
Submitted:
12 September 2025
Posted:
16 September 2025
Read the latest preprint version here
Preprints on COVID-19 and SARS-CoV-2
Abstract
This study provides a comprehensive overview of SARS-CoV-2 genomic surveillance in Central America and the Dominican Republic from February 2020 to January 2023, highlighting the collaborative efforts of the Pan American Health Organization (PAHO), and the Council of Ministers of Health of Central America (COMISCA). A total of 26, 595 sequences from the GISAID database were analyzed, correlating findings with key events reported by participating entities. The genomic analysis reveals significant co-circulation of variants, with notable lineage diversity observed throughout the pandemic. Variants of concern (VOC) like Alpha, Gamma, Delta and Omicron were identified alongside variants of interest (VOI) like Lambda and Mu. The emergence of recombinant lineages further illustrates the ongoing evolution of the virus and its spread across the region, underscoring the interconnectedness of Central America and the Dominican Republic. The collaborative model facilitated broader sequencing coverage, enabling more extensive surveillance than individual countries could achieve alone. Despite the successes of regional collaborations, challenges remain, particularly regarding sequencing capacity in countries impacted by socioeconomic inequalities. Addressing these gaps is essential to enhance public health responses to current and future pandemics.
Keywords:
COVID‐19
; SARS‐CoV‐2
; coronavirus
; genomic surveillance
1. Introduction
In late 2019, numerous cases of pneumonia with an unknown etiology were detected in the city of Wuhan, China. The causal agent was identified as a novel type of coronavirus and was named SARS-CoV-2 [1]. The World Health Organization (WHO) officially designated the illness caused by SARS-CoV-2 as COVID-19 (coronavirus disease 2019). Subsequently, in March 2020 this emerging outbreak was declared a global pandemic [1,2].
As an RNA virus, SARS-CoV-2 undergoes frequent mutations as part of its replication cycle. The high replication rates during the pandemic resulted in the emergence of new genotypes [3]. The emergence of variants that had likely a greater effect in public health led the WHO to categorize them based on shared attributes and required public health actions. These categories include variants of concern (VOC), variants of interest (VOI) and variants under monitoring (VUM) [4]. Notably, variants that were initially classified as VOC, like Alpha, Beta, Gamma, Delta and Omicron, were all reported in Central America [5,6]. Additionally, several lineages were first observed in countries within Central America and the Dominican Republic [7,8,9,10]. These findings underscore the critical importance of genomic surveillance in this region to monitor the spread and evolution of the virus effectively.
The COVID-19 pandemic emphasized the significance of genomic surveillance, shedding light on the challenges associated with its implementation in the various public health systems. Consequently, since the beginning of the pandemic, government entities have made substantial investments towards the advancement and implementation of whole genome sequencing (WGS) [11,12].
The implementation of an integrated genomic surveillance system during the pandemic has proven to be crucial for public health understanding and decision-making. This system has been instrumental in monitoring the real-time evolution of SARS-CoV-2 through the open exchange of data and online platforms like GISAID (Global Initiative on Sharing All Influenza Data, https://gisaid.org) [11,12]. By identifying and categorizing the various lineages of circulating viral diversity, this system has enabled the early detection of emerging variants before their widespread dissemination. Moreover, this monitoring made possible the linking of cases to clusters, tracing the origins of outbreaks, and identifying both national and international transmission routes. The knowledge gained about the different mutations present in circulating viruses was essential for the development of vaccines, treatments and diagnostic tests, as well as monitoring the impact of control measures [11,12].
In Central America and the Dominican Republic, the progress made in genomic surveillance was strengthened by an effective collaboration model between the Pan American Health Organization (PAHO) and the Council of Ministers of Health of Central America (COMISCA). This collaboration aimed at enhancing capacities in the various countries of Central America and the Dominican Republic during the COVID-19 pandemic.
Thus, this study aimed to highlight the milestones in the strengthening of the genomic surveillance systems in the sub regions and to conduct a descriptive analysis of the lineages and circulating variants in Central America and the Dominican Republic from February 2020 to January 2023. This analysis is based on diverse SARS-CoV-2 genomes available in the GISAID database, sourced from material sequenced and shared by the public health laboratories within the subregion.
2. Materials and Methods
2.1. Data Acquisition
In order to conduct a comprehensive analysis of SARS-CoV-2 genomic surveillance in Central America and the Dominican Republic, datasets containing whole genome sequences and accompanying metadata were acquired from the GISAID repository, accessible at https://gisaid.org (metadata is provided in Table S1). These datasets were comprised of information voluntarily shared by participating countries.
2.2. Timeline of Key Events
Key events leading to enhancing capacities in the various countries of Central America and the Dominican Republic during the COVID-19 pandemic were provided by PAHO (https://www.paho.org/en), the COMISCA Executive Secretary (SE-COMISCA) (https://www.sica.int/comisca/inicio), Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud (INCIENSA) (https://www.inciensa.sa.cr) and Instituto Conmemorativo Gorgas de Estudios de la Salud (ICGES) (https://www.gorgas.gob.pa).
The timeline plot was generated using ggplot2 v3.4.4 R package. The complete R script is publicly accessible in GitHub (https://github.com/sofiaherreraa/SARS-CoV-2_CADR).
2.3. COVID-19 Epidemiological Data
COVID-19 case numbers and total deaths for each country were obtained from daily reports provided by Our World in Data [13]. Sequenced SARS-CoV-2 samples for each country were sourced from the GISAID platform (https://gisaid.org), with data collection limited to samples collected up until January 31st, 2023.
A dataset was constructed comprising the monthly proportions of SARS-CoV-2 lineages, total reported cases and deaths, and a temporal index for analysis. To reduce noise from lineages with minimal fluctuation, the variance of each lineage’s proportion over time was calculated, and only those with a variance greater than 0.001 were retained. Two regression models were then implemented to assess the association between lineage proportions and epidemiological outcomes: a Poisson regression model for monthly case counts and another for death counts, each incorporating time and the selected lineage proportions as predictors. Overdispersion was assessed by calculating the ratio of the sum of squared Pearson residuals to the residual degrees of freedom. If this ratio exceeded 1.5, a negative binomial regression was used instead of Poisson regression to account for extra variability. All model coefficients, standard errors, z-values, and p-values are presented in Supplementary Table S2. Normalized case/death counts were scaled using min–max normalization (0–1 range). The secondary axis represents the normalized values, aligned to facilitate visual comparison with lineage proportions.
All statistical analyses and visualizations, including descriptive trends, regression models, and the generation of Supplementary Table S2, were performed using R (version 3.4.4), primarily with the ggplot2 package. The complete R script is publicly accessible in GitHub (https://github.com/sofiaherreraa/SARS-CoV-2_CADR).
2.4. Analysis of Circulating Variants
To assess the enhancing capacities in the various countries of Central America and the Dominican Republic during the COVID-19 pandemic throughout the period of study, genomic sequences deposited in GISAID from each country were analyzed longitudinally. The initial dataset comprised a total of 26,595 sequences, covering the period from February 2020 to January 2023. These sequences were sourced from eight countries (COMISCA members) within the region, specifically: Costa Rica, Panama, Guatemala, Dominican Republic, Belize, Nicaragua, El Salvador, and Honduras.
The Pango lineage database was used to analyze the frequency of lineages among countries [10,14]. The analysis focused on identifying and characterizing the predominant lineages circulating in these regions. Specifically, the top 14 most prevalent lineages were individually assessed. Additional exploratory analyses were conducted using the top 30, 50, and 100 lineages (Figure S1); however, these broader selections did not provide added interpretive value and introduced considerable noise, leading to a focus on the 14 most representative ones for clarity and relevance.
To further investigate the relevance of these lineages within the context of Central America and the Dominican Republic, an overall threshold of 35 sequences (n > 35) per month was applied across the full regional dataset to determine which lineages were assessed individually. Lineages not meeting this threshold were grouped under the category “others.” This cutoff was selected based on internal comparisons of different thresholds (n = 35, 50, 75, and 100; data not shown) to optimize clarity and ensure representativeness. Lower thresholds led to the inclusion of many low-frequency lineages with limited interpretive value, while higher thresholds excluded lineages that were relevant in some months but not consistently abundant. The final set of lineages identified through this overall threshold was then used consistently across the individual country panels. Any lineages not included in the overall panel were grouped as “others” in the country-specific figures to maintain comparability and minimize noise.
All plots were generated using ggplot2 v3.4.4 R package. The complete R scripts are publicly accessible on GitHub (https://github.com/sofiaherreraa/SARS-CoV-2_CADR).
2.5. Phylogenetic Analysis
Sequences were selected based on a minimum coverage threshold of 28,000 (approximately 95% of the full SARS-CoV-2 genome length) and less than 6% ambiguous bases (Ns) (N), to ensure high-quality assemblies and minimize biases associated with incomplete or low-confidence genomes. This filtering yielded a dataset of 25,185 sequences.
To reduce computational load while maintaining representative temporal coverage, a stratified sampling was then conducted annually from February 2020 to January 2023, using Subsampler v1.1.0 [15]. This sampling approach resulted in a total of 2,487 sequences, equivalent to an average sampling rate of 9.875% per year.
Sequence alignment was performed using MAFFT v7.471 [16], followed by a maximum likelihood-based phylogenetic analysis with IQ-TREE v1.6.12 [17], employing the GTR+F+R2 substitution model selected by ModelFinder [18]. The resulting phylogenetic tree was visualized using iTOL v4 [19], incorporating metadata.
3. Results
3.1. Implementation of Genomic Surveillance and Sequencing Capacity
From February 2020 to January 2023, genomic surveillance efforts in Central America and the Dominican Republic identified multiple SARS-CoV-2 lineages and variants. A timeline of key events is illustrated in Figure 1, to call out the processes implemented to enhance genomic surveillance capacities in the various countries of Central America and the Dominican Republic during the COVID-19 pandemic. In January 2021, the local laboratory capabilities in the selected countries, INCIENSA in Costa Rica and ICGES in Panama, were assessed. Based on the assessment, it was identified that INCIENSA and ICGES required additional sequencing equipment, supplies, reagents and staff to support the laboratories [20,21]. Additionally, during this process, REDLAB (the Network of National Public Health Laboratories of Central America and the Dominican Republic) reached an agreement with its laboratory members to participate in this activity, which involved sending out SARS-CoV-2 positive samples for sequencing. As seen in Figure 1, shipment of these samples began in February 2021 up until January 2023, where some countries shipped to Panama (ICGES) and others to Costa Rica (INCIENSA).
To describe the SARS-CoV-2 genomic surveillance in Central America and the Dominican Republic, Table 1 presents statistical COVID-19 data from the beginning of the pandemic up until January 31st, 2023. Nicaragua shows the highest percentage of sequenced COVID-19 cases; however, this data may be biased due to the country’s surveillance strategies. Therefore, it will not be considered for comparative analyses. Belize also shows a relatively higher percentage of sequenced COVID-19 cases compared to other evaluated countries. This discrepancy is likely influenced by Belize’s relatively small population of approximately 400 thousand inhabitants [22].
To ensure quality in downstream analyses, retrieved sequences were filtered by completeness and ambiguity thresholds. In most countries, over 90% of the sequences met the quality criteria (≥95% genome coverage and <6% ambiguous bases), except for Nicaragua, where only 72% of sequences passed the filters, potentially reflecting differences in sequencing quality or protocols.
Note: Values marked with an asterisk (*) refer to sequences collected within the time window from February 1st, 2020 to January 31st, 2023.
Costa Rica and Panama have higher percentage of sequenced COVID-19 cases (0.84% and 0.64%) compared to the other countries in the region. Conversely, Honduras has a significantly lower percentage of sequenced cases at 0.06%. The Dominican Republic, El Salvador and Guatemala have similar percentages and are slightly lower than those reported for Costa Rica and Panama.
3.2. Circulating Variants and Lineage
The data illustrated in Figure 2 highlights the distribution of circulating SARS-CoV-2 variants over time in each country. At the start of the pandemic, the Dominican Republic, Belize, El Salvador, and Honduras had minimal to no sequenced SARS-CoV-2 cases. A noticeable increase in sequencing activity was observed from early 2021 onward.
Figure 2 displays that the same SARS-CoV-2 lineages were detected across several countries during overlapping time periods. Early in the pandemic, despite limited sequencing in some countries, the B.1 lineage was reported throughout the region. In mid-2021, Delta sub-lineages AY100 and AY113 [23] were present across the included countries. The Omicron sub-variant BA.1.1 [24] was identified in samples from Central America and the Dominican Republic between December 2021 and January 2022.
To illustrate the temporal dynamics of circulating SARS-CoV-2 lineages in Central America and the Dominican Republic, Figure 3 presents a composite area plot showing the monthly proportion of lineages at the regional level and in each country. The number of lineages included was filtered using an overall threshold of more than 35 sequences, as lower-frequency lineages provided limited additional interpretive value. The same color palette and lineage classification used in the regional summary were applied to the country-level plots to ensure consistency in interpretation.
As shown in Figure 3, no lineage exceeded 35 occurrences in February 2020. Between March 2020 and February 2021, at least one lineage consistently surpassed this threshold, representing approximately 20% of sequenced samples each month.
Between March 2021 and December 2021, the number of lineages exceeding 35 occurrences increased significantly. By September 2021, Delta sublineages AY.100 and AY.113 comprised more than 50% of the circulating lineages; with Delta’s dominance maintained in October and November 2021 with the rise of new sublineages [23]. However, in January, February, and March 2022, lineage diversity with more than 35 occurrences was significantly reduced due to the dominance of the Omicron subvariant BA.1.1 [24], which had been prevalent since December 2021, and accounted for at least 50% of cases during the first three months of 2022. From April 2022 to November 2022, lineage diversity exceeding 35 occurrences rose again. In December 2022, the recombinant lineage XBB.1 [25] emerged, comprising more than 50%, resulting in a decline in overall lineage diversity. By January 2023, three Omicron lineages predominated: XBB.1, XBB.1.5 and BQ1.1, with the first two being recombinant lineages [25].
3.3. Phylogenetic Analysis of SARS-CoV-2 Lineages
Figure 4 illustrates the evolution of different variants throughout the study period in Central America and the Dominican Republic, presenting temporal and country-specific information. In 2020, no clear predominance of any variant was observed; however, variants such as Gamma and Alpha, along with closely related lineages, were present to some extent. By 2021, Gamma, Alpha and Delta were present in significant proportions, with Delta showing a clear predominance. Omicron was also present to a lesser extent. In 2022, Omicron became the dominant variant, while Delta was present in smaller amounts.
3.4. Associations between Lineage Dynamics and Epidemiological Trends
Figure 5 overlays lineage proportions with normalized monthly COVID-19 cases (panel B) and deaths (panel A). Cases and deaths were normalized using min–max scaling (0–1 range) and plotted on a secondary y-axis to facilitate From 2022 onwards, certain lineages such as BA.1.1, BA.2, and their sublineages showed marked increases in prevalence concurrent with rises in reported case numbers. Conversely, the monthly death counts exhibited a downward trend during the same period. Negative binomial regression models were fitted to assess associations between lineage proportions and epidemiological metrics. While a few lineages demonstrated statistically significant associations—such as BA.2.9 with case counts (p = 0.028) and XBB.1 with death counts (p = 0.015)—the overall explanatory power of individual lineages was limited. Full regression outputs, including coefficients and significance values, are provided in Table S2.
4. Discussion
Over the years, outbreaks like severe acute respiratory syndrome (SARS) (2002-2023), influenza (2009) and Ebola (2013-2014) have highlighted the need for genomic surveillance to support public health surveillance systems [26]. Genomics played a key role in combating the COVID-19 pandemic by enabling to track the SARS-CoV-2 variants and their spread, as well as the development of molecular diagnostics techniques, therapeutics and vaccines, and strengthen or ease social distancing measures; all based on information derived from whole-genomes sequences [27].
The recognition of the importance of genomic surveillance during the COVID-19 pandemic led to organizations such as PAHO [20] and COMISCA [21] to enhance genomic surveillance efforts across Central America and the Dominican Republic. Developing bioinformatics analysis capabilities is a key goal of these strengthening programs. As of 2025, many countries are still in the early stages of developing these skills.
In this area, INCIENSA in Costa Rica and ICGES in Panama were key players, as they led the development of genomic sequencing of SARS-CoV-2 virus in the region as illustrated in Figure 1 [21]. These efforts led to enhanced sequencing capacities among Central America and the Dominican Republic. As demonstrated in Figure 2, several countries with little to no sequenced samples throughout 2020 experienced a significant increase in sequencing activities in 2021, with many maintaining this progress into 2022.
As illustrated in Table 1, Nicaragua and Belize exhibit the highest percentages of sequenced COVID-19 cases. Nevertheless, as previously mentioned, Nicaragua’s data might be biased due to the country’s surveillance strategies and was not considered for comparative purposes.
In contrast, Belize’s elevated percentage of sequenced COVID-19 cases can be attributed to its substantially smaller population compared to the other countries analyzed in this study. Belize’s population is approximately 11 times smaller than that of Panama, which has the next fewest inhabitants after Belize [22].
Now that the outliers have been excluded, a clearer understanding of the sequencing capacities in Central America and the Dominican Republic can be achieved. Further analysis of Table 1 reveals Costa Rica and Panama have higher percentages of sequenced COVID-19 cases (0.84% and 0.64%, respectively) compared to the other countries in the region. This is to be expected, as both countries led the development of genomic sequencing of SARS-CoV-2 virus in the region; consequently, the necessary equipment and reagents were established locally [21]. In contrast, countries like Dominican Republic, El Salvador, Guatemala and Honduras were required to ship their samples to Panama or Costa Rica for sequencing, as illustrated in Figure 1, which accounted for their lower percentage of sequenced COVID-19 cases. Additionally, Honduras demonstrates a significantly lower percentage (0.06%) than the other countries shipping samples. This can be correlated with limited testing capacity, exacerbated by the impacts of Hurricanes Eta and Iota and the simultaneous dengue epidemic affecting the country [28].
Research has demonstrated that sequencing at least 5% of all positive tests allows the detection of emerging strains with prevalence between 0.1% to 1.0% [29]. However, as shown in Table 1, none of the countries achieved this threshold. In fact, only 6.8% of the countries worldwide (13 out of 189) managed to sequence 5% or more of their total confirmed cases, while 45.5% (86 out of 189) sequenced less than 0.5% of confirmed cases [30]. The observed scenario, where most countries in Central America and the Dominican Republic sequenced fewer than 0.5% of cases reflects a broader trend. Over half of low- and middle-income countries sequenced less than 0.5% of their cases during the first two years of the pandemic. This underscores the global disparities in SARS-CoV-2 genomic surveillance [30].
Despite these limitations in volume, the quality of the sequences was generally high. Across countries, the average completeness exceeded 98%, and most sequences met the quality threshold of ≥95% coverage and <6% ambiguities. While Nicaragua showed a slightly lower proportion (72.2%) of high-quality sequences, the remaining countries consistently surpassed 90%, suggesting that although sequencing efforts were limited in quantity, they were robust in technical execution.
Understanding the genomic landscape of SARS-CoV-2 is crucial for tracking its evolution and informing public health responses. The analysis of genotypes and their circulation patterns provides insights into how different variants emerge and spread within populations. It is important to emphasize the synchronous circulation of the different lineages across the region, highlighting a remarkably active transmission dynamic. Such transmission dynamics have been documented not only among Latin American countries [31,32,33] but also across other continents [34,35] and globally, largely facilitated by international travel as part of modern globalization [36].
These dynamics underscore the necessity for countries to establish robust communication channels not only with their neighboring nations but also with countries that have high levels of travel and trade connections. Effective monitoring of circulating strains requires cross-border collaboration to track and respond to emerging variants, especially in regions with significant interconnectedness. The frequency of different lineages over time and their geographic distribution, can be observed in Figure 2. In early stages of the pandemic, the B.1 lineage was detected throughout Central America and Dominican Republic. By early 2021, A 2.5 also emerged across these countries. Mid- 2021, saw the rise of Delta sublineages AY.100 and AY.113 [23], while early 2022, marked the widespread presence of Omicron sublineages, with BA.1.1 [2], becoming the first predominant sub lineage. This omicron trend continued through 2022 and early 2023, with recombinant Omicron sublineages such as XBB.1 [25], becoming prominent by late 2022 and early 2023. The synchronous presence of recombinant lineages, such as XBB.1, further underscores the regional interconnectedness and transmission dynamics, highlighting the need to have capacity to detect the locally generated new variants.
The mentioned lineage diversity and evolution over time can be further analyzed in Figure 3 and Figure 4. Throughout 2020 predominant lineages in Central America and the Dominican Republic included A.2, A.2.4 and B.1, which are the only lineages that exhibited more than 35 cases until December 2020. The B.1.1.7 lineage, classified as Alpha and a VOC was first detected in the region in late 2020, aligning with its initial report [37]. Although Alpha became more prevalent in the first half of 2021, it failed to achieve dominance [38].
In mid-2021, along with B.1.1.7, the P.1 lineage classified as Gamma and a VOC, also emerged as a predominant variant [39]. Additionally, Lambda (C.37) and Mu (B1.621), both designated as VOIs, were also predominant variants in the second quarter of 2021, alongside P.1, which continued to play a crucial role [40,41].
Delta sublineages (VOC) first became predominant in mid-2021, rapidly replacing variants like Gamma and other variants [42]. From mid to late 2021, Delta variants achieved significant dominance, as illustrated in Figure 3. Among these, AY.100 and AY113 emerged as the most prevalent Delta sublineages in late 2021, with their highest peak in September 2022 accounting for around 60% of the sequenced cases. However, other sublineages - including AY.25.1, B.1.617.2, AY.119, AY.3, AY.4 and AY.43 - also contributed to greater lineage diversity during this period [23,37]. A similar scenario was reported in Mexico [2]. Figure 4 further highlights the dominance of these Delta variants throughout 2021.
By December 2021, Omicron, classified as a VOC, represented around 40% of cases with BA.1.1 as the most frequent sub-variant [24]. By January 2022, Omicron was now responsible for around 90% of the cases, with BA.1.1 accounting for around 60% of the cases. This displacement of variant Delta with Omicron was widely documented, including Europe and the United States of America [43,44,45,46].
In 2022, the dominance of the Omicron (illustrated in Figure 4), resulted in a marked decline in lineage diversity characterized by more than 35 occurrences, as noted in Figure 3. This variant rapidly spread across all countries in Central America and the Dominican Republic, establishing itself as the predominant strain in the region. The sustained dominance of Omicron continued through 2022 and into early 2023. However, it is important to highlight that in early 2022 the lineage diversity with lineages surpassing more than 35 cases was very low. However, as the months progressed this diversity increased, peaking at the beginning of the second semester of 2022, as depicted in Figure 3.
During the SARS-CoV-2 pandemic, there was a significant co-circulation of variants within the same geographic location, this favored coinfections that led to recombination processes [7,47,48]. Several recombinant lineages have been reported for SARS-CoV-2. In Central America and the Dominican Republic, five recombinant lineages were predominant as seen in Figure 3. The first recombinant lineage in the region to exceed 35 occurrences was XB, with parental lineages B.1.634 and B.1.631. This lineage emerged in the United Stated but spread across North and Central America [7,47]. XB peaked in the region during June and July 2021.
The second recombinant lineage to surpass 35 occurrences was XAM (BA.1.1/BA.2.9) [48], which was most prevalent in the second quarter of 2022. It was primarily reported in Panama and the United States, but occurrences were also noted in other countries like the Dominican Republic [7,48]. The third recombinant to reach more than 35 occurrences was XAF, which emerged in Costa Rica and was parented by BA.1 and BA.2 [7]. Although it was predominantly found in the United States and Costa Rica [7], our data indicates it was also significantly present in Panama. Like XAM, XAF peaked in the second quarter of 2022.
In late 2022 and early 2023, the recombinant lineages XBB.1 and XBB.1.5 became highly predominant accounting for more than 50% of cases in Central America and the Dominican Republic. XBB.1 was parented by BJ.1 and BA.2.75 and was reported in India in August 2022, and quickly became predominant in Asia [49], later spreading to America that same year as illustrated in Figure 3. In contrast, XBB.1.5 was first detected in United States in October 2022 and evolved from XBB. It accounted for approximately 50% of cases in the United States and was recognized as a VOC [50,51].
The trends illustrated in Figure 5 reveal a noticeable decoupling between the temporal dynamics of SARS-CoV-2 lineages and epidemiological outcomes. The introduction and rapid expansion of Omicron sublineages (e.g., BA.1.1, BA.2, XBB.1.5) in early 2022 coincided with a marked increase in reported case numbers (Figure 5B), whereas COVID-19-related deaths continued to decline steadily during the same period (Figure 5A). This divergence was supported by a low overall correlation (r = 0.10) between the average monthly lineage proportions and total deaths. Additionally, although regression models using negative binomial distributions identified weak but statistically significant associations—such as BA.2.9 with decreased case counts (p = 0.028) and XBB.1 with reduced mortality (p = 0.0148)—no individual lineage accounted for a substantial portion of the variability in either trend (Supplementary Table S2). These patterns suggest that factors beyond lineage predominance, such as increased vaccination coverage, accumulated population immunity, or changes in clinical management and public health strategies, likely had a more prominent role in shaping the epidemic curve during the Omicron-dominant period.
The partnership between PAHO, COMISCA and the participant countries in Central America and the Dominican Republic was key to building regional capacity. It enabled countries without laboratories to access genomic surveillance and participate in sequencing efforts. This combined effort fosters the democratization of genomic tools in low- and middle-income countries, enabling them to contribute with data that supports evidence-based decision-making both at regional and global levels.
However, challenges remain, particularly in sequencing capacity in countries like Honduras impacted by socioeconomic inequalities and limited access to public health systems [52]. Addressing these gaps is essential to ensure robust responses to future pandemics or outbreaks. Additionally, several countries presented prolonged gaps in sequencing activity, particularly during the early stages of the pandemic. These gaps may be attributed to logistical constraints, limited laboratory infrastructure, lack of trained personnel, or resource limitations in public health systems. For example, Honduras and El Salvador reported very few sequences during key periods [28]. These disparities not only reflect structural inequalities across the region but also limit the representativeness of regional genomic trends. Similar gaps have been reported globally, approximately 63% of SARS-CoV-2 sequences lacked age or sex information and over 95% lacked clinical data such as vaccination status or symptom history [53].
Furthermore, genomic surveillance data plays a crucial role in informing public health decisions, including the development of appropriate tests, vaccines and treatment strategies [52]. This highlights the necessity for continued regional cooperation to effectively manage viral evolution and expand surveillance efforts to encompass other pathogens and antimicrobial resistance, which has seen a notable rise in recent years [52,54]. Investing in genomic surveillance will enhance our understanding of viral circulation dynamics and health systems impact. It also strengthens infectious disease control strategies and supports real-time intervention management [54]. Sustained genomic surveillance is vital for preparedness against future health threats, emphasizing the need for ongoing collaboration among countries.
Vaccination efforts across Central America and the Dominican Republic may have influenced the dynamics of lineage circulation [6]. Although this study did not assess vaccination coverage by country, it is plausible that higher vaccination rates helped reduce the spread or persistence of certain variants. Conversely, delays in vaccine rollout could have provided favorable conditions for the expansion of specific lineages. Future analyses integrating vaccination timelines and coverage data with genomic trends would provide valuable insights into the relationship between immunization and variant dynamics [55].
The genomic surveillance data obtained during the study period supported public health decision-making processes in the region. For instance, the detection of specific lineages contributed to the revision of travel advisories and the adjustment of entry requirements in some countries [36]. Additionally, the sequencing results provided evidence to refine vaccination strategies and prioritize populations at higher risk [11]. Although the short-term efforts were successful, a long-term surveillance strategy is essential. This includes sustained funding, regional laboratory networks, workforce training, and integration of genomic data into routine public health response systems [26,30].
Regional mobility likely played a critical role in the introduction and spread of SARS-CoV-2 lineages [36]. While the study did not include travel history metadata, the emergence of similar lineages in neighboring countries within a short time frame suggests transboundary movement of the virus [31]. Countries with stronger international connectivity, such as Panama and Costa Rica, may have served as primary entry points for founder lineages [56]. Additionally, the lack of detected VOCs, VOIs or VUMs in some countries may reflect gaps in sequencing coverage rather than true absence of circulation [30]. This underscores the need to integrate mobility data and improve coordinated genomic surveillance across borders.
Moreover, irregular migration may have contributed to viral spread through unconventional routes. One such situation is the Darién Gap, a dense jungle corridor between South America and Panama. In 2022, the Panamanian government reported over 250,000 migrants crossing this area under precarious conditions, potentially increasing the region’s epidemiological connectivity [57]. In addition, seasonal and binational migration among Indigenous populations, such as the Ngäbe–Buglé communities residing along the Panama–Costa Rica border, may also play a role in regional disease dynamics. These communities frequently migrate for agricultural work, particularly during Costa Rica’s coffee harvest, and often settle in towns near production areas. Many Ngäbe individuals are binational citizens who regularly access health services in Costa Rica, following routes that overlap with known northbound migratory pathways. These movements underscore the need for integrated, cross-border genomic surveillance strategies that account for the health vulnerabilities of highly mobile and underserved populations [58].
While several genomic surveillance studies have been conducted in Latin America [59,60,61,62], to our knowledge, this is the first study to describe the regional characteristics of the SARS-CoV-2 genome in Central America and the Dominican Republic. These findings provide critical public health insights by underscoring the need for equitable access to sequencing technologies and sustained regional collaboration. Such efforts are essential to effectively monitor viral evolution and inform evidence-based interventions in a region historically constrained by limited resources and infrastructure.
This study has several limitations. It relies on publicly available data with limited metadata, assumes that sequenced genomes accurately represent local and regional contexts, and does not account for country-specific factors such as diagnostic and surveillance investments, containment measures, or other relevant country-level indicators. Nonetheless, integrating regional genomic surveillance into national health systems could improve outbreak response times, enhance the detection of emerging variants, and enable more efficient allocation of scarce healthcare resources.
5. Conclusions
This overview of SARS-CoV-2 genomic surveillance in Central America and Dominican Republic highlights the critical role of the collaborative efforts between PAHO and COMISCA in enhancing regional sequencing capacity. By improving laboratory resources and fostering cooperation, countries effectively monitored key variants, including Delta and Omicron, demonstrating synchronous transmission dynamics across the region. The rise of recombinant variants emphasizes the ongoing evolution of the virus and the need to sustain genomic monitoring.
This collaborative model provides a robust framework for future public health crises, ensuring readiness to respond to emerging threats. Moving forward, countries should continue to build on these achievements, addressing opportunities for improvement in the sequencing capacity and promoting further collaborations to safeguard public health in the region.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org, Table S1: SARS-CoV-2 genomic surveillance datasets from the GISAID repository, covering Central America and the Dominican Republic from February 2020 to February 2023, Table S2: Regression Coefficients from Negative Binomial Models Assessing the Association Between Monthly SARS-CoV-2 Lineage Proportions and Reported Cases and Deaths in Central America and the Dominican Republic, Feb2020–Jan2023, Figure S1: Complementary Figure 1. Individual SARS-CoV-2 sequences obtained by country in Central America and Dominican Republic from February 2020 to January 2023. Panels show the top 30, 50 and 100 most prevalent lineages individually labeled, all the remaining lineages were labeled as other.
Autor Contributions: Conceptualization, Alexander Martínez, Estela Cordero-Laurent, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Jairo Mendez-Rico, Juliana Leite, Leticia Franco, Lionel Gresh, Monica Barahona, Naomi Iihoshi and Priscila Born; methodology, Alexander Martínez, Daniel Ulate, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Sofia Herrera Agüero; formal analysis, Alexander Martínez, Daniel Ulate, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Sofia Herrera Agüero; investigation, Alexander Martínez, Daniel Ulate, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Sofia Herrera Agüero; resources, Sofia Herrera Agüero, Aldo Sosa., Alexander Martínez, Ambar Moreno, César Roberto Conde Pereira, Claudia Gonzalez , Claudio Soto Garita, Daniel Ulate, Estela Cordero-Laurent, Hebleen Brenes, Isaac Miguel Sánchez, Jairo Mendez-Rico, Jessica Góndola, Jose Arturo Molina-Mora, Juliana Leite, Leticia Franco, Linda Mendoza, Lionel Gresh, Lucia De La Cruz, Mitzi Castro Paz, Monica Barahona, Naomi Iihoshi, Oris Chavarria, Priscila Born, Ruby Melany Aguillón, Ruth Carolina Vasquez Cordova, Selene Gonzalez, Sofia Carolina Alvarado Silva, Xochitl Sandoval López, Yvonne Imbert, Francisco Duarte-Martínez; data curation, Alexander Martínez, Daniel Ulate, Francisco Duarte-Martínez, Jose Arturo Molina-Mora; writing—original draft preparation, Sofia Herrera Agüero; writing—review and editing, Sofia Herrera Agüero, Alexander Martínez, Jose Arturo Molina-Mora, Francisco Duarte-Martínez; visualization, Alexander Martínez, Daniel Ulate, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Sofia Herrera Agüero; supervision, Alexander Martínez, Estela Cordero-Laurent, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Jairo Mendez-Rico, Juliana Leite, Leticia Franco, Lionel Gresh, Monica Barahona, Naomi Iihoshi and Priscila Born; project administration, Alexander Martínez, Estela Cordero-Laurent, Francisco Duarte-Martínez, Jose Arturo Molina-Mora, Jairo Mendez-Rico, Juliana Leite, Leticia Franco, Lionel Gresh, Monica Barahona, Naomi Iihoshi and Priscila Born; funding acquisition, Alexander Martínez, Jose Arturo Molina-Mora, Francisco Duarte-Martínez, Jairo Mendez-Rico, Juliana Leite, Leticia Franco, Lionel Gresh, Monica Barahona, Naomi Iihoshi and Priscila Born. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by: the University of Costa Rica, project “C0196 Bioinformatics and artificial intelligence protocol to support laboratory-based epidemiological surveillance of the SARS-CoV-2 virus by identifying genomic and clinical-demographic patterns in Costa Rica (2020-2024)”; the US Centers for Disease Control and Prevention Cooperative Agreement Number NU2HGH000022 “Enhancing Global Health Security: Expanding Efforts and Strategies to Protect and Improve Public Health Globally” awarded to Executive Secretary of the Council of Health Ministers of Central America and the Dominican Republic and Cooperative Agreement Number NU2HGH000086 “Shoulder to Shoulder: Accompanying Ministries of Health in Central America in Strengthening Local and Global Health Security” awarded to the Pan American Health Organization; and the Wellcome Trus grant number 223610/B/21/Z. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the US Centers for Disease Control and Prevention or the Department of Health and Human Services.
Informed Consent Statement
Informed consent was not required for this study due to its retrospective design, and primarily because the samples were processed in accordance with national legislation and institutional requirements as part of laboratory-based epidemiological surveillance for pathogens of public health significance.
Data Availability Statement
The original data presented in the study are openly available in SARS-CoV-2_CADR repository at https://github.com/sofiaherreraa/SARS-CoV-2_CADR/tree/main. Additionally, whole genome sequence datasets and the associated metadata are accessible through the GISAID repository at https://gisaid.org.
Acknowledgments
We wish to acknowledge the laboratories that provided positive SARS-COV-2 samples, as well as PAHO and COMISCA for their invaluable support. We are grateful for the work of public health laboratories and other institutions that generated SARS-CoV-2 genomic sequences, in particular, the Medical Laboratory Services (Belize), Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud (Costa Rica), Laboratorio Nacional de Salud Publica Dr. Defilló (Dominican Republic), Laboratorio Nacional de Salud Publica “Dr. Max Bloch” (El Salvador), Laboratorio Nacional de Salud (Guatemala), Laboratorio Nacional de Vigilancia de la Salud (Honduras), Centro Nacional de Diagnostico y Referencia (Nicaragua) and Instituto Conmemorativo Gorgas de Estudios de la Salud (Panama).Their cooperation and contribution were crucial to the successful completion of our research.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| COMISCA | Council of Ministers of Health of Central America |
| COVID-19 | Coronavirus disease 2019 |
| GISAID | Global Initiative on Sharing All Influenza Data |
| ICGES | Instituto Conmemorativo Gorgas de Estudios de la Salud |
| INCIENSA | Instituto Costarricense de Investigación y Enseñanza en Nutrición y Salud |
| PAHO | Pan American Health Organization |
| RNA | Ribonucleic acid |
| SARS | Severe acute respiratory syndrome |
| SE-COMISCA | COMISCA Executive Secretary |
| VOC | Variants of concern |
| VOI | Variants of interest |
| VUM | Variants under monitoring |
| WGS | Whole genome sequencing |
| WHO | World Health Organization |
References
- Zhou, P.; Yang, X. Lou; Wang, X. G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H. R.; Zhu, Y.; Li, B.; Huang, C. L.; Chen, H. D.; Chen, J.; Luo, Y.; Guo, H.; Jiang, R. Di; Liu, M. Q.; Chen, Y.; Shen, X. R.; Wang, X.; Zheng, X. S.; Zhao, K.; Chen, Q. J.; Deng, F.; Liu, L. L.; Yan, B.; Zhan, F. X.; Wang, Y. Y.; Xiao, G. F.; Shi, Z. L. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomedica 2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G. F.; Shi, W. The Emergence, Genomic Diversity and Global Spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Tracking SARS-CoV-2 variants, 2024.
- Pan American Health Organization. Update on the emergence of SARS-CoV-2 Omicron sublineages and recombination events (18 November 2022), 18 November 1066.
- Pan American Health Organization. Epidemiological Update: SARS-CoV-2 variants in the Region of the Americas (1 December 2021), 1 December 1066.
- Singh, P.; Sharma, K.; Shaw, D.; Bhargava, A.; Negi, S. S. Mosaic Recombination Inflicted Various SARS-CoV-2 Lineages to Emerge into Novel Virus Variants: A Review Update. Indian Journal of Clinical Biochemistry 2023, 38, 418–425. [Google Scholar] [CrossRef]
- Molina-Mora, J. A.; Cordero-Laurent, E.; Godínez, A.; Calderón-Osorno, M.; Brenes, H.; Soto-Garita, C.; Pérez-Corrales, C.; Drexler, J. F.; Moreira-Soto, A.; Corrales-Aguilar, E.; Duarte-Martínez, F. SARS-CoV-2 Genomic Surveillance in Costa Rica: Evidence of a Divergent Population and an Increased Detection of a Spike T1117I Mutation. Infection, Genetics and Evolution 2021, 92, 104872. [Google Scholar] [CrossRef] [PubMed]
- Molina-Mora, J. A. Insights into the Mutation T1117I in the Spike and the Lineage B.1.1.389 of SARS-CoV-2 Circulating in Costa Rica. Gene Rep 2022, 27, 101554. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E. C.; O’Toole, Á.; Hill, V.; McCrone, J. T.; Ruis, C.; du Plessis, L.; Pybus, O. G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat Microbiol 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Oude Munnink, B. B.; Worp, N.; Nieuwenhuijse, D. F.; Sikkema, R. S.; Haagmans, B.; Fouchier, R. A. M.; Koopmans, M. The next Phase of SARS-CoV-2 Surveillance: Real-Time Molecular Epidemiology. Nat Med 2021, 27, 1518–1524. [Google Scholar] [CrossRef]
- Stockdale, J. E.; Liu, P.; Colijn, C. The Potential of Genomics for Infectious Disease Forecasting. Nat Microbiol 2022, 7, 1736–1743. [Google Scholar] [CrossRef]
- Mathieu, E.; Ritchie, H.; Rodés-Guirao, L.; Appel, C.; Giattino, C.; Hasell, J.; Macdonald, B.; Dattani, S.; Beltekian, D.; Ortiz-Ospina, E.; Roser, M. Coronavirus Pandemic (COVID-19). https://ourworldindata.org/covid-cases.
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J. T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; Yeats, C.; du Plessis, L.; Maloney, D.; Medd, N.; Attwood, S. W.; Aanensen, D. M.; Holmes, E. C.; Pybus, O. G.; Rambaut, A. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol 2021, 7. [Google Scholar] [CrossRef]
- Alpert, T.; Brito, A. F.; Lasek-Nesselquist, E.; Rothman, J.; Valesano, A. L.; MacKay, M. J.; Petrone, M. E.; Breban, M. I.; Watkins, A. E.; Vogels, C. B. F.; Kalinich, C. C.; Dellicour, S.; Russell, A.; Kelly, J. P.; Shudt, M.; Plitnick, J.; Schneider, E.; Fitzsimmons, W. J.; Khullar, G.; Metti, J.; Dudley, J. T.; Nash, M.; Beaubier, N.; Wang, J.; Liu, C.; Hui, P.; Muyombwe, A.; Downing, R.; Razeq, J.; Bart, S. M.; Grills, A.; Morrison, S. M.; Murphy, S.; Neal, C.; Laszlo, E.; Rennert, H.; Cushing, M.; Westblade, L.; Velu, P.; Craney, A.; Cong, L.; Peaper, D. R.; Landry, M. L.; Cook, P. W.; Fauver, J. R.; Mason, C. E.; Lauring, A. S.; St. George, K.; MacCannell, D. R.; Grubaugh, N. D. Early Introductions and Transmission of SARS-CoV-2 Variant B.1.1.7 in the United States. Cell 2021, 184, 2595–2604e13. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Minh, B. Q.; Schmidt, H. A.; Chernomor, O.; Schrempf, D.; Woodhams, M. D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B. Q.; Wong, T. K. F.; von Haeseler, A.; Jermiin, L. S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v4: Recent Updates and New Developments. Nucleic Acids Res 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Pan American Health Organization. Evaluation of the Pan American Health Organization Response to COVID-19 2020–2022. Volume I. Final Report, 2023. [CrossRef]
- COMISCA. Acuerdo Cooperativo Salud Global COMISCA/CDC apoya el fortalecimiento al Laboratorio INCIENSA, 1278.
- U.S. Census Bureau. International Database (IDB), 2023.
- Taboada, B.; Zárate, S.; García-López, R.; Muñoz-Medina, J. E.; Sanchez-Flores, A.; Herrera-Estrella, A.; Boukadida, C.; Gómez-Gil, B.; Mojica, N. S.; Rosales-Rivera, M.; Salas-Lais, A. G.; Gutiérrez-Ríos, R. M.; Loza, A.; Rivera-Gutierrez, X.; Vazquez-Perez, J. A.; Matías-Florentino, M.; Pérez-García, M.; Ávila-Ríos, S.; Hurtado, J. M.; Herrera-Nájera, C. I.; Núñez-Contreras, J. de J.; Sarquiz-Martínez, B.; García-Arias, V. E.; Santiago-Mauricio, M. G.; Martínez-Miguel, B.; Enciso-Ibarra, J.; Cháidez-Quiróz, C.; Iša, P.; Wong-Chew, R. M.; Jiménez-Corona, M. E.; López, S.; Arias, C. F. Dominance of Three Sublineages of the SARS-CoV-2 Delta Variant in Mexico. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA.1) and Sub-variants (BA.1.1, BA.2, and BA.3) of SARS-CoV-2 Spike Infectivity and Pathogenicity: A Comparative Sequence and Structural-based Computational Assessment. J Med Virol 2022, 94, 4780–4791. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; Nao, N.; Itakura, Y.; Deguchi, S.; Suzuki, R.; Wang, L.; Begum, M. M.; Kita, S.; Yajima, H.; Sasaki, J.; Sasaki-Tabata, K.; Shimizu, R.; Tsuda, M.; Kosugi, Y.; Fujita, S.; Pan, L.; Sauter, D.; Yoshimatsu, K.; Suzuki, S.; Asakura, H.; Nagashima, M.; Sadamasu, K.; Yoshimura, K.; Yamamoto, Y.; Nagamoto, T.; Schreiber, G.; Maenaka, K.; Ito, H.; Misawa, N.; Kimura, I.; Suganami, M.; Chiba, M.; Yoshimura, R.; Yasuda, K.; Iida, K.; Ohsumi, N.; Strange, A. P.; Takahashi, O.; Ichihara, K.; Shibatani, Y.; Nishiuchi, T.; Kato, M.; Ferdous, Z.; Mouri, H.; Shishido, K.; Sawa, H.; Hashimoto, R.; Watanabe, Y.; Sakamoto, A.; Yasuhara, N.; Suzuki, T.; Kimura, K.; Nakajima, Y.; Nakagawa, S.; Wu, J.; Shirakawa, K.; Takaori-Kondo, A.; Nagata, K.; Kazuma, Y.; Nomura, R.; Horisawa, Y.; Tashiro, Y.; Kawai, Y.; Irie, T.; Kawabata, R.; Motozono, C.; Toyoda, M.; Ueno, T.; Hashiguchi, T.; Ikeda, T.; Fukuhara, T.; Saito, A.; Tanaka, S.; Matsuno, K.; Takayama, K.; Sato, K. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants; 2023; Vol. 14. [CrossRef]
- Gardy, J. L.; Loman, N. J. Towards a Genomics-Informed, Real-Time, Global Pathogen Surveillance System. Nat Rev Genet 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Saravanan, K. A.; Panigrahi, M.; Kumar, H.; Rajawat, D.; Nayak, S. S.; Bhushan, B.; Dutt, T. Role of Genomics in Combating COVID-19 Pandemic. Gene, 20 May 2022. [Google Scholar] [CrossRef]
- Durón, R. M.; Sánchez, E.; Choi, J. N.; Peralta, G.; Ventura, S. G.; Soto, R. J.; Rodríguez, G.; Ahrens, C.; Farach, E.; Figueroa, J.; Pineda, G.; Romero, A.; Rodríguez, O.; Discua, D.; Salgado, J. Honduras: Two Hurricanes, COVID-19, Dengue and the Need for a New Digital Health Surveillance System. J Public Health (Bangkok) 2021, 43, e297–e298. [Google Scholar] [CrossRef]
- Vavrek, D.; Speroni, L.; Curnow, K. J.; Oberholzer, M.; Moeder, V.; Febbo, P. G. Genomic Surveillance at Scale Is Required to Detect Newly Emerging Strains at an Early Timepoint. 2021. [CrossRef]
- Brito, A. F.; Semenova, E.; Dudas, G.; Hassler, G. W.; Kalinich, C. C.; Kraemer, M. U. G.; Ho, J.; Tegally, H.; Githinji, G.; Agoti, C. N.; Matkin, L. E.; Whittaker, C.; Kantardjiev, T.; Korsun, N.; Stoitsova, S.; Dimitrova, R.; Trifonova, I.; Dobrinov, V.; Grigorova, L.; Stoykov, I.; Grigorova, I.; Gancheva, A.; Jennison, A.; Leong, L.; Speers, D.; Baird, R.; Cooley, L.; Kennedy, K.; de Ligt, J.; Rawlinson, W.; van Hal, S.; Williamson, D.; Singh, R.; Nathaniel-Girdharrie, S. M.; Edghill, L.; Indar, L.; St. John, J.; Gonzalez-Escobar, G.; Ramkisoon, V.; Brown-Jordan, A.; Ramjag, A.; Mohammed, N.; Foster, J. E.; Potter, I.; Greenaway-Duberry, S.; George, K.; Belmar-George, S.; Lee, J.; Bisasor-McKenzie, J.; Astwood, N.; Sealey-Thomas, R.; Laws, H.; Singh, N.; Oyinloye, A.; McMillan, P.; Hinds, A.; Nandram, N.; Parasram, R.; Khan-Mohammed, Z.; Charles, S.; Andrewin, A.; Johnson, D.; Keizer-Beache, S.; Oura, C.; Pybus, O. G.; Faria, N. R.; Stegger, M.; Albertsen, M.; Fomsgaard, A.; Rasmussen, M.; Khouri, R.; Naveca, F.; Graf, T.; Miyajima, F.; Wallau, G.; Motta, F.; Khare, S.; Freitas, L.; Schiavina, C.; Bach, G.; Schultz, M. B.; Chew, Y. H.; Makheja, M.; Born, P.; Calegario, G.; Romano, S.; Finello, J.; Diallo, A.; Lee, R. T. C.; Xu, Y. N.; Yeo, W.; Tiruvayipati, S.; Yadahalli, S.; Wilkinson, E.; Iranzadeh, A.; Giandhari, J.; Doolabh, D.; Pillay, S.; Ramphal, U.; San, J. E.; Msomi, N.; Mlisana, K.; von Gottberg, A.; Walaza, S.; Ismail, A.; Mohale, T.; Engelbrecht, S.; Van Zyl, G.; Preiser, W.; Sigal, A.; Hardie, D.; Marais, G.; Hsiao, M.; Korsman, S.; Davies, M. A.; Tyers, L.; Mudau, I.; York, D.; Maslo, C.; Goedhals, D.; Abrahams, S.; Laguda-Akingba, O.; Alisoltani-Dehkordi, A.; Godzik, A.; Wibmer, C. K.; Martin, D.; Lessells, R. J.; Bhiman, J. N.; Williamson, C.; de Oliveira, T.; Chen, C.; Nadeau, S.; du Plessis, L.; Beckmann, C.; Redondo, M.; Kobel, O.; Noppen, C.; Seidel, S.; de Souza, N. S.; Beerenwinkel, N.; Topolsky, I.; Jablonski, P.; Fuhrmann, L.; Dreifuss, D.; Jahn, K.; Ferreira, P.; Posada-Céspedes, S.; Beisel, C.; Denes, R.; Feldkamp, M.; Nissen, I.; Santacroce, N.; Burcklen, E.; Aquino, C.; de Gouvea, A. C.; Moccia, M. D.; Grüter, S.; Sykes, T.; Opitz, L.; White, G.; Neff, L.; Popovic, D.; Patrignani, A.; Tracy, J.; Schlapbach, R.; Dermitzakis, E.; Harshman, K.; Xenarios, I.; Pegeot, H.; Cerutti, L.; Penet, D.; Stadler, T.; Howden, B. P.; Sintchenko, V.; Zuckerman, N. S.; Mor, O.; Blankenship, H. M.; de Oliveira, T.; Lin, R. T. P.; Siqueira, M. M.; Resende, P. C.; Vasconcelos, A. T. R.; Spilki, F. R.; Aguiar, R. S.; Alexiev, I.; Ivanov, I. N.; Philipova, I.; Carrington, C. V. F.; Sahadeo, N. S. D.; Branda, B.; Gurry, C.; Maurer-Stroh, S.; Naidoo, D.; von Eije, K. J.; Perkins, M. D.; van Kerkhove, M.; Hill, S. C.; Sabino, E. C.; Pybus, O. G.; Dye, C.; Bhatt, S.; Flaxman, S.; Suchard, M. A.; Grubaugh, N. D.; Baele, G.; Faria, N. R. Global Disparities in SARS-CoV-2 Genomic Surveillance. Nat Commun 2022, 13. [Google Scholar] [CrossRef]
- Mir, D.; Rego, N.; Resende, P. C.; Tort, F.; Fernández-Calero, T.; Noya, V.; Brandes, M.; Possi, T.; Arleo, M.; Reyes, N.; Victoria, M.; Lizasoain, A.; Castells, M.; Maya, L.; Salvo, M.; Schäffer Gregianini, T.; Mar da Rosa, M. T.; Garay Martins, L.; Alonso, C.; Vega, Y.; Salazar, C.; Ferrés, I.; Smircich, P.; Sotelo Silveira, J.; Fort, R. S.; Mathó, C.; Arantes, I.; Appolinario, L.; Mendonça, A. C.; Benítez-Galeano, M. J.; Simoes, C.; Graña, M.; Motta, F.; Siqueira, M. M.; Bello, G.; Colina, R.; Spangenberg, L. Recurrent Dissemination of SARS-CoV-2 Through the Uruguayan–Brazilian Border. Front Microbiol 2021, 12. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Muñoz, M.; Florez, C.; Gomez, S.; Rico, A.; Pardo, L.; Barros, E. C.; Hernández, C.; Delgado, L.; Jaimes, J. E.; Pérez, L.; Teherán, A. A.; Alshammary, H. A.; Obla, A.; Khan, Z.; Dutta, J.; van de Guchte, A.; Gonzalez-Reiche, A. S.; Hernandez, M. M.; Sordillo, E. M.; Simon, V.; van Bakel, H.; Llewellyn, M. S.; Ramírez, J. D. SARS-CoV-2 Spread across the Colombian-Venezuelan Border. Infection, Genetics and Evolution 2020, 86. [Google Scholar] [CrossRef]
- Patiño, L. H.; Ballesteros, N.; Muñoz, M.; Ramírez, A. L.; Luna, N.; Castañeda, S.; Gutierrez-Marin, R.; Mendoza-Ibarra, J. A.; Rodriguez, R.; Bohada, D. P.; Ramírez, J. D.; Paniz-Mondolfi, A. Mu SARS-CoV-2 (B.1.621) Variant: A Genomic Snapshot across the Colombian-Venezuelan Border. J Med Virol 2023, 95. [Google Scholar] [CrossRef]
- Hodcroft, E. B.; Zuber, M.; Nadeau, S.; Vaughan, T. G.; Crawford, K. H. D.; Althaus, C. L.; Reichmuth, M. L.; Bowen, J. E.; Walls, A. C.; Corti, D.; Bloom, J. D.; Veesler, D.; Mateo, D.; Hernando, A.; Comas, I.; González-Candelas, F.; González-Candelas, F.; Goig, G. A.; Chiner-Oms, Á.; Cancino-Muñoz, I.; López, M. G.; Torres-Puente, M.; Gomez-Navarro, I.; Jiménez-Serrano, S.; Ruiz-Roldán, L.; Bracho, M. A.; García-González, N.; Martínez-Priego, L.; Galán-Vendrell, I.; Ruiz-Hueso, P.; De Marco, G.; Ferrús, M. L.; Carbó-Ramírez, S.; D’Auria, G.; Coscollá, M.; Ruiz-Rodríguez, P.; Roig-Sena, F. J.; Sanmartín, I.; Garcia-Souto, D.; Pequeno-Valtierra, A.; Tubio, J. M. C.; Rodríguez-Castro, J.; Rabella, N.; Navarro, F.; Miró, E.; Rodríguez-Iglesias, M.; Galán-Sanchez, F.; Rodriguez-Pallares, S.; de Toro, M.; Escudero, M. B.; Azcona-Gutiérrez, J. M.; Alberdi, M. B.; Mayor, A.; García-Basteiro, A. L.; Moncunill, G.; Dobaño, C.; Cisteró, P.; García-de-Viedma, D.; Pérez-Lago, L.; Herranz, M.; Sicilia, J.; Catalán-Alonso, P.; Muñoz, P.; Muñoz-Cuevas, C.; Rodríguez-Rodríguez, G.; Alberola-Enguidanos, J.; Nogueira, J. M.; Camarena, J. J.; Rezusta, A.; Tristancho-Baró, A.; Milagro, A.; Martínez-Cameo, N. F.; Gracia-Grataloup, Y.; Martró, E.; Bordoy, A. E.; Not, A.; Antuori-Torres, A.; Benito, R.; Algarate, S.; Bueno, J.; del Pozo, J. L.; Boga, J. A.; Castelló-Abietar, C.; Rojo-Alba, S.; Alvarez-Argüelles, M. E.; Melon, S.; Aranzamendi-Zaldumbide, M.; Vergara-Gómez, A.; Fernández-Pinero, J.; Martínez, M. J.; Vila, J.; Rubio, E.; Peiró-Mestres, A.; Navero-Castillejos, J.; Posada, D.; Valverde, D.; Estévez-Gómez, N.; Fernandez-Silva, I.; de Chiara, L.; Gallego-García, P.; Varela, N.; Moreno, R.; Tirado, M. D.; Gomez-Pinedo, U.; Gozalo-Margüello, M.; Eliecer-Cano, M.; Méndez-Legaza, J. M.; Rodríguez-Lozano, J.; Siller, M.; Pablo-Marcos, D.; Oliver, A.; Reina, J.; López-Causapé, C.; Canut-Blasco, A.; Hernáez-Crespo, S.; Cordón, M. L. A.; Lecároz-Agara, M. C.; Gómez-González, C.; Aguirre-Quiñonero, A.; López-Mirones, J. I.; Fernández-Torres, M.; Almela-Ferrer, M. R.; Gonzalo-Jiménez, N.; Ruiz-García, M. M.; Galiana, A.; Sanchez-Almendro, J.; Cilla, G.; Montes, M.; Piñeiro, L.; Sorarrain, A.; Marimón, J. M.; Gomez-Ruiz, M. D.; López-Hontangas, J. L.; González Barberá, E. M.; Navarro-Marí, J. M.; Pedrosa-Corral, I.; Sanbonmatsu-Gámez, S.; Pérez-González, C.; Chamizo-López, F.; Bordes-Benítez, A.; Navarro, D.; Albert, E.; Torres, I.; Gascón, I.; Torregrosa-Hetland, C. J.; Pastor-Boix, E.; Cascales-Ramos, P.; Fuster-Escrivá, B.; Gimeno-Cardona, C.; Ocete, M. D.; Medina-Gonzalez, R.; González-Cantó, J.; Martínez-Macias, O.; Palop-Borrás, B.; de Toro, I.; Mediavilla-Gradolph, M. C.; Pérez-Ruiz, M.; González-Recio, Ó.; Gutiérrez-Rivas, M.; Simarro-Córdoba, E.; Lozano-Serra, J.; Robles-Fonseca, L.; de Salazar, A.; Viñuela-González, L.; Chueca, N.; García, F.; Gómez-Camarasa, C.; Carvajal, A.; de la Puente, R.; Martín-Sánchez, V.; Fregeneda-Grandes, J. M.; Molina, A. J.; Argüello, H.; Fernández-Villa, T.; Farga-Martí, M. A.; Domínguez-Márquez, V.; Costa-Alcalde, J. J.; Trastoy, R.; Barbeito-Castiñeiras, G.; Coira, A.; Pérez-del-Molino, M. L.; Aguilera, A.; Planas, A. M.; Soriano, A.; Fernandez-Cádenas, I.; Pérez-Tur, J.; Marcos, M. Á.; Moreno-Docón, A.; Viedma, E.; Mingorance, J.; Galán-Montemayor, J. C.; Parra-Grande, M.; Stadler, T.; Neher, R. A. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Serwin, K.; Aksak-Was˛, B.; Parczewski, M. Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Arora, P.; Mrig, S.; Goldust, Y.; Kroumpouzos, G.; Karadağ, A. S.; Rudnicka, L.; Galadari, H.; Szepietowski, J. C.; Di Lernia, V.; Goren, A.; Kassir, M.; Goldust, M. New Coronavirus (Sars-Cov-2) Crossing Borders beyond Cities, Nations, and Continents: Impact of International Travel. Balkan Med J 2021, 38, 205–211. [Google Scholar] [CrossRef]
- Choi, J. Y.; Smith, D. M. SARS-CoV-2 Variants of Concern. Yonsei Med J 2021, 62, 961–968. [Google Scholar] [CrossRef]
- Zárate, S.; Taboada, B.; Muñoz-Medina, J. E.; Iša, P.; Sanchez-Flores, A.; Boukadida, C.; Herrera-Estrella, A.; Selem Mojica, N.; Rosales-Rivera, M.; Gómez-Gil, B.; Salas-Lais, A. G.; Santacruz-Tinoco, C. E.; Montoya-Fuentes, H.; Alvarado-Yaah, J. E.; Molina-Salinas, G. M.; Espinoza-Ayala, G. E.; Enciso-Moreno, J. A.; Gutiérrez-Ríos, R. M.; Loza, A.; Moreno-Contreras, J.; García-López, R.; Rivera-Gutierrez, X.; Comas-García, A.; Wong-Chew, R. M.; Jiménez-Corona, M.-E.; del Angel, R. M.; Vazquez-Perez, J. A.; Matías-Florentino, M.; Pérez-García, M.; Ávila-Ríos, S.; Castelán-Sánchez, H. G.; Delaye, L.; Martínez-Castilla, L. P.; Escalera-Zamudio, M.; López, S.; Arias, C. F. The Alpha Variant (B.1.1.7) of SARS-CoV-2 Failed to Become Dominant in Mexico. Microbiol Spectr 2022, 10. [Google Scholar] [CrossRef]
- Stefanelli, P.; Trentini, F.; Guzzetta, G.; Marziano, V.; Mammone, A.; Schepisi, M. S.; Poletti, P.; Grané, C. M.; Manica, M.; del Manso, M.; Andrianou, X.; Ajelli, M.; Rezza, G.; Brusaferro, S.; Merler, S.; Di Martino, A.; Ambrosio, L.; Lo Presti, A.; Fiore, S.; Fabiani, C.; Benedetti, E.; Di Mario, G.; Facchini, M.; Puzelli, S.; Calzoletti, L.; Fontana, S.; Venturi, G.; Fortuna, C.; Marsili, G.; Amendola, A.; Stuppia, L.; Savini, G.; Picerno, A.; Lopizzo, T.; Dell’Edera, D.; Minchella, P.; Greco, F.; Viglietto, G.; Atripaldi, L.; Limone, A.; D’Agaro, P.; Licastro, D.; Pongolini, S.; Sambri, V.; Dirani, G.; Zannoli, S.; Affanni, P.; Colucci, M. E.; Capobianchi, M. R.; Icardi, G.; Bruzzone, B.; Lillo, F.; Orsi, A.; Pariani, E.; Baldanti, F.; Molecolare, U. V.; Gismondo, M. R.; Maggi, F.; Caruso, A.; Ceriotti, F.; Boniotti, M. B.; Barbieri, I.; Bagnarelli, P.; Menzo, S.; Garofalo, S.; Scutellà, M.; Pagani, E.; Collini, L.; Ghisetti, V.; Brossa, S.; Ru, G.; Bozzetta, E.; Chironna, M.; Parisi, A.; Rubino, S.; Serra, C.; Piras, G.; Coghe, F.; Vitale, F.; Tramuto, F.; Scalia, G.; Palermo, C. I.; Mancuso, G.; Pollicino, T.; Di Gaudio, F.; Vullo, S.; Reale, S.; Cusi, M. G.; Rossolini, G. M.; Pistello, M.; Mencacci, A.; Camilloni, B.; Severini, S.; Di Benedetto, M.; Terregino, C.; Monne, I.; Biscaro, V. Co-Circulation of SARS-CoV-2 Alpha and Gamma Variants in Italy, February and March 2021. Eurosurveillance 2022, 27. [Google Scholar] [CrossRef]
- Oróstica, K. Y.; Mohr, S. B.; Dehning, J.; Bauer, S.; Medina-Ortiz, D.; Iftekhar, E. N.; Mujica, K.; Covarrubias, P. C.; Ulloa, S.; Castillo, A. E.; Daza-Sánchez, A.; Verdugo, R. A.; Fernández, J.; Olivera-Nappa, Á.; Priesemann, V.; Contreras, S. Early Mutational Signatures and Transmissibility of SARS-CoV-2 Gamma and Lambda Variants in Chile. Sci Rep 2024, 14. [Google Scholar] [CrossRef]
- Rahimi, F.; Kamali, N.; Bezmin Abadi, A. T. The Mu Strain: The Last but Not Least Circulating Variant of Interest’ Potentially Affecting the COVID-19 Pandemic. Future Virology, 1 January 2022. [Google Scholar] [CrossRef]
- Giovanetti, M.; Fonseca, V.; Wilkinson, E.; Tegally, H.; San, E. J.; Althaus, C. L.; Xavier, J.; Nanev Slavov, S.; Viala, V. L.; Ranieri Jerônimo Lima, A.; Ribeiro, G.; Souza-Neto, J. A.; Fukumasu, H.; Lehmann Coutinho, L.; Venancio Da Cunha, R.; Freitas, C.; Campelo De A E Melo, C. F.; Navegantes De Araújo, W.; Do Carmo Said, R. F.; Almiron, M.; De Oliveira, T.; Coccuzzo Sampaio, S.; Elias, M. C.; Covas, D. T.; Holmes, E. C.; Lourenço, J.; Kashima, S.; De Alcantara, L. C. J. Replacement of the Gamma by the Delta Variant in Brazil: Impact of Lineage Displacement on the Ongoing Pandemic. Virus Evol 2022, 8. [Google Scholar] [CrossRef]
- Fall, A.; Eldesouki, R. E.; Sachithanandham, J.; Morris, C. P.; Norton, J. M.; Gaston, D. C.; Forman, M.; Abdullah, O.; Gallagher, N.; Li, M.; Swanson, N. J.; Pekosz, A.; Klein, E. Y.; Mostafa, H. H.; Feinstone, W. H. The Displacement of the SARS-CoV-2 Variant Delta with Omicron: An Investigation of Hospital Admissions and Upper Respiratory Viral Loads. 2022. [CrossRef]
- Wang, Y.; Long, Y.; Wang, F.; Li, C.; Liu, W. Characterization of SARS-CoV-2 Recombinants and Emerging Omicron Sublineages. Int J Med Sci 2023, 20, 151–162. [Google Scholar] [CrossRef]
- Lee, W. L.; Armas, F.; Guarneri, F.; Gu, X.; Formenti, N.; Wu, F.; Chandra, F.; Parisio, G.; Chen, H.; Xiao, A.; Romeo, C.; Scali, F.; Tonni, M.; Leifels, M.; Chua, F. J. D.; Kwok, G. W.; Tay, J. Y.; Pasquali, P.; Thompson, J.; Alborali, G. L.; Alm, E. J. Rapid Displacement of SARS-CoV-2 Variant Delta by Omicron Revealed by Allele-Specific PCR in Wastewater. Water Res 2022, 221. [Google Scholar] [CrossRef]
- Andre, M.; Lau, L. S.; Pokharel, M. D.; Ramelow, J.; Owens, F.; Souchak, J.; Akkaoui, J.; Ales, E.; Brown, H.; Shil, R.; Nazaire, V.; Manevski, M.; Paul, N. P.; Esteban-Lopez, M.; Ceyhan, Y.; El-Hage, N. From Alpha to Omicron: How Different Variants of Concern of the SARS-Coronavirus-2 Impacted the World. Biology (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Shiraz, R.; Tripathi, S. Enhanced Recombination Among SARS-CoV-2 Omicron Variants Contributes to Viral Immune Escape. 2022. [CrossRef]
- Paulino-Ramírez, R.; Pham, K.; Breban, M. I.; Peguero, A.; Jabier, M.; Sánchez, N.; Eustate, I.; Ruiz, I.; Grubaugh, N. D.; Hahn, A. M. Genome Sequence of a Recombinant SARS-CoV-2 Lineage XAM (BA.1.1/BA.2.9) Strain from a Clinical Sample in Santo Domingo, Dominican Republic. Microbiol Resour Announc 2023, 12. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A. D.; Liu, M.; Wang, M.; Yu, J.; Valdez, R.; Lauring, A. S.; Sheng, Z.; Wang, H. H.; Gordon, A.; Liu, L.; Ho, D. D. Alarming Antibody Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286e8. [Google Scholar] [CrossRef]
- Parums, D. V. Editorial: The XBB.1.5 (‘Kraken’) Subvariant of Omicron SARS-CoV-2 and Its Rapid Global Spread. Medical Science Monitor, 2023. [Google Scholar] [CrossRef]
- Vogel, L. What to Know about Omicron XBB.1.5. CMAJ. Canadian Medical Association Journal 2023, 195, E127–E128. [Google Scholar] [CrossRef]
- Brito, A. F.; Semenova, E.; Dudas, G.; Hassler, G. W.; Kalinich, C. C.; Kraemer, M. U. G.; Ho, J.; Tegally, H.; Githinji, G.; Agoti, C. N.; Matkin, L. E.; Whittaker, C.; Howden, B. P.; Sintchenko, V.; Zuckerman, N. S.; Mor, O.; Blankenship, H. M.; Oliveira, T. de; Lin, R. T. P.; Siqueira, M. M.; Resende, P. C.; Vasconcelos, A. T. R.; Spilki, F. R.; Aguiar, R. S.; Alexiev, I.; Ivanov, I. N.; Philipova, I.; Carrington, C. V. F.; Sahadeo, N. S. D.; Gurry, C.; Maurer-Stroh, S.; Naidoo, D.; von Eije, K. J.; Perkins, M. D.; Kerkhove, M. van; Hill, S. C.; Sabino, E. C.; Pybus, O. G.; Dye, C.; Bhatt, S.; Flaxman, S.; Suchard, M. A.; Grubaugh, N. D.; Baele, G.; Faria, N. R. Global Disparities in SARS-CoV-2 Genomic Surveillance. , 2021. 26 August. [CrossRef]
- Wheeler, N. E.; Price, V.; Cunningham-Oakes, E.; Tsang, K. K.; Nunn, J. G.; Midega, J. T.; Anjum, M. F.; Wade, M. J.; Feasey, N. A.; Peacock, S. J.; Jauneikaite, E.; Baker, K. S. Innovations in Genomic Antimicrobial Resistance Surveillance. The Lancet Microbe, 1 December 2023. [Google Scholar] [CrossRef]
- Jena, D.; Ghosh, A.; Jha, A.; Prasad, P.; Raghav, S. K. Impact of vaccination on SARS-CoV-2 evolution and immune escape variants. Vaccine 2024, 42(21), 126153. [Google Scholar] [CrossRef]
- Franco, D.; Gonzalez, C.; Abrego, L. E.; Carrera, J. P.; Diaz, Y.; Caicedo, Y.; et al. Early transmission dynamics, spread, and genomic characterization of SARS-CoV-2 in Panama. Emerg Infect Dis 2021, 27(2), 612–615. [Google Scholar] [CrossRef]
- Datos Abiertos de Panamá. Migración - Irregulares en tránsito por Darién por país 2022 (16 de enero de 2023). 2022.
- Cumbrera, A.; Calzada, J. E.; Chaves, L. F.; Hurtado, L. A. Spatiotemporal analysis of malaria transmission in the autonomous indigenous regions of Panama, Central America, 2015–2022. Trop Med Infect Dis 2024, 9(4), 90. [Google Scholar] [CrossRef]
- Molina-Mora, J. A.; Reales-González, J.; Camacho, E.; Duarte-Martínez, F.; Tsukayama, P.; Soto-Garita, C.; Brenes, H.; Cordero-Laurent, E.; Ribeiro dos Santos, A.; Guedes Salgado, C.; Santos Silva, C.; Santana de Souza, J.; Nunes, G.; Negri, T.; Vidal, A.; Oliveira, R.; Oliveira, G.; Muñoz-Medina, J. E.; Salas-Lais, A. G.; Mireles-Rivera, G.; Sosa, E.; Turjanski, A.; Monzani, M. C.; Carobene, M. G.; Remes Lenicov, F.; Schottlender, G.; Fernández Do Porto, D. A.; Kreuze, J. F.; Sacristán, L.; Guevara-Suarez, M.; Cristancho, M.; Campos-Sánchez, R.; Herrera-Estrella, A. Overview of the SARS-CoV-2 Genotypes Circulating in Latin America during 2021. Front Public Health 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Concha-Toloza, M.; González, L. C.; Estrella, A. H. H.; Porto, D. F. Do; Campos-Sánchez, R.; Molina-Mora, J. A. Genomic, Socio-Environmental, and Sequencing Capability Patterns in the Surveillance of SARS-CoV-2 in Latin America and the Caribbean up to 2023. , 2024. 15 November. [CrossRef]
- Lira-Morales, J. D.; López-Cuevas, O.; Medrano-Félix, J. A.; González-Gómez, J. P.; González-López, I.; Castro-Del Campo, N.; Gomez-Gil, B.; Chaidez, C. Genomic Surveillance of SARS-CoV-2 in México: Three Years since Wuhan, China’s First Reported Case. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro Dias, M. F.; Andriolo, B. V.; Silvestre, D. H.; Cascabulho, P. L.; Leal da Silva, M. Genomic Surveillance and Sequencing of SARS-CoV-2 across South America. Revista Panamericana de Salud Pública 2023, 47, 1. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Timeline of processes implemented to strengthen genomic surveillance of SARS-CoV-2 in Central America and the Dominican Republic.
Figure 1.
Timeline of processes implemented to strengthen genomic surveillance of SARS-CoV-2 in Central America and the Dominican Republic.
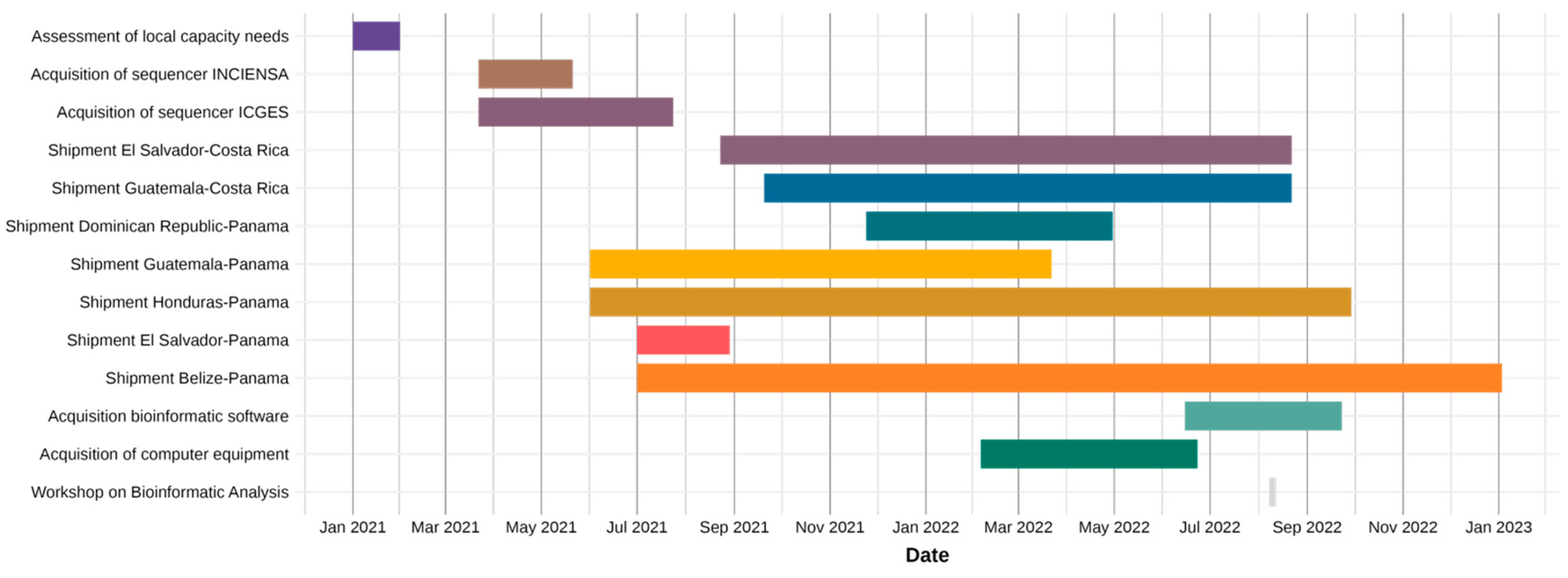
Figure 2.
Individual SARS-CoV-2 sequences obtained by country in Central America and Dominican Republic from February 2020 to January 2023. The top 14 most prevalent lineages were individually labeled, and all the remaining lineages were labeled as other.
Figure 2.
Individual SARS-CoV-2 sequences obtained by country in Central America and Dominican Republic from February 2020 to January 2023. The top 14 most prevalent lineages were individually labeled, and all the remaining lineages were labeled as other.
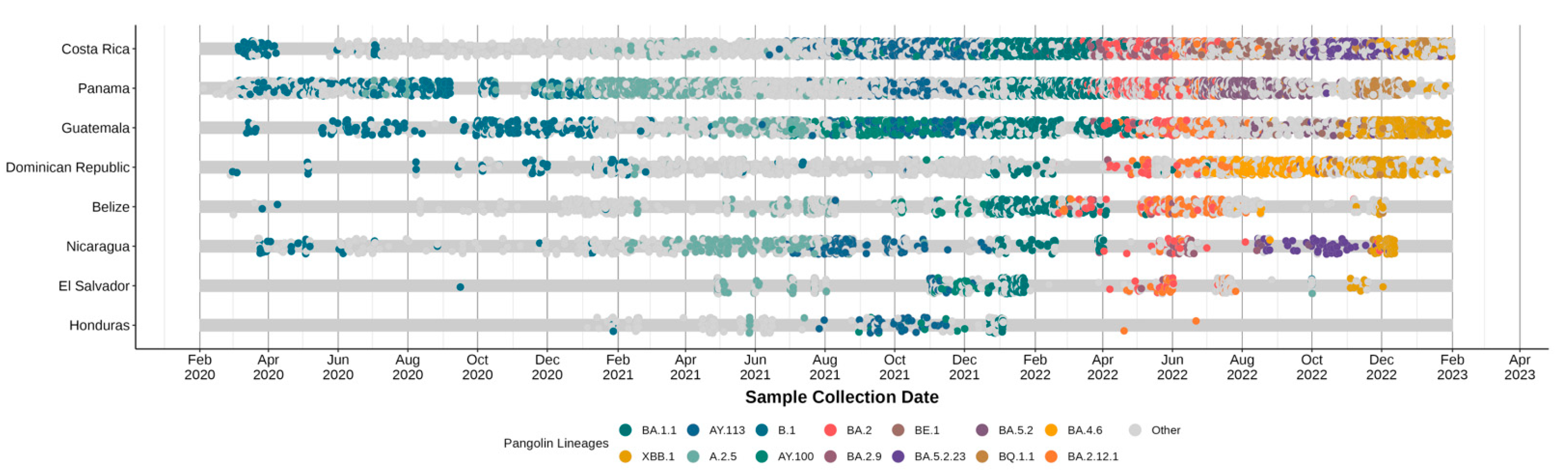
Figure 3.
Monthly relative frequency of SARS-CoV-2 lineages circulating in Central America and the Dominican Republic from February 2020 to January 2023. (a) Overall lineage distribution in the region. Lineages with a cumulative frequency greater than 35 sequences were shown individually; less frequent lineages (n ≤ 35) were grouped under “Others”. Percentages represent the relative frequency of each lineage per month. (b–i) Country-specific lineage distributions. The lower panels display the monthly distribution of lineages in each country. Only months with available data are shown, with blank spaces indicating the absence of sampling. The color palette and lineage grouping are consistent with the overall chart, including the “Others” category.
Figure 3.
Monthly relative frequency of SARS-CoV-2 lineages circulating in Central America and the Dominican Republic from February 2020 to January 2023. (a) Overall lineage distribution in the region. Lineages with a cumulative frequency greater than 35 sequences were shown individually; less frequent lineages (n ≤ 35) were grouped under “Others”. Percentages represent the relative frequency of each lineage per month. (b–i) Country-specific lineage distributions. The lower panels display the monthly distribution of lineages in each country. Only months with available data are shown, with blank spaces indicating the absence of sampling. The color palette and lineage grouping are consistent with the overall chart, including the “Others” category.
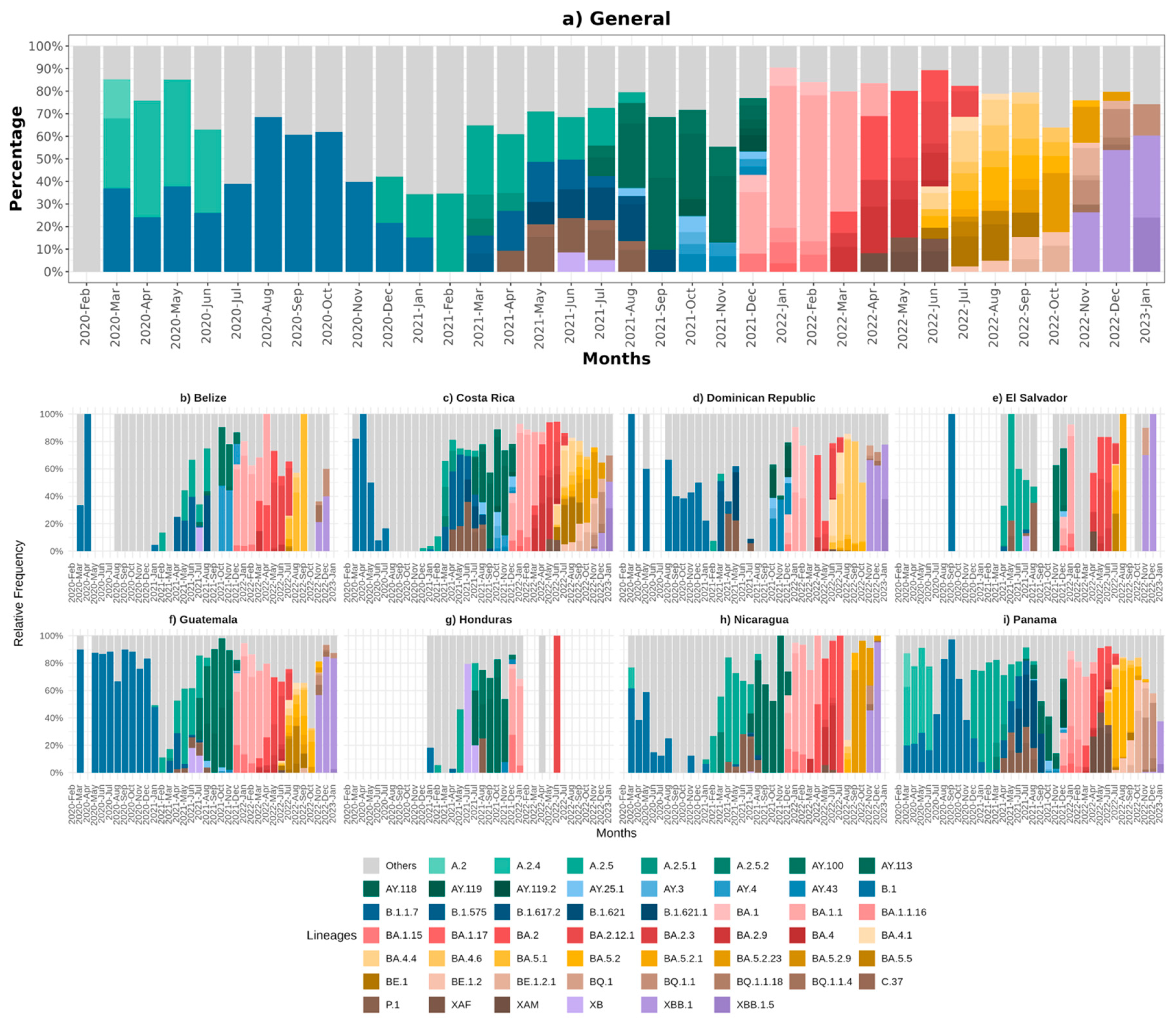
Figure 4.
Max lik Phylogenetic tree of SARS-CoV-2 sequences from Central America and the Dominican Republic, spanning February 2020 to January 2023.
Figure 4.
Max lik Phylogenetic tree of SARS-CoV-2 sequences from Central America and the Dominican Republic, spanning February 2020 to January 2023.
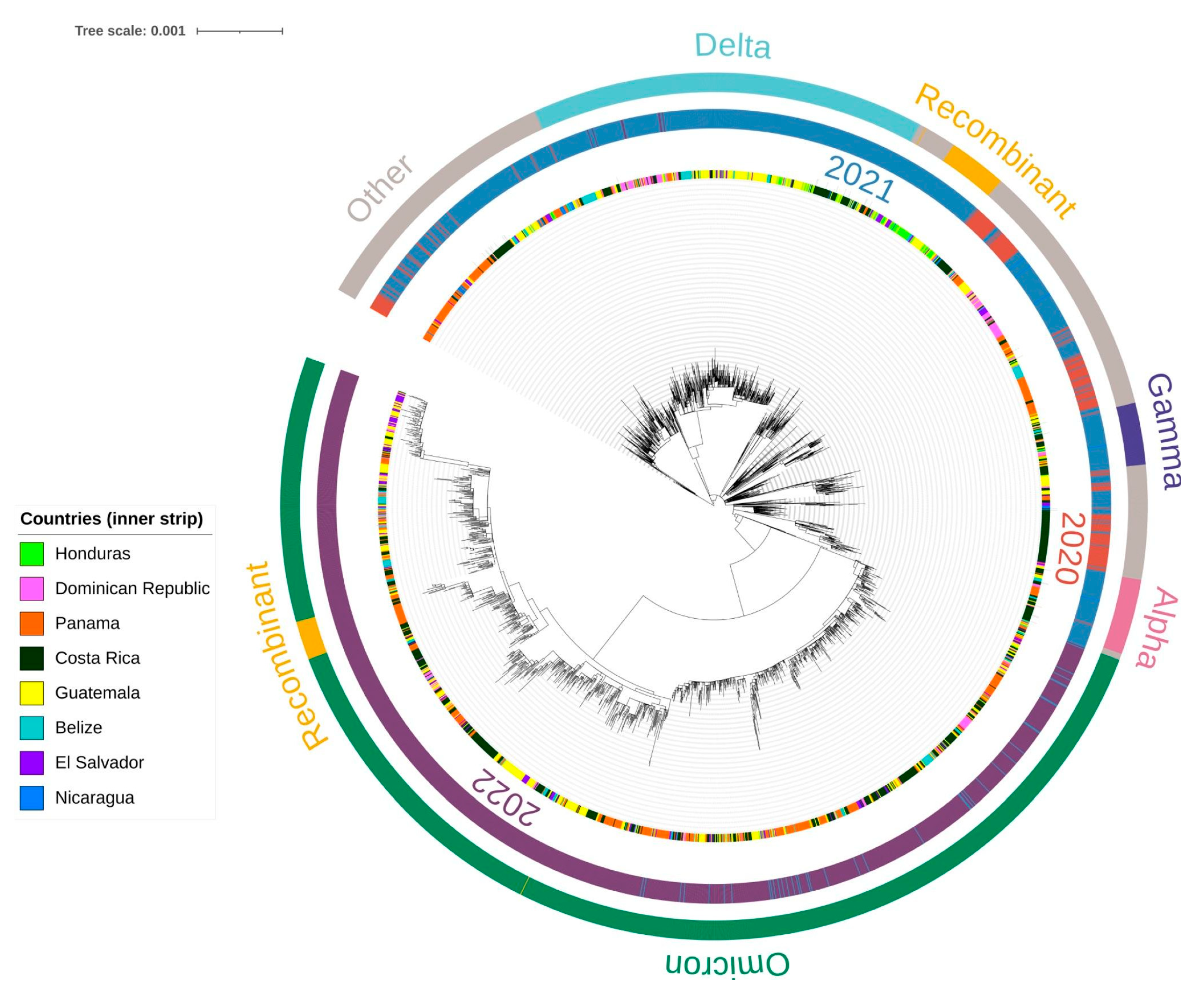
Figure 5.
Temporal dynamics of SARS-CoV-2 lineage proportions with COVID-19 statistical curves in Central America and the Dominican Republic. (a) Monthly proportions of SARS-CoV-2 lineages detected by genomic surveillance are shown as stacked areas. The black dashed line represents the normalized total monthly COVID-19 deaths (secondary y-axis), facilitating comparison between lineage prevalence and mortality trends over time. (b) Same as (a), but the black dashed line depicts the normalized total monthly reported COVID-19 cases instead of deaths. Lineage proportions are calculated as the fraction of total cases sequenced each month. Colors correspond to specific viral lineages.
Figure 5.
Temporal dynamics of SARS-CoV-2 lineage proportions with COVID-19 statistical curves in Central America and the Dominican Republic. (a) Monthly proportions of SARS-CoV-2 lineages detected by genomic surveillance are shown as stacked areas. The black dashed line represents the normalized total monthly COVID-19 deaths (secondary y-axis), facilitating comparison between lineage prevalence and mortality trends over time. (b) Same as (a), but the black dashed line depicts the normalized total monthly reported COVID-19 cases instead of deaths. Lineage proportions are calculated as the fraction of total cases sequenced each month. Colors correspond to specific viral lineages.
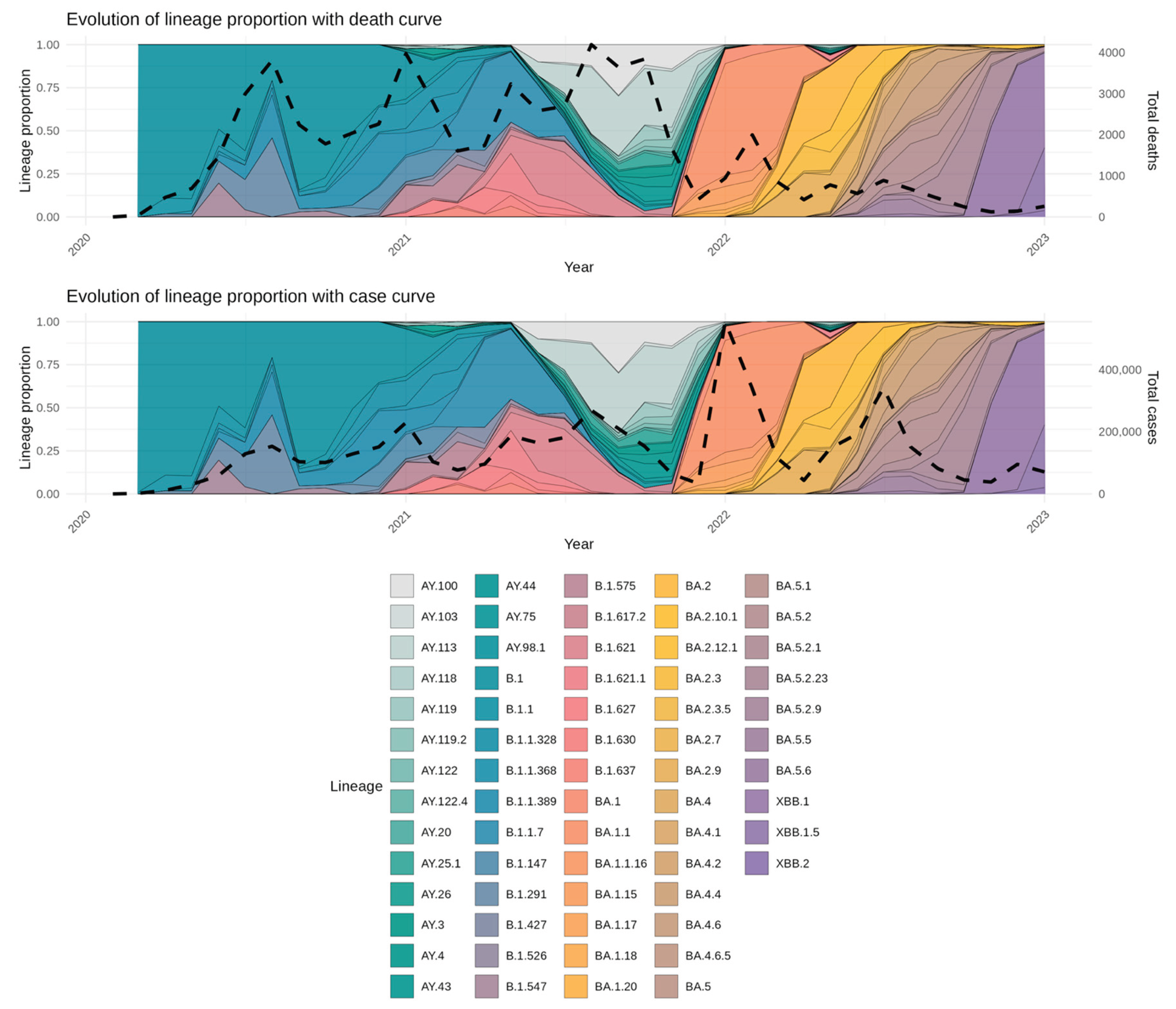

Table 1.
COVID-19 statistical data in Central America and the Dominican Republic from the beginning of the pandemic to January 31st, 2023.
Table 1.
COVID-19 statistical data in Central America and the Dominican Republic from the beginning of the pandemic to January 31st, 2023.
| Country | COVID-19 Cases per Million | Total deaths | Total reported COVID-19 cases |
Total sequenced SARS-CoV-2 Samples |
Percentage of Sequenced COVID-19 Cases (%) |
Genomes Retrieved from GISAID* | Completeness Average (%)* | Sequences with ≥95% Coverage and <6% Ambiguities* | Sequences that met the quality criteria (%)* |
|---|---|---|---|---|---|---|---|---|---|
| Belize | 174,223.1 | 688 | 70,610 | 1,183 | 1.68 | 1,071 | 99.37 | 970 | 90.57 |
| Costa Rica | 228,954.6 | 9,158 | 1,186,176 | 9,960 | 0.84 | 9,921 | 99.35 | 9,865 | 99.44 |
| Dominican Republic | 58,793.97 | 4,384 | 660,187 | 2,628 | 0.40 | 2,585 | 99.46 | 2,345 | 90.72 |
| El Salvador | 31,845.41 | 4,230 | 201,785 | 914 | 0.45 | 633 | 99.48 | 616 | 97.31 |
| Guatemala | 68,736.55 | 20,092 | 1,226,529 | 4,623 | 0.38 | 4,504 | 99.32 | 4,053 | 89.99 |
| Honduras | 45,103.27 | 11,104 | 470,556 | 287 | 0.06 | 233 | 99.43 | 225 | 96.57 |
| Nicaragua | 2,240.661 | 245 | 15,569 | 1,065 | 6.84 | 1,064 | 98.79 | 768 | 72.18 |
| Panama | 23,3567.4 | 8,596 | 1,029,701 | 6,613 | 0.64 | 6,584 | 99.28 | 6,343 | 96.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



