
Submitted:
05 May 2025
Posted:
06 May 2025
You are already at the latest version
Abstract
CD38, a nicotinamide adenine dinucleotide (NAD+) glycohydrolase, increases in old murine macrophages after infection compared to young controls. We aimed to determine whether the increase of CD38 in old murine macrophages after infection is directly associated with enhanced inflammation induced by the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) when compared to young controls. Additionally, we determined the effects of a specific CD38 inhibitor (78c) on CD38; NAD+; IL-1, IL-6, TNF- expressions; and anti-oxidative responses in old murine macrophages induced by oral pathogens. Results showed that old murine macrophages significantly enhanced CD38 and reduced NAD+ levels 24 h after Aa or Pg infection compared to young controls. This enhanced CD38 in old murine macrophages was not directly correlated with the degree of inflammation in macrophages. Inhibition of CD38 by 78c reduced CD38, enhanced NAD+ levels, attenuated IL-1b, IL-6, TNF-a proinflammatory cytokine levels, decreased NAPDH oxidase 1 (Nox1) expression, and enhanced expressions of anti-oxidative enzymes [superoxide dismutase1 (Sod1), glutathione peroxidase 4 (Gpx4), Peroxiredoxin 1 (Prdx1), thioredoxin reductase 1 (Txnrd1), and catalase (Cat)] in old murine macrophages infected with Aa or Pg. These results suggest that inhibition of CD38 by 78c is a promising therapeutic strategy to treat aging-associated periodontitis.
Keywords:
CD38
; NAD+
; aging
; oxidative stress
; cytokines
; periodontitis
1. Introduction
Cluster of Differentiation 38 (CD38) is a type II transmembrane ecto-enzyme ubiquitously expressed in most tissues and cells in mice and humans [1]. Predominantly, CD38 is highly expressed in B cells, plasma cells, natural killer cells, dendritic cells, T cells, monocytes, macrophages, neutrophils, and hematopoietic stem cells [1]. CD38 is a nicotinamide adenine dinucleotide (NAD+) glycohydrolase, which breaks down NAD+ and generates nicotinamide (NAM), ADP-ribose (ADPR), and cyclic ADP-ribose (cADPR) [1,2,3,4]. NAD+ has limited ability to diffuse through cell membrane barriers and plays an important role in activating NAD+-dependent signaling pathways in different subcellular compartments [5]. NAD+ can be reduced to NADH via dehydrogenases, and NAD+ can be phosphorylated to NADP+ via NAD+ kinases [6]. The NAD+/NADH couple regulates cellular energy metabolism, glycolysis, and mitochondrial oxidative phosphorylation. In contrast, NADP+/NADPH maintains redox homeostasis and supports the biosynthesis of fatty and nucleic acids [6]. Additionally, NAD+ serve as a substrate for other NAD+ consuming enzymes, including sirtuins and poly-(ADP-Ribose) polymerases (PARPs) [7]. Sirtuins are NAD+-dependent histone deacetylases, which regulate diverse cellular processes including cellular metabolism, mitochondrial homeostasis, autophagy, DNA repair, apoptosis, oxidative stress, and inflammatory response [8,9]. Whereas PARPs catalyze the covalent attachment of monomers or polymers of ADP-ribose units on a variety of amino acid residues on target proteins. PARPs play roles in DNA damage detection and repair, genomic stability, programmed cell death, and inflammation [10,11].
Individuals over 60 years of age account for 11% of world population in 2016 and it is projected to reach 22% by 2050 [12]. In the aged population, a decline occurs in the NAD+ level [13,14,15]. In contrast, an enhancement occurs in the CD38 levels during aging [16,17], which may be associated with increased aging-related inflammation through a process called inflammaging [18,19]. Increased CD38 during aging leads to further NAD+ depletion. Notably, decreased NAD+ in the aging population affects many aging-associated immune dysfunctions, including mitochondrial dysfunction, intracellular accumulation of oxidative damaged macromolecules (DNA, lipids, and proteins), dysregulated energy metabolism, impaired cellular “waste disposal”, impaired adaptive stress response, compromised DNA repair, dysregulated neuronal Ca2+ handling, stem cell exhaustion, and inflammation [13]. Therefore, the decline of NAD+ contributes to the pathogenesis of various of aging-associated diseases, including infection, neurodegenerative diseases [13,14,15], cancer [2], and type II diabetes [2,14,20]. Hence, CD38 has become a therapeutic target for treating these aging-associated diseases [1,2,3,4,13,14,21].
Aging is associated with development of many diseases, including periodontal disease, which is associated with comorbid systemic diseases, poor physical functioning, inflammatory dysregulation, and limited ability to self-care in frail older populations [22]. A previous study [23] showed that young mice do not develop measurable periodontal bone loss unless heavily infected with human periodontal pathogens. In contrast, old mice displayed significantly increased periodontal bone loss, accompanied by elevated expression of proinflammatory cytokines (IL-1β, TNF-α, and IL-17A) and innate immune receptors involved in the induction or amplification of inflammation [including toll-like receptor 2 (TLR2), CD14, CD11b, CD18, and complement C5a] [23].
Oral bacterial pathogens, including Aggregatibacter actinomycetemcomitans (Aa, a major oral pathogen associated with 90% of localized aggressive periodontitis and 30% to 50% of severe adult periodontitis [24] and Porphyromonas gingivalis (Pg, another major oral pathogen in the initiation and development of severe forms of chronic periodontal disease [25,26]) activate TLRs and their downstream signaling pathways [27,28,29,30,31], including NF-κB, PI3K, and MAPKs [including extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinase (JNK), and p38 MAPK] [32,33], leading to the production of proinflammatory cytokines [including IL-1β, IL-6, ΤNF-α, and receptor activator of NF-κB ligand (RANKL)] . These proinflammatory mediators subsequently cause periodontal tissue damage and alveolar bone loss.
Additionally, oral bacterial pathogens can modulate the generation of reactive oxygen species (ROS) in specific cell types by activating NADPH oxidases (NOXs), which play roles in host defense to eliminate infected bacterial pathogens [25,26,34,35]. However, excessive ROS causes oxidative stress, contributing to mitochondrial dysfunction in aging [36]. The innate immune response also possesses many anti-oxidative enzymes, [including superoxide dismutase1 (Sod1), glutathione peroxidase 4 (Gpx4), Peroxiredoxin 1 (Prdx1), thioredoxin reductase 1 (Txnrd1), and catalase (Cat)], which reduce oxidative stress induced by bacterial pathogens [6]. NADP+/NADPH serve as coenzymes in the anti-oxidative response to maintain cellular redox homeostasis[6,37]. NAD+ depletion with aging caused mitochondrial dysfunction, a decline in energy production and accumulation of ROS that produce high oxidative stress [5].
A previous study [16] showed that old (18-month-old) murine bone marrow-derived monocytes and macrophages (BMMs) displayed higher CD38 protein levels when stimulated by various doses (0.5 to 50 ng/ml) of bacterial lipopolysaccharide (LPS) for 20 h compared to young (3-month-old) mice controls. However, it was not clear if the enhanced CD38 levels in old murine BMMs were directly correlated to enhanced proinflammatory cytokine levels in old murine BMMs. Additionally, it was not clear if inhibition of CD38 by a CD38 specific inhibitor (78c) in old murine BMMs could attenuate proinflammatory cytokine levels, enhance NAD+ expression, and reduce the oxidative stress induced by oral pathogens. In the present study, we first compared CD38 and NAD+ levels in young vs. old murine BMMs with or without infection with the oral pathogens Aa or Pg. Next, we determined if the CD38 protein expression was directly correlated with the activated NF-κB, PI3K, and MAPKs protein levels or the enhanced proinflammatory cytokine (IL-1β, IL-6, ΤNF-α) levels in old murine BMMs when compared to young controls. Finally, we evaluated the effects of a CD38 specific inhibitor (78c) in CD38 and NAD+ levels, proinflammatory cytokine expression, and oxidative stress in old murine BMMs induced by oral pathogens.
2. Results
2.1. Old Murine BMMs Exhibited Significantly Higher CD38 Protein and Lower NAD+ Expressions After Infection with the oral Pathogens Aa or Pg Compared with Young Controls
First, we compared CD38 protein levels in young vs. old murine BMMs with or without infection with the oral pathogens Aa or Pg. As shown in Figure 1 (A&B), CD38 protein levels were undetectable in uninfected young and old murine BMMs. In contrast, 24 h after Aa or Pg infection, old murine BMMs exhibited significantly higher CD38 protein levels compared with young controls (**p<0.01, ***p<0.001). Compared with young controls, old murine BMMs displayed an average of 3.1-fold and 2.3-fold of CD38 protein levels after Aa or Pg infection, respectively. Accordingly, in uninfected young or old murine BMMs, the NAD+ levels were similar between young and old murine BMMs. In contrast, in old murine BMMs infected with Aa or Pg for 24 h, the NAD+ levels were significantly lower in old murine BMMs than young controls (*p<0.05, ***p<0.001). Compared with young controls, the NAD+ levels were reduced about 43.6% and 42.3% in old murine BMMs infected with Aa or Pg, respectively. These results support that old murine BMMs expressed abnormally higher CD38 protein levels and lower NAD+ levels after infection with the oral pathogens Aa or Pg compared with young controls.
2.2. The Abnormal High CD38 Protein Level in Old Murine BMMs After Infection with the Oral Pathogens Aa or Pg Was Not Directly Correlated with the Level of Immune Responses in Old Murine BMMs Compared with Young Control
To determine if old murine BMMs displayed an abnormally high immune response to oral pathogenic infection that contributes to the high CD38 expression in old murine BMMs after infection with an oral pathogen, we quantified the protein levels of CD38 and some protein kinases (including NF-κB, PI3K, and MAPKs) by Western blot in old and young murine BMMs at various time points (1, 2, 4, 6, 8, and 24 h) after infection with Aa or Pg. As shown in Figure 2 (A&B), the old murine BMMs displayed a delayed activation in p-NF-κBp65, p-PI3K, p-ERK, p-JNK, and p-p38 MAPK compared with young controls. The activation of NF-κB, PI3K, and MAPKs occurred at an early time point (1 h) in young murine BMMs after Aa or Pg infection. In contrast, old murine BMMs displayed weak activations of p-NF-κBp65, p-PI3K, p-ERK, p-JNK, and p-p38 MAPK at all time points (1h to 8h) except 24 h after Aa or Pg infection. Although old murine BMMs displayed a delayed immune response to Aa or Pg infection, the old murine BMMs still expressed higher detectable CD38 protein levels at 8 h after bacterial infection compared with young controls (Figure 2 A&B). Although the old murine BMMs exhibited higher CD38 protein expression at 24 h than young controls, the old murine BMMs showed similar levels of p-NF-κBp65, p-PI3K, p-ERK, p-JNK, and p-p38 at 24 h following Aa or Pg infection compared with young controls (Figure 2C-H, ns: no significance). Additionally, quantification of the levels of IL-1β, IL-6, and TNF-α pro-inflammatory cytokines (Figure 3) showed that old murine BMMs displayed similar levels of IL-1β, IL-6, and TNF-α 24 h after infection with Aa or Pg compared with controls in young BMMs (Figure 3A-C). These results support the conclusion that the abnormally high CD38 expression in old murine BMMs was not directly correlated with the activation of NF-κB, PI3K, and MAPKs protein kinases, nor the IL-1β, IL-6, and TNF-α pro-inflammatory cytokine levels in old murine BMMs.
2.3. Inhibition of CD38 by 78c Suppressed CD38, NF-κB, PI3K, and MAPKs Protein Kinases, Enhanced NAD+, and Attenuated IL-1β, IL-6, and TNF-α pro-Inflammatory Cytokines Levels in Old Murine BMMs Infected with the Oral Pathogens Aa or Pg.
Previously, our study [38] demonstrated that inhibition of CD38 by 78c reduced CD38, attenuated the activation of NF-κB, PI3K, and MAPKs induced by the oral pathogens Aa or Pg in murine BMMs derived from TALLYHO/JngJ mice (type 2 diabetic mice). We hypothesized that treatment with 78c would also suppress CD38 and the activation of NF-κB, PI3K, and MAPKs induced by the oral pathogens Aa or Pg in old murine BMMs. As shown in Figure 4(A&B), treatment with 78c (10 µM) reduced CD38, p-NF-κBp65, p-PI3K, p-ERK, p-JNK, and p-p38 MAPK induced by Aa or Pg. Accordingly, we observed reductions of IL-1β, IL-6, and TNF-α pro-inflammatory cytokines in cells treated with 78c compared with controls. Treatment with 78c (1.25 µM, 2.5 µM, 5 µM, and 10 µM) in old murine BMMs reduced IL-1β by 50.1%, 63.9%, 88.7%, and 96.5%, respectively, as induced by Aa, and attenuated IL-1β by 73.7%, 87.3%, 89.6%, and 95.6%, respectively, as induced by Pg (Figure 4C). Treatment with 78c (1.25 µM, 2.5 µM, 5 µM, and 10 µM) also reduced IL-6 by 40.6%, 53.2%, 70.9%, and 88.1%, respectively, as induced by Aa, and attenuated IL-6 by 35.2%, 68.4%, 82.2%, and 90.4%, respectively, as induced by Pg (Figure 4D). Additionally, Treatment with 78c (1.25 µM, 2.5 µM, 5 µM, and 10 µM) reduced TNF-α by 36.7%, 44.3%, 65.7%, and 75.4%, respectively, as induced by Aa, and attenuated TNF-α by 21.4%, 36.8%, 47.1%, and 64.1%, respectively, as induced by Pg (Figure 4E). In uninfected old murine BMMs, treatment with 78c (5 µM and 10 µM) enhanced NAD+ by 1.2-fold and 1.3-fold, respectively (Figure 4F). The NAD+ levels declined about 82.8% in cells treated with vehicle and infected with Aa and decreased about 42.9% in cells treated with vehicle and infected with Pg as compared with the NAD+ levels in cells treated with vehicle without bacterial infection (Figure 4F). Treatment with 78c (1.25 µM, 2.5 µM, 5 µM, and 10 µM) enhanced NAD+ by 1.9-fold, 2.7-fold, 4.3-fold, and 6.6-fold, respectively, in old murine BMMs infected with Aa and enhanced NAD+ by 1.3-fold, 2.1-fold, 2.4-fold, and 3.0-fold, respectively, in old murine BMMs infected with Pg (Figure 4F). These data support that inhibition of CD38 by 78c suppressed CD38 and prevented the decline of NAD+ induced by oral pathogens. Additionally, inhibition of CD38 by 78c suppressed the activation of NF-κB, PI3K, and MAPKs as induced by the oral pathogens Aa or Pg, subsequently attenuated IL-1β, IL-6, and TNF-α pro-inflammatory cytokines as induced by oral pathogens.
2.4. Inhibition of CD38 by 78c Reduced Oxidative Stress in Old Murine BMMs Infected with Oral Pathogens
Since excessive ROS causes oxidative stress, contributing to mitochondrial dysfunction in aging [36], and NADP+/NADPH serve as coenzymes in the anti-oxidative response to maintain cellular redox homeostasis [6,37], we hypothesized that inhibition of CD38 by 78c in old murine BMMs could increase NAD+ and subsequently reduce oxidative stress induced by oral pathogens. As shown in Figure 5A, treatment with 78c (10 µM) for 24 h significantly reduced ROS in old murine BMMs either with or without infection of Aa (*p<0.05) compared with vehicle controls. Treatment with 78c (1.25 to 10 µM) for 24 h also significantly reduced ROS in old murine BMMs infected with Pg (*p<0.05, **p<0.01, *** p<0.01). To determine how 78c regulates oxidative response and affects aging-associated immune responses in old murine BMMs, we quantified the mRNA levels of CD38, Nox1, anti-oxidative enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat), and NAD+ consuming enzymes (Sirt1 and Parp1) in old murine BMMs that were either left uninfected or were infected with Aa or Pg for 8 h. Treatment with 78c (5 and 10 µM) reduced CD38 mRNA levels by 44.5% and 61.2% as induced by Aa, respectively, and reduced CD38 mRNA levels by 28.5% and 63.2% as induced by Pg, respectively (Figure 5B). Treatment with 78c dose-dependently reduced Nox1 as induced by Aa or Pg compared with controls (Figure 5C). Additionally, treatment with 78c dose-dependently enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat) mRNA levels in old murine BMMs (Figure 3D-H). Furthermore, treatment with 78c does-dependently enhanced two NAD+ consuming enzymes (Sirt1 and Parp1) mRNA levels in old murine BMMs either uninfected or infected with Aa or Pg compared with controls (Figure 5I&J). Western blot protein assay also showed that treatment with 78c (10 µM) in old murine BMMs suppressed Nox1 protein levels at 1, 2, 4, and 6 h after Aa infection compared with controls (Figure 6A), and reduced Nox1 protein expressions at 1, 2, and 4 h after Pg infection compared with controls (Figure 6 B). After treatment with 78 c (10 µM) for 24 h, CD38 protein levels were reduced in old murine BMMs infected with Aa or Pg compared with controls (Figure 6C). At 24 h after treatment, Nox1 protein levels were similar between 78c-treated cells and vehicle-treated cells. We observed enhanced protein levels of anti-oxidant enzymes (Sod1, Gpx4, Prdx1,Txnrd1, and Cat) in uninfected old murine BMMs or Aa or Pg-infected BMMs compared with controls (Figure 6C). These results support that 78c attenuated oxidative stress by inhibiting Nox1 mRNA and protein expressions, while enhancing anti-oxidant enzyme mRNA and protein expressions. Additionally, we also observed enhanced NAD+ consuming enzymes (Sirt1 and Parp) proteins in 78c (10 µM)-treated uninfected old murine BMMs or Aa or Pg infected BMMs compared with controls (Figure 6C).
3. Discussion
Periodontitis is an inflammatory bone loss disease. Oral bacterial pathogens not only induce the generation of IL-1β, IL-6, and TNF-α, but also RANKL, the major osteoclast differentiation factor [39]. RANKL binds with its receptor RANK, which promote osteoclast precursors (monocytes and macrophages) to differentiate and fuse to form multinucleated osteoclasts, leading to alveolar bone loss. In the current study, we demonstrated that old murine BMMs exhibited an abnormal immune response to infection with the oral pathogens Aa or Pg, including a delayed activation of NF-κB, PI3K, and MAPKs protein kinases, an enhanced CD38 expression, and a reduced NAD+ expression compared with controls in young murine BMMs (Figure 1, Figure 2A&B). Our study is in accordance with a previous study [16], which showed that old murine BMMs displayed higher CD38 protein expressions when stimulated by LPS for 20 h compared with young mice controls. Previously, Chini et al. [40] showed that inducing senescence in human umbilical vain endothelial cells (HUVECs) by DNA damage through exposure to X-ray irradiation or gamma irradiation enhanced markers for senescence, including p21, p16Ink4a, and PAI1. However, CD38 mRNA was not induced by these treatments [40]. Instead, Chini et al. [40] discovered that senescent cells induced by either X-ray irradiation or gamma irradiation secreted higher inflammatory cytokines, including IL6, IL-8, and monocyte chemoattractant protein -1 (MCP1) than controls. When murine BMMs were incubated with conditioned media derived from senescent cells induced by either X-ray irradiation or gamma irradiation, the mRNA and protein levels of CD38 were induced [40]. In the current study, uninfected old murine BMMs and young murine BMMs expressed undetectable CD38 protein levels and similar NAD+ levels (Figure 1A-C). After infection with the oral pathogens Aa or Pg for 24 h, CD38 protein levels were increased in both old and young murine BMMs (Figure 2A-C), supporting that CD38 is an inflammatory marker. However, we did not observe significant differences between the old murine BMMs and young murine BMMs in the activation of NF-κB, PI3K, and MAPKs protein kinases or IL-1β, IL-6, TNF-α cytokine expression 24 h after infection with Aa or Pg (Figure 1C-H, Figure 2A-C). This finding suggests that the high CD38 expression in old murine BMMs after infection with oral pathogens was not directly correlated with the activation of NF-κB, PI3K, and MAPKs protein kinases nor the amount of pro-inflammatory cytokine (IL-1β, IL-6, TNF-α) released in old murine BMMs.
Our previous study [38] demonstrated that treatment with 78c suppressed NF-κB, PI3K, and MAPKs protein kinases induced by the oral pathogens Aa or Pg in murine BMMs derived from TALLYHO/JngJ mice (type 2 diabetic mice). In accordance with our previous study, treatment with 78c also reduced the activation of NF-κB, PI3K, and MAPKs protein kinases and attenuated the levels of the pro-inflammatory cytokine (IL-1β, IL-6, TNF-α) that were induced by the oral pathogens Aa or Pg in old murine BMMs (Figure 4A-E). Because NADPH oxidase (Nox) activation is associated with sensing the molecular signatures of microbial pathogens by TLRs [35] and ROS-mediated cellular signal pathways interplay with TLRs down-stream signaling pathways (including NF-κB, PI3K, and MAPKs protein kinases) [41], inhibition of NF-κB, PI3K, and MAPKs protein kinases by 78c could reduce Nox1 expression induced by oral pathogens (Figure 6A-B). The enhancement of anti-oxidant enzymes (including Sod1, Gpx4, Prdx1,Txnrd1, and Cat) in murine BMMs treated with 78c (Figure 5D-H, Figure 6C) was caused by the increase of NAD+ levels in 78c-treated cells (Figure 4F). As NADPH/NADP+ are involved in maintaining redox homeostasis by turning O2- into H2O2 by Sod1 and subsequently turning H2O2 into H2O by other anti-oxidant enzymes (including Gpx4, Prdx1, Txnrd1, and Cat) [6,37], inhibition of the degradation of NAD+ by CD38 in 78c-treated murine BMMs could enhance NAD+ and subsequently increase these anti-oxidant enzyme expressions. Our findings are in accordance with a prior study [42], which demonstrated that inhibition CD38 by apigenin (a flavonoid with CD38 inhibitory activity) ameliorated oxidative stress by enhancing Sod and Gpx expressions in skeletal muscle of aged mice.
Additionally, our previous study [38] demonstrated that treatment with 78c suppressed osteoclastogenesis and bone resorption induced by RANKL. Mechanistically, we demonstrated that treatment with 78c reduced podosome (basic cell adhesion unit) components, (including PI3K, Pyk2, Src, F-actin, integrins, paxillin, and talin) induced by RANKL. Treatment with 78c also attenuated osteoclastogenic factors, including the nuclear factor of activated T-cells cytoplasmic calcineurin-dependent 1 (Nfatc1), cathepsin K (Ctsk), acid phosphatase 5 (Acp5), osteoclast stimulatory transmembrane protein (Ocstamp), and dendritic cell-specific transmembrane protein (Dcstamp), induced by RANKL. Therefore, treatment with 78c could potentially alleviate inflammatory bone loss in patients with periodontitis.
Previous studies [9,43] demonstrated that sirtuins play roles in extending the lifespan of organisms. Numerous studies reported that the SIR2, the first identified sirtuin protein in yeast, extended the lifespan in yeast [44], C. elegans [45], and Drosophila [46]. The Sirt1 is the most studied and the mammalian closest ortholog to SIR2. Sirt1 expression declines with aging in animals and human tissues [47]. In contrast, over-expression of Sir1 in the brain extended the lifespan of mice [48]. Over-expression of Sirt1 in pancreatic β cells of mice also improved glucose tolerance and enhanced glucose-stimulated insulin secretion in mice at 3 to 8 m of age compared with controls [49]. In the current study, treatment with 78c enhanced NAD+ (Figure 4F) and subsequently increased Sirt1 mRNA and protein expressions in old murine BMMs either uninfected or infected with oral pathogens (Figure 5I and Figure 6C), supporting that treatment with 78c is a promising therapeutic approach to treat aging-associated periodontitis, which can enhance NAD+ and Sirt1, maintain mitochondrial homeostasis and metabolic function, and promote longevity. In response to the increase of NAD+ in 78c-treated cells, we also observed enhanced Parp1 mRNA (Figure 5J) and Parp protein levels (Figure 6C) in old murine BMMs treated with 78c. This enhanced Parp could assist in repairing damaged DNA in cells. The Sirtuins and Parp can cleave NAD+ and release nicotinamide (NAM) [5,37]. NAM can be recycled by
the enzyme nicotinamide phosphoribosyltransferase (NAMPT)
to
nicotinamide mononucleotide (NMN) and subsequently be synthesized to NAD+ by NMN adenylyltransferases
(NMNATs) via the salvage pathway [5,37]. We observed that treatment with 78c (5 or 10 µM) displayed higher NAD+ levels in old murine BMMs infected with Pg compared with controls (Figure 4F). This could be caused by enhanced Sirt1 and Parp levels in 78c-treated cells, which could in turn promote the regeneration of NAD+ by the salvage pathway.
Previously, accumulated evidence suggests that NAD+ levels decline with aging at a systemic level in diverse organisms, including rodents and humans, contributing to the development of many aging-associated diseases [14,50,51]. These enhanced NAD+ levels in the aging population are associated with chronic inflammation in aging patients, called inflammaging [18,19]. In the current study, the mice were bred in a specific pathogen-free condition, and the mice were relatively healthy without inflammation. Therefore, we did not detect CD38 protein levels in uninfected murine BMMs, and uninfected old murine BMMs expressed similar levels of NAD+ compared with young controls. Because human bodies are exposed to varieties of pathogens and aging patients often have comorbidity with various chronic inflammation (including atherosclerosis, cardiovascular events, cancer, autoimmune diseases), aging patients could have high levels of CD38+ and reduced levels of NAD+ compared with young controls.
Scaling and root surface debridement are the traditional “gold standard” treatment for stage I-III periodontitis. There are still patients or sites that show poor response to non-surgical periodontal treatment and long-term supportive maintenance efforts. This could be due to sustained dysbiosis, bacteria invasion to periodontal tissues, or a non-resolving chronic inflammatory response. Previous studies [52,53] demonstrated that treatment with 78c in aged mice reversed age-related NAD+ decline and increased lifespan and health span of naturally aged mice. Treatment with 78c improved several physiological and metabolic aging parameters, including glucose tolerance, muscle function, exercise capacity, and cardiac function in mouse natural and accelerated aging models [52,53]. Our previous study [38] demonstrated that inhibition of CD38 by 78 attenuated IL-1β, IL-6, TNF-α pro-inflammatory cytokine expressions induced by the oral pathogens Aa or Pg in murine BMMs derived from TALLYHO/JngJ mice (type 2 diabetic mice). Additionally, treatment with 78c reduced osteoclastogenesis and bone resorption induced by RANKL (Figure 7) [38]. In the current study, we also showed that treatment with 78c in old murine BMMs inhibited NF-κB, PI3K, and MAPKs protein kinases as induced by the oral pathogens Aa or Pg, and subsequently alleviated IL-1β, IL-6, TNF-α pro-inflammatory cytokine expressions, and Nox1 mRNA and protein expressions. Additionally, treatment with 78c suppressed CD38 and enhanced NAD+ levels, and subsequently increased the mRNA and protein levels of anti-oxidant enzymes (Sod1, Gpx4, Prdx1,Txnrd1, and Cat) in old murine BMMs either uninfected or infected with the oral pathogens Aa or Pg. Our and other peoples’ studies support that inhibition of CD38 by 78 could serve as an adjunctive therapy for aging-associated periodontitis to inhibit periodontal inflammation, attenuate osteoclastogenesis and alveolar bone resorption, alleviate oxidative stress, and prolong the health span of human beings.
The current study has some limitations. Although we showed that old murine BMMs displayed higher CD38 protein levels after infection with the oral pathogens Aa or Pg, it was not clear about the mechanisms that were associated with the increase of CD38 in old murine BMMs. Future studies should determine why old murine BMMs express higher CD38 compared to young controls after bacterial infection. Additionally, aging patients have various age-associated diseases (including atherosclerosis, neurodegenerative diseases, autoimmune diseases, and type II diabetes). We only determined CD38 expression in response to the oral pathogens Aa or Pg infection. Future studies should determine if old murine BMMs displayed higher CD38 in response to other stimuli and if inhibition of CD38 by 78c could reduce CD38, alleviate inflammation, and reduce oxidative stress induced by other stimuli. Furthermore, since we only conducted in vitro studies, future in vivo studies need to determine if treatment with 78c could alleviate periodontal inflammation, attenuate alveolar bone loss, and reduce oxidative stress in old animals with periodontitis.
4. Materials and Methods
4.1. Animals, and Reagents
Old (18-month-old) and young (2-month-old) male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Young female and male mice were bred to generate 2 to 3-month-old young control mice. Mice were housed under a 12 h light/12 h dark cycle in specific pathogen-free conditions and had free access to food and water. All animal-related work was conducted in accordance with the guidelines laid down by the National Institutes of Health (NIH) in the United States regarding the usage of animals for experimental procedures and approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (IACUC-2021-01287). 78c was purchased from Tocris Bioscience (Minneapolis, MN, USA), dissolved in dimethyl sulfoxide (DMSO) as a 10 mM stock solution, and stored at −20 °C. An equal volume of DMSO (as compared to 10 mM 78c) was diluted in cell culture media and served as a vehicle control.
4.2. Generation of L929 Conditioned Media
L929 (mouse fibroblast cell line) was purchased from the American Type Cell Culture Collection (ATCC, Manassas, VA, USA). L929 cells (5 × 105) were plated in a T75 flask and cultured for six days in 30 mL of Dulbeco’s Modified Eagle Medium (DMEM, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS). The cell culture media was filtered through a 0.22 μM filter, aliquoted, and stored at −80 °C as L929 conditioned media (containing macrophage colony-stimulating factor, M-CSF).
4.3. Generation of Bone Marrow-Derived Monocytes and Macrophages (BMMs)
Murine bone marrow cells were harvested from old (18-month old) or young (2- to 3-month-old) male C57BL/6J mice by flushing bone marrow cells from the tibia and femur using 10 mL cell culture media with a 10 mL syringe and 27 gauge needle (Becton Dickinson, Franklin Lakes, New Jersey, USA). The cell culture media is complete minimal essential media (MEM)-α (ThermoFisher Scientific) supplemented with 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin. To remove tissue debris, the bone marrow cells were filtered through a 40 μM nylon cell strainer (ThermoFisher Scientific). Then, murine bone marrow cells were cultured in a complete MEM-α media supplement with 20% L929 conditioned media for three days. The attached bone marrow-stromal cells were discarded. The suspended bone marrow cells were transferred to new cell culture plates and cultured in complete MEM-α media supplement with 20% L929 conditioned media for another seven days until cells were differentiated into attached BMMs. Previous studies [54,55] showed that the adherent bone marrow-derived macrophages were usually better than 90% pure using macrophage markers such as CD11b and F4/80. One day before infection, the attached murine BMMs were lifted by treating with 10 mM EDTA and were plated in new cell culture dishes in MEM-α media with 5% FBS and 20% L929 conditioned media without antibiotics.
4.4. Bacterial Culture
Oral bacterial pathogens Aggregatibacter actinomycetemcomitans (Aa, ATCC 43718) and Porphyromonas gingivalis (Pg, ATCC 33277) were originally obtained the American Type Culture Collection. Aa was cultured in brain–heart infusion broth (Fisher Scientific, Suwanee, GA, USA) at 37 °C with 10% CO2. Pg was cultured to the early exponential phase in tryptic soy broth (Becton Dickinson, Sparks, MD, USA) supplemented with yeast extract (Becton Dickinson, 1 mg/mL), menadione (Chem-Implex Int’l Inc., Wood Dale, IL, USA, 1μg/mL), and hemin (Millipore Sigma, St. Louis, MO, USA, 5 μg/mL) at 37 °C under anaerobic conditions and harvested as previously described [26,56,57]. Briefly, the cell pellets of Pg or Aa were washed and resuspended with PBS before infection. Pg concentration was determined using a Klett-Summerson photometer (Bel-Art, Wayne, NJ, USA), followed by serial dilution and plating on tryptic soy agar plates supplemented with yeast extract (Becton Dickinson, 5 mg/mL), menadione (Chem-Implex Int’l Inc., Wood Dale, IL, USA, 1μg/mL), hemin (Millipore Sigma, St. Louis, MO, USA, 5 μg/mL), and sheep blood (Hemostat Laboratories, Dixon, CA) at 37 °C under anaerobic conditions. The K value 1.0 was equal to about 1.0 × 109 CFU/mL of Pg. Aa bacterial concentration was determined by measuring bacterial optical density at 600 nm followed by serial dilution and plating on brain heart infusion agar plates (Fisher Scientific). OD600 = 1 was equal to about 3 × 108 CFU/mL of Aa. Agar plate counts (CFU/mL) were used for both bacteria to calculate the multiplicity of infection (MOI), and MOI 20 was used for Pg or Aa infection of murine BMMs to detect noticeable cytokine expressions in the BMMs. A control group of cells were not infected with bacteria.
4.5. NAD+ Assay
The NAD+ levels were determined using a NAD+/NADH cell-based assay kit according to the manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI, USA). The NAD+ levels were calibrated by cell growth and viability determined using a CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA).
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
IL-1β levels in cell lysates, IL-6, and TNF-α protein levels in cell culture media of BMMs were quantified using ELISA kits (R&D Systems, Minneapolis, MN, USA). The concentration of cytokines was normalized by protein concentration, which was determined using a DC protein Assay Kit (Bio-Rad Laboratories, Hercules, CA, USA) in cell lysates (100 μL RIPA cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA)/well in a 12-well cell culture plate).
4.7. RNA Extraction and Real-Time PCR
According to the manufacturer’s instructions, total RNA was isolated from murine bone marrow cells using TRIZOL (ThermoFisher Scientific). Complementary DNA was synthesized using a TaqMan reverse transcription kit (Life Technologies, Carlsbad, CA, USA) using the total RNA (1 μg). Real-time PCR was performed using a StepOnePlus Real-Time PCR System (Life Technologies). PCR conditions used were as follows: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The following amplicon primers were obtained from Life Technologies: CD38 (Mm00483143_m1), Nox1 (Mm00549170_m1), Sod1 (Mm01344233_g1), Gpx4 (Mm04411498_m1), Prdx1(Mm01621996_s1), Txnrd1 (Mm00443675_m1), Cat (Mm00437992_m1), Sirt1 (Mm01168521_m1), Parp1 (Mm01321084_m1), and β-actin (Mm02619580_g1). Amplicon concentration was determined using threshold cycle values compared with standard curves for each primer. Sample mRNA levels were normalized to an endogenous control β-actin expression and were expressed as fold changes compared with control groups.
4.8. Western Blot Analysis
Per the manufacturer’s guidance, protein was extracted from murine BMMs using a RIPA cell lysis buffer (Cell Signaling Technology). Total protein (25 μg) was loaded on 10% Tris-HCl gels, electro-transferred to nitrocellulose membranes, blocked, and incubated overnight at 4 °C with primary antibodies. The antibodies to CD38, p-PI3K, p-ERK, p-JNK, p-p38, p-NF-κB p65, Sod1, Gpx4, Trdx1,Txnrd1(Trxr1), Cat, Sirt1, Parp, and pan-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). An antibody to Nox1 was obtained from Invitrogen (Carlsbad, CA, USA). All primary antibodies were diluted in ratios of 1:500 or 1:1000. After washing, the nitrocellulose membranes were incubated at room temperature for one hour with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology) and developed using SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific). Digital images and protein densitometry were analyzed with a G-BOX chemiluminescence imaging system (Syngene, Frederick, MD, USA).
4.9. Statistical Analysis
Data were checked for normality using a QQ plot. The data were analyzed using a one-way ANOVA with Dunnett’s or Tukey’s multiple comparisons tests. All statistical tests were performed using GraphPad Prism software (Version 10.4.0, GraphPad Software Inc., La Jolla, CA, USA). Values are expressed as means ± standard error of the means (SEM) of three independent experiments. A p-value of 0.05 or less was considered significant.
Author Contributions
Conceptualization, H.Y., Ö.Y; methodology, K.C, N.C., B.W., Ö.Y, and H.Y.; investigation, K.C, N.C., B.W., Ö.Y., W.H., and H.Y.; writing –original draft preparation, K.C., H.Y.; writing-review and editing, K.C, N.C., B.W., Ö.Y., W.H., and H.Y.; funding acquisition, H.Y., Ö.Y., W.H.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Institutes of Health (grant numbers R21DE030865, R21DE033977, and UL1 TR001450). Additionally, this work was partially supported by a grant from Center for Healthy Aging at the Medical University of South Carolina.
Institutional Review Board Statement
This study was approved by IACUC by Medical University of South Carolina (Charleston, SC USA) under protocol IACUC-2021-01287.
Informed Consent Statement
Not Applicable
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
We thank. R.J. Lambert at the Writing Center at the Medical University of South Carolina for manuscript review and assistance. Figure 7 was generated using BioRender.
Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- Piedra-Quintero, Z.L.; Wilson, Z.; Nava, P.; Guerau-de-Arellano, M. CD38: An Immunomodulatory Molecule in Inflammation and Autoimmunity. Frontiers in immunology 2020, 11, 597959. [Google Scholar]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Frontiers in immunology 2019, 10, 1187. [Google Scholar] [CrossRef]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Benzi, A.; Grozio, A.; Spinelli, S.; Sturla, L.; Guse, A.H.; De Flora, A.; Zocchi, E.; Heeren, J.; Bruzzone, S. Role of CD38 in Adipose Tissue: Tuning Coenzyme Availability? Nutrients 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal transduction and targeted therapy 2021, 6, 2. [Google Scholar]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxidants & redox signaling 2019, 30, 251–294. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mechanisms of ageing and development 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB reports 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes & development 2017, 31, 101–126. [Google Scholar]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes & development 2020, 34, 341–359. [Google Scholar]
- Kanasi, E.; Ayilavarapu, S.; Jones, J. The aging population: demographics and the biology of aging. Periodontology 2000 2016, 72, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell metabolism 2019, 30, 630–655. [Google Scholar] [PubMed]
- Verdin, E. NAD⁺ in aging, metabolism, and neurodegeneration. Science (New York, N.Y.) 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proceedings of the National Academy of Sciences of the United States of America 2015, 112, 2876–2881. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nature metabolism 2020, 2, 1284–1304. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell metabolism 2016, 23, 1127–1139. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. The journals of gerontology. Series A, Biological sciences and medical sciences 2014, 69 Suppl 1, S4-9.
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nature reviews. Endocrinology 2018, 14, 576–590. [Google Scholar]
- Zapata-Pérez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD(+) homeostasis in human health and disease. EMBO molecular medicine 2021, 13, e13943. [Google Scholar] [CrossRef]
- Peclat, T.R.; Shi, B.; Varga, J.; Chini, E.N. The NADase enzyme CD38: an emerging pharmacological target for systemic sclerosis, systemic lupus erythematosus and rheumatoid arthritis. Current opinion in rheumatology 2020, 32, 488–496. [Google Scholar] [CrossRef]
- Clark, D.; Kotronia, E.; Ramsay, S.E. Frailty, aging, and periodontal disease: Basic biologic considerations. Periodontology 2000 2021, 87, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Hosur, K.B.; Domon, H.; Hajishengallis, G. Periodontal inflammation and bone loss in aged mice. Journal of periodontal research 2010, 45, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Raja, M.; Ummer, F.; Dhivakar, C.P. Aggregatibacter actinomycetemcomitans - a tooth killer? J Clin Diagn Res. 2014, 8, ZE13–16. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cellular microbiology 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.S.; Atanasova, K.R.; Lee, J.; Diamond, G.; Deguzman, J.; Hee Choi, C.; Yilmaz, Ö. Opportunistic Pathogen Porphyromonas gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Frontiers in cellular and infection microbiology 2017, 7, 291. [Google Scholar] [CrossRef]
- Cai, J.; Chen, J.; Guo, H.; Pan, Y.; Zhang, Y.; Zhao, W.; Li, X.; Li, Y. Recombinant fimbriae protein of Porphyromonas gingivalis induces an inflammatory response via the TLR4/NF-κB signaling pathway in human peripheral blood mononuclear cells. International journal of molecular medicine 2019, 43, 1430–1440. [Google Scholar] [CrossRef]
- Hodgkinson, C.P.; Laxton, R.C.; Patel, K.; Ye, S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 2008, 28, 2275–2281. [Google Scholar] [CrossRef]
- Kanaya, S.; Nemoto, E.; Ogawa, T.; Shimauchi, H. Porphyromonas gingivalis fimbriae induce unique dendritic cell subsets via Toll-like receptor 2. Journal of periodontal research 2009, 44, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, Y.; Cui, Q.; Xu, J.; Tang, Z.; Wang, Y.; He, C.; Wang, X. Toll-like receptor 4 plays a key role in advanced glycation end products-induced M1 macrophage polarization. Biochemical and biophysical research communications 2020, 531, 602–608. [Google Scholar] [CrossRef]
- Lima, H.R.; Gelani, V.; Fernandes, A.P.; Gasparoto, T.H.; Torres, S.A.; Santos, C.F.; Garlet, G.P.; da Silva, J.S.; Campanelli, A.P. The essential role of toll like receptor-4 in the control of Aggregatibacter actinomycetemcomitans infection in mice. Journal of clinical periodontology 2010, 37, 248–254. [Google Scholar] [CrossRef]
- Watanabe, K.; Yilmaz, O.; Nakhjiri, S.F.; Belton, C.M.; Lamont, R.J. Association of mitogen-activated protein kinase pathways with gingival epithelial cell responses to Porphyromonas gingivalis infection. Infection and immunity 2001, 69, 6731–6737. [Google Scholar] [CrossRef]
- Yilmaz, O.; Jungas, T.; Verbeke, P.; Ojcius, D.M. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infection and immunity 2004, 72, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. International journal of molecular sciences 2011, 12, 334–352. [Google Scholar] [PubMed]
- Ogier-Denis, E.; Mkaddem, S.B.; Vandewalle, A. NOX enzymes and Toll-like receptor signaling. Seminars in immunopathology 2008, 30, 291–300. [Google Scholar] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 2020, 5, 227. [Google Scholar]
- Lory, W.; Chowdhury, N.; Wellslager, B.; Pandruvada, S.; Huang, Y.; Yilmaz, Ö.; Yu, H. CD38 Inhibitor 78c Attenuates Pro-Inflammatory Cytokine Expression and Osteoclastogenesis in Macrophages. Cells 2024, 13. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone research 2013, 1, 11–26. [Google Scholar]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochemical and biophysical research communications 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative medicine and cellular longevity 2016, 2016:4350965.
- Wang, D.; Yang, Y.; Zou, X.; Zhang, J.; Zheng, Z.; Wang, Z. Antioxidant Apigenin Relieves Age-Related Muscle Atrophy by Inhibiting Oxidative Stress and Hyperactive Mitophagy and Apoptosis in Skeletal Muscle of Mice. The journals of gerontology. Series A, Biological sciences and medical sciences 2020, 75, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Tabibzadeh, S. Signaling pathways and effectors of aging. Frontiers in bioscience (Landmark edition) 2021, 26, 50–96. [Google Scholar] [CrossRef] [PubMed]
- Stumpferl, S.W.; Brand, S.E.; Jiang, J.C.; Korona, B.; Tiwari, A.; Dai, J.; Seo, J.G.; Jazwinski, S.M. Natural genetic variation in yeast longevity. Genome research 2012, 22, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Ludewig, A.H.; Izrayelit, Y.; Park, D.; Malik, R.U.; Zimmermann, A.; Mahanti, P.; Fox, B.W.; Bethke, A.; Doering, F.; Riddle, D.L.; et al. Pheromone sensing regulates Caenorhabditis elegans lifespan and stress resistance via the deacetylase SIR-2.1. Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 5522–5527. [Google Scholar] [CrossRef]
- Banerjee, K.K.; Ayyub, C.; Ali, S.Z.; Mandot, V.; Prasad, N.G.; Kolthur-Seetharam, U. dSir2 in the adult fat body, but not in muscles, regulates life span in a diet-dependent manner. Cell reports 2012, 2, 1485–1491. [Google Scholar] [CrossRef]
- Cho, S.H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. The Journal of neuroscience : the official journal of the Society for Neuroscience 2015, 35, 807–818. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell metabolism 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Moynihan, K.A.; Grimm, A.A.; Plueger, M.M.; Bernal-Mizrachi, E.; Ford, E.; Cras-Méneur, C.; Permutt, M.A.; Imai, S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell metabolism 2005, 2, 105–117. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell metabolism 2015, 22, 31–53. [Google Scholar] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends in cell biology 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Peclat, T.R.; Thompson, K.L.; Warner, G.M.; Chini, C.C.S.; Tarragó, M.G.; Mazdeh, D.Z.; Zhang, C.; Zavala-Solorio, J.; Kolumam, G.; Liang Wong, Y.; et al. CD38 inhibitor 78c increases mice lifespan and healthspan in a model of chronological aging. Aging cell 2022, 21, e13589. [Google Scholar] [CrossRef]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell metabolism 2018, 27, 1081–1095.e1010. [Google Scholar] [CrossRef] [PubMed]
- Manzanero, S. Generation of mouse bone marrow-derived macrophages. Methods in molecular biology (Clifton, N.J.) 2012, 844, 177–181. [Google Scholar] [PubMed]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Current protocols in immunology 2008, Chapter 14, 14.11.11-14.11.14. [Google Scholar] [CrossRef]
- Spooner, R.; DeGuzman, J.; Lee, K.L.; Yilmaz, O. Danger signal adenosine via adenosine 2a receptor stimulates growth of Porphyromonas gingivalis in primary gingival epithelial cells. Molecular oral microbiology 2014, 29, 67–78. [Google Scholar] [CrossRef]
- Wellslager, B.; Roberts, J.; Chowdhury, N.; Madan, L.; Orellana, E.; Yilmaz, Ö. Porphyromonas gingivalis activates Heat-Shock-Protein 27 to drive a LC3C-specific probacterial form of select autophagy that is redox sensitive for intracellular bacterial survival in human gingival mucosa. bioRxiv : the preprint server for biology 2024.
Figure 1.
Old murine BMMs exhibited significantly higher CD38 protein and lower NAD+ expressions after infection with either Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) than young controls. Young and old murine BMMs were either uninfected or infected with an oral bacterial pathogen Aa or Pg (MOI 20) for 24 h. (A) Protein levels of CD38 and pan-actin in cell lysate were determined by Western Blot. (B) Protein densitometry of CD38 were quantified compared with control actin expression (n=3). (C) NAD+ levels in murine BMMs with or without bacterial infection (n=4). Statistics were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (*p<0.05, **p<0.01, ***p<0.001).
Figure 1.
Old murine BMMs exhibited significantly higher CD38 protein and lower NAD+ expressions after infection with either Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) than young controls. Young and old murine BMMs were either uninfected or infected with an oral bacterial pathogen Aa or Pg (MOI 20) for 24 h. (A) Protein levels of CD38 and pan-actin in cell lysate were determined by Western Blot. (B) Protein densitometry of CD38 were quantified compared with control actin expression (n=3). (C) NAD+ levels in murine BMMs with or without bacterial infection (n=4). Statistics were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (*p<0.05, **p<0.01, ***p<0.001).
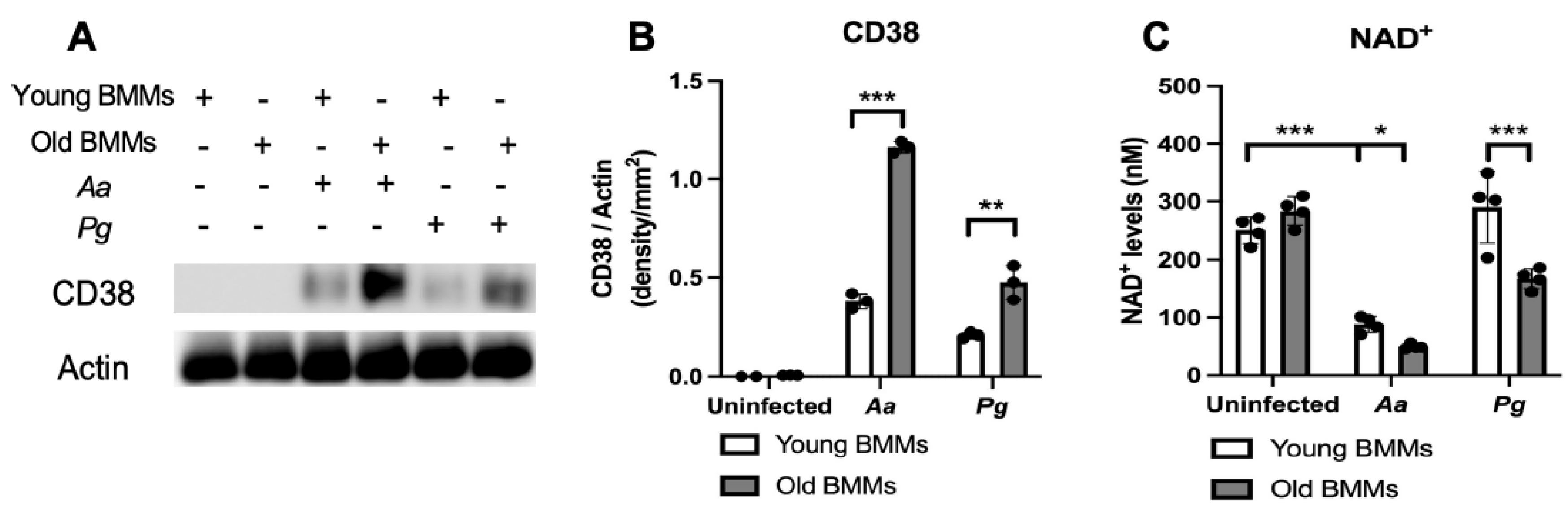
Figure 2.
Old murine BMMs displayed delayed immune responses to infection with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) compared to young controls. Young and old murine BMMs were either uninfected or infected with an oral bacterial pathogen Aa or Pg for 1 to 24 h. (A) Protein levels of CD38, p-NFκBp65, p-PI3K, p-ERK, p-JNK, p-p38, and pan-actin in cell lysate were determined by Western Blot. Protein densitometry of p-NFκBp65 (D), p-PI3K (E), p-ERK (F), p-JNK (G), and p-p38 (H) 24 h after bacterial infection were evaluated. Statistics were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (n=3, ns: no significance).
Figure 2.
Old murine BMMs displayed delayed immune responses to infection with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) compared to young controls. Young and old murine BMMs were either uninfected or infected with an oral bacterial pathogen Aa or Pg for 1 to 24 h. (A) Protein levels of CD38, p-NFκBp65, p-PI3K, p-ERK, p-JNK, p-p38, and pan-actin in cell lysate were determined by Western Blot. Protein densitometry of p-NFκBp65 (D), p-PI3K (E), p-ERK (F), p-JNK (G), and p-p38 (H) 24 h after bacterial infection were evaluated. Statistics were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (n=3, ns: no significance).
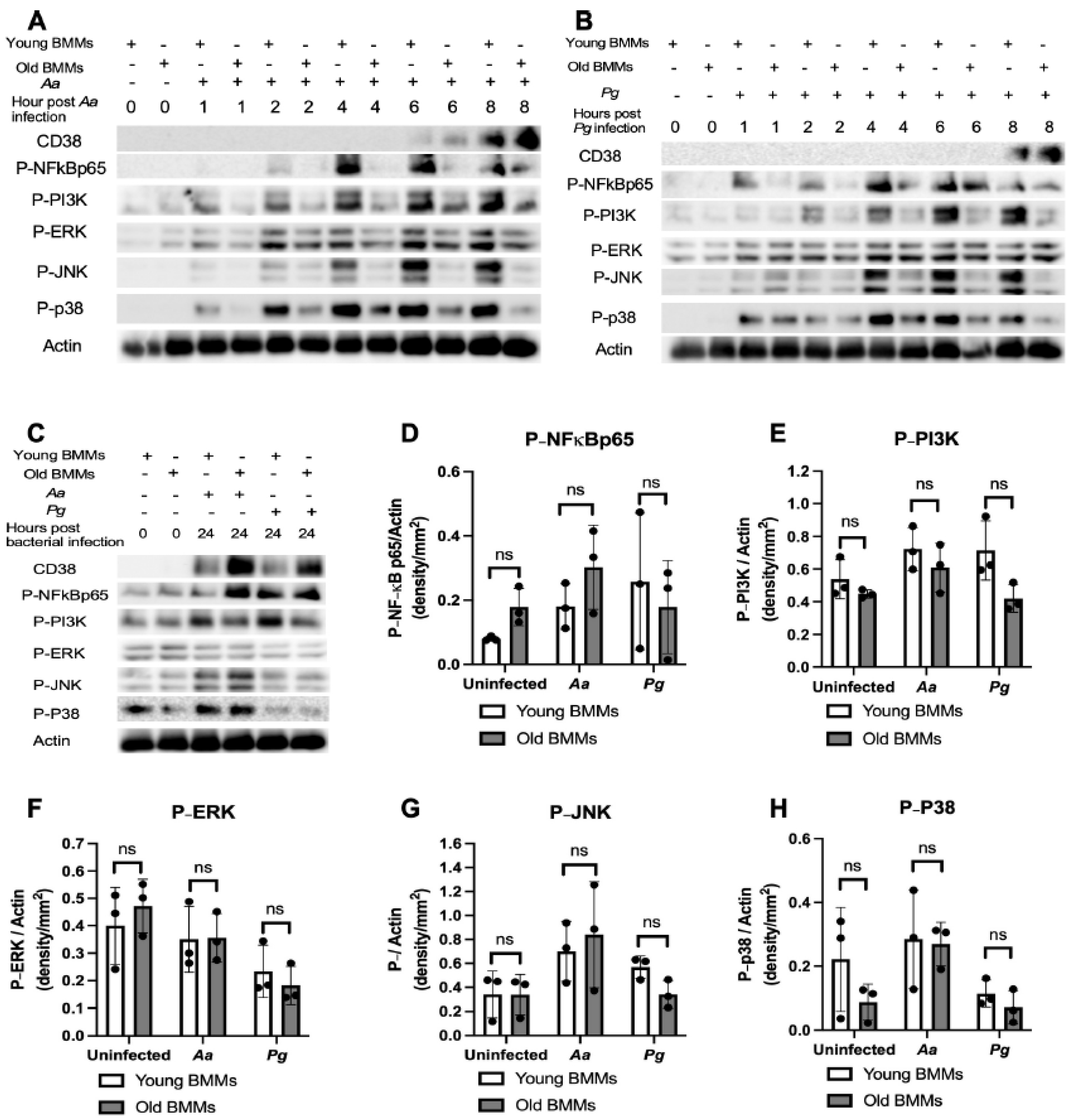
Figure 3.
Old murine BMMs displayed similar IL-1β, IL-6, and TNF-α cytokine levels 24 h after infection with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) compared to young controls. Young and old murine BMMs were either uninfected or infected with an oral bacterial pathogen Aa or Pg (MOI 20) for 24 h. Cytokine levels of IL-1β (A), IL-6 (B), and TNF-α (C) were quantified by ELISA and calibrated by protein expression in cell lysate. Statistics were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (n=5, ns: no significance).
Figure 3.
Old murine BMMs displayed similar IL-1β, IL-6, and TNF-α cytokine levels 24 h after infection with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg) compared to young controls. Young and old murine BMMs were either uninfected or infected with an oral bacterial pathogen Aa or Pg (MOI 20) for 24 h. Cytokine levels of IL-1β (A), IL-6 (B), and TNF-α (C) were quantified by ELISA and calibrated by protein expression in cell lysate. Statistics were analyzed by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (n=5, ns: no significance).
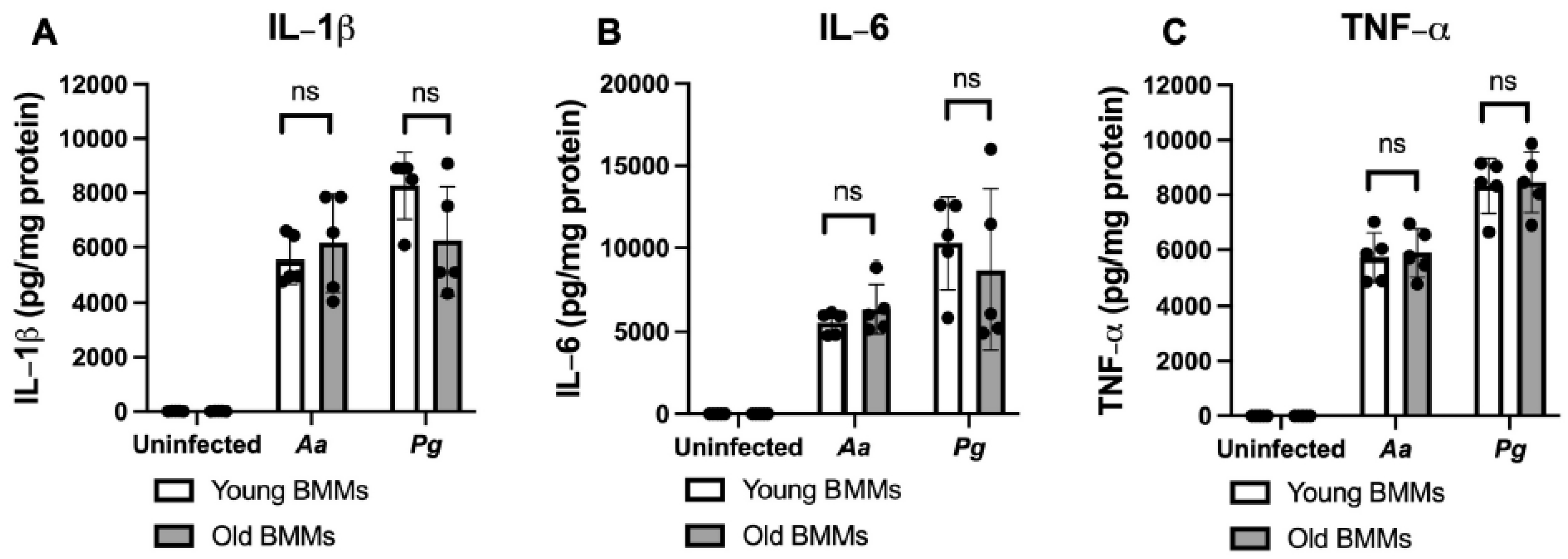
Figure 4.
Inhibition of CD38 by 78c suppressed CD38, NFκB, PI3K, and MAPKs; attenuated IL-1β, IL-6, and TNF-α pro-inflammatory cytokine levels; and enhanced NAD+ in old murine BMMs after infection with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg). Old murine BMMs were treated with vehicle or 78c (10 µM) with or without infection with the oral bacterial pathogen Aa or Pg for 1 to 8 h. Protein levels of CD38, p-NFκBp65, p-PI3K, p-ERK, p-JNK, p-p38, and pan-actin in cell lysate induced by Aa (A) or Pg (B) were determined by Western Blot. Old murine BMMs were treated with vehicle (diluted DMSO) or 78c (1.25 to 10 µM) with or without infection of Aa or Pg for 24 h. Cytokine levels of IL-1β (C), IL-6 (D), and TNF-α (E) were quantified by ELISA and calibrated by protein expression in cell lysate. (F) NAD+ levels were measured and calibrated by cell growth and viability. Statistics were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=4, *p<0.05, ***p<0.001).
Figure 4.
Inhibition of CD38 by 78c suppressed CD38, NFκB, PI3K, and MAPKs; attenuated IL-1β, IL-6, and TNF-α pro-inflammatory cytokine levels; and enhanced NAD+ in old murine BMMs after infection with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg). Old murine BMMs were treated with vehicle or 78c (10 µM) with or without infection with the oral bacterial pathogen Aa or Pg for 1 to 8 h. Protein levels of CD38, p-NFκBp65, p-PI3K, p-ERK, p-JNK, p-p38, and pan-actin in cell lysate induced by Aa (A) or Pg (B) were determined by Western Blot. Old murine BMMs were treated with vehicle (diluted DMSO) or 78c (1.25 to 10 µM) with or without infection of Aa or Pg for 24 h. Cytokine levels of IL-1β (C), IL-6 (D), and TNF-α (E) were quantified by ELISA and calibrated by protein expression in cell lysate. (F) NAD+ levels were measured and calibrated by cell growth and viability. Statistics were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (n=4, *p<0.05, ***p<0.001).
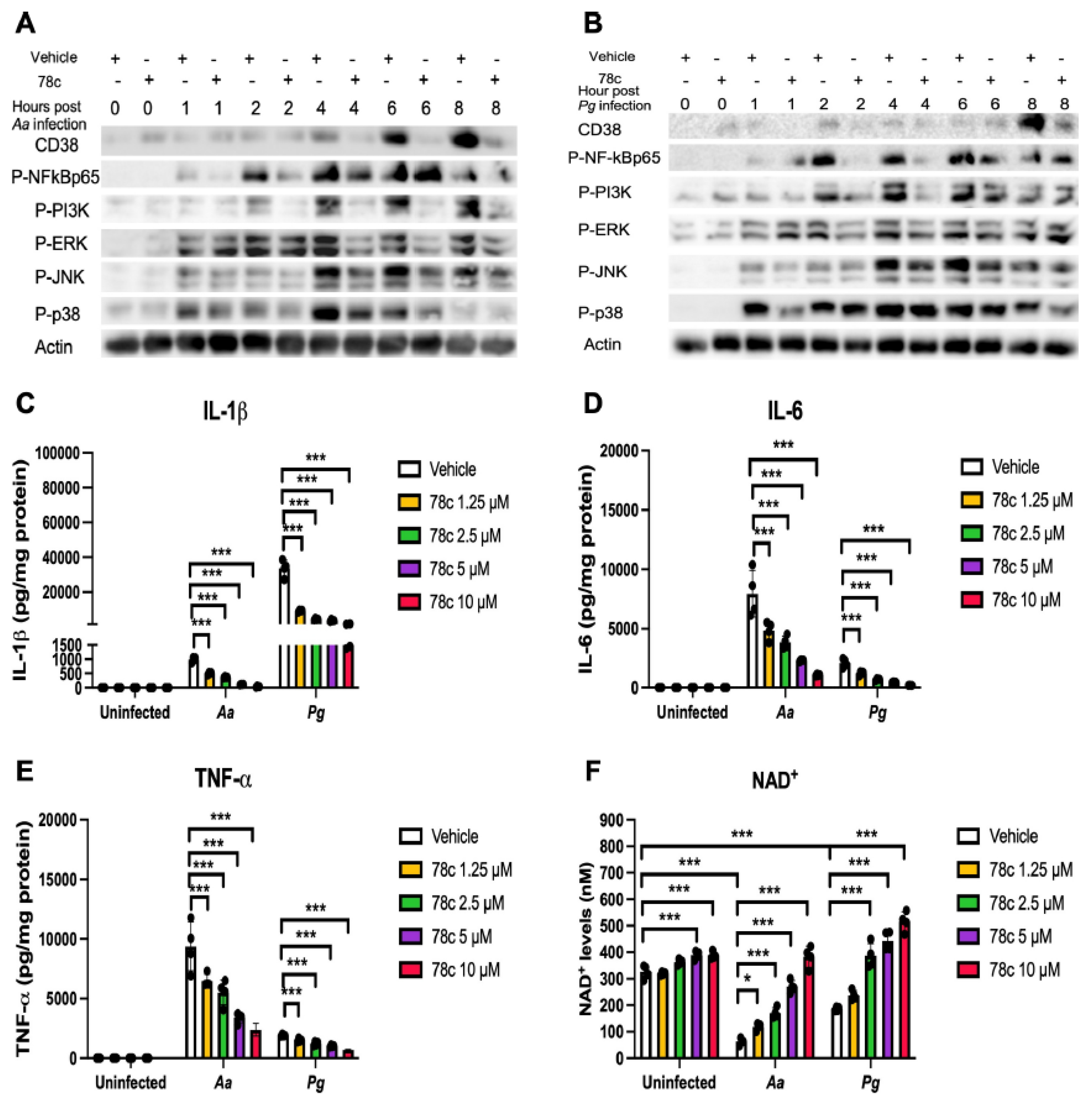
Figure 5.
Inhibition of CD38 by 78c reduced reactive oxygen species (ROS) and Nox1, but enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat), Sirt1, and Parp1 mRNA levels in old murine BMMs either uninfected or infected with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg). Old murine BMMs were treated with vehicle or 78c (1.25 to 10 µM) with or without infection with either the oral bacterial pathogen Aa or Pg for 24h (ROS study) or 8 h (RT-qPCR study). (A) ROS was detected by measuring fluorescence in cells using a CellROXTM Green reagent and calibrated by cell growth and viability (n=4). (B) CD38 mRNA, (C) Nox1 mRNA, (D) Sod1 mRNA, (E) Gpx4 mRNA, (F) Prdx1 mRNA, (G) Txnrd1 mRNA, (H) Cat mRNA, (I) Sirt1 mRNA, and (J) Parp1 mRNA levels were quantified using RT-PCR and normalized by β-actin expression (n=3). Statistics were analyzed using an ordinary one-way ANOVA with Dunnett’s multiple comparisons test (* p < 0.05, ** p < 0.01, *** p < 0.001).
Figure 5.
Inhibition of CD38 by 78c reduced reactive oxygen species (ROS) and Nox1, but enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat), Sirt1, and Parp1 mRNA levels in old murine BMMs either uninfected or infected with either the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg). Old murine BMMs were treated with vehicle or 78c (1.25 to 10 µM) with or without infection with either the oral bacterial pathogen Aa or Pg for 24h (ROS study) or 8 h (RT-qPCR study). (A) ROS was detected by measuring fluorescence in cells using a CellROXTM Green reagent and calibrated by cell growth and viability (n=4). (B) CD38 mRNA, (C) Nox1 mRNA, (D) Sod1 mRNA, (E) Gpx4 mRNA, (F) Prdx1 mRNA, (G) Txnrd1 mRNA, (H) Cat mRNA, (I) Sirt1 mRNA, and (J) Parp1 mRNA levels were quantified using RT-PCR and normalized by β-actin expression (n=3). Statistics were analyzed using an ordinary one-way ANOVA with Dunnett’s multiple comparisons test (* p < 0.05, ** p < 0.01, *** p < 0.001).
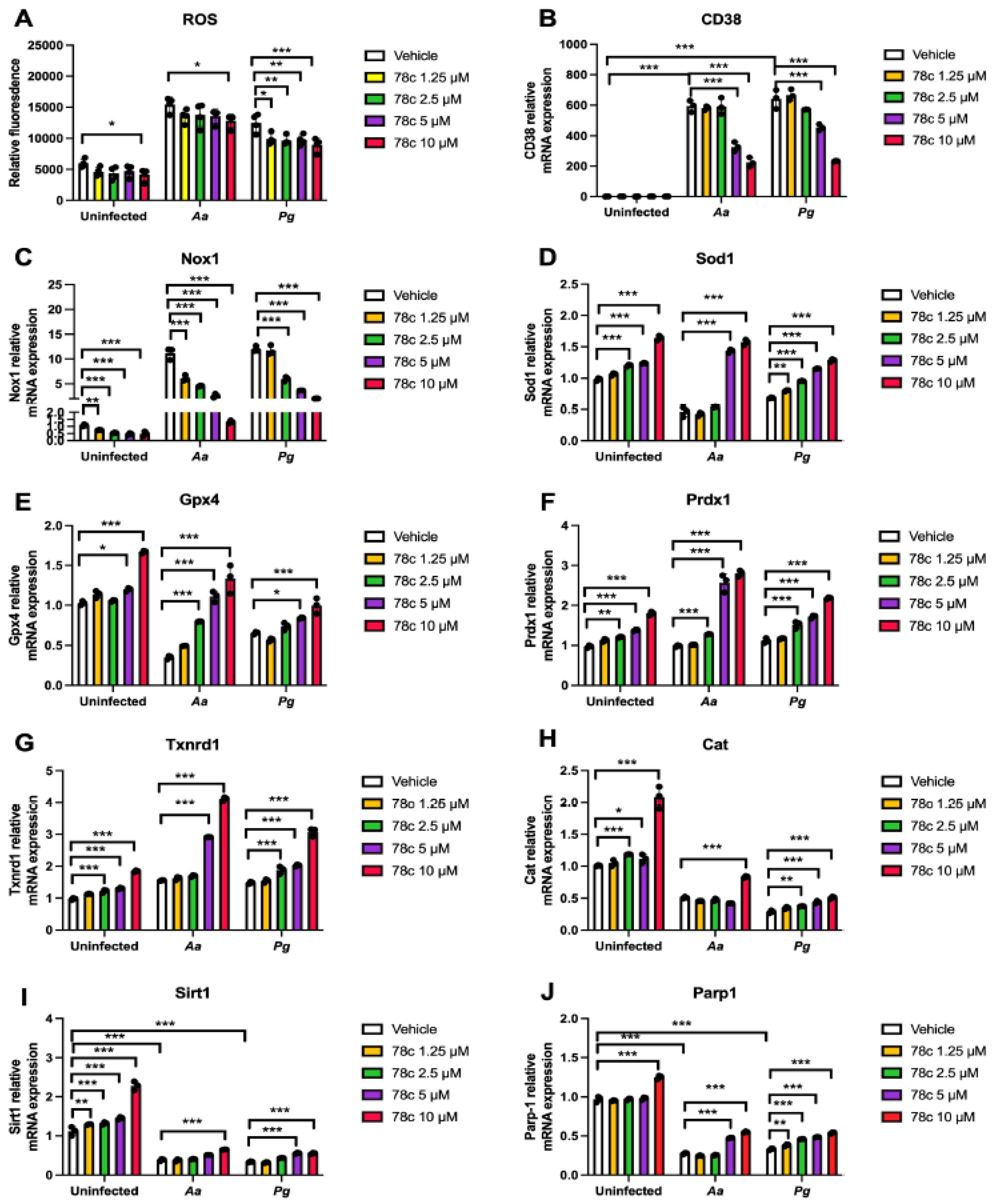
Figure 6.
Inhibition of CD38 by 78c inhibited Nox1, but enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat), Sirt1, and Parp1 protein levels in old murine BMMs either uninfected or infected with the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg). Old murine BMMs were treated with vehicle or 78c (10 µM) with or without infection of either of the oral bacterial pathogens Aa or Pg for various time points (1, 2, 4, 8, or 24h. Nox1 and pan-actin protein expressions in old murine BMMs infected with Aa (A) or Pg (B) were evaluated by Western blot. CD38, Nox1, Sod1, Gpx4, Prdx1, Txnrd1, Cat, Sirt1, Parp, and pan-actin protein expression (C) in old murine BMMs either uninfected or infected with Aa or Pg for 24h were evaluated by Western blot.
Figure 6.
Inhibition of CD38 by 78c inhibited Nox1, but enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat), Sirt1, and Parp1 protein levels in old murine BMMs either uninfected or infected with the oral pathogens Aggregatibacter actinomycetemcomitans (Aa) or Porphyromonas gingivalis (Pg). Old murine BMMs were treated with vehicle or 78c (10 µM) with or without infection of either of the oral bacterial pathogens Aa or Pg for various time points (1, 2, 4, 8, or 24h. Nox1 and pan-actin protein expressions in old murine BMMs infected with Aa (A) or Pg (B) were evaluated by Western blot. CD38, Nox1, Sod1, Gpx4, Prdx1, Txnrd1, Cat, Sirt1, Parp, and pan-actin protein expression (C) in old murine BMMs either uninfected or infected with Aa or Pg for 24h were evaluated by Western blot.
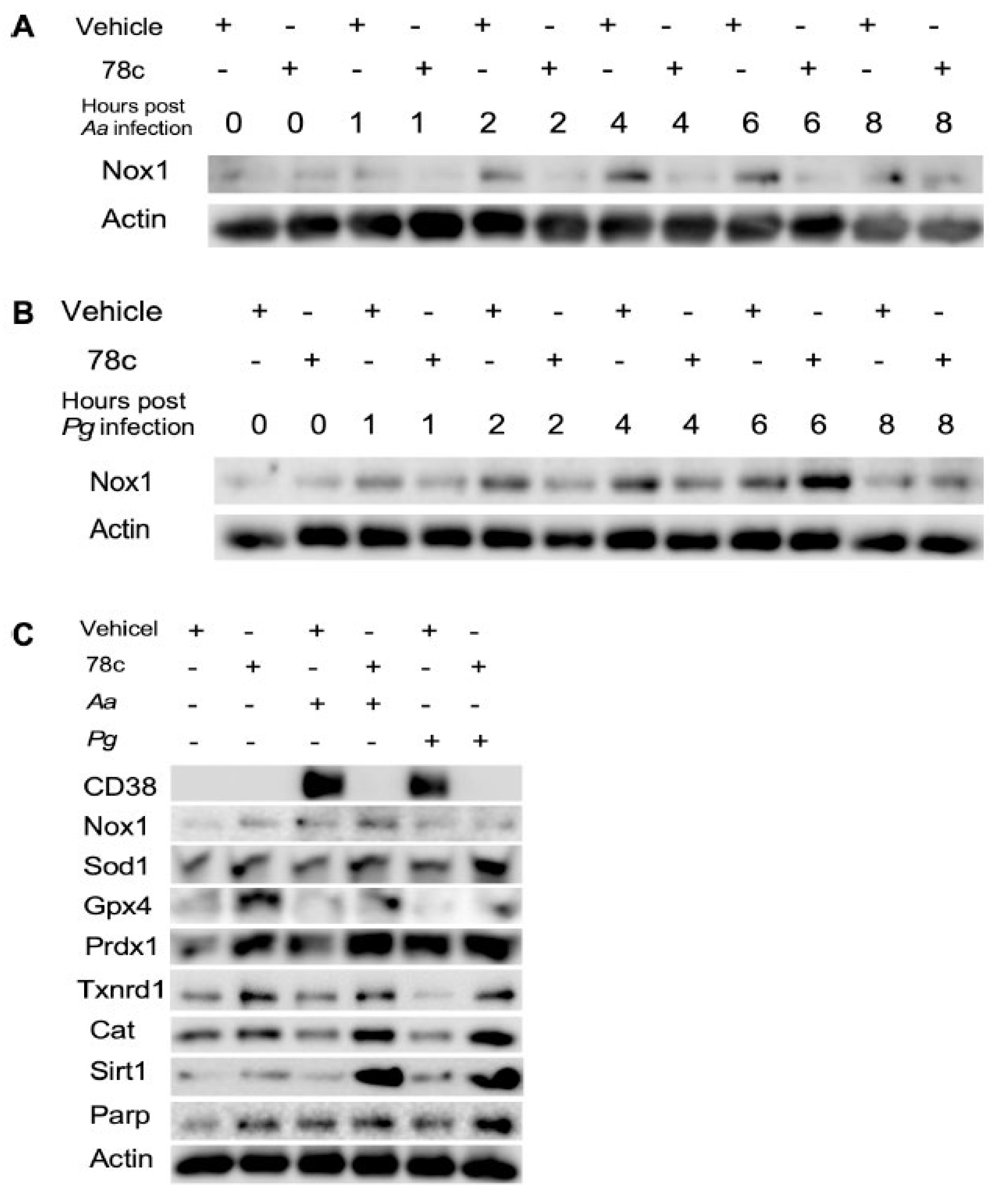
Figure 7.
Roles of a CD38 inhibitor (78c) in treating aging-associated with periodontitis. Treatment with 78c suppressed podosome component (PI3K, Pyk2, Src, F-actin, integrins, paxillin, and talin) expressions, subsequently inhibiting osteoclastogenesis and bone resorption. Treatment with 78c inhibited NFκB, PI3K, and MAPKs protein kinases induced by oral bacterial pathogens, suppressing IL-1β, IL-6, TNF-α, and inflammation. Treatment with 78c reduced Nox1 expression, increased NAD+, and enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat) expression, subsequently reduce oxidative stress and maintain mitochondrial homeostasis. Treatment with 78c increased NAD+ consuming enzymes (Sirt1 and Parp) expressions, which subsequently promote longevity and the repair of DNA damages.
Figure 7.
Roles of a CD38 inhibitor (78c) in treating aging-associated with periodontitis. Treatment with 78c suppressed podosome component (PI3K, Pyk2, Src, F-actin, integrins, paxillin, and talin) expressions, subsequently inhibiting osteoclastogenesis and bone resorption. Treatment with 78c inhibited NFκB, PI3K, and MAPKs protein kinases induced by oral bacterial pathogens, suppressing IL-1β, IL-6, TNF-α, and inflammation. Treatment with 78c reduced Nox1 expression, increased NAD+, and enhanced anti-oxidant enzymes (Sod1, Gpx4, Prdx1, Txnrd1, and Cat) expression, subsequently reduce oxidative stress and maintain mitochondrial homeostasis. Treatment with 78c increased NAD+ consuming enzymes (Sirt1 and Parp) expressions, which subsequently promote longevity and the repair of DNA damages.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



