Submitted:
02 May 2025
Posted:
07 May 2025
You are already at the latest version
Abstract
Single-cell analysis shows variations in the expression profile of differentiating cell types and sub-populations. Single cell proteomics (SCP) has emerged as a promising technology defining the cellular proteomics and their functions at a single cell level. SCP is a cutting-edge area of molecular biology focuses on the study of proteins within individual cells. In SCP, absolute quantification of single-cell proteins and highly multiplexed protein measurements is possible with the innovative techniques such as, Single Cell ProtEomics by Mass Spectrometry (SCOPE, SCOPE2), Nanodroplet Processing in One Pot for Trace Samples (nanoPOTS) and Mass Cytometry (CyTOF), Imaging Mass Cytometry (IMC). Recent advancement in SCP is achieved with the development of new methods including miniaturized sample preparation, instrumentation advancements, optimized chromatographic techniques, and expansion of bioinformatics approaches. Integration of single cell multi-omics such as, single cell proteomics (SCP), single cell genomics (SCG), single cell RNA sequencing (scRNA-seq), single cell metabolomics (SCM) and single cell epigenomics (SCEpi) allows comprehensive understanding of gene regulation and cellular behavior of complex biological systems. SCP is crucial for studying complex biological systems and cellular heterogeneity, hence provides valuable insights of tumor heterogeneity, disease mechanisms, therapeutic development, hematopoiesis, neurobiology, cellular development and developmental biology.
Keywords:
Flow cytometery (FCM)
; Imaging Mass Cytometry (IMC)
; Mass Cytometry (CyTOF)
; Mass spectrometry (MS)
; Nano-flow liquid chromatography mass spectrometry (nLC-MS)
; Nanodroplet Processing in One Pot for Trace Samples (nanoPOTS)
; Single cell proteomics (SCP)
; Single Cell ProtEomics by Mass Spectrometry (SCoPE)
Article Highlights
- Single cell proteomics (SCP) is a promising technology defining the cellular proteomics at a single cell level.
- Absolute quantification of single-cell proteins is possible with the recent techniques.
- Innovative techniques such as, SCOPE, SCOPE2, nanoPOTS, CyTOF and IMC are used for SCP analysis.
- Multi-omics integration of SCP, SCG, scRNA-seq, SCM and SCEpi provide deep understanding of complex biological systems.
- SCP provides valuable insight of tumor heterogeneity, disease mechanisms, therapeutic development, neurobiology, and cellular and developmental biology.
Expert Opinion
Single-cell analysis shows variations in the expression profile of differentiating cell types and sub-populations originating from heterogeneous samples. In addition to the well-established techniques, single cell genomics (SCG) and single cell transcriptomics (SCT), recently single cell proteomics (SCP) has emerged as a promising technology for analyzing the entire set of proteins (proteome) in a sample. Single cell proteomics (SCP) has emerged as a promising technology defining the cellular proteomics and their functions at a single cell level. SCP is a cutting-edge technology enables a deeper understanding of cellular heterogeneity for studying the complex biological systems. Indeed, proteins and their posttranslational modifications are crucial of cellular functionality, identity and diversity. Therefore, SCP is crucial for defining the cellular proteomics and their functions at a single cell level for understanding cellular response. SCP is a cutting-edge area of molecular biology and analytical chemistry that focuses on the study of proteins within individual cells, rather than averaging protein data from a population of cells. Many biological systems are highly heterogeneous, consisting of intermixed subpopulations, different cell types and tissue substructures. The first technological advancement in the field of SCP has achieved with the development of Single Cell ProtEomics by Mass Spectrometry (SCoPE) platform to allow the detection of over 1000 proteins in a single sample. SCP currently lacks a state-of-the-art method for the protein identification and quantification. Despite this, absolute quantification of single-cell proteins and highly multiplexed protein measurements is possible with the recent technological advancements using innovative techniques such as, Single Cell ProtEomics by Mass Spectrometry (SCoPE, SCoPE2), Nanodroplet Processing in One Pot for Trace Samples (nanoPOTS), Mass Cytometry (CyTOF) and Imaging Mass Cytometry (IMC). Recently, substantial development of nano-flow liquid chromatography mass spectrometry (nLC-MS) and advancement of data analysis algorithms allows identification and quantification of low abundant proteins in SCP. Moreover, advancement in SCP is being achieved with the development of new technologies including miniaturized sample preparation, instrumentation advancements, optimization of chromatographic techniques, ultra-sensitive MS instrumentation, refined acquisition of mass spectra, and expansion of bioinformatics approaches. Further, integration of single cell multi-omics such as, single cell proteomics (SCP), single cell genomics (SCG), single cell RNA sequencing (scRNA-seq), single cell metabolomics (SCM) and single cell epigenomics (SCEpi) allows comprehensive understanding of gene regulation and cellular behavior of complex biological systems. Technological advancements in SCP analysis with the development of microfluidics, nLC-MS/MS, proteoCHIP, SCBC, scWB system, CycMIST, LCM-nanoPOTS, SCCP, iProChip–DIA offers highly sensitivity protein detection at the single-cell level. Moreover, automation, miniaturization of platforms, development of hybrid technologies, robust development of bioinformatics and computational tools, and advanced statistical methods improves SCP. Future prospects of SCP can be driven by integration of AI and ML to understand the complex proteomic data. Importantly, SCP is crucial for studying complex biological systems and cellular heterogeneity, hence provides valuable insights of tumor heterogeneity, disease mechanisms, therapeutic development, hematopoiesis, neurobiology, cellular development and developmental biology. In this review, we discussed about the recent advancements in SCP including different techniques and tools available/ developed for the SCP analysis. Moreover, we also discussed the achievement and limitation of available methods, along with the recent advancements and future perspective of this emerging technology.
Introduction
Single-cell analysis shows the variations in the expression profile of differentiating cell types and sub-populations originating from heterogeneous samples. In addition to the well-established techniques, single cell genomics (SCG) and single cell transcriptomics (SCT), recently single cell proteomics (SCP) has emerged as a promising technology for analyzing the entire set of proteins (proteome) in a sample [1,2,3,4]. Indeed, proteins and their posttranslational modifications are crucial of cellular functionality, identity and diversity. Therefore, SCP is crucial for defining the cellular proteomics and their functions at a single cell level for understanding cellular response. SCP is a cutting-edge area of molecular biology and analytical chemistry that focuses on the study of proteins within individual cells, rather than averaging protein data from a population of cells [5]. Many biological systems are highly heterogeneous, consisting of intermixed subpopulations, different cell types and tissue substructures. Hence, SCP enables a deeper understanding of cellular heterogeneity crucial for studying complex biological systems [6]. In addition, SCP identifies and characterizes rare cell types or cellular (sub) populations in an unbiased way which would remain unnoticed in classical bulk analyses. Moreover, SCP provides valuable insights of tumor heterogeneity, hematopoiesis, neurobiology, cellular development and developmental biology [7,8,9].
Although, SCP has emerged as a promising technology, however identification of low protein quantities (picogram) at single cell is challenging. In addition, lack of protein amplification methods (similar to the PCR) further make difficulties in the detection of low abundant proteins. However, traditional proteomics methods like mass spectrometry (MS) and western blotting for single-cell analysis is technically challenging. The first technological advancement in the field of SCP has achieved with the development of Single Cell ProtEomics by Mass Spectrometry (SCoPE) platform to allows the detection of over 1000 proteins in a single sample [10]. SCP currently lacks a state-of-the-art method for the protein identification and quantification. Despite this, absolute quantification of single-cell proteins and highly multiplexed protein measurements is possible with the recent technological advancements using innovative techniques such as, Single Cell ProtEomics by Mass Spectrometry (SCoPE, SCoPE2), Nanodroplet Processing in One Pot for Trace Samples (nanoPOTS), Mass Cytometry (CyTOF) and Imaging Mass Cytometry (IMC) [10,11,12,13,14]. Recently, substantial development of nano-flow liquid chromatography mass spectrometry (nLC-MS) and advancement of data analysis algorithms allows identification and quantification of low abundant proteins in SCP [15]. Moreover, further expansion of SCP is being achieved with the development of new technologies including, miniaturized sample preparation, instrumentation advancements, optimization of chromatographic techniques, ultra-sensitive MS instrumentation, refined acquisition of mass spectra, and expansion of bioinformatics approaches [16,17,18]. In this review, we discussed about the recent advancements in SCP including different techniques and tools available/ developed for the SCP analysis. Moreover, we also discussed the achievement and limitation of available methods, along with the recent advancements and future perspective of this emerging technology.
Methods of SCP
To date, there are two main approaches available for analyzing the proteome at single-cell level, namely (i) mass spectrometry (MS) based methods and (ii) antibody-based methods. MS based methods enabled protein identification and quantification including protein post-translational modifications (PTMs), whereas antibody-based methods are developed to analyze the amounts and types of proteins. The detailed description of these methods is discussed below-
[1] Mass Spectrometry (MS)-Based Methods
Typically, MS is a sensitive and non-targeted method, optimized for complete proteome or bulk analysis of the cells. Recently, MS-based proteomics has been further extended for SCP. Miniaturized sample preparation techniques in the form of microwell plates, such as nanoPOTS have enabled proteomic of single cells. However, major bottleneck of these techniques is the limited throughput in terms of number of wells available for sampling and duration of sample analysis [12]. In response to these challenges, multiplexes SCP, Single-Cell ProtEomics by Mass Spectrometry (SCoPE and SCoPE2) were developed. These methods allow the identification of thousands of proteins using isobaric tandem mass tags (TMT) labelling and liquid chromatography-tandem mass spectrometry (LC-MS/MS) [10,11].
- I-Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS)
The first breakthrough in SCP was the development of an innovative method named, Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) [10]. SCoPE-MS investigated the proteome changes during stem cells differentiation into single embryo and embryonic stem cells, and quantify over thousands of proteins in differentiating mouse embryonic stem (ES) cells. SCoPE-MS is broadly applicable for proteome profiling of single cell types with significantly better and faster identification than label-free methods. This method uses isobaric tandem mass tags (TMT) labelling to multiplex single-cell proteomics enabling profiling and quantification of thousands of proteins in individual cells [19]. Further, multiplexing is improved by combining upto 18 cells using TMTpro into single sample to generate richer fragmentation spectra leading to significant increase in peptide and protein identifications [10].
The generalized workflow of SCoPE-MS is single cell isolation, protein extraction and digestion, TMT labeling, mass spectrometry analysis and data analysis. SCoPE-MS mechanically lyses single cells through sonication or freeze-thaw cycles, which reduces protein loss during surfactant cleanup procedures. In addition, sample loss is further minimized by reducing sample volume and sample transfers steps. Moreover, multiplexed detection is carried out using TMT, which labels the samples with different combination of isotopes to distinguish them in tandem MS. Simultaneously, SCoPE-MS involve multiplexing (10-plex TMT) to permits TMT labelling for mixed sample of single-cell, reference cell and carrier cells. Further, the TMT tagged proteome of these mixed cell population is analyzed together by liquid chromatography tandem LC-MS/MS. Peptides ions selected in MS1 are fragmented and passed to MS2 for the quantification and identification. The peptide mixtures are then analyzed with enhanced peptide identification and optimization by LC-MS/MS. The addition of carrier and reference cells increase the sample throughput, provides sufficient peptide ions and improves peptide identification in SCoPE-MS platform. SCoPE generates the protein profile of all the cells taken for the analysis (Figure 1).
- II-Single-Cell ProtEomics by Mass Spectrometry (SCoPE-MS) SCoPE2
Further advancement of SCoPE is the generation of SCoPE-MS (SCoPE2). SCoPE2 is designed to increase the sensitivity, accuracy, and throughput of single-cell protein analysis by optimizing the experimental conditions and data analysis protocols. Notably, SCoPE2 includes state-of-the-art methods including, minimal ProteOmic sample preparation (mPOP), and high-throughput automation [11,20]. These improvements enabled the measurement of over 3000 proteins by SCoPE2, as compared to the identification of 1000 proteins by SCoPE [11,21]. SCoPE2 represents a significant advancement in SCP, enabling detailed and comprehensive study of cellular heterogeneity and protein function. Indeed, SCoPE2 offers several technological advancements over SCoPE in terms of sample preparation, heterogenous samples analysis, multiplexing, and improved protein detection and quantification. However, the generalized workflow for SCoPE2 is similar to SCoPE (Figure 1). Improved sample preparation techniques used in SCoPE2 minimize the sample loss, hence making this technique critical for SCP of samples available in small amount. Moreover, SCoPE2 improves protein detection of low abundant proteins, and provides more accurate and reliable quantification of proteins in different cell types or conditions. Simultaneous TMT labelling of multiple single cells allows analysis of proteins from many cells, thus increasing throughput and efficiency.
- III-Nanodroplet Processing in One Pot for Trace Samples (nanoPOTS)
To overcome the issue of sample loss and sensitive detection of single cell proteins via. SCoPE or SCoPE2, an automated and miniaturized sample preparation methods, nanoPOTS was developed [12]. This cutting-edge microfluidic technology uses microwell plates to prepare single-cell samples for highly sensitive and precise protein analysis of very small biological samples. This chip-based nanodroplet platform uses micro-fabricated nanowell array chips designed to accommodate nanoliter volumes of sample for the analysis. Further, a nanoliter robotic pipetting system ensures minimal sample loss with high efficiency. In nanoPOTS, all the processes including, sample preparation, protein extraction, alkylation, and digestion are performed onto a nanowell-patterned single chip [12]. Further, label-free MS analysis is used for improved peptide separation, ionization, proteins detection and quantification. The label-free analysis obviates the use of chemical labels, which may contribute chemical impurities that interfere with sample separation or MS analysis. The ultrasensitive label-free liquid chromatography coupled with ultra-sensitive tandem mass spectrometry (LC-MS/MS) provides comprehensive proteomic profiles [22]. Later, second generation nanoPOTS, termed nested nanoPOTS (N2) chip was developed to quantify upto 1,500 proteins from 100 individual cells [23]. The N2 chip is designed to accommodate 243 cells (total 27 nested nanowell, each contain 9 nanowells) on a single chip to achieve ten-fold sample processing and protein identification and quantification than nanoPOTS (Figure 2). Both, nanoPOTS and nanoPOTS2 provide several advantages including, miniaturized sample preparation in nanowells to minimizes sample loss, suitable for large-scale high-throughput analysis, and offer high sensitivity for the identification and quantification of thousands the proteins from very small samples.
[2] Antibody-Based Techniques
Antibody based SCP uses antibodies for targeted protein detection and quantification in single cells. The main idea is to use specific antibodies that bind to proteins of interest in a single cell and measure their abundance through various techniques, such as, Flow Cytometry (FCM), Mass Cytometry (CyTOF) and Imaging Mass Cytometry (IMC). FCM is a traditional method uses fluorescent-tagged antibodies to measure proteins in single-cell suspensions. CyTOF an advancement of FCM uses metal-tagged antibodies and MS for the simultaneous analysis of a larger number of proteins. IMC combines laser ablation with mass cytometry to achieve spatial imaging of tissue sections. Typically, these methods allow the identification of thousands of proteins using specific antibodies.
- I-Flow Cytometry (FCM)
Cytometry-based single-cell methods, flow cytometry (FCM) is a powerful technique used in SCP to analyze and sort individual cells based on their protein expression profiles. FCM is a standard profiling technology that offers analysis of thousands of cells per second, using quantification of fluorescence emissions by fluorescent antibodies bound to single cells [14]. In FCM based SCP (FCM-SCP), cells from the tissue are separated using micro-dissection and are sorted by fluorescence-activated cell sorting (FACS). Further, cell lysis, digestion is performed, and peptide labelling is achieved using fluorescently-tagged antibodies. Later, multiplexd sample is prepared by mixing the peptide from each sample, which is further injected into LS-MS/MS for enhanced peptide identification and quantification to provide single cell proteome profile (Figure 3). FCM-SCP is a high throughput method, enable analysis of thousands of cells per second, making it suitable for large-scale studies [24]. It also facilitates multiplexing to allow simultaneous measurement of multiple proteins in each cell using different fluorescent tags. In addition, it offers single cell resolution for detailed analysis of cellular heterogeneity within complex tissues. Nevertheless, FCM-SCP typically requires large sample volume (10-100 μL) and restricts the characterization of cell morphology [25]. To overcome these limitations, imaging flow cytometry (IFC) microsystem was developed by integrating FCM and high-resolution imaging microscopy to enable high throughput analysis of SCP with better spatial resolution [26,27]. Later, multi-parametric IFC using multi-color fluorescence and bright-field analysis has been developed to enable high resolution analysis (60,000cells/s) of cellular proteins [28]. This technique uses blur-free images with high sensitivity for sub-cellular analysis and high throughput localization of proteins.
- II-Mass Cytometry (CyTOF)
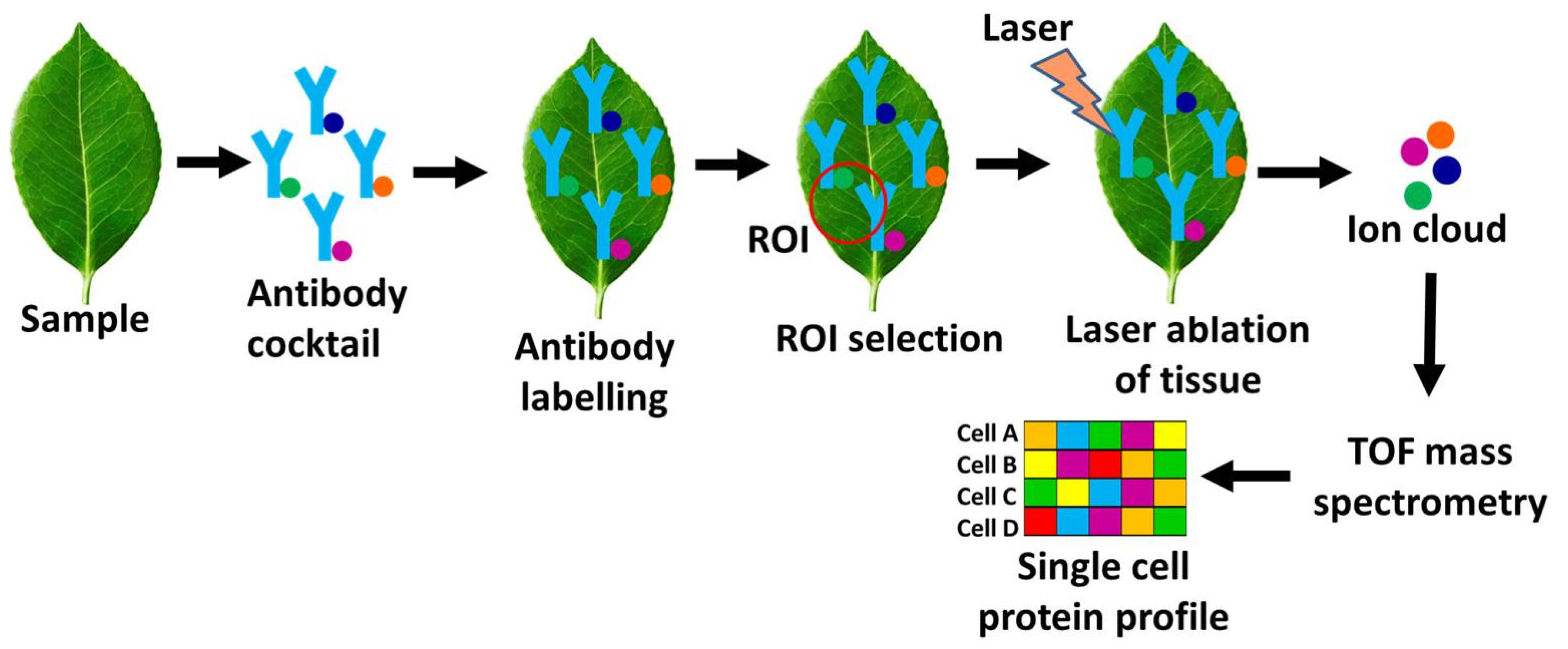
FCM is a traditional method commonly uses fluorescent-tagged antibodies to bind with the protein of interest on the cells surface or within it, in single cell suspension. One of the main limitations of FCM is spectral overlaps of signals retrieved from fluorescent labels. Further, combining traditional FCM and MS, a hybrid technology termed as mass cytometry/ cytometry by time of flight (CyTOF) was developed to achieve highly multiplexed SCP analysis [13]. This method offers unparalleled insights into the complexity of biological systems. CyTOF is based on the concept of using heavy-metal isotopes to label antibodies rather than fluorescent tags. The use of metal tags allows the combination of several antibodies in a single experiment, without significant spillover between detector channels. In CyTOF, antibodies are conjugated with exogenous isotopically pure elements to indirectly label the target proteins. These antibodies bind to the proteins of interest on the cell surface or within it and permit further detection by mass spectrometry. Unlike fluorescent tags used in traditional FCM, metal isotopes used in CyTOF avoid spectral overlap and allows simultaneous measurement of several proteins. The labeled cells are introduced into the mass cytometer to atomized and ionized them into ion clouds. Further, the ion clouds are analyzed by time-of-flight mass spectrometry to provide the detailed insights of protein expression profiles of individual cells (Figure 4). CyTOF is also employed to identify the key transcription factors and cell surface markers in single or hematopoietic cell [13,29]. In addition, CyTOF is used for the simultaneous analysis of cell surface markers and myogenic transformation factors to resolve the heterogeneity of myogenic compartments and myogenic progressions in skeletal muscle cells [30]. Later, using CyTOF, a barcoding system was developed to facilitate high-dimensional single-cell CRISPR screening [31]. This method generated more than 100 protein barcodes (Pro-codes) or encoding triplet of linear epitopes for subsequent CyTOF analyses to enable the detection of total 364 Pro-code populations by using just 14 antibodies. Recently, CyTOF was utilized to study the changes in immune reconstitution at different phases of human cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation [32]. In general, CyTOF is more advantageous than FCM, provides precise quantitative measurement of protein at the single-cell level, revealing cellular heterogeneity. In addition, CyTOF facilitate multiplexed analyses for simultaneously measurement of several proteins without spectral overlap.
- [III] Imaging Mass Cytometry (IMC)
Recent innovations in mass cytometry include the development of imaging mass cytometry (IMC) combining mass cytometry with spatial imaging. IMC is a cutting-edge technique which, in addition to the protein’s quantification, also permits visualization and distribution of proteins within tissues for comprehensive understanding of cellular interactions and microenvironments. Since, FCM and CyTOF acquire the loss of crucial spatial information during processing the single-cell suspension, the single-cell analysis of tissue samples, including frozen tissue sections and formalin-fixed paraffin-embedded (FFPE) tissues has been formulated by IMC [33,34]. Single cell IMC provides spatially-resolved analysis for investigating phenotypic heterogeneity and tumor architecture. IMC allows tissues staining with multiplexed metal isotope-tagged antibodies to enable the simultaneous detection of a broader range of protein markers. IMC applies a focused laser to ablate the tissues or tissue sections, vaporized them by pulsed laser, and transfer the ionized metal tags to ICP-MS for mass cytometry analysis. Data collected following the analysis is used to reconstruct high-resolution images, showing the spatial distribution and abundance of various proteins within the tissue (Figure 5). Recently, advanced image analysis software and machine learning algorithms have been developed to automate the quantification and interpretation of IMC data. IMC has been also utilized efficiently to examine the single-cell phenotype of breast cancer tissues [35]. Notably, the information undetectable in 2D IMC, a 3D IMC was developed with high spatial resolution to study the tumor invasion [36]. Further, the integration of IMC with high definition single cell analysis (HD-SCA) assays has been used to characterize rare tumor cells [37]. In addition, using HD-SCA, an in-depth phenotyping of a liquid biopsy has been carried out for subcellular localization of markers within circulating tumor cells (CTCs). In general, IMC can simultaneously detect and visualize multiple protein markers within a single tissue section. It also provides precise quantification of protein levels and spatial information to study the relationships between different cell types and their microenvironments.
Integration with Other Omics Approaches
The integration of SCP with other omics technologies such as single cell genomics (SCG), single cell RNA sequencing (scRNA-seq), single cell metabolomics (SCM) and single cell epigenomics (SCEpi) is an exciting frontier in systems biology. Combining single cell multi-omics provide comprehensive understanding of gene regulation and cellular behavior to enhance our understanding of complex biological systems [38]. Integration of multi-omics approach reveals better insights into its potential implications for cellular processes, post-translational modifications, cellular activity and biological mechanism as compared to the single omics approach [39]. SCP has been integrated with several single cell techniques-
- Single-Cell Proteomics and single cell transcriptomics integration
Integration of SCP and scRNA-seq provide better understanding on the gene expression at the protein level. Many cellular processes are regulated at the level of protein modification, localization, and interactions, which are not captured by scRNA-seq alone. Hence, integration of SCP and scRNA-seq provides an important insight on post-transcriptional regulation, protein degradation, and translation efficiency [40]. Moreover, correlated gene expression with protein abundance, provides a holistic view of cellular regulation [41].
- 2.
- Single-Cell Proteomics and single cell Epigenomics integration
Epigenomics focuses on the chemical changes to DNA and histone proteins (such as DNA methylation, histone methylation/ acetylation/ phosphorylation etc.) affecting gene expression without altering the underlying DNA sequence. Epigenetic modifications play a key role in cell differentiation, development and reprogramming. Integration of epigenomics with proteomics offers understanding on changes in the chromatin landscape influencing protein expression and cellular behavior at single-cell level [42]. Combining epigenomic and proteomic provide an important insights into how epigenetic changes drive the synthesis of specific proteins and guiding cell fate during development, disease, stem cell differentiation and cancer progression [43].
- 3.
- Single-Cell Proteomics and single cell Metabolomics integration
Integration of proteomics and metabolomics is important for elucidating the molecular mechanism. Combining metabolomics with proteomics can establish direct link between protein expression and cellular metabolic activity. Integration of single cell proteomics and single cell metabolomic offers the exploration of metabolic flux in real-time, especially in response to environmental or therapeutic stimuli to provide deeper insights into the metabolic reprogramming [44]. This integration can also be applied to drug discovery, particularly in understanding changes in protein machinery of the cell and its metabolic state, revealing new therapeutic targets.
- 4.
- Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq)
Cellular Indexing of Transcriptomes and Epitopes (CITE-seq) is a cutting-edge technique that combines single-cell RNA sequencing (scRNA-seq) with antibody-based proteomic profiling. The goal of CITE-seq is to measure both the transcriptome (gene expression) and the surface protein markers on the same individual cell. CITE-seq allows comprehensive understanding of cellular heterogeneity and the relationship between gene expression and cell surface protein markers [45]. CITE-seq uses oligonucleotide-tagged antibodies specific to cell surface markers (e.g., CD markers) to identify the expression of cell surface proteins. CITE-seq can measure multiple surface proteins simultaneously, providing a broad and deep analysis of cellular features. Profiling of both transcriptomic and proteomic data in CITE-seq allows improved identification of specific cell types, their states and interactions [46]. In addition, CITE-Seq can be adapted to detect small molecules, and for RNA interference, CRISPR, and other gene editing techniques. CITE-Seq expands its potential for protein detection and measurement by using largenumber of antibodies to detect cell surface proteins. Recently, multi-omics analysis such as, CITE-seq and nanoSPLITS integrate proteomics workflow with well-established single-cell RNA sequencing technologies has enabled bi-modal analysis [47].
Technological Advancements and Future Directions
Technological advancements in SCP analysis is achieved with the development of microfluidics to enables the isolation and manipulation of individual cells in a controlled environment. Microfluidics and automation improve sample preparation by reducing sample loss and increasing high throughput analysis. Moreover, advancements in MS with the development of nLC-MS/MS, allows improved proteins profiling at single-cell level [48]. Furthermore, with the development of innovative antibody-based methods like single-cell antibody-based assays (CyTOF or FCM) and proximity extension assays (PEA) highly sensitivity protein detection at the single-cell level is achieved. In addition, development of new sample preparation techniques, such as nanoPOTS and proteoCHIP furher improves the efficiency and accuracy of protein analysis at the single-cell level [49].
CyTOF is one of the most widely used approache used for targeted SCP analysis, providing exploration of brain cell phenotype, immune cell differentiation, cell heterogeneity and cancer signaling [50,51]. Moreover, CyTOF in combination with cell imaging has been applied successfully to explore the subcellular distribution of proteins in individual cells during cellular processes, immune cell function and immune-therapeutic applications [52]. Recently, single-cell CyTOF has been used to investigate heterogeneity in non-small cell lung cancer cell (NSCLC) lines [53]. In addition, a single-cell barcode chip (SCBC) approach has been employed to explore cellular interactions and communications by measuring secretory proteins [54]. SCBC analysis has emerged as a promising tool to study phenotypic markers, functional heterogeneity, signaling pathways, cell–cell interaction, cellular communication and immune cell responses [55,56]. Single cell–resolution western blotting (scWB) system has been used to investigate the heterogeneity of circulating tumor cells (CTCs) in cancer patients [57]. The scWB approach has also demonstrated its ability to interrogate the cellular heterogeneity and monitor single-cell differentiation of neural stem cells [58]. On the other hand, droplet microfluidics is used to detect surface receptors such as CD3 and CD19 markers from single-cell for cancers diagnosis, and for the measurement of receptor tyrosine kinase activity in lung cancer cells [59,60]. Notably, surface-enhanced Raman spectroscopy (SERS) based microfluidic assay was used to monitor MCSP, MCAM, and LNGFR surface markers in CTCs [61]. Meanwhile, CycMIST platform providing highly multiplexed profiling facilitate investigation of transcription factors, surface markers, and signaling proteins for Alzheimer’s disease (AD) [62]. Meanwhile, nanoPOTS integrated with LCM have been used to differentiate the cell specific proteomes in human cancer tissues, rat brain, and mouse uterus [63]. Particularly, the updated version of nanoPOTs, N2 has been used to identify cell-type-specific surface markers, to uncover cellular heterogeneity and cellular microenvironments [23]. Recently, LCM-nanoPOTS has been modified with a hanging drop format to find tissue specific or cell-type specific biomarkers of mouse uterine tissues [64]. Indeed, single-cell chemical proteomics (SCCP) derived from SCoPE-MS has been developed to uncover heterogeneous responses of adenocarcinoma cells, drug resistance, and diverse cellular phenotypes upon drug treatments [65]. Moreover, iProChip–DIA demonstrated important drug targets, biomarkers and surface proteins associated with the NSCLC pathway from lung cancer cell [66]. In addition, recent progress in SCP technologies has enabled the 3D mapping of proteome expression to explore single-cell heterogeneity, protein translocation events, and cell–cell interactions [18].
These advancements offer crucial insight into the mechanisms of tumor metastasis, disease progression and treatment response, and accelerate the therapeutic applications of SCP. In addition, future prospects of SCP are driven by technological advancements including, robust development of bioinformatics and computational tools, and statistical methods, for interpreting the complex data generated by SCP. Further advancements in SCP include automation, miniaturization of platforms, hybrid technologies, and integration with single cell omics data. Combining proteomics with imaging technologies like immune-histochemistry or super-resolution microscopy could provide detailed insights into cellular function. Additionally, the integration of artificial intelligence (AI) and machine learning (ML) can be helpful for the identification of complex proteomic data.
Applications
SCP is an emerging field providing insights into cellular heterogeneity, disease mechanisms, and biological processes. Some of the key applications of SCP include
- ▪
- Cancer research: SCP holds immense promise for cancer research, particularly in understanding cancer progression, tumor heterogeneity, prediction of drug resistance and identification of new biomarkers and novel therapeutic targets.
- ▪
- Stem cell and developmental biology: SCP profiling of individual stem cells provides insights into cell differentiation and tissue regeneration for understanding developmental biology.
- ▪
- ▪ Neurobiology: SCP is important to study neurons or glial cells to explore proteome change during brain function, neurological disorders and neurodegenerative diseases.
- ▪
- Developmental biology: SCP explores cell differentiation and development to understand early embryonic development or organogenesis.
- ▪
- Cellular heterogeneity: SCP is important for understanding the cellular heterogeneity of complex tissues like tumors.
- ▪
- Biomarker discovery: SCP enables the discovery of biomarkers for specific cell types, states, or disease.
- ▪
- Drug response profiling: SCP is useful for monitoring the effects of drugs on individual cells, identifying drug-resistant cell populations, and discovering drug mechanisms in real time.
- ▪
- Immunology: SCP aid understanding the immune system at unprecedented resolution, identifying unique protein signatures in different immune cells, and monitoring immune responses during infection or vaccination.
- ▪
- Neurodegenerative diseases: SCP allows examination of protein misfolding, aggregation, and cellular stress responses in individual neurons in Alzheimer’s, Parkinson’s, and others neurological diseases.
Conclusions
SCP is a cutting-edge technology defining the cellular proteomics and their functions at a single cell level to enables a deeper understanding of cellular heterogeneity for studying the complex biological systems. Absolute quantification of highly multiplexed protein measurements is achieved with the recent technological advancements using innovative techniques such as, SCOPE, SCOPE2, nanoPOTS, CyTOF and IMC. Moreover, advancement in SCP is being achieved with the development of new technologies including miniaturized sample preparation, instrumentation advancements, optimization of chromatographic techniques, ultra-sensitive MS instrumentation, refined acquisition of mass spectra, and expansion of bioinformatics approaches. Integration of SCP with SCG, scRNA-seq, SCM and SCEpi develop a hybrid technology to provide comprehensive understanding of cellular behavior. Technological advancements in SCP analysis with the development of microfluidics, nLC-MS/MS, proteoCHIP, SCBC, scWB system, CycMIST, LCM-nanoPOTS, SCCP, iProChip–DIA offers highly sensitivity protein detection at the single-cell level. Moreover, automation, miniaturization of platforms, development of hybrid technologies, robust development of bioinformatics and computational tools, and advanced statistical methods improves SCP. Future prospects of SCP can be driven by integration of AI and ML to understand the complex proteomic data. Importantly, SCP provides valuable insights of tumor heterogeneity, disease mechanisms, therapeutic development, hematopoiesis, neurobiology, cellular development and developmental biology. In conclusion, SCP offers a unique perspective to deepen our understanding on molecular biology.
Author Contributions
Vikram Singh is the sole contributor to the article.
Disclosure Statement
The author declare that he does not have any competing interests.
Declaration of Interest
Author has no conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Paolillo C, Londin E, Fortina P. Single-Cell Genomics. Clin Chem, 65(8), 972-985 (2019). [CrossRef]
- He S, Wang LH, Liu Y et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol, 21(1), 294 (2020). [CrossRef]
- Emara S, Amer S, Ali A, Abouleila Y, Oga A, Masujima T. Single-Cell Metabolomics. Adv Exp Med Biol, 965, 323-343 (2017).
- Wen L, Tang F. Single cell epigenome sequencing technologies. Mol Aspects Med, 59, 62-69 (2018). [CrossRef]
- Vistain LF, Tay S. Single-Cell Proteomics. Trends Biochem Sci, 46(8), 661-672 (2021).
- Mund A, Coscia F, Kriston A et al. Deep Visual Proteomics defines single-cell identity and heterogeneity. Nat Biotechnol, 40(8), 1231-1240 (2022). [CrossRef]
- Wilson NK, Kent DG, Buettner F et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell, 16(6), 712-724 (2015). [CrossRef]
- Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR. Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat Neurosci, 24(4), 584-594 (2021). [CrossRef]
- Vegliante R, Pastushenko I, Blanpain C. Deciphering functional tumor states at single-cell resolution. Embo j, 41(2), e109221 (2022). [CrossRef]
- Budnik B, Levy E, Harmange G, Slavov N. SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biology, 19(1), 161 (2018). [CrossRef]
- Specht H, Emmott E, Petelski AA et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol, 22(1), 50 (2021). [CrossRef]
- Zhu Y, Piehowski PD, Zhao R et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10-100 mammalian cells. Nat Commun, 9(1), 882 (2018). [CrossRef]
- Bandura DR, Baranov VI, Ornatsky OI et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem, 81(16), 6813-6822 (2009). [CrossRef]
- Robinson JP, Roederer M. HISTORY OF SCIENCE. Flow cytometry strikes gold. Science, 350(6262), 739-740 (2015). [CrossRef]
- Ctortecka C, Hartlmayr D, Seth A et al. An Automated Nanowell-Array Workflow for Quantitative Multiplexed Single-Cell Proteomics Sample Preparation at High Sensitivity. Mol Cell Proteomics, 22(12), 100665 (2023). [CrossRef]
- Kim S, Kamarulzaman L, Taniguchi Y. Recent methodological advances towards single-cell proteomics. Proc Jpn Acad Ser B Phys Biol Sci, 99(8), 306-327 (2023). [CrossRef]
- Nalla LV, Kanukolanu A, Yeduvaka M, Gajula SNR. Advancements in Single-Cell Proteomics and Mass Spectrometry-Based Techniques for Unmasking Cellular Diversity in Triple Negative Breast Cancer. PROTEOMICS – Clinical Applications, 19(1), e202400101 (2025). [CrossRef]
- Petrosius V, Schoof EM. Recent advances in the field of single-cell proteomics. Transl Oncol, 27, 101556 (2023). [CrossRef]
- Huang B, Wu H, Bhaya D et al. Counting low-copy number proteins in a single cell. Science, 315(5808), 81-84 (2007). [CrossRef]
- Specht H, Harmange G, Perlman DH et al. Automated sample preparation for high-throughput single-cell proteomics. bioRxiv, 399774 (2018).
- Petelski AA, Emmott E, Leduc A et al. Multiplexed single-cell proteomics using SCoPE2. Nat Protoc, 16(12), 5398-5425 (2021). [CrossRef]
- Cong Y, Liang Y, Motamedchaboki K et al. Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal Chem, 92(3), 2665-2671 (2020). [CrossRef]
- Woo J, Williams SM, Markillie LM et al. High-throughput and high-efficiency sample preparation for single-cell proteomics using a nested nanowell chip. Nat Commun, 12(1), 6246 (2021). [CrossRef]
- Lohani V, A.R A, Kundu S, Akhter MDQ, Bag S. Single-Cell Proteomics with Spatial Attributes: Tools and Techniques. ACS Omega, 8(20), 17499-17510 (2023). [CrossRef]
- Golden JP, Kim JS, Erickson JS et al. Multi-wavelength microflow cytometer using groove-generated sheath flow. Lab Chip, 9(13), 1942-1950 (2009). [CrossRef]
- Stavrakis S, Holzner G, Choo J, deMello A. High-throughput microfluidic imaging flow cytometry. Curr Opin Biotechnol, 55, 36-43 (2019). [CrossRef]
- Miura T, Mikami H, Isozaki A, Ito T, Ozeki Y, Goda K. On-chip light-sheet fluorescence imaging flow cytometry at a high flow speed of 1 m/s. Biomed Opt Express, 9(7), 3424-3433 (2018). [CrossRef]
- Holzner G, Mateescu B, van Leeuwen D et al. High-throughput multiparametric imaging flow cytometry: toward diffraction-limited sub-cellular detection and monitoring of sub-cellular processes. Cell Reports, 34(10), 108824 (2021). [CrossRef]
- Bendall SC, Davis KL, Amir el AD et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell, 157(3), 714-725 (2014). [CrossRef]
- Porpiglia E, Samusik N, Ho ATV et al. High-resolution myogenic lineage mapping by single-cell mass cytometry. Nat Cell Biol, 19(5), 558-567 (2017). [CrossRef]
- Wroblewska A, Dhainaut M, Ben-Zvi B et al. Protein Barcodes Enable High-Dimensional Single-Cell CRISPR Screens. Cell, 175(4), 1141-1155.e1116 (2018). [CrossRef]
- Stern L, McGuire H, Avdic S et al. Mass Cytometry for the Assessment of Immune Reconstitution After Hematopoietic Stem Cell Transplantation. Front Immunol, 9, 1672 (2018). [CrossRef]
- Chang Q, Ornatsky O, Hedley D. Staining of Frozen and Formalin-Fixed, Paraffin-Embedded Tissues with Metal-Labeled Antibodies for Imaging Mass Cytometry Analysis. Curr Protoc Cytom, 82, 12.47.11-12.47.18 (2017). [CrossRef]
- Catena R, Montuenga LM, Bodenmiller B. Ruthenium counterstaining for imaging mass cytometry. J Pathol, 244(4), 479-484 (2018). [CrossRef]
- Jackson HW, Fischer JR, Zanotelli VRT et al. The single-cell pathology landscape of breast cancer. Nature, 578(7796), 615-620 (2020). [CrossRef]
- Kuett L, Catena R, Özcan A et al. Three-dimensional imaging mass cytometry for highly multiplexed molecular and cellular mapping of tissues and the tumor microenvironment. Nat Cancer, 3(1), 122-133 (2022). [CrossRef]
- Gerdtsson E, Pore M, Thiele JA et al. Multiplex protein detection on circulating tumor cells from liquid biopsies using imaging mass cytometry. Converg Sci Phys Oncol, 4(1) (2018). [CrossRef]
- Flynn E, Almonte-Loya A, Fragiadakis GK. Single-Cell Multiomics. Annu Rev Biomed Data Sci, 6, 313-337 (2023). [CrossRef]
- Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Experimental & Molecular Medicine, 52(9), 1428-1442 (2020). [CrossRef]
- Jha PK, Valekunja UK, Ray S, Nollet M, Reddy AB. Single-cell transcriptomics and cell-specific proteomics reveals molecular signatures of sleep. Commun Biol, 5(1), 846 (2022). [CrossRef]
- Ali A, Davidson S, Fraenkel E et al. Single cell metabolism: current and future trends. Metabolomics, 18(10), 77 (2022). [CrossRef]
- Vandereyken K, Sifrim A, Thienpont B, Voet T. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet, 24(8), 494-515 (2023). [CrossRef]
- Bi H, Weng X. Single-Cell Epigenomics and Proteomics Methods Integrated in Multiomics. Fundamental Research, (2024). [CrossRef]
- Orsburn BC. Metabolomic, Proteomic, and Single-Cell Proteomic Analysis of Cancer Cells Treated with the KRAS(G12D) Inhibitor MRTX1133. J Proteome Res, 22(12), 3703-3713 (2023).
- Stoeckius M, Hafemeister C, Stephenson W et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods, 14(9), 865-868 (2017). [CrossRef]
- Kumar S, Orcales F, Shih BB et al. Cellular indexing of transcriptomes and epitopes (CITE-Seq) in hidradenitis suppurativa identifies dysregulated cell types in peripheral blood and facilitates diagnosis via machine learning. Res Sq, (2024). [CrossRef]
- Fulcher JM, Markillie LM, Mitchell HD et al. Parallel measurement of transcriptomes and proteomes from same single cells using nanodroplet splitting. Nat Commun, 15(1), 10614 (2024). [CrossRef]
- Zheng R, Matzinger M, Mayer RL, Valenta A, Sun X, Mechtler K. A High-Sensitivity Low-Nanoflow LC-MS Configuration for High-Throughput Sample-Limited Proteomics. Analytical Chemistry, 95(51), 18673-18678 (2023). [CrossRef]
- Hartlmayr D, Ctortecka C, Seth A, Mendjan S, Tourniaire G, Mechtler K. An automated workflow for label-free and multiplexed single cell proteomics sample preparation at unprecedented sensitivity. bioRxiv, 2021.2004.2014.439828 (2021). [CrossRef]
- Zhang T, Warden AR, Li Y, Ding X. Progress and applications of mass cytometry in sketching immune landscapes. Clin Transl Med, 10(6), e206 (2020). [CrossRef]
- Lun XK, Szklarczyk D, Gábor A et al. Analysis of the Human Kinome and Phosphatome by Mass Cytometry Reveals Overexpression-Induced Effects on Cancer-Related Signaling. Mol Cell, 74(5), 1086-1102.e1085 (2019). [CrossRef]
- Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer, 21(6), 345-359 (2021). [CrossRef]
- Neuperger P, Szalontai K, Gémes N et al. Single-cell mass cytometric analysis of peripheral immunity and multiplex plasma marker profiling of non-small cell lung cancer patients receiving PD-1 targeting immune checkpoint inhibitors in comparison with platinum-based chemotherapy. Front Immunol, 14, 1243233 (2023). [CrossRef]
- Kravchenko-Balasha N, Shin YS, Sutherland A, Levine RD, Heath JR. Intercellular signaling through secreted proteins induces free-energy gradient-directed cell movement. Proc Natl Acad Sci U S A, 113(20), 5520-5525 (2016). [CrossRef]
- Xue Q, Bettini E, Paczkowski P et al. Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J Immunother Cancer, 5(1), 85 (2017). [CrossRef]
- Su Y, Wei W, Robert L et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A, 114(52), 13679-13684 (2017). [CrossRef]
- Sinkala E, Sollier-Christen E, Renier C et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat Commun, 8, 14622 (2017). [CrossRef]
- Hughes AJ, Spelke DP, Xu Z, Kang CC, Schaffer DV, Herr AE. Single-cell western blotting. Nat Methods, 11(7), 749-755 (2014). [CrossRef]
- Shahi P, Kim SC, Haliburton JR, Gartner ZJ, Abate AR. Abseq: Ultrahigh-throughput single cell protein profiling with droplet microfluidic barcoding. Sci Rep, 7, 44447 (2017). [CrossRef]
- Ramji R, Wang M, Bhagat AA et al. Single cell kinase signaling assay using pinched flow coupled droplet microfluidics. Biomicrofluidics, 8(3), 034104 (2014). [CrossRef]
- Reza KK, Dey S, Wuethrich A et al. In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment. ACS Nano, 15(7), 11231-11243 (2021). [CrossRef]
- Yang L, Ball A, Liu J et al. Cyclic microchip assay for measurement of hundreds of functional proteins in single neurons. Nat Commun, 13(1), 3548 (2022). [CrossRef]
- Piehowski PD, Zhu Y, Bramer LM et al. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-μm spatial resolution. Nat Commun, 11(1), 8 (2020). [CrossRef]
- Kwon Y, Piehowski PD, Zhao R et al. Hanging drop sample preparation improves sensitivity of spatial proteomics. Lab Chip, 22(15), 2869-2877 (2022). [CrossRef]
- Végvári Á, Rodriguez JE, Zubarev RA. Single-Cell Chemical Proteomics (SCCP) Interrogates the Timing and Heterogeneity of Cancer Cell Commitment to Death. Anal Chem, 94(26), 9261-9269 (2022). [CrossRef]
- Gebreyesus ST, Siyal AA, Kitata RB et al. Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat Commun, 13(1), 37 (2022). [CrossRef]
Figure 1.
Single-cell proteomics with Single-cell ProtEomics by mass spectrometry (SCoPE-MS) to generate proteome profile of a single cell. The heterogeneous cells are sorted into three groups, single-cell, reference cells, and carrier cells, with a cell number ratio of 1:5:100, respectively. Addition of carrier and reference cells increase the sample throughput, provides sufficient peptide ions and improves peptide identification in SCoPE-MS platform. Next, cells are lysed using a freeze-heat cycle or sonication and digested. Further, tandem mass tags (TMTs) multiplex labeling is performed for all the cells. All TMT tagged cells are mixed together and proteome of these mixed cell population is analyzed together by liquid chromatography tandem mass spectroscopy (LC-MS/MS). Peptides ions selected in the first MS analysis (MS1) are fragmented and passed to the second MS analysis (MS2) for quantification and identification. The LC-MS/MS analysis generates the single cell protein profile for all the analyzed cells. Both, SCoPE and SCoPE2 are suitable for large-scale high-throughput analysis, and offer highly sensitive protein identification and quantification.
Figure 1.
Single-cell proteomics with Single-cell ProtEomics by mass spectrometry (SCoPE-MS) to generate proteome profile of a single cell. The heterogeneous cells are sorted into three groups, single-cell, reference cells, and carrier cells, with a cell number ratio of 1:5:100, respectively. Addition of carrier and reference cells increase the sample throughput, provides sufficient peptide ions and improves peptide identification in SCoPE-MS platform. Next, cells are lysed using a freeze-heat cycle or sonication and digested. Further, tandem mass tags (TMTs) multiplex labeling is performed for all the cells. All TMT tagged cells are mixed together and proteome of these mixed cell population is analyzed together by liquid chromatography tandem mass spectroscopy (LC-MS/MS). Peptides ions selected in the first MS analysis (MS1) are fragmented and passed to the second MS analysis (MS2) for quantification and identification. The LC-MS/MS analysis generates the single cell protein profile for all the analyzed cells. Both, SCoPE and SCoPE2 are suitable for large-scale high-throughput analysis, and offer highly sensitive protein identification and quantification.
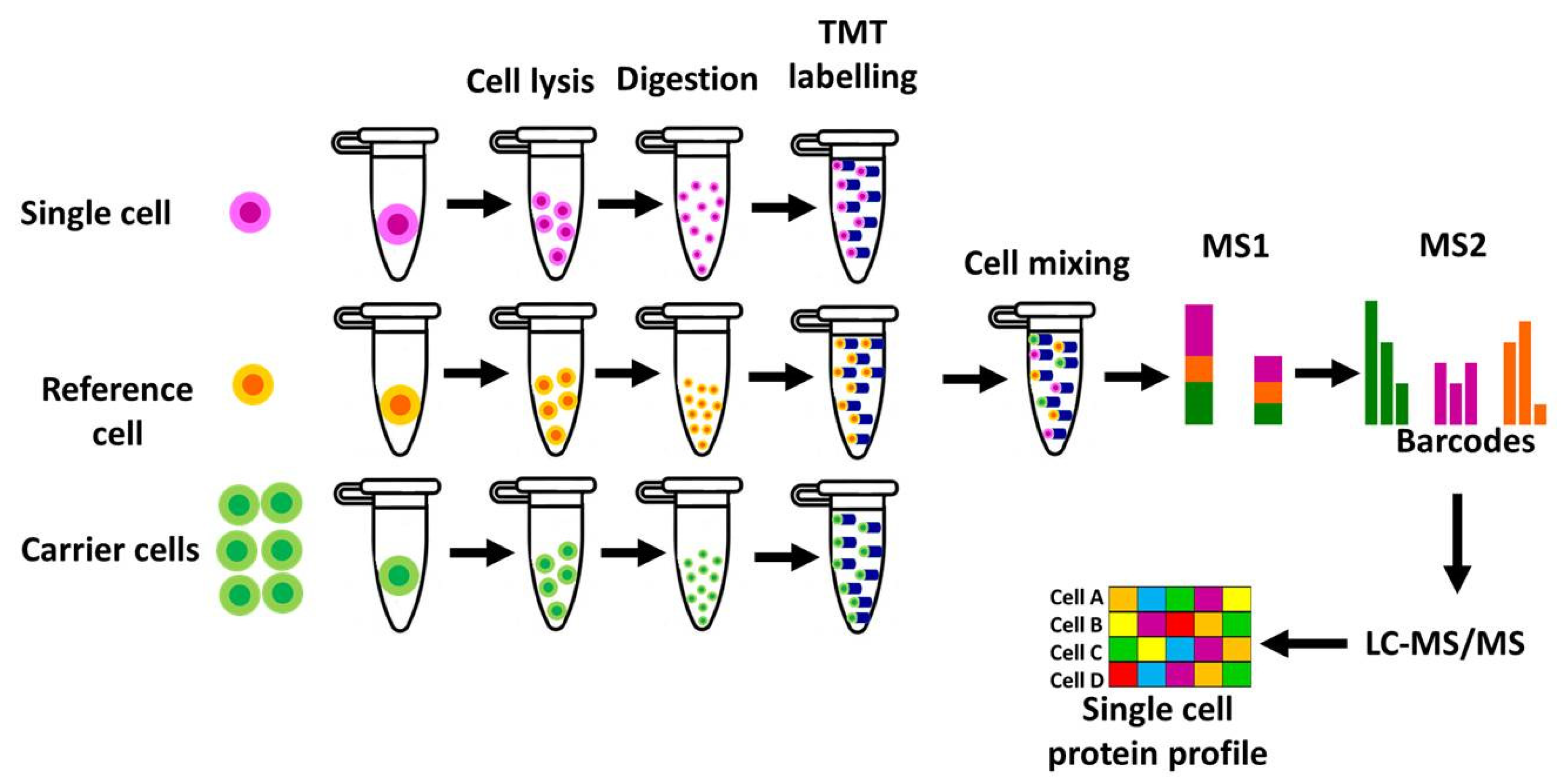
Figure 2.
Single-cell proteomics with Nanodroplet Processing in One Pot for Trace Samples (NanoPOTS) to generate proteome profile of a single cell. This cutting-edge microfluidic technology uses microwell plates to prepare single-cell samples for highly sensitive and precise protein analysis of very small biological samples. This chip-based nanodroplet platform uses micro-fabricated nanowell array chips designed to accommodate nanoliter volumes of sample for the analysis. All the processes including, sample preparation, protein extraction, alkylation, and digestion are performed onto a nanowell-patterened single chip. Nested NanoPOTS (N2) chip is designed to accommodate 243 cells (total 27 nested nanowell, each contain 9 nanowells) on a single chip to achieve ten-fold sample processing and protein identification and quantification than NanoPOTS. Peptides mixtures in nine wells are collected together and analyzed for enhanced peptide identification and optimization by nLC-MS/MS. The label-free MS analysis is used for improved peptide separation, ionization, proteins detection and quantification to provides comprehensive proteomic profiles of all the analyzed cells. Both, NanoPOTS and NanoPOTS2 includes, miniaturized sample preparation in nanowells to minimizes sample loss, suitable for large-scale high-throughput analysis, and offer high sensitivity for the identification and quantification of thousands the proteins from very small samples.
Figure 2.
Single-cell proteomics with Nanodroplet Processing in One Pot for Trace Samples (NanoPOTS) to generate proteome profile of a single cell. This cutting-edge microfluidic technology uses microwell plates to prepare single-cell samples for highly sensitive and precise protein analysis of very small biological samples. This chip-based nanodroplet platform uses micro-fabricated nanowell array chips designed to accommodate nanoliter volumes of sample for the analysis. All the processes including, sample preparation, protein extraction, alkylation, and digestion are performed onto a nanowell-patterened single chip. Nested NanoPOTS (N2) chip is designed to accommodate 243 cells (total 27 nested nanowell, each contain 9 nanowells) on a single chip to achieve ten-fold sample processing and protein identification and quantification than NanoPOTS. Peptides mixtures in nine wells are collected together and analyzed for enhanced peptide identification and optimization by nLC-MS/MS. The label-free MS analysis is used for improved peptide separation, ionization, proteins detection and quantification to provides comprehensive proteomic profiles of all the analyzed cells. Both, NanoPOTS and NanoPOTS2 includes, miniaturized sample preparation in nanowells to minimizes sample loss, suitable for large-scale high-throughput analysis, and offer high sensitivity for the identification and quantification of thousands the proteins from very small samples.
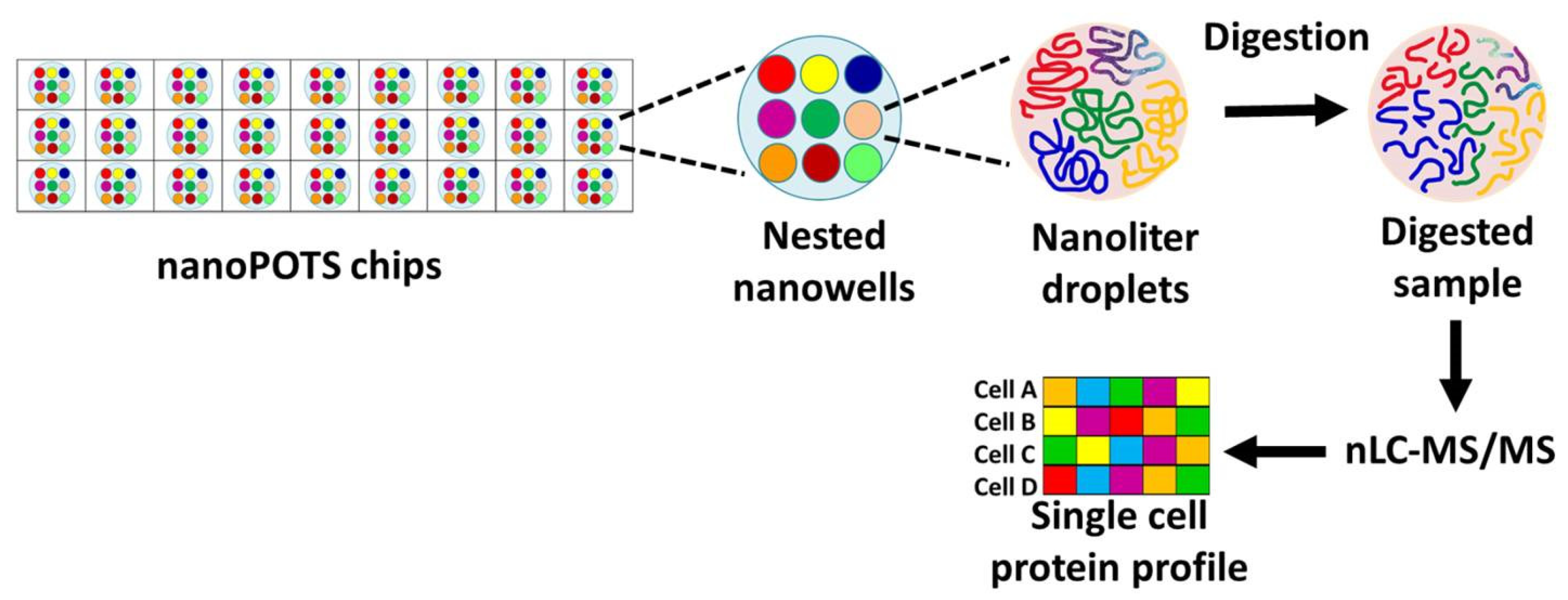
Figure 3.
Flow cytometery (FCM) based single-cell proteomics (SCP), FCM-SCP is developed to generate the proteome profile of a single cell. FCM-SCP is a high throughput method, enable analysis of thousands of cells per second, making it suitable for large-scale studies. In FCM, cells from the tissue are separated using micro-dissection and are sorted by fluorescence-activated cell sorting (FACS). Further, cell lysis, digestion, and peptide labelling are performed using fluorescently-tagged antibodies. The multiplexd sample is prepared by mixing the peptide from each sample, which is injected into LS-MS/MS for enhanced peptide identification and quantification. Notably, LC-MS/MS analysis generates the single cell protein profile of all the analyzed cells.
Figure 3.
Flow cytometery (FCM) based single-cell proteomics (SCP), FCM-SCP is developed to generate the proteome profile of a single cell. FCM-SCP is a high throughput method, enable analysis of thousands of cells per second, making it suitable for large-scale studies. In FCM, cells from the tissue are separated using micro-dissection and are sorted by fluorescence-activated cell sorting (FACS). Further, cell lysis, digestion, and peptide labelling are performed using fluorescently-tagged antibodies. The multiplexd sample is prepared by mixing the peptide from each sample, which is injected into LS-MS/MS for enhanced peptide identification and quantification. Notably, LC-MS/MS analysis generates the single cell protein profile of all the analyzed cells.
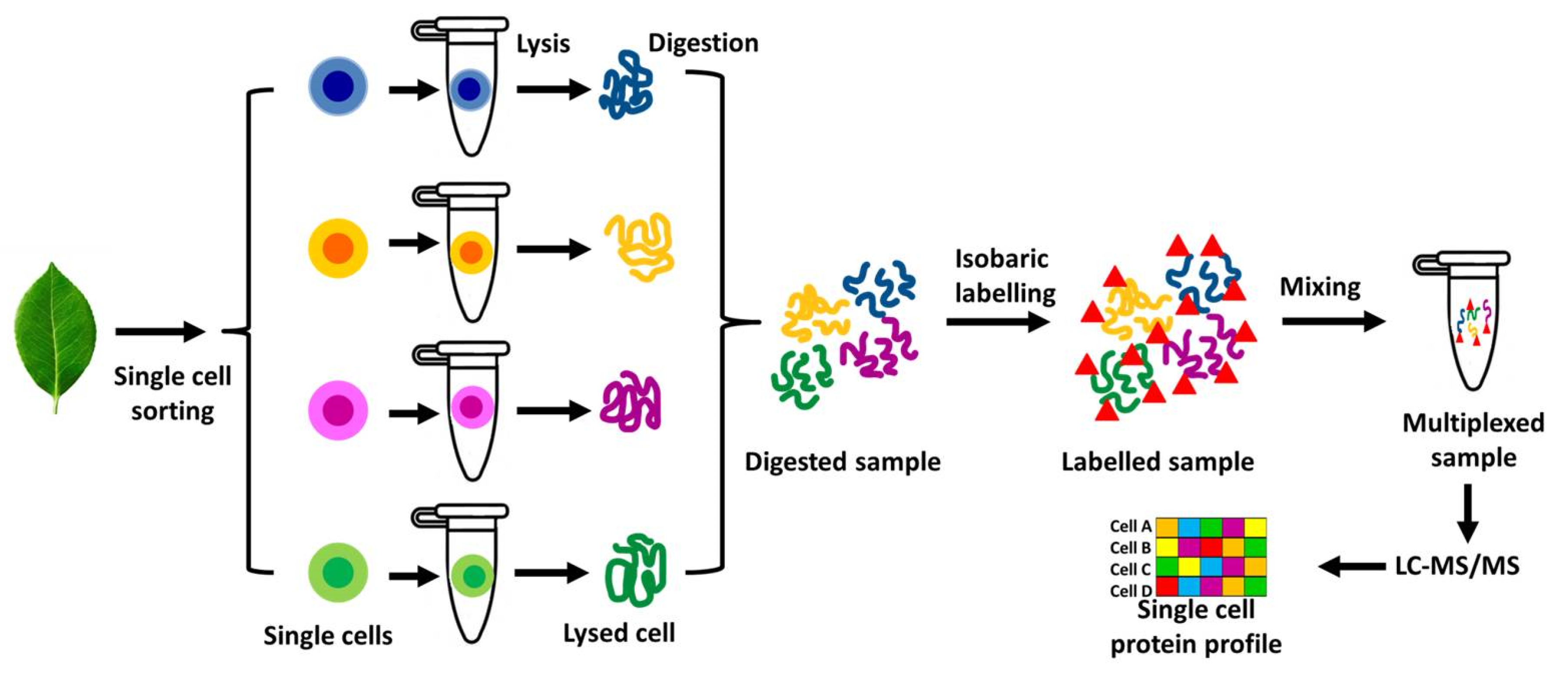
Figure 4.
Single-cell proteomics with Mass Cytometry (CyTOF) to generate proteome profile of a single cell. CyTOF is based on the concept of using heavy-metal isotopes conjugated with exogenous isotopically pure elements to indirectly label the target proteins. The single-cell suspension is incubated with a cocktail of antibodies conjugated to heavy metal isotopes to allows antibody binding to specific epitope of each cell. Further, the antibody conjugated single cells are introduced into the inductively coupled plasma (ICP) for droplet nebulization to create single-cell droplets. Subsequently, the droplets are atomized, ionised to vaporize the sample into ion clouds. Further, the ion clouds are analyzed by time-of-flight (TOF) mass spectrometry to enables the detailed insights into the protein expression profiles of individual cells.
Figure 4.
Single-cell proteomics with Mass Cytometry (CyTOF) to generate proteome profile of a single cell. CyTOF is based on the concept of using heavy-metal isotopes conjugated with exogenous isotopically pure elements to indirectly label the target proteins. The single-cell suspension is incubated with a cocktail of antibodies conjugated to heavy metal isotopes to allows antibody binding to specific epitope of each cell. Further, the antibody conjugated single cells are introduced into the inductively coupled plasma (ICP) for droplet nebulization to create single-cell droplets. Subsequently, the droplets are atomized, ionised to vaporize the sample into ion clouds. Further, the ion clouds are analyzed by time-of-flight (TOF) mass spectrometry to enables the detailed insights into the protein expression profiles of individual cells.

Figure 5.
Single-cell proteomics with Imaging Mass Cytometry (IMC) to generate proteome profile of a single cell. In IMC analysis, tissue of interest is sliced and slide mounted. The samples in the slide is incubated with a cocktail of antibodies conjugated to heavy metal isotopes to allows antibody binding with specific epitope of each cell. Regions of interest (ROI) are selected and ablated by laser to vaporized them by pulsed laser, and generated ion clouds are transferred to inductively coupled plasma (ICP)-mass spectroscopy (ICP-MS) for mass cytometry analysis. The MS analysis generates the single cell protein profile for all the analyzed cells.
Figure 5.
Single-cell proteomics with Imaging Mass Cytometry (IMC) to generate proteome profile of a single cell. In IMC analysis, tissue of interest is sliced and slide mounted. The samples in the slide is incubated with a cocktail of antibodies conjugated to heavy metal isotopes to allows antibody binding with specific epitope of each cell. Regions of interest (ROI) are selected and ablated by laser to vaporized them by pulsed laser, and generated ion clouds are transferred to inductively coupled plasma (ICP)-mass spectroscopy (ICP-MS) for mass cytometry analysis. The MS analysis generates the single cell protein profile for all the analyzed cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



