Submitted:
18 May 2025
Posted:
19 May 2025
You are already at the latest version
Abstract
The traditional view of the genome has largely centered on protein-coding and non-coding regions, as these parts show clear observable functions. The non-coding sequences previously considered “junk DNA” have been extensively studied for the last few decades and are recognized to encode regulatory elements. This shift is expanding our understanding of genome complexity and its hidden potential. The non expressing sequences have not received much attention. To our best knowledge, we demonstrated for the first time that naturally non-expressing intergenic DNA sequences from Escherichia coli and Saccharomyces cerevisiae can be synthetically expressed to produce functional molecules. Leveraging this insight and recognizing the limited innovation in current drug discovery efforts, often relying on derivatives of existing or repurposed drugs — we propose a next-generation, first-in-class drug discovery platform that harnesses the vast, untapped genomic landscape—comprising non-expressed DNA sequences, non-translating sequences, and retired gene elements—through the integration of Artificial Intelligence (AI) and Quantum Computing (QC). The AI would enable high-throughput prediction of functional molecules from this untapped genomic reservoir, while QC would provide unprecedented molecular simulation capabilities to identify optimal molecules for specific targets. We are on the cusp of a transformative opportunity to recode naturally occurring DNA sequences from model organisms into a novel therapeutic pipeline and integrate Artificial Intelligence, Quantum Computing, and deep genome science, for the first time, to drive next-generation drug discovery. This integrative approach of reprogramming latent genomic code paves the way for building a deep genome foundry leading to first-in-the-class drug discovery pipeline.
Keywords:
dark genome
; synthetic biology
; therapeutic
; drug discovery
Introduction
The canonical understanding of the genome has long focused on protein-coding and RNA coding regions guided by the assumption that evolutionary selection has optimized the use of functional sequences. The non protein coding DNA sequences that were historically considered junk, proved to be an important reservoir of RNA coding genetic elements. However, a fundamental question remains: Why did nature select only a specific subset of DNA sequences for transcription? Was this the result of an exhaustive sampling of all possible genomic combinations and selecting the best candidates for becoming genes? Do vast regions of the genome—largely silent—harbour untapped expression potential?
To probe this hypothesis, we asked: Can the non-utilized or ‘non-expressed’ regions of the genome be artificially expressed to produce novel peptides or proteins? If expressed, can the lab-derived molecules demonstrate structural stability and biological functionality? We hypothesized that diverse genomic sequences, including those not traditionally associated with gene expression, harbor untapped potential for novel biological functions beyond what is currently known.
The first proof-of-concept for the above was presented by Dhar et al 2009, where functional peptides were successfully expressed from the intergenic regions of Escherichia coli—genomic areas not known to express under natural conditions. The results demonstrated that latent coding capacity exists within non-expressing regions, and that artificial expression can lead to biologically active molecules. This work, along with 15 years of subsequent research, has consistently demonstrated that the delivery of therapeutic molecules is not a one-time observation but a reproducible success. Given the vast search space and the boundless potential for discovering novel therapeutics, there is a compelling need for deeper scientific inquiry and greater industrial investment.
Existing predictive models focus on translational area, and are inadequate . They fail to capture the complex interplay between genetics, immune responses, and metabolic factors. These models are unable to account for the multi-dimensional nature of the biological data that influences clinical outcomes. As a result, currently, for many indications the diagnostics and therapeutics are suboptimal.
The vast expanse of the genome that lies beyond conventionally annotated protein-coding regions—often dismissed as non-functional or “junk”—represents a huge untapped reservoir for pharma industry. This unused genomic information encompassing non-expressed DNA sequences (antisense, reverse ORFs, intergenic, and repetitive DNA sequences), non-translatable sequences (ribosomal RNA, tRNA, introns and non-coding RNAs (ncRNAs) and retired gene sequences (pseudogenes) has remained underexplored by traditional drug discovery efforts (Figure 1)
The convergence of Artificial Intelligence (AI) and quantum computing is poised to revolutionize drug discovery by addressing the limitations of traditional approaches that are time-consuming, costly, and prone to high failure rates. AI enables rapid analysis of vast biological datasets, identification of novel drug targets, and predictive modeling of molecular interactions, thereby significantly accelerating lead identification and optimization. Simultaneously, quantum computing offers a fundamentally new computational paradigm capable of simulating quantum mechanical behaviors of molecules with far greater accuracy than classical systems. This is particularly critical for understanding protein folding, reaction mechanisms, and drug-target binding affinities—core challenges in pharmaceutical development. Together, AI and quantum computing have the potential to drastically reduce development timelines, improve success rates, and unlock novel therapeutics, ushering in a new era of precision and efficiency in drug discovery.
The current limitation of predictive models stems from their inability to integrate high-dimensional, non-linear data that includes complex interplay of omics (e.g,genetic, immune, and metabolic). Traditional computational methods employing high performance computing (HPC) tools, struggle to process this data effectively, missing patterns and correlations that could significantly improve prediction accuracy. Quantum computing, with its ability to analyse complex, multi-dimensional datasets, presents an innovative solution to this problem. By leveraging quantum algorithms working in tandem with existing HPC algorithms, we can uncover the complex hidden relationships of this vast unapped reservoir with kown omics data, enabling more accurate and holistic predictions. Quantum computing could revolutionize this by processing high-dimensional, non-linear biological data, uncovering patterns missed by traditional models. Recent advancements in quantum hardware suggest that quantum algorithms could excel in pattern recognition, discovery, and large dataset analysis. By developing these algorithms now, India can be ready and capitalize on quantum computing when it becomes commercially available.
There are no existing pipelines available to assist exploration of the vast non-expressed, non-translatable genome and resurrection of retired genes that can pave way for effective therapeutics. This represents a paradigm shift—an opportunity to build a radically novel and distinct first-in-the-class drug discovery pipeline that mainstream pharmaceutical strategies have yet to recognize. Unlocking the untapped genomic code not only expand the druggable universe but also accelerate the development of first-in-class therapeutics for currently untreatable diseases.
Here we propose an integrated platform that leverages less explored sequences along with Artificial Intelligence and Quantum Computing to discover and optimize novel therapeutic molecules. While AI would identify functional elements and prioritizes drug candidates from complex genomic data, Quantum computing would generate models of molecular interactions with high precision. This combined approach accelerate drug discovery and broaden therapeutic possibilities beyond traditional methods of drug discovery. To enable this, we envision development of a Deep Genome Foundry that generates and integrate data from classic bioinformatics, AI and Quantum Computing, for delivering more effective therapeutic solutions.
Methods
1. Identifying Novel Genome elements
The first step is to use classic bioinformatics methods to systematically identify and catalogue non-expressing DNA sequences, non-translating RNA sequences and gene fossils (pseudogenes). These include non-expressed, non-translational sequences and pseudogenes across model organisms (E. coli, S. cerevisiae, D. melanogaster, C. elegans and H. sapiens).
Genome annotations are cross-referenced with NCBI’s Gene Expression Omnibus and Non Redundant Protein Databases to exclude known expressed genes. Sequences with lowest global sequence similarity scores are considered and added to the database for further analysis.
2. RNA and Protein Prediction
Candidate gene sequences are transcribed and translated in silico to generate RNA, peptide and protein sequence equivalents with structural, interaction and putative function features using classic bioinformatics and AI-based tools. Properties such as charge, hydrophobicity, stability, solubility, and secondary structure are computed, along with toxicity and immunogenicity, using standard bioinformatics tools like alphafold, and Toxinpred.
3. Target-Based Virtual Screening
To evaluate therapeutic potential, candidate molecules are subjected to target-based virtual screening. A curated set of disease-relevant protein targets (e.g., kinases, GPCRs, enzymes, or viral proteins) are used as a reference. Molecular docking simulations are performed using AutoDock Vina and Glide, with binding affinities and interaction profiles assessed. Top-ranked peptide-target complexes are selected for further refinement.
4. Molecular Dynamics (MD) and Quantum-Enhanced Simulations
Selected docked complexes are sent for molecular dynamics (MD) simulations using GROMACS to assess interaction stability, conformational changes, and thermodynamic profiles over nanosecond to microsecond timescales. In parallel, quantum computing-based simulation modules (e.g., Qiskit Chemistry or Orca) are applied to high-priority complexes to model quantum-level interactions, electron distributions, and reaction energies for enhanced precision in lead optimization.
5. In Vitro Validation and Functional Assays
Peptides or proteins with favorable in-silico profiles were chemically synthesized or expressed in suitable systems (e.g., E. coli, HEK293, or cell-free systems). Cell-based assays (e.g., proliferation, cytotoxicity, reporter assays) were are performed to determine biological activity, with relevant cell lines. Expression levels are studied using Western blotting methods, flow cytometry, and data is analyzed.
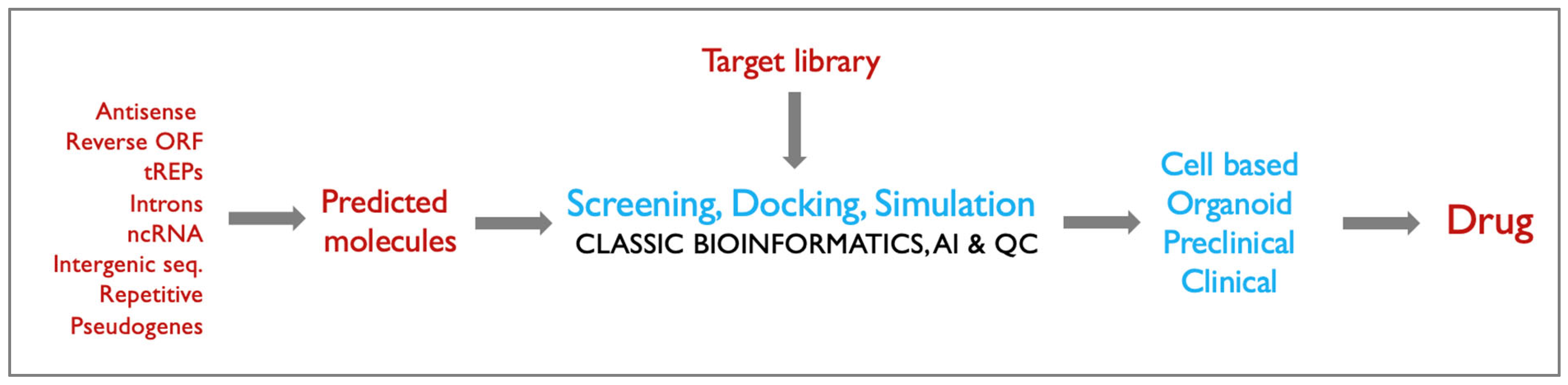
As an extension of the previous work, validated hits can be further tested in more complex models to assess relevance in disease pathophysiology, specificity and safety. . Promising candidates can be progressed to preclinical animal studies e.g., murine or zebrafish models to evaluate efficacy, pharmacokinetics, biodistribution, and toxicology. Lead molecules demonstrating significant efficacy and safety in preclinical models will be advanced to clinical trials (Figure 2).
Results
Our study is the first one to demonstrate and provide evidence for harnessing the non-expressing genomic reservoir to accelerate drug discovery.
(a) Proof-of-the-concept: Six unique intergenic sequences, each exceeding 100 base pairs in length and lacking any prior evidence of transcriptional activity, were randomly selected. The potential intergenic DNA sequences were PCR-amplified and cloned into the pBAD202/D-TOPO expression vector. Upon induction, all six synthetically engineered genes demonstated both transcription and translation activity. Western blot analysis confirmed successful expression of his-tagged intergenic sequence derived proteins. Notably, expression of one novel protein (Eka1) led to marked growth inhibition in E. coli, an effect that was completely reversed when cells were cultured in inducer-free conditions. Computational modeling predicted that two of the six de novo proteins adopt stable globular tertiary structures. This is the first report demonstrating the artificial synthesis of computationally predicted, non-native proteins synthesized from non-expressing genomic elements (Dhar et al 2009).
(b) Anti-Malaria: Using S. cerevisiae intergenic sequences, a library of synthetic peptides was computationally generated and screened for similarity to natural ligands of key Plasmodium falciparum invasion proteins: EBA-175, MSP-1(19), and AMA-1. Molecular docking simulations revealed favourable binding of selected peptides to their targets, suggesting potential to inhibit parasite invasion (Joshi et al 2013). Peptides were chemically synthesized and tested against clinical strains of the parasite in the infected blood cell culture. Experimental studies showed that more than 60% of the parasites were unable to enter infected red blood cells (personal communication, Dr Shailja Singh, JNU). A follow up study led to the identification and characterization of four novel microRNAs—ast-mir-2502, ast-mir-2559, ast-mir-3868, and ast-mir-9891 in Anopheles stephensi, from over 3,000 transcriptome sequences. Computational analyses predicted 26 potential gene targets involved in essential processes like gametogenesis, morphogenesis, protein translation, and signal transduction. The findings offer foundational insights into miRNA-mediated gene regulation in mosquitoes and suggest new molecular targets for vector control (Krishnan et al 2015).
(c) Anti-Alzheimers’: A library of 2,500 intergenic sequences was screened for open reading frames, resulting in 424 novel peptides with no known similarity to existing proteins. Using I-TASSER, secondary and tertiary structures of these peptides were predicted and virtually screened against Beta-secretase 1 (BACE1), a key target in Alzheimer’s disease. Docking studies using PatchDock and FireDock revealed peptides with strong binding affinities and favorable interactions at the BACE1 active site. Lead peptides exhibited optimal molecular weight (500–5000 Da) and functional sites such as N-glycosylation and phosphorylation, suggesting potential for enhanced bioavailability and further chemical modification via click chemistry. While this represents an early-stage finding, it demonstrated therapeutic potential of mining genomic dark matter for developing molecules against neurodegenerative diseases (Raj et al 2015).
As a key outcome of this study, two peptides—ECOI2 and ECOI3—exhibited notable inhibitory activity against BACE1. In a FRET-based enzymatic assay, ECOI2 achieved up to 86.7% inhibition of BACE1 activity at a concentration of 1 µM, while ECOI3 displayed moderate inhibition. Treatment of SH-SY5Y neuroblastoma cells with ECOI2 led to a marked reduction in BACE1 protein levels, as evidenced by Western blot analysis, without affecting BACE1 mRNA expression—suggesting a post-translational mode of regulation. ELISA quantification further demonstrated that ECOI2 significantly decreased the levels of amyloidogenic Aβ1–40 and Aβ1–42 peptides. Notably, MTT assays confirmed the non-cytotoxic nature of both peptides in SH-SY5Y cells. Collectively, these findings position ECOI2 as a potent and biologically active BACE1 inhibitor, warranting further preclinical evaluation in relevant animal models (Verma et al 2023).
(d) Anti-Leishmania: Transfer RNAs (tRNAs), primarily recognized for their role in protein synthesis, have not been explored much as peptide-encoding molecules due to intrinsic translational limitations at the ribosomal interface. Considering that tRNAs have not undergone translation throughout evolutionary history, we asked whether chemically synthesized peptides derived from tRNA sequences (tREPs) could exhibit functional activity. To explore this, 87 Escherichia coli tRNAs were computationally translated into peptide sequences, yielding 29 structurally stable candidates. Of these, chemically synthesized tREP-18 exhibited potent antileishmanial activity against Leishmania donovani Ag83 promastigotes (IC₅₀ = 22.13 nM) and the PKDL clinical isolate BS12 (IC₅₀ = 18 nM), with minimal toxicity to J774.A1 macrophages (CC₅₀ = 275 µM). Cell viability and LDH assays revealed dose- and time-dependent cytotoxicity. AFM and SEM analyses showed membrane disruption and cytoskeletal damage in treated parasites. This study reports the first functional tRNA-derived peptide with antiparasitic efficacy, establishing tREPs as a novel class of bioactive molecules and opening new avenues in drug discovery and synthetic biology.
(e) Vaccines: We investigated the potential of tRNA-encoded peptides (tREPs) as a novel source for developing epitope-based vaccines against viral pathogens. Leveraging the growing recognition of functional synthetic peptides derived from non-expressing sequences, we developed a comprehensive computational pipeline that integrates curated data sources and standard prediction tools to identify and rank candidate epitopes from tRNA sequences. For each viral target, the top-ranking epitope—predicted to bind specific HLA molecules—was subjected to 200 ns molecular dynamics (MD) simulations and binding free energy analyses to assess stability and interaction strength. Our results highlight two promising tREP-derived epitopes: RRHIDIVV for Mamastrovirus 3 and IMVRFSAE for Norovirus GII, both demonstrating favorable binding and structural stability. These findings suggest that tREPs offer an untapped molecular reservoir for vaccine design. By demonstrating antiviral potential in tRNA-derived peptides, our work opens a novel path toward rational, computation-enabled vaccine development for expanding this unexplored class of molecules (Shanthappa et al 2024)
(f) Additional evidences A large number of ORFs have been predicted from intergenic and antisense sequences of E. coli. Out of these, a subset was selected based on coding potential, conservation, and structural predictions. Several peptides were predicted to affect bacterial growth, stress response, or metabolism—indicating potential regulatory or antimicrobial properties (Varughese et al 2016).
Antisense proteins: We examined full-length hypothetical genes located on antisense strands in both forward and reverse orientations. Sequences containing in-frame stop codons upon computational translation were excluded from further analysis. Our findings revealed untapped genomic potential in the form of full-length antisense and reverse antisense proteins in E. coli (0.7% and 5.1%), S. cerevisiae (0.15% and 0.5%), and D. melanogaster (0.2% and 2.1%), respectively. Predicted physicochemical properties indicated that many of these peptides could adopt stable structures with functional relevance. Subcellular localization predictions suggested diverse cellular roles, with some proteins showing potential for secretion. Functional annotations linked many candidates to enzymatic or transporter activity (Garg & Dhar 2023a).
Reverse proteins: Reverse proteins are full-length translational equivalents derived by reading existing protein-coding sequences in the reverse direction, specifically in the -1 frame. We systematically explore reverse proteins across E. coli, S. cerevisiae, and D. melanogaster, uncovering their structural, functional, and interaction profiles. Reverse proteins were computationally predicted to encode enzymes such as oxidoreductases, lyases, transferases, hydrolases, and ATP synthases. Subcellular localization predictions further indicate that reverse proteins may play compartment-specific roles within the cell. Our work uncovers an unrecognized layer of genomic coding potential, offering a platform for the discovery of ‘first-in-the-class’ functional proteins. Although this study focused on full-length sequences, extending the framework to include partial-length reverse proteins could dramatically expand the synthetic proteome landscape. While bidirectional expression from a single locus may seem non-intuitive in natural systems, synthetically engineering reverse- proteins introduces exciting opportunities to enrich genomic, transcriptomic, and proteomic datasets and unlock new applications in delivering novel therapeutic peptides and proteins (Nayak & Dhar 2023a).
Intronic proteins: Full-length intronic sequences from S. cerevisiae, C. elegans, and D. melanogaster were computationally translated to explore their potential as novel proteins with defined structural, physicochemical, and functional properties. This work revealed that intron-derived proteins, long thought to be non-coding byproducts of gene architecture, may actually fold into stable, structured entities. Ramachandran plot analysis confirmed that the majority of residues in these proteins occupy energetically favorable regions, supporting their potential to form biologically relevant conformations. Subcellular localization predictions mapped these intronic proteins to distinct compartments—including the nucleus, mitochondria, and cytoplasm—with several candidates exhibiting features of secretory proteins. Functional annotations suggested roles as membrane transporters, DNA-binding proteins, and enzymes, with predicted metal ion interactions involving calcium, zinc, and manganese—characteristics often seen in therapeutic targets (Garg & Dhar, 2023b).
Noncoding proteins: The non-coding RNA comprises a group of RNA molecules that do not encode proteins but play important regulatory roles within the cell. We computationally translated “ncRNA” sequences from C. elegans, D. melanogaster, A. thaliana, and H. sapiens into putative proteins, analyzing their structural, physicochemical, and functional properties. Using I-TASSER, predicted proteins showed stable folding with favorable Ramachandran plots and physicochemical profiles suitable for cellular environments. Functional predictions revealed diverse enzymatic activities, including hydrolases, oxidoreductases, and kinases, along with roles as transporters, signaling molecules, and membrane proteins. Many proteins were localized to key cellular compartments such as the cytoplasm and nucleus. These findings suggest that “ncRNA-derived proteins” (non-coding proteins, NCPs) represent a novel class of biomolecules with potential applications in drug discovery (Nayak & Dhar 2023b).
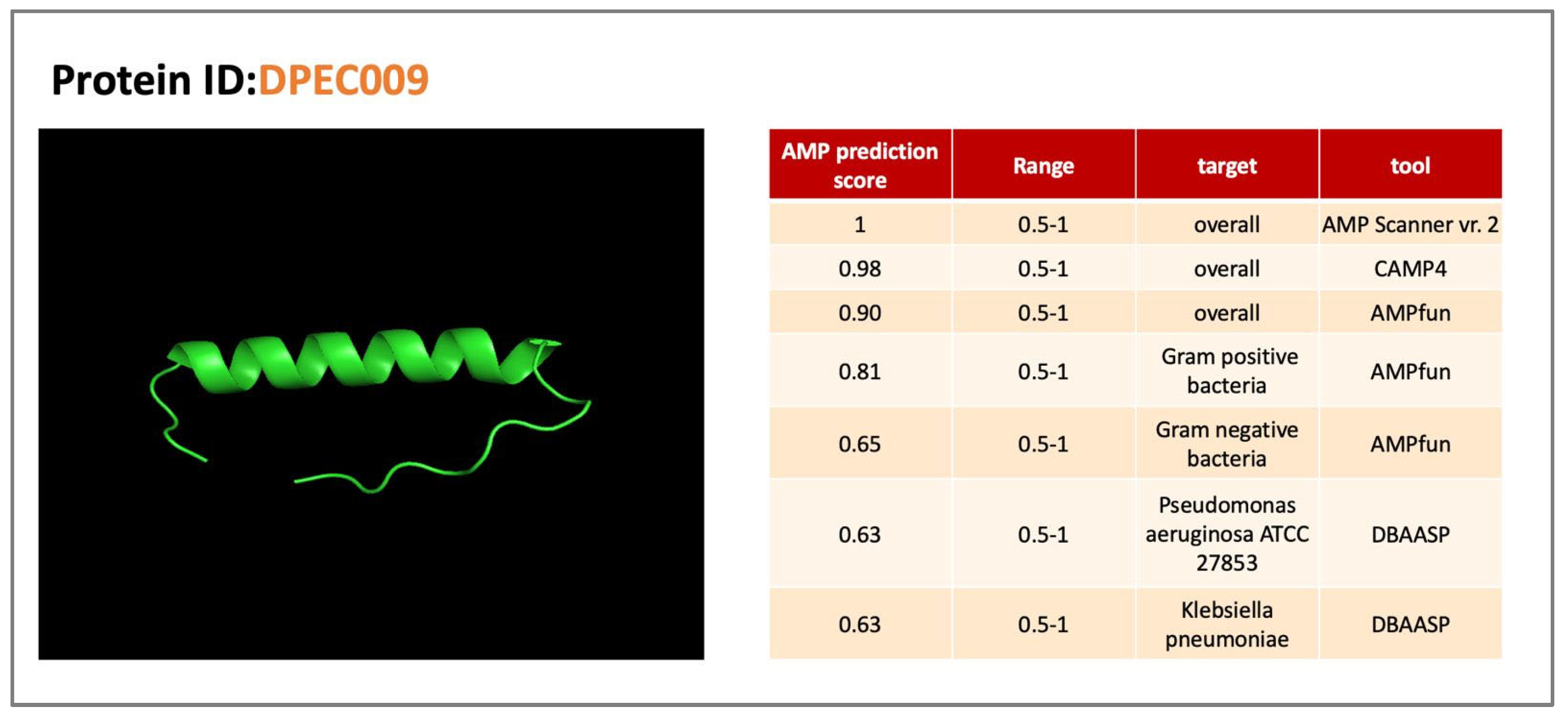
As an extension of above work, we performed a study of proteins derived from intergenic sequences of E. coli and found strong evidence of antimicrobial properties against gram negative and gram-positive bacteria
Figure 3.
Predicted Antimicrobial peptide from E.coli intergenic sequence.
Discussion
The future is poised to witness a profound shift in the way we conceive, discover, and develop medicines—driven by a confluence of synthetic biology, artificial intelligence, and quantum computing. At the heart of such transformation lies the recoded genomic elements as a latent resource for entirely new classes of bioactive molecules with a potential of accelerating drug discovery in an unprecedented manner
Traditionally, pharmaceutical discovery has relied on well-annotated genes, known protein families, and disease-associated biomarkers. The non expressed, non-translated and retired genome elements —when activated through synthetic recoding—represents an untapped universe of circular-RNA/t-RNA molecules, proteins and peptides that are evolutionarily silent but structurally and functionally viable. These molecules are not constrained by homology, historical annotation, or evolutionary bias, making them ideal candidates for first-in-class drugs with novel mechanisms of action and possibility of overcoming microbial resistance.
The grand challenge, however, lies in the sheer complexity of navigating this genomic terra incognita. Here, artificial intelligence becomes indispensable. Deep learning models, especially transformer-based architectures and generative AI, can be trained on known protein structures and expression patterns to predict which dark genomic sequences are likely to fold into stable, soluble, and bioactive proteins. Reinforcement learning can guide sequence optimization for enhanced binding, minimal toxicity, or cell permeability. Importantly, these models can computationally screen millions of synthetic candidates leading to a narrow and well defined set of experiments for discovering their therapeutic potential.
While genome-level information provides the static blueprint of an organism, omics approaches dynamically decode how this blueprint is interpreted, expressed, and modulated across cellular systems, offering a multilayered view of biological complexity.
Quantum computing could revolutionize this by processing high-dimensional, non-linear biological data, uncovering patterns missed by traditional models. Recent advancements in quantum hardware suggest that quantum algorithms could excel in pattern recognition, biomarker discovery, and large dataset analysis. By developing these algorithms now, India can be ready and capitalize on quantum computing when it becomes commercially available.
However, AI alone is not sufficient. The fine-grained molecular dynamics of protein–protein interactions, binding energy calculations, and quantum tunneling effects—especially relevant for enzymatic inhibition and signal transduction—require computational frameworks beyond classical limitations. This is where quantum computing enters the equation. Quantum simulations can model the behavior of electrons and atoms with exquisite precision, enabling the identification of subtle, low-energy conformations that govern biological activity. When applied to dark genome-derived molecules, quantum tools can reveal binding pockets, transition states, and folding landscapes that are otherwise computationally intractable.
Together, AI and quantum computing can create a closed-loop platform: AI would help in predicting promising dark genome derived molecules, while quantum simulations can validate their interactions with disease targets. This iterative cycle forms a self-optimizing, intelligent drug discovery engine, capable of navigating unannotated genomic space and delivering novel therapeutics at unprecedented speed and scale.
The pharmaceutical implications are enormous, enabling the precision-first drug design, where molecules are not merely screened but engineered for context-specific activity, such as tumor microenvironment acidity, pathogen-specific receptors, or neurochemical signatures. Second, it opens the door to programmable therapeutics—dark genome-derived molecules that can be customized for individual patients based on their unique genomic architecture.
For the pharmaceutical industry, this marks a disruptive inflection point. The traditional pipeline—target identification, lead discovery, optimization, and clinical trials—is linear, costly, and failure-prone. In contrast, the dark genome platform powered by AI and quantum technologies offers a fast, data-rich, and adaptive pipeline. In the broader context, the ability to mine a single genome for thousands of potential RNA and Peptide based therapeutics democratizes drug discovery. Academic labs, biotech startups, and emerging economies could harness this strategy without requiring large compound libraries or decades of gene annotation. With open-source AI models and cloud-based quantum computing becoming increasingly accessible, the barriers to innovation are being rapidly dismantled.
A Global Centre of Excellence focused on building a deep genome foundry this theme will lead to the development of first-in-the-class molecules, revolutionizing disease treatment, and address pharma’s innovation bottleneck. By uniting synthetic biology, machine learning, and quantum technology, the hub can unlock unprecedented molecular engineering capabilities, generate new industries, and position itself as a global leader in next-generation precision medicine and post-genomic innovation.
In summary, integration of recoded genomic elements, AI, and quantum computing marks a new epoch in biomedicine. It is not merely an expansion of existing capabilities—it is a fundamental redefinition of drug discovery efforts. The pharmaceutical industry, long in search of a disruptive breakthrough, may well find its next renaissance not in foreign compounds or rare organisms, but in the silent, overlooked regions in our own DNA backyard !
Competing Interest
The authors declare no competing interest.
Acknowledgement
We gratefully acknowledge the support of RIKEN Genomic Sciences Centre (Yokohama), Jawaharlal Nehru University (New Delhi), CVJ Centre for Synthetic Biology (Kochi), MIT Vishwaprayag University (Maharashtra), and Thrafford Lifescience (Noida) in supporting this work and enabling this publication. PKD would like to warmly acknowledge the efforts of Mr Sarangdhar Nayak (JNU) in contributing Figure 3 of this paper. This concepts presented in this paper reflects the shared effort and collective insights of all co-authors.
References
- Chakrabarti, A., Kaushik, M., Khan, J., et al. (2022). tREPs – a new class of functional tRNA encoded peptides. ACS Omega, 7(22), 18361–18373. [CrossRef]
- Dhar, P. K., Nanduri, B., et al. (2009). Synthesizing non-natural parts from natural genomic template. Journal of Biological Engineering, 3, 2. [CrossRef]
- Garg, M., & Dhar, P. K. (2023a). Repurposing the Dark Genome I: Antisense Proteins. bioRxiv. [CrossRef]
- Garg, M., & Dhar, P. K. (2023b). Repurposing The Dark Genome. III - Intronic Proteins. bioRxiv. [CrossRef]
- Joshi, M., Kundapura, S. V., Poovaiah, T., Ingle, K., & Dhar, P. K. (2013). Discovering novel anti-malarial peptides from the not-coding genome—A working hypothesis. Current Synthetic and Systems Biology, 1(1).
- Krishnan, R., Kumar, V., Ananth, V., et al. (2015). Computational identification of novel microRNAs and their targets in the malarial vector Anopheles stephensi. Systems and Synthetic Biology Journal, 9, 11–17.
- Nayak, S., & Dhar, P. K. (2023a). Repurposing the Dark Genome II – Reverse Proteins. bioRxiv. [CrossRef]
- Nayak, S., & Dhar, P. K. (2023b). Repurposing the Dark Genome IV – Noncoding Proteins. bioRxiv. [CrossRef]
- Raj, N., Helen, A., Manoj, N., et al. (2015). In silico study of peptide inhibitors against BACE. Systems and Synthetic Biology Journal, 9, 67–72.
- Shidhi, P. R., Suravajhala, P., Nayeema, A., et al. (2015). Making novel proteins from pseudogenes. Bioinformatics, 31(1), 33–39. [CrossRef]
- Varughese, D., Nair, A. S., & Dhar, P. K. (2017). Function annotation of novel peptides generated from the non-expressing genome of Drosophila melanogaster. Bioinformation, 13(1), 17–20.
- Verma, N., Manvati, S., & Dhar, P. K. (2023). Harnessing Escherichia coli’s Dark Genome to Produce Anti-Alzheimer Peptides. bioRxiv. [CrossRef]
Figure 1.
Conceptual framework depicting the de novo origin of next-gen therapeutics.
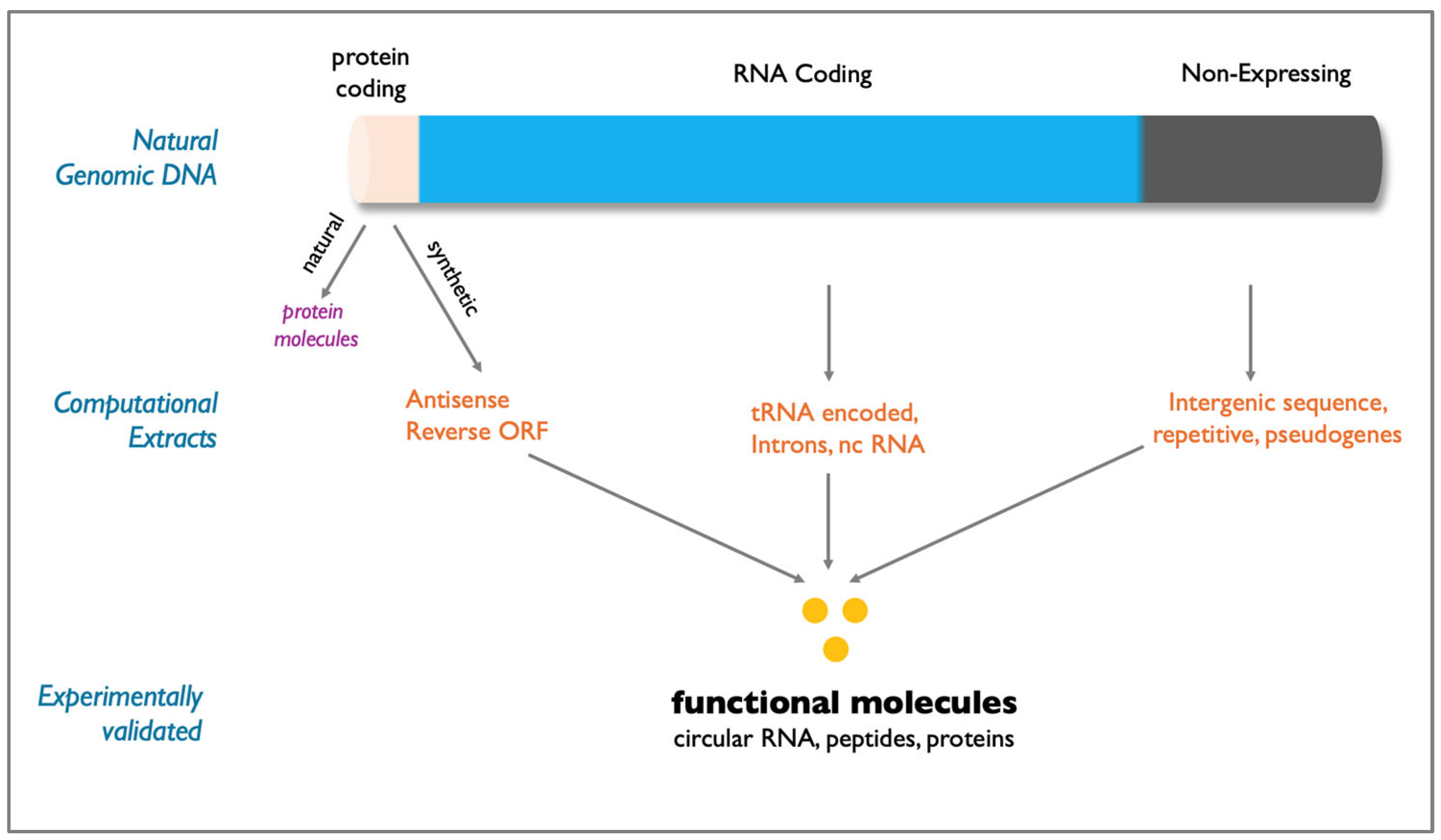
Figure 2.
The Work Flow of the novel drug discovery pipeline.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



