
Submitted:
20 May 2025
Posted:
21 May 2025
You are already at the latest version
Abstract
Horizontal trafficking of subcellular components, such as nucleic acids, proteins, and membrane fragments, is utilized by tumor cells to facilitate tumor cell proliferation and survival. Conventionally, tumor cells have been known to undergo long-range transfer through the import and export of extracellular vesicles and exosomes. However, other means of intercellular transfer are also employed by tumor cells. These trafficking methods can facilitate changes in anti-tumor immunity and distribute oncogenic protein variants to nearby cells to provide a hospitable tumor microenvironment. The molecular mechanisms that drive many of these cell trafficking mechanisms are conserved, relying on de novo synthesis of filamentous actin. However, the delineation between these processes is not yet known. This review will highlight four recently characterized and underappreciated near cell trafficking mechanisms: (i) trogocytosis, (ii) entosis, (iii) cell fusion, and (iv) tunneling nanotubes/microtubes utilized by tumor cells to promote a hospitable microenvironment.
Keywords:
trogocytosis
; enosis
; cell fusion
; nanotubes
; cell-cell trafficking
1. Introduction
Horizontal integration of cellular components originating from an unrelated cell is a staple biological process that is well characterized in other organisms, such as horizontal gene transfer in bacteria first characterized in 1928 by Griffith [1]. Other model organisms utilized in laboratory organisms also undergo horizontal trafficking, such as the formation of thin, tubular trafficking networks known as cytonemes in drosophila melanogaster [2]. In mammals, Chargaff and West recorded the first instance of transfer of subcellular compartments between two cells in the form of platelet-derived extracellular vesicles [3].
Cancers are dynamic diseases that can adapt to their surrounding microenvironments in order to survive and proliferate. Although it is accepted that external factors could contribute to the increase in tumor mutational burden, this accumulation only affected a small subset of cells that would ultimately clonally expand to form a tumor. Characterization of the downstream consequences of cell-cell trafficking within the context of cancer only began as recently as 2006 [4]. Further studies have demonstrated how intercellular transfer of DNA, RNA, and protein alter the interplay between tumor and other resident tumor microenvironment (TME) cells [5,6,7,8]. This review compiles the molecular mechanisms that drive four unconventional near-cell trafficking methods and the outcomes of each method in reshaping tumor plasticity, dampening anti-tumor immunity, and increasing the tumor mutational burden (Figure 1).
2. An Overview of Contact Dependent, Cell-Cell Trafficking Methods
2.1. Trogocytosis
2.1.1. A Historical Overview of the Biological Functions of Trogocytosis in Physiology and Disease
The contact-dependent transfer of plasma membrane from a donor cell to a recipient cell is known as trogocytosis, was first described by Cone, Sprent, and Marchalonis in 1972 [9,10]. Trogocytic interactions were first characterized as a unidirectional transfer of membrane-bound proteins and lipids from an antigen presentation cell to a lymphocyte and was demonstrated to be a critical process in CD4+ and CD8+ T cell recognition of antigens [11,12]. Other essential physiological functions have been implicated with trogocytosis, such as the spermicidal function of vaginal neutrophils [13]. Neuronal circuit maturation is also dependent on microglia trogocytosis between presynaptic boutons and axons on nerve cells [14]. In contrast, pathogens can also harness trogocytosis to avoid selective pressure originating from the host environment [15,16]. E. histolytica parasites trogocytose membrane fragments of human cells and led to a sharp influx of Ca2+ into the human cells, resulting in lytic death [15].
2.1.2. Trogocytosis Within the Tumor-Immune Microenvironment
In cancers, trogocytosis alters the transcriptome of cells within the tumor-immune microenvironment (TIME). Trogocytosis between tumor and immune cells can occur in both directions and leads to decreased immune cell sensitization through the transfer of functional immune checkpoint proteins, increased lymphocyte fratricide and “self” peptides [17,18,19,20,21]. In contrast, tumor-immune cell trogocytosis can also enhance the cytotoxic metabolism of effector immune cells, leading to enhanced tumor clearance [22]. Recently, the transfer of genomic DNA was also implicated in trogocytic interactions between tumor and immune cells; though it is currently unknown whether this transfer is a direct consequence of trogocytosis or driven by different interactions that share conserved pathways with [17,20,21].
2.2. Entosis
The term “entosis” was first coined by Overholtzer et al. in 2007 where they described this process as a nonapoptotic cell death process that is initiated by loss of adherence to the extracellular matrix [23]. This phenomenon was unique as it involved the invasion of one living cell into another, resulting in the formation of a “cell-in-cell” structure (CIC) [23]. These CIC structures can refer to one or multiple individual cells invading a single host [24]. The engulfed cell, commonly referred to as the “loser”, can encounter one of several fates. Death by lysosomal acidification of the entotic vacuole containing the “loser” cell is driven by the LC3-mediated recruitment of autophagy-related proteins and is the most frequently observed outcome [25]. However, it is possible for engulfed cells to escape their host or even undergo cell division while still entrapped [23]. In the context of cancer, the significance of entosis is multifaceted as it has a role in both pro- and anti-tumor processes. On the one hand, entosis may limit tumor metastasis by eliminating cancer cells that break free from the surrounding extracellular matrix [23,25,26]. However, researchers have observed a correlation between the number of entotic cells and severity of disease across multiple types of cancer [27,28].
2.3. Cell Fusion
Cell fusion refers to the process by which two cells in contact with each other merge surface membranes and combine into a single daughter cell. In normal biology, there are several process involving cell fusion that occur in mammals including the fusion of sperm and egg, fusion of myoblasts into myotubes during muscle development, fusion of macrophages to form multinucleated giant cells, and the fusion of monocytic cells to form osteoclasts [29,30,31,32]. In cancer, cell fusion is thought to be a relatively rare event with researchers estimating that 0.0066 and 6.5% of cancer cells undergo this process in vitro [33]. Although rare, cell fusion is believed to have a significant role in shaping tumor metastasis, drug resistance and heterogeneity. Researchers have shown that cell fusion in cancer can lead to parasexual recombination of genetically distinct cancer cells, leading to intratumoral heterogeneity [34].
2.4. Tunneling Nanotubes
Tunneling nanotubes (TNTs) and tumor microtubes (TMs) are three-dimensional, hollow, and tubular structures that connect between two cells that are similar to cytonemes found in d.melanogaster [35]. They are classified based on tube length and thickness. TNTs and TMs allow for the transfer of subcellular components and are most commonly observed in neurons, astrocytes, and glial cells and are essential in the maintenance of the neuronal networks of the central nervous system in normal physiology [36]. The main structural component of TNTs and TMs are filamentous actin (F-actin) fibers coated with cell membrane. TNTs and TMs can traffic functional pieces of subcellular components including cell membrane fragments, mitochondrial DNA (mtDNA), and protein[6,7,37]. In solid cancers, structures to TNTs and TMs were first characterized in 2004 by Rustom et al. in a rat adrenal tumor and have since been discovered to be utilized by cancer cells to alter gene and protein expression in the TME of human breast, ovarian, cervical, lung, and pancreatic cancers [38]. Formation of TNTs are not limited between two cancer cells and have been shown to form between a tumor cell and a T cell [7]. Formation of TNTs are also observed to lead to the increased expression of epithelial-to-mesenchymal transition (EMT) related proteins in cancer cells [39]. Taken together, TNTs promote tumor development and progression through the shuttling of functional DNA, organelles, and oncogenic proteins in order to elevate tumor cell respiration and mutational burden.
3. Mechanisms of Cell-Cell Trafficking
3.1. Mechanisms of Trogocytosis
3.1.1. Solid Tumor Trogocytosis
Very little is known about the mechanisms that drive trogocytosis. Within the context of cancer, most studies investigated the immunomodulatory effects of trogocytosis in tumor-immune cell synapse rather than physiological pathways that induce trogocytosis. Trafficking via trogocytosis occurs in both directions, where the tumor cell is able to act as both a donor and a recipient cell [17,18,20,21,40,41]. However, it is currently unknown whether this “swap” occurs concomitantly or if they are two separate trogocytic interactions. The initiation of trogocytosis is dependent on physical contact between two cells [12]. It does not occur between two cell populations when separated by a barrier such as a transwell in in vitro cocultures [18,20,21].
Trogocytosis is also associated with elevated or aberrant phosphatidylinositol 3-kinase (PI3K) activity [42]. Overactivation of PI3K results in hyperproliferation in cancer cells, increasing cancer cell growth rate and apoptotic resistance. Studies assessing the immunomodulatory effects of trogocytosis have shown that Wortmannin and Latrunculin A/B by inhibiting F-actin synthesis [42]. However, the conclusions drawn from studies using these inhibitors to prevent tumor cell acquisition of lymphocyte protein via trogocytosis is controversial, as expression of PI3K is necessary for cell maintenance and survival (Figure 2).
3.1.2. Liquid Tumor Trogocytosis
In contrast, trogocytic interactions between leukemia cells, a bone marrow-derived liquid tumor, and natural killer (NK cells) were found to be driven by SLAM-receptor signaling [18]. SLAM receptors are type I glycoproteins that are expressed on a subset of immune cells including activated T cells, B cells, macrophages, and dendritic cells [43]. However, it is still unknown what specific downstream effectors of SLAM receptors drive trogocytosis. Should the mechanism be similar to immune cell trogocytosis of solid tumor cells, trogocytosisdriven by NK cells may be driven by SLAM receptor recruitment of PAK interacting protein (β-PIX), a guanine nucleotide exchange factor specific for downstream Rho/Rho-associated coiled-coil containing kinase (ROCK) mediated F-actin polymerization [44]. This hypothesis would be consistent with the findings that treatment with latrunculin A also impeded Chimeric Antigen Receptor T cell (CAR-T) trogocytosis of multiple myeloma cells (Figure 2) [45].
3.2. Mechanisms of Entosis
3.2.1. ROCK Signalling Pathway
Entosis is a complicated process that is primarily driven by the internalized cell, rather than the engulfing cell, which distinguishes it from other processes like phagocytosis (Figure 3). Mechanotransduction and differences in the stiffness of the membranes of the host and invading cell are critical for entosis to occur. Differences in the activation of myosin II specifically has been shown to promote entosis as the cell with higher myosin II activity are more likely to invade their neighbors [46]. This invasion is primarily driven by ROCK signaling [23]. More specifically, Cdc42, Rho (RhoA, RhoB and RhoC) and Rac (Rac1, Rac2 and Rac3) are the most critical small GTPase members of the Rho family when it comes to driving entosis [47,48]. Activation of the ROCK signaling pathway leads to a higher concentration of active Rho in the distal end of the invading cell, opposite of its point of contact with the host. The resulting mechanical tension promotes cellular invasion and subsequent entosis [48,49].
3.2.2. External Drivers of Entosis
Loss of adhesion of cells from the extracellular matrix has been associated with entosis since the process was originally discovered [23]. In normal tissues, cells that detach from the matrix typically undergo a programmed cell death pathway such as anoikis or apoptosis. This likely explains why the most common place to find CIC structures is in the fluid exudates of cancer patients (urine, bile, ascites, pleural fluid, etc.) (Overholtzer and Brugge 2008, Nature Reviews Mol. Cell Biol.). However, cancer cells can overcome this by altering integrin expression, upregulating pro-survival pathways like PI3K/Akt, and altering their metabolism [50]. After detaching from the basement membrane, free floating cells must form adherens junctions with the host cell for entosis to begin [23,51,52,53]. After adherens junctions are established, an imbalance of actin-myosin contractions between the cells promotes invasion of the internalizing cell and entosis occurs [46,48].
Another external factor that can influence entosis is nutrient deprivation. Researchers have shown that under these conditions the engulfing cell is able to harvest nutrients, such as amino acids, from the invader, ultimately supporting the engulfer’s survival [54] . Glucose deprivation appears to have the most profound effect on rates of entosis. For example, Hamann et al. have shown that increased activity of the starvation-induced kinase AMPK can skew a cell towards an “invader” phenotype [55]. This may lead to increased competition between cancer cells, such that engulfing cells may be able to promote their own survival through scavenging nutrients from invading cells under starvation conditions.
Ultraviolet radiation (UV) is also capable of inducing entosis. In 2021 Chen et al. showed that when exposed to a high dose of UV (100J/m2), 35% of MCF7 human breast cancer cells underwent entosis [56]. This study demonstrated that UV-induced entosis was regulated by c-Jun N-terminal kinase (JNK) and p38 stress-activated kinases in the internalized cell [56]. Ingesting other cells seems to provide a survival advantage to the host cell when challenged by UV radiation, indicating that nutrients scavenged from an internalized cell can provide some protection from various stressors to the host [56]. Currently, it remains unknown whether non-transformed cells in vivo exhibit the same increase of entosis in response to UV radiation. This remains an important field of study as UV induced entosis or normal cells could play a role in early cancer cell transformation and genomic instability.
3.3. Mechanisms of Cell Fusion
The process of cell fusion can be broken down into five major steps: cell priming, chemotaxis, adhesion, fusion, and post-fusion (Figure 4) [57]. In general, these steps are critical in order for the fusing cells to overcome the barrier posed by the integrity of the phospholipid bilayer membranes of the cells’ surfaces. Cell fusion is important for several biological processes in normal physiology including fertilization, muscle development, skeletal remodeling through the generation of multinucleated osteoclasts, and placentation [58,59]. However, when this process becomes dysregulated it can lead to the development of cancer [60,61].
3.3.1. Priming
To undergo cell fusion, first the cells must go through a priming phase that prepares the machinery necessary for successful fusion to occur. This involves making modifications to the lipid composition of the cell membrane by translocating the inner-leaflet lipids including phosphatidylserine to the cell surface [57]. During fertilization, it is believed that exposed phosphatidylserine (PS) localized to the anterior acrosomal region of sperm is critical for the membrane destabilization process that allows penetration of the zona pellucida [57,62]. Next, the architecture of the cell membrane is altered to make it more fluid through the depletion of cholesterol and filipin in an albumin dependent manner [63]. While the significance of PS in driving cancer cell fusion is not well understood, early studies have shown that it is likely necessary [33,64,65].
3.3.2. Chemotaxis
The role of chemotaxis in driving cancer cell fusion has not been studied, however sperm, myoblasts, macrophages, and other cells rely on the secretion of chemoattractants to find competent fusion partners [57]. There are many chemoattractants that can facilitate cell fusion, however the most relevant to cancer are platelet-derived growth factor (PDGF), tumor necrosis factor alpha (TNF-α), epidermal growth factor (EGF), and interleukin-4 (IL-4) [66,67,68,69,70,71]. More research is needed to determine if these chemokines play a role in promoting the fusion of cancer cells.
3.3.3. Adherence
For cells to undergo cell fusion they must be in physical contact with one another. There are several cell surface adhesion proteins that promote adhesion and recognition between the two cells including cadherins, tetraspanins, integrins, and immunoglobulin super family members.
Cadherins that participate in homotypic cell-cell contact have been shown to have a role in many types of cell fusion. Researchers have shown that treatment of macrophages with anti-E-cadherin antibodies blocks the formation of multinucleated giant cells (MGC) [72]. MGCs are formed through the fusion of macrophages and play an important role in bone remodeling and innate immunity [73]. During osteoclastogenesis, E-cadherin expression by osteoclasts can influence gene expression to promote migration and fusion [74]. While E-cadherin expression generally suppresses tumor formation in normal cells, its dysregulation has been shown to lead to carcinogenesis [75]. It is possible that dysregulation of E-cadherin can drive cancer cell fusion and malignancy, however a direct connection between E-cadherin and cancer cell fusion has yet to be demonstrated.
Tetraspanins are cell surface molecules that can interact with other cell surface proteins like integrins and Ig super family proteins. In mouse models, researchers have shown that the tetraspanins CD9 and CD81 are needed to facilitate fusion of the egg and sperm [76,77,78]. Neutralization of CD9 in macrophages also disrupts the formation of osteoclasts through fusion [79]. In contrast, CD9 and CD81 can limit the fusion of cells in certain circumstances as CD9 and CD81 deficient mice have increased numbers of MGCs during inflammation [80]. Tetraspanins are also known to associate with certain integrins to promote cell fusion. For example, β1 integrin is thought to encourage myoblast and myotube fusion by organizing and controlling the expression of fusion proteins including CD9 [81].
3.3.4. Fusogens and Membrane Pore Formation
The most widely accepted model of cell fusion pore formation begins with the formation of a hemifusion intermediate in which the outer lipid layer of the surface membranes of each fusing cell are the first to merge with each other. During the hemifusion intermediate stage, the outermost leaflets of opposed membranes are connected while the inner leaflets remain intact [82]. Next, tension in the diaphragm formed by the inner leaflets promotes their merging and the subsequent formation of a fusion pore [83]. After the initial pore is formed, it expands and allows the mixing of intracellular contents in a single body. Under normal conditions, the plasma membrane of cells will not spontaneously fuse together due to the high energy required to overcome the forces opposing cell fusion. However, a class of proteins known as “fusogens” have been shown to lower the energy barrier by altering the lipid bilayer and facilitate fusion [84,85]. Fusogens play an important role in mediating viral entry into host cells [83]. They are typically homo- or hetero-oligomeric complexes composed of transmembrane glycoproteins that can be activated by changes in pH [8,86]. Identifying fusongens in mammalian cells has remained challenging with the exception of Myomaker (Mymk) and Myomerger (Mymg). When they are expressed endogenously, these two fusogens have been shown to drive the fusion of activated muscle cells [87,88,89,90]. To date, however, Mymk and Mymg have yet to be definitively implicated in driving cell fusion in any form of cancer.
Syncytin-1, which is a membrane glycoprotein encoded by the human endogenous retrovirus element HER-W, is similar in structure to HIV Env fusogen and is believed to play a role in aberrant cancer cell fusion [57]. In normal cells, syncytin-1 and its homolog syncytin-2 drive the formation of the placental syncytiotrophoblast [91]. However, aberrant expression of syncytin-1 has been implicated in promoting cell fusion in endometrial carcinoma and it may have a tumorigenic role in non-small cell lung cancer (NSCLC), breast cancer, neuroblastoma and testicular cancer [92,93,94,95]. Syncytin-1 promotes cell fusion by binding to the receptors ASCT1 or ASCT2, which are also overexpressed in some cancers[96,97].
3.3.5. Post-Fusion Recovery
After fusion, cells must undergo a period of resetting to either prevent themselves from undergoing another fusion event or prepare to fuse again [31]. One of the first problems the new cell must address is the surplus of plasma membrane that can result in suboptimal cell surface tension [57]. Myoferlin is a member of the ferlin family of proteins and it is highly expressed in myoblasts undergoing [98]. Myoferlin is responsible for promoting the endocytic recycling of excess plasma membrane in an EHD2-dependent manner [99]. The anti-apoptosis proteins Bcl-2 and c-Flip are also believed to be upregulated in recently-fused cells to prevent unwanted cell death [100,101].
3.4. Mechanisms of Tunneling Nanotubes and Tumor Microtubes
3.4.1. Differences Between TNTs and TMs
Cancer cells form TNTs and TMs with cell types including immune cells and other cancer cells. TMs and TNTs are similar in function and are differentiated through their size and width. TMs are typically longer (500 µM) and have larger bore size (1-2 µM) compared to TNTs (100 µM length and less than 1 µM width) [39,102,103]. Additionally, branching and multiple connections are more frequently observed in TNTs. TMs and TNTs are both sheathed in the host cell membrane [103]. Due to their thinness, TNTs have also been observed to be more fragile compared to TMs, which can survive multiple washes and buffer replacements during immunofluorescence labeling [7,104]. Both have been observed to form between cancer cells in vitro and in vivo [6,36,105]. TMs can oftentimes be visualized using brightfield or phase contrast microscopy whereas TNTs typically require higher fidelity imaging techniques such as scanning electron microscopy (SEM) [7]. However, TMs and TNTs are functionally similar and are both utilized by tumor cells to adjoin with other cells for the purposes of trafficking functional fragments of protein, nucleic acids, and subcellular structures.
3.4.2. The Molecular Mechanisms of TNT and TM Formation and Structure
In normal physiology, expression of p53 is essential for TNT formation between astrocytes [106]. However, a hallmark of many tumors is the loss of p53 function, signifying that tumors may rely on an alternative mechanism to drive the formation of TNTs/TMs [107]. Additionally, it is also currently unknown whether the mechanisms that drive TMs and TNT formation are conserved. Formation of both TNTs and TMs in tumors are driven by actin polymerization [38,106,108,109]. Treatment with drugs that promote F-actin depolymerization such as Latrunculin B also prevent TNT and TMs formation and promote the collapse of preexisting TNTs and TMs [104].
Tumor necrosis factor ɑ-induced protein (TNFAIP2) has been implicated in TNT formation through its interaction with RalA, a GTPase belonging to the Ras superfamily of proteins [110,111,112]. RalA interacts with guanosine-5’-triphosphate (B-GTP), a binding interaction catalyzed by 3-phosphoinositide-dependent kinase 1 (PDK1) [113,114]. The RalA/B-GTP complex then binds to RalA Binding Protein 1 (RALBP1), activating Rac1/RhoA-mediated actin polymerization [115]. The exocyst complex, an octomeric protein complex consisting of Exo70, Exo80, Sec3, Sec5, Sec6, Sec8, Sec10, and Sec15, has also been implicated in the formation of nanotubes [110,116]. It is believed that RhoA-mediated F-actin synthesis downstream of the exocyst complex, which itself is a downstream effector of TNFAIP2-RalA interactions, can also induce nanotube formation. Direct inhibition of TNFAIP2 via RNA interference (RNAi) and inhibition of TNFAIP2 and Ral interactions led to decreased de novo TNT synthesis [117,118].
Connections formed between a TNT or TM and a cell rely on the expression of Connexin 43 (Cx43), a protein that forms gap junctions [119,120]. Gap junctions formed as a result of Cx43 expression facilitate the movement of cellular components into and out of the TNTs and TM [121]. Cx43 may also further induce TNT and TM formation, as its expression also stimulates PI3K signaling, leading to further downstream actin polymerization. Beyond p53 and PI3K, overexpression of the mutant KRASG12D/G13D oncogene has led to increased formation of TNTs and TMs [6]. These signaling pathways also stimulate TNFAIP2 function, signifying that there is no one master regulator of TNT and TM formation as a potential therapeutic target.
To summarize, many of the mechanisms that drive TNT and TM formation are regulated by de novo F-actin synthesis, similar to phagocytosis and entosis (Figure 5) [23,24,122]. Although TNT and TM development does not require physical contact between two cells, they are considered to be very fragile and cannot be formed through physical barriers or pores [7]. For example, Saha et al. in 2022 investigated the transfer of mtDNA from T cells and breast tumor cells and established that TNTs failed to form between T cells and cancer cells when separated by a transwell [7]. This study also demonstrated the fragility of the nanotubes as evidenced by images captured via field emission scanning electron microscopy (FESEM) [7]. TNTs and TMs are more prone to breakage resulting in the incomplete transfer of components between two cells when compared to transport with nanotubes shorter in length [7,39,102,103]. The current hypothesis of the molecular mechanisms that drive TNT formation is that cooperative interaction between TNFAIP2 and RalA drives the recruitment of the exocyst complex, which in turn elevates de novo synthesis of long, thin F-actin filopodia connecting two or more cells. Although it is unknown whether TNFAIP2 also drives the formation of TMs, they are similar structurally and in terms of function when compared to TNTs, suggesting that the pathways that drive their formation are conserved.
3.5. Similarities Between the Mechanisms That Drive Phagocytic Cup Formation and Other Cell-Cell Trafficking
3.5.1. Recognition of Target Cells and Particles Flagged for Phagocytosis
Phagocytosis is a programmed cellular function that results in the engulfment and complete lysis of a cell or particle. Although phagocytosis is not considered a method of cell-cell trafficking, there are similarities between the mechanisms that drive phagocytosis and the cell-cell trafficking methods previously highlighted.
Professional phagocytes mainly consist of innate immune cells such as macrophages, neutrophils, and monocytes [123]. They are recruited to the site of trauma over short- and long-range through chemoattractants, including complement activators C3a and C5a, which are secreted by tissue resident cells [124]. Professional phagocytes express unique “eat-me” or phagocytic receptors that recognize pathogen associated molecular patterns (PAMPs) and/or opsonin receptors that detect antibodies, extracellular proteins, and other “tags” that signal the target cell for engulfment. Professional phagocytes also express “don’t-eat-me” receptors such as signal regulatory protein ɑ (SIRPɑ), which can attenuate phagocytic activity upon recognition of ligands such as CD47 [125,126].
3.5.2. Formation of the Phagocytic Cup Scaffolding
The process of phagocytosis formally initiates upon phagocytic or opsonin receptor binding to an “eat-me” ligand [127]. A commonly recognized ligand is PS, a typical marker for cells undergoing apoptosis [128,129]. Upon ligand recognition, depolymerization of the phagocyte’s cortical actin cytoskeleton occurs at the site of receptor-ligand interaction [130,131]. Subsequently, an arm-like membrane protrusions known as pseudopod then begins to protrude outwards at the site of cytoskeletal disruption [132]. The formation of pseudopodia is universally driven by the nucleation of actin monomers via Arp2/3 activation to form and lengthen strands of F-actin. Induction of Arp2/3 in phagocytosis varies (Figure 6A). For example, FcγR recognition of opsonins activates Arp2/3 via induction WASP and N-WASP while phagocytic receptor recognition of C3a/C5a induces Arp2/3 via the Rho/Rac signaling cascade [124,133].
Upon reaching the target cell, the pseudopod then diverts at the base of the target cell into two pseudopodia, leading to the formation of the nascent end of an invagination of the phagocyte cell membrane known as the phagocytic cup (Figure 2B) [134,135]. Further development of the phagocytic cup occurs as F-actin and pseudopodia form adjacent to the side of the target cell as a result of actin nucleation via sustained Arp2/3 signaling and lengthening by myosin II motor proteins (Figure 6C) [136,137]. Concurrent with pseudopodia extension, a F-actin also forms perpendicular to the base of the phagocytic cup (Figure 6D) [137]. This contractile ring then migrates from the nascent to cortical end of the phagocytic cup as pseudopodia lengthening occurs, squeezing out any extra-particle fluids and ensuring that the pseudopodia conform to the surface of the target cell [138]. The contractility of the ring is primarily driven by the enzyme myosin light chain kinase, which powers the myosin II and IXb motor proteins to continually contract and relax the ring (Figure 6E) [136,137,139]. The transition between the phagocytic cup to a complete phagosome occurs when the cortical end of the target cell becomes fully engulfed upon the successful fusion of the pseudopodia alongside the polymerization of F-actin via increased PI3K signaling (Figure 6F) [140]. The phagosome is then endocytosed by the phagocyte and fused to endosomes and lysosomes to undergo lysis [141].
3.5.3. Conservation Between the Mechanism of Phagocytic Cup Formation and Other Methods of Cell-Cell Trafficking
As described in the above sections, the signaling cascade pathways described in each cell-cell trafficking mechanism all rely on actin polymerization to form pseudopodia or invadopodia, with the exception of cell fusion. There are some differences in upstream inducers of each cell-cell trafficking mechanism, such as TNFAIP2-mediated formation of TNTs and TMs [117]. However, activation of these pathways all ultimately feed into some form of Cdc42/Rho/Rac-mediated nucleation of actin monomers and subsequent F-actin polymerization. While some obvious differences between mechanisms can be observed, such as the need for full cell-cell contact in entosis versus two cells only needing to be nearby for the formation of TNTs and TMs, those differences are increasingly unclear when comparing the mechanisms of trogocytosis and entosis, for example.
It is believed by some that trogocytosis is the result of an incomplete phagocytic cup formation. Krendel & Gauthier visualized the “wringing” of a target particle driven by many of the same myosin motors found on the F-actin ring that initially forms at the base of a phagocytic cup and defined this partial transfer as trogocytosis [135]. The contractile forces generated by this ring is hypothesized to have partially sheared the target particle due to an excess of protrusive and contractile forces. These contractile forces were also paired with the early termination of filopodia formation, essentially resulting in an early termination or closure of what would normally be a phagosome (Figure 6G) [135]. However, it is important to note that the definition of trogocytosis is specifically the partial transfer of membrane or membrane proteins. Krendel & Gauthier, only observed this interaction while using a fluorescent PAGE bead as a surrogate for the donor cell, which ultimately can only determine that there was the partial transfer of a particle and not transfer of membrane or functional membrane proteins [135]. Until this mechanism is shown between two living cells, it cannot be construed as the definitive mechanism for trogocytosis.
4. Outcomes of Cell-Cell Trafficking Events in the Context of Cancer
4.1. Trogocytosis
4.1.1. Reprogramming Antitumor Immunity to Alter Tumor Survivability
Trogocytic interactions within the TIME can alter tumor cell survival and influence transcriptomic changes. Poupot et al. were the first to observe lymphocyte trogocytosis of tumor cells in 2005 and concluded that immune cells, specifically γδ-T cells, could trogocytose anaplastic lymphoma cells in vitro [142]. This interaction also resulted in a subset of memory γδ-T cells that could bind to and trogocytose the anaplastic lymphoma cells and increased sensitization of cytotoxic γδ-T cells [142]. Most notably, trogocytosis did not occur between cocultured allogeneic PBMCs from two different patients, thus highlighting how immune cell trogocytosis of tumor cells could increase immune cell function [142]. Trogocytosis may also lead to a direct increase in the cytotoxic capabilities of the trogocyte. For example, NK cells displayed an increased trogocytosis of HER2 from trastuzumab-treated HER2+ breast cancer cells [22]. Acquisition of HER2 by NK cells resulted in increased expression of CD107a/LAMP1, resulting in increased cytoxic function of NK cells [22]. In addition, NK cells may also obtain TYRO3 via trogocytosis to achieve the same effect [143].
In contrast, trogocytosis can also decrease antitumor immunity. Hasim et al. reported in 2022 that NK cells could exogenously express functional PD1 protein after undergoing trogocytosis with double-positive PD1+/PD-L1+ leukemia cells [18]. Tumor cells can also obtain immunoregulatory proteins CTLA4 and Tim3 from immune cells [17]. In both of these cases, the transferred proteins were still functional post trogocytosis and led to a dampened immune response in vivo [17,18]. The transfer of genomic DNA has also been attributed with the process of trogocytosis, though it is currently unknown whether this transfer occurs as a consequence of trogocytosis or is a separate trafficking mechanism that occurs concomitantly [17,20,21]. Trogocytosis of tumor specific antigens (TSA) may also lead to increased immune cell fratricide [19]. In an in vitro and in vivo model of melanoma, T cells obtained class I peptide-MHC (pMHC) from conventional dendritic cells (cDCs) containing a neoantigen produced by melanoma cells [19]. Lymphoid trogocytosis of cDCs led to increased T cell fratricide by CD8+ T cells primed against the trogocytosed pMHC complex, leading to decreased immune response [19].
4.1.2. Trogocytosis-Mediated Impediment of Recombinant Therapeutics
CAR-T cells, a type of T cell with T cell receptors that were engineered ex vivo to detect specific target antigens expressed on tumor cells, are a promising novel immunotherapy [144]. However, despite an initial increase in tumor cell death, recurrence is common post CAR-T cell immunotherapy and stems from the reduced expression of the neoantigen recognized by the CAR [144]. Although the loss of the target antigen is often permanent in recurring tumors, CAR-T cell trogocytosis of tumor cells contributes to a mutable reduction of target antigen expression, resulting in increased CAR-T cell fratricide and decreased antitumor immunity [144,145]. Expression of the target antigen by the cancer cell was not accounted for in the 2022 Pagliano et al. study, a reduction in one specific TSA may not be expected as conventional T cells can be stimulated by a multitude of antigen peptides [19,144,145]. In contrast, CAR-T cells are typically primed against a smaller subset of neoantigens that also may not account for the heterogeneity of the tumor, suggesting that significantly reduced antigen abundance on tumor cells occurs primarily during CAR-T cell trogocytosis [146].
Both conventional and CAR-T cells undergo trogocytosis during TCR/CAR recognition of a neoantigen by an antigen presenting cell (APC), indicating that trogocytosis is associated with an inherent process required for T cell activation [45]. Zhou et al. were able to attenuate CAR-T cell trogocytosis in 2023 by fusing the C terminal domain of CTLA4 to the cytoplasmic tail of the CAR. Endocytosis of CTLA4 is an intrinsic and continuous function of a T cell and leads to the rapid degradation of and recycling of CTLA4 protein [45]. By fusing the C terminal domain of the CAR to CTLA4, the CAR is endocytosed concomitantly with CTLA4, leading to increased recycling of both receptors [45]. This decreased binding of CAR to a ligand led to decreased trogocytosis and less T cell fratricide [45].
In contrast to the findings regarding the immunomodulation, a mechanism by which T cell trogocytosis induces stimulation of adaptive immunity has recently been characterized [147]. CD28 is a receptor protein that recognizes B7 ligands (CD80/86) expressed on APCs [148]. Costimulatory signaling of CD28-B7 is dependent on TCR recognition of MHC peptides [148]. In an article authored in 2023, Xu et al. outlines a mechanism by which T cells can trogocytose CD80/86 ligands in a CD28-dependent fashion [147]. This interaction did lead to the depletion of CD80/86 expression on the APC, which decreased the activation of naive T cells; however, increased autostimulation was observed in T cells [147]. CD28-mediated trogocytosis also occurs independently of TCR-recognition of MHC peptides [147]. Although this study did not investigate trogocytosis induced autostimulation within the context of cancer, it indicates that CD28 trogocytosis of CD80/86 allows for the clonal expansion of specific T cell populations with TCRs primed against a potent neoantigen, which could lead to increased tumor clearance. Reduced availability of CD80/86 on APCs would also decrease CTLA4-CD80/86 interactions, which leads to naive T cell exhaustion and inactivation of the TCR. In contrast, a consequence of increased autostimulation of a small population subset of T cells may also lead to decreased antitumor immunity similar to the limitations in neoantigen recognition observed in CAR-T cells [45,144]. Supporting this hypothesis, blockade of CD28 or CTLA4 decreased regulatory T cell (TReg) autostimulation, suggesting that antitumor immunity may be impeded [147]. However, when combined with therapeutic strategy of fusion the cytoplasmic domains of a CAR and CTLA4, harnessing CD28-mediated trogocytosis of CD80/86 may induce increased clonal expansion of CAR T cells effective against the tumor while decreasing CAR-mediated trogocytic acquisition of MHC peptides, thereby decreasing fratricide.
4.2. Entosis
The role of entosis in tumor development and progression has not been fully characterized. This is largely due to the multifaceted role of entosis in tumor evolution where there is evidence that it can both promote and suppress malignancy. It is believed that entosis can act as a tumor suppressor by eliminating cancer cells that detach from the basement membrane [23,25,149]. At the same time, however, entosis can promote cancer progression by contributing to genomic instability and aneuploidy of host cells, supplying nutrients to malignant hosts and shielding invading cells from both immune surveillance and anti-cancer drugs [54,150,151,152,153]. Given that increased entosis correlates with increased stage of disease and worse patient outcome, it is thought that entosis is generally beneficial for tumor growth [28,150]. Instances of a cell-in-cell phenotype have been observed in many different types of malignancies including small cell lung carcinoma, ductal breast carcinoma and other breast tumors, renal cell carcinoma, colorectal cancer, and pancreatic ductal adenocarcinoma to name a few [154,155,156,157,158,159]. Interestingly, though we will not be going into further detail here, entosis has also been implicated in the pathogenesis of non-cancerous diseases such as microcephaly [160].
Aneuploidy, otherwise known as an imbalanced number of whole chromosomes or chromosomal arms, is known to be a major driver of tumor development and progression [161,162]. Entosis is known to contribute to aneuploidy in human breast tumors by disrupting normal cytokinesis of the host cell [150]. This can occur when an engulfed cell blocks the cleavage furrow of its dividing host. Instead of undergoing normal cell division, it is possible for the host cell to integrate the internalized cell’s nucleus resulting in a binucleated cell [150]. If the binucleated cell attempts to undergo division again, it often promotes aneuploidy through uneven chromosome distribution and breakage in the daughter cells [163]. In normal cells, p53 and other tumor suppressors help maintain the stability of the genome and prevent aberrant chromosome numbers [164]. However, in cancer it is common for these critical regulators to be lost, allowing aneuploidy because of entosis to perpetuate and contribute to more severe malignancy [165].
Approximately 50% of engulfed cells undergo lysosomal degradation by their host, allowing the engulfing cell can scavenge nutrients such as glucose and amino acids from the corpse of the loser cell [25,54,55]. It is believed that this may be a mechanism by which cancer cells can persist in a high-stress, low-nutrient setting such as the TME. In fact, Hamann et al. have shown that when placed in starvation conditions, MCF-7 cells that ingest their neighbors proliferate 10-fold more frequently than their non-entotic counterparts [55]. In the context of tumor development, entosis could serve as a mechanism by which less-fit individuals are sacrificed to redistribute nutrients to the fittest cells, thus ensuring the survival of the cancer population. This suggests that cells may have some ability to sense fitness through points of contact like adherens junctions, which remains an important topic for further studies.
One of the more novel ways in which entosis contributes to tumor progression is through promoting drug resistance. Drug resistance remains a significant limiting factor when it comes to achieving cures for cancer patients. Although many types of cancers are initially sensitive to certain anti-cancer drugs and therapies, as the disease progresses, they often acquire resistance to these interventions and become more severe [166]. Recently, researchers have shown that entosis can contribute to this problem by allowing cancer cells to shield themselves from anti-cancer therapies by “hiding” inside other cells [51,153,167]. Several different anti-cancer drugs have been implicated in causing the formation of CICs including FOLFOX and FOLFRI regimens, taxanes like Paclitaxel, and nintedanib. In addition to causing drug resistance, the induction of entosis through anti-cancer treatments has been shown to promote aneuploidy in drug-resistant populations [167].
4.3. Cell Fusion
Genomic instability is a characteristic of many types of cancers [168]. Cell fusion can contribute to this problem by causing polyploidy, or the addition of an extra set of chromosomes, in the final fused cell [60]. The new hybrid cell may acquire the biological characteristics of its parent cells or develop new ones that can promote stronger colony formation, migration, proliferation, and survival abilities [169,170].
Cell fusion has also been shown to play an important role in facilitating metastasis to distant organs. Metastasis is a complex process that requires a cancer cell to overcome many barriers they don’t typically face in the primary site such as detaching from their point of origin, surviving in circulation, invading through layers of tissue, and surviving in an unfamiliar microenvironment [61]. Migratory bone marrow-derived cells, such as macrophages, have been shown to participate in cell fusion with tumor cells in mouse models of melanoma and breast cancer and are theorized to impart the characteristics needed to successfully metastasize on the resulting daughter cell [169,171]. It is also possible that fusion between a cancer and non-cancerous cell in the TME could result in a daughter cell that is better adapted for surviving in a new environment compared to its parent cells.
Cancer stem cells (CSCs) are a relatively rare subset of cancer cells that act as a reservoir of self-sustaining cells and are capable of self-renewing and maintaining the tumor [172]. CSCs have been identified in a wide range of both liquid and solid tumors including multiple myeloma, pancreatic cancer, breast cancer, leukemia and glioblastoma to name a few [173,174,175,176,177]. Several groups have suggested that cell fusion may be a mechanism by which CSCs are created, whether through fusion between somatic and stem cells or normal cells and tumor cells [178,179,180,181,182]. Cell fusion often results in daughter cells that are more tumorigenic than their parent cells, sometimes exhibiting CSC characteristics [178,183]. These fusion events could also help explain intratumoral heterogeneity. The daughter cell that results from cell fusion can have a unique combination of traits from its parent cells that may result in a novel CSC phenotype that otherwise would not have arisen in the tumor.
Drug resistance is another significant challenge to overcome when treating cancer. Researchers have shown that cell fusion can contribute to drug resistance, either through the fusion of a non-resistant and resistant cell or even non-resistant tumor cells with bone marrow derived cells (BMDCs) [184]. Recently, it was even shown that fusion between cancer cells and nearby muscle cells enhances the drug resistance of prostate tumors [64]. A possible mechanism for how cell fusion can contribute to drug resistance is through the formation of polyploid giant cancer cells when exposed to certain chemotherapies [185,186]. It has also been theorized that mitochondrial abnormalities as a result of cytoplasmic fusion may also contribute to drug resistance, however more research is needed to confirm this notion [187,188].
4.4. Tunneling Nanotubes and Microtubes
4.1.1. Transfer of Nucleic Acids
Within the context of cancers, TNTs and MTs are distinct from previous trafficking methods in that it only requires two cells to be nearby and does not require physical contact [36]. However, the transfer of cellular components when facilitated by TNTs and MTs extends beyond membrane proteins and fragments, such as the acquisition of mitochondria and mtDNA [7,106,108]. Regulation of mtDNA is crucial for maintaining cellular respiration and metabolism. Through TNT- and TM-mediated shuttling, mitochondria and mtDNA can be transferred between two tumor cells, resulting in a tumor cell with increased proliferative capabilities. Additionally, Tumor cells can also siphon mitochondria from other cells within the TME such as mesenchymal stromal cells and platelets, leading to increased tolerance of reactive oxygen species (ROS), an inducer of DNA damage and cellular stress [189,190].
Tumor infiltrating lymphocytes (TILs) are another population which can have their mitochondria and mtDNA hijacked by a tumor cell, resulting in decreased T cell oxygen consumption rate [7]. Fragmentation of mitochondria via antimycin A or rotenone did not decrease the rate of transfer, but did render any transferred fragments unviable [7,191]. Clinically, direct targeting of any aberrant mitochondrial fragments is not therapeutically possible, as the origins of the trafficked mitochondria would need to be ascertained and targeted against [7]. Additionally, any developed therapies would potentially need to be localized, as systemic therapies may ablate cells distal to the tumor, such as other mesenchymal stromal cells not located near the TME.
TNT-mediated trafficking of nucleic acids is not limited to mtDNA, as the transfer of microRNAs (miRNA), a type of long non-coding RNA, are also observed to be trafficked by nanotubes. miR-19A is an oncogenic driver miRNA [192,193]. One of its oncogenic functions include the negative modulation of MHC Class I gene expression, which has been implicated in antitumor immunity. Transfer of miR-19A was observed between two osteoblast cell lines as well as between an osteoblast cell and a non-immortalized normal cell in vitro [193]. Other oncogenic miRNAs such as miR-132 and miR-155 are also transferred via tumor TNT networks and resulted in the oncogenic transcriptional reprogramming of angiogenic and proliferation pathways [192,194]. Viral RNA can also be transferred by nano and microtubes, which contributes to the spread of viral pathogens. This has been observed across multiple species, including human PR8-influenza and HMPV RNA viruses [195,196]. Although the transfer of cancer-causing viral RNA, such as human papillomavirus and epstein-barr virus, has not yet been implicated in malignancies, these observations suggest that replication and dispersion may also be facilitated by TNTs and TMs.
4.1.2. Transfer of Functional Proteins
While much of the current focus of TNTs and TMs is on alteration of cellular respiration via the transfer of mitochondria, an often-underappreciated facet trafficking via TNTs and TMs is the transfer of functional oncogenic proteins. For example, Desir et al. observed TNT-mediated trafficking of mutant KRASG12D and KRASG13D, two constitutively active variants commonly observed in pancreatic, lung, and colorectal cancers, using in vitro cocultures between two distinct colorectal cancer (CRC) cell lines [6]. Increased TNT formation was also associated with elevated KRASG12D/G13D expression [6]. Transfer of both KRASG12D/G13D mutants occurred when the mutation was endogenously expressed by the tumor cell, indicating that the formation and subsequent transfer via TNTs is an innate function of the tumor cell. Transferred KRASG12D/G13D was still functional and increased the expression of phosphorylated ERK (pERK), a downstream effector of activated KRAS. Increased TNT formation was observed in cells that received the mutant KRAS variants and also altered cell morphology, leading to decreased recipient cell size as early as 48 hours post-coculture [6]. Taken altogether, these findings indicate the expansion of a TNT network contributes to the formation of a heterogenous TME and may originate from a small number of oncogene-expressing tumor progenitor cells that then traffic oncogenic protein to induce aberrant signaling in recipient cells.
5. Conclusions
The contact dependent processes of intracellular trafficking of materials such as trogocytosis, entosis, cell fusion, and tunneling nanotubes are starting to be recognized as important mechanisms by which cancer cells can mold the TIME to their advantage. These events can promote improved tumor cell immune evasion, drug resistance, intratumoral heterogeneity, metastasis, and many other pro-tumor processes that can lead to more severe disease outcomes. As the field of immunotherapy moves forward, it is critical that these contact-dependent trafficking events be researched in further detail. The discovery of how trogocytosis can decrease the efficacy of CAR-T cell therapy or the ability of cancer cells to avoid anti-cancer drugs by hiding in their neighbors through entosis are just a few examples of how these processes will have serious implications for the next generation of immunotherapies. Additionally, through their contributions to genomic instability, contact-dependent trafficking may play an unexpected role in initiating tumorigenesis. Further research is needed to fully elucidate the role these processes play, both in creating cancer cells and destroying them.
Author Contributions
writing—original draft preparation, H.Q.M, A.S.; writing—review and editing, H.Q.M., A.S., A.L.M.B; All authors have read and agreed to the published version of the manuscript.
Acknowledgements
We would like to thank L. Strickland, P. Xie and J. Shin for their review and feedback during the preparation of this manuscript for submission.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TME | Tumor microenvironment |
| TIME | Tumor immune microenvironment |
| CIC | Cell-in-cell |
| TNT | Tunneling nanotubes |
| TM | Tumor Microtubes |
| F-actin | Filamentous actin |
| mtDNA | Mitochondrial DNA |
| EMT | Epithelial-to-mesenchymal transition |
| PI3K | Phosphatidylinositol 3-kinase |
| NK | Natural killer |
| β-PIX | PAK interacting protein |
| CAR-T | Chimeric antigen receptor T cell |
| ROCK | Rho-associated coiled-coil containing kinase |
| UV | Ultraviolet radiation |
| JNK | c-Jun N-terminal kinase |
| PS | Phosphatidylserine |
| PDGF | Platelet-derived growth factor |
| TNF-α | Tumor necrosis factor alpha |
| EGF | Epidermal growth factor |
| IL-4 | Interleukin-4 |
| MGC | Multinucleated giant cells |
| Mymk | Myomaker |
| Mymg | Myomerger |
| NSCLC | Non-small cell lung cancer |
| SEM | Scanning electron microscopy |
| TNFAIP2 | Tumor necrosis factor ɑ-induced protein |
| B-GTP | Guanosine-5’-triphosphate |
| PDK1 | 3-phosphoinositide-dependent kinase 1 |
| RALB | PalA Binding Protein 1 |
| RNAi | RNA interference |
| Cx43 | Connexin 43 |
| FESE | Field emission scanning electron microscopy |
| PAMP | Pathogen associated molecular patterns |
| SIRPα | Signal regulatory protein α |
| TSA | Tumor specific antigens |
| p-MHC | Peptide major histocompatibility complex |
| cDC | Conventional dendritic cell |
| APC | Antigen presenting cell |
| Treg | Regulatory T cell |
| CSC | Cancer stem cell |
| BMDC | Bone marrow derived cell |
| ROS | Reactive oxygen species |
| TILs | Tumor infiltrating lymphocytes |
| miRNA | microRNA |
| CRC | Colorectal cancer |
| pERK | Phosphorylated ERK |
References
- Dröge, M.; Pühler, A.; Selbitschka, W. Horizontal gene transfer as a biosafety issue: a natural phenomenon of public concern. J Biotechnol 1998, 64, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Weber, F.A.; Kornberg, T.B. Cytonemes: cellular processes that project to the principal signaling center in Drosophila imaginal discs. Cell 1999, 97, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J Biol Chem 1946, 166, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, K.; Yang, M.; Jiang, P.; Xu, M.; Yamamoto, N.; Tsuchiya, H.; Tomita, K.; Moossa, A.R.; Bouvet, M.; Hoffman, R.M. Development of real-time subcellular dynamic multicolor imaging of cancer-cell trafficking in live mice with a variable-magnification whole-mouse imaging system. Cancer Res 2006, 66, 4208–4214. [Google Scholar] [CrossRef]
- Cheng, X.T.; Xie, Y.X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 2018, 14, 1472–1474. [Google Scholar] [CrossRef]
- Desir, S.; Wong, P.; Turbyville, T.; Chen, D.; Shetty, M.; Clark, C.; Zhai, E.; Romin, Y.; Manova-Todorova, K.; Starr, T.K.; et al. Intercellular Transfer of Oncogenic KRAS via Tunneling Nanotubes Introduces Intracellular Mutational Heterogeneity in Colon Cancer Cells. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nature Nanotechnology 2022, 17, 98–106. [Google Scholar] [CrossRef]
- White, J.M. Membrane fusion. Science 1992, 258, 917–924. [Google Scholar] [CrossRef]
- Cone, R.E.; Sprent, J.; Marchalonis, J.J. Antigen-binding specificity of isolated cell-surface immunoglobulin from thymus cells activated to histocompatibility antigens. Proc Natl Acad Sci U S A 1972, 69, 2556–2560. [Google Scholar] [CrossRef]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat Immunol 2003, 4, 815. [Google Scholar] [CrossRef]
- Huang, J.-F.; Yang, Y.; Sepulveda, H.; Shi, W.; Hwang, I.; Peterson, P.A.; Jackson, M.R.; Sprent, J.; Cai, Z. TCR-Mediated Internalization of Peptide-MHC Complexes Acquired by T Cells. Science 1999, 286, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Hudrisier, D.; Joly, E. Plasma membrane nibbling: all lymphocytes do it, but why. ELSO Gaz 2002, 9, 1–5. [Google Scholar]
- Olivera-Valle, I.; Latorre, M.C.; Calvo, M.; Gaspar, B.; Gómez-Oro, C.; Collazos, A.; Breton, A.; Caballero-Campo, P.; Ardoy, M.; Asensio, F.; et al. Vaginal neutrophils eliminate sperm by trogocytosis. Hum Reprod 2020, 35, 2567–2578. [Google Scholar] [CrossRef]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications 2018, 9, 1228. [Google Scholar] [CrossRef]
- Ralston, K.S.; Solga, M.D.; Mackey-Lawrence, N.M.; Somlata, ᅟ. ; Bhattacharya, A.; Petri Jr, W.A. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature 2014, 508, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Steele, S.; Radlinski, L.; Taft-Benz, S.; Brunton, J.; Kawula, T.H. Trogocytosis-associated cell to cell spread of intracellular bacterial pathogens. Elife 2016, 5, e10625. [Google Scholar] [CrossRef]
- Shin, J.H.; Jeong, J.; Maher, S.E.; Lee, H.W.; Lim, J.; Bothwell, A.L.M. Colon cancer cells acquire immune regulatory molecules from tumor-infiltrating lymphocytes by trogocytosis. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Hasim, M.S.; Marotel, M.; Hodgins, J.J.; Vulpis, E.; Makinson, O.J.; Asif, S.; Shih, H.Y.; Scheer, A.K.; MacMillan, O.; Alonso, F.G.; et al. When killers become thieves: Trogocytosed PD-1 inhibits NK cells in cancer. Sci Adv 2022, 8, eabj3286. [Google Scholar] [CrossRef]
- Pagliano, O.; Morrison, R.M.; Chauvin, J.M.; Banerjee, H.; Davar, D.; Ding, Q.; Tanegashima, T.; Gao, W.; Chakka, S.R.; DeBlasio, R.; et al. Tim-3 mediates T cell trogocytosis to limit antitumor immunity. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Marcarian, H.Q.; Sivakoses, A.; Arias, A.M.; Ihedioha, O.; Bishop, M.C.; Lee, B.R.; Bothwell, A.L.M. Renal cancer cells acquire immune surface protein through trogocytosis and horizontal gene transfer. bioRxiv 2024. [Google Scholar] [CrossRef]
- Sivakoses, A.; Marcarian, H.Q.; Arias, A.M.; Lam, A.R.; Ihedioha, O.C.; Santamaria, J.A.; Gurtner, G.C.; Bothwell, A.L.M. Triple Negative Breast Cancer Cells Acquire Lymphocyte Proteins and Genomic DNA During Trogocytosis with T Cells. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kataoka, T.R.; Hirata, M.; Kawaguchi, K.; Nishie, M.; Haga, H.; Toi, M. Trogocytosis-mediated expression of HER2 on immune cells may be associated with a pathological complete response to trastuzumab-based primary systemic therapy in HER2-overexpressing breast cancer patients. BMC Cancer 2015, 15, 39. [Google Scholar] [CrossRef]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef]
- Garanina, A.S.; Kisurina-Evgenieva, O.P.; Erokhina, M.V.; Smirnova, E.A.; Factor, V.M.; Onishchenko, G.E. Consecutive entosis stages in human substrate-dependent cultured cells. Scientific Reports 2017, 7, 12555. [Google Scholar] [CrossRef]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cibas, E.S.; Huang, H.; Hodgson, L.; Overholtzer, M. Induction of entosis by epithelial cadherin expression. Cell Research 2014, 24, 1288–1298. [Google Scholar] [CrossRef]
- Schenker, H.; Büttner-Herold, M.; Fietkau, R.; Distel, L.V. Cell-in-cell structures are more potent predictors of outcome than senescence or apoptosis in head and neck squamous cell carcinomas. Radiation Oncology 2017, 12, 21. [Google Scholar] [CrossRef]
- Schwegler, M.; Wirsing, A.M.; Schenker, H.M.; Ott, L.; Ries, J.M.; Büttner-Herold, M.; Fietkau, R.; Putz, F.; Distel, L.V. Prognostic Value of Homotypic Cell Internalization by Nonprofessional Phagocytic Cancer Cells. Biomed Res Int 2015, 2015, 359392. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Saeki, Y. Osteoclast cell fusion: mechanisms and molecules. Mod Rheumatol 2008, 18, 220–227. [Google Scholar] [CrossRef]
- Vjugina, U.; Evans, J.P. New insights into the molecular basis of mammalian sperm-egg membrane interactions. Front Biosci 2008, 13, 462–476. [Google Scholar] [CrossRef]
- Rochlin, K.; Yu, S.; Roy, S.; Baylies, M.K. Myoblast fusion: When it takes more to make one. Developmental Biology 2010, 341, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Helming, L.; Gordon, S. The molecular basis of macrophage fusion. Immunobiology 2007, 212, 785–793. [Google Scholar] [CrossRef]
- Dittmar, T.; Hass, R. Intrinsic signalling factors associated with cancer cell-cell fusion. Cell Commun Signal 2023, 21, 68. [Google Scholar] [CrossRef]
- Miroshnychenko, D.; Baratchart, E.; Ferrall-Fairbanks, M.C.; Velde, R.V.; Laurie, M.A.; Bui, M.M.; Tan, A.C.; Altrock, P.M.; Basanta, D.; Marusyk, A. Spontaneous cell fusions as a mechanism of parasexual recombination in tumour cell populations. Nat Ecol Evol 2021, 5, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J Cell Sci 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Jana, A.; Ladner, K.; Lou, E.; Nain, A.S. Tunneling Nanotubes between Cells Migrating in ECM Mimicking Fibrous Environments. Cancers 2022, 14, 1989. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One 2012, 7, e33093. [Google Scholar] [CrossRef]
- Gondois-Rey, F.; Miller, T.; Laletin, V.; Morelli, X.; Collette, Y.; Nunes, J.; Olive, D. CD47-SIRPalpha Controls ADCC Killing of Primary T Cells by PMN Through a Combination of Trogocytosis and NADPH Oxidase Activation. Front Immunol 2022, 13, 899068. [Google Scholar] [CrossRef]
- Karczmarczyk, A.; Chojnacki, M.; Paziewska, M.; Karp, M.; Skórka, K.; Zaleska, J.; Purkot, J.; Własiuk, P.; Giannopoulos, K. HLA-G can be transfered via trogocytosis from leukemic cells to T cells in chronic lymphocytic leukemia. Hum Immunol 2024, 85, 111178. [Google Scholar] [CrossRef] [PubMed]
- Aucher, A.; Magdeleine, E.; Joly, E.; Hudrisier, D. Capture of plasma membrane fragments from target cells by trogocytosis requires signaling in T cells but not in B cells. Blood 2008, 111, 5621–5628. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, M.A.; Mor, A. The SLAM family receptors: Potential therapeutic targets for inflammatory and autoimmune diseases. Autoimmun Rev 2018, 17, 674–682. [Google Scholar] [CrossRef]
- Detre, C.; Keszei, M.; Romero, X.; Tsokos, G.C.; Terhorst, C. SLAM family receptors and the SLAM-associated protein (SAP) modulate T cell functions. Semin Immunopathol 2010, 32, 157–171. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, H.; Fang, S.Y.; Chow, R.D.; Tang, K.; Majety, M.; Bai, M.; Dong, M.B.; Renauer, P.A.; Shang, X.; et al. CTLA-4 tail fusion enhances CAR-T antitumor immunity. Nat Immunol 2023, 24, 1499–1510. [Google Scholar] [CrossRef]
- Wan, Q.; Liu, J.; Zheng, Z.; Zhu, H.; Chu, X.; Dong, Z.; Huang, S.; Du, Q. Regulation of myosin activation during cell-cell contact formation by Par3-Lgl antagonism: entosis without matrix detachment. Mol Biol Cell 2012, 23, 2076–2091. [Google Scholar] [CrossRef] [PubMed]
- Kianfar, M.; Balcerak, A.; Chmielarczyk, M.; Tarnowski, L.; Grzybowska, E.A. Cell Death by Entosis: Triggers, Molecular Mechanisms and Clinical Significance. International Journal of Molecular Sciences 2022, 23, 4985. [Google Scholar] [CrossRef]
- Gaptulbarova, K.А.; Tsydenova, I.A.; Dolgasheva, D.S.; Kravtsova, E.A.; Ibragimova, M.K.; Vtorushin, S.V.; Litviakov, N.V. Mechanisms and significance of entosis for tumour growth and progression. Cell Death Discovery 2024, 10, 109. [Google Scholar] [CrossRef]
- Purvanov, V.; Holst, M.; Khan, J.; Baarlink, C.; Grosse, R. G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion. eLife 2014, 3, e02786. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef]
- Durgan, J.; Florey, O. Cancer cell cannibalism: Multiple triggers emerge for entosis. Biochim Biophys Acta Mol Cell Res 2018, 1865, 831–841. [Google Scholar] [CrossRef]
- Yamada, S.; Nelson, W.J. Localized zones of Rho and Rac activities drive initiation and expansion of epithelial cell–cell adhesion. Journal of Cell Biology 2007, 178, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Brugge, J.S. The cell biology of cell-in-cell structures. Nat Rev Mol Cell Biol 2008, 9, 796–809. [Google Scholar] [CrossRef]
- Krajcovic, M.; Krishna, S.; Akkari, L.; Joyce, J.A.; Overholtzer, M. mTOR regulates phagosome and entotic vacuole fission. Mol Biol Cell 2013, 24, 3736–3745. [Google Scholar] [CrossRef]
- Hamann, J.C.; Surcel, A.; Chen, R.; Teragawa, C.; Albeck, J.G.; Robinson, D.N.; Overholtzer, M. Entosis Is Induced by Glucose Starvation. Cell Rep 2017, 20, 201–210. [Google Scholar] [CrossRef]
- Chen, R.; Ram, A.; Albeck, J.G.; Overholtzer, M. Entosis is induced by ultraviolet radiation. iScience 2021, 24, 102902. [Google Scholar] [CrossRef]
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of mammalian cell fusion. Adv Exp Med Biol 2011, 713, 33–64. [Google Scholar] [CrossRef]
- Whitlock, J.M.; Leikina, E.; Melikov, K.; De Castro, L.F.; Mattijssen, S.; Maraia, R.J.; Collins, M.T.; Chernomordik, L.V. Cell surface-bound La protein regulates the cell fusion stage of osteoclastogenesis. Nature Communications 2023, 14, 616. [Google Scholar] [CrossRef]
- Goh, Q.; Millay, D.P. Requirement of myomaker-mediated stem cell fusion for skeletal muscle hypertrophy. Elife 2017, 6. [Google Scholar] [CrossRef]
- Bastida-Ruiz, D.; Van Hoesen, K.; Cohen, M. The Dark Side of Cell Fusion. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.A.; Gadella, B.M. Bicarbonate-induced membrane processing in sperm capacitation. Theriogenology 2005, 63, 342–351. [Google Scholar] [CrossRef]
- Harrison, R.A.; Ashworth, P.J.; Miller, N.G. Bicarbonate/CO2, an effector of capacitation, induces a rapid and reversible change in the lipid architecture of boar sperm plasma membranes. Mol Reprod Dev 1996, 45, 378–391. [Google Scholar] [CrossRef]
- Uygur, B.; Leikina, E.; Melikov, K.; Villasmil, R.; Verma, S.K.; Vary, C.P.H.; Chernomordik, L.V. Interactions with Muscle Cells Boost Fusion, Stemness, and Drug Resistance of Prostate Cancer Cells. Mol Cancer Res 2019, 17, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: a mechanism of breast cancer metastasis. The FASEB Journal 2015, 29, 4036–4045. [Google Scholar] [CrossRef]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef]
- Lafreniere, J.F.; Mills, P.; Bouchentouf, M.; Tremblay, J.P. Interleukin-4 improves the migration of human myogenic precursor cells in vitro and in vivo. Experimental Cell Research 2006, 312, 1127–1141. [Google Scholar] [CrossRef]
- Robertson, M.J.; Manley, T.J.; Donahue, C.; Levine, H.; Ritz, J. Costimulatory signals are required for optimal proliferation of human natural killer cells. J Immunol 1993, 150, 1705–1714. [Google Scholar] [CrossRef]
- Torrente, Y.; El Fahime, E.; Caron, N.J.; Del Bo, R.; Belicchi, M.; Pisati, F.; Tremblay, J.P.; Bresolin, N. Tumor necrosis factor-alpha (TNF-alpha) stimulates chemotactic response in mouse myogenic cells. Cell Transplant 2003, 12, 91–100. [Google Scholar] [CrossRef]
- Allen, D.L.; Teitelbaum, D.H.; Kurachi, K. Growth factor stimulation of matrix metalloproteinase expression and myoblast migration and invasion in vitro. Am J Physiol Cell Physiol 2003, 284, C805–815. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Muneyuki, Y.; Takezawa, Y.; Kino-Oka, M.; Saito, A.; Sawa, Y.; Taya, M. Synergic stimulation of laminin and epidermal growth factor facilitates the myoblast growth through promoting migration. J Biosci Bioeng 2009, 108, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.L.; Mikhailenko, I.; Tondravi, M.M.; Keegan, A.D. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: contribution of E-cadherin. J Leukoc Biol 2007, 82, 1542–1553. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, K.; Vanoppen, M.; Rose, C.D.; Matthys, P.; Wouters, C.H. Multinucleated Giant Cells: Current Insights in Phenotype, Biological Activities, and Mechanism of Formation. Front Cell Dev Biol 2022, 10, 873226. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, C.; Harrison, R.E. E-cadherin is important for cell differentiation during osteoclastogenesis. Bone 2016, 86, 106–118. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit Rev Oncol Hematol 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Le Naour, F.; Rubinstein, E.; Jasmin, C.; Prenant, M.; Boucheix, C. Severely reduced female fertility in CD9-deficient mice. Science 2000, 287, 319–321. [Google Scholar] [CrossRef]
- Miyado, K.; Yamada, G.; Yamada, S.; Hasuwa, H.; Nakamura, Y.; Ryu, F.; Suzuki, K.; Kosai, K.; Inoue, K.; Ogura, A.; et al. Requirement of CD9 on the egg plasma membrane for fertilization. Science 2000, 287, 321–324. [Google Scholar] [CrossRef]
- Rubinstein, E.; Ziyyat, A.; Prenant, M.; Wrobel, E.; Wolf, J.P.; Levy, S.; Le Naour, F.; Boucheix, C. Reduced fertility of female mice lacking CD81. Dev Biol 2006, 290, 351–358. [Google Scholar] [CrossRef]
- Ishii, M.; Iwai, K.; Koike, M.; Ohshima, S.; Kudo-Tanaka, E.; Ishii, T.; Mima, T.; Katada, Y.; Miyatake, K.; Uchiyama, Y.; et al. RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J Bone Miner Res 2006, 21, 965–976. [Google Scholar] [CrossRef]
- Takeda, Y.; Tachibana, I.; Miyado, K.; Kobayashi, M.; Miyazaki, T.; Funakoshi, T.; Kimura, H.; Yamane, H.; Saito, Y.; Goto, H.; et al. Tetraspanins CD9 and CD81 function to prevent the fusion of mononuclear phagocytes. J Cell Biol 2003, 161, 945–956. [Google Scholar] [CrossRef]
- Schwander, M.; Leu, M.; Stumm, M.; Dorchies, O.M.; Ruegg, U.T.; Schittny, J.; Müller, U. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev Cell 2003, 4, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Pérot, P.; Montgiraud, C.; Lavillette, D.; Mallet, F. A Comparative Portrait of Retroviral Fusogens and Syncytins. In Cell Fusions: Regulation and Control, Larsson, L.-I., Ed.; Springer Netherlands: Dordrecht, 2011; pp. 63–115. [Google Scholar]
- Sapir, A.; Avinoam, O.; Podbilewicz, B.; Chernomordik, L.V. Viral and developmental cell fusion mechanisms: conservation and divergence. Dev Cell 2008, 14, 11–21. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Zimmerberg, J.; Kozlov, M.M. Membranes of the world unite! J Cell Biol 2006, 175, 201–207. [Google Scholar] [CrossRef]
- Jahn, R.; Lang, T.; Südhof, T.C. Membrane fusion. Cell 2003, 112, 519–533. [Google Scholar] [CrossRef]
- Earp, L.J.; Delos, S.E.; Park, H.E.; White, J.M. The many mechanisms of viral membrane fusion proteins. Curr Top Microbiol Immunol 2005, 285, 25–66. [Google Scholar] [CrossRef]
- Gibbs, D.E.; Pyle, A.D. Muscle fusogens go viral for gene delivery to skeletal muscle. Cell 2023, 186, 2041–2043. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; O'Rourke, J.R.; Sutherland, L.B.; Bezprozvannaya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 2013, 499, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.E.; Goh, Q.; Kurosaka, M.; Gamage, D.G.; Petrany, M.J.; Prasad, V.; Millay, D.P. Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat Commun 2017, 8, 15665. [Google Scholar] [CrossRef]
- Leikina, E.; Gamage, D.G.; Prasad, V.; Goykhberg, J.; Crowe, M.; Diao, J.; Kozlov, M.M.; Chernomordik, L.V.; Millay, D.P. Myomaker and Myomerger Work Independently to Control Distinct Steps of Membrane Remodeling during Myoblast Fusion. Developmental Cell 2018, 46, 767–780.e767. [Google Scholar] [CrossRef]
- Chen, C.P.; Chen, L.F.; Yang, S.R.; Chen, C.Y.; Ko, C.C.; Chang, G.D.; Chen, H. Functional characterization of the human placental fusogenic membrane protein syncytin 2. Biol Reprod 2008, 79, 815–823. [Google Scholar] [CrossRef]
- Strick, R.; Ackermann, S.; Langbein, M.; Swiatek, J.; Schubert, S.W.; Hashemolhosseini, S.; Koscheck, T.; Fasching, P.A.; Schild, R.L.; Beckmann, M.W.; et al. Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-beta. J Mol Med (Berl) 2007, 85, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fu, Y.; Xia, X.; Zhang, X.; Xiao, K.; Zhuang, X.; Zhang, Y. Knockdown of SP1/Syncytin1 axis inhibits the proliferation and metastasis through the AKT and ERK1/2 signaling pathways in non-small cell lung cancer. Cancer Medicine 2019, 8, 5750–5759. [Google Scholar] [CrossRef]
- Chen, Y.C.; Gonzalez, M.E.; Burman, B.; Zhao, X.; Anwar, T.; Tran, M.; Medhora, N.; Hiziroglu, A.B.; Lee, W.; Cheng, Y.H.; et al. Mesenchymal Stem/Stromal Cell Engulfment Reveals Metastatic Advantage in Breast Cancer. Cell Rep 2019, 27, 3916–3926.e3915. [Google Scholar] [CrossRef]
- Gimenez, J.; Montgiraud, C.; Oriol, G.; Pichon, J.P.; Ruel, K.; Tsatsaris, V.; Gerbaud, P.; Frendo, J.L.; Evain-Brion, D.; Mallet, F. Comparative methylation of ERVWE1/syncytin-1 and other human endogenous retrovirus LTRs in placenta tissues. DNA Res 2009, 16, 195–211. [Google Scholar] [CrossRef]
- Marin, M.; Lavillette, D.; Kelly, S.M.; Kabat, D. N-linked glycosylation and sequence changes in a critical negative control region of the ASCT1 and ASCT2 neutral amino acid transporters determine their retroviral receptor functions. J Virol 2003, 77, 2936–2945. [Google Scholar] [CrossRef]
- Sun, T.; Xiao, X. Targeting ACAT1 in cancer: from threat to treatment. Frontiers in Oncology, 2024. [Google Scholar] [CrossRef]
- Doherty, K.R.; Cave, A.; Davis, D.B.; Delmonte, A.J.; Posey, A.; Earley, J.U.; Hadhazy, M.; McNally, E.M. Normal myoblast fusion requires myoferlin. Development 2005, 132, 5565–5575. [Google Scholar] [CrossRef]
- Doherty, K.R.; Demonbreun, A.R.; Wallace, G.Q.; Cave, A.; Posey, A.D.; Heretis, K.; Pytel, P.; McNally, E.M. The endocytic recycling protein EHD2 interacts with myoferlin to regulate myoblast fusion. J Biol Chem 2008, 283, 20252–20260. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Huppertz, B. The paradox of caspase 8 in human villous trophoblast fusion. Placenta 2010, 31, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ka, H.; Hunt, J.S. FLICE-inhibitory protein: expression in early and late gestation human placentas. Placenta 2006, 27, 626–634. [Google Scholar] [CrossRef]
- Austefjord, M.W.; Gerdes, H.H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun Integr Biol 2014, 7, e27934. [Google Scholar] [CrossRef]
- Ady, J.W.; Desir, S.; Thayanithy, V.; Vogel, R.I.; Moreira, A.L.; Downey, R.J.; Fong, Y.; Manova-Todorova, K.; Moore, M.A.; Lou, E. Intercellular communication in malignant pleural mesothelioma: properties of tunneling nanotubes. Front Physiol 2014, 5, 400. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.; Gondaliya, P.; Patel, T. Tunneling Nanotube-Mediated Communication: A Mechanism of Intercellular Nucleic Acid Transfer. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death & Differentiation 2011, 18, 732–742. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discovery 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Wittig, D.; Wang, X.; Walter, C.; Gerdes, H.H.; Funk, R.H.; Roehlecke, C. Multi-level communication of human retinal pigment epithelial cells via tunneling nanotubes. PLoS One 2012, 7, e33195. [Google Scholar] [CrossRef]
- Bukoreshtliev, N.V.; Wang, X.; Hodneland, E.; Gurke, S.; Barroso, J.F.; Gerdes, H.H. Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett 2009, 583, 1481–1488. [Google Scholar] [CrossRef]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat Cell Biol 2009, 11, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Hanna, S.J.; McCoy-Simandle, K.; Miskolci, V.; Guo, P.; Cammer, M.; Hodgson, L.; Cox, D. The Role of Rho-GTPases and actin polymerization during Macrophage Tunneling Nanotube Biogenesis. Scientific Reports 2017, 7, 8547. [Google Scholar] [CrossRef]
- Ohno, H.; Hase, K.; Kimura, S. M-Sec: Emerging secrets of tunneling nanotube formation. Commun Integr Biol 2010, 3, 231–233. [Google Scholar] [CrossRef]
- Lopez, J.A.; Kwan, E.P.; Xie, L.; He, Y.; James, D.E.; Gaisano, H.Y. The RalA GTPase is a central regulator of insulin exocytosis from pancreatic islet beta cells. J Biol Chem 2008, 283, 17939–17945. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Rusanescu, G.; Hou, W.; Schaffhausen, B.; Feig, L.A. PDK1 mediates growth factor-induced Ral-GEF activation by a kinase-independent mechanism. Embo j 2002, 21, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Goldfinger, L.E.; Ptak, C.; Jeffery, E.D.; Shabanowitz, J.; Hunt, D.F.; Ginsberg, M.H. RLIP76 (RalBP1) is an R-Ras effector that mediates adhesion-dependent Rac activation and cell migration. J Cell Biol 2006, 174, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; Yoo, J.-S.; Hazuka, C.D.; Peterson, K.E.; Hsu, S.-C.; Scheller, R.H. Subunit structure of the mammalian exocyst complex. Proceedings of the National Academy of Sciences 1997, 94, 14438–14443. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, D.; Li, X. The role of mitochondrial transfer via tunneling nanotubes in the central nervous system: A review. Medicine (Baltimore) 2024, 103, e37352. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Luh, F.; Ho, Y.-S.; Yen, Y. Exosomes: a review of biologic function, diagnostic and targeted therapy applications, and clinical trials. Journal of Biomedical Science 2024, 31, 67. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Lurtz, M.M.; Louis, C.F. Intracellular calcium regulation of connexin43. Am J Physiol Cell Physiol 2007, 293, C1806–1813. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nature Communications 2018, 9, 3846. [Google Scholar] [CrossRef]
- Park, H.; Cox, D. Cdc42 regulates Fc gamma receptor-mediated phagocytosis through the activation and phosphorylation of Wiskott-Aldrich syndrome protein (WASP) and neural-WASP. Mol Biol Cell 2009, 20, 4500–4508. [Google Scholar] [CrossRef]
- Brown, G.C. Cell death by phagocytosis. Nat Rev Immunol 2024, 24, 91–102. [Google Scholar] [CrossRef]
- Cockram, T.O.J.; Dundee, J.M.; Popescu, A.S.; Brown, G.C. The Phagocytic Code Regulating Phagocytosis of Mammalian Cells. Front Immunol 2021, 12, 629979. [Google Scholar] [CrossRef]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Lovewell, R.R.; Patankar, Y.R.; Berwin, B. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 2014, 306, L591–603. [Google Scholar] [CrossRef]
- Taban, Q.; Mumtaz, P.T.; Masoodi, K.Z.; Haq, E.; Ahmad, S.M. Scavenger receptors in host defense: from functional aspects to mode of action. Cell Commun Signal 2022, 20, 2. [Google Scholar] [CrossRef]
- Reddien, P.W.; Cameron, S.; Horvitz, H.R. Phagocytosis promotes programmed cell death in C. elegans. Nature 2001, 412, 198–202. [Google Scholar] [CrossRef]
- Johnsen, H.L.; Horvitz, H.R. Both the apoptotic suicide pathway and phagocytosis are required for a programmed cell death in Caenorhabditis elegans. BMC Biology 2016, 14, 39. [Google Scholar] [CrossRef]
- Carlier, M.-F.; Laurent, V.; Santolini, J.; Melki, R.; Didry, D.; Xia, G.-X.; Hong, Y.; Chua, N.-H.; Pantaloni, D. Actin Depolymerizing Factor (ADF/Cofilin) Enhances the Rate of Filament Turnover: Implication in Actin-based Motility. Journal of Cell Biology 1997, 136, 1307–1322. [Google Scholar] [CrossRef]
- Rodal, A.A.; Tetreault, J.W.; Lappalainen, P.; Drubin, D.G.; Amberg, D.C. Aip1p Interacts with Cofilin to Disassemble Actin Filaments. Journal of Cell Biology 1999, 145, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.A.; Aderem, A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J Exp Med 1996, 184, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Dart, A.E.; Donnelly, S.K.; Holden, D.W.; Way, M.; Caron, E. Nck and Cdc42 co-operate to recruit N-WASP to promote FcγR-mediated phagocytosis. J Cell Sci 2012, 125, 2825–2830. [Google Scholar] [CrossRef]
- Omer, S.; Li, J.; Yang, C.X.; Harrison, R.E. Ninein promotes F-actin cup formation and inward phagosome movement during phagocytosis in macrophages. Mol Biol Cell 2024, 35, ar26. [Google Scholar] [CrossRef]
- Krendel, M.; Gauthier, N.C. Building the phagocytic cup on an actin scaffold. Curr Opin Cell Biol 2022, 77, 102112. [Google Scholar] [CrossRef]
- Zhang, W.; Robinson, D.N. Balance of actively generated contractile and resistive forces controls cytokinesis dynamics. Proc Natl Acad Sci U S A 2005, 102, 7186–7191. [Google Scholar] [CrossRef]
- Zang, J.H.; Cavet, G.; Sabry, J.H.; Wagner, P.; Moores, S.L.; Spudich, J.A. On the role of myosin-II in cytokinesis: division of Dictyostelium cells under adhesive and nonadhesive conditions. Mol Biol Cell 1997, 8, 2617–2629. [Google Scholar] [CrossRef]
- Araki, N. Role of microtubules and myosins in Fc gamma receptor-mediated phagocytosis. FBL 2006, 11, 1479–1490. [Google Scholar] [CrossRef]
- Xu, Y.; Pektor, S.; Balkow, S.; Hemkemeyer, S.A.; Liu, Z.; Grobe, K.; Hanley, P.J.; Shen, L.; Bros, M.; Schmidt, T.; et al. Dendritic cell motility and T cell activation requires regulation of Rho-cofilin signaling by the Rho-GTPase activating protein myosin IXb. J Immunol 2014, 192, 3559–3568. [Google Scholar] [CrossRef]
- Schlam, D.; Bagshaw, R.D.; Freeman, S.A.; Collins, R.F.; Pawson, T.; Fairn, G.D.; Grinstein, S. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through Rac/Cdc42 GTPase-activating proteins. Nature Communications 2015, 6, 8623. [Google Scholar] [CrossRef]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev 2016, 273, 156–179. [Google Scholar] [CrossRef]
- Poupot, M.; Pont, F.; Fournié, J.J. Profiling blood lymphocyte interactions with cancer cells uncovers the innate reactivity of human gamma delta T cells to anaplastic large cell lymphoma. J Immunol 2005, 174, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Ma, R.; Li, Z.; Mansour, A.G.; Teng, K.Y.; Chen, L.; Zhang, J.; Barr, T.; Caligiuri, M.A.; Yu, J. Hijacking TYRO3 from Tumor Cells via Trogocytosis Enhances NK-cell Effector Functions and Proliferation. Cancer Immunol Res 2021, 9, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Du, Y.; Li, G.; Yu, M.; Hu, H.; Pan, C.; Wang, D.; Shi, Z.; Yan, X.; Li, X.; et al. Trogocytosis of CAR molecule regulates CAR-T cell dysfunction and tumor antigen escape. Signal Transduct Target Ther 2023, 8, 457. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: promising targets for cancer therapy. Signal Transduction and Targeted Therapy 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Dennett, P.; Zhang, J.; Sherrard, A.; Zhao, Y.; Masubuchi, T.; Bui, J.D.; Chen, X.; Hui, E. CTLA4 depletes T cell endogenous and trogocytosed B7 ligands via cis-endocytosis. J Exp Med 2023, 220. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7–CD28 superfamily. Nature Reviews Immunology 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Qian, Y.; Shi, Y. Natural killer cells go inside: Entosis versus cannibalism. Cell Research 2009, 19, 1320–1321. [Google Scholar] [CrossRef]
- Krajcovic, M.; Johnson, N.B.; Sun, Q.; Normand, G.; Hoover, N.; Yao, E.; Richardson, A.L.; King, R.W.; Cibas, E.S.; Schnitt, S.J.; et al. A non-genetic route to aneuploidy in human cancers. Nat Cell Biol 2011, 13, 324–330. [Google Scholar] [CrossRef]
- Fais, S. Cannibalism: a way to feed on metastatic tumors. Cancer Lett 2007, 258, 155–164. [Google Scholar] [CrossRef]
- Gutwillig, A.; Santana-Magal, N.; Farhat-Younis, L.; Rasoulouniriana, D.; Madi, A.; Luxenburg, C.; Cohen, J.; Padmanabhan, K.; Shomron, N.; Shapira, G.; et al. Transient cell-in-cell formation underlies tumor relapse and resistance to immunotherapy. eLife 2022, 11, e80315. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhang, Y.; Li, S.; Sun, F.; Wang, G.; Yang, T.; Wei, D.; Guo, L.; Xiao, H. Induction of entosis in prostate cancer cells by nintedanib and its therapeutic implications. Oncol Lett 2019, 17, 3151–3162. [Google Scholar] [CrossRef]
- Brouwer, E.; Stegeman, C.A.; Huitema, M.G.; Limburg, P.C.; Kallenberg, C.G. T cell reactivity to proteinase 3 and myeloperoxidase in patients with Wegener's granulomatosis (WG). Clin Exp Immunol 1994, 98, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Abodief, W.T.; Dey, P.; Al-Hattab, O. Cell cannibalism in ductal carcinoma of breast. Cytopathology 2006, 17, 304–305. [Google Scholar] [CrossRef]
- Dziuba, I.; Gawel, A.M.; Tyrna, P.; Machtyl, J.; Olszanecka, M.; Pawlik, A.; Wójcik, C.; Bialy, L.P.; Mlynarczuk-Bialy, I. Homotypic Entosis as a Potential Novel Diagnostic Marker in Breast Cancer. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Kong, Y.; Liang, Y.; Wang, J. Foci of Entotic Nuclei in Different Grades of Noninherited Renal Cell Cancers. IUBMB Life 2015, 67, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, E.; Düssmann, H.; Salvucci, M.; Cavanagh, B.L.; Van Schaeybroeck, S.; Longley, D.B.; Martin, S.J.; Prehn, J.H.M. TRAIL signaling promotes entosis in colorectal cancer. J Cell Biol 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xu, R.; Zhang, H.; Xue, X.; Ruze, R.; Chen, Y.; Yin, X.; Wang, C.; Zhao, Y. Cell-in-Cell-Mediated Entosis Reveals a Progressive Mechanism in Pancreatic Cancer. Gastroenterology 2023, 165, 1505–1521.e1520. [Google Scholar] [CrossRef]
- Sterling, N.A.; Cho, S.H.; Kim, S. Entosis implicates a new role for P53 in microcephaly pathogenesis, beyond apoptosis. Bioessays 2024, 46, e2300245. [Google Scholar] [CrossRef]
- Sdeor, E.; Okada, H.; Saad, R.; Ben-Yishay, T.; Ben-David, U. Aneuploidy as a driver of human cancer. Nat Genet 2024, 56, 2014–2026. [Google Scholar] [CrossRef]
- Lakhani, A.A.; Thompson, S.L.; Sheltzer, J.M. Aneuploidy in human cancer: new tools and perspectives. Trends Genet 2023, 39, 968–980. [Google Scholar] [CrossRef]
- Zhang, T.; Lv, L.; Huang, Y.; Ren, X.; Shi, Q. Chromosome nondisjunction during bipolar mitoses of binucleated intermediates promote aneuploidy formation along with multipolar mitoses rather than chromosome loss in micronuclei induced by asbestos. Oncotarget 2016, 8. [Google Scholar]
- Talos, F.; Moll, U.M. Role of the p53 family in stabilizing the genome and preventing polyploidization. Adv Exp Med Biol 2010, 676, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun Signal 2024, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, I.; Potapov, A.; Ignatova, N.; Bugrova, M.; Shchechkin, I.; Lukina, M.; Shimolina, L.; Kolesnikova, E.; Shirmanova, M.; Zagaynova, E. Cell hiding in colorectal cancer: correlation with response to chemotherapy in vitro and in vivo. Sci Rep 2024, 14, 28762. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Zhang, L.N.; Huang, Y.H.; Zhao, L. Fusion of macrophages promotes breast cancer cell proliferation, migration and invasion through activating epithelial-mesenchymal transition and Wnt/β-catenin signaling pathway. Arch Biochem Biophys 2019, 676, 108137. [Google Scholar] [CrossRef]
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zänker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin Exp Metastasis 2011, 28, 75–90. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer Stem Cells—Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Research 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Matsui, W.; Huff, C.A.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, C.; Brandi, J.; Cecconi, D. Pancreatic cancer stem cells: Perspectives on potential therapeutic approaches of pancreatic ductal adenocarcinoma. World J Stem Cells 2018, 10, 172–182. [Google Scholar] [CrossRef]
- Zhang, X.; Powell, K.; Li, L. Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers 2020, 12, 3765. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Staicu, G.-A.; Sevastre, A.-S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma Stem Cells—Useful Tools in the Battle against Cancer. International Journal of Molecular Sciences 2022, 23, 4602. [Google Scholar]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N Engl J Med 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Dittmar, T.; Hass, R. Extracellular Events Involved in Cancer Cell–Cell Fusion. International Journal of Molecular Sciences 2022, 23, 16071. [Google Scholar] [PubMed]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- He, X.; Li, B.; Shao, Y.; Zhao, N.; Hsu, Y.; Zhang, Z.; Zhu, L. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer 2015, 15, 24. [Google Scholar] [CrossRef]
- Merle, C.; Lagarde, P.; Lartigue, L.; Chibon, F. Acquisition of cancer stem cell capacities after spontaneous cell fusion. BMC Cancer 2021, 21, 241. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, Y.; Park, S.C.; Eun, J.R.; Nguyen, N.T.; Tschudy-Seney, B.; Jung, Y.J.; Theise, N.D.; Zern, M.A.; Duan, Y. CD34(+) Liver Cancer Stem Cells Were Formed by Fusion of Hepatobiliary Stem/Progenitor Cells with Hematopoietic Precursor-Derived Myeloid Intermediates. Stem Cells Dev 2015, 24, 2467–2478. [Google Scholar] [CrossRef]
- Luo, F.; Liu, T.; Wang, J.; Li, J.; Ma, P.; Ding, H.; Feng, G.; Lin, D.; Xu, Y.; Yang, K. Bone marrow mesenchymal stem cells participate in prostate carcinogenesis and promote growth of prostate cancer by cell fusion in vivo. Oncotarget 2016, 7. [Google Scholar]
- Nagler, C.; Hardt, C.; Zänker, K.S.; Dittmar, T. Co-cultivation of murine BMDCs with 67NR mouse mammary carcinoma cells give rise to highly drug resistant cells. Cancer Cell Int 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.J.; Naghdloo, A.; Prabakar, R.K.; Kamal, M.; Cadaneanu, R.; Garraway, I.P.; Lewis, M.; Aparicio, A.; Zurita-Saavedra, A.; Corn, P.; et al. Polyploid cancer cells reveal signatures of chemotherapy resistance. Oncogene 2025, 44, 439–449. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp Biol Med (Maywood) 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Xiang, W.; Xue, B.Z.; Wang, Y.H.; Yi, D.Y.; Jiang, X.B.; Zhao, H.Y.; Fu, P. Cell fusion in cancer hallmarks: Current research status and future indications. Oncol Lett 2021, 22, 530. [Google Scholar] [CrossRef]
- Guan, F.; Wu, X.; Zhou, J.; Lin, Y.; He, Y.; Fan, C.; Zeng, Z.; Xiong, W. Mitochondrial transfer in tunneling nanotubes—a new target for cancer therapy. Journal of Experimental & Clinical Cancer Research 2024, 43, 147. [Google Scholar] [CrossRef]
- Solimando, A.G.; Di Palma, F.; Desantis, V.; Vacca, A.; Svelto, M.; Pisani, F. Tunneling nanotubes between bone marrow stromal cells support transmitophagy and resistance to apoptosis in myeloma. Blood Cancer Journal 2025, 15, 3. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Liu, S.; Xiao, J.; He, Y.; Shao, Z.; Zhang, Y.; Cai, X.; Xiong, L. Tunneling Nanotube-Mediated Mitochondrial Transfer Rescues Nucleus Pulposus Cells from Mitochondrial Dysfunction and Apoptosis. Oxid Med Cell Longev 2022, 2022, 3613319. [Google Scholar] [CrossRef]
- Lu, J.J.; Yang, W.M.; Li, F.; Zhu, W.; Chen, Z. Tunneling Nanotubes Mediated microRNA-155 Intercellular Transportation Promotes Bladder Cancer Cells' Invasive and Proliferative Capacity. Int J Nanomedicine 2019, 14, 9731–9743. [Google Scholar] [CrossRef]
- Harvey, J.D.; Jena, P.V.; Baker, H.A.; Zerze, G.H.; Williams, R.M.; Galassi, T.V.; Roxbury, D.; Mittal, J.; Heller, D.A. A Carbon Nanotube Reporter of miRNA Hybridization Events In Vivo. Nat Biomed Eng 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhu, Q.; Lu, L.; Liu, Y. MiR-132 inhibits migration and invasion and increases chemosensitivity of cisplatin-resistant oral squamous cell carcinoma cells via targeting TGF-β1. Bioengineered 2020, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kim, J.H.; Ranjan, P.; Metcalfe, M.G.; Cao, W.; Mishina, M.; Gangappa, S.; Guo, Z.; Boyden, E.S.; Zaki, S.; et al. Influenza virus exploits tunneling nanotubes for cell-to-cell spread. Scientific Reports 2017, 7, 40360. [Google Scholar] [CrossRef]
- Cifuentes-Muñoz, N.; Branttie, J.; Slaughter, K.B.; Dutch, R.E. Human Metapneumovirus Induces Formation of Inclusion Bodies for Efficient Genome Replication and Transcription. J Virol 2017, 91. [Google Scholar] [CrossRef]
Figure 1.
Graphical abstract depicting the four major contact-dependent cell trafficking processes (Trogocytosis, Entosis, Cell Fusion, and Tunneling Nanotubes) and the potential ways they can influence tumorigenesis and progression. Lines indicate tumor-promoting characteristics that have been previously demonstrated for each process.
Figure 1.
Graphical abstract depicting the four major contact-dependent cell trafficking processes (Trogocytosis, Entosis, Cell Fusion, and Tunneling Nanotubes) and the potential ways they can influence tumorigenesis and progression. Lines indicate tumor-promoting characteristics that have been previously demonstrated for each process.
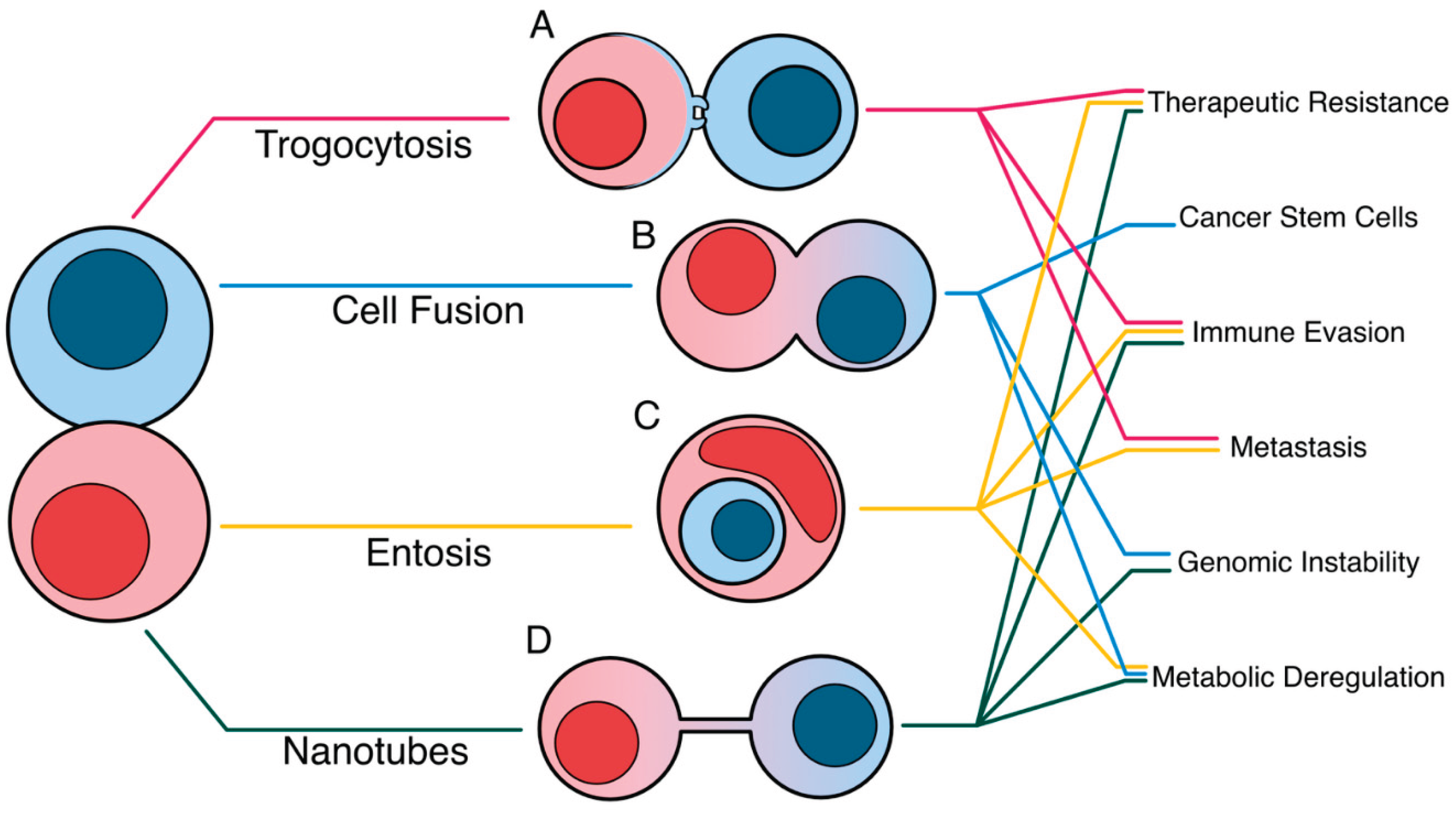
Figure 2.
Overview of the mechanisms of Trogocytosis. Trogocytosis is the transfer of membrane fragment from a donor cell to a recipient cell. When these cells come into contact with each other, F-actin polymerization (grey circles) engages and results in the transfer of functional proteins from the donor cell, such as PD-1. This transfer can result in immunomodulatory effect. Inhibition with PI3K inhibitor wortmannin or F-actin polymerization inhibitor Latrunculin A/B inhibits trogocytosis. Upstream inhibitors of PI3K-mediated ROCK F-actin polymerization, such as SLAM inhibitors may also inhibit trogocytosis.
Figure 2.
Overview of the mechanisms of Trogocytosis. Trogocytosis is the transfer of membrane fragment from a donor cell to a recipient cell. When these cells come into contact with each other, F-actin polymerization (grey circles) engages and results in the transfer of functional proteins from the donor cell, such as PD-1. This transfer can result in immunomodulatory effect. Inhibition with PI3K inhibitor wortmannin or F-actin polymerization inhibitor Latrunculin A/B inhibits trogocytosis. Upstream inhibitors of PI3K-mediated ROCK F-actin polymerization, such as SLAM inhibitors may also inhibit trogocytosis.
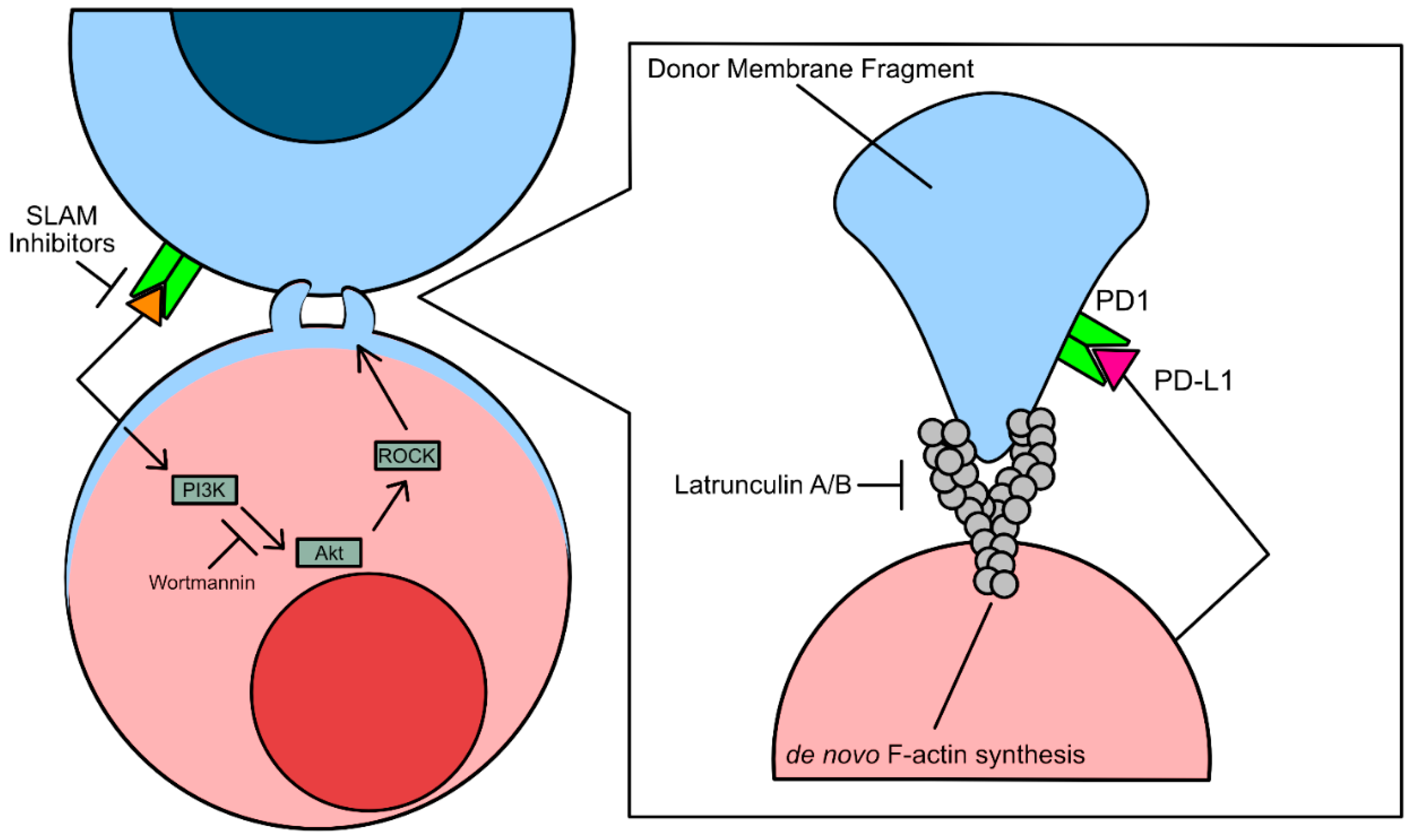
Figure 3.
Overview of the mechanisms of entosis. Entosis is primarily initiated by the detachment of an adherent cell from the surrounding extracellular matrix. When the detached cell encounters an adherent neighbor, there is a difference in stiffness between them. The invading cell is typically stiffer than the adherent cell, which helps facilitate its engulfment. ROCK signaling in the invading cell results in mechanical tension that promotes cellular invasion and entosis. After the loser cell is fully engulfed, it can undergo one of three possible outcomes. The invading cell may divide, be killed through lysosomal degradation, or it can escape its host cell.
Figure 3.
Overview of the mechanisms of entosis. Entosis is primarily initiated by the detachment of an adherent cell from the surrounding extracellular matrix. When the detached cell encounters an adherent neighbor, there is a difference in stiffness between them. The invading cell is typically stiffer than the adherent cell, which helps facilitate its engulfment. ROCK signaling in the invading cell results in mechanical tension that promotes cellular invasion and entosis. After the loser cell is fully engulfed, it can undergo one of three possible outcomes. The invading cell may divide, be killed through lysosomal degradation, or it can escape its host cell.
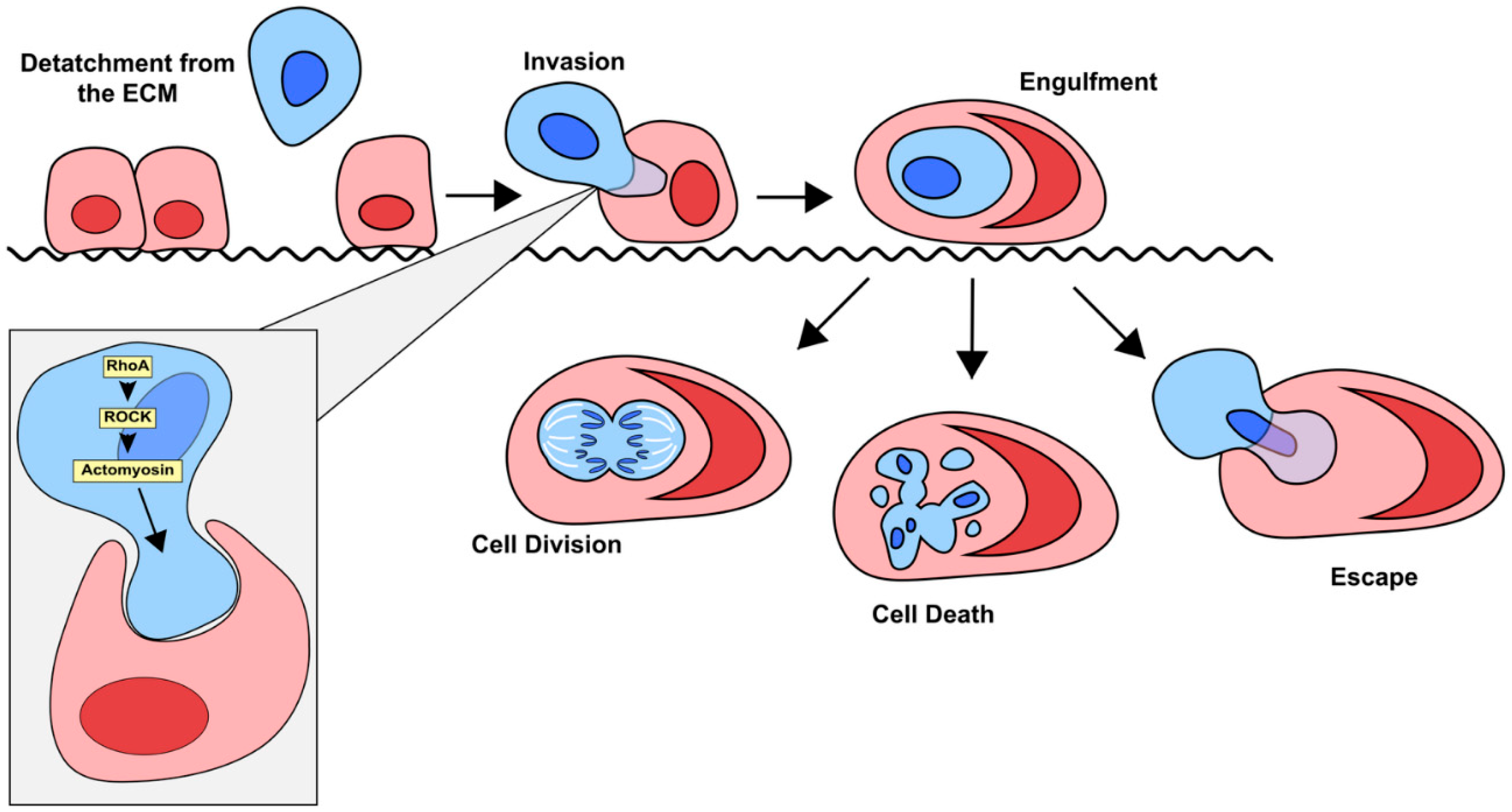
Figure 4.
Overview of the mechanisms of cell fusion. Cell fusion initially begins with a priming phase, in which the fusing cells alter the architecture of their cell membrane and prepare the machinery necessary for fusion to successfully occur. Next, there is a phase of chemotaxis in which chemoattractant signaling between the two fusing cells promotes their migration towards each other. Once the cells are physically in contact, they adhere to one another using cell adhesion molecules such as E-cadherin, tetraspanins, and integrins. After adherence, the cells begin to form a fusion pore. The most popular models suggest that as the cell membranes fuse, they form a hemifusion intermediate in which only the surface leaflets of the bilipid membranes are fused. Tension as a result of this intermediate state results in the fusion of the inner leaflets and the subsequent formation of a membrane pore. This pore then expands to complete the fusion of the two cells into a single body. After fusion, the fused cell then undergoes a recovery phase in which the excess membrane is removed. After fusion, the cell may then exit its fusogenic state or repeat the process with another fusion partner.
Figure 4.
Overview of the mechanisms of cell fusion. Cell fusion initially begins with a priming phase, in which the fusing cells alter the architecture of their cell membrane and prepare the machinery necessary for fusion to successfully occur. Next, there is a phase of chemotaxis in which chemoattractant signaling between the two fusing cells promotes their migration towards each other. Once the cells are physically in contact, they adhere to one another using cell adhesion molecules such as E-cadherin, tetraspanins, and integrins. After adherence, the cells begin to form a fusion pore. The most popular models suggest that as the cell membranes fuse, they form a hemifusion intermediate in which only the surface leaflets of the bilipid membranes are fused. Tension as a result of this intermediate state results in the fusion of the inner leaflets and the subsequent formation of a membrane pore. This pore then expands to complete the fusion of the two cells into a single body. After fusion, the fused cell then undergoes a recovery phase in which the excess membrane is removed. After fusion, the cell may then exit its fusogenic state or repeat the process with another fusion partner.
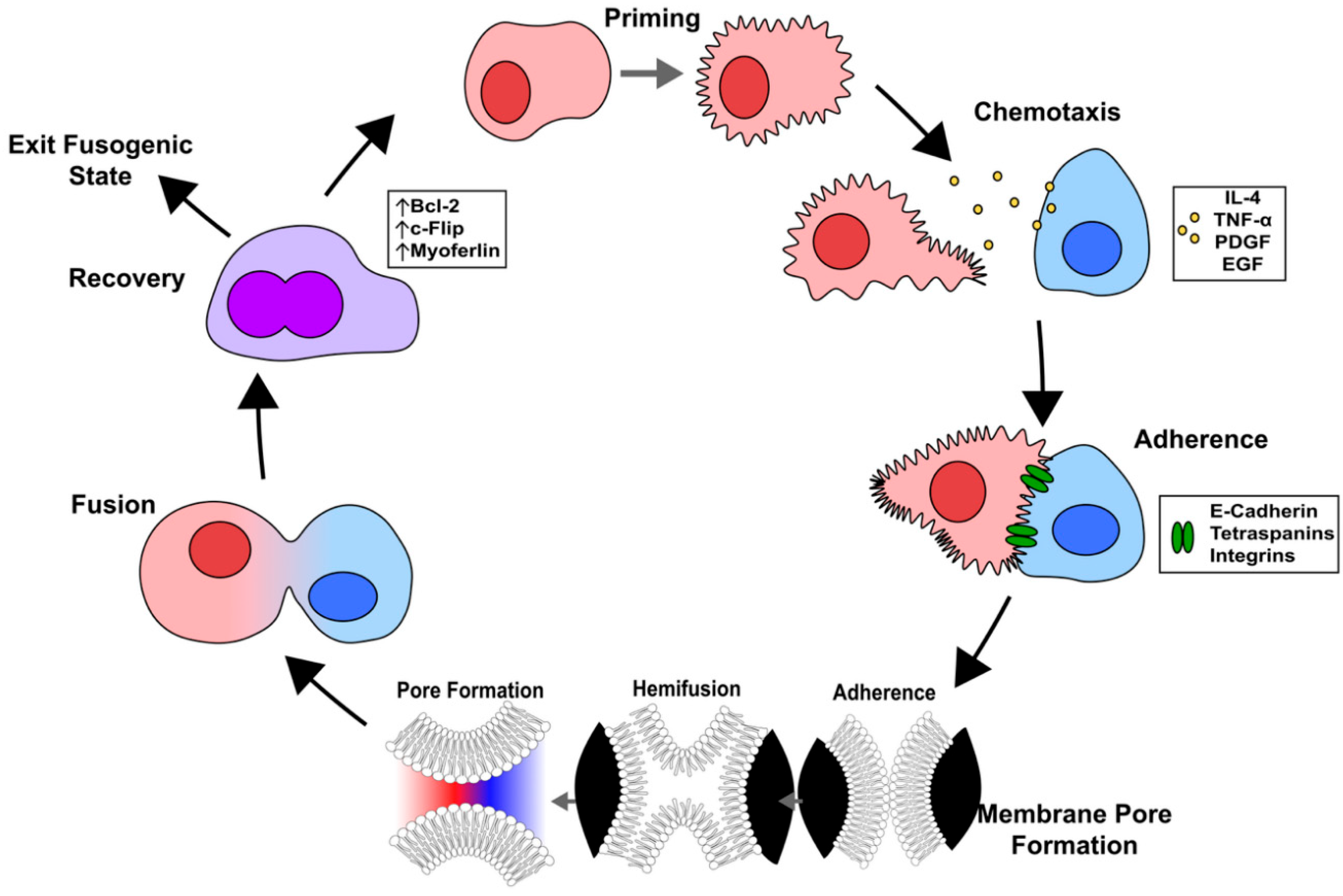
Figure 5.
Overview of the mechanisms of TNT and MT formation. (A) TNTs and MTs form as a result of TNFAIP/RalA protein-protein interactions leading to the recruitment of the exocyst complex proteins. Downstream PI3K and ROCK-mediated F-actin polymerization induces nanotube development. Connexin 43 gap junctions facilitate cargo entrance and exit at either end of the TNT or MT. (B) The cargo of TNTs and MTs can consist of nucleic acids, mitochondrial fragments, and functional proteins. Trafficked cargo can influence the gene and protein expression of the target cells.
Figure 5.
Overview of the mechanisms of TNT and MT formation. (A) TNTs and MTs form as a result of TNFAIP/RalA protein-protein interactions leading to the recruitment of the exocyst complex proteins. Downstream PI3K and ROCK-mediated F-actin polymerization induces nanotube development. Connexin 43 gap junctions facilitate cargo entrance and exit at either end of the TNT or MT. (B) The cargo of TNTs and MTs can consist of nucleic acids, mitochondrial fragments, and functional proteins. Trafficked cargo can influence the gene and protein expression of the target cells.
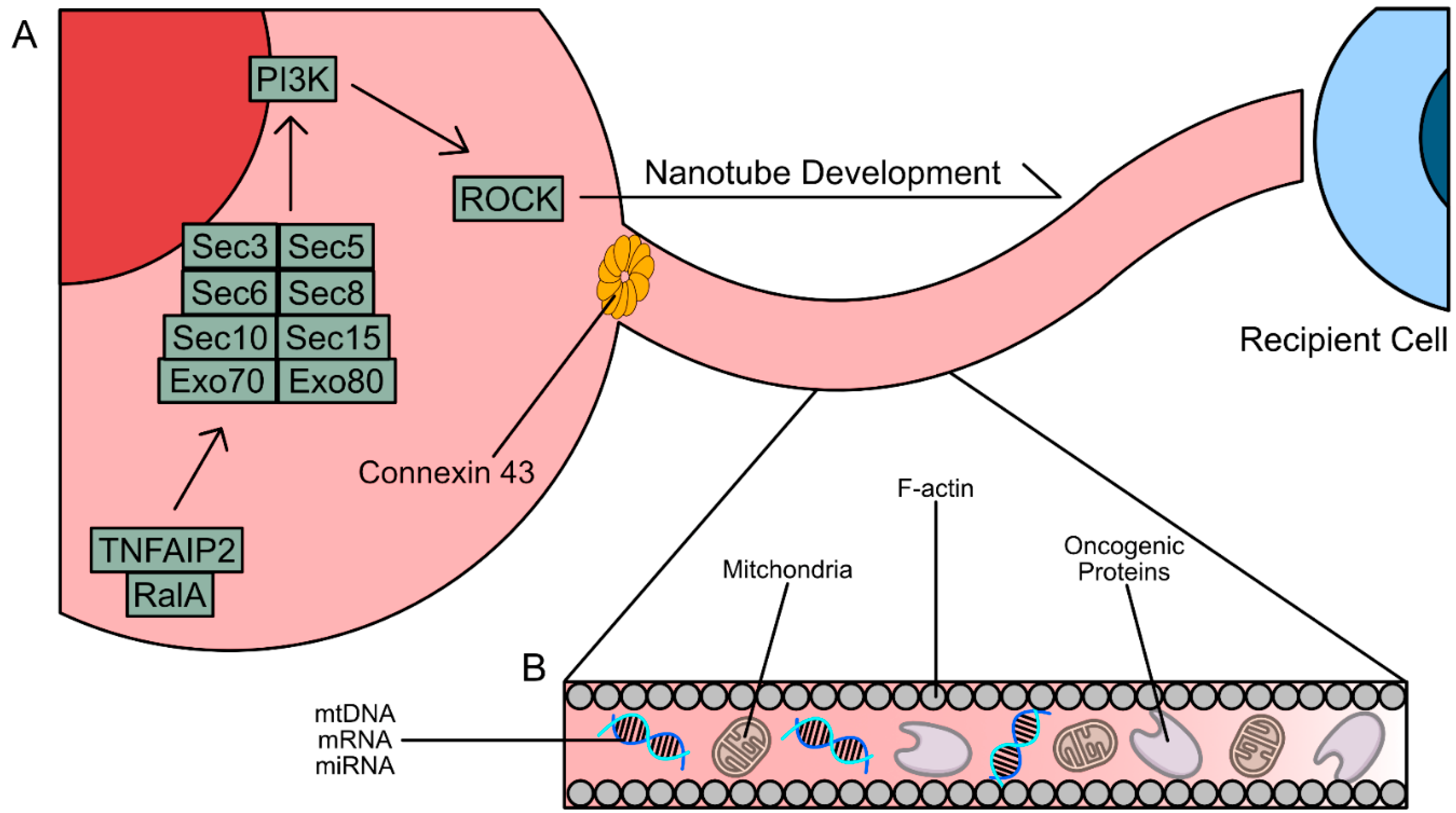
Figure 6.
Overview of the mechanisms driving phagocytic cupping. Molecular steps of phagocytic cup formation. (A) Extension of filopodia (pink) to the base of the target cell (blue). (B & C) Divergence of the filopodia into two segments after reaching the base of the target cell. (D & E) Formation of a contractile ring driven by myosin II and IxB. The contractile ring moves up the phagocytic cup scaffolding laterally. A top-down viewpoint schematic is illustrated in the orange box and the direction of the contractile forces is illustrated with blue arrows. (F) Closure of the phagocytic cup to complete phagosome formation prior to internalization. (G) Illustration of a potential trogocytosis mechanism, where closure of the phagocytic cup seen in (F) occurs prematurely due to excess forces exerted by the contractile ring.
Figure 6.
Overview of the mechanisms driving phagocytic cupping. Molecular steps of phagocytic cup formation. (A) Extension of filopodia (pink) to the base of the target cell (blue). (B & C) Divergence of the filopodia into two segments after reaching the base of the target cell. (D & E) Formation of a contractile ring driven by myosin II and IxB. The contractile ring moves up the phagocytic cup scaffolding laterally. A top-down viewpoint schematic is illustrated in the orange box and the direction of the contractile forces is illustrated with blue arrows. (F) Closure of the phagocytic cup to complete phagosome formation prior to internalization. (G) Illustration of a potential trogocytosis mechanism, where closure of the phagocytic cup seen in (F) occurs prematurely due to excess forces exerted by the contractile ring.
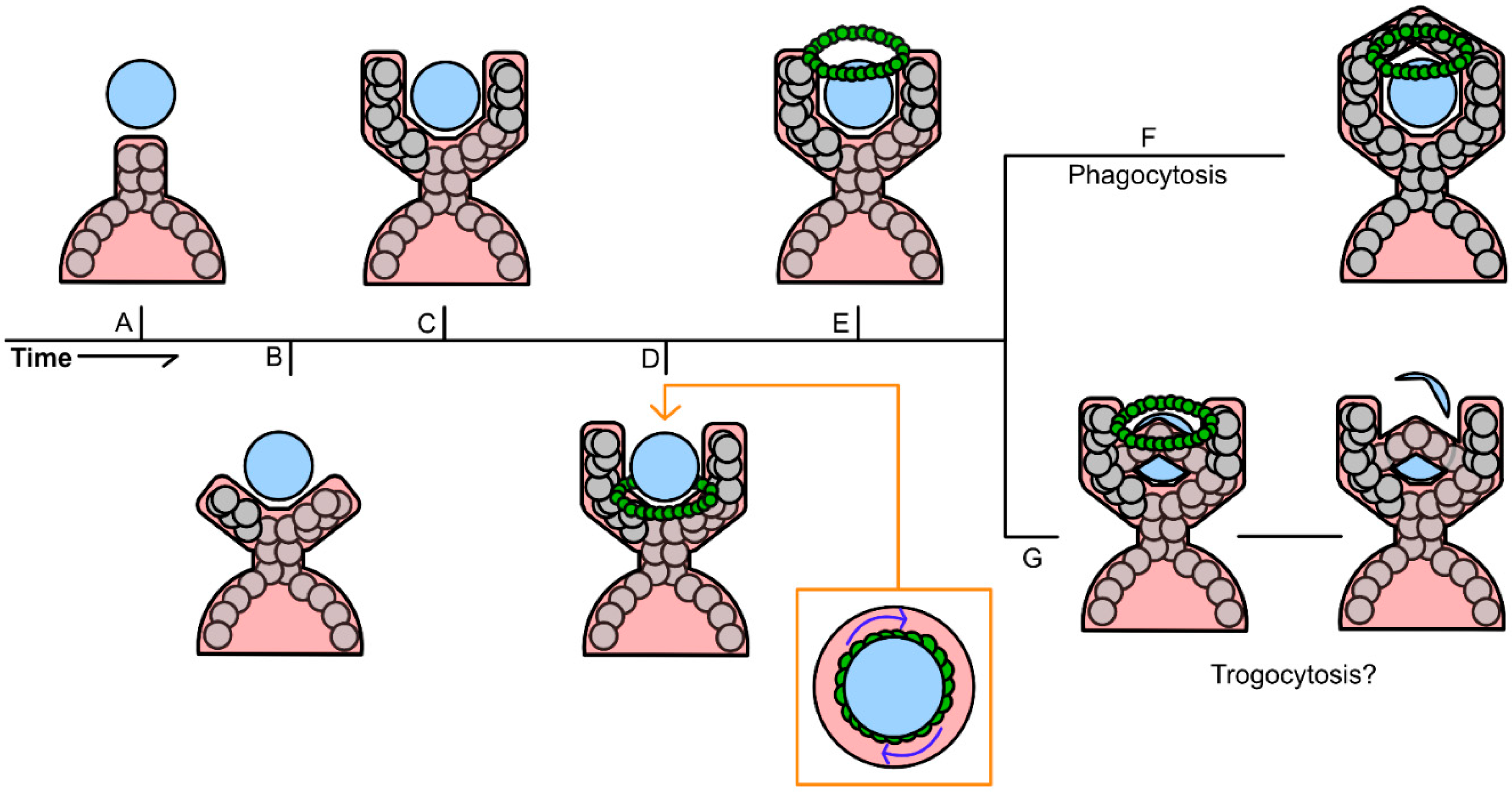
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



