
Submitted:
29 April 2025
Posted:
30 April 2025
You are already at the latest version
Abstract
The increasing contamination of water sources by pesticides, particularly malathion, poses a challenge for conventional treatment methods, while the global energy crisis highlights the need for sustainable alternatives such as hydrogen. Photocatalytic water splitting is a promising method for hydrogen production, but its efficiency is hindered by poor charge separation, limited light absorption, and slow reaction rates. This study explores TiO₂-based nanocomposites, specifically P25-reduced graphene oxide (rGO) modified with varying MoS₂ loadings (1%, 3%, 5%, and 10%), to simultaneously enhance pollutant degradation and hydrogen evolution. rGO improves charge carrier separation, increases surface area, and facilitates electron transport, while MoS₂ serves as a co-catalyst that promotes charge transfer and provides active sites for hydrogen evolution reactions. The nanocomposites were synthesized and characterized by X-ray diffraction (XRD), Raman spectroscopy, transmission electron microscopy (TEM), UV-Vis diffuse reflectance spectroscopy (DRS), and photoluminescence (PL) to evaluate structural, morphological, and optical properties. Photocatalytic degradation of malathion was analyzed under simulated solar irradiation using UV-Vis spectroscopy and gas chromatography-mass spectrometry (GC-MS), while hydrogen production was assessed in an aqueous suspension with a sacrificial agent, with hydrogen evolution quantified via gas chromatography (GC-TCD). The results demonstrated that the synergistic incorporation of rGO and MoS₂ into P25 TiO₂ significantly improved photocatalytic performance, where rGO enhanced charge separation and electrical conductivity, while MoS₂ acted as an electron acceptor and catalytic site for hydrogen generation. Under optimized conditions, nearly 100% degradation of malathion was achieved within 2 hours, and hydrogen evolution rates approached 6000 µmol g⁻¹ h⁻¹. An optimal MoS₂ loading maximized efficiency, though excessive amounts led to charge recombination. This study highlights the potential of P25-rGO-MoS₂ nanocomposites for scalable applications in water treatment and hydrogen production, contributing to sustainable and cost-effective photocatalytic technologies.
Keywords:
photodegradation
; photocatalytic hydrogen evolution
; malathion
1. Introduction
Environmental contamination caused by pesticides has become a growing concern due to their widespread use in agriculture and their persistence in water bodies [1]. Among these contaminants, malathion, an organophosphate pesticide, is particularly problematic due to its toxicity, bioaccumulation potential, and resistance to natural degradation [2,3]. Traditional water treatment methods, such as adsorption, coagulation, and biological degradation, often failed to remove malathion efficiently, requiring the development of alternative approaches [4]. At the same time, the global energy crisis and the urgent need for sustainable fuel sources had driven increasing interest in hydrogen production as an environmentally friendly alternative to fossil fuels [5,6,7]. Photocatalytic water splitting had emerged as a promising method for producing hydrogen in a clean and renewable manner [8,9]. However, challenges related to inefficient charge separation, limited light absorption, and low reaction rates had hindered its large-scale application [10]. To address these issues, the present study explored the use of TiO₂-based nanocomposites, specifically P25-graphene nanocatalysts modified with different MoS₂ loadings (1%, 3%, 5%, and 10%), for two key applications: the photocatalytic degradation of malathion under simulated solar irradiation and the photocatalytic production of hydrogen from water. The incorporation of graphene into P25 TiO₂ had previously been shown to improve charge carrier separation, enhance surface area, and facilitate electron transport, leading to increased photocatalytic efficiency [11,12,13]. Additionally, MoS₂, a transition metal dichalcogenide with a narrow bandgap (~1.8 eV) and high electron mobility, had demonstrated potential as a co-catalyst in photocatalysis by promoting charge transfer and providing active sites for hydrogen evolution reactions [14,15]. However, the influence of MoS₂ loading on the performance of P25-graphene nanocomposites had not been systematically evaluated for simultaneous pollutant degradation and hydrogen production.
In this study, a series of P25-graphene-MoS₂ nanocomposites were synthesized and characterized to investigate their structural, morphological, and optical properties. X-ray diffraction (XRD) was used to confirm phase composition, while Raman spectroscopy provided insight into structural interactions between P25, graphene, and MoS₂. Transmission electron microscopy (TEM) allowed for the characterization of dispersion and morphology, and UV-Vis reflectance spectroscopy (DRS) was employed to assess optical absorption properties. Photoluminescence (PL) spectroscopy was further used to evaluate charge carrier recombination behavior. The photocatalytic degradation of malathion was carried out under simulated solar irradiation, and the degradation efficiency was monitored using gas chromatography mass-spectrometry (GC-MS). Additionally, photocatalytic hydrogen production experiments were conducted in an aqueous suspension with sacrificial reagents, and hydrogen evolution rates were quantified using gas chromatography (GC-TCD).
The results demonstrated that the incorporation of graphene and MoS₂ into P25 TiO₂ significantly enhanced photocatalytic activity for both malathion degradation and hydrogen evolution. As expected [16], the presence of graphene facilitated charge separation and improved electrical conductivity, reducing electron-hole recombination. MoS₂ further contributed to the process by acting as an electron acceptor and providing catalytic sites for hydrogen generation. An optimal MoS₂ loading was identified, beyond which excessive MoS₂ led to charge recombination and a decline in performance. These findings highlighted the importance of nanocomposite composition in optimizing photocatalytic efficiency and provided valuable insights into the design of multifunctional photocatalysts for environmental remediation and sustainable energy production. This study not only demonstrated the dual functionality of P25-graphene-MoS₂ nanocomposites but also provided a comprehensive understanding of the synergistic effects between the individual components. By evaluating the impact of MoS₂ loading, this work contributes to the development of cost-effective and scalable photocatalysts with potential applications in water treatment and renewable hydrogen production. The findings underscore the potential of engineered nanomaterials in addressing critical environmental and energy challenges, paving the way for further advancements in photocatalysis-based technologies.
2. Materials and Methods
2.1. Materials
All chemicals were used as received without further purification, and solutions were prepared using deionized water (Milli-Q water, 18.2 MΩcm⁻¹ at 25 °C). The TiO₂ used in this study was Aeroxide(R) P25 (99.5% purity), provided by Thermo Scientific Chemicals. Reduced graphene oxide (rGO, purified powder), MoS2 (powder, <2mm, 98%), N-Methyl-2-pyrrolidone (ReagentPlus®, 99%), and Malathion (TraceCERT® standard, 99.5%) were provided by Sigma Aldrich (Milwaukee, WI, USA). Sodium sulfite (Na2SO3) anhydrous for analysis EMSURE® Reag. Ph Eur, and sodium sulfide nonahydrate (≥99.99% trace metals basis), used as sacrificial reagents, were provided by Sigma Aldrich (Milwaukee, WI, USA). Hydrogen peroxide (H₂O₂, 30% w/w), methanol (CH₃OH, HPLC grade, >99.9%), and 0.45 μm syringe filters were also obtained from Sigma Aldrich (Milwaukee, WI, USA). Sodium hydroxide (NaOH, pellets, ACS Cerfified) and hydrochloric acid (HCl, ACS, 36.5-38%) were provided by Fisher Chemical (Waltham, MA, USA).
2.2. Preparation of TiO₂-rGO composite
The synthesis of the TiO₂-rGO composite was conducted via a hydrothermal method, modifying the approach reported by Perera et al [17]. To ensure a well-defined composition, the weight ratio (w/w) of rGO to TiO₂ (P25) was set at 3%. In a single-step sonication process, rGO was dispersed in 30 mL of deionized water containing 10.5 g of NaOH. This solution was subjected to ultrasonication at 40 kHz for 1 hour to achieve a uniform dispersion of rGO. Simultaneously, the TiO₂ powder was incorporated into the mixture, ensuring homogeneous suspension. After sonication, the dispersion was subjected to magnetic stirring at 500 rpm for 1 hour. Subsequently, the resulting TiO₂-rGO suspension was transferred into a 100 mL Teflon-lined stainless-steel autoclave and subjected to a hydrothermal treatment at 120 °C for 24 hours under static conditions. This process facilitated the incorporation of rGO onto the TiO₂ surface while preserving the structural integrity of both materials. Upon completion of the hydrothermal reaction, the obtained gray gel was carefully collected and washed with a 0.1 M HCl solution (prepared using concentrated HCl, 35%, Fisher Scientific). The sample was continuously stirred overnight at room temperature (~25 °C) to ensure thorough removal of NaOH. To purify the material, sequential washing steps were performed. The product was washed five times with deionized water, followed by a final rinse with ethanol to enhance dispersion and minimize agglomeration. Centrifugation (6000 rpm, 10 min) was employed between each washing step to facilitate solid-liquid separation and ensure the removal of unreacted precursors and residual ionic species. Following purification, the material was dried in a vacuum oven at 80 °C for 12 hours. Finally, to enhance crystallinity and optimize the physicochemical properties of the composite, the TiO₂-rGO material was annealed at 300 °C for 60 minutes in an air atmosphere. This thermal treatment was aimed at improving the interaction between TiO₂ and rGO while maintaining the integrity of the composite structure.
2.3. Preparation of MoS2@TiO₂-rGO catalysts
The incorporation of exfoliated MoS₂ into the previously synthesized TiO₂-rGO composite was carried out via a solution-based self-assembly method. Exfoliated MoS₂ nanosheets were obtained via liquid-phase exfoliation (LPE) in anhydrous N-methyl-2-pyrrolidone (NMP) [18,19]. The exfoliation process involved ultrasonication at 40 kHz for 6 hours in an ice-cooled bath. After sonication, the dispersion was centrifuged at 3000 rpm for 30 minutes, and the supernatant, containing well-dispersed MoS₂ nanosheets, was collected and filtered to remove unexfoliated bulk material. The resulting MoS₂ suspension was washed with ethanol four times and then redispersed in deionized water (pH 7) at a concentration of 1 mg/mL under continuous stirring for 1 hour before use. To incorporate exfoliated MoS₂ into the TiO₂-rGO matrix, 0.5 g of TiO₂-rGO was dispersed in 50 mL of deionized water under magnetic stirring at 600 rpm for 1 hour. Subsequently, a MoS₂ suspension (1 mg/mL) was added dropwise in the appropriate amount to obtain composites containing 1%, 3%, 5%, and 10% MoS₂ by weight relative to TiO₂-rGO, while maintaining continuous stirring. The resulting mixture was then subjected to ultrasonication at 40 kHz for 30 minutes to ensure homogeneous distribution and to enhance the interaction between the MoS₂ nanosheets and the TiO₂-rGO hybrid structure. Following sonication, the dispersion was stirred for 4 hours at room temperature to enhance adhesion and allow van der Waals interactions between MoS₂ and rGO layers. After stirring, the MoS₂-TiO₂-rGO composite was collected via centrifugation at 8000 rpm for 10 minutes, washed three times with ethanol/water (1:1 v/v), and dried in a vacuum oven at 80 °C for 12 hours. To improve the structural integrity and enhance the electronic coupling between the components, the dried composite was subjected to a mild thermal annealing step at 250 °C for 2 hours under an inert nitrogen atmosphere. This step facilitated the reduction of residual oxygen functional groups in rGO and improved the contact between MoS₂ and the TiO₂-rGO surface without inducing phase transitions.
2.4. Characterization Techniques
A comprehensive set of characterization techniques was employed to evaluate the structural, morphological, optical, and chemical properties of the synthesized catalysts. The specific surface area and porosity of the materials were determined using Brunauer-Emmett-Teller (BET) analysis on a Micromeritics ASAP 2020 instrument, measuring nitrogen adsorption-desorption isotherms at 77 K. The morphological features and microstructural details of the composites were analyzed by Field Emission Scanning Electron Microscopy (FESEM) using an JEOL IT-500HR instrument. Additionally, High-Resolution Transmission Electron Microscopy (HRTEM) was conducted on a JEOL JEM 3000F microscope operating at 300 kV. The crystalline phases of the synthesized catalysts were characterized by X-ray Diffraction (XRD) on a Bruker D8 Advance diffractometer, operating at 40 kV and 40 mA. Raman spectroscopy was performed on a DXR Thermo Raman Microscope, employing a 532 nm laser source with a power setting of 5 mW and a resolution of 5 cm⁻¹. The chemical states were analyzed using X-ray Photoelectron Spectroscopy (XPS) on an ESCALAB 220i-XL spectrometer, by using non-monochromatic Mg Kα radiation of a twin anode at 20 mA and 12 kV. The bandgap estimation was obtained by UV-Vis Diffuse Reflectance Spectroscopy (UV-Vis DRS) on a Perkin Elmer Lambda 850 UV-Vis Spectrophotometer. Photoluminescence (PL) spectroscopy was carried out using an Edinburgh FS900 Fluorescence Spectrometer to evaluate charge carrier recombination and photogenerated electron-hole interactions. The photocatalytic degradation of malathion was characterized by UV-Vis spectroscopy on a Perkin Elmer Lambda 850 UV-Vis Spectrophotometer. To characterize the degradation intermediates, gas chromatography-mass spectrometry (GC-MS) analysis was performed using a Shimadzu GC 2010 Plus-QP2020 system. The separation of organic compounds was achieved using a 30 m × 0.25 mm i.d. capillary column (Rtx-5MS, Restek Corporation, Bellefonte, PA, USA) with helium (99.999% purity) as the carrier gas.
2.5. Photocatalytic Degradation Experiments
Photodegradation experiments were conducted using a 20-ppm solution of malathion mixed with 1.1 g/L of the selected catalyst. The pH of the resulting solution was adjusted to 7 using either sodium hydroxide (NaOH) or hydrochloric acid (HCl). To ensure adsorption/desorption equilibrium between the catalyst and the contaminant, the suspension was stirred in the dark for 30 minutes. Following this equilibration period, a 1 mL aliquot of a 0.01% hydrogen peroxide (H₂O₂) solution was added, and the system was continuously aerated to maintain sufficient oxygen levels. The reaction mixture was then exposed to a solar simulator equipped with dual white light bulbs (100 W, 6300 lx). Upon initiating irradiation, the experiment proceeded for 60 minutes at a controlled temperature of 22°C, with 5 mL samples collected at 10-minute intervals. Each collected sample was passed through a 0.45 μm membrane filter to remove any catalyst residues and subsequently analyzed using UV-visible spectroscopy.
For a detailed investigation of the photodegradation byproducts by GC-MS, aliquots were collected at specific time points throughout the reaction. Each sample was prefiltered to remove any residual catalyst particles and subsequently extracted using dichloromethane as the sole organic solvent via liquid-liquid extraction. The organic phase was separated and dried over anhydrous sodium sulfate to eliminate traces of water. The dried extract was then partially concentrated under reduced pressure to enhance analyte detection, avoiding complete solvent removal. The concentrated dichloromethane solution was directly used for analysis by GC-MS. A 5 µL aliquot of the sample was injected for chromatographic separation and mass spectral identification. To ensure analytical reliability, four injections were performed for each time point: three sample replicates and one blank. Helium served as the carrier gas to ensure optimal chromatographic resolution.
2.6. Photocatalytic Hydrogen Production Experiments
To investigate the photocatalytic production of hydrogen by water splitting, a carefully designed experimental setup was employed. The procedure involved dispersing 50 mg of the selected catalyst in 100 mL of deionized water within a 250 mL quartz reaction vessel. To enhance electron transfer efficiency, sacrificial electron donor solutions were incorporated, consisting of sodium sulfite (Na₂SO₃) at a concentration of 0.03 M and sodium sulfide (Na₂S) at 0.5 M [20]. The reaction system was maintained at a controlled temperature of 20 °C and purged with nitrogen (N₂) gas for 30 minutes to remove dissolved oxygen and other interfering gases, ensuring an inert environment. Next, the reaction mixture was exposed to a controlled light source to initiate photocatalytic activity. The irradiation was carried out using a UV-Vis light system with an intensity of 120 mW cm⁻². To examine the influence of different energy levels on hydrogen production, irradiation was conducted at specific wavelengths of 220 nm, 320 nm, 400 nm, 500 nm, 600 nm, and 700 nm using appropriate cut-off optical filters. The experiment was performed over a duration of 2 hours, allowing sufficient time for the photocatalytic splitting of water molecules and the subsequent evolution of hydrogen gas. The generated hydrogen was collected and analyzed quantitatively by gas chromatography (GC) coupled to a thermal conductivity detector (TCD). A Perkin-Elmer Clarus 600 chromatograph was used for precise measurement of hydrogen concentration.
3. Results and Discussion
3.1. Characterization of the Catalysts
Figure 1 shows SEM images of the synthesized materials at increasing magnifications. The P25-rGO support (Figure 1a,b) exhibits an interconnected network of fibrous TiO₂ structures with lengths of several hundred nanometers and diameters below 20 nm. These nanofibers form a porous and open framework that facilitates mass transport and light penetration. The reduced graphene oxide (rGO) component is not distinguishable at this scale due to its low contrast and highly exfoliated nature, but it is expected to interweave throughout the fibrous TiO₂ matrix, supporting structural cohesion and charge transport. In the MoS₂-modified catalyst (Figure 1c), corresponding to 5 wt% MoS₂@P25-rGO, small and dispersed bright domains can be observed along the TiO₂ fibers (highlighted by yellow arrows), which are attributed to the localized deposition of MoS₂ nanosheets. These features suggest a good distribution of MoS₂ without bulk aggregation, maintaining the structural integrity of the fibrous support.
The different composites were also characterized by HRTEM (see Figure 2). Figure 2a shows a dense arrangement of TiO₂ nanofibers, with rGO sheets visible as faint, translucent layers enveloping the oxide structures. Figure 2b provides a higher-resolution view of an individual TiO₂ fiber, where lattice fringes are clearly observed. The inset highlights an interplanar spacing of ca. 0.35 nm, corresponding to the (101) plane of anatase TiO₂, consistent with XRD analysis. Figure 2c shows a HRTEM image of a single-layer MoS₂ nanosheet. The atomically resolved honeycomb pattern indicates high structural quality and confirms the presence of monolayer MoS₂. The corresponding SAED pattern (inset) reveals a hexagonal diffraction arrangement, characteristic of the 2H-phase of MoS₂. The clear spots and absence of diffuse rings confirm high crystallinity and minimal structural defects.
The crystalline phase composition of the prepared catalysts was examined by XRD (see Figure 3). Pure P25 TiO₂ exhibits the most characteristic reflections of anatase TiO₂ at 25.5°, 38°, 48.2°, 54.4°, assigned to the (101), (004), (200), (105) planes of anatase, along with a rutile peak at ca. 27.7° [21]. The TiO₂-rGO composite shows a virtually identical diffraction pattern to P25, indicating that the TiO₂ retained its crystalline structure after the graphene incorporation. Notably, no distinct new peaks attributable to graphene are observed; any potential (002) graphitic peak (~23°) is broadened or overlapped by the strong TiO₂ (101) peak [21]. This is expected given the low loading and exfoliated nature of rGO, which lacks long-range order in stacking. Upon adding MoS₂, the composite XRD patterns still predominantly display TiO₂ reflections, but new low-angle peaks appear. In particular, a faint diffraction peak appears around 13–14° in the 5%MoS₂@TiO₂-rGO sample (see asterisk), which is the (002) basal plane of hexagonal MoS₂ [21]. An additional minor peak at ca. 33° can be discerned, matching the (100) plane of MoS₂ [22] (see asterisk). The presence of these MoS₂ reflections confirms the successful incorporation of crystalline MoS₂ in the TiO₂-rGO matrix. Importantly, no significant shifts in the TiO₂ peak positions are detected upon MoS₂ or rGO addition, suggesting that Mo and S are not substituting into the TiO₂ lattice but rather MoS₂ and rGO form an intimate heterostructure on the TiO₂ surface. The combination of TiO₂ and MoS₂ diffraction features, with no extra impurity phases, evidences the formation of the intended composite.
UV-Vis DRS was used to assess the optical absorption properties and bandgap energies of the catalysts (Figure 4). Pristine P25 TiO₂ shows a strong absorption edge in the UV region (ca. 390 nm), corresponding to a bandgap of about 3.22 eV (consistent with anatase TiO₂) [23]. Incorporation of rGO extends the absorption into the visible range (the TiO₂-rGO sample appears darker), with a red-shifted absorption edge. Tauc plot analysis (Figure 4) indicates a reduced bandgap of ~2.95 eV for TiO₂-rGO, implying that the introduction of rGO facilitates visible-light absorption. This bandgap narrowing can be attributed to the electronic interaction between TiO₂ and the conductive rGO, which may introduce mid-gap states and promote the formation of an adsorption tail in the band structure. Upon loading 5% MoS₂ onto TiO₂-rGO, the absorption edge shifts further into the visible (up to ca. 455–460 nm), yielding an estimated optical bandgap of ca. 2.72 eV for the 5%MoS₂@TiO₂-rGO composite [23]. The progressive red-shift in the absorption onset from 3.22 eV (TiO₂) to 2.72 eV (MoS₂@TiO₂-rGO) confirms that the synergy of rGO and MoS₂ effectively extends the light-harvesting range of TiO₂ into the visible spectrum. This behavior is consistent with MoS₂ acting as a narrow-bandgap sensitizer (2H-MoS₂ has a much smaller bandgap of ~1.2–1.8 eV) and rGO acting as a photosensitizer and electron conduit [23]. The black-colored MoS₂ nanosheets strongly absorb visible light and, when coupled with TiO₂, enable the heterostructure to use a greater portion of the solar spectrum [23]. In addition, intimate contact between TiO₂ and MoS₂ (and rGO) can create sub-band-gap states or band bending at the interface, further contributing to the observed bandgap reduction [24,25]. The enhanced visible-light absorption evidenced by DRS directly correlates with improved photocatalytic activity under solar irradiation – by harvesting more photons in the visible range, the MoS₂@TiO₂-rGO catalyst can generate more charge carriers for pollutant degradation and H₂ evolution compared to pure TiO₂ [26].
Photoluminescence spectroscopy was used to investigate the recombination behavior of photogenerated charge carriers in the photocatalysts. Figure 5 displays the room-temperature PL emission spectra (λexc = 380 nm) for P25-rGO and its composites containing different MoS₂ loadings (1%, 3%, 5%, and 10%). The P25-rGO sample exhibits a strong and broad emission band in the UV-visible range, reflecting a high rate of radiative recombination of electron-hole pairs in the absence of additional charge separation pathways [27]. Upon incorporation of MoS₂, the PL intensity generally decreases, indicating improved charge separation due to the synergistic effects of MoS₂ and rGO [28]. The quenching trend follows the order: P25-rGO > 1%MoS₂@P25-rGO > 10%MoS₂@P25-rGO > 3%MoS₂@P25-rGO > 5%MoS₂@P25-rGO, with the 5%MoS₂@P25-rGO composite showing the lowest PL intensity among all tested materials. Interestingly, the composite with 10% MoS₂ exhibits a higher PL intensity than those with 3% and 5%, suggesting that excessive MoS₂ content may not be beneficial. This could be due to agglomeration of MoS₂ layers or shielding effects that interfere with light absorption and charge transfer processes. Therefore, beyond an optimal loading, MoS₂ may hinder rather than enhance photocatalytic performance [28]. Overall, the PL quenching confirms that moderate MoS₂ incorporation enhances charge carrier separation, while excessive loading could counteract this benefit. The significant PL reduction observed in 5%MoS₂@P25-rGO points to an optimal interfacial configuration between TiO₂, rGO, and MoS₂ that favors efficient charge extraction and transport [22,27,28]. These results are consistent with the photocatalytic activity trends, as will be discussed in a later section, where the 5%MoS₂ composite also displayed the highest performance in both malathion degradation and hydrogen evolution, confirming that suppressed electron-hole recombination is a key factor in the enhanced reactivity of these ternary composites.
Raman spectroscopy was employed to investigate the structural features and component interactions within the 5%MoS₂@TiO₂-rGO photocatalyst. Figure 6 displays the Raman spectra of individual and composite materials: (a) TiO₂-P25, (b) rGO, (c) MoS₂, and (d) the ternary nanocomposite 5%MoS₂@TiO₂-rGO. In the spectrum of pristine TiO₂-P25 (Figure 6a), three characteristic vibrational modes of the anatase phase are clearly observed at approximately 398 cm⁻¹ (B₁g), 518 cm⁻¹ (A₁g + B₁g), and 640 cm⁻¹ (Eg) [21]. The spectrum of rGO (Figure 6b) exhibits two prominent and broad peaks centered at ca. 1345 cm⁻¹ (D band) and 1590 cm⁻¹ (G band). The G band arises from the E₂g vibrational mode of sp² carbon atoms (graphitic domains), while the D band originates from defect-activated breathing modes in disordered sp² structures [21]. The observed intensity ratio (ID/IG ≈ 0.8–1.0) suggests a partially reduced graphene oxide with residual structural defects and oxygenated functionalities, being this an expected outcome of mild reduction protocols. In the MoS₂ spectrum (Figure 6c), the in-plane E₂g¹ mode (ca. 379 cm⁻¹) and the out-of-plane A₁g mode (ca. 404 cm⁻¹) characteristic of the 2H phase of MoS₂ are clearly observed [27]. The band separation (~25 cm⁻¹) is consistent with few-layer MoS₂, as larger separations are typical in thinner nanosheets due to decreased interlayer interactions [22]. A weak overtone near 990 cm⁻¹, attributed to the 2A₁g mode, further supports the presence of multilayer character. The composite 5%MoS₂@TiO₂-rGO (Figure 6d) presents vibrational features from all three components. The anatase TiO₂ bands (black asterisks), the rGO D and G bands (red asterisks), and the MoS₂ peaks (green asterisks) are all clearly visible, confirming the coexistence of each constituent in the hybrid structure [27]. Importantly, no new bands or significant peak shifts are observed, suggesting that no undesirable side reactions (e.g., Mo oxidation, Ti–C bonding, or carbide formation) occurred during synthesis. These results validate the structural integrity of the ternary composite and the successful assembly of TiO₂, rGO, and MoS₂ without phase degradation. The presence of well-defined and distinct vibrational signatures from each component further implies favorable interfacial contact, which may facilitate charge separation and transport, being critical factors in enhancing photocatalytic activity.
The specific surface areas of the synthesized materials were determined via nitrogen adsorption–desorption measurements using the Brunauer–Emmett–Teller (BET) method. As summarized in Table S1, the commercial P25 TiO₂ sample exhibited a surface area of 48 m²/g, consistent with its well-established properties. Upon incorporation of reduced graphene oxide (rGO), the surface area increased substantially to 483 m²/g, reflecting the textural contribution of rGO sheets, which help prevent TiO₂ agglomeration and promote a more open porous structure. Further addition of exfoliated MoS₂ led to a progressive rise in surface area, with values of 492, 496, 503, and 521 m²/g for the composites containing 1%, 3%, 5%, and 10% MoS₂, respectively. This trend suggests that the introduction of layered MoS₂ contributes to additional mesoporosity and helps maintain a high surface-to-volume ratio in the hybrid system. Interestingly, however, as will be discussed in subsequent sections, the composite with 10% MoS₂, despite having the highest BET surface area, exhibited inferior photocatalytic performance in both malathion degradation and hydrogen evolution. This highlights that surface area alone is not the determining factor for photocatalytic efficiency. At higher MoS₂ contents, excessive coverage or restacking of MoS₂ layers may hinder light absorption or block active sites, disrupting the optimal heterojunction structure necessary for efficient charge separation and transfer [29].
To investigate the surface chemical composition and oxidation states of the elements present in the 5%MoS₂@TiO₂-rGO composite, high-resolution X-ray photoelectron spectroscopy (XPS) analyses were performed (Figure 7). The spectra confirm the presence of all key elements: Ti, O, C, Mo, and S. As shown in Figure 7a, the high-resolution Ti 2p spectrum reveals two well-defined peaks at 458.8 eV and 464.3 eV, corresponding to Ti 2p₃/₂ and Ti 2p₁/₂, respectively, which are characteristic of Ti⁴⁺ in TiO₂ [30,31]. A minor shoulder at ca. 459.3 eV may indicate surface heterogeneity or electronic interactions with MoS₂ or rGO [32], but no significant signal is observed at lower binding energies to suggest the presence of Ti³⁺ species, confirming that the TiO₂ structure remains predominantly in the fully oxidized state [30]. The O 1s spectrum (Figure 7b) shows a major peak at 529.8 eV attributed to lattice oxygen (Ti–O–Ti) and a secondary component at 531.0 eV, which corresponds to surface hydroxyl groups, adsorbed water, or oxygenated species on rGO [33]. These surface oxygen functionalities are often associated with enhanced photocatalytic activity, as they can facilitate charge separation and radical formation [33]. The C 1s spectrum (Figure 7c) displays a dominant signal at 284.6 eV due to sp²-hybridized carbon atoms in the graphene lattice (C=C), along with minor peaks at 287.2 eV and 289.3 eV that can be assigned to carbonyl (C=O) and carboxyl (O–C=O) groups, respectively [34,35]. The relatively low intensity of these oxidized carbon species confirms the successful partial reduction of graphene oxide to rGO, while the residual functional groups are beneficial for improving interfacial bonding and electron transfer between components [36]. The Mo 3d spectrum (Figure 7d) exhibits two main peaks located at 228.9 eV (Mo 3d₅/₂) and 232.2 eV (Mo 3d₃/₂), characteristic of Mo⁴⁺ in MoS₂ [30,37]. No additional peaks are detected in the higher binding energy range (233–235 eV), ruling out the presence of significant amounts of oxidized Mo⁶⁺ species such as MoO₃ [37]. In the same region, a broad feature at 226.4 eV is assigned to the S 2s signal [38], further supporting the existence of sulfide species (S²⁻) in the MoS₂ lattice [30,38]. Altogether, the XPS results confirm the integration of TiO₂, MoS₂, and rGO into a ternary heterostructure with minimal chemical perturbation and strong interfacial interactions. The preservation of the oxidation states of Ti⁴⁺ and Mo⁴⁺, along with the partial reduction of rGO, is consistent with the enhanced photocatalytic behavior observed in degradation and hydrogen evolution experiments.
3.2. Photocatalytic Degradation of Malathion
To establish the optimal reaction conditions, a series of preliminary experiments were carried out using the most active material, 5%MoS₂@TiO₂-rGO, as the reference (see Figure S1). These studies focused on evaluating the influence of key operational parameters, such as catalyst loading, initial pH of the solution, and the presence or absence of irradiation and oxygen on the degradation of malathion. The outcomes not only allowed us to determine the ideal experimental conditions for maximum photocatalytic efficiency but also served to confirm the photocatalytic origin of the degradation process through control experiments. These optimized parameters were subsequently applied in the evaluation of the remaining catalysts to ensure consistent and comparable performance assessments. Figure S1a shows the effect of catalyst loading (from 0.4 to 1.8 g/L) on the photodegradation efficiency of malathion after 2 hours of UV-visible irradiation. An increase in catalyst loading led to improved degradation up to an optimal concentration of 1.0 g/L, where the degradation reached nearly 100%. This enhancement is attributed to the increased number of active sites and photon absorption capacity. However, beyond this concentration, the degradation efficiency decreased significantly. At 1.6 and 1.8 g/L, degradation dropped to around 65% and 50%, respectively. This decline is likely due to increased turbidity and light scattering at higher catalyst concentrations, which reduce light penetration and active photon flux within the suspension. Figure S1b shows the influence of the solution pH on photocatalytic degradation efficiency. Experiments were performed over a pH range of 4 to 10, keeping all other conditions constant. The photocatalytic activity showed a marked dependence on pH, with maximum degradation (ca. 100%) occurring at neutral to slightly acidic conditions (pH 6–7). Below this range, especially at pH 4, degradation efficiency decreased sharply (~60%), likely due to reduced malathion adsorption or catalyst surface protonation. In alkaline media (pH > 8), the degradation also decreased, possibly due to hydroxide ion competition or destabilization of reactive oxygen species. These results suggest that the surface charge of the photocatalyst and the speciation of malathion both influence the reaction kinetics, and that near-neutral conditions are ideal for optimal degradation. To confirm the photocatalytic nature of the malathion degradation process, a series of control tests were conducted (Figure S1c). These included: (i) photolysis (irradiation without catalyst), (ii) catalysis (catalyst in dark), and (iii) photocatalysis under anoxic conditions (argon-purged system). The results clearly show that significant degradation occurred only under full photocatalytic conditions (light + catalyst + air), where the malathion concentration dropped steadily over time, reaching almost complete mineralization within 120 minutes. In contrast, all control conditions showed minimal activity: photolysis and catalysis resulted in only minor losses (<15%), and the anoxic photocatalytic test demonstrated reduced efficiency, highlighting the essential role of dissolved oxygen as an electron acceptor in the generation of reactive oxygen species (ROS).
The photocatalytic activity of the synthesized materials was evaluated under UV-visible irradiation. The performance of the different catalysts—P25-rGO and MoS₂-modified composites with 1%, 3%, 5%, and 10% MoS₂—was compared under previously optimized reaction conditions (1.0 g/L catalyst loading and pH 7). As shown in Figure 8, all MoS₂-containing composites outperformed the P25-rGO sample, demonstrating the beneficial effect of MoS₂ addition. Among the tested materials, 5%MoS₂@P25-rGO exhibited the highest degradation rate, achieving near-complete removal of malathion within 120 minutes. This enhanced activity is attributed to the synergistic interaction between TiO₂, rGO, and MoS₂, which promotes charge separation and broadens light absorption. The 3%MoS₂ and 1%MoS₂ composites also showed significant improvements compared to P25-rGO, but to a lesser extent. Interestingly, the 10%MoS₂@P25-rGO catalyst exhibited slightly lower activity than the 3% and 5% counterparts, likely due to excessive MoS₂ loading that can shield the active surface or induce recombination centers, corroborating the previously discussed BET and PL results.
To better understand the degradation mechanism of malathion using the 5%MoS₂@P25-rGO composite, a series of scavenger experiments were carried out to identify the main reactive species involved (see Figure S2). The addition of 1,4-benzoquinone (BQ), a selective quencher of superoxide radicals (·O₂⁻) [39], resulted in a pronounced decrease in degradation efficiency, strongly suggesting that ·O₂⁻ species play a central role in the photocatalytic process [39]. In contrast, the use of EDTA-Na₂, a hole (h⁺) scavenger [40], led to negligible inhibition, indicating that direct oxidation by photogenerated holes is not the primary degradation pathway [40]. Similarly, the addition of tert-butanol (t-BuOH), a hydroxyl radical (·OH) scavenger [41,42], caused only moderate suppression, pointing to a secondary contribution of ·OH radicals [41,42]. These findings are consistent with a mechanism in which photoexcited electrons, generated upon UV-visible irradiation of TiO₂, are efficiently transferred to MoS₂ and/or rGO, reducing adsorbed O₂ molecules to form superoxide radicals. The layered structure and intimate contact among TiO₂, MoS₂, and rGO facilitate efficient charge separation and migration across the heterostructure, possibly through a Type-II or Z-scheme charge transfer mechanism. MoS₂, with its suitable conduction band position, acts as an electron acceptor and stabilizer, while rGO provides a rapid electron transport pathway [15,43]. The result is enhanced generation of ·O₂⁻ species, which act as the dominant oxidizing agents responsible for the breakdown of malathion.
A detailed GC-MS analysis was conducted to elucidate the photocatalytic degradation pathway of malathion under UV-visible irradiation using the most active catalyst, 5%MoS₂@P25-rGO (Figure 9). Reaction aliquots were collected at different irradiation times and analyzed to identify intermediate products based on their mass-to-charge (m/z) ratios. The parent compound, malathion (m/z = 330), was progressively decomposed through a sequence of hydrolytic and oxidative transformations. Five main degradation pathways (A–E) were proposed based on the detected fragments and their temporal evolution, as illustrated in Figure 9. In Pathway A, hydrolysis of ester bonds and ring opening lead to the formation of lower-mass products [44]. Pathways B and C involve oxidative desulfuration and P–S bond cleavage, producing fragments such as m/z 303, 302, 214, and 156 [45,46,47,48]. Pathway D comprises further oxidation and sulfur removal, generating species at m/z 317, 270, and 224 [44], while Pathway E involves oxidative demethylation and side-chain fragmentation, yielding intermediate ions like m/z 287, 241, and 133 [49]. The presence of low molecular weight fragments (m/z 128, 126, 137) indicates the occurrence of advanced oxidation processes, suggesting partial mineralization of malathion into CO₂ and H₂O, consistent with the mineralization trends observed in other TiO₂-based systems. Importantly, the formation of intermediates such as m/z 214 and 156 supports the predominant role of superoxide radicals (·O₂⁻) as oxidative agents, consistent with the radical trapping experiments discussed previously [50,51]. The heterostructure of TiO₂, MoS₂, and rGO favors efficient charge separation and facilitates electron transfer to molecular oxygen, sustaining the generation of reactive oxygen species (ROS). MoS₂, due to its narrow bandgap and appropriate conduction band alignment, acts as an effective electron sink, while rGO enhances electron transport and surface dispersion. This synergistic configuration promotes a Z-scheme or Type II-like mechanism that enhances photoinduced redox activity [52]. Altogether, the results demonstrate that the 5%MoS₂@P25-rGO catalyst enables efficient and multi-step degradation of malathion via concurrent hydrolytic and oxidative pathways, ultimately leading to detoxification of the pollutant and partial mineralization under mild conditions.
To ensure the practical viability of the developed photocatalysts, long-term operational stability and reusability were also evaluated. In this context, a recyclability study was conducted using the most active material, 5%MoS₂@P25-rGO, to assess its performance over successive degradation cycles. As shown in Figure S3, the photocatalyst maintained nearly constant activity throughout ten consecutive runs, with only a slight decline of approximately 4.7% in degradation efficiency. This stability underscores the structural robustness and chemical durability of the MoS₂-rGO-TiO₂ heterojunction, confirming its suitability for repeated use in aqueous photocatalytic systems under UV-visible light irradiation.
Based on all the results presented above, a plausible mechanism for the photocatalytic degradation of malathion has been proposed, as illustrated in Figure 10. The electronic band structure and the migration direction of photogenerated charge carriers were estimated using the Mulliken electronegativity approach [53,54] in conjunction with the experimentally determined bandgap values (see Figure 4). The conduction and valence band edge potentials were calculated according to the following equations:
where X is the absolute electronegativity of the semiconductor, EC=4.50 eV is the energy of free electrons on the hydrogen scale [55], Eg is the optical bandgap, and ECB and EVB are the potentials of the conduction and valence bands, respectively. Based on this model, the calculated band edge positions for P25-rGO are ECB=−0.165 eV and EVB=+2.785 eV, while for MoS₂ they are ECB=−0.405 eV and EVB=+2.045 eV. Under visible light irradiation, TiO₂ (P25) is largely inactive due to its wide bandgap (~3.2 eV) [56]. However, MoS₂ and rGO, with narrower bandgaps, can absorb visible photons and become photoexcited [57]. In the case of MoS₂, visible light promotes electrons from the valence band to the conduction band, leaving behind holes. These photoexcited electrons, due to the more negative conduction band of MoS₂ (–0.405 eV) relative to P25-rGO (–0.165 eV), can transfer to the TiO₂–rGO interface, where they are readily scavenged by molecular oxygen dissolved in the medium. This reduction leads to the formation of superoxide radicals (·O₂⁻), which are highly reactive and capable of oxidizing malathion. Simultaneously, holes remaining in MoS₂ and photoinduced holes in rGO may weakly contribute to oxidation, although scavenger experiments indicate that their role is secondary (see Figure S2). Instead, hydroxyl radicals (·OH), generated from water or hydroxide oxidation by valence band holes in TiO₂, provide an additional oxidative pathway. The high surface area of rGO facilitates these processes by providing a large number of adsorption and reaction sites, while also improving charge mobility and suppressing recombination via rapid electron conduction [58]. The dominant degradation route, as supported by radical quenching experiments and GC-MS analysis, is thus initiated by ·O₂⁻ radicals attacking the ester and phosphorothioate bonds in malathion, leading to a stepwise oxidative fragmentation into less toxic and lower-molecular-weight intermediates. This mechanism is fully consistent with the observed suppression of activity upon addition of 1,4-benzoquinone (a ·O₂⁻ scavenger), as well as with the enhanced photocatalytic activity shown by the 5%MoS₂@P25-rGO composite compared to binary or unmodified systems. The proposed mechanism involves a type-II heterojunction [59] in which MoS₂ and rGO sensitize the composite to visible light [14], and the hierarchical structure promotes directional charge transfer from MoS₂ to TiO₂–rGO [60]. This configuration enables the generation of reactive oxygen species—mainly superoxide and, to a lesser extent, hydroxyl radicals—which drive the oxidative degradation of malathion under solar-like irradiation conditions.
ECB = X – EC – 0.5Eg
EVB = ECB + Eg
3.3. Photocatalytic Hydrogen Production
As performed for the malathion photodegradation studies, a similar approach was employed to determine the optimal conditions for photocatalytic hydrogen production (Figure S4). The influence of catalyst loading on the photocatalytic hydrogen production performance of the most active nanocomposite, 5%MoS₂@P25-rGO, was first investigated (Figure S4a). The hydrogen evolution rate increased with catalyst concentration up to an optimal loading of 1.0 g L⁻¹, reaching a maximum yield of nearly 6000 µmol g⁻¹ h⁻¹. Beyond this point, the activity decreased, likely due to excessive light scattering, increased turbidity, and agglomeration of photocatalyst particles, which limit light penetration and reduce the number of accessible active sites. At lower catalyst dosages, the lower availability of surface-active regions similarly limits the overall rate of hydrogen generation. The effect of solution pH on hydrogen production was subsequently evaluated (Figure S4b). The system exhibited optimal performance under neutral conditions (pH = 7), where both charge carrier separation and proton availability are favorably balanced. In strongly acidic environments (pH = 4), the excessive concentration of H⁺ ions can hinder charge mobility and promote recombination. Conversely, under alkaline conditions (pH = 10), the reduced proton concentration limits the supply of reactants necessary for H₂ evolution, leading to a significant drop in photocatalytic efficiency [61]. To confirm the photocatalytic nature of the observed hydrogen generation, control experiments were carried out under different conditions (Figure S4c). Negligible H₂ evolution was detected in the absence of either the catalyst or light, confirming that both components are essential for the reaction to proceed. These results validate that the process is strictly photo-driven and demonstrate the synergy between MoS₂, TiO₂, and rGO in facilitating efficient light-induced hydrogen evolution.
Figure 11 shows the wavelength-dependent hydrogen production profiles, clearly highlighting the superior photocatalytic activity of MoS₂-modified P25-rGO composites relative to the unmodified P25-rGO system. As a function of the incident photon energy, all catalysts show enhanced activity within the 300–500 nm range, where the absorption of UV and visible light is most efficient. Among the materials studied, the 5%MoS₂@P25-rGO nanocomposite exhibits the highest hydrogen evolution rate, reaching nearly 6000 µmol g⁻¹ h⁻¹ at 500 nm. This outstanding activity can be rationalized by the formation of an efficient heterojunction between TiO₂, MoS₂, and rGO, which synergistically enhances charge separation, interfacial charge transfer, and visible-light absorption. TiO₂ serves as a stable wide-bandgap photocatalyst with strong UV absorption, while rGO provides a conductive platform that facilitates electron mobility, reduces charge recombination, and promotes light harvesting through its extended π-conjugated system [62]. The incorporation of MoS₂ introduces a narrow-bandgap semiconductor with well-known catalytic activity for the hydrogen evolution reaction (HER), acting as a co-catalyst that offers abundant active edge sites and lowers the overpotential required for proton reduction [63]. The observed activity trend—5%MoS₂ > 3%MoS₂ > 10%MoS₂ > 1%MoS₂ > P25-rGO—clearly indicates that a moderate MoS₂ loading is optimal for balancing these effects. At 1% MoS₂, the number of catalytically active sites is likely insufficient to significantly improve HER kinetics, while an excessive amount of MoS₂ (e.g., 10%) could lead to detrimental effects such as nanoparticle agglomeration, light shielding, and partial coverage of TiO₂ or rGO surfaces, thereby impeding photon absorption and reducing charge accessibility. Additionally, the decline in activity observed beyond 500 nm is consistent with the intrinsic bandgap limitations of the semiconductor components, as the photon energy becomes inadequate to excite electrons from the valence to the conduction band. These findings support that the careful modulation of MoS₂ content within the ternary composite is essential to maximize photocatalytic efficiency, and they emphasize the importance of interfacial engineering, band alignment, and light absorption optimization in designing next-generation nanostructured materials for sustainable hydrogen production.
The long-term performance and mechanistic aspects of the photocatalytic hydrogen evolution reaction (HER) using MoS₂@P25-rGO composites were further examined through a series of complementary experiments, including transient photocurrent measurements, scavenger assays, and recyclability tests. The photocurrent responses under chopped light illumination (Figure 12) provide direct insight into the efficiency of photogenerated charge separation and transport. The 5%MoS₂@P25-rGO catalyst exhibited the highest photocurrent density (~5 μA), followed by 3%MoS₂@P25-rGO, 10%MoS₂@P25-rGO, 1%MoS₂@P25-rGO, and finally the unmodified P25-rGO. This order of photocurrent response precisely mirrors the trend observed in photocatalytic hydrogen production (Figure 11), and also aligns with the photocatalytic activity for malathion degradation discussed in earlier sections. These consistent trends across multiple techniques confirm that the enhanced photoactivity of the 5%MoS₂ composite is directly linked to its superior charge separation and transport characteristics, which are facilitated by the synergistic interactions between MoS₂, P25, and rGO.
To further clarify the mechanistic pathway of HER, radical scavenger experiments were conducted using EDTA-Na₂, a known hole scavenger (see Figure S5) [40]. The addition of EDTA resulted in a significant enhancement in H₂ production across the tested wavelengths compared to the control without scavenger. This suggests that photogenerated holes act as recombination centers or engage in parallel oxidative reactions, and that their suppression enables a higher fraction of electrons to participate in proton reduction. These results support the hypothesis that MoS₂ not only facilitates electron transfer but also serves as an efficient co-catalyst for proton reduction, with rGO acting as an electron mediator that enhances interfacial conductivity [15,64].
In terms of practical application, the recyclability of the 5%MoS₂@P25-rGO photocatalyst was evaluated over ten consecutive HER cycles (Figure S6). The system retained 91.9% of its initial hydrogen production capacity after ten uses, with a performance drop of only 8.1%. This photostability underscores the structural robustness of the heterostructure and the durability of the active sites, confirming the feasibility of this material for long-term solar hydrogen generation. The strong interfacial bonding among MoS₂, TiO₂, and rGO components likely prevents leaching or deactivation, maintaining catalytic integrity over multiple uses. Taken together, these findings demonstrate the strong correlation between photocatalytic performance, charge transport efficiency, and material stability, positioning 5%MoS₂@P25-rGO as a promising candidate for sustainable hydrogen production.
Based on the previously discussed results, a plausible mechanism has been proposed to explain the photocatalytic hydrogen evolution activity, consistent with the experimental observations (see Figure 13). The outstanding H₂ production performance of the 5%MoS₂@P25-rGO photocatalyst arises from the interplay between its three constituents—TiO₂ (P25), MoS₂, and reduced graphene oxide (rGO)—which together form a hierarchical heterostructure capable of efficient light absorption, charge separation, and catalytic functionality under visible-light irradiation. Among them, MoS₂ acts as the primary absorber of visible light. Upon irradiation, electrons are promoted from its valence band (VB) to its conduction band (CB), leaving behind photogenerated holes. TiO₂ (P25), with a wider bandgap (~3.2 eV), is less responsive to visible light; however, the incorporation of MoS₂ and rGO into the structure red-shifts the optical absorption of the composite, allowing some activation of TiO₂ under solar-simulated conditions. Moreover, interfacial interactions can induce localized mid-gap states, enhancing visible-light response. Band edge calculations based on Mulliken electronegativity theory suggest that the CB potential of MoS₂ (–0.405 eV vs NHE) is more negative than that of TiO₂ (–0.165 eV), while TiO₂ has a more positive VB (+2.785 eV), making it a potent oxidant. This band alignment favors a directional flow of charge carriers: electrons generated in TiO₂ or MoS₂ transfer toward MoS₂ and rGO, while holes accumulate on TiO₂ [65]. Additionally, rGO acts as a conductive electron mediator that bridges MoS₂ and TiO₂, facilitating ultrafast charge transfer and delocalization, while also serving as a high-surface-area scaffold for active site dispersion [36]. This spatial charge separation is further evidenced by the strong quenching of photoluminescence (PL) in the composite and its enhanced transient photocurrent response, which indicate suppressed electron-hole recombination. The 5%MoS₂@P25-rGO composite shows the highest photocurrent density and the lowest PL intensity among all tested samples, consistent with its superior H₂ production rates. At the MoS₂ surface, electrons reduce protons (H⁺) from the solution to generate H₂, taking advantage of the abundant and catalytically active edge sites on MoS₂. Meanwhile, the holes in TiO₂ oxidize sacrificial agents added to the solution, preventing recombination and sustaining the redox cycle. Scavenger experiments confirm that hole consumption significantly enhances H₂ evolution, highlighting the importance of maintaining separate pathways for electrons and holes. This mechanism is coherent with that proposed for malathion degradation, where the same spatial charge separation and vectorial charge migration were identified. In the absence of oxygen, electrons that would otherwise reduce O₂ (to form ·O₂⁻ for oxidative degradation) are now fully available for proton reduction, thus explaining the high H₂ evolution rates. The rGO sheets not only improve conductivity and dispersibility of MoS₂, but also ensure intimate contact among the components, which is essential for maintaining an efficient interfacial electric field and continuous charge flow [66,67]. Overall, the 5%MoS₂@P25-rGO catalyst operates via a cooperative mechanism that combines light absorption, charge generation, and catalytic functionality across its components. The result is a system capable of exploiting a broad portion of the solar spectrum while maintaining low recombination losses and high redox activity—delivering significant hydrogen generation rates and demonstrating its promise for sustainable solar fuel applications.
4. Conclusions
In this study, a series of MoS₂-decorated TiO₂-rGO ternary nanocomposites were successfully synthesized and extensively characterized to evaluate their dual functionality in photocatalytic malathion degradation and hydrogen evolution under UV-visible irradiation. The hybrid photocatalysts were prepared by integrating exfoliated MoS₂ nanosheets with TiO₂ nanoparticles supported on reduced graphene oxide, creating intimate heterojunctions designed to enhance light absorption, charge separation, and surface reactivity. Among the catalysts tested, the composite containing 5 wt% MoS₂ exhibited the highest photocatalytic performance in both applications, confirming the critical importance of compositional balance and interfacial engineering in these systems. Comprehensive structural, morphological, and electronic analyses revealed that all nanocomposites maintained the anatase phase of TiO₂ while incorporating well-dispersed rGO and MoS₂. Raman spectroscopy and XRD confirmed the phase purity and successful integration of all components without the formation of undesirable secondary phases. HRTEM and SEM analyses illustrated the morphological coherence and homogeneous dispersion of the layered materials, while BET analysis demonstrated a marked increase in surface area with the inclusion of rGO and MoS₂. Interestingly, despite having the highest surface area, the 10% MoS₂-loaded composite underperformed in both degradation and hydrogen evolution, indicating that excessive MoS₂ can hinder photocatalytic efficiency by introducing recombination centers or limiting photon penetration due to light shielding or overcoating of active sites. Optical studies provided further insights into the behavior of the ternary systems. UV-Vis DRS demonstrated a systematic red shift and bandgap narrowing with increasing MoS₂ content, enhancing visible-light absorption. PL measurements revealed a significant suppression of electron-hole recombination, particularly in the 5% MoS₂@TiO₂-rGO sample, suggesting a highly efficient charge separation mechanism facilitated by the synergistic roles of rGO as an electron mediator and MoS₂ as an electron acceptor and catalytic site. XPS confirmed the stability of the Ti⁴⁺ and Mo⁴⁺ oxidation states, as well as interfacial electronic interactions indicative of covalent or electrostatic coupling between the components. Photocatalytic degradation of malathion revealed clear performance trends across the different composites, with the 5% MoS₂ catalyst achieving nearly complete degradation within 60–90 minutes. GC-MS analysis identified multiple intermediate products and proposed five principal degradation pathways involving hydrolysis, desulfuration, demethylation, and oxidative ring-opening, supported by radical scavenger experiments which established superoxide radicals (·O₂⁻) as the main active species. The optimal photocatalytic performance was linked to the ability of the heterostructure to promote efficient interfacial charge separation and favor a Z-scheme or Type-II electron transfer mechanism. Additionally, recyclability tests confirmed that the best-performing catalyst retained more than 95% of its activity over ten consecutive cycles, underscoring its structural stability and potential for real-world environmental applications. In the hydrogen evolution reaction (HER), similar trends were observed. The photocatalytic H₂ production peaked at 5% MoS₂ loading, achieving yields of nearly 6000 µmol·g⁻¹·h⁻¹ under neutral pH and optimal catalyst dosage (1.0 g·L⁻¹). Photocurrent measurements, radical trapping studies, and recyclability tests supported the same conclusions drawn for malathion degradation: that effective charge separation and transport are crucial for photocatalytic efficiency, and that the ternary synergy among TiO₂, MoS₂, and rGO plays a pivotal role. Notably, the catalyst’s photocurrent generation profile mirrored both HER and pollutant degradation efficiencies, providing strong evidence of consistent structure–function relationships.
Altogether, this work demonstrates that careful modulation of MoS₂ content in TiO₂-rGO nanocomposites results in significantly improved photocatalytic activity for both pollutant removal and renewable energy production. The 5% MoS₂@TiO₂-rGO nanocomposite represents an optimal configuration for exploiting visible-light-induced processes via enhanced interfacial charge transfer, light harvesting, and active site accessibility. These findings not only highlight the versatility of MoS₂-based ternary photocatalysts but also offer valuable design principles for developing next-generation multifunctional materials aimed at environmental remediation and sustainable hydrogen production.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. Evaluation of the initial concentration of 5%MoS2@P25-rGO on the photodegradation of malathion (a); effect of pH on the photocatalytic activity of the 5%MoS2@P25-rGO catalyst for photodegradation of malathion (b); and control experiments for 5%MoS2@P25-rGO with malathion, under irradiation (c); Figure S2. Photodegradation of malathion by 5%MoS2@P25-rGO in the presence of different scavengers; Figure S3. Recyclability of 5%MoS2@P25-rGO after 10 consecutive catalytic cycles of photodegradation of malathion under irradiation. Figure S4. Evaluation of the initial concentration of 5%MoS2@P25-rGO on the efficiency of hydrogen production (a); effect of pH on the photocatalytic activity of the 5%MoS2@P25-rGO catalyst for hydrogen production (b); and control experiments for 5%MoS2@P25-rGO on the efficiency of hydrogen production (c); Figure S5. Hydrogen production via water splitting using 5%MoS2@P25-rGO under irradiation, and also in the presence of EDTA-Na2; Figure S6. Recyclability of 5%MoS2@P25-rGO after 10 consecutive catalytic cycles of hydrogen production, under irradiation at 500 nm; Table S1. BET surface area of the synthesized materials.
Author Contributions
Conceptualization, F.M., C.M. and A.M.; methodology, F.M.; formal analysis, A.M. and F.M.; investigation, A.M., C.M-P., L.S-V., E.R., E.N., P.S., J.D., M.C., P.B., C.M., F.M., resources, F.M., C.M., M.C.; writing—original draft preparation, A.M. and F.M.; writing—review and editing, A.M., F.M., C.M.; supervision, F.M.; project administration, F.M. and M.C.; funding acquisition, F.M., A.M., C.M., M.C. and J.D. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from NSF Center for the Advancement of Wearable Technologies-CAWT (Grant 1849243), from the Consortium of Hybrid Resilient Energy Systems CHRES (DE-NA0003982), and from the Spanish Ministry of Economy and Competitiveness, under NanoCat-Com Project (PID2021-124667OB-I00), are gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data is contained in the article and is available from the corresponding authors on reasonable request.
Acknowledgments
The facilities provided by the National Center for Electron Microscopy (ICTS—ELECMI) at Complutense University of Madrid (Spain), and by the Materials Characterization Center at University of Puerto Rico are gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ahmad, M.F.; Ahmad, F.A.; Alsayegh, A.A.; Zeyaullah, M.; AlShahrani, A.M.; Muzammil, K.; Saati, A.A.; Wahab, S.; Elbendary, E.Y.; Kambal, N.; et al. Pesticides Impacts on Human Health and the Environment with Their Mechanisms of Action and Possible Countermeasures. Heliyon 2024, 10, e29128. [Google Scholar] [CrossRef] [PubMed]
- Jensen, I.M.; Whatling, P. Malathion. In Hayes’ Handbook of Pesticide Toxicology; Elsevier, 2010; pp. 1527–1542 ISBN 978-0-12-374367-1.
- Wang, T.; Zhou, Y.; Wang, L.; Sui, J.; Chen, F.; Yang, J.; Chen, S.; Cui, X.; Yang, Y.; Zhang, W. Assessing the Biotic and Abiotic Degradation of Malathion in the Environment: Current Strategies and Advances. Journal of Environmental Chemical Engineering 2025, 13, 115429. [Google Scholar] [CrossRef]
- Khan, A.U.; Khan, A.N.; Waris, A.; Ilyas, M.; Zamel, D. Phytoremediation of Pollutants from Wastewater: A Concise Review. Open Life Sciences 2022, 17, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Qazi, U.Y. Future of Hydrogen as an Alternative Fuel for Next-Generation Industrial Applications; Challenges and Expected Opportunities. Energies 2022, 15, 4741. [Google Scholar] [CrossRef]
- Jeje, S.O.; Marazani, T.; Obiko, J.O.; Shongwe, M.B. Advancing the Hydrogen Production Economy: A Comprehensive Review of Technologies, Sustainability, and Future Prospects. International Journal of Hydrogen Energy 2024, 78, 642–661. [Google Scholar] [CrossRef]
- Atilhan, S.; Park, S.; El-Halwagi, M.M.; Atilhan, M.; Moore, M.; Nielsen, R.B. Green Hydrogen as an Alternative Fuel for the Shipping Industry. Current Opinion in Chemical Engineering 2021, 31, 100668. [Google Scholar] [CrossRef]
- Fajrina, N.; Tahir, M. A Critical Review in Strategies to Improve Photocatalytic Water Splitting towards Hydrogen Production. International Journal of Hydrogen Energy 2019, 44, 540–577. [Google Scholar] [CrossRef]
- Gunawan, D.; Zhang, J.; Li, Q.; Toe, C.Y.; Scott, J.; Antonietti, M.; Guo, J.; Amal, R. Materials Advances in Photocatalytic Solar Hydrogen Production: Integrating Systems and Economics for a Sustainable Future. Advanced Materials 2024, 2404618. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, B.; Jayarama, A.; Rao, A.S.; Nagarkar, S.S.; Dutta, A.; Duttagupta, S.P.; Prabhu, S.S.; Pinto, R. Challenges in Photocatalytic Hydrogen Evolution: Importance of Photocatalysts and Photocatalytic Reactors. International Journal of Hydrogen Energy 2024, 81, 1442–1466. [Google Scholar] [CrossRef]
- Tang, B.; Chen, H.; Peng, H.; Wang, Z.; Huang, W. Graphene Modified TiO2 Composite Photocatalysts: Mechanism, Progress and Perspective. Nanomaterials 2018, 8, 105. [Google Scholar] [CrossRef]
- Padmanabhan, N.T.; Thomas, N.; Louis, J.; Mathew, D.T.; Ganguly, P.; John, H.; Pillai, S.C. Graphene Coupled TiO2 Photocatalysts for Environmental Applications: A Review. Chemosphere 2021, 271, 129506. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Zang, C.; Huang, Z.; Zhang, H.; Ren, L.; Qi, X.; Zhong, J. Enhanced Photocatalytic Activities of Three-Dimensional Graphene-Based Aerogel Embedding TiO 2 Nanoparticles and Loading MoS 2 Nanosheets as Co-Catalyst. International Journal of Hydrogen Energy 2014, 39, 19502–19512. [Google Scholar] [CrossRef]
- Panchal, D.; Sharma, A.; Pal, S. Engineered MoS2 Nanostructures for Improved Photocatalytic Applications in Water Treatment. Materials Today Sustainability 2023, 21, 100264. [Google Scholar] [CrossRef]
- Balan, B.; Xavier, M.M.; Mathew, S. MoS2 -Based Nanocomposites for Photocatalytic Hydrogen Evolution and Carbon Dioxide Reduction. ACS Omega 2023, 8, 25649–25673. [Google Scholar] [CrossRef]
- Fu, S.; Zhang, H.; Tielrooij, K.-J.; Bonn, M.; Wang, H.I. Tracking and Controlling Ultrafast Charge and Energy Flow in Graphene-Semiconductor Heterostructures. The Innovation 2025, 6, 100764. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.D.; Mariano, R.G.; Vu, K.; Nour, N.; Seitz, O.; Chabal, Y.; Balkus, K.J. Hydrothermal Synthesis of Graphene-TiO2 Nanotube Composites with Enhanced Photocatalytic Activity. ACS Catal. 2012, 2, 949–956. [Google Scholar] [CrossRef]
- Karim, S.S.; Sudais, A.; Shah, M.S.; Farrukh, S.; Ali, S.; Ahmed, M.; Salahuddin, Z.; Fan, X. A Contemplating Review on Different Synthesis Methods of 2D-Molybdenum Disulfide (MoS2) Nanosheets. Fuel 2023, 351, 128923. [Google Scholar] [CrossRef]
- Gupta, A.; Arunachalam, V.; Vasudevan, S. Liquid-Phase Exfoliation of MoS2 Nanosheets: The Critical Role of Trace Water. J. Phys. Chem. Lett. 2016, 7, 4884–4890. [Google Scholar] [CrossRef] [PubMed]
- Machín, A.; Cotto, M.C.; Márquez, F.; Díaz-Sánchez, J.; Polop, C.; Morant, C. Hydrogen Production and Li-Ion Battery Performance with MoS2-SiNWs-SWNTs@ZnONPs Nanocomposites. Nanomaterials 2024, 14, 1911. [Google Scholar] [CrossRef]
- Gao, W.; Wang, M.; Ran, C.; Li, L. Facile One-Pot Synthesis of MoS2 Quantum Dots–Graphene–TiO2 Composites for Highly Enhanced Photocatalytic Properties. Chem. Commun. 2015, 51, 1709–1712. [Google Scholar] [CrossRef]
- Gao, Y.; Zheng, Y.; Chai, J.; Tian, J.; Jing, T.; Zhang, D.; Cheng, J.; Peng, H.; Liu, B.; Zheng, G. Highly Effective Photocatalytic Performance of {001}-TiO2 /MoS2 /RGO Hybrid Heterostructures for the Reduction of Rh B. RSC Adv. 2019, 9, 15033–15041. [Google Scholar] [CrossRef]
- Chang, Z.; Sun, X.; Liao, Z.; Liu, Q.; Han, J. Design and Preparation of Polyimide/TiO2@MoS2 Nanofibers by Hydrothermal Synthesis and Their Photocatalytic Performance. Polymers 2022, 14, 3230. [Google Scholar] [CrossRef]
- Liu, C.; Wang, L.; Tang, Y.; Luo, S.; Liu, Y.; Zhang, S.; Zeng, Y.; Xu, Y. Vertical Single or Few-Layer MoS2 Nanosheets Rooting into TiO2 Nanofibers for Highly Efficient Photocatalytic Hydrogen Evolution. Applied Catalysis B: Environmental 2015, 164, 1–9. [Google Scholar] [CrossRef]
- Asadinamin, M.; Živković, A.; De Leeuw, N.H.; Lewis, S.P. Role of Interfacial Morphology in Cu2 O/TiO2 and Band Bending: Insights from Density Functional Theory. ACS Appl. Mater. Interfaces 2024, 16, 35781–35792. [Google Scholar] [CrossRef]
- Kite, S.V.; Kadam, A.N.; Sathe, D.J.; Patil, S.; Mali, S.S.; Hong, C.K.; Lee, S.; Garadkar, K.M. Nanostructured TiO2 Sensitized with MoS2 Nanoflowers for Enhanced Photodegradation Efficiency toward Methyl Orange. ACS Omega 2021, 6, 17071–17085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qi, H.; Zhang, L.; Wang, Y.; Zhong, L.; Zheng, Y.; Wen, X.; Zhang, X.; Xue, J. A Unique RGO Aerogel/TiO2/MoS2 Composite Photocatalyst with a 3D Sandwich Network for the Removal of Organic Dyes by the Cooperative Action of Adsorption and Photocatalysis 2021.
- Tien, T.-M.; Chen, E.L. A Novel MoS2/TiO2/Graphene Nanohybrid for Enhanced Photocatalytic Hydrogen Evolution under Visible Light Irradiation. Catalysts 2023, 13, 1152. [Google Scholar] [CrossRef]
- Machín, A.; Morant, C.; Soto-Vázquez, L.; Resto, E.; Ducongé, J.; Cotto, M.; Berríos-Rolón, P.J.; Martínez-Perales, C.; Márquez, F. Synergistic Effects of Co3O4-gC3N4-Coated ZnO Nanoparticles: A Novel Approach for Enhanced Photocatalytic Degradation of Ciprofloxacin and Hydrogen Evolution via Water Splitting. Materials 2024, 17, 1059. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M. Practical Surface Analysis; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Zhu, L.; Lu, Q.; Lv, L.; Wang, Y.; Hu, Y.; Deng, Z.; Lou, Z.; Hou, Y.; Teng, F. Ligand-Free Rutile and Anatase TiO2 Nanocrystals as Electron Extraction Layers for High Performance Inverted Polymer Solar Cells. RSC Adv. 2017, 7, 20084–20092. [Google Scholar] [CrossRef]
- Xing, M.; Shen, F.; Qiu, B.; Zhang, J. Highly-Dispersed Boron-Doped Graphene Nanosheets Loaded with TiO2 Nanoparticles for Enhancing CO2 Photoreduction. Sci Rep 2014, 4, 6341. [Google Scholar] [CrossRef]
- Fan, C.; Chen, C.; Wang, J.; Fu, X.; Ren, Z.; Qian, G.; Wang, Z. Black Hydroxylated Titanium Dioxide Prepared via Ultrasonication with Enhanced Photocatalytic Activity. Sci Rep 2015, 5, 11712. [Google Scholar] [CrossRef]
- Park, S.-K.; Yu, S.-H.; Pinna, N.; Woo, S.; Jang, B.; Chung, Y.-H.; Cho, Y.-H.; Sung, Y.-E.; Piao, Y. A Facile Hydrazine-Assisted Hydrothermal Method for the Deposition of Monodisperse SnO2 Nanoparticles onto Graphene for Lithium Ion Batteries. J. Mater. Chem. 2012, 22, 2520–2525. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.-M. The Reduction of Graphene Oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Gillespie, P.N.O.; Martsinovich, N. Origin of Charge Trapping in TiO2 /Reduced Graphene Oxide Photocatalytic Composites: Insights from Theory. ACS Appl. Mater. Interfaces 2019, 11, 31909–31922. [Google Scholar] [CrossRef]
- Fominski, V.; Demin, M.; Nevolin, V.; Fominski, D.; Romanov, R.; Gritskevich, M.; Smirnov, N. Reactive Pulsed Laser Deposition of Clustered-Type MoSx (x ~ 2, 3, and 4) Films and Their Solid Lubricant Properties at Low Temperature. Nanomaterials 2020, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Tai, G.; Zeng, T.; Yu, J.; Zhou, J.; You, Y.; Wang, X.; Wu, H.; Sun, X.; Hu, T.; Guo, W. Fast and Large-Area Growth of Uniform MoS2 Monolayers on Molybdenum Foils. Nanoscale 2016, 8, 2234–2241. [Google Scholar] [CrossRef] [PubMed]
- Fónagy, O.; Szabó-Bárdos, E.; Horváth, O. 1,4-Benzoquinone and 1,4-Hydroquinone Based Determination of Electron and Superoxide Radical Formed in Heterogeneous Photocatalytic Systems. Journal of Photochemistry and Photobiology A: Chemistry 2021, 407, 113057. [Google Scholar] [CrossRef]
- Shah, B.R.; Patel, U.D. Mechanistic Aspects of Photocatalytic Degradation of Lindane by TiO2 in the Presence of Oxalic Acid and EDTA as Hole-Scavengers. Journal of Environmental Chemical Engineering 2021, 9, 105458. [Google Scholar] [CrossRef]
- Dong, Y.; Feng, C.; Jiang, P.; Wang, G.; Li, K.; Miao, H. Simple One-Pot Synthesis of ZnO/Ag Heterostructures and the Application in Visible-Light-Responsive Photocatalysis. RSC Adv. 2014, 4, 7340–7346. [Google Scholar] [CrossRef]
- Tizaoui, C.; Grima, N.M.; Derdar, M.Z. Effect of the Radical Scavenger T-Butanol on Gas–Liquid Mass Transfer. Chemical Engineering Science 2009, 64, 4375–4382. [Google Scholar] [CrossRef]
- Han, B.; Hu, Y.H. MoS2 as a Co-catalyst for Photocatalytic Hydrogen Production from Water. Energy Science & Engineering 2016, 4, 285–304. [Google Scholar] [CrossRef]
- Li, W.; Zhao, Y.; Yan, X.; Duan, J.; Saint, C.P.; Beecham, S. Transformation Pathway and Toxicity Assessment of Malathion in Aqueous Solution during UV Photolysis and Photocatalysis. Chemosphere 2019, 234, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Cui, Y.; Xu, W.; Kong, L.; Zuo, Y.; Yuan, L.; Chen, W. Degradation of Malathion in the Solution of Acetyl Peroxyborate Activated by Carbonate: Products, Kinetics and Mechanism. Journal of Hazardous Materials 2021, 407, 124808. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Huang, M.; Zhang, J.; Yan, W. Effect of Dielectric Barrier Discharge Plasma on the Degradation of Malathion and Chlorpyrifos on Lettuce. J Sci Food Agric 2021, 101, 424–432. [Google Scholar] [CrossRef]
- Dar, M.A.; Kaushik, G. Biodegradation of Malathion in Amended Soil by Indigenous Novel Bacterial Consortia and Analysis of Degradation Pathway. Soil Systems 2023, 7, 81. [Google Scholar] [CrossRef]
- Pergal, M.V.; Kodranov, I.D.; Pergal, M.M.; Gašić, U.; Stanković, D.M.; Petković, B.B.; Manojlović, D.D. Degradation Products, Mineralization, and Toxicity Assessment of Pesticides Malathion and Fenitrothion. Water Air Soil Pollut 2020, 231, 433. [Google Scholar] [CrossRef]
- Li, W.; Yan, X.; Gao, C.; Duan, J.; Beecham, S. A Consecutive Chlorination and Alkaline Hydrolysis Process for Rapid Degradation and Detoxication of Malathion in Aqueous Solution. Chemical Engineering Journal 2020, 392, 123793. [Google Scholar] [CrossRef]
- Wu, S.; Quan, X. Design Principles and Strategies of Photocatalytic H2 O2 Production from O2 Reduction. ACS EST Eng. 2022, 2, 1068–1079. [Google Scholar] [CrossRef]
- Dvoranová, D.; Barbieriková, Z.; Brezová, V. Radical Intermediates in Photoinduced Reactions on TiO2 (An EPR Spin Trapping Study). Molecules 2014, 19, 17279–17304. [Google Scholar] [CrossRef]
- Liu, J.; Jin, Y.; Lei, B.; Zhao, X.; Huang, Y.; Zhang, L.; Zhu, Y. Studies on Electronic Structure and Optical Properties of MoS2/X (X = WSe2, MoSe2, AlN, and ZnO) Heterojunction by First Principles. Catalysts 2024, 14, 678. [Google Scholar] [CrossRef]
- Prabavathi, S.L.; Saravanakumar, K.; Nkambule, T.T.I.; Muthuraj, V.; Mamba, G. Enhanced Photoactivity of Cerium Tungstate-Modified Graphitic Carbon Nitride Heterojunction Photocatalyst for the Photodegradation of Moxifloxacin. J Mater Sci: Mater Electron 2020, 31, 11434–11447. [Google Scholar] [CrossRef]
- Jourshabani, M.; Shariatinia, Z.; Badiei, A. Synthesis and Characterization of Novel Sm2O3/S-Doped g-C3N4 Nanocomposites with Enhanced Photocatalytic Activities under Visible Light Irradiation. Applied Surface Science 2018, 427, 375–387. [Google Scholar] [CrossRef]
- Cao, J.; Li, X.; Lin, H.; Chen, S.; Fu, X. In Situ Preparation of Novel p–n Junction Photocatalyst BiOI/(BiO)2CO3 with Enhanced Visible Light Photocatalytic Activity. Journal of Hazardous Materials 2012, 239–240, 316–324. [Google Scholar] [CrossRef]
- Amano, F.; Yamamoto, A.; Kumagai, J. Highly Active Rutile TiO2 for Photocatalysis under Violet Light Irradiation at 405 Nm. Catalysts 2022, 12, 1079. [Google Scholar] [CrossRef]
- Prabhakarrao, N.; Chandra, M.R.; Rao, T.S. Synthesis of Zr Doped TiO2/Reduced Graphene Oxide (rGO) Nanocomposite Material for Efficient Photocatalytic Degradation of Eosin Blue Dye under Visible Light Irradiation. Journal of Alloys and Compounds 2017, 694, 596–606. [Google Scholar] [CrossRef]
- Chen, D.; Zou, L.; Li, S.; Zheng, F. Nanospherical like Reduced Graphene Oxide Decorated TiO2 Nanoparticles: An Advanced Catalyst for the Hydrogen Evolution Reaction. Sci Rep 2016, 6, 20335. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Wang, S.; Yang, F.; Zhou, W. Mesoporous Black TiO2/MoS2/Cu2S Hierarchical Tandem Heterojunctions toward Optimized Photothermal-Photocatalytic Fuel Production. Chemical Engineering Journal 2022, 427, 131830. [Google Scholar] [CrossRef]
- Quan, Q.; Xie, S.; Weng, B.; Wang, Y.; Xu, Y. Revealing the Double-Edged Sword Role of Graphene on Boosted Charge Transfer versus Active Site Control in TiO2 Nanotube Arrays@RGO/MoS2 Heterostructure. Small 2018, 14, 1704531. [Google Scholar] [CrossRef]
- Fang, M.; Wang, B.; Wan, G.; Cao, H.; Wang, C.; Cao, P.; Han, S.; Zhu, D.; Liu, W. Boosted Scavenger-Free Photocatalytic H2O2 Production over Alkali-Doped Poly(Heptazine Imide) under Controlled Solution Conditions. Chemical Engineering Journal 2024, 494, 152969. [Google Scholar] [CrossRef]
- Bitsos, D.R.; Salepis, A.; Orfanos, E.; Coutsolelos, A.G.; Kosheleva, R.I.; Mitropoulos, A.C.; Ladomenou, K. Exploring Metal- and Porphyrin-Modified TiO2-Based Photocatalysts for Efficient and Sustainable Hydrogen Production. Inorganics 2025, 13, 121. [Google Scholar] [CrossRef]
- Gnanaseelan, N.; Latha, M.; Mantilla, A.; Sathish-Kumar, K.; Caballero-Briones, F. The Role of Redox States and Junctions in Photocatalytic Hydrogen Generation of MoS2-TiO2-rGO and CeO2-Ce2Ti3O8.7-TiO2-rGO Composites. Materials Science in Semiconductor Processing 2020, 118, 105185. [Google Scholar] [CrossRef]
- Li, H.; Yu, K.; Li, C.; Tang, Z.; Guo, B.; Lei, X.; Fu, H.; Zhu, Z. Charge-Transfer Induced High Efficient Hydrogen Evolution of MoS2/Graphene Cocatalyst. Sci Rep 2015, 5, 18730. [Google Scholar] [CrossRef] [PubMed]
- Hezam, A.; Alkanad, K.; Bajiri, M.A.; Strunk, J.; Takahashi, K.; Drmosh, Q.A.; Al-Zaqri, N.; Krishnappagowda, L.N. 2D/1D MoS2 /TiO2 Heterostructure Photocatalyst with a Switchable CO2 Reduction Product. Small Methods 2023, 7, 2201103. [Google Scholar] [CrossRef] [PubMed]
- Szkoda, M.; Ilnicka, A.; Trzciński, K.; Zarach, Z.; Roda, D.; Nowak, A.P. Synthesis and Characterization of MoS2-Carbon Based Materials for Enhanced Energy Storage Applications. Sci Rep 2024, 14, 26128. [Google Scholar] [CrossRef] [PubMed]
- Rana, K.; Kaur, H.; Singh, N.; Sithole, T.; Siwal, S.S. Graphene-Based Materials: Unravelling Its Impact in Wastewater Treatment for Sustainable Environments. Next Materials 2024, 3, 100107. [Google Scholar] [CrossRef]
Figure 1.
Field Emission Scanning Electron Microscopy (FESEM) image of P25-rGO at different magnification (a, b); and 5%MoS2@P25-rGO (c). Yellow arrows in c highlights dispersed MoS₂ nanosheets.
Figure 1.
Field Emission Scanning Electron Microscopy (FESEM) image of P25-rGO at different magnification (a, b); and 5%MoS2@P25-rGO (c). Yellow arrows in c highlights dispersed MoS₂ nanosheets.

Figure 2.
High-resolution transmission electron microscopy (HRTEM) images of the synthesized materials: (a) P25-rGO composite; (b) P25-rGO at higher magnification, with the inset highlighting lattice fringes corresponding to an interplanar spacing of approximately 0.35 nm, assigned to the (101) plane of anatase TiO₂; and (c) a monolayer MoS₂ sheet, with the inset displaying the selected area electron diffraction (SAED) pattern characteristic of hexagonal 2H-MoS₂.
Figure 2.
High-resolution transmission electron microscopy (HRTEM) images of the synthesized materials: (a) P25-rGO composite; (b) P25-rGO at higher magnification, with the inset highlighting lattice fringes corresponding to an interplanar spacing of approximately 0.35 nm, assigned to the (101) plane of anatase TiO₂; and (c) a monolayer MoS₂ sheet, with the inset displaying the selected area electron diffraction (SAED) pattern characteristic of hexagonal 2H-MoS₂.

Figure 3.
XRD patterns of commercial TiO2-P25 (a); rGO (b); exfoliated MoS2 (c); and 5%MoS2@P25-rGO (d).
Figure 3.
XRD patterns of commercial TiO2-P25 (a); rGO (b); exfoliated MoS2 (c); and 5%MoS2@P25-rGO (d).
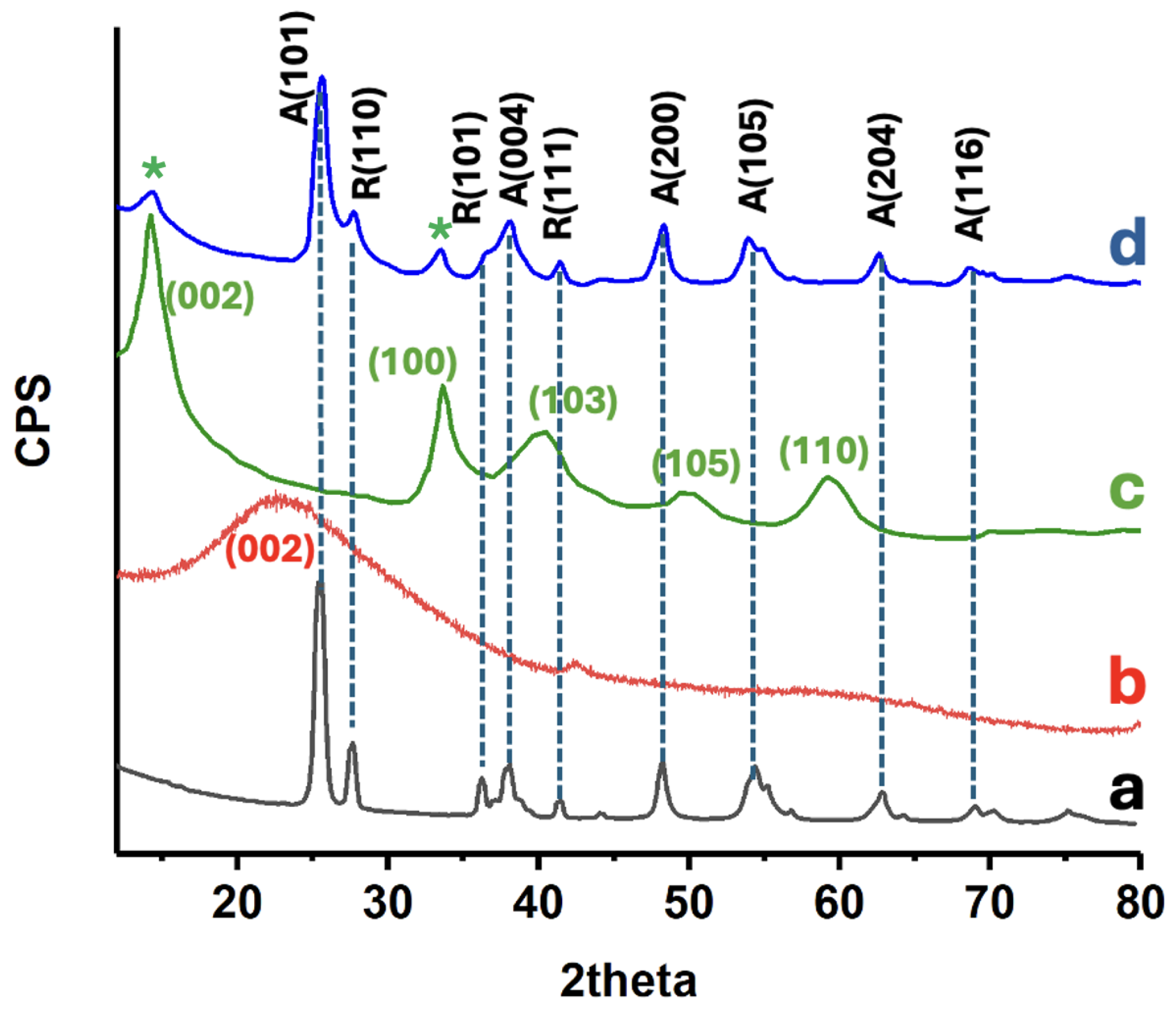
Figure 4.
Tauc plots of (αhν)2 versus energy (eV), and determination of the bandgap energy of TiO2 (P25) (a); TiO2-rGO (b); and 5%MoS2@TiO2-rGO (c).
Figure 4.
Tauc plots of (αhν)2 versus energy (eV), and determination of the bandgap energy of TiO2 (P25) (a); TiO2-rGO (b); and 5%MoS2@TiO2-rGO (c).
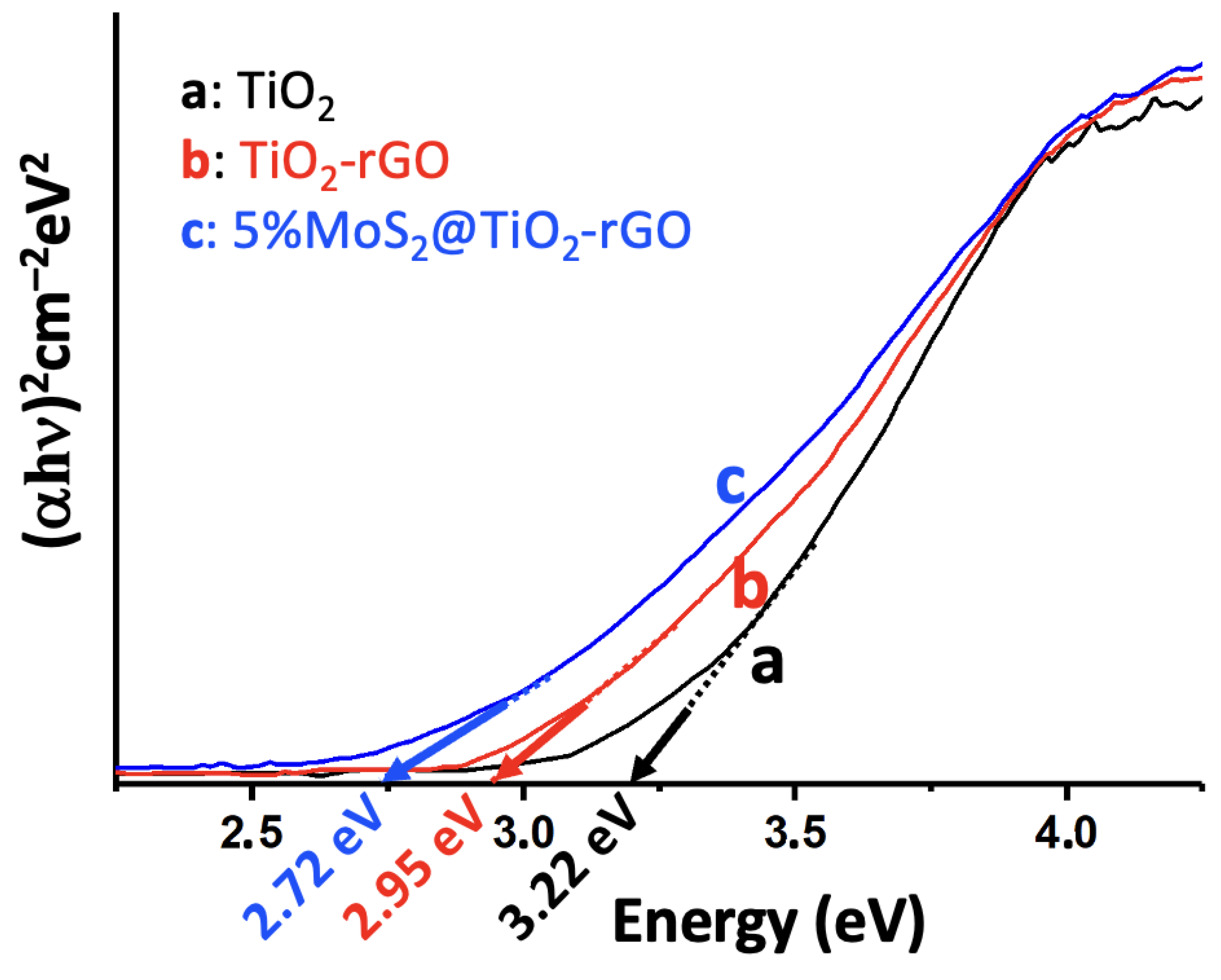
Figure 5.
PL spectra of P25-rGO, 1%MoS2@P25-rGO, 3%MoS2@P25-rGO, 5%MoS2@P25-rGO, and 10%MoS2@P25-rGO.
Figure 5.
PL spectra of P25-rGO, 1%MoS2@P25-rGO, 3%MoS2@P25-rGO, 5%MoS2@P25-rGO, and 10%MoS2@P25-rGO.
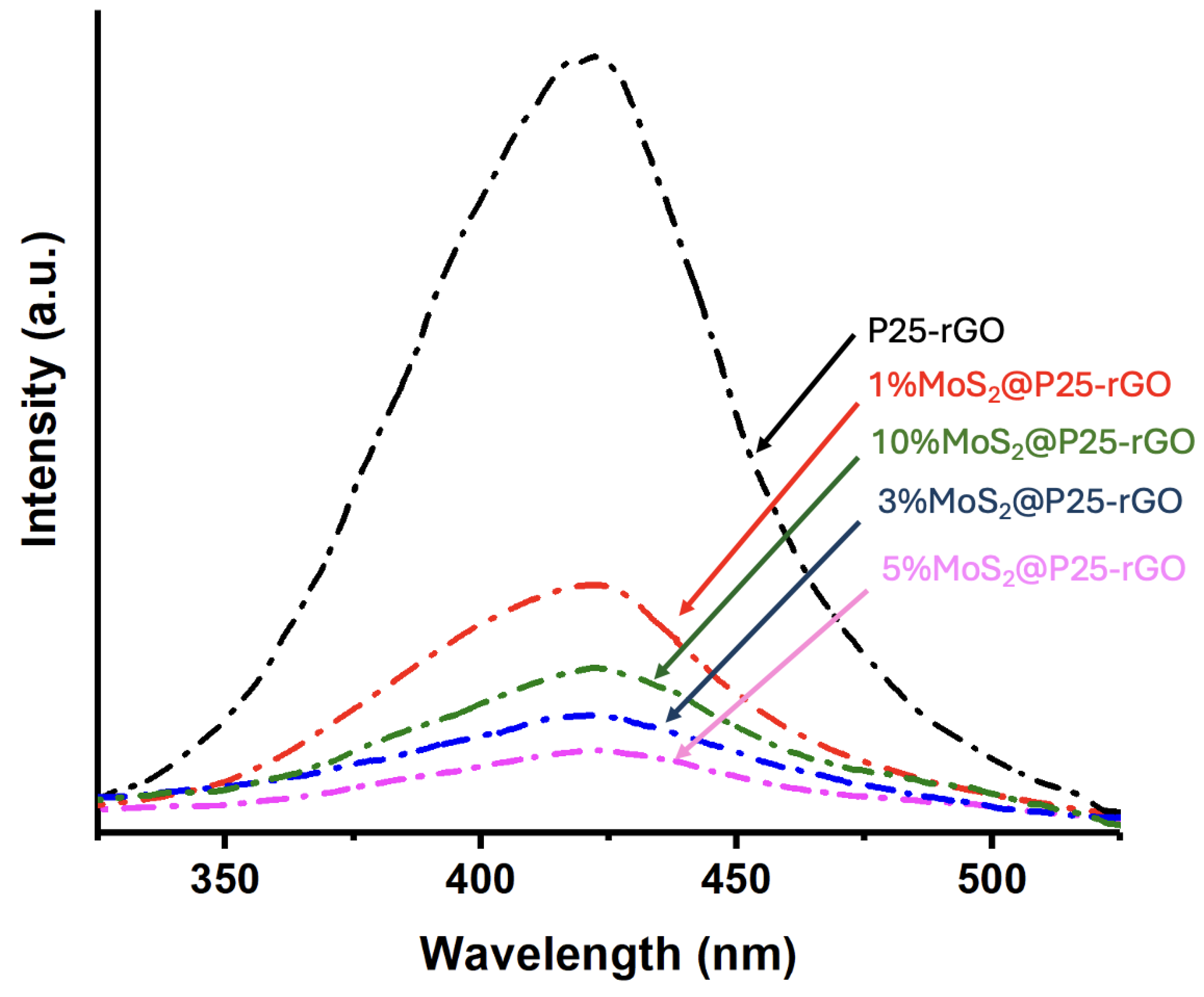
Figure 6.
Raman spectra of TiO2-P25 (a); rGO (b); Exfoliated MoS2 (c); and 5%MoS2@TiO2-rGO (d).
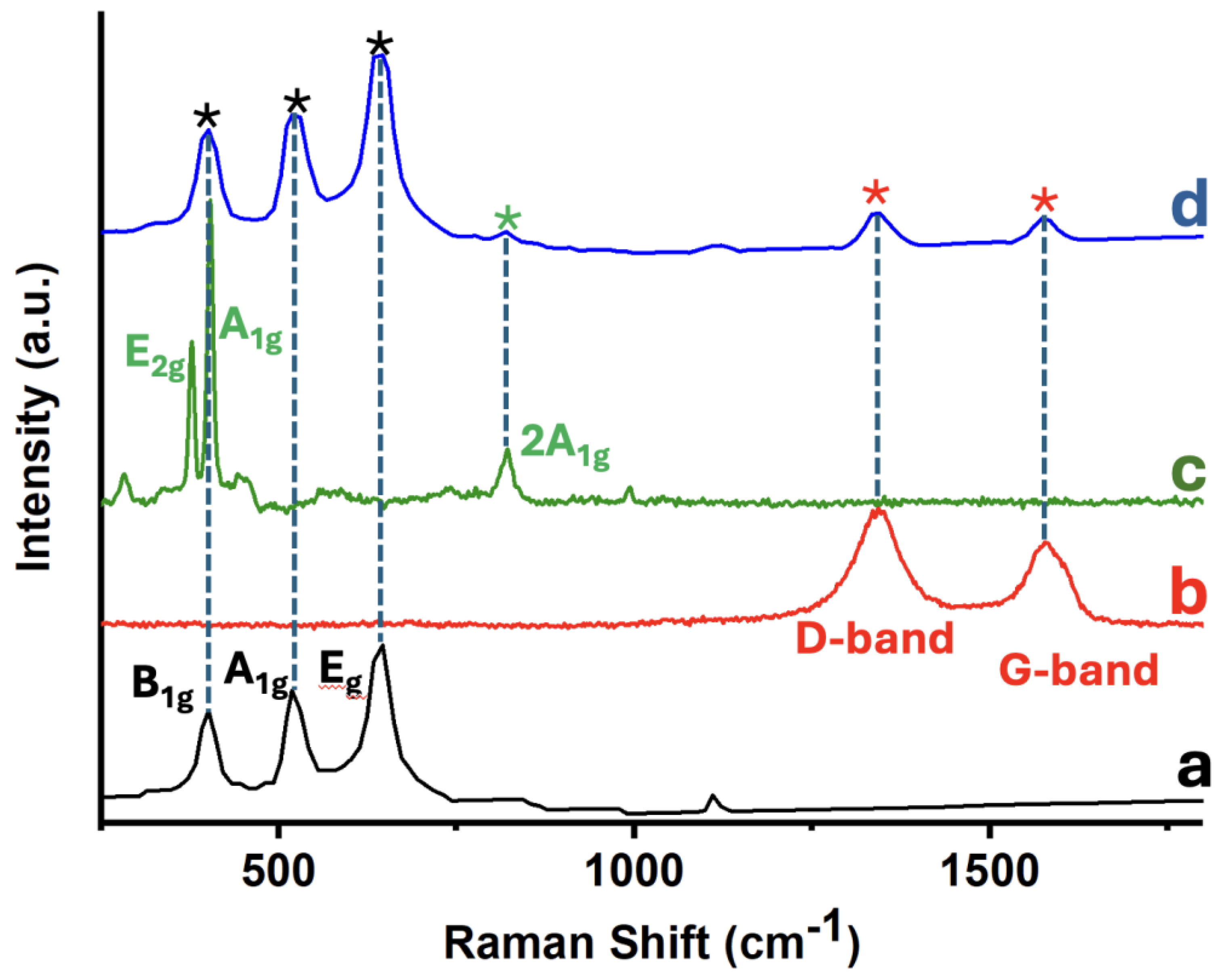
Figure 7.
XPS core level spectra for 5%MoS₂@TiO₂-rGO: Ti2p (a); O1s (b); C1s (c); and Mo3d-S2s (d).
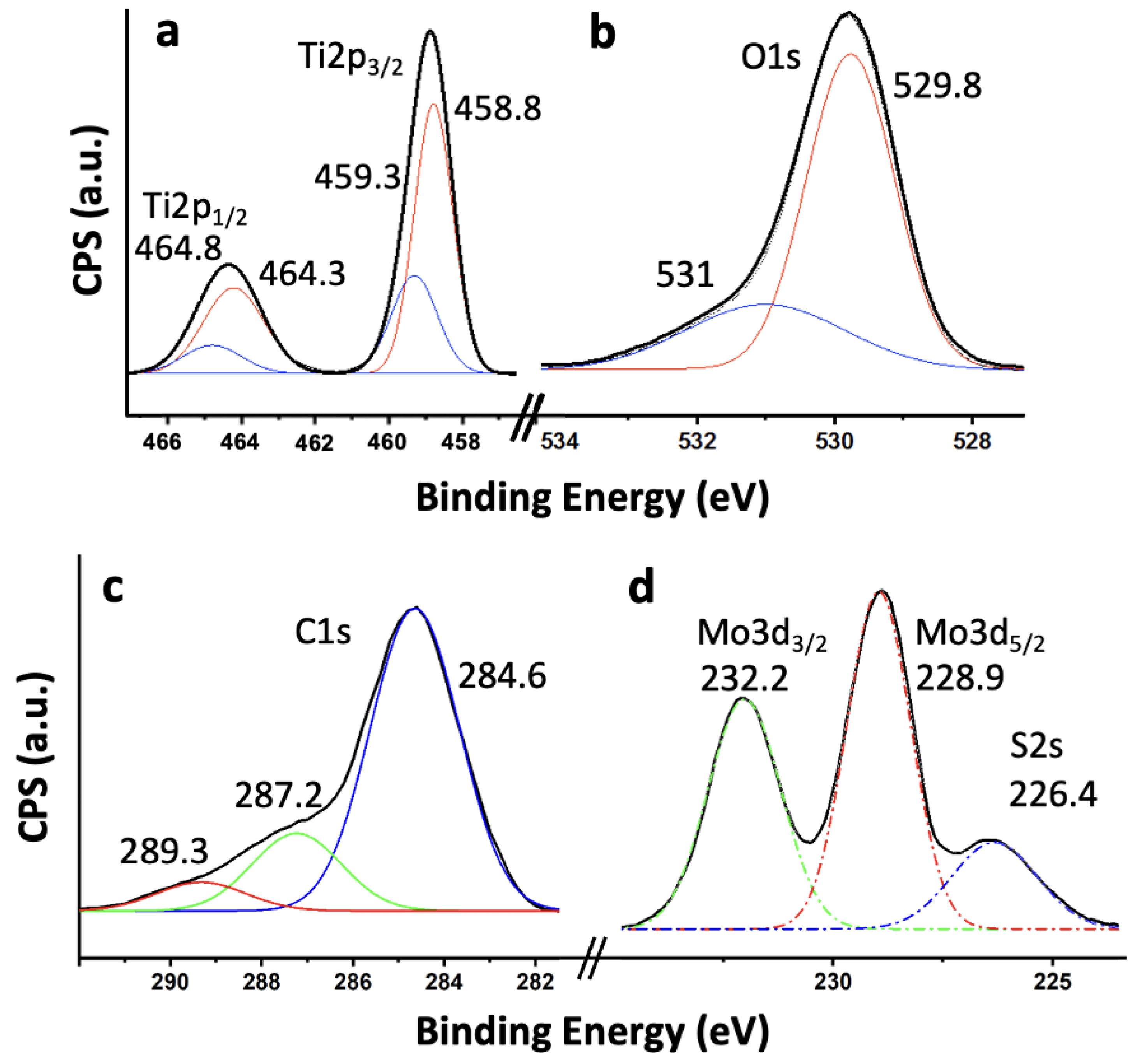
Figure 8.
Photodegradation rate of malathion as a function of time by the different synthesized catalysts.
Figure 8.
Photodegradation rate of malathion as a function of time by the different synthesized catalysts.
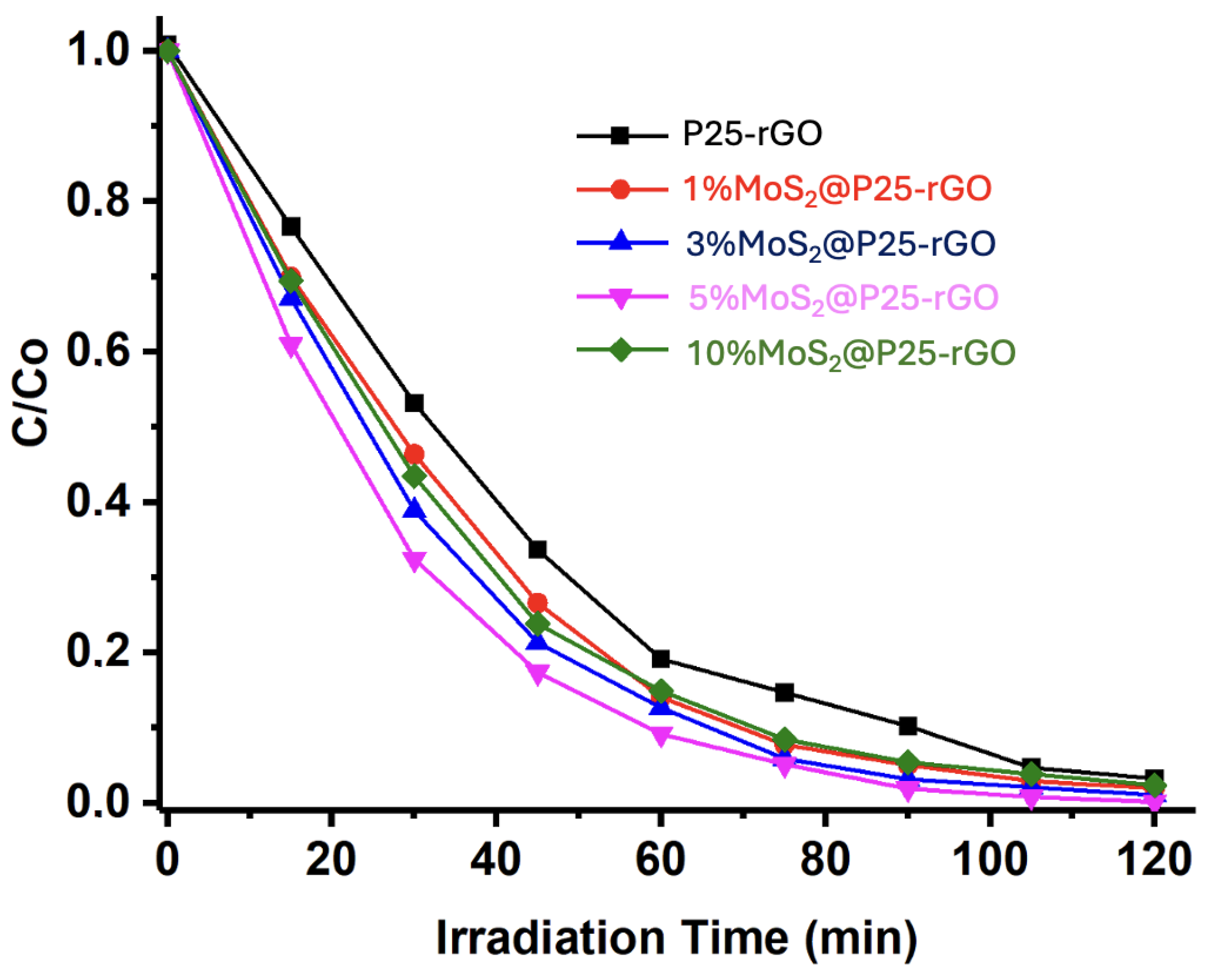
Figure 9.
Proposed photocatalytic degradation pathways of malathion (m/z 330) under UV-visible irradiation using the 5%MoS₂@P25-rGO catalyst, based on GC-MS analysis. Five main degradation routes (Pathways A–E) were identified, involving hydrolysis, desulfuration, C–O and P–S bond cleavage, oxidative demethylation, and ring-opening reactions.
Figure 9.
Proposed photocatalytic degradation pathways of malathion (m/z 330) under UV-visible irradiation using the 5%MoS₂@P25-rGO catalyst, based on GC-MS analysis. Five main degradation routes (Pathways A–E) were identified, involving hydrolysis, desulfuration, C–O and P–S bond cleavage, oxidative demethylation, and ring-opening reactions.
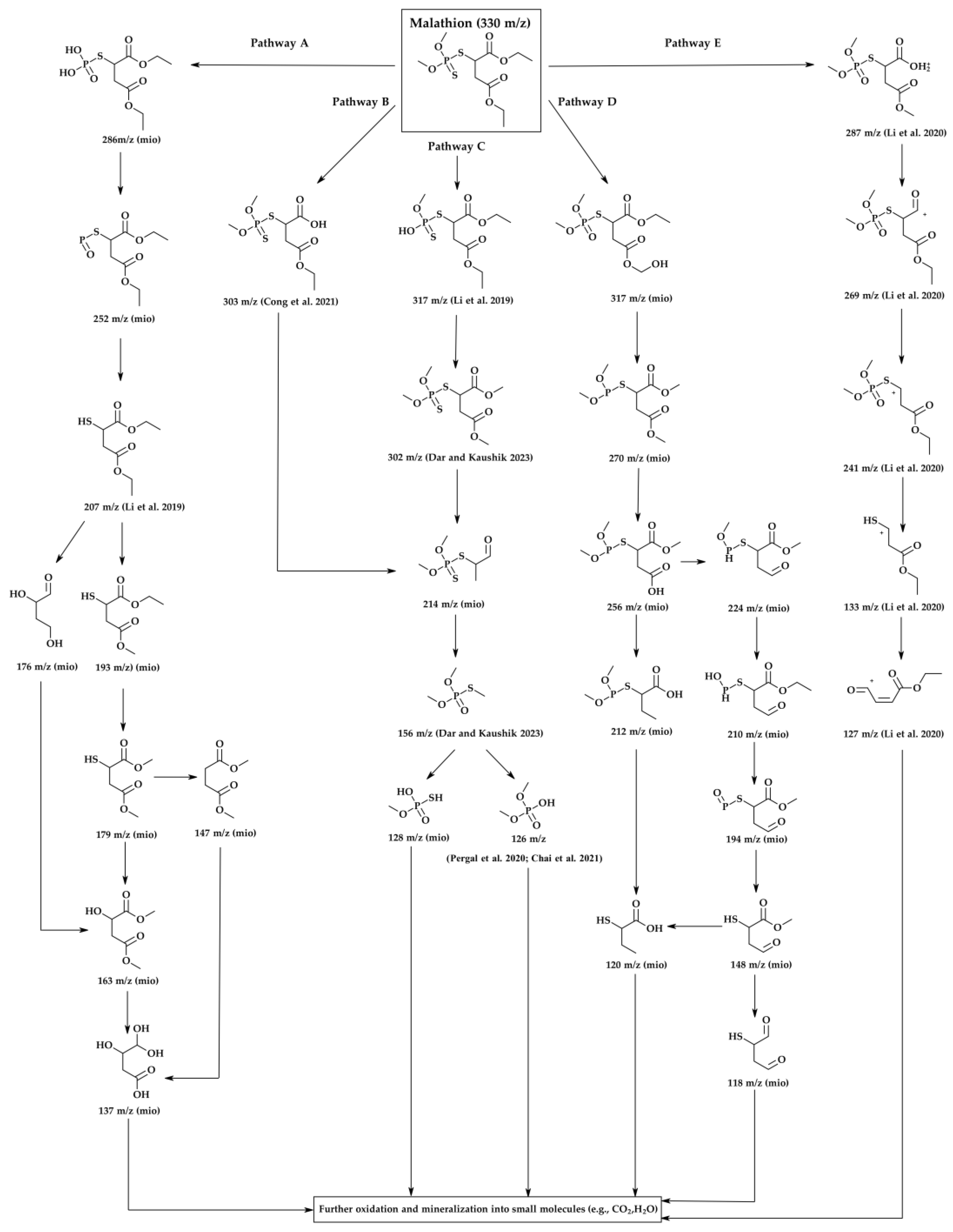
Figure 10.
Schematic diagram of the proposed mechanism for the photodegradation of malathion, using the 5%MoS2@P25-rGO catalyst under irradiation.
Figure 10.
Schematic diagram of the proposed mechanism for the photodegradation of malathion, using the 5%MoS2@P25-rGO catalyst under irradiation.
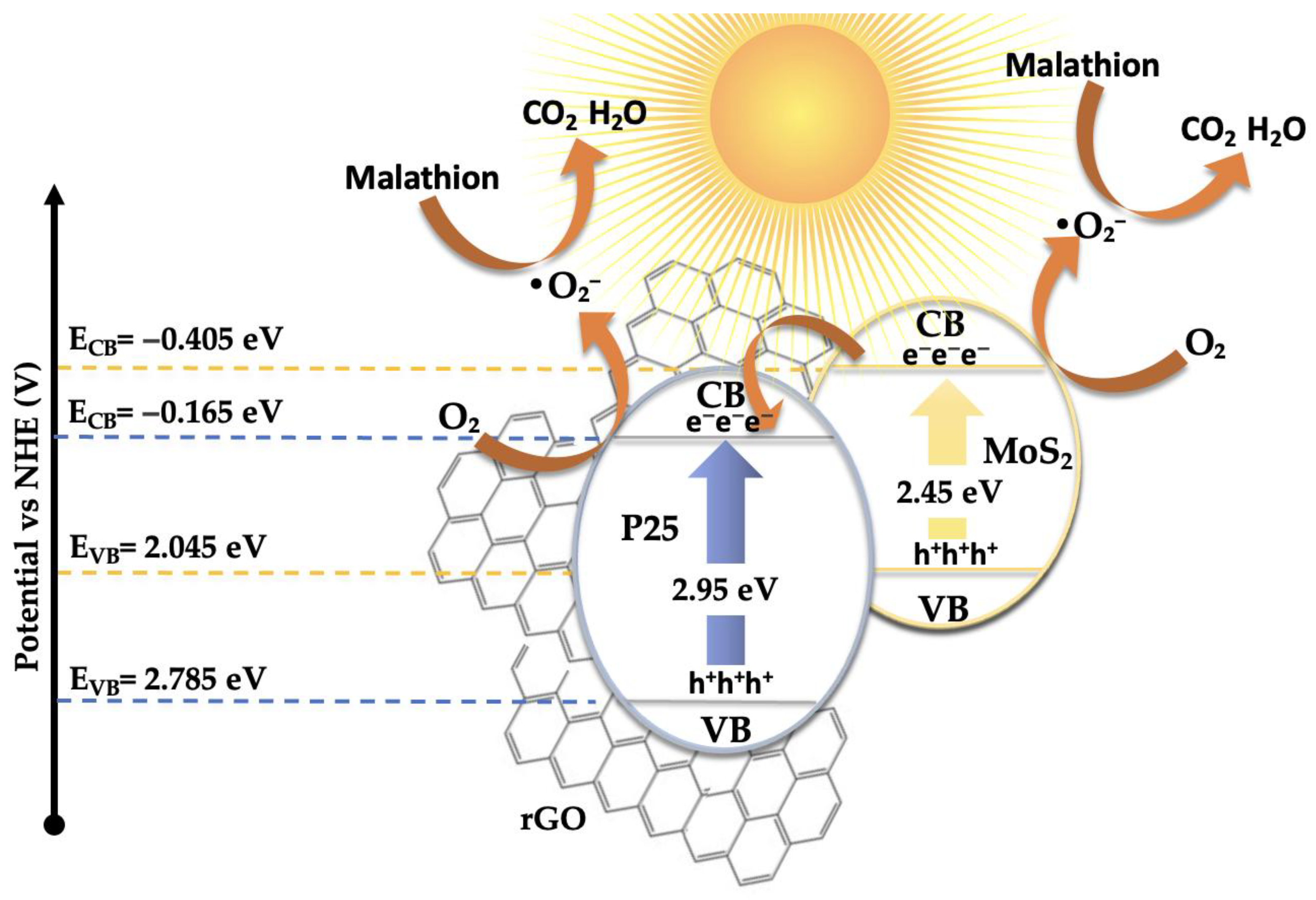
Figure 11.
Hydrogen production profiles of the synthesized catalysts under irradiation at different wavelengths.
Figure 11.
Hydrogen production profiles of the synthesized catalysts under irradiation at different wavelengths.
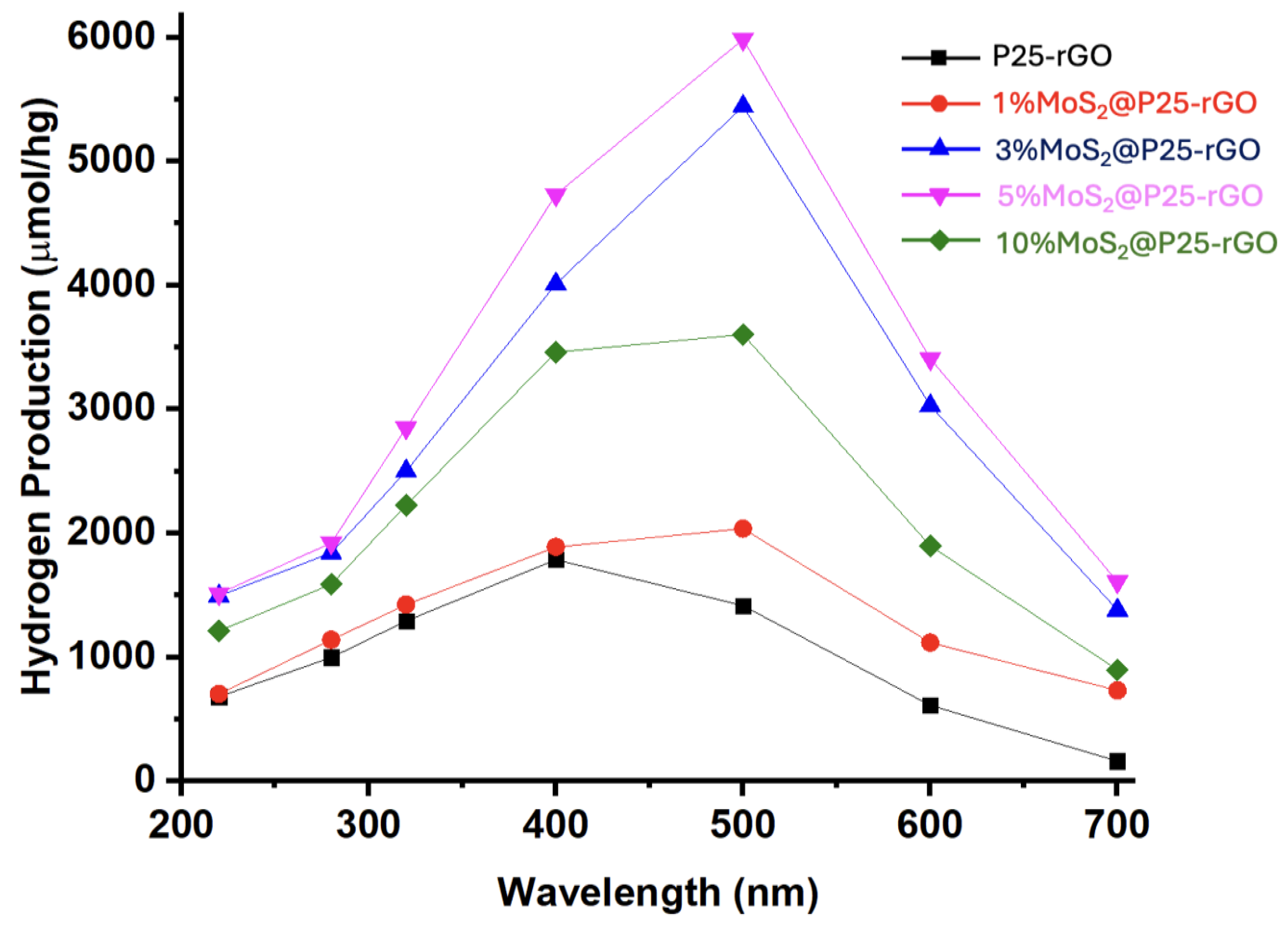
Figure 12.
Transient photocurrent response in the light on−off processes of the synthesized catalysts under irradiation at 500 nm.
Figure 12.
Transient photocurrent response in the light on−off processes of the synthesized catalysts under irradiation at 500 nm.
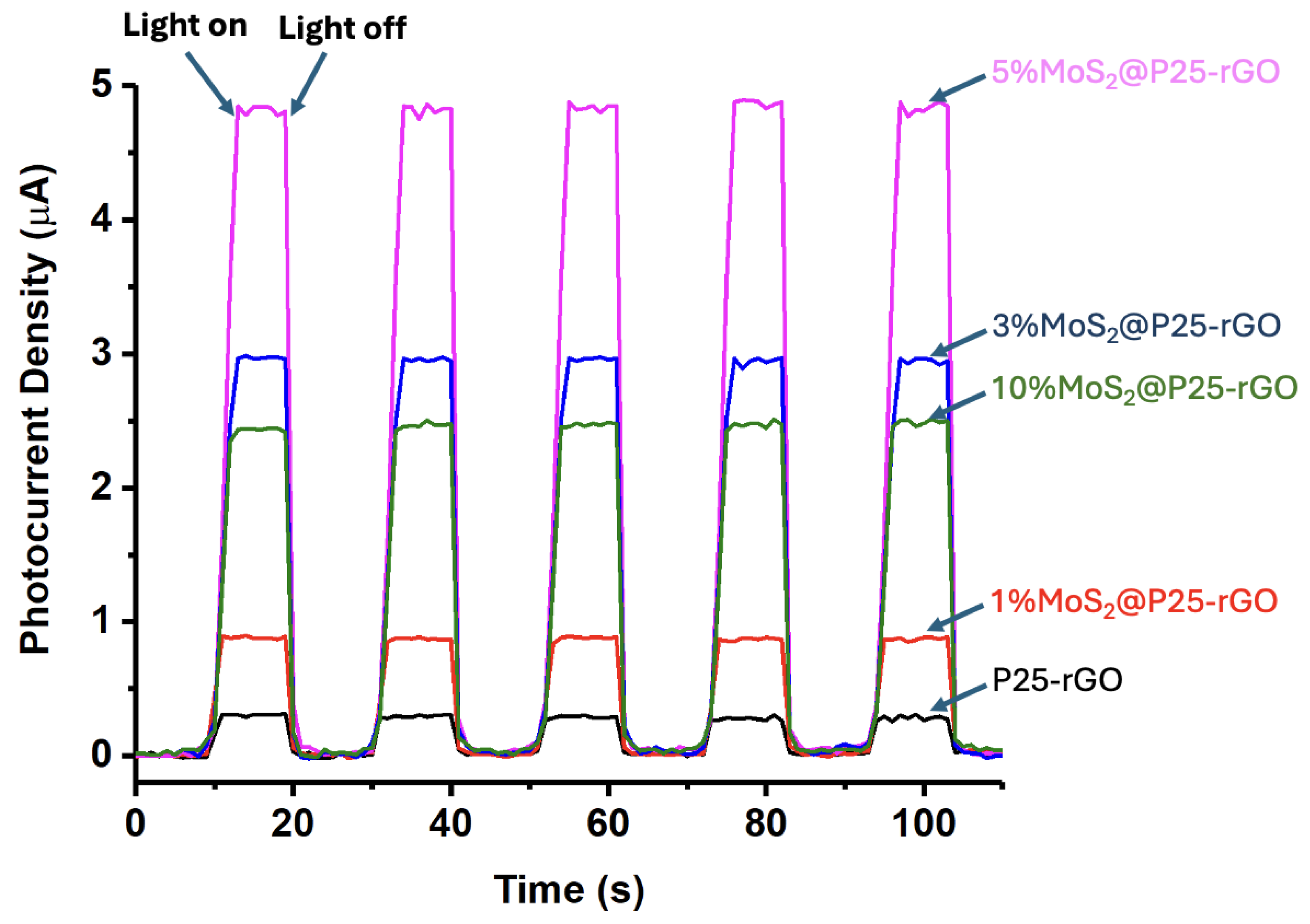
Figure 13.
Schematic diagram of the proposed mechanism for hydrogen production under irradiation.
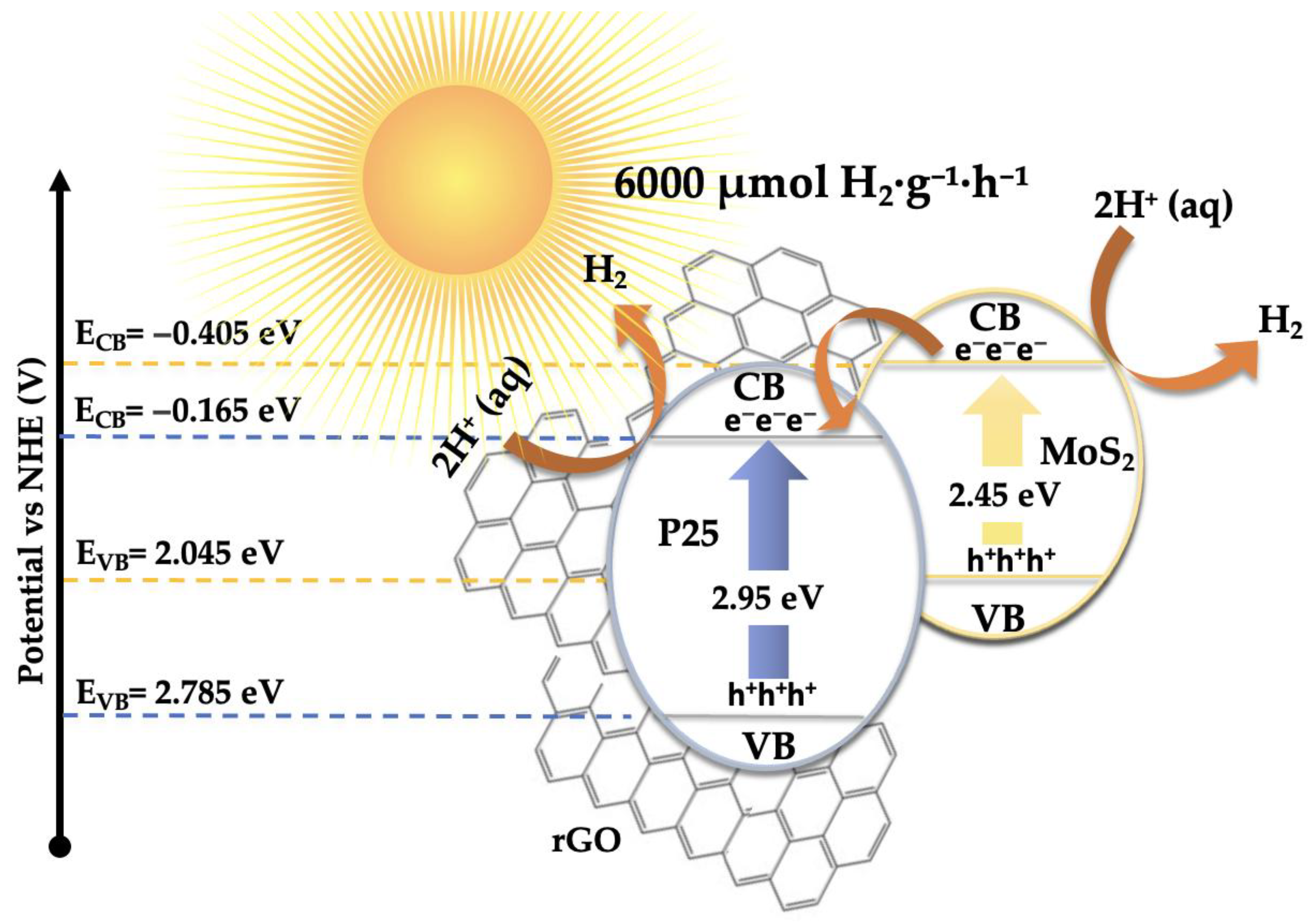
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



