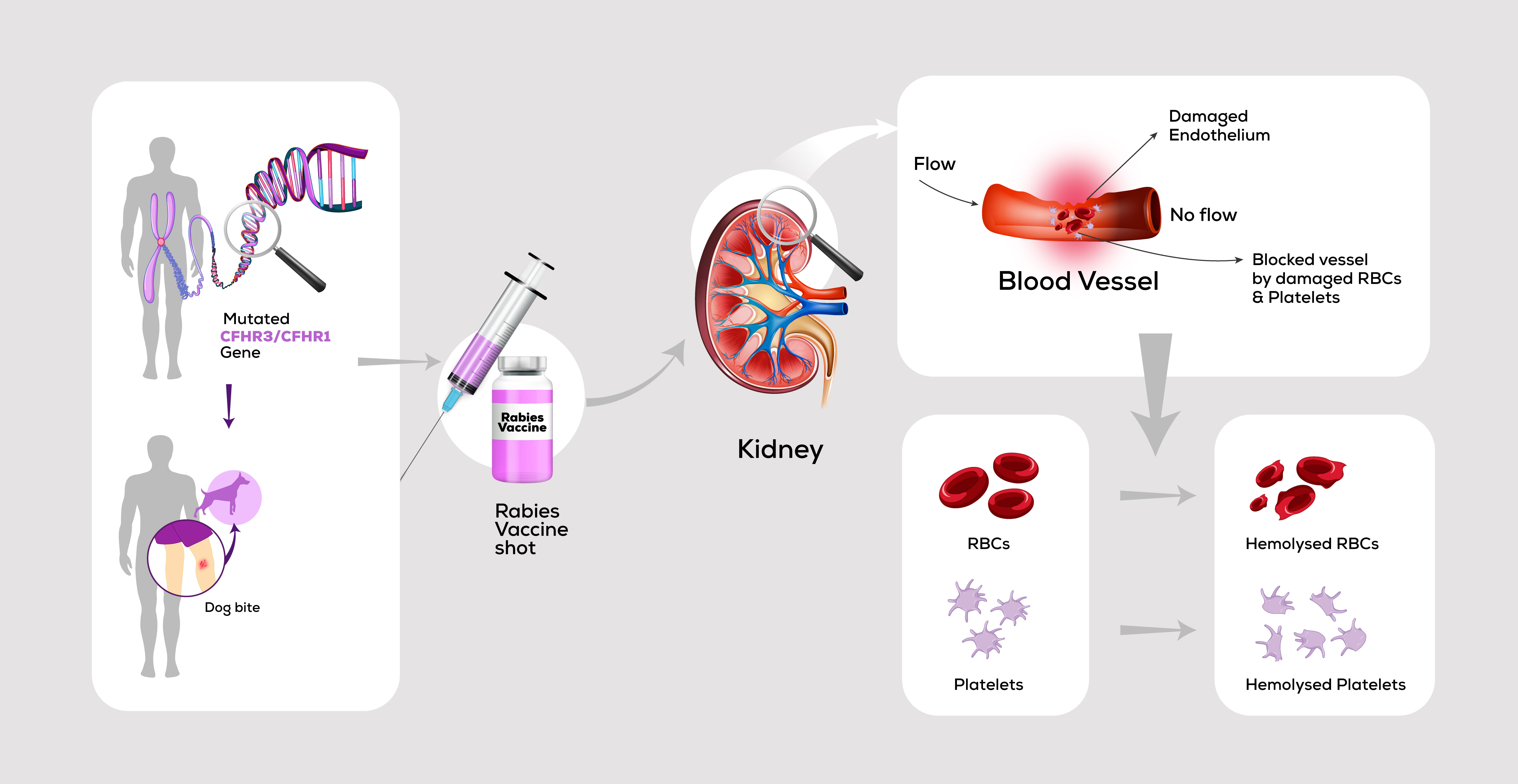
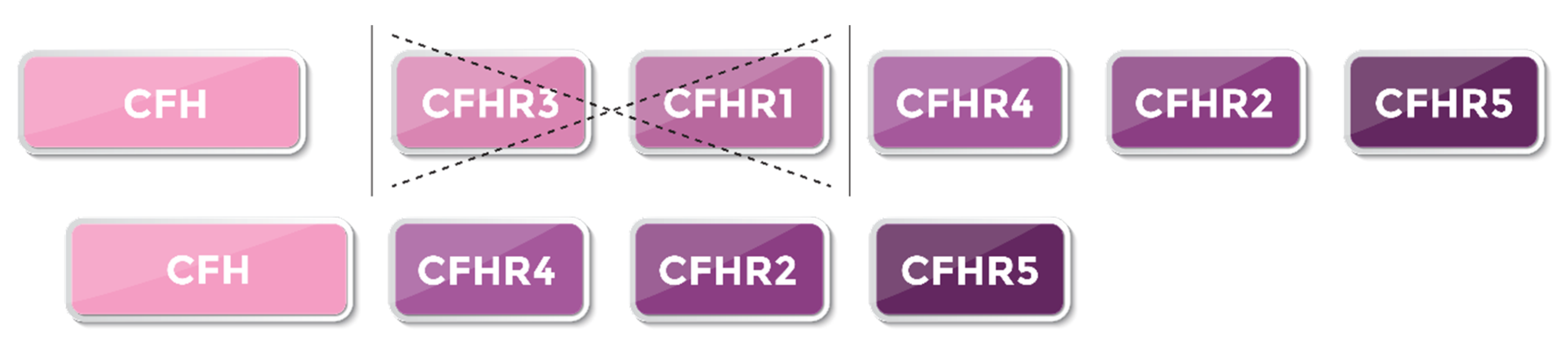
WES report showed a heterozygous mutation with large deletions encompassing CHFR3/CHFR1 genes in the long arm of Chromosome 1 spanning Chr1:196743954- 196801180, sparing CFH. (Figure 4)
Figure 2.
.
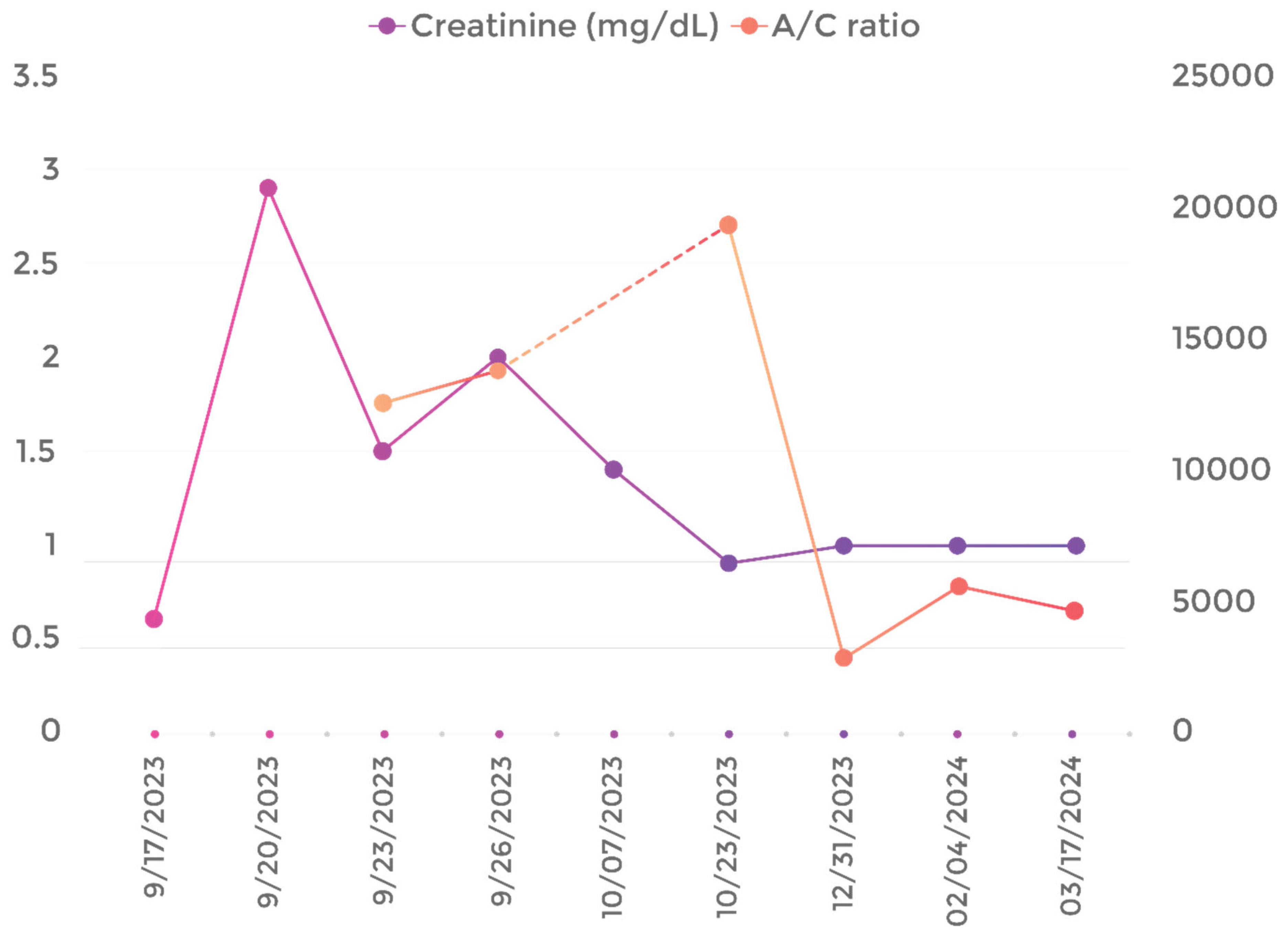
Figure 3.
.
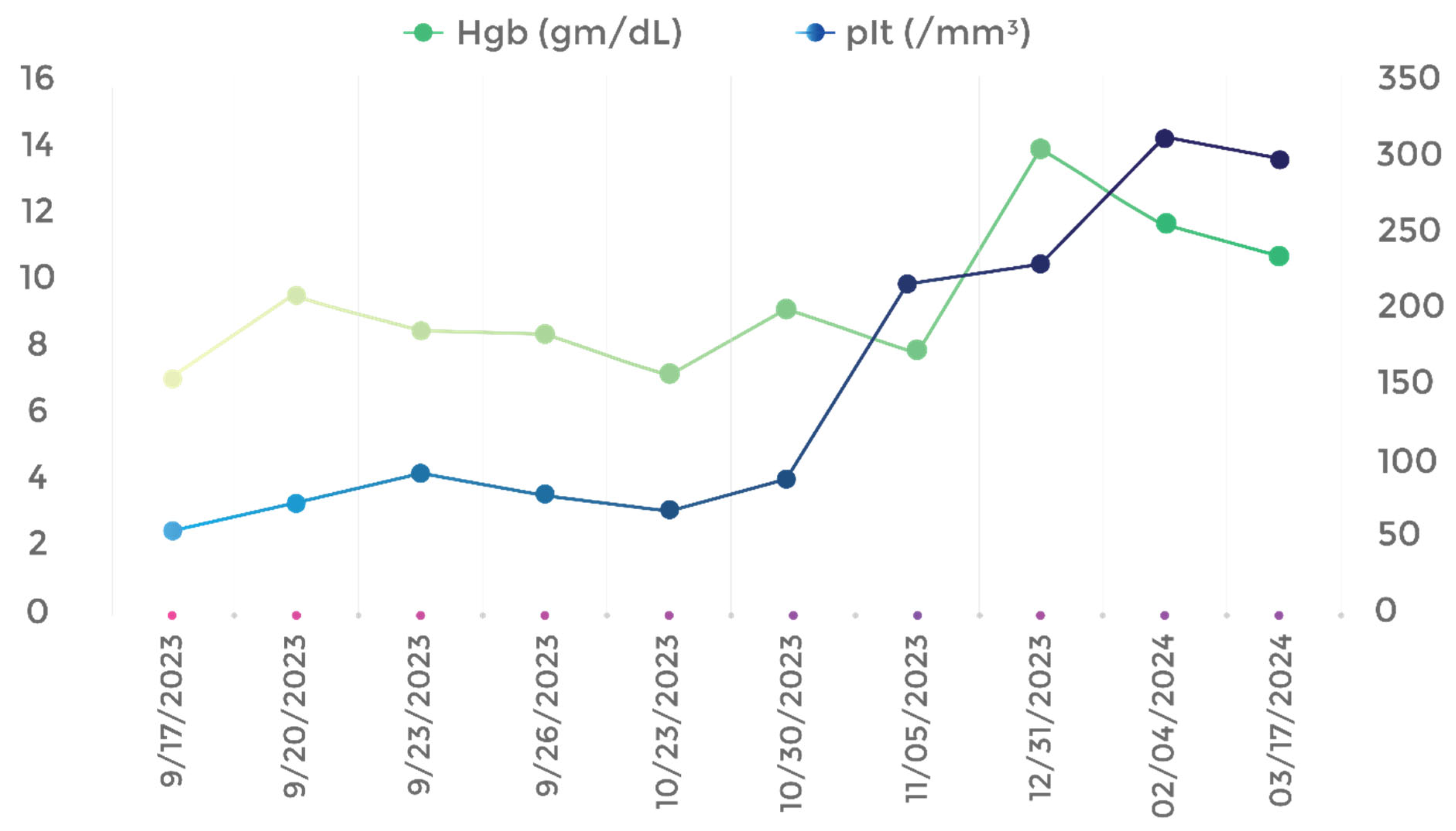
Figure 4.
.
Discussion:
Post vaccination endotheliopathy occurs as a consequence of activation of two pathways, inflammatory and microthrombotic.
The release of inflammatory cytokines, such as IL-6, tumor necrosis factors and interferons, occurring concomitantly with platelet activation and exocytosis of unusually large von Willebrand factor (ULVWF) multimers which become anchored to endothelial cells and recruit activated platelets. The latter results in formation of microthrombi strings which trigger disseminated intravascular microthrombosis , which is the underlying pathology of endotheliopathy-associated vascular microthrombotic disease (EA-VMTD).[5]
Needless to mention, the protective value of rabies vaccine far outweighs its adverse effects. Yet, some serious adverse effects were reported, including a case of TTP in a previously healthy man [6], TTP in a known case of SLE [7] , a case of glomerulonephritis back in 1981 , and to our knowledge, this is the first aHUS case to be reported post- rabies vaccination.
TTP was also reported in 3 post influenza,1 post pneumococcal and 1 post H1N1 vaccinations. [8]
It was also highlighted during the COVID-19 era, post mRNA vaccination against SARS- CoV-2, in a genetically susceptible individual, a heterozygous carrier of a rare pathogenic variation in CFH (c.3096C>A, p.C1032X). [9]
After searching the CDC WONDER for data on adverse reactions of rabies vaccines, thrombocytopenia accounted for 0.17%, thrombocytopenic purpura 0.02%, AKI in 0.03% and abnormal urine analysis, comprising albuminuria and granular casts in 0.05% of cases [Vaccine Adverse Event Reporting System (VAERS) using CDC WONDER Online Database; available from https://wonder.cdc.gov/vaers.html] Thrombotic microangiopathy (TMA) is a condition characterised by Coomb’s negative haemolytic anemia and thrombocytopenia along with microvascular thrombosis due to abnormalities in the wall of arterioles and capillaries, resulting in ischemic organ damage. It comprises thrombotic thrombocytopenic purpura (TTP), Drug-induced TMA, Metabolism- mediated TMA and Haemolytic uremic syndrome.
The latter is then classified into HUS with coexisting conditions, infection-induced HUS , the commonest of which is STEC, Cobalamin C defect–HUS, DGKE–HUS and atypical HUS (aHUS), which is now known as complement mediated HUS. [10]
Atypical HUS represents 5 -10% of HUS in children , and is associated with a mortality rate of 20–25%, [1] 40-60% relapse rate, [11] and a morbidity of 48%, as pediatric patients typically progress to end stage renal disease (ESRD) at the first episode or within less than 1 year.
Genetic variants in complement regulatory proteins (CRPs) account for 50% to 60% of all aHUS cases, with approximately 30–50% having no known identifiable mutation. [1]
This dysregulation follows mutations in genes coding for complement regulatory proteins, such as factor H (CFH), factor I (CFI), membrane cofactor protein (MCP), complement 3 (C3), factor B (CFB), or thrombomodulin (THBD), or by the presence of anti-CFH antibodies with consequent hyperactivation of the complement system. [10]
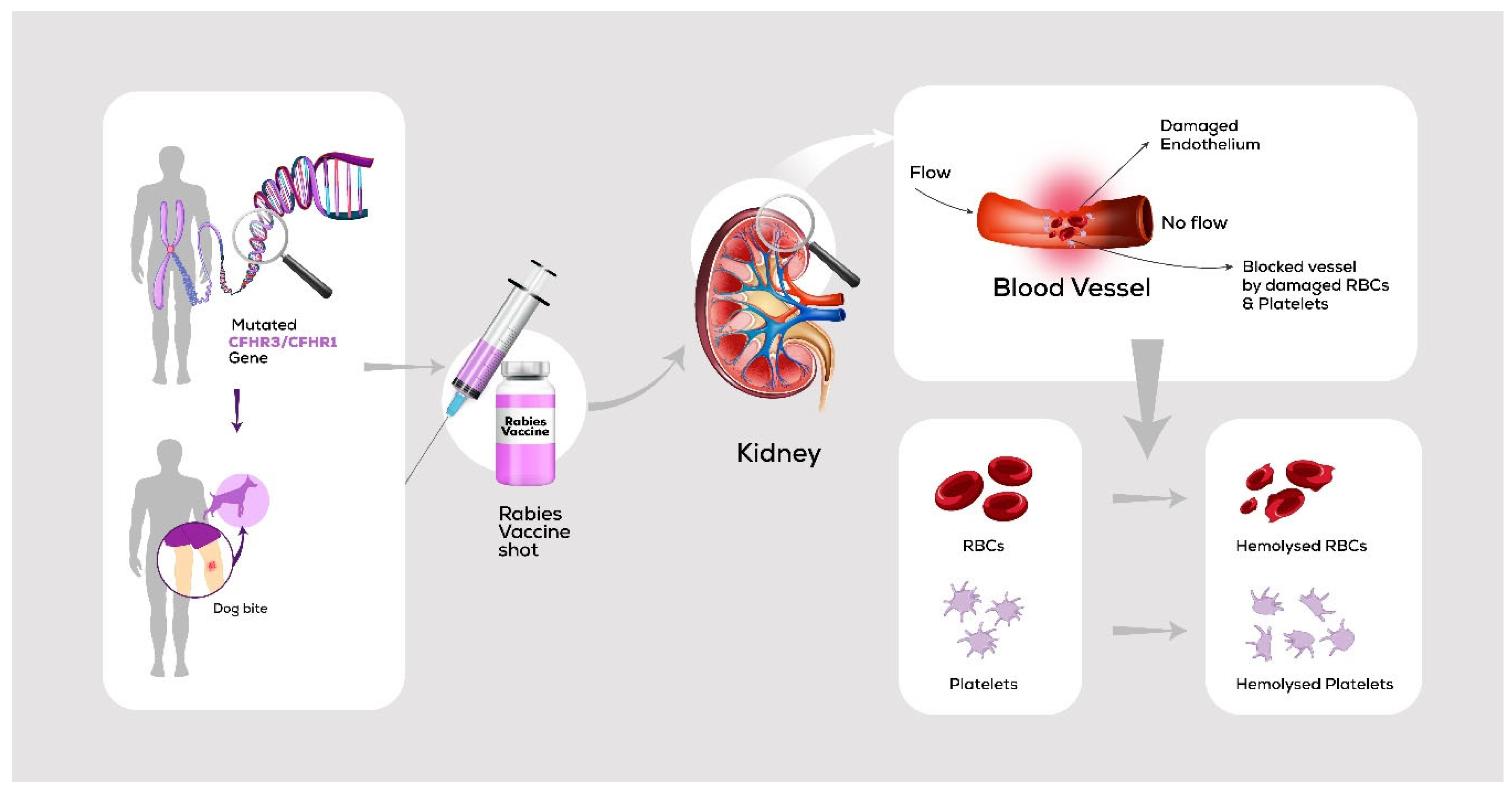
Loirat Ch. et al, stated that anti-CFH antibodies account for approximately 6% of aHUS, mainly in children between 7-11 years of age,[11] while in India, anti-FH antibody-associated aHUS constitutes around 50% of pediatric patients between 5– 15 years old [12] the presence of which would interfere with CFH binding to C3b, hence, uncontrolled C3 convertase activity, C3 consumption and MAC production, resulting in endothelial cell dysfunction and microvascular thrombi formation, with the kidneys being the target organ. (Figure 5)
Figure 5.
.
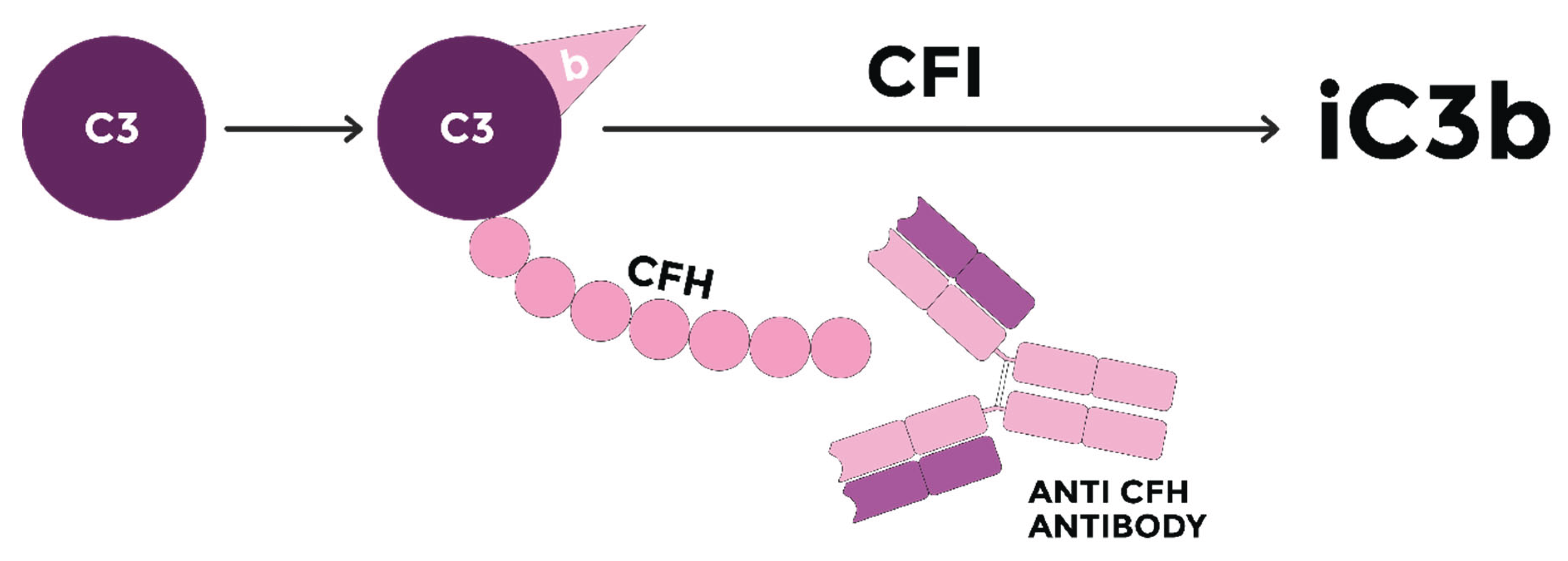
Ninety percent of patients with anti CFH-antibodies have a complete deficiency of CFHR1 and CFHR3 associated to a homozygous deletion of CFHR1 and CFHR3 , suggesting that this deletion has a pathogenic role in the development of anti-CFH autoantibodies[11]
Which is in concordance with the WES results of the aforementioned case; a heterozygous mutation with large deletions encompassing CHFR3/CHFR1 genes in the long arm of Chromosome.
A literature by J. A. Ayres et al. suggests that there are alterations in the immune response of those subjected to anti-rabies vaccination, guided by elevated levels of the pro-inflammatory cytokines IFN-γ, IL-2, and IL-10, which might explain the molecular pathogenesis of the aforementioned case.[13]
Furthermore, IFN-γ, and IL-10 were also found significantly elevated in those subjected to psychological stress.
Hence, the condition could be multifactorial, and the vaccine might not be solely incriminated in blowing of the aforementioned clinical picture.
Physical and psychological stress induced by the dog bite and the vaccine injection might be contributing factors. However, further studies are required to confirm and possibly elucidate the exact underlying molecular pathogenesis responsible for the occurrence of such unusual post-vaccination sequelae.
Management of such cases can be particularly challenging in developing countries.
As stated by Bagga A. et al, the standard treatment of atypical HUS without anti-FH antibodies lies in C5 blockage by Eculizumab, the first successful terminal complement inhibitor available to pediatric aHUS patients. However, since it’s not readily available in developing countries due to its high cost, Plasma exchange (PEX) forms the basis of management in such countries. Eculizumab is reconsidered if there’s lack of remission despite 7–10 days of PEX, in life-threatening features (seizures, cardiac dysfunction), complications due to PEX or vascular access, and in inherited defect in complement regulation.
While in anti-FH antibody-associated HUS, patients are successfully managed given the combination of prompt PEX and immunosuppressive therapy in both, developed and developing countries.
Recommendations for Eculizumab in such cases are the same as in atypical HUS without anti-FH antibodies
It’s also recommended to monitor antibody titres during first 12-24 months to predict relapse [12]
Our case highlights the possibility of occurrence of atypical HUS following Rabies vaccination in immune susceptible individuals, with the possibility of involvement of IFN-γ, IL-2, and IL-10 in the pathogenesis.
Acknowledgments
I would like to express my sincere gratitude towards our Nephrology team; namely Professor Dr Fatina Fadel, Dr Eman Abubakr, Dr Sherouk Abdelrahman and Dr Mohamed Thabet for contributing in the management and follow up of the case to date, and towards our Hematology team, Dr Marwa Abdelhady and Dr Mai Mohamed who have also contributed in the initial management of the case. I would also like to express my gratitude towards Dr Antoine Abdelmassih, for his invaluable guidance and for teaching me the basics of scientific research.
References
- Raina, R.; et al. Pediatric atypical hemolytic uremic syndrome advances. Cells, 2021; 1–21. [Google Scholar]
- Liuba, P.; et al. Residual adverse changes in arterial endothelial function and LDL oxidation after a mild systemic inflammation induced by influenza vaccination. Ann. Med. 2007, 39, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Abdelmassih, A. F. , Kamel, A., Mishriky, F. & Ismail, H. Is it infection or rather vascular inflammation ? Game-changer insights and recommendations from patterns of multi-organ involvement and affected subgroups in COVID-19. 2020. [Google Scholar] [CrossRef]
- AbdelMassih, A.; et al. Is the heparin-induced thrombocytopenia-like syndrome associated with ChAdOx vaccine related to the vaccine itself or to an autoimmune reaction to severe acute respiratory syndrome 2 coronavirus: insights and implications from previous reports in infected. New Microbes New Infect. 2021, 41, 100884. [Google Scholar] [CrossRef] [PubMed]
- Chang, J. C. & Hawley, H. B. Vaccine-associated thrombocytopenia and thrombosis: Venous endotheliopathy leading to venous combined micro- macrothrombosis. Med. 2021; 57. [Google Scholar]
- Kadikoylu, G. , Yavasoglu, I. & Bolaman, Z. Rabies vaccine-associated thrombotic thrombocytopenic purpura. Transfus. Med. 2014, 24, 428–429. [Google Scholar] [PubMed]
- Cui, Y. , Wei, J. & Peng, X. Case Report: Rabies Vaccine-Induced Thrombotic Thrombocytopenic Purpura in a Patient With Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 1–5. [Google Scholar]
- Gonzalez-Mancera, M. S. & Gonzalez, J. M. In reply to: Cd4+cd8+ double- positive t-lymphocytes: Pitfalls. Turkish J. Hematol. 2020, 37, 217–218. [Google Scholar]
- Rysava, R.; et al. Atypical hemolytic uremic syndrome triggered by mRNA vaccination against SARS-CoV-2: Case report. Front. Immunol. 2022, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Spasiano, A.; et al. Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation? Int. J. Mol. Sci. 2023; 24. [Google Scholar]
- KAVUKÇU, S. , İRKEN, G., OLGUN, N. & GÜÇLÜ, A. Atypical hemolytic uremic syndrome. Pediatr. Int. 1995, 37, 638–641. [Google Scholar]
- Bagga, A.; et al. Hemolytic uremic syndrome in a developing country: Consensus guidelines. Pediatr. Nephrol. 2019, 34, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J. A. , Barraviera, B., Calvi, S. A., Carvalho, N. R. & Peraçoli, M. T. S. Antibody and cytokine serum levels in patients subjected to anti-rabies prophylaxis with serum-vaccination. J. Venom. Anim. Toxins Incl. Trop. Dis. 2006, 12, 435–455. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



